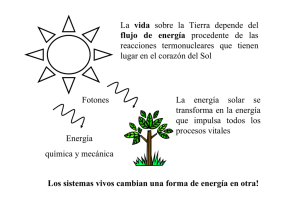
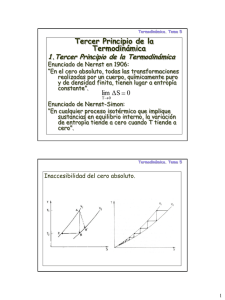
material postitulacion - Facultad de Ciencias Exactas, Físicas y
Anuncio

M.A.Perillo Biofisica-Quimica ISBN 987-9406 Ed. Universitas 2002 “A la única autoridad a que debéis rendiros es a la de la búsqueda del conocimiento. Si opináis que lo que os digo os estimula, tomadlo y perfeccionadlo y si no, combatidlo con fuerza, mas no lo destruyáis, quizás todo posea algo de verdad” Sócrates, 410 a.c. Dra María Angélica PERILLO Inv.CONICET Prof.Adjunta, FCEFyN, UNC POSTITULACION EN CIENCIAS NATURALES FACULTAD DE CIENCIAS EXACTAS FÍSICAS Y NATURALES UNIVERSIDAD NACIONAL DE CORDOBA i M.A.Perillo Biofisica-Quimica INDICE El curso: i- Objetivo ii- Actividades iii- Evaluación iv- Bibliografía 1. TERMODINÁMICA Consignas de trabajo Introducción Vocabulario termodinámico Clasificación de los sistemas según su interacción con el entorno Cambios de estado Energia, calor y trabajo Primera ley Naturaleza molecular de la energía interna Segunda ley Combinación de la primera y de la segunda ley de la termodinámica Principios de termodinámica estadística Problemas para resolver Bibliografía específica Unidad 1 iv 1 1 1 1 2 2 3 6 7 11 14 17 20 23 2. FUERZAS INTERMOLECULARES.ESTRUCTURAS DE AUTOAGREGACIÓN Consignas de trabajo Sustancias anfipáticas y autoagregación Estructuras de autoagregación Fuerzas intermoleculares Efecto hidrofobico: Fuerza impulsora de la autoagregación Condiciones termodinámicas para la autoagregación Restricciones geométricas para la autoagregación: Trabajo practico n° 1: Determinacion de la concentracion micelar critica (cmc) Cuestionario de orientación Biomembranas: Cuestionario de orientación para análisis de textos seleccionados: iLa formación de membranas celulares iiFluidez de las membranas celulares Bibliografía específica Unidad 2 24 24 24 25 25 25 27 28 29 30 30 3. CINÉTICA Y MODULACIÓN DE LA ACTIVIDAD ENZIMÁTICA Consignas de trabajo Cinética y catálisis Un catalizador es.. Catálisis enzimática Variación de la velocidad en función del pH, para una reacción enzimática Dependencia típica de la velocidad de una reacción enzimática como función del sustrato Cuestionario de orientación para análisis de textos seleccionados. i- Biocatalizadores ii- Modulación de la actividad enzimática en medios heterogéneos Bibliografía específica Unidad 3 32 32 32 33 34 35 35 38 4. AUTOORGANIZACIÓN ESPACIAL Y TEMPORAL Consignas de trabajo 40 40 31 39 Termodinámica de sistemas abiertos Termodinámica del equilibrio y apartada del mismo Termodinámica lineal del desequilibrio Termodinámica no lineal del desequilibrio Sistemas complejos Diagramas de fase Teorías del caos Catástrofes en reacciones químicas Problemas filosóficos asociados a la teoría de catástrofes “El oregonator” De la oscilación al caos Es caos determinista o azar? Rutas del caos El caos Las enzimas caóticas o Trabajo practico n 2: Una demostración simple de autorganización: el experimento de la botella azul Análisis de textos seleccionados Bibliografía específica Unidad 4 40 42 44 44 46 47 48 49 50 52 56 57 60 63 65 66 ii M.A.Perillo Biofisica-Quimica PREFACIO La Biofísica-química es la disciplina que trata de explicar los procesos fundamentales de la vida en base a leyes físicas. A principios del siglo XX era aceptado que los sistemas cerrados tienden a adquidir estados de máximo desorden en su evolución hacia estados de equilibrio. Este axioma, conocido como la segunda ley de la termodinámica, parecía ser violado por los seres vivos, los cuales, en un mundo que tiende a desorganizarse, siguen el curso opuesto. Erwin Schroedinger, en su libro “Qué es la vida?” publicado en 1945, explica que los seres vivos siempre se las arreglan para aumentar su organización a expensas de la desorganización del medio, lo cual da como resultado un aumento global del desorden y por lo tanto no se viola la segunda ley. Esto es posible debido a que son sistemas abiertos, capaces de intercambiar materia y energía con su entorno. Algunos consideran a este libro como el comienzo de la biofísica-química. Qué estudia la biofísica-química?: la formación de estructuras de autoagregación, biomembranas, canales, estructura de proteínas, interacción lípido-proteínas, modulación de la actividad de proteínas, fenómenos de autoorganización espacio-temporal en sistemas complejos, etc. El análisis de estos problemas siempre involucra conceptos energéticos. En el presente curso desarrollaremos conceptos básicos sobre: i) termodinámica de sistemas químicos cerrados, con el objetivo de comprender y predecir la posibilidad de la evolución de los mismos hacia estados de equilibrio; ii) restricciones termodinámicas y geométricas para la formación espontánea de agregados multimoleculares de relevancia biológica tales como las biomembranas; iii) las características dinámicas de la estructura de membrana y su importancia en la modulación de la actividad de proteínas que interactúan con ella (como ejemplo se analizarará la interacción enzima-membrana); iv) evolución espontánea de sistemas abiertos hacia estados organizados espacial mente (por ej. patrones espaciales en colonias de levaduras) y temporalmente (relojes químicos) . Si bien las ecuaciones matemáticas estarán presentes, éstas serán utilizadas como expresión cualitativa de los fenómenos que describen. Será dará importancia al significado de la ecuación y no a su deducción o al detalle de su estructura. Sólo cuando la comprensión de los conceptos lo haga necesario, se realizarán cálculos matemáticos. Las guías de estudio al final de cada unidad destacan cuáles son los conceptos fundamentales de cada tema y sirven para la autoevaluación por parte de los alumnos. Agradezco al Dr Daniel García y a la Bióloga Julieta Sánchez, con quienes comparto diariamente el trabajo de laboratorio, por su colaboración en la búsqueda bibliográfica para la confección de este material y en la elaboración de las guías de análisis de textos. Y a Uds, los valientes alumnos, los invito a animarse a mirar una ecuación y encontrar en ella un concepto. Sé que para muchos éstas no resultan muy amigables. Permítanme acompañarlos en este camino. El objetivo vale la pena... será una puerta abierta hacia la complejidad de la naturaleza. Dra María Angélica Perillo iii M.A.Perillo Biofisica-Quimica BIOFISICA QUIMICA OBJETIVO Se espera que al finalizar este curso los alumnos sean capaces de: Comprender los conceptos fundamentales y las implicancias biotecnológicas de la modulación, evolución y autoorganización de sistemas bioquímicos dinámicos. CONTENIDOS UNIDAD 1. TERMODINÁMICA Sistemas: abiertos, cerrados, aislados. Estados. Cambios de estados. Variables y funciones de estado. Energética: Calor y trabajo. Cambios isotérmicos, adiabáticos, reversibles, irreversibles. Leyes de la termodinámica. Funciones termodinámicas de estado. Dependencia de ∆U y ∆H con la temperatura. Termoquímica. Entropía. Eficiencia de Carnot, eficiencia de procesos biológicos. Equilibrio material: Equilibrio de reacción: Trabajo máximo. Funciones de Hemholz y de Gibbs. Relaciones de Maxwell. Termodinámica química. Actividad biológica: la termodinámica del ATP. Otros compuestos con alto potencial de transferencia de grupo. Acoplamientos. Principios de termodinámica estadística. Distribución de Boltzmann. Configuraciones posibles. Configuraciones más probables. Fluctuaciones. UNIDAD 2. FUERZAS INTERMOLECULARES.ESTRUCTURAS DE AUTOAGREGACIÓN. Restricciones geométricas y entrópicas. Interacción lípido-lípido, lípido-proteína, proteína-proteína. Membranas biológicas. Modulación de la organización de membranas. Adaptaciones a las condiciones del medio ambiente. Modelos artificiales de biomembranas. Liposomas en la tecnología de alimentos y en la tecnología farmacéutica. UNIDAD 3. CINÉTICA Y MODULACIÓN DE LA ACTIVIDAD ENZIMÁTICA. Conceptos de cinética química. Factores que afectan la velocidad de reacciones. Catalizadores biológicos. Enzimas, ribozimas. Activación e inhibición. Inducción y represión. Modulación de la actividad enzimática en medios heterogéneos. Ejemplos con sustratos solubles y sustratos organizados. Enzimas encapsuladas. UNIDAD 4. AUTOORGANIZACIÓN ESPACIAL Y TEMPORAL. Introducción a la termodinámica de los sistemas abiertos. Sistemas alejados del equilibrio. Equilibrio local. Sistemas disipativos. Relaciones entre fuerzas y flujos. Estado estacionario en sistemas lineales: principio de reciprocidad de Onsager. Velocidad de producción de entropía. Estabilidad del equilibrio. Estabilidad del estado estacionario. Sistemas bioquímicos que presentan comportamiento complejo. Autocatálisis, reacciones oscilantes, biestabilidad. Evolución de la complejidad biológica. Teoría del caos. Fractales. MODALIDAD DE TRABAJO: 1- Seminarios de discusión del contenido del material bibliográfico y de trabajos científicos; 2- Resolución de problemas; 3- Trabajos de laboratorio: a) Estructuras de autoorganización y concentracion micelar critica. b) Autoorganización espacio-temporal EVALUACION: evaluaciones conceptuales durante las clases. Guías de problemas e informes de trabajos de laboratorio, los cuales deberán ser presentados el día del examen final. Este último consistirá en un cuestionario y problemas sobre los contenidos discutidos en clase (a libro abierto). CRONOGRAMA: Unidad 1: primera clase Unidades 2 y 3: segunda clase Unidad 4: tercera clase Examen: cuarta clase. BIBLIOGRAFÍA GENERAL Aon,M.A. and Cortassa,S. Dynamic biological organization. Fundamentals as applied to cellular systems. Chapman and Hall, London, UK. 1997. iv M.A.Perillo Biofisica-Quimica Atkins,P.W. Fisicoquimica. Ed. Addison-Wesley Iberoamericana, S.A. Wilmington, Delaware, USA.1991 Israelachvili,J.N. Intermolecular and surface forces. Academic Press, New York. 1989. Maggio B. Introducción a la biofísico-química. Ed.Gonzalez Truccone. Córdoba, Argentina. 1987. Mahan, B. Química, Curso Universitario. Fondo Educativo Interamericano S.A., Moore W.J. Química física. Ed.URMO, S.A.,Bilbao, Espana.1977. Olivares, C.M. Tecnologias emergentes de computo. Teoria del caos. Alfaomega Grupo Editor, S.A., Mexico. 1997. Peacocke,A.R. An introduction to the physical chemistry of biological organization. Oxford Sci.Pub. Clarendon Press, Oxford. 1989. v M.A.Perillo UNIDAD 1. Biofisica-Quimica TERMODINÁMICA CONSIGNAS DE TRABAJO: - Repase los siguientes temas: gases ideales, ecuación general de estado, Ley de Boyle y ley de Guy-Lussac. Representación gráfica en el plano P vs V de cambios de estado de gases ideales. Sistema Internacional de unidades, múltiplos y submúltiplos. En caso necesario recurra a textos de Quimica General o de Física. - Realice el estudio independiente del texto que se presenta más abajo (encontrará una síntesis de ecuaciones termodinámicas relevantes en el Anexo I). Intente responder las preguntas insertas en el mismo y resolver los problemas que se detallan al final de esta unidad. Tanto las preguntas como los problemas serán discutidos durante la primera clase. 1. INTRODUCCIÓN La termodinámica estudia los fenómenos de intercambio de energía que acompañan a los procesos físicos y químicos. Esta ciencia trata sobre las propiedades macroscópicas, observables, de la materia, sin hacer suposiciones sobre su naturaleza atómica. Se basa en tres leyes, dos de las cuales son inmediatamente aplicables a nuestra experiencia: 1. La energía del universo es constante. 2. La entropía del universo está en aumento. Estas leyes no se derivan sino que se deducen de experimentos. Su universalidad ha sido demostrada. En esta unidad discutiremos de qué manera esta ciencia nos permite predecir que: los sistemas cerrados, capaces de sufrir cambios reversibles, evolucionan hacia el estado de equilibrio termodinámico, de mínima energía y de máximo desorden y los sistemas abiertos, dinámicos, complejos y no lineales evolucionan hacia estados estacionarios y el hacia el orden a través de las fluctuaciones. El razonamiento termodinámico puede emplearse, por ejemplo, para que nos revele si es posible, en principio, ir de un estado particular de los reactantes a otro estado particular de los productos de una reacción, pero no puede revelarnos si ese cambio puede ser realizado en un tiempo finito. Esto último es un problema de la cinética química, sobre lo cual trataremos en la tercera unidad. 2. VOCABULARIO TERMODINÁMICO A continuación se definirán los siguientes conceptos: sistema, entorno, universo, clasificación de los sistemas según su interacción con el entorno, estados de un sistema, variables de estado, funciones de estado, cambios de estado, estado de equilibrio, estado estacionario. La parte del mundo físico que se aisla para su estudio se denomina sistema. El sistema se relaciona con su entorno. sistema + entorno = universo 1 M.A.Perillo Biofisica-Quimica CLASIFICACIÓN DE LOS SISTEMAS SEGÚN SU INTERACCION CON EL ENTORNO INTERCAMBIO SISTEMA ENERGIA CALOR TRABAJO MATERIA Aislado No No No No Adiabático Si No Si No Cerrado Si Si Si No Abierto Si Si Si Si * calor y trabajo son dos formas de intercambio de energía: más adelante se explica cuál es la diferencia entre ellas. El estado del sistema se define por medio de las variables de estado: Presión (P), Temperatura (T), Volumen (V), composición (xi) . FUNCION DE ESTADO ----- independiente del camino ---- por ej. CAMBIO DE ENERGÍA , o forma de intercambio NO SON FUNCIONES DE ESTADO ------- dependen del camino ------ por ej.CALOR Y TRABAJO o forma de intercambio Por ser una función de estado E es una diferencial exacta ∆E ESTADO 1 ESTADO 2 E′ ∆E1,2 = ∫dE + ∫dE’ = E2-E1+E1-E2 = 0 CAMBIOS DE ESTADO Los cambios de estado pueden ser: reversibles o irreversibles. Un cambio reversible transcurre por medio de una sucesión de infinitos estados de equilibrio (en infinitos pasos). En la práctica esto no es posible, por lo cual los procesos reales son irreversibles. En el caso en que alguna de las variables que definen el estado del sistema permanezca constante, el cambio de estado se denomina: Isotérmico (a T=cte) 2 M.A.Perillo Biofisica-Quimica Isobárico (a P=cte) Isocórico (a V=cte) Adiabático (a Q=cte) ESTADO DE EQUILIBRIO Un estado de equilibrio es aquel en el cual las propiedades macroscópicas del sistema tales como su temperatura, densidad y composición química, son bien definidas y no cambian con el tiempo. 1. Se establece en sistemas cerrados. 2. El equilibrio en los sistemas moleculares es dinámico, y es una consecuencia de la igualdad de las velocidades de reacciones opuestas. 3. El sistema pasa espontáneamente hacia el estado de equilibrio. Si un sistema, inicialmente en equilibrio, es perturbado por algún cambio en su medio ambiente, él reacciona de un modo tal que vuelve a su estado de equilibrio (Principio de Le Chatelier). 4. La naturaleza y las propiedades de un estado de equilibrio son las mismas, independientemente de cómo es alcanzado. 5. La condición de un sistema en equilibrio representa una transacción entre dos tendencias opuestas: la exigencia de las moléculas a asumir el estado de energía más bajo, y la tendencia hacia el caos molecular, o la entropía máxima. Un ejemplo de equilibrio físico: equilibrio líquido-vapor Un líquido a temperatura de ebullición relativamente baja, colocado en un recipiente abierto a la atmósfera, se evaporará por completo. Esto se debe a que la fracción de moléculas que poseen un valor mínimo de energía E para superar las fuerzas de interacción intermoleculares de la fase líquida es proporcional a e-E/kT (ley de distribución de MaxwellBoltzmann). Siempre que la temperatura permanezca constante, la fracción de moléculas líquidas que tiene energía suficiente para evaporarse permanece igual, y la evaporación continúa hasta que no queda líquido. En un recipiente cerrado: el líquido comienza a evaporarse a una velocidad que es determinada principalmente por la fracción de las moléculas que tienen energía cinética suficiente para vencer a las fuerzas atractivas y dejar la superficie. Inicialmente, la velocidad de condensación es cero, porque no hay moléculas en el vapor. Mientras la temperatura permanece constante, la evaporación continúa a una velocidad constante, y aumenta el número de moléculas que hay en la fase vapor. Mientras, la velocidad de condensación comienza a crecer, porque a medida que la presión de vapor crece, el número de moléculas gaseosas que chocan y reingresan a la superficie del líquido también aumenta. La velocidad de condensación eventualmente llega a ser igual a la velocidad de evaporación. En ese momento el número de moléculas que entran y salen del vapor por unidad de tiempo son iguales, y en consecuencia la presión del vapor deja de aumentar y permanece constante. Si se deja al sistema en reposo a una temperatura fija, la evaporación y la condensación continúan a velocidades iguales, y la presión del vapor quedad sin cambio. Esta es la situación de equilibrio entre las dos fases. Observe que a partir de que se alcanza el equilibrio, la evaporación y la condensación no se detienen, sino que la constancia de la presión de vapor en el equilibrio es una consecuencia de estos procesos que se producen a velocidades iguales. Es por esto que decimos que el equilibrio es de naturaleza dinámica. 3 M.A.Perillo Biofisica-Quimica Evaporación Velocidad Condensación Tiempo Problema: Suponga que el sistema líquido-vapor está en equilibrio dentro de un cilindro cerrado por un pistón movible. Qué ocurriría si se levanta el pistón y aumenta el volumen del cilindro, manteniendo constante la temperatura? Respuesta: disminuye la presión del vapor, el sistema se aparta del equilibrio, hay menos moléculas en la fase vapor por unidad de volumen por lo cual disminuye el número de colisiones con la superficie líquida por unidad de tiempo y la velocidad de condensación decrece momentáneamente. La velocidad de evaporación no cambia porque no cambió la temperatura y porque la expansión no altera al estado líquido. Consecuentemente, el sistema responderá aumentando el número de moléculas en la fase vapor hasta que las velocidades de evaporación y de condensación vuelvan a igualarse, y el equilibrio de fase sea restaurado. Un ejemplo de equilibrio químico: Las reacciones químicas, como los cambios de fase, pueden ser reversibles. Como consecuencia hay condiciones de presión y temperatura bajo las cuales los reaccionantes y los productos coexisten en el equilibrio. Consideremos la descomposición térmica del carbonato de calcio: CaCO3(s) CaO(s) + CO2 (g) [1] Realizando la reacción en un recipiente abierto, que permita que el dióxido de carbono sea arrastrado, se puede efectuar la conversión completa de la sal. Por su parte el óxido de calcio reacciona con el dióxido de carbono, y si la presión del CO2 es suficientemente alta, el óxido puede ser convertido totalmente en carbonato. CaO(s) + CO2 (g) CaCO3(s) [2] Estas dos reacciones pueden ser consideradas como un único proceso químico reversible: CaCO3(s) CaO(s) + CO2 (g) Si comenzamos con CaCO3 al cual calentamos en un recipiente cerrado, comenzará a descomponerse formando CaO y CO2 (reacción [1]). A medida que se acumula CO2 su presión aumenta y comienza a ocurrir la reaccion [2], a una velocidad que aumenta a medida que se acumula más CO2. Cuando las velocidades de las dos reacciones opuestas se igualan, la presión de CO2 permanece constante, es decir no hay ambio neto en la concentración de este compuesto, por lo cual se deduce que el sistema alcanzó el equilibrio. 4 M.A.Perillo Biofisica-Quimica Experimento: si a un sistema conteniendo CaCO3, CaO y CO2 en equilibrio le agregamos una 14 cantidad infinitamente pequeña de CO2 donde el carbono sea C (un isótopo radiactivo del elemento carbono), si no se modifican la presión o la temperatura, el estado de equilibrio no será modificado sin embargo, después de una tiempo, se encontrará que parte del CaCO3 sólido contine 14 C. Cómo explica este resultado? La constante de equilibrio Se puede demostrar experimentalmente que la presión de CO2 en equilibrio con CaCO3 y CaO sólidos es un función únicamente de la temperatura. Una vez que el sistema alcanzó el equilibrio, se puede agregar o retirar sólidos, mientras algo de sólido esté presente en el sistema, la presión de CO2 permanecerá constante. Es decir que para caracterizar el estado de equilibrio sólo se necesita indicar la presión de CO2. Si en la reacción química intervienen varios reactivos o productos gasesos o disueltos, hay una cantidad infita de conjuntos de presiones o concentraciones de reactantes y productos que pueden existir en el equilibrio. Sin embargo existe una relación sencilla entre las concentraciones o presiones de los reactivos en equilibrio, llamada constante de equilibrio. Dada la reacción donde a moles del compuesto A reaccionan con b moles de B para producir c moles de C y d moles de D según: aA + bB cC + dD se demuestra experimentalmente que las concentraciones de equilibrio de reactantes y productos están relacionadas por el requisito de que la función: [C]c .[D]d Keq= [A]a . [B]b sea una constante fija, cuyo valor dependa de la temperatura y de la identidad de los reactantes y de los productos. ESTADO ESTACIONARIO En un estado estacionario, al igual que en el estado de equilibrio, las propiedades macroscópicas del sistema tales como su temperatura, densidad y composición química, son bien definidas y no cambian con el tiempo. 1. Se establece en sistemas abiertos. 2. El equilibrio en los sistemas moleculares es dinámico, y es una consecuencia de la igualdad de las velocidades de reacciones o flujos opuestos. Ejemplo de sistema físico en estado estacionario: Suponga un tanque, con una entrada y una salida para líquido, lleno de agua hasta un cierto nivel. Si la velocidad de ingreso de agua es igual a la velocidad de egreso, el nivel del agua se mantendrá constante en función del tiempo. Esto ocurre con el agua de los refrigerantes que se utilizan en los procesos de destilación y de calentamiento a reflujo. En ellos, el agua circula entrando por abajo y saliendo por arriba con la misma velocidad; esto permite que el refrigerante esté siempre lleno de agua. Puede dibujarlo? Ejemplo de sistema químico en estado estacionario: Reacciones acopladas: las siguientes reacciones están acopladas por medio de un compuesto común a ambas. 1 A B [a] 2 5 M.A.Perillo Biofisica-Quimica B+C 3 D [b] 4 La reacción [a] es reversible, sin embargo no puede alcanzar el equilibrio dado que el producto de la misma es continuamente consumido en la reacción [b], con la cual está acoplada. Lo mismo ocurre con la reacción [b], donde la concentración de uno de sus reactante es continuamente aportado a la misma por medio de la ocurrencia de la reacción [a]. Si se alcanza la condición en que la velocidad de producción de B iguala a la velocidad de consumo de B, la concentración de esta sustancia permanecerá constante en función del tiempo y en estas condiciones se habrá alcanzado el estado estacionario en la concentración de B. Escriba una expresión matemática que refleje esta situación. Tenga en cuenta que B se forma por medio de las reacciones 1 y 4, y que desaparece por las reacciones 2y 3. Para que esto sea posible: i) deberá haber un suministro continuo de A y una extracción continua de D. Esto ocurre en los ciclos metabólicos celulares. Discútalo y de ejemplos. ii) si no es posible aportar A y extraer D, la concentración de A deberá ser muy grande y la de D muy pequeña, lo cual sólo será posible en los momentos iniciales de la reacción (a tiempos suficientemente cortos como para que la cantidad de A consumido sea despreciable con respecto la cantidad inicial de A y que D no se haya acumulado suficientemente como para que la velocidad de la reacción 4 se vuelva significativa). Para recordar: Este enunciado corresponde al concepto de velocidad inicial, utilizado en cinética enzimática. La ecuación de Michaelis-Menten, la cual relaciona velocidad de reacción con la concentración de sustrato, sólo es aplicable si se trabaja a velocidad inicial, es decir: si la velocidad de una reacción catalizada por enzimas se mide a un tiempo suficientemente largo como para que se haya alcanzado el estado estacionario en la concentración de complejo enzima-sustrato, pero no tan largo como para que la condición de estado estacionario no se haya perdido por un excesivo consumo de sustrato. 3. PRIMERA LEY DE LA TERMODINÁMICA Ley de conservación de la energía: “La energía del universo se conserva” Atención: el universo es un sistema aislado La energía se presenta bajo las formas de calor y trabajo. INCOHERENTE _ CALOR COHERENTE + _ Energía molecular e interacciones (dQ) + TRABAJO (dw) SISTEMA 6 M.A.Perillo Biofisica-Quimica Entonces, la 1ra ley puede expresarse como: dE = dQ + dw Donde dE, dQ y dw representan los cambios de energía interna, calor y trabajo respectivamente. En el caso de reacciones nucleares debemos hablar de “Ley de conservación de la masa y de la energía”. En este caso se debe considerar la ley de Einstein de interconversión de materia y energía: 2 E = m.c NATURALEZA MOLECULAR DE LA ENERGIA INERNA (E) E = Etr + Erot + E vibr + Eel + Eintermol + Erep Etr : energía translacional = 3/2 R.T (para un gas) 900 cal/mol Erot : energía rotacional = 3/2 R.T (molécula no lineal) 600-900 cal/mol R.T (molécula lineal) E vibr : energía vibracional; E vibr =f(T) es función de T; sólo a T=0 E vibr =0) Eel : energía electrónica Eel = Eeq - E∞ Eintermol : energía intermolecular líquidos: 3000-5000 cal/mol gas a 1atm/25°C = 1–10 cal/mol; a 40 atm/25°C= 40-400 cal/mol Erep :energía de la masa en reposo de electrones y nucleos Convenciones de signos +q: calor tomado por el sistema desde el entorno -q: calor cedido por el sistema al entorno +w: trabajo realizado por el entorno sobre el sistema -w: trabajo realizado por el sistema sobre el entorno ENERGIA, CALOR Y TRABAJO Algunas ecuaciones útiles para calcularlos en distintos tipos de cambios de estado A continuación se deducen ecuaciones que permiten calcular cambios de energía interna, calor y trabajo para un sistema que sufre distintos tipos de cambios de estado. El proceso detallado está ejemplificado para el caso de un proceso irreversible isocórico (a). Intente seguir un razonamiento similar a los fines de comprender las deducciones correspondientes a las otras condiciones experimentales (no es necesario que lo haga). El objetivo es mostrar: i) el significado de los cambios de energía interna (∆E) y de entalpía (∆H) ii) que los valores de ∆E y ∆H serán distintos de cero sólo si hay un cambio de temperatura. 7 M.A.Perillo iii) Biofisica-Quimica Que los valores de ∆E y ∆H serán los mismos independientemente del camino que se siga para realizar el cambio de estado (reversible o irreversible), dado que son funciones de estado iv) Que los valores de q y w serán distintos, dependiendo del camino que se siga para realizar el cambio de estado (reversible o irreversible), dado que NO son funciones de estado Suponemos que el sistema está constituido por un gas y que sólo puede realizar trabajos de expansión y de compresión. Recordemos: Un gas ideal: 1- consta de moléculas de volumen puntal, sin interacción entre ellas. 2- cumple con la ecuación general de estado P.V = n.R.T 3- dw = -d(P.V) (trabajo representa el area bajo la curva de un gráfico P vs V) a) PROCESO IRREVERSIBLE ISOCÓRICO Isocórico se refiere a un proceso que transcurre a V=constante (Vfinal = Vinicial), por lo tanto: ∆V = Vfinal – Vinicial = 0 De acuerdo a la definición de trabajo dada más arriba: ∆w = -P∆V (a presión constante), siendo ∆V=0, resulta ∆w=0. Dado que el cambio de energía interna es la suma de calor más trabajo: dE = dq + dw siendo ∆w=0, queda dE = dq (el cambio de energía interna resulta ser igual sólo a la cantidad de calor intercambiada por el sistema con el entorno, en condiciones de volumen constante, por lo cual se le da un nombre especial: qv dqv = n.Cv.dT La cantidad de calor absorbido o cedido por el sistema depende de la cantidad de materia (masa expresada en número de moles (n) o en gramos (m)), de la sustancia de que se trata (implícita en la capacidad calorífica a volumen constante Cv) y del cambio de temperatura (dT) De lo expuesto se deduce que: El cambio de energía interna equivale al cambio de calor a volumen constante. dE = qv dE = Cv.dT Cv = (dE/dT)V Si se calienta un sistema, sin permitir que cambie su volumen, se produce un movimiento incoherente (aumento de la agitación molecular) también expresado como un aumento de la energía térmica 8 M.A.Perillo Biofisica-Quimica b) PROCESO IRREVERSIBLE ISOBÁRICO dw ≠ 0 dE = dq + dw dqp = dE – dw dqp = dE + P.dV dqp = dH El cambio de entalpía (∆H) equivale al cambio de calor presión constante. dH = CP.dT CP = (dH/dT)P Se produce: movimiento coherente (dw) e incoherente (dq) con aumento de energía térmica y producción de trabajo El cálculo de trabajo realizado por o sobre el sistema (área bajo la curva del gráfico P vs V) es sencillo cuando el proceso se realiza en forma irreversible a P=cte dado que el área resultante es la de un cuadrilátero, la cual se calcula como lado x lado (en este caso ∆Px∆V). Ver gráfico I. Cuando, el proceso es reversible (a través de una sucesión de infinitos estados de equilibrios), la forma del área es más compleja y su cálculo requiere sumar las areas de infinitos cuadriláteros cada uno de los cuales tendrá valores de ∆P que van cambiando a medida que cambia el volumen y valores de ∆V infinitamente pequeños. La operación matemática que permite realizar este cálculo de área es la integración y el símbolo que la representa es ∫. Ver gráfico II. Presión Presión I II P1 P1 P2 P2 V1 V2 Volumen V1 V2 Volumen c) PROCESO REVERSIBLE ISOTÉRMICO ∆T = 0 dE = n.Cv.dT ∆E = 0 ∆E = q + w = 0 q = -w dwrev = -PdV P.V = n.R.T P= n.R.T / V dwrev = -n.R.T dV/V wrev = -n.R.T ∫ dV/V = -n.R.T (ln V2 – ln V1) = n.R.T (ln V1 – ln V2) -q = wrev = n.R.T. ln(V1/V2) d) PROCESO REVERSIBLE ISOBÁRICO ∆E = q + w dw = -P∫dV = -P (Vf – Vi) dE = Cv ∫dT qp = Cp.dT e) PROCESO REVERSIBLE ISOCÓRICO ∆V = 0 w=0 E = qv = n.Cv.∆T 9 M.A.Perillo Biofisica-Quimica f) PROCESO REVERSIBLE ADIABATICO dq = 0 dE = Cv.dT -PdV = Cv dT dE = dw P= n.R.T / V -n.R.T.dV/V = Cv.dT -n.R ∫ dV/V = Cv ∫ dT/T -n.R (lnV2 – ln V1) = Cv (lnT2 – lnT1) n.R (lnV1 – ln V2) = Cv (lnT2 – lnT1) n.R.ln(V1/V2) = Cv.ln(T2/T1) (V1/V2)n.R = (T2/T1)Cv n.R / Cv = γ -1 T2/T1 = P2.V2/P1.V1 P2.V2/P1.V1 = (V2/V1)γ-1 en condiciones adiabáticas, la ley de Boyle se expresa como P.Vγ=cte , donde γ= Cp/Cv además Cp-Cv=R g) EXPANSIÓN ADIABÁTICA EN EL VACÍO q=0 w=0 ∆E = 0 dE = dq + dw dE = Cv.dT + (dE/dV)T. ∆V dE = (∂E/∂T)V.dT + (∂E/∂V)T.dV (∂E/∂T)V= Cv (∂E/∂V)T=Presión interna Fluido perfecto: (∂E/∂V)T=0 (presión interna es nula ∴ no hay interacciones intermoleculares) Ahora podemos ampliar nuestra definición de gas ideal: Un gas ideal: 1- consta de moléculas de volumen puntal, sin interacción entre ellas. 2-cumple con la ecuación general de estado P.V = n.R.T 3-su presión interna es nula (∂E/∂V)T=0 i) En un gráfico P vs V indicar el trabajo de expansión y compresión irreversibles y reversibles. Comparar los resultados obtenidos. ii) En un gráfico P vs V indicar el camino que sigue el sistema durante un cambio isotérmico y uno adiabático. iii) Comparar las cantidades de trabajo realizado en la expansión reversibles isotérmico y adiabático, de V1 a V2. Cómo funciona la primera ley?. Por ejemplo: Si en el interior de nuestro sistema, contenido en un recipiente cerrado de paredes rígidas y adiabáticas ocurre una reacción química exotérmica, la liberación de calor producirá un aumento de la temperatura del solvente, dado que: 10 M.A.Perillo i) Biofisica-Quimica por ser las paredes rígidas no puede ocurrir cambio de volumen; por lo tanto ∆V=0 y, en consecuencia, w=0; ii) por ser las paredes adiabáticas, el calor liberado por la reacción no puede ser disipado hacia el entorno entonces el solvente recibe un q>0 iii) por ∆E=q+w= q + 0; entonces ∆E>0; siendo ∆E=n.Cv.∆T entonces resulta ∆T>0. Si las paredes fueran elásticas, el calor hubiera producido una expansión (aumento de volumen ∆V>0). Esto representa trabajo realizado por el sistema sobre el entorno: dw=-PdV, es decir dw<0. (revise la convención de los signos de la página 7). Entonces: dE=+q-w, dependiendo de cuánto se expanda el sistema podría ocurrir que todo el calor generado se use para producir trabajo de expansión (q=w) en ese caso ∆E=0. Siendo ∆E=n.Cv.∆T=0, entonces resultaría ∆T=0. Por lo tanto no habría aumento de temperatura. Si las paredes, aún sin ser elásticas, permitieran el intercambio de calor con el entorno, el calor que recibe el solvente (+q) podría disiparse al entorno (-q) y en consecuencia siendo el cambio neto de calor dq=0, y no habiendo trabajo (dw=0), resultaría ∆E = q+w = 0+0 = n.Cv.∆T = 0 por lo tanto ∆T=0. En este caso tampoco habría aumento de temperatura. Conclusiones: la energía fluye bajo dos formas interconvertibles (calor y trabajo) las cuales, si es posible se intercambian con el entorno. La energía no se crea ni se destruye. De donde provino el calor inicial?. De la ruptura de enlaces ocurrida durante la reacción química. Entonces, el solvente representaba el entorno de la reacción. El solvente recibió calor que fue previamente energía química contenida en los enlaces de los compuestos reaccionantes. Este calor (que es una forma de energía), según el tipo de relación que existía entre el solvente y su entorno (el exterior del recipiente), quedó en el solvente induciendo un aumento de su temperatura, se disipó al entorno o se transformó en trabajo (otra forma de energía). 4. SEGUNDA LEY DE LA TERMODINÁMICA La 1ra Ley nos asegura que Esist + Eentorno = constante, pero no establece si un fenómeno puede ocurrir en cualquier dirección (es reversible), ni cuál es la dirección del cambio espontáneo. Es decir: cuál es la flecha del tiempo? c) a) b) Frío d) q Caliente q Frío Caliente 11 M.A.Perillo Biofisica-Quimica Cualquiera de estos cambios están permitidos por la 1ra Ley, pero nuestra experiencia nos indica que sólo a) y c) pueden ocurrir espontáneamente. ENUNCIADO SEGÚN KELVIN-PLANK Hay un límite para convertir calor en trabajo. No es posible transformar todo el calor absorbido en trabajo Fuente de calor q w=q Máquina cíclica viola la 2da Ley Eficiencia= trabajo producido/ calor consumido = -w/q Fuente de calor a TC ∆E = q + w qC - qF - w = 0 qC -W Máquina térnica e = 1- qF/qC -qF [ec.1] ∴sólo si qF=0 e=1 Fuente fría a TF PRINCIPIO DE CARNOT: e = (qC – qF)/qC ninguna máquina térmica puede ser más eficiente que una máquina térmica reversible. Qué es el ciclo de Carnot reversible? Es un proceso cíclico que sufre un sistema cerrado, que incluye cuatro pasos reversibles: a) expansión isotérmica, b) expansión adiabática, c) compresión isotérmica y d) compresión adiabática (con la cual se alcanza el estado inicial). iv) Intente representar los cambios descriptos para el ciclo de Carnot en un gráfico P vs V. Para cada etapa se aplica: dE = dq + dW CvdT =dq + nRT dV/V (a) (b) y dividiendo m.a.m. por T CvdT/T =dq/T + nR dV/V [ec.2] (c) Sumamos las expresiones similares a [ec.2] para cada uno de las etapas del ciclo, y tenemos en cuenta que: • E y V son funciones de estado, por lo tanto las integrales cíclicas de los términos (a) y (c) son iguales a 0. • El término (b) puede escribirse como o∫ dqrev/T = 0 12 M.A.Perillo Biofisica-Quimica o∫ dqrev/T = ∆S = 0 Se deduce: (válido para un sistema cerrado y proceso reversible) Donde ∆S representa el cambio de entropía y se calcula como ∆S = dqrev/T ∆S es una función de estado, en consecuencia adquiere el mismo valor para un proceso sea reversible o irreversible, pero sólo cuando el proceso es reversible ∆S se puede calcular como dqrev/T. A partir del análisis del ciclo de Carnot reversible se deduce qF/qc = TF/Tc, y reemplazando en ec.1 se deduce: e = 1 – TF/TC [ec.3] • El máximo valor que puede adoptar e (eficiencia máxima) es emáx= 1 • Eso sólo sería posible a TF=0 • pero TF=0 no es posible ∴ e=1 no es posible v) vi) Analice la ec.3 y explique qué combinación de temperaturas permite aumentar la eficiencia de una máquina térmica? Investigue en textos de Química Biológica cuál es la eficiencia de algún proceso celular y compare ese valor con el de máquinas a vapor (10-40%). CICLO DE CARNOT IRREVERSIBLE El ciclo de Carnot irreversible es otro modelo y permitió deducir que en un proceso irreversible ∆S > 0 Este ciclo consiste en cuatro etapas: a) expansión isotérmica reversible, b) expansión adiabática reversible, c) compresión adiabática irreversible y d) compresión adiabática reversible. Siendo ∆S un función de estado, la sumatoria de los cambios de entropía alrededor del ciclo también resulta: o∫ dqirrev/T = ∆S = 0 Para las etapas adiabáticas reversibles (b y d) ∆S=0. Luego la suma de ∆S para las etapas a y c también deberá anularse. Siendo ∆Sa>0 se deduce que ∆Sc >0. P. De esta forma se estableció que: ∆S>0 para todo proceso irreversible en un sistema aislado vi) vii) La etapa b es adiabática, por lo tanto es un proceso donde el intercambio de calor del sistema con el entorno es nulo. Cómo se interpreta este resultado? Podemos aplicar ∆S=q/T, para calcular ∆S de la etapa c? Porqué? 13 M.A.Perillo Biofisica-Quimica Generalizando para todo cambio espontáneo de un sistema aislado : ∆S≥0. Siendo el universo un sistema aislado, se puede escribir: ∆Suniverso ≥0 ∆Suniverso = ∆Ssistema + ∆Sentorno ≥ 0 [ec.4] - El signo igual se aplica a procesos reversibles y el mayor a procesos irreversibles. - La ec.4 no se opone a la posibilidad de que, en un proceso irreversible, ocurra un disminución o un cambio nulo en sistema, siempre que ese efecto sea compensado por un cambio opuesto en el entorno, y viceversa. El segundo principio, en su versión original, describe la evolución espontánea de un sistema aislado (que no intercambia materia ni energía con el exterior) y establece que en el curso de esa evolución la energía del sistema, si bien permanece constante, se transforma en parte en calor. Éste no puede a su vez transformarse en otra forma de energía, por lo que al cabo de cierto tiempo el sistema llega al equilibrio termodinámico, estado final en el que ninguna transformación de energía es ya posible. Este segundo principio puede ser formulado también a través de una magnitud abstracta, llamada entropía. La entropía, según el segundo principio, sólo puede crecer en el desarrollo de cualquier transformación de energía, de forma que, transcurrido un tiempo suficientemente largo, alcanza un valor máximo que caracteriza el estado de equilibrio termodinámico. vii) viii) ix) Grafique la variación de la entropía en función del tiempo. Cómo cambian el orden (o el desorden) del sistema durante su evolución hacia el equilibrio termodinámico. Revise los conceptos de equilibrio térmico, mecánico y material (químico y de fases) en textos de química general o de física. Desorden y orden corresponden pues, respectivamente, a probabilidad e improbabilidad, o a entropía y su contrario, neguentropía. Esto lo demuestra la termodinámica estadística (ver más adelante). COMBINACIÓN DE LA PRIMERA Y DE LA SEGUNDA DE LA TERMODINÁMICA A pesar que la Primera Ley de la Termodinámica establece que la energía invariablemente se conserva, nosotros frecuentemente nos preocupamos por la disponibilidad de energía. Esta inconsistencia aparente se debe al hecho que la energía térmica ambiental, en la cual “la energía térmica de alta calidad” y muchas otras formas de energía tienden a degradarse, posee escaso potencial de trabajo. Por lo tanto, es la energía libre y no la energía total (las diversas formas de energías de la 1ra Ley) lo que normalmente intentamos conservar. 14 M.A.Perillo Biofisica-Quimica La combinación de la primera y de la segunda ley permiten arribar a una expresión de energía libre: dA = d(E – T. S) ≤ -SdT - PdV dG = d(H – T.S) ≤ -SdT + VdP ∆A = ∆E – T. ∆S = 0 a T y V constantes, sistema cerrado ∆G = ∆H – T. ∆S = 0 a T y P constantes, sistema cerrado ∆A: energía libre de Hemholdz ∆G: energía libre de Gibbs Condición de equilibrio para sistema cerrado a T Y V constantes y que realice sólo trabajo -P∆V (expansión o compresión) ∆A=0 Condición de equilibrio para sistema cerrado a T Y P constantes y que realice sólo trabajo -P∆V (expansión o compresión) ∆G=0 x) Supongamos un cambio de fase irreversible a T y P, y conociendo que ∆Suniv=∆Ssistema + ∆Sentorno y que ∆S=qrev/T, intente derivar la ecuación que sigue: ∆Suniverso = -∆Gsistema/T xi) Grafique el cambio de energía libre a T y P constantes en función del tiempo para un sistema cerrado que evoluciona hacia el equilibrio. Compare con el gráfico de ∆S vs tiempo que realizó anteriormente. EQUILIBRIO MATERIAL dG= TdS + PdV + Σµi dni Σµi dni = 0 dG= TdS + PdV + ΣΣαµiα dniα ΣΣαµiα dniα = 0 donde µI α= (∂Gα/∂ni) T,P,nj≠i (POTENCIAL QUÍMICO del compuesto j en la fase α, siendo (condición de equilibrio de reacción) (condición de equilibrio de fases) constantes P, T y moles de los demás compuestos) • El potencial químico ∆µ representa el cambio de energía libre por unidad de masa. • ∆µ es la fuerza impulsora para la transferencia de materia 15 M.A.Perillo Biofisica-Quimica EQUILIBRIO DE FASES ======== µI α =µI β = µI γ =..... En el equilibrio de fases, el potencial químico de cada compuesto i es igual en todas las fases que coexisten en equilibrio) EQUILIBRIO DE REACCION K = (Aa.Bb) / (Cc . Dd) Para una reacción química aA + bB ====== cC +dD Siendo A, B, C y D las concentraciones molares, a,b,c y d los coeficientes estequiométricos y K la constante de equilibrio de la reacción. Cuando la relación de concentraciones de reactantes y productos elevados a un número igual su respectivo coeficiente estequiométrico da como resultado un valor equivalente a K, el sistema está en equilibrio, de lo contrario su valor será Q. ∆G0=-R.T.ln K ∆G0 = energía libre estándar (P=1 atm y concentraciones 1 M) El cambio de energía libre ∆G permite predecir la dirección de cambio espontáneo de una reacción química. ∆G=∆Go + R.T.lnQ [3] Si las concentraciones de A,B, C y D son 1M, entonces Q=1. Dado que ln 1= 0 reemplazando en la ecuación [3] ∆G=∆Go + R.T.ln 1 en consecuencia ∆G=∆Go + 0 ∆G=∆Go si Q=K ∴ ∆G=0 sistema en equilibrio Si Q>K ∆G>0 reacción se desplaza hacia el lado de los reactantes para alcanzar el equilibrio. Si Q<K ∆G<0 reacción se desplaza hacia el lado de los productos para alcanzar el equilibrio. 16 M.A.Perillo Biofisica-Quimica PRINCIPIOS DE TERMODINÁMICA ESTADÍSTICA El camino que conduce al equilibrio termodinámico o hacia la entropía máxima es un camino hacia la desorganización o el desorden progresivo. Puede considerarse también, por ejemplo, un sistema constituido por la superficie del suelo y una piedra situada libremente a cinco metros por encima, se dirá -con razón- que es una configuración muy improbable. Improbable pero no imposible: al lanzar una piedra hacia arriba, existe un punto sobre el suelo en el que la misma va a inmovilizarse antes de volver a caer hacia la tierra. En ese punto, la energía total del sistema es energía potencial; la piedra inicia su caída y a medida que va cayendo, esa energía potencial va transformándose en energía cinética, para terminar en calor cuando se da contra el suelo. Esa caída de la piedra es un viaje hacia el equilibrio termodinámico, hacia la entropía máxima, pero también hacia un estado cada vez más probable: no hace falta decir que encontrar piedras en el suelo, al costado del camino, es bastante más común que encontrar una libremente suspendida a tres metros por encima de la cabeza. De este modo, si se plantea en términos de probabilidades, los procesos espontáneos que llevan a los sistemas aislados hacia el equilibrio termodinámico consisten en una sucesión de estados cada vez más probables. El objetivo de la termodinámica estadística es deducir las propiedades macroscópicas de la materia a partir de propiedades de las moléculas que componen el sistema. Estado temodinámico: macroestado Estado cuántico: microestado Es necesario determinar la configuración del sistema. Esto es especificar la cantidad de moléculas ni que hay en cada estado de energía εi. PREGUNTAS QUE SE DEBEN RESPONDER: • Cuantas configuraciones son posibles? • Existen configuraciones más probables? En caso afirmativo determinar el peso estadístico de cada una (W i) (atención: no confundir peso estadístico con trabajo). CONDICIONES QUE SE DEBEN CUMPLIR: Σ ni.ei = E W = Σni = N N! n1!.n 2!.n3!..ni! donde: E=energía total del sistema; N= no total de moléculas del sistema. ni = no de moléculas en el estado i. 17 M.A.Perillo Biofisica-Quimica Cómo distribuimos 6 moléculas entre 6 estados posibles?. 6 6 6 5 5 4 4 4 3 3 2 2 • 5 1 ••••• W1 = 1 6! =6 5!.1! • 3 • • • • • W2 = •••• •• 2 1 6! = 60 3!.2!.1! W3 = 6! = 15 2!.4! CONCLUSIÓN: La distribución central presenta el mayor peso estadístico W 2. DISTRIBUCiÓN DE BOLTZMANN: a T=cte el nivel más poblado es el de menor εi ni * e = N − εi o k .T ∑e − * El denominador (q) indica el n de estados temodinámicamente accesibles a la temperatura T. i εi k .T * A T=0, q=1 ∴sólo estado basal (ε0) es posible. * Si T↑ ∴q ↑ i q se denomina función de partición molecular (no confundir con calor). ESTADÍSTICA DEL CAMBIO DE ENERGÍA INTERNA a) modificación de los niveles de energía disponibles b) cambio en el número de moléculas en cada nivel dE = 1 1 .∑ ni .dε i + .∑ ε i .dni N i N i El sistema absorbe calor Cambia dni inicial dQ = final 1 .∑ ε i .dni N i El entorno hace trabajo sobre el sistema Cambia dεi inicial dw = final 1 .∑ ni .dε i N i Note que la absorción de calor induce un aumento en la dispersión de moléculas entre los mismos estados iniciales, mientras que trabajo modifica la distribución de energía de los niveles termodinámicamente accesibles. 18 M.A.Perillo Biofisica-Quimica ESTADÍSTICA DEL CAMBIO DE ENTROPÍA dS = dq 1 = .∑ ε i .dni T T .N i ∆S=k.ln∆W Ley de Boltzmann La absorción de calor aumenta la dispersión de moléculas entre los distintos estados porque el estado más desordenado es el más probable (mayor W). La ley de Boltzmann establece la correlación entre entropía y desorden. ni / N wi pεi εi εi Trabajo vs energía de niveles individuales Fracción de moléculas en nivel i vs energía de niveles individuales E εi Probabilidad de fluctuaciones en la energía interna vs energía de niveles individuales xii) Interprete los gráficos precedentes. 19 M.A.Perillo Biofisica-Quimica CUESTIONARIOS DE ORIENTACIÓN Y PROBLEMAS PARA RESOLVER CONSIGNA DE TRABAJO: - Intente responder el siguientes cuestionario después de haber realizado el estudio independiente del material precedente. Las respuestas serán discutidas en la primera clase. Los problemas serán resueltos en la primera clase. PRIMERA LEY DE LA TERMODINAMICA 1. Qué estudian la termodinámica clásica y la estadística respectivamente? 2. Defina: sistema, entorno, universo. 3. Cómo se define el estado del sistema? 4. Indique cómo se denominan los sistemas de acuerdo a su relación con el entorno. 5. Defina cambios de estado de tipo: isotérmico, adiabático, isocórico, isobárico, reversible, irreversible. 6. Defina función termodinámica de estado (FTE) y dé cuatro ejemplos. Volumen es una FTE?. 7. Defina gas ideal. 8. Deduzca las leyes de Boyle y de Gay-Lussac a partir de la ecuación general de estado de gases ideales. 9. En un plano P vs V grafique cambios de tipo isotérmico, isobárico, isocórico, adiabático. En el mismo tipo de gráfico represente un ejemplo de trabajo reversible e irreversible. Utilice gráficos distintos para cada condición. 10. Enuncie la primera ley de la termodinámica. Indique sus limitaciones. 11. Qué es la energía interna? 12. Sobre la base de conceptos de termodinámica estadística indique las diferencias entre calor y trabajo. 13. Qué representan los signos “+” y “-” en relación a las cantidades de calor y trabajo que un sistema intercambia con el entorno? 14. Un gas perfecto sufre un proceso de expansión a presión constante. Aumenta o disminuye su energía interna? JSR. 15. Clasifique las siguientes cantidades como extensivas o intensivas e indique las unidades SI para cada una: a) densidad; E; c) H; d) Cp; e) calor específico; f) Cv. 16. Diga si las siguientes afirmaciones son verdaderas o falsas? a) ∆H es una función de estado b) Un proceso en el cual la temperatura final es igual a la temperatura inicial debe ser un proceso isotérmico c) Para un sistema cerrado en reposo en ausencia de campos externos E=q+w d) E permanece constante en todo proceso cíclico en un sistema cerrado. e) ∆E=0 en todo proceso cíclico. f) ∆T=0 para todo proceso adiabático en un sistema cerrado. g) Un proceso termodinámico queda definido al especificar el estado inicial y el estado final del proceso. h) Si un sistema cerrado en reposo y en ausencia de campos externos sufre un proceso adiabático con w=0 entonces la temperatura del sistema debe permanecer constante. i) El valor del trabajo de expansión y de compresión es usualmente despreciable en sólidos y líquidos. 17. En un gráfico P vs V indicar el camino que sigue el sistema durante un cambio isotérmico y uno adiabático. 18. Comparar las cantidades de trabajo realizado en cambios reversibles isotérmico y adiabático. 19. Represente los cambios descriptos para el ciclo de Carnot reversible en un gráfico P vs V. 20. Un mol de gas ideal se expande desde V1=1 litro hasta V2= 1.5 litros, a presión constante de 1 atm. Suponga que el sistema es aislado. Diga JSR si: a) Cambia la temperatura del gas? (utilice un gráfico P-V para orientarse) b) Cambia la energía interna? (recuerde que E=q+w ; w=-P∆V; E=nCp∆T). 20 M.A.Perillo Biofisica-Quimica c) Cuál será el efecto que esto tendrá a nivel molecular? (revise los conceptos sobre naturaleza molecular de la energía interna de la pág.18 del apunte) 21. Indique cualitativamente cuáll sería la cantidad de trabajo que se realiza en un proceso adiabático a volumen constante. 22. suponga un sistema constituido por un gran número de moléculas cuyas energías se distribuyen de acuerdo a la ley de Bolzmann, si al comparar la energía de los estados 1 y 2 resulta que Energía 1 > Energía 2, cuál de los dos estados tendrá mayor número de moléculas a una dada T?; JSR. SEGUNDA LEY DE LA TERMODINAMICA 23. Qué indica la flecha del tiempo y cuál es FTE la que la representa? 24. Describa entropía y su relación en el orden del sistema 25. Defina entropía de acuerdo a la termodinámica clásica y a la estadística. 26. Cuál de los siguientes enunciados puede probarse a partir de la segunda ley de la temodinámica? a) en un sistema cerrado, el equilibrio corresponde a la posición de máxima entropía del sistema; b) la entropía de un sistema aislado debe permanecer constante; c) en un sistema encerrado entre paredes impermeables y adiabáticas, la entropía es máxima en el equilibrio; d) la entropía de un sistema cerrado nunca puede disminuir; e) la entropía de un sistema aislado nunca puede disminuir. 27. Defina eficiencia termodinámica. 28. Describa cualitativamente como evaluaría la eficiencia celular para la utilización de glucosa y para la fotosíntesis. 29. Analice la ecuación 3 de la pag.4 y explique qué combinación de temperaturas permite aumentar la eficiencia de una máquina térmica? 30. Es posible alcanzar un eficiencia del 100% en la utilización de energía para producir trabajo? JSR. 31. El ciclo de Carnot irreversible consiste en cuatro etapas: a) expansión isotérmica reversible, b) expansión adiabática reversible, c) compresión adiabática irreversible y d) compresión adiabática reversible. - La etapa c es adiabática, por lo tanto es un proceso donde el intercambio de calor del sistema con el entorno es nulo, sin embargo ∆S >0. a) Cómo se deduce este resultado?. b) Cómo se interpreta este resultado?. c) cuál es la generalización que se hizo a partir de este resultado. d) Podemos aplicar ∆S=q/T, para calcular ∆S de la etapa c? Porqué? 32. Grafique la variación de la entropía en función del tiempo para un sistema cerrado. 33. El hecho que en un proceso irreversible se espere que ∆S>0 obliga a que el sistema se desordene?. 34. Cómo cambian el orden (o el desorden) del sistema durante su evolución hacia el equilibrio termodinámico. 35. Explique los conceptos de equilibrio térmico, mecánico y material (químico y de fases). 36. Cuál es la FTE que representa la energía útil para producir trabajo? 37. Grafique el cambio de energía libre a T y P constantes en función del tiempo para un sistema cerrado que evoluciona hacia el equilibrio. Compare con el gráfico de ∆S vs tiempo que realizó anteriormente. PROBLEMAS (serán resueltos en clase) Tema: 1ra Ley de la Termodinámica 1. Calor (Q) y trabajo (W) no son funciones de estado. Sin embargo, sus cambios o la combinacion de sus cambios se usan para representar variaciones de funciones de estado (dE=dQ+dW; dS=dQ/T; dH=dE+pdV). Porqué? 2. Un adulto típico ingiere 2200 Kcal/día. Muestre que un adulto consume energía aproximadamente a la misma velocidad que un foquito de 100 W. 21 M.A.Perillo Biofisica-Quimica 3. Suponga que 0.1 moles de un gas perfecto con Cv =1.5.R (independientemente de la temperatura), sufre el proceso ciclico reversible 1 2 3 4 que se muestra en la figura, donde P o V. Calcule q, w y ∆E para cada etapa y para el ciclo completo. Dibuje las isotermas. Obtenga los valores de P, V y T a partir del gráfico o, si es necesario, calcúlelos a partir de la ecuación general de estado para gases ideales. P (atm) 3.00 1.00 2 3 1 4 1000 2000 V (ml) Tema: 2da Ley de la Termodinámica 1. Suponga una máquina de vapor que trabaja con una fuente caliente a 800°C y una fría a 0°C. a) Calcule el máximo rendimiento posible. b) Si qc es 1000J, calcule el máximo valor de –w y el mínimo valor de –qf. 2. Para cada uno de estos procesos, deducir si ∆S y∆Suniv son negativos, cero o positivos: a) Fusión reversible de benceno sólido a 1 atm en el punto de fusión normal; b) fusión reversible de hielo a 1 atm y 0oC; c) expansión reversible adiabática de un gas perfecto; d) expansión reversible isotérmica de un gas perfecto; e) expansión adiabática de un gas perfecto en el vacío (experimento de Joule); f) calentamiento reversible de un gas perfecto a presión constante P; g) enfriamiento reversible de un gas perfecto a volumen constante; h) combustión del benceno en un recipiente con paredes adiabáticas. 3. El calor molar de vaporizacion del Ar en el punto normal de ebullición (87,3 K) es 1,56 Kcal/mol. A) Calcule ∆S cuando se vaporiza 1 mol de Ar a 87,3 K y 1 atm. B) Calcule ∆S cuando se condensan 5 g de Ar (g) a líquido a 87,3 K y 1 atm. 4. 200 g de oro (Cp=0.0313 cal/gC) a 120oC se depositan sobre 25 g de agua a 10oC, y el sistema alcanza el equilibrio en un recipiente adiabático. Calcule a) la temperatura final; b) ∆S Au; c) ∆S agua d) ∆S Au + ∆S agua. 5. Cuál de los siguientes enunciados puede probarse a partir de la segunda ley de la temodinámica? a) en un sistema cerrado, el equilibrio corresponde a la posición de máxima entropía del sistema; b) la entropía de un sistema aislado debe permanecer constante; c) en un sistema encerrado entre paredes impermeables y adiabáticas, la entropía es máxima en el equilibrio; d) la entropía de un sistema cerrado nunca puede disminuir; e) la entropía de un sistema aislado nunca puede disminuir. Tema: Energía libre 1. Indique si ∆A y ∆G de los siguientes procesos son positivos, cero o negativos: a) fusión reversible de benceno sólido 1 atm en el punto de fusión normal; b) fusión reversible de hielo a 1 atm y 0 C; c) expansión reversible adiabática de un gas perfecto; d) expansión adiabática de un gas perfecto en el vacío; e) expansión reversible isotérmica de un gas perfecto; f) expansión adiabática de un gas perfecto en el vacío g) estrangulamiento adiabático de un gas perfecto; h) calentamiento reversible de un gas perfecto a presión constante P; i) enfriamiento 22 M.A.Perillo Biofisica-Quimica reversible de un gas perfecto a volumen constante. (Recuerde que todo proceso reversible transcurre a través de una sucesión de infinitos estados de equilibrio). 2. Diga cuál sustancia de los siguientes pares tiene mayor potencial químico (mayor energía libre por unidad de moles): a) H2O(l) a 25° C y 1 atm, H2O(g) a 25°C y 1 atm; b) H2O(s) a 0°C y 1 atm, H2O (l) a 0°C y 1 atm; c) H2O(s) a -5° C y 1 atm, H2O (l) sobreenfriada a –5°C y 1 atm d) C6H12O6 (s) a 25°C y 1 atm, C6H12O6 (ac) en una solución acuosa insaturada a 25°C y 1 atm. e) C6H12O6(s) a 25° C y 1 atm; C6H12O6 en una solución acuosa saturada a 25°C y 1 atm. f) C6H12O6 (s) a 25°C y 1 atm; C6H12O6 en una solución acuosa supersaturada a 25°C y 1 atm. 7. Escriba la condición de equilibrio químico y la constante equilibrio de N2 + 3H2 → 2NH3 en un sistema cerrado. 8. Para cada uno de los siguientes procesos señale si alguna de las diferencias ∆E, ∆H, ∆S, ∆G debe ser cero. a) un gas no ideal que recorre un ciclo de Carnot. b) quemado de hidrógeno en un calorímetro adiabático de volumen constante. c) fusión del hielo a 0°C y 1 atm. BIBLIOGRAFÍA ESPECIFICA UNIDAD 1. Atkins,P.W. Fisicoquimica. Ed. Addison-Wesley Iberoamericana, S.A. Wilmington, Delaware, USA.1991 Israelachvili,J.N. Intermolecular and surface forces. Academic Press, New York. 1989. Maggio B. Introducción a la biofísico-química. Ed.Gonzalez Truccone. Córdoba, Argentina. 1987. Moore W.J. Química física. Ed.URMO, S.A.,Bilbao, Espana.1977. 23 M.A.Perillo Biofisica-Quimica UNIDAD 2. FUERZAS INTERMOLECULARES Y ESTRUCTURAS DE AUTOAGREGACIÓN CONSIGNAS DE TRABAJO: Repase los conceptos de: magnitudes escalares y vectoriales, módulo, dirección y sentido de un vector, proyección de un vector sobre los ejes de un sistema cartesiano, conceptos generales sobre electrostática (fuerza eléctrica, dipolo, momento dipolar, polarización); enlaces (covalentes, iónicos), interacción por puente de hidrógeno, interacciones de London-van der Waals. En caso necesario recurra a textos de Quimica General o de Física. - Realice el estudio independiente del texto que se presenta más abajo. Intente responder las preguntas insertas en el mismo. Las respuestas a las mismas serán discutidas durante la primera clase - Lea los tres trabajos de divulgación científica que se adjuntan en el Anexo I. e intente responder a los cuestionarios sobre biomembranas que se encuentran al final de esta unidad. - Lea atentamente la guía del Trabajo Práctico No 1. o Durante la segunda clase se discutirán los trabajos y se realizará el T.P N 1. - SUSTANCIAS ANFIPÁTICAS Y AUTOAGREGACIÓN Sustancias anfipáticas son aquellas que poseen por lo menos dos zonas de polaridad opuesta y orientada asimétricamente en su estructura química. Cuando en un sistema acuoso se incrementa la concentración de una sustancia anfipática, varias propiedades como la presión osmótica (Π), la conductividad (c) y la tensión superficial (γ) varían con la concentración del anfifilo según lo indicado en la Fig.2.1. Región hidrofílica monómero micela Región hidrofóbica Π γ Concentración monómero y micela c γ C monómero micela Π CMC CMC Concentración anfifilo Total agregado de anfifilo Fig. 2.1 24 M.A.Perillo Biofisica-Quimica En soluciones diluidas de sustancia anfipática estos parámetros (Π, γ, C) varían en función de la concentración según lo indicado en la Fig. 2.1 de manera similar a soluciones de electrolitos simples, pero existe una región o valor de concentración en la cual estos parámetros cambian bruscamente. Esta región de concentración en la cual ocurre el fenómeno se denomina concetración micelar crítica (CMC). La explicación del fenómeno a nivel molecular se puede resumir didiendo que en la zona de CMC se equilibrio entre el anfifilo en su forma de monómero y un agregado multimolecular denominado micela. Una micela es una estructura formada por agregación de varios monómeros de anfifilo de tal forma en que se satisfacen las interacciones hidrofílicas y el efecto hidrofóbico de tal forma que la micela reduce el área de contacto de las zonas no polares del anfifilo con la fase acuosa mientras que que al mismo tiempo permanecen solvatados los grupos hidrofílicos. La forma de representar la fase micelar se esquematiza en la Fig. 2.1. La figura muestra que la concentración de monómero aumenta hasta alcanzar un valor máximo y al agregar más moléculas anfipáticas comienza a formarse la fase micelar. La concentración máxima de monómero en solución en equilibrio con la fase micelar corresponde a la CMC. ESTRUCTURAS DE AUTOAGREGACIÓN Las membranas biológicas naturales así como los modelos artificiales de membrana (liposomas, micelas, capas monomoleculares) provienen de la autoagregación de moléculas anfipáticas, también llamadas anfifilícas. i) Qué es una molécula anfipática? De ejemplos. Las fuerzas que mantienen juntas a estas moléculas en la estructura de autoagregación no son covalentes ni iónicas fuertes (tales como las que caracterizan a los enlaces químicos) sino fuerzas más debiles como van der Waals, puente de hidrógeno, hidrofóbicas y fuerzas electrostáticas débiles. Fuerzas intermoleculares Los grupos hidrofílicos se reconocen por su tendencia a ser solubles en agua y por repelerse mutuamente en agua. Los grupos hidrofóbicos son capaces de formar puntes de hidrógeno y cuando se ponen en contacto con el agua inducen la formación de “cajas de solvente” a su alrededor. Estas estructuras no son rígidas sino lábiles y los puentes de hidrógeno que las estabilizan no son más fuertes que los que se encuentran en el agua pura, pero las moléculas de agua que forman estas “cajas” están más ordenadas que aquellas que se encuentran en el seno del líquido. ii) La reorientación de agua alrededor de solutos o superficies no polares es entrópicamente desfavorable. Porqué? EFECTO HIDROFOBICO: Fuerza impulsora de la autoagregación La formación de cajas de solvente alrededor de grupos hidrofóbicos rompe la estructura del agua e impone una nueva y más ordenada estructura. Además, aún cuando la interacción de las moléculas no polares es en realidad atractiva (∆H<0) debido a las fuerzas de dispersión, la interacción agua-agua es mucho más atractiva. 25 M.A.Perillo Biofisica-Quimica La baja solubilidad en agua de las moléculas no polares y la naturaleza entrópica de la energía libre de solubilización se conoce como efecto hidrofóbico. Fig.2.2 El alcance de la interacción electrostática disminuye a media que aumenta el exponente al cual está elevado r (la distancia de separación entre las dos moléculas) según la siguiente secuencia: carga-carga>carga-dipolo> dipolo-dipolo. 26 M.A.Perillo Biofisica-Quimica Condiciones termodinámicas para la autoagregación k .T X . log N = cte ; N N αk.T µ No = µ∞o + p N µ = µ N = µ No + N=1,2,3... [2] donde N es el número de agregación, X es la actividad de las moléculas incorporadas en el agregado de número de agregación o N; µ N es la parte estándar del potencial químico por molécula en el agregado y depende de la energía libre por molécula en el seno de la o solución ( µ ∞ ), de la fuerza de la interacción intermolecular (≈α) y de la dimensionalidad del agregado (1/p); k es la constante de Boltzman y T la temperatura absoluta. i) La estructura de autoagregación que se forma será la compatible con un valor de µ No menor al potencial químico del monómero y la concentración de monómero a la cual se forma será la que satisfaga la condición de que µ No alcance un mínimo. Porqué? ii) Cómo se denomina a la concentración de monómero a la cual aparecen agregados? iii) Cuál es la dimensionalidad de los siguientes modelos de membrana: a) capa monomolecular en la interfase agua-aire, b) liposoma, c) micela 27 M.A.Perillo Biofisica-Quimica Fig.2.3 Restricciones geométricas para la autoagregación Tomado de Israelachvili, 1989. Las propiedades de empaquetamiento de la moléculas anfipáticas dependen de factores termodinámicos acoplados a la geometría molecular. Las restricciones impuestas sobre la curvatura intrínseca de la interfase membrana-agua (también llamada curvatura espontánea derivada de la forma molecular está representada por el parámetro crítico de empaquetamiento (Pc). Pc=v/ (a0.l) donde v= volumen del grupo polar, a0=area que se correlaciona con la mínica energía por molécula en el agregado y l= longitud de la cadena hidrocarbonada. El tipo de estructura de autoagregación que se forma puede ser predicha a partir del valor que adopta Pc: micela (Pc<0.5), bicapas (0.5<Pc<1) y fases invertidas tales como la hexagonal II o la cúbica (Pc>1). Diferentes tipos de lípidos pueden acomodarse según sus geometrías a los fines de empaquetarse en una biomembrana, ajustándose el tipo de curvatura adoptada por la monocapa externa (curvatura positiva) y por la monocapa interna (curvatura negativa). De esta forma compensan la tensión de curvatura de la bicapa. 28 M.A.Perillo Biofisica-Quimica TRABAJO PRACTICO N° 1 DETERMINACION DE LA CONCENTRACION MICELAR CRITICA (CMC) CONSIGNA DE TRABAJO: Lea atentamente esta guía. El T.P. será realizado en la clase no2. INTRODUCCION a) El fundamento del TP consiste en que un colorante como el azul de bromo timol en agua debido a su baja solubilidad en medios de alta constante dieléctrica tiene valores muy bajos de coeficiente de absorción, pero es muy soluble en medios de baja polaridad tales como el corazón hidrofóbico de una micela y desarrolla un color azul obteniéndose buenos valores de Absorbancia (A) en esos medios. Cuando en la solución no existe una fase micelar los valores de A serán bajos y constantes. Los valores de A aumentarán significativamente cuando en la solución se forme la fase micelar ya que el colorante particiona a la región no polar de la micela. PARTE PRACTICA MATERIALES 1) Colorante: Azul de Bromo Timol 3.3 % P/V en metanol. Diluir 1/100 en agua antes de usar 2) Detergentes: a) Cetrimida (detergente catiónico al 5.46 % P/V (PM = 364.45). Determinación de CMC de Cetrimida a) a temperatura ambiente b) a 90°C INDICACIONES a) Una vez agregados todos los reactivos, se espera 30 minutos a temperatura ambiente y se leer la A a 485 nm. CETRIMIDA Tubo N° Blanco 1 2 3 4 5 6 7 8 9 10 Cetrimida Colorante 5.46% (ml) Ml 0,00 0,2 0,10 0,2 0,20 0,2 0,30 0,2 0,35 0,2 0,40 0,2 0,45 0,2 0,50 0,2 0,60 0,2 0,70 0,2 0,80 0,2 Agua ml 2,80 2,70 2,60 2,50 2,45 2,40 2,35 2,30 2,20 2,10 2,00 Volumen final 3,0 3,0 3,0 3,0 3,0 3,0 3,0 3,0 3,0 3,0 3,0 Concentración A A mM (T amb) (90°C) Una vez obtenidos los valores de A a temperatura ambiente, graficar la A en función de la concentración de detergente expresada en mM. El punto de inflexión corresponde a la CMC. 29 M.A.Perillo Biofisica-Quimica CUESTIONARIO DE ORIENTACIÓN CONSIGNA DE TRABAJO: Intente responder el cuestionario sobre “Fuerzas intermoleculares y estructuras de autoagregación” después de haber realizado el estudio independiente del material precedente. Las respuestas serán discutidas en la segunda clase. - Lea al menos uno de los trabajos de divulgación científica del Anexo I (a convenir con el profesor), e intente responder al cuestionario correspondiente. Los trabajos serán discutidos en la segunda clase. La formación de membranas celulares Lodish H. y Rothman J. (1979) Investigación y Ciencia, Vol.30, pág.20 Fluidez de las membranas celulares Gómez-Fernández J. y Goñi F. (1983) Investigación y Ciencia, Vol.79, pág.14 - FUERZAS INTERMOLECULARES Y ESTRUCTURAS DE AUTOAGREGACION 1. Defina magnitudes vectoriales y escalares. De ejemplos. 2. Qué es momento dipolar. En cuál de las categorías indicadas en el punto anterior incluiría al momento dipolar? 3. En qué se diferencian los enlaces: a) homonucleares y heteronucleares; b) polar y no polar. Cómo se correlacionan las propiedades a y b entre si? 4. Cómo influye la polaridad de los enlaces individuales sobre la polaridad de una molécula? 5. Qué es una molécula anfipática? 6. Qué es una molécula zwitteriónica? 7. Indique tipos de interacciones electrostáticas intermoleculares capaces de estabilizar estructuras de autoagregación. 8. Qué es el alcance de una interacción electrostática y de qué depende? 9. Ordene los distintos tipos de interacciones electrostáticas en orden decreciente según la energía y el alcance de las mismas. 10. Qué es la caja de solvente de una molécula en solución? 11. Defina efecto hidrofóbico. 12. La orientación de agua alrededor de solutos o superficies no polares es entrópicamente desfavorable. Porqué? 13. Explique porqué un fenómeno de autoagregación de moléculas anfipáticas no viola el segundo principio de la termodinámica. 14. Cómo se denomina a la concentración de monómero a la cual aparecen agregados multimoleculares? BIOMEMBRANAS Cuestionario de orientación para análisis de textos seleccionados. Lea los trabajos de divulgación científica del Anexo I y conteste a las siguentes preguntas: La formación de membranas celulares Lodish H. y Rothman J. (1979) Investigación y Ciencia, Vol.30, pág.20 1) ¿Cuáles son las principales propiedades funcionales de una membrana celular? 2) ¿Qué entiende por asimetría estructural y funcional en membranas? 3) ¿Además de la membrana plasmática, qué otros orgánulos celulares poseen membranas? 4) ¿Qué tipo de moléculas orgánicas forma parte de una membrana celular? 5) ¿Qué es una molécula anfipática? ¿Por qué moléculas de este tipo son capaces de organizarse espontáneamente cuando son dispersadas en soluciones acuosas? 6) Tanto lípidos como proteínas son capaces de moverse en la membrana ¿qué tipo de movimientos conoce para ambos casos? 30 M.A.Perillo Biofisica-Quimica 7) ¿Cómo se clasifican las proteínas según su grado de asociación con la membrana? ¿Qué rol desempeñan en estas asociaciones los aminoácidos que las constituyen, en cuanto a su naturaleza hidrofílica o hidrofóbica? 8) ¿Cómo transcurre el proceso general a través del cual se forma completamente una proteína integral de membrana, desde su síntesis hasta su inserción en la membrana? 9) ¿Dónde son sintetizados principalmente los fosfolípidos formadores de membranas y cómo se mantiene la asimetría de la misma? Fluidez de las membranas celulares Gómez-Fernández J. y Goñi F. (1983) Investigación y Ciencia, Vol.79, pág.14 1) ¿Cómo define fluidez de la membrana? 2) ¿Cómo influyen las características de las cadenas hidrocarbonadas de los fosfolípidos componentes de membrana sobre la fluidez de la misma? 3) ¿Cómo afecta la temperatura a la fluidez? ¿Qué es la temperatura de transición de fase (Tc)? 4) ¿Por qué razón las membranas celulares no suelen presentar transiciones de fase altamente cooperativas? (qué es cooperatividad?) 5) Explique brevemente en qué consisten y para qué se emplean en el estudio de membranas las siguientes técnicas: - Espectrometría de resonancia de espin electrónico. - Calorimetría diferencial de barrido. - Difracción de rayos X. - Espectroscopía de polarización de fluorescencia. - Espectrometría de resonancia magnética nuclear. 6) ¿Cómo afecta a la fluidez de la membrana su interacción con colesterol y con proteínas? 7) ¿Qué es adaptación homeoviscosa? BIBLIOGRAFÍA ESPECIFICA UNIDAD 2 Israelachvili,J.N. Intermolecular and surface forces. Academic Press, New York. 1989. Maggio B. Introducción a la biofísico-química. Ed.Gonzalez Truccone. Córdoba, Argentina. 1987. Perillo, M.A. y cols. Guía de Trabajos Prácticos de Biofísica-Química para Ciencias Biológicas, FCEFyN, Univ.Nac.Córdoba. Guía de T.P. de Biofísica, Fac.Cs.Químicas, UNC. Schifter, I. La ciencia del caos. http:/omega.ilce.edu.mx:3000/sites/ciencia. Textos seleccionados Lodish H. y Rothman J. (1979) La formación de membranas celulares. Investigación y Ciencia, Vol.30, pág.20 Gómez-Fernández J. y Goñi F. (1983) Fluidez de las membranas celulares. Investigación y Ciencia, Vol.79, pág.14 31 M.A.Perillo Biofisica-Quimica UNIDAD 3. CINÉTICA Y MODULACIÓN DE LA ACTIVIDAD ENZIMÁTICA. CONSIGNAS DE TRABAJO: - Repase los conceptos de cinética química. En caso necesario recurra a textos de Quimica General. - Realice el estudio independiente del texto que se presenta más abajo. Intente responder las preguntas insertas en el mismo. Las respuestas a las mismas serán discutidas durante la segunda clase - Lea los dos trabajos de divulgación científica y los tres trabajos científicos originales que se adjuntan en el Anexo II e intente responder a los cuestionarios sobre biocatalizadores que se encuentra al final de esta unidad. En el caso de los trabajos en inglés sólo intente interpretar los resultados mostrados en las figuras seleccionadas indicadas en la guía de estudio correspondiente. Durante la segunda clase se discutirán los trabajos. La catálisis es una rama de la cinética química que estudia el conjunto de procedimientos y conocimientos que permiten que la velocidad con la que trascurre una reacción se incremente insitu. Para una reacción química del tipo A+B C+D la velocidad de la reacción puede representarse como donde presentan la variación de la concentración de A, B, C o D respecto del tiempo y el signo (-) representa la desaparición de reactivos (A o B) y el signo (+) la aparición de productos (C o D). En la mayoría de las reacciones, la velocidad de transformación es proporcional a la concentración de reactivos elevados a una potencia; por ejemplo para la reacción A+B Productos V α [A]p[B]q , o V = k[A]p[B]q Donde: k = constante de proporcionalidad (constante de velocidad); p y q = órdenes parciales de reacción; p + q = n = orden global de reacción Los órdenes de velocidad pueden ser enteros, fraccionarios, positivos, negativos, o aun cero. En general este orden no está relacionado con la estequiometría de la reacción, sino más bien con el mecanismo de la misma. Para que una reacción química se lleve a cabo, es necesario suministrar una cierta cantidad de energía a las moléculas de reactivo. Esto puede ser 32 M.A.Perillo Biofisica-Quimica representado de la manera siguiente para la reacción anterior Fig.3.1 Diagrama de energía potencial para una reacción exotérmica. La barrera energética que separa los reactivos de los productos se denomina energía de activación. La velocidad de reacción depende de esa energía de activación a través de la constante de velocidad (k). Esta constante de velocidad depende también de la temperatura y la forma matemática de representarla es a través de la llamada ley de Arrhenius. donde: k = constante de velocidad k0 = factor preexponencial Ea = energía de activación R = constante de los gases ideales T = Temperatura en grados K Un catalizador: 1- modifica la velocidad de la transformación. 2- no se considera ni reactivo ni producto en la reacción 3- es una sustancia que sin estar permanentemente involucrada en la reacción, incrementa la velocidad con la que una transformación química se aproxima al equilibrio. 4- altera el mecanismo de reacción así como la velocidad total de la misma. 5- se regenera en el último paso de la reacción. 6- modifica el mecanismo de la reacción, llevando a ésta por un camino que requiere menor energía de activación. Figura 3.2 Curva de la energía potencial a lo largo de la coordenada de la reacción para un proceso catalítico heterogeneo. 33 M.A.Perillo Biofisica-Quimica Al introducir el catalizador (K), A y B interaccionan con él, si el catalizador es heterogéneo (sólido), se dice que A y B se adsorben en la superficie, formando un complejo superficial ABK inestable (línea punteada). Este complejo superficial reaccionará al suministrarle energía de manera que formará los productos que aún quedan fijos (línea de guiones) sobre la superficie. Para sacar los productos adsorbidos, es necesario otra pequeña energía que nos conduce al estado final productos +K (doble línea). i) ii) iii) iv) v) vi) Qué estudia la cinética química? Cómo define matemáticamente la velocidad de una reacción química? En qué unidades se mide? Defina orden de reacción y energía de activación. De qué depende la constante de velocidad de una reacción? Qué es y como actúa un catalizador? El ∆G de la reacción 2H2 + O2 ==== H2O es negativo, sin embargo si se mezclan H2 y O2 a temperatura ambiente no se observa la producción de H2O. Puede explicar estos hallazgos? CATÁLISIS ENZIMÁTICA La mayoría de las reacciones que ocurren en los sistemas vivos son catalizadas por proteínas conocidas con el nombre de enzimas. Cientos de enzimas han sido aisladas y probablemente existen cientos de miles en la naturaleza. Su estructura es muy compleja, y puede ser representada como se muestra en la Fig. 3.3. Fig.3.3. Estructura cristalina de la carboxipeptidasa A. Las enzimas reciben su nombre en función de su actividad específica, así, por ejemplo, la enzima “ureasa” cataliza con eficiencia la hidrólisis de la urea, las proteasas actúan sobre las proteínas, las amidasas sobre las amidas, etc. La mayoría de las enzimas desde el punto de vista químico son proteínas, pero pueden asociarse con substancias no proteínicas, llamadas coenzimas o grupos prostéticos, que son esenciales para la acción de la enzima. A veces las enzimas son inactivas catalíticamente, si no se encuentran en presencia de ciertos iones metálicos. No toda la molécula de proteína presenta actividad catalítica, sino únicamente una región relativamente pequeña, la cual se denomina centro activo. Además de las enzimas, los ribozimas y algunos anticuerpos pueden ser catalizadores biológicos. 34 M.A.Perillo Biofisica-Quimica Los mecanismos de reacción de las enzimas son muy complejos, implicando un número de etapas elementales cada una de las cuales puede incluir interacciones complejas entre varios grupos de las moléculas de la enzima y el sustrato. En las reacciones catalizadas por enzimas las velocidades de reacción, así como los mecanismos se ven afectados por cambios en la concentración, el pH y la temperatura. Figura 3.4 Variación de la velocidad en función del pH, para una reacción enzimática. En la elucidación de los mecanismos de reacción de las enzimas, y principalmente aquellas que involucran un ion-metálico o metaloenzimas, se requiere conocer: 1) La afinidad de los reactantes, las coenzimas y los cofactores; 2) Las constantes de velocidad para cada paso; 3) Las relaciones geométricas tridimensionales entre los reactantes, las coenzimas en relación a los sitios catalíticamente importantes de la enzima; y 4) el mecanismo de cada paso, es decir los arreglos atómicos y electrónicos. El mecanismo químico está determinado por el tipo de rompimiento y formación de enlaces que lleva a cabo la enzima. Se ha propuesto que las enzimas contienen en sus “centros activos” ácidos (AH) o bases (B) de manera que se puede calcular su fuerza ácida o básica. Así la pepsina, enzima que cataliza la hidrólisis de ciertos enlaces pépticos en el estómago, tiene un valor de pK de 2.2. El único grupo orgánico que puede dar este valor es el COOH-. También hay enzimas con caracter básico como la quimotripsina y la colinesterasa con un pK=7.2. Las velocidades de las reacciones catalizadas por enzimas son en general de primer orden respecto a la enzima. Sin embargo, es frecuente encontrar una dependencia de la concentración del sustrato (sobre el que actúa la enzima), como se muestra en la figura 5. La velocidad varía linealmente con la concentración de sustrato a concentraciones bajas (primer orden respecto al sustrato) y se hace independiente de la concentración de éste (orden cero) a concentraciones elevadas. Este tipo de comportamiento fue explicado por Michaelis y Menten en función del mecanismo siguiente: 1. Interacción de la enzima con el substrato (reactivo), para formar un complejo intermediario 11 k1 E1+1S1 1ES xxxk-1 2. Descomposición del complejo intermediario para dar los productos y regenerar la enzima k2111 ES E+P E es la enzima, S el sustrato, ES un complejo y P es el producto. Aplicando el tratamiento cinético denominado del estado estacionario, en el que se asume que la concentración del complejo intermediario es constante obtenemos k1[E] S - k_1 [ES] - k2[ES] = 0 I 35 M.A.Perillo Biofisica-Quimica Si la concentración total de la enzima [E]o es igual a la suma de la concentración en enzima libre [E], más la concentración de enzima que forma el complejo [ES]: [E]o = [E] + [ES] II Introduciendo [E] de la ecuación II en la ecuación I, tenemos: k1 ([E]o - [ES]) [S] - (k_1 + k2)[ES] = 0 de donde podemos obtener la concentración de enzima que está formando el complejo Se asume que la etapa determinante de la reacción es la descomposición del complejo, entonces la velocidad de la reacción es v=k2[ES] en donde se substituye [ES] que rearreglando nos da: Ecuación de Michaelis-Menten III K M= donde Km se denomina constante de Michaelis. De la ecuación III se deduce que cuando [S] es suficientemente pequeña, IV lo que indica primer orden respecto a la concentración de sustrato. Por el contrario, cuando [S] es mucho mayor que Km, Velocidad máxima (Vmax) Vmax = k2 [Eo] V y la cinética es orden cero respecto al sustrato. En ambos casos el orden es 1 para la concentración de enzima. La ecuación III da cuenta entonces del comportamiento observado en la Fig. 5.3. 36 M.A.Perillo Biofisica-Quimica Fig. 3.5 Dependencia típica de la velocidad de una reacción enzimática en función del sustrato S. La velocidad se mide en unidades de masa de producto formado por min. La actividad de una enzima se mide en masa de producto formado por unidad de tiempo por unidad de masa de enzima (si E está pura) o masa de tejido o volumen de preparación (si E es parte de una mezcla; por ej. siestá presente en un homogenato de tejido o suero). V=µmol de P/min; A=µmol de P /min/ml Las velocidades catalizadas por enzimas pasan por un máximo al ir subiendo la temperatura. A 60°C por ejemplo, muchas propiedades de la enzima se alteran, algunas de ellas irreversiblemente. Estos cambios se conocen con el nombre de desnaturalización y son los responsables del decrecimiento de la actividad de la enzima. TABLA 1. Efecto catalítico de algunas enzimas para diferentes reacciones. E Reacción Catalizador T° C k k0 Kcal/mol Hidrólisis de la urea “ Hidrólisis de trifosfato de adenosina “ Descomposición del H2O2 “ H3O+ ureasa 62.0 20.8 7.4X10-7 5.0x106 1.8X1010 1.7x1013 H3O+ 40.0 4.7x10-6 2.4x109 21.2 miosina Fe++ catalasa 25.0 22.0 22.0 8.2x106 56 3.5x107 21.1 10.1 01.7 1.6x1022 1.8x109 6.4x108 24.6 06.8 En la tabla 1 se muestran valores de las constantes de velocidad, energías de activación y factores preexponenciales de tres reacciones catalizadas por enzimas y se incluyen a título de comparación los valores de otros catalizadores. vii) viii) ix) x) xi) Grafique la variación de la concentración de E, S , ES y P en función del tiempo. En el gráfico anterior indique el momento en se alcanza el estado estacionario de [ES]. Cuál es su importancia (vea ec.I) Para una reacción catalizada por enzimas diga: a) cómo varía la velocidad en función del tiempo, b) indique el orden de reacción respecto a [S] para el caso en que si [S]<<KM (velocidad inicial) y [S]>>KM (Velocidad máxima). Una de las pruebas bioquímicas que se realizan para diagnosticar hepatitis se basa en la determinación de la cantidad de varias enzimas en el suero del paciente (GPT, GOT, fosfatasa alcalina). En qué región de la curva de velocidad vs [S] se debe trabajar? (vea ec.V y Fig.5.3) a) Qué es actividad enzimática, b) en qué unidades se mide y c) cuál es su relación con cantidad de enzima?. 37 M.A.Perillo Biofisica-Quimica BIOCATALIZADORES Cuestionario de orientación para análisis de textos seleccionados. Lea los trabajos de divulgación científica del Anexo II y conteste a las siguentes preguntas: Estabilidad de los biocatalizadores Plou, FJ; Alcalde, M y Ballesteros, A. (1999) Investigación y Ciencia. 273: 46-55. 1) ¿Cuáles son las diferencias entre catalizadores biológicos y no biológicos? (también vea Tabla 1 del texto precedente). 2) De ejemplos de la utilización de enzimas en procesos industriales. 3) ¿Qué factores afectan la estabilidad de las enzimas? 4) ¿Cuales son las fuerzas responsables de mantener la estructura de las enzimas? 5) La energía libre del sistema (G) esta relacionada con la estabilidad de una enzima. Se relaciona son la entalpía (H) y la entropía (S) a traves de la fórmula: G = H- TS Si se incuba una proteína en agua y al cabo de un tiempo se mide un aumento de entalpía (H). La enzima se está: a) Desnaturalizando, b) Plegando. Explique. 6) Indique en cada caso si el tipo de mecanismo corresponde a una desnaturalización, a una inactivación o a ambas. a) Exposición de todos los aminoácidos de la proteína al solvente, b) Eliminación de la coenzima del centro activo, c) Ruptura de puentes de hidrógeno, d) Agregación de enzimas, e) Reducción de enlaces disulfuros. 7) Las enzimas capaces de operar en condiciones extremas se pueden obtener de: a)Microorganismos, b) Ingeniería Genética. Ejemplifique 8) ¿Qué estrategias de inmovilización de enzimas se han desarrollado hasta ahora? 9) Además de la inmovilización como tratamiento para mantener la estabilidad de los biocatalizadores, se realizan otros tratamientos, como la modificación química de las enzimas y el agregado de sustancias al medio donde esta se encuentra. Explique en que consisten. 10) Marque con una cruz las moléculas orgánicas que pueden actuar como biocatalizadores a) Proteínas, b) ADN, c) Lípidos, d) Anticuerpos, e) ARN, f) Glúcidos. Modulación de la actividad enzimática 1. Perillo, M.A., Guidotti, A., Costa, E., Yu, R.K. and Maggio, B. 1994. Molec.Membr.Biol. 11,119. 2. Perillo, M.A., Yu, R.K. and Maggio, B. 1994. Biochim.Biophys.Acta. 1193, 155. 3. Sánchez, J.M. and Perillo M.A. 2002. Colloids & Surfaces B:Biointerfaces. 24,21. Qué factores pueden modular la actividad de una enzima en un medio heterogéneo? Nota: Para responder a esta pregunta observe Fig.5 trabajo 1; Fig.5 trabajo 2 y Tabla 2 trabajo 3. 38 M.A.Perillo Biofisica-Quimica BIBLIOGRAFIA ESPECÍFICA UNIDAD 3 Blanco A. 2000. Química Biológica. 7ª edición , El Ateneo, Bs.As. Nelson, D.L. y Cox, M.M. 2000. Leningher. Principles of Biochemistry. 3ª Edición. Worth Pulishers, New York. Arce A., Perillo M.A. y cols. Guía de T.P. de Química Biológica, FCEFyN, UNC. Schifter, I. La ciencia del caos. http:/omega.ilce.edu.mx:3000/sites/ciencia. Textos seleccionados Plou, FJ; Alcalde, M y A. Ballesteros. (1999) Estabilidad de los biocatalizadores. Investigación y Ciencia. 273: 46-55. Perillo, M.A., Guidotti, A., Costa, E., Yu, R.K. and Maggio B. (1994). Modulation of phospholipases A2 and C activities against dilauroylphosphoylcholine in mixed monolayers with semisynthetic derivatives of ganglioside and sphingosine. Molecular Membrane Biology. 11, 119. Perillo, M.A., Yu, R.K. and Maggio B. (1994). Modulation of the activity of Clostridium perfringens neuraminidase by the molecular organization of gangliosides in monolayers. Biochemical et Biophysical Acta, 1193, 155. Sanchez J.M. and Perillo M.A. (2002). Membrane adsorption or penetration differentially modulates β-galactosidase activity against soluble substrates. Colloids and Surfaces, 24,21. 39 M.A.Perillo UNIDAD 4. Biofisica-Quimica AUTOORGANIZACIÓN ESPACIAL Y TEMPORAL CONSIGNAS DE TRABAJO: - Lea el texto que sigue basado principalmente en los trabajos de Madressi, R y de Perillo MA citados en la bibliografia. Identifique los siguientes conceptos: sistema dinámico, espacio de fases, atractor, sensibilidad a las condiciones iniciales, caos determinista, bifurcación, catástrofe, Oregonator, exponente de Lyapunov, autoorganización espacio temporal, fractales. - Lea los trabajos de divulgación científica del Anexo III y responda a los cuestionarios correspondientes al final de la unidad. - Lea la guía de T.P. n 2; la parte experimental será desarrollada en la tercera clase. o TERMODINAMICA DEL EQUILIBRIO Y APARTADA DEL MISMO. La mecánica clásica newtoniana considera a las ecuaciones del movimiento como invariantes con respecto a la inversión del tiempo, por lo cual permite reconstruir la historia de un proceso retrocediendo en el tiempo mediante la inversión de su signo, t por -t. Esto sugiere una ambigua atemporalidad en la física, muy poco intuitiva frente al mundo que rodea al ser humano. La termodinámica del equilibrio introdujo en la física explicaciones para los procesos reversibles a través de la interpretación probabilística de la entropía formulada por Boltzmann. Es la solución que halla que al final de un proceso espontáneo, el sistema ha tendido al estado de mayor probabilidad. La segunda ley, en la cual t ya no es igual a -t, constituye una ley de desorganización progresiva, de pérdida de memoria, de destrucción de la información previa y de olvido de las condiciones iniciales. La entropía mide (según la visión clásica) la tendencia a la uniformidad, la probabilidad termodinámica creciente y la pérdida de organización. EN SINTESIS Si el segundo principio de la termodinámica rigiera absoluta e implacablemente, las perspectivas de futuro del Universo no serían nada alentadoras. Al igual que la piedra suspendida en el aire, el Universo habría empezado en un nivel de entropía muy bajo, correspondiente a un «orden» inicial, para llegar a la muerte térmica al cabo de un tiempo suficientemente largo. No hay modo de saber si esto es cierto o no hasta que llegue el momento fatal, puesto que se ignora si el Universo es un sistema abierto o aislado. Sí es claro, en cambio, que el segundo principio, al hacer referencia a sistemas aislados y en equilibrio, no es compatible con la descripción de los sistemas vivos, abiertos por excelencia. Si se hace la prueba de aislar un ser vivo, privándolo del intercambio de materia y energía con su entorno, por ejemplo, encerrar un pajarito en un cubo de cristal perfectamente hermético, se comprobará el sistema se dirigirá inexorablemente a su estado de equilibrio termodinámico, es decir a la muerte biológica. Para los seres vivos se necesita pues una termodinámica de no equilibrio para sistemas no aislados. Esa termodinámica tiene en cuenta casos particulares en los que sistemas abiertos pueden alcanzar una situación estable de no equilibrio llamada «estado estacionario». La situación estable es posible porque el sistema, al ser abierto, puede enviar al entorno toda la entropía que en su interior se produce y mantener así su propia entropía constante. En otras palabras, el sistema establece una suerte de pacto con el entorno, se adapta a él y permanece estable en el estado estacionario, sin avanzar hacia el equilibrio termodinámico. Cualquier perturbación fortuita que tienda a desplazarlo del estado estacionario es resistida; el 40 M.A.Perillo Biofisica-Quimica sistema, por así decir, es capaz de «absorber» esas perturbaciones azarosas (llamadas «fluctuaciones»). Las mismas no tienen pues oportunidad de progresar, de amplificarse, y por lo tanto no alteran el comportamiento del sistema. Lejos del equilibrio se presentan casos de inestabilidad, en los cuales las fluctuaciones sí pueden resultar decisivas. Lo que ocurre es que el estado estacionario compatible con las condiciones del entorno deja de ser único, situación que se expresa matemáticamente en las ecuaciones que describen la evolución del sistema: las mismas se vuelven no lineales, es decir que tienen más de una solución. Llevado esto a una gráfica (ver figura), aparecen puntos críticos, llamados bifurcaciones, donde la evolución futura del sistema deja de ser única, depende de una perturbación ínfima (antes irrelevante) y es por ende incierta: varias soluciones son posibles, pero sólo una se convertirá en realidad. ¿Cuál de ellas? Eso lo decide el azar. Ilya Prigogine ha llamado a este fenómeno «orden por fluctuaciones» Veamos el siguiente experimento ideado por Knudsen: 1 Recipiente a temperatura T1 Capilar por el cual se llega al equilibrio 2 Recipiente con T2=T1, sin diferencia de caminos libres medios moleculares con el izquierdo Recipiente refrigerado a T2, moléculas de bajo camino libre medio Recipiente a temperatura T1>>T2 Capilar por el cual se llega al desequilibrio Fig.4.1 Se estudiaron las características especiales del flujo por un capilar por el cual pasaban moléculas gaseosas(Fig.4.1). Sin alterar la temperatura (estado 1, arriba) se establecía un equilibrio y alternado las temperaturas de los dos recipientes interconectados (estado 2, abajo) se acumulaban más moléculas en el sector frío. Pese a que se trata de un sistema cerrado, al ser no-adiabático sus dos subsistemas no están en el equilibrio sino en estado estacionario de desequilibrio. Si se sigue bajando la temperatura a la derecha, se exagera el desequilibrio y la distinta influencia de las fuerzas de unión intermoleculares; debido a esto las moléculas gaseosas empezarán a sufrir una transición de fase de licuación al acceder a valores críticos. Muchos otros fenómenos de la física ocurren en el desequilibrio. Para el estado 2, el orden del gas en el recipiente frío, es mayor que en el recipiente caliente. En el estado 1, el gas muestra diS = 0 (conservación de entropía) y en el estado 2, diS > 0 (creación de entropía). Obsérvese que lo que impide el equilibrio es el permanente desequilibrio entre T1 y T2. Donde se detecta irreversibilidad en este experimento, tiene que haber creación de entropía. - xiii) Cómo define al estado estacionario? En qué condiciones puede alcanzarse? - xiv) Qué es un estado de equilibrio? Cuáles sistemas pueden alcanzar el equilibrio? - xv) Cuáles son las semejanzas y diferencias entre estado estacionario y estado de equilibrio? - xvi) En cuál de estos tipos clasificaría el estado de: a) el nivel del agua del tanque de su casa, b) un ser vivo, c) la reacción de disociación de un ácido debil en agua, d) la concentración de complejo enzimasustrato (E-S) en una reacción enzimática (E + S ===== ES ------- E + P (para más detalles ver unidad 3) 41 M.A.Perillo Biofisica-Quimica Balance de entropía S. Si el subíndice e significa flujo entre el entorno y el sistema cerrado noadiabático y el subíndice i indica generación de entropía debido a irreversibilidades, dSsistema ≥ q/T = deS ≤ dSsistema = diS + deS con diS > 0 (segunda ley) (1) donde el caso límite diS = 0 (conservación de entropía) corresponde a condiciones reversibles y el caso diS > 0 (creación de entropía) corresponde a condiciones irreversibles. Con estas últimas condiciones, se puede dar que dSsistema < 0 si tanto |deS| > diS como deS < 0 (“disipativo”) Se aclara que no hay ley física alguna que restrinja el signo de deS. Así se puede generar validamente un flujo negativo de entropía mientras los ordenamientos locales estén “robando” orden de su entorno. En el caso del estado 2 del experimento de Knudsen, el subsistema gaseoso se ordena a la derecha exportando desorden del entorno, tensando grados de libertad moleculares y eventualmente generando calor por cambio de fase (principio de Le Chatelier). Este marco conceptual acepta que dSsistema > 0, pero no alcanza a explicar otros tipos de orden que aparecen en presencia de fuertes condiciones de no equilibrio como las cadenas tróficas, el acople de reacciones exo- y endotérmicas. Predice el devenir de un organismo vivo, incluso su cerebro si lo tuviere, en los últimos momentos cuando muere y se descompone, pero no en la etapa importante de su progresiva madurez. TERMODINAMICA LINEAL DEL DESEQUILIBRIO. a. CERCANIA A UN ESTADO ESTACIONARIO El equilibrio termodinámico de un sistema cerrado es una idealización. Los parámetros termodinámicos sufren inevitables oscilaciones alrededor del equilibrio. Se las denomina fluctuaciones. En un sistema abierto, la relajación al equilibrio nunca se completa del todo. Más bien se tiende en ese caso a una relajación hacia un estado estacionario. Este presenta iguales características: es una idealización. Onsager, en la segunda década del siglo XX, planteó una nueva termodinámica de sistemas abiertos en condiciones de linealidad por cercanía a un estado estacionario. En esas condiciones los componentes no-lineales decaen y se pueden ignorar y así rige la siguiente expresión lineal para la velocidad de creación de entropía por unidad de volumen. k diS/dt = Σ Ji Xi Aparecen fuerzas impulsoras Xi y flujos entrópicos Ji generalizados. Aquí se llaman fuerzas impulsoras a los gradientes de temperatura, concentración, fuerza, etc. y flujos (por ejemplo el efecto difusotérmico a discutir) a los efectos asociados de flujos de energía, masa o cantidad de movimiento que surgen por la presencia de gradientes. A partir de sus planteos despejó -junto con otros autores como Prigogine, Ono, etc. - el principio de mínima disipación de energía (Onsager). Es aplicable a los estados estacionarios de no-equilibrio. El concepto de orden de dichos estados estacionarios expresa que el orden k está definido por el número de fuerzas impulsoras esclavizadas por ligaduras entre las n fuerzas impulsoras actuantes. Por ejemplo, si se produce un efecto térmico en la Fig. 4.1, estado 2, bajando más la temperatura T2 a la 42 M.A.Perillo Biofisica-Quimica derecha, se le conjuga al efecto térmico un efecto difusivo, generando un efecto único, un flujo “conjugado” único, denominado difusotérmico. En este caso, k vale 1; n es el número de fuerzas impulsoras totales (2), n-k el número de fuerzas impulsoras libres y k el número de fuerzas impulsoras esclavizadas. Se generan así n-k flujos que decaen al acercarse al estado estacionario. Esto siempre en condiciones alejadas con respecto a una transición de fase. Cuando hay orden cero (k = 0), se está en el caso límite de un estado estacionario que coincide con un equilibrio alejado de las transiciones de fase, motivo por el cual el estado no está sujeto a restricciones externas ni a ligaduras. Al no haber ningún flujo, la variación de la entropía vale diS = 0. El gas, Fig. 4.1, estado 1, posee todos sus eventuales grados de libertad en plena vigencia y en forma irrestricta. Cualquier perturbación microscópica que lo aleja del equilibrio, es vencida o amortiguada tensando algunos de esos grados de libertad y se relaja de nuevo hacia el caso límite, el equilibrio. Fig.4.2 Variación de la entropía del sistema en función del tiempo (Tomado de Peacocke, 1989). S: entropía total del sistema Ssist. = Si + Se Se: entropía debida a los intercambios con el entorno Si: entropía debida a los procesos irreversibles internos del sistema. P:velocidad de producción de entropía. La columna a representa a un sistema cerrado que evoluciona hacia el equilibrio termodinámico. La columna b representa un sistema abierto que evoluciona hacia un estado estacionario. El equilibrio se alcanza cuando S es máxima y P=0. En el estado estacionario S aumenta en función del tiempo a una velocidad constante y distinta de cero (mínima velocidad de producción de entropía. b. CERCANIA A UNA TRANSICION DE FASE. En condiciones cercanas a una transición de fase, resulta 0< k ≤ n (ya sea clásica o de Onsager). Algunos de los grados de libertad ya están tensados por esa cercanía y sin embargo, los cálculos indican que las perturbaciones o fluctuaciones pequeñas alrededor del estado estacionario, si aparecen, no progresan ni trascienden macroscópicamente. En cambio, se relajan hacia el estado estacionario. Estos estados estacionarios muestran, con su mínima creación de entropía, que el sistema no se halla en la situación de evolucionar hacia condiciones aún más entrópicas, ya que no existen en sus cercanías. 43 M.A.Perillo Biofisica-Quimica Este análisis es válido sólo para condiciones de Onsager (sistema lineal) cercanas al equilibrio, situaciones donde se verifica una dependencia lineal entre las fuerzas impulsoras libres y los flujos conjugados. El intento de explicar los sistemas complejos del segundo párrafo (más arriba) con esta ayuda tiene el límite recién mencionado, ya que en su contexto no aparecen predicciones sobre otras transiciones de fase en pleno desequilibrio. La termodinámica lineal del desequilibrio sigue siendo gobernada por el mismo “principio de orden de Boltzmann” de la termodinámica clásica, que se puede interpretar así: en el desorden máximo también surge un cierto “orden”, ya que todo el sistema está uniformemente desordenado y casi todos los grados de libertad microscópicos se hallan activos en el estado estacionario. En el equilibrio, todos. TERMODINAMICA NO-LINEAL DEL DESEQUILIBRIO. La termodinámica clásica está muy fuertemente condicionada por el hecho de estar restringida al equilibrio, fuera del cual solamente rige la cinética. La termodinámica de Onsager está muy fuertemente restringida porque las fluctuaciones no- lineales decaen y se pueden ignorar cerca del equilibrio, lo cual no es cierto lejos de él. A partir de ciertos valores críticos del apartamiento con respecto al equilibrio, las ecuaciones cinéticas pasan a ser no-lineales. Aparecen soluciones múltiples. Hay un nuevo principio de orden, diferente del “orden” de Boltzmann: principio denominado “orden por fluctuaciones” propuesto por la escuela belga de Prigogine. En el “orden” de Boltzmann, que garantice la aparición del desorden generalizado, las perturbaciones/fluctuaciones provocan transición de fase del equilibrio cuando el sistema se halla en sus cercanías o provocan atenuación en la dirección de alguna transición de fase del equilibrio cuando el sistema no lo está. Se comparan ahora estas nuevas fluctuaciones en pleno desequilibrio con las anteriormente descriptas. Difieren en que las nuevas no muestran posibilidad matemática de autoatenuarse si están lejos de una transición de fase del desequilibrio. Como antes, provocan una transición de fase del desequilibrio si están en sus cercanías y varían también en su dirección. El orden de magnitud de estas fluctuaciones supera al de las fluctuaciones atenuables de Onsager. Las de Onsager son fluctuaciones cercanas al equilibrio, por ejemplo las fuerzas impulsoras locales que establecen un pequeño valor por encima de 0ºC que permiten que funda el hielo, en contacto con agua líquida. Las aquí mencionadas son fluctuaciones importantes por presencia de una mayor energía ambiental. Cerca de una transición de fase del desequilibrio en que ha de aparecer un nuevo orden (por ejemplo, con predictibilidad mayor de la ubicación de partículas en el espacio) se aprecia una paulatina coherencia colectiva de trayectorias. o SISTEMAS COMPLEJOS UN SISTEMA DINÁMICO, COMPLEJO, ALEJADO DEL EQUILIBRIO Y NO LINEAL PUEDE EVOLUCIONAR HACIA LA AUTOORGANIZACIÓN ESPACIAL Y TEMPORAL. LOS DIAGRAMAS DE FASES Cómo se representa la dinámica de un sistema en forma geométrica. El sistema dinámico está caracterizado por dos elementos: un estado y una dinámica. El primero representa las variables que lo caracterizan y el segundo es una regla que describe la forma en que el estado evoluciona, por ejemplo con el tiempo, para lo cual los científicos utilizan una construcción en un espacio matemático llamado de fases. La Fig.4.3 muestra el diagrama de fases que representa el movimiento de un péndulo que se balancea en ausencia de fricción.en el cual las variables son posición (L) y velocidad (V). El 44 M.A.Perillo Biofisica-Quimica péndulo sigue una órbita o camino en el espacio de fases bidimensional. Después de ser lanzado, la oscilación infinita describe una trayectoria que tiene forma de elipse Fig.4.3 Movimiento de un péndulo SIN fricción en el espacio de fases: V = velocidad, L = posición. El péndulo sigue una órbita elíptica. La elipse se llama ciclo límite y es un atractor Después de cierto tiempo, el conjunto de puntos iniciales contenidos en el cuadrado señalado en la figura se vuelve a encontrar dentro de otro cuadrado de área idéntica al primero. Ahora supongamos que el péndulo oscila experimentando una resistencia a su movimiento, por ejemplo, la que ocasiona la fricción de sus partes con el aire. Esto produce que al cabo de un tiempo pierda su impulso y termine por detenerse; en el espacio de fases la dinámica quedará descrita por una espiral, la cual se va contrayendo sobre sí misma hasta terminar en un punto fijo (un atractor), cuando llega al equilibrio. Fig. 4.4 Movimiento de un péndulo CON fricción en el espacio de fases. V = velocidad, L = posición. En los dos casos, el comportamiento de los sistemas es predecible cuando la amplitud del movimiento es moderada. El espacio de fases puede tener más de dos dimensiones y en esos casos la dinámica del sistema es mucho más compleja y aparecen los atractores llamados extraños. ATRACTORES SORPRENDENTES: ver más adelante. i) ii) Qué es un atractor? Qué tipo de atractores conoce? 45 M.A.Perillo TEORÍAS DEL CAOS – Biofisica-Quimica Ciclones y mariposas “Una mariposa aletea en la selva amazónica y pone en marcha sucesos que terminarán produciendo, algunos días después, un ciclón en el Caribe.” De acuerdo a la mecánica newtoniana, el mundo es un gigantesco mecanismo regido por «leyes naturales» eternas e inmutables. Esas leyes, que se expresan mediante ecuaciones matemáticas (las ecuaciones del Edward Lorenz movimiento), determinan que en circunstancias idénticas resulten siempre cosas idénticas, y si las circunstancias cambian ligeramente, el resultado cambiará también en forma proporcionalmente pequeña. Esta última propiedad se verifica fácilmente, por ejemplo, al disparar un proyectil: si se apunta una fracción de milímetro más abajo o más arriba, la diferencia en la trayectoria será igualmente minúscula y no impedirá que el tirador dé en el blanco. Pero esto no ocurre siempre: en muchos casos una variación ínfima en las condiciones iniciales puede amplificarse dramáticamente a medida que avanza el tiempo. Esta característica, propia de muchos sistemas dinámicos de cualquier naturaleza (físicos, químicos, biológicos, etc.), se llama precisamente sensibilidad a las condiciones iniciales. Se comprende sin dificultad los aprietos en que este hecho pone a la mecánica clásica diseñada por Sir Isaac y otros: el comportamiento futuro de esa clase de sistemas deja de ser predecible, las ecuaciones de Newton ya no son capaces de indicar qué pasará con un sistema semejante en un momento dado. Esos sistemas, sensibles a las condiciones iniciales, presentan lo que se ha llamado un comportamiento caótico. Laplace dio sin nombrarlo una definición sintética del determinismo: debemos considerar, decía, «el estado presente del Universo como el efecto de su estado anterior, y como causa de su estado futuro». En otras palabras, dado el estado de un sistema en un instante preciso, para cada uno de los momentos anteriores o ulteriores hay un único estado de ese sistema compatible con el primero. Sin duda esto implica que la totalidad de su historia podría ser reconstruida o predicha conociendo todos los datos relativos a un instante cualquiera de esa historia. La expresión «caos determinista» con que se designa a este tipo de comportamientos de un sistema no hace, en definitiva, sino dar cuenta de esa reconciliación entre lo impredecible y lo determinado. No es un asunto menor, sobre todo si se considera que implica reconocer que hay muchos fenómenos en la naturaleza que son, a la vez, transparentes y opacos para el conocimiento humano. El tiempo irreversible Una de las curiosidades de las leyes naturales es que en ellas el tiempo no tiene una dirección definida: tanto da si el tiempo avanza o retrocede, y por lo tanto los procesos resultan perfectamente reversibles. Obsérvese, en particular, la segunda ley de Newton, donde la fuerza resulta del producto de la masa por la aceleración: F = m x a. La aceleración es, a su vez, velocidad sobre tiempo (v/t), y la velocidad es distancia sobre tiempo (d/t). La aceleración es, en consecuencia, distancia sobre tiempo al cuadrado (d/t²), y la segunda ley de Newton puede escribirse entonces: F = m x d/t². La variable tiempo (t) se halla pues elevada al cuadrado, lo que significa que cualquiera sea su signo, positivo o negativo, el resultado no varía. Dicho de otro modo, la trayectoria del movimiento es idéntica en una dirección o la otra. Conocidas las condiciones iniciales de un sistema -su estado en un instante cualquiera-, las ecuaciones del movimiento proporcionan una trayectoria única y permiten reconstruir toda su historia y todo su futuro. Es como si se filmara un péndulo de movimiento perpetuo: no se puede saber en qué sentido se pasa la película. Pero la intuición o la experiencia personal indican otra cosa: el tiempo no es reversible. Algo aparentemente tan trivial como la mezcla de agua con tinta roja lo muestra claramente; de nada sirve seguir revolviendo la mezcla con la esperanza de que el agua y la tinta vuelvan a separarse espontáneamente. El proceso es irreversible. El tiempo tiene una dirección o, como se dice 46 M.A.Perillo Biofisica-Quimica también, una «flecha». La «flecha del tiempo» fue introducida en física por el segundo principio de la termodinámica (ver Unidad 1) Hasta aquí, la vida del sistema sigue siendo previsible y tranquila. Pero esto es así siempre y cuando el sistema no se halle demasiado alejado del equilibrio termodinámico. En caso contrario, las cosas cambian radicalmente. Bifurcaciones Lejos del equilibrio se presentan casos de inestabilidad, en los cuales las fluctuaciones sí pueden resultar decisivas. Lo que ocurre es que el estado estacionario compatible con las condiciones del entorno deja de ser único, situación que se expresa matemáticamente en las ecuaciones que describen la evolución del sistema: las mismas se vuelven no lineales, es decir que tienen más de una solución. Llevado esto a una gráfica (ver Fig.4.11), aparecen puntos críticos, llamados bifurcaciones, donde la evolución futura del sistema deja de ser única, depende de una perturbación ínfima (antes irrelevante) y es por ende incierta: varias soluciones son posibles, pero sólo una se convertirá en realidad. ¿Cuál de ellas? Eso lo decide el azar. El nombre de «bifurcación» dado a esos puntos críticos expresa bien la situación: se llega a un estado de incertidumbre, donde varias sendas se abren y no es posible saber de antemano cuál de ellas habrá de ser seguida por el sistema. Lo que ocurre en una bifurcación recuerda la situación de sensibilidad a las condiciones iniciales: basta con apartarse una distancia tan débil como se quiera de la bifurcación para ser precipitado en una dinámica que se aleja para siempre de la misma. Es algo así como encontrarse en el ojo de un ciclón: en ese lugar reina la calma, la armonía; pero un mínimo apartamiento significa ser devorado por la turbulencia. Según Jorge Wagensberg «entre dos bifurcaciones reinan las leyes deterministas, pero en la inmediata vecindad de tales puntos críticos reina el azar. CATÁSTROFES EN REACCIONES QUÍMICAS En la segunda mitad del siglo 20 tuvo lugar un cambio distintivo en el pensamiento científico. Este cambio estuvo asociado con la aparición de nuevos conceptos en ciencia que involucraban el estudio integral (global) de los problemas considerados, en contraste el método de análisis derivado de Descartes (dividiendo el problema en partes). Estas nuevas teorías fueron: • Teoría de los sistemas (Ludwing von Bertalanffy) • Cibernética (Norbert Wiener) • Sinergética o teorías de la bifurcación (teoría de los fenómenos cooperativos) (Herman Haken) • Teoría de catástrofes (René Thom) Todas estas teorías enfatizan el carácter no lineal de los fenómenos que examinan. Anteriormente se acostumbraba a linealizar las respuestas a los fines de encontrar la descripción de los fenómenos no lineales, probablemente debido a la falta de métodos matemáticos apropiados. Los primeros problemas no lineales fueron estudiados en el campo de la hidrodinámica y de la relatividad general, los cuales produjeron un avance notable en la teoría de ecuaciones diferenciales no lineales. Posteriormente se desarrollaron nuevas herramientas matemáticas por medio de las teorías de solitones, atractores extraños y teoría de fractales. Catástrofe: fenómeno consistente en la pérdida de estabilidad de un estado otrora estable de un sistema, seguido por la rápida transición hacia otro estado estable del sistema bajo las nuevas condiciones. • La pérdida de la estabilidad es consecuencia del cambio lento y continuo de los parámetros que determinan el estado del sistema • La transición hacia el nuevo estado es generalmente abrupta Prigogine comenzó con el estudio de reacciones químicas en condiciones alejadas del equilibrio termodinámico. Él descrubrió la aparición de estructuras espaciales y oscilantes en sistemas homogéneos y la generación de estados turbulentos en reactores de flujo. Estos resultados son 47 M.A.Perillo Biofisica-Quimica de vital importancia para las teorías de reactores químicos y para la investigación de organismos vivos, y forman la base de la dilucidación teóricas de los problemas relacionados con la diversificación de tejidos y la explicación de relojes biológicos. PROBLEMAS FILOSÓFICOS ASOCIADOS A LA TEORÍA DE CATÁSTROFES La descripción de la realidad en la teoría de catástrofes está limitada a algunas formas de cosas y de fenómenos. • describe aquellos fenómenos cuya forma es resistente a perturbaciones, es decir: estructuralmente estable • describe cambios en la forma que tienen lugar como consecuencia de la variación continua en los parámetros de control • enfatiza cambios en la forma que sean cualitativos y estructuralmente estables Esto se relaciona con el “concepto” de Platón y la “forma” de Aristóteles. Según Platón el concepto básico es una idea y las cosas son imitación de las ideas. Según Aristóteles materia y forma existen combinadas en una entidad. La forma es un componente esencial y permanente de las cosas, es cognoscible. La idea de que los cambios cualitativos en el sistema son debido a lentos cambios cuantitativos en los parámetros de control se relaciona con Hegel y Engels. Según Hegel en el cambio de fase del agua, que él analiza, temperatura y presión son los parámetros de control y densidad es el parámetro de estado factible de ser medido. Según Engels “las constantes físicas son nada más que nombres de puntos nodales en los cuales cambios cuantitativos causan cambios cualitativos en el estado de un sistema; en los puntos nodales cantidad se vuelve cualidad. En la teoría de catástrofes el nodo en el que ocurre esa transición corresponde al conjunto de catástrofe (conjunto de valores de los parámetros de control) en el cual el sistema es sensible (susceptible a perturbaciones). El conjunto de catátrofe divide los estados del sistema; la transición entre esos estados constituye la catástrofe. TEORÍA DE CATÁSTROFES EN QUÍMICA En años recientes, se han descripto numerosas reacciones químicas que exhiben una dinámica compleja. La reacción de Belousov-Zhabotinskii (BZ), fue descubierta por Belousov en 1950. Esta reacción implica la oxidación de ácido cítrico por bromato en medio ácido, en precencia de Br- y Ce(IV)-Ce(III) como catalizadores. Belousov observó oscilaciones en las concentraciones de algunos componentes (Br-, Ce(III), Ce(IV), a pesar de la homogeneización de la mezcla de reacción. Zhabotinskii le agregó un indicador (ferroína: FeSO4 + o-fenantrolina) lo cual permitió visualizar las oscilaciones con una claridad marcadamete superior. En la reacción de BZ es posible observar varios estados dinámicos de interés: • estado de equilibrio, • estado periódico temporal (oscilaciones en la concentraciones en función del tiempo), • estado periódico espacial, (oscilaciones en la concentraciones en función del espacio) • estado estacionario (estructuras disipativas), • estado periódico espacio-temporal (ondas químicas propagándose) • estado turbulento (oscilaciones caóticas, estructuras espaciales estocásticas, ondas químicas estocásticas). Estas periodicidades espaciales o temporales, en un comienzo fueron atribuidas a inhomogeneidades (partículas de polvo, burbujas de gas liberado, agitación insuficiente de la solución), suponiendo que en sistemas verdaderamente homogéneos esto no podía ocurrir. Esta opinión provenía del paradigma profundamente enraizado en la química que considera que: “los sistema se aproximan al estado de equilibrio monotónicamente, debido a que esto resulta del requerimiento termodinámico de la disminución de la energía libre del sistema como un todo”. Este concepto cambió a partir de los resultados sobre estudios de sistemas alejados del 48 M.A.Perillo Biofisica-Quimica equilibrio; de acuerdo con la segunda ley de la termodinámica la aparición de orden bajo condiciones de homogeneidad sólo puede tener lugar en sistemas abiertos. Estructuras no homogéneas pueden aparecer satisfaciendo el requerimiento termodinámico de la disminución monotónica en la energía libre del sistema como un todo, dado que éstas aparecen como resultado de reacciones colaterales. Por medio del cambio en las condiciones iniciales, el sistema puede pasar desde el estado estacionario hacia cualquiera de los estados mencionados; otras transiciones también son permitidas. La mezcla de reacción es el sistema; el estado del sistema puede ser representado por las variables de estado; otras variables, llamadas parámetros de control son modificadas continuamente y un cambio en el estado del sistema es examinado. Un cambio abrupto en el estado del sistema ocurre para algunos valores de los parámetros de control. Por ejemplo: un cambio continuo en la concentración de uno de los componentes del sistema puede inducir un cambio desde el estado estacionario hacia un estado oscilatorio. MODELO SIMPLIFICADO DE LA REACCIÓN DE BELOUSOV-ZHAVOTINSKII “EL OREGONATOR” Las cinco reacciones más importantes en el modelo de Fields-Körös-Noyes para el mecanismo de la reacción BZ son las siguientes: BrO3- + Br- 2H+ HBrO2 + HOBr (determina velocidad de la reacción) (A) HBrO2 + Br- + H+ 2HOBr 2Ce3+ + BrO3- + 3H+ 2Ce4+ H2O + 2HBrO2 (B) (producción autocatalítica de HBrO2; © no es reaacción elemental) BrO3- 2HBrO2 4Ce4+ + BrCH(COOH)2 + 2H2O A + Y X + P X + Y 2P A + X 2X + 2Z 2X A + P Z fY + + HOBr + H (D) (determina aumento de [HBrO2] 4Ce3+ + Br- + HCOOH + CO2 + 5H+ (E) A = BrO3X= HBrO2 P= HOBr Y= BrZ= Ce4+ En este modelo se considera que la reacción BZ transcurre en una solución agitada, a T y P cte, en un sistema cerrado, que los cambios en [BrO3-] son despreciables y que [HOBr] no afecta la cinética de la reacción. El ácido bromoso (HBrO2) es producido autocatalíticamente y su aumento es limitado por la reacción de desproporción (D). 49 M.A.Perillo Biofisica-Quimica Fig. 4.5 Estructuras de autoagreación espacial en la reacción de BelousovShabotinsky. DE LA OSCILACIÓN AL CAOS La estrategia para alejar la reacción del equilibrio consiste en realizarla en un sistema abierto, es decir, en el que se intercambia materia y energía con el medio circundante. Para ello es necesario modificar el sistema de reacción, para lo cual se alimenta en forma independiente y controlada los reactivos, que llegan a un dispositivo en el cual se mezclan y reaccionan, mientras que paulatinamente se extraen los productos de la transformación. La alimentación continua de reactivos y la extracción simultánea de la mezcla de reacción impiden que el sistema alcance el equilibrio. Además, al alimentar los reactivos en forma independiente es posible modificar las concentraciones individuales de cada uno de los participantes y también, si se varía la velocidad de alimentación al reactor, se altera en forma controlada el tiempo en el cual los reactivos están en contacto entre sí, llamado el tiempo de residencia o de contacto. El análisis cuantitativo de lo que sucede en el reactor se lleva a cabo introduciendo diminutos electrodos sensibles a la variación de las concentraciones de las especies de cerio en sus estados oxidado y reducido, así como otros que son sensibles a la concentración del ion bromuro. Mediante experimentos con diversas condiciones iniciales se obtienen datos sobre la dinámica del sistema. En la Fig.4.6a se grafica en el eje horizontal el tiempo de reacción y en el vertical el valor del potencial medido con el electrodo: en el caso de la figura a), el flujo total de reactivos es de un mililitro por minuto, en el caso b) es de 4.0. 50 M.A.Perillo Biofisica-Quimica Fig. 4.6a Oscilaciones de la reacción B- Z en función del flujo de reactivos, a) 1 ml/minuto; b) 4.0 ml/minuto. Resultados experimentales de K Graziani, J. Hudson y R. Schmitz de la Universidad de Illinois, EUA, 1977 obtenidos al variar el tiempo de residencia; con un electrodo de platino se midió en forma continua el cambio en el potencial de óxido- reducción de la reacción y con ello se obtuvo una estimación de la relación de iones cerio (III) / (IV). De los resultados experimentales es evidente que para unas condiciones se presentan oscilaciones regulares, mientras que en otras las oscilaciones son mucho más complejas. Los autores de este trabajo. Se había demostrado la extrema sensibilidad del comportamiento de la reacción a las condiciones iniciales. Pequeñas modificaciones en las mismas hacían oscilar la reacción con periodos diferentes. Se demostró, por otra parte, la presencia de duplicaciones periódicas, así como de intermitencia del tipo I que, como recordamos en la ecuación logística, son rutas hacia el caos. Además, Graziani y colaboradores demostraron más adelante que bajo ciertas condiciones de reacción, las oscilaciones se ven interrumpidas abruptamente y la reacción entra en un estado no oscilatorio. Por último, una prueba adicional de la presencia del caos resultó cuando se comprobó la existencia de un atractor extraño. En la Fig.4.6b se muestra la simulación que se obtuvo por computadora a partir de datos experimentales, empleando el esquema reaccional antes mencionado. 51 M.A.Perillo Biofisica-Quimica Fig. 4.6b El atractor extraño de la reacción B- Z Diagrama de fases y en el que se grafican los valores en escala logarítmica de tres de los componentes del sistema: el ácido bromomalónico (g) el ion cerio-IV (b) y el ion bromuro (m). Si el sistema tuviese un comportamiento periódico, sería de esperarse que el atractor generado fuese de ciclo limite, pero éste no es el caso ES CAOS DETERMINISTA O AZAR? Hoy en día se ha llegado a la conclusión de que una ley puramente determinista puede manifestarse por fenómenos totalmente aleatorios. Esta característica está dada por el carácter no lineal de las ecuaciones matemáticas que modelan el sistema físico. Dado que estas ecuaciones no permiten una solución analítica exacta los científicos han tenido dificultades para construir teorías que permitan su predicción. Mucho se ha avanzado gracias al uso de las computadoras, que han permitido al matemático explorar e identificar pautas de comportamiento indispensables. Esta nueva aproximación, en la que interviene una combinación de “experimentos” numéricos y de análisis matemático, ha dado origen a un campo llamado dinámica no lineal. Quienes trabajan en él usan el término caos para referirse al comportamiento irregular e impredecible, aunque determinista, de los sistemas no lineales. Un primer ejemplo de un sistema no lineal, muy sencillo, que exhibe una transición de un comportamiento regular a uno caótico es el generado por la llamada ecuación logística. Para profundizar en ella sólo necesitamos papel, lápiz y una calculadora sencilla. El primer paso para desarrollar la ecuación consiste en definir una variable X; para aquellos que no les gusta trabajar con una simple letra, podemos sugerir que asocien la letra con una idea menos abstracta, como una población de insectos, la cantidad de una sustancia que reacciona en un vaso o bien, si se desea, el número de personas de una población que han oído un chiste (el lector podrá darle a X el sentido que su imaginación le dicte). Así, el sistema está descrito por una sola variable X, es decir que para definir toda la evolución del mismo, bastará conocer el valor que toma X en cierto instante dado. Para simplificar aún más las cosas diremos que la variable tiempo sólo puede tener valores enteros. El modelo deberá entonces ser capaz de predecir el valor de Xsig a partir del último calculado, es decir, el valor de X; veamos cómo sucede esto: Xsig = K (l - X) X (ecuación 1) Xsig será el siguiente valor calculado a partir de X; K es una constante que será detallada más, adelante. Supongamos que X vale 0.04 y K, 2.7. Calculemos el valor de Xsig: 52 M.A.Perillo Biofisica-Quimica Xsig= 2.7 (1 - 0.04) 0.04 el resultado es 0.10368. Si ahora repetimos la operación, pero empleando X =0.1, el resultado será 0.2430 y así continuamos resolviendo la ecuación, aumentando en cada caso el valor de X hasta llegar a 1. Se grafica en el eje horizontal los valores de X empleados y en el vertical los que resultan de estas simples operaciones. En la Fig.4.7 se muestra el resultado de nuestro cálculo. Esta ecuación, descrita por P. Verhulst en el siglo pasado, fue propuesta para modelar el crecimiento de una población de insectos en un medio ambiente limitado. La gráfica nos indica que cuando la densidad de población es inicialmente pequeña, las generaciones siguientes serán más numerosas, pero sólo hasta un límite, a partir del cual el crecimiento disminuye. La sobrepoblación, el incremento en la mortalidad, la competencia por el alimento y muchos otros factores hacen que la población disminuya, como se observa en la presencia de valores altos de X. Se asume que la población no sigue un crecimiento exponencial incontrolable, como lo había predicho el economista inglés T. Malthus en 1798, basándose en una ecuación del tipo: Xsig =K X (ecuación 2) De acuerdo con su modelo, Malthus predecía que si no se ponía un “freno moral” (matrimonios aplazados y continencia sexual) no tardaría mucho para que la humanidad agotara la cantidad de alimentos disponibles. La ecuación 2 describe un crecimiento lineal, mientras que la ecuación 1 genera un arco parabólico que empieza a elevarse desde el origen, llega a un máximo y desciende simétricamente hasta alcanzar nuevamente el cero. La constante K hace que la altura del máximo se modifique, hecho que se puede demostrar si graficamos la ecuación empleando un valor diferente de K. Fig. 4.7 Forma típica de la relación entre X y Xsig, descrita por la ecuación logística, K = 2.7. Hagamos una pequeña modificación a nuestro cálculo: con el mismo valor de K = 2.7, usemos inicialmente el de X =0.04. El valor de Xsig que se obtiene se sustituye como nueva X y se obtiene otra Xsig; repitamos la operación sesenta veces (en matemáticas a esto se le llama iterar la ecuación). Veamos cómo se obtienen los dos primeros valores: 53 M.A.Perillo Biofisica-Quimica El primer resultado es 0.10368, como ya lo vimos antes; ahora calculemos con él el siguiente: Xsig= 2.7 (1-0.10368) 0.10368 que nos produce 0.25091. A partir de la iteración número dieciocho el producto es 0.6296. Si se da cualquier otro valor inicial entre cero y uno y se itera, se obtendrá el mismo valor final 0.6296. La Fig.4.8 muestra una gráfica de las operaciones realizadas al iterar la ecuación; en el eje vertical están dispuestos los valores de X y en horizontal los de Xsig. Una manera sencilla de realizar las iteraciones sin necesidad de calcularlas con una máquina, la presentamos en esta misma Fig.4.8 Hemos trazado una recta punteada que parte del origen, cuya expresión equivale a X = Xsig. Supongamos que partimos del valor inicial 0.04; si trazamos una línea vertical hasta tocar la curva y luego una horizontal que toque la recta punteada, tendremos el valor de Xsig y de esa manera repetimos la operación tal y como se describe mediante los trazos con flecha realizados en la figura. Como se puede observar, se llega a un punto fijo en el cual convergen todas las trayectorias generadas por las iteraciones. Figura 4.8 Iteración de la ecuación logística para K = 2.7. Presencia de un atractor. En la Fig.4.9 se muestra el mismo desarrollo de la ecuación descrita, salvo que hemos modificado el valor de K a 3.15. En este caso el punto fijo deja de ser estable y a pesar de que inicialmente atrae las órbitas hacia él, se genera una bifurcación en la que aparecen dos puntos estables (marcados como X1 y X2 en la gráfica) que, independientemente del valor inicial escogido, “atraen” todas las órbitas que se generen por la iteración de la ecuación. Debemos aclarar que en dinámica se llama bifurcación al cambio en el número de soluciones posibles para una ecuación cuando se varía un elemento, en este caso, el valor de K. Hagamos otro análisis de la ecuación empleando ahora K = 3.53. Los resultados están representados de una manera diferente en la Fig.4.10; ahora en el eje horizontal está anotado el número de iteraciones de la ecuación y en el vertical el valor resultante de la variable X. El periodo dos se ha bifurcado y ahora existe un ciclo de cuatro valores. En lugar de continuar describiendo el comportamiento de la ecuación para diferentes valores individuales de K, presentamos en la Fig.4.11 una vista 54 M.A.Perillo Biofisica-Quimica general del modelo por medio de un diagrama de bifurcación en el cual en el eje horizontal se disponen los diferentes valores de K de 3.5 hasta 4 y en el eje vertical los de Xsig normalizados de cero a uno; cada valor de la figura se ha obtenido iterando la ecuación más de cien veces. La gráfica de la dinámica irregular de la ecuación logística nos da una imagen del caos. Como muchos de los sistemas no lineales que existen en la naturaleza, el modelo matemático exhibe un comportamiento que parece azaroso a pesar del hecho de que las ecuaciones que describen su comportamiento son enteramente deterministas. Los resultados nos enseñan que si K se sitúa entre uno y tres, pensando en el modelo de crecimiento poblacional de los insectos, los valores iniciales evolucionan a una población que alcanza un equilibrio. Figura 4.9 Iteración de la ecuación logística para K= 3.15. Presencia de dos atractores con valores Xl y X2. Figura 4.10 Valores de X para 60 iteraciones de la ecuación logística para K = 3.53. Ciclo de cuatro valores. 55 M.A.Perillo Biofisica-Quimica LAS RUTAS DEL CAOS Al incrementarse K entre 3 y 4 la dinámica cambia en forma notable y aparecen bifurcaciones de orden 2 que son reemplazadas por ciclos en los que se alternan cuatro valores, que luego serán de 8, 16, 32, 64. Este proceso, al que se le suele llamar duplicación periódica, es una secuencia que antecede el periodo caótico y también se le denomina bifurcación en forma de tenedor. Este mecanismo de duplicación periódica ha sido muy estudiado ya que representa una de las rutas hacia el caos y es común en muchos sistemas dinámicos reales. En la Fig.4.11 se observa que las regiones de comportamiento caótico se ven interrumpidas por intervalos de comportamiento periódico, uno de ellos se presenta cuando el valor de K es 3.569, otro mucho más prolongado ocurre en las vecindades de K 3.83 pero aquí se presenta un ciclo de tres valores estables al inicio del periodo, que permanece durante un corto lapso para luego desencadenar nuevamente un comportamiento caótico. Un esquema de este mecanismo se muestra en la figura 6. Este último proceso en cual las bifurcaciones son impares representa otra de las rutas hacia el caos y se le conoce como intermitencia del tipo I, fenómeno que posteriormente estudiaremos en sistemas dinámicos naturales. Fig. 4.11 Diagrama de bifuraciones de la ecuación logística cuando K varía de 3.5 a 4. En el eje horizontal se encuentra el valor de K, en el vertical los de X. Figura 4.12 Detalle del diagrama de bifuraciones en la ecuación logística para el ciclo de periodo tres. 56 M.A.Perillo Biofisica-Quimica EL CAOS ¿Qué pasa cuando el valor de K es mayor de cuatro? La situación cambia en forma importante. Considérese la Fig.4.13, en la cual gráficamente presentamos los resultados de la ecuación para cinco valores de K que van desde 4.0001 a 4.0005 con un valor inicial de X = 0.4. En el eje horizontal se representa el número de iteraciones que llegan hasta 200 y en el vertical el valor siguiente de X. Como se observa, cuatro de las cinco ecuaciones se salen rápidamente de la escala negativa en las primeras 200 iteraciones, pero el orden que siguen es totalmente inesperado. La primera que se sale de la escala es la que tiene el valor de 4.0004, luego sigue la de 4.0003, y después la de 4.0005. En la iteración 200 sólo la que emplea el valor de K = 4.0002 sigue en el rango entre cero y uno, hemos entrado en el régimen caótico en el cual la periodicidad es difícil de predecir. Volvamos un momento al ejemplo del biólogo que sigue el crecimiento de la población de insectos: supongamos que cada iteración de la ecuación representa un año, por tanto, sólo uno de los sistemas representados en la figura anterior será estable por dos centurias, lo cual deja más que satisfecho a nuestro científico con su predicción. Sin embargo, si cada iteración representa una semana, el periodo de estabilidad deducido será mucho menor. Fig. 4.13 Iteración de la ecuación logística para cinco valores de K mayores de cuatro. Xinicial: 0.4 EL NÚMERO 4.669 201 609 102 990 9... Una conclusión interesante que resulta del estudio de la “inocente” ecuación logística es que las bifurcaciones periódicas son una de las rutas hacia el caos, pero lo notable de este hecho es que dicho mecanismo es válido para cualquier ecuación que tenga un solo valor K. La universalidad de este proceso se reproduce sin importar la física detallada o la descripción del modelo teórico que se estudie. Más aún, la dinámica de fenómenos que transitan de la estabilidad al caos por el mecanismo de la bifurcación se realiza de una manera que puede evaluarse cuantitativamente, lo cual fue descubierto por M. Feigenbaum entre otros, un físico estadounidense que analizó los datos de la famosa ecuación con su calculadora. 57 M.A.Perillo Biofisica-Quimica Hagamos un esquema para entender lo que Feigenbaum encontró, véase la Fig.4.14. En la parte superior del diagrama hemos anotado un eje de valores para X que va de cero a uno, tal como lo hicimos con la ecuación logística. Los símbolos K1 hasta K6 representan valores de la constante en los cuales se sucede una bifurcación. Si vemos la secuencia de arriba hacia abajo se nota que cada punto es reemplazado por dos “gemelos”. Según el primer número universal descubierto por Feigenbaum, la distancia entre los números gemelos es 2.5029078750958...veces más pequeña que la que existe entre los puntos que les dieron origen. Cada secuencia de puntos que se origina una línea abajo reproduce el modelo global de la que le antecede, salvo que están más juntos. Feigenbaum encontró además que el cociente de diferencias en los valores de K requeridos para llevarse a cabo una bifurcación, por ejemplo (K3 - K2 / K4 - K3), es constante y vale 4.66201... para cualquier sistema no lineal unidimensional. Más adelante veremos que en los fenómenos físicos reales el paso a la dinámica caótica se lleva a cabo por un mecanismo similar al descrito teóricamente por las observaciones de Feigenbaum. Figura 4.14 Bifurcaciones periódicas de la ecuación logística. TODO DEPENDE DEL INICIO Una conclusión muy importante que se deriva del estudio del caos fue claramente descrita por Henri Poincaré en 1908. Este notable matemático francés, empleando las herramientas del cálculo, escribió la siguiente conclusión de sus trabajos sobre las ecuaciones que describen la evolución temporal de varios sistemas: “una causa muy pequeña, que se nos escapa, determina un efecto notable que no podemos ver y decimos entonces que tal efecto se debe al azar.” Al referirse a la definición de las condiciones iniciales que deben emplearse (por ejemplo, en nuestro caso sería el valor dado a X en la ecuación logística), nos dice que por mas que se trate de precisarlas, éstas serán siempre aproximadas y: [...] puede suceder que pequeñas diferencias en las condiciones iniciales engendren unas aún mayores en el fenómeno final, un pequeño error en las primeras produciría uno enorme sobre las segundas. La predicción se vuelve imposible y tenemos el fenómeno fortuito. Para ver en la práctica lo dicho por Poincaré debemos recurrir nuevamente a nuestra ecuación. Empleando una K = 4.00 se calculan los resultados de las iteraciones con dos valores iniciales ligeramente diferentes entre sí, por ejemplo X = 0.4000 por una parte y X = 0.4001 por la otra. (El cuadro de la página siguiente muestra algunos de los valores que se obtienen.) 58 M.A.Perillo Biofisica-Quimica Para la sexta iteración la diferencia ha llegado al cuarto decimal, pero en la 21 está ya en el primero, es decir que cualquier efecto, no importa lo pequeño que sea, alcanza proporciones macroscópicas y la diferencia en los resultados crece exponencialmente. Una manera de obtener una medida de la sensibilidad de un sistema a las condiciones iniciales fue descrita por el matemático ruso A. M. Lyapunov (1857-1918). Supongamos que partimos de la ecuación logística dando dos valores iniciales que difieren muy poco entre sí, por ejemplo, X y X + ε, e iteramos la ecuación n veces. Lyapunov encontró que la divergencia puede caracterizarse aproximadamente por una fórmula que nos dice que el valor de la diferencia iterada n veces es aproximadamente igual a ε enλ. Núm. iteraciones 0 6 21 Valores de Xsig 0.4000 0.025466 0.774403 0.40001 0.025261 0.554030 Diferencia -0.000010 0.000205 0.220373 La letra λ es conocida como el exponente de Lyapunov, y nos da una velocidad promedio a la cual se separan las dos trayectorias. Si el exponente es negativo, las trayectorias poco separadas en el inicio tenderán a converger y la evolución no será caótica. Por el contrario, si el exponente es positivo las trayectorias divergen y la evolución es sensible a las condiciones iniciales y por tanto es caótica. En la Fig.4.15 el exponente se gráfica como función del valor K de la ecuación logística para valores entre 2.8 y 4.0. Si comparamos esta figura con la número 4.11 observamos que el signo del exponente se correlaciona muy bien con el comportamiento del sistema, es decir, que los valores positivos corresponden a las bifurcaciones, mientras que los negativos se asocian al comportamiento periódico en la región entre 3.5 y 4 del valor de K. Figura 4.15 El exponente de Lyapunov vs. K para la ecuación logística. 59 M.A.Perillo Biofisica-Quimica Gráfico de Lyapunov Fig.4.16 El gráfico de Lyapunov se construye ubicando en el plano x-y los valores de dos parámetros de control del sistema dinámico. En el plano se ubica el punto correspondiente dándosele un color que representa el valor del exponente de Lyapunov. Valores negativos representan comportamientos predecibles (por ej.oscilatorios). Valores positivos representan comportamientos caóticos. En este caso la escala de colores usada fue: gradiente desde el azul al negro= λ>0 Ecuación logística (i) Derivada primera de (i) Exponente de Lyapunov El gráfico de Lyapunov permite apreciar la sensibilidad de un sistema complejo a pequeñas fluctuaciones en los valores de los parámetros de control. Se pone de manifiesto el concepto de linea de frontera del caos a partir de la posibilidad de que una sutil variación en los parámetros de control puede inducir un cambio cualitativo en el comportamiento dinámico del sistema. LAS ENZIMAS CAÓTICAS Si las reacciones catalíticas que se llevan a cabo en un reactor industrial pueden presentar, bajo ciertas condiciones, dinámicas complejas, ¿qué pasa en el complejo reactor catalítico que es nuestro cuerpo. Por lo general, la composición química de un adulto se mantiene constante, y si bien uno renueva constantemente sus estructuras, las reacciones que se producen en su seno son necesariamente alimentadas por sustancias que la persona toma del exterior. Estas sustancias son el oxígeno y los alimentos. En condiciones extremas, los alimentos en presencia del oxígeno se queman para darnos productos termodinámicamente estables, como el bióxido de carbono y el agua. Pero en nuestro cuerpo reaccionan a la temperatura ambiente con el oxigeno en presencia de catalizadores muy eficientes: las enzimas. Por acción de ellas los animales, por ejemplo, transforman el oxígeno y la mayoría de los alimentos en gas carbónico y agua, pero este paso se acompaña de otro en el cual parte de las sustancias se convierten en los materiales de los cuales están constituidos los seres, que no son el producto final que dicta la termodinámica del proceso. Obviamente estas últimas (llamados productos metaestables), se convertirán en los productos más estables, lo cual permitirá la producción de trabajo o simplemente de calor. Si todas las reacciones que conducen al equilibrio termodinámico están acompañadas de un aumento de la entropía del sistema, es decir, de una degradación del orden que le caracteriza, podría parecer contradictorio el hecho de que se generen estructuras tan ordenadas como las que poseen los seres vivientes. Durante mucho tiempo prevaleció la idea de que las reacciones bioquímicas inevitablemente convergían de inmediato a un estado termodinámicamente estacionario y que éste era único. Gracias a los trabajos de Prigogine, se demostró que en un sistema abierto que intercambia energía y materia con el medio exterior, en el caso de que los suministros externos sean suficientemente grandes, el sistema puede tender hacia un régimen constante que no es precisamente el del equilibrio. Se trata de un estado estacionario no equilibrado y se relaciona con lo que se ha dado en llamar estructuras disipativas, es decir, estructuras que se crean y se mantienen gracias a intercambio de energía con el mundo exterior en condiciones de no equilibrio. Ya hemos mencionado que la reacción de Belousov-Shabotinsky presenta estructura temporal y espacial. 60 M.A.Perillo Biofisica-Quimica Veamos ahora uno de los famosos ciclos de reacción que se llevan a cabo en el nivel bioquímico en muchos sistemas biológico, llamado ciclo del ácido cítrico o ciclo de Krebs Fig. 4.17 Diagrama simplificado del ciclo de Krebs. Los componentes principales son: C = citrato, A = cis-aconiato, I = isocianato, K = alfa cetoglutárico, S5 = succinil coenzima A, S6 = succinato, F = fumarato, M = malato, 0 = oxaloacetato. El acetato entra al ciclo como acetil coenzima A (paso 1 y reacciona con el oxaloacetato y agua para formar el citrato. El ciclo de Krebs (Fig.4.17) acoplado a la “cadena respiratoria” es uno de los mecanismos más importantes que tienen los organismos vivos, pues regula el metabolismo de los carbohidratos y de los ácidos grasos en las células, además de ser fundamental en los procesos biosintéticos. La reacción neta consiste en la oxidación completa de dos grupos carboxílicos (CH3COOH) a dióxido de carbono y agua. Durante el transcurso de la transformación se generan doce enlaces fosfato de alta energía (ATP). La mayoría de esas moléculas de ATP provienen de una reacción de oxidación de la especie nicotinamida-adenina dinucleótido (NADH), catalizada por enzimas del grupo de las deshidrogenasas organizadas vectorialmente en la membrana interna de la mitocondria celular. En este vasto y complejo mecanismo intervienen otros reactivos y productos no incluidos en el esquema de la reacción de oxidación del NAD. Un modelo sencillo de la reacción es el siguiente: O2 + 2H+ + 2 NADH → 2H2O + NAD+ La reacción consiste en la oxidación del NADH con la consecuente reducción del oxígeno molecular en presencia de un ácido, representado por la especie H+. La enzima interviene en su forma oxidada y reducida, asociada en ambos casos con el ion Fe. En 1965, muy poco tiempo después de haber sido descubierta la reacción de Belousov, un grupo japonés encabezado por I. Yamazaki informó que bajo ciertas condiciones experimentales, la concentración de oxígeno medido presentaba oscilaciones periódicas. También los daneses L. Olsen y H. Degn estudiaron las oscilaciones de esta reacción bioquímica. Comprobaron que al disminuir paulatinamente la cantidad inicial de enzima que se añadía a la reacción, había un punto en donde era posible obtener un comportamiento caótico, como lo llamaron en un artículo publicado en 1977. Se propuso un modelo cinético que, mediante simulación computacional, era capaz de reproducir los resultados obtenidos en el laboratorio y dibujar el tipo de atractor extraño que se generaba. He aquí un modelo simplificado del mecanismo de reacción: 61 M.A.Perillo Biofisica-Quimica A + B + X → 2X 2X → 2Y A +B + Y → 2X X → P Y → Q (R1) (R2) (R3) (R4) (R5) Fig.4.18 Esquema del ciclo de la oxidación de la NAHD. En este caso, A representa la cantidad de oxígeno proveniente de la fase gaseosa que se disuelve en la solución reaccionante, B representa la cantidad de NADH, X e Y son especies intermedias que no aparecen en los productos finales, mientras que P y Q son dichos productos. Hemos de aclarar que en la práctica se ha constatado que la reacción tiene más de veinte pasos, muchos de ellos aún no conocidos con suficiente detalle. La dinámica de la reacción no es tan sencilla como aparenta, por lo que en la Fig.4.18 se presenta un diagrama para aclarar el proceso. Note que las tres primeras reacciones son autocatalíticas, y que en la primera y en la tercera se genera el mismo producto, X, pero con reactivos diferentes en cada caso. Así encontramos dos subsistemas que operan entrelazados a manera de interruptores químicos; por tanto es de esperar que bajo ciertas circunstancias las oscilaciones actúen en forma sincronizada, otras veces en forma alternada y lo más importante, que en algunas ocasiones entren en un estado caótico. También se describió el ”oscilador glicolítico” correspondiente a la dinámica de las especies químicas que participan en la glicólisis anaerobia durante la cual, en el citosol celular la glucosa es finalmente oxidada a piruvato. (se detallará en clase). También los conocidos ritmos circadianos, asociados a nuestros ciclos de sueño y vigilia son otra manifestación de organización temporal. Puede citar otros ejemplos de fenómenos de autoorganización espacial o temporal? 62 M.A.Perillo Biofisica-Quimica TRABAJO PRACTICO No 2 Basado en Adamcikova L. and Sevcik P. (1998) Journal of Chemical Education, Vol.75, p.1580 UNA DEMOSTRACIÓN SIMPLE DE AUTORGANIZACIÓN: EL EXPERIMENTO DE LA BOTELLA AZUL La siguiente experiencia involucra la reducción alcalina por glucosa del azul de metileno. Una vez que la solución se deja reposar, ésta se torna incolora indicando que el colorante ha sido reducido por la glucosa. La agitación posterior del recipiente que la contiene causará que retorne el color propio del colorante a medida que el azul de metileno sea oxidado a su forma azul por el oxígeno atmosférico. La formación de patrones de color característicos aparecen cuando NaOH, glucosa y azul de metileno son mezclados en una cápsula de Petri. Esta cápsula puede ser montada sobre un retroproyector para proyectar la imagen y analizar la formación de dichos patrones. MATERIALES: • Cápsula de Petri • 1 M NaOH • 0,5 M de glucosa (o galactosa, xilosa, arabinosa o manosa) • 1 mM de azul de metileno o azul de toluidina. PROCEDIMIENTO: Mezclar 5 ml de NaOH, 1,6 ml de Glucosa, 0,5 ml de colorante y 2,9 ml de agua. Colocar parte de la mezcla en la cápsula de Petri hasta alcanzar una profundidad aproximada de 2-3 mm. Esta permanecerá azul por 3-4 min.; luego aparecerán y empezarán a crecer pequeñas regiones incoloras, formando estructuras en mosaico. Si la cápsula es cubierta por un vidrio, no se verá ninguna formación de patrones en la mezcla de reacción. Agitando la cápsula se producirá una coloración azul homogénea. Si dejamos reposar unos minutos, el color azul revertirá los patrones iniciales. Este proceso puede ser repetido varias veces. Los patrones son transitorios porque el sistema evoluciona hacia el equilibrio termodinámico. Estos patrones desaparecen a las 2-3 hs, cuando se alcanza un color violeta homogéneo. Un interesante cambio de color puede también ser observado cuando se agrega un indicador tal como la fenolftaleína. Trabajos Prácticos En los trabajos prácticos se demostró la evolución de sistemas hacia estados estables con la ocurrencia de dos tipos de fenómenos de autoagregación espacial. a) Describa cada uno de los experimentos. b) Indique el tipo de estructura de autoagregación que se obtuvo en cada caso. c) Para cada uno de ellos indique si se trató de un sistema abierto o cerrado; d) Indique si se alcanzó un estado estacionario o de equilibrio. e) Indique los factores que permiten y que inducen la autoagregación en cada caso. 63 M.A.Perillo Biofisica-Quimica CUESTIONARIO DE ORIENTACIÓN UNIDAD 4 CONSIGNA DE TRABAJO: - - Intente responder el cuestionario sobre “Termodinámica de sistemas abiertos” y “Augoorganización espacial y temporal” después de haber realizado el estudio independiente del material precedente. Las respuestas serán discutidas en la tercera clase. Lea los trabajos de divulgación científica del Anexo III, e intente responder al cuestionario correspondiente. Los trabajos serán discutidos en la tercera clase. TERMODINÁMICA DE SISTEMAS ABIERTOS 1. Indique las similitudes y diferencias entre estado de equilibrio y estado estacionario. 2. En cuál de estos tipos clasificaría el estado de: a) el nivel constante del agua de un recipiente donde la cantidad agua que ingresa y que egresa por unidad de tiempo es misma, b) un ser vivo, c) la reacción de disociación de un ácido debil en agua a partir del momento en que las concentraciones de todas las especies químicas en solución se hacen constantes. Explique. 3. Explique el efecto difusotérmico 4. Qué es una estructura disipativa? 5. Explique porqué la existencia de sistemas organizados, tales como los seres vivos, no violan la segunda ley de la termodinámica. 6. Cómo varían entropía (S) y velocidad de producción de entropía (P) a medida que un sistema se aproxima hacia: a) un estado de equilibrio, b) un estado estacionario. AUTOORGANIZACIÓN ESPACIAL Y TEMPORAL 1. Defina sistemas lineales y no lineales. 2. Indique qué es la dinámica de un sistema? 3. Qué tipos de dinámica puede adoptar un sistema? 4. Qué es un espacio de fases? 5. Qué es un atractor? Indique tipos de atractores y la relación de cada uno con un tipo de dinámica del sistema. 6. Cuáles son las propiedades de los sistemas capaces de exhibir fenómenos de autoorganización espacial y/o temporal. 7. Cuáles son las propiedades de los sistemas que pueden exhibir dinámicas caóticas. 8. Diferencie caos determinista y azar. De ejemplos. 9. De ejemplos de sistemas con distintos niveles jerárquicos de organización. 10. Qué son propiedades emergenes? 11. En qué consiste el procedimiento matermático llamado iteración? De un ejemplo. ANÁLISIS DE TEXTOS SELECCIONADOS Termodinámica de la evolución biológica, Lurié D. Y Wagensberg J. (1979). Investigación y Ciencia. Vol. 30, p.102. 1) Compare la evolución de la distribución de temperaturas en una barra aislada en todo su contorno excepto en los extremos (pág.105) con la imagen termodinámica de la evolución de un ser vivo (pág.109) (la linea recta oblicua representa. 2) Identifique en los termogramas de la pág.107 (gráfico superior), los momentos de crecimiento y diferenciación de un anfibio y de una rata. Cómo explica el ascenso y el descenso en la producción de calor? Compare la parte descendente de esos gráficos con la curva superior del gráfico de Evolución del balance entrópico de la pág.108. 3) Observe el gráfico inferior de la pag.109. A medida que el comportamiento del sistema se aleja del equilibrio, aumenta la probabilidad de sufrir bifurcaciones, es decir de reestructurarse, evolucionar hacia nuevos estados. 4) Observe la aparición de autoorganización temporal en la producción de calor en las colonias de Flavobacterium marino como consecuencia del funcionamiento coherente del ciclo glicolítico en agregados celulares. Se aprecia como picos pequeños en el gráfico de la 64 M.A.Perillo Biofisica-Quimica derecha, que aparecen como estructura fina superpuesta a los picos grades del gráfico de la izquierda. La proximidad física de las células permite que éstas se sincronicen. El lenguaje de los fractales. Jürgens, H.; Peitgen, Heinz-Otto y Saupe, D. (1990). Investigación y Ciencia Vol.69, p.46. 1) 2) 3) 4) Qué es la geometría fractal? Cuál es su diferencia con la geometría euclideana? Cuál es la característica fundamental de los fractales? Qué es la invariancia bajo escala o autosimilitud? Ejemplifique. Cuál es la relación entre caos y fractales? BIBLIOGRAFÍA ESPECIFICA UNIDAD 4. TERMODINAMICA DE SISTEMAS ABIERTOS Barral, R. y von der Becke, C. Biotermodinámica del cerebro. http://www.chez.com/vonvon Peacocke,A.R. An introduction to the physical chemistry of biological organization. Oxford Sci.Pub. Clarendon Press, Oxford. 1989. Schifter, I. La ciencia del caos. http:/omega.ilce.edu.mx:3000/sites/ciencia. SISTEMAS COMPLEJOS Adamcikova L. and Sevcik P. (1998) Journal of Chemical Education, Vol.75, p.1580. Madressi, R. Teorías del caos, orden, desorden, flecha del tiempo. Insomnia no3 Perillo M.A. Guía de Trabajos Prácticos de Biofísica Química, FCEFyN, UNC. Olivares, C.M. Tecnologias emergentes de computo. Teoria del caos. Alfaomega Grupo Editor, S.A., Mexico. 1997. Textos seleccionados Lurié D. Y Wagensberg J. Termodinámica de la evolución biológica, Investigación y Ciencia, Vol.30, p.102 (1979). Jürgens, H.; Peitgen, Heinz-Otto y Saupe, D. El lenguaje de los fractales. Investigación y Ciencia Vol.69, p.46. (1990). 65