Capítulo 1
Anuncio

mecánica estadística
Principios Fundamentales
Capítulo 1
2014
Objetivo de la mecánica estadística
Predecir el comportamiento macroscópico
de un sistema, en base de las propiedades
microscópicas de las partículas que lo
componen.
La estructura de la mecánica estadística está
basada en dos postulados:
La equiprobabilidad de todos los estados accesibles
a un sistema aislado
La definición estadística de entropía.
Niveles de
Energía
Estructura
Molecular
Mecánica
Cuántica
Electrónicos
Vibracionales
Rotacionales
+
correcciones
Propiedades
MACROSCOPICAS
Termodinámica
Estadística
Los observables termodinámicos macroscópicos son promedios de las
propiedades moleculares
La termodinámica estadística
ayuda a reducir el numero de
grados de libertad del sistema
1 mol gas
ideal
~1024 variables
Conjunto hipotético de un número muy grande N
de sistemas idénticos, réplica de uno original.
El formalismo estadístico
está basado en el concepto
de colectivo
N→∞
Los miembros del colectivo se definen
especificando
alguna
de
las
variables
termodinámicas (número de partículas, Energía,
Volumen, Temperatura, etc ...)
Arbitrariamente grande
Especificación del estado de un sistema
El estudio de las partículas atómicas ha demostrado que
cualquier sistema formados por tales partículas puede
describirse mediante las “Leyes de la mecánica”
Cada estado cuántico de un sistema aislado está asociado a
un valor definido de su energía y se denomina nivel energético.
Normalmente existe solo un estado cuántico posible
correspondiente a su energía inferior; a este estado se le llama
estado fundamental del sistema. Además hay infinitos estados
posibles con mayores energías a estos y se les denomina estados
excitados del sistema. (Esto es válido para todos los sistemas,
aunque sean muy complejos).
Consideremos
el
lanzamiento
de
una
moneda.
Para
este
experimento, se tienen dos
eventos o sucesos posibles:
SELLO (S)
CARA (C)
Si lanzamos 3 veces la
moneda, el número de
resultados está dado por la
expresión:
c
c
c
s
3
N =2 =8
s
c
s
c
c
s
s
c
s
s
8 resultados
posibles.
De esta forma, la probabilidad de obtener uno de los anteriores resultados es:
1
p=
8
Así, la probabilidad de obtener el arreglo ccs, es de 1/8, la cual es la misma
probabilidad para el arreglo csc, o para el arreglo sss.
{
MICROESTADOS
CCC SCC
CCS
SCS
CSC
SSC
CSS
SSS
Cada arreglo en particular
conoce como MICROESTADO
se
Un microestado es cada uno de los posibles estados en los que puede
encontrarse el sistema estudiado
Ejemplos:
(i) Spin aislado
r
σ
M
E
1
+1
μ0
-μ0H
2
-1
-μ0
+μ0H
µ0 es el momento magnético y H el campo magnético.
(ii) Sistema ideal
de N spines
E=-MH
µ0 es el momento magnético y H el
campo magnético.
r
σ1 σ2 σ3 σ4
M
E
1
+ + + +
4 μ0
-4 μ0H
2
3
4
5
+
+
+
-
2 μ0
2 μ0
2 μ0
2 μ0
-2 μ0H
-2 μ0H
-2 μ0H
-2 μ0H
0
0
0
0
0
0
0
0
0
0
0
0
-2 μ0
-2 μ0
-2 μ0
-2 μ0
2 μ0H
2 μ0H
2 μ0H
2 μ0H
-4 μ0
4 μ0H
+
+
+
+
+
+
+
+
+
6 + + - 7 + - + 8 + - - +
9
- + + 10 - + - +
11 - - + +
12
13
14
15
+ - - - + - - - + - - - +
16 -
-
-
-
Postulado fundamental
Una visión realista de un sistema macroscópico es aquella
en que el sistema realiza transiciones extremadamente
rápidas y azarosas entre sus estados cuánticos. Cualquier
tipo de medición macroscópica detecta solamente un
promedio de las propiedades de miríadas de estados
cuánticos.
Un sistema macroscópico aislado muestrea
todo estado cuántico permitido con igual
probabilidad.
PRIMER POSTULADO
DE LA MECÁNICA
ESTADÍSTICA
“La
probabilidad
de
obtener
cualquier microestado es la misma
para todos los microestados en un
sistema en equilibrio”.
Definimos MACROESTADO, como el conjunto de microestados iguales. En
nuestro experimento con la moneda, existen 4 macroestados: ccc, sss, ccs, ssc.
SEGUNDO POSTULADO
DE LA MECÁNICA
ESTADÍSTICA
“El macroestado más probable es el
que tenga el mayor número de
microestados para un sistema en
equilibrio”.
QUÍMICA FÍSICA AVANZADA
Conjunto Microcanónico y Ergodicidad
Un conjunto estadístico (statistical ensemble) es una colección de un
número muy grande de réplicas del sistema bajo estudio.
Un conjunto estadístico de sistemas aislados se denomina conjunto
microcanónico (microcanonical ensemble).
N,V,E
N,V,E
N,V,E
N,V,E
I
...
II
III
N
Numero de partículas constante: N
Volumen constante: V
Energía constante: E
La probabilidad de encontrar un observable A con un valor entre A y
A+dA, P(A) dA, podrá calcularse simplemente como (cada estado tiene el
mismo peso):
N
P ( A ) dA = lim
N0 → ∞
N0
dA
donde N(dA) es el número de sistemas en los que A toma el valor entre A
y A+dA y N0 es el número total de sistemas del conjunto. Como P(A) está
normalizada de forma que:
∫ P ( A ) dA = 1
entonces el promedio de A sobre el conjunto microcanónico, A ,
será:
A = ∫ A P ( A ) dA
Se denomina Ergódico a todo sistema que cumple con:
t
A = lim
t→∞
∫ A r ( t ), p ( t )d t
0
Postulado de Gibbs
El promedio temporal para un propiedad macroscópica
de un miembro del colectivo es igual al valor medio de la
propiedad en el colectivo
Eliminación de la variable temporal
La termodinámica estadística no
necesita el concepto de tiempo
Sistemas en Equilibrio
Ligaduras, equilibrio e irreversibilidad
Las restricciones externas (a escala macroscópica) actúan como
ligaduras que restringen los estados en que puede encontrarse el
sistema. Por lo tanto el número de estados accesibles Ω, dependen de
las ligaduras del sistema.
Vf
Vi
Vi<Vf
El sistema tiende a variar hasta que finalmente alcanza el
equilibrio, en la que se encuentra con igual probabilidad de
estar en cualquiera de sus estados accesibles.
Si se elimina alguna ligadura original, el número de estados
accesibles es mucho mayor que el número de estados
accesibles original.
Ω f ≥ Ωi
(i) Caso especial en que Ωf=Ωi
La situación de equilibrio del sistema queda sin perturbación después
de eliminar las ligaduras.
(ii) Caso normal en que Ωf>Ωi
Inmediatamente después de sacar la legadura, la probabilidad de
encontrarse en uno u otro estado accesible, es la misma, pero la
probabilidad de encontrarse en cualquiera de los (Ωf-Ωi) es cero. Esta
distribución de probabilidad NO uniforme no corresponde a una
situación de equilibrio.
Una vez sacada la ligadura, la probabilidad de que vuelva a su
estado inicial es casi cero (que se encuentren todas las partículas
en Vi). Se tiene que:
Ω f >> Ωi
Entonces esto demuestra que es muy raro que todas las
partículas vuelvan a Ωi :
Ωi
Pi =
Ωf
Diremos, entonces, que un proceso es irreversible si la situación
inicial de un conjunto de sistemas aislados que han sufrido este
proceso no puede restablecerse imponiendo simplemente una
ligadura.
Leyes de la Termodinámica (Repaso)
Ley cero (o principio 0) de la Termodinámica
Si dos sistemas están por separado en equilibrio con un tercero, entonces
también deben estar en equilibrio entre ellos.
Si tres o mas sistemas están en contacto térmico y todos juntos en equilibrio,
entonces cualquier par está en equilibrio por separado.
Primera Ley de la Termodinámica
El trabajo necesario para cambiar el estado de un sistema aislado depende
únicamente de los estados inicial y final, y es independiente del método usado para
realizar el cambio.
El cambio de la energía interna en una transformación adiabática es ∆ U = W.
Sistema termodinámico
típico mostrando la
entrada desde una fuente
de calor (caldera) a la
izquierda y la salida a un
disipador de calor
(condensador) a la
derecha. El trabajo se
extrae en este caso por
una serie de pistones.
La conservación
de energía será:
∆U = Q − W
Definición de calor: La cantidad de calor Q absorbido por un sistema es el cambio
en su energía interna que no se debe al trabajo.
Segunda Ley de la Termodinámica
La base de esta ley es el
hecho de que si mezclamos
partes iguales de dos gases
nunca los encontraremos
separados de forma
espontánea en un instante
posterior.
Enunciado de Clausius: No
hay ninguna transformación
termodinámica cuyo único
efecto sea transferir calor de
un foco frío a otro caliente.
Enunciado de Kelvin: No hay
ninguna transformación
termodinámica cuyo único
efecto sea extraer calor de
un foco y convertirlo
totalmente en trabajo.
Segunda Ley de la Termodinámica
Es imposible construir un
dispositivo que opere,
según un ciclo, y que no
produzca otros efectos,
además de la
transferencias de calor de
un cuerpo fío para un
cuerpo caliente.
dS ≥
δQ
T
EL PERPETUUM MOBILE
¡¡¡¡¡¡¡¡¡¡NO EXISTE!!!!!!!!!!!
Segunda Ley de la Termodinámica
Principio de máxima entropía
Existe una función de estado de los parámetros extensivos de
cualquier sistema termodinámico, llamada entropía S, con las
siguientes propiedades:
los valores que toman las variables extensivas son los
que maximizan S consistentes con los parámetros
externos.
la entropía de un sistema compuesto es la suma de las
entropías de sus subsistemas.
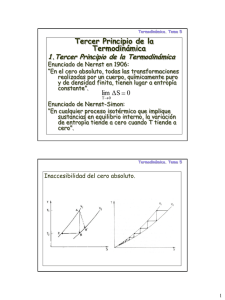
Tercera Ley de la Termodinámica
Teorema de Nerst: Una reacción química entre fases puras cristalinas que
ocurre en el cero absoluto no produce ningún cambio de entropía.
Enunciado de Nerst-Simon: El cambio de entropía que resulta de cualquier
transformación isoterma reversible de un sistema tiende a cero según la
temperatura se aproxima a cero.
lim S = 0
T →0
Enunciado de Planck: Para T → 0, la entropía de cualquier sistema en
equilibrio se aproxima a una constante que es independiente de las demás
variables termodinámicas.
Teorema de la inaccesibilidad del cero absoluto: No existe ningún proceso
capaz de reducir la temperatura de un sistema al cero absoluto en un
número finito de pasos.
Definición microscópica de entropía.
Boltzmann propuso la definición microscópica de entropía:
S = k B ln Ω
donde kB es la constante de Boltzmann (kB = 1.3807×10−23 J / K ) y Ω es el
número de estados accesibles del sistema.
La entropía es proporcional al desorden.
Cuando T→ 0, los miembros del conjunto tienden a poblar únicamente
el nivel de energía fundamental. Si éste es único (no degenerado), Ω = 1
y la entropía es cero.
La entropía es siempre mayor o igual a a cero (S ≥ 0).
Con el postulado fundamental de uniformidad probabilística para los
estados accesibles y la definición microscópica de entropía, quedan
establecidas las bases de la mecánica estadística.
Capacidad Calorífica
Llamamos capacidad calorífica de un sólido al
calor necesario para elevar en un grado la temperatura de
una determinada cantidad de material (se mide en Joule/ºC
o J/K):
Es frecuente utilizar la capacidad calorífica molar
“c = C/n” (J/ºC mol o J/ K mol), en la que la cantidad de
materia considerada es un mol, mientras que en la
definición de calor específico se suprime la dependencia
con la masa total involucrada (J/ ºC kg o J/ K kg).
Ley de Dulong y Petit (1819)
En la teoría clásica de los calores específicos, la
energía media a una temperatura dada se calcula teniendo
en cuenta todas las energías posibles y calculando el valor
promedio mediante la estadística de Boltzmann. Al hacer el
cálculo de la energía media se llega a la conclusión de que,
a cada grado de libertad de las partículas del sistema le
corresponde una energía media kBT/2 (este es el llamado
principio de equipartición de la energía). Así, en los gases
monoatómicos, donde cada partícula tiene tres grados de
libertad, la energía media por partícula sería 3kBT/2.
La energía interna por mol sería: U(T) = NA3kBT/2,
donde NA es el número de Avogadro.
En un sólido, si se consideran las vibraciones
independientes de los átomos, todos con la misma frecuencia
y considerando tres grados de libertad, se obtiene una energía
molar promedio de valor U(T)=3NAkBT, de la que se deduce
un calor específico molar de:
cV=3NAkB= 3R =
24.9 J/(mol K).
En 1819 Dulong y Petit descubrieron que la mayor parte de los
sólidos presentaban una capacidad calorífica molar a temperatura ambiente de
aproximadamente 25 J/mol K.
Dicho valor coincidía con el predicho por el modelo anterior, lo cual
supuso un triunfo de la teoría cinética clásica.
Sin embargo, pronto se observaron desviaciones a
temperaturas más bajas, y se constató que dicha constante
constituía en realidad un límite superior: por debajo de cierta
temperatura característica de cada sólido, llamada
temperatura de Debye (θD), la capacidad calorífica disminuye
y tiende a cero proporcionalmente al cubo de la temperatura
para las temperaturas próximas a cero, como muestra la
figura.
Cv
Modelo de Einstein (1907)
Se puede explicar este comportamiento a bajas
temperaturas usando mecánica cuántica. La
primera explicación fue dada por Eintein en
1907. El consideró al solido como un conjunto
de osciladores armónicos cuánticos que vibran a
la misma frecuencia ω0. La teoría cuántica nos
dice que los niveles de energía de un oscilador
es dado por:
εi = (i + ½) hω0
donde i = 0,1,2…, y h es la contante de Planck.
Por conveniencia, podemos tomar el cero de
energía de tal modo que cada oscilador puede
tomar energías n ℏw0, con n = 0, 1, 2... (en lugar
de (n + 1/2 ) ℏω0).
ω0
Aplicación del Formalismo
Capitulo Anterior
Microcanónico
del
Si U es la energía total del sistema, hay en total U/ ℏ w0 cuantos.
El número de estados accesibles del sistema, Ω, será igual al
número de maneras de distribuir los U / ℏ w0 cuantos entre los 3N
osciladores. Esto puede calcularse fácilmente considerando una
colección de 3N + U / ℏ w0 objetos (osciladores + cuantos)
El número de permutaciones de todos estos objetos entre sí es
(3N + U/ℏw0 – 1)! Pero si permutamos barras entre sí o círculos
entre sí, no obtenemos estados diferentes y entonces tenemos:
Usando la fórmula de Stirling para grandes números:
ln M ! ≈ M ln M − M
obtenemos (poniendo U0 = 3 Nℏω0 ):
Ésta es la ecuación fundamental del sistema, pues a través de las
relaciones entre la entropía y las demás cantidades
termodinámicas es posible calcular otros observables. Por
ejemplo:
De aquí se obtiene la energía media por oscilador,
<ε >= U / 3N, como:
La cantidad ΘE = ℏw0 /kB se denomina temperatura de Einstein
del cristal y generalmente es del orden de magnitud de la
temperatura de fusión del sólido
Luego derivando respecto a la temperatura obtenemos
el calor especifico por mol:
El termino
Einstein θE.
se la define como la temperatura de
La formula de Einstein nos da la Ley de Dounlog y Petit
CV ─>3R cuando T ─> ∞ y CV ─> 0 cuando T ─> 0
Modelo polimérico de una banda elástica
Cada monómero, de longitud a, puede tener 4 orientaciones:
en +x, –x, a las que les asignamos la energía cero, y en +y, –y, a
las que les asignamos una energía ε que tiene en cuenta las
interacciones con los otros polímeros del manojo (la banda se
considera mucho más larga que ancha y de un grosor pequeño).
Energía = 0
Energía = ε
Calculemos la entropía del polímero S, en función de la energía
interna del sistema U, del número N de monómeros de la cadena y
de las coordenadas Lx, Ly del extremo de la cadena.
Sean: N+x , N–x, N+y, N–y el número de monómeros orientados
en las direcciones permitidas. Entonces podemos escribir las
siguientes relaciones entre las variables:
y el número de monómeros en cada dirección puede expresarse
como:
El número de configuraciones del polímero consistentes con las
coordenadas dadas Lx, Ly y con una dada energía U, es:
Aproximación de Stirling
Aplicando la aproximación de Stirling, la entropía resulta ser:
Ahora, por el primer principio de la termodinámica dU = ∂W + ∂Q,
y como ∂Q = 0 por estar el sistema aislado, tenemos:
por lo que las tensiones pueden escribirse como:
Entonces, expresando la derivada parcial de la entropía respecto
de Ly de la forma:
que es la ecuación de estado “mecánica” de la banda.
Conclusiones
El principio fundamental de equiprobabilidad de los estados
accesibles de un sistema aislado en equilibrio es la base de la
mecánica estadística.
La definición microscópica de entropía, S=kBlnΩ, permite obtener
las cantidades termodinámicas de interés, mediante un simple
conteo de estados consistentes con los vínculos impuestos.
La remoción de un vínculo (o restricción) hace que la entropía
aumente hasta alcanzar un nuevo equilibrio, el que está
caracterizado por S = máximo.
Dicha definición, que constituye el segundo postulado, es
consistente con las leyes de la termodinámica.