Facultad De Ciencias Químicas LICENCIATURA DE QUÍMICA INDUSTRIAL MANUAL DE TÉCNICAS EN QUÍMICA ORGÁNICA L.Q.I. Ma. Raquel Cruz de León M.C. Blanca Esthela Neri Segura M.C. Nancy Adriana Pérez Rodríguez M.C. Claudia Celeste Velázquez González EDICIÓN 2007 ISBN-978-970-694-470-2 No. Registro: 03-2007-111412042700-01 INDICE PRÓLOGO 2 EVALUACIÓN DEL CURSO 3 CONTENIDO DEL REPORTE 4 EL DIARIO DE LABORATORIO 5 EXTRACCIÓN 9 CRISTALIZACIÓN 22 DESTILACIÓN SIMPLE Y FRACCIONADA A PRESIÓN NORMAL Y PRESIÓN REDUCIDA 34 DESTILACIÓN POR ARRASTRE CON VAPOR 54 ANÁLISIS ELEMENTAL CUALITATIVO 65 ANÁLISIS FUNCIONAL ORGÁNICO 77 CROMATOGRAFÍA 96 PUNTOS DE FUSIÓN Y EBULLICIÓN 110 INDICE DE REFRACCIÓN 128 APÉNDICE A REGLAMENTO GENERAL DE LABORATORIOS 139 APÉNDICE B DISPOSICIÓN DE RESIDUOS 142 1 PRÓLOGO Pensando en la mejora continua, la Academia de Química Orgánica actualizó este manual, cuya primera edición fue elaborada por los maestros; Q.F.B. Gloria Esquivel Zamora, M.C. Ma. Martha E. Luna Saucedo, Dr. Javier Macossay Torres, M.C. Blanca Esthela Neri Segura, M.C. Nancy Adriana Pérez Rodríguez, y Dra. Norma Nelly Treviño Flores. Esta cuarta edición fue revisada por la M.C. Claudia Celeste Velásquez González. En esta cuarta actualización se ha incluido algún material o reactivo que se requiere para la realización de las prácticas, el cambio de la tercera edición consideraba la secuencia que realmente se lleva a cabo después de efectuar una síntesis orgánica. El manual está dirigido tanto a los alumnos que por primera vez cursan un laboratorio de química orgánica, como a los que habiéndolo cursado requieren aplicar alguna de las técnicas aquí presentadas. Cada técnica está fundamentada lo más claramente posible, considerando un conocimiento mínimo de la química orgánica y de la química en general por parte del estudiante. Cada práctica incluye el objetivo a cumplir, el fundamento teórico en el que se basa, descrito someramente, la descripción del equipo, material y reactivos a utilizar, el procedimiento detallado a seguir, el correspondiente diagrama de flujo y referencias bibliográficas. El formato presentado en el procedimiento está inspirado en las nuevas tendencias mundiales sobre criterios y certificaciones de calidad y consiste en pasos numerados y el diagrama de flujo correspondiente. El objetivo es presentar un texto de laboratorio que permita realizar una práctica con el menor número de dudas y/o errores. Se exhorta a que se aproveche cada experimento, leyendo, antes de llegar al laboratorio, la práctica a realizar y cubriendo los requisitos o tareas que el maestro indique. Las técnicas presentadas son básicas en un laboratorio de química orgánica y serán de utilidad en trabajos de investigación superior y por supuesto en la práctica profesional. Se incluye un apéndice para la disposición de los residuos generados en cada una de las prácticas, de acuerdo al Reglamento General de Laboratorios de la Facultad de Ciencias Químicas. El trabajo experimental es indispensable en química orgánica, es fuente de conocimiento, comprobación y descubrimiento. Para el químico es, o debería ser, una fuente de placer en el proceso de aprendizaje. El Licenciado en Química Industrial es un científico, esto obliga a cuestionar, no a seguir recetas, sino a observar, analizar y concluir. 2 EVALUACIÓN DEL CURSO LABORATORIO DE QUÍMICA ORGÁNICA I 1. TRABAJO PRÁCTICO 50% 1.1. Reportes 10% 1.2. Diario de Laboratorio 15% 1.2.1. Propiedades generales de las sustancias a utilizar 1.2.2. Esquema del procedimiento 1.2.3. Observaciones 1.3. Aspectos de Laboratorio 1.3.1. Puntualidad 1.3.2. Seguridad 1.3.3. Orden 1.3.4. Limpieza 1.3.5. Montaje de equipo 1.4. Examen diario 2. EXÁMENES 5 5 5 10% 2 2 2 2 2 15% 50% 2.1. Examen de medio Término 25% 2.2. Examen Final 25% 3 CONTENIDO DEL REPORTE 1. PORTADA Datos de Ia institución, personales y título del trabajo. 2. OBJETIVO Meta final de la práctica. 3. FUNDAMENTO Extensión y complemento del fundamento incluido en el manual. 4. PROCEDIMIENTO GENERAL Procedimiento realmente llevado a cabo. 5. RESULTADOS Observaciones, cálculos, datos de constantes físicas, tablas de resultados. 6. CONCLUSIONES Se establece una relación entre lo indicado en el objetivo, fundamento y lo reportado en Ia sección de resultados. 7. REFERENCIAS BIBLIOGRAFICAS Autor (apellido, nombre) nombre del Iibro o título del artículo, nombre de Ia revista (si es el caso), editorial, país, volumen, año (entre paréntesis), página o páginas consultadas. 4 EL DIARIO DE LABORATORIO La comunicación escrita es el método más importante por la cual los químicos transmiten su trabajo a la comunidad científica. Esto empieza con los registros en el diario de laboratorio. El diario de laboratorio es, sin exagerar, la herramienta más importante con que el químico puede contar. Un buen desarrollo de éste va a determinar el éxito de la investigación efectuada. Sin él no podemos asegurar un trabajo reproducible, efectivo y confiable; su importancia está subrayada por aquel viejo proverbio “vale más la más pálida tinta que la más brillante memoria”. Todo químico deberá estar convencido que su trabajo será doblemente productivo si hace un uso adecuado de este valioso recurso. El diario de laboratorio es la fuente de información utilizada para preparar artículos científicos y especialmente importante para respaldar una patente. Los aspectos generales sobre como mantener un diario de laboratorio son: • Se debe utilizar un diario de pasta dura, las hojas deben estar permanentemente unidas, no deben emplearse cuadernos que tengan hojas fácilmente desprendibles. • Cada página debe ser numerada en orden consecutivo. • Se debe dejar al inicio del diario unas hojas en blanco para hacer un índice de su contenido. • La escritura debe hacerse con tinta a prueba de agua, nunca se debe usar lápiz o marcador pues la humedad y el tiempo llegan a borrar las notas. • Si la página no se llenó completamente se debe usar una “X” en el espacio vacío. • Nunca se debe borrar; si se ha cometido un error éste debe tacharse con una línea horizontal y luego escribir lo correcto. • No se debe saltar páginas; esto es incorrecto. • El registro no debe ser ambiguo. Se debe cuidar la gramática y ortografía. • En los laboratorios de investigación industrial se requiere su firma, así como la de un testigo, porque el diario puede ser usado como documento legal. • Siempre se debe escribir y organizar su trabajo para que alguien mas pueda repetir su experimento sin confusión o duda. Un registro completo y legible es un factor clave. • El diario debe ser su amigo, su confidente de trabajo. Los componentes claves de un registro en el diario de laboratorio son: 1. 2. 3. 4. La fecha de ejecución del experimento El título del experimento Propósito del experimento (objetivo) La ecuación completa y balanceada de la reacción, cuando aplique. 5 5. La lista de los compuestos químicos que se van a utilizar o producir durante el experimento, que incluya los nombres de los compuestos, la estructura y peso molecular, las constantes físicas más comunes como, punto de fusión y ebullición, índice de refracción, densidad, toxicidad, etc. Para esto se consultan manuales. 6. Un esquema detallado del procedimiento que va a seguirse antes de llegar al laboratorio. Este procedimiento esquemático describirá en forma muy directa los pasos a realizar, facilitará el trabajo y ayudará a saber anticipadamente el material y reactivos que se usarán por lo que el trabajo experimental será más efectivo. (ver esquema 1) 7. El procedimiento que usted realmente haya hecho y no lo que su texto o esquema del procedimiento de laboratorio dice que debió hacer. 8. Todas las observaciones, (cambios de color, incrementos o descensos de temperatura, explosiones, etc.); esto, de preferencia, debe ir acompañado de una explicación o hipótesis razonable de porqué ocurrieron dichos fenómenos. 9. Referencias bibliográficas sobre el producto o procedimiento. 10. La firma de la persona que llevó a cabo el experimento y la de un testigo, si se requiere. 11. Los cálculos para determinar el porcentaje de rendimiento, cuando aplique. CANTIDAD (g o moles) EXPERIMENTAL % RENDIMIENTO = X 100 CANTIDAD (g o moles) TEÓRICA 12. Los cálculos para determinar el porcentaje de recuperación, en el caso de una recristalización o extracción. CANTIDAD (g o moles) EXPERIMENTAL % RECUPERACIÓN = X 100 CANTIDAD (g o moles) TEÓRICA 6 ESQUEMA 1 EJEMPLO DEL ESQUEMA DE UN PROCEDIMIENTO Extracción de ácido benzóico disuelto en cloroformo 7 ESQUEMA 1 Continuación 8 PRÁCTICA No. 1 EXTRACCIÓN I. OBJETIVO Aplicar el proceso de extracción líquido-líquido para aislar o separar, eficientemente, un compuesto orgánico de una mezcla, además de la extracción sólido-líquido para el aislamiento de metabolitos secundarios a partir de zacate de limón . II. FUNDAMENTO La transferencia de un soluto desde una mezcla hacia un solvente es llamada extracción. El soluto es extraído, de la mezcla donde se encuentra, por un solvente inmiscible con la mezcla, mediante el proceso de distribución. Cuando la mezcla se agita con el solvente elegido, el soluto se distribuirá entre las dos fases, llegando al equilibrio cuando las dos fases se hayan separado. Csm Css Donde: Csm y Css son concentraciones, en g/L, del soluto en la mezcla original y en el solvente extractor, respectivamente. En ese momento la relación de las concentraciones del soluto en cada fase está definida por una constante. Esta constante llamada coeficiente de distribución o de partición, K, está definida por: K= Css Csm El coeficiente de distribución tiene un valor constante para cada soluto considerado y depende de la naturaleza del solvente utilizado en cada caso. La relación de las concentraciones del soluto en ambas fases es proporcional a la relación de las solubilidades respectivas. Sss K= Ssm Donde: Sss y Ssm son las solubilidades del soluto en el solvente extractor y en la mezcla original, respectivamente. 9 Para un caso en que la mezcla original sea líquida se tiene que: gss / Vs Sss K= = Ssm Donde: Vs = Vm = gss = gsm = y: K = gsm /Vm Volumen del solvente extractor Volumen de la mezcla original gramos de soluto extraídos con el solvente gramos de soluto remanentes en la mezcla original gss Vm gsm Vs A partir de la fórmula anterior se puede deducir que es más conveniente dividir el solvente extractor en varias porciones que hacer una sola extracción con todo el solvente. Para ejemplificar lo anterior podemos suponer un sistema con un coeficiente de distribución de 10; dicho sistema consiste de 5.0 g de un compuesto orgánico disuelto en 100 mL de agua. Compararemos la efectividad de 3 extracciones, cada una con 50 mL de éter, contra una sola extracción con 150 mL de éter. En la primera extracción con 50 mL de éter, la cantidad extraída a la capa etérea (x) se determina por el siguiente cálculo: Css K= 10 = Csm Csm Csm = ( ( x 50 5 -x 100 ) ) (1) Despejando de la ecuación (1) resulta: 100x 10 = (5-x) (50) 10 = 100x 10 250 – 50x 2500 – 500x = 100x x = 2500 / 600 = 4.166g En la segunda extracción de la solución acuosa, que ahora contiene 0.83 g de soluto (5 – 4.166), utilizando otros 50 mL de éter la cantidad extraída, x´, será calculada de la ecuación (2). Css K= 10 = CCsm sm = ( ( x´ 50 ) ) (2) 0.834 – x´ 100 Despejando de la ecuación (2) resulta: 100 x´ 10 = 50 (0.834 – x) Despejando x´ = 0.695 De una manera similar puede demostrarse que en la tercera extracción con otros 50 mL de éter se remueven 0.1156 g de soluto (x”) a la fase etérea, dejando 0.02 g de soluto en la fase acuosa. Sumando la cantidad extraída de soluto en las fases etéreas (4.166+0.695+0.1156) se tienen 4.976 g de soluto extraído. Si la extracción se ejecuta usando una cantidad equivalente de éter (150 mL) en una sola extracción, la cantidad extraída de soluto (y) será: Css K= 10 = CCsm sm Csm = ( ( y 150 5-y 100 ) ) (3) Despejando de la ecuación (3) resulta: 100 y 10 = 150 (5 – y) 11 y = 4.687 Analizando los resultados se tiene que se extrajeron 0.289 g más de soluto en las tres extracciones, utilizando porciones pequeñas de éter, que efectuando una sola extracción con el total del volumen. Algunos de los disolventes más utilizados son: éter etílico, benceno, tolueno, éter de petróleo, cloruro de metileno, cloroformo, acetato de etilo y butanol. Hay diferentes métodos de extracción pero los más utilizados son: el sólido-líquido y el líquido-líquido. EXTRACCIÓN SÓLIDO - LÍQUIDO La forma más simple para efectuar una extracción sólido-líquido es agitar el sólido, con un solvente dado, en un matraz Erlenmeyer y a continuación decantar o filtrar el solvente, sin embargo, se prefiere realizar esta extracción de una forma continua, utilizando un equipo conocido como extractor Soxhlet (ver figura 1). condensador vapor película porosa para retener el sólido tubo sifón tubo lateral vertical matraz de destilación solvente de extracción Extracción con Equipo Soxhlet FIGURA 1 EXTRACCIÓN LÍQUIDO - LÍQUIDO El embudo de separación (ver figura 2) es muy efectivo para realizar extracciones líquido-líquido a niveles semimicro y macro escala. Utilizando el embudo, los procesos 12 de agitación y separación se efectúan dentro del embudo, en una etapa y por una sola vez. Para llenar el embudo de separación usualmente se soporta sobre un anillo metálico tal y como lo muestra la figura 2. Dado que el embudo es muy fácil de romper, se recomienda que el anillo metálico esté cubierto con tres trozos, pequeños y abiertos, de manguera de hule, a su alrededor. El tapón deberá removerse cuando se drene Anillo metálico Separación de fases FIGURA 2 Se agregan las dos soluciones al embudo, y se maneja tal y como se indica en la figura 3. Es esencial sujetar firmemente el tapón debido a que los dos solventes inmiscibles generan una presión cuando son mezclados, esta presión puede forzar al tapón a salir disparado del embudo. La presión resulta de la suma de las presiones parciales de los solventes cuando son agitados juntos, estando en equilibrio ambos vapores con la solución. El problema de la presión llega a ser especialmente grande cuando se realizan extracciones con bicarbonato de sodio, donde las impurezas acídicas reaccionan con éste para producir dióxido de carbono, causando un incremento en la presión dentro del embudo de separación. Para liberar la presión generada, el embudo es invertido y ventilado abriendo lentamente la llave tal y como se muestra en la figura 3. 13 PELIGRO liberación de gases no dirigir la salida del embudo hacía alguna persona sujetar al mismo tiempo el tapón y la llave de paso firmemente fases líquidas sujetar aquí firmemente Manejo del embudo de extracción durante la agitación de las fases FIGURA 3 EXTRACCIÓN A MICROESCALA A nivel microescala también se pueden realizar extracciones líquido-líquido, en donde se trabaja con mínimas cantidades de sustancia y material pequeño y de fácil adquisición, dentro de las ventajas que se tienen al trabajar con pequeñas cantidades de sustancia, se encuentran: la disminución del costo de la práctica y del riesgo de accidentes por el manejo de reactivos, el poder almacenar los residuos en un espacio más pequeño o tratarlos, de manera económica, para hacerlos inocuos o menos tóxicos. AGENTES DE SECADO Al realizar la separación de las fases, la fase orgánica contiene trazas de humedad por lo que debe ser secada antes de realizar cualquier otra operación, ya que pequeñas cantidades de humedad inhiben la cristalización de algunos sólidos o, algunos líquidos, cuando destilan en presencia de agua, reaccionan con ésta, hidrolizándose, o destilando junto con el agua a una temperatura distinta de sus puntos de ebullición (ver Práctica No. 5, Destilación por arrastre con vapor). Las sales inorgánicas anhídras, como los sulfatos de sodio, calcio y magnesio, son las más utilizadas como agentes de secado, estos compuestos forman hidratos insolubles, removiendo el agua de la fase orgánica.1 Hay tres requisitos básicos para un agente de secado; (1) No reaccionar con la sustancia a secar (2) No disolverse en la sustancia a secar, y (3) Ser fácil y completamente separable de la fase líquida seca. 14 III. EQUIPO • Plancha de calentamiento • Campana de humos IV. MATERIAL IV. 1. Extracción líquido-líquido • • • • • • • • • • • • • • • Aro metálico Soporte metálico Embudo de filtración rápida Embudo de separación de 100 mL (ver figura 2) Frasco de tapón esmerilado de boca ancha Tubo de ensayo de 13 x 100 Agitador de vidrio Papel filtro Pizeta Probeta de 100 mL Matraz Erlenmeyer de 250 mL Vaso de precipitado de 250 mL Vaso de precipitado de 100 mL Vidrio de reloj Perlas de ebullición o pedacería de vidrio IV. 2. Extracción con equipo Soxhlet • • • • • • • • • • • • • • Equipo Soxhlet Tijeras Balanza granataria Matraz bola de 500 mL Mangueras Manta de calentamiento o plancha de calentamiento Soporte metálico Cartucho de papel para Soxhlet Vaso de precipitado de 400 mL Agitador de vidrio o teflón Sistema de destilación simple o rotavapor Vial o frasco de 25 mL con tapa Probeta de 10 mL Pinzas para el Soxhlet 15 V. REACTIVOS V. 1. Extracción líquido-líquido • • • • Ácido benzoico en cloroformo al 2 % P/V Solución de hidróxido de sodio al 20 % P/V Cloruro de calcio o sulfato de sodio anhidros Ácido clorhídrico concentrado V. 2. Extracción con equipo Soxhlet • • • • 250 mL de cloruro de metileno Perlas de ebullición Zacate de limón (6.5 g) Sulfato de sodio anhidro VI. PROCEDIMIENTO VI. 1. Extracción líquido-líquido 1. 2. 3. 4. 5. 6. 7. 8. Medir en la probeta de capacidad de 100 mL, 30 mL de la solución de ácido benzoico en cloroformo al 2% P/V. Transferir la solución del paso 1 al embudo de separación, el cual deberá estar soportado sobre el aro metálico. Medir en la probeta usada en el paso 1, 20 mL de solución de hidróxido de sodio al 20% P/V. Transferir la solución de hidróxido de sodio al 20 % P/V al embudo de separación. Tapar el embudo de separación y proceder a agitar vigorosamente como se muestra en la figura 3 por 1 o 2 minutos. El embudo de separación debe manejarse con ambas manos; con una se sujeta el tapón (asegurándolo con el dedo índice) y con la otra se manipula la llave. Se invierte el embudo y se abre la llave para eliminar la presión generada en el interior, se cierra la llave. Repetir 2 veces más el paso 5. Colocar el embudo en su posición normal (ver figura 2) sobre el aro metálico. Destapar y dejar reposar hasta que sea nítida la separación entre las dos fases. Nota: En caso de que se haya formado una emulsión durante este paso, ésta puede romperse de las siguientes maneras: a) comunicar un movimiento de giro suave al líquido manteniendo la posición normal del embudo de separación; b) agitar vigorosamente la capa emulsionada con una varilla de vidrio; c) saturar la capa acuosa con sal común, lo cuál hace disminuir la solubilidad en agua de la 16 9. 10. 11. 12. 13. 14. 15. 16. 17. 18. 19. 20. mayor parte del soluto y de los disolventes orgánicos, esto se conoce como efecto salino. Separar ambas fases, sacar la inferior por la llave y la superior por la boca para prevenir posibles contaminaciones. Utilizar el vaso de 250 mL para ir recolectando la fase acuosa. Transferir la fase orgánica de nuevo al embudo de separación. Si están identificadas plenamente las fases continuar en el paso 12, si hay dudas continuar en el paso 11. Identificar las fases ensayando la solubilidad en agua. Colocar 1 mL de agua en un tubo de ensayo de 13 x 100 y dejar caer unas gotas de la fase a identificar, si se observa inmiscibilidad de las mismas, entonces es la fase orgánica, en cambio si hay miscibilidad entre las fases es la fase acuosa. Repetir los pasos 3 al 10 dos veces más para completar 3 extracciones. Transferir la fase orgánica a un matraz Erlenmeyer de 250 mL y agregar 0.1 g de agente de secado (cloruro de calcio ó sulfato de sodio anhidros) para eliminar el agua residual. Transferir la fase orgánica seca a un frasco de tapón esmerilado y etiquetarlo como fase orgánica de la práctica de extracción. Neutralizar la fase acuosa (recolectada en el paso 9) con ácido clorhídrico concentrado al cambio del papel tornasol. En este punto el ácido benzoico comenzará a precipitar. Preparar el embudo de filtración rápida colocando el papel filtro. Filtrar. Secar en la estufa, a 90 °C, el ácido benzoico, colocando el papel filtro sobre un vidrio de reloj. Deseche el filtrado en el drenaje, asegurándose que esté neutro. Guarde los cristales secos para la práctica de recristalización. VI. 2. Extracción con equipo Soxhlet 1. 2. 3. 4. Cortar el zacate de limón en trozos de aproximadamente 1 cm. Pesar 6.5 g aproximadamente de zacate de limón cortado. Armar un equipo Soxhlet (ver figura 1). Colocar el zacate en un cartucho para Soxhlet y cubrir la boca del cartucho con un trozo de papel filtro. 5. Colocar el cartucho en la cámara de extracción. 6. Agregar 250 mL de cloruro de metileno al matraz bola. 7. Calentar el sistema y extraer por 1 hora. 8. Desmontar el equipo. 9. Secar el extracto con sulfato de sodio anhidro. 10. Decantar. Guardar el extracto en frasco de capacidad de 250mL para práctica de destilación simple. 17 VII. DIAGRAMA DE FLUJO VII. 1. Extracción líquido-líquido 3 2 1 Medir 30 mL de la solución de ácido benzoico en cloroformo Transferir al embudo de separación la solución del ácido benzoico 1 6 3 2 5 Repetir el paso 5, dos veces más. Dejar reposar hasta que se separen ambas fases 6,7,8 7 4 Tapar el embudo de separación y agitar vigorosamente. Liberar la presión generada en él. 5 Transferir la solución de hidróxido de sodio al embudo de separación donde está la solución del ácido benzoico 4 8 Separar ambas fases, sacando la inferior por la llave y la superior por la boca 9 Recolectar la fase acuosa en un vaso de 250 mL y la fase orgánica regresarla al embudo de separación 9,10 3 NO Medir 10 mL de la solución de hidróxido de sodio al 20 % 10 Repetir pasos 3 a 8 dos veces más Se efectuaron pasos del 3 al 8 tres veces 12 SI 11 Transferir la fase orgánica a un matraz Erlenmeyer de 250 mL y agregar 0.1 g de agente de secado 13 Está identificada la fase orgánica NO 9 SI 10 Colocar 1 mL de agua en un tubo de ensayo; agregar unas gotas de la fase a identificar. Si esta fase es inmiscible en el agua se trata de la fase orgánica 11 12 Una vez seca la fase orgánica transferirla a un frasco de tapón esmerilado y etiquetarlo 14 13 Neutralizar la fase acuosa, recolectada en el paso 9 con ácido clorhídrico concentrado 15 14 18 15 Guardar el precipitado en un frasco de boca ancha y etiquetarlo FIN 14 Filtrar el ácido benzóico precipitado y desechar el filtrado neutro al drenaje. 16, 17, 19 18, 20 VII. 2. Extracción con equipo Soxhlet 2 1 Cortar y pesar el zacate de limón 3 Armar el Soxhlet equipo 3 1, 2 Colocar el zacate en el cartucho. Colocar el cartucho en la cámara de extracción 4, 5 6 5 Desmontar el equipo. Secar el extracto con sulfato de sodio anhidro 4 Calentar el sistema y extraer por una hora . 7 Agregar 250 mL de cloruro de metileno al matraz bola 6 8, 9 7 Decantar el solvente y guardarlo. Ver destilación simple FIN 10 Pasos correspondientes al VI. PROCEDIMIENTO 19 VIII. BIBLIOGRAFÍA 1. Pavia, Donald L., Lampman, Gary M., Kriz Jr., George S.; Introduction to Organic Laboratory Techniques, a Contemporary Approach, tercera edición, Ed. Saunders College Publishing, 1988. 2. Mayo, Dana W., Weston P., Charles, Pike, Ronald M. y Trumper, Peter K., Microscale Organic Laboratory with Multistep and Multiscale Syntheses, tercera edición, John Wiley & Sons, Inc., 1993. 20 CUESTIONARIO 1.- ¿Qué es y como funciona un extractor sohxlet? 2.- ¿En qué casos conviene emplear el método de extracción continua? 3.- Explique brevemente en que consiste separar solutos por medio de la extracción con disolventes. 4.- Nombre y estructura del aceite esencial aislado por medio de extracción contínua 5.- Ejemplos de aplicaciones de la extracción líquido-líquido 21 PRÁCTICA No. 2 CRISTALIZACIÓN I. OBJETIVO Aplicar los conceptos teóricos y prácticos de la técnica de cristalización y aplicarlos para purificar un compuesto orgánico. II. FUNDAMENTO Los compuestos orgánicos que son sólidos a temperatura ambiente normalmente son purificados por cristalización. En general, esta técnica involucra disolver el material a purificar en un solvente (ó mezcla de solventes) caliente y enfriar la solución lentamente. El material disuelto tiene una menor solubilidad a temperaturas bajas, por lo cual precipitará cuando la solución sea enfriada. Este fenómeno es llamado precipitación si el crecimiento del cristal es rápido y no selectivo, y es llamado cristalización si el proceso es relativamente lento y selectivo. La cristalización es un proceso en equilibrio y produce material muy puro. Inicialmente, un cristal “semilla” se forma, creciendo en una forma reversible capa por capa, “seleccionando” de la solución las moléculas correctas (moléculas del mismo compuesto). En contraste, en el proceso de precipitación se forma muy rápido la estructura cristalina, atrapando así impurezas. En general, se debe de evitar un enfriamiento muy lento o muy rápido. El primer problema por resolver al efectuar una cristalización es escoger el solvente en el cual el material a purificar muestre el comportamiento adecuado. Idealmente, el material debe ser poco soluble a temperatura ambiente pero bastante soluble en el punto de ebullición del solvente. Solvente pobre muy soluble a cualquier temperatura gramos solubles Solvente adecuado muy soluble a temperaturas altas poco soluble a temperatura ambiente Solvente pobre poco soluble a cualquier temperatura Temperatura Gráfica de solubilidad versus temperatura FIGURA 1 22 Como se puede observar en la gráfica de la figura 1, la curva de solubilidad para un solvente utilizado en cristalización debe de tener una pendiente pronunciada. Esto significa que el material será poco soluble a temperatura ambiente y muy soluble a temperaturas elevadas. Mientras que una pendiente pequeña no resultará en un cambio significativo en la solubilidad del material cuando existan cambios de temperatura. La solubilidad de compuestos orgánicos es una función de las polaridades tanto del solvente como del soluto. Una escala conveniente para medir la polaridad de un solvente se basa en la constante dieléctrica ε que es una medida para separar cargas opuestas. En general a mayor valor de ε mayor será la polaridad del solvente. Si el soluto es muy polar, se necesitará un solvente muy polar, si el material no es polar, se tendrá que hacer uso de un solvente no polar, manteniéndose la regla de “lo similar disuelve a lo similar”. En la tabla 1 se muestran las solventes orgánicos comunes. ε y otras propiedades de algunos Tabla 1 Propiedades de diferentes solventes Solvente Constante Dieléctrica p. eb. (°C) p. f. (°C) Solubilidad en Agua Flamabilidad 100 100.5 189 153 81.6 65 78 56 118 61 35 80 80.7 0 7 18.5 <0 <0 <0 <0 <0 17 <0 <0 5 6.5 + + + + + + + + + Ligeramente - + + + + + + + + ++ + ++ ε Agua Ácido metanoico Dimetilsulfóxido N,N-Dimetilformamida Acetonitrilo Metanol Etanol Acetona Acido Acético Cloroformo Éter dietílico Benceno Ciclohexano 81 58 48 38 38 32 26 21 6 5 4 2 2 La efectividad de la cristalización depende de la diferencia en solubilidad del material en el solvente caliente y en el solvente frío, lo que se busca es que la sustancia deseada cristalice al enfriar pero no las impurezas. 23 La solución que se obtiene de una cristalización se llama licor madre; una desventaja de este método de purificación es que se pierde mucho material de interés durante el proceso. En general, una cristalización es exitosa solamente si existe una pequeña cantidad de impurezas. Si existen dos sustancias con solubilidades similares, presentes en cantidades similares, éstas no podrán ser separadas. Selección del Solvente Idealmente, un buen solvente para cristalización debe disolver muy poco material de interés a bajas temperaturas, pero mucho material cuando está caliente. Sin embargo, en la práctica esto es más complicado de lo que parece. Frecuentemente, se selecciona un solvente de cristalización experimentando con varios solventes en pequeñas cantidades en tubos de ensayo. Este procedimiento de prueba y error es muy común cuándo uno trabaja con compuestos nuevos. Si los compuestos son conocidos, se pueden consultar tablas o manuales para obtener información respecto al solvente adecuado para cristalización. Al escoger un solvente, se debe tener cuidado en no escoger un solvente con punto de ebullición mayor al punto de fusión del compuesto a cristalizar, esto provocaría una fusión de compuesto y posible separación del solvente; esto haría que al enfriar el solvente, el material de interés se mantenga como una aceite o solidifique, pero no cristalice. Un criterio adicional para la selección del solvente es su volatilidad. Un solvente con bajo punto de ebullición puede ser separado de los cristales por evaporación sin dificultad. (ver tabla 1). III. EQUIPO • • • • • • Balanza analítica Plancha de calentamiento y agitación Manta de calentamiento Reóstato Agitador magnético Bomba de vacío de agua IV. MATERIAL • Embudo de filtración rápida de cuello corto • Vaso de precipitado de 250 mL 24 • • • • • • • • • • • • • • • • • • • • • • • • • Etiquetas chicas Papel aluminio Agitador de vidrio Matraz de fondo plano de 2 bocas de 50 o 100 mL Tapón para matraz bola Condensador Embudo de adición Vaso de precipitado de 150 mL Papel filtro para filtración rápida y vacío Probeta de 100 mL Mechero Espátula Pipeta serológica de 1 mL y 10 mL Tubos de ensayo de 13 x 100 (10) Vidrio de reloj Frasco de seguridad Frasco de boca ancha Embudo Buchner Matraz Quitasato Mangueras Trampa para vacío o sustituto Pinzas para tubo de ensayo Pinzas para soporte Tripié Tela de alambre V. REACTIVOS • Muestra (proveniente de práctica de extracción) • Carbón activado • Solventes: Agua destilada Metanol Etanol 95% Ácido acético Acetona Éter etílico Cloroformo Isopropanol 25 VI. PROCEDIMIENTO Elección del Solvente 1. Preparar 8 cápsulas de papel aluminio. 2. Pesar en una balanza analítica 8 porciones de aproximadamente 25 mg del sólido a cristalizar utilizando cada una de las cápsulas hechas con papel de aluminio. 3. Colocar cada porción del sólido a cristalizar en 8 tubos de ensaye de 13 x 100. 4. Etiquetar cada tubo con el nombre del solvente que se va a probar. 5. Colocar un vaso de precipitado de 250 mL con agua en una plancha de calentamiento y calentarla hasta ebullición. 6. Manejar cada uno de los tubos de ensaye por separado. 7. Añadir al tubo de ensaye 0.5 mL del solvente a probar, mediante una pipeta serológica de 1 mL; si hay disolución total a temperatura ambiente, descartar dicho solvente para la cristalización. Continuar en el paso 12, si no hay disolución continuar en el siguiente paso. 8. Colocar el tubo de ensaye conteniendo solvente en el vaso de precipitado con agua. En el caso de que el solvente sea agua o ácido acético se puede calentar cuidadosamente, directo con el mechero. 9. Llevar a ebullición el solvente. No permitir evaporación total. 10. Observar el tubo; si el sólido se ha disuelto, esperar a que se enfríe a temperatura ambiente y observar si cristaliza, de ser así puede ser considerado como solvente de cristalización; continuar en el paso 13. Si no se disolvió o si precipita antes de enfriar, continuar en el siguiente paso. 11. Agregar porciones de 0.1 mL de solvente (máxima cantidad de solvente a agregar 2 mL) y calentar hasta observar si hay disolución y agitar, si hay cristalización al enfriar, continuar en el paso 13. Si no hay disolución, continuar en el siguiente paso. 12. Desechar (en el depósito de residuos de solventes orgánicos) el solvente. 13. Calcular la solubilidad (mg/mL) para cada solvente en su temperatura de ebullición correspondiente y seleccionar el mejor solvente de cristalización. Nota: El solvente más adecuado es aquel que presente el mayor poder de solubilidad en su temperatura de ebullición y el soluto cristalice adecuadamente al enfriarse el solvente. Cristalización 14. Pesar la cantidad de muestra que se va a cristalizar en un matraz bola de 2 bocas de capacidad adecuada. 15. Calcular la cantidad de solvente requerida de acuerdo a la cantidad de soluto a cristalizar. 16. Añadir la mitad de la cantidad requerida del solvente elegido y núcleos de ebullición al matraz bola. Agregar un exceso para evitar la cristalización en embudo. 26 17. Sostener el matraz bola con unas pinzas conectadas a un soporte metálico. 18. Conectar al matraz bola un condensador a reflujo con agua refrigerante circulando y un embudo de adición. (Ver figura 2). Reflujo con adición FIGURA 2 19. Colocar un baño de agua en una plancha de calentamiento e introducir el matraz bola en el baño o bien colocar el matraz en una manta de calentamiento. En el caso de que el solvente sea agua o ácido acético se puede calentar cuidadosamente, directamente con el mechero. 20. Calentar a ebullición la mezcla contenida en el matraz. Agregar lentamente el resto de la cantidad del solvente elegido a través del embudo de adición, antes de agregar toda la cantidad de solvente, observar: si no hay disolución completa, continuar en el paso 21; si hay disolución total, agregar un 15 % de exceso de solvente y continuar en el paso 23. 21. Agregar lentamente el resto de la cantidad del solvente elegido a través del embudo de adición. 22. Continuar con el calentamiento a ebullición hasta disolución del sólido. Agregar un 15 % de exceso de solvente. 23. Desconectar el embudo de adición del matraz y agregar una pequeña cantidad (la punta de una espátula) de carbón activado a la mezcla. 24. Precalentar el embudo de filtración rápida haciéndole pasar solvente caliente y filtrar la disolución en caliente. 25. Recibir el filtrado caliente en un vaso de precipitado limpio de capacidad adecuada y permitir que el filtrado se enfríe hasta temperatura ambiente. Si los cristales no aparecen seguir con el paso 26, si aparecen seguir con el paso 27. 27 26. Inducir la cristalización raspando las paredes del recipiente utilizando un varilla de vidrio o agregando un cristal del soluto (tomarlo de los tubos de ensayo en donde se realizaron las pruebas) o enfriando la solución en un baño de agua con hielo. 27. Filtrar a vacío la solución que contiene a los cristales utilizando el embudo Buchner (ver figura 3). Ver referencia 1. 28. Lavar los cristales usando pequeñas cantidades de solvente frío para remover el licor madre adherido a la superficie de los cristales. 29. Secar los cristales en el papel filtro. 30. Pesar los cristales y calcular el porciento de recuperación del sólido purificado. 31. Colocar los cristales con todo y papel filtro en un frasco de boca ancha, para ser utilizado en la práctica de punto de fusión. EMBUDO BUCHNER VACIO MATRAZ QUITASATO TRAMPA Filtración a vacío con matraz quitasato y embudo Buchner FIGURA 3 28 VI. DIAGRAMA DE FLUJO Elección del Solvente 2 1 Colocar 25 mg del sólido en cada uno de 8 tubos de ensayo. Etiquetar cada tubo con el nombre del solvente a probar 3 Colocar un vaso de precipitado con agua en una plancha de calentamiento y calentarla hasta ebullición Manejar cada uno de los tubos de ensayo por separado 6 5 1,2,3,4 4 SI EL SOLVENTE ES INFLAMABLE NO Agregar a cada uno de los tubos de ensayo 0.5 mL del solvente a probar. SE DISUELVE EL SOLUTO 6 SI 7 10 NO 5 7 6 Calentar cuidadosamente directo con el mechero Colocar cada uno de los tubos de ensayo conteniendo solvente en el vaso de precipitado con agua caliente. 8 8 Calentar cada uno de los tubos de ensayo conteniendo solvente, hasta ebullición 9 8 8 9 Agregar porciones de 0.1 mL de solvente (agregar como máximo 2 mL) y calentar hasta observar disolución 13 11 NO Observar si hay disolución y anotar observaciones en el diario del laboratorio SE DISUELVE EL SOLUTO SI 10 10 29 10 11 Esperar a que enfríe a temperatura ambiente el solvente y observar si cristaliza el soluto CRISTALIZA EL SOLUTO SI Considerar a este solvente como posible solvente de cristalización 10 10 NO FIN 13 12 Descartar al solvente probado como solvente de cristalización Calcular la solubilidad (mg/mL) para cada solvente y seleccionar el mejor solvente de cristalización 1 2 13 14 Cristalización 14 15 Pesar la cantidad de muestra que se va a cristalizar en un matraz bola de 2 bocas de capacidad adecuada y calcular la cantidad requerida de solvente 19 14, 15 Calentar a ebullición la mezcla contenida en el matraz bola. En el caso de que el solvente sea agua o ácido acético se puede calentar directo con el mechero 20 19, 20 16 Añadir la mitad de la cantidad requerida del solvente elegido y núcleos de ebullición al matraz bola Sostener el matraz bola con unas pinzas conectadas a un soporte metálico 17 16 18 17 Colocar un baño de agua en una plancha de calentamiento e introducir el matraz bola en el baño o bien colocar el matraz en una manta de calentamiento Conectar al matraz bola un condensador a reflujo y un embudo de adición 18 19 30 20 21 Agregar lentamente el solvente elegido a través del embudo de adición Continuar con el calentamiento a ebullición, antes de agregar todo el solvente, observar 21 22 24 Precalentar el embudo de filtración rápida y filtrar la disolución en caliente. Recibir el filtrado caliente en un vaso de precipitado 24, 25 HAY DISOLUCIÓN Agregar el resto de la cantidad del solvente elegido. Continuar con el calentamiento hasta disolución del sólido. Agregar un 15 % de exceso de solvente Agregar un 15 % de exceso de solvente. Desconectar el embudo de adición del matraz bola y Agregar una pequeña cantidad de carbón activado 23 4 HAY CRISTALIZACIÓN SI 26 23 20, 23 Filtrar a vacío utilizando un embudo Buchner (ver figura 3) 27 NO 29 Inducir la cristalización, raspando con una varilla de vidrio las paredes del matraz o agregando un cristal del soluto o enfriando en baño de agua y hielo SI 22 25 27 23 23 25 Permitir que el filtrado se enfríe hasta temperatura ambiente NO 28 Secar, pesar, calcular el porciento de recuperación del sólido purificado y guardar los cristales en un frasco de boca ancha limpio y etiquetado Lavar los cristales que se quedan en el papel filtro con pequeñas cantidades de solvente frío 28 29, 30, 31 26 FIN 26 - Pasos correspondientes al VI. Procedimiento 31 VIII. BIBLIOGRAFÍA 1. Pavia, Donald L., Lampman, Gary M., Kriz Jr., George S., Introduction to Organic Laboratory Techniques, A Contemporary Approach, tercera edición, Saunders College Publishing, 1988. 2. Hart, R.D. Schuetz, Organic Chemistry, A Short Course, quinta edición, Hougton Mifflin Company, 1978. 3. Popp, H.P. Schultz, Organic Chemical Preparations, W.B. Saunders Company, 1964. 4. Domínguez, Xorge A., Domínguez, S. Xorge Alejandro, Química Orgánica Experimental, Editorial Limusa, primera edición, 1982. 32 CUESTIONARIO 1.- ¿Cuáles fueron los disolventes usados para hacer la cristalización? 2.- ¿Qué características presentan el par de disolventes? 3.- ¿Por qué es importante mantener el volumen de la solución durante el calentamiento? 4.- ¿Qué diferencias encuentra entre la sustancia pura y la sustancia sin purificar? 33 PRÁCTICA No. 3 DESTILACIÓN SIMPLE Y FRACCIONADA A PRESIÓN NORMAL Y PRESIÓN REDUCIDA I. OBJETIVO Aprender la técnica de la destilación simple y fraccionada tanto a presión normal como presión reducida, así como adquirir el criterio para aplicar estas técnicas y saber montar correctamente el equipo correspondiente. II. FUNDAMENTO II.1. Destilación La destilación es un método para purificar sustancias que son líquidas a temperatura ambiente. Este proceso consiste en que una sustancia es llevada, por medio de calentamiento, a la fase vapor, para luego ser condensada cuando se hace pasar por un medio refrigerante. La técnica es útil para purificar un líquido o bien para separar una mezcla de líquidos cuando los componentes tienen diferentes puntos de ebullición y siguen un comportamiento ideal, de acuerdo a la ley de Raoult, es decir, no forman azeótropos. El químico puede disponer de cuatro métodos básicos de destilación que son: destilación simple, destilación fraccionada, destilación a presión reducida (fraccionada y simple) y destilación por arrastre con vapor. II. 2. Puntos de ebullición El punto de ebullición es la constante física en la cual se fundamenta la destilación. Cuando un líquido es calentado, su presión de vapor se incrementa hasta igualar la presión atmosférica, en este punto la temperatura se eleva hasta alcanzar un valor constante y se observa que el compuesto ebulle. La temperatura a la cual la presión de vapor del líquido se iguala a la presión atmosférica se conoce como punto normal de ebullición y es una constante característica del compuesto en cuestión. La relación entre la presión aplicada y la temperatura de ebullición de un líquido está dada en la siguiente gráfica (ver figura 1). 34 Peb. a:760 mm de Hg 760 presión de vapor (mm de Hg) Peb. a: 100 mm de Hg 100 temperatura Relación entre presión de vapor y temperatura FIGURA 1 Ya que la ebullición depende de la presión del sistema, es importante registrar la presión barométrica a la cual se lleva a cabo la destilación. Como una regla, no muy exacta, el punto de ebullición disminuye 0.5 °C por cada 10 milímetros de descenso en la columna de Hg cuando se trabaja a una presión cercana a la atmosférica, y 10 ºC por cada 10 mm de descenso en la columna de Hg cuando se trabaja a presiones cercanas a cero. Una mayor aproximación del cambio del punto de ebullición con la variación de la presión puede ser obtenida por el uso de un nomograma. (ver figura 2). La manera de usar el nomograma presentado en la figura 2, es la siguiente: suponer que la temperatura de ebullición reportada es 100 oC a 1 mm, para determinar la temperatura de ebullición a 18 mm, conectar 100 oC en la columna A a 1 mm (columna C) con una regla transparente de plástico y observar donde intersecta esta línea con la columna B (alrededor de 280 oC). Este valor podría corresponder al punto de ebullición normal, luego, conectar 280 oC (columna B) con 18 mm (columna C) y observar donde intersecta la línea con la columna A (151 oC). La temperatura de ebullición aproximada será 151 oC a 18 mm. II. 3. Destilación Simple Cuando un líquido puro es calentado en el matraz destilador (ver figura 3) el vapor de éste se eleva hasta llegar a ponerse en contacto con el bulbo del termómetro donde se establecerá un equilibrio líquido-vapor, ya que el vapor que llega se condensa, en ese instante, la temperatura permanece constante y el vapor que pasa por el refrigerante se 35 condensa. En este proceso la temperatura no varía porque la composición de la fase líquida y la del vapor en equilibrio es la misma en todo momento. PUNTO DE EBULLICIÓN OBSERVADO A “P” mm o C A PUNTO DE EBULLICIÓN CORREGIDO A 760 mm o PRESIÓN “P” mm 0. 1 C C B Nomograma de alineamiento presión -temperatura FIGURA 2 Cuando una mezcla de líquidos es calentada, la temperatura por lo general no permanece constante, sino que varía todo el tiempo, esto se debe a que en una mezcla la composición del vapor en equilibrio con la solución calentada, no es la misma como es en el caso de un líquido puro. Lo anterior puede ser fácilmente deducido de la gráfica mostrada en la figura 4. En esta figura se muestra un diagrama de fases ilustrativo del comportamiento de una mezcla de dos componentes (A y B). En el diagrama, las líneas horizontales representan temperaturas constantes, la curva superior la composición de la fase vapor y la inferior la del líquido. 36 termómetro adaptador para termómetro Condensador o refrigerante cabeza de destilación Tubo de salida o adaptador para vacío matraz destilador matraz colector salida de agua entrada de agua Equipo para Destilación Simple FIGURA 3 P.eb. A VAPOR TEMPERATURA X t Y P.eb. B LÍQUIDO 100% A W COMPOSICIÓN Z 100% B Diagrama de fases FIGURA 4 37 Para la línea horizontal t habrá dos intersecciones, una con la curva de composición del líquido, y la otra con la línea de composición del vapor que está en equilibrio a dicha temperatura. La intersección X con la curva del líquido nos proporciona la composición de la solución W la intersección Y con la curva del vapor nos da la composición de la solución Z. Una mezcla A y B de composición W tendrá el siguiente comportamiento al ser calentada. La temperatura del líquido aumentará hasta alcanzar su punto de ebullición en la línea horizontal t la cual corresponde a la línea WX y comienza a vaporizar a lo largo de la línea XY, el vapor tendrá entonces una composición Z; en otras palabras, el primer vapor que destila de la mezcla no contiene solo B, es más rico en B que la mezcla original pero tiene una cantidad apreciable de A desde el inicio de la destilación. El resultado es que no es posible separar completamente una mezcla por destilación simple. Existen dos casos en los cuales es posible obtener una separación aceptable de los compuestos relativamente puros. En el primer caso, los componentes deberán tener una diferencia en sus puntos de ebullición mayor a 100 ºC. Un segundo caso es cuando B tiene una concentración en la mezcla menor que 10 %. Cuando la diferencia de puntos de ebullición no es muy grande y se requiera una alta pureza, se debe emplear la destilación fraccionada. La posición del bulbo de mercurio del termómetro es básica para obtener una correcta lectura de la temperatura de ebullición del líquido, el termómetro debe colocarse en la corriente de vapor (ver figura 3). En el matraz destilador se coloca la mezcla a destilar, cuidando de no rebasar la tercera parte del volumen del mismo, pues es necesario guardar un espacio suficiente para que el líquido ebulla, tampoco debe usarse un volumen muy bajo porque esto dificulta la destilación, ya que el líquido solamente refluiría, por la gran diferencia de tamaño de la muestra y el matraz. Cuando se calienta el sistema, la temperatura del líquido se incrementa hasta alcanzar el punto de ebullición, en ese momento un anillo del condensado (anillo de reflujo) asciende y toca el bulbo del termómetro, esto hace que la columna de mercurio se eleve bruscamente llegando a un punto estacionario, o sea, el punto de ebullición, después el vapor pasa al refrigerante y condensa. Para la destilación podemos emplear dos tipos de condensadores: el condensador de agua y el condensador de aire. El condensador de agua es el mostrado en la figura 3 (éste tiene una chaqueta de enfriamiento). Sin embargo, cuando el líquido que va a ser destilado, tiene un alto punto de ebullición (mayor de 180 ºC a la presión de trabajo) se debe emplear un condensador de aire, el cual no tiene chaqueta de enfriamiento (o simplemente no se recircula agua en el condensador mostrado en la figura 3). Esto, entre otras razones, es porque si se emplea un condensador de agua la alta temperatura del vapor caliente 38 puede causar un choque térmico con la temperatura del refrigerante y éste podría romperse. II. 4. Destilación Simple a Presión Reducida La destilación a presión reducida se usa cuando los compuestos que se requiere purificar presentan puntos de ebullición muy elevados (arriba de 200 ºC) o cuando estos se descomponen al alcanzar su punto de ebullición a la presión atmosférica. Por ejemplo un líquido que tenga un punto de ebullición de 200 ºC a presión normal, tendrá un punto de ebullición de 90 ºC a 20 mm de Hg. El nomograma de la figura 2, puede ser usado para determinar cual va a ser la nueva temperatura de ebullición a una presión dada para cualquier líquido no asociado (que no forma puentes de hidrógeno). La técnica a presión reducida se efectúa empleando casi el mismo equipo de la destilación simple (ver figura 3) con cuatro variantes, una es la intercalación de un tubo de Claisen (ver figura 8) (donde se coloca un burbujeador) entre el matraz destilador y la cabeza de destilación; otra es el uso de un recolector de fracciones llamado “vaquita” (ver figura 5); otro recolector de fracciones es el que se presenta en la figura 6, los cuales son muy útiles para recolectar fracciones a diferentes intervalos de temperatura, sin tener que detener la destilación para cambiar el matraz colector; otra variante con respecto al equipo para destilación simple, es el uso de un manómetro (hay de varios tipos: digitales, de aguja, etc. en la figura 7 se muestran algunos de mercurio); el uso de un sistema que proporcione el vacío necesario para trabajar es, por último, la cuarta diferencia. Este último puede ser una trampa de agua, una bomba de agua para vacío o una bomba de aceite para alto vacío. La trampa puede proporcionar hasta 400 mm de Hg de presión interna, mientras que la bomba de aceite puede dar menos de 0.5 mm de Hg. Las bombas de agua pueden alcanzar hasta 25 mm de Hg. En la figura 8 se muestra un equipo para destilación simple a presión reducida donde se muestran todas las diferencias mencionadas anteriormente, excepto los colectores de destilado (ver figuras 5 y 6) los cuales serían colocados en lugar del matraz colector mostrado en la figura 8. Colector de fracciones (“vaquita”) FIGURA 5 39 vacío aire Matraz recolector de fracciones en una destilación a presión reducida FIGURA 6 Manómetros o vacuómetros FIGURA 7 En la figura 8 vemos las diferentes partes de que consta el equipo, el tubo A puede suprimirse, empleando en su lugar agitación magnética. Las mangueras para conectar el equipo al sistema de vacío deberán ser de pared gruesa, además, se debe revisar cuidadosamente el material de vidrio, cualquier fisura puede provocar una implosión. Cuando se trabaja a bajas presiones (con bomba de vacío) se hace necesaria una buena trampa para proteger la bomba así como lubricar las juntas esmeriladas con grasa de silicona para alto vacío. VI. 5. Destilación Fraccionada Es frecuente que un componente en una mezcla, no solo es la mayor parte de una muestra sino que también tiene un punto de ebullición muy cercano al componente de interés. En este caso, una destilación simple, no efectuará ninguna separación. 40 pinzas para control de vacío C adaptador para termómetro llave de paso B vacío A tubo T o Y manómetro Trampa para presiones moderadamente bajas tubo Claisen adaptador para vacío tubo generador de burbujas burbujador alternativo agua matraz recolector Equipo para destilación simple a presión reducida FIGURA 8 Como ya sabemos, el condensado inicial, será enriquecido con el componente más volátil. Si este condensado inicial C1 (ver figura 3) es redestilado, el condensado inicial de este segundo destilado C2 será más enriquecido del componente más volátil, al mismo tiempo, el matraz destilador, se enriquecerá del componente menos volátil. Por una continua repetición de este proceso (una serie de destilaciones simples), podremos llegar a separar dicha mezcla, obviamente esto consumiría mucho tiempo y sería muy tedioso. Afortunadamente, usando una columna de fraccionamiento sobre el matraz destilador (ver figura 9) es posible efectuar la misma separación en una sola operación. 41 Existen varios tipos de columnas de fraccionamiento que se pueden emplear, como las mostradas en la figura 10, las columnas A y B (ésta última es plateada para mantener mejor el calor durante la destilación) pueden ser empacadas con cualquier material, la C es del tipo Vigreux, y la D es una columna tipo Hempel. Columna de fraccionamiento Equipo de Destilación Fraccionada FIGURA 9 La columna Vigreux tiene pequeños dientes internos hacia abajo, la Hempel es del tipo empacada con pedacería de vidrio o con anillos o hélices de acero inoxidable. Ambos tipos de columnas presentan una gran área de superficie donde el equilibrio líquidovapor se establece un número muy grande de veces, esto equivaldría a un número igual de destilaciones simples, en un mismo proceso. A B C D Columnas para Destilación Fraccionada FIGURA 10 42 Cuando el matraz destilador es calentado, el vapor es enriquecido en el componente más volátil, este vapor se mueve y llega a la parte inferior de la columna donde entra en contacto con el empaque, aquí el líquido formado es de nuevo calentado con los vapores que vienen del matraz destilador y vuelven a ascender con una composición más rica en el más volátil, el menos volátil retorna al destilador efectuándose una segunda destilación. El vapor en ascenso encontrará por supuesto mas empaque y esta condensación de vapores, calentados por otros vapores, continuará una secuencia de redestilaciones sucesivas hasta alcanzar la parte superior de la columna. De esta manera, el vapor que llega a la cabeza de destilación consiste de un componente puro. La eficiencia de una columna, es dada por el número total de platos teóricos2. Es importante aclarar que los equipos de destilación mostrados aquí son solo un ejemplo de los muchos diseños que se encuentran en el mercado. Existen equipos para destilación simple y fraccionada para trabajo a microescala (ver figura 11), así mismo existen diseños especiales que se usan en cierto tipo de industrias (ver figura 12), los arreglos, dependiendo del tipo y cantidad de muestra son muy variados (ver figura 13). Sin embargo, el funcionamiento de todos ellos se basan en el mismo concepto que se ha explicado en las secciones anteriores. Equipo de destilación usado en microescala FIGURA 11 43 vacío aire Equipo de destilación fraccionada a presión reducida FIGURA 12 Equipos de destilación para diferentes aplicaciones FIGURA 13 III. EQUIPO • Plancha de calentamiento con agitación o bien manta de calentamiento con reóstato y agitador magnético 44 • Bomba de vacío o trampa de vacío según la presión a que se debe llevar a cabo la destilación. • Manómetro • Baño de enfriamiento • Bomba de recirculación IV. MATERIAL • Equipo Exelo o Corning (matraz destilador, refrigerante, cabeza de destilación, matraz colector y adaptador para vacío) • Termómetro • Vaso de precipitado de 600 mL para baño María (si no hay manta de calentamiento) • Soportes (2) • Espátula • Pinzas para soporte (4) • Mangueras de hule (2) • Vaquita con 3 matraces de 25 mL unión esmerilada (ver figura 5) • Barrita magnética • Matraz quitasato • Mangueras de pared gruesa (2) • Trampa (para agua o solventes) para proteger la bomba de vacío • Unión T de vidrio • Llave de Mohr para controlar la presión • Tubo de Claisen • Vaso Dewar (termo) • Núcleos de ebullición V. REACTIVOS • • • • • Muestras a destilar (ver extracción contínua, propanol− 2 octanol) Vaselina y/o grasa silicona para alto vacío Dióxido de carbono sólido (sí se trabaja al alto vacío) Acetona (sí se trabaja al alto vacío) Sulfato de magnesio o de sodio anhidro VI. PROCEDIMIENTO VI. 1. Destilación a Presión Normal 1. 2. 3. Lavar y secar perfectamente el equipo de vidrio que se va a emplear (ver figura 3). Revisar el material de vidrio para detectar fisuras. Secar el líquido a destilar añadiendo, con una espátula sulfato de sodio anhidro y decantarlo en el matraz destilador, cuidando que no se llene más de la mitad. Armar el equipo de vidrio para destilación simple (ver figura 3) 45 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. 14. 15. 16. 17. Lubricar las juntas esmeriladas con vaselina. Sujetar el matraz destilador y el refrigerante con pinzas. Sujetar el colector de tal manera que pueda ser fácilmente separado. Agregar al matraz de destilación algunas perlas (núcleos) de ebullición. Nota: Si se va a usar agitación magnética, no se agregan las perlas de ebullición. Pesar el matraz de destilación con las perlas de ebullición. Pesar la muestra líquida a destilar y transferirla al matraz de destilación. Pesar el matraz de destilación con las perlas de ebullición y la muestra. Anotar los datos de los pesos en el diario de laboratorio. Hacer pasar el agua por el refrigerante, cuidando que la manguera de entrada de agua se conecte de tal manera que el agua entre en contra de la fuerza de gravedad. Calentar el contenido del matraz de destilación empleando un baño de agua o de aceite o bien una manta de calentamiento, según sea el punto de ebullición del compuesto a destilar, dicho calentamiento debe ser tal que el destilado se obtenga con una velocidad no mayor a una gota por segundo. Nota: Es muy importante mantener el sistema abierto a la atmósfera cuando se trabaja a presión normal pues si esto no se hace, se provoca una explosión. Recoger el destilado en el matraz colector previamente pesado. Medir y anotar el intervalo de temperatura al cual destila la muestra. Nota: No calentar el matraz a sequedad. Pesar los matraces de destilación y colector y anotar los datos en el diario. Guardar el destilado en un frasco adecuado, debidamente etiquetado. VI. 2. Presión Reducida (400 mm. de Hg o mayores) 1. Efectuar los pasos 1 y 2 del procedimiento anterior VI. 1. Si la muestra líquida a destilar contiene un solvente volátil eliminarlo previamente, efectuando todos los pasos indicados en VI. 1. 2. Ensamblar el aparato de la figura 8, sin incluir el burbujeador. Para conectar el adaptador de vacío a la trampa (usar una trampa de agua) y el manómetro se deben usar mangueras de pared gruesa. 3. Lubricar las juntas esmeriladas con grasa silicona. 4. Sujetar el matraz destilador y el refrigerante con pinzas. 5. Sujetar el colector tipo “vaquita” (pesar previamente todos los matraces) de tal manera que pueda ser fácilmente separado. Si no se dispone del colector tipo “vaquita” usar un matraz como se muestra en la figura 8 y suspender el vacío cada que se reciba una nueva fracción. 6. Efectuar los pasos 6 a 12 del procedimiento anterior VI. 1. 7. Abrir las pinzas en el control de vacío C. 8. Abrir la trampa de vacío para conectar el sistema de vacío 9. Cerrar las pinzas del control de vacío C cuidando de que el líquido no ebulla ni se vomite a los matraces colectores y abrir la llave de paso B (para conectar el manómetro) hasta alcanzar la presión deseada en el manómetro. 10. Calentar hasta que el líquido destile. 46 11. Colectar las diferentes fracciones de acuerdo a su rango de ebullición, rotando cuidadosamente la unión esmerilada de la vaquita, cuidando que la presión interna del sistema no varíe. Nota: No calentar el matraz a sequedad. 12. Al terminar la destilación se abren las pinzas del control de vacío C y se retiran los matraces de la vaquita, la agitación y el calentamiento. 13. Pesar los matraces de destilación y colector y anotar los datos en el diario. 14. Guardar las fracciones destiladas en frascos debidamente etiquetados. VI. 3. Alto Vacío 1. Efectuar los pasos 1 y 2 del procedimiento VI. 1. Si la muestra líquida a destilar contiene un solvente volátil eliminarlo previamente, efectuando todos los pasos indicados en VI. 1. 2. Ensamblar el aparato de la figura 8. (con la opción de usar un colector como el de las figuras 5 o 6). En lugar de la trampa mostrada en la figura 8 usar la que se indica en los pasos 3 y 4. 3. Preparar una mezcla de hielo seco machacado, y acetona en un vaso Dewar. 4. Introducir la trampa de agua y solventes en el vaso Dewar (un vaso térmico). 5. Efectuar los pasos 6 a 12 del procedimiento VI. 1. 6. Calentar ligeramente el matraz destilador mediante una manta de calentamiento conectada a un reóstato. Si no se usa el burbujeador, colocar en el matraz destilador una barra magnética y encender la plancha de agitación. 7. Conectar la bomba de alto vacío con las pinzas de control de presión abierta C. 8. Cerrar las pinzas del control de vacío C cuidando de que el líquido no ebulla ni se vomite a los matraces colectores y abrir la llave de paso B (para conectar el manómetro) hasta alcanzar la presión deseada en el vacuómetro. 9. Continuar el calentamiento gradualmente con mucha precaución hasta que destilen las diferentes fracciones de la muestra. 10. Efectuar los pasos 11 a 14 del procedimiento VI. 2. VI. 4. Destilación fraccionada 1. 2. 3. 4. 5. 6. 7. 8. 9. Lavar y secar perfectamente el equipo de vidrio que se va a emplear (ver figura 9) Secar el líquido a destilar añadiendo con una espátula sulfato de sodio anhidro y decantarlo en el matraz destilador, cuidando que no se llene más de la mitad. Empacar la columna de fraccionamiento con el material seleccionado. Armar el equipo de vidrio usado para destilación fraccionada (ver figura 9). Efectuar los pasos 4 a 13 del procedimiento VI. 1. Recoger en diferentes colectores (previamente pesados) las fracciones destiladas dependiendo del intervalo de ebullición. Medir y anotar el intervalo de temperatura al cual destila cada fracción. Nota: No calentar el matraz a sequedad. Pesar los matraces de destilación y colectores y anotar los datos en el diario. Guardar las fracciones destiladas en un frasco adecuado debidamente etiquetado. 47 VII. DIAGRAMA DE FLUJO VII. 1. Destilación a Presión Normal 1 2 3 Lavar y secar perfectamente el equipo de vidrio que se va a emplear (ver figura 3) Revisar el material de vidrio para detectar fisuras Secar el líquido a destilar con sulfato de sodio anhidro y decantarlo en el matraz destilador 1 1 2 6 5 4 Sujetar el colector de tal manera que pueda ser fácilmente separado. Lubricar las juntas esmeriladas con vaselina. Sujetar el matraz destilador y el refrigerante con pinzas Armar el equipo de vidrio para la destilación simple (ver figura 3) 6 3 4,5 7 8 9 Agregar al matraz de destilación algunos núcleos de ebullición, si es que no se va a usar agitación magnética Pesar el matraz de destilación con las perlas de ebullición, la muestra líquida a destilar y el matraz de destilación con las perlas de ebullición y la muestra Anotar los datos de los pesos en el diario de laboratorio. Hacer pasar agua por el refrigerante 7 8,9,10 12 11 Recoger el destilado en el matraz colector previamente pesado. Medir y anotar el intervalo de temperatura al cual destila la muestra. Nota: No calentar el matraz a sequedad Destilar a una velocidad no mayor de una gota por segundo. Mantener el sistema de destilación abierto 13 11,12 10 Calentar el contenido del matraz de destilación, según sea el punto de ebullición del compuesto a destilar 13 13 14,15 48 13 Pesar los matraces de destilación y colector y anotar los datos en el diario. Guardar el destilado en un frasco adecuado, debidamente etiquetado FIN 16,17 VII. 2. Presión Reducida (400 mm. de Hg o mayores) 2 1 Efectuar los pasos 1 a 3 del diagrama de flujo VII. 1. SI La muestra líquida contiene un solvente volátil 1 Destilar solvente volátil efectuando todos los pasos 4 a 13 del diagrama anterior VII. 1. 1 NO 3 5 4 Sujetar el colector (pesar previamente) de tal manera que pueda ser fácilmente separado. Lubricar las juntas esmeriladas con grasa de silicona. Sujetar el matraz destilador y el refrigerante con pinzas 5 Ensamblar el aparato de la figura 8, sin el burbujeador. Para conectar el adaptador de vacío a la trampa y el manómetro usar mangueras de pared gruesa. 3,4 2 6 7 8 Efectuar los pasos 6 a 9 del diagrama de flujo VII. 1. Abrir las pinzas en el control de vacío C (ver figura 8) Abrir la trampa de vacío para conectar el sistema de vacío 6 7 9 8 49 9 10 11 Cerrar las pinzas del control de vacío C cuidando de que el líquido no ebulla a saltos ni se vomite al matraz colector Abrir la llave de paso B hasta alcanzar la presión deseada en el manómetro Calentar con precaución hasta que el líquido destile. 9 9 14 10 12 13 Colectar las fracciones de acuerdo a su rango de ebullición, cuidando que la presión interna del sistema no varíe Abrir las pinzas del control de vacío C al terminar la destilación. Retirar el matraz colector, la agitación y el calentamiento Pesar los matraces de destilación y colector y anotar los datos en el diario. 13 11 12 15 Guardar las fracciones destiladas en frascos debidamente etiquetados FIN 14 VII. 3. Alto Vacío 2 1 Efectuar los pasos 1 a 3 del diagrama de flujo VII. 1. SI La muestra líquida contiene un solvente volátil 1 Destilar solvente volátil efectuando todos los pasos 4 a 13 del diagrama anterior VII. 1. 1 NO 5 4 3 Introducir la trampa de agua y solventes en el vaso Dewar Preparar una mezcla de hielo seco machacado, y acetona en un vaso Dewar. Ensamblar el aparato de la figura 8. (con la opción de usar un colector como el de las figuras 5 o 6) 4 6 3 2 50 6 7 8 Efectuar los pasos 6 a 9 del diagrama de flujo VII. 1. Calentar ligeramente el matraz destilador mediante una manta de calentamiento Agitar la muestra a destilar mediante agitador magnético 5 6 11 10 Abrir la llave de paso B hasta alcanzar la presión deseada en el vacuómetro Cerrar las pinzas en C cuidando de que el líquido no ebulla a saltos ni se vomite a los matraces colectores 8 12 9 Conectar la bomba de alto vacío manteniendo las pinzas de control de presión C abierta 7 8 13 Continuar el calentamiento gradualmente con mucha precaución hasta que destile la muestra 6 Efectuar los pasos 13 a 16 del diagrama de flujo VII. 2. FIN 10 9 VII. 4. Destilación fraccionada 1 2 3 Lavar y secar perfectamente el equipo de vidrio que se va a emplear (ver figura 9) Secar el líquido a destilar con sulfato de sodio anhidro. Decantar en el matraz destilador Empacar la columna de fraccionamiento con el material seleccionado 1 2 3 6 5 4 Recoger en diferentes colectores (previamente pesados) las fracciones destiladas Efectuar los pasos 5 a 11 del diagrama de flujo VII. 1. Armar el equipo de vidrio usado para destilación fraccionada (ver figura 9) 6 7 5 4 51 7 8 9 Medir y anotar el intervalo de temperatura al cual destila cada fracción Pesar los matraces de destilación y colectores y anotar los datos en el diario Guardar las fracciones destiladas en un frasco adecuado etiquetado. 7 8 Pasos correspondientes al VI. PROCEDIMIENTO 9 FIN VIII. BIBLIOGRAFÍA 1. Pavia, Donald L. Lampam, Gary M. y Kriz Jr, Goerge S., Introduction to Organic Laboratory Techniques a Contemporary Approach; tercera edición, Saunders College Publishing, 1988. 2. Krubsack, Arnold J., Experimental Organic Chemistry, Allyn and Bacon Inc., Boston, 1973. 52 CUESTIONARIO 1.- ¿Cómo 2.- ¿Qué varía el punto de ebullición de un líquido con la presión externa? . es la destilación y cuáles son sus ventajas? 3.- Dé las diferencias entre una destilación a presión reducida y una destilación a presión normal, y si alguna de las dos necesita agitación, explicar el por qué. 4.- Razón por la cual se deben de utilizar perlas de ebullición en la técnica de destilación. 53 PRÁCTICA No. 4 DESTILACIÓN POR ARRASTRE CON VAPOR I. OBJETIVO Aislar compuestos orgánicos de interés a partir de productos naturales o mezclas de reacción complejas utilizando la técnica de destilación por arrastre con vapor. II. FUNDAMENTO Las destilaciones simples y fraccionadas se aplican para separar (o purificar) mezclas de líquidos completamente solubles. Sin embargo, cuando los líquidos son inmiscibles, estos pueden ser también destilados. Una mezcla de líquidos inmiscibles ebullirá a una temperatura más baja que los puntos de ebullición de los componentes individuales. Cuando se utiliza vapor de agua para proveer una de las fases inmiscibles, se le conoce al proceso como destilación por arrastre con vapor. Las sustancias inestables ó de alto punto de ebullición pueden ser separadas de la mezcla líquida a temperaturas menores a los 100 oC, evitando así la descomposición del material deseado. Cuando las sustancias son calentadas en combinación con el agua, los vapores de ambas se mezclan en la fase gaseosa, codestilando de esta forma, siempre y cuando dichas sustancias presenten una mínima presión de vapor a 100 oC. El líquido de interés se separa de la fase acuosa, después de enfriar, debido a su inmiscibilidad. La destilación por arrastre con vapor es ampliamente utilizada en el aislamiento de líquidos y sólidos de fuentes naturales, o para remover productos de mezclas complejas de reacción. Si dos líquidos A y B son completamente solubles y no existe interacción, forman una solución ideal y siguen la ley de Raoult: Líquidos miscibles: P total = PAoNA + PBoNB Donde: P total - Presión total PAo - Presión parcial de A PBo - Presión parcial de B NA - Fracción molar de A NB - Fracción molar de B La cual nos indica que la composición del vapor dependerá de las presiones de vapor y las fracciones mol de cada componente de la mezcla. En contraste, cuando dos líquidos son inmiscibles producen una mezcla heterogénea, donde cada uno ejerce su propia presión de vapor independiente del resto de los componentes, dando la siguiente ecuación: Líquidos inmiscibles: P total = PAo + PBo 54 Las presiones de vapor de los líquidos puros se suman, a una temperatura dada, para obtener la presión total de la mezcla. Cuando la presión total es igual a 760 mm Hg, se obtiene la ebullición de la mezcla. La composición del vapor en una mezcla inmiscible, se define como: moles A / moles B = PAo / PBo Una mezcla de líquidos ebulle cuando la suma de sus presiones parciales se iguala a la presión atmosférica (760 mm Hg a nivel del mar). La mezcla de dos líquidos inmiscibles ebulle a una temperatura más baja que los puntos de ebullición de cada componente. Por lo tanto una mezcla de dos líquidos, en donde uno de éstos es el agua, ebullirá siempre a una temperatura menor de 100 OC, ya que el segundo líquido estará aportando algo de presión de vapor a la presión total del sistema y consecuentemente los 760 mm Hg necesarios para ebullir se alcanzarán antes de los 100 OC que necesita el agua para ebullir cuando destila sola. En una mezcla de líquidos miscibles, la composición del vapor depende de las cantidades relativas de A y B presentes, por lo que la composición del vapor cambia durante la destilación. En contraste, la composición de vapor con líquidos inmiscibles es independiente de la cantidad de A y B, explicando así el porqué la composición de vapor se mantiene constante durante el proceso de destilación. Los métodos utilizados para destilación por arrastre con vapor en el laboratorio son dos. El primer método llamado “método de vapor en vivo” utiliza vapor de agua que pasa hacia el matraz conteniendo la mezcla que contiene el o los compuestos de interés. Es el método más ampliamente utilizado, especialmente con substancias de alto peso molecular y sólidos volátiles. 55 agua agua agua Muestra a destilar Equipo de destilación por arrastre, método de vapor en vivo FIGURA 1 El segundo método conocido como “método directo”, es más sencillo desde el punto de vista experimental. En este método se genera vapor de agua in situ calentando el matraz que contiene el compuesto y agua. A medida que el vapor codestila con el compuesto de interés, se le agrega agua en pequeñas cantidades desde el embudo de separación. Este método es útil para mezclas de líquidos volátiles o pequeñas cantidades de material, pero no se utiliza para separaciones de sustancias a partir de materiales sólidos. 56 AGUA AGUA AGUA Y MEZCLA POR DESTILAR AGUA Equipo de destilación por arrastre, método directo FIGURA 2 El método de vapor en vivo tiene una gran aplicación para separar los aceites esenciales de productos naturales. Las esencias y aromas que presentan las plantas se deben a aceites esenciales, los cuales han sido apreciados desde la antigüedad. Entre algunos aceites importantes, comercialmente, se incluyen los de almendra, anís, canela, clavo, comino, eucalipto, ajo, jazmín, pimienta, rosa y madera de sándalo, entre otros. Estos aceites son utilizados, comúnmente, por sus olores y sabores agradables, en perfumes, inciensos, esencias, etc. Estos compuestos se encuentran principalmente en los espacios intercelulares en tejidos vegetales; comúnmente se encuentran almacenados en las semillas ó flores. Muchos aceites esenciales pueden ser aislados por destilación por arrastre con vapor. Otros métodos para aislar este tipo de materiales son por extracción ó ejerciendo presión. Los aceites esenciales pueden contener, además de ésteres, los cuáles son responsables de olores y sabores característicos, mezclas complejas de hidrocarburos, alcoholes y compuestos carbonílicos. Una de las opciones para esta práctica es aislar el eugenol a partir de clavo, del cual es el principal constituyente; este compuesto presenta el olor característico del clavo y tiene un sabor pungente; se utiliza como un antiséptico bucal y analgésico. 57 En cuanto al método directo de destilación por arrastre con vapor éste se puede aplicar en mezclas líquidas en donde uno de los componentes es relativamente volátil e insoluble o al menos ligeramente soluble en agua. Esta situación se presenta con frecuencia al terminar una reacción química donde al final se tienen mezclados sustratos sin reaccionar, catalizadores, subproductos indeseables y el producto de interés. Este último puede ser separado fácilmente de la mezcla de reacción por destilación por arrastre, usando el método directo. Esto no se aplica cuando hay sólidos en la mezcla de reacción. III. EQUIPO • Balanza granataria • Plancha de calentamiento IV. MATERIAL • • • • • • • • • • • • • • • • • • Matraz de 3 bocas de 250 mL Embudo de separación de 125 mL Adaptador (cabeza de destilación) Unión Claisen (opcional) Espátula Termómetro Condensador Adaptador para termómetro Vaso de precipitado 100 mL (2) Vaso de precipitado de 250 mL Soporte universal (2) Tripié Mechero Tela de asbesto Anillo para soporte Pinzas para soporte (mínimo 3) Mangueras (2) Perlas de ebullición V. REACTIVOS • • • • • Clavos de olor o anis estrella o canela, etc (5 g) Agua desmineralizada Diclorometano (35 mL) Sulfato de sodio anhidro Grasa de silicona o vaselina 58 PROCEDIMIENTO VI. 1. MÉTODO DE VAPOR EN VIVO 1. Ensamblar el equipo para destilación por arrastre con vapor (ver figura 1). Colocar las pinzas de preferencia en todas las uniones (adaptador al matraz, condensador al matraz, embudo al matraz). Lubricar las uniones esmeriladas con una pequeña cantidad de grasa de silicona o vaselina. 2. Pesar 5 g de clavos y agregarlos a un matraz de 3 bocas de 250 mL a través de una de las bocas del matraz. 3. Calentar a ebullición el agua contenida en el matraz generador de vapor, mediante el mechero. 4. Recolectar el destilado hasta obtener 100 mL, o bien hasta que no salga turbio (tener cuidado que el contenido del matraz no se vomite hacia el condensador). 5. Tomar nota (en el diario del laboratorio) de la temperatura de destilación y del olor del destilado. 6. Enfriar el destilado a temperatura ambiente. 7. Transferir el destilado al embudo de separación de 125 mL y extraerlo 3 veces con porciones de 10 mL de diclorometano (revisar práctica de extracción, asegurarse de no tirar la fase orgánica). 8. Transferir la fase orgánica a un vaso de precipitado de 100 mL. Secar con una pequeña cantidad de sulfato de sodio anhidro. 9. Decantar la solución (a un matraz de destilación de 100 mL previamente pesado) para separar el agente de secado del solvente. 10. Destilar el diclorometano en un baño de vapor de agua o en una plancha de calentamiento (de preferencia en la campana de extracción). (ver procedimiento de destilación simple, práctica No. 5). El matraz de destilación debe de contener un residuo incoloro ó ligeramente amarillento, el cual es “aceite de clavo”. 11. Pesar el matraz de destilación conteniendo el residuo y por diferencia calcular el peso del aceite destilado. Determinar el porcentaje de contenido de aceite en el clavo, anotar los datos en el diario del laboratorio. VI. 2. MÉTODO DIRECTO 1. 2. 3. Ensamblar el equipo para destilación por arrastre con vapor (método de vapor in situ, ver figura 2). Colocar las pinzas de preferencia en todas las uniones (adaptador al matraz, condensador al matraz, embudo al matraz). Lubricar las uniones esmeriladas con una pequeña cantidad de grasa de silicona o vaselina. Pesar 10 g de la muestra a destilar y agregarla junto con 100 mL de agua en un matraz de 3 bocas de 250 mL a través de una de las bocas del matraz. (quitar el embudo de separación). Añadir 100 mL de agua al embudo de separación y colocarlo de nuevo a una de las bocas del matraz de destilación. 59 4. Calentar la suspensión a ebullición cuidadosamente, mediante el mechero (tener cuidado que el contenido del matraz no ebulla hacia el condensador) hasta obtener 100 mL de destilado, o bien hasta que éste no salga turbio. 5. Mantener el nivel original del líquido en el matraz de destilación durante la destilación, agregando agua del embudo de separación lentamente (lo necesario para mantener el nivel original del líquido en el matraz de destilación. 6. Tomar nota (en el diario del laboratorio) de la temperatura de destilación y del olor del destilado. 7. Enfriar el destilado a temperatura ambiente. 8. Transferir el destilado al embudo de separación de 125 mL y extraerlo 3 veces con porciones de 10 mL de un solvente adecuado (ver procedimiento de extracción práctica No. 1, asegurarse de no tirar la fase orgánica). 9. Transferir la fase orgánica a un vaso de precipitados de 100 mL. Secar con una pequeña cantidad de sulfato de sodio anhidro. 10. Decantar la solución (a un vaso de precipitado de 100 mL previamente pesado) para separar el agente de secado del solvente. 11. Destilar (ver práctica No. 5) el solvente en baño de agua en la campana de extracción. 12. Pesar el matraz de destilación conteniendo el residuo y por diferencia calcular el peso de la muestra y determinar el porcentaje de rendimiento, anotar los datos en el diario del laboratorio. 60 VII. DIAGRAMA DE FLUJO VII. 1. MÉTODO VAPOR EN VIVO 1 3 2 Ensamblar equipo de destilación por arrastre (ver figura No. 1). Lubricar las uniones Pesar 5 g de clavos de olor y transferirlos a un matraz 3 bocas de 250 mL 4 5 Enfriar el destilado a temperatura ambiente Tomar nota de la temperatura de destilación y olor del destilado 6 7 5 Extraer 3 veces el destilado con porciones de 10 mL de solvente 7 12 4 Transferir fase orgánica a un vaso de precipitado de 100 mL 7 Decantar el destilado a un matraz de destilación previamente pesado 10 8 10 11 Armar el equipo de destilación para separar el solvente Recolectar el destilado hasta obtener 100 mL de una suspensión turbia 9 8 Transferir el destilado al embudo de separación de 125 mL 3 2 1 6 Calentar a ebullición el agua contenida en el matraz generador de vapor Secar con sulfato de sodio anhidro durante 3 minutos 9 8, 9 14 13 Pesar el matraz de destilación conteniendo el residuo Calcular porcentaje de contenido de aceite en el clavo 11 Pasos correspondientes al VI. 1. PROCEDIMIENTO FIN 11 61 VII. 2. MÉTODO DIRECTO 1 3 2 Ensamblar equipo de destilación por arrastre (ver figura No. 2). Lubricar las uniones Agregar 10 g de la muestra y 100 mL de agua al matraz 250 mL 3 bocas 4 5 Agregar agua al matraz para mantener el nivel original del líquido Calentar la suspensión a ebullición cuidadosamente 5 7 Transferir el destilado al embudo de separación de 125 mL 8 8 14 Secar con sulfato de sodio anhídro. Decantar el VIII. destilado BIBLIOGRAFÍA 7 10 Extraer 3 veces el destilado con porciones de 10 mL de solvente 9 13 Enfriar el destilado a temperatura ambiente 4 11 Transferir fase orgánica a un vaso de precipitado de 100 mL 3 9 Colectar 100 mL de destilado, el cual será una suspensión turbia 6 12 Colocar el embudo en una de las bocas del matraz de destilación 4 8 Tomar nota en el diario de laboratorio de la temperatura de destilación 3 2 1 3.-6 Añadir 100 mL de agua al embudo de separación 15 Efectuar una destilación simple para separar el solvente de extracción 9, 10 Pasos correspondientes al VI. 2. PROCEDIMIENTO Calcular porcentaje de contenido de aceite en el clavo 11 12 FIN 62 VIII. BIBLIOGRAFÍA 1. Pavia, Donald L., Lampman, Gary M., Kriz Jr., George S.; Introduction to Organic Laboratory Techniques, a Contemporary Approach, tercera edición, Ed. Saunders College Publishing, 1988. 2. Hart, H. y Schuetz, R. D.; Organic Chemistry, A Short Course, quinta edición, Houghton Mifflin Company, 1978. 3. Popp, F. D. y Schultz, H. P.; Organic Chemical Preparations, W. B. Saunders Company, 1964. 63 CUESTIONARIO 1.- Describa cuál es el aspecto que presentan los diferentes extractos obtenidos. 2.- ¿Qué características de una sustancia la hacen susceptible de ser aislada por el método de destilación por arrastre con vapor? 3.- Propiedades y características de los aceites esenciales aislados en esta práctica. 4.- Describir la Ley de las presiones parciales de Dalton 64 PRÁCTICA No. 5 ANÁLISIS ELEMENTAL CUALITATIVO I. OBJETIVO Determinar en una muestra orgánica la presencia de los elementos: azufre, nitrógeno, cloro, bromo y yodo. II. FUNDAMENTO Establecer la presencia de azufre, nitrógeno y los halógenos (F, Cl, Br y I) en un compuesto orgánico (o mezcla) proporciona al químico una gran ayuda en la investigación de la identidad de una sustancia comercial o completamente nueva. Si el químico detecta estos elementos, en la muestra, puede tener una idea del grupo funcional que posee el compuesto y por lo tanto de la familia a la que pertenece. La muestra se funde con sodio metálico para transformarla en iones los cuales se detectan mediante pruebas específicas. Compuesto orgánico con C, H, N, S y/o X Na flama NaCN, Na2S, NaX, NaOH PRECAUCIONES El éxito del análisis dependerá, en gran parte, de tomar en cuenta las siguientes precauciones: • Algunas clases de compuestos orgánicos, tales como, los nitroalcanos, azidas orgánicas, diazoésteres, sales de diazonio y algunos polihaluros alifáticos (cloroformo, tetracloruro de carbono), reaccionan explosivamente con el sodio caliente, siempre deberán usarse lentes de seguridad cuando se realicen estas descomposiciones. • Si al agregar la muestra al sodio fundido se escucha un estallido o se produce una explosión (generalmente muy pequeña), se debe interrumpir la manipulación y se reducirán 0.5 g la muestra con zinc, para esto, la muestra se calienta suavemente a ebullición con 5 mL de ácido acético glacial y 0.5 g de zinc en polvo. Después que 65 se ha disuelto la mayor parte del zinc, la mezcla se evapora a sequedad y se funde el residuo, por el procedimiento indicado previamente. • El calentamiento con el sodio debe ser al rojo vivo, para asegurar que toda la muestra haya fundido, si queda muestra sin fundir puede dar lugar a reacciones de interferencia al hacer las pruebas. Es esencial usar un exceso de sodio ya que si están presentes azufre y nitrógeno se puede producir tiocianato de sodio (NaCNS), que dará coloración roja con el fierro (III), pero no el azul de Prusia, dado que no habrá iones cianuro libres. Con el exceso de sodio el tiocianato formado se descompone para dar iones cianuro y sulfuro. NaSCN + 2 Nao NaCN + Na2S • Evitar que quede muestra sin fundir en el tubo, ya que, en la solución final puede causar interferencia. • La solución que se obtenga, al seguir el procedimiento, debe ser incolora, para que no interfiera con el color que se debe observar en cada prueba, no se recomienda el uso de carbón activado pues adsorbería gran cantidad de los iones que se han obtenido. • Seguir al pie de la letra los procedimientos, sobre todo en el caso de ajustes de pH, cantidad y concentración de reactivos, esto ahorra tiempo, dinero y esfuerzo. • Cuando el compuesto es de alto peso molecular, por ejemplo, la difenilamina (169 g/mol), la proporción del nitrógeno es de un 8 % lo cual hace que se produzcan muy pocos iones cianuro (la cantidad de muestra que se funde es de 3-5 mg). Se puede solucionar este problema efectuando 3 fusiones, juntando las soluciones y concentrando. Cuando no se tiene idea del tipo de muestra, pero se supone, por otras evidencias, que puede estar el nitrógeno presente, y no da positiva la prueba, se deben considerar dos hipótesis: primera, que puede estar mal hecha la fusión y segunda que la muestra es de alto peso molecular. • Cuando se encuentra presente el azufre se debe ajustar la cantidad de reactivo de fierro (II) para la determinación de nitrógeno. Dado que el sulfuro ferroso es poco soluble, para asegurar que existen suficientes iones fierro (II), tanto para los iones cianuro como para el sulfuro, la cantidad de sulfato ferroso debe incrementarse en un 50 %. Cuando se añade el ácido sulfúrico para disolver los óxidos de fierro, debe haber un residuo de sulfuro ferroso en el fondo del tubo. Este residuo no interfiere con la prueba para nitrógeno. • Destruir el sodio en exceso con etanol. Durante esta reacción se produce hidrógeno gas, que se puede inflamar en la boca del tubo, se debe apagar este pequeño incendio cubriendo la boca del tubo con una tela de asbesto o un vidrio de reloj. La ecuación de la correspondiente reacción es: 66 CH3 CH2 O- Na+ + 1/2 H2 ↑ (gas) CH3CH2OH + Nao Reacciones de identificación ♦ Determinación de azufre: Na2S + Pb(OAc)2 PbS + 2NaOAc negro ♦ Determinación de nitrógeno 6 NaCN + FeSO4 Na4 [Fe(CN)6] Na4 [Fe(CN)6] + 2 Fe2 (SO4)3 + Na2SO4 Fe4 [Fe(CN)6]3 + Na2 SO4 Azul de Prusia ♦ Determinación de halógenos en general NaX + AgNO3 AgX ↓ + NaNO3 ♦ Reacción del ion yoduro con nitrito de sodio 2NaI + 2NaNO2 + 4CH3 COOH CHCl3 I2 + 2NO + 4CH3COONa + 2H2O violeta ♦ Reacción del ion bromuro con agua de cloro 2Br - + Cl2 CHCl3 Br2 + - 2Cl rojo ♦ Reacción del ion cloruro con nitrato de plata NaCl + AgNO3 AgCl ↓ + NaNO3 blanco 67 III. EQUIPO • Balanza analítica IV. MATERIAL • • • • • • • • • • • • • • • Tubos de ensayo, de vidrio tipo Pyrex, 13 x 100 Vaso de precipitado de 100 o 250 mL Tripié Tela de asbesto Mechero Cerillos Manguera Pinza para tubo Espátula Embudo de filtración rápida, tallo corto Gotero Papel filtro Papel aluminio Papel Hydrión (pH 1-14) Papel tornasol V. REACTIVOS • • • • • • • • • • • • • • • Sodio metálico Etanol Acido acético glacial Acetato de plomo 1 % Sulfato ferroso amónico (cristales) Fluoruro de potasio al 30 % Ácido sulfúrico al 30 % Ácido sulfúrico al 10 % Ácido sulfúrico concentrado Ácido nítrico 10 % Nitrato de plata 2 % Cloroformo Nitrito de sodio 20 % Agua de cloro o una solución de hipoclorito de sodio estabilizado, como el chlorox. Persulfato de potasio, sólido 68 VI. PROCEDIMIENTO VI. 1. Fusión con sodio 1. 2. 3. 4. 5. 6. 7. 8. 9. Lavar y secar perfectamente un tubo de ensayo. Cortar un trozo de sodio de aproximadamente 4 mm3 y colocarlo en el tubo. Precaución: El sodio es tóxico, no debe tomarse con las manos, desprende hidrógeno en contacto con el agua, o cualquier sustancia que tenga hidrógenos acídicos. Pesar de 5-10 mg de muestra sólida o, si es líquida, utilizar 1 gota de muestra. Colocar el sólido en la espátula o el líquido en un gotero. Calentar el sodio en el tubo de ensayo hasta que se forme una esfera. plateada, retirar del mechero y añadir la muestra, observar la reacción. Seguir calentando, hasta el rojo vivo, por 7 minutos. Dejar enfriar y añadir 1 mL de etanol para eliminar el sodio en exceso. Después de que desaparezca el burbujeo, evaporar el etanol a sequedad. Introducir el tubo caliente en un vaso que contenga 15 mL de agua desmineralizada. Filtrar la solución, el tubo se rompe en contacto con el agua y los fragmentos de vidrio y otras partículas sólidas se eliminan de esa manera. Si la solución es transparente continuar en el paso 10, si no, continuar en el paso 2. VI. 2. Determinación de azufre 10. Acidular, con ácido acético, 2 mL de la solución obtenida en la fusión. 11. Añadir unas gotas de la solución de acetato de plomo. 12. Observar, si se forma un precipitado negro, reportar azufre positivo. VI. 3. Determinación de nitrógeno 13. Ajustar el pH de 1 mL de la solución de la fusión, a 13, utilizar papel pH 1-14. Si el pH es 14, acidificar con ácido acético glacial. 14. Agregar 2 gotas de una solución saturada de sulfato ferroso amónico (recientemente preparada, colocando unos cristales de la sal en un tubo y añadiendo 1 mL de agua). 15. Agregar 2 gotas de una solución de fluoruro de potasio al 30 %. 16. Hervir la solución, cuidadosamente, por 30 segundos. 17. Acidular la solución caliente con ácido sulfúrico al 10 % (precaución), gota a gota, agitando después de cada adición, hasta que se disuelva el hidróxido de fierro formado (una gota puede ser suficiente). 18. Reporte nitrógeno positivo si aparece una coloración y/o precipitado azul de Prusia. 69 19. Realizar de nuevo la prueba si no se observa la coloración azul, si la muestra contiene nitrógeno y no aparece el color azul, consultar al maestro. VI. 4. Determinación de halógenos en general 20. Acidular 2 mL de la solución de la fusión, con ácido nítrico 10 %. 21. Hervir suavemente durante 3 minutos para eliminar, si los hay, el ácido sulfhídrico y el cianhídrico. 22. Añadir unas gotas de nitrato de plata al 2 %. 23. Observar, si aparece un precipitado denso, color blanco o crema, reportar prueba de halógenos positiva y continuar en el paso 24, si no aparece precipitado dar por terminado el procedimiento. VI. 5. Determinación de cloro, bromo y yodo 24. Acidular 10 mL de la solución de la fusión, con ácido sulfúrico al 30 %. 25. Hervir 3 minutos y enfriar. 26. Investigar si hay yodo, añadiendo 0.5 mL de cloroformo a 1 mL de la solución y unas gotas de la solución de nitrito de sodio. 27. Reportar yodo positivo si aparece un color púrpura. 28. Si hay yodo presente, tratar el resto de la solución con nitrito de sodio y extraer con cloroformo hasta que desaparezca la coloración. 29. Hervir la solución durante un minuto y enfriar. 30. Añadir 0.5 mL de cloroformo a 1 mL de la solución anterior y dos gotas de agua de cloro, asegurarse que la solución esté ácida antes de añadir el agua de cloro. 31. Reportar bromo positivo si aparece una coloración café. 32. Diluir el resto de la solución hasta 60 mL. 33. Añadir 2 mL de ácido sulfúrico concentrado y 0.5 g de persulfato de potasio. 34. Hervir la solución 5 minutos. 35. Enfriar y añadir 3 gotas de nitrato de plata. 36. Reportar cloro positivo si aparece un precipitado blanco. 70 VII. DIAGRAMA DE FLUJO VII. 1. Fusión con sodio 3 2 1 Lavar y secar perfectamente un tubo de ensayo Usar lentes de seguridad en todo momento Cortar un trozo de sodio de 4 mm3 y colocarlo en el tubo 2 1 6 4 5 Calentar el sodio en el tubo de ensayo hasta que se forme una esfera plateada Colocar el sólido en la espátula o el líquido en un gotero o pipeta 4 5 7 Retirar del mechero y añadir la muestra, observar la reacción Seguir calentando, hasta el rojo vivo, por 7 minutos 5 6 11 12 8 9 NO 7 Permitir que desaparezca el burbujeo de la reacción del sodio y luego evaporar el etanol a sequedad 8 SI 2 Dejar enfriar y añadir 1 mL de etanol para eliminar el sodio en exceso 10 Introducir el tubo caliente en un vaso que contenga 15 mL de agua desmineralizada Filtrar la solución para separar los fragmentos de vidrio y otras partículas sólidas 3 9 8 Solución incolora Pesar de 5-10 mg de muestra sólida, si es líquida, utilice 1 gota de muestra 1 Procedimiento VII. 2. Procedimiento VII. 1. 71 VII. 2. Determinación de azufre 2 1 Añadir unas gotas de la solución de acetato de plomo Acidular, con ácido acético, 2 mL de la solución obtenida en la fusión precipitado negro N O4 11 10 3 4 SI Reportar prueba de azufre positiva Reportar prueba de azufre negativa 12 1 Procedimiento VII. 3. VII. 3. Determinación de nitrógeno 3 2 1 Agregar 2 gotas de una solución saturada de sulfato ferroso amónico Ajustar el pH de 1 mL de la solución de la fusión, a 13. Si tiene pH 14, acidificar con ácido acético glacial. Agregar 2 gotas de fluoruro de potasio al 30 %. Hervir la solución por 30 s 15,16 14 13 Reportar prueba de nitrógeno positiva 4 Acidular la solución caliente con ácido sulfúrico al 10 % gota a gota, hasta que se disuelva el hidróxido de fierro formado Precipitado o coloración azul de Prusia SI 18 Consultar al maestro 1 19 Procedimiento VII. 4. NO 1 17 Procedimiento VII. 4. 72 VII. 4. Determinación de halógenos en general 2 1 Acidular 2 mL de la solución de la fusión, con ácido nítrico 10 % 3 Hervir suavemente durante 3 minutos para eliminar, si los hay, el ácido sulfhídrico y el cianhídrico 20 Añadir unas gotas de nitrato de plata al 2 % 22 21 5 4 Reportar prueba halógenos positiva de precipitado denso, color blanco o crema, SI Observar, si aparece un precipitado 23 23 NO 1 FIN Procedimiento VII. 5. VII. 5. Determinación de cloro, bromo y yodo 1 NO 2 Acidular 10 mL de la solución de la fusión, con ácido sulfúrico al 30%. Hervir 3 minutos y enfriar Investigar si hay yodo, añadiendo 0.5 mL de cloroformo a 1 mL de la solución y unas gotas de la solución de nitrito de sodio 24,25 26 4 Reportar prueba yodo negativa 5 de 4 Color púrpura 3 SI Reportar yodo positivo. Tratar el resto de la solución con nitrito de sodio. Extraer con cloroformo hasta que desaparezca la coloración 28 73 6 5 Hervir la solución durante un minuto y enfriar 29 8 NO A Añadir 1 mL de la solución anterior a 0.5 mL de cloroformo y dos gotas de agua de cloro, asegurarse que la solución esté ácida antes de añadir el agua de cloro Coloración café SI 30 7 8 9 Diluir Diluir el resto de la solución proveniente del paso 5 de este diagrama hasta 60 mL Reportar prueba de bromo positiva Reportar prueba de bromo negativa 31 32 10 11 Añadir 2 mL de ácido sulfúrico concentrado y 0.5 g de persulfato de potasio 12 Hervir la minutos solución Enfriar y añadir 3 gotas de nitrato de plata 5 34 35 33 13 Reportar cloro positivo Precipitado blanco SI 36 14 FIN NO Reportar negativo cloro 74 VIII. BIBLIOGRAFÍA 1. Shriner, R. L., Fuson R. C. y Curtin D. Y.; Identificación Sistemática de Compuestos Orgánicos. Ed. Limusa, Capítulo 5, p. 75. 1979. 2. Pavia, Donald L., Lampman, Gary M., Kriz Jr., George S.; Introduction to Organic Laboratory Techniques, a Contemporary Approach, tercera edición, Ed. Saunders College Publishing, 1988. 3. Vogel. A.I.; Practical Organic Chemistry, tercera edición, Ed. Longman. 1977. 75 CUESTIONARIO 1.- En la manipulación del sodio metálico , por qué debe evitarse la presencia de agua? 2.- Cuando se ha encontrado azufre en la disolución alcalina, éste debe eliminarse antes de ensayar la presencia de halógenos porque …… 3.- Mencione 5 propiedades y/o características de las sustancias, que contribuyan a su identificación cualitativa elemental. 4.- ¿Cuál es el principio en el que se basa el análisis cualitativo elemental orgánico por el método de fusión alcalina?. 76 PRÁCTICA No. 6 ANÁLISIS FUNCIONAL ORGÁNICO I. OBJETIVO Conocer la importancia que tiene el análisis funcional orgánico en la investigación de la fórmula estructural de los compuestos orgánicos, así como realizar en el laboratorio las pruebas de clasificación más comunes en los compuestos orgánicos e Identificar los grupos funcionales presentes en una muestra problema. II. FUNDAMENTO La reactividad de los compuestos orgánicos depende de los grupos funcionales presentes en las moléculas, en la presente práctica se estudiarán el comportamiento de algunas familias de compuestos orgánicos, como: hidrocarburos (alcanos, alquenos, alquinos, aromáticos), alcoholes, aldehídos y cetonas, frente a algunos reactivos específicos. II. 1. Pruebas de Clasificación para Hidrocarburos II. 1.1. Reacción con ácido sulfúrico concentrado a) Los alcanos son inertes ante esta prueba, y lo que se observa es la no miscibilidad del alcano y el ácido sulfúrico concentrado. b) Los alquenos y alquinos forman sulfatos y bisulfatos, observándose la disolución del ácido sulfúrico o bien la formación de una emulsión. AE R - CH = CH2 + H2SO4 R - CH - CH3 OSO3H Donde: AE - Reacción de adición electrófila c) Algunos alquenos y alquinos se polimerizan cuando son tratados con ácido sulfúrico, en este caso se observa la formación de una fase sólida. R2C = CHR + H + H2SO4 R2C - CHR – CR2 - CH2R A + R2C - CH2R + n (R2C = CHR) R2C = CHR A H2SO4 B R2 C - CH - (CR2 - CHR) n - CR2 - CH2R + H O R SO3H + B 77 d) Los compuestos aromáticos con grupos donadores de electrones o grupos con heteroaátomos (O, N, S) forman ácidos sulfónicos y se observa la disolución del ácido sulfúrico. CH3 CH3 SO3H + 2 H2SO4 SEAr CH3 CH3 Donde: SEAr - Reacción de sustitución electrófila aromática II. 1. 2. Reacción con ácido sulfúrico fumante Los hidrocarburos aromáticos insolubles en ácido sulfúrico concentrado reaccionan, con el ácido sulfúrico fumante, formando ácidos sulfónicos. SO3H + SO3 Benceno H2SO4 SSE Ar EAr Ácido bencensulfónico II. 1. 3. Reacción con cloroformo / tricloruro de aluminio Los compuestos aromáticos que no tengan grupos funcionales fuertemente electronegativos como, nitros, carboxilos, cianos, etc. reaccionan con tricloruro de aluminio y cloroformo dando una variedad de colores. Los compuestos alifáticos insolubles en ácido sulfúrico no dan color o solo un amarillo muy pálido. Los colores típicos producidos son los que se presentan en la tabla No. 1. 78 Tabla No. 1 Colores de algunos compuestos ante la prueba con cloroformo / tricloruro de aluminio COMPUESTO COLOR Benceno Anaranjado a Rojo Haluros de Acido Anaranjado a Rojo Naftaleno Azul Bifenilo Púrpura Fenantreno Púrpura Antraceno Verde Colores parecidos se obtienen cuando se sustituye el cloroformo por tetracloruro de carbono. Con el tiempo los colores cambian a varios tonos de café. La reacción origina especies como el ion trifenilmetilcarbonio, los cuales quedan en solución como sales de tetracloroaluminato (AlCl4 ); son estos iones trifenilmetilcarbonio los responsables de los colores observados. C AlCl4 Ion trifenilmetilcarbonio Estos iones se forman por tres reacciones sucesivas de alquilación de Friedel – Crafts junto con una reacción de desproporcionación. 79 La prueba se debe efectuar con cuidado pues el tricloruro de aluminio es un ácido muy corrosivo, venenoso y que reacciona violentamente con el agua. Por lo que es muy importante que tanto el material a usar como los reactivos estén exentos de humedad ya que ésta inhibe la reacción. II. 1. 4. Reacción con bromo en cloroformo Este reactivo se usa extensamente para averiguar la presencia de un enlace olefínico o acetilénico. El resultado de esta prueba debe relacionarse con el resultado de la prueba con permanganato de potasio para obtener una interpretación correcta. El cloroformo es un buen disolvente para el bromo y para muchos compuestos orgánicos, pero no disuelve el bromuro de hidrógeno. Por lo tanto el desprendimiento de bromuro de hidrógeno se acepta como evidencia de que la reacción es de sustitución en lugar de adición. Cuando se emplea para revelar la presencia de insaturaciones, este reactivo puede conducir a conclusiones erróneas por dos razones: La primera es que no todos los compuestos olefínicos absorben bromo, la presencia de grupos electronegativos o muy voluminosos sobre los átomos de carbono de un enlace etilénico hace que la adición sea lenta y en casos extremos inhibe la reacción, las siguientes reacciones mostrarán este caso. Br CH = CH2 + Br2 Rápida CH - CH2 Br Br2/CHCl3 NO HAY REACCIÓN HOOC COOH CH = CH - CO2H + Br 2 Br Lenta CH - CH - CO2H Br Φ Φ Br2/CHCl3 NO HAY REACCIÓN ΦΦ = Φ Fenil Una prueba positivao para insaturación es aquella en la cual el color del bromo desaparece sin que se desprenda bromuro de hidrógeno. 80 La desaparición del color del bromo, acompañada del desprendimiento de bromuro de hidrógeno, indica que ha ocurrido sustitución (por radicales libres o electrófila) y es característico de muchos compuestos. A ésta categoría pertenecen los alcanos, hidrocarburos aromáticos y algunos otros compuestos como los que se pueden enolizar. CH3 - CH2 - CH2 - CH2 - CH3 + Br2 Luz u.v. CH3 - CH2 - CH2 - CH – CH3 + HBr SRL Br Donde SRL – Reacción por radicales libres OH OH OH Br 2 + 2Br2 AlCl3 + 2 HBr + SEAr Br II. 1. 5. Reacción con permanganato de potasio Una solución de permanganato de potasio se decolora con compuestos que tienen enlaces etilénicos y acetilénicos, este ensayo se conoce como la prueba de Baeyer para insaturaciones. En solución acuosa diluida y fría, el producto principal de la reacción del permanganato de potasio sobre una olefina es el glicol. Si se calienta la mezcla de reacción se efectúa una oxidación adicional que conduce finalmente a la ruptura de la cadena de carbón. Los enlaces acetilénicos usualmente se rompen por oxidación y producen ácidos. Esta prueba es general, en particular es útil con compuestos tales como el difeniletileno, tetrafeniletileno y dibromuro de difeniletileno, que no decoloran la solución de bromo en cloroformo CH = CH + 4 H 2O + 2 KMnO4 3 CH - CH OH OH + 2 KOH + 2 MnO2 (Sólido café) 81 Br Br Br C = C 3 + 4 H2O + 2 KMnO4 Br C - C OH OH + + 2 KOH + 2MnO2 (sólido café) R–C C – R´ + 2 KMnO4 R – COO - + R´ - COO - + 2 MnO2 Café La prueba de Baeyer, aunque tiene mayor aplicación que la prueba de bromo para compuestos insaturados, a su vez presenta interferencias. Todas las sustancias fácilmente oxidables dan positiva esta prueba. Los compuestos carbonílicos que decoloran las soluciones de bromo, generalmente dan negativa la prueba de Baeyer. La acetona es un buen ejemplo, aunque decolora rápidamente las soluciones de bromo, se puede usar como disolvente en la prueba de Baeyer, los aldehidos dan pruebas de Baeyer positiva, sin embargo, muchos de ellos tales como el benzaldehido y el formaldehido, no decoloran la solución de bromo. Otros tipos de compuestos pueden contener cantidades pequeñas de impurezas que pueden decolorar las soluciones de permanganato. Por esta razón, la decoloración de una o dos gotas de la solución de permanganato no siempre puede aceptarse como prueba positiva. Los compuestos carbonílicos que decoloran la solución de bromo, generalmente dan negativa la prueba de Baeyer. II. 1. 6. Prueba con sodio metálico Los compuestos con triple enlace terminal pueden ser distinguidos de alquenos y otros alquinos basándose en la acidez del hidrógeno terminal. Una de las pruebas usadas es la reacción con sodio. Hay 2 tipos generales de compuestos orgánicos que reaccionan con sodio desprendiendo hidrógeno. Compuestos neutros que contengan átomos de hidrógeno que se puedan sustituir con facilidad, los grupos funcionales que contengan una átomo de hidrógeno unido a oxígeno, nitrógeno o azufre, pueden reaccionar con sodio desprendiendo hidrógeno: 2 R-OH + 2 Na 2 R-O- Na+ + H2 Los alquinos terminales también dan positiva la reacción con sodio. CH 2 RC CH + 2 Na CH + 2 Na - - Na+ C C Na+ + H2 2R - C - C Na + + H2 82 Se debe estar seguro que la muestra no contenga agua pues aún la presencia de trazas de humedad produce la reacción con el sodio. H2O + Na NaOH + ½ H2 Esta prueba es de utilidad en alcoholes de peso molecular intermedio, es decir, aquellos que contienen de 3 a 8 átomos de carbono. Los alcoholes sencillos solo con dificultad pueden obtenerse en condiciones anhidras. Los alcoholes de peso molecular elevado reaccionan lentamente con sodio, y a menudo el desprendimiento de gas es tan lento que hace que la prueba sea de poco valor. Para evitar la interferencia de humedad en el ensayo, si la muestra es sólida, se debe secar por 30 minutos a 120 °C y sí es líquida el secado será con sulfato de sodio anhidro. Generalmente la prueba se lleva a cabo añadiendo una pequeña cantidad de sodio a la muestra problema “TOMAR PRECAUCIONES”. El sodio es muy tóxico y debe manejarse con cuidado. Se deben utilizar siempre pinzas y nunca se debe tomar directamente con los dedos. Se seca con papel filtro y se corta con una navaja o cuchillo afilado. Las cortezas y los residuos de sodio se deben destruir, tratándolos con un exceso de etanol. EVITAR CONTACTO CON AGUA. Siempre que se trabaje con sodio SE DEBEN UTILIZAR LENTES DE SEGURIDAD. II. 1. 7. Uso de otros iones metálicos Otra prueba para alquinos terminales con producción de acetiluros insolubles es usando iones metálicos: R CH + + Cu(NH3)2 R + Cu + NH4 + NH 3 Ag + NH4 + NH3 Rojo R CH + + Ag(NH3)2 R + blanco II. 2. Alcoholes II. 2. 1. Prueba de Lucas Los alcoholes secundarios y terciarios reaccionan con ácido clorhídrico en presencia de cloruro de zinc produciendo cloruros de alquilo insolubles en el medio, evidentemente sólo se aplica a los alcoholes que son solubles en el reactivo, esto limita la prueba, en general, a los alcoholes monofuncionales de menos de 6 átomos de carbono y algunos compuestos polifuncionales que contengan el grupo hidroxilo. 83 ZnCl2 R2CHOH + HCl R2CHCl + H2O Líquido Insoluble R3COH + HCl ZnCl2 R3CCl + H2O Líquido Insoluble Como es de esperarse en estas reacciones, los efectos de la estructura sobre la reactividad están íntimamente relacionados. Así los alcoholes primarios no reaccionan apreciablemente a temperaturas ordinarias con el ácido clorhídrico-cloruro de cinc; por una parte el ion cloruro es un agente nucleófilo demasiado débil para efectuar una reacción de desplazamiento concertado y por otra parte, el ion carbonio primario es demasiado inestable para servir como intermediario en el mecanismo. Los alcoholes terciarios reaccionan tan rápidamente con el ácido clorhídrico concentrado, que el halogenuro de alquilo se puede observar, después de unos cuantos segundos a temperatura ambiente, como turbidez o segunda fase. La acidez del medio se aumenta por la adición del cloruro de zinc anhidro (un fuerte ácido de Lewis) y la velocidad de la reacción aumenta aún más. Los alcoholes secundarios tienen una reactividad intermedia entre los alcoholes primarios y terciarios, aunque no se ven afectados apreciablemente por el ácido clorhídrico solo, reaccionan bastante rápido en presencia de cloruro de zinc; la apariencia turbia de la mezcla se observa en 5 minutos y en 10 minutos se puede ver una capa bien definida. II. 2. 2. Reacción con nitrato cérico. La prueba del nitrato cérico se realiza en alcoholes y fenoles con menos de diez átomos de carbono. Una prueba positiva para alcoholes queda indicada por un cambio en el color del reactivo que pasa de amarillo a rojo. R - CH2OH + [(NH4)2Ce4+ (NO3)6] HNO3 R - CH = O + Ce3+ Rojo R - CH2OH 2 Ce4+ + e R - CH = O + 2 H + + 2 e 2 Ce 3+ 84 R - CH2OH + 2 Ce 4+ R - CH = O + 2H+ + 2 Ce3+ Rojo Amarillo Cuando las moléculas son más grandes, el color que se produce es insuficiente para hacer útil la prueba. Los aldehidos, cetonas, ácidos, halogenuros de alquilo, ésteres y otros compuestos comunes que sólo contienen carbono, hidrógeno, oxígeno y halógenos, no interfieren. II. 3. Derivados Carbonílicos II. 3. 1. Prueba con 2,4-dinitrofenilhidracina La reacción entre los derivados carbonílicos y la 2,4-dinitrofenilhidracina producen 2,4dinitrofenilhidrazonas que son sólidos insolubles. NO2 C + O2N O NH - NH2 + H NO2 C = N - NH + H2O NH2 NO2 2,4-dinitrofenilhidrazona En ocasiones el producto inicial es aceitoso y, al reposar, se vuelve cristalino. Sin embargo, algunas cetonas dan dinitrofenilhidrazonas que son aceites. Por ejemplo, la metil-n-octilcetona, la di-n-amilcetona y sustancias similares, no forman dinitrofenilhidrazonas sólidas. El color de una 2,4-dinitrofenilhidrazona puede dar indicación acerca de la estructura de los aldehídos o cetonas de los que deriva. Las dinitrofenilhidrazonas de aldehidos o cetonas en las que el grupo carbonilo no está conjugado con otro grupo funcional, son amarillas. La conjugación con un doble enlace carbono-carbono o con un anillo fácilmente se puede determinar por un examen del espectro ultravioleta. Este desplazamiento también es responsable de un cambio de color del amarillo al rojo anaranjado. Entonces, en general puede suponerse que una dinitrofenilhidrazona amarilla no está conjugada. Sin embargo, un color anaranjado o rojo debe interpretarse con precaución, ya que puede deberse a la contaminación con una impureza (por ejemplo, la misma 2,4-dinitrofenilhidrazina es roja-anaranjada). III. EQUIPO No se requiere 85 IV. MATERIAL • • • • • • • • • Gradilla Tubos de ensayo 13 x 100 (10) y 18 x 150 (2) Espátula Gotero Pipetas de 1 y 5 mL Pizeta Mechero Termómetro Pinzas para tubo de ensayo V. REACTIVOS • • • • • • • • • • • • • • • • • • • • • Permanganato de potasio (1 %) Bromo en cloroformo (2 %) Tricloruro de aluminio anhidro (o sublimado) Reactivo de ácido clorhídrico-cloruro de cinc Carburo de calcio Cloruro de cobre (I) amoniacal Nitrato de plata amoniacal Nitrato cérico amonico Cloroformo Dioxano Tolueno Naftaleno Antraceno Fenantreno Acetona Benzaldehido Hexano u otro alcano disponible Ciclohexeno u otro alqueno disponible Anhidrido crómico Ácido sulfúrico Ácido sulfúrico fumante, opcional 86 VI. PROCEDIMIENTO Al efectuar las siguientes pruebas y concluir con seguridad el tipo de grupo funcional presente (o ausente) en la muestra, no continuar realizando el resto de las pruebas. Al finalizar cada una de las pruebas siguientes, realizar el mismo ensayo pero en lugar de muestra desconocida use una muestra testigo (compuesto que contenga el mismo grupo funcional por identificar). Aunque ya se haya identificado el grupo funcional de la muestra desconocida, realizar todos los ensayos usando muestras testigo. VI. 1. Reacción con ácido sulfúrico concentrado 1. 2. 3. 4. Colocar aproximadamente 2 mL de ácido sulfúrico concentrado en un tubo de ensayo de 13 x 100 seco. Agregar 0.5 mL de la muestra y agitar vigorosamente con precaución. Observar si hay disolución, separación de fases, cambio de color, desprendimiento de humos, desprendimiento de calor, etc. Anotar observaciones en el diario del laboratorio. VI. 2. Reacción con ácido sulfúrico fumante 5. 6. 7. Colocar 2 mL de ácido sulfúrico fumante en un tubo de ensayo seco. Agregar 0.5 mL de la muestra, tapar y agitar vigorosamente (precaución). Observar y anotar en el diario. Nota: Únicamente los hidrocarburos aromáticos se disuelven desprendiendo calor, pero sin que se carbonice la muestra. completamente VI. 3. Prueba con cloroformo y tricloruro de aluminio 8. Colocar 2 mL de cloroformo seco (secado con sulfato de sodio anhidro) en un tubo de ensayo. 9. Añadir 0.1 mL (o 30 mg) de la(s) muestra(s) problema(s) al tubo de ensayo conteniendo el cloroformo. 10. Mezclar perfectamente e inclinar el tubo de ensayo hasta humedecer la pared. 11. Añadir 0.5 a 1.0 g de tricloruro de aluminio anhidro puro, procurar que algo de polvo se pegue en la pared lateral del tubo de ensayo. 12. Observar el color del polvo que está sobre la pared lo mismo que el color de la solución. 13. Anotar observaciones en el diario. Nota: Si el tricloruro de aluminio está hidratado o presenta una coloración amarilla deberá sublimarse antes de agregar la solución problema. Si este es el caso seguir el procedimiento VI. 3. 1. 87 VI. 3. 1. Purificación del tricloruro de aluminio 1. 2. 3. Colocar 1 g de tricloruro de aluminio en un tubo de ensayo de 18 x 150. Inclinar el tubo de ensayo y calentar. El tricloruro de aluminio sublimará y se depositará en las paredes del tubo. Retirar el fuego y antes de enfriar dejar caer sobre el sublimado unas gotas de la solución de cloroformo con la muestra. VI. 4. Prueba con bromo en cloroformo 14. 15. 16. 17. 18. Colocar 30 a 50 mg (o 0.2 mL) del compuesto problema en un tubo de ensayo. Añadir 2 mL de cloroformo al tubo de ensayo anterior Adicionar una solución de bromo al 2 % en cloroformo gota a gota (agitando) hasta que persista el color del bromo por un minuto. Colocar en la boca del tubo de ensayo una tira de papel tornasol azul previamente humedecido. Observar sí se presenta un cambio de color o sí se desprenden humos blancos. Nota: Si se requieren más de 2 gotas de la solución de bromo para hacer que la coloración permanezca al menos 1 minuto la prueba es positiva VI. 5. Prueba con permanganato de potasio 19. 20. 21. 22. Disolver 30 a 50 mg (o 0.2 mL) de la muestra en 2 mL de agua o etanol en un tubo de ensayo. Añadir una solución de permanganato de potasio al 1 %, gota a gota con agitación. Observar la decoloración del permanganato de potasio y la aparición del precipitado café de dióxido de manganeso. Anotar observaciones en el diario de laboratorio. Nota: La prueba es positiva si se decoloran por lo menos 3 gotas de permanganato de potasio. Si se presenta una reacción débil, puede deberse a la presencia de impurezas. VI. 6. Prueba para Alquinos terminales 23. 24. 25. 26. Colocar 5 mL de solución cuprosa amoniacal y de solución de nitrato de plata amoniacal por separado en dos tubos de ensayo. Añadir 0.1 mL (ó 30 mg) de la(s) muestra(s) problema(s) a cada tubo de ensayo. Observar si se formó un precipitado. Anotar observaciones en el diario Nota: Se debe tener precaución durante el ensayo pues los acetiluros de cobre y plata son muy explosivos cuando se secan. Los acetiluros residuales se destruyen por calentamiento con ácido nítrico diluido. No guardar los acetiluros en la gaveta pues al secarse pueden explotar. 88 VI. 6. 1. Prueba de Control (Preparación de acetileno) 27. 28. 29. 30. 31. 32. 33. 34. 35. 36. Armar el aparato mostrado en la figura 1 Colocar 3 g de carburo de calcio en trozos en el matraz de destilación. Colocar en el embudo 10 mL de agua desmineralizada. Abrir la llave del embudo dejando gotear lentamente el agua. Colocar en un tubo de ensayo 5 mL de nitrato de plata amoniacal. Colocar en un tubo de ensayo 5 mL de cloruro de cobre (I) amoniacal. Colocar en un tubo de ensayo bromo en cloroformo Colocar en un tubo de ensayo solución diluída de permanganato de potasio. Pasar una corriente de acetileno (aproximadamente durante un minuto) en los 4 tubos de ensayo (18 x 150) de los pasos 31 a 34. Anotar observaciones en el diario Equipo para la preparación de acetileno FIGURA 1 VI. 7. Prueba de Alcoholes VI. 7. 1. Prueba de Lucas 37. 38. 39. 40. 41. Colocar 1 mL de muestra problema en un tubo de ensayo Añadir 10 mL del reactivo de ácido clorhídrico-cloruro de cinc. La temperatura debe mantenerse entre 26-27 °C. Tapar el tubo y agitar la mezcla contenida en el tubo de ensayo. Reposar la mezcla. Observar si hubo formación del cloruro de alquilo en un lapso de 10 minutos. Nota: Ya que la prueba de Lucas depende de la aparición del cloruro de alquilo como una segunda fase líquida, evidentemente solo es aplicable a los alcoholes que son solubles en el reactivo. Esto limita la prueba, en general, a los alcoholes monofuncionales con menos de 6 átomos de carbono. VI. 7. 2. Prueba con nitrato cérico VI. 7. 2. 1. Compuestos solubles en agua 42. Mezclar perfectamente 0.5 mL del reactivo de nitrato cérico con 3 mL de agua destilada en un tubo de ensayo. 43. Añadir 4 a 5 gotas del compuesto que se va a ensayar. 89 44. 45. 46. Tapar el tubo y agitar la mezcla. Observar el cambio de color. Anotar observaciones en el diario. VI. 7. 2. 2. Compuestos insolubles en agua 47. 48. 49. Colocar 0.5 mL de nitrato cérico en un tubo de ensayo, Agregar al tubo de ensayo 3 mL de dioxano. Si se forma un precipitado, añadir agua (de 3 a 4 gotas), agitando, hasta que la solución esté transparente. Añadir de 4 o 5 gotas del compuesto que se va a investigar, agitar y observar cambios de color. VI. 8. Prueba de aldehídos y cetonas 50. 51. 52. 53. 54. 55. 56. Colocar 1 o 2 gotas del compuesto que se va a investigar en un tubo de ensayo. Disolver en 2 mL de etanol al 95 %. Agregar 3 mL del reactivo de 2,4-dinitrofenilhidrazina. Agitar vigorosamente. Observar si se forma inmediatamente un precipitado, si se forma continuar en el paso 56. Si no se forma continuar en el paso 55. Reposar durante 15 minutos Anotar observaciones en el diario. 90 VII. DIAGRAMA DE FLUJO Nota 1: Al efectuar las siguientes pasos del diagrama y concluir con seguridad el tipo de grupo funcional presente (o ausente) en la muestra, no continuar realizando el resto de los pasos. Nota 2: Al finalizar cada una de las pruebas para identificar un grupo funcional en particular, realizar el mismo ensayo pero en lugar de muestra desconocida use una muestra testigo (compuesto que contenga el mismo grupo funcional por identificar). Aunque ya se haya identificado el grupo funcional de la muestra desconocido, realizar todos los ensayos usando muestras testigo. 1 2 Agregar 0.5 mL de muestra y agitar vigorosamente Colocar en un tubo de ensayo 2 mL H2SO4 concentrado 6 1 5 Observar y anotar en el diario 7 7 2 Agregar 0.5 mL del muestra, tapar y agitar vigorosamente Colocar en un tubo de ensayo 2 mL de CHCl3 seco 8 Añadir 0.1 mL (30 mg) de muestra, mezclar e inclinar el tubo hasta humedecer la pared 9,10 Adicionar gota a gota y agitando una solución de bromo al 2% en CHCl3 hasta que persista el color del bromo por 1 min 13 16 Coloque en la boca del tubo de ensayo una tira de papel indicador previamente humedecido 17 4 Colocar 2 mL de H2SO4(SO3) en un tubo de ensayo 5 9 Añadir 0.5-1.0 g de AlCl3 procurando se pegue en la pared lateral del tubo de ensayo. 11 10 11 12 3,4 6 8 7 3 Observar y anotar en el diario Añadir 30 a 50 mg (o 0.2 mL) de muestra a 2 mL de CHCl3 14,15 14 Observar y tomar nota del color del polvo que está sobre la pared lo mismo que el color de la solución 12,13 15 Observar si hubo cambio de color, si es necesario sople a través de la boca del tubo para revelar la presencia de HBr 18 Observar si se desprenden humos blancos (HBr) 18 16 91 16 17 Colocar en un tubo de ensayo 2 mL de H2O o EtOH y disolver 30 a 50 mg (o 0.2 mL) de muestra 18 Añadir una solución de KMnO4 al 1 % gota a gota con agitación 20 19 Se considera positiva la prueba si se decoloran por lo menos 3 gotas de KMnO4 y hay aparición de un precipitado café de MnO2 21 20 21 Colocar 5 mL de una solución cuprosa de cloruro amoniacal. y añadir (0.1 mL, 30 mg) la muestra problema. Observar si se forma un precipitado 23,24,25 22 Colocar en un tubo de ensayo 5 mL de plata amoniacal y añadir (0.1 mL) la muestra. Observar si se forma un precipitado el cual se destruye con HNO3 26 Observar y anotar en el diario 22 23,24,25 22 23 Anotar todas las observaciones en el diario 19 24 Armar un aparato como el de la figura 1 y colocar en el matraz 3 g de carburo de calcio en trozos Colocar en el embudo 10 mL de H2 O desmineralizada, la cual se deja gotear lentamente al matraz 27,28 25 26 27 Añadir 10 mL de reactivo HCl-ZnCl2 a temperatura entre 2627 °C a la muestra Colocar 1 mL de muestra en un tubo de ensayo 37 38 28 29 Tapar el tubo y agitar, luego reposar la mezcla 39,40 29,30 Efectuar los pasos 11 a 22 de este diagrama solo que en lugar de la muestra problema usar el acetileno generado por el brazo lateral de la figura 1 31-36 Anotar todas las observaciones en el diario 30 41 92 30 32 31 Colocar 0.5 mL del reactivo de nitrato cérico en un tubo de ensayo Añadir 4 o 5 gotas del compuesto a ensayar Mezclar el nitrato cérico con 3 mL de agua 42 42 43 34 35 Añadir 3 mL de dioxano. Si se forma un precipitado agregar 3 o 4 gotas de agua 33 Colocar 0.5 mL del reactivo de nitrato cérico en un tubo de ensayo Tapar el tubo y agitar Anotar observaciones en el diario 44-46 47 48 36 38 37 Agitar hasta que la solución esté transparente 48 Añadir 2 mL de etanol al 95 % y agitar hasta disolución 51 40 49 41 42 Agitar la mezcla y observar el cambio de color Añadir 4 o 5 gotas del compuesto a ensayar Colocar 1 o 2 gotas del compuesto a investigar en un tubo de ensayo Agregar 3 mL del reactivo de 2,4-dinitrofenilhidracina y agitar vigorosamente, observar 49 52-54 44 NO Hay formación de precipitado SI 40 Anotar todas las observaciones en el diario 50 43 49 Reposar durante 15 min hasta observar si hay formación de un precipitado 45 55 Anotar las observaciones en el diario Pasos correspondientes al VI. PROCEDIMIENTO FIN 56 93 VIII. BIBLIOGRAFÍA 1. Shriner, R. L., Fuson, R. C. y Curtin D. Y., Identificación Sistemática de Compuestos Orgánicos. Ed. Limusa. p. 40 y 41, 1979. 94 CUESTIONARIO 1.- Investigue diferentes reacciones que permiten la identificación de alcoholes, indicando la reacción característica. 2.- El grupo carbonilo se encuentra presente tanto en aldehídos como cetonas, de que forma se pueden diferenciar estos grupos funcionales experimentalmente. 3.- Indique la reacción de formación de las fenilhidrazonas producidas al utilizar el reactivo 2,4DNFH. 4.- Describa la reacción de alquilación de Friedel-Crafts 95 PRÁCTICA No. 7 CROMATOGRAFÍA I. OBJETIVO Aplicar los fundamentos teóricos de esta importante técnica de separación, purificación e identificación y desarrollar en el laboratorio las técnicas cromatográficas más utilizadas. II. FUNDAMENTO La palabra cromatografía fue originalmente empleada por Tswett en 1906, para describir las bandas coloreadas observadas cuando una solución, conteniendo pigmentos de plantas se pasó a través de una columna empacada con un adsorbente. En la actualidad, dicho término engloba una gran variedad de técnicas, que son ampliamente usadas con propósitos tanto analíticos como de separación. La cromatografía se clasifica en cromatografía de adsorción y cromatografía de partición de acuerdo al fenómeno involucrado en la separación. Todos los métodos cromatográficos operan bajo el principio de que los componentes de una mezcla se van a distribuir desigualmente entre dos fases. La fase móvil es generalmente un líquido o un gas que fluye continuamente sobre una fase estacionaria que puede ser un líquido o un sólido. Los componentes individuales de la mezcla tienen diferentes afinidades por la fase móvil y la fase estacionaria, de esta manera se establece un equilibrio entre ambas fases y los constituyentes de dicha mezcla son selectivamente separados temporalmente de la fase móvil, por la unión con la fase estacionaria. Como cada componente tiene un coeficiente de distribución diferente entre las dos fases, estos se separan en regiones llamadas bandas migratorias (o manchas), esta separación se debe a que algunos componentes se unen mas fuertemente con la fase estacionaria que los demás y por lo tanto éstos se mueven mas lentamente en la dirección del flujo de la fase móvil. Las fuerzas atractivas que se involucran en estas adsorciones selectivas, son las mismas fuerzas que causan las interacciones atractivas entre las moléculas como son: las interacciones electrostáticas, las dipolo-dipolo, los puentes de hidrógeno y las fuerzas de Van der Waals. Los métodos cromatográficos usados por los químicos modernos pueden ser clasificados por la naturaleza de la fase móvil y la fase estacionaria, por ejemplo, cromatografía en columna, cromatografía de gases (GC), cromatografía de líquidos de alta resolución (HPLC) y cromatografía en capa fina (TLC) involucran una interacción 96 líquido-sólido o gas-sólido, (cromatografía de adsorción). La cromatografía de gaslíquido es un tipo de cromatografía de gases que involucra una interacción entre una fase móvil gaseosa y una fase estacionaria líquida adsorbida sobre un sólido inerte (cromatografía de partición). La cromatografía en papel pertenece también a este último tipo. II. 1. Términos empleados en cromatografía de adsorción Soluto: La sustancia que se va a separar. Adsorbente: La sustancia finamente dividida la cual constituye la fase estacionaria en la cromatografía de adsorción. Origen o línea base: Punto en el cual la muestra es aplicada. Desarrollo: El paso de un líquido a través del adsorbente para efectuar la separación de la muestra. Eluente: Es la fase móvil que efectúa la separación del soluto, éste puede ser un líquido puro, una mezcla de líquidos o un gas. Eluato: Es la mezcla de la fase móvil que efectuó la separación junto con los solutos al final de la cromatografía. Revelado: Cuando los componentes de la muestra no son coloreados (es decir, visibles a simple vista) el cromatograma es visualizado por el empleo o aplicación de luz ultravioleta o algún reactivo, pueden ser vapores de yodo; a éstos se les conoce como reveladores universales. Rf o factor de retención: Es la relación de la distancia recorrida por el soluto o componente (medida desde el punto de aplicación hasta en el centro geométrico de la mancha correspondiente) y la distancia recorrida por el frente del eluente. II. 2. Cromatografía en capa fina TLC (thin layer chromatography) Fue en 1938 cuando, por primera vez, se describió la cromatografía en capa fina en la literatura, pero hasta 1960, se introdujo ampliamente su uso en los laboratorios de investigación en los Estados Unidos. La gran aceptación de esta técnica es debida a su versatilidad ya que puede emplearse tanto como una técnica rápida de análisis (en cinco minutos y con unos cuantos microgramos se puede obtener una información completa de la muestra), así como para conocer condiciones de reacción, en tiempos cortos, por comparación de los productos 97 con los reactivos de los que se parten, otro uso es la selección del eluente adecuado para cromatografía en columna, etc. II. 2. 1. Adsorbentes Existen una gran cantidad de adsorbentes disponibles comercialmente, ordenados en forma decreciente de polaridad tenemos: alúmina (Al2O3), gel de sílice (ácido silícico SiO2 xH2O), florisil (silicato de magnesio activado), celulosa, carbohidratos y algunos otros de menor uso. En el laboratorio se utilizan por lo general, gel de sílice y alúmina, ambos son sólidos polares. El gel de sílice es una sustancia ligeramente ácida por lo que se emplea para separar compuestos neutros o ácidos. La alúmina es una sustancia altamente activa y fuertemente adsorbente, comercialmente se puede adquirir en tres formas: ácida, básica y neutra. Las alúminas ácida y básica son usadas para separar compuestos ácidos y básicos respectivamente. La forma neutra se usa para separar compuestos sensibles al pH. El grado de actividad de los adsorbentes es disminuido por la presencia de pequeñas cantidades de agua, por eso el porcentaje de humedad va a ser una de las variables que se debe considerar al efectuar una cromatografía de adsorción. Otro factor a considerar es el área específica, altas áreas específicas aseguran equilibrios rápidos y buenas separaciones. Las áreas requeridas son del orden de cientos de metros cuadrados por gramo. La fuerza de adsorción de un compuesto orgánico depende no solamente de la polaridad del adsorbente y de su naturaleza, sino también de la naturaleza de los grupos funcionales presentes en la molécula. En la cromatografía en fase normal, el material adsorbente de la fase estacionaria es polar y el solvente usado como fase móvil es no polar. En estas condiciones, los grupos funcionales polares (ácidos carboxílicos, alcoholes, aminas) son mas fuertemente retenidos que los grupos menos polares (éteres, ésteres, haluros de alquilo); ver la figura 1. Incremento de adsorción sobre fases estacionarias no polares -CO2H, -OH, -NH2, -SH, -CHO, - C=O, -CO2R, -OCH3, -C=C-, -X Incremento de adsorción sobre fases estacionarias polares Orden de adsorción en fases estacionarias FIGURA 1 98 En cromatografía en fase reversa el material de empaque para la fase estacionaria consiste en perlas de vidrio recubiertas con una película de hidrocarburo no polar y mezclas de agua y solventes orgánicos son empleados como fase móvil; bajo estas condiciones las moléculas orgánicas no polares son fuertemente atraídas por la fase estacionaria no polar y los solutos polares son más atraídos por la fase móvil. El orden de elución es el reverso de la fase normal y muchas veces este método puede separar mezclas que por fase normal no se logra. II. 2.2. Eluente o fase móvil Elegir el eluente o fase móvil es otro proceso importante en la cromatografía. La elección de éste se lleva a cabo considerando los siguientes criterios (la cantidad de muestra requerida es mínima): 1) Un eluente efectivo debe disolver al soluto pero no debe competir con él por los sitios de la fase estacionaria 2) Si el soluto no es soluble en el eluente éste puede quedar permanentemente adsorbido en la fase estacionaria. 3) El eluente no debe ser tan polar porque el soluto se moverá tan rápidamente que no tendrá oportunidad de establecer los equilibrios necesarios para la separación. La diferente habilidad de los eluentes para mover un soluto dado se denomina poder de elución. En la tabla 1 se muestra el orden de polaridad, con base en su constante dieléctrica, de algunos solventes en cromatografía en fase normal, éste por supuesto es el inverso para la cromatografía en fase reversa. Comunmente para escoger el eluente, se marcan puntos (con un lápiz) en una placa cromatográfica de TLC separados dos centímetros en forma alternada (ver figura 2) y sobre los puntos se aplica una pequeña cantidad de muestra en solución. Se deja secar la muestra y enseguida se aplica el primer eluente con un capilar normal. Si después de que seca el eluente se aprecia la muestra en el origen, debe aplicarse en el siguiente punto un eluente de mayor polaridad y así sucesivamente hasta encontrar el que presente un mayor número de círculos concéntricos. Se da el caso que el soluto no corra por la baja polaridad del eluente pero el siguiente lo extienda demasiado, se debe entonces usar una mezcla de los dos eluentes. 99 TABLA 1 Constantes dieléctricas de algunos solventes SOLVENTE CONSTANTE DIELÉCTRICA 1.9 2.0 2.0 2.2 2.3 2.4 3.4 4.3 4.8 6.0 10.0 12.3 18.3 20.1 20.7 24.3 32.6 37.0 78.5 Hexano Éter de petróleo Ciclohexano Tetracloruro de carbono Benceno Tolueno Tricloroetileno Éter dietílico Cloroformo Acetato de etilo Cloroetano Piridina 2-Propanol 1-Propanol Acetona Etanol Metanol Acetonitrilo Agua Mejor eluente a b c Cromatografía en capa fina FIGURA 2 100 La cromatografía en capa fina, puede iniciarse preparando los platos o placas cromatográficas, el adsorbente es usualmente alúmina o gel de sílice en suspensión acuosa mezclada con una pequeña cantidad de aglutinante, suele emplearse almidón o sulfato de calcio. Las placas son recubiertas con una delgada capa de 250 micras de espesor, luego son secadas en una estufa a 110 oC durante 1 hora, para eliminar el agua presente, pues esta disminuye la actividad del adsorbente. Las placas pueden ser de vidrio o de plástico duro. También se venden ya preparadas. Una vez que la placa esté lista, con un lápiz se marca el origen y se aplica la muestra (ver figura 3) con un microcapilar. El cromatograma se desarrolla en una cuba cromatográfica a la que previamente se le añadió un eluente adecuado; precaución, la altura del eluente no debe rebasar el origen (ver figura 3). Se deja correr hasta que el eluente alcance un centímetro antes de la altura tope de la placa, se puede marcar previamente esa marca con un lápiz. Para visualizar las bandas, si los compuestos son incoloros y presentan fluorescencia, se emplea la lámpara de luz ultravioleta, cuando esto no funciona, se puede emplear vapores de yodo (se introduce la cromatoplaca en un recipiente con cristales de yodo) o bien se emplea un reactivo específico aplicado con un aspersor sobre la placa. Una vez detectados los componentes, se calcula el Rf (ver figura 3). Frente del solvente c 2 tiempo Separación de componentes b Rf(componente 1) = a/c Rf(componente 2) = b/c 1 a Origen o línea base Cromatografía en capa fina FIGURA 3 II. 3. Cromatografía en columna Esta técnica también es una forma de cromatografía de adsorción líquido-sólido y constituye una poderosa herramienta en la síntesis orgánica, pues tanto en la industria como en la investigación, frecuentemente es necesario separar los componentes de una mezcla de reacción para ser usados posteriormente en otras rutas sintéticas. La cromatografía en columna es idónea para este propósito. Cuando el propósito es separar una mezcla se le conoce como técnica preparativa. 101 Es importante mencionar que los principios teóricos vistos para TLC se aplican de igual manera para cromatografía en columna. La selección del mejor eluente y adsorbente se puede efectuar por medio de cromatografía en capa fina. Básicamente una cromatografía en columna consiste en lo siguiente: la muestra a separar se disuelve en un solvente adecuado y es colocada en la parte superior de una columna empacada con un sólido adsorbente finamente dividido que sirve como fase estacionaria (ver figura 4). Luego se hace pasar el eluente (fase móvil) a través de la columna. Los componentes de la mezcla son inicialmente adsorbidos por la fase estacionaria en la parte superior de la columna, pero luego el eluente los mueve hacia abajo, a diferentes velocidades, dependiendo de su afinidad por el material adsorbente. Un componente débilmente adsorbido viajará más rápido que uno fuertemente unido a la fase estacionaria. Eluente Arena a Adsorbent e Arena Algodón Columna cromatográfica FIGURA 4 Cuando los componentes son coloreados, se observan diferentes bandas a medida que éstas se separan por medio de efecto del eluente y los diferentes tiempos de retención de cada soluto. Cada fracción es entonces separada en recipientes diferentes. En cuanto a la cantidad de adsorbente usada para empacar la columna varía de acuerdo a la diferencia en los coeficientes de distribución y las polaridades de los componentes individuales del sistema cromatográfico. Para separaciones simples es posible usar 10 g de adsorbente por 1g de muestra, pero si los componentes tienen polaridades semejantes, la relación debe incrementarse de 100-200 :1. Una proporción 25 :1 es un buen punto de partida en esos casos. Como regla general la relación de altura a diámetro de una columna empacada debe ser 8 :1. 102 II. 3. 1. Proceso de empacado Antes de iniciar el proceso de empacado se debe tener bien claro que la eficiencia de la separación va a depender de lo bien que este proceso sea llevado a cabo, si llegan a formarse fracturas en el adsorbente por causa de un empaque deficiente, es conveniente volver a empezar. Existen dos formas de empacar una columna: el método en seco y el método en húmedo; en el primero se va añadiendo el adsorbente seco sobre el eluente que se encuentra llenando la columna. En el segundo se prepara una suspensión del adsorbente en el eluente seleccionado y se añade a la columna que debe estar llena sólo a la mitad de su capacidad. El método en húmedo se requiere cuando se usa como adsorbente gel de sílice. La columna debe ser de vidrio con llave de teflón o bien provista de un pedazo de manguera con una llave de Mohr para controlar la salida del solvente. Para empacar la columna por el método en seco se llena con el eluente, se introduce un trozo de lana de vidrio o de algodón hasta acomodarlo en el fondo de la columna, sacando las burbujas de aire, luego se pone un cm de arena (ver figura 4) procurando un nivel uniforme en ésta, para asegurar un empaque uniforme del adsorbente y bandas bien definidas de los solutos. Al empacar, se debe mantener ligeramente abierta la llave para desalojar las burbujas de aire, pero nunca se deja que el nivel del eluente baje hasta el adsorbente porque se van a producir fracturas y hay que volver a empezar. Es necesario también pegar suavemente en la columna con un lápiz recubierto de manguera de hule para uniformizar el nivel del sólido. Al terminar de llenar se coloca otro centímetro de arena, la cual tiene como función prevenir el disturbio del adsorbente al añadir el eluente durante el proceso cromatográfico. II. 3. 2. Proceso de elución Para iniciar la cromatografía de la muestra, ésta se disuelve en una mínima cantidad de solvente y se añade a la columna que previamente ha sido desalojada del eluente hasta el nivel de la parte superior de la arena. Se deja adsorber la muestra y se lava con un poco de eluente, después se llena la columna con más líquido. La elución de la muestra puede ser simple o fraccionada (llamada también técnica de gradiente). En la elución simple se usa un solo eluente durante toda la separación. Este procedimiento funciona bien para la separación de mezclas de dos o tres componentes con polaridades semejantes. La técnica de elución fraccionada es la más adecuada. En ésta se usa, una serie de eluentes con incrementos en polaridad para llevar a cabo la elución de la mezcla en la columna. 103 La elución se inicia con un eluente no polar como hexano o éter de petróleo para separar la primera banda con los componentes menos polares. Los componentes de mayor polaridad, quedan adsorbidos en la fase estacionaria y para moverlos se hace un incremento gradual en la polaridad del eluente para que las bandas individuales se separen y no coeluyan. Por ejemplo, si se parte de hexano, se va añadiendo 5 % de cloroformo y 95 % de hexano, luego se incrementa el cloroformo a 10, 15, 20, 40, 80 % disminuyendo el hexano. De esta manera las diferentes bandas migran, hasta abandonar la columna. Como una regla general se tiene que añadir tres veces el volumen de la columna antes de cambiar al siguiente nivel de polaridad. Cuando las diferentes bandas son coloreadas, las fracciones se colectan en varios recipientes (tubos de ensayo o matraces Erlenmeyer). Luego se elimina el solvente para recuperar los solutos. Desafortunadamente muchos compuestos orgánicos son incoloros y a simple vista no se pueden diferenciar las bandas para colectarlas. En estos casos es posible usar detectores electrónicos que en la mayoría de los casos no están disponibles en laboratorios comunes; por ello, la ayuda que presta la cromatografía en capa fina, para monitorear las distintas bandas, es de un gran valor. • La necesidad de efectuar separaciones más rápidas y de mayor resolución ha requerido el desarrollo de técnicas mas elaboradas. Tal es el caso de la cromatografía de gases (CG) o la cromatografía de alta presión o HPLC (high performance liquid chromatography) pero la descripción de ésta cae fuera de los alcances del presente manual. III. MATERIAL • • • • • • • • • • • • • • • • Tubos capilares Mechero Vasos de precipitado de 100mL o cuba cromatográfica Vidrio de reloj Regla de 30 cm Probeta de 25 mL Lápiz Placas para TLC Papel filtro # 41 y papel para cromatografía Tubo de ensayo Pipetas de 10 mL Gotero Pipetas de 1mL Hilo Cinta adhesiva Tijeras 104 IV. REACTIVOS • Eluentes: éter etílico, acetona, hexano, metanol, etanol, butanol-acetato de etilo (4 : 2) • Colorantes vegetales V. PROCEDIMIENTO V.1. Cromatografía en capa fina V. 1. 1. Separación de los componentes de los colorantes vegetales 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. 14. 15. Marcar con un lápiz puntos separados dos centímetros en una placa cromatográfica de TLC (ver figura 2). Aplicar una pequeña cantidad de la muestra en solución, con un microcapilar, procurando no romper la capa de adsorbente y que la mancha sea de un diámetro no mayor a 1 mm. Dejar secar la muestra y aplicar el primer eluente (menos poloar) con un capilar normal. Si después de que seca el eluente se aprecia la muestra en el origen, aplicar en el siguiente punto un eluente de mayor polaridad y así sucesivamente. Elegir el eluente adecuado con base a donde haya mayor separación de círculos concéntricos. Marcar el origen en otra placa, midiendo un centímetro desde la base de la placa (ver figura 3). Aplicar la muestra con un microcapilar sobre el origen. Medir 3 mL del eluente seleccionado. Añadir el eluente a la cuba y mantenerla cerrada. Cortar un papel filtro del tamaño de la cuba e introducirlo, dando vuelta alrededor de las paredes internas, dejando una ranura para poder observar. Introducir la placa en la cuba. El eluente no debe tocar el origen. Desarrollar el cromatograma. Sacar la placa cuando falte 1 cm para llegar a la altura máxima y poner una marca (ver figura 3). Dejar secar la placa. Medir la distancia recorrida por el eluente. Localizar las manchas y medir las distancias recorridas, desde el origen hasta el centro geométrico de las mismas. Calcular los Rf para cada una de ellas (ver figura 3). V. 1. 2. Separación de mezcla de tintas o colorantes en papel 1. Recortar el papel cromatográfico en forma de rectángulo, aproximadamente de 10 x 3 cm. 2. Elegir el eluente adecuado de acuerdo a la sección V.1.1. pasos del 1 al 4. 3. Marcar el origen con un lápiz midiendo 1 cm a la derecha del papel y 1 cm de la base del papel hacia arriba. 105 4. Colocar la muestra en solución mediante un capilar estirado en el primer punto, cuidar que el diámetro de la mancha no sea mayor de 1 a 2 mm. 5. Medir 3 mL del eluente seleccionado. 6. Añadir el eluente a la cuba y mantenerla cerrada. 7. Cortar un papel filtro del tamaño de la cuba e introducirlo, dando vuelta alrededor de las paredes internas, dejando una ranura para poder observar. 8. Introducir la placa en la cuba. El eluente no debe tocar el origen. 9. Desarrollar el cromatograma. 10. Sacar la placa cuando falte 1 cm para llegar a la altura máxima y poner una marca (ver figura 3). 11. Dejar secar la placa. Medir la distancia recorrida por el eluente. 12. Localizar las manchas y medir las distancias recorridas, desde el origen hasta el centro geométrico de las mismas. 13. Calcular los Rf para cada una de ellas (ver figura 3). VI. DIAGRAMA DE FLUJO VI. 1. Cromatografía en capa fina VI. 1. 1. Separación de los componentes de colorantes vegetales 2 1 3 Marcar en una placa de TLC los puntos con un lápiz (ver. Fig. 2), y aplicar la muestra mediante un microcapilar en varios puntos Dejar secar la muestra y aplicar los eluentes a probar con un capilar. Elegir el eluente adecuado 1,2 3,4 5 6 Cortar un papel filtro del tamaño de la cuba e introducirlo dando vuelta, cubriendo las paredes de la cuba Marcar el origen en otra placa, midiendo un centímetro desde la base de la placa (ver figura 3). 5 4 Medir 3 mL del eluente seleccionado y añadir el eluente a la cuba. 7,8 Aplicar la muestra con un microcapilar estirado sobre el origen. 6 9 7 106 7 9 8 Introducir la placa en la cuba. El eluente no debe tocar el origen. De-sarrollar el cromato-grama Sacar la placa cuando falte 1 cm para llegar a la altura máxima y poner una marca (ver figura 3). 10,11 Dejar secar la placa. Medir la distancia recorrida por el eluente 13 12 Procedimiento VI. 1. 2. 11 10 Calcular los Rf para cada uno de los componentes detectados en el cromatograma. Localizar las manchas y medir las distancias recorridas, desde el centro geométrico al origen 14 15 VI. 1. 2. Separación de mezcla de tintas y/o colorantes en papel 1 2 Recortar un rectángulo de 10 x 3 cm del papel comatográfico 1 3 Elegir el eluente adecuado de acuerdo a la sección V.1.1. pasos del 1 al 4 2 Marcar el origen en el papel cromatográfico, midiendo un centímetro de la base del papel (ver figura 3) 3 4 107 4 5 Aplicar la muestra con un microcapilar estirado sobre el origen 6 Medir 3 mL del eluente seleccionado y añadir el eluente a la cuba Cortar un papel filtro del tamaño de la cuba e introducirlo dando vuelta, cubriendo las paredes de la cuba 4 5,6 7 9 8 Dejar secar la placa. Medir la distancia recorrida por el eluente 11 7 Sacar la placa cuando falte 1 cm para llegar a la altura máxima y poner una marca (ver figura 3) Introducir la placa en la cuba. El eluente no debe tocar el origen. Desarrollar el cromatograma 10 8,9 11 10 Localizar las manchas y medir las distancias recorridas, desde el centro geométrico al origen Calcular los Rf para cada uno de los componentes detectados en el cromatograma 12 FIN 13 Pasos correspondientes al VI. PROCEDIMIENTO VIII. BIBLIOGRAFÍA 1. Krubsack Arnold J., Experimental Organic Chemistry, Allyn and Bacon Inc. Boston, 1973. 2. Gilberts, Martin Roberts, Experimental Organic Chemistry, Saunders College Publisher, 1994. 108 CUESTIONARIO 1.- Dé la definición de cromatografía. Clasificación de la cromatografía. Ejemplos 2.- ¿Qué significa R.f. y para qué sirve conocerlo? 3.- La cromatografía en capa fina, sus características y aplicaciones. 4.- Eluentes, soportes y reveladores más comunes para cromatografía en capa fina. 5.- Factores que influyen en una separación por cromatografía en capa fina. 109 PRÁCTICA No. 8 PUNTOS DE FUSIÓN Y EBULLICIÓN I. OBJETIVOS Determinar experimentalmente el punto de fusión y ebullición y valorarlos como información indispensable en la caracterización de una sustancia pura, adicionalmente, practicar y dominar las técnicas de medición de puntos de fusión de sólidos orgánicos y puntos de ebullición de líquidos orgánicos utilizando diferentes métodos y equipos para establecer el grado de pureza del material sobre la base de los intervalos de fusión y ebullición. II. FUNDAMENTO II.1. Punto de Fusión El índice de pureza primario, usado en química orgánica, para compuestos cristalinos, es el punto de fusión. Se puede definir como, la temperatura a la cual un sólido se transforma en líquido a presión atmosférica. El punto de fusión de una sustancia cristalina pura es una propiedad física de la sustancia. Dado que la presión de vapor de un sólido es baja, el punto de fusión es poco sensible a los cambios de presión (dentro de límites razonables). El punto de fusión puede usarse para identificar una sustancia dada. La indicación de pureza se puede dar en dos formas, primero, a mayor pureza del material, mas alto su punto de fusión, segundo, a mayor pureza el intervalo de fusión es más pequeño. Con respecto a la relación entre el punto de fusión y la estructura del compuesto orgánico, la generalización de que a mayor fuerza intermolecular presente el compuesto, mayor será su punto de fusión, para compuestos de similar peso molecular, tiene muchas excepciones. Lo que en la mayoría de los casos se cumple es que una estructura más simétrica produce un mayor punto de fusión (para compuestos con similares pesos moleculares). En general, la adición de pequeñas cantidades de una impureza a una sustancia pura causará una disminución en el punto de fusión, proporcional a la cantidad de impureza añadida. En la figura 1 se observa el comportamiento usual del punto de fusión de diversas mezclas de dos sustancias A y B. Las curvas superiores indican la temperatura a la cual toda la mezcla se encuentra en estado líquido. La curva inferior indica la 110 temperatura a la cual se observa el inicio de la fusión. Si se parte de la sustancia pura A, el punto de fusión disminuirá al ir añadiendo la impureza B. Al tener una mezcla particular se alcanza un mínimo en la temperatura de fusión y a partir de ahí aumenta hasta llegar al punto de fusión de la sustancia pura B. Pf A Líquido A + B Pf B Fusión completa Primera gota de líquido Temperatur a Intervalo Sólido A + B 0%B 0%A Punto de fusión – Curva de composición FIGURA 1 En la figura 1, la distancia vertical entre la curva cóncava y la convexa representa el intervalo de fusión. Podemos considerar una mezcla conteniendo la sustancia A con una cantidad relativamente pequeña de B, en esta mezcla el punto de fusión disminuirá y el intervalo de fusión aumentará. Por lo tanto para mezclas conteniendo pequeñas cantidades de impurezas (menos de 15 %), el intervalo de fusión indica la pureza del material. Una sustancia que funde en un intervalo pequeño debe ser pura, sin embargo, en el punto mínimo de la curva punto de fusión contra composición (figura 1), la mezcla forma un eutéctico, que también funde en un intervalo pequeñísimo. No todas las mezclas binarias forman eutécticos y algunas forman mas de uno. La figura 2 es un diagrama de fases que describe el comportamiento del sistema A-B durante la fusión. 111 B A tA tB Solución líquida A+B tM Solución líquida + sólido A Temperatur a Solución líquida + sólido B tC Sólido A + B M % mol A 100 80 60 40 20 0 % mol B 0 20 40 60 80 100 Diagrama de fases para la fusión de un sistema de dos componentes FIGURA 2 Si A es una sustancia pura, funde exactamente al llegar a su punto de fusión tA; Esto está representado por el punto A en el lado izquierdo del diagrama. Cuando B es una sustancia pura, funde a tB, su punto de fusión está representado por el punto B, al lado derecho del diagrama. En las mezclas de A + B el comportamiento es diferente. Si consideramos una mezcla de 80% A y 20% B (en porcentaje molar), el punto de fusión de esta mezcla está dado por tM en el punto M del diagrama. La adición de B a A ha bajado el punto de fusión de A de tA a tM; Esto también ha expandido el intervalo de fusión, la temperatura tM corresponde al límite superior del intervalo de fusión. La disminución del punto de fusión de A por la adición de B se lleva a cabo de la siguiente manera: A tiene el punto de fusión mas bajo en el diagrama de fase y al calentarse comienza a fundir y el sólido B se empieza a disolver en el líquido A que se formó; cuando el sólido B se disuelve en el líquido A el punto de fusión baja. Para comprender esto consideremos el punto de fusión en la dirección opuesta; cuando un líquido a alta temperatura se enfría llegará al punto donde solidifica (se congela), la temperatura a la cual se congela es idéntica a su punto de fusión; recordaremos que el punto de congelación de un líquido se puede bajar por la adición de una impureza. Dado que el punto de fusión y el punto de congelamiento son idénticos, una disminución en el punto de congelamiento corresponde a una disminución en el punto 112 de fusión; a mayor cantidad de impureza añadida mayor es la disminución en el punto de fusión, sin embargo, hay un límite, no podemos disolver una cantidad infinita de impureza en un líquido, en algún momento el líquido se satura. La solubilidad de B en A tiene un límite y este se alcanza precisamente en C, el punto eutéctico. El punto de fusión de la mezcla no puede ser menor que tC, la temperatura de fusión del eutéctico. II. 1. 1. Precauciones en la Determinación del Punto de Fusión Para llevar a cabo la determinación de punto de fusión se debe tomar en cuenta lo siguiente: • Las sustancias deben estar completamente secas y pulverizadas. • Cada determinación se debe hacer sobre muestra nueva, el punto de fusión varía cuando la muestra ya ha sido fundida. • El material de vidrio, sobre todo en el caso de capilares preparados estirando vidrio, debe ser lavado para evitar la acción del álcali en la superficie del vidrio, que puede catalizar reacciones como la condensación aldólica de aldehídos y cetonas, las mutarrotaciones de los azúcares y sus derivados, la descarboxilación de ciertos ácidos y algunas transposiciones moleculares. La limpieza se hace lavando el material con una solución diluida de un detergente neutro, después con ácido clorhídrico diluido (10%) y, finalmente, con agua destilada. • Los aparatos deberán estar calibrados. En el caso de la calibración del termómetro se deberá hacer en el sistema donde se va a emplear, sea tubo de Thiele o MeltTemp; El aparato Electrothermal ya está calibrado; éste se calibra anualmente enviándolo al proveedor o bien, mediante un equipo especial (kit de calibración), por el responsable del laboratorio. • Para la calibración del termómetro haremos la determinación del punto de fusión de las sustancias dadas en la tabla 1; éstas deben ser puras y estar finamente divididas. Con los datos obtenidos y los puntos de fusión reportados en la bibliografía se hará una curva de calibración (figura 3) colocando en el eje de las ordenadas los puntos de fusión observados y en el de las abscisas los puntos de fusión corregidos (reportados o reales). Se traza la curva OA entre los puntos. En las determinaciones subsecuentes, el punto de fusión observado (B) se proyecta horizontalmente hasta la curva, se hace descender verticalmente hasta obtener el valor corregido (C). • Los métodos de determinación del punto de fusión en esta práctica son para sustancias que funden en un intervalo de 25 - 300 oC, para puntos de fusión mas 113 altos se recomiendan métodos especiales, entre ellos, el uso de la barra de Maquenne1. • En el método del Tubo de Thiele se utilizan líquidos como el agua, la glicerina o el ácido sulfúrico, dependiendo del intervalo de fusión de la muestra o muestras. La glicerina es muy higroscópica y puede absorber tanta agua que hierve antes de su punto de ebullición (290 oC d.) por lo tanto se debe guardar siempre en un recipiente cerrado. El ácido sulfúrico es sumamente agresivo y debemos manejarlo con mucha precaución y de ser posible sustituirlo por otro líquido. La forma del tubo de Thiele permite corrientes de convección en el líquido, que mantienen una distribución de la temperatura bastante uniforme a través de todo el líquido TABLA 1 Estándares para Punto de Fusión Puntos de Fusión (oC) Compuestos Punto de fusión observado hielo acetanilida benzamida urea ácido succínico ácido 3,5-dinitrobenzoico 0 115 128 132 189 205 A B C Punto de fusión corregido Curva de calibración FIGURA 3 II. 2. Punto de Ebullición 114 Cuando un líquido se calienta, su presión de vapor se incrementa hasta el punto donde se iguala a la presión aplicada (usualmente la presión atmosférica). En este punto se observa que el líquido ebulle. El punto de ebullición normal se mide a 760 mm de Hg (1 atmósfera). A una presión aplicada menor, la presión de vapor necesaria para ebullir disminuye y el líquido hervirá a menor temperatura. La relación entre la presión aplicada y la temperatura de ebullición de un líquido se determina mediante su comportamiento presión de vapor - temperatura, la figura 4 representa idealmente, una curva típica de esta relación. mm Hg Presión de vapor 760 100 p. de eb. a 100 mm de Hg p. de eb. a 760 mm de Hg (1 atm) temperatura Curva ideal de presión de vapor vs. temperatura para un líquido FIGURA 4 Dado que los puntos de ebullición son muy sensibles a los cambios de presión, es muy importante registrar la presión barométrica si la determinación se lleva a una elevación significativamente mayor o menor que el nivel del mar. Cuando use una bomba de vacío o un aspirador de agua se debe medir la presión de trabajo. Como una regla general, el punto de ebullición de muchos líquidos disminuye 0.5 oC por cada 10 mm de Hg de disminución, en la presión, cuando se está cerca de los 760 mm Hg. A presiones mas bajas, disminuye 10 oC cada vez que la presión se baje a la mitad de la anterior. Por ejemplo, si el punto de ebullición observado es 150 oC a 10 mm de Hg de presión, el punto de ebullición será de 140 oC a 5 mm de Hg de presión. Para estimados más exactos podemos hacer uso de un nomograma2. Con respecto a la relación entre el punto de ebullición y la estructura de un compuesto orgánico, se puede decir que a mayor peso molecular mayor punto de ebullición para compuestos con similares fuerzas intermoleculares. Para compuestos de similar peso molecular el punto de ebullición será mayor para aquellos que presenten fuerzas intermoleculares más intensas. 115 Para determinar el punto de ebullición podemos hacerlo de dos maneras, cuando se tiene suficiente muestra para hacer una destilación, se puede tomar la temperatura de equilibrio marcada por el termómetro, este método requiere gran cantidad de muestra (5-10 mL), el otro método, el que efectuaremos en esta práctica, es a escala semimicro, solo se requiere utilizar una o dos gotas del líquido. Tomar en cuenta las precauciones consideradas en el punto de fusión lo mismo con respecto a la calibración y si es necesario, corregir, por el efecto de temperatura en el vástago del termómetro3. III. EQUIPO • • • Electrothermal Serie IA9000, Aparato Digital para punto de fusión Aparato para punto de fusión Fisher-Johns Aparato para punto de fusión Melt-Temp IV. MATERIAL • • • • • • • • • • • • Soporte Pinzas para soporte Mechero Manguera Tubo de Thiele Gotero Tubo Durham Capilares Termómetro Cubreobjetos Algodón Ligas V. REACTIVOS • • • • Sustancias para calibración (Tabla 1) Glicerina Sustancias sólidas orgánicas (de preferencia aquellas con Pf menor de 140°C) Sustancias líquidas (metanol, etanol) VI. PROCEDIMIENTO VI. 1. Determinación del Punto de Fusión VI. 1. 1. Punto de fusión por el método del tubo de Thiele 116 1. Preparar un tubo de Thiele, figura 5, llenándolo con glicerina, aproximadamente 1 cm arriba de la salida al “brazo” del tubo de Thiele. 2. Colocar el termómetro en un tapón monohoradado que se ha preparado con una abertura en forma de cuña en un lado del tapón, para evitar diferencias de presión, ver figura 6. 3. Sellar uno de los extremos de un capilar utilizando el mechero. 4. Introducir el sólido seco y pulverizado al tubo capilar en una cantidad que forme una columna no mayor de 1 o 2 mm. Esto se hace presionando suavemente, la abertura del capilar sobre la muestra pulverizada. Para que la muestra llegue al lado sellado del tubo, se deja caer éste (con el lado cerrado hacia abajo) dentro de un tubo de vidrio de unos 60 cm de largo que se sostiene verticalmente sobre la mesa. Cuando el capilar golpea la superficie de la mesa los cristales bajan al fondo del tubo capilar, si es necesario se repite el procedimiento. Por seguridad no golpee el capilar sobre la mesa para hacer que baje el sólido, ya que el tubo es muy frágil y puede romperse causándole una herida. 5. Unir el capilar al termómetro mediante una banda de hule (se puede cortar un pequeño pedazo de manguera formando una banda de hule resistente). Cuidar que la banda quede fuera del baño de glicerina. Colocar el capilar de manera que la muestra este lo mas cerca posible del bulbo del termómetro ver figura 5. Cuidar que la glicerina no entre al capilar. 6. Armar el equipo como se muestra en la figura 5. 7. Iniciar el calentamiento colocando el mechero bajo el brazo del tubo de Thiele como en la figura 5. Nota: El calentamiento debe ser lo más uniforme posible y el aumento en temperatura debe ser de 2 oC/min. 8. Observar y tomar nota de la temperatura a la que aparece la primera gota de líquido y la temperatura a la que funde completamente la muestra, esto es el intervalo de fusión. También observar si hay cambios en el color, en el aspecto de los cristales, si se desprenden gases o cualquier otro indicio de descomposición. Repetir dos veces más la determinación y promediar los resultados. Tapón monohoradado Liga tubo capilar Muestra Tubo Thiele 117 Tapón monohoradado con abertura en forma de cuña FIGURA 6 VI. 1. 2. Punto de Fusión por el método del Melt-Temp 9. Conectar el aparato Melt-Temp, ver figura 7. Debe tener instalado un termómetro calibrado para ese aparato en particular. El termómetro debe unirse con resina epoxi, si se quiere seguir trabajando con el sistema calibrado en otra ocasión. 10. Preparar un capilar con muestra, como en el paso 4. Colocarlo en las guías para los capilares, que están colocadas en la zona de calentamiento, junto al termómetro. 11. Encender el aparato y regular la velocidad de calentamiento mediante el control de transformador variable. La temperatura no debe aumentar a mas de 2 oC /min. 12. Observar la muestra, a través del ocular (lupa), y estar observando la temperatura en el termómetro. Registrar la temperatura inicial y final de la fusión. El aparato debe dejarse enfriar antes de una nueva determinación. 118 Aparato Melt-Temp FIGURA 7 VI. 1. 3. Punto de Fusión por el método del Fisher-Johns 13. Conectar el aparato calibrado, ver figura 8. 14. Colocar la muestra pulverizada entre dos cubreobjetos, o trozos de cubreobjetos, a manera de “sandwich” (debe ser una capa delgada de polvo). 15. Colocar el sandwich sobre la depresión de la platina de aluminio. 16. Encender el aparato y regular la velocidad de calentamiento mediante el control de transformador variable. 17. Observar a través de la lupa la muestra, y leer la temperatura a la que aparece la primera gota de líquido y la temperatura a la que todo el sólido se ha fundido. 18. Anotar los datos del intervalo de fusión en el diario de laboratorio. 119 Aparato de Fisher-Johns FIGURA 8 VI. 1. 4. Punto de Fusión por el método del Electrothermal 19. Colocar el electrothermal4 sobre la mesa y conectarlo según la figura 9. 20. Aflojar los tornillos mostrados en la figura 10, para mover el brazo, hacer el ajuste y volver a apretar. 21. Encender la fuente de poder (ON en la figura 9) y permitir que el aparato adquiera la temperatura ambiente. Se recomienda esperar 30 minutos. 22. Preparar el capilar con la muestra según el paso 4 del método del Tubo de Thiele. Colocar el capilar en el lugar para muestras, hay tres lugares, en los dos restantes debe colocar dos capilares más, con muestra o vacíos. 23. Establecer el “SET POINT” o temperatura base, unos 10 oC abajo del punto de fusión esperado (si la muestra es conocida). Suponiendo que la muestra funde a 117 oC, el set point será 107 oC. Esta temperatura se establece como “SET POINT”, presionando la tecla, con la flecha hacia arriba, 11 veces (10 grados por cada vez que se presiona) nos dará 110 oC, presionamos la tecla, con la flecha hacia abajo, 3 veces (menos 1 grado por cada vez que la presionemos) y obtenemos los 107 oC que se necesita. Si se comete un error se presiona la tecla CLEAR y se vuelve a empezar. 24. Llegar al set point presionando la tecla GOTO. Se iluminarán las cuatro barras L.E.D. al ir subiendo la temperatura. Para revisar el set point se puede presionar la tecla que tiene la flecha hacia arriba y aparecerá (por un segundo) la temperatura que hemos fijado. Durante este paso, la temperatura sube rápidamente, cuando se alcanza el set point se enciende el círculo a la izquierda del GOTO y se oirán 3 “beeps” para indicar que se ha llegado a la temperatura base (“SET POINT”). 25. Presionar GOTO para iniciar la rampa de calentamiento, con una velocidad de 1 oC/min (esta velocidad es la que permite el aparato), se encenderá el círculo al lado derecho de la flecha del GOTO. 26. Observar la muestra, a través del ocular (que puede ser ajustado girándolo), y la temperatura inicial y final de la fusión. 27. Anotar los datos en el diario de laboratorio. Dejar enfriar antes de otra determinación. 120 Selector de voltaje (120 V o 240 V) Fusible Fuente de poder Instrumento Control de fuente principal Electrothermal FIGURA 9 Pinzas Tornillo Movimiento y ajuste del brazo FIGURA 10 121 VI. 2. Determinación del Punto de Ebullición 28. 29. 30. 31. 32. 33. 34. 35. 36. 37. Preparar un tubo de Thiele con glicerina (ver pasos 1 y 2). Sellar por un extremo, un capilar con un mechero. Colocar en un tubo Durham una o dos gotas de líquido. Introducir el capilar sellado, “boca abajo”, dentro del tubo Durham. Utilizando una banda de hule unir el tubo Durham con el termómetro. Ver figura 11. Colocar el termómetro en el Tubo de Thiele, cuidar que la glicerina no entre al tubo Durham. Calentar el Tubo de Thiele (ver paso 7). Calentar hasta que salga una corriente continua de burbujas del capilar (registrar esta temperatura como t1). Retirar el mechero, y observar cuando el líquido entra al capilar (registrar esta temperatura como t2). Reportar el intervalo de temperatura de ebullición t1 - t2. Tubo Durham, con la muestra y el capilar sellado, unido al termómetro FIGURA 11 122 VII. DIAGRAMA DE FLUJO VII. 1. Punto de Fusión VII. 1. 1. Punto de Fusión por el Método del Tubo de Thiele 1 2 Llenar el tubo de Thiele, con glicerina, 1 cm arriba de la salida al brazo del tubo 3 Colocar el termómetro en un tapón monohoradado con un corte en forma de cuña Sellar un tubo capilar con el mechero 3 2 1 5 6 4 Calentar el tubo de Thiele con el mechero en el brazo lateral del tubo. Controlar la velocidad de calentamiento a 72 o C/min 7 Unir el capilar al termómetro, mediante una banda de hule y armar equipo figura 5. Observar y registrar la temperatura inicial y final de la fusión. Anotar los datos en el diario de laboratorio muestra final Introducir el sólido al capilar 4 5,6 8 NO Ejecutar los pasos del 3 al 7 de este diagrama de fllujo SI 8 FIN VII. 1. 2. Punto de Fusión por el Método del Melt-Temp 1 2 Conectar el Melt-temp. Debe tener instalado un termómetro calibrado en ese aparato 9 3 Preparar un capilar con muestra y colocarlo en las guías para muestra 10 Encender el aparato y regular la velocidad de calentamiento a 2 o C/min. 11 4 123 4 5 Observar la muestra y el termómetro y registrar la temperatura inicial y final de la fusión. Anotar los datos en el diario de laboratorio NO muestra final Dejar enfriar el aparato y repetir los pasos 2 al 4 de este diagrama SI 12 FIN VII. 1. 3. Punto de Fusión por el Método del Fisher-Johns 1 2 Calibrar el aparato. Conectarlo y observar que la toma de corriente es especial 3 Preparar un “sandwich” de la muestra pulverizada, entre dos cubreobjetos 4 5 Anotar los datos en el diario de laboratorio 18 15 14 13 6 Colocar el “sandwich” sobre la depresión de la platina de aluminio Observar a través de la lupa el intervalo de fusión. Encender el aparato y regular la velocidad de calentamiento 16 17 7 Muestra final NO Repetir los pasos del 2 al 6 FIN 18 SI FIN 124 VII. 1. 4. Punto de Fusión por el Método del Electrothermal 2 1 Conectar el aparato y mover los tornillos para ajustar el brazo 3 Encender la fuente de poder y esperar 30 minutos 19,20 6 Preparar el capilar con la muestra 22 21 5 Después de alcanzar el SET POINT volver a presionar GOTO para iniciar la rampa de calentamiento 4 Establecer el SET POINT utilizando las flechas y luego presionar GOTO Colocar el capilar en su lugar y otros dos capilares para ocupar los sitios vacíos 22 23,24 25 7 8 Observar y registrar las temperaturas inicial y final SI Muestra final Anotar los datos en el diario de laboratorio 27 26 NO 9 Seguir los pasos del 3 al 8 de este diagrama FIN VII. 2. Punto de Ebullición. Método del Tubo de Thiele 3 2 1 Preparar un tubo de Thiele con glicerina 28 Sellar por un extremo, un capilar. Colocar en un tubo Durham una o dos gotas de líquido Introducir el capilar sellado, “boca abajo”, dentro del tubo Durham 31 29,30 4 125 4 5 Utilizar una banda de hule para unir el tubo Durham con el termómetro. Colocar el termómetro en el tubo de Thiele, cuidar que la glicerina no entre al tubo Durham 32,33 6 Calentar el tubo de Thiele. Calentar hasta que salga una corriente continua de burbujas del capilar, registrar esta temperatura como t1. Retirar el mechero, y observar cuando el líquido entra al capilar, anotar esta temperatura como t2. 36 34,35 7 Anotar los resultados en el diario de laboratorio SI Muestra final 37 NO 8 FIN Nota: Repetir los pasos del 2 al 7 de este diagrama Pasos correspondientes al VI. PROCEDIMIENTO VIII. BIBLIOGRAFÍA 1. Fuson, Reynold; Curtin David; Shriner, Ralph; Identificación Sistemática de Compuestos Orgánicos, cuarta Reimpresión, Ed. Limusa, México p.40, 41 y 43, 1979. 2. Pavia, Donald L., Lampam, Gary M. y Kriz Jr, Goerge S., Introduction to Organic Laboratory Techniques a Contemporary Approach; tercera edición, Saunders College Publishing, 1988. 3. Electrothermal Serie IA9000. aparato digital para punto de fusión, Manual de funcionamiento. 126 CUESTIONARIO 1.- Cuál es la utilidad del Punto de fusión en de muestra orgánica. 2.- Factores que determinan la fusión de un sólido. 3.- ¿Qué es una Mezcla eutéctica?. 4.- Haga un listado de 10 compuestos orgánicos líquidos e indique su Punto de ebullición 5.- Como se corrige el punto de ebullición a diferente presión. 127 PRÁCTICA No. 9 ÍNDICE DE REFRACCIÓN I. OBJETIVO Determinar experimentalmente el índice de refracción de diferentes muestras de líquidos orgánicos. II. FUNDAMENTO El índice de refracción (n) es una propiedad física muy útil, apoya la identificación de sustancias y, además, puede ser usada como criterio de pureza de un líquido ya que la medición es muy sensible a pequeñas impurezas que contenga el líquido analizado. Cuando un rayo de luz incide en la superficie de un líquido, sufre cambios en velocidad y en dirección, en el límite de interfase, estos cambios dependen de la temperatura y de la longitud de onda de la luz. El índice de refracción de un líquido es precisamente la relación entre la velocidad con que viaja el rayo de luz en el aire con respecto a aquella con que viaja el rayo de luz en el medio. También se puede expresar el índice de refracción como la relación del seno del ángulo de incidencia de un rayo de luz en el aire, con respecto al seno del ángulo de refracción en el líquido (ver figura 1). Lo anterior también se cumple si el medio es un sólido transparente. El índice de refracción es denotado por la letra n utilizando un superíndice que indica a qué temperatura se realizó la medición y un subíndice para indicar la longitud de onda de la luz monocromática usada en la medición. Usualmente la luz usada es la correspondiente a la llamada “línea D del sodio” que corresponde a 589 nm (1 nm = 10-9 m); en este caso una letra D aparecerá como subíndice. Vaire = c naire c Vmedio = nmedio Donde: c = velocidad de la luz en el vacío Vaire = velocidad de la luz en el aire Vmedio = velocidad de la luz en el medio (líquido o sólido) naire = 1 128 Por lo tanto, el índice de refracción (n) de un sólido o líquido sería: n = Vaire n= Vmedio sen θ sen φ Donde: θ - ángulo de incidencia del rayo de luz en el aire φ - ángulo de refracción del rayo de luz en el medio (líquido o sólido) Vaire θ Vmedio φ El Índice de refracción FIGURA 1 Es mucho más común medir el índice de refracción respecto a algún medio distinto del vacío; con este objeto se emplea a menudo el aire como referencia. Afortunadamente, el cambio en el índice de refracción del aire con la temperatura y la presión es muy pequeño. Un índice de refracción nD medido respecto al aire, con la línea D del sodio puede convertirse en nvacío con la ecuación: nvacío = 1.00027 nD No es posible medir directamente los ángulos de incidencia y de refracción, por lo que se han desarrollado sistemas ópticos especiales que dependen del ángulo crítico de reflexión en el límite del líquido con un prisma de vidrio de índice de refracción conocido. El ángulo crítico se define como el ángulo de refracción en un medio cuando el ángulo de la radiación incidente es de 90 grados. El refractómetro tipo Abbe opera sobre este principio. Las ventajas de este refractómetro son: 129 • Puede determinar los nD para una gran variedad de muestras líquidas y sólidas debido a su amplio intervalo de trabajo 1.30 a 1.71 nD. • Para iluminar la muestra se puede usar una fuente de luz blanca ya que el sistema da el índice de refracción para la línea D del sodio. • Solo se requiere unas gotas de líquido • Tiene incorporado un control de temperatura de los prismas y de la muestra • Los prismas compensadores de Amici permiten la determinación de la dispersión específica7. Una de las desventajas que presenta este refractómetro es la de no poder usarse con muestras que dañan el material de los prismas, como líquidos corrosivos tales como ácidos y bases, o con aquellos que afectan la resina que fija los prismas a la estructura metálica. III. EQUIPO • Refractómetro tipo Abbe, marca Milton Roy, modelo ABBE-3L, con termómetro acoplado (0 - 100 oC) • Baño de control de temperatura constante IV. MATERIAL • • • • • Goteros de plástico o pipetas beral Pizeta (1) Algodón (1 trozo) Tubos de ensayo de 13X 100 o 18x150 Vidrio para calibración (nD20=1.5125) V. REACTIVOS • Agua destilada • Muestras líquidas (tolueno, isopropanol, nitrobenceno, 1-butanol, benzaldehído, ciclohexanona, propanol) ciclohexano, VI. PROCEDIMIENTO 1. 2. 3. Encender el sistema de control de temperatura del refractómetro. Esperar a que el termómetro marque 20 ± 0.2 oC. Encender la lámpara moviendo el control de encendido hacia arriba. (ver figura 2) 130 OCULAR CONTROL DE ENCENDIDO Vista lateral izquierda FIGURA 2 4. 5. Levantar el prisma superior del refractómetro (ver figura 6). Limpiar* los prismas utilizando un trozo de algodón impregnado con agua desmineralizada o isopropanol, secar los prismas. 6. Colocar en el centro del prisma inferior 2 o 3 gotas de agua desionizada o destilada, utilizando una gotero de plástico (no usar aplicadores de vidrio o metal). Asegurar que la cantidad de muestra usada llene completamente el espacio entre los dos prismas. 7. Bajar el prisma superior y observar por el ocular (ver figura 2); identificar las 2 zonas que aparecen en el campo visual (ver figura 4). 8. Girar el control manual (ver figura 6) hasta obtener una línea clara y definida 9. Girar el control manual hasta que la línea límite (1 en la figura 4) coincida con la intersección o retículo (2 en la figura 4) de las líneas cruzadas de la zona de refracción. 10. Mover y sostener el control de encendido hacia abajo para observar a través del ocular las escalas de medición (ver figura 5). 11. Leer el índice de refracción en la correspondiente escala y anotarlo hasta la milésima cifra en el diario del laboratorio. 12. Repetir la medición del índice de refracción 4 veces más centrando la línea límite de reflexión desde arriba y desde abajo de las líneas en cruz. La precisión del valor del índice debe ser ± 0.0001. Se debe obtener un valor de 1.333. Si se obtiene un valor diferente a 1.333 continuar en el paso 18. Termina verificación. 131 DETERMINACIÓN DEL nD DE MUESTRAS LIQUIDAS 13. Separar y limpiar los prismas utilizando un trozo de algodón impregnado con isopropanol o agua desmineralizada, secar los prismas. 14. Colocar en el centro del prisma inferior 2 o 3 gotas del líquido problema, utilizando un gotero de plástico (no usar aplicadores de vidrio o metal). Asegurar que la cantidad de muestra usada llene completamente el espacio entre los dos prismas. 15. Ejecutar los pasos 7 a 11 16. Repetir la medición del índice de refracción 4 veces más centrando la línea límite de reflexión desde arriba y desde abajo de las líneas en cruz. Calcular el coeficiente de variación de la medición y anotarla en el diario de laboratorio. 17. Ejecutar los pasos 12 a 16 para otros 3 líquidos problema. Continuar en el paso 29. CALIBRACION 18. Encender el sistema de control de temperatura del refractómetro. 19. Esperar a que el termómetro marque 20 ± 0.2 oC. 20. Encender la lámpara moviendo el control de encendido hacia arriba. (ver figura 2) 21. Colocar en el centro del prisma inferior 1 gota de 1-bromonaftaleno, utilizando un gotero de plástico (no usar aplicadores de vidrio o metal). 22. Colocar el vidrio de calibración sobre la gota del 1-bromonaftaleno con el extremo pulido hacia la iluminación. 23. Mover el vidrio de calibración para dispersar completamente el área de contacto. No permitir exceso de 1-bromonaftaleno en los bordes. 24. Colocar y sostener el control de encendido de la lámpara hacia abajo y usar el control manual para fijar la escala de índices al valor grabado en el vidrio de calibración (nD = 1.5125). Enfocar el ocular para obtener una mejor definición del retículo y la escala. 25. Mover el control de encendido hacia arriba y colocar el brazo de la lámpara (ver figura 6) para lograr un mejor contraste y definición en la línea límite de reflexión (1 en la figura 4). 26. Girar el disco compensador (ver figura 6) hasta que la sección acromática de la línea límite de reflexión esté centrada en la marca del retículo vertical. 27. Usar el control manual para centrar la línea límite de reflexión exactamente en el retículo. 28. Colocar y sostener el control de encendido de la lámpara hacia abajo y anotar el valor del índice de refracción. Debe coincidir con el valor grabado en el vidrio de calibración. 29. Repetir la medición del paso 4 veces más centrando la línea límite de reflexión desde arriba y desde abajo de las líneas en cruz. La precisión del valor del índice debe ser ± 0.0001. Continuar en el paso 13. Si el valor del índice no coincide con el esperado, continuar en el paso 30. 132 30. Insertar una llave inglesa, mover el tornillo (ver figura 3) que se encuentra al lado del control manual y ajustar la escala al valor esperado del vidrio de calibración. 31. Finalizar el procedimiento *Nota: El sello de los prismas es una resina epoxi, por lo cual para limpiar los prismas no deben usarse los siguientes solventes: N,N-Dimetilformamida, fenoles, cresoles, soluciones de ácido acético, N,N-dimetilacetamida, tetrahidrofurano, acetato de metilo, acetato de vinilo, thiner. CONTROL MANUAL Vista lateral derecha FIGURA 3 TORNILLO Vista lateral derecha FIGURA 3 Vista de la reflexión total FIGURA 4 133 1. Escala de índice de refracción 2. Escala de sólidos totales disueltos 3. Retículo dual Vista de escalas FIGURA 5 1. 2. 3. 4. 5. 6. 7. 8. Lámpara de iluminación Caja del prisma superior Prisma de medición Caja del prisma inferior Bisagra Disco compensador Termómetro Tubo de entrada de agua Componentes del refractómetro FIGURA 6 134 VII. DIAGRAMA DE FLUJO Verificación 2 1 Encender el sistema de control de temperatura del refractómetro y esperar a que el termómetro marque 20 ± 0.2 oC 1,2 3 Encender la lámpara moviendo el control de encendido hacia arriba. Levantar el prisma superior del refractómetro, limpiar y secar los prismas 6 5 Escribir el valor nD, hasta la milésima cifra, en el diario del laboratorio. Repetir la medición del nD 4 veces más centrando la línea límite de reflexión desde arriba y desde abajo de las líneas en cruz. 11,12 Colocar en el prisma inferior 2 o 3 gotas de agua desionizada. 6 3,4,5 Girar el control manual hasta que la línea límite coincida con la intersección de las líneas cruzadas en la zona de refracción. Mover el con-trol de encendido hacia abajo y observar las escalas de 8,9,10 medición 4 Bajar el prisma superior y observar por el ocular. Identificar las 2 zonas que aparecen en el campo visual. Girar el control manual hasta obtener una línea clara y definida 7 Determinación del nD de muestras líquidas 7 SI 20 nD = 1.333 8 Separar y limpiar los prismas con un trozo de algodón impregnado con un solvente adecuado. Secar los prismas Colocar en el prisma inferior 2 o 3 gotas del líquido problema 14 NO 13 18 10 FIN 9 Ejecutar los pasos 7 a 9 de este diagrama para las demás muestras líquidas 16,17 Ejecutar los pasos 4 a 6 de este diagrama 15 135 calibración 11 13 12 Ejecutar el paso 2 de este diagrama Colocar en el prisma inferior una gota de 1bromonaftaleno 18, 19,20 Colocar sobre el 1-bromo Naftaleno el vidrio de calibración con la parte pulida hacia la iluminación 21 22 15 14 Mover vidrio de calibración para dispersar completamente el 1bromonaftaleno; no permitir exceso de éste en los bordes. 23 Mantener el control de encendido de la lámpara hacia abajo y usar el control manual para fijar la escala de índices al valor grabado en el vidrio de calibración (nD = 1.5125). 18 19 Usar el control manual para centrar la línea límite de reflexión exactamente en el retículo. 27 16 24 Girar el disco compensador hasta que la sección acromática de la línea límite de reflexión esté centrada en la marca del retículo vertical. 26 20 Enfocar el ocular para obtener una mejor definición del retículo y la escala. 24 17 Mover el control de encendido hacia arriba y colocar el brazo de la lámpara para lograr un mejor contraste y definición en la línea límite de reflexión 25 21 Colocar y sostener el control de encendido de la lámpara hacia abajo y anotar el valor del índice de refracción. Debe coincidir con el valor grabado en el vidrio de calibración. Repetir la medición nD 4 veces más centrando la línea límite de reflexión desde arriba y desde abajo de las líneas en cruz. 29 NO 20 nD = 1.5125 22 SI 23 28 22 Insertar una llave inglesa, mover el tornillo que se encuentra al lado del control manual y ajustar la escala al valor esperado del vidrio de calibración. 30 23 Ejecutar los pasos 7 a 10 de diagrama de determinación de muestras. 29 Pasos correspondientes al VI. PROCEDIMIENTO 136 VIII. BIBLIOGRAFIA 1. Fuson, Reynold; Curtin David; Shriner, Ralph; Identificación Sistemática de Compuestos Orgánicos, cuarta reimpresión, Limusa, México 1979. 2. Scoog, D.A. y West, D.M.; Análisis Instrumental, segunda edición, Nueva McGraw Hill, 1990. 3. Weissberger, A.; Physical Methods of Organic Analysis, Vol. 1, Part 2, Capítulo 18. 4. Lagowsky, Joseph J.; Laboratory Experiments in Chemistry, D. Van Nostrand, 1977. 5. Vogel, Arthur I.; Elementary Practical Organic Chemistry, Part 2, Qualitative Organic Analysis, Longman, segunda Edición, 1966. 6. Pavia, Donald L. Lampam, Gary M. y Kriz Jr, Goerge S.; Introduction to Organic Laboratory Techniques a Contemporary Approach; tercera edición, Saunders College Publishing, 1988. 7. Fuson, Reynold; Curtin David; Shriner, Ralph; Identificación Sistemática de Compuestos Orgánicos, cuarta reimpresión, Limusa, México, p. 65-66, 1979. 137 CUESTIONARIO 1.- Elabore una lista con las muestras proporcionadas indicando su índice de refracción. 2.- ¿Que es índice de refracción y como se determina? 3.- Escriba al menos tres diferentes tipos de refractómetros que se pueden utilizar para la determinación de índice de refracción 138 APÉNDICE A FACULTAD DE CIENCIAS QUÍMICAS REGLAMENTO GENERAL DE LABORATORIOS 1. Es responsabilidad de la administración de cada laboratorio mantenerlo en condiciones adecuadas de Seguridad, Salud y Orden. 2. Antes de iniciar las prácticas, el maestro inspeccionará las condiciones físicas del laboratorio y, de encontrar situaciones que representen riesgo grave, deberá reportar dicha situación al Jefe de Laboratorio y/o asistente o auxiliar del mismo, para que sea corregida, en caso de que no exista la posibilidad de atención inmediata, la práctica será suspendida. 3. Si durante la práctica surgiera una condición que ponga en riesgo grave la Seguridad y Salud de las personas, equipos, materiales o instalaciones se procederá a suspender la práctica debiendo informar de la situación al Jefe de Laboratorio y/o al asistente o auxiliar del mismo, elaborando el reporte por escrito correspondiente. 4. Los alumnos solo podrán trabajar en el horario asignado a su práctica, registrado en el Departamento de Servicios Escolares, con la presencia del maestro titular. En ausencia del maestro, la práctica no podrá ser realizada. En caso de requerirse sesión (es) extraordinaria (s), el maestro deberá solicitar por escrito la autorización de la (s) misma (s) al Jefe de Laboratorio y/o al asistente del mismo y éste otorgará el permiso acorde con la disponibilidad de las instalaciones. 5. Se deberá cumplir y respetar la calendarización de prácticas fijada y autorizada por la Jefatura de la Carrera. Así mismo, se deberá efectuar las prácticas establecidas por sesión, permitiéndose cuando sea necesario, a juicio del maestro, efectuar cambios en la programación de las mismas, notificando por escrito al jefe de laboratorio y/o asistente o auxiliar, y este otorgará la autorización acorde a la disponibilidad del laboratorio. 6. El maestro deberá cumplir con el uso del equipo de protección personal básico de laboratorio; también es responsabilidad del maestro verificar que antes de iniciar la práctica, todos los alumnos cuenten con su equipo de protección personal básico: lentes de seguridad, bata larga de algodón, pantalón de algodón (preferentemente) y zapato cerrado, debiendo encontrarse el equipo en buenas condiciones. El alumno que no cumpla con los requisitos anteriores; no podrá realizar la práctica. 7. El maestro deberá asegurarse que los alumnos utilicen adecuadamente el equipo de protección personal durante el desarrollo de la práctica. El maestro llevará un registro de los alumnos que sean observados sin usar su equipo de protección personal o usándolo de maneras inadecuada, cada registro contará como una falta al Reglamento General de Laboratorios. La acumulación de 4 faltas al 139 Reglamento General de Laboratorios, implica una suspensión para el alumno de la práctica en el semestre y la no-acreditación de la misma. 8. En lo referente al abastecimiento, consumo y desecho de reactivos o sustancias, se deberá cumplir con las siguientes disposiciones: Los reactivos son proporcionados por la institución, por lo que se pide a los administradores de los laboratorios, maestros y alumnos, hacer uso racional de los mismos, utilizando solo lo necesario, evitando el desperdicio. Cumplir con el procedimiento para el almacenaje y disposición de sustancias químicas, el cual se encuentra publicado en los laboratorios de la institución e incluido en todos los manuales de prácticas. 9. El maestro deberá permanecer en el laboratorio durante todo el desarrollo de la práctica. 10. Es necesario por procedimiento de riesgo de asistencia, que el maestro permanezca en un lugar visible. 11. Por razones de seguridad y orden esta prohibido en el laboratorio: • Correr. • Utilizar lenguaje obsceno o palabras altisonantes. • Hacer bromas. • Introducir radiograbadoras, audífonos o radios. • Ingerir alimentos o bebidas. • Fumar. • Ingreso de personas ajenas a la institución o al grupo que desarrolla la práctica. • Uso de zapatos de tacones de altura superior a 4cm. o zapato abierto. • Cabello largo (las personas con esta característica deberán recoger su cabello y sujetarlo adecuadamente, como medida de prevención para evitar contacto con el fuego o sustancias peligrosas). • Uso de shorts o bermudas. • Y en general todo acto y/o conducta que incite al desorden. Cualquier violación a lo establecido en este punto como una falta al Reglamento General de Laboratorios. 12. Toda persona tiene la obligación de reportar por escrito los actos y/o condiciones inadecuadas al responsable inmediato superior, utilizando para ello el formato de "Informe de Actos y Condiciones". 13. Todo alumno que sufra una lesión deberá reportarla al maestro encargado de la práctica y de no encontrarse éste, deberá dirigirse con el Jefe de Laboratorio y/o asistente del mismo. 140 14. Todo empleado que sufra una lesión deberá reportarla a su jefe inmediato. 15. Todo accidente ocurrido en los laboratorios deberá ser atendido para su control, por la primera persona capacitada y enterada de la situación. 16. Todo accidente ocurrido en los laboratorios deberá ser investigado acorde con lo establecido en el Procedimiento de Investigación de Accidentes de la Facultad. 17. Al término de la práctica el maestro será responsable de supervisar que los alumnos ordenen y limpien su lugar de trabajo, asegurando que el laboratorio sea entregado a la administración del laboratorio, en las mismas condiciones que lo recibieron, para asegurarse de que se cumplan con éste lineamiento, maestro y auxiliar (o jefe de laboratorio) realizarán un recorrido de inspección, registrando la misma en el formato correspondiente. 18. La persona que se presente bajo el influjo de alcohol o drogas, que incurra en actos de violencia, daño a la propiedad intencional, negligencia o tome objetos o valores sin autorización, será reportado de manera inmediata ante la H. Comisión de Honor y Justicia de la Junta Directiva de la Facultad de Ciencias Químicas, quien tomará las acciones correspondientes al caso. 19. El Reglamento General de Laboratorio en su totalidad, es aplicable a maestros y en general a todo personal integrante de la institución. 20. Todo lo no contemplado en el Reglamento, será válido por el comité de Seguridad y Salud de la Institución. 141 APÉNDICE B DISPOSICIÓN DE RESIDUOS ETIQUETAS DE LOS CONTENEDORES • • • • • • • • • • CONTENEDOR A Solución salina PH 6 – 8 • Sales inorgánicas • Ácidos inorgánicos Ácidos orgánicos Bases inorgánicas CONTENEDOR B Sólidos inorgánicos Sales inorgánicas CONTENEDOR D Tóxicos e inflamables • Solventes orgánicos • Bases orgánicas y amidas Solventes orgánicos CONTENEDOR G Combinaciones • orgánicas sólidas CONTENEDOR E Muy tóxico Cancerígeno orgánico • • • • • CONTENEDOR C Tóxicos e inflamables Solventes orgánicos Bases orgánicas y aminas Solventes orgánicos halogenados CONTENEDOR F Sales de metales preciosos CONTENEDOR H Oxidantes PRÁCTICA No. 1 EXTRACCIÓN Reactivo y/o Producto NaOH 20% ,Neutralizar con H2SO4 20% HCl concentrado, neutralizar con NaOH 20% Na2SO4 CaCl2 Cloruro de metileno destilado Zacate de limón extraído Contenedor A A B B Entregar a auxiliar Bote de basura 142 PRÁCTICA No. 2 CRISTALIZACIÓN Reactivo y/o Producto Metanol Etanol Ac. Acético, Neutralizar con NaOH 20% Acetona Éter etílico Éter de petróleo Cloroformo Agua- etanol Contenedor C C A C C C D C PRÁCTICA No. 3 DESTILACIÓN SIMPLE Y FRACCIONADA A PRESIÓN NORMAL Y PRESIÓN REDUCIDA Reactivo y/o Producto Muestras destiladas Acetona MgSO4 Na2SO4 Contenedor Entregar al auxiliar C B B PRÁCTICA No. 4 DESTILACIÓN POR ARRASTRE CON VAPOR Reactivo y/o Producto Diclorometano Na2SO4 Clavos de olor Fase acuosa Contenedor D B Bote de basura drenaje 143 PRÁCTICA No. 5 ANÁLISIS ELEMENTAL CUALITATIVO Reactivo y/o Producto Sodio metálico Acetato de plomo 1% Sulfuro de plomo Sulfato ferroso amónico Soluciones ácidas, neutralizar Soluciones alcalinas, neutralizar Haluros de plata Cloroformo Agua de cloro (Chlorox) Persulfato de potasio Azul de Prusia Contenedor Entregar al auxiliar E orgánico E inorgánico B A A F D C B E orgánico PRÁCTICA No. 6 ANÁLISIS FUNCIONAL ORGÁNICO Reactivo y/o Producto Ác. Sulfúrico, neutralizar Ác. Sulfúrico fumante, neutralizar Sulfato de sodio Tricloruro de aluminio y cloroformo (líquido) Tricloruro de aluminio y cloroformo (sólido) Bromo en cloroformo , prueba positiva Bromo en cloroformo , prueba negativa Permanganato, prueba positiva Permanganato, prueba negativa Soln. de acetiluros con Ác. Nítrico, neutralizar Ác. Clorhídrico/Cloruro de Zinc, neutralizar Nitrato cérico 2,4-dinitrofenilhidrazina, prueba positiva, sólido 2,4-dinitrofenilhidrazina, prueba negativa Contenedor C C B D E orgánico D H B H AoF A E orgánico o H G E orgánico o H PRÁCTICA No. 7 144 CROMATOGRAFÍA Reactivo y/o Producto Éter etílico Acetona Hexano Metanol Etanol Butanol-acetato de etilo Contenedor C C C C C C PRÁCTICA No. 8 PUNTOS DE FUSIÓN Y EBULLICIÓN Reactivo y/o Producto Muestras – problema Contenedor Entregar al auxiliar PRÁCTICA No. 9 ÍNDICE DE REFRACCIÓN Reactivo y/o Producto Muestras - problema Contenedor Entregar al auxiliar 145
 0
0
Anuncio
Documentos relacionados
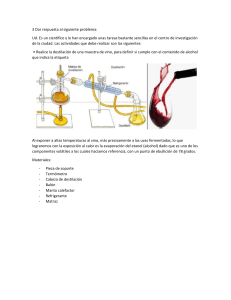








Descargar
Anuncio
Añadir este documento a la recogida (s)
Puede agregar este documento a su colección de estudio (s)
Iniciar sesión Disponible sólo para usuarios autorizadosAñadir a este documento guardado
Puede agregar este documento a su lista guardada
Iniciar sesión Disponible sólo para usuarios autorizados