Document
Anuncio

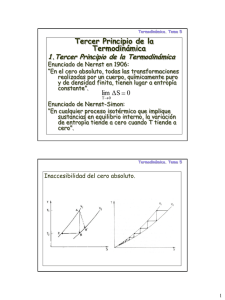
Termodinámica, cinética y transporte químico en sistemas geológicos Introducción Termodinámica: Rama de las ciencias físicas que estudia los diferentes niveles energéticos y las transferencias de energía entre sistemas así como entre diferentes estados de la materia (líq., sol., gas.). Es una poderosa herramienta aplicable a diversos problemas del mundo físico, dentro de campos tan diversos como la Física, Química, Biología y Ciencias de la Tierra (Geología, Oceanografía, Meteorología). Aplicación de la Termodinámica a las diferentes reacciones químicas que tienen lugar en los diversos ambientes geológicos Termodinámica Química Físico Química Escala Campos de Estudio Organización del Sistema Termodinámica Clásica Termodinámica Química Estado de Equilibrio Estado Estacionario Termodinámica Irreversible Macroscópica Estado Transiente Dinámica de Fluidos Microscópica Atómica Cinética Química Mecánica Cuántica Fenómenos de transporte Mecánica Estadística La Termodinámica Clásica está basada en el estado de equilibrio y procesos reversibles enfoque macroscópico (sólo mediciones de T, p, etc.) no se requiere un conocimiento de la estructura molecular o atómica!!!! transformaciones físicas y reacciones químicas redistribuciones atómicas y moleculares que conllevan cambios energéticos medibles a escala macroscópica Algunos ejemplos de transformaciones físicas y reacciones químicas: • Transformación [H2Osólida H2Olíquida]. En esta reacción, el agua presenta por lo menos dos formas distintas o fases que cambian espontáneamente dependiendo de las condiciones ambientales. • Transformación [Cdiamante Cgrafito]. Este es un tipo de cambio espontáneo e irreversible en condiciones ambientales. • Reacción [4Fe3O4 + O2 = 6Fe2O3]. En condiciones oxidantes, la magnetita se transforma espontáneamente en hematita. • Reacción [CaCO3 + H2CO3 = Ca2+ + 2HCO3]. La disolución de calcita se produce espontáneamente en zonas kársticas. • Reacción [CaAl2Si2O8 + Na+ + Si4+ = NaAlSi3O8 + Ca2+ +Al3+]. La sustitución iónica de calcio por sodio ocurre espontáneamente en la serie de las plagioclasas al disminuir la temperatura en un magma en cristalización. Estos sencillos ejemplos nos permitirán tener una noción general de los cambios físicos y químicos, para poder entender ejemplos posteriores en que los cambios energéticos no son tan obvios Mediante el estudio de los cambios energéticos asociados a las reacciones químicas, podremos responder las siguientes preguntas de VITAL INTERÉS en Geología: • Qué factores controlan los cambios (reacciones) que observamos en la naturaleza? • Porqué algunas reacciones van en dos direcciones mientras que otras tan solo lo hacen en una una? • Qué minerales pueden coexistir en equilibrio bajo determinadas condiciones de Temperatura, Presión y Composición? [p,T,X] [mxs] • Qué condiciones de equilibrio pueden estabilizar una determinada asociación mineral? [mxs] [p,T,X] Termodinámica y Procesos Geológicos • Procesos Ígneos - cristalización de magmas (equilibrio de fases) - geotermobarometría • Procesos Hidrotermales - solubilidad de minerales, estudio de inclusiones fluidas • Procesos Sedimentarios y Supérgenos - meteorización química - diagénesis • Procesos Metamórficos - transformaciones en estado sólido por variaciones en p,T (reacciones producto del metamorfismo dinámico o de contacto) Cinética química: es la rama de la físicoquímica que estudia los procesos físicos y químicos tomando en cuenta la perspectiva temporal. A diferencia de la termodinámica, que estudia estado iniciales y finales y se formula a partir del concepto de equilibrio, la cinética química incorpora la variable tiempo. Las escalas de tiempo involucradas en el estudio de la cinética de procesos geológicos están comprendidas entre 10-12 segundos (movimiento molecular) y 1015 segundos (100 millones de años, escalas de tiempo relevantes para la tectónica de placas). Existen, entre ambos extremos, 27 órdenes de magnitud, dentro del cual podemos distinguir procesos geoquímicas tales como: • • • • • • • • • • • • • Formación de complejos moleculares Fenómenos de adsorción de complejos en superficies minerales Reacción en las superficies Flujo de masa Fraccionamiento isotópico Nucleación y crecimiento cristalino Oxidación, reducción Diagénesis Generación de magmas Ciclos geoquímicas Subducción Convención del manto Erosión, etc. La Cinética Química se ocupa, a diferencia de la termodinámica, de los estados intermedios (desequilibrio) y es inherentemente dependiente del camino que siguen los procesos enfoque microscópico y fenomenológico se requiere un conocimiento de la estructura molecular o atómica permite monitorear la evolución de los sistemas y llegar estimar propiedades macroscópicas observables (composición, T, P, V y tiempo) Debido a la naturaleza temporal de los procesos geológicos y la dificultad de determinar el equilibrio en sistemas naturales, la cinética es más “realista” que la termodinámica Fenómenos de transporte químico (teoría de transporte): corresponde al transporte físico de componentes químicos. Incorpora, además del factor tiempo, el factor “cuán lejos” los componentes químicos se desplazan en función de tiempo, algo que la cinética química por sí sola no logra capturar. La teoría de transporte químico se basa en el concepto de conservación de la energía, la masa y el momento. Nos permite estudiar procesos tales como la difusión de elementos químicos en minerales, magmas, soluciones, gases, etc., y evaluar pérdidas/ganancias de componentes químicos que puedan afectar el equilibrio del sistema. Entre otras aplicaciones: • • • Transporte y difusión de elementos químicos en materiales geológicos Evaluar el cierre de sistema isotópicos y geocronológicos Dispersión de contaminantes en el medio ambiente La teoría de transporte químico puede ser abordado desde dos perspectivas: - Enfoque macroscópico: relaciona los flujos de materia con fuerzas termodinámicas (gradientes de T, P, masa, etc). Se utilizan métodos de la física clásica. - Enfoque microscópico: describe los procesos de transporte desde una escala atómica. Requiere del uso de métodos de la física estadística y la mecánica cuántica. Termodinámica = cuán estable? Cinética= cuánto tiempo? Fenómenos de transporte = cuán lejos en cuánto tiempo? Termodinámica: Conceptos Fundamentales Sistema termodinámico es un porción de materia separada del resto del universo observable por límites bien definidos. Nota: los sistemas se definen según la conveniencia del observador, y los límites del sistema se eligen para permitir al investigador aplicar la Termodinámica de acuerdo a sus necesidades. Universo (Ω) Sistema Límites del sistema Sistema Abierto permite el intercambio tanto de materia como de energía a través de sus fronteras. Materia Sistema Abierto Fuente térmica Sistema Cerrado permite el intercambio de energía pero no de masa a través de los límites del sistema. Sistema Cerrado Fuente térmica Sistema Aislado no permite el intercambio ni de materia ni energía a través de sus límites. Lo que está contenido dentro del sistema mantiene constante su energía y composición. Sistema Aislado Fuente térmica Estados de equilibrio, estacionario y transiente Consideremos un conducto volcánico poco profundo que está conectado a dos fuentes térmicas T1 (superficie) y T2 (cámara magmática): T1 0 Txn = T1 xn v Q = "k!T T1 < T2 xi xo Txo = T2 T2 T = T (t , x) & 'T # dT = T&dt + $ ! dx % 'x "t Estado Estacionario Suponiendo que el conducto es homogéneo y los reservorios térmicos suficientemente grandes, la temperatura decrece linealmente a través de éste. La temperatura cambia con la posición pero no con el tiempo. & 'T # T& = $ ! =0 % 't " x & 'T # $ ! >0 % 'x "t Estado Transiente Supongamos ahora que la fuente térmica (el magma) finalmente cristaliza y se enfría. Pasamos de un estado estacionario a uno transiente. ya que la temperatura en el conducto decaerá en función de distancia y el tiempo. & 'T # T& = $ ! >0 % 'T " x & 'T # $ ! >0 % 'x "t Estado de Equilibrio Finalmente, se logra el estado de equilibrio térmico en el conducto cuando la temperatura se hace constante. & 'T # & 'T # T& = $ ! =0=$ ! % 't " x % 'x "T Resumiendo, las ecuaciones que explican el flujo de calor por conducción en el conducto (1 dimensión) son las siguientes: • Estado transiente • Estado estacionario • Estado de equilibrio "c #T #2T = k 2 = k$ 2T #t #x ! "c #T =0 #t ! "c #T #T = k$ 2T = =0 #t #x k! 2T = 0 !T = cte !x ! Extrapolando ahora el concepto de equilibrio térmico en la corteza al problema de “equilibrio termodinámico”: • En un sistema termodinámico en equilibrio, ninguna de sus propiedades varía con respecto al tiempo o distancia. • La presencia de gradientes físicos o químicos altera el estado de equilibrio. • Después de la perturbación el sistema vuelve a su estado de equilibrio o de mínima energía (o potencial mínimo). Consideremos un ejemplo mecánico del estado de equilibrio: A A Energía potencial B B (estado de mínima energía) ΣFi = 0 ΣΓ i = 0 Haciendo la analogía a una reacción química donde A = reactivos y B = productos: A mghA reactivos ΔE B Potencial gravitatorio Potencial químico mghB productos análogo Los sistemas químicos tienden a minimizar su contenido energético Estable Cada sustancia o grupo de sustancias se encuentran en su estado más estable. Equilibrio Metaestable Una sustancia o grupo de sustancias no han logrado el estado de equilibrio, pero no muestran variación en el tiempo. Ej. Sobreenfriamiento del H2O: el agua líquida puede enfriarse bajo los 0 [ºC] sin que se forme hielo. Sin embargo, cualquier perturbación del estado de equilibrio “metaestable” del agua (introducir un sólido o mover el agua) conllevará al congelamiento inmediato del sistema. B A C D A = equilibrio metaestable B = inestable C = inestable D = equilibrio estable Procesos reversibles e irreversibles Procesos Irreversibles Son todos aquellos procesos naturales que ocurren en una sola dirección, y que se llevan a cabo en etapas finitas (Δ). Si el proceso es una reacción química, es importante mencionar que el término “irreversible” se aplica para un determinado “set” de condiciones (ej. T,p). Ej. transformación de diamante en grafito es irreversible a condiciones ambientales. Sin embargo, el proceso es imperceptible a escala de tiempo humana debido a la lentísima cinética de la reacción. Diamante a (p,T) atmosférica: Diamante Grafito (irreversible) Grafito Procesos Reversibles Son todos aquellos procesos “ideales” que ocurren en etapas infinitesimales de tal forma que el sistema se mantiene en equilibrio en cada una las etapas parciales. La importancia de este concepto radica en que los cambios en las propiedades termodinámicas en procesos reversibles pueden ser fácilmente calculados. Además, si se puede probar que los cambios en las propiedades son independientes de la trayectoria (es decir, dependen sólo de los estados inicial y final), entonces el cambio calculado en condiciones reversibles es idéntico para el mismo proceso realizado en forma irreversible. La reversibilidad y el equilibrio están íntimamente relacionados: A dζ = n& A = n& B = B dn A dnB = =0 dt dt dζ = " n& A = n& B = " dn A dnB = !0 dt dt (equilibrio estático) (equilibrio dinámico) Variables y funciones de estado Las variables de estado corresponden a propiedades medibles de la materia que describen el estado macroscópico de un sistema. Las variables de estado determinan el estado presente del sistema, independientemente de la trayectoria o camino para llegar a él (p,T,V,ρ,η, etc). Las variables de estado dependen únicamente del estado de equilibrio y no de la manera en que el sistema cambia de un estado a otro!! variables intensivas variables de estado variables extensivas Variables Extensivas Dependen de la masa total del sistema. El valor de cualquier propiedad extensiva se obtiene sumando los valores de la misma en todas las partes del sistema. Ej. volumen (V), número de moles (n). Variables Intensivas Son independientes de la masa total del sistema. Se miden en cualquier parte del sistema, y tienen un valor uniforme en cualquier punto del sistema en equilibrio. Ej. temperatura (T), presión (p). S i VT = ΣVi νE nT = Σni pT = pi = …= pn Subdivisión de un sistema S en “i” partes νI TT = Ti = ...= Tn Toda variable extensiva (νE) se transforma en intensiva (νI) al dividir por otra variable extensiva (νE’): vE vI = vE ' Las variables de estado definen funciones de estado, que controlan las variaciones entre estados durante un determinado proceso. Una función de estado z definida por z = f (x,y) y su por su diferencial dz debe cumplir los siguientes requisitos matemáticos: • El diferencial total dz debe ser exacto, de acuerdo al test de Euler. & 'z # & 'z # dz = $ ! dx + $$ !! dy = M ( x, y )dx + N ( x, y )dy % 'x " y % 'y " x Si M y N son funciones conocidas, dz es un diferencial exacto si: & 'M $$ % 'y # & 'N # !! = $ ! " x % 'x " y • Para cualquier proceso z1(x,y) z2(x,y), el cambio Δz es independiente de la trayectoria. De esta forma, puede ser calculado sólo en base a los estados iniciales y finales del sistema: 2 "z = z2 ( x, y) # z1 ( x, y) = $ dz( x, y) 1 • El cambio neto en un ciclo es siempre cero: ! ! dz( x, y) = 0 Ejemplos • las funciones más utilizadas en Termodinámica (energía interna U, entalpía H, entropía S, energía libre de Gibbs G) son todas funciones de estado, independientes de la trayectoria. Haciendo un análogo mecánico, son equivalentes a fuerzas conservativas tales como la gravedad. • el calor Q y el trabajo W no son funciones de estado, sino funciones de trayectoria. Sus diferenciales son inexactos, e integrados en un ciclo no se anulan. Análogamente, son equivalentes a fuerzas disipativas tales como el roce. Fases y Componentes El estado de los sistemas termodinámicos está definido por la magnitud de sus variables extensivas e intensivas. Sin embargo, es necesario definir ciertos términos que se refieren a las características de la materia que componen el sistema: Fase (P) Parte de un sistema que es físicamente homogénea, con límites claros y definidos y, por lo tanto, mecánicamente separable de las otras fases adyacentes. En la naturaleza existen tres tipos de fases: sólidas, líquidas y gaseosas. De esta forma, los sistemas se dividen en homogéneos (1 fase) y heterogéneos (2 o más fases). La forma, orientación y posición de la fase con respecto a las otras es irrelevante, por lo que una fase puede aparecer en varias posiciones dentro de un sistema. Ej. Los minerales que conforman un granito (Qz, Pl, K-Feld, Msc) constituyen fases individuales de ese sistema, ya que son idealmente homogéneos y mecánicamente separables entre sí. Sin embargo, el conjunto de gases individuales (CO2, SO2, Cl2, etc) que conforman una fumarola volcánica constituyen una sola fase gaseosa. Componente (c) Es un término utilizado para describir la composición química de un sistema, y corresponde al mínimo número de entidades químicas independientes necesarias para especificar la composición de todas las fases presentes en el sistema. Ej. En un sistema simple definido por la disolución de sal en agua líquida en equilibrio con vapor de agua (interfase océano-atmósfera), tendremos dos fases (líq+vap) y dos componentes (NaCl+H2O). Por otra parte, el componente único SiO2 describe la composición de todas las fases naturales de la sílice (cuarzo, cristobalita, tridimita, etc.). Grados de libertad (f) Corresponde al número de variables intensivas (p,T principalmente) que podemos variar de tal manera de mantener el número de fases constante en un sistema en equilibrio. Por ejemplo, si tenemos tres fases que coexisten en equilibrio a (p,T) fija, basta con que variemos levemente las condiciones de (p,T) para cambiar el equilibrio de las fases. Las fases, componentes y grados de libertad se relacionan mediante la “regla de la fases de Gibbs”: f +P =c+2 Primera Ley de la Termodinámica 1. Ley Cero y Escalas de T y p Si una sustancia 1 cuya función f (p1,V1) define una temperatura T1, y que está en equilibrio térmico con una sustancia 2, tendremos: f (p1,V1) = f (p2,V2) Y si la sustancia 1 también está en equilibrio térmico con una tercera sustancia: f (p1,V1) = f (p3,V3) Entonces, se concluye que: f (p2,V2) = f (p3,V3) Ley Cero “Si dos cuerpos están en equilibrio térmico con un tercero, entonces los tres cuerpos estarán en equilibrio térmico entre sí”. Una consecuencia de la “ley cero” permite deducir que si las tres sustancias están en equilibrio térmico, se puede decir que poseen una propiedad en común. A esta propiedad o función se la denomina “temperatura”, que es una propiedad intensiva, puesto que no depende de la cantidad de sustancia considerada. Por ende, la temperatura es la propiedad que se utiliza para determinar si un sistema está en equilibrio térmico con otro(s). De esta forma, es posible determinar una escala de temperatura, y uno de los grandes triunfos de la termodinámica fue la demostración que existe un cero absoluto de temperatura (ver Tercera Ley). No obstante hay que recordar que existen diferentes tipos de escalas de temperatura: Escala Kelvin de Temperatura La escala Kelvin tiene el cero absoluto 0[K] y un valor de 273.16 [K] para el punto triple del sistema H2O, en el cual coexisten en equilibrio 3 fases: hielo, agua y vapor de agua. El punto de fusión del hielo (o congelación del agua) es 273.15 [K], mientras que la temperatura de ebullición del agua (o condensación del vapor) es de 373.15 [K]. Escala Celsius de Temperatura Esta escala da un valor de 0 [ºC] para el punto de fusión de hielo (273.15[K]) y un valor de –273.15 [ºC] para el cero absoluto (0 [K]). En la escala Celsius existen 100 grados de diferencia entre los puntos de congelación y ebullición del agua a 1 [atm] de presión. Se puede establecer entonces la siguiente ecuación de conversión entre escalas: T [K] = t [ºC] + 273.15 (1) • Es de vital importancia recordar que todas las relaciones termodinámicas utilizan la temperatura en escala Kelvin, de modo que la condición “estándar” de temperatura es 278.15 [K] (25 [ºC]). p [atm] 100 [ºC] 1 Diagrama de Fases del H2O hielo agua vapor T [K] 273.15 273.16 373.15 Escalas de Presión La presión se define como fuerza por unidad de superficie (p = F/s). La fuerza (F) se expresa en Newton (1 Newton [N] = 1 [kg.m.s-2]) , y la superficie en metros cuadrados. La unidad SI de presión es el Pascal, definido por: 1 Pascal [Pa] = 1 [N.m-2] Otras unidades: 1 bar [bar] = 105 [Pa] 1 atmósfera [atm] = 1.01325 [bar] = 760 [mmHg] = 760 [Torr] Las unidades de presión más usadas en Ciencias de la Tierra: - el giga Pascal: 1[GPa] = 109 [Pa] - el kilo bar: 1 [kbar] = 1000 [bar] = 0.1 [GPa] 2. Energía, Trabajo y Calor El concepto de energía implica la capacidad de un cuerpo para producir trabajo. Por ejemplo, si levantamos un objeto desde el suelo y lo ponemos sobre una mesa, hemos realizado un trabajo físico y hemos consumido energía al realizarlo. La energía que hemos gastado no se ha perdido, sino que ha sido transferida al objeto, lo que se refleja en una mayor energía potencial de éste respecto de su posición anterior. De esta forma, la energía y el trabajo están íntimamente relacionadas, ya que podemos usar nuestra energía para producir trabajo, y aplicar trabajo para incrementar la energía. No obstante, la generación de trabajo no es la única manera de cambiar la energía de un sistema y, por otro lado, un cambio de energía de un sistema no siempre se traduce en la producción de trabajo. Podemos además cambiar la energía del objeto en cuestión calentándolo o enfriándolo. De este modo, debemos considerar tanto el trabajo (en todas sus formas) como el calor, como dos formas consistentes de producir cambios energéticos. Existen diferentes maneras de producir trabajo en un sistema, pero desde el punto de vista termodinámico vamos a considerar el trabajo sólo como producto de los cambios de presión (p) y volumen (V). Los conceptos de energía, trabajo y calor son de importancia fundamental en Termodinámica y sus definiciones deben entenderse a cabalidad: Energía Interna (U) De un punto de vista microscópico, la energía interna de un sistema corresponde a la energía total del sistema, contenida en los átomos de éste. Puede definirse como un “contenido energético promedio por átomo”, de acuerdo a la siguiente ecuación: U = 0,5 f k T (2) U es la energía interna o contenido energético promedio por átomo, f son los grados de libertad disponibles para “invertir” energía en un átomo (f = 6 para los cristales), k es la Constante de Boltzmann y T la temperatura absoluta. El estado energético macroscópico del sistema estará dado entonces por el número de átomos (moles), la presión y la temperatura. Esta magnitud absoluta es difícil de cuantificar, por lo que se miden los cambios en la energía interna. La energía interna es función de estado, y se mide en Joules [J]. Trabajo (W) En Termodinámica, trabajo se define como “cualquier cantidad que fluye a través de la frontera de un sistema durante un cambio de estado y que puede usarse por completo para elevar un cuerpo del entorno”. Además: • el trabajo sólo aparece en las fronteras o límites del sistema • el trabajo sólo aparece durante un cambio de estado • el trabajo se manifiesta por su efecto sobre el entorno • la cantidad de trabajo es igual a mgh, con m: masa; g: aceleración de gravedad; h: altura de elevación del objeto (unidad: Joule [J]; 1 [J] = 1 [Nm]). Matemáticamente, el diferencial del trabajo es inexacto (notación dW ), dependiente de la trayectoria y su integral a través de un ciclo no es cero. Por ende, el trabajo no es una función de estado sino de trayectoria: ! f W= " dW i (3) Wciclo = # dW " 0 ! ! Convención de signos para el trabajo (W) y el calor (Q): “Científica” Q+ “Ingenieril” Q- Sistema Q+ W+ W- Q- Sistema WW+ Nosotros usaremos la convención “científica” para el trabajo y el calor, en la que todos los aportes de calor y trabajo desde el ambiente al sistema son positivos, y las pérdidas negativas. La convención “ingenieril” utiliza signo positivo para el trabajo realizado por el sistema, ya que una máquina térmica es positiva si realiza trabajo. Antes de continuar con la definición de calor, vamos a centrarnos en el trabajo del tipo PV, es decir, presión-volumen. Usaremos un esquema de pistón-cilindro con gas, que permite visualizar cambios de volumen en forma sencilla: Pext2 2 pext1 2 1 Pint2 V2 1 pint1,V1 Δl T = cte 1 Las dos fuerzas que tienden a mover el pistón están equilibradas en la posición 1 (Pext1 = Pint1). Si soltamos los pestillos del pistón, el pistón se moverá hasta que la presión interna se equilibre con la presión externa, al fijar los pestillos en la posición 2. De esta forma, durante la transición 1 2, Pint > Pext, hasta llegar a la posición 2, donde Pint2 = Pext2. Si el pistón está bien lubricado, podemos ignorar los efectos de la fricción, por lo que la historia presión-volumen del cambio 1 2 se puede visualizar en la siguiente gráfica: P posición 1 (Pint1 = 20) 20 Isoterma posición 1 (Pint1 = 20) 10 pex t V1 V2 V La Pext durante la expansión es constante, ya que está fijada por la masa del pistón. El trabajo producido durante la expansión corresponderá a la fuerza que ejerce el pistón (F) por la distancia: Wexp = F Δl Wexp = - (Pext A) Δl = - Pext ΔV Wexp = - Pext ΔV (4) A es el área del pistón y Δl la distancia que recorre. El trabajo de expansión (4) tiene signo negativo ya que el sistema produce un trabajo sobre el ambiente. Sin embargo, si añadimos otro peso más sobre el pistón para la siguiente expansión, pudiera ocurrir que tenemos un peso total demasiado grande como para permitir que el pistón alcance la posición 2, quedando en una posición intermedia entre 1 y 2. Si sacamos luego el peso, tendríamos un modelo de expansión como el siguiente: Pp pos 1 20 Pext’ 10 Pext V En este caso, el trabajo total será mayor que en la primera expansión, sumando ambas áreas. Si continuamos realizando el mismo proceso de expansión cada vez en mayor cantidad de etapas (recordar que cada etapa es un estado de equilibrio!), nos estaremos aproximando a un límite de trabajo máximo obtenible con la expansión del gas. De esta forma, el trabajo máximo (Wmáx) de expansión se logrará cuando tomemos para cada expansión parcial un ΔV infinitesimalmente pequeño: V2 n Wmáx = " lim % P#V = " & PdV #V $0 (5) V1 i=1 En este caso no habrá distinción entre Pext y Pint puesto que presentarán diferencial infinitesimales, ya que nuestro modelo de expansión máxima está compuesta por una sucesión continua de estado de equilibrio. Debido a estas ! condiciones, podemos decir que el trabajo máximo de expansión es trabajo reversible. Hay que destacar el signo negativo para el Wmáx, ya que es trabajo realizado por el sistema sobre el ambiente: V2 P Wmáx = " # PdV = "P(V2 " V1 ) 201 V1 10 2 ! V1 V2 V Considerando que el trabajo máximo se logra en condiciones reversibles (ideales), el trabajo realizado durante expansiones irreversibles (o reales) es menor al trabajo máximo reversible. Podemos entonces expresar en forma general el trabajo de expansión como: V2 W " # $ PdV (W ≤ Wmáx) (6) V1 El mismo razonamiento realizado anteriormente puede realizarse para obtener la expresión del trabajo de compresión (signo +). Llevando al límite infinitesimal cada ! compresión (ΔV0), la suma de éstas corresponderá al trabajo mínimo ejercido por el ambiente sobre el sistema. Por ende, el trabajo asociado a compresiones reales irreversibles será mayor al trabajo mínimo reversible: V2 W" # PdV (W ≥ Wmáx) (7) V1 Calor (Q) ! En Termodinámica se define el calor como una cantidad que fluye a través de la frontera de un sistema durante un cambio de estado, en virtud de una diferencia de temperatura entre el sistema y sus entorno, fluyendo de un punto de mayor temperatura a otro de temperatura menor. Además: • • • • el calor sólo aparece en la frontera del sistema el calor sólo aparece durante un cambio de estado el calor se manifiesta por un efecto sobre el entorno el calor es igual al número de gramos de agua del entorno que aumentan su temperatura en un grado, partiendo de una temperatura específica bajo una presión específica (unidades: Joules [J] o Calorías [cal]; 1 [cal] = 4.184 [J]) Al igual que el trabajo, el calor no es función de estado: f Q= " dQ i ! ! Qciclo = # dW " 0 (8) Haciendo una equivalencia con la expresión de trabajo (6), el calor asociado a procesos reversibles e irreversibles se puede definir como: Z2 ! Q # " TdZ (9) Z1 El calor asociado procesos reversibles será máximo, mientras que el calor asociado a procesos naturales o irreversibles será menor. El calor se relaciona entonces con la temperatura T y una propiedad Z, que más adelante definiremos como “entropía”. El Calor y el Trabajo no son entidades separadas, sino que son formas de energía que pueden ser transferidas en diferentes vías. Veamos esto mediante una analogía práctica: consideremos el agua contenida en una laguna muy profunda, de manera que la cantidad de agua sea muy grande, pero finita y medible, que corresponderá a la energía interna U del sistema: lluvia dWr+ dQin+ dWe evaporación dU dQout - U El agua puede ser extraída o añadida al pozo por escorrentía (“calor”) o por precipitación/evaporación (“trabajo”). El balance diferencial de masa (o de “agua”) para el sistema será el siguiente: dU = (dQin - dQout) + (dWr – dWe) (10) ΔU = (Qin – Qout) + (Wr – We) (11) Integrando, donde ΔU representa el cambio en el nivel del pozo (“cambio energético”). Una vez que el agua ha entrado al pozo, pierde su identidad como agua de escorrentía (“calor”) o agua de lluvia (“trabajo”), ya que el pozo simplemente contendrá agua (“energía”). Mediante este simple modelo, podemos enunciar ahora la Primera Ley. 3. Primera Ley de la Termodinámica Si U es el contenido energético del sistema, y éste puede ganar o perder energía únicamente por flujo de calor Q o trabajo W, entonces la Primera Ley de la Termodinámica se expresa, en sus formas diferencial e integrada, como: dU = dQ + dWr (12) ΔU = Q + W (13) La Primera Ley de la Termodinámica es equivalente a la Ley de Conservación de la Energía: la energía total de todos los procesos siempre se conserva. Como U es función de estado (depende sólo de sus estados inicial y final), la energía interna es cero para un proceso cíclico: ! dU = 0 (14) Los reservorios geotérmicos proveen un ejemplo natural de transformación que involucra flujo de calor y trabajo. Consideremos un reservorio geotérmico en profundidad de T~200 [ºC] y p~100 [bar]. Si el fluido migra a través de fracturas hasta la superficie, la presión y temperatura decaerán por expansión. El vapor puede ser utilizado en superficie para producir trabajo en una turbina, y así reconvertir la energía mecánica en energía eléctrica, por ejemplo. Durante la producción de energía en la turbina, el calor se pierde irreversiblemente a través de la líneas y tuberías de la planta, por lo que sólo parte de la energía disponible en el reservorio (energía interna) podrá ser utilizada y transformada. El trabajo realizado por el vapor (sistema) sobre la turbina (ambiente) será menor al trabajo máximo reversible. Si el sistema fuera perfectamente aislado, y además pudiera realizar trabajo, el flujo de calor asociado al proceso sería cero (dQ = 0), y toda la energía se podría transformar en trabajo, sin pérdida de calor. Este tipo de sistemas se denominan adiabáticos, y el trabajo realizado bajo estas condiciones se transforma en una función de estado (dU = dW). Superficie libre para realizar trabajo Sistema Aislado Si consideramos la energía interna (U) como una función de la temperatura (T) y el volumen (V), su diferencial total será: & 'U # & 'U # dU = $ ! dT + $ ! dV % 'T "V % 'V " T (15) Combinando con la Primera Ley, se obtiene: dU = dQ + dW = dQ ± ( & 'U # & 'U # pdV = $ ! dT + $ ! dV % 'T "V % 'V " T (16) Si consideramos un cambio de estado a volumen constante, dV = 0: & 'U # dU = dQV = $ ! dT % 'T "V (17) La ecuación (16) relaciona el calor transferido desde el entorno (dQV) con el aumento de temperatura (dT) del sistema a volumen constante. Tanto dQV como dT son fácilmente medibles, y el cuociente dQV/dT define la capacidad calorífica a volumen constante (Cv): CV ( dQV & 'U # =$ ! dT % 'T "V (18) Como la energía interna de un sistema es una propiedad extensiva de estado, la capacidad calorífica CV también lo es. La capacidad calorífica por mol (C) es una propiedad intensiva tabulada en tablas de datos termodinámicos. Puede definirse en términos sencillos como “cantidad de energía necesaria para elevar 1 grado de temperatura 1 gramo de sustancia”. Ahora bien, la expresión anterior (17) nos permite expresar el cambio finito en la energía interna (ΔU) integrando la capacidad calorífica con respecto a la temperatura: T2 "U = ! C dT V (19) T1 Las dos últimas ecuaciones expresan la variación en la energía durante transformaciones a V = cte, y pueden ser aplicadas a cualquier sistema: gases, líquidos, sólidos, mezclas, etc. Obsérvese en la ec.16 que la energía interna y el calor tienen el mismo signo; por ende, si el calor fluye hacia el entorno (-), ΔU también será negativo y la energía del sistema disminuirá. Por el contrario, si el calor fluye desde el entorno al sistema (+), la energía aumentará. Además, como CV es siempre positivo, cualquier aumento o descenso el la temperatura se reflejará en un aumento o descenso de la energía, a V = cte. Aunque la ec.18 es general para cualquier proceso a V = cte, en la práctica esto presenta ciertos problemas al trabajar sólo con líquidos y sólidos. Debido a la baja compresibilidad de sólidos y líquidos, cualquier aumento de temperatura a volumen constante producirá un fuerte incremento en la presión. Desde un punto de vista experimental, los procesos a V = cte. son útiles para aquellos sistemas que son, al menos en parte, gaseosos (ej. fumarolas volcánicas). Ejemplo: calcular el cambio de energía interna (ΔU) y el calor absorbido (QV) para la transformación a volumen constante de 1 mol de helio, desde 25 a 35 [ºC]. Considere CV = 1.5 R (con R = constante de los gases = 8.314 [J/Kmol]). T2 "U = 45 # C dT = 1.5R# dt = (1.5)(8.314)(20) ! 250 [J/Kmol] V T1 25 ΔU = Q + p ΔV , ΔU = QV = 250 [J/Kmol] Entalpía (H) Consideremos ahora un proceso llevado a cabo a presión constante. Si solamente existe trabajo del tipo PV, entonces el cambio en la energía interna es: reordenando, ΔU = U2 – U1 = Q – P(V2 – V1) (20) (U2 + PV2) - (U1 + PV1) = Qp (21) Qp es función de estado, ya que está determinado por las variables de estado U, P y V. Definimos así una nueva función de estado de gran importancia en Termodinámica, llamada entalpía (H): H ≡ U + PV (22) La entalpía del sistema es una propiedad extensiva de estado. Combinando las ecuaciones 21 y 22 podemos expresar el calor transferido (liberado o absorbido) a presión constante como una variación de entalpía o “delta H”, o “calor de reacción” (ΔH, en Joules): ΔH = H2 – H1 = Qp (23) Si el sistema absorbe calor desde el ambiente al sistema (Q>0) se producirá un aumento de la entalpía del sistema (H2>H1), por lo que ΔH>0 (signo +). Si el sistema libera calor hacia el ambiente (Q<0), la entalpía del sistema disminuirá respecto de su estado inicial (H2<H1), por lo que ΔH<0. Podemos definir entonces, de un punto de vista energético, 2 tipos de procesos: procesos endotérmicos (ΔH>0): absorben calor. Ej. - fusión de los casquetes polares por calentamiento global - fusión de rocas corticales profundas - fusión de basaltos lunares por impacto meteoríticos - deshidratación de micas durante metamorfismo progradante - ebullición de fases volátiles en intrusivos procesos exotérmicos (ΔH<0): liberan calor. Ej. - cristalización de minerales en un magma - enfriamiento de una lava en superficie - consolidación de intrusivos - hidratación de la anhidrita a yeso - condensación de gases en cráteres volcánicos Como H es función de estado, su diferencial dH es exacta. Escogiendo T y P como variables convenientes (H = f (T,P)), entonces su diferencial total puede expresarse como: # "H & # "H & dH = % ( dT + % ( dP $ "T ' P $ "P 'T (24) Para una transformación a presión constante, dP = 0: ! & 'H # dH = dQ p = $ ! dT % 'T " p (25) La expresión (25) relaciona el calor transferido desde el entorno con el aumento de temperatura del sistema. Podemos definir entonces la capacidad calorífica a presión constante (Cp) como el cuociente: Cp ( dQ p & 'H # =$ ! dT % 'T " p (26) Para un cambio finito de estado a presión constante, la variación en la entalpía se puede calcular a partir de la integral: T2 "H = ! C dT p (27) T1 A modo de resumen, podemos decir que el calor liberado o absorbido por el sistema, Q, es directamente proporcional a la variación en temperatura, ΔT, que éste experimenta (ver ecs. 18 y 26), siendo la capacidad calorífica la constante de proporcionalidad, C: Q ∝ ΔT , Q = C ΔT La expresión anterior es equivalente a la ley de Fourier de conducción del calor, donde el flujo de calor es proporcional al gradiente de temperatura mediante la conductividad térmica kappa: J = "k#T En geología, las diferencias entre CV y CP son generalmente pequeñas. Sin embargo, preferiremos ! la utilización de la capacidad calorífica a presión constante, ya que se deriva de la entalpía, que es una medida del calor del sistema. Para intervalos de temperatura pequeños y substancias simples, CP puede considerarse constante para efectos de integración. No obstante, es común su dependencia con la temperatura para minerales. Los valores de CP se obtienen entonces experimentalmente para diferentes temperaturas y se ajustan a una serie de potencias de la forma: Cp = a + bT + cT2 + dT3 … (28) Ejemplo Para la plata (Ag), Cp = (23.43 + 0.00628 T). Calcular la variación de entalpía si se calientan 3 moles de plata desde 25 [ºC] hasta su punto de fusión a 961 [ºC], bajo una presión de 1 [atm]. A p = cte, para 1 [mol], con T1 = (273.15 + 25) y T2 = (273.15 + 961) T2 "H = # T1 T2 C p dT = # T1 1 (23.43 + 0.00628T )dT = 23.43(T2 ! T1 ) + 0.00628(T22 ! T12 ) 2 ΔH = 26430 [J/mol], y para 3 moles ΔH = 79290 [J] Aplicación de la Primera Ley de la Termodinámica a las reacciones químicas: Calor de Reacción La entalpía (H), al igual que la energía interna (U), no es una cantidad absoluta. En la práctica, no podemos medir la entalpía intrínseca de una sustancia, sino el variación en la entalpía (es decir en la energía) asociada a un determinado cambio (reacción química, transformación física, etc.). Con el propósito de asignar valores de entalpía a las sustancias de interés geológico (minerales, fases volátiles, especies diluidas, etc) , definiremos la entalpía de formación en condiciones estándar o “ΔH de formación” o “calor de formación”, y se denota ΔHfº. El “estado estándar” se refiere a la forma más estable de una sustancia, a una temperatura y presión de referencia. Por convención, la presión estándar por lo general es 1 [bar] y la temperatura estándar se considera 298.15 [K] (equivalente a 25 [ºC]). Los valores tabulados de ΔHfº para los distintos minerales y especies en solución pueden ser consultados en cualquier texto o base de datos de Termodinámica Geológica. A las entalpías estándar para los distintos elementos de la tabla periódica se les asigna arbitrariamente un valor cero. De esta forma, ha sido posible determinar experimentalmente el “calor o entalpía de formación” de compuestos puros (ej. del cuarzo, anhidrita, pirita, etc.), datos que después pueden ser combinados en reacciones químicas entre compuestos, con el objeto de determinar el calor (energía) involucrado en la reacción, y el signo de éste (absorbido o liberado). Tomemos como ejemplo la formación de agua líquida (H2O(l)), un compuesto puro, a partir de los elementos que lo componen: H2(g) + (1/2)O2 = H2O(l) ΔHfº = -285.83 [kJ/mol] Dicha reacción es fuertemente exotérmica, es decir libera calor al ambiente (signo negativo). La entalpía absoluta del agua pura, una medida de su “estado energético”, es menor que la de los reactivos, por lo que la tendencia natural al combinar hidrógeno y oxígeno es formar agua para reducir la “energía” del sistema a un estado mínimo. Ahora bien, podemos combinar los calores de reacción de las sustancias puras para reacciones más complejas, que involucran la participación de varias sustancias puras. Tomemos como ejemplo la hidratación de corindón (Al2O3(cx)) para formar gibbsita (Al[OH]3(cx)): (1/2)Al2O3 + (3/2)H2O(l) = Al[OH]3 Queremos calcular el calor de reacción (o “delta H de reacción”) a partir de los calores de formación en condiciones estándar de los compuestos puros: ΔHfº (Al2O3) = -1675.70 [kJ/mol] ΔHfº (Al[OH]3) = -1293.13 [kJ/mol] ΔHfº (H2O) = -285.83 [kJ/mol] Aplicaremos entonces la Ley de Hess, que puede ser expresada matemáticamente como “la diferencia de entalpías de formación entre los productos y los reactivos”: "H reacción = ! í "H i º f ,i (29) i donde νi son los coeficientes estequiométricos de cada especie en la reacción y ΔHfºi los calores de formación para cada una de las especies. El signo de los coeficientes estequiométricos es positivo para los productos y negativo para los reactivos. Entones, para la reacción en cuestión tenemos: ΔHrº = 2 ΔHfº (Al[OH]3) - 1/2 ΔHfº (Al2O3) - 3/2 ΔHfº (H2O) ΔHrº = (-1293.13) - 1/2 (-1675.70) - 3/2 (-285.83) = -26.535 [kJ/mol] Entonces, si hidratamos un cristal de corindón, se liberarán 26.53 [kJ] por cada mol de gibbsita formada. La reacción es exotérmica. Segunda Ley de la Termodinámica 1. Semántica En el tema anterior hemos discutido y analizado los cambios de energía de un sistema en base a sus transferencias de energía (en forma de calor y trabajo) desde y hacia el entorno. Sin embargo, nos acecha la siguiente pregunta: desde un punto de vista energético, qué determina que un proceso tenga lugar o no, y en qué dirección ocurre espontáneamente? La Segunda Ley de la Termodinámica es uno de los postulados empíricos más fascinantes de toda la ciencia. Ésta es aplicable directamente a la mayoría de los procesos fisicoquímicos, aunque muchos han tratado de aplicarla en campos tan disímiles como la biología de a evolución y la teoría económica. Históricamente, la Segunda Ley nació de la búsqueda de una función de estado que describiera la tendencia de todos los procesos a cambiar en alguna dirección específica. Por ejemplo, el agua siempre fluye de mayor a menor presión, las rocas fracturadas siempre ruedan de un potencial gravitatorio alto a uno más bajo, las sales disueltas siempre difunden de soluciones más concentradas a menos concentradas y el calor fluye siempre de alta a baja temperatura. Todos los procesos naturales son unidireccionales, y la Segunda Ley expresa esa tendencia. La Primera ley de la Termodinámica no dice nada respecto de la direccionalidad en los procesos. Sólo exige que la energía del universo permanezca constante antes y después del proceso, por lo que la Primera Ley se cumple cualquiera sea la dirección de la transformación. El “actor principal” de la Segunda Ley es una nueva función de estado denominada entropía por Clausius (del griego “transformación”), que mide la tendencia al flujo espontáneo de calor, y que, al ser combinada con la Primera Ley, permite obtener una serie de poderosas expresiones cuantitativas que describen la dirección espontánea de las reacciones químicas y procesos físicos. Esta tendencia unidireccional de los procesos naturales se expresa mediante la Segunda Ley, que posee varias definiciones. Gran parte de las definiciones clásicas tienen que ver con la eficiencia de máquinas térmicas ideales o reales, ya que fueron postuladas durante el siglo IXX, en plena revolución industrial. • La más popular es la definición de Clausius: “Es imposible para un sistema, operando en un ciclo y conectado a una sola fuente térmica, producir trabajo sobre el ambiente”. T1 T1 máq. W--- máq. (T1> T2) W--- posible imposible T2 • Otra definición rompe con el mito de la “máquina 100% eficiente”. Cada máquina pierde inevitablemente energía durante la conversión de calor en trabajo, la cual se disipa y no está disponible para ser convertida: El calor no puede ser extraído de un determinado cuerpo y ser transformado enteramente en trabajo. Como habíamos visto con anterioridad, el trabajo máximo que podía lograrse en un sistema es trabajo reversible; sin embargo, la reversibilidad es característica de los sistemas ideales, y no se logra en los sistemas reales. Por ende, el trabajo real es siempre menor al trabajo reversible. De la misma forma, el calor real es siempre menor al calor reversible (calor máximo). Los procesos irreversibles (reales) causan un “degradación” de la energía (hay menos disponibilidad de efectuar trabajo). • Un enfoque completamente distinto de la Segunda Ley fue enunciado en el Siglo XX, mediante la aplicación de la Mecánica Estadística a la Termodinámica. Usando un enfoque molecular en vez de macroscópico, Boltzmann y otros demostraron la relación existente entre la dirección espontánea de los procesos y la probabilidad: Cuando un sistema evoluciona en libertad cambiará, generalmente, hacia una condición de máxima probabilidad. Dicho enunciado permite hacer la conexión entre (i) la energía distribuida a escala molecular y atómica, y la (ii) energía distribuida a escala macroscópica. Se ha demostrado que este postulado es equivalente a los dos mencionados anteriormente. 2. Entropía Nuestro siguiente paso consiste en expresar matemáticamente la Segunda Ley de la Termodinámica y definir una nueva función de estado, la entropía, que expresará la tendencia al flujo espontáneo de calor. En todos los cursos de Fisicoquímica surge la pregunta: ¿Qué es la entropía? El estudiante rara vez considera satisfactoria la explicación del profesor acerca de este concepto, ya que tiende a pensar en la entropía como un concepto palpable físicamente (ejemplo: “desorden”). La dificultad surge por dos razones. Primero, debe admitirse que la entropía es algo menos palpable que una cantidad de trabajo o calor. Segundo, la pregunta es vaga en sí misma. Para evitar tales tribulaciones, en una primera aproximación cambiaremos la pregunta qué es la entropía? por la pregunta ¿cómo cambia la entropía bajo determinadas circunstancias? Si sabemos cómo se comporta la entropía como función de estado, sabremos algo acerca de lo que “es”. Más adelante relacionaremos la entropía con la “aleatoriedad” en una distribución de partículas, que corresponde a la definición estadística. En Termodinámica clásica, la entropía está definida por la siguiente ecuación diferencial: dS = dQrev T (30) La entropía S es una función de estado definida por la razón entre el calor y la temperatura, en condiciones reversibles, es decir, ideales. Integrando a ! través de un ciclo reversible, la entropía es siempre cero: " dS = " dQrev =0 T (31) Por otra parte, para procesos cíclicos en condiciones irreversibles (o reales): ! " dS = " dQrev <0 T (32) Tenemos entonces que para un ciclo irreversible, la integral de línea en trayectoria cerrada es menor que para un ciclo reversible: ! irrev rev 2 1 ! " dQrev <0 T 2 1 ! " dQrev =0 T Tales diferencias pueden expresarse mediante la desigualdad de Clausius, una de las consecuencias más importantes y útiles de la Segunda Ley. Consideremos el proceso cíclico anterior, con una etapa reversible y otra irreversible: irrev 2 1 rev " dQciclo = T 2 " 1 dQirrev + T 2 1 # dS > # ! 1 2 1 " 2 dQrev <0# T 2 " 1 dQirrev $ T 1 " dS < 0 2 dQirrev dQirrev " dS > T T dS > ! dQirrev T Desigualdad de Clausius “Cualquier proceso natural (irreversible) se efectúa con un aumento en la ! entropía del sistema” Esta relación tiene gran importancia, ya que nos permitirá determinar la dirección de los procesos espontáneos. Podemos escribir ahora una forma más general de la Segunda Ley de la Termodinámica, válida para sistemas aislados (OJO!): dS " Por ende: dQ T (33) - procesos reversibles (ideales): dStotal = 0 ! - procesos irreversibles (reales): dStotal > 0 (generan entropía!) dStotal = dSsistema + dSambiente (34) Formas de calcular la entropía: ΣS • de una transformación química, a T = cte.: ΔSº = • de una transformación física, a T = cte.: ΔSº = Qrev/T Unidades de la entropía S: ºfinal - ΣS ºinicial [Joule/mol/K] Clausius resumió entonces la Primera y Segunda Ley en la siguiente frase: “La energía del Universo es constante y la entropía tiende a un máximo” La Segunda Ley de la Termodinámica y la Desigualdad de Clausius han sido usadas con frecuencia para interpretar la entropía como una medida del desorden y la aleatoriedad. El ejemplo clásico consiste en dos gases que se mezclan en condiciones adiabáticas a V = cte. Después de mezclarse en forma espontánea, el sistema está más “desordenado” que antes (ha ganado entropía, o energía por el hecho que el estado de desorden es el estado de máxima probabilidad para ese caso). Sin embargo, existen numerosos procesos geológicos espontáneos que muestran, contrariamente, un incremento en el “orden”, y que tienden a confundir la aplicación del concepto de “aumento de entropía o desorden de los procesos naturales” (“todos los procesos generan S”): - Los magmas cristalizan espontáneamente, incrementando su ordenamiento molecular. - El agua se congela espontáneamente a bajas temperaturas, aumentando su orden. - El Sistema Solar evolucionó espontáneamente a partir de una nebulosa desornada a un sistema planetario ordenado. - La evolución de la Tierra y sus organismos puede considerarse como un proceso de ordenamiento de moléculas y átomos libre a estructuras más complejas y ordenadas. Por ende, tomar literalmente la segunda ley en términos de orden/desorden y asociar aumento o conservación de entropía (dS ≥ 0) a TODOS los procesos naturales llevará inevitablemente a conclusiones erróneas. Esto tiene que ver con la definición del los límites del sistema. Todos los procesos anteriores son reales, irreversibles, e intercambian entropía con el ambiente (son sistemas cerrados y/o abiertos), por lo que la Segunda Ley no puede ser aplicada directamente a partir de su definición, que es para sistema aislados (eq. 33). En todos los casos anteriores, los sistemas han perdido energía (perdido entropía) por el hecho de no ser perfectamente aislados. 3. Variaciones de Entropía Recordando la Primera Ley, tenemos: dU = dQrev + dW = dQrev – pdV dQrev = dU + pdV , y dividiendo por T dQrev/T = (1/T)dU + (p/T)dV dS = (1/T)dU + (p/T)dV (35) La ecuación (35) relaciona la variación en la entropía dS con variaciones en la energía y volumen, dU y dV, y con la presión y temperatura del sistema. Según esta expresión, la entropía puede variar de dos formas independientes: variando la energía interna o el volumen. A V=cte (dV=0), la entropía aumenta, y si U = cte (dU=0), un aumento de volumen conlleva un aumento de entropía. Variación con la Temperatura y el Volumen Debido a la dificultad de medir la energía del sistema, expresaremos la entropía en función de T y V, o de T y p. Si consideramos la entropía como dependiente de la temperatura y el volumen, S = S (T,V), su diferencial total será: dS = (∂S/∂T)V dT + (∂S/∂V)T dV (36) Queremos obtener una expresión simple para (∂S/∂T)V. Si se expresa dU en términos de dT y dV: dU = (∂U/∂T)V dT + (∂U/∂V)T dV dU = Cv dT + (∂U/∂V)T dV , reemplazando en (35) dS = (1/T) [(∂U/∂T)V dT + (∂U/∂V)T dV] + (p/T)dV dS = (1/T) (∂U/∂T)V dT + (1/T) [(∂U/∂V)T + p]dV (37) Comparando (37) con (36): (∂S/∂T)V = (1/T) (∂U/∂T)v (∂S/∂T)V = Cv/T (38) Ahora queremos obtener una expresión simple para (∂S/∂V)T : (∂S/∂V)T = (1/T) [(∂U/∂V)T + p] (∂U/∂V)T = T(∂S/∂V)T – p , despejando (∂U/∂V)T , derivando con respecto a T a V=cte (∂ 2U/∂V∂T) = (∂ S/∂V)T +T (∂ 2S/∂V∂T) – (∂ p/∂T)V (39) Diferenciando Cv = (∂U/∂T)v = T(∂S/∂T)V , con respecto a V con T=cte : (∂ 2U/∂T∂V) = 0 + T(∂ 2S/∂T∂V) (40) Igualando las ecs. (39) y (40): (∂ 2U/∂V∂T) = (∂ 2U/∂T∂V) (∂ S/∂V)T + T(∂ 2S/∂V∂T) – (∂ p/∂T)V = T(∂ 2S/∂T∂V) (∂ S/∂V)T = (∂ p/∂T)v (41) Ahora podemos escribir una expression simplificada para S=S(T,V): dS = (Cv/T) dT + (∂p/∂T)V dV (42) Para un Gas Ideal, pV = nRT, y (∂ p/∂T)v = nR/V dS = (Cv/T)dT + (nR/V)dV (43) Si integramos (43) considerando que Cv = cte en el intervalo de temperatura, tenemos: S2 T2 V2 1 1 " dS = Cv " T dT + nR " V dV S1 T1 V1 ΔS = Cv ln(T2/T1) + nR ln(V2/V1) (44) ! De la ec. (44) podemos deducir que ΔS = 0 sólo si T y V son constantes. Variación con la Temperatura y la Presión Si consideramos la entropía como dependiente de la temperatura y la presión, S = S (T,p), su diferencial total será: dS = (∂S/∂T)p dT + (∂S/∂p)T dp En condiciones reversibles: (45) dU = TdS – pdV (46) H = U + pV dH = dU + pdV + VdP Igualando (46) y (47): dH – pdV –Vdp = dU (47) dH – pdV –Vdp = TdS – pdV dH = TdS – Vdp Despejando dS: (48) dS = (1/T)dH – (V/T)dp , y reemplazando dH por su diferencial total: dS = (1/T) [(∂H/∂T)p dT + (∂H/∂p)T dp] – (V/T)dP dS = (1/T) (∂H/∂T)p dT + (1/T) [(∂H/∂p)T – V]dp (49) Comparando (49) con (45): (∂S/∂T)p = (1/T) (∂H/∂T)p = (Cp/T) , ya que Cp = (∂H/∂T)p = T (∂S/∂T)p (∂S/∂p)T = (1/T)[ (∂H/∂p)T – V] Despejando ahora (∂H/∂p)T : (∂H/∂p)T = T(∂S/∂p)T + V , ahora derivamos con respecto a T (∂ 2H/∂p∂T) = (∂S/∂p)T + T(∂ 2S/∂p∂T) + (∂V/∂T)p (50) Derivando luego Cp con respecto a p: (∂Cp/∂T) = (∂ 2H/∂T∂p) = T(∂ 2S/∂T∂p) (51) Igualando (50) y (51): T(∂ 2S/∂T∂p) = (∂S/∂p)T + T(∂ 2S/∂p∂T) + (∂V/∂T)p (∂S/∂p)T = - (∂V/∂T)p (52) Entonces, ahora tenemos las expresiones para definir la entropía en función de T y p: dS = (∂S/∂T)p dT + (∂S/∂p)T dp dS = (Cp/T)dT - (∂V/∂T)pdP (53) Para un Gas Ideal, pV = nRT, y (∂p/∂T)v = nR/V dS = (Cp/T)dT - (nR/p)dp (54) Si integramos (54) considerando que Cp = cte en el intervalo de temperatura, tenemos: S2 T2 " dS = C " p S1 T1 p 2 1 1 dT # nR " dp T p1 p ΔS = Cp ln(T2/T1) - nR ln(p2/p1) (55) ! 5. La Segunda Ley: Adiábata del Manto Terrestre Deseamos encontrar una expresión que nos permita calcular la estructura térmica de la Tierra, es decir, encontrar una función del tipo T = T(z), sin tener que recurrir a resolver la ecuación de conducción del calor. Para ello utilizaremos expresiones derivadas de la Segunda Ley de la Termodinámica. Para ello, tenemos que definir primero el coeficiente de expansión térmica α, el cual expresa cuanto varía el volumen de un sólido al calentarse a presión constante: α = (1/Vo)(∂V/∂T)p (56) Donde Vo es el volumen inicial y (∂V/∂T)p el cambio en volumen. La variación de la entropía con la temperatura y la presión es (ec. 53): dS = (Cp/T)dT - (∂V/∂T)pdp Reemplazando (56) en (53): dS = (Cp/T)dT - αVodp (57) Sin embargo, ya que la densidad de la corteza varía (por ejemplo, entre la corteza continental, oceánica y el manto), debemos expresar (57) en función de la densidad ρ. Si ρ =(1/V), masa unitaria por simplicidad, V =(1/ρ), entonces (57) es equivalente a: dS = (Cp/T)dT – (α/ρ) dp (58) Los gradientes de un sistema en convección, como es el caso del manto terrestre, son cercanos a adiabáticos (sistema aislado cuyos límites permiten realizar trabajo, pero el sistema está cerrado a la transferencia de masa y calor). Si consideramos condiciones adiabáticas (dQ = 0), entonces dS = 0: (dT/dp) = (αT)/(ρCp) (59) Combinando (59) con la presión litostática (dp/dz) = ρg (con z = profundidad y g = aceleración de gravedad), tenemos: (dT/dz) = (α gT)/Cp (60) Los valores típicos para tales constantes en el manto superior son aproximadamente: α = 3x10-5 [K-1] Cp = 1 [kJ/kg/K] T ≈ 1573 [K] g = 10 [m/s2] Reemplazando las constantes, se tiene que el gradiente geotérmico del manto es del orden de 5x10-4 K/m (0.5 ºC/km): dT/dz = 0.5 K/km Adiábata del manto (= gradiente geotérmico del manto) Línea gruesa gris (~0.5 ºC/km) Tercera Ley de la Termodinámica + Equilibrio y Espontaneidad 1. Tercera Ley de la Termodinámica La dependencia de la entropía con la temperatura puede de definirse de manera similar a la entalpía. En condiciones reversibles: 0 T2 T2 0 T1 "S = "S + Cp dT T # T1 El calor específico de las sustancias sólidas tiende a converger a cero: ! Cp muscovita pirofilita [J.mol-1.K-1] T[K] 0 250 500 lim C p = 0 # lim $ T "0 T "0 Y reversiblemente: ! Cp dt = 0 T dS = dQ rev/T = CpdT/T = (Cp/T)dT lim dS = 0 T "0 “la entropía de una sustancia pura perfectamente cristalina es CERO en el cero absoluto de temperatura (S0K = 0)” ! 0 K = -273 ºC → en el cero absoluto, no existe movimiento de los átomos, y por lo tanto no existe desorden ni entropía Esto nos permite ahora calcular entropías absolutas de la siguiente manera: T 0 ST02 = S0K + Cp dT S 0K T " ! ! 2. Equilibrio y espontaneidad La Primera Ley nos da información respecto del balance de energía en las transformaciones físicas y reacciones químicas (la energía se conserva!!), mientras que la Segunda Ley, a través de la entropía, nos permite saber si los procesos generan o no entropía (dS ≥ 0). A partir de la Desigualdad de Clausius, enunciaremos la “Condición General de Equilibrio y Espontaneidad”: dS ≥ dQ/T TdS – dQ ≥ 0 (TdS – dQirrev > 0 y TdS – dQrev = 0) De la Primera Ley, tenemos que: dU = dQ – pdV dQ = dU + pdV Reemplazando la Primera Ley en la Desigualdad de Clausius, obtenemos la Condición General de Equilibrio y Espontaneidad: TdS – dU – pdV ≥ 0 = 0 : proceso en equilibrio (reversible) > 0 : proceso irreversible espontáneo Vamos a trabajar esta ecuación para: - sistema aislado - temperatura constante - temperatura y presión constantes (util!) 2.1 Sistema Aislado Ya hemos visto este caso. Para sistema aislados y adiabáticos, tenemos que no existe flujo de calor (dQ = 0) y a V = cte, la ecuación anterior se reduce: TdS ≥ 0 ΔS = 0 ΔS > 0 proceso en equilibrio (condiciones reversibles) proceso espontáneo, irreversible (genera entropía) 2.2 Sistema a Temperatura Constante Tenemos que la Condición de Equilibrio y Espontaneidad es la siguiente: TdS – dU – pdV ≥ 0 Si diferenciamos la temperatura por la entropía (TS), tenemos: d(TS) = SdT + TdS d(TS) = TdS Reemplazando en la Condición: , a T = cte , dT = 0 d(TS) – dU – pdV ≥ 0 , y reagrupando – [dU – d(TS)] – pdV ≥ 0 dA Definimos así una nueva función de estado llamada Energía Libre de Helmholtz (A). Esta función de estado NO TIENE mayor relevancia en Ciencias de la Tierra (pocos procesos ocurren sólo a T=cte), pero sí en física, química, bioquímica e ingeniería química: A ≡ U – TS 2.3 Sistema a Temperatura y Presión Constante (** importante) La Condición de Equilibrio y Espontaneidad es: TdS – dU – pdV ≥ 0 a T = cte: a p = cte: TdS = d(TS) pdV = d(pV) d(TS) – dU – d(pV) ≥ 0 – [dU + d(pV) – d(TS)] ≥ 0 [dU + d(pV) – d(TS)] ≤ 0 dG Donde dG es el diferencial de una nueva función de estado que definiremos como Energía Libre de Gibbs (G). La Energía Libre de Gibbs (G) es la función de estado más útil de la Termodinámica en Ciencias de la Tierra, ya que comprende en forma implícita la energía interna U, la entalpía H y la entropía S. A la vez, es el resultado de la combinación de la Primera Ley con la Desigualdad de Clausius. Se define formalmente como: G ≡ U + pV – TS O en forma alternativa: G ≡ H – TS Volviendo a la expresión de Equilibrio y Espontaneidad para G (a p y T ctes), tenemos que: ΔG ≤ 0 Definimos entonces, a presión y temperatura constantes: procesos en equilibrio físico y químico, cuando ΔG = 0 procesos irreversibles, espontáneos, cuando ΔG < 0 procesos no-espontáneos, cuando ΔG > 0 Ahora podemos responder las preguntas que nos hicimos empezando el curso: ¿ Bajo qué condiciones los productos de una reacción estarán en equilibrio? ¿Si no están en equilibrio, en qué dirección se lleva a cabo la reacción? - para reacción que está en equilibrio, la energía libre de Gibbs de los reactivos (Gr) es igual a la de los productos (Gp) - las reacciones químicas ocurrirán espontáneamente en la dirección de la menor energía libre de Gibbs G Gr Gp G ΔG = 0 Gr Gp ΔG < 0 Unidades de la energía libre de Gibbs (molar): [Joule/mol] Lo que nos interesa, al igual que con la entalpía H, es el cambio de energía libre de Gibbs, o “Delta G”( ΔG). Existen, al igual que para ΔH, varias “jerarquías” para el Delta G de una reacción: ΔG ΔGr ΔGºr ΔGºf ΔG: delta G (simplemente, “variación en la energía libre de Gibbs”) ΔGr: delta G de reacción (variación de G para una reacción química) ΔGºr: delta G de reacción en condiciones estándar ΔGºf: delta G de formación en condiciones estándar Para una reacción química, al igual que con el “delta H”, el delta G (ΔG)ven condiciones estándar se calcula mediante la Ley de Hess en base a los calores de formación estándar de las sustancias más simples, los cuales se obtienen por calorimetría y están tabulados. Además, se pueden calcular mediante la siguiente ecuación fundamental: ΔGr = ΔHr -TΔSr T, p, ctes Ejemplo Para la reacción de hidratación de la anhidrita para formar yeso, decidir si la reacción es endotérmica o exotérmica, y si es espontánea o no: CaSO4 (anhidrita) + 2H2O(l) = CaSO4.2H2O ΔHºf (anh) = -1434,11 kJ/mol ΔHof (agua) = -285,830 kJ/mol ΔHºf (yeso) = -2022,63 kJ/mol Sºf (anh) = 106,7 J/mol/K Sºf (agua) = 69,91 J/mol/K Sºf (yeso) = 194,1 J/mol/K (yeso) ΔGºf (anh) = -1321,79 kJ/mol ΔGºf (agua) = -237,129 kJ/mol ΔGºf (yeso) = -1797,28 [kJ/mol]. ΔHºr = (-2022,63) – (-1434,11 + (2)-285,830) = - 16,86 kJ (exotérmica) ΔSºr = 194,1 - 106,7- 2(69,91) = - 52,42 J/K = - 0,05242 kJ/K Ahora podemos calcular ΔGºr de dos formas: ΔGºr = ΔHºr - TΔSºr = - 16,86 – (298)(- 0,05242) = - 16,86 +15,62 = - 1,24 kJ ΔGor = -1797,28 – (-1321,79 + (2).-237,129) = - 1,23 kJ (la reacción es espontánea) Conclusión, el yeso es más estable que la anhidrita en presencia de agua, en condiciones de superficie terrestre (298 K, 1 bar). Es decir, la anhidrita se combina espontáneamente con el agua para formar yeso, liberando energía. 2.4 Variación de G con la variables de estado De tal forma como U = U(T,V) y H = H(T,p), la energía libre de Gibbs G es función de T y p, G = G(T,p). El diferencial total de G es: dG(T,p) = (∂G/∂T)pdT + (∂G/∂p)Tdp Diferenciando la definición de energía libre de Gibbs: G = U + pV – TS dG = dU + pdV + Vdp –TdS – SdT y dU = TdS- pdV , entonces dG = TdS – pdV + pdV + Vdp –TdS – SdT dG = – SdT + Vdp Comparando ahora ambos diferenciales de G: dG(T,p) = (∂G/∂T)pdT + (∂G/∂p)Tdp -S V (∂G/∂T)p = - S G (∂G/∂p)T = V G -S T V p Este resultado es importante: - Aquellos minerales (fases) con mayor entropía serán más estables a mayor T - Aquellos minerales (fases) con menor volumen serán más estables a mayor P 2.5 Dependencia del ΔG con la Temperatura G = U – TS + pV G = H – TS G = H + T (∂G/∂T)p dividiendo por T G/T = H/T + (∂G/∂T)p despejando (∂G/∂T)p (∂G/∂T)p – G/T = - H/T T(∂(G/T)/∂T)p (∂(G/T)/∂T)p = - H/T2 (∂(ΔG/T)/∂T)p = - ΔH/T2 Ecuación de Gibbs-Helmholtz La ecuación de Gibbs-Helmholtz nos permite calcular el valor de “delta G” de una reacción o cambio físico a temperaturas distintas a las standard, y a partir de datos standard. En su forma integrada definida: T 0 "G0T "G 0298K "H298K = # $ dT T 298K 298K T 2 Suponiendo ΔH constante para el intervalo de temperatura, y p = cte =1 bar. ! 2.6 Las 4 ecuaciones fundamentales de la Termodinámica Corresponden a las expresiones diferenciales de las cuatro funciones de estado más importantes en Termodinámica (U, H, A, y G), en función de las variables de estado. Pueden obtenerse fácilmente mediante la siguiente regla nemotécnica: T (+) U H p (-) V (+) A G S (-) dU = – pdV + TdS dH = Vdp + TdS dA = – pdV – SdT dG = VdP – SdT Estas ecuaciones son importantes, ya que nos permiten obtener las Relaciones de Maxwell de la Termodinámica, en base a sus derivadas cruzadas: – (∂p/∂S)V = (∂T/∂V)S (∂V/∂S)p = (∂T/∂p)V (∂p/∂T)V = (∂S/∂V)T (∂V/∂T)p = – (∂S/∂p)T