Pdf profesor. - Futuremedicos.com
Anuncio

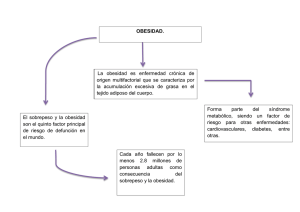
Obesidad y diabetes Homeostasis energética Peso corporal • Dr. José G. Castaño Aumento Ingesta Energía Disminución Gasto Energía Ingestión de: Actividad física Proteínas Termogénesis inducida por la dieta Grasas Metabolismo Basal Carbohidratos El peso corporal se mantiene por una compleja red de mecanismos que aseguran un aporte energético constante para mantener las funciones celulares Bioquímica nutricional elemental Energía Alimentos Termogénesis inducida por dieta (5-10%) Tejido adiposo Almacen TG Termogénesis, respuesta al frío. UCPs: desacoplamiento OXPHOS “Exceso” ATP “Trabajo” Metabolismo basal (60%) Glucógeno Reservas en Músculo e Hígado OXPHOS Actividad Física Clasificación de la Obesidad. CLASIFICACIONES DE LA OBESIDAD SEGUN GARROW GRADO IMC Grado 0: Normopeso 20 - 24.9 Grado I: Sobrepeso 25 - 29.9 Grado II: Obesidad 30 - 39.9 Obesidad 40 Grado III: mórbida SEGUN AMERICAN HEART ASSOCIATION GRADO IMC RIESGO Clase 0 Normal 20 - 24.9 Muy bajo Clase I obesidad leve 25 - 29.9 Bajo obesidad Clase II 30 - 34.9 Moderado moderada Clase III obesidad severa 35 - 39.9 Alto obesidad 40 Muy alto Clase IV mórbida Epidemia de obesidad en el mundo • En la última decada la incidencia de obesidad ha aumentado en 1/3. • 154 millones de personas sufren obesidad en el mundo • 300,000 individuos mueren por obesidad en USA por año. • Niños y adolescentes forman parte de la epidemia de obesidad. El consumo energético viendo la televisión es un 12% menor que durante el sueño. • Obesidad es un grave riesgo de enfermedad y un factor de riesgo alto para diabetes tipo 1 Interación de genes y medio GENES MEDIO 51 años del descubrimiento de la estructura del DNA, 1953 “Los genes no son solamente el soporte de la herencia. Son esquisitos mecanismos para traducir la experiencia en acción”. “ Sus acciones no son fijas en absoluto, por la forma en que se enciende o apaga su actividad transcripcional en respuesta a instrucciones externas. Tasa de mutación espontánea 0.5% /por millón de años En 10,000 años de evolución En 10,000 años ~ 0.005% cambios GRANDES cambios en dieta y estilo de vida “ Son instrumentos para extraer información del ambiente, cada minuto, cada segundo, el modelo de genes que se expresa está cambiando en nuestras células. Los Genes son los mecanismos de la experiencia.” Matt Ridley 2003 Papel de la dieta en la evolución humana • La adaptación y supervivencia de una especie tiene dos componentes energéticos: Energía que se toma y energía que se gasta en el mantenimiento y la progresión de la especie • Energía de mantenimiento es la energía requerida para mantener a un animal vivo en el día a día. • Energía productiva asociada con la producción y crianza de los descendientes. Para los mamíferos, como nosotros, esta energía extra debe cubrir los costes de la maternidad durante el embarazo y la lactancia. Papel de la dieta en la evolución humana • La bipedestación: evolutivamente supone un ahorro energético frente a andar a 4 patas (marcha normal). Eso suposo un cambio notable en nuestro esqueleto óseo. – A medida que el continente africano se volvió más seco, los bosques fueron desapereciendo (sabanas), haciendo que las fuentes alimenticias estuvieran más dispersas. – La bipedestación fue una de las primeras estrategias en la evolución humana, dado que permitía una reducción importante en la cantidad de calorías gastadas en conseguir alimentos que estaban dispersos y a grandes distancias Papel de la dieta en la evolución humana • S. Boyd Eaton y Melvin J. Konner (1985) argumentaban que la prevalencia en las sociedades modernas de --obesidad, hipertension, enfermedad coronaria y diabetes, --es consecuencia de un falta de concordancia entre los hábitos alimenticios modernos y el tipo de dieta que nuestra especie adoptó evolutivamente durante la etapa como cazadores y recolectores. • William R. Leonard (2002) argumenta que los humanos han evolucionado no para subsistir en una dieta simple, Dieta Paleolítica, sino para ser comedores flexibles con dieta variada. Esto tiene importancia en el actual debate sobre la dieta Papel de la dieta en la evolución humana • GRANDES CEREBROS Y HOMBRES HAMBRIENTOS: El siguiente cambio evolutivo más importante fue el aumento de tamaño del cerebro. 8-10% H. erectus Desarrolla la cultura de cazadores y recolectores 20-25% 16X músculo 2 Papel de la dieta en la evolución humana: fuera de Africa • La evolución de H. erectus en Africa hace 1.8 millones de años marcó el tercer gran paso de la evolución de la humanidad: el movimiento inicial de los hominidos fuera de Africa. • Lo que un animal come dicta cuanto territorio necesita. Se ha estimado que el H. erectus con su aumento de tamaño y consumo energético necesitaba 10 veces más territorio que el Australopithecus. • La marcha más al Norte determinó un aumento del gasto energético por los cambios clímáticos. H. Neandertaliensis necesitaba un mínimo de 4000 Kcal/día para sobrevivir Hipótesis del gen ahorrador. The “thirsty” gene Papel de la dieta en la evolución humana: cocinado, agricultura y ganadería • El cocinado de los alimentos, junto con el desarrollo de la agricultura y posteriormente de la ganadería, aumentaron la disponibilidad de alimentos y el enrequecimiento nutritivo de los mismos. • En menor volumen, mayor poder energético y nutricional. En la actualidad eso se lleva al máximo con suplementos líquidos o barras de comida rápida. • Como nuestros antepasados, tratamos de conseguir la mayor capacidad nutritiva de nuestros alimentos con el menor volumen posible y con el menor gasto energético Factores que controlan la obesidad • Unos 200 genes, de los cuales 5-10 son los más importantes, constituyen una red de mecanismos fisiológicos redundantes que hemos adquirido durante la evolución para controlar nuestra alimentación. Esta maquinaria producía una gran eficiencia en la carga de reservas energéticas (grasas) ante una fuente rica de alimentos, puesto que no siempre era accesible esa fuente (no todos los días, no siempre cerca). • Esa misma red la tenemos ahora funcionante, esos genes ahorradores, que nos dieron una ventaja evolutiva, son los que ahora al tener comida Modelos monogénicos de obesidad en ratón Mutación Obeso Diabetes/Fat Agouti Genotipo ob/ob db/db ratón, fa/fa rata Ay/a, AVY/a Gen leptina Teoría Lipostática 1953 Un factor circulante inhibe la ingesta y el acúmulo de tejido adiposo. Receptor leptina Proteína Agouti Fat fat/fat CpE: mutación Carboxipeptidasa E defecto en procesamiento de COOH de hormonas peptídicas Tubby tub/tub Tubby (Proteínas G señalización? Degeneración de retina y coclear, además de diabetes, obesidad y resistencia a insulina.) • ratón Ob/ob mutación espontánea que produce obesidad • Leptina (ob/ob) gen clonado en 1994 3 PAPEL DE LA LEPTINA Antecedentes •Leptina. Hormona adipostática con poder saciante descubierta a finales de 1994 Teoría Lipostática de Kennedy: factor humoral procedente del tejido adiposo, que a través de su acción hipotalámica, informa al SNC sobre el grado de adiposidad, modulando así el balance energético • La lesión del núcleo ventromedial del HT (centro de la saciedad) produce hiperfagia y obesidad Hervey: experimentos de parabiosis entre una rata obesa por una lesión hipotálamo ventromedial y una normal; el animal sano muere por caquexia • El animal obeso debe producir un “factor anorexígeno” circulante al que el obeso no es sensible debido a la lesión en Hipotálamo. Hausverger: el ⇑ peso en ratones obesos (ob/ob) se puede prevenir mediante parabiosis con un ratón normal. Coleman : demostró la existencia de un factor saciante transferible, que actuaría a nivel central, cuya ausencia (ratón ob/ob) o falta de actividad (ratón db/db) sería responsable (en parte) de las alteraciones fenotípicas observadas en los modelos de obesidad genética. RECEPTORES DE LA LEPTINA (Ob-R) •Proteína de membrana homóloga al receptor de la familia de citoquinas de clase 1 (incluyendo receptores para IL6, LIF, GCSF y glicoproteína 13041) •Localización de los receptores: •Tej. Periféricos: tej. adiposo, pulmón, riñón, hígado, músculo, testículos, islotes pancreáticos, células hematopoyéticas. •Cerebrales: plexo coroideo HipoTálamo (núcleo arcuato, PV y VM: regulación del balance energético) Ratón obeso dominante AGOUTI amarillo y ratón negro delgado no-agouti • en la piel da color amarillo de la piel • en cerebro MC4R da lugar a obesidad LA MOLECULA: péptido de 167 aa , con una secuencia señal de 21 aa que se escinde antes de pasar al torrente circulatorio. •La proteína madura tiene 146 aa y 16 kDa con una estructura terciaria similar a las citoquinas clásicas de hélice larga como la IL-2 •Pocas diferencias interespecies: la leptina humana tiene una homología del 84% con la del ratón y del 83% con la de rata. RITMO CIRCADIANO: relacionado con la pauta de ingesta: secreción pulsátil regulada por insulina y otras hormonas. •No conocemos el mecanismo responsable de los picos, pero parece regulado por: - horas luz/oscuridad - ingesta - horas de sueño NIVELES SÉRICOS: NORMOPESO: 1-15 ng/ml IMC> 30: >30 ng/ml El tratamiento con leptina a los deficientes elimina la obesidad ALTERACION GENETICA DE LEPTINA Y/O RECEPTOR EN HUMANOS RARO •La mutación del Ob-R causa la aparición precoz de obesidad en ratones (insensibilidad a la leptina, transtornos metabolicos, obesidad mórbida, alteraciones neuroendocrinas). •Las mutaciones del receptor son MUY RARAS EN HUMANOS Producción de proteína agouti AGRP AVY/a (Agouti Related Protein) antagoniza α-MSH - LEPTOS = DELGADO Ratón Agouti a/a 4 Pro-opiomelanocortina (POMC) α-MSH induce pigmentación en ratón AVY/a Un único polipéptido que se procesa por proteólisis dando lugar a las diferentes polipéptidos Tratado con MSH Sin tratar α-MSH induce pigmentación más marcada en el ratón Ay γ-melanocortina α-melanocortina β-endorfina β-melanocortina Ablación del gen de POMC da lugar a obesidad Mutaciones en POMC. Ejemplo • Mutación en gen humano POMC (AR) cursa - pelo rojo - hiperfagia - obesidad • Mutación puntual Arg(R) por Gly(G) produce un defecto en el procesamiento dando una proteína de fusión: β-MSH/β-endorfina ratón POMC knockout - obesidad - trastorno de pigmentación BG Challis et al Human Molecular Genetics 11: 1997, 2002 H Krude et al, Nature Genetics 19; 155,1998 Fenotipos de los ratones Knockout de los receptores de melanocortina MC4-R y MC3-R MC4-R -/- obeso, hiperfagia, hiperglicemia, hiperinsulinemia, aumento longitud del cuerpo MC3-R -/- aumento de masa de tejido graso, aumento de eficiencia alimentaria, disminución de actividad MC3-R -/- MC4-R -/- más obesos y mas masa adiposa que MC4-R -/- Obesidad mórbida asociada con mutaciones en el gen humano MC4-R • >4% de los niños con obesidad mórbida tienen una mutación en un alelo del receptor MC4-R •Es la causa monogénica más común de obesidad en humanos • Fenotipo: - hiperfagia comienza a ~ los 8 meses - Tendencia a estatura alta - hiperinsulinemia - aumento de la densidad mineral ósea No hay evidencia en humanos de mutaciones en MC3-R relacionadas con obesidad o diabetes tipo 2 5 Mutaciones en el receptor MC4-R Conclusión: el sistema de la Melanocortina juega un papel esencial en la homeostasis energética. Knock out ratón : POMC KO ratón - obeso MC3-R KO ratón - obeso MC4-R KO ratón - obeso Mutaciones Humanas : Mutaciones POMC (recesiva) Mutaciones MC4-R obesidad mórbida mutaciones en un alelo ¿Qué datos nos dan información sobre obesidades genéticas? • Niños que están por encima del percentil 97% en BMI (IMC) para su altura, o 97% para su edad, son susceptibles de estudios genéticos, en especial si tienen hermanos obesos. • Evaluación sospecha clínica de la obesidad genética: – Niveles de leptina en plasma (niveles muy bajos indican mutación en leptina, niveles muy altos indican mutación en receptor de leptina). – Niveles de insulina y pro-insulina (niveles altos de proinsulina pueden indicar defecto en los enzimas de procesamiento de la insulina). – Color de pelo (familias pelirojas, Modulación hipotalámica de la toma de alimentos Orexigénico NPY (H. Arcuato) AGRP(H. Arcuato) Galanina MCH (H. lateral) Orexinas (H. lateral) Opioides Ï Ingesta alimentos Ï tono Parasimpático (Ï insulina) Ð tono simpático (Ð gasto) Ï aumento ingesta grasa Anorexigénico POMC Î α−MSH (H. Arcuato) CART (H. Arcuato) CRH (H. PVN) TRH (H, PVN) Insulina (páncreas) GIP-1 Serotonina - obeso ~5% niños con tienen Regulación saciedad/hambre en SNC z Factores de saciedad en SNC: z POMC/Alpha-MSH, α-melanocyte stimulating hormone CART, transcrito regulado por cocaína y anfetaminas CCK-PZ, colecistoquinina-pancreozimina GLP-I, glucagon-like peptide I Serotonina z Factores de hambre en SNC: AGRP, agouti-related peptide NP-Y, Neuropeptido-Y Galanina Orexinas A and B MCH Proyecciones desde el núcleo arcuato NPY/AGRP y POMC/CART Aumento ingesta Disminución ingesta Ð Ingesta alimentos Ð tono Parasimpático (Ð insulina) Ï tono simpático (Ï gasto) Ï oxidación de grasas Disminución Ingesta Nature 404, 661 - 671 (06 April 2000) 6 Control de neuronas secundarias en hipotálamo Anorexia Proyección neuronas NPY del núcleo arcuato Bulimia Axones CRH OXI orexinas TRH MCH Nature 404, 661 - 671 (06 April 2000) Proyección desde hipotálamo lateral: Melanin Concentrating Hormone y Respuesta a ingesta de alimentos Orexinas Post-absorción Post-ingestión Cognitivos Disminución Gasto energético Sensoriales Sistema nervioso Autónomo Vago Alimento Temprano Tardío Saciedad Bulimia. Aumento de Ingesta Control del apetito y del peso Regulación a corto plazo de la alimentación Factores olfativos,visuales y emocionales, factores cognitivos superiores Hipotálamo núcleo arcuato C RH Catabólica + •Percepción del sabor •Cantidad de comida •Saciedad o aumento de apetitoregulada por nutrientes y por señales nerviosas y peptídicas derivadas del GI y del páncreas. •Las señales que emanan del GI y de los almacenes de energía (adiposo) son recibidas e integradas en diversos circuitos neuronales en el hipotálamo y en tronco del encéfalo. + α-M SH PO MC Leptina Receptor _ AG RP TR H NP -Y Calor α−M S H T3 UC P3 Calor Leptina Tracto Solitario Vago M C 4-R Alimento + Sistema nervioso simpático Vago Señales agudas (Aferentes) Catecolaminas ++ α2-A R _ Insulina Glucosa Adiposo marrón U CP 1 Sustratos Insulina Aumento mobilización grasas tejido adiposo blanco Aumento de FFA y oxidación por músculo en ejercicio M C 4-R antagonistas PO MC Señales crónicas (Aferentes) Hipotálamo Núcleo lateral Saciedad Ha mbre Núcleo Anabólica + TS H Hipotálamo Núcleos paraventricular/ventromedial _ R esistina U CP2 LPL + Lipostato _ HSL FFA Adiposo Blanco 7 Cross-Talk de la regulación del apetito ¿Qué péptidos son importantes para suprimir el apetito? Ratones K.O ALTA Baja Leptina LEPTINA Apetito Orexigénico Saciedad Anorexigénico Nucleo Arcuato Hipotálamo NPY POMC AGRP LHA Orexinas MCH endorfinas X α CART PVN OXI CRH, TRH IL-1β MSH MC4 X APETITO INGESTA PESO CORPORAL SNS gasto energético Péptido Leptina* Insulina Obesidad leve (específica cerebro) Serotonina Obesidad 5-HT2C POMC* DIANAS Obesidad MC4* KO DROGAS Obesidad •*Mutaciones en humanos dan Obesidad temprana CRH No efecto CCK No efecto GLP-1 MC-4 mutaciones receptor (5%) Obesidad leve La obesidad humana es poligénica Bombesina POMC mutaciones en procesa miento (pelirojos) No efecto Ahima & Osei. Trends in Mol Med 2001; 7 :205 ¿Qué péptidos son importantes para aumentar el apetito? Ratones K.O Péptido KO Obesidad Péptidos que aumentan la cantidad de comida por toma TG sobrexpresion NPY obesos↓ pérdida de peso, Normal No efecto* AGRP Obesidad MCH ↓pérdida de peso Galanina No efecto Opioides obesidad moderada ?? Orexinas No efecto GABA No efecto • Neuropéptido Y (NPY) • Peptido YY (PYY) • Pancreatic Polypeptide (PP) Todos estos péptidos para que ejerzan su efecto tiene que administrarse directamente en el SNC y no periféricamente como los que disminuyen la cantidad de comida por toma. Lo que indica que los controladores entran en el circuito a diferentes niveles. Ahima & Osei. Trends in Mol Med 2001; 7 :205 Péptidos que afectan a nutrientes específicos Nutriente Aumenta Disminuye Grasa Galanina Opioides CCK, Enterostatina CRH Vasopresina Carbohidratos NPY Insulina CCK Proteína GHRH Glucagon Sodio Angiotensina Consecuencias de la obesidad • OBESIDAD favorece • • • • • Diabetes tipo 2 Hipertension Dislipemia Enf. Cardiovasculares Enf. degenerativas 8 Tipos de diabetes y edad de diagnóstico Diabetes • Diabetes primaria – Tipo I – IDDM / Juvenil – 10%. – Tipo II – NIDDM /Adulto – 80%. – MODY – 5% maturity onset Genética – Diabetes Gestacional Tipo 2 Tipo I MODY • Diabetes secundaria – Destrucción de islotes. MIDD – Infecciosa – rubeola congénita, CMV. – Pancreatitis/tumores/Hemocromato sis. E d i tí Patogénesis de diabetes tipo I Factor genético HLA-DR3/DR4 5 10 15 20 25 30 35 40 45 50 55 60 65 70 75 80 85 90 Edad del diagnóstico Patogenésis de la diabetes tipo II Ambientales? virus..?? Insulitis autoinmune Defecto genético célula ß Secrección anormal Factores Ambientales Obesidad Resistencia a insulina Destrucción cél. ß Déficit relativo insulina Deficiencia insulina severa Agotamiento cel. ß DM tipo I Defectos genéticos en la diabetes tipo2. MODY: Maturity Onset Diabetes of the Young 11% MODY MODYX 14 % Glucokinasa (MODY2) 75% Factores de Transcripción <1% 3% <1% 69% 3% IPF1 NeuroD1 HNF1α HNF4α HNF1β (MODY3) (MODY1) (MODY5) (MODY4) DM tipo II IDDM Glucokinasa (MODY2) • Rara en enfermos diabéticos hiperglicemia incidental en niños • Glucosa en ayuno persistentemente elevada desde el nacimiento (5.5-9 mmol/l) • Poco aumento (< 3mmol/l) en sobrecarga oral de glucosa • No obeso • Frecuentemente asintómatico • TESTAR A LOS PADRES! 9 Dos subtipos de MODY Glucokinasa y factores de transcripción HNF1α (MODY3) • Causa más común de MODY • Puede ser equivocadamente diagnosticada de tipo 1 • Se desarrolla entre 12-30 años • Glucosa en ayunas puede ser normal al principio • Gran aumento (>5mmol/l) en sobrecarga oral de glucosa • Glicemia aumenta con la edad • Glucosuria y normalmente no obesos • Padres y abuelos normalmente diabéticos Factor Transcripción (HNF-1α) 20 16 Glucosa (mmol/l) 12 Glucokinasa 8 . Normal 4 0 0 20 40 60 80 100 Edad (años) Pearson, et al Diferenciación clínica de los tipos de diabetes Independiente Insulina MODY Tipo 2 Tipo I Si No Padres afectados 1 Comienzo < 25 años 1-2 Si +/- Obesidad Acanthosis Si no usual Si + + + - Nigricans Autoanticuerpos ICA, IA2 or GAD No Peptido C Si 0-1 Secrección anormal Factores Ambientales Obesidad Resistencia a insulina Déficit relativo insulina Si (>95%) Si (alto) No claro (0-1 nmol/L) (>1nmo Normal Defecto genético célula ß - (<0.3 nmol/L) Lípidos Patogenésis de la diabetes tipo II +/- + + No Diabetes 20 HDL baja TG altos Norma GENÉTICA DE LA DIABETES TIPO 2 Estudios en gemelos - 90% de concordancia en monozigotos - 15% de concordancia en dizigotos Estudios familiares: Agotamiento cel. ß • 15% de los hijos diabéticos tienen un progenitor diabético Resistencia a la insulina: Defectos en el receptor y post-receptor Aumento producción de glucosa Tejidos periférico (músculo, adiposo Uso insuficiente de la glucosa Aumento glucosa X Hígado • 50% de los hijos son diabéticos con 2 progenitores diabéticos • 15% serán diabéticos si tienen un hermano diabético Páncreas Ninguna correlación con los genes de HLA IDDM Causas de hiperglicemia en diabetes tipo 2 - 25% de los diabéticos tienen un familiar diabético - En la diabetes tipo 2: DM tipo II X Trastorno secrección de insulina 10 Cross- talk del metabolismo de lípidos y carbohidratos Múltiples factores llevan al agotamiento de la función de células β GK Resistina Obesidad Resistencia insulina Glicación de proteínas: Hb1Ac Cél - β “Lipotoxicidad” disfunción (elevación de FFA, TG) ⇑¿compensa? “Ayuno” ⇑OX FFA ⇑ Cetogénes is ⇑ ⇑ ⇓ Glut 4 Unger RH, Orci L. Biochim Biophys Acta. 2002;1585:202212. Nature Medicine 10, 355 - 361 (2004) ⇑ ⇓ ⇑ ⇓ Glut 4 Obesidad Resistencia periférica insulina PPAR PPAR ¿Cómo dar cuenta de esta orquestación en diferentes tejidos ? TF y coactivadores RXR Orquestación de la respuesta multitisular. Papel de los diferentes TF PPAR α, γ, δ en tejidos ⇑ RXR Hiperglicemia CBP SRC RXR PPAR Coactivador: PGC1-α Mediadores HAT Acetilación de histonas dismiuye su carga positiva y abre nucleosomas para que entre RNApol II y aumenta transcripción Programas Transcripcionales tisulares regulados por un coactivador: PPARγ coactivador PGC-1α ⇑Gluconeogenesis ⇑OX FFA ⇑ PEPCK ⇑ ⇑ G-6-Pasa Cetogénes ⇑OXPHOSis GR PPARα ⇑Mito biogen FOXO-1 HNF-4α ⇑ PGC-1α cAMP PPARγ, PPARα, T3R PGC-1α Mitocondria Biogénesis Termogénesis UCP1 Modificación cromatina RXR HAT: histona acetil-transferasa. PPAR Coactivador: PGC1-α Integración a nivel muscular. Acción de PGC1-α Aumento de Fibras Tipo I, Biogénesis Mito Ca2+ ⇑ PGC-1α contracción CREB Ayuno: glucagón, β-adrenérgicos Diabetes tipo 1 Diabetes tipo 2 ⇓ 20% PGC-1α Diabetes tipo 2 11 Diabetes tipo 2 complicaciones ¿Qué es el síndrome metabólico? •Resistencia a la insulina –Síndrome de resistencia a la insulina •Estilo de vida: obesidad – Síndrome metabólico Síndrome metabólico, Resistencia Insulina, y Ateroesclerosis Hiperinsulinemia/hiperproinsuline mia Resistencia insulina Intoleranc ia Glucosa Aumento de triglicérido s Disminución de colesterol HDL LDL aumentada Aterosclerosis Enfermedad cardiovascular Hipertensión Disfunción endotelial Alteració n toleranci a Glucosa • Hipertensión • Resistencia a Insulina + hiperglicemia • Estado Proinflamatorio • Estado Protrombótico Obesidad: El tejido adiposo contribuye a al síndrome metabólico • Adiponectina (⇓ ⇓ en obesos) (Proteína 30kDa) relacionada con el complemento. Secreción estimulada por la insulina. Efectos: – Implicada en la regulación de la distribución de nutrientes – Antiinflamatorio y antiaterogénico • Niveles circulantes ⇓ en obesos • ¿¿Clave de la conexión obesidad/ aterosclerosis?? • Angiotensinógeno (⇑ en obesos) • • Complicaciones Incapacidad Hiperglicemia Retinopatia – ÇTG, Çapo B, ⇑ LDL, ÈHDL • Inhibidor 1 del activador del plasminógeno (PAI-1) (⇑ ⇑ en Historia Natural de la diabetes tipo 2 Genéticos Comienz o diabete s • Dislipidemia aterógenicas Sustrato de la renina en el sistema renina-angiotensina regulador de la tensión arterial. Niveles de angiotensinógeno ⇑ en obesos ¿¿ factor clave en la asociación obesidad/HTA ?? Aumento de PAI-1, TNFα Disminución de Adiponectina MacFarlane S et al. J Clin Endocrinol Metab. 2001;86:713-718. Ambiente • nutrición • obesidad • ejercicio Componentes del síndrome metabólico Resistencia Insulina Hiperinsulinemia Nefropatía Neuropatía Hipertensión Aumento HDL-C Ateroesclerosis Aumento TG Muerte Ceguera Fracaso Renal Enf. coronaria Amputación miembro obesos) Inhibe la transformación de plasminógeno en plasmina, capaz de degradar los coágulos de fibrina y por tanto promueve la coagulación. Niveles ⇑ ⇑ en obesos ¿determinante de la alta incidencia de tromboembolias? Tratamiento de la obesidad • Debería ser realizado por un equipo multidisciplinario compuesto por médicos, nutricionistas, kinesiólogos y psicólogos. 1.-Dieta. 2.-Modificaciones conductales. 3.-Ejercicio. 4.-Fármacos. 12 Efedrina y cafeína Farmacología Obesidad • Anorexigenos catecolamínicos y serotoninérgico s. • Efedrinacafeína. • Fluoxetina. • Beta 3 adrenérgicos. • Sibutramina Sibutramina • Inhibe la recaptación de serotonina y epinefrina, aumentando la saciedad e incrementando el gasto energético por aumento de la termogénesis. O O Orlistat • Inhibe la lipasa pancreática que disminuye la hidrólisis de triglicéridos en el intestino y la absorción de grasas y vitaminas liposolubles. • • • • Aumentan gasto energético Aumentan ritmo cardíaco 25 a 40 % de pérdida de peso OJO HIPERTENSION Fluoxetina (Prozac) • Inhibe la recaptación de serotonina en sinaptosomas y en plaquetas • Anti-depresivo y anti-bulímico Tratatamiento de Diabetes tipo 2 • Anti-diabéticos orales 1. Sulfonilureas y meglitinides – aumentan niveles de insulina 2. Metformin – inhibe gluconeogenesis y glicogenolisis hepática y mejoran sensibilidad a insulina 3. Thiazolidinedionas – ligandos de PPARγ suprimen las expresión de determinados genes y bajan triglicéridos 13 Prevención de diabetes tipo 2 por Troglitazona Fac c i ón de pac i ent es c on di abet es 0.6 0.5 0.4 0.3 Placebo 0.2 Troglitazona 0.1 0.0 0 10 P=0.005 TRIPOD=Troglitazone in the Prevention of Diabetes. Buchanan TA. ADA. 2001. 20 30 40 Meses de ensayo 50 60 Tratatamiento de Diabetes tipo 2 • Anti-diabéticos orales 1. Sulfonilureas y meglitinides – aumentan niveles de insulina 2. Metformin – inhibe gluconeogenesis y glicogenolisis hepática y mejoran sensibilidad a insulina 3. Thiazolidinedionas – ligandos de PPARγ suprimen las expresión de determinados genes y bajan triglicéridos 4. Acarbosa – reducción absorción de carbohidratos. Glucobay 5 Terapia combinada es lo mejor Muchas preguntas por resolver??? ¿Alguna pregunta? ¿Alguna contradicción? Si la Troglitazona, es un ligando de PPARγ, y activa la captación de LDL Oxidada ( ⇑ CD36) ¿no aumentaríamos la incidencia de ateroesclerosis en diabéticos al tratarlos con esta droga? 14