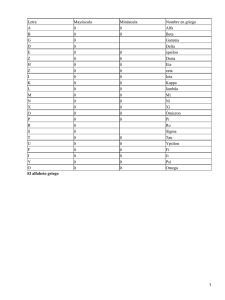
Traducido del inglés al español - www.onlinedoctranslator.com Hindawi Publishing Corporation Revista internacional sobre la enfermedad de Alzheimer Volumen 2012, ID del artículo 731526,13páginas doi:10.1155/2012/731526 Artículo de revisión Estructura y patología de la proteína Tau en la enfermedad de Alzheimer Michala Kolarova,1, 2Francisco García-Sierra,3Ales Bartos,1, 4 Jan Ricny,1y Daniela Ripova1 1Laboratorio de Bioquímica y Fisiopatología Cerebral y Centro de Enfermedad de Alzheimer, Centro Psiquiátrico de Praga, Ústavnı́ 91, 181 03 Praga 8, República Checa 2Tercera Facultad de Medicina, Universidad Carolina de Praga, Ruská 87, 100 00 Praga 10, República Checa Departamento de Biología Celular, Centro de Investigación y Estudios Avanzados, Instituto Politécnico Nacional, 3 Avenida Instituto Politécnico Nacional 2508, 07360 Ciudad de México, DF, México Departamento de Neurología, Tercera Facultad de Medicina, Hospital Universitario Královské Vinohrady, Universidad Carolina de Praga, 4 Šrobárova 50, 100 34 Praga 10, República Checa La correspondencia deberá dirigirse a Michala Kolarova,[email protected] Recibido el 19 de enero de 2012; revisado el 28 de marzo de 2012; aceptado el 29 de marzo de 2012 Editor académico: David Blum Copyright © 2012 Michala Kolarova et al. Este es un artículo de acceso abierto distribuido bajo la Licencia Creative Commons Attribution, que permite el uso, la distribución y la reproducción sin restricciones en cualquier medio, siempre que se cite correctamente el trabajo original. La enfermedad de Alzheimer (EA) es el tipo más común de demencia. En relación con la tendencia mundial de prolongar la vida humana y el creciente número de ancianos en la población, la EA se convierte en uno de los problemas socioeconómicos y de salud más graves de la actualidad. La proteína tau promueve el ensamblaje y estabiliza los microtúbulos, lo que contribuye al funcionamiento adecuado de la neurona. Las alteraciones en la cantidad o la estructura de la proteína tau pueden afectar su papel como estabilizador de los microtúbulos, así como algunos de los procesos en los que está implicada. Los mecanismos moleculares que rigen la agregación de tau están representados principalmente por varias modificaciones postraduccionales que alteran su estructura y estado conformacional. Por lo tanto, la fosforilación anormal y el truncamiento de la proteína tau han ganado atención como mecanismos clave que convierten a la proteína tau en una entidad patológica. Se han documentado evidencias sobre la importancia clinicopatológica de la tau fosforilada y truncada durante la progresión de la EA, así como su capacidad para ejercer citotoxicidad cuando se expresa en modelos celulares y animales. Este artículo describe la estructura y función normales de la proteína tau y sus principales alteraciones durante su agregación patológica en la EA. 1. Introducción La enfermedad de Alzheimer (EA) es el tipo más común de demencia que se caracteriza por el deterioro de la memoria y la alteración de diversas capacidades cognitivas. En asociación con la tendencia global de prolongar la vida humana y el creciente número de ancianos en la población humana, la EA se convierte en uno de los problemas socioeconómicos y de salud más importantes de la actualidad. La EA y las tauopatías relacionadas se caracterizan histopatológicamente por una neurodegeneración lenta y progresiva, que se asocia principalmente con la acumulación ensamblaje de la tubulina en microtúbulos, uno de los principales componentes del citoesqueleto neuronal que define la morfología normal y proporciona soporte estructural a las neuronas [2]. La unión de la tubulina a la tau está regulada por su estado de fosforilación, que normalmente está regulado por la acción coordinada de las quinasas y fosfatasas sobre la molécula de tau [3,4]. En condiciones patológicas, como el caso de la EA, no solo la fosforilación anormal de la proteína tau disminuye su capacidad de unión a la tubulina, lo que conduce a la desorganización de los microtúbulos, sino que también esta proteína se autopolimeriza y se agrega en forma de NFT [5,6]. intracelular de proteína tau que conduce a los llamados ovillos neurofibrilares (NFT) y otras inclusiones que contienen tau modificada [1]. La proteína tau se descubrió a mediados de la 2. El gen Tau década de 1970 del siglo XX al estudiar los factores necesarios para El gen tau humano se encuentra a más de 100 kb en el brazo largo la formación de microtúbulos. La proteína tau promueve del cromosoma 17 en la posición de banda 17q21 y contiene 16 Revista internacional sobre la enfermedad de Alzheimer exones. El exón 1 es parte del promotor y se transcribe pero no se 50 N-terminal traduce. Los exones 1, 4, 5, 7, 9, 11, 12 y 13 son exones constitutivos. N1 Los exones 2, 3 y 10 se empalman alternativamente y se manifiestan 100 N2 en el cerebro adulto. El exón 2 puede aparecer solo, pero el exón 3 150 nunca aparece independientemente del exón 2 [7]. En el sistema nervioso central, el empalme alternativo de los exones 2, 3 y 10 da ]. R1 La proteína Tau pertenece a un grupo de proteínas denominadas 250 P2 expresan de manera diferencial durante el desarrollo del cerebro [7 3. Estructura y función de la proteína Tau 200 P1 como resultado la aparición de seis isoformas de tau que se 306VQIVYK 311 275VQIIN K280 R2 R3 350 400 R4 Proteínas Asociadas a Microtúbulos (MAP), que en común son resistentes 300 C-terminal al calor y se ven limitadamente afectadas por el tratamiento ácido sin perder su función [8]. Esta propiedad observada en la proteína tau se debe a un contenido muy bajo de estructura secundaria. De hecho, varios estudios biofísicos revelaron que la proteína tau es una proteína prototípica “desplegada de forma nativa” [9–11]. Dado que las proteínas desordenadas tienden a ser muy flexibles y tienen conformaciones variables, hasta ahora no han sido susceptibles de análisis estructural mediante cristalografía. Por lo tanto, la espectroscopia de resonancia magnética nuclear es el único método plausible que permite una descripción de sus conformaciones y dinámicas con alta resolución [12]. Ahora es posible obtener la asignación completa de la estructura principal de la proteína tau de 441 residuos (la isoforma de tau más larga X = AA básica (+) X = AA polar sin carga (hidrofílico) X = AA no polar (hidrofóbico) X = AA ácido (−) Cifra1: secuencia de aminoácidos de la isoforma tau más larga (441 aminoácidos). N1 y N2: secuencias polipeptídicas codificadas por los exones 2 y 3; P1 y P2: regiones ricas en prolina; R1–R4: dominios de unión a microtúbulos codificados por los exones 9–12;275VQIINK280y 306VQIVYK311: secuencias conβ-estructura (modificada por [13]). encontrada en el sistema nervioso central humano;Figura 1). Esto permite investigar la estructura y la dinámica de la proteína soluble de longitud completa y determinar los residuos involucrados en la se expresan de forma diferencial durante el desarrollo del cerebro. interacción entre tau y microtúbulos con una resolución de residuo único Por ejemplo, solo una isoforma de tau, caracterizada por 3R y sin [13]. insertos N-terminales, está presente durante las etapas fetales, Seis isoformas de la proteína tau difieren según el contenido de tres (3R) o cuatro (4R) dominios de unión a tubulina (repeticiones, R) de 31 o 32 aminoácidos en la parte C-terminal de la proteína tau y mientras que las isoformas con uno o dos insertos N-terminales y 3o 4R se expresan durante la edad adulta [7]. La proteína tau está presente en mayor medida en los axones de las una (1N), dos (2N) o ninguna inserción de 29 aminoácidos cada una neuronas, pero también se encuentra en los oligodendrocitos. Otra en la porción N-terminal de la molécula. Estas isoformas, que varían proteína de unión a microtúbulos, denominada MAP2, se encuentra en el en tamaño de 352 a 441 residuos de aminoácidos, están compartimento somatodendrítico de las neuronas, mientras que la MAP4 relacionadas con la presencia o ausencia de secuencias codificadas es mucho más ubicua [17]. por los exones 2, 3 o 10. La inclusión de la región de repetición imperfecta que codifica el exón 10 conduce a la expresión de tau que contiene cuatro repeticiones de unión a microtúbulos (MTBR) (tau 4R: 0N4R, 1N4R, 2N4R), mientras que la exclusión del exón 10 da como resultado productos de empalme que expresan tau con tres MTBR (tau 3R: 0N3R, 1N3R, 2N3R) [7,14].Estas seis isoformas también se denominanτ3 litros, τ3S,τ3,τ4 litros,τ4S, yτ4 [15]. El análisis de la secuencia primaria demuestra que la proteína tau consiste en una porción ácida de la mitad del extremo N-terminal seguida de una región rica en prolina y la cola C-terminal, que es la parte básica de la proteína. Las secuencias polipeptídicas codificadas por los exones 2 y 3 añaden acidez a la proteína tau, mientras que el exón 10 codifica una secuencia cargada positivamente que contribuye al carácter básico de la proteína tau. Por otro lado, la región N-terminal tiene un punto isoeléctrico (pI) de 3,8 seguida del dominio rico en prolina, que tiene un pI de 11,4. La región Cterminal también está cargada positivamente con un pI de 10,8. En otras palabras, la proteína tau es más bien un dipolo con dos dominios de carga opuesta, que pueden ser modulados por modificaciones postraduccionales [16]. Debido a que cada una de estas isoformas tiene funciones fisiológicas específicas, 3.1. El dominio de proyección y su interacción con otras moléculas. Las dos secuencias de 29 aminoácidos codificadas por los exones 2 y 3 dan longitudes diferentes a la parte N-terminal de la proteína tau. La parte N-terminal se conoce como dominio de proyección, ya que se proyecta desde la superficie de los microtúbulos donde puede interactuar con otros elementos del citoesqueleto y la membrana plasmática neuronal. De hecho, los dominios de proyección de la proteína tau determinan el espaciamiento entre los microtúbulos en el axón y pueden aumentar el diámetro axonal [7,18]. Las neuronas periféricas suelen proyectar un axón muy largo y de gran diámetro. Este tipo de neuronas contiene una secuencia tau N-terminal adicional codificada por el exón 4A y, por lo tanto, genera una isoforma tau específica llamada “tau grande” [7,18–20]. En cuanto a las interacciones con otros componentes del citoesqueleto, la proteína tau se une a los filamentos de espectrina y actina, lo que puede permitir que los microtúbulos estabilizados por tau se interconecten con neurofilamentos que restringen la flexibilidad de las redes de microtúbulos. Otra molécula que interactúa con la proteína tau es una isomerasa cis/trans de peptidil-prolil Pin 1. Isomeriza solo motivos de fosfoserina/treonina-prolina y se une a 9730, 2012, 1, Descargado de https://onlinelibrary.wiley.com/doi/10.1155/2012/731526 por Cochrane Perú, Wiley Online Library el [17/02/2025]. Consulte los Términos y condiciones (https://onlinelibrary.wiley.com/terms-and-conditions) en Wiley Online Library para conocer las reglas de uso; los artículos de acceso abierto se rigen por la Licencia Creative Commons aplicable. 2 Fosfotau La proteína tau después de su fosforilación en Thr231residuo. La isomerización induce cambios conformacionales que hacen que la Vesícula proteína tau sea accesible para la fosfatasa proteica (PP) 2A, lo que a su vez conduce a la desfosforilación de la proteína tau. La proteína Pin 1 regula las funciones de la proteína tau y la APP y es importante Kinesina para la protección contra la degeneración que ocurre durante el proceso de envejecimiento. La actividad de Pin 1 disminuye por − oxidación en la enfermedad de Alzheimer [21]. Además, la proteína tau a través de su dominio de proyección N-terminal puede interactuar con elementos membranosos intracelulares como las Tauro proteína Microtúbulo mitocondrias [22] y la membrana plasmática neuronal [23]. En el citosol de las neuronas, los depósitos de proteína tau en forma + fosforilada o desfosforilada se mantienen en equilibrio mediante acciones coordinadas de quinasas y fosfatasas, respectivamente. Varios estudios en líneas celulares revelaron que la proteína tau unida a la membrana plasmática se desfosforila [24,25]. La proteína tau se une a través de su región rica en prolina a los dominios de homología Src 3 (SH3) de varias proteínas, incluida Fyn, una tirosina Cifra2: Función normal de la proteína tau. La proteína tau estabiliza los microtúbulos a través de cuatro dominios de unión a la tubulina (recuadros azules) en el caso de la isoforma más larga. La unión de la proteína tau a los microtúbulos se mantiene en equilibrio mediante acciones coordinadas de quinasas y fosfatasas. La fosforilación de tau (bolas rosas) regula su actividad quinasa de la familia Src. La asociación de tau y Fyn depende del para unirse a los microtúbulos y puede afectar el transporte axonal. La estado de fosforilación de tau, porque la PHFtau insoluble aislada proteína tau puede inhibir el transporte dirigido por el extremo positivo de las del cerebro con enfermedad de Alzheimer no se une al dominio SH3 vesículas a lo largo de los microtúbulos por la quinesina. de Fyn [26]. Recientemente se ha demostrado que Fyn desempeña un papel en el tráfico de proteínas [27]. Por ejemplo, Fyn puede aumentar la expresión superficial de la proteína precursora amiloide La proteína aumenta la tasa de polimerización de los microtúbulos y (APP) a través de la fosforilación de tirosina [28]. El tráfico de la al mismo tiempo inhibe su tasa de despolimerización [37]. Las proteína tau a la membrana plasmática es un proceso bidireccional, repeticiones de 18 aminoácidos se unen a los microtúbulos a través ya que el aumento de la fosforilación de tau inducido por la de una matriz flexible de sitios débiles distribuidos. La forma adulta inhibición de PP2A reduce significativamente la proporción de tau de tau promueve el ensamblaje de microtúbulos de manera más asociada a la membrana. La relocalización activa de tau en activa que las formas fetales [14,38]. Curiosamente, la parte más respuesta a cambios en la fosforilación sugiere un posible papel de potente que induce la polimerización de los microtúbulos es la esta proteína en las vías de señalización intracelular [29,30]. interregión entre las repeticiones 1 y 2 (interregión R1-R2) y, más Recientemente se ha demostrado que la tau se une a la Fyn en las específicamente, el péptido275KVQIINKK280dentro de esta secuencia espinas dendríticas, y esta interacción regula la señalización del [7,39]. Esta interregión R1-R2 es exclusiva de la proteína tau 4R, receptor de ácido N-metil-D-aspártico (NMDA) [31]. La tau patológica específica de adultos y responsable de la diferencia en las afinidades puede participar en la localización de la quinasa Fyn en el de unión entre la proteína tau 3R y la proteína tau 4R [7,35]. compartimento postsináptico, donde fosforila las subunidades Evidencias recientes apoyan un papel del MTBR en la modulación del NMDAR, lo que provoca un aumento de la entrada de Ca2+ estado de fosforilación de la proteína tau. Se ha demostrado una conductancia y conduce a excitotoxicidad [32].En vivoSe ha unión directa y competitiva entre esta región (residuos 224-236 demostrado que la tau interactúa directamente con los receptores según la numeración de la isoforma más larga) y el microtúbulo por ionotrópicos de glutamato [33]. En los oligodendrocitos, la un lado y la misma región con la PP2A por otro lado [40].Como asociación de tau con Fyn regula el crecimiento del proceso consecuencia, los microtúbulos podrían inhibir la actividad de PP2A citoplasmático [34]. La interacción alterada de la quinasa Fyn y la al competir por la unión a tau en el MTBR. proteína tau hiperfosforilada conduce a la hipomielinización y la desmielinización evolutiva de los axones [34]. Todas estas evidencias Los microtúbulos contribuyen a diversos procesos celulares indican que el estado fosforilado de la proteína tau no sólo afecta la como la morfogénesis celular, la división celular y el tráfico estabilidad de los microtúbulos sino que también produce intracelular [41,42]. En las células, los microtúbulos pueden cambiar alteraciones en la plasticidad neuronal. su longitud a través de la inestabilidad dinámica [43]. Pueden servir como pistas para el transporte de orgánulos mediado por proteínas 3.2. El dominio asociado a los microtúbulos.La proteína Tau se une a los microtúbulos a través de algunos dominios repetidos (R1–R4) (codificados por los exones 9–12) ubicados en el extremo C de la molécula (Figura 2) [35]. Cada repetición consta de tramos de 18 residuos altamente conservados que se repiten de forma imperfecta tres veces en la proteína tau fetal y cuatro veces en la forma específica del adulto [35]. Las repeticiones están separadas entre sí por regiones espaciadoras de 13 o 14 residuos. La función principal de tau, antes mencionada como promotor de la polimerización de la tubulina, depende principalmente del MTBR [35,36]. Se ha informado quein vitrotau motoras dependientes de microtúbulos, como la kinesina motora dirigida al extremo positivo y sus parientes, o la dineína motora dirigida al extremo negativo [44,45]. Estos motores pueden transportar sus cargas, por ejemplo, mitocondrias [46,47], lisosomas [48], peroxisomas [49], y vesículas endocíticas o exocitóticas [50] hacia la periferia celular o de regreso hacia el centro organizador de microtúbulos (MTOC), respectivamente. Se ha demostrado que la proteína tau afecta el transporte axonal [17,51,52]. La proteína tau altera el tráfico intracelular debido a su fuerte unión a los microtúbulos y probablemente separa las cargas de la kinesina. Sin embargo, la proteína tau no tiene influencia en la velocidad 9730, 2012, 1, Descargado de https://onlinelibrary.wiley.com/doi/10.1155/2012/731526 por Cochrane Perú, Wiley Online Library el [17/02/2025]. Consulte los Términos y condiciones (https://onlinelibrary.wiley.com/terms-and-conditions) en Wiley Online Library para conocer las reglas de uso; los artículos de acceso abierto se rigen por la Licencia Creative Commons aplicable. 3 Revista internacional sobre la enfermedad de Alzheimer Revista internacional sobre la enfermedad de Alzheimer de kinesina con cargas [52]. Esto implica que la fosforilación de tau debería jugar un papel importante porque esta modificación regula la afinidad de tau por los microtúbulos. hace que la proteína tau pierda su actividad biológica. En cuanto a la posible propensión de la proteína tau a ser fosforilada, se informó que la variante más larga de la proteína tau (441 aminoácidos) contiene alrededor de 80 sitios potenciales de fosforilación de serina 4. Patología de Tau En la enfermedad de Alzheimer, el papel normal de la proteína tau es o treonina [7]. La mayoría de estos sitios potenciales se encuentran en las proximidades del MTBR en la región rica en prolina y en el extremo C-terminal de la molécula de proteína tau [16,74] con ineficaz para mantener el citoesqueleto bien organizado en el proceso excepción de Ser262, Ser293, Ser324, y Ser356 axonal porque esta proteína pierde su capacidad de unirse a los (motivo KXGS) en los dominios R1, R2, R3 y R4 [75,76]. En la enfermedad, la fosforilación anormal de tau podría ser, aunque no mutuamente excluyentes, el resultado de la regulación positiva de la(s) quinasa(s) tau o de la regulación negativa de la(s) fosfatasa(s) tau [62,74]. Se han evaluado varias de estas enzimas y las quinasas que se cree que desempeñan el papel más importante en la fosforilación de tau en el cerebro incluyen GSK-3β, quinasa dependiente de ciclina 5 (cdk5), proteína quinasa dependiente de AMPc (PKA) y quinasa II dependiente de calcio/calmodulina (CaMK-II) [77] GSK-3βPuede desempeñar un papel importante en la regulación de la fosforilación de tau tanto en condiciones fisiológicas como patológicas. GSK-3β ¿Puede fosforilar tau en Ser?199, El231, Ser396, Ser400, Ser404, y Ser 413en vivoy in vitro(numerados según la isoforma tau más larga), residuos que en su mayoría están fosforilados en PHF-tau [78]. La fosforilación antes mencionada en Thr231provoca un cambio conformacional local que permite el acceso de GSK-3βu otras quinasas para fosforilar aún más la proteína tau. Por otro lado, un efecto complementario y opuesto es el de PP1, PP2A, PP2B y PP2C, que pueden desfosforilar la proteína tau.in vitro[79Se ha descubierto que la actividad de PP2A está reducida en áreas seleccionadas del cerebro de pacientes con EA [4]. En general, la fosfoproteína tau está al menos tres a cuatro veces más hiperfosforilada en el cerebro de pacientes con EA que en el cerebro de individuos mayores sin demencia [80]. microtúbulos. Este comportamiento anormal es promovido por cambios conformacionales y plegamientos incorrectos en la estructura normal de tau [53–55] que conduce a su agregación aberrante en estructuras fibrilares dentro de las neuronas de individuos dementes [56–58]. Por lo tanto, la mayoría de los grupos alterados de proteína tau en la enfermedad se redistribuyen y se agregan tanto en el compartimento somatodendrítico como en los procesos aislados de las neuronas afectadas. Las alteraciones en la cantidad o la estructura de la proteína tau pueden afectar la estabilización de los microtúbulos y otros procesos relacionados con esta proteína [59,60]. Por ejemplo, la sobreexpresión o la mala localización que aumentan la concentración intracelular de tau pueden inhibir el transporte dirigido al extremo positivo de las vesículas a lo largo de los microtúbulos por la kinesina, de modo que el transporte dirigido al extremo negativo por la dineína se vuelve más dominante [17]. La inhibición del transporte al extremo positivo del microtúbulo ralentiza la exocitosis y afecta la distribución de las mitocondrias, que se agrupan cerca del MTOC. La ausencia de mitocondrias y retículo endoplásmico en las regiones periféricas de los axones podría producir una disminución del metabolismo de la glucosa y los lípidos y de la síntesis de ATP y una pérdida de Ca2+homeostasis [61] que conduce a un proceso de degeneración distal conocido como “muerte retrógrada” de los axones [ 62]. Además, la proteína tau fosforilada tiene afinidad por la kinesina y, por lo tanto, se transporta a los sitios distales del neuropilo. Esto puede explicar la observación de que la patología de los ovillos en la A nivel celular, la fosforilación anormal de tau introduce enfermedad de Alzheimer parece iniciarse distalmente y luego se alteraciones en varios procesos que están regulados directamente propaga de manera retrógrada al pericarion. Este proceso puede ser un por la organización adecuada de la red de microtúbulos. En una mecanismo para proteger la estabilidad de los microtúbulos al neurona madura normal, la tubulina está presente en un exceso de transportar la tau hiperfosforilada más rápidamente a otras ubicaciones más de diez veces la de tau, y por lo tanto, prácticamente toda la celulares donde la tau puede formar agregados [51]. proteína tau está limitada a microtúbulos en la célula [81,82]. En las neuronas afectadas por la EA, la tau citosólica anormalmente Los mecanismos por los cuales la proteína tau se convierte en una entidad fosforilada (P-tau de la EA) no se une a la tubulina ni promueve el no funcional son motivo de debate. Se propone que las modificaciones ensamblaje de microtúbulos [83–85]. En cambio, esta proteína postraduccionales anormales son la causa principal de este fracaso.63,64]. En inhibe el ensamblaje y altera la organización de los microtúbulos.83] . Además, se informó que la proteína tau anormalmente fosforilada separa la tau normal de los microtúbulos hacia la fase citosólica [83], hasta el 40% de la tau hiperfosforilada anormalmente en el cerebro de pacientes con EA está presente en el citosol y no está polimerizada en filamentos helicoidales apareados (PHF) ni formando NFT [80]. La AD P-tau también elimina las otras dos MAP neuronales principales, MAP1 y MAP2, de la red de microtúbulos [86 ]. Esta característica tóxica de la AD P-tau parece deberse únicamente a su estado de fosforilación anormal porque la desfosforilación de la AD Ptau rescata a esta proteína para que realice sus tareas normales [84]. Mediante el uso de anticuerpos monoclonales dependientes de la fosforilación contra tau y espectrometría de masas, se informó que al menos 39 sitios fosforilados en la molécula de tau están asociados con PHF nativo aislado del cerebro de pacientes con EA [ 87]. este sentido, la fosforilación anormal (hiperfosforilación), la acetilación, la glicación, la ubiquitinación, la nitración, la escisión proteolítica (truncamiento), los cambios conformacionales y algunas otras modificaciones [53, 65–73Se ha propuesto que la fosforilación, acetilación y truncamiento anormales de la proteína tau causan la pérdida de la función normal y la aparición de características patológicas de la proteína tau. En las próximas secciones, centraremos nuestro interés en describir la evidencia que respalda la fosforilación, acetilación y truncamiento anormales de la proteína tau como cambios importantes durante el procesamiento patológico de la proteína tau en la enfermedad de Alzheimer. 4.1. La hiperfosforilación de la proteína Tau.La fosforilación de tau regula su actividad para unirse a los microtúbulos y estimular su ensamblaje como se describió anteriormente. Se requiere un nivel normal de fosforilación para la función óptima de tau, mientras que el estado hiperfosforilado 9730, 2012, 1, Descargado de https://onlinelibrary.wiley.com/doi/10.1155/2012/731526 por Cochrane Perú, Wiley Online Library el [17/02/2025]. Consulte los Términos y condiciones (https://onlinelibrary.wiley.com/terms-and-conditions) en Wiley Online Library para conocer las reglas de uso; los artículos de acceso abierto se rigen por la Licencia Creative Commons aplicable. 4 En cuanto a laen el lugarAgregación de tau hiperfosforilada, se ha generado una gran cantidad de evidencia a lo largo de los años para identificar la tau fosforilada anormalmente como el componente principal de las características neuropatológicas distintivas que definen la EA [6,15,65,88–90]. Se ha observado que la tau hiperfosforilada es el componente principal de los PHF y filamentos rectos (SF), NFT, hilos de neuropilo (NT) y neuritas distróficas asociadas a placa en el cerebro de casos de EA [81,91]. La densidad de NFT distribuida a lo largo del hipocampo, la corteza entorinal y el neocórtex se ha correlacionado con el grado de demencia en este trastorno [92]. Además, la acumulación más temprana de tau en el hipocampo de pacientes con EA, antes de la formación de NFT, se ha considerado como un material granular difuso inmunorreactivo a los anticuerpos tau dependientes de la fosforilación [93–95]. Sin embargo, en la formación anormal de PHF, las moléculas de tau pueden seguir diferentes alteraciones a partir de las cuales la fosforilación anormal (aunque esto puede no ser esencial) provoca un plegamiento incorrecto y cambios conformacionales que fortalecen su agregación anormal [79,96]. Estudios recientes demostraron que la hiperfosforilación de tau La hiperfosforilación puede hacer que este residuo esté disponible para una acetilación posterior, lo que perjudicaría aún más la unión a los microtúbulos y/o promovería la agregación de tau, además de impulsar alteraciones patológicas de tau. Aunque la acetilación de proteínas se ha estudiado ampliamente en el contexto de las histonas y la transcripción genética, los enfoques proteómicos han identificado proteínas acetiladas en el citoplasma y otros orgánulos [ 103]. Un estudio reciente sugiere que la acetilación de Lys280puede ser un paso intermedio en la formación de enredos [102]. Lis acetilada280se asoció principalmente con ovillos neurofibrilares intracelulares en comparación con preovillos o ovillos fantasmas extracelulares en todas las etapas de Braak [73,102]. Lis acetilada280 También se colocaliza con epítopos antitau específicos de los extremos N y C. Esto indica que está presente en los ovillos neurofibrilares antes del truncamiento posterior de la proteína tau [ 102]. Las enzimas que añaden un grupo acetilo a la proteína se denominan histona acetiltransferasa (HAT) o lisina acetiltransferasa. De las cuatro clases principales de HAT, p300/CBP (proteína de 300 kDa y proteína de unión a CREB) y pCAF (factor asociado a p300 y CBP) están presentes exclusivamente en los metazoos [104]. Las enzimas que ocurre antes de su escisión [97,98] y que la escisión de tau tiene eliminan un grupo acetilo de la proteína se denominan histona lugar antes de la formación de NFT [99]. En unin vitroModelo de desacetilasa (HDAC) o lisina desacetilasa. Existen tres clases de HDAC. Las apoptosis neuronal inducida por etanol, la hiperfosforilación de tau actividades de las HDAC de las clases I y II (HADC1–11) dependen del zinc ocurre antes de la escisión de tau [98,100]. En conjunto, estos como cofactor; las actividades de las HDAC de clase III (sirtuinas) resultados pueden indicar que la fosforilación anormal es un evento dependen de los niveles relativos de NAD+ y NADH [105,106]. De los siete clave que desencadena la agregación patológica de tau en la EA. miembros de las sirtuinas de mamíferos (SIRT1–7), SIRT1 es la más estudiada y está fuertemente implicada en enfermedades relacionadas con el envejecimiento, incluida la EA [107]. Los niveles de SIRT1 se 4.2. La acetilación de la proteína Tau.El mecanismo que lleva a la proteína reducen en los cerebros con EA y la reducción se correlaciona con la tau soluble normal a hiperfosforilarse y desprenderse de los acumulación de agregados de tau hiperfosforilados [108]. Se descubrió microtúbulos para formar inclusiones de tau sigue siendo desconocido y que SIRT1 reduce AβGeneración mediante la activación de la las modificaciones postraduccionales distintas de la fosforilación podrían transcripción de un gen que codificaalfa-secretasa [109]. La deficiencia de regular la función y la agregación de la proteína tau. Cabe destacar que SIRT1 también podría exacerbar la acumulación de Aβ, lo que podría la acetilación reversible de la lisina ha surgido como una posible aumentar aún más la acetilación y la fosforilación de tau. Dado que una modificación reguladora implicada en la EA y otros trastornos disminución de la actividad de SIRT1 puede tener efectos claramente neurodegenerativos. Estudios recientes demuestran que la acetilación de nocivos para la salud neuronal, las estrategias terapéuticas destinadas a la proteína tau es una modificación postraduccional que puede regular la aumentar la actividad de las sirtuinas en el cerebro con enfermedad de función normal de la proteína tau [73,101,102]. Dado que la acetilación Alzheimer justifican una mayor investigación. neutraliza las cargas en el dominio de unión a los microtúbulos, la acetilación aberrante podría interferir con la unión de tau a los microtúbulos, lo que lleva a una disfunción de tau, y sugiere un papel en la agregación patológica de tau en la EA y tauopatías relacionadas [73]. Aumento de la acetilación de tau en Lys280Podría perjudicar las interacciones de tau con los microtúbulos y proporcionar mayores reservas de tau citosólica disponible para la agregación patológica de PHF [39,101]. En consonancia con esto, Lys280, ubicada en la región interrepeticiones (275VQIINKK280), se identificó previamente como uno de los tres residuos de lisina más críticos para modular las interacciones taumicrotúbulos [39]. La acetilación de agregados de tau se asoció con inclusiones de tau hiperfosforiladas y ThS-positivas tanto en modelos de ratones Tg como en tauopatías humanas [101]. Esto implica que la regulación negativa de la función de tau podría ocurrir a través de eventos de fosforilación y acetilación solos o en combinación. La molécula de proteína tau contiene muchos sitios de fosforilación, como se mencionó anteriormente, y la mayoría de ellos se encuentran en 4.3. La agregación de la proteína Tau in vitro.La molécula de tau tiene largas extensiones de regiones cargadas positiva y negativamente que no son propicias para la asociación hidrofóbica intermolecular [81,110]. Elβ-La estructura en tau monomérica se concentra solo en R2 (exón 10) y R3 (exón 11), que pueden autoensamblarse por sí solos en filamentos [111] y se combina con heparina como inductor artificial [112]. Evidenciain vitroHa revelado que la autoagregación de tau en filamentos se ve inhibida por la presencia de extremos N y C intactos, que se encuentran sobre el MTBR y evitan la interacción entre estos dominios pegajosos [15]. La fosforilación anormal de las regiones flanqueantes N-terminal y C-terminal puede inducir una conformación estructural relajada en la molécula de tau que desenganche ambos extremos de la región MTBR. Esta situación permite la autointeracción entre estos dominios pegajosos en la formación de PHF/SF (Figura 3) [15]. regiones que flanquean la repetición de unión a microtúbulos [74], en el que Lys280se encuentra. Por lo tanto, tau Otras modificaciones, como la desamidación, podrían facilitar la polimerización de la proteína tau. Curiosamente, varias 9730, 2012, 1, Descargado de https://onlinelibrary.wiley.com/doi/10.1155/2012/731526 por Cochrane Perú, Wiley Online Library el [17/02/2025]. Consulte los Términos y condiciones (https://onlinelibrary.wiley.com/terms-and-conditions) en Wiley Online Library para conocer las reglas de uso; los artículos de acceso abierto se rigen por la Licencia Creative Commons aplicable. 5 Revista internacional sobre la enfermedad de Alzheimer Revista internacional sobre la enfermedad de Alzheimer N2 P1 N1 N-terminal PPPPPP N1 N2 275VQIINK280 P2 R1 R2 306VQIVYK311 N-terminal R3 P1 P2 R1R2R3 R4 306VQIVYK311 R4 ∼10 P C-terminal 275VQIINK280 PAG PAG PAG C-terminal GSK-3β CDK5 PAG PAG PAG acampar PPPPPPP Formación de filamentos Cifra3: Fosforilación de la proteína tau. La proteína tau se autoensambla principalmente a través de los dominios de unión a microtúbulos/repetición R3 en las proteínas tau 3R y a través de R3 y R2 en las proteínas tau 4R (R2 (275VQIINK280) y R3 (306VQIVYK311) tenerβ-estructura). Las regiones N-terminal y C-terminal de las repeticiones son inhibidoras. La hiperfosforilación de tau neutraliza estos dominios inhibidores básicos, lo que permite la interacción tau-tau (sitios de fosforilación indicados por Ps violeta) (modificado por [15]). Años más tarde, se demostró que la desamidación ocurre en tau obtenida de PHF [113]. Debido a que se necesita una alta concentración de proteína tau para polimerizar [114], algunos sugieren que otros compuestos, que actúan como cofactores, podrían ser necesarios para facilitar el autoensamblaje de la proteína tau [115–117]. Agregados fibrilares solubles y de alto orden como los NFT [124 –126]. En ratones transgénicos que sobreexpresan tau, la mayoría de las alteraciones cognitivas observadas surgieron en etapas de aparición profunda de agregados multiméricos de tau y antes de la formación de NFT [126]. Independientemente del estado de fosforilación de la proteína tau, se encontró que los sulfoglicosaminoglicanos (sGAG), una clase de moléculas polianiónicas, facilitan la polimerización de la proteína tau.in vitro[115,116]. Además, estos sGAG se encontraron junto con tau en NFT, cuando se analizó la patología tau-neurofibrilar en el cerebro de casos de EA [115,116].In vitroLos paradigmas de polimerización de tau también han utilizado ácido araquidónico como inductor polianiónico [118], lo que da como resultado tasas de formación de filamentos más altas. Otros polianiones nativos, como la región rica en ácido glutámico presente en la región C-terminal de la tubulina, también pueden facilitar la agregación de la proteína tau. Esta agregación requiere la presencia del tercer motivo de unión a la tubulina de la molécula de tau [115]. La oxidación es otro proceso que facilita la agregación de la proteína tau. Debido a que las moléculas de tau 3R contienen solo una cisteína, la oxidación de la cisteína produce enlaces disulfuro y, por lo tanto, el autoensamblaje de la proteína tau [119]. No se presenta en moléculas de tau 4R con dos cisteínas, que pueden formar enlaces disulfuro intramoleculares [119]. A pesar dein vitroSe ha demostrado que los polímeros tau formados se forman mediante espectroscopia, dispersión láser y microscopía electrónica [120–123], hallazgos recientes demuestran que se pueden formar oligómeros de tau prefibrilaresin vitropor reticulación inducida por luz de tau con benzofenona-4-maleimida (B4M) [123]. También se observaron estos oligómeros de tau.en el lugarEn las primeras etapas de la EA, cuando se evaluó un anticuerpo monoclonal y específico contra estas entidades oligoméricas de tau en el cerebro de los casos de EA [123 Se informa que las especies oligoméricas de proteína tau tienen una mayor toxicidad a lo largo del tiempo. 4.4. El truncamiento de la proteína Tau.La escisión proteolítica de la proteína tau, como un mecanismo alternativo que interviene en la agregación anormal de tau, fue propuesta tempranamente por el grupo de Whischik en la Universidad de Cambridge después de un extenso análisis bioquímico de la estructura mínima de los PHF [69, 127,128]. El componente mínimo de los PHF, denominado núcleo PHF, estaba compuesto principalmente por un fragmento de tau que solo contenía la región MTBR y terminaba en la posición Glu.391 Hasta el día de hoy, la identificación de la enzima que produce esta escisión proteolítica es incierta. Sin embargo, se ha demostrado la presencia de este truncamiento asociado a la patología neurofibrilar en el cerebro de pacientes con EA [129,130]. Además, desdein vitro paradigmas de polimerización, las construcciones tau que carecen de la cola carboxi se ensamblan mucho más rápido y en mayor medida que las tau de longitud completa [131]. A pesar de estas evidencias tempranas, durante un tiempo no se prestó atención a la proteólisis de tau, y su contribución a la enfermedad fue incierta. Nuevos hallazgos muestran una proteólisis aberrante en el cerebro de los casos de EA asociada con la muerte celular programada [132, 133]. Se dedicaron estudios posteriores a investigar la contribución de la apoptosis y las caspasas asociadas al proceso neurodegenerativo subyacente a la EA. En este sentido, se observó que las células apoptóticas proliferaban en áreas del cerebro afectadas por la acumulación fibrilar de proteína tau y amiloide.β depósitos [134–136]. Al mismo tiempo, se informó de un aumento de la expresión de varias enzimas de la familia de las caspasas en el cerebro de los casos de EA [99,137,138]. 9730, 2012, 1, Descargado de https://onlinelibrary.wiley.com/doi/10.1155/2012/731526 por Cochrane Perú, Wiley Online Library el [17/02/2025]. Consulte los Términos y condiciones (https://onlinelibrary.wiley.com/terms-and-conditions) en Wiley Online Library para conocer las reglas de uso; los artículos de acceso abierto se rigen por la Licencia Creative Commons aplicable. 6 Las caspasas son proteasas de cisteína que escinden el residuo de ácido aspártico en la secuencia de consenso canónica DXXD en el lado carboxilo de la molécula. Estas enzimas participan en una cascada proteolítica que conduce a la muerte celular por apoptosis. encontrado en casos con una mayor densidad de NFT compuestos por las dos variantes de tau truncada [163]. En el hipocampo de pacientes con EA, se ha descrito que la maduración de los NFT no está sincronizada. Por lo tanto, estas La principal caspasa asesina en las neuronas es la caspasa 3 [139]. estructuras tienen diferentes etapas de procesamiento de la Los miembros de la familia de las caspasas desempeñan un papel proteína tau [163]. Se informó que diferentes poblaciones de NFT en fundamental en Aβ- apoptosis neuronal inducida [140] y se activan la misma área del hipocampo eran mutuamente excluyentes cuando en neuronas apoptóticas en la EA [141]. Se sabía que la proteína tau estaban compuestas por Asp421- o Glu391-tau truncada sin contiene varios sitios canónicos para la escisión de la caspasa [142, colocalización en ningún punto durante la maduración de los NFT [ 143], del cual se obtiene un residuo susceptible en Asp421Se informó 163]. Durante la progresión de la enfermedad, Asp421-El que se había escindidoin vitropor caspasa 3 [72]. La escisión en Asp truncamiento es un evento temprano que precede al segundo 421liberó un péptido discreto (Ser422-Leu441) que es capaz de formar truncamiento del extremo C en el Glu391, siendo estas últimas las un anfipáticoalfa-hélice [144]. Proteína Tau truncada en Asp421se que se producen en etapas intermedias y avanzadas de la evolución ensambla más fácilmente que la molécula de longitud completa [72, de los NFT [163]. Un informe reciente indica que la proteína tau en 144]. Cuando un péptido sintético que comprendía el fragmento los NFT puede estar doblemente sujeta a proteólisis apoptótica y después de la escisión de la caspasa se añadió nuevamente a la proteosomal, ya que se encontró una fuerte ubiquitinación en Asp. molécula de tau en un paradigma de polimerización, se inhibió el 421- tau truncada asociada a la patología neurofibrilar en la EA [165]. ensamblaje de esta proteína. En la enfermedad, se produce el truncamiento de la proteína Se ha propuesto que, mediante la combinación de anticuerpos tau en Asp.421Se corroboró en asociación con la patología que mapean diferentes regiones de la molécula de tau, se produce neurofibrilar mediante el uso del anticuerpo monoclonal Tau-C3, una vía continua y específica de cambios conformacionales y que reconoce específicamente este sitio de escisión generado por la truncamiento de la proteína tau durante la maduración de los NFT. actividad de la caspasa 3 [72,145]. Curiosamente, la fosforilación de Estos anticuerpos son, concretamente, dependientes de la la proteína tau en el residuo Ser422Parecía prevenir la escisión conformación y la fosforilación y reconocen sitios de truncamiento [ proteolítica de tau en Asp421[146]. Después del truncamiento en Asp 66,67,145]. 421Se ha informado que se produce otra escisión de la proteína tau Estos estudios propusieron que no solo el número de NFT sino en Glu391Este estado es reconocido por el anticuerpo MN423, que también el estado de proteólisis del extremo C que está asociado indica las transiciones a ovillos “tardíos” [56,67,145Se ha informado con cambios conformacionales (modificación estructural a lo largo que se produce otro truncamiento en el extremo N de la proteína de la molécula de tau) define la progresión de la EA [166]. Todos tau en el residuo Asp13, que en este caso es producida por la estos hallazgos en conjunto pueden respaldar la relevancia del actividad de la caspasa 6 [147]. A pesar de lain vitroSi bien se ha truncamiento de la proteína tau como un evento patogénico y un demostrado que este truncamiento en el extremo N es importante marcador confiable tanto para el diagnóstico como para la para favorecer la agregación de tau, su significado patológico y su orientación terapéutica en la EA. aparición en el cerebro de pacientes con EA aún están lejos de demostrarse. Se ha evaluado el efecto patológico de la proteína tau truncada en el extremo C sobre el funcionamiento normal de las células en 5. Conclusión cultivos celulares y modelos animales transgénicos. Utilizando Se acepta en gran medida que la manifestación clínica de la células neuronales y no neuronales, la sobreexpresión de la proteína demencia en la EA se debe a la pérdida neuronal que se produce en tau truncada produce varias alteraciones en la organización y las áreas del cerebro asociadas con las funciones cognitivas de los funcionamiento de orgánulos membranosos, como las mitocondrias pacientes. Se informa que las inclusiones fibrilares son responsables y el retículo endoplasmático. Incluso se han descrito algunos de la muerte celular. Sin embargo, han surgido discrepancias en los ejemplos de muerte celular por mecanismos apoptóticos [148–156]. estudios que demuestran que el deterioro cognitivo en modelos En animales transgénicos, los roedores portadores de tau truncada animales ocurre antes de la formación inicial de estructuras han desarrollado alteraciones en el rendimiento cognitivo asociadas fibrilares. La extrapolación de estos resultados al inicio real de la con la muerte neuronal y la agregación anormal de tau escindida [ enfermedad en humanos todavía se considera inexacta para 100,157–162]. algunos investigadores. En este sentido, una serie de informes que Finalmente, el papel anormal del truncamiento de la proteína tau y su significado patológico en la EA ha sido demostrado por estudios clinicopatológicos donde se analizó la aparición de tau truncada asociada a estructuras fibrilares durante el desarrollo de la demencia [130, 145,163 ]. Estos estudios corroboran la importancia de la proteína tau truncada en ambos sitios Asp421y Glu391. H. Braak y E. Braak describieron una correlación positiva de estos eventos con la progresión neuropatológica de la enfermedad [164] y se demostró una relación con la gravedad clínica de la demencia [130,163]. Además, la presencia de la apolipoproteína-E (mi4) La variante alélica fue analizan el cerebro de pacientes con EA coinciden en que la agregación fibrilar de tau es el mejor correlacionador con el inicio y la progresión de la demencia. Se acepta mayoritariamente que las modificaciones postraduccionales anormales, es decir, la hiperfosforilación, la acetilación, la glicación, la nitración, el truncamiento y otras, son responsables de la estructura alterada de tau en la EA. Algunos de estos eventos se han organizado secuencialmente durante la formación de NFT y la evolución de la enfermedad. Se ha demostrado la validación a niveles clinicopatológicos con la carga de tau anormalmente fosforilada y truncada en poblaciones de casos de EA. En particular, la fosforilación, acetilación y truncamiento anormales son 9730, 2012, 1, Descargado de https://onlinelibrary.wiley.com/doi/10.1155/2012/731526 por Cochrane Perú, Wiley Online Library el [17/02/2025]. Consulte los Términos y condiciones (https://onlinelibrary.wiley.com/terms-and-conditions) en Wiley Online Library para conocer las reglas de uso; los artículos de acceso abierto se rigen por la Licencia Creative Commons aplicable. 7 Revista internacional sobre la enfermedad de Alzheimer Revista internacional sobre la enfermedad de Alzheimer Además, se respaldan como eventos patológicosin vitroExperimentos que demuestran que estas modificaciones aumentan la fibrilación de tau e inducen toxicidad celular.in vitroLos animales transgénicos que portan estas formas alteradas de la proteína tau también desarrollan alteraciones cognitivas. Creemos que resolver la génesis de los cambios conformacionales de la proteína tau promovidos por estas modificaciones postraduccionales y su papel en la fibrilación en la enfermedad son logros importantes para evaluar el potencial de las terapias dirigidas a la proteína tau. Además, la determinación precisa de la proteína tau alterada en el líquido cefalorraquídeo y otros fluidos corporales puede proporcionar una mejor expectativa para predecir la aparición y evolución de la demencia. Expresiones de gratitud Este trabajo fue financiado por el Proyecto de Excelencia en Investigación Básica (No. P 304/12/G069) de la Agencia de Subvenciones de la República Checa. G.-S. Francisco recibió apoyo de la subvención CONACyT-México CB-152535. Referencias [1] RB Maccioni, JP Muñoz y L. Barbeito, "Las bases moleculares de la enfermedad de Alzheimer y otros trastornos neurodegenerativos".Archivos de investigación médica, vol. 32, no. 5, págs. 367–381, 2001. [2] KS Kosik, “La biología molecular y celular de tau”,Patología cerebral, vol. 3, no. 1, págs. 39–43, 1993. [3] EM Mandelkow, J. Biernat, G. Drewes, N. Gustke, B. Trinczek y E. Mandelkow, “Dominios Tau, fosforilación e interacciones con microtúbulos”,Neurobiología del envejecimiento, vol. 16, no. 3, págs. 355–363, 1995. [4] F. Liu, K. Iqbal, I. Grundke-Iqbal, S. Rossie y CX Gong, “Desfosforilación de tau por la proteína fosfatasa 5: deterioro en la enfermedad de Alzheimer”,Revista de química biológica, vol. 280, núm. 3, págs. 1790–1796, 2005. [5] J. Avila, “Tau quinasas y fosfatasas: comentario”,Revista de medicina celular y molecular, vol. 12, no. 1, págs. 258– 259, 2008. [6] K. Iqbal e I. Grundke-Iqbal, “Degeneración neurofibrilar de Alzheimer: importancia, etiopatogenia, terapéutica y prevención: serie de revisión de Alzheimer”,Revista de medicina celular y molecular, vol. 12, no. 1, págs. 38–55, 2008. [7] N. Sergeant, A. Delacourte y L. Buée, "La proteína tau como biomarcador diferencial de tauopatías"Acta bioquímica y biofísica, vol. 1739, núm. 2, págs. 179–197, 2005. [8] DW Cleveland, SY Hwo y MW Kirschner, “Propiedades físicas y químicas del factor tau purificado y el papel de tau en el ensamblaje de microtúbulos”,Revista de Biología Molecular, vol. 116, no. 2, págs. 227–247, 1977. [12] HJ Dyson y PE Wright, “Proteínas intrínsecamente no estructuradas y sus funciones”,Reseñas de Nature Biología celular molecular, vol. 6, no. 3, págs. 197–208, 2005. [13] MD Mukrasch, S. Bibow, J. Korukottu et al., "Polimorfismo estructural de Tau de 441 residuos con resolución de residuo único".PLoS Biología, vol. 7, no. 2, ID de artículo e1000034, 2009. [14] M. Goedert y R. Jakes, “Expresión de isoformas separadas de la proteína tau humana: correlación con el patrón tau en el cerebro y efectos sobre la polimerización de la tubulina”,La revista EMBO, vol. 9, no. 13, págs. 4225–4230, 1990. [15] ADC Alonso, T. Zaidi, M. Novak, I. Grundke-Iqbal y K. Iqbal, “La hiperfosforilación induce el autoensamblaje deτ en marañas de filamentos helicoidales emparejados/filamentos rectos”, Actas de la Academia Nacional de Ciencias de los Estados Unidos de América, vol. 98, no. 12, págs. 6923–6928, 2001. [16] N. Sergeant, A. Bretteville, M. Hamdane et al., “Bioquímica de Tau en la enfermedad de Alzheimer y trastornos neurológicos relacionados”,Revisión experta de la proteómica, vol. 5, no. 2, págs. 207– 224, 2008. [17] A. Ebneth, R. Godemann, K. Stamer et al., “La sobreexpresión de la proteína tau inhibe el tráfico dependiente de kinesina de vesículas, mitocondrias y retículo endoplásmico: implicaciones para la enfermedad de Alzheimer”,Revista de biología celular, vol. 143, no. 3, págs. 777–794, 1998. [18] D. Couchie, C. Mavilia, IS Georgieff, RKH Liem, ML Shelanski y J. Nunez, “Estructura primaria de la proteína tau de alto peso molecular presente en el sistema nervioso periférico”, Actas de la Academia Nacional de Ciencias de los Estados Unidos de América, vol. 89, núm. 10, págs. 4378–4381, 1992. [19] IS Georgieff, RKH Liem, D. Couchie, C. Mavilia, J. Nunez y ML Shelanski, “Expresión de tau de alto peso molecular en los sistemas nerviosos central y periférico”, Revista de ciencia celular, vol. 105, no. 3, págs. 729–737, 1993. [20] M. Goedert, MG Spillantini y RA Crowther, “Clonación de una gran proteína asociada a microtúbulos tau característica del sistema nervioso periférico”,Actas de la Academia Nacional de Ciencias de los Estados Unidos de América, vol. 89, no. 5, págs. 1983–1987, 1992. [21] M. Balastik, J. Lim, L. Pastorino y KP Lu, “Pin1 en la enfermedad de Alzheimer: ¿múltiples sustratos, un mecanismo regulador?”Acta bioquímica y biofísica, vol. 1772, núm. 4, págs. 422–429, 2007. [22] D. Jung, D. Filliol, M. Miehe y A. Rendon, “Interacción de las mitocondrias cerebrales con microtúbulos reconstituidos a partir de tubulina cerebral y MAP2 o TAU”,Motilidad celular y citoesqueleto, vol. 24, no. 4, págs. 245–255, 1993. [23] R. Brandt, J. Léger y G. Lee, “Interacción de tau con la membrana plasmática neural mediada por el dominio de proyección aminoterminal de tau”,Revista de biología celular, vol. 131, núm. 5, págs. 1327–1340, 1995. [24] M. Arrasate, M. Pérez y J. Avila, “La desfosforilación de Tau en el sitio Tau-1 se correlaciona con su asociación a la membrana celular”, [9] M. Von Bergen, S. Barghorn, J. Biernat, EM Mandelkow y E. Mandelkow, Investigación neuroquímica, vol. 25, no. 1, págs. 43–50, 2000. “La agregación de Tau está impulsada por una transición de una [25] T. Maas, J. Eidenmüller y R. Brandt, “La interacción de tau con la corteza de la membrana neural está regulada por la fosforilación en sitios que se modifican en filamentos helicoidales pareados”,Revista de química biológica, vol. 275, núm. 21, págs. 15733–15740, 2000. estructura de espiral aleatoria a una estructura de lámina beta”,Acta bioquímica y biofísica, vol. 1739, núm. 2, págs. 158–166, 2005. [10] TC Gamblin, “Potenciales relaciones estructura/función de los elementos estructurales secundarios predichos de tau”,Acta bioquímica y biofísica, vol. 1739, núm. 2, págs. 140–149, 2005. [11] S. Jeganathan, M. Von Bergen, EM Mandelkow y E. Mandelkow, “El carácter nativo desplegado de Tau y su agregación a filamentos helicoidales pareados similares a los de Alzheimer”, Bioquímica, vol. 47, núm. 40, págs. 10526–10539, 2008. [26] CH Reynolds, CJ Garwood, S. Wray et al., “La fosforilación regula las interacciones de tau con los dominios de homología Src 3 de la fosfatidilinositol 3-quinasa, fosfolipasa Cgamma1, Grb2 y quinasas de la familia Src”,Revista de química biológica, vol. 283, núm. 26, págs. 18177–18186, 2008. 9730, 2012, 1, Descargado de https://onlinelibrary.wiley.com/doi/10.1155/2012/731526 por Cochrane Perú, Wiley Online Library el [17/02/2025]. Consulte los Términos y condiciones (https://onlinelibrary.wiley.com/terms-and-conditions) en Wiley Online Library para conocer las reglas de uso; los artículos de acceso abierto se rigen por la Licencia Creative Commons aplicable. 8 [27] A. Baba, K. Akagi, M. Takayanagi, JG Flanagan, T. Kobayashi y M. Hattori, “La tirosina quinasa Fyn regula la expresión superficial de la efrina unida a glicosilfosfatidilinositol a través de la modulación del metabolismo de la esfingomielina”, Revista de química biológica, vol. 284, núm. 14, págs. 9206–9214, 2009. [28] HS Hoe, SS Minami, A. Makarova et al., “Modulación de Fyn de los efectos de Dab1 sobre la proteína precursora amiloide y el procesamiento del receptor apoe 2”,Revista de química biológica, vol. 283, núm. 10, págs. 6288–6299, 2008. [29] G. Lee, R. Thangavel, VM Sharma et al., “Fosforilación de tau por fyn: implicaciones para la enfermedad de Alzheimer”,Revista de neurociencia, vol. 24, no. 9, págs. 2304–2312, 2004. [30] AM Pooler, A. Usardi, CJ Evans, KL Philpott, W. Noble y DP Hanger, “La asociación dinámica de tau con las membranas neuronales está regulada por la fosforilación”,Neurobiología del envejecimiento, vol. 33, núm. 2, págs. 431.e27–431.e38, 2012. [31] LM Ittner, YD Ke, F. Delerue et al., “La función dendrítica de tau media media amiloide-βtoxicidad en modelos de ratón con enfermedad de Alzheimer”,Celúla, vol. 142, núm. 3, págs. 387–397, 2010. [32] SM Pritchard, PJ Dolan, A. Vitkus y GVW Johnson, “La toxicidad de tau en la enfermedad de Alzheimer: recambio, objetivos y posibles terapias”,Revista de medicina celular y molecular, vol. 15, núm. 8, págs. 1621–1635, 2011. [33] GP Cardona-Gómez, C. Arango-Davila, JC Gallego-Gómez, A. Barrera-Ocampo, H. Pimienta y LM García-Segura, “El estrógeno disocia Tau y la subunidad del receptor del ácido alfa-amino-3-hidroxi-5-metilisoxazol-4-propiónico en el hipocampo postisquémico”.Informe neurológico, vol. 17, no. 12, págs. 1337–1341, 2006. [34] C. Klein, EM Kramer, AM Cardine, B. Schraven, R. Brandt y J. Trotter, “El proceso de crecimiento de los oligodendrocitos se promueve mediante la interacción de la quinasa fyn con la proteína citoesquelética tau”,Revista de neurociencia, vol. 22, no. 3, págs. 698–707, 2002. [35] R. Brandt y G. Lee, “Organización funcional de la proteína tau asociada a microtúbulos. Identificación de regiones que afectan el crecimiento, la nucleación y la formación de haces de microtúbulos in vitro”,Revista de química biológica, vol. 268, núm. 5, págs. 3414–3419, 1993. [36] JW Mandell y GA Banker, “Un gradiente espacial de fosforilación de la proteína tau en axones nacientes”,Revista de neurociencia, vol. 16, no. 18, págs. 5727–5740, 1996. [37] DN Drechsel, AA Hyman, MH Cobb y MW Kirschner, “Modulación de la inestabilidad dinámica del ensamblaje de la tubulina por la proteína tau asociada a los microtúbulos”,Biología molecular de la célula, vol. 3, no. 10, págs. 1141–1154, 1992. [38] KA Butner y MW Kirschner, “La proteína Tau se une a los microtúbulos a través de una matriz flexible de sitios débiles distribuidos”,Revista de biología celular, vol. 115, no. 3, págs. 717– 730, 1991. [39] BL Goode y SC Feinstein, “Identificación de un nuevo dominio de unión y ensamblaje de microtúbulos en la región interrepetición regulada por el desarrollo de tau”,Revista de biología celular, vol. 124, núm. 5, págs. 769–781, 1994. [42] HV Goodson, C. Valetti y TE Kreis, “Motores y tráfico de membranas”,Opinión actual en biología celular, vol. 9, no. 1, págs. 18–28, 1997. [43] CM Waterman-Storer y ED Salmon, “Dinámica de los microtúbulos: la cinta de correr vuelve a aparecer”,Biología actual, vol. 7, no. 6, págs. R369–R372, 1997. [44] ST Brady y AO Sperry, “Diversidad bioquímica y funcional de los motores de microtúbulos en el sistema nervioso”, Opinión actual en neurobiología, vol. 5, no. 5, págs. 551–558, 1995. [45] J. Lippincott-Schwartz, NB Cole, A. Marotta, PA Conrad y GS Bloom, “La kinesina es el motor del tráfico de membrana del Golgi al RE mediado por microtúbulos”,Revista de biología celular, vol. 128, no. 3, págs. 293–306, 1995. [46] RL Morris y PJ Hollenbeck, “La regulación del transporte mitocondrial bidireccional está coordinada con el crecimiento axonal”,Revista de ciencia celular, vol. 104, no. 3, págs. 917– 927, 1993. [47] Y. Tanaka, Y. Kanai, Y. Okada et al., “La interrupción dirigida de la cadena pesada de kinesina convencional de ratón, kif5B, da como resultado una agrupación perinuclear anormal de mitocondrias”,Celúla, vol. 93, no. 7, págs. 1147–1158, 1998. [48] PJ Hollenbeck y JA Swanson, “Extensión radial de los lisosomas tubulares de macrófagos soportados por kinesina”, Naturaleza, vol. 346, núm. 6287, págs. 864–866, 1990. [49] EAC Wiemer, T. Wenzel, TJ Deerinck, MH Ellisman y S. Subramani, “Visualización del compartimento peroxisomal en células de mamíferos vivos: comportamiento dinámico y asociación con microtúbulos”,Revista de biología celular, vol. 136, no. 1, págs. 71–80, 1997. [50] SJ Scales, R. Pepperkok y TE Kreis, “La visualización del transporte de ER a Golgi en células vivas revela un modo de acción secuencial para COPII y COPI”,Celúla, vol. 90, no. 6, págs. 1137–1148, 1997. [51] I. Cuchillo-Ibanez, A. Seereeram, HL Byers et al., “La fosforilación de tau regula su transporte axonal al controlar su unión a la kinesina”,Revista FASEB, vol. 22, no. 9, págs. 3186–3195, 2008. [52] B. Trinczek, A. Ebneth, EM Mandelkow y E. Mandelkow, “Tau regula la unión/desprendimiento pero no la velocidad de los motores en el transporte dependiente de microtúbulos de vesículas y orgánulos individuales”,Revista de ciencia celular, vol. 112, núm. 14, págs. 2355–2367, 1999. [53] RW Carrell y B. Gooptu, “Cambios conformacionales y enfermedades: serpinas, priones y Alzheimer”,La opinión actual en biología estructural, vol. 8, no. 6, págs. 799–809, 1998. [54] N. Fox, RJ Harvey y MN Rossor, “Plegamiento de proteínas, fenómenos de nucleación y neurodegeneración retardada en la enfermedad de Alzheimer”,Reseñas en Neurociencias, vol. 7, no. 1, págs. 21–28, 1996. [55] BT Hyman, JC Augustinack y M. Ingelsson, “Cambios transcripcionales y conformacionales de la molécula tau en la enfermedad de Alzheimer”,Acta bioquímica y biofísica, vol. 1739, núm. 2, págs. 150–157, 2005. [56] F. García-Sierra, N. Ghoshal, B. Quinn, RW Berry y LI Bı́nder, “Cambios conformacionales y truncamiento de la proteína tau [40] E. Sontag, V. Nunbhakdi-Craig, G. Lee et al., “Interacciones moleculares entre la proteína fosfatasa 2A, tau y microtúbulos. Implicaciones para la regulación de la fosforilación de tau y el desarrollo de tauopatías”,Revista de química biológica, vol. 274, núm. 36, págs. 25490–25498, 1999. [57] N. Ghoshal, F. García-Sierra, Y. Fu et al., "Tau-66: evidencia de una nueva conformación tau en la enfermedad de Alzheimer".Revista de neuroquímica, vol. 77, núm. 5, págs. 1372–1385, 2001. [41] DG Drubin y WJ Nelson, “Orígenes de la polaridad celular”,Celúla, vol. 84, no. 3, págs. 335–344, 1996. [58] N. Ghoshal, F. Garcı́a-Sierra, J. Wuu et al., “Los cambios conformacionales de Tau corresponden a alteraciones de la durante la evolución de la maraña en la enfermedad de Alzheimer”, Revista de la enfermedad de Alzheimer, vol. 5, no. 2, págs. 65–77, 2003. 9730, 2012, 1, Descargado de https://onlinelibrary.wiley.com/doi/10.1155/2012/731526 por Cochrane Perú, Wiley Online Library el [17/02/2025]. Consulte los Términos y condiciones (https://onlinelibrary.wiley.com/terms-and-conditions) en Wiley Online Library para conocer las reglas de uso; los artículos de acceso abierto se rigen por la Licencia Creative Commons aplicable. 9 Revista internacional sobre la enfermedad de Alzheimer Revista internacional sobre la enfermedad de Alzheimer “La memoria en el deterioro cognitivo leve y la enfermedad de Alzheimer”Neurología experimental, vol. 177, no. 2, págs. 475– 493, 2002. [59] EM Mandelkow, K. Stamer, R. Vogel, E. Thies y E. Mandelkow, “Obstrucción de axones por tau, inhibición del tráfico axonal y privación de sinapsis”,Neurobiología del envejecimiento, vol. 24, núm. 8, págs. 1079–1085, 2003. [60] NE LaPointe, G. Morfini, G. Pigino et al., “El extremo amino de tau inhibe el transporte axonal dependiente de kinesina: implicaciones para la toxicidad del filamento”,Revista de investigación en neurociencia, vol. 87, no. 2, págs. 440–451, 2009. [61] AH Futerman y GA Banker, “La economía del crecimiento de las neuritas: la adición de una nueva membrana a los axones en crecimiento”, Tendencias en neurociencias, vol. 19, no. 4, págs. 144–149, 1996. [62] JQ Trojanowski y VMY Lee, “Fosforilación de filamentos helicoidales emparejados de tau en lesiones neurofibrilares de la enfermedad de Alzheimer: centrándose en las fosfatasas”,Revista FASEB, vol. 9, no. 15, págs. 1570–1576, 1995. [63] L. Martin, X. Latypova y F. Terro, “Modificaciones postraduccionales de la proteína tau: implicaciones para la enfermedad de Alzheimer”,Neuroquímica Internacional, vol. 58, no. 4, págs. 458– 471, 2011. [64] C. Soto, “Alzheimer y enfermedad priónica como trastornos de la conformación proteica: implicaciones para el diseño de nuevos enfoques terapéuticos”,Revista de medicina molecular, vol. 77, no. 5, págs. 412–418, 1999. [65] I. Grundke-Iqbal, K. Iqbal y YC Tung, “Fosforilación anormal de la proteína asociada a los microtúbulosτ (tau) en la patología del citoesqueleto de Alzheimer”,Actas de la Academia Nacional de Ciencias de los Estados Unidos de América, vol. 83, núm. 13, págs. 44913–4917, 1986. [66] S. Mondragón-Rodríguez, G. Basurto-Islas, LI Binder y F. Garcı́a-Sierra, “Cambios conformacionales y escisión: ¿son responsables de la agregación de tau en la enfermedad de Alzheimer?”Neurología del futuro, vol. 4, no. 1, págs. 39–53, 2009. [67] LI Binder, AL Guillozet-Bongaarts, F. Garcia-Sierra y RW Berry, “Tau, ovillos y enfermedad de Alzheimer”, Acta bioquímica y biofísica, vol. 1739, núm. 2, págs. 216–223, 2005. [68] B. Kuhla, C. Haase, K. Flach, HJ Lüth, T. Arendt y G. Münch, “Efecto de la pseudofosforilación y la reticulación por peroxidación lipídica y precursores de productos finales de glicación avanzada sobre la agregación de tau y la formación de filamentos”,Revista de química biológica, vol. 282, núm. 10, págs. 6984– 6991, 2007. [69] CM Wischik, M. Novak, PC Edwards, A. Klug, W. Tichelaar y RA Crowther, “Caracterización estructural del núcleo del filamento helicoidal emparejado de la enfermedad de Alzheimer”, Actas de la Academia Nacional de Ciencias de los Estados Unidos de América, vol. 85, núm. 13, págs. 4884–4888, 1988. [70] G. Perry, P. Mulvihill, VA Fried, HT Smith, I. Grundke-Iqbal y K. Iqbal, “Propiedades inmunoquímicas de los conjugados de ubiquitina en los filamentos helicoidales pareados de la enfermedad de Alzheimer”,Revista de neuroquímica, vol. 52, núm. 5, págs. 1523– 1528, 1989. [71] MR Reynolds, RW Berry y LI Binder, “Nitración específica del sitio y puente oxidativo de ditirosina de la τproteína por peroxinitrito: implicaciones para la enfermedad de Alzheimer”,Bioquímica, vol. 44, núm. 5, págs. 1690–1700, 2005. [72] TC Gamblin, F. Chen, A. Zambrano et al., “Escisión de tau por caspasa: vinculación de ovillos neurofibrilares y amiloide en “Enfermedad de Alzheimer”,Actas de la Academia Nacional de Ciencias de los Estados Unidos de América, vol. 100, núm. 17, págs. 10032–10037, 2003. [73] SW Min, SH Cho, Y. Zhou et al., “La acetilación de tau inhibe su degradación y contribuye a la tauopatía”, Neurona, vol. 67, no. 6, págs. 953–966, 2010. [74] L. Buée, T. Bussière, V. Buée-Scherrer, A. Delacourte y PR Hof, “Isoformas de la proteína Tau, fosforilación y papel en trastornos neurodegenerativos”,Reseñas de investigaciones sobre el cerebro, vol. 33, no. 1, págs. 95–130, 2000. [75] G. Drewes, B. Trinczek, S. Illenberger et al., “Proteína asociada a microtúbulos/quinasa reguladora de afinidad de microtúbulos (p110(mark)). Una nueva proteína quinasa que regula las interacciones de microtúbulos y la inestabilidad dinámica mediante fosforilación en el sitio específico de Alzheimer serina 262”,Revista de química biológica, vol. 270, núm. 13, págs. 7679– 7688, 1995. [76] CA Dickey, A. Kamal, K. Lundgren et al., “El complejo HSP90CHIP de alta afinidad reconoce y degrada selectivamente las proteínas cliente tau fosforiladas”,Revista de investigación clínica, vol. 117, no. 3, págs. 648–658, 2007. [77] CX Gong y K. Iqbal, “Hiperfosforilación de la proteína tau asociada a microtúbulos: un objetivo terapéutico prometedor para la enfermedad de Alzheimer”,Química medicinal actual, vol. 15, núm. 23, págs. 2321–2328, 2008. [78] F. Liu, T. Zaidi, K. Iqbal, I. Grundke-Iqbal, RK Merkle y CX Gong, “Papel de la glicosilación en la hiperfosforilación de tau en la enfermedad de Alzheimer”,Cartas de la FEBS, vol. 512, núm. 1–3, págs. 101–106, 2002. [79] J. Avila, JJ Lucas, M. Pérez y F. Hernández, “Papel de la proteína tau tanto en condiciones fisiológicas como patológicas”, Reseñas fisiológicas, vol. 84, no. 2, págs. 361–384, 2004. [80] E. Kopke, YC Tung, S. Shaikh, CA Del Alonso, K. Iqbal e I. GrundkeIqbal, “Proteína tau asociada a microtúbulos. Fosforilación anormal de un conjunto de filamentos helicoidales no apareados en la enfermedad de Alzheimer”,Revista de química biológica, vol. 268, núm. 32, págs. 24374–24384, 1993. [81] K. Iqbal, F. Liu, CX Gong, AC del Alonso e I. Grundke-Iqbal, “Mecanismos de neurodegeneración inducida por tau”, Acta neuropatológica, vol. 118, no. 1, págs. 53–69, 2009. [82] S. Khatoon, I. Grundke-Iqbal y K. Iqbal, “Niveles de tan normal y anormalmente fosforilado en diferentes compartimentos celulares y regionales de cerebros con enfermedad de Alzheimer y de control”,Cartas de la FEBS, vol. 351, no. 1, págs. 80–84, 1994. [83] ADC Alonso, T. Zaidi, I. Grundke-Iqbal y K. Iqbal, “Papel de la tau anormalmente fosforilada en la descomposición de los microtúbulos en la enfermedad de Alzheimer”,Actas de la Academia Nacional de Ciencias de los Estados Unidos de América, vol. 91, no. 12, págs. 5562–5566, 1994. [84] B. Li, MO Chohan, I. Grundke-Iqbal y K. Iqbal, “Alteración de la red de microtúbulos por tau hiperfosforilada anormalmente en Alzheimer”,Acta neuropatológica, vol. 113, no. 5, págs. 501–511, 2007. [85] JZ Wang, CX Gong, T. Zaidi, I. Grundke-Iqbal y K. Iqbal, “Desfosforilación de filamentos helicoidales emparejados de Alzheimer por la proteína fosfatasa-2A y -2B”,Revista de química biológica, vol. 270, núm. 9, págs. 4854–4860, 1995. [86] ADC Alonso, I. Grundke-Iqbal, HS Barra y K. Iqbal, “Fosforilación anormal de tau y el mecanismo de degeneración neurofibrilar de Alzheimer: secuestro de proteínas asociadas a microtúbulos 1 y 2 y desmontaje de microtúbulos por la tau anormal”,Actas de la 9730, 2012, 1, Descargado de https://onlinelibrary.wiley.com/doi/10.1155/2012/731526 por Cochrane Perú, Wiley Online Library el [17/02/2025]. Consulte los Términos y condiciones (https://onlinelibrary.wiley.com/terms-and-conditions) en Wiley Online Library para conocer las reglas de uso; los artículos de acceso abierto se rigen por la Licencia Creative Commons aplicable. 10 Academia Nacional de Ciencias de los Estados Unidos de América, vol. 94, no. 1, págs. 298–303, 1997. [87] DP Hanger, HL Byers, S. Wray et al., “Nuevos sitios de fosforilación en Tau del cerebro de Alzheimer respaldan un papel de la caseína quinasa 1 en la patogénesis de la enfermedad”,Revista de química biológica, vol. 282, núm. 32, págs. 23645–23654, 2007. [88] J. Biernat, N. Gustke, G. Drewes, EM Mandelkow y E. Mandelkow, “La fosforilación de Ser262 reduce fuertemente la unión de tau a los microtúbulos: distinción entre inmunorreactividad similar a PHF y unión a microtúbulos”, Neurona, vol. 11, no. 1, págs. 153–163, 1993. [89] JT Du, CH Yu, LX Zhou et al., “La fosforilación modula la conformación local y la capacidad de autoagregación de un péptido de la cuarta repetición de unión al microtúbulo tau”, Revista FEBS, vol. 274, núm. 19, págs. 5012–5020, 2007. [90] VMY Lee, BJ Balin, L. Otvos y JQ Trojanowski, “A68: una subunidad principal de filamentos helicoidales pareados y formas derivatizadas de tau normal”,Ciencia, vol. 251, núm. 4994, págs. 675– 678, 1991. [91] I. Grundke-Iqbal, K. Iqbal y M. Quinlan, “Proteína tau asociada a microtúbulos. Un componente de los filamentos helicoidales emparejados de Alzheimer”,Revista de química biológica, vol. 261, núm. 13, págs. 6084–6089, 1986. [92] PV Arriagada, JH Growdon, ET Hedley-Whyte y BT Hyman, “Los ovillos neurofibrilares, pero no las placas seniles, son paralelos a la duración y la gravedad de la enfermedad de Alzheimer”,Neurología, vol. 42, no. 3, págs. 631–639, 1992. [93] C. Bancher, C. Brunner, H. Lassmann et al., “Acumulación de fosforilaciones anormalesτ“Precede a la formación de ovillos neurofibrilares en la enfermedad de Alzheimer”,Investigación del cerebro, vol. 477, no. 1-2, págs. 90–99, 1989. [94] E. Braak, H. Braaak y EM Mandelkow, “Una secuencia de cambios en el citoesqueleto relacionados con la formación de ovillos neurofibrilares y hebras de neuropilo”,Acta neuropatológica, vol. 87, no. 6, págs. 554–567, 1994. [95] F. García-Sierra, JJ Hauw, C. Duyckaerts, CM Wischik, J. Luna-Muñoz y R. Mena, “La extensión de la patología neurofibrilar en las neuronas de la vía perforante es el determinante clave de la demencia en los muy ancianos”,Acta neuropatológica, vol. 100, no. 1, págs. 29–35, 2000. [101] TJ Cohen, JL Guo, DE Hurtado et al., “La acetilación de tau inhibe su función y promueve la agregación patológica de tau”,Comunicaciones de la naturaleza, vol. 2, no. 1, artículo 252, 2011. [102] DJ Irwin, TJ Cohen, M. Grossman et al., “Tau acetilada, una nueva firma patológica en la enfermedad de Alzheimer y otras tauopatías”,Cerebro, vol. 135, no. 3, págs. 807–818, 2012. [103] C. Choudhary, C. Kumar, F. Gnad et al., “La acetilación de lisina se dirige a los complejos proteicos y co-regula las principales funciones celulares”, Ciencia, vol. 325, núm. 5942, págs. 834–840, 2009. [104] RH Goodman y S. Smolik, “CBP/p300 en el crecimiento, transformación y desarrollo celular”,Genes y desarrollo, vol. 14, núm. 13, págs. 1553–1577, 2000. [105] MC Haigis y LP Guarente, “Las sirtuinas de los mamíferos: funciones emergentes en la fisiología, el envejecimiento y la restricción calórica”, Genes y desarrollo, vol. 20, no. 21, págs. 2913–2921, 2006. [106] S. Michan y D. Sinclair, “Sirtuinas en mamíferos: conocimientos sobre su función biológica”,Revista bioquímica, vol. 404, núm. 1, págs. 1–13, 2007. [107] L. Gan y L. Mucke, “Caminos de convergencia: sirtuinas en el envejecimiento y la neurodegeneración”,Neurona, vol. 58, no. 1, págs. 10–14, 2008. [108] C. Julien, C. Tremblay, V. Émond et al., "La reducción de Sirtuin 1 es paralela a la acumulación de tau en la enfermedad de Alzheimer". Revista de neuropatología y neurología experimental, vol. 68, no. 1, págs. 48–58, 2009. [109] G. Donmez, D. Wang, DE Cohen y L. Guarente, “SIRT1 suprimeβ -producción de amiloide mediante la activación de laalfa- gen secretasa ADAM10”,Celúla, vol. 142, no. 2, págs. 320–332, 2010. [110] GC Ruben, K. Iqbal, I. Grundke-Iqbal, HM Wisniewski, T. L. Ciardelli y JE Johnson, “La proteína tau asociada a los microtúbulos forma un polímero helicoidal de triple cadena de izquierda a derecha”,Revista de química biológica, vol. 266, núm. 32, págs. 22019–22027, 1991. [111] M. Von Bergen, P. Friedhoff, J. Biernat, J. Heberle, EM Mandelkow y E. Mandelkow, “Asamblea deτLa proteína en filamentos helicoidales emparejados de Alzheimer depende de un motivo de secuencia local (306VQIVYK311) que formaβestructura," Actas de la Academia Nacional de Ciencias de los Estados Unidos de América, vol. 97, no. 10, págs. 5129–5134, 2000. [96] KS Kosik, CL Joachim y DJ Selkoe, “Proteína tau asociada a microtúbulos (τ) es un componente antigénico importante de los filamentos helicoidales pareados en la enfermedad de Alzheimer”. Actas de la Academia Nacional de Ciencias de los Estados Unidos de América, vol. 83, núm. 11, págs. 4044–4048, 1986. [112] M. Arrasate, M. Pérez, R. Armas-Portela y J. Ávila, “Polimerización de péptidos tau en estructuras fibrilares. El efecto de las mutaciones de FTDP-17”,Cartas de la FEBS, vol. 446, no. 1, págs. 199–202, 1999. [97] S. Mondragón-Rodrı́guez, G. Basurto-Islas, I. Santa-Maria et al., "La escisión y los cambios conformacionales de la proteína tau siguen la fosforilación durante la enfermedad de Alzheimer".Revista internacional de patología experimental, vol. 89, no. 2, págs. 81– 90, 2008. [113] A. Watanabe, K. Takio y Y. Ihara, “Desamidación y formación de isoaspartato en tau untada en filamentos helicoidales pareados: propiedades inusuales del dominio de unión a microtúbulos de tau”,Revista de química biológica, vol. 274, núm. 11, págs. 7368–7378, 1999. [98] M. Saito, G. Chakraborty, RF Mao, SM Paik, C. Vadasz y M. Saito, “Fosforilación y escisión de Tau en la neurodegeneración inducida por etanol en el cerebro de ratón en desarrollo”, Investigación neuroquímica, vol. 35, no. 4, págs. 651–659, 2010. [114] RA Crowther, OF Olesen, R. Jakes y M. Goedert, “Las repeticiones de unión [99] TT Rohn, RA Rissman, MC Davis, YE Kim, CW Cotman y E. Head, “Activación de la caspasa-9 y escisión de tau por caspasa en el cerebro con enfermedad de Alzheimer”,Neurobiología de la enfermedad, vol. 11, no. 2, págs. 341–354, 2002. [115] M. Pérez, JM Valpuesta, M. Medina, E. Montejo De Garcini y J. Ávila, “Polimerización deτen filamentos en presencia de heparina: la secuencia mínima requerida paraτ- τ interacción,"Revista de neuroquímica, vol. 67, no. 3, págs. 1183–1190, 1996. [100] Q. Zhang, X. Zhang y A. Sun, “La tau truncada en D421 está asociada con la neurodegeneración y la formación de ovillos en el cerebro de modelos transgénicos de Alzheimer”,Acta neuropatológica, vol. 117, núm. 6, págs. 687–697, 2009. de microtúbulos de la proteína tau se ensamblan en filamentos como los que se encuentran en la enfermedad de Alzheimer”,Cartas de la FEBS, vol. 309, no. 2, págs. 199–202, 1992. [116] M. Goedert, R. Jakes, MG Spillantini, M. Hasegawa, M. J. Smith y RA Crowther, “Ensamblaje de proteína tau asociada a microtúbulos en filamentos similares al Alzheimer inducidos 9730, 2012, 1, Descargado de https://onlinelibrary.wiley.com/doi/10.1155/2012/731526 por Cochrane Perú, Wiley Online Library el [17/02/2025]. Consulte los Términos y condiciones (https://onlinelibrary.wiley.com/terms-and-conditions) en Wiley Online Library para conocer las reglas de uso; los artículos de acceso abierto se rigen por la Licencia Creative Commons aplicable. 11 Revista internacional sobre la enfermedad de Alzheimer Revista internacional sobre la enfermedad de Alzheimer por glicosaminoglicanos sulfatados”,Naturaleza, vol. 383, núm. 6600, enfermedad,"Revista de ciencia celular, vol. 113, no. 21, págs. 3737– págs. 550–553, 1996. 3745, 2000. [117] T. Kampers, P. Friedhoff, J. Biernat, EM Mandelkow y [132] TT Rohn, RA Rissman, E. Head y CW Cotman, “Activación de la caspasa en E. Mandelkow, “El ARN estimula la agregación de la proteína tau asociada a el cerebro con enfermedad de Alzheimer: tortuosa y tortuosa”,Noticias los microtúbulos en filamentos helicoidales emparejados similares al y perspectivas sobre medicamentos, vol. 15, no. 9, págs. 549–557, Alzheimer”,Cartas de la FEBS, vol. 399, no. 3, págs. 344–349, 1996. [118] DM Wilson y LI Binder, “Los ácidos grasos libres estimulan la polimerización de tau y amiloideβPéptidos: evidencia in vitro de un efector común de patogénesis en la enfermedad de Alzheimer”,Revista Americana de Patología, vol. 150, núm. 6, págs. 2181–2195, 1997. [119] S. Barghorn y E. Mandelkow, “Hacia un esquema unificado para la agregación de tau en filamentos helicoidales emparejados de Alzheimer”,Bioquímica, vol. 41, núm. 50, págs. 14885–14896, 2002. [120] TC Gamblin, RW Berry y LI Binder, “Polimerización de Tau: función del extremo amino”,Bioquímica, vol. 42, núm. 7, págs. 2252–2257, 2003. [121] J. Kuret, CN Chirita, EE Congdon et al., "Pathways of tau fibrillization",Acta bioquímica y biofísica, vol. 1739, núm. 2, págs. 167–178, 2005. [122] ME King, TC Gamblin, J. Kuret y LI Binder, “Ensamblaje diferencial de isoformas de tau humana en presencia de ácido araquidónico”,Revista de neuroquímica, vol. 74, núm. 4, págs. 1749–1757, 2000. [123] KR Patterson, C. Remmers, Y. Fu et al., “Caracterización de oligómeros de tau prefibrilares in vitro y en la enfermedad de Alzheimer”, Revista de química biológica, vol. 286, núm. 26, págs. 23063–23076, 2011. [124] K. Santacruz, J. Lewis, T. Spires et al., “Medicina: la supresión de tau en un modelo de ratón neurodegenerativo mejora la función de la memoria”, Ciencia, vol. 309, núm. 5733, págs. 476–481, 2005. [125] A. Sydow, A. Van Der Jeugd, F. Zheng et al., “Los defectos inducidos por tau en la plasticidad sináptica, el aprendizaje y la memoria son reversibles en ratones transgénicos después de desactivar el mutante tau tóxico”,Revista de neurociencia, vol. 31, no. 7, págs. 2511– 2525, 2011. [126] Z. Berger, H. Roder, A. Hanna et al., “Acumulación de especies tau patológicas y pérdida de memoria en un modelo condicional de tauopatía”,Revista de neurociencia, vol. 27, núm. 14, págs. 3650– 3662, 2007. [127] CM Wischik, M. Novak, HC Thogersen et al., “Aislamiento de un fragmento de tau derivado del núcleo del filamento helicoidal emparejado de la enfermedad de Alzheimer”,Actas de la Academia Nacional de Ciencias de los Estados Unidos de América, vol. 85, no. 12, págs. 4506–4510, 1988. 2002. [133] SM De La Monte, YK Sohn y JR Wands, “Correlaciones de la apoptosis mediada por p53 y Fas (CD95) en la enfermedad de Alzheimer”,Revista de Ciencias Neurológicas, vol. 152, no. 1, págs. 73–83, 1997. [134] C. Stadelmann, TL Deckwerth, A. Srinivasan et al., “Activación de la caspasa-3 en neuronas individuales y gránulos autofágicos de degeneración granulovacuolar en la enfermedad de Alzheimer: evidencia de muerte celular apoptótica”,Revista Americana de Patología, vol. 155, núm. 5, págs. 1459–1466, 1999. [135] TT Rohn, E. Head, JH Su et al., “Correlación entre la activación de la caspasa y la formación de ovillos neurofibrilares en la enfermedad de Alzheimer”,Revista Americana de Patología, vol. 158, no. 1, págs. 189–198, 2001. [136] Z. Nagy y MM Esiri, “Expresión de proteínas relacionadas con la apoptosis en el hipocampo en la enfermedad de Alzheimer”,Neurobiología del envejecimiento, vol. 18, no. 6, págs. 565–571, 1997. [137] H. Guo, S. Albrecht, M. Bourdeau, T. Petzke, C. Bergeron y AC LeBlanc, “Caspasa-6 activa y tau escindida por caspasa-6 en hilos de neuropilo, placas neuríticas y ovillos neurofibrilares de la enfermedad de Alzheimer”,Revista Americana de Patología, vol. 165, no. 2, págs. 523–531, 2004. [138] JH Su, M. Zhao, AJ Anderson, A. Srinivasan y CW Cotman, “Expresión de caspasa-3 activada en cerebros de Alzheimer y de control envejecidos: correlación con la patología de Alzheimer”, Investigación del cerebro, vol. 898, no. 2, págs. 350–357, 2001. [139] V. Cryns y J. Yuan, “Proteasas por las que morirse”,Genes y desarrollo, vol. 12, núm. 11, págs. 1551–1570, 1998. [140] FG Gervais, D. Xu, GS Robertson et al., “Participación de las caspasas en la escisión proteolítica de la proteína amiloide de Alzheimer”.β Proteína precursora y amiloidogénica Aβformación de péptidos”, Celúla, vol. 97, no. 3, págs. 395–406, 1999. [141] G. Smale, NR Nichols, DR Brady, CE Finch y W. E. Horton, “Evidencia de muerte celular apoptótica en la enfermedad de Alzheimer”,Neurología experimental, vol. 133, no. 2, págs. 225– 230, 1995. [142] RA Rissman, WW Poon, M. Blurton-Jones et al., “La escisión de tau por caspasa es un evento temprano en la patología de la maraña de la enfermedad de Alzheimer”,Revista de investigación clínica, vol. 114, no. 1, págs. 121–130, 2004. [128] M. Novak, J. Kabat y CM Wischik, “Caracterización molecular de la unidad tau mínima resistente a la proteasa del filamento helicoidal apareado de la enfermedad de Alzheimer”,La revista EMBO, vol. 12, no. 1, págs. 365–370, 1993. [143] CW Cotman, WW Poon, RA Rissman y M. Blurton-Jones, “El papel de la escisión de tau por caspasa en la neuropatología de la enfermedad de Alzheimer”,Revista de neuropatología y neurología experimental, vol. 64, no. 2, págs. 104–112, 2005. [129] R. Mena, PC Edwards, CR Harrington, EB Mukaetova-Ladinska y CM Wischik, “Estadificación del ensamblaje patológico de la proteína tau truncada en filamentos helicoidales apareados en la enfermedad de Alzheimer”,Acta neuropatológica, vol. 91, no. 6, págs. 633–641, 1996. [144] RW Berry, A. Abraha, S. Lagalwar et al., “Inhibición de la polimerización de [130] F. Garcı́a-Sierra, CM Wischik, CR Harrington, J. Luna-Muñoz y R. Mena, “Acumulación de proteína tau truncada en el extremo C asociada con la vulnerabilidad de la vía perforante en etapas tempranas de patología neurofibrilar en la enfermedad de Alzheimer”,Revista de neuroanatomía química, vol. 22, no. 1-2, págs. 65–77, 2001. [131] A. Abraha, N. Ghoshal, TC Gamblin et al., “C-terminal Inhibición del ensamblaje de tau in vitro y en el Alzheimer tau por su fragmento de escisión de caspasa carboxiterminal”, Bioquímica, vol. 42, núm. 27, págs. 8325–8331, 2003. [145] AL Guillozet-Bongaarts, F. Garcia-Sierra, MR Reynolds et al., “Truncamiento de Tau durante la evolución de los ovillos neurofibrilares en la enfermedad de Alzheimer”,Neurobiología del envejecimiento, vol. 26, núm. 7, págs. 1015– 1022, 2005. [146] AL Guillozet-Bongaarts, ME Cahill, VL Cryns, MR Reynolds, RW Berry y LI Binder, “La pseudofosforilación de tau en la serina 422 inhibe la escisión de la caspasa: evidencia in vitro e implicaciones para la formación de ovillos in vivo”, Revista de neuroquímica, vol. 97, no. 4, págs. 1005–1014, 2006. 9730, 2012, 1, Descargado de https://onlinelibrary.wiley.com/doi/10.1155/2012/731526 por Cochrane Perú, Wiley Online Library el [17/02/2025]. Consulte los Términos y condiciones (https://onlinelibrary.wiley.com/terms-and-conditions) en Wiley Online Library para conocer las reglas de uso; los artículos de acceso abierto se rigen por la Licencia Creative Commons aplicable. 12 [147] PM Horowitz, KR Patterson, AL Guillozet-Bongaarts et al., “Cambios [162] PJ McMillan, BC Kraemer, L. Robinson, JB Leverenz, tempranos en la terminal N y escisión de tau por caspasa-6 en la M. Raskind y G. Schellenberg, “El truncamiento de tau en E391 promueve cambios patológicos tempranos en ratones transgénicos”, Revista de neuropatología y neurología experimental, vol. 70, núm. 11, págs. 1006–1019, 2011. enfermedad de Alzheimer”,Revista de neurociencia, vol. 24, núm. 36, págs. 7895–7902, 2004. [148] B. Bandyopadhyay, G. Li, H. Yin y J. Kuret, “Agregación de tau y toxicidad en un modelo de cultivo celular de tauopatía”, Revista de química biológica, vol. 282, núm. 22, págs. 16454–16464, 2007. [149] N. Canu, L. Dus, C. Barbato et al., “Escisión y desfosforilación de Tau en neuronas granulares cerebelosas en proceso de apoptosis”, Revista de neurociencia, vol. 18, no. 18, págs. 7061–7074, 1998. [163] G. Basurto-Islas, J. Luna-Muñoz, AL Guillozet-Bongaarts, LI Binder, R. Mena y F. Garcı́a-Sierra, “La acumulación de tau escindida por ácido aspártico 421 y ácido glutámico 391 en ovillos neurofibrilares se correlaciona con la progresión de la enfermedad de Alzheimer”,Revista de neuropatología y neurología experimental, vol. 67, núm. 5, págs. 470–483, 2008. A. Cattaneo, “Sobreexpresión de fragmentos de tau del núcleo PHF del [164] H. Braak y E. Braak, “Estadificación neuropatológica de los cambios relacionados con el Alzheimer”,Acta neuropatológica, vol. 82, no. 4, págs. 239–259, 1991. Alzheimer: implicaciones para la hipótesis del truncamiento de tau”, [165] F. García-Sierra, JJ Jarero-Basulto, Z. Kristofikova, E. Majer, [150] L. Fasulo, M. Ovecka, J. Kabat, A. Bradbury, M. Novak y Investigación sobre el Alzheimer, vol. 2, no. 5, págs. 195–200, 1996. [151] L. Fasulo, G. Ugolini, M. Visintin et al., “La proteína tau asociada a los microtúbulos neuronales es un sustrato para la caspasa-3 y un efector de la apoptosis”,Revista de neuroquímica, vol. 75, no. 2, págs. 624–633, 2000. [152] L. Fasulo, G. Ugolini y A. Cattaneo, “Efecto apoptótico de la tau escindida por caspasa-3 en neuronas del hipocampo y su potenciación por la mutación N279K de tau FTDP”,Revista de la enfermedad de Alzheimer, vol. 7, no. 1, págs. 3–13, 2005. [153] W. Chun y GVW Johnson, “El papel de la fosforilación y escisión de tau en la muerte de células neuronales”,Fronteras en biociencias, vol. 12, no. 2, págs. 733–756, 2007. [154] TA Matthews-Roberson, RA Quintanilla, H. Ding y GVW Johnson, “Las neuronas corticales inmortalizadas que expresan tau escindida por caspasa están sensibilizadas a la muerte celular inducida por estrés del retículo endoplásmico”.Investigación del cerebro, vol. 1234, núm. C, págs. 206–212, 2008. [155] RA Quintanilla, TA Matthews-Roberson, PJ Dolan y GVW Johnsion, “La expresión de tau escindida por caspasa induce disfunción mitocondrial en neuronas corticales inmortalizadas: implicaciones para la patogénesis de la enfermedad de Alzheimer”,Revista de química biológica, vol. 284, núm. 28, págs. 18754– 18766, 2009. [156] RA Quintanilla, PJ Dolan, YN Jin y GVW Johnson, “Tau truncada y Aβ deterioran cooperativamente las mitocondrias en las neuronas primarias”,Neurobiología del envejecimiento, vol. 33, núm. 3, págs. 619.e25–619.e35, 2012. [157] P. Filipcik, M. Cente, G. Krajciova, I. Vanicky y M. Novak, “Las neuronas corticales e hipocampales de ratas transgénicas tau truncada expresan múltiples marcadores de neurodegeneración”,Neurobiología celular y molecular, vol. 29, no. 6-7, págs. 895–900, 2009. [158] P. Delobel, I. Lavenir, G. Fraser et al., “Análisis de la fosforilación y truncamiento de tau en un modelo de ratón de tauopatía humana”,Revista Americana de Patología, vol. 172, no. 1, págs. 123–131, 2008. [159] A. De Calignon, LM Fox, R. Pitstick et al., “La activación de la caspasa precede y conduce a los ovillos”,Naturaleza, vol. 464, núm. 7292, págs. 1201–1204, 2010. [160] P. Koson, N. Zilka, A. Kovac et al., “Los niveles de expresión de tau truncada determinan la duración de vida de un modelo de rata de tauopatía sin causar pérdida neuronal ni correlacionarse con la carga de ovillos neurofibrilares terminales”,Revista Europea de Neurociencia, vol. 28, no. 2, págs. 239–246, 2008. [161] M. Cente, P. Filipcik, M. Pevalova y M. Novak, “La expresión de una proteína tau truncada induce estrés oxidativo en un modelo de tauopatía en roedores”,Revista Europea de Neurociencia, vol. 24, no. 4, págs. 1085–1090, 2006. LI Binder y D. Ripova, “La ubiquitina está asociada con el truncamiento temprano de la proteína tau en el ácido aspártico421 durante la maduración de los ovillos neurofibrilares en la enfermedad de Alzheimer”, Patología cerebral, vol. 22, no. 2, págs. 240–250, 2012. [166] F. Garcı́a-Sierra, S. Mondragón-Rodrı́guez y G. Basurto-Islas, “Truncamiento de la proteína tau y su significado patológico en la enfermedad de Alzheimer”,Revista de la enfermedad de Alzheimer , vol. 14, no. 4, págs. 401–409, 2008. 9730, 2012, 1, Descargado de https://onlinelibrary.wiley.com/doi/10.1155/2012/731526 por Cochrane Perú, Wiley Online Library el [17/02/2025]. Consulte los Términos y condiciones (https://onlinelibrary.wiley.com/terms-and-conditions) en Wiley Online Library para conocer las reglas de uso; los artículos de acceso abierto se rigen por la Licencia Creative Commons aplicable. 13 Revista internacional sobre la enfermedad de Alzheimer