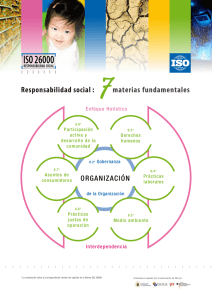
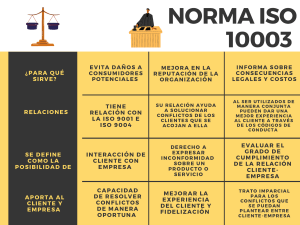
ISO 14.971:2009 Medical Devices – Application of risk management to medical devices ISO 14971:2007, corrected version 2007-10-01 Objetivos • • • • Conocer el Alcance de la Gestión de Riesgos Estructura - Requisitos Beneficios Relación con la ISO 13.485 ®2012 Det Norske Veritas – Gestión de Riesgos ISO 14971 – LYDSAC 26 October 2012 Slide 2 ISO 14971:2009 – Alcance (Scope) Especifica un proceso para el fabricante para identificar los peligros asociados con los dispositivos médicos, incluyendo los dispositivos para Diagnostico In Vitro (IVD), para estimar y evaluar los riesgos asociados, para controlar estos riesgos y para monitorear la eficacia de los controles ®2012 Det Norske Veritas – Gestión de Riesgos ISO 14971 – LYDSAC 26 October 2012 Slide 3 ISO 14971:2009 – Alcance (Scope) Los requisitos son aplicables a todas las etapas del ciclo de vida del dispositivo médico No aplica a la toma de decisiones clínicas No especifica los niveles de riesgo aceptables Esta norma internacional no requiere que el fabricante tenga un sistema de Gestión de Calidad implementado. Aunque, la Gestión de Riesgos puede ser una parte integrante del Sistema de Gestión de Calidad. ®2012 Det Norske Veritas – Gestión de Riesgos ISO 14971 – LYDSAC 26 October 2012 Slide 4 Definiciones Daño – Lesión física o deterioro de la salud de las personas, o daño a la propiedad o al medioambiente. Peligro – Potencial fuente de daño. Situación Riesgosa – Circunstancia en la cual las personas, la propiedad o el medio ambiente están expuestas a uno o mas peligros. ®2012 Det Norske Veritas – Gestión de Riesgos ISO 14971 – LYDSAC Rev.0 26 October 2012 Slide 5 Definiciones Severidad – Medida de las posibles consecuencias de un peligro. Riesgo – Combinación de la probabilidad de ocurrencia de daño y el grado de severidad de tal daño. Riesgo residual – Riesgo que permanece después de haber tomado las medidas de control de riesgos. ®2008 Det Norske Veritas – Gestión de Riesgos ISO 14971 – 081230 Rev.0 26 October 2012 Slide 6 Definiciones Uso previsto– Finalidad Prevista Uso del producto, proceso o servicio , que se pretende de acuerdo a las especificaciones, instrucciones y la información provista por el fabricante. Dispositivo Médico – Cualquier instrumento, aparato, implemento, máquina, dispositivo, implante, reactivos o calibrador in vitro, software, material o cualquier otro ítem, similar o de uso relacionado que el fabricante pretender para ser utilizado solo o en combinación para seres humanos para uno o más de los siguientes propósitos: ®2012 Det Norske Veritas – Gestión de Riesgos ISO 14971 – LYDSAC 26 October 2012 Slide 7 Dispositivo médico (Cont.) Diagnóstico, prevención, monitoreo o tratamiento, o alivio de enfermedades. Diagnóstico, monitoreo, tratamiento, alivio o compensación de una lesión. Investigación, reemplazo. Modificación o soporte de la anatomía o de un proceso fisiológico. Soporte o sostenimiento de la vida. Control de la concepción Desinfección de dispositivos médicos Proveer información para propósitos médicos por medio de examinaciones in vitro de muestras obtenidas del cuerpo humano. Y que no realiza su función principal en o sobre el cuerpo humano por medios farmacológicos, inmunológicos o metabólicos, pero que puede ser apoyado en su función por tales medios. ®2008 Det Norske Veritas – Gestión de Riesgos ISO 14971 – 081230 Rev.0 26 October 2012 Slide 8 Proceso de gestión de riesgos Seguimiento Producción Análisis Post producción Control Evaluación Gestión de riesgo – Aplicación sistemática de políticas, procedimientos y practicas de gestión a las tareas de analizar, estimar, controlar y monitorear riesgos. ®2012 Det Norske Veritas – Gestión de Riesgos ISO 14971 – LYDSAC 26 October 2012 Slide 9 Responsabilidades – Alta Dirección Asegurar la provisión de recursos adecuados Asegurar la asignación de personal calificado. Definir y documentar su política para determinar criterios para la aceptabilidad de riesgos , teniendo en cuenta Normas Internacionales, regulaciones nacionales o internacionales, información disponible, “estado del arte”. Revisar la adecuación del proceso de gestión de riesgos a intervalos planificados para asegurar la continua eficacia del proceso de gestión de riesgos y documentar cualquier decisión y acción tomada ®2012 Det Norske Veritas – Gestión de Riesgos ISO 14971 – LYDSAC 26 October 2012 Slide 10 Política de Aceptabilidad de Riesgo Para determinar criterios de aceptabilidad y guía para desarrollo de producto Aplicación de normas Internacionales, regulaciones, información disponible, “Estado del Arte” ALARP ( as low as reasonably practicable) TAN BAJO COMO SEA RAZONABLEMENTE POSIBLE ®2012 Det Norske Veritas – Gestión de Riesgos ISO 14971 – LYDSAC 26 October 2012 Slide 11 Calificación del personal Conocimiento y experiencia apropiado a las tareas asignadas Conocimiento y experiencia del: - Dispositivo medico o similares - Del uso previsto - Tecnologías involucradas - Técnicas de gestión de riesgos Registros ®2012 Det Norske Veritas – Gestión de Riesgos ISO 14971 – LYDSAC 26 October 2012 Slide 12 Archivo de gestión de riesgos Conjunto de registros y otros documentos que se producen por un proceso de gestión de riesgos. Debe ser establecido y mantenido por el fabricante Debe proveer trazabilidad para cada peligro identificado a : - Análisis de riesgo - Evaluación de riesgo - Implementación y verificación de las medidas de control - La evaluación de cualquier riesgo residual ®2012 Det Norske Veritas – Gestión de Riesgos ISO 14971 – LYDSAC 26 October 2012 Slide 13 Análisis de riesgo - Documentación Descripción e identificación del dispositivo Uso previsto Características cuantitativas y cualitativas que pueda afectar a la seguridad ( límites ) Lista de peligros conocidos o previsibles (normal y de fallo) Estimación de riesgos Persona y organización Alcance y fecha Cumplimiento es verificado por inspección del archivo de gestión de riesgo ®2008 Det Norske Veritas – Gestión de Riesgos ISO 14971 – 081230 Rev.0 26 October 2012 Slide 14 Técnicas de análisis de riesgos E S T R U C T U R A •HACCP • Hazop • FMEA • Árbol de fallas (FTA) • Análisis de Riesgo Preliminar (PHA) • Evaluación de Riesgos por Grupo • Técnica de "¿Qué pasa si..? (SWIF) • Listas de verificación • Evaluación Simple de Riesgos Minutos ®2012 Det Norske Veritas – Gestión de Riesgos ISO 14971 – LYDSAC TIEMPO 26 October 2012 Semanas Slide 15 Diseño inherente a seguridad Eliminar el riesgo en particular Reducir la probabilidad de ocurrencia del daño Reducir la severidad ®20012 Det Norske Veritas – Gestión de Riesgos ISO 14971 – LYDSAC 26 October 2012 Slide 16 Medidas protectoras Uso de corte automático o válvulas de seguridad Uso de alarmas acústicas o visuales para alertar al operador de las condición riesgosa ®2012 Det Norske Veritas – Gestión de Riesgos ISO 14971 – LYDSAC 26 October 2012 Slide 17 Valoración del riesgo residual Control para determinar si las medidas llevan al riesgo a un valor aceptable Los mismos criterios definidos en el plan de gestión de riesgos Riesgos residuales no aceptables, aplicar medidas adicionales hasta reducir hasta valores aceptables. Riesgos aceptables, decidir sobre que riesgo debe ser revelado y que información es necesaria incluir en los documentos acompañantes. ®2012 Det Norske Veritas – Gestión de Riesgos ISO 14971 – LYDSAC 26 October 2012 Slide 18 Análisis riesgo/ Beneficios Riesgo inaceptable y medidas de control de riesgos no practicas Justificar aceptabilidad del riesgo basado en el beneficio Recopilar y revisar datos y literatura para determinar si los beneficios compensan el riesgo residual Individuos experimentados y con conocimiento especifico Ensayos clínicos En otras palabras….es la justificación y/o DDJJ del Fabricante ®2012 Det Norske Veritas – Gestión de Riesgos ISO 14971 – LYDSAC 26 October 2012 Slide 19 Directiva 93/42 de Dispositivos Médicos s/enmienda 2007/47:EC Anexo IX Clasificación Es la responsabilidad del fabricante clasificar el Dispositivo Médico basado en el USO PREVISTO. Clasificación basada en el Riesgo Clase I (incluye esterilización y medición) Clase IIa Clase II b Clase III Requisitos Esenciales • Los REQUISITOS ESENCIALES constituyen el conjunto total de requisitos que deben cumplirse para todo tipo de Dispositivo Médico, independientemente de su clasificación • Donde se documentan? En el Check List de Requisitos Esenciales Requisitos Esenciales Se puede demostrar CONFORMIDAD a través de: • LAS NORMAS ARMONIZADAS • ENSAYOS / TESTEOS • INVESTIGACION CLINICA • EVALUACION CLINICA Y DATOS CLINICOS • ANALISIS DE RIESGOS (ISO 14.971) Guía de Clasificación GUIA MEDDEV 2.1/4 Independientemente de la clase del dispositivo, todos los dispositivos deben: - Cumplir los requisitos esenciales, incluidos los requisitos relativos a la información a suministrar por el fabricante (Anexo I de la Directiva 93/42/CEE); - Estar sujetos a las obligaciones establecidas por el sistema de dispositivo de vigilancia médica. http://ec.europa.eu/consumers/sectors/medical4 1 rev 9 classification en.pdf devices/files/meddev/2 Condiciones • Contratos por 5 años • Auditorías y Evaluaciones de Conformidad de CE Marking realizadas por Equipos de Expertos Interdisciplinarios • Cumplimiento y conformidad con • los Requisitos Esenciales Declaración CE y Marcado CE en los Productos una vez aprobados por DNV Reunir todas las normas "armonizadas" aplicables Establecer un SGC Según las normas ISO 13485 y 14971 y los requerimientos esenciales (Enfoque de Procesos) Establecer el análisis de riesgo para cada producto (Enfoque de Producto) ¿Nivel de riesgo residual aceptable? NO Se adoptan las medidas remediadoras SI Con los datos del Análisis de Riesgo se realizan todos los ensayos que solicitan las normas para validar diseño del producto y del proceso ¿Resultados satisfactorios? NO Ajustes de corrección sobre procesos y productos SI Se confecciona el listado de requerimientos esenciales Se arma el file técnico Se redacta la Nota de Conformidad Certificación del sistema proceso / producto ante DNV PROCESO DE OBTENCIÓN DEL "CE MARKING"