1. Introducción:
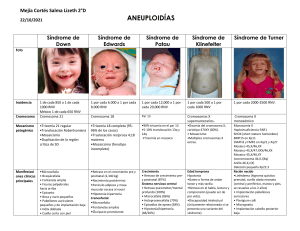
El síndrome de Edwards o trisomía 18, descrito en 1 960 por Edwards
y col (1), es la segunda trisomía autosómica más frecuente en recién
nacidos (2). Su incidencia es de 1/3 000 - 1/8 000 nacidos vivos, pero
su incidencia real depende del diagnóstico prenatal. (3,4). Es más
frecuente en madres de edad avanzada (2,5), es un síndrome
polimórfico, que se caracteriza por anomalías congénitas múltiples (6);
siendo las más frecuentes el retraso del crecimiento prenatal y
posnatal, retraso psicomotor y mental severo, alteraciones del tono
muscular, anomalías cráneo faciales, anomalías en extremidades,
hipoplasia ungueal, esternón corto, malformaciones urogenitales,
gastrointestinales, del sistema nervioso central y, especialmente,
cardiopatías congénitas. La supervivencia media es de 2,5 a 70 días
entre el primer y cuarto mes de vida (2) y entre 90 y 95% muere en el
primer año de vida (3). Los casos con supervivencia mayor de un año
suelen sobrevivir más tiempo (2% hasta los 5 años de vida), siendo
excepcionales los casos que llegan a la adolescencia (2). Debido a la
poca frecuencia de pacientes con este síndrome y más aún, con larga
supervivencia
2. Objetivos:
La trisomía se define por tener un cromosoma adicional agregado a un
par de cromosomas en este caso es al par dieciocho. El diagnóstico
habrá de hacerse mediante el estudio cromosómico. La trisomía del
cromosoma 18, aparece con mayor frecuencia en madres de edad
avanzada, a partir de los 35 años la frecuencia aumenta
progresivamente desde 1/2.500 hasta 1/500 a los 43 años.
En mujeres de más 35 años de edad, o con un hijo anterior con
trisomía 18 debe hacerse diagnóstico prenatal
mediante amniocentesis (procedimiento obstétrico mediante el cual se
extrae una pequeña cantidad de líquido amniótico para su posterior
análisis).
Síntomas:
Poco peso al nacimiento
Deformaciones en el cráneo y rostro, como la microcefalia, mandíbula
pequeña, orejas pequeñas y desubicadas, boca pequeña o paladar
ojival
Tórax en forma de quilla
Criptorquidia
Uñas poco desarrolladas
Pies convexos
Deficiencia del crecimiento
Forma anormal del cráneo y características faciales
Manos apretadas
Pies inferiores del eje de balancín
Anormalidades cardiacas y renales
En los exámenes efectuados en la mujer embarazada puede aparecer
un útero más grande de lo normal y una mayor cantidad de líquido
amniótico. Tras el nacimiento también se ha constatado la presencia de
una placenta inusualmente pequeña.
Otros síntomas que se aprecian en el recién nacido son:
Problemas oculares, como hendidura en el iris, coloboma o cataratas.
Diástasis de rectos (del músculo recto abdominal).
Malformaciones esqueléticas como la escoliosis, hipoplasia o aplasia.
Hernia umbilical o inguinal.
Cardiopatías como comunicación interauricular o intraventricular o
conducto arterial persistente. Las cardiopatías están presentes en el
90% de los casos.
Problemas renales como el riñón poliquístico, riñón en herradura o
hidronefrosis (acumulación anormal de orina)
3. Marco Teórico:
El síndrome de Edwards, descrito en 1960, es un síndrome
polimalformativo debido a la existencia de tres cromosomas 18. Su
incidencia es de 1/3.000-1/8.000 nacidos vivos1, dependiendo de la
realización de diagnóstico prenatal
El 95 % de los casos corresponde a trisomía completa3,4. Aparece con
mayor frecuencia en mujeres (4:1) y está asociado a edad avanzada de
la madre4. Presenta numerosas anomalías congénitas1–3,5. Las más
frecuentes son el retraso del crecimiento prenatal y posnatal, retraso
psicomotor y mental, alteraciones del tono muscular, anomalías
craneofaciales, anomalías en extremidades, hipoplasia ungueal,
esternón corto, malformaciones urogenitales, gastrointestinales, del
sistema nervioso central y, especialmente, cardiopatías congénitas (90
%)1,3,4.
El 90–95 % muere en el primer año de vida2,3,6–9. Son excepcionales los
casos que llegan a la adolescencia1,4,9.
Se presenta el caso de una paciente con trisomía 18 completa y
supervivencia superior a los 15 años. Nacida a término mediante
cesárea por depresión neonatal, presentaba rasgos dismórficos
múltiples.
El cariotipo demostró en todas las metafases estudiadas una trisomía
18 (47,XX, +18). El estudio radiológico mostraba cardiomegalia y
afectación perihiliar, costillas adelgazadas y subluxación coxofemoral
bilateral. La ecografía cerebral presentaba discreta dilatación de los
ventrículos laterales. La ecografía renal fue normal. En el estudio
cardiológico
se
evidenció ductus permeable,
comunicación
interventricular restrictiva, ausencia del septo membranoso y coartación
de aorta.
Durante su infancia presentó hiperreactividad bronquial, rinitis
obstructiva, infecciones urinarias, bronquitis, neumonías de repetición,
crisis hipertensivas y cuadros convulsivos. Se detectó divertículo de
Meckel, que precisó intervención, y tejido pancreático ectópico. Sufrió
fractura de fémur derecho y de ambas clavículas. Ha sido marcado su
retraso ponderoestatural, psicomotor y mental. Hace 3 años se hizo
necesaria la implantación de gastrostomía.
La paciente fue examinada en los primeros meses de vida y se indicó
tratamiento fisioterápico en nuestro Servicio. Debido a sus múltiples
complicaciones y reagudizaciones se deterioró su estado general, lo
que imposibilitó las salidas del domicilio de la niña de forma regular y se
perdió el seguimiento por parte del servicio de Rehabilitación. En 2004,
en relación con un reingreso hospitalario, la paciente fue nuevamente
remitida a nuestro servicio.
Actualmente presenta pobre conexión al medio, tetraparesia espástica
y múltiples deformidades ortopédicas secundarias, destacando su
escoliosis (fig. 1), miembros en flexión y pies equinovaro-aductos. La
niña no presenta dolor y su equilibrio articular permite el aseo adecuado.
No tolera lecho postural y sólo se logra sedestación en silla durante
unos períodos de tiempo muy limitados.
4. Conclusiones:
En los casos de T18 la mortalidad in útero y neonatal es alta, las
características clínicas in útero y en recién nacidos han sido
bien descritas. Dado que son pocos los casos que superan los 5
años el fenotipo aún está por establecerse. En la paciente aquí
reportada se encontraron hallazgos en la cavidad oral no
descritos en la literatura.
5.Bibliografía:
Baty B, Blackburn B, Carey J. Natural history of trisomy 18 and 13: I.
Growth, physical assessment, medical histories, survival and
recurrence risk. Am J Med Genet. 1994;49:175-88. • Baty B, Jorde L,
Blackburn B, Carey J. Natural history of trisomy 18 and 13: II.
Psychomotor development. Am J Med Genet 1994;49:189-94. •
Boghosian-Sell L, Mewar R, Harrison W, Shapiro RM, Zackai EH,
Carey J et al. Molecular mapping of the Edwards syndrome to two
noncontiguous regions on chromosome 18. Am J Hum Genet.
1994;55:476-83. • Carey J. Health supervision and anticipatory
guidance for children with genetic disorders. Pediatr Clin N Am.
1992;39(1):25-53.
 0
0