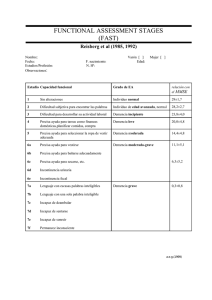
Capitulo de muestra
Anuncio

Unidad 5 Problemas del sistema nervioso 5.1 Dolor en la cara J. M. Garzón Hernández ¿De qué hablamos? Introducción El dolor facial es un problema de salud frecuente en la consulta de Atención Primaria (AP), siendo su origen muy diverso. Entre las principales causas destacan las alteraciones musculoesqueléticas, las neurovasculares y los trastornos dentales. Un buen número de las entidades que causan dolor facial se tratan en diferentes temas de esta guía, por lo que en este capítulo nos centraremos fundamentalmente en los problemas más relevantes que cursan con dolor facial crónico: el dolor facial atípico, la neuralgia del trigémino y la disfunción de la articulación temporomandibular (DATM). Clasificación De las clasificaciones que existen del dolor facial nos referiremos aquí a la determinada por la duración del cuadro, y que presenta las siguientes características: • Dolor facial agudo (< 3 meses): – Causa infecciosa, traumática o tumoral. – Inicio rápido y autolimitado. – Área facial delimitada. – Buena respuesta terapéutica. • Dolor facial crónico (> 3 meses): – Origen idiopático. – Intensidad del dolor desproporcionada a los hallazgos. – Área facial imprecisa. – Mala respuesta terapéutica. Epidemiología básica La neuralgia del trigémino afecta anualmente a 4-5 de cada 105 personas, y es más frecuente en mujeres (3:2). Las formas idiopáticas o esenciales se dan en personas mayores de 50-60 años (el 90 % de los casos comienzan después de los 40 años). Por debajo de esa edad, hay que sospechar formas sintomáticas o secundarias. La DATM ocurre en aproximadamente el 10 % de la población mayor de 18 años, es principalmente una enfermedad de adultos jóvenes y de mediana edad, no habitual en niños o ancianos, y es aproximadamente dos veces más común en mujeres que en hombres. El dolor facial atípico es más frecuente en las mujeres de 30 a 50 años de edad. Tabla 1. Causas de dolor facial Trastornos dentales Sinusitis aguda/crónica Otitis externa o media DATM Cefaleas Trastornos del SNC Neuralgia del trigémino ¿Qué lo puede ocasionar? Causas En la tabla 1 se exponen las causas del dolor facial, y en la tabla 2 se detallan las más frecuentes del dolor facial crónico. ¿Qué tenemos que hacer? Anamnesis Es fundamental realizar una buena historia clínica y una detallada exploración física, ya que el diagnóstico, en la mayoría de los casos, es clínico, pudiendo alcanzarse con estas dos herramientas en el 70 % de los casos. • Antecedentes familiares: tumores, neuralgia del trigémino. • Antecedentes personales: sinusitis, alergias, cefaleas, hipertensión arterial (HTA), trastornos neurológicos (incluyendo esclerosis múltiple), trastornos de ansiedad o del humor, traumatismos, historia dental, estado general, apetito, pérdida de peso, tratamientos farmacológicos previos. • Enfermedad actual: es fundamental el análisis de las características del dolor: inicio, duración, localización, evolución, causas desencadenantes o atenuantes. También es imprescindible interrogar sobre síntomas acom- Neuralgia del glosofaríngeo Tumor cerebral Traumatismo craneal Tabla 2. Causas más frecuentes de dolor facial crónico Infarto talámico Causas Dolor facial atípico o persistente idiopático Dolor facial atípico o persistente idiopático 33 Dolor de origen ocular DATM 25 Odontalgia atípica 25 Patología región orofacial 16 Dolor torácico isquémico DATM: disfunción de la articulación temporomandibular; SNC: sistema nervioso central. Porcentaje (%) DATM: disfunción de la articulación temporomandibular. 397 5.1 Dolor en la cara pañantes como cefalea, dolores o ruidos articulares, vértigos, síntomas oculares, apetito, peso, estado de ánimo, estrés, etc. Los criterios diagnósticos específicos de la neuralgia del trigémino esencial o primaria son los siguientes: – Crisis paroxísticas de dolor facial o frontal cuya duración oscila entre unos segundos y 2 minutos. – El dolor presenta las características siguientes: distribución en el territorio de las ramas del nervio trigémino (oftálmica, maxilar o mandibular). La afectación de la rama oftálmica es infrecuente. Fundamentalmente en labios, encías, carrillos y mentón. Repentino, intenso, agudo, superficial y punzante o quemante. Duración de pocos segundos a 1-2 minutos. Se produce por estimulación de zonas «gatillo». El comienzo suele ser repentino y las crisis suelen durar varias semanas o meses con remisión espontánea durante largo tiempo pero que puede recidivar. – En períodos intercrisis el paciente está asintomático. – No existe déficit neurológico, incluyendo la sensibilidad. – Los ataques son estereotipados en cada paciente. – Deben excluirse, mediante la historia clínica, la exploración física y las pruebas complementarias, otras causas de dolor facial (figura 1). • Diferencias clínicas entre la neuralgia del trigémino primaria y secundaria (tabla 3). La neuralgia del trigémino en una persona joven o de aparición bilateral debe hacer pensar en una esclerosis múltiple. La pérdida sensitiva en el territorio del trigémino siempre debe hacer pensar en una neuralgia del trigémino secundaria a otro problema como esclerosis múltiple, accidente cerebrovascular agudo (ACVA), tumores, infecciones. • En la neuralgia del glosofaríngeo (muy rara) el dolor es similar al del trigémino pero se localiza en la fosa amigdalar, mitad de la faringe, y se puede irradiar al oído. El dolor puede ser desencadenado por la deglución o la tos. La edad de inicio es entre 40-60 años. • Bajo la denominación de dolor facial atípico o dolor facial persistente idiopático se incluyen aquellos pacientes cuyo dolor no se adapta a las características de los otros procesos, ya sea en duración, localización o factores desencadenantes. Suele ser de origen desconocido, sobrepasa el territorio del trigémino y no presenta zonas gatillo. Exploración física • Exploración general: incluyendo peso, índice de masa corporal (IMC) y presión arterial (PA). • Exploración craneocervical: – Asimetrías faciales (tumefacciones, desviaciones, etc.). – Alteraciones sensitivas (indica patología orgánica subyacente). – Alteraciones de la estática cervical. – Presencia o no de puntos gatillo en la cara. – Inspección y palpación de las arterias temporales y de los globos oculares. • Exploración de la articulación temporomandibular (ATM), (indicando al paciente, mientras se realiza la palpación bimanual de ambas articulaciones, que abra y cierre la boca y que realice movimientos laterales). Inspección, palpación dental y de la oclusión de los mismos. • Exploración del sistema neuromuscular de la cara: – Inspección de los músculos: sobre todo los maseteros (inervados por el trigémino). – Palpación de los músculos maseteros para descartar hipertrofias musculares localizadas, del temporal, pterigoideos externo e interno, trapecio y esternocleidomastoideo, en búsqueda de puntos dolorosos o espasmos musculares. Exploraciones complementarias Iniciales • Analítica: hemograma, velocidad de sedimentación globular (VSG) (muy elevada en la arteritis de la temporal), proteinograma. • Radiografías: craneofacial (senos paranasales), base de cráneo y columna cervical (temas 24.9 y 24.5). Posteriores • Analítica: factor reumatoide (se recomienda en el estudio de la DATM rebelde al tratamiento). • Radiografía de la ATM. • Tomografía computarizada (TC) craneofacial o resonancia magnética (RM): siendo esta última la exploración más eficaz para descartar una neuralgia sintomática o secundaria, con una sensibilidad del 98 % y una especificidad del 96 %. Tabla 3. Diferencias entre la neuralgia del trigémino primaria y secundaria 398 Primaria Secundaria Etiología Desconocida Lesión compresiva Edad > 50 años < 50 años Afectación Unilateral Unilateral o bilateral Calidad dolor Paroxismos Continuo Puntos gatillo Frecuente Infrecuente Déficit neurológico No Frecuente Respuesta a fármacos Buena Escasa Dolor de cabeza Dolor periorbitario intenso con síntomas vegetativos Dolor de cuello, mandíbula de tipo isquémico Dolor unilateral continuo Sinusitis Tema 5.2 Cefalea en racimos Tema 3.2 Tema 2.1 Dolor ocular Sí No Seguimiento en APS Sí Responde Tto. en APS No Carotidinia Cuello Derivación a neurología Hipersensibilidad en el lado del dolor y carótida prominente Neuralgia del glosofaríngeo Neuralgia del trigémino < 40 años déficit sensitivo intercrisis dudas diagnósticas Garganta Sí Dolor de localización predominante Sd. bulbar lateral Sí Derivación urgente al hospital Dolor facial atípico Sí Derivación a Cirugía maxilofacial Derivación a reumatología Diagnóstico de confirmación: Biopsia arteria temporal Tto: Prednisona 60 mg 1 mes Descenso gradual según VSG/PCR mensual Sospecha de arteritis temporal Incremento VSG y PCR Anemia Sí No ¿Responde? Bloqueo Anquilosis No Derivación a odontología Dolor dental atípico Ruidos Dinámica alterada Patología ATM Sí No Dolor con la masticación Se asocia a cefalea frontotemporal y ocasionalmente fiebre o claudicación mandibular No Dolor quemante, de hormigueo o descarga eléctrica, pueden desencadenarlo roces suaves. Dolor neuropático Afectación hemicara Sí No Alta sensibilidad termoalgésica en hemicuerpo contralateral No No Dolor con sintomatología y localización características Mandibular Sí APS: atención primaria de salud; ATM: articulación temporomandibular; PCR: proteína C reactiva; TCE: traumatismo craneoencefálico; VSG: velocidad de sedimentación globular. Figura 1. Algoritmo de actuación en el dolor de la cara. Tema 12.4 Tema 11.15 Dolor de oído Tema 2.11 TCE Dolor dental Tema 2.4 Dolor en la cara 5.1 Dolor en la cara 399 5.1 Dolor en la cara ¿Qué propuesta haremos? Errores más frecuentes Tratamiento Neuralgia del trigémino Farmacológico Es el tratamiento de primera elección. Se realiza con fármacos anticomiciales, cuya eficacia suele reducirse con el paso del tiempo. En períodos intercrisis de más de 6 meses se puede valorar retirar el fármaco, haciéndolo siempre de forma gradual. • Carbamazepina: fármaco de elección (50-70 % de eficacia, 20 % de recaídas y 5-10 % de reacciones adversas); dosis de inicio 100 mg/24 h/v.o., aumentando 100 mg cada 24-48 h, si es preciso, hasta 800-1.200 mg/día repartidos en 3-4 dosis, 1 hora antes de las comidas. Después de un período de estabilidad, reducir la dosis hasta la mínima necesaria para mantener al paciente sin crisis. – Efectos secundarios: somnolencia, mareo, ataxia, náuseas y raramente agranulocitosis. No debe iniciarse en los casos de anemia, plaquetopenia o leucopenia. – Control analítico previo (hemograma, función hepática, renal) y electrocardiograma, mensual los primeros 3 meses y posteriormente cada 6-12 meses, para detectar precozmente efectos secundarios que, aunque raros y, en general, idiosincrásicos (insuficiencia hepática o renal, alteraciones de la conducción cardíaca o discrasias sanguíneas) pueden obligar a retirar el fármaco. – Después de una primera crisis, si el paciente queda asintomático con el tratamiento farmacológico, intentar suprimirlo después de 8 semanas sin síntomas. Repetir el tratamiento si reaparece el dolor. • Gabapentina: buenos resultados en el tratamiento del dolor neuropático, aunque los estudios en neuralgia del trigémino son escasos y de pequeña potencia. De todas formas se recomienda como fármaco de segunda línea si falla o no se tolera la carbamazepina. Dosis progresivas hasta 900-3.600 mg/día/v.o. en tres tomas. – Efectos secundarios: mareo, somnolencia y cansancio. En determinados casos refractarios puede asociarse con la carbamazepina. Consejos prácticos • No infravalorar el dolor referido por el paciente. • No olvidar que el dolor facial puede ser la expresión de un proceso subyacente grave. • Fomentar normas de autocuidado en la DATM. • Realizar estudio de neuroimagen en toda neuralgia del trigémino secundaria o sintomática. • En el dolor facial atípico o persistente idiopático no utilizar analgésicos. Usar amitriptilina en dosis bajas: 25-100 mg en dosis única nocturna. 400 • Pensar que la sinusitis crónica es la causa más frecuente de dolor facial. • Infravalorar el componente depresivo del dolor facial crónico (puede conducir al suicidio). • No derivar para estudio una neuralgia del trigémino con alteraciones sensitivas en períodos intercrisis. • Confundir la neuralgia del trigémino con la cefalea en racimos. • Otros fármacos: la pregabalina y la lamotrigina pudieran plantearse en caso de resistencia o intolerancia a los anteriores. Tratamiento quirúrgico Es necesario en el 25-50 % de los pacientes con neuralgia del trigémino primaria. • Técnicas percutáneas: presentan un porcentaje inferior de éxito que la técnica descompresiva y un mayor índice de recidivas que obligan a reintervenir. De elección en pacientes debilitados y/o ancianos: – Termocoagulación percutánea o gangliólisis por radiofrecuencia. – Microcompresión percutánea del ganglio de Gasser. – Inyección de glicerol en la cavidad de Meckel. • Descompresión microvascular: actúa sobre anomalías vasculares que afectan a la raíz del trigémino (en una parte importante de los pacientes la causa es la compresión de la raíz del nervio por la arteria cerebelosa superior o una vena). Alivio del dolor en 70-90 % de los casos. Indicada en pacientes con buen estado general. Disfunción de la articulación temporomandibular Tratamiento no farmacológico • Normas de autocuidado: – Reducir la tensión emocional y el estrés. – Disminuir el bruxismo mediante aparato ortésico dental de colocación nocturna. – No forzar la apertura máxima de boca (bostezar, comer una pieza grande de fruta, etc.). – Corregir posturas y hábitos incorrectos (no apretar ni rechinar los dientes, no masticar chicle, etc.) • Fisioterapia: calor, ejercicios musculares, férulas oclusales. Tratamiento quirúrgico En caso de existir bloqueo articular agudo o anquilosis. Tratamiento farmacológico • Analgésicos/antiinflamatorios no esteroideos (AINE): paracetamol 0,5-1 g/8 h/v.o.; ibuprofeno 600 mg/8 h/ v.o.; diclofenaco 50 mg/8 h/v.o. • Relajantes musculares: diazepam 5 mg/día/v.o. en dosis nocturna (máximo 10-14 días). 5.1 Dolor en la cara Derivación Neuralgia del trigémino • Paciente menor de 40 años con neuralgia del trigémino de reciente comienzo. • En caso de duda diagnóstica o necesidad de tratamiento quirúrgico. • Fracaso de la respuesta a carbamazepina (dosis máxima: 1.200 mg/día). • Si hay déficit sensitivo, en períodos intercrisis, en el territorio facial afectado, por sospecha de neuralgia del trigémino secundaria a causa orgánica. Bibliografía recomendada 1. Sánchez MJ, Serrano C, Del Barrio MT. Dolor facial (I y II). Medicine. 2007;09:4548-57. 2. Quail G. Atypical facial pain—a diagnostic challenge. Aust Fam Physician. 2005;34(8):641-5. 3. Wiffen PJ, McQuay HJ, Moore RA. Carbamazepina para el dolor agudo y crónico (Revisión Cochrane traducida). En: La Biblioteca Cochrane Plus, número 3, 2008. Oxford, Update Software Ltd. Disponible en: http://www.update-software.com. (Traducida de The Cochrane Library, Issue. Chichester, UK: John Wiley & Sons, Ltd.). [Resumen] [Texto completo en Biblioteca Cochrane]. Disfunción de la articulación temporomandibular • Si fracasan los tratamientos no farmacológico y farmacológico. • Si existen dudas diagnósticas o se precisan pruebas complementarias especiales (TC de la ATM). • Presencia de bloqueo articular o anquilosis. 4. Hentschel K, Capobianco DJ, Dodick DW. Facial pain. Neurologist. 2005;11(4):244-9. 401 5.2 Dolor de cabeza B. Barragán Camín ¿De qué hablamos? Definición Se denomina cefalea a toda aquella sensación molesta o desagradable localizada en la parte superior de la cabeza, desde la región suboccipital a la zona orbitaria. Epidemiología básica En Atención Primaria (AP) la cefalea supone el 25 % de consultas de causa neurológica. Se calcula una prevalencia de cefalea en población adulta del 56 % (42 % en cefalea tensional, que es la más frecuente en todas las edades, 11 % en migraña y 3-5 % en cefalea crónica diaria). Hay que tener en cuenta el elevado porcentaje de personas con cefalea que no acuden al médico. Tanto la cefalea tensional como la migraña son más frecuentes en mujeres, en proporción 3:1 y 2-3:1, respectivamente. Cefaleas primarias Migraña (con aura o sin aura) Cefalea tensional Cefalea en racimos y otras cefaleas autonómicas del trigémino (incluye hemicránea paroxística nocturna) Otras cefaleas: asociadas a tos, ejercicio, actividad sexual, etc. Cefaleas secundarias Traumatismo craneal o cervical Trastornos vasculares craneales o cervicales (incluye arteritis de Horton) Trastorno intracraneal no vascular Fármacos: ingesta/supresión Infección (intracraneal o sistémica) Trastorno de la homeostasis (incluye hipertensión arterial) Clasificación Alteraciones de las estructuras faciales o craneales Sigue vigente la clasificación de la Sociedad Internacional de Cefaleas (IHS), en la actualización de 2004 (tabla 1). Se distinguen dos grandes grupos de cefaleas: primarias (90-95 % del total) y secundarias. La suma de cefalea tensional y migraña representa el 80 % de las cefaleas primarias, y pueden coexistir (cefalea mixta). Trastornos psiquiátricos ¿Qué lo puede ocasionar? Causas Cefaleas primarias • Cefalea tensional: cefalea mal definida, generalmente holocraneal u occipital, descrita como un «casco» que oprime la cabeza. Afecta preferentemente a las edades medias de la vida. Es de intensidad leve o moderada, y por ello no interfiere en las actividades de la vida diaria ni el sueño. No empeora con la actividad física, pero sí lo suele hacer a medida que transcurre el día. Puede durar de 30 minutos a 7 días, y acompañada o no de fotofobia y fonofobia, pero nunca de náuseas (a no ser que se cronifique). Su etiopatogenia es compleja, ya que intervienen la contractura de la musculatura pericraneal y el estrés, pero también una disminución del umbral del dolor. • Migraña: cefalea recurrente, generalmente hemicraneal y pulsátil, de intensidad moderada o grave, altamente invalidante. Dura entre 4 y 72 horas y se acompaña de, al menos, uno de los siguientes síntomas: fotofobia, fonofobia, náuseas y vómitos. El dolor empeora con la actividad física, incluso con el movimiento de la cabeza (signo del traquetreo) y suele aliviarse reduciendo los estímulos ambientales (luz, ruido) y con el sueño. Puede haber hipersensibilidad del cuero cabelludo, que persiste aun después de la cefalea. Es raro que debute después de los 40 años. Se trata de una cefalea neurovascular, con cierta predisposición genética. En ocasio- 402 Tabla 1. Clasificación de las cefaleas (IHS, 2004) Neuralgias craneales y causas centrales de dolor facial (incluye neuralgia del trigémino y migraña oftalmopléjica) Cefaleas no clasificables o inespecíficas nes, pueden identificarse factores desencadenantes: estrés (el más frecuente), factores hormonales (menstruación, embarazo, anticonceptivos orales), alteraciones del sueño (por defecto y por exceso: «migraña del fin de semana»), ayuno prolongado, alcohol, cambios atmosféricos, estímulos sensoriales intensos y algunos alimentos (la lista propuesta es extensa, pero sólo hay evidencia en el caso de los alimentos ricos en glutamato: «síndrome del restaurante chino»). Los factores hormonales, citados anteriormente, tienen un efecto paradójico: pueden empeorar o mejorar la migraña. Puede haber síntomas inespecíficos antes y después de la cefalea, constituyendo las fases de pródromos y de la llamada «resaca» de la migraña: bostezos, astenia, irritabilidad, apatía. Un 20 % de las migrañas se acompañan de síntomas neurológicos reversibles, de instauración siempre progresiva, conocidos como aura. Los más frecuentes son los visuales (escotomas, fotopsias, ceguera transitoria), seguidos de los sensitivos y las alteraciones del lenguaje. El aura suele preceder a la cefalea, con un intervalo libre inferior a 1 hora, aunque ambas pueden coincidir. Suele durar unos 5-20 minutos, y en cualquier caso siempre menos de 1 hora. Auras más prolongadas, seguidas de cefalea no migrañosa o migrañas de más de 72 horas de duración (estatus migrañoso) exigen descartar de forma urgente isquemia cerebral. Un aura típica sin cefalea se considera una forma atípica de migraña que plantea problemas diagnósticos si se trata de la primera crisis. En niños, la migraña suele ser bilateral y más breve, y se han identificado cuadros de dolor abdominal recurrente, vómitos de repetición y vértigo paroxístico como precursores de la migraña. 5.2 Dolor de cabeza Tabla 2. Criterio de abuso de analgésicos Ácido acetilsalicílico/paracetamol/AINE: 15 días o más al mes durante más de 3 meses Triptanes/opioides/ergotamínicos/combinaciones: 10 días o más al mes durante más de 3 meses AINE: antiinflamatorios no esteroideos. • Cefalea crónica diaria: generalmente derivada de una cefalea tensional o de una migraña. Se presenta un mínimo de 15 días al mes, durante un mínimo de 3 meses. Puede asociarse a abuso de analgésicos o a ansiedad y/o depresión. La cefalea por abuso de analgésicos (tabla 2) es, en realidad, una cefalea secundaria, pero se desarrolla en personas con cefalea primaria y se relaciona con la automedicación. Es reversible al dejar los analgésicos (el paciente vuelve a su patrón previo de cefalea al cabo de unos 2 meses). No se conoce la razón, pero no ocurre si el paciente toma esos mismos analgésicos para patologías diferentes de la cefalea. • Cefalea en racimos (cluster headache): es la más dolorosa de las cefaleas primarias, y la única que predomina en varones, con una proporción 3-5:1. Se inicia sobre los 30 años de edad, y consiste en, al menos, cinco episodios de cefalea unilateral en el territorio de la primera rama del trigémino (sobre todo en el periorbitario, pero también en la región temporal). Dura 15 a 180 minutos, y provoca intensa agitación psicomotriz, acompañada de uno o más de los siguientes síntomas autonómicos, homolaterales al dolor: inyección conjuntival y/o lagrimeo, obstrucción nasal y/o rinorrea, edema palpebral, sudoración de cara y frente, miosis y/o ptosis palpebral. Las crisis pueden aparecer distintas veces al día (hasta 8 veces), varios días seguidos, generalmente a la misma hora (en fase REM del sueño, tanto de noche como durante la siesta). El dolor despierta al paciente. El alcohol y los fármacos vasodilatadores pueden precipitar las crisis. Un 10 % corresponden a la forma crónica, con racimos de más de 1 año sin remisión, o con remisiones inferiores a 1 mes. Un 3-5 % corresponden a formas secundarias a procesos intracraneales. • Hemicránea crónica paroxística: se parece a la cefalea en racimos, pero es más breve (hasta 45 min) y predomina en mujeres jóvenes. Puede llegar a 30 ataques diarios, y no tiene predominio horario. Es característica la buena respuesta a indometacina. • Otras cefaleas primarias: asociadas a maniobras de Valsalva (sobre todo a la tos), al esfuerzo físico y a la actividad sexual. Todas ellas exigen descartar lesión intracraneal estructural. Cefaleas secundarias La clasificación de la IHS (v. tabla 1) incluye tipos muy diversos, entre los cuales destacan: • Arteritis de la temporal o de Horton: vasculitis sistémica de vasos de grueso y mediano calibre. Es más frecuente en mujeres y prácticamente siempre a partir de los 50 años. Provoca una cefalea frontotemporal intensa irradiada a la mandíbula. Los pulsos temporales pueden estar indurados o abolidos, o ser dolorosos a la palpación. Se puede asociar a fiebre, síndrome tóxico, hipersensibilidad del cuero cabelludo, claudicación mandibular (40 %), polimialgia reumática (50 %) y neuritis óptica isquémica. El riesgo de ceguera irreversible obliga a una confirmación diagnóstica urgente para iniciar el tratamiento de forma precoz. • Cefalea por fármacos (tabla 3). • Cefalea por hipertensión arterial (HTA): hay que tener en cuenta que sólo aparece en la HTA grave. Suele ser bilateral y pulsátil, y empeora con la actividad física y las maniobras de Valsalva. ¿Qué tenemos que hacer? Anamnesis El diagnóstico de la cefalea es clínico, basado en la anamnesis y la exploración física (figura 1). Tabla 3. Fármacos de uso en Atención Primaria que pueden provocar cefalea Acetazolamida Estrógenos Omeprazol Aciclovir Griseofulvina Paroxetina Antagonistas del calcio Hidralazina Pentoxifilina Antihistamínicos Isoniazida Progestágenos AINE Litio Quinolonas Atenolol Metildopa Ranitidina Captopril Metoprolol Rifampicina Carbimazol Metronidazol Sildenafilo Cimetidina Morfina y derivados Teofilina Codeína Nitratos Tetraciclinas Disulfiram Nitrofurantoína Trimetoprima-sulfametoxazol AINE: antiinflamatorios no esteroideos. 403 5.2 Dolor de cabeza Dolor de cabeza Cefalea de Horton Alteración pulsos temporales Tema 11.15 Traumatismo craneoencefálico Tema 15.1 PA elevada Tema 5.1 Dolor facial Tema 11.1 Dolor cervical Tema 12.1 Alteraciones de la visión Bajo Derivar a urgencias hospital No El cuadro clínico hace sospechar una cefalea de causa secundaria a otro proceso Cefalea progresiva no filiada Cambio de características Debut > 40 años Desencadenada con cambios posturales, maniobra de Valsalva o ejercicio físico No No ¿Presenta crisis similares recurrentes? Episodios repetidos de corta duración 15-180 min > 15 Derivación urgente/preferente a Neurología Predominio femenino. Jóvenes o mediana edad. Predominio unilateral y pulsátil Agravada por actividad física diaria Duración 4-72 h intensidad moderadagrave, inhibe o incapacita. Factores desencadenantes característicos Síntomas asociados: náuseas, vómitos fotofobia, fonofobia Alto ¿Qué impacto tiene la cefalea en la vida del paciente, en orden a interferir en sus actividades? Cefalea mal definida, opresiva. Mediana o moderada intensidad. Puede inhibir, pero no incapacitar sus actividades. Localización variable, predominantemente bilateral 3.ª década de la vida de 30 min a 7 d de duración síntomas asociados leves o ausentes. No agravada por actividad física rutinaria Cefalea tensional Focalidad neurológica (rigidez de nuca, etc.) Cefalea intensísima de inicio brusco. Afectación del estado general papiledema en fondo de ojo ¿Cuántos días al mes tiene cefalea? < 15 No Predominantemente masculina Orbitaria, supraorbitaria o temporal. Síntomas vegetativos homolaterales (inyección conjuntival, lagrimeo, rinorrea, miosis, ptosis, edema palpebral) Ambos sexos. Todas las edades Duración > 4 h resistente al tto. antecedentes de cefalea tensional o migraña abuso analgésicos para cefalea y/o lesión previa cabeza/cuello Cumple criterios abuso analgésico Cefalea en racimos (cluster headache) No Síntomas reversibles de focalidad neurológica instauración progresiva en más de 4 min y de duración < 1 h Migraña con aura No Migraña sin aura Categorizar el grado de gravedad basándose en incapacidad funcional, duración de los síntomas, latencia al cenit de la cefalea Tto. en APS Tto. y profilaxis en APS Cefalea crónica diaria por abuso de analgésicosa Cefalea crónica diaria Leve Moderada-grave o intolerancia a los AINE Tratamiento Tto. en APS Duración > 72 h Estado migrañoso Tto. y profilaxis en APS Valorar ITC a neurología Derivar a urgencias hospital Figura 1. Algoritmo diagnóstico de las cefaleas. a Utilización de analgésicos y/o AINE durante ≥ 15 días al mes durante más de 3 meses. • Antecedentes familiares: hay un riesgo 2-4 veces mayor de migraña en familiares de primer grado. • Antecedentes personales: edad, sexo, profesión, consumo y supresión de fármacos y otras sustancias (como el alcohol). Tiene especial importancia el patrón de consumo de analgésicos, que puede cronificar la cefalea. • Enfermedad actual (tablas 4 y 5): localización, forma de instauración, tipo de dolor, frecuencia, intensidad, duración, síntomas asociados, factores desencadenantes, y factores que agravan o alivian la cefalea. Es imprescindible buscar los llamados signos de alarma (tabla 6), 404 que obligan a descartar de forma urgente patología grave. Exploración física Exploración general que incluya la temperatura y la presión arterial, una exploración neurológica y el fondo de ojo para descartar un edema de papila. En las cefaleas primarias la exploración es básicamente normal. Según la sospecha diagnóstica puede estar indicado explorar la columna cervical o alguna estructura facial. En pacientes 5.2 Dolor de cabeza Tabla 4. Criterios diagnósticos de migraña sin aura (IHS, 2004) Cinco o más episodios de cefalea, en los que se ha descartado cefalea secundaria, que cumplan los siguientes tres criterios: Cada crisis dura 4-72 h (sin tratamiento o con tratamiento ineficaz) Al menos dos de los siguientes: pulsátil, unilateral, intensidad moderada/alta, empeora con la actividad física diaria o impide realizarla Al menos uno de los siguientes: náuseas, vómitos, fotofobia, fonofobia mayores de 50 años, siempre hay que palpar las arterias temporales, que en la arteritis de la temporal son induradas, dolorosas o incluso no palpables. Exploraciones complementarias No suelen ser necesarias. Hay que realizar pruebas de imagen, ya sea una tomografía computarizada (TC) o una resonancia magnética (RM) si se sospecha un proceso intracraneal. Si se trata de un debut de cefalea a partir de los 50 años, solicitar una determinación de la velocidad de sedimentación globular (VSG) para descartar arteritis de la temporal. ¿Qué propuesta haremos? Tabla 5. Criterios diagnósticos de migraña con aura (IHS, 2004) Dos o más episodios de cefalea, en los que se ha descartado cefalea secundaria, que cumplan los siguientes tres criterios: Aparición de aura: síntomas neurológicos reversibles visuales, sensitivos o alteraciones del lenguaje (se excluyen los síntomas motores) Al menos dos de los siguientes: los síntomas visuales son homónimos y/o los síntomas sensitivos son unilaterales; al menos uno de los síntomas del aura se desarrolla durante más de 5 min o varios de ellos se suceden durante más de 5 min; cada síntoma dura 5-60 min La cefalea comienza durante el aura o en los 60 min posteriores a ella Tabla 6. Signos de alarma ante una cefalea Cefalea progresiva de inicio reciente y no filiada Cefalea que debuta en mayores de 50 años Cefalea desencadenada por maniobras de Valsalva, ejercicio físico o cambios posturales Cefalea ya conocida que cambia de características Cefalea intensa de inicio brusco Focalidad neurológica, a excepción del aura migrañosa Afectación del estado general Alteración de la conciencia o trastorno del comportamiento Tratamiento No farmacológico • Evitar en lo posible el estrés, valorando técnicas de relajación, y procurar ritmos regulares de comida y sueño. • En la cefalea tensional no hay datos concluyentes sobre la eficacia de técnicas como la acupuntura, el yoga o la manipulación espinal. • En la migraña se recomienda acostarse en un lugar aislado y a oscuras. • Un paño húmedo y frío en la región frontotemporal puede aliviar tanto la migraña como la cefalea tensional. • Evitar la ingesta de alcohol (importante en migraña y, sobre todo, en la cefalea en racimos) y de cafeína. • En caso de cefalea recurrente es muy útil elaborar un diario de cefaleas, ya que permite identificar los diferentes tipos que pueden coexistir en un mismo paciente, así como los factores desencadenantes y la posible indicación de tratamiento profiláctico. Farmacológico Como norma general, evitar combinaciones de analgésicos en dosis bajas y asociaciones de analgésicos con cafeína y codeína, que pueden contribuir a cronificar la cefalea. Cefalea tensional • El tratamiento sintomático de elección son los antiinflamatorios no esteroideos (AINE), en las mismas dosis que en la migraña. El tratamiento profiláctico (tabla 7) está indicado en caso de consumo de analgésicos más de 8 días al mes, y se retira de forma progresiva 4-6 meses después de la mejoría clínica. Estado migrañoso (migraña que dura más de 72 h) Aura atípica (que dura más de 1 h o con síntomas motores) Aura seguida de cefalea no migrañosa Aura no seguida de cefalea, en pacientes que no han tenido nunca migraña con aura Aura en pacientes que toman anticonceptivos hormonales, en pacientes que no han tenido nunca migraña con aura Migraña • Se debe recomendar tomar analgésicos en dosis óptimas (es frecuente la infradosificación) y en fases precoces de la cefalea. El tratamiento no es eficaz para el aura, sólo para la cefalea. • En crisis moderadas son de elección los AINE orales, sobre todo en formulaciones de absorción rápida (1.000 mg de ácido acetilsalicílico, 600 mg de ibuprofeno, 500 mg de 405 5.2 Dolor de cabeza Tabla 7. Tratamiento profiláctico de las cefaleas Tipo de cefalea Tratamiento profiláctico Cefalea tensional Amitriptilina (primera elección): 25-75 mg/día en dosis única nocturna. Más eficaz que los inhibidores selectivos de la recaptación de serotonina Migraña Betabloqueantes (primera elección): propranolol 40-240 mg/día, atenolol 50-100 mg/día, nadolol 20-120 mg/día, metoprolol 50-200 mg/día Antagonistas de los canales del calcio: flunaricina 5-10 mg/día, verapamilo 240-360 mg/día Amitriptilina (primera elección si hay abuso de analgésicos o cefalea mixta): 20-75 mg/día en dosis única nocturna Naproxeno: 500-1.000 mg (migraña menstrual) Topiramato: 50-150 mg/día. Útil si patología asociada (epilepsia, trastorno bipolar) Cefalea en racimos Asociar siempre prednisona 40-80 mg/día o dexametasona 8 mg/día durante 7 días y en pauta descendente durante 3 semanas (eficaz a corto plazo) y verapamilo 240-360 mg/día o litio 400-1.200 mg/día o topiramato 200-400 mg/día (profilaxis de mantenimiento) naproxeno, 25 mg de dexketoprofeno o 50 mg de diclofenaco). También es útil el metamizol, a dosis de 1.000 mg. Todos ellos pueden repetirse cada 8 horas, a excepción de naproxeno, que se administra cada 12 horas. • En crisis graves hay tres opciones: esos mismos grupos de fármacos por vía i.m. (75 mg de diclofenaco, 30 mg de ketorolaco o 2.000 mg de metamizol), los triptanes y los ergóticos. • Los triptanes (tabla 8) tienen una acción vasoconstrictora, y están contraindicados en caso de enfermedad coronaria, cerebrovascular, HTA no controlada, arteriopatía periférica, enfermedad de Raynaud, insuficiencia hepática o renal, embarazo y lactancia. En niños mayores de 12 años se puede utilizar sumatriptán nasal. Los principales efectos secundarios de los triptanes son el dolor torácico y las palpitaciones. Son más caros y tienen menor experiencia de uso que los ergóticos, pero suelen ser preferibles a ellos. Entre el uso de un triptán y de un ergótico siempre debe transcurrir un mínimo de 24 horas. Existen presentaciones tipo «flas» que son mejor toleradas en caso de náuseas. En un 40-60 % de los casos son eficaces (mejoran el dolor en un plazo de 2 h), pero hay un 30-40 % de recidivas. Si una dosis de triptán no es eficaz, se puede repetir a las 2 horas pero no se deben emplear más dosis en 24 horas. Si un triptán no es eficaz, no invalida al resto del grupo. Por su mecanismo de acción, deben administrarse con precaución en caso de tratamiento con inhibidores selectivos de la recaptación de serotonina. Hay que reducir la dosis de rizatriptán a la mitad en caso de tratamiento con propranolol. • Los ergóticos son fármacos de segunda línea, en pacientes con crisis esporádicas. No son recomendables como fármacos de inicio por su fenómeno de rebote y efectos secundarios. Se relacionan con cierta frecuencia con la automedicación y el abuso de analgésicos, favorecido por su frecuente asociación a cafeína. Tienen las mismas contraindicaciones que los tripta- Tabla 8. Triptanes comercializados en España Triptán Dosis habitual (mg) Administración Dosis máxima (mg/día) Sumatriptán 50-100 10-20 6 v.o. intranasal s.c. 200d 40d 12d Naratriptán 2,5 v.o. 5d Zolmitriptán 2,5-5 0 5 v.o. intranasal 10a Rizatriptán 10 v.o. 20b Almotriptán 12,5-25 0 v.o. 50c Eletriptán 40 v.o. 80d Frovatriptán 2,5 v.o. 5d a 406 Dosis máxima: 5 mg/día en insuficiencia hepática grave y algunos tratamientos concomitantes. b Si tratamiento concomitante con propranolol, o insuficiencia hepática/renal, reducir la dosis a 5 mg/día. c Dosis 12,5 mg/día si insuficiencia renal grave. d En insuficiencia renal, 20 mg oral y máximo 40 mg/día. 5.2 Dolor de cabeza nes. Su principal efecto secundario son las náuseas y los vómitos. Se dispone de ergotamina (1-2 mg vía oral o 2 mg vía rectal, que tiene mejor absorción) y dihidroergotamina (1 mg vía oral). Se puede repetir el 50 % de esas dosis cada media hora, hasta un máximo de 6 mg/día. • Además del analgésico, se debe administrar siempre un antiemético, aunque el paciente no tenga náuseas (metoclopramida 10 mg vía oral o i.m., domperidona 10 a 30 mg vía oral o 60 mg vía rectal), ya que mejora la absorción del tratamiento. Hay que valorar también la administración de un ansiolítico de semivida larga (5 mg de diazepam o de clorazepato dipotásico). • Por lo que respecta al tratamiento profiláctico (v. tabla 7), está indicado en los siguientes casos: ineficacia o contraindicación del tratamiento sintomático; interferencia importante de la migraña en las actividades de la vida diaria, tres o más crisis al mes; incapacidad psicológica de afrontar las crisis, y migraña hemipléjica o con aura prolongada. Debe intentarse la monoterapia. Se evalúa su eficacia (reducción de crisis al 50 %) a las 6 a 8 semanas, y debe mantenerse entre 6 y 12 meses. Cefalea crónica diaria • Investigar y tratar la ansiedad y/o depresión y el abuso de analgésicos, para recuperar el patrón de cefalea original. • En caso de abuso de analgésicos, la medida fundamental es suprimirlos, así que hay que avisar al paciente de que al principio va a empeorar. • Se debe ofrecer un tratamiento para la cefalea de rebote, que no suele durar más de 2 semanas. Los AINE son de primera elección, en las mismas dosis que en el tratamiento sintomático pero restringiendo su uso a un máximo de dos veces por semana. Valorar añadir un antiemético y/o un ansiolítico. Si el fármaco implicado en el abuso es un AINE, elegir la amitriptilina. Si era un ergótico, un opiáceo, cafeína o una benzodiazepina, pautar prednisona oral en dosis iniciales de 1 mg/kg/día y reducir en unos 15 días. Valorar el ingreso hospitalario en casos de alta dependencia, consumo de psicofármacos, trastorno mental o fracaso en intentos previos. Tras la fase de cefalea de rebote, seleccio- Consejos prácticos • No confundir la resaca de la migraña con un fracaso del tratamiento. • Los triptanes y los ergóticos, por su mecanismo vasoconstrictor, no deben administrarse hasta que el aura haya desaparecido por completo. • Los anticonceptivos orales que contienen estrógenos están contraindicados en migrañas con aura (y en pacientes con migrañas sin aura y dos o más factores de riesgo vascular), por el riesgo de isquemia cerebral. La alternativa son los anticonceptivos que sólo contienen progestágenos. Errores más frecuentes • Menospreciar las quejas del paciente con cefalea. • No interrogar sobre el abuso de analgésicos (causa más frecuente de falta de respuesta al tratamiento). • Olvidar la posibilidad de una cefalea en racimos ante una migraña que dura menos de 2 horas. • Tratar cada episodio de cefalea como algo aislado. • Repetir analgésicos que no funcionaron en episodios anteriores de migraña. • No informar al paciente en caso de cefalea recurrente y no explorar sus dudas y sus temores. • Sobreestimar los defectos de refracción y las sinusitis como causa de cefalea. • Elaborar una dieta estricta en caso de migraña: sólo debe recomendarse de forma sistemática evitar el consumo de alcohol y glutamato. nar el tratamiento profiláctico que corresponda, según el patrón original de cefalea. Cefalea en racimos • El tratamiento sintomático consiste en sumatriptán 6 mg s.c. o 20 mg inhalado, pudiendo repetirse otra vez en 24 horas, y/o la administración de oxígeno al 100 % durante 15 minutos, ambos con un grado de recomendación A. • Zolmitriptán ha demostrado una eficacia inferior. El tratamiento profiláctico (v. tabla 7) está indicado siempre, y debe combinar dos fármacos: uno de eficacia a corto plazo y otro de mantenimiento. En casos rebeldes existen tratamientos quirúrgicos aplicados a las ramas sensitivas del trigémino, pero hay que tener en cuenta los efectos secundarios y la posibilidad de recurrencias. Hemicránea crónica paroxística • Indometacina 75-150 mg/día durante 3 meses. En ocasiones hay que valorar mantenerla de forma indefinida en dosis bajas. Arteritis de la temporal • Prednisona en dosis de 40-60 mg/día. Empezar a reducir la dosis pasadas 4 semanas y mantener 5-10 mg/día durante 1-2 años. Derivación Urgencias del hospital • Signos de alarma (v. tabla 6) o antecedente traumático. Neurología • Cefalea primaria no filiada o falta de respuesta al tratamiento (sintomático o profiláctico). • Recordar también los casos en que hay que valorar el ingreso hospitalario para el tratamiento de una cefalea por abuso de analgésicos. Psiquiatría • Trastornos de ansiedad o depresión que así lo requieran. 407 5.2 Dolor de cabeza ¿Se ha resuelto del todo? Pronóstico y seguimiento La migraña es una enfermedad crónica, y por ello requiere un programa de visitas de control periódicas hasta que el paciente esté estable. Tiende a mejorar con la edad. ¿Podemos hacer alguna cosa más? Bibliografía recomendada 1. Hijas F, Sánchez-Migallón MJ, Andrés del Barrio MT, Yusta A. Cefalea. Medicine. 2007;9:4465-72. 2. Andrés del Barrio MT, Sánchez MJ, Serrano C, Yusta A. Migraña. Medicine. 2007;9:4480-7. 3. Steiner TJ, Paemeleire K, Jensen R, Valade D, Savi L, Laínez MJA, et al. European principles of management of common headache disorders in primary care. J Headache Pain. 2007;8(suppl 1). Disponible en: http://www.ehf-org.org/Documents/principles_of_ management_headache_disorders.pdf Actividades preventivas • En cefaleas recurrentes o crónicas, evitar la automedicación para prevenir la cefalea por abuso de analgésicos. Educación sanitaria Consultar la Guía práctica de la salud, p. 27 y 28. 4. Calero S, Castillo J, Martínez JM, García ML, Moreno P, Tranche S (coordinador). Cefaleas. FMC. Protocolo 3/2008. 5. Headache Classification Subcommittee of the Internacional Headache Society. The International Classification of Headache Disorders. 2.ª ed. Cephalalgia. 2004;24(1):1-151. 6. Jiménez PE, Marsal C. Cefalea. Med Clin (Barc) 2006;127:744-8. 7. Grupo de estudio de la cefalea de la Sociedad Española de Neurología. Actitud diagnóstica y terapéutica en la cefalea. Mateos V, Díaz S, Huerta M, Porta J, Pozo P. editores. Sociedad Española de Neurología. Recomendaciones 2006. Disponible en: http:// cefaleas.sen.es/profesionales/recomendaciones2006.htm 408 5.3 Parálisis facial M. R. Alonso Martín ¿De qué hablamos? Introducción La parálisis facial es la debilidad o pérdida de función de la musculatura inervada por el nervio facial (VII par craneal). Esto da lugar a una disminución parcial o completa de la motilidad voluntaria de la cara, que puede ser unilateral o bilateral. Conceptos iniciales El nervio facial es mixto. La función motora es la principal, siendo la responsable de inervar los músculos relacionados con la expresión de la cara. Tiene un componente sensitivo responsable de la sensibilidad del conducto auditivo externo, del gusto en los dos tercios anteriores de la lengua y de la secreción de la glándula lacrimal. Clasificación La clasificación inicial de las parálisis faciales hace referencia al nivel en donde se localiza la lesión causante. Cabe distinguir las siguientes: • Parálisis facial periférica: debida a la afectación del nervio facial a cualquier nivel desde su núcleo, que está situado en la protuberancia, hasta sus fibras más periféricas, intracraneales o extracraneales. La parálisis de Bell o parálisis facial idiopática es la forma más frecuente de este grupo (del 60 al 75 %). • Parálisis facial central: es debida a una lesión superior al núcleo, en la corteza o zonas subcorticales del cerebro. La tabla 1 recoge la localización de las lesiones en las parálisis centrales y periféricas y sus posibles causas. Epidemiología La parálisis de Bell o parálisis facial idiopática presenta una incidencia anual variable según los países, siendo, en general, de 11-40/105 habitantes. Afecta a ambos sexos por igual y puede aparecer a cualquier edad, con una incidencia máxima entre los 15 y los 45 años de edad. En el embarazo el riesgo aumenta tanto en el tercer trimestre como en la primera semana posparto. La diabetes mellitus se halla presente en el 5-10 % de los pacientes con parálisis facial unilateral idiopática. Las recurrencias homolaterales o contralaterales afectan a un 8-10 % de los pacientes. ¿Qué lo puede ocasionar? Causas En la tabla 1 se enumeran las causas de las parálisis centrales y periféricas. La causa de la parálisis de Bell se desconoce en las periféricas, si bien desde el aislamiento del ácido desoxirribonucleico (ADN) del virus del herpes simple (VHS-1) en el líquido endoneural del nervio facial de personas con esta parálisis, la activación del VHS-1 es ampliamente aceptada como la causa de la mayoría de los casos de este tipo de parálisis. La inflamación del nervio, y la consecuente compresión e isquemia dentro del canal facial, sería el mecanismo fisiopatológico desencadenado por esta activación viral. Diagnóstico diferencial En la parálisis central o supranuclear, a diferencia de la periférica o nuclear, los músculos frontal y orbicular del ojo no están afectados debido a que los músculos faciales superiores están inervados por vías corticobulbares procedentes de la corteza motora de ambos hemisferios cerebrales. Éstas se reúnen en el núcleo del nervio por lo que las lesiones a este nivel o inferior sí afectan a la movilidad de los músculos referidos, originando la frente sin arrugas, el fenómeno de Bell y el signo de la pestaña. Por el contrario, los músculos de la parte inferior de la cara están inervados únicamente por el hemisferio opuesto por lo que la parálisis se da tanto en la afectación central como en la periférica. En la figura 1 se desarrolla el algoritmo diagnóstico de la parálisis facial. ¿Qué tenemos que hacer? Anamnesis • Antecedentes personales: heridas penetrantes de parótida y oído medio, barotrauma, fracturas faciales o del hueso temporal y base del cráneo; yatrogenia (cirugía), traumatismos durante el parto, infecciones, enfermedades metabólicas (en la diabetes mellitus la incidencia de parálisis facial es cuatro veces superior), sarcoidosis, embarazo, hipertensión arterial, posibles mordeduras de garrapata (la borreliosis causa de parálisis facial). • Enfermedad actual: los aspectos claves que deben tenerse en cuenta para poder diferenciar las parálisis idiopáticas de las secundarias son la forma de inicio, la evolución y los síntomas prodrómicos. Estos últimos, como la sensación de entumecimiento o dolor retroauricular o hiperacusia, son característicos de la parálisis de Bell. Ésta suele tener una instauración brusca y es máxima a las 48-72 horas, con un curso progresivo y máxima paresia en las primeras 3 semanas y una recuperación total o parcial en 6 meses y con o sin afectación del gusto en los dos tercios anteriores de la lengua y disminución de la secreción lagrimal y/o salival. Un comienzo prolongado, una evolución lentamente progresiva, la ausencia de recuperación o una posterior recaída sugieren un proceso orgánico subyacente. Asimismo, los antecedentes de parálisis recurrentes y síntomas asociados como cefalea o afectación de otros pares craneales sugieren causas orgánicas de la parálisis facial. La asociación de fiebre, artritis, eritema migratorio en las extremidades o tronco y parálisis facial hacen sospechar la enfermedad de Lyme (Borrelia burgdorferi) y obliga a buscar la picadura de una garrapata. 409 5.3 Parálisis facial Tabla 1. Parálisis facial. Localización de la lesión y causas PARÁLISIS FACIAL CENTRAL Localización de la lesión Síntomas acompañantes Causas Corteza cerebral y regiones subcorticales Hemiparesia espástica unilateral a la parálisis facial Infartos y hemorragias corticales y subcorticales Tumores PARÁLISIS FACIAL PERIFÉRICA Localización de la lesión Síntomas acompañantes Causas Protuberancia Parálisis VI par Hemiparesia contralateral Nistagmo Hipostesia termoanalgésica contralateral Congénita (enfermedad de Möebius) Vascular Esclerosis múltiple Tumor (metástasis) Encefalitis Siringomielia Esclerosis lateral amiotrófica Ángulo pontocerebeloso o meato auditivo interno Afectación de los pares V, VII, IX, X y XI Pérdida de gusto y salivación Pérdida de lagrimeo Hiperacusia Neurinoma del acústico Meningiomas Colesteatoma Tumor del glomus Carcinomatosis meníngea Canal facial Pérdida de gusto y salivación Preservación del lagrimeo Parálisis de Bell Fractura de peñasco Otitis, mastoiditis Síndrome de Ramsay-Hunt (herpes zoster) Neoplasias VIH Borreliosis Diabetes mellitus Síndrome de Guillain-Barré Agujero estilomastoideo y trayecto extracraneal Preservación del gusto, salivación y lagrimeo Afectación parcial de grupos musculares Edema facial, lengua plegada Sarcoidosis Tumores de parótida Síndrome de Melkersson-Rosenthal Traumatismos y cirugía VIH: virus de la inmunodeficiencia humana. Exploración física Se debe valorar el grado de parálisis, según los hallazgos encontrados, para poder valorar la evolución. La Academia Americana de Otorrinolaringología ha adoptado el sistema de gradación de la parálisis facial propuesto por House y Brackmann, que se muestra en la tabla 2. • Exploración del nervio facial: – Parálisis de los músculos de la frente (no tiene arrugas, lo que es indicativo de afección periférica). – Al intentar cerrar los párpados, la hendidura palpebral queda abierta y permite ver la esclerótica, debido al ascenso del globo ocular (signo de Bell). Este signo permite orientar el diagnóstico de parálisis periférica. – La afectación mínima orbicular se detecta por el «signo de la pestaña» al indicar al paciente que cierre fuertemente los párpados, protruye la pestaña del lado afectado. – Disminución o ausencia del parpadeo del ojo del lado 410 afectado, eversión del párpado inferior por pérdida de tono (afección del orificio lacrimal), lagrimeo. – Asimetría de la cara con desviación de la comisura bucal hacia el lado sano, descenso de la comisura bucal, pérdida de saliva en el lado afectado y dificultad para silbar. – Exploración del gusto, que se realiza habitualmente sólo por anamnesis. • Exploración de otros pares craneales y neurológica general básica. Buscando afectación de otros pares craneales (trigémino, glosofaríngeo, hipogloso) o la asociación de paresias en extremidades que nos indicarán una causa probablemente central. • Exploración de la boca y valoración de la salivación. La presencia de lengua escrotal fisurada, edema facial y parálisis recurrente hace sospechar el síndrome de Melkersson-Rosenthal, donde los episodios de parálisis facial suelen comenzar antes de los 20 años. • Exploración ocular. Alteración del lagrimeo, úlceras corneales. 5.3 Parálisis facial Parálisis facial Inspección: los músculos frontales y orbiculares están paréticos: frente sin arrugas, fenómeno de Bella, signo de la pestañab Sí Parálisis periférica Normal Negativos Antecedentes personales Otoscopia Traumatismos, cirugía, infecciones, diabetes, sarcoidosis, embarazo, HTA Vesículas: Ramsay-Hunt Valorar ITC a ORL Rinne (+) Weber (+) del lado afecto Otitis media, mastoiditis ITC preferente a ORL Pérdida lagrimeo Neurinoma Meningioma Colesteatoma Tumor glomus Carcinomatosis meníngea Palpación cuello Normal Parálisis central Inicio brusco y signos de enfermedad aguda Audiometría: explor. diapasones Rinne (–) Weber (–) Actuación específica al antecedente No Anormal Sí No Vascular Infección SNC Debut tumoral Enfermedad desmielinizante: EM, ELA Siringobulbia Tumor Derivación urgente al hospital ITC urgente/ preferente a neurología Tumores Adenopatías Parálisis periférica primaria. Parálisis de Bell Tto. en APS ITC urgente/ preferente a ORL Revisión a la semana. Gradación: completa/incompleta Buena respuesta al tto. en la 4.ª sem Recuperación en la 6.ª sem Ausencia de recidiva homolateral No Sí Seguimiento en APS Figura 1. Actitud ante un paciente con parálisis facial. a Signo de Bell. Al intentar cerrar el párpado, la hendidura palpebral queda abierta y sólo se ve la esclerótica. Signo de la pestaña. Al indicar al paciente que cierre fuertemente los párpados, las pestañas del lado afectado protruyen. Permite detectar afectación mínima orbicular. APS: atención primaria de salud; ELA: esclerosis lateral amiotrófica; EM: esclerosis múltiple; HTA: hipertensión arterial; ITC: interconsulta; ORL: otorrinolaringología. b • Otoscopia: – El objetivo es descartar patología del oído medio. – La presencia de vesículas herpéticas en el conducto auditivo externo indica un síndrome de Ramsay-Hunt (herpes zoster), no obstante en más de la mitad de las ocasiones existe una neuralgia de distribución metamérica o disestesias, sin la presencia de vesículas, que pueden no aparecer o retrasar su aparición (Zoster sine herpete). – Una hipoacusia sensorial unilateral con otoscopia normal indica lesión en el conducto auditivo interno o en ángulo pontocerebeloso. – La presencia de una poliposis o la existencia de granulación en el canal auditivo puede sugerir un colesteatoma o una otitis externa maligna. • Exploración de la región cervical: – Especialmente de la región parotídea (tumores). – Buscar adenopatías satélites de procesos tumorales o infecciosos. Exploraciones complementarias El diagnóstico inicial de parálisis facial es clínico. Las pruebas complementarias pueden ayudar a graduar la gravedad de la lesión en cuanto al pronóstico y a confirmar si se sospecha una causa distinta a la idiopática. Iniciales • Estudio de laboratorio: es de utilidad limitada. Pueden ser de ayuda: hemograma, velocidad de sedimentación globular (VSG), glucosa (debido al riesgo relativo mayor entre diabéticos frente a los no diabéticos de desarrollar una parálisis de Bell), electrólitos, creatinina y alanino aminotransferasa (ALT). 411 5.3 Parálisis facial Tabla 2. Clasificación de House-Brackmann de gradación de la parálisis facial para valorar su evolución (Academia Americana de Otorrinolaringología) Grado Definición I. Normal Función normal del facial en todas las áreas II. Ligera disfunción Ligera debilidad que se hace evidente en la exploración. En reposo: simetría y tono normales. Movimiento de la frente casi normal III. Disfunción moderada Diferencia obvia, aunque no desfigurante, entre ambas mitades; no hay deterioro funcional; sincinesia motora pero no grave, contracturas, espasmo hemifacial o ambos. En reposo: tono y simetría normal. Movimiento: poco a ningún movimiento de frente; capacidad para mover los ojos con un esfuerzo intenso con asimetría evidente. Sincinesia evidente aunque no desfigurante, contracturas y espasmo hemifacial o ambas pertenecen al grado III, independientemente del grado de actividad motora IV. Disfunción moderadamente grave Existe debilidad evidente, asimetría desfigurante o ambas. En reposo: simetría y tono normal. Movimiento: no hay movimiento de frente; incapacidad para cerrar los ojos con máximo esfuerzo; si la simetría, el espasmo o ambos son lo suficientemente intensos como para interferir la función, se considerará del grado IV independientemente del grado de actividad motora V. Disfunción grave Movimiento apenas perceptible. En reposo: posible asimetría con caída del ángulo de la boca. Las sincinesias, las contracturas y el espasmo hemifacial están, por lo general, ausentes VI. Parálisis facial completa Hay pérdida de tono; asimetría; sin movilidad; no hay sincinesia, ni contractura, ni hemiespasmo facial Posteriores • Test de Schirmer: para el estudio de la lacrimación (tema 12.7). • Serología (tema 23.15): si la historia clínica sugiere su presencia, solicitar serología de herpes, virus de la inmunodeficiencia humana (VIH), enfermedad de Lyme, sífilis. • Electromiografía y electroneurografía (tema 25.15): el conocimiento del grado de denervación tiene valor pronóstico, pero no están bien definidas sus indicaciones. Se realizará entre el tercero y el decimocuarto día de la aparición de la parálisis. Una denervación mayor del 95 % en las primeras 2 semanas tiene mal pronóstico. Después de 3-4 semanas de evolución, la utilidad principal es el seguimiento de los pacientes a largo plazo; si a los 3 meses no han aparecido potenciales de renervación, hay que considerar la posibilidad de una causa secundaria. La mayoría de las parálisis faciales que se atienden en Atención Primaria (AP) corresponden a casos idiopáticos con afectación incompleta e inicio de la recuperación en las 3 semanas del inicio del cuadro; estos casos no requieren exploraciones complementarias. Las parálisis con afectación completa o que no muestren recuperación clínica en 3 semanas deberían ser evaluadas mediante estudios electromiográficos (EMG). • Audiometría (tema 25.9): la presencia de una hipoacusia sensoperceptiva puede hacer pensar en un neurinoma extratemporal. La presencia de sordera de conducción obliga a descartar causas de la parálisis en el oído medio (infecciones, colesteatoma). • Tomografía computarizada cervicofacial y craneal, y RM (temas 24.23 y 24.24): para descartar patología traumática otológica, infecciosa o tumoral (ángulo ponto-bulbo-cerebeloso). 412 ¿Qué propuesta haremos? Tratamiento El tratamiento de la parálisis facial idiopática o de Bell puede iniciarse en AP. El 70 % de los pacientes presentan recuperación total espontánea. No farmacológico • Protección del ojo: lágrimas artificiales durante el día y pomadas lubricantes oculares durante la noche; gafas de protección solar durante el día y cierre palpebral nocturno, con gasas y esparadrapo. El paciente debe notificar si tiene dolor o secreción ocular. • Masaje facial: tres veces al día durante 10 minutos para evitar la atrofia muscular. La estimulación eléctrica de los músculos paralizados está contraindicada porque retrasa la regeneración nerviosa. Farmacológico • El uso de corticosteroides en la parálisis facial periférica sigue siendo controvertido, aunque se utilizan ampliamente. En diversos estudios, incluyendo un metaanálisis, se plantea que el tratamiento con corticosteroides es beneficioso al disminuir las secuelas; pero la revisión Cochrane (Salinas et al., 2005) concluye que las pruebas disponibles de los ensayos controlados aleatorios no muestran con claridad un beneficio significativo. Un estudio reciente (Sullivan et al., 2007) demostró un beneficio claro (recuperación funcional) del tratamiento con corticosteroides a los 3 meses (83 % recuperaciones en el grupo tratado con prednisolona comparado con el 63 % en el resto) y a los 9 meses (94 frente al 81 %). 5.3 Parálisis facial Consejos prácticos • La parálisis del nervio facial es un síntoma, no un diagnóstico. Se debe averiguar la etiología de la parálisis facial antes de establecer un diagnóstico. • Una vez establecido el diagnóstico de parálisis facial, es importante hacer una gradación con fines pronósticos (completa/incompleta). • La parálisis de Bell suele cursar con pródromos como sensación de entumecimiento, dolor retroauricular y disgeusia. • El signo de Bell permite diferenciar la parálisis facial periférica de la de origen central. • Si se formula el diagnóstico provisional de parálisis de Bell y no se produce la resolución de los síntomas, debemos reconsiderar el diagnóstico. Ante el diagnóstico erróneo de una parálisis de facial idiopática o de Bell, los tumores son la etiología más frecuente. • En las parálisis recurrentes, sobre todo homolaterales, debe descartarse la existencia de una causa tumoral y en personas jóvenes, la esclerosis múltiple. Se suele utilizar prednisona en dosis de 1 mg/kg/día durante 5-10 días y luego se reduce gradualmente en 5 días. No existen evidencias claras para apoyar el tratamiento con corticoides en niños. • El tratamiento con aciclovir o valaciclovir (se valora que el VHS podría producir casos de parálisis de Bell) ya sea solo o combinado con corticosteroides, ha dado lugar también a resultados dispares en la parálisis facial idiopática. Los resultados de la revisión Cochrane del año 2005 concluyen que se necesitan más datos para poder realizar una recomendación definitiva con respecto al efecto del aciclovir o el valaciclovir sobre la parálisis facial. El aciclovir, en un estudio reciente (Sullivan et al., 2007), en monoterapia no demostró eficacia alguna ni tampoco beneficio adicional al combinarlo con la prednisolona. En caso de utilizar estos fármacos: aciclovir en dosis de 800 mg/5 veces al día o valaciclovir 1 g/8 h durante 7 días. • En casos de sincinesias y hemiespasmos posparalíticos, valorar infiltraciones con toxina botulínica A (GR A). Derivación Preferente • Otorrinolaringología – Sospecha de patología tumoral. – Formas secundarias a patología otológica. – En la parálisis de Bell, si hay una mala respuesta al tratamiento después de 2 semanas del inicio, se debe derivar al otorrinolaringólogo (ORL). • Neurología – Sospecha de patología tumoral. – Parálisis recidivante homolateral, parálisis de comienzo gradual (semanas), parálisis bilateral. – Dudas sobre el diagnóstico. Errores más frecuentes • No diferenciar las parálisis centrales de las periféricas. • No distinguir las parálisis faciales secundarias de las idiopáticas, un 20 % tienen causas subyacentes que deben de ser manejadas adecuadamente. • No realizar una correcta protección ocular. ¿Se ha resuelto del todo? Complicaciones más frecuentes • Úlcera corneal; paresia permanente; contractura; reinervación anómala: sincinesias (cerrar el párpado al mover el labio), lágrimas de cocodrilo (lagrimeo al masticar por reinervación errónea), espasmo hemifacial. • La cirugía rehabilitadora y el reentrenamiento neuromuscular podrían ser de ayuda para paliar parte de estas complicaciones. Pronóstico y seguimiento • La gravedad de una parálisis facial periférica puede estimarse clínicamente. El sistema de gradación adoptado por la Academia Americana de Otorrinolaringología (tabla 2) permite registrar de una manera objetiva la gravedad de la lesión porque el pronóstico de la recuperación está relacionado con el grado de lesión (grado de recomendación A). Se distinguirían dos tipos: – Parálisis facial incompleta (se correspondería con los grados I-II-III y IV): pronóstico de recuperación a las 3 semanas 94 %. La presencia de una parálisis incompleta en la primera semana es el signo pronóstico más favorable. – Parálisis facial completa (grados V-VI ): pronóstico de recuperación a las 3 semanas 60 %. La posibilidad de realizar una contracción voluntaria, aunque sea mínima, implica cierto grado de recuperación, mientras que la abolición completa de la motilidad facial sería indicador de mal pronóstico. • El 80 % de las parálisis de Bell se recuperan perfectamente en 3 semanas, si esto no ocurre, es improbable que lo haga hasta las 4-6 semanas con la regeneración del nervio, y a los 6 meses, los que no recuperaron, permanecerán con secuelas. • El pronóstico del síndrome de Ramsay-Hunt es significativamente peor. • La edad superior a 60 años, la presencia de dolor intenso, alteración del gusto, parálisis completa y la asociación con otras patologías (diabetes, hipertensión arterial) o el embarazo, son factores de mal pronóstico. • Pauta de visitas a realizar en una parálisis facial: – Primera visita a la semana (aplicar el sistema de gradación, completa o incompleta). 413 5.3 Parálisis facial – Segunda visita a las 3 semanas: tendría que haber una mejora evidente; si no fuese así, realizar pruebas electromiográficas. – Tercera visita a las 6 semanas: debe haber recuperación total y si no fuese así, derivar. 2. Allen D, Dunn L. Aciclovir o valaciclovir para la parálisis de Bell (parálisis facial idiopática) (Revisión Cochrane traducida). En: Biblioteca Cochrane Plus, 2005, número 4. Oxford: Update Software Ltd. Disponible en: URL: http://www.update-software.com. (Traducida de The Cochrane Library, 2005 Issue 4. Chichester, UK: John Wiley & Sons, Ltd.) ¿Podemos hacer alguna cosa más? Educación sanitaria Explicar el buen pronóstico de la mayoría de los casos de parálisis de Bell, ya que los pacientes suelen pensar que tienen un accidente vascular o un tumor, por ello son necesarios los seguimientos continuados hasta su recuperación, prestándole apoyo psicológico si es necesario. Indicarle que proteja el ojo para prevenir cualquier daño permanente. Consultar en Guía práctica de la salud, p. 29. 3. Santos-Lasaosa S, López del Val J, Ortells M, Escalza I, Navas I. Parálisis facial periférica: etiología, diagnóstico y tratamiento. Rev Neurol. 2000;31:14-6. 4. Sullivan FM, Swan IR, Donnan PT, Morrison JM, Smith BH, McKinstry B, et al. Early treatment with prednisolone or acyclovir in Bell’s palsy. N. Engl J Med. 2007;357(16):1598-607. 5. Serrano B, Novell T. Parálisis facial. AMF 2006;2(10):575-82. 6. Bell’s Palsy. Clinical Knowledge Summaries. 2008. Disponible en: http://www.cks.nhs.uk/bells_palsy#-370056 7. Quant EC, Jeste SS, Muni RH, Cape AV, Bhussar MK, Peleg AY. The benefits of steroids versus steroids plus antivirals for treatment of Bell’s palsy: a meta-analysis. BMJ. 2009;339:b3354. 8. de Almeida JR, Al Khabori M, Guyatt GH, Witterick IJ, Lin VY, Ned- Bibliografía recomendada zelski JM, Chen JM. Combined corticosteroid and antiviral treatment for Bell palsy: a systematic review and meta-analysis. JAMA. 2009;302(9):985-93. 1. Salinas RA, Álvarez G, Ferreira J. Corticosteroides para la parálisis 9. Madhok V, Falk G, Fahey T, Sullivan FM. Prescribe prednisolone de Bell (parálisis facial idiopática) (Revisión Cochrane traducida). alone for Bell’s palsy diagnosed within 72 hours of symptom on- En: Biblioteca Cochrane Plus, 2005, número 3. Oxford: Update Software Ldt. (Resumen). Texto completo disponible en: URL: http://www.cochrane.org/reviews/en/ab001942.html 414 set. BMJ. 2009;338:b255. 10. Tiemstra JD, Khatkhate N. Bell’s palsy: diagnosis and management. Am Fam Physician. 2007;76(7):997-1002. 5.4 Mareo y vértigo M. J. Salazar Scheifler ¿De qué hablamos? Introducción La sensación de «mareo» es uno de los motivos de consulta más frecuentes en Atención Primaria (AP) (2-5 % en todas las edades y 7 % en > 60 años). Este término engloba un sinfín de situaciones diversas, que hay que intentar diferenciar, en primer lugar, mediante la definición más clara posible de los síntomas del paciente. Conceptos iniciales • Mareo: término impreciso usado habitualmente por el paciente para tratar de describir síntomas subjetivos, como la sensación de desmayo, vacío, falta de estabilidad o equilibrio e incluso el vértigo (es el equivalente del término inglés dizziness). El mareo es referido, con frecuencia, como una alteración del equilibrio. Este último es el estado mediante el cual se conserva la postura del cuerpo contra las fuerzas de la gravedad. Los mecanismos más importantes que intervienen en el mantenimiento de la postura en equilibrio son tres: visual, vestibular y propioceptivo, que están sometidos a una continua autorregulación. Una lesión en cualquiera de éstos puede manifestarse como un trastorno del equilibrio, quejándose el paciente de mareo o falta de estabilidad. La lesión del sistema vestibular provoca, sobre todo, sensación de vértigo. • Inestabilidad: dificultad para mantener la postura y/o la marcha, necesitando puntos de apoyo. • Vértigo: ilusión de movimiento. Sensación subjetiva de giro o desplazamiento del propio cuerpo o del entorno respecto de éste. Indica disfunción del sistema vestibular. • Presíncope, desfallecimiento, vahído o prelipotimia: sensación inminente de ir a «perder la conciencia». • Síncope: pérdida brusca de conciencia, acompañada de pérdida del tono muscular, con recuperación espontánea (tema 3.16). • Debilidad: pérdida de la fuerza muscular. • Drop attack: caída brusca sin pérdida de conciencia por pérdida de fuerza momentánea. • Cinetosis: malestar experimentado con el movimiento rítmico o pendular, así como con giros o cambios bruscos de velocidad (aceleración y desaceleración horizontal, vertical o rotatorio) al ir en coche, barco, etc. (v. Vértigo fisiológico). • Oscilopsia: imposibilidad de leer a la vez que se gira la cabeza rítmicamente, de un lado a otro. Ocurre por lesión vestibular, sobre todo bilateral, por afectación del reflejo oculovestibular. Clasificación del vértigo 1. Vértigo fisiológico Se produce cuando el cerebro se enfrenta a un desequilibrio en una de las siguientes situaciones: • La cinetosis, que ocurre cuando una hiperestimulación vestibular se acompaña de cierta estabilidad visual, como en los viajes en coche, avión, barco, etc. Suele acompañarse de náuseas, vómitos, sudor frío, sialorrea, etc., y la susceptibilidad individual a padecerlo es muy variable, produciéndose habituación hacia las 72 horas. • También puede ocurrir cuando hay cierta estabilidad vestibular y disbalance visual: cines con imágenes estereoscópicas, utilización de gafas nuevas. • Cuando se pierden las referencias visuales estables (plano horizontal) en el llamado vértigo de las alturas. 2. Vértigo patológico Puede ser de dos tipos: • Vértigo periférico: La causa (tabla 1) afecta al laberinto vestibular o al nervio vestibular. El vértigo periférico suele ser brusco, acompañándose de síntomas vegetativos (náuseas y vómitos). En general (salvo el vértigo paroxístico posicional benigno [VPPB]), presenta un síndrome vestibular completo (nistagmo, signo de Romberg «presente», prueba de Barany «positiva») y armónico (nistagmo hacia el lado sano y signo de Romberg, prueba de Barany y marcha hacia el lado de la lesión) (tabla 2). Puede acompañarse o no de síntomas cocleares (hipoacusia y acúfenos), pero nunca de síntomas neurológicos. Es siempre temporal y empeora con los Tabla 1. Causas del vértigo Vértigo periférico Fisiológico: cinetosis Canalolitiasis o cupulolitiasis (40 %) Traumatismo craneal (TCE) y/o cervical agudo (conmoción laberíntica) Infecciones (neuronitis vestibular [40 %], laberintitis aguda) Cambio brusco de presión (barotrauma, levantadores de peso) Fármacos: AAS, aminoglucósidos, anticonvulsivos, diuréticos, litio, AINE, quinina, cisplatino, imipramina Tóxicos: alcohol, arsénico, benceno, monóxido de carbono Hydrops endolinfático (enfermedad de Ménière) (10 %) Tumores: neurinoma VIII par, meningioma, colesteatoma Posquirúrgicos: poslaberintectomía, posresección neurinoma VIII Vértigo central Enfermedad cerebrovascular aguda (3 %): troncoencéfalo o cerebelo Esclerosis múltiple Migraña basilar Tumores de la fosa posterior (meningiomas, meduloblastomas, gliomas, metástasis, etc.) AAS: ácido acetilsalicílico; AINE: antiinflamatorios no esteroideos. 415 5.4 Mareo y vértigo Tabla 2. Diagnóstico diferencial entre vértigo periférico y central Periférico Central Vértigo Brusco Temporal Rotatorio, bien definido Vómitos frecuentes Influido por la postura Sin clínica neurológica asociada Síndrome vestibular completo y armónico (no siempre) Insidioso (excepto ACVA y migraña) Continuo Mal definido Raro, salvo en ACVA Espontáneo, no influido por la postura Con clínica neurológica asociada Síndrome vestibular incompleto y no armónico Nistagmo espontáneo Horizontal-rotatorio Unidireccional Se suprime por fijación visual Concomitante al vértigo Hacia el lado sano Rotatorio, horizontal o vertical puro Bidireccional No se suprime Puede ser aislado Hacia el lado de la lesión Nistagmo provocado Tiene período de latencia (3-10 s) Es fatigable Reproducible Cumple la ley de Alexander Se agota No tiene período de latencia No se fatiga No reproducible No cumple la ley de Alexander No se agota ACVA: accidente cerebrovascular agudo. movimientos de cabeza. Una persona que está «mareada» todo el día y se mueve de un lado a otro no tiene vértigo periférico. En el nistagmo del vértigo periférico, la fase rápida bate hacia el lado sano y empeora al mirar hacia el lado de la sacudida rápida, disminuyendo al mirar hacia el otro lado (ley de Alexander), y en el de los centrales, bate al lado enfermo y no cumple la ley de Alexander (es de gran ayuda para diferenciar los vértigos periféricos de los centrales) (v. tabla 2). • Vértigo central Afecta a estructuras centrales (troncoencéfalo [TE], cerebelo y corteza cerebral) (v. tabla 1). El vértigo central puede ser brusco (casos de accidente isquémico transitorio [AIT] o accidente cerebrovascular agudo [ACVA] instaurado), aunque generalmente es subagudo y mantenido, no se suele acompañar de hipoacusia (salvo afectación aislada de la arteria auditiva interna), no se modifica con los cambios de postura y siempre se acompaña de otros síntomas y signos neurológicos del territorio vertebrobasilar (diplopía, disartria, ataxia, caídas bruscas [drop attack], paresias o parestesias, hemihipoestesia, etc.). El síndrome vestibular es incompleto (signo de Romberg negativo, en la prueba de Barany los «dedos índices» pueden elevarse o separarse, o desviarse sólo uno), disarmónico y desproporcionado (v. tabla 2). Estos pacientes no suelen poder caminar. Es menos frecuente que el periférico, pero hay que recordar que, hasta un 25 % de los pacientes con factores de riesgo cardiovascular (FRCV) que acuden a urgencias con vértigo grave, nistagmo e inestabilidad pueden presentar un vértigo central por un infarto cerebeloso. El nistagmo es de características centrales (v. tabla 2). 416 ¿Qué lo puede ocasionar? Causas La causa más frecuente del cajón de sastre que es el término mareo es el vértigo periférico (44 %), y dentro de éste, el vértigo posicional paroxístico benigno (VPPB) (el 25 % de las causas de mareo). Le siguen en frecuencia los multicausales (26 %), los psiquiátricos (16 %), los de causa desconocida (13 %), el vértigo central (11 %) y el de causa cardiovascular (6-14 %). A mayor edad de los pacientes, mayor porcentaje de causas cardiovasculares, de vértigo de tipo central y mareos multicausales. Todos los cuadros genéricamente denominados mareos pueden encuadrarse en cuatro grandes grupos diagnósticos: psicógeno, presíncope o desfallecimiento (tema 3.16), inestabilidad o trastorno de la marcha (tema 5.10) y vértigo (figura 1). Mareo mal definido o de tipo psicógeno Sensación de tener la cabeza como hueca, embotada, de ir flotando; como si se fueran a caer, cosa que casi nunca ocurre. Aparece frecuentemente en ambientes cerrados y/o llenos de gente y en situaciones estresantes. Puede acompañarse de angustia o tristeza, insomnio, falta de energía, etc. No desaparece con el decúbito (a diferencia del presíncope). La exploración física es normal. Aparece con más frecuencia en los trastornos por ansiedad generalizados (TAG, fobias y crisis de pánico), depresión y en los trastornos somatomorfos (trastorno de somatización, hipocondría, trastorno de conversión). No Seguimiento en APS Neuritis vestibular Sí Mejoría Tto. en APS, revisión 48 h Sí No Tema 5.2 Neurolaberintitis vírica Laberintitis Neurinoma del n. acústico No Derivación urgente a ORL Infarto laberíntico Traumatismo Sí Inicio súbito Migraña ITC ordinaria a ORL Enfermedad de Ménière Fístula perilinfática Enfermedad autoinmunitaria Hipoacusia neurosensorial unilateral o asimétrica Espontáneo recurrente ITC a neurología Esclerosis múltiple, accidente isquémico transitorio Sí Maniobra de Dix-Hallpikec Positiva Tema 5.10 Tema 3.16 Tema 6.1 Maniobra de Epleyf. Ejercicios de rehabilitación Brant-Daroff Vértigo posicional benigno canal semicircular posterior Nistagmo periféricoe Posicional recurrente Vértigo posicional benigno canales semicirculares horizontal y superior No Inestabilidad Trastorno de la marcha Desfallecimiento Presíncope Trastorno psicógeno Crisis breves, recurrentes, que se desencadenan con movimientos de la cabeza y ceden con el reposo Negativa Focalidad neurológica o nistagmo centrald Pérdida del control postural. Aparece al deambular Mejora sentado Breve, síntomas vegetativos, sensación de pérdida inminente de la conciencia Focalidad neurológica o nistagmo centrald Crisis de 20 min a 24 h de duración, recurrentes y con síntomas asociados Sí Continuo, diario, exploración anodina. Poco definido «cabeza hueca» b Fármacos que pueden ocasionar mareos: AAS, aminoglucósidos, quinina, cisplatino, anticonvulsivos, diuréticos, litio, AINE, imipramina. Tóxicos que ocasionan mareos: alcohol, arsénico, benceno, monóxido de carbono. c Maniobra o test posicional de Dix-Hallpike: cambiar al paciente de sentado a decúbito supino con la cabeza 45º bajo el plano de la camilla y girada hacia el lado afectado. Vértigo a los 3-5 s de latencia. d Nistagmo central: cualquier dirección, sin latencia, no fatigable, inarmónico y puede ser aislado. e Nistagmo periférico: horizontal o rotatorio, con latencia, fatigable, armónico, concomitante al vértigo. f Maniobra de Epley (para vértigo posicional benigno/canales semicirculares posteriores): mantener posición Dix-Hallpike 2-3 s. Girar lentamente 90º hacia el lado contralateral y mantener 1-2 s. Girar al paciente a decúbito lateral hacia el lado contralateral, manteniendo cabeza rotada 45º y conservando esta posición 3-4 s. Para finalizar, se sienta al paciente suavemente. Repetir en consultas sucesivas si persiste la sintomatología. AAS: ácido acetilsalicílico; APS: atención primaria de salud; ITC: interconsulta; ORL: otorrinolaringología. a No Hipoacusia neurosensorial unilateral o asimétrica Audición normal Figura 1. Algoritmo diagnóstico del mareo y del vértigo. Derivación urgente al hospital Infarto/ hemorragia cerebelosos. Infarto troncoencefálico Esclerosis múltiple Sí Signos de alarma Espontáneo, agudo, prolongado Crisis de más de 24 h de duración, vértigo intenso, empeora con movimento. Desequilibrio Suspender AP de fármacosa, tóxicosb Ilusión de movimiento. Vértigo Anamnesis. Duración No Descripción subjetiva de la sensación experimentada por el paciente de inestabilidad con síntomas acompañantes Mareo 5.4 Mareo y vértigo 417 5.4 Mareo y vértigo Desfallecimiento o presíncope Sensación de ir a perder el conocimiento, de ir a desmayarse, cosa que puede ocurrir (síncope), si la duración de la causa que lo provocó (p. ej., arritmia), dura lo suficiente como para afectar al flujo cerebral o si no se toman las medidas oportunas (tumbarse y elevar las piernas) en el caso del presíncope vasovagal. Son características, durante el episodio, la hipotensión, la bradicardia o la taquicardia con pulso filiforme y palidez. Es un cuadro brusco, en general breve, de recuperación rápida y en el caso del vasovagal, con pródromos: visión borrosa y sudoración (tema 3.16). Inestabilidad Sensación de caminar de manera inestable, de perder el equilibrio. También se llama desequilibrio. Consiste en la pérdida del control postural en bipedestación o al caminar, con tendencia a caer, referido sobre todo a las piernas y el tronco. Suele ser continuo, casi nunca en rachas o a brotes. No hay sensación de desplazamiento. Desaparece al sentarse. Pueden ocasionar sensación de mareo y se descubren en la exploración física (hay que hacer andar siempre a los pacientes). Son causa de inestabilidad los trastornos de la marcha (tema 5.10) y el déficit sensorial múltiple (DSM). Este último es un cuadro de inestabilidad que, cuando es leve, los pacientes lo refieren como mareo. Suele ser debido a diversos déficit sensoriales. Es frecuente en ancianos debido a la suma de varios factores: déficit auditivo (presbiacusia y/o tapones de cerumen), asociado con frecuencia a un déficit visual (cataratas, degeneración macular asociada a la edad [DMAE], etc.), a la frecuente polimedicación (hipotensores, ansiolíticos; los pacientes que toman de forma continuada más de 4 fármacos tienen muy aumentado el riesgo de caídas) y a trastornos osteoarticulares (artrosis). Además, se pueden sumar enfermedades extrapiramidales (enfermedad de Parkinson) y/o neuropatías periféricas (diabetes mellitus, alcoholismo crónico). Los pacientes no pueden realizar movimientos adaptativos rápidos. Tienen miedo a caerse y aparece al caminar y, sobre todo, en ambientes desconocidos y/o faltos de luz, mejorando al apoyarse o sentarse. Vértigo Es una ilusión de movimiento. Sensación de rotación o movimiento de objetos o de uno mismo en el espacio («como de ir en un tiovivo o en un barco»). Debido a un desorden del sistema vestibular, por asimetría de la actividad neuronal derecha e izquierda, causada por la afectación unilateral de alguno de los siguientes niveles: aparato vestibular (oído interno), nervio vestibular, núcleo vestibular o cerebelo. La destrucción bilateral (p. ej., toxicidad por gentamicina) puede producir desequilibrio, pero no vértigo. Diagnóstico diferencial de los vértigos 1. Vértigos periféricos Se clasifican en función de si presentan o no síntomas cocleares (hipoacusia y/o acúfenos) (v. figura 1). 418 Vértigos periféricos sin síntomas cocleares • Vértigo posicional paroxístico benigno (VPPB): es el más frecuente (17-42 %) de todos los vértigos. Se caracteriza por crisis de vértigo breves, de segundos de duración (paroxístico), recurrentes, precipitados por cambios en la posición de la cabeza en relación a la gravedad: según el canal semicircular afectado se provocan al mirar hacia arriba, tumbarse o levantarse de la cama, postura del dentista o peluquería o bien al girarse hacia los lados en la cama. Aparece en rachas que pueden durar varias semanas, para remitir espontáneamente (35-50 %) con una media de resolución en 2-6 semanas. Se presenta, por lo general, a partir de los 50-60 años, aumentando su frecuencia con la edad (pico a los 60-70 años), y es más frecuente en mujeres. A veces, se acompaña de náuseas y vómitos, pero nunca de hipoacusia ni de síntomas neurológicos. En algunos pacientes puede quedar una cierta inestabilidad postural intercrisis, por un tiempo limitado. Se cree que son debidos a los movimientos de los otolitos (cristales de carbonato cálcico) en los canales semicirculares. Puede ocurrir espontáneamente (> 50 %) sin causa identificable o después de un traumatismo craneal (TCE) (conmoción laberíntica), neuronitis vestibular, infecciones del oído medio y más raramente en el curso de enfermedades del oído interno (laberintitis), poscirugía del oído medio y tras encamamientos prolongados. Los elementos claves para el diagnóstico son la anamnesis y la prueba de provocación de Dix-Hallpike (figura 2). Si el resultado de ésta es negativo pero el cuadro clínico revaluado sigue siendo sugestivo de VPPB, estará indicada la práctica de la maniobra de Pagnini (figura 3) que es específica cuando el problema radica en el canal semicircular horizontal. Si finalmente ésta es también negativa, se puede repetir la maniobra de Dix-Hallpike en otra visita. De persistir la negatividad de las pruebas, habrá que buscar otras causas de vértigo. La exploración otológica y neurológica son normales. • Neuronitis vestibular: consiste en un episodio brusco de vértigo, de 2-7 días de duración, autolimitado, que empeora con cualquier tipo de movimiento (obliga al paciente a permanecer inmóvil) y persiste en reposo. Se suele acompañar de síntomas vegetativos intensos: náuseas y vómitos, de nistagmo espontáneo, de tipo periférico, que le impide fijar la vista y al igual que el anterior, el paciente no tiene síntomas cocleares, ni neurológicos. Con frecuencia despierta con un ataque de vértigo, náuseas y vómitos, y tras un acmé de 2-3 días, va remitiendo (compensación central) en 2-3 semanas, quedando cierta inestabilidad postural (sobre todo en ancianos, en los que puede persistir hasta meses). Finalmente remite el nistagmo. En el 20 % de los pacientes puede recurrir (aunque menos intenso) o quedar un VPPB residual (15-25 %) a los meses o años del ataque inicial. Es frecuente en jóvenes, y en el 25 % de los casos 5.4 Mareo y vértigo A 1 2 3 B Figura 3. Prueba de provocación de Pagnini. (1) El paciente se en- Figura 2. Maniobra de Dix-Hallpike: (A) El examinador se sitúa a la derecha del paciente, rotando su cabeza 45º hacia la derecha, para alinear su CSP derecho con el plano sagital del cuerpo. (B) El examinador mueve al paciente, que está con los ojos abiertos, de la posición sentada a la de decúbito, al borde de la camilla, con el oído derecho hacia abajo, se gira el cuello hasta que la barbilla está al frente y se vuelve a sentar al paciente. Se repite hacia el otro lado. El examinador anotará las características del nistagmo y del vértigo (latencia y duración). Será positivo si desencadena el vértigo y el nistagmo al excitar el CSP del lado afectado. CSP: canal semicircular posterior. Adaptado de: Bhattacharyya: Otolaryngol Head Neck Surg. 2008;139(5) Sup 4: S47–S81. existe un antecedente de infección «vírica» del tracto respiratorio. En la exploración el paciente se mantiene estable con los ojos abiertos, pero puede desviarse hacia el lado afectado con los ojos cerrados (prueba de Romberg +), y el nistagmo es de características periféricas. En personas con FRCV hay que hacer el diagnóstico diferencial con un ACVA cerebeloso o con AIT de la rama vestibular anterior de la arteria vertebrobasilar (que puede debutar con un cuadro vertiginoso, brusco, de unos minutos de duración). Vértigos periféricos con síntomas cocleares (hipoacusia y/o acúfenos) • Laberintitis aguda: clínica de vértigo cataclísmico con sordera. Suele durar 1-3 semanas. El vértigo se suele recuperar (por compensación central), pero la sordera no se recupera, pudiendo quedar una sensación de desequilibrio y acúfenos, sobre todo en las de origen bacteriano. Se acompaña de otros síntomas de la enfermedad causante, que suelen dominar el cuadro clínico: infecciones bacterianas (otitis, meningitis, mastoiditis, lúes), víricas (sarampión, parotiditis), fármacos y colesteatoma. cuentra tumbado en posición neutra (decúbito supino). Se gira la cabeza del paciente rápidamente hacia la derecha (2) y se examina el nistagmo. El nistagmo, que es horizontal, suele ser geotrófico (el componente rápido bate hacia el oído que está en posición inferior), o menos frecuentemente, ageotrófico (bate hacia el lado superior, tanto al girar hacia un lado como hacia el otro). El lado afectado se presume aquel hacia el que bate el nistagmo con más intensidad. Se vuelve la cabeza a la posición de inicio y se gira rápidamente hacia la izquierda (3) examinando de nuevo el nistagmo. Adaptado de: Bhattacharyya: Otolaryngol Head Neck Surg. 2008;139(5)Sup 4: S47–S81. • Vértigo de Ménière: episodios de vértigo brusco, no desencadenados por la postura, que suelen durar 2-3 horas (mínimo 20 min), recurrentes, que se acompañan de hipoacusia, acúfenos (tipo caracola) y sensación de plenitud en el oído, con distorsión en la percepción de los ruidos. Con frecuencia los pacientes presentan náuseas y vómitos. Los brotes suelen aparecer con una frecuencia de 6 a 12 por año y los períodos de remisión pueden durar días, meses o incluso años. Al inicio se suele recuperar la función, tanto coclear como vestibular, por lo que la exploración intercrisis puede ser normal. Con el tiempo suelen desaparecer las crisis vertiginosas (aunque los pacientes pueden referir un cierto desequilibrio nocturno), quedando como secuela una hipoacusia, que puede ser progresiva (primero se pierden las frecuencias bajas). Puede detenerse en cualquier estadio, pero si progresa, en los últimos estadios padecerán sordera con sonido distorsionado y acúfenos continuos. Tiene una evolución fluctuante, progresiva e impredecible y es frecuente la bilateralidad (30 %). En el 60-80 % de los casos se resuelven los síntomas, menos la pérdida auditiva, en 5-10 años. La prevalencia es de 6/1.000 en la población general y aparece hacia la cuarta-sexta década de la vida. Se produce por un aumento de la presión de la endolinfa, con edema endolinfático (hydrops). Para el diagnóstico se necesitan, al menos, dos episodios de vértigo y la confirmación audiométrica: hipoacusia sensorial con reclutamiento (inicialmente para las frecuencias graves) (tema 25.9). • Neurinoma del acústico: es una tumoración (generalmente schwanoma) de la rama vestibular del VIII par en el conducto auditivo interno. Sus manifestaciones 419 5.4 Mareo y vértigo se encuentran a caballo entre los periféricos y los centrales. La clínica se inicia con una hipoacusia neurosensorial, con o sin acúfenos, seguida de vértigo mantenido (20 %) (a diferencia del síndrome de Ménière, en que el vértigo es paroxístico y desde el inicio), pero sobre todo, de inestabilidad (50 %), ya que al ser de lenta progresión, da tiempo a que desarrolle compensación central. A medida que se extiende el neurinoma, comienzan a aparecer síntomas neurológicos de vecindad: parálisis facial (VII), anestesia corneal (V), y tardíamente ataxia (ángulo pontocerebeloso). Puede formar parte de la neurofibromatosis de von Recklinghausen, pudiendo ser bilateral. • Fístula perilinfática: aparece tras un trauma penetrante en oído medio, barotrauma o trauma acústico intenso, fractura temporal, cirugía ótica, etc. Cursa con vértigo de tipo periférico, náuseas y vómitos, acúfenos e hipoacusia inmediatos al traumatismo o en relación a cambios de presión en el oído. A diferencia del VPPB, se provoca por cambios de presión, no de la postura de la cabeza. Un estímulo sonoro intenso o la maniobra de Valsalva puede desencadenar la clínica (signo de Tulio). Audiometría: hipoacusia neurosensorial o mixta. Otros vértigos periféricos • Traumatismos craneoencefálicos (TCE): – Con fractura del temporal: la lesión vestibular periférica puede ser permanente y se acompaña de vértigo, sordera y parálisis facial unilateral. Aparece otorrea hemorrágica. – Sin fractura temporal o conmoción laberíntica (TCE leve): confusión o desorientación, pérdida de conciencia inferior a 30 minutos, amnesia postraumática inferior a 24 horas u otros signos focales transitorios. Suele asociarse a cefalea (60 %), irritabilidad, insomnio, dificultad de concentración (38 %), memoria y atención, mareo (35-75 %) y/o vértigo. El vértigo postraumático sin fractura temporal puede ser de dos tipos: – Vértigo agudo postraumático: inmediato al traumatismo, de características periféricas, que empeora con los cambios de postura. Mejora en pocos días y la mayoría de los pacientes estarán asintomáticos en 1-3 meses. – Vértigo posicional postraumático: es el más frecuente, aparece a los días o semanas del traumatismo, en forma de ataques de vértigo, cortos y súbitos, desencadenados con los movimientos de la cabeza. La mayoría remiten a los 3 meses y todos a los 2 años. • Fármacos y tóxicos: – Aminoglucósidos: más que vértigo provocan ataxia y desequilibrio por afectación vestibular bilateral y casi siempre hipoacusia, que puede ser irreversible. La estreptomicina y la gentamicina son los que más afectan al aparato vestibular. Los aminoglucósidos tópicos para tratamiento de la otitis externa están contraindicados en personas con perforación del tímpano. 420 Los diuréticos (furosemida y ácido etacrínico) potencian el efecto tóxico de los aminoglucósidos. – Anticonvulsivos: pueden provocar vértigo y nistagmo reversible si se supera el rango tóxico. – Salicilatos: el vértigo y los acúfenos son uno de los primeros síntomas de la intoxicación por salicilatos. Son reversibles a los 2-3 días de la retirada del fármaco. – Cisplatino: tiene toxicidad auditiva y vestibular. – Alcohol: tiene un efecto mixto provocando un vértigo posicional por toxicidad directa sobre la cúpula e inestabilidad y ataxia por disfunción cerebelosa. Los niveles de alcoholemia se relacionan bien con la presencia de nistagmo. 2. Vértigos de origen central Las lesiones se encuentran en la vía vestibular central: troncoencéfalo, cerebelo, conexiones vestibulocerebelosas y de forma excepcional, en las estructuras supratentoriales: tálamo y corteza cerebral. Entre todas las causas de vértigo, la más frecuente, dentro de los centrales, es el vértigo migrañoso (14 %); el debido a isquemia cerebrovascular supone el 5 % de las causas, y los tumores menos del 1 %. Sin embargo, el vértigo aparece en un 75-100 % de los ACVA del territorio vertebrobasilar (20 % del total de los ACVA). El vértigo central, salvo los debidos a migraña o a ACVA, que son bruscos, es de inicio lento e insidioso y de curso mantenido. El nistagmo es de características centrales (tabla 2). Se acompaña siempre de otros síntomas neurológicos. Puede encontrarse en las siguientes situaciones: ACVA del troncoencéfalo o del cerebelo • Isquemia transitoria vertebrobasilar: se debe sospechar en todo paciente con FRCV que presente un vértigo brusco de minutos de duración (menos de 30 min), que repite varias veces al día y/o varios días seguidos, máxime si se acompaña de otros síntomas neurológicos vertebrobasilares: afectación de pares craneales (diplopía, disartria, disfagia), ataxia, etc., y que no se afecta por la postura. Tampoco se acompaña de hipoacusia. El nistagmo es de características centrales. • Infarto isquémico de la arteria cerebelosa posterosuperior (síndrome de Wallenberg o síndrome bulbar lateral): debuta con un vértigo intenso asociado a vómitos. Se acompaña de hemianestesia facial homolateral y hemianestesia corporal contralateral disociada (térmica y dolorosa), disartria, disfonía, disfagia, nistagmo, síndrome de Claude-Bernard-Horner (ptosis, miosis y enoftalmos) homolateral y síndrome cerebeloso con dismetría de extremidades homolateral y ataxia (hacia el lado afectado). • Infarto isquémico de la arteria cerebelosa anteroinferior (síndrome bulboprotuberancial lateral): el vértigo se acompaña de sordera neurosensorial brusca homolateral o bilateral, nistagmo horizontal-rotatorio al lado sano, síntomas y signos cerebelosos: ataxia, dismetría, e hipoestesia facial homolateral con pa- 5.4 Mareo y vértigo rálisis facial o sin ella, y con hipoestesia corporal contralateral o sin ella. • Infarto isquémico de la arteria laberíntica (infarto laberíntico): cuadro brusco de sordera, vértigo y nistagmo, indistinguible de una laberintitis aguda. El nistagmo, de tipo central, es más intenso al mirar hacia el oído afectado. Se debe sospechar en personas con FRCV y ausencia de otros síntomas (infección, etc.). • Infarto isquémico cerebeloso: los pacientes suelen presentar intensa ataxia (incluso sentados), vértigo, dismetría y hemianopsia contralateral. • Hemorragia cerebelosa: se suele acompañar de cefalea intensa «occipital», mareo con vómitos profusos y ataxia (marcha sin ayuda imposible). Con frecuencia rigidez de nuca, parálisis facial y de la mirada conjugada. Suele evolucionar rápidamente a tetraparesia y coma. • Disección de la arteria vertebral: después de maniobras quiroprácticas o práctica deportiva. Cursa con cervicalgia (de nuca a hombros), vértigo, diplopía y parestesias faciales unilaterales. Puede preceder a la aparición de un ACVA en varios días. Esclerosis múltiple Es una enfermedad degenerativa de la sustancia blanca que evoluciona a brotes, con exacerbaciones y remisiones espontáneas y formas progresivas (20 %). En cada uno de los brotes puede desarrollar síntomas diferentes: el primer síntoma suele ser la paresia de extremidades inferiores (40 %), vértigo (como primer síntoma en el 5 % de los casos y en algún momento de su evolución en un 30-40 %), neuritis retrobulbar, espasticidad, trastornos vesicales, ataxia, oftalmoplejía internuclear con oscilopsia (por afectación del fascículo longitudinal posterior), etc. Los síntomas tienen una duración de varios días (semanas y a veces hasta meses). En los brotes aparecen nuevos síntomas o recidivan los anteriores. Suele iniciarse en la segunda o tercera década con predominancia femenina (2/1). De causa desconocida, se acepta una base autoinmunitaria, de antígeno desconocido, aunque se cree que es un virus y que ocurre en personas predispuestas genéticamente. Migraña basilar Puede cursar con aura en forma de síntomas neurológicos vertebrobasilares (vértigo, escotomas, hemianopsia homónima, diplopía, disartria, ataxia, parestesias simultáneas bilaterales, acúfenos, hipoacusia) que preceden a la cefalea (la cefalea pulsátil típica suele ser occipital y con fonofobia) o no, y puede ocurrir sin ésta (equivalente migrañoso, frecuente en niños) y cuando ocurren, suelen durar unos 30-60 minutos y son reversibles. La cefalea, si aparece, suele ser nucal y de unas 24 horas de duración. Un 15-20 % de las personas migrañosas tiene episodios de vértigo; el cuadro es indistinguible del VPPB, salvo por la historia de migraña. El vértigo paroxístico benigno de la infancia ha sido considerado un equivalente migrañoso de jaqueca de tipo basilar que se manifiesta en la infancia. Suele aparecer en los primeros 4 años de vida. Los ataques de vérti- go duran unos segundos y se desencadenan por cambios posturales, sobre todo en posición horizontal. Los antecedentes familiares de jaqueca son frecuentes. Tumores del cerebelo o troncoencéfalo Muy poco frecuentes: meduloblastoma en niños, gliomas o metástasis en adultos. Se acompañan generalmente de otra clínica neurológica (ataxia, alteración de los movimientos oculares, etc.) y de datos de hipertensión endocraneal, aunque a veces, al inicio, pueden quejarse sólo de cefalea de reciente comienzo y vértigo mantenido. Epilepsia del lóbulo temporal Estas crisis de vértigo pueden aparecer como crisis parciales simples, formar parte de una crisis parcial compleja o preceder como un «aura» a una crisis generalizada y se suelen acompañar de otros síntomas como convulsiones, alucinaciones o ilusiones sensoriales, fenómenos psíquicos, etc. Síndrome de Arnold-Chiari Herniación congénita de las amígdalas, asociado a menor o mayor descenso cerebral. Los síntomas suelen comenzar en la edad adulta. Presentan un nistagmo vertical (down beat) y oscilopsia en esa dirección. La cefalea tusígena es común. ¿Qué tenemos que hacer? Anamnesis Es la parte más importante para la orientación diagnóstica (entrevista clínica semidirigida, sistemática y estructurada) con el fin de encuadrar el mareo en uno de los cuatro grandes grupos diagnósticos: psicógeno, cardiovascular, inestabilidad o vértigo. • Antecedentes personales: historia de patología ótica, visual o neurológica previa. Episodios de migraña. Antecedentes de depresión o ansiedad. Traumatismo craneal o barotrauma previo. Existencia de FRCV. Fármacos (hipotensores, antiarrítmicos, antidiabéticos orales, vestibulotóxicos). Tóxicos: alcohol, tabaco, otras drogas. • Enfermedad actual: – Cualidad del síntoma: tener la cabeza como hueca (psicógeno), desfallecimiento o sensación de ir a perder el conocimiento (presíncope), inestabilidad o desequilibrio (déficit sensorial múltiple), giro de objetos o de uno mismo (vértigo). – Frecuencia, duración y curso del síntoma: un mareo breve y con recuperación completa es probable que sea un presíncope (cardiovascular), mientras que si es de larga duración, será más probable psicógeno. Un vértigo que dura segundos es probable que sea un VPPB; si minutos, un AIT o migraña; si horas, síndrome de Ménière; si días, neuronitis o laberintitis, y más continuado, central; en forma de crisis aisladas o recurrentes (vértigo periférico), o más mantenido (vértigo central). 421 5.4 Mareo y vértigo – Factores desencadenantes y posición previa: vértigo con cambios de postura de la cabeza (vértigo posicional benigno), mareo al levantarse (hipotensión ortostática), en ambientes cerrados (psicógeno), al caminar (inestabilidad), traumatismo (conmoción laberíntica). – Síntomas acompañantes: hipoacusia, acúfenos, cortejo vegetativo en el vértigo periférico, clínica neurológica asociada en el vértigo central, hipoacusia en el síndrome de Ménière. Exploración física Las maniobras de mayor rentabilidad diagnóstica para el mareo son tres: • La toma de presión arterial (PA) en decúbito y de pie (5 min en decúbito y 1 min de pie) para la hipotensión ortostática. • Maniobra de Dix-Hallpike (sensibilidad 82 %). • Búsqueda de la presencia de nistagmo para diferenciar el vértigo periférico del central. General y cardiovascular Coloración de piel y mucosas. Auscultación cardíaca (soplos, frecuencia cardíaca, ritmo) y de los troncos supraaórticos y exploración de pulsos. PA en ambos brazos, y en decúbito y de pie; existe hipotensión ortostática si se objetiva una caída de la presión arterial sistólica (PAS) > 20 mmHg y/o la presión arterial diastólica (PAD) > 10 mmHg al adoptar el paciente la posición vertical 1 minuto después de haber permanecido 5 minutos en decúbito. Otorrinolaringología (ORL) • Otoscopia (colesteatoma, vesículas de herpes zoster). • Audición: voz y acumetría con diapasón (tema 2.12). Neurológica • Nivel de conciencia y funciones superiores. Habla. • Visión (optotipos), pupilas y pares craneales (examinar los movimientos oculares y comprobar la sensibilidad de la cara y córnea (una anestesia corneal puede revelar un tumor del ángulo pontocerebeloso). • Fuerza y sensibilidad de las extremidades. • Pruebas de coordinación cerebelosas: pruebas dedonariz, talón-rodilla, y marcha (tema 5.10). • Estudio del nistagmo: es un indicador muy útil de disfunción vestibular y sirve para diferenciar el vértigo periférico del de origen central (tabla 2). Consta de un movimiento lento y otro rápido que es el que define su dirección. Puede ser espontáneo o provocado. El espontáneo es difícil de visualizar y nos podemos ayudar de las gafas de fenzel (suprimen la fijación de la mirada). El provocado lo podemos desencadenar girando la cabeza del paciente rápidamente en el plano horizontal con los ojos abiertos o bien con la maniobra de DixHallpike (figura 2). • Tests posicionales o vestibulares: – Prueba de Romberg: Se mantiene al paciente de pie con los pies juntos y los ojos cerrados. Si hay afectación del sistema pro- 422 pioceptivo, caerá nada más cerrar los ojos. Si la afectación es vestibular, presentará lateropulsión hacia el lado afectado tras unos segundos. Si es cerebelosa, no podrá guardar el equilibrio, ni con los ojos abiertos, ni cerrados. – Marcha en estrella: Se le hace dar al paciente cinco pasos adelante y cinco atrás varias veces, y si la lesión es vestibular, éste se desviará hacia el lado afectado. – Test de Dix-Hallpike: Consiste en provocar el nistagmo en pacientes afectados de vértigo posicional benigno (figura 2). – Test de los índices de Barany: El examinador se sitúa delante del paciente, con los brazos extendidos y los dedos en dirección al paciente. Éste hará coincidir sus índices con los del examinador y, una vez hecho, se le indicará que cierre los ojos. Si los índices se desvían hacia un lado, indicará afectación laberíntica del mismo lado (figura 4). En el vértigo central puede elevarse, separarse o desviarse sólo un índice. – Reflejo oculovestibular: Se le da al paciente a leer un texto a la vez que se le hace girar la cabeza rítmicamente de un lado a otro. Si hay afectación vestibular no podrá leer (oscilopsia), sobre todo si ésta es bilateral. Exploraciones complementarias En caso de VPPB no habría que hacer más pruebas complementarias. Su diagnóstico se basa en la anamnesis y exploración física (recomendación clase C). Iniciales • Laboratorio: hemograma (anemia), glucemia (hipoglucemia), Cr, Na, K (alteraciones hidroelectrolíticas), ALT (hepatotoxicidad). • Electrocardiograma (ECG): en ocasiones puede dar la clave de un mareo (p. ej., bloqueo auriculoventricular avanzado o completo). Hay que realizarlo ante todo mareo con sensación de desfallecimiento o de ir a perder la conciencia y ante cualquier mareo de causa no aclarada. • Audiometría: si se aprecia hipoacusia (tema 25.9.). Figura 4. Test índice de Barany. 5.4 Mareo y vértigo Posteriores Se pedirán o realizarán por el especialista correspondiente, en función de la clínica y patología de sospecha: • Test de función vestibular (nistagmografía, tests calóricos): en VPPB si el nistagmo es atípico, sospecha de otra patología vestibular, falta de respuesta a las maniobras de reposición canalicular (figuras 5 y 6) o frecuentes recurrencias (grado C). • Potenciales evocados, resonancia magnética, tomografía computarizada (TC), electroencefalograma (EEG), ecocardiograma, Holter, mesa basculante. • Tests de evaluación psiquiátrica: útiles en el mareo de causa desconocida, sobre todo el de larga duración, así como en el estudio del síncope recurrente, vértigo recurrente o de larga evolución y de etiología desconocida (tema 21.41). 1 2 1 3 2 Figura 6. Maniobra de Semont para CSP derecho. (1) El paciente se sienta derecho, y gira la cabeza 45º hacia la izquierda, moviéndosele rápidamente a la posición de tumbado de lado (2). Se le mantiene así unos 30 segundos y es rápidamente llevado a la posición contraria, también tumbado de lado, sin permanecer en la de sentado y sin cambiar la orientación de la cabeza respecto a los hombros (3). Se mantiene otros 30 segundos y el paciente recupera gradualmente la posición inicial de sentado. CSP: canal semicircular posterior. Adaptado de: Bhattacharyya: Otolaryngol Head Neck Surg. 2008;139(5)Sup 4: S47–S81. ¿Qué propuesta haremos? 3 Tratamiento 5 4 Figura 5. Maniobra de Epley o procedimiento de reposición canalicular de partículas (PRC) para el CSP derecho. (1) El paciente se encuentra sentado con la cabeza girada 45º hacia el oído afectado (el que resultó positivo en la maniobra de Dix-Hallpike), en este caso el derecho. (2) El paciente es tumbado rápidamente con la cabeza colgando, postura que se mantiene unos 30 s. (3) La cabeza se gira 90º hacia el otro lado (oído sano) y se mantiene 20 s. (4) Se continúa rotando la cabeza otros 90º, teniendo que mover el paciente también el cuerpo a la postura de decúbito lateral izquierdo. Esta posición se mantiene otros 30 s. (5) Se lleva entonces al paciente a la posición de sentado, completando así la maniobra. En total la maniobra debe llevar menos de 5 min. Si ha sido efectiva, no volverá a aparecer vértigo, ni nistagmo. CSP: canal semicircular posterior. Adaptado de: Bhattacharyya: Otolaryngol Head Neck Surg. 2008;139(5)Sup 4: S47–S81. No farmacológico • En el caso del vértigo periférico, y sobre todo en el VPPB, lo primero es tranquilizar al paciente y explicarle el origen benigno de su trastorno, que lo más probable es que desaparezca o mejore en poco tiempo. • Habrá que informar que hay casos de mayor duración y explicar los síntomas de alarma: neurológicos (diplopía, disartria, pérdida de fuerza, etc.) o hipoacusia. Se le indicará que evite las posturas provocadoras del vértigo y actividades en las que el riesgo de caídas sea elevado, hasta que desaparezcan los síntomas. Y considerar los riesgos de actividades como conducir, usar escaleras, andamios y máquinas peligrosas (valorar la incapacidad transitoria [IT]) e incluso nadar. • En el VPPB, al ser un proceso tan agudo, no suele necesitar medicación, en todo caso, antieméticos, por las náuseas y vómitos. En el resto de los vértigos se pueden ofrecer, además, sedantes vestibulares, pero teniendo en cuenta que el tratamiento farmacológico y el reposo han de ser breves. • Si no hay recuperación satisfactoria, pasar a un programa de ejercicio vestibular (la compensación central es más rápida y completa cuanto antes se muevan los pacientes). Hay que iniciarlo, una vez pasado el episodio agudo, tan pronto como se pueda (hacia el tercer día, una vez controlados los síntomas). • Ejercicios de rehabilitación vestibular: Pueden disminuir los síntomas y la discapacidad en el tratamiento de los vértigos periféricos (hay de modera- 423 5.4 Mareo y vértigo da a fuerte evidencia de que la rehabilitación vestibular es una forma segura y efectiva de abordar los trastornos vestibulares periféricos unilaterales, salvo en el caso del VPPB en que son de elección las maniobras de reposición canalicular) y parece que tienen también utilidad en los de origen central, siendo más efectivos que la medicación. Los más utilizados son los de Brandt-Daroff (figura 7). Estos ejercicios deben realizarse en tres ciclos al día de cinco repeticiones cada uno y durante 2 semanas. El paciente se sienta en la cama con los ojos cerrados y se le indica que se deje caer hacia un lado y hacia el otro. En cada posición se descansa unos segundos hasta que desaparece paulatinamente el vértigo. • Procedimientos de reposición canalicular (PRC): En el VPPB es de elección el PRC de Epley o la maniobra de liberación (ML) de Semont, que se basan en movimientos que tratan de desplazar los otolitos del canal semicircular (CS) al utrículo, donde son inactivos. – Maniobra posicional de Epley (figura 5): En caso de bilateralidad se hará la maniobra hacia el lado con mayor clínica y/o nistagmo más intenso. Si ha sido efectiva, no volverá a aparecer vértigo, ni nistagmo. Hay evidencia de que la maniobra de Epley es un tratamiento seguro y efectivo para el VPPB del canal posterior. No hay suficiente evidencia de que provea una resolución de los síntomas a largo plazo. La eficacia de una única maniobra es del 78 %. Puede recurrir en un 10-20 % de los casos en 1-2 semanas (resolución del 96 % tras repetición; NTT 1,3-3,7). El número de veces que habrá que realizarla dependerá de la persistencia de los síntomas, así como de sus recurrencias. Contraindicación: casos de grave artrosis cervical o estenosis carotídea significativa. – Maniobra de Semont (figura 6): Tiene la ventaja de que con ésta la hiperextensión del cuello es menor y de que lo puede hacer el propio paciente en casa. En el caso de que persistan los síntomas a pesar de las maniobras de reposición anteriormente descritas, se pueden utilizar ejercicios de rehabilitación laberíntica. – Maniobra de Lempert (figura 8) para el VPPB del canal horizontal (10-17 % de los VPPB). En caso de no haber respuesta, valorar el tratamiento quirúrgico por ORL (obliteración del canal o neurectomía vestibular). • En el síndrome de Ménière, si no hay mejoría, se valorará el tratamiento quirúrgico por ORL (sección de la rama vestibular del VIII par, gentamicina transtimpánica, shunt endolinfático y si ya no queda audición, laberintectomía). • En el déficit sensorial múltiple: intentar corregir los déficit sensoriales que se pueda (audición y visión). Eliminar las barreras arquitectónicas en el hogar y hacer que éste sea luminoso. Evitar la polimedicación. Enseñar al paciente a levantarse del suelo y a usar bastón o andador. Efectuar ejercicios de rehabilitación del equilibrio (temas 22.5 y 22.6) y/o corregir los trastornos articulares que lo alteren. Farmacológico En el VPPB no hay evidencias que apoyen la recomendación del empleo de fármacos (son menos efectivos que las maniobras de reposición), salvo antieméticos si hay vómitos intensos o sensación nauseosa residual y para efectuar las maniobras de Epley o Semont. En la fase aguda de cualquier otro tipo de vértigo se recomienda reposo y, si es muy intenso, se utilizarán fár- 1 4 2 3 Posición 1 5 7 6 Posición 2 Posición 4 Posición 3 Figura 7. Ejercicios posicionales de Brandt-Daroff. 424 Figura 8. Maniobra de Lempert: cuando se ha determinado que el VPPB es del CSH derecho, el paciente es llevado a través de una serie de pasos de giros de 90º del paso 1 al 5, estando en cada posición de 10 a 30 s. De la posición 5 pasa a decúbito supino para pasar rápidamente a la posición de sentado. CSH: canal semicircular horizontal. Adaptado de: Fife TD. Neurology. 2008;70:2067-74. 5.4 Mareo y vértigo macos antivertiginosos (sedantes vestibulares) durante el menor tiempo posible (3-7 días). Respecto al síndrome de Ménière no hay evidencia de que el tratamiento sintomático sea beneficioso durante el episodio agudo. Si predominan los vómitos, administrar un antiemético (metoclopramida 10 mg/8-12 h v.o., i.m. o i.v. o domperidona 10 mg/6 h v.o. o rectal). Si los síntomas son muy graves, se puede requerir ingreso para tratamiento i.v. de sedantes vestibulares, hidratación y nutrición. • Sedantes vestibulares: Bloquean la acción de los neurotransmisores que intervienen en la transmisión nerviosa del órgano y vía vestibular. Entre ellos cabe destacar los siguientes: – Antihistamínicos (sobre todo los de primera generación): sus efectos antivertiginosos son debidos al bloqueo de los receptores histamínicos y colinérgicos, teniendo también efecto sedante. Efectos secundarios anticolinérgicos, sedación y afectación de la función psicomotora. También se pueden usar como profilaxis de la cinetosis. Dimenhidranato: 50-100 mg/4-6 h v.o. o rectal, o 25-50 mg i.m. Hidroxicina: 25 mg/6-8 h v.o. Cinarizina 15 mg /4-6 h v.o. Flunarizina 10 mg cada 24 horas. Son antihistamínicos y antagonistas del calcio que por su débil acción antidopaminérgica pueden producir síntomas parkinsonianos, sobre todo en ancianos. – Neurolépticos: actividad anti-H1 y antiemética. Cuidado con sus efectos secundarios: déficit cognitivo, distonía aguda, hiperprolactinemia. Sulpiride en dosis 50-300 mg/día v.o. o i.m., máximo 4-6 semanas. Tietilperacina en dosis de 6,5 mg/8-12 h v.o. o rectal. – Benzodiazepinas: en dosis bajas son sedantes vestibulares, pero sus efectos secundarios (adicción, trastornos de memoria, riesgo de caídas, interferencia para la compensación central, etc.) hacen poco recomendable su uso. Lorazepam 1 mg/12 h (podría usarse la vía sublingual en un ataque agudo de vértigo) o diazepam 5 mg/12 h. – Anticolinérgicos: la escopolamina es raramente usada como profilaxis de la cinetosis. Dosis profiláctica: 0,5 mg/3 h. Para tratar crisis de cinetosis intensa 0,2 mg i.m. Tiene efectos secundarios anticolinérgicos pero es menos sedativo y afecta menos a la función psicomotora que los antihistamínicos (para personas que tienen que estar alertas). – Vasodilatadores (betahistina): faltan estudios para valorar su eficacia en el vértigo. Es un análogo de la histamina (VD): precaución si hay historia de asma o de ulcus. Derivación La mayoría de los mareos se pueden tratar en AP (84 %) y es en este ámbito en el que hay que detectar los que hay que derivar. Consejos prácticos • La anamnesis y la exploración física llevan al diagnóstico de la causa del mareo en la mayoría de los casos, por lo que es muy importante ser riguroso y metódico en su estudio, ya que de aquéllos se van a derivar el resto de las exploraciones y la toma de decisiones. • Todo mareo de causa desconocida debe ser revaluado y vigilado en el tiempo. • La mayoría de los pacientes podrán ser tratados en AP, pero existen situaciones de riesgo: mareos que se acompañen de síntomas neurológicos, cardiológicos o desencadenados con el ejercicio o un vértigo brusco sin relación con los movimientos de la cabeza. • Se recomienda investigar la posible existencia de VPPB en ancianos con historia de caídas recurrentes, ya que en ellos también es frecuente, aunque no debe olvidarse la comorbilidad y los fármacos. • El tratamiento sintomático del vértigo ha de ser breve, y sólo si éste es muy intenso, con el objeto de que se movilice lo antes posible. • Tras un ataque de vértigo (salvo en el VPPB), se recomienda una audiometría (para asegurar que la función coclear es normal). Urgente Clínica de mareo o vértigo asociada a una de las siguientes situaciones: • Hipotensión aguda: hemorragia, deshidratación, fármacos. • Alteración del ECG: isquemia, bloqueo AV completo, taquiarritmias, QT alargado. • Signos y/o síntomas neurológicos acompañantes o ha habido en las 24 horas previas (descartar ACVA). Ordinaria o preferente • Cardiología: – Enfermedad cardíaca y/o anomalías ECG asociadas. • ORL: – Vértigo con hipoacusia. – Vértigo de causa desconocida, recurrente o crónico, descartados trastornos psiquiátricos. Errores más frecuentes • Atribuir un mareo a cervicoartrosis o a la edad avanzada. • Achacar un vértigo a una otitis. Una otitis media aguda no produce vértigo si no se acompaña de una laberintitis y una otitis crónica tampoco lo hace, salvo que se complique con un hydrops endolinfático o con un colesteatoma. • No hay excusas para no hacer un test posicional (Dix-Hallpike) en ancianos. • No explorar la marcha. Es el mejor predictor de un vértigo central: son incapaces de dar más que un par de pasos sin caer, o incluso ni eso. • Dar sedantes vestibulares a un VPPB. • No advertir a los pacientes de la existencia de recurrencias. 425 5.4 Mareo y vértigo – Para el diagnóstico diferencial del vértigo periférico y central cuando no está claro. • Neurología: – Síntomas y/o signos neurológicos no agudos. – Vértigo recurrente de causa desconocida (realizado estudio psiquiátrico y ORL). Bibliografía recomendada 1. Hillier SL, Holohan V. Vestibular rehabilitation for unilateral peripheral vestibular dysfunction. Cochrane Database of Systematic Reviews 2007, Issue 4. Art. No.: CD005397. DOI: 10.1002/ 14651858.CD005397.pub2. ¿Se ha resuelto del todo? 2. Bhattacharyya N, Baugh RF, Orvidas L, Barrs D, Bornston LJ, Cass S, et al. Clinical practice guideline: benign paroxysmal positional vertigo. Otolaryngol Head Neck Surg. 2008;139(5)Suppl 4: Pronóstico y seguimiento S47-81. 3. Fife TD, Iverson DJ, Lempert T, FurmanJ, Tusa RJ, Balow RW, et al. Dependerá fundamentalmente de la causa principal que motive el mareo. • El pronóstico del mareo, en general, y del vértigo en particular, es bueno. La tasa de mortalidad no difiere del grupo de control ajustado para la edad. No obstante, hay que recordar que puede ser debido a causas graves, que precisen actuación inmediata, sobre todo los de causa cerebrovascular y cardíaca, que se asocian a mayor morbilidad por caídas, sobre todo en ancianos, y que repercute en la calidad de vida de los pacientes. • Todo mareo (de cualquiera de los cuatro bloques diagnósticos) no diagnosticado hay que seguirlo en el tiempo (control evolutivo) y si repite, revaluar desde el principio. • Un mareo prolongado en el tiempo, que no desarrolle síntomas y/o signos neurológicos, difícilmente será de origen central. ¿Podemos hacer alguna cosa más? Educación sanitaria Consultar en Guía práctica de la salud, p. 31. 426 Practice Parameter: Therapies for Benign Paroxysmal Positional Vertigo: An Evidence Based Review. Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2008;70:2067-74. Disponible en: http://www. neurology.org/cgi/reprint/70/22/2067 4. Corell M, Alonso I. Vértigo posicional paroxístico benigno. AMF. 2007;3(9):503-50. 5. Santos S. Síndrome vestibular periférico. Rev Med Univ. Navarra. 2003;47(4):38-50. 6. Trinidad G, Pons MA, Trinidad G, Pantoja C, Mora E, Blasco A. Vértigo posicional. ¿Síntoma, signo o enfermedad? Acta Otorrinolaringol Esp. 2008;59:21-9. 7. Labuguen RH. Initial evaluation of vertigo. Am Fam Physician. 2006;73(2):244-51. 8. Kanagalingam J, Hajioff D, Bennett S. Vertigo. BMJ. 2005;330 (7490):523. 5.5 Convulsiones F. Fiol Gelabert ¿De qué hablamos? Introducción Tradicionalmente entendemos por convulsión o crisis convulsiva al conjunto de síntomas y signos como la pérdida de conocimiento y movimientos involuntarios resultado de la expresión clínica de una descarga neuronal excesiva. La causa más frecuente de convulsiones repetidas es la epilepsia pero convulsión y epilepsia no son términos superponibles. El concepto de epilepsia requiere la recurrencia de las crisis convulsivas. Pero tampoco son sinónimos de epilepsia las crisis convulsivas repetidas en el contexto de enfermedades agudas como síndrome febril, encefalitis, traumatismos craneales y síncopes prolongados no epilépticos. Definición • Crisis epiléptica (CE). Recientemente, la Liga Internacional contra la Epilepsia (ILAE) y el International Bureau for Epilepsia (IBE), proponen la siguiente definición de CE: acontecimiento transitorio de signos y/o síntomas debidos a una actividad neuronal cerebral anormal excesiva o síncrona. Dependiendo del área cerebral afectada, las manifestaciones de la crisis serán motoras, sensitivas, psíquicas o mixtas. • La epilepsia se define como trastorno del cerebro caracterizado por una predisposición para generar crisis epilépticas y por las consecuencias neurobiológicas, cognitivas, psicológicas y sociales de esta condición. Si bien la definición tradicional exigía la ocurrencia de, al menos, dos CE no provocadas para confirmar la existencia de epilepsia, la ILAE ha propuesto como nueva definición de epilepsia cuando al menos se da una CE (no necesariamente no provocada) junto a una alteración duradera en el cerebro que aumenta la probabilidad de futuras CE. • Síndrome epiléptico. Entidad consistente en crisis epilépticas recurrentes y caracterizadas por una edad de presentación, una serie definida de síntomas y signos, un perfil temporal, unos factores desencadenantes específicos, una causa determinada y, con frecuencia, un pronóstico conocido. Clasificación Crisis epilépticas (tabla 1) Desde 1989 se acepta la clasificación revisada de la ILAE (v. tabla 1). El criterio básico de la cual es la distinción entre crisis parciales y crisis generalizadas. Las primeras muestran un inicio parcial de semiología variable (las descargas neuronales están localizadas en un área focal del cerebro) y, de forma secundaria, pueden generalizarse. Las segundas son aquellas en las que tanto en la clínica como en el electroencefalograma (EEG) puede demostrarse un inicio focal y la descarga neuronal es bilateral, síncrona y simétrica en los dos hemisferios desde el principio. Tabla 1. Clasificación de las crisis epilépticas Crisis parciales Crisis parciales simples (sin afectación de la conciencia) Con síntomas motores Con síntomas sensoriales (crisis somatosensoriales, visuales, auditivas, olfativas, gustativas, vertiginosas) Con síntomas o signos vegetativos Con síntomas psíquicos Crisis parciales complejas (con afectación de la conciencia) Comienzo con crisis parcial simple seguido por afectación de la conciencia Comienzo con afectación de la conciencia Crisis parciales secundariamente generalizadas Parcial simple con generalización secundaria Parcial compleja con generalización secundaria Parcial simple con evolución a compleja y generalización secundaria Crisis generalizadas No convulsivas Ausencias típicas Crisis atónicas Convulsivas Mioclonías Clónicas Tónicas Tonicoclónicas Atónicas Crisis inclasificables Las crisis parciales pueden ser simples, sin alteración de conciencia, o complejas, con alteración de conciencia. • Las crisis parciales simples consisten en sensaciones o percepciones anormales de tipo visual, sensitivo, psíquico, olfatorio o en una actividad motora (movimientos clónicos, posturas tónicas). • Las crisis parciales complejas se caracterizan por mirada ausente y la realización de actos más o menos complejos (automatismos manuales, movimientos de deglución, chupeteo) y amnesia de lo sucedido durante el período que dura la crisis y el período poscrítico. Las crisis parciales complejas son las más frecuentes entre los adultos y orientan más a una patología focal, como puede ser la tumoral. Las crisis generalizadas primarias suelen iniciarse en la infancia o adolescencia. Síndromes epilépticos (tabla 2) Epidemiología La epilepsia es una enfermedad frecuente, con una prevalencia de 5-8/1.000 habitantes y una incidencia de 30-50/100.000 habitantes/año. La incidencia varía notablemente con la edad, es máxima en la infancia y la adolescencia, decrece en los individuos jóvenes y vuelve a aumentar en los sujetos mayores de 60 años. 427 5.5 Convulsiones Tabla 2. Clasificación de los síndromes epilépticos Epilepsias localizadas (focales) Idiopáticas Epilepsia benigna de la infancia con puntas centrotemporales Epilepsia de la infancia con paroxismos occipitales Epilepsia primaria de la lectura Sintomáticas Epilepsia parcial continua progresiva de la infancia (síndrome de Kojewnikow) Síndromes caracterizados por crisis con modos específicos de precipitación Epilepsias del lóbulo temporal Epilepsias del lóbulo frontal Epilepsias del lóbulo parietal Epilepsias del lóbulo occipital Criptogénicas Epilepsias del lóbulo temporal Epilepsias del lóbulo frontal Epilepsias del lóbulo parietal Epilepsias del lóbulo occipital Epilepsias o síndromes generalizados Idiopáticos Convulsiones neonatales benignas familiares Convulsiones neonatales benignas Epilepsia mioclónica benigna de la infancia Ausencia infantil Ausencia juvenil Epilepsia con crisis de gran mal al despertar Otras epilepsias generalizadas idiopáticas Epilepsias con crisis precipitadas por modos de activación específicos Criptogénicos o sintomáticos Síndrome de West o espasmos infantiles Síndrome de Lennox-Gastaut Epilepsia con crisis mioclónicas astáticas Epilepsia con ausencias mioclónicas Sintomáticos Etiología no especificada Encefalopatía mioclónica temprana Encefalopatía infantil temprana con brotes de supresión Síndromes específicos Epilepsias o síndromes sin determinar si son generalizados o focales Con crisis generalizadas y focales Crisis neonatales Epilepsia mioclónica grave de la infancia Epilepsia con punta-onda continuada durante el sueño lento Afasia epiléptica adquirida (síndrome de Landau-Kleffner) Sin claras crisis generalizadas o focales Síndromes especiales Convulsiones febriles Crisis aisladas o estado de mal epiléptico aislado Crisis en el seno de una alteración metabólica o tóxica 428 ¿Qué lo puede ocasionar? Causas Los factores que pueden provocar crisis epilépticas se exponen en las tablas 3 y 4. Diagnóstico diferencial El diagnóstico diferencial debe establecerse con trastornos que pueden presentarse de forma paroxística simulando o provocando una convulsión pero que no son de origen epiléptico: síncope, accidente isquémico transitorio (AIT), migraña acompañada, hipoglucemia, etc. (figura 1). ¿Qué tenemos que hacer? Anamnesis El diagnóstico de una crisis convulsiva es fundamentalmente clínico. La anamnesis constituye el pilar fundamental en el diagnóstico de las CE. Ésta debe estructurarse de la siguiente manera: • Antecedentes familiares: investigación de enfermedades neurológicas familiares, y de antecedentes de epilepsia o enfermedades asociadas a epilepsia. • Antecedentes personales: hacer énfasis en la historia perinatal, crisis convulsivas previas, antecedentes cardiovasculares, déficit neurológicos, cefalea, neoplasias previas, diabetes, fiebre, fármacos recibidos, tóxicos (alcohol, drogas). • Enfermedad actual: la anamnesis de un paciente que ha sufrido una posible crisis debe responder a tres cuestiones: si se trata realmente de una crisis convulsiva, el tipo de crisis que ha presentado y la causa de ésta. Para ello es indispensable interrogar minuciosamente al paciente, con la colaboración de algún testigo de la crisis, sobre todo lo que pueda recordar de lo sucedido antes, durante y después de la crisis. Las características clínicas que sugieren que una crisis convulsiva puede ser de origen epiléptico se hallan en la tabla 5. Exploración física • Constantes vitales: presión arterial, pulso, frecuencia respiratoria y temperatura. • Exploración general y neurológica detalladas, incluido el fondo de ojo. Exploraciones complementarias Iniciales • Glucemia capilar: en un paciente con una primera CE o epilepsia de inicio reciente, no se ha demostrado la utilidad de la determinación sistemática de glucosa y electrolitos, si bien no se puede excluir la posibilidad de que en raros casos aporte información útil para el tratamiento del paciente. Se recomienda la realización de 5.5 Convulsiones Tabla 3. Factores que pueden provocar crisis epilépticas Factores genéticos y relacionados con el nacimiento Influencia genética (idiopática, criptogenética), cromosomopatías, infecciones, anoxia, traumatismo en el parto Heredofamiliar Neurofibromatosis, esclerosis tuberosa, síndrome de Sturge-Weber Enfermedades infecciosas Meningitis, encefalitis, abscesos Tóxicos Metales, CO, alcohol, fármacos (antidepresivos) y abstinencia de fármacos Enfermedades cardiovasculares Hemorragia subaracnoidea, trombosis de senos venosos, encefalopatía hipertensiva, síncope prolongado Alteraciones nutricionales o metabólicas Hiponatremia, hipocalcemia, deshidratación, hipoglucemia, aminoacidopatías, deficiencia vitamínica, porfiria aguda intermitente Neoplasias Intracraneales primarias, metástasis, linfoma, leucemia, malformaciones vasculares Otros Traumatismos craneales Crisis febriles Tabla 4. Causas más probables de epilepsia según la edad de comienzo Lactancia (0-2 años) Mal desarrollo congénito, lesión cerebral perinatal, trastorno metabólico, déficit de vitamina B6, fenilcetonuria Infancia (2-10 años) Anoxia perinatal, lesión cerebral, infecciones, idiopática Adolescencia (10-18 años) Idiopática, traumatismo, alcohol, neoplasia, drogadicción Edad media (35-60 años) Alcohol, neoplasia, traumatismo, enfermedad vascular, drogadicción Senectud (> 60 años) Enfermedad vascular, neoplasia, enfermedad degenerativa hemograma, glucemia y electrolitos en los pacientes adultos o en niños con signos o síntomas sugestivos de una alteración de este tipo como vómitos, diarrea, deshidratación o disminución persistente del nivel de conciencia. Tampoco se ha demostrado la utilidad de la realización sistemática de un cribado toxicológico: indicado únicamente en caso de sospecha de exposición a drogas. • Electrocardiograma (ECG): en busca de ritmos embolígenos o hallazgos sugestivos de isquemia cardíaca aguda (temas 25.1 y 25.2). Posteriores • Tomografía computarizada (TC): imprescindible en muchos casos en el diagnóstico de urgencias (cambios neurológicos focales, fiebre o trauma craneal). • Resonancia magnética (RM) cerebral: prueba de imagen estructural de primera elección. • EEG: en el momento de la crisis, si es posible, si no intercrítico. Puede ayudar a confirmar el diagnóstico de epilepsia, facilitar su clasificación y revelar cambios que aumenten la sospecha de alguna lesión estructu- ral subyacente. Pero un EEG normal no excluye el diagnóstico de epilepsia (entre un 10-20 % de los pacientes con crisis epilépticas no muestra anomalías EEG entre las crisis aunque se realicen tres EEG repetidos, pero en este grupo con frecuencia se detectan anormalidades con un EEG de sueño o con deprivación) y un 2-3 % de la población presenta alteraciones en el EEG sin padecer epilepsia. En las epilepsias generalizadas la sensibilidad es mayor que en las epilepsias parciales, y la correcta utilización de estimulaciones específicas es más eficaz que la reiteración de EEG (tema 25.16). • Monitorización vídeo-EEG (MVEEG): la de larga duración ha demostrado ser útil como instrumento diagnóstico en pacientes con trastornos paroxísticos intermitentes difíciles de registrar, así como en los casos en los que, existiendo alta sospecha clínica de epileptogenicidad, no se encuentra evidencia de actividad crítica o intercrítica con los registros EEG convencionales. Una segunda utilidad de la MVEEG es la clasificación del tipo de CE en pacientes epilépticos conocidos. Cabe destacar como limitación su alto coste económico. ¿Qué propuesta haremos? Tratamiento Urgente durante la crisis • Evitar que el paciente se lastime apartando de su alrededor los objetos que puedan dañarle, que se muerda la lengua o haga aspiraciones (tubo de Guedel). Comprobar la glucemia capilar, la no existencia de bradicardia extrema y la saturación de oxígeno en sangre arterial (SaO2), si es factible. Una vez cedida la crisis, se colocará al paciente en posición de seguridad (tema 21.10). • Si la crisis cede espontáneamente, no administrar fármacos. En caso de duración superior a 5 minutos o ante una nueva crisis, administrar lentamente diazepam 2 mg/min/i.v. hasta el cese de la crisis o hasta un máximo 429 430 Sí Sí Sí Derivación urgente al hospital Bajar fiebre, Alcoholismo: tiamina, glucosa Datos de enf. aguda o desencadenante: Sd. febril, TCE, intoxicación etílica, hipoglucemia Historia detallada Exploración física Pruebas iniciales Sí Derivación urgente al hospital Sd. febril, TCE o focalidad neurológica No Sí ITC ordinaria a neurología No Paciente > 18 años Sí Características que sugieren origen epiléptico: inicio súbito, pérdida de conciencia, estado poscrítico Historia detallada Exploración física Exploración neurológica Fondo de ojo Primer episodio No Valorar iniciar tto. o ITC preferente a neurología si hay una segunda crisis o riesgo para actividad habitual Seguimiento en APS Valorar posible abandono de la medicación, Desencadenantes: fiebre, falta de sueño, alcohol Valorar niveles de fármacos Sí Epilepsia diagnosticada y crisis previas similares No Presenciada Evitar que el paciente se lastime apartando de su alrededor los objetos que puedan dañarle, que se muerda la lengua o haga aspiraciones (tubo de Guedel). AIT: accidente isquémico transitorio; AP: Atención Primaria; APS: atención primaria de salud; ITC: interconsulta; TC: tomografía computarizada; TCE: traumatismo craneoencefálico. a Figura 1. Algoritmo de actuación en las convulsiones en el adulto. Derivación urgente al hospital en transporte medicalizado No Cede la crisis Tras 10 min repetir dosis diazepam No Cede la crisis Tto. con diazepam No Tras 5 min cede espontáneamente Medidas de protección físicaa Sí Convulsión en el adulto Sí ITC urgente a neurología No Normal Descartar convulsión secundaria (neoplasia, enf. vascular), TC preferente Factor precipitante: alcohol, hipoglucemia o cocaína Descartar síncope, AIT, migraña acompañada 5.5 Convulsiones 5.5 Convulsiones Tabla 5. Características clínicas que sugieren que una crisis convulsiva puede ser de origen epiléptico Crisis parcial que precede al episodio convulsivo (aura) Estado poscrítico: estado confusional prolongado (dificultad en reconocer a personas o ambientes familiares) inmediatamente después del episodio Automatismos durante el episodio de pérdida de conciencia (chupeteo, parpadeo) Déficit neurológico después de la crisis (afasia, hemiparesia) de 30 mg. También puede administrarse por vía rectal (10-20 mg en adultos, 0,5 mg/kg en niños). Una alternativa al diazepam es midazolam 0,1 mg/kg/i.v. Si no cede, derivar al paciente con urgencia al hospital. Si se sospecha etilismo, administrar 100 mg/i.v. o i.m. de tiamina más 50 ml i.v. de glucosa al 50 %. • En el paciente no conocido como epiléptico o cuando no se pueda asegurar un período de observación para comprobar su recuperación, se aconseja su traslado a un servicio de urgencias hospitalario. Siempre que se derive al hospital, debe hacerse en las mejores condiciones posibles (informe asistencial, decúbito lateral izquierdo, tubo de Guedel, vía venosa) y no dejar al paciente solo. • Convulsiones en el niño (tema 13.8). No urgente Primera crisis • Se buscará un factor precipitante, como alcohol, hipoglucemia, tóxicos. En caso de presentar nueva crisis convulsiva, el paciente será evaluado para descartar una epilepsia. • Si mientras espera a ser visitado por el neurólogo las crisis se van repitiendo o bien suponen un riesgo para la actividad habitual del paciente, podrá iniciarse un tratamiento farmacológico siempre y cuando se haya podido realizar una orientación diagnóstica. Epilepsia ya conocida • Valorar si ha existido abandono de la medicación o hay otros factores desencadenantes (fiebre, estrés, falta de sueño, alcohol). • Si se ha olvidado una dosis del fármaco, se puede tomar dosis doble. • Si es preciso, solicitar valores plasmáticos del medicamento para evaluar la necesidad de aumentar la dosis (tema 23.19). za de epilepsia, no hay evidencia de que una crisis de breve duración pueda producir daño cerebral, y por los posibles efectos adversos de los FAE. La decisión debe hacerse de forma individualizada, por ejemplo podría indicarse el tratamiento en caso de existir un importante riesgo laboral. Después de la segunda crisis el riesgo de padecer una tercera es superior al 65 % por lo que está indicado iniciar el tratamiento farmacológico. • Debe iniciarse siempre como monoterapia (tablas 6 a 9). El 70 % de los pacientes responden a ésta. En los que continúan con crisis está indicado incrementar a la máxima dosis tolerada. Si no responden o aparecen efectos secundarios, cambiar a otro fármaco en monoterapia. La asociación de fármacos no potencia de forma marcada su efectividad pero sí puede potenciar su toxicidad. En casos refractarios se emplean combinaciones de fármacos. El tratamiento con FAE de segunda generación suele ser coste efectivo en pacientes que no se controlan o sufren efectos adversos con FAE clásicos, cuando éstos están contraindicados y si hay riesgo de interacciones farmacocinéticas. En ancianos se ha demostrado la efectividad de los fármacos de segunda generación por su menor interacción farmacológica y su inferior tasa de efectos secundarios, especialmente gabapentina y lamotrigina. Derivación Urgente • En una crisis observada si el paciente no es conocido como epiléptico o cuando no se pueda asegurar un período de observación para comprobar su recuperación. • Si en la exploración se objetiva algún signo de gravedad, como fiebre o focalidad neurológica, o bien existen antecedentes de traumatismo craneal o tratamiento anticoagulante. Ordinaria o preferente • En caso de primera crisis generalizada: si no cumplen los criterios para derivar a urgencias, solicitar visita a neurología para valoración. Los pacientes mayores de 18 años tienen mayor riesgo de tumor o enfermedad cerebrovascular. • En pacientes ya diagnosticados, derivarlos si no tienen un buen control de sus crisis epilépticas. También se recomienda una revisión anual en neurología a los pacientes con buen control. ¿Se ha resuelto del todo? Pronóstico y seguimiento Tratamiento farmacológico Inicio del tratamiento con fármacos antiepilépticos (FAE) • En la primera crisis no se recomienda iniciarlo: cerca del 30-50 % de los pacientes presentarán una nueva crisis, pero dada la dificultad del diagnóstico de certe- El objetivo del tratamiento crónico de la epilepsia es mantener al enfermo libre de crisis con los mínimos efectos secundarios y la máxima reinserción sociolaboral posible, lo que se consigue en un 70-80 % de los casos. • Una vez iniciado el tratamiento, puede que se sigan repitiendo episodios de crisis epilépticas, en cuyo caso 431 5.5 Convulsiones Tabla 6. Antiepilépticos y dosis habituales Adulto (mg/d) Niño (mg/kg/d) Fenobarbital (PB) 120-250 4-6 Fenitoína (DPH) 300-400 4-7 Fármaco Primidona (PRM) 750-1.500 10-25 Carbamazepina (CBZ) 600-1.200 20-30 Ácido valproico (VPA) 1.000-3.000 10-60 Clonazepam (CZ) 1,5-20 0,01-0,3 Etosuximida (ETX) 250-1.000 20-40 1.000-2.000 40-80 Lamotrigina (LMT) Vigabatrina (VIG) 100-400 3-6 Gabapentina (GPN) 900-1.800 10-45 Oxcarbazepina (OXC) 900-1.200 30 Tiagabina (TGB) Topiramato (TPM) Levetiracetam (LTC) Clobazam (CLB) 15-60 15-30 400-1.000 3-9 1.000-3.000 20-60 20-40 0,5-2 Pregabalina (PGB) 150-600 5-10 Zonisamida (ZNS) 300-500 4-8 Tabla 7. Fármacos de elección según el tipo de crisis Tipo de crisis Primera elección Segunda elección Parciales VPA, CBZ, DPH, GPN, LMT, OXC, TPM, LTC PGB, TBG Generalizadas convulsivas VPA, LMT, TPM, LTC Felbamato Mioclónica juvenil VPA, LMT, LTC LMT, TPM Ausencias VPA, ETX, LMT – Espasmos infantiles VIG, VPA ACTH, prednisona ACTH: hormona adrenocorticotrópica; CBZ: carbamazepina; DPH: fenitoína; ETX: etosuximida; GPN: gabapentina; LMT: lamotrigina; LTC: levetiracetam; OXC: oxcarbazepina; PGB: pregabalina; TBG: tiagabina; TPM: topiramato; VIG: vigabatrina; VPA: ácido valproico. hay que asegurarse, en primer lugar, de que el paciente toma adecuadamente la medicación, y en segundo lugar, hay que investigar la posible existencia de interacciones con otros fármacos (tabla 9). • Los valores plasmáticos sirven de guía, pero no deben ser un elemento definitivo para impedir el aumento de la medicación por encima de los valores séricos establecidos para la población general. En ocasiones, el control es difícil de conseguir a pesar de un buen cumplimiento del paciente. Si se utilizan las dosis máximas de un fármaco sin conseguir el control, debe plantearse el cambio de medicación o la asociación de varios anticonvulsivos; esto debe ser valorado por el neurólogo. No existen datos que apoyen el uso sistemático de determinaciones de niveles de antiepilépticos para la optimización de los tratamientos en FAE clásicos. En algunas situaciones clínicas sería útil la monitorización 432 plasmática de los FAE para evaluar una posible toxicidad y sus causas, así como las de pérdida de eficacia e identificar incumplimientos terapéuticos. • Debe considerarse la supresión del tratamiento con FAE en pacientes libres de CE durante 2 años, ya que el riesgo de recurrencia es razonablemente bajo para muchos de ellos y la prolongación del tratamiento apenas consigue disminuir el riesgo de recurrencia. La supresión de la medicación antiepiléptica no modifica el pronóstico a largo plazo de la epilepsia. El riesgo de recurrencia tras la retirada de la medicación en pacientes libres de CE durante 2 años o más es del 30-40 % en estudios con niños y adultos. La decisión de suspender el tratamiento debe consensuarse entre el médico y el paciente o sus familiares después de proporcionar información sobre los riesgos y beneficios de la supresión del tratamiento. 5.5 Convulsiones Tabla 8. Efectos secundarios de los antiepilépticos Antiepilépticos Efectos secundarios Ácido valproico Sedación, alteraciones cognitivas, alteraciones de conducta, asterixis y temblor, alteración del apetito, aumento de peso, vómitos, diarreas, pancreatitis, hepatopatía, alopecia, trombocitopenia Carbamazepina Sedación, alteraciones cognitivas, trastornos de conducta, asterixis, arritmias, bloqueo cardíaco, neuropatías, anorexia, aumento de peso, hepatopatía, dermatitis, hiponatremia, macrocitosis, agranulocitosis, SIADH Clobazam Sedación, ataxia, depresión respiratoria Etosuximida Discinesias, alteraciones psíquicas, exantema, agranulocitosis, lupus eritematoso, anorexia Felbamato Anemia aplásica mortal, hepatotoxicidad grave Fenitoína Sedación, hipertricosis e hirsutismo, alteraciones cognitivas, hepatopatía, neuropatías, asterixis, adenopatías generalizadas, dermatitis, hipotensión, bradicardia y bloqueo cardíaco, déficit de ácido fólico, lupus eritematoso, anemia aplásica y megaloblástica Fenobarbital Sedación, alteraciones cognitivas, trastornos de conducta, asterixis, hepatopatía, estreñimiento, exantemas, dermatitis, osteomalacia, déficit de ácido fólico, anemia megaloblástica, agranulocitosis, capsulitis del hombro Gabapentina Somnolencia, mareo, depresión, nistagmo, alteraciones psíquicas, cansancio Lamotrigina Diplopía, vértigo, ataxia, somnolencia, exantema, depresión, cuadros psicóticos, síndrome de Stevens-Johnson Levetiracetam Somnolencia, cefalea, astenia, cuadros catarrales, alteraciones del comportamiento, leucopenia y anemia leves Oxcarbazepina Sedación, ataxia, diplopía, náuseas, exantema, hiponatremia Pregabalina Sedación, mareo, aumento de peso, edema periférico, temblor Primidona Trastornos cognitivos/conducta, ataxia, diplopía, vértigo, neuropatías, anorexia, vómitos, dermatitis, hiperplasia gingival, hombro congelado, déficit de ácido fólico, macrocitosis Tiagabina Mareo, temblor, dificultad de concentración, irritabilidad, depresión, psicosis Topiramato Pérdida de peso, cefalea, nistagmo, temblor, somnolencia, depresión, cálculos renales, miopía, glaucoma Vigabatrina Sedación, vértigo, ataxia, temblor, cuadros psicóticos, depresión, aumento de peso, restricción del campo visual Zonisamida Exantema, anhidrosis, hipertermia, insomnio, depresión SIADH: síndrome de secreción inapropiada de ADH. ¿Podemos hacer alguna cosa más? Actividades preventivas • Prevención primaria: mejorar la asistencia al parto, prevención de accidentes. • Prevención secundaria: evitar factores desencadenantes y llevar una vida sana (hábitos regulares de sueño y abstención del consumo de alcohol, cocaína, cannabis u otras drogas). No practicar deportes de riesgo potencial e intentar seguir un tipo de vida normal evitando la sobreprotección. • Permiso de conducir: en una primera crisis, si el paciente no es conductor profesional, se aconsejará no conducir durante 6 meses (tiempo para esclarecer el diagnóstico). Los conductores profesionales no están autorizados a conducir vehículos durante 1 año. En el caso de epilepsia puede obtenerse una licencia no profesional para la conducción de vehículos tras 1 año sin crisis. Es excepcional plantearse una licencia profesional. • La mujer epiléptica puede emplear anticonceptivos orales (ACO). Es preferible tomar fármacos que no interaccionen con los ACO (tabla 10), pero si es necesario emplearlos, utilizarlos con, al menos, 50 µg de estradiol. • Gestación: en caso de embarazo en una paciente epiléptica, no suspender la medicación. La gestación se considera de alto riesgo por la mayor incidencia de problemas en el feto, posibles efectos teratógenos de los fármacos y riesgo de aumento de las crisis. Sin embargo, el 90 % de los embarazos en mujeres epilépticas llegan a buen término. El ácido valproico y la carbamazepina se asocian con una mayor incidencia de defectos del desarrollo del tubo neural. También se debe aconsejar tomar 5 mg de ácido fólico 1 mes antes del embarazo y durante éste. Los recién nacidos de madres en tratamiento con antiepilépticos deben recibir 1 mg de vitamina K i.m. o i.v., y las madres suelen recibir 10 mg/día/v.o. de vitamina K durante el último mes de gestación. • Vida laboral: la capacidad para trabajar dependerá de 433 5.5 Convulsiones Tabla 9. Interacciones farmacológicas de los antiepilépticos Antiepilépticos Interacciones farmacológicas Carbamazepina Reduce las concentraciones plasmáticas de teofilina, anticoagulantes orales, haloperidol, corticosteroides, anticonceptivos orales, rifampicina, cloranfenicol, doxiciclina, fármacos antiepilépticos Aumenta el metabolismo de la tiroxina Aumentan su acción: cimetidina, danazol, dextropropoxifeno, diltiazem, verapamilo, eritromicina, isoniazida Su uso junto con IMAO puede producir crisis hipertensiva Fenitoína Disminuye los efectos de: corticosteroides, dicumarol, digoxina, doxiciclina, estrógenos, haloperidol, metadona, anticonceptivos orales, dopamina, furosemida, levodopa, sulfonilureas, barbitúricos, carbamazepina, diazóxido, ácido fólico, teofilina, antiácidos, nitrofurantoína Aumentan sus efectos terapéuticos: alopurinol, cloramfenicol, cimetidina, diazepam, isoniazida, trimetroprima, ácido valproico, salicilatos, imipramina Fenobarbital Puede disminuir los efectos de: anticonceptivos orales, corticosteroides, dicumarol, digoxina Puede potenciar los efectos indeseables de los antidepresivos tricíclicos. El ácido valproico aumenta los valores séricos de fenobarbital Gabapentina Lamotrigina No administrar junto con antiácidos Los fármacos inductores o inhibidores enzimáticos pueden influir sobre el metabolismo de lamotrigina No altera la eficacia de los anticonceptivos orales, aunque éstos pueden hacer necesario incrementar las dosis del fármaco Levetiracetam No interacciona con otros fármacos antiepilépticos. Tampoco se han observado interacciones clínicamente significativas con anticonceptivos orales, digoxina, ni con warfarina Oxcarbazepina Interacciona con anticonceptivos orales. No interacciona con eritromicina, cimetidina, ni warfarina. Carbamazepina, fenitoína y fenobarbital reducen la disponibilidad del metabolito activo de la oxcarbazepina Pregabalina No se han observado interacciones clínicamente significativas Primidona Carbamazepina, fenitoína y ácido valproico incrementan la conversión de primidona a fenobarbital Tiagabina No modifica los niveles de los otros fármacos antiepilépticos, tampoco de los anticonceptivos orales. Los fármacos antiepilépticos inductores enzimaticos sí que provocan disminución de la concentración y de la semivida de la tagabina Los inhibidores enzimáticos del citocromo P450 no alteran la farmacocinetica de la tiagabina Topiramato Disminuye los niveles de digoxina y anticonceptivos orales Administrado con fármacos antiepilépticos inductores se debe incrementar la dosis del topiramato casi al doble Valproato Potencia los efectos de IMAO, otras sustancias depresoras del sistema nervioso central, anticoagulantes Su uso concomitante con clonazepam puede inducir crisis de ausencia IMAO: inhibidores de la monoaminooxidasa. Consejos prácticos • Lo más importante es no establecer nunca el diagnóstico de epilepsia sin una clínica convincente, aunque el EEG presente signos irritativos. • Tener siempre presente los cuadros de origen psiquiátrico. • Recordar que diversos anticomiciales pueden disminuir la eficacia de los anticonceptivos orales. • Recordar a los padres de los niños con convulsiones febriles la importancia de reducir la fiebre bañándoles con agua templada o toallas mojadas, y la utilidad del diazepam rectal durante la crisis. 434 Errores más frecuentes • Diagnosticar una epilepsia por una única crisis convulsiva. • Ante episodios raros, recurrentes y paroxísticos, no pensar en crisis epilépticas parciales como posible causa de aquéllos. • En adultos que han presentado una pérdida de conciencia, no descartar la crisis convulsiva como causa de aquélla. • En el estudio de una pérdida de conciencia, no preocuparse de interrogar a los testigos para poder diagnosticar o descartar una posible crisis epiléptica. • En un adulto que ha presentado una primera crisis convulsiva, no hacer un estudio de imagen craneoencefálico. 5.5 Convulsiones Tabla 10. Efectos de los fármacos antiepilépticos sobre la concentración de los anticonceptivos orales Efecto significativo Efecto no significativo Carbamazepina Ácido valproico Fenitoína Benzodiazepinas Fenobarbital Etosuximida Bibliografía recomendada 1. Commission on classification and terminology of the International League Against Epilepsy. Proposal for revised classification of epi- Oxcarbazepina Gabapentina Primidona Lamotriginab Topiramatoa Levetiracetam Tiagabina Vigabatrina Pregabalina Zonisamida a En monoterapia en dosis ⱕ 200 mg/d no parece interferir significativamente con los ACO. b Los ACO reducen las concentraciones plasmáticas de lamotrigina en más del 50 %. ACO: anticonceptivos orales. lepsies and epileptic syndromes. Epilepsia. 1989;30:389-99. 2. Theodore WH, Porter RJ. Epilepsia: 100 principios básicos. 3.ª ed. Barcelona: IATROS; 1996. 3. Arroyo S, Campistol J, Comes E, Fossas P, Martínez I, Padró LL, et al. El tratamiento de las epilepsias. Guía terapéutica de la Sociedad Catalana de Neurología; 2004. Disponible en: http://www.scn.es/ form/guiasterap/Guia-ter-EPILEPSIA-2004.pdf 4. Cubero P, Castillo J, Comas B, Fiol F, Frontera G, García ML, et al. Guía de actualización clínica en neurología. Barcelona: semFYC ediciones, 2004. 5. Valiente ML, Gutiérrez A, Valiente MT. Crisis epilépticas. AMF. 2008;4(2):71-9 6. Enfermedades del sistema nervioso. Epilepsia. Medicine. 2007; 9:4814-9. 7. Guía andaluza de epilepsia 2009. Sociedad Andaluza de epilepsia. Disponible en: http://www.guiasalud.es/home.asp 8. The epilepsies: The diagnosis and management of the epilepsies in adults and children in primary and secondary care. NICE clinical la frecuencia de las crisis. Ciertas profesiones no pueden realizarse ya que, por sus características, exigen unas determinadas restricciones (conductor profesional, piloto, policía, etc.). Guideline 20, Oct. 2004. Disponible en: www.nice.org.uk. Educación sanitaria Es conveniente dar apoyo escrito a las personas que han presentado convulsiones o tienen epilepsia (Guía práctica de la salud, p.32). 435 5.6 Temblor B. Casabella Abril ¿De qué hablamos? Introducción El temblor es un movimiento involuntario anómalo, característicamente rítmico y oscilatorio, de una o varias partes del cuerpo alrededor de un punto fijo o eje. Epidemiología Es el trastorno del movimiento más frecuente. En pacientes seniles el temblor esencial (TE) es 10 veces más frecuente que la enfermedad de Parkinson (EP). La prevalencia del TE en mayores de 40 años es del 0,4-6 %, con una media de edad de inicio de 35-45 años; aumentando hasta llegar a ser casi del 5 % en mayores de 65 años. La prevalencia de la EP es del 0,16 % (1 % en mayores de 65 años). Clasificación Es útil clasificar el temblor, según la situación en que aparece o predomina, en reposo o con la acción. • Temblor de reposo: aparece en situación de relajación muscular. El prototipo es el temblor parkinsoniano. • Temblor de acción: cabe distinguir: – Temblor postural o de actitud: surge cuando se mantiene una postura voluntaria contra la gravedad (p. ej., brazos extendidos al frente). Aquí se encuadra el temblor esencial. – Temblor de intención: aparece al finalizar un movimiento preciso, al acercarse al objetivo (p. ej., con la maniobra dedo-nariz). El prototipo sería el temblor cerebeloso. ¿Qué lo puede ocasionar? Causas Los mecanismos que lo originan no son bien conocidos. Recientemente, algunas investigaciones sugieren que el cerebelo se encuentra funcional y anatómicamente alterado en el temblor esencial. Predominio del temblor de reposo La primera sospecha diagnóstica es un parkinsonismo (EP, síndrome parkinsoniano inducido por fármacos, parkinsonismo plus). • Enfermedad de Parkinson Debuta habitualmente en la quinta-sexta década de la vida. Es el trastorno neurodegenerativo más frecuente después de la enfermedad de Alzheimer (EA). El temblor es el signo físico inicial en el 70 % de los casos. Suele ser de inicio unilateral en manos (como «contar monedas», con frecuencia de oscilación de 3-7 Hz) y progresa por la extremidad inferior del mismo lado. Posteriormente tiende a la bilateralidad. La anamnesis y la exploración física siguen siendo la 436 principal fuente de información para un diagnóstico preciso, ya que éste sigue siendo clínico. Deben cumplirse dos de los cuatro signos mayores clásicos, durante más de 1 año: – Temblor de reposo. – Hipertonía en rueda dentada (rigidez con resaltes al realizar la flexoextensión repetida del antebrazo sobre el brazo). – Bradicinesia. – Inestabilidad postural. Los signos menores que apoyan el diagnóstico son: hipersialorrea, disfagia, depresión, demencia subcortical, alteraciones del habla (disartria, tartamudeo, disfonía), micrografía, disautonomía (hipotensión ortostática, disfunción vesical y/o sexual, estreñimiento) y dermatitis seborreica. La buena respuesta al tratamiento con levodopa sería un criterio que apoya con claridad la existencia de EP. Recientemente técnicas complementarias en el estudio de la EP como el DaTSCAN, una solución inyectable que contiene el principio activo ioflupano (I-123), que ayuda a detectar la pérdida de células nerviosas que liberan dopamina, o el estudio de la vía simpática cardíaca mediante gammagrafía cardíaca con metayodobencilguanidina (MIBG), han ayudado en el diagnóstico de este síndrome. • Parkinsonismo farmacológico Es la segunda causa más frecuente de síndrome parkinsoniano (10-30 %). Puede aparecer tras meses o años de haber iniciado el consumo del fármaco (tabla 1). Los criterios diagnósticos son los siguientes: – Presencia de dos o más de los signos mayores de parkinsonismo. – Ausencia de síntomas parkinsonianos antes de la exposición al fármaco responsable y desaparición o mejoría al suspender el fármaco responsable. Es sugerente de parkinsonismo farmacológico: inicio con temblor de reposo bilateral, temblor mandibular aislado, asociación de temblor postural y de reposo, presencia de discinesia orolingual (sin levodopa) y escasa evolutividad. • Otros parkinsonismos sintomáticos o secundarios Son raros, pueden desarrollarse en relación con traumatismos craneoencefálicos repetidos, tumores cerebrales, infartos subcorticales, hidrocefalia normotensa, degeneración hepatocerebral crónica. • Parkinsonismo plus o asociado a otras enfermedades neurodegenerativas Son enfermedades neurodegenerativas que en su evolución asocian rigidez y temblor y que pueden simular una EP: parálisis supranuclear progresiva, atrofia multisistémica, degeneración corticobasal, enfermedad de Creutzfeldt-Jakob, enfermedad de Alzheimer, etc. Predominio del temblor de acción • Temblor de intención Aparece al finalizar un movimiento de precisión (dedonariz) y si se asocian otros signos (nistagmo, marcha atáxica ampliando la base de sustentación, hipotonía, 5.6 Temblor Tabla 1. Fármacos y tóxicos causantes o agravantes del temblor Temblor de reposo o parkinsoniano Calcioantagonistas (cinaricina, flunaricina) Fenotiazinas y análogos (clorpromazina, flufenazina, olanzapina, tioproperazina, trifluoperazina y otros) Butirofenonas (haloperidol y otros) Tioxantenos (tiotixeno y otros) Benzamidas (sulpirida y otros) Otros neurolépticos Antieméticos (metoclopramida y otros) Antihipertensivos (reserpina, metildopa, captopril, amlodipino, nifedipino, verapamilo) Antiarrítmicos (amiodarona) Tóxicos (monóxido de carbono, plomo, magnesio, pesticidas) Otros (litio, loracepam, diazepam, clordiazepóxido, buspirona, cimetidina, isoniazida, valproato sódico, fluvoxamina, fluoxetina, difenhidramina) Aumento del temblor postural fisiológico Hormonas tiroideas Amiodarona (hipertiroidismo) Betaadrenérgicos (salbutamol, efedrina y otros) Xantinas (teofilina, cafeína) Antidepresivos tricíclicos e ISRS Alcoholismo (abstinencia, neuropatía) Drogas (nicotina, anfetaminas, cocaína y otras) Otros (corticosteroides y ciclosporina) Temblor de intención Hidantoínas disdiadococinesia) apunta a una afectación cerebelosa o de sus conexiones. • Temblor al realizar una actividad concreta Como al escribir o al ponerse de pie. Algunos autores lo consideran variantes del TE. Temblor postural Aparece al mantener una postura antigravitatoria (p. ej., brazos extendidos al frente), y se divide en: • Temblor fisiológico sintomático o exagerado por situaciones fisiológicas pasajeras (frío, fatiga muscular, estrés emocional) o patológicas: – Abstinencia alcohólica: temblor matutino (por deprivación nocturna) en manos, lengua, párpados que se hace generalizado y a veces con mioclonías en el delirium tremens plenamente instaurado. – Trastorno agudo por ansiedad (crisis de pánico, hiperventilación) o crónico («desde siempre», manos frías y sudorosas). El temblor aislado como cuadro conversivo histérico es raro; si se presenta suele ser de postura y sugestionable. – Hipertiroidismo: bilateral y simétrico, fino y rápido, predomina en manos (sudorosas pero calientes, a diferencia del trastorno por ansiedad) y puede generalizarse si hay disfunción grave. – Otras: hipoglucemia, fármacos (v. tabla 1), feocromocitoma. Si el temblor no es evidente durante la exploración, pensar en un fenómeno episódico y hacer hincapié en los síntomas acompañantes (nerviosismo, palpitaciones y sensación de hambre en la hipoglucemia, escalofríos por hipertermia, cefalea y crisis hipertensiva en el feocromocitoma, etc.). • Temblor esencial Es monosintomático. Es el temblor más común en el anciano (el término «temblor senil» ya no se utiliza) pero puede aparecer a cualquier edad (suele comenzar a partir de la cuarta o quinta década de la vida). En la mitad de los casos hay algún familiar de primer grado afectado (temblor esencial familiar) lo que sugiere un patrón de herencia autosómica dominante. Su fisiopatología todavía se desconoce. Su frecuencia de oscilación es de 5-7 Hz (algo superior al de la EP). Aumenta con el nerviosismo y el ejercicio físico. Suele suprimirse con dosis moderadas de alcohol. Las localizaciones más frecuentes son las siguientes: extremidades superiores, cabeza, mandíbula, lengua, voz; y con menos frecuencia, tronco o extremidades inferiores. Al inicio suele ser algo asimétrico (más evidente sobre la mano dominante), pero es típicamente bilateral y distal. • Temblor postural sintomático o secundario Posee las características del TE pero se asocia a otra enfermedad neurológica: neuropatía periférica, EP avanzada (se asocian el temblor postural y el de reposo como «contar monedas»), distonía de torsión, traumatismo craneoencefálico, enfermedad de motoneurona y neurolúes. Si el temblor es de gran amplitud («aleteo de ave» o «de esgrimista»), debe pensarse en esclerosis múltiple o en una enfermedad de Wilson. Diagnóstico diferencial • El temblor no debe confundirse con otras hipercinesias, como los tics, mioclonías, discinesias o flapping (tema 5.7) y la epilepsia parcial (tema 5.5). El temblor es un movimiento claramente rítmico y los otros no. 437 5.6 Temblor • La presencia de rigidez, bradicinesia, predominio de temblor de reposo, evolución invalidante y respuesta favorable a la levodopa apoyan el diagnóstico de EP, a diferencia del TE (figura 1). • Otras enfermedades neurológicas que pueden confundirse con una EP son: atrofia multisistémica, parálisis supranuclear progresiva, parkinsonismo vascular, hidrocefalia normotensa y enfermedad por cuerpos de Lewy. Tabla 2. Anamnesis del temblor Edad de inicio Localización inicial y amplitud Evolución: variabilidad en el mismo día, en el tiempo Circunstancias que desencadenan, mejoran o agravan: fármacos, actividades concretas, estrés, alcohol Síntomas neurológicos acompañantes: parkinsonismo, depresión, deterioro cognitivo, focalidad, parestesias, hipoestesia, debilidad ¿Qué tenemos que hacer? Grado de molestia o incapacitación funcional: física (actividades instrumentales y básicas como beber, afeitarse, hablar, escribir, vestirse, etc.) y psíquica Anamnesis • Antecedentes familiares: TE, EP o enfermedad de Wilson. • Antecedentes personales: endocrinometabólicos (hipertiroidismo, diabetes mellitus, hipoglucemia, hipercalcemia), psiquiátricos (ansiedad, abuso de sustancias), hepatopatía y nefropatía (encefalopatía, neuropatía). Fármacos y tóxicos (v. tabla 1). • Enfermedad actual: anamnesis del temblor (tabla 2). Exploración física En realidad se inicia ya al entrar el paciente en la consulta (temblor al darle la mano), sigue durante la entrevista (gestualidad, parpadeo, temblor de voz, etc.) y continúa con una secuencia de maniobras sencillas que pueden efectuarse en 2-3 minutos (figura 2): • En bipedestación relajada y ojos cerrados, se observa si aparece temblor (al juntar los pies se valora también el signo de Romberg); se levantan los brazos sobre la horizontal para objetivar si hay debilidad. Tras la maniobra dedo-nariz se observa la marcha. Finalmente, se valora la estabilidad postural mediante el «pull-test» o test del empujón: se avisa al paciente de que le daremos un leve empujón hacia atrás y que intente frenarse (normalidad = sin paso atrás; inestabilidad leve = 1 o 2 pasos atrás; inestabilidad franca = más de 2 pasos o «caería»). Temblor “debilidad” Dismetría (dedo-nariz) Marcha Inestabilidad postural Entrevista Exploración física Hipertonía Maniobra de distracción 10’’ Bradicinesia 10-20’’ 5-10’’ Escritura y dibujo 1’ Escribir nombre o firma mano D. e I. Lento y rápido 1’ 1’ Temblor (También familiar sospechoso) Figura 2. Exploración del temblor. 438 10-15’’ 10’’ Niños ITC a neurología Esclerosis en placas Jóvenes/adultos Cambiar fármaco, abordar alcoholismo Tumores de fosa posterior No Sospecha patología cerebelosa o sus conexiones Sí Fármacos (hidantoínas) o consumo de alcohol Temblor intencional Sí Evidente sólo al llegar al objetivo Enf. vascular cerebral Ancianos Control y seguimiento en APS Descartar fenómeno episódico: hipoglucemia, escalofríos, trast. ansiedad o temblor fisiológico: frío, emoción, fatiga muscular Temblor no evidenciado durante la exploración Tto. en APS ITC a endocrinología Sí Tto. en APS Otras causas Tto. en APS Fármacos y tóxicosb Tto. en APS Temblor esencial Sí Enf. de Parkinson avanzada Distonía de torsión Neuropatías Neurolúes TCE Se asocia a otras alteraciones neurológicas No Síntoma único Aumenta con nerviosismo Disminuye con la ingesta de alcohol Oscilaciones regulares menos rápidas Se modifica al mantener la postura Sd. parkinsoniano (farmacológico)a Temblor fisiológico exagerado Trast. ansiedad (tema 6.1) Hipertiroidismo (tema 14.4) Tto. en APS Alcoholismo (tema 7.3) Temblor fisiológico Leve Oscilaciones cortas, rápidas e irregulares ITC a neurología Enf. de Parkinson Disminuye o cede ¿Temblor de reposo? ITC a neurología Esclerosis en placas Enf. de Wilson «Aleteo de ave» Oscilaciones amplias e irregulares Temblor postural Aumenta Fármacos que producen o agravan el temblor de reposo: calcioantagonistas, fenotiazinas, haloperidol, metoclopramida, reserpina, metildopa, captopril, amiodarona, mexilatina, procaína, litio, lorazepam, diazepam, clordiazepóxido, buspirona, cimetidina, isoniazida, valproato, fluvoxamina, fluoxetina, y difenhidramina. b Fármacos que aumentan el temblor postural fisiológico: extractos tiroideos, amiodarona, salbutamol, efedrina, teofilina, cafeína, antidepresivos tricíclicos, nicotina, anfetaminas, cocaína, corticoides y ciclosporina. APS: atención primaria de salud; ITC: interconsulta; TCE: traumatismo craneoencefálico. a Figura 1. Algoritmo diagnóstico del temblor. No Temblor presente al realizar movimientos de precisión No Temblor 5.6 Temblor 439 5.6 Temblor • En sedestación sobre la camilla: se valora la existencia de hipertonía (si es necesaria una maniobra de distracción, se hace dibujar una letra en el aire con el brazo no evaluado), exaltación del reflejo glabelar o signo de Myerson (golpear lentamente con un dedo la glabela o protuberancia frontal media. El paciente con enfermedad de Parkinson pestañea repetida y sincrónicamente con cada percusión; las personas normales dejan de pestañear tras las primeras percusiones), y el reflejo plantar. • En sedestación sobre la silla, se valora la bradicinesia: inexpresividad facial, parpadeo escaso (normal: 15 a 20 veces por minuto), lentitud/«claudicación» en la abertura y cierre rápidos de dedos índice y pulgar en pinza o en el punteado de pies con el talón fijo. Hay que explorar cada extremidad por separado. Completar la exploración física general, en especial la temperatura axilar y la palpación del tiroides. En la figura 1 se expone un algoritmo diagnóstico que, pasando por la exploración física, relaciona el tipo de temblor con la sospecha etiológica. Exploraciones complementarias Iniciales Si no hay una sospecha etiológica clara: hemograma, glucemia, función renal y hepática, ionograma (Na, K, Ca), TSH y T4 libre. Posteriores Ante una sospecha etiológica: neurolúes (serología específica); feocromocitoma (catecolaminas en orina); enfermedad de Wilson (ceruloplasmina, cupremia/uria); neuropatía periférica, enfermedad de motoneurona (EMG-ENG). Ni las pruebas de laboratorio ni las técnicas de neuroimagen aportan datos relevantes al diagnóstico de la EP ni del TE, pero sirven para descartar comorbilidad. ¿Qué propuesta haremos? Tratamiento Paciente No farmacológico • Temblor esencial Se tratará farmacológicamente sólo si molesta o repercute psicológica o funcionalmente para las actividades de la vida diaria. Tranquilizar, aclarando que no padece una EP y comentar la posibilidad de que exista un factor familiar (TE familiar). Explicar que el estrés, estimulantes como la cafeína o la nicotina y el cansancio pueden agravarlo y que se dispone de fármacos que lo alivian si es necesario. • Enfermedad de Parkinson Es fundamental el tratamiento rehabilitador con terapia ocupacional para mantener el mayor grado de autonomía y mejorar la calidad de vida. Permite retrasar complicaciones como las rigideces permanentes y el deterioro cognitivo. Ajustes en el hogar: sillas, inodoro, utensilios de cocina. Uso de bastón. Alimentación rica en fibra, asegurar hidratación correcta. La cirugía (talamotomía, estimulación del tálamo) se reserva para el temblor incapacitante rebelde a fármacos. Farmacológico • Temblor esencial El propranolol produce una mejoría significativa en el 50-70 % de los casos (tabla 3). El temblor específico de la escritura y el aislado de la voz tiene respuesta escasa al tratamiento. Si hay poca repercusión funcional, se Tabla 3. Tratamiento farmacológico en el temblor esencial Primera elección Segunda elección Tercera elección Nombre genérico Propranolol (comp. 10 y 40 mg) Primidona (comp. 250 mg) Fenobarbital (comp. 100 mg) Posología Inicial: 10-40 mg/8-12 h v.o. Media: 80-120 mg/día Máxima: 160-320 mg/día (opción: dar la forma retard de 160 mg en una sola dosis diaria) Primidona: Inicial: 1/4-1/8 de comp. noche v.o. Media: 250 mg/día Máxima: 750 mg/día Fenobarbital: 100 mg noche Metoprolol 50-100 mg/12 h (primera elección en asmáticos) Nadolol 40-80 mg/24 h Clonazepam: 0,5-1 mg/12-24 h Lorazepam: 2 mg/12 h Diazepam: 5-10 mg/12 h Gabapentina: 1.200 mg/día Recomendaciones ECG previo Incremento 20-40 mg semanales Monitorización de la presión arterial y la frecuencia cardíaca Las dosis inferiores de cada rango son preferibles en ancianos Primidona: incremento, cada 3 días, de inicio subir 1/4-1/8 de comp. diario (evitar mareo + ataxia) Fenobarbital: en ancianos tiene mejor perfil de efectos secundarios que primidona ECG: electrocardiograma. 440 Temblor ortostático: clonazepam Temblor leve-moderado: ocasionalmente lorazepam, diazepam 5.6 Temblor puede usar el propranolol en una sola dosis (40-120 mg), 2 horas antes de posibles situaciones de estrés, como hablar en público. En asmáticos se dará en su lugar metoprolol. El atenolol es poco eficaz. La ineficacia del propranolol presupone la del resto de betabloqueantes. Los nuevos fármacos antiepilépticos constituyen una posibilidad en el tratamiento del temblor esencial refractario. El fármaco de segunda línea sería la primidona pero puede producir efectos adversos que pueden ser peores que el propio temblor; destacan los efectos neurotóxicos, como sedación, mareo, vértigo, fatiga o ataxia, que pueden aparecer incluso con dosis muy pequeñas; se debe empezar por dosis bajas de 50 mg/día e ir subiendo lentamente hasta una dosis máxima de 250 mg/día. Sin embargo, parece más aconsejable derivar al neurólogo al paciente que no mejora con dosis intermedias de propranolol (120-180 mg/día). • Síndrome parkinsoniano Si es secundario a fármacos, hay que proceder a la retirada progresiva de éstos. Si no se puede retirar la medicación causal, se puede prescribir el antiparkinsoniano anticolinérgico: biperideno 1 mg/12 h y aumentar según respuesta (máximo 12 mg/día en 3 tomas), teniendo en cuenta que no se debe utilizar en mayores de 70 años. – Para establecer el tratamiento de la EP es recomendable realizar una evaluación inicial por un neurólogo y combinar fisioterapia y psicoterapia con el tratamiento farmacológico. El manejo posterior es conveniente que lo dirija el especialista en colaboración con el equipo de Atención Primaria (AP). En la EP el tratamiento farmacológico debe retrasarse hasta que los síntomas provoquen incapacidad, para demorar los efectos secundarios y la pérdida de eficacia. – La combinación de levodopa/carbidopa es el tratamiento de elección en la EP, iniciando con dosis de 100/25 mg/día para ir aumentando progresivamente hasta el control sintomático (máx. 400-800/100-200 mg/día). La levodopa es poco eficaz en el control del temblor de la EP. Se debe administrar 30 minutos antes de las comidas y esperar entre 3-6 meses para valorar su eficacia. Si produce náuseas, asociar domperidona (20-30 mg/ día antes de las comidas). Nunca suspender bruscamente la levodopa porque se puede producir un cuadro muy grave de rigidez-inmovilidad. – Con la falta de respuesta a dosis altas de levodopa/carbidopa cabe añadir un agonista dopaminérgico como la bromocriptina en dosis de 5-10 mg/8 h; iniciando con 1,25 mg/24 h con incrementos semanales de 1,25 mg. Otros agonistas dopaminérgicos utilizados en la EP son lisurida, pergolida y pramipexol. Al asociar estos coadyuvantes hay que reducir en un 20-30 % la dosis anterior de levodopa. Todos ellos deben iniciarse con dosis muy bajas y aumentar la dosis lentamente (conviene revisar detenidamente la ficha técnica). La pergolida se puede asociar raramente a fibrosis retroperitoneal y pleural y a disfunción valvular cardíaca (en tratamientos prolongados se aconseja realizar una revisión ecocardiográfica). El tratamiento con levodopa pierde eficacia con el tiempo, disminuyendo al 50 % a los 5 años del inicio, apareciendo fluctuaciones y discinesias. Por ello, en menores de 65 años, se aconseja comenzar con agonistas dopaminérgicos. – Cuando se presente pérdida de eficacia de la levodopa, manifestada por fluctuaciones sintomatológicas (on/off) y por el acortamiento del efecto, se puede recurrir a la administración de la levodopa en forma retard o en intervalos más frecuentes. Si persisten las fluctuaciones motoras, se puede recurrir a los inhibidores de la catecol-O-metiltransferasa (COMT) periférica (entacapona) que se asocia siempre a la levodopa, disminuyendo las dosis de ésta, pero no están claras todavía sus posibles ventajas clínicas y su relación coste/beneficio. – Los anticolinérgicos (biperideno, trihexifenidilo, prociclidina) pueden tener un efecto aditivo al de la levodopa y, en las fases precoces, pueden ser efectivos sobre el temblor, pero debido a sus efectos secundarios importantes y a que no deben emplearse en ancianos, son fármacos actualmente de segunda línea en la EP. – La infiltración local de toxina botulínica es una opción en los casos de temblores localizados incapacitantes. Familia Información compartida con la que recibirá el paciente. Para la EP, véase la Guía práctica de la salud (GPS), p. 33. Para el TE, véase el anexo 1. Comunidad En el tratamiento integral del paciente con Parkinson y de su familia tienen un papel relevante las asociaciones de enfermos de Parkinson. Éstas permiten encauzar el problema por parte del enfermo y de su familia, y constituyen una fuente de recursos indispensables en el tratamiento de esta enfermedad crónica y muy incapacitante. Derivación Neurología • Sospecha de EP, para confirmar el diagnóstico, que es clínico y comenzar la pauta del tratamiento farmacológico. • Presencia de demencia, alucinaciones visuales vívidas, incontinencia urinaria o caídas hacia atrás, o de focalidad neurológica no explicada: signo de Babinski, prueba de Romberg positiva, prueba dedo-nariz patológica. Todo ello puede alertar sobre la existencia de un parkinsonismo secundario no farmacológico (hidrocefalia normotensiva, enfermedad cerebrovascular) o de un parkinsonismo plus (enfermedad de Alzheimer o enfermedad con cuerpos de Lewy, parálisis supranuclear progresiva, atrofia multisistémica). • Paciente diagnosticado de TE que no mejora con el tratamiento o, aunque mejore, aparece un parkinsonismo progresivo. 441 5.6 Temblor Anexo 1. Hoja informativa para el paciente con temblor esencial El temblor es un movimiento rítmico anormal de una parte del cuerpo (manos, cabeza, voz, etc.). La causa más frecuente de temblor es el llamado temblor esencial. Es una enfermedad benigna de carácter hereditario. Puede dificultar las tareas de la vida diaria como comer, beber, coser, etc., aunque es muy raro que sea invalidante. ¿Qué puede hacer? El temblor esencial debe ser diagnosticado por su médico de familia. Hay otras enfermedades, menos comunes, que también producen temblor y que van a necesitar un tratamiento distinto. Es muy importante evitar el uso de tabaco, estimulantes y moderar el consumo de alcohol y ciertos medicamentos (p. ej., abuso de Ventolín®). Aunque note mejoría del temblor al tomar alcohol, este efecto es pasajero y engañoso ya que al final el temblor empeorará. Puede ser útil mantener los antebrazos y muñecas apoyados cuando quiera comer/beber utilizando una cuchara, un vaso y no llenarlos del todo. Engrosar los mangos de los cubiertos con cilindros de espuma también puede ayudar a reducir el temblor de manos. ¿Cuándo consultar a su médico? Si a pesar del tratamiento y de evitar la cosas que agravan el temblor, éste va a más. claros cuadros psicóticos. En muchas ocasiones, debidos a efectos secundarios farmacológicos. Se aconseja reducir las dosis de los fármacos por este orden: anticolinérgicos, amantadina, selegilina, agonistas dopaminérgicos y levodopa. En caso de persistir y no poder reducir más los fármacos antiparkinsonianos, se puede asociar un neuroléptico, de forma temporal, que tenga pocos efectos extrapiramidales como quetiapina (dosis bajas por la noche). Pronóstico y seguimiento • El TE no acorta la expectativa de vida, suele ser lentamente progresivo y raramente resulta muy incapacitante (TE grave o maligno). El tratamiento suele ser efectivo mientras se mantenga. • La EP tiene una evolución progresiva y a la larga invalidante a pesar del tratamiento específico. A los 10 años de evolución, más del 60 % de los pacientes tienen una incapacidad motora manifiesta. La media para presentar incapacidad para caminar es de 14 años en las personas no tratadas farmacológicamente. La levodopa retrasa la evolución natural en unos 3-5 años con clara mejoría de la calidad de vida (tabla 4). • La mayor parte de temblores inducidos por fármacos se resuelven, a veces al cabo de varios meses de abandonar el tratamiento. Si no tolera la medicación, aparecen efectos secundarios o síntomas nuevos. ¿Podemos hacer alguna cosa más? Actividades preventivas Salud mental Presencia de trastorno depresivo que no mejora con la mejoría sintomática del Parkinson. Hay que tener en cuenta que los antidepresivos inhibidores selectivos de la recaptación de serotonina (ISRS) pueden empeorar la sintomatología como el temblor y la rigidez. Rehabilitación Excepto en fases iniciales, la terapia ocupacional es una parte importante del tratamiento de estos pacientes para mejorar sus habilidades físicas y psíquicas, evitar rigideces permanentes y caídas, y mejorar su autoestima. • Racionalizar el uso de fármacos inductores de parkinsonismo y temblor. • Consejos y adaptaciones para la prevención de caídas, sobre todo si existe parkinsonismo. Tabla 4. Estadios evolutivos en la enfermedad de Parkinson, según Hoehn y Yahr Estadio 0 Ausencia de signos de la enfermedad 1 Enfermedad sólo unilateral 1,5 ¿Se ha resuelto del todo? Complicaciones • Caídas, especialmente en el parkinsonismo. Capsulitis del hombro derivadas de la inmovilidad y las caídas. • Síntomas depresivos y/o aislamiento social cuando el temblor es intenso. Los ISRS están contraindicados con los inhibidores de la MAO-B irreversibles dopaminérgicos selectivos que se emplean en el tratamiento de la EP, como son la selegilina y la rasagilina por el peligro de síndrome serotoninérgico y crisis hipertensiva. • Es frecuente el desarrollo de alucinaciones e incluso 442 2 2,5 Enfermedad unilateral y axial Enfermedad bilateral con sintomatología leve sin trastornos de los reflejos posturales Enfermedad bilateral con sintomatología leve con recuperación en el test del empujón 3 Enfermedad bilateral con sintomatología leve-moderada y trastorno de los reflejos posturales, físicamente el paciente es independiente 4 Incapacidad importante derivada de la enfermedad pero aún es capaz de caminar sin ayuda 5 Sin ayuda, el paciente está en la silla de ruedas o en la cama 5.6 Temblor Educación sanitaria Consejos prácticos • El temblor debería dejar de ser un problema inadvertido, infravalorado e infratratado. Raramente precisa una valoración urgente. • Pensar que un mismo paciente puede tener diferentes tipos de temblor. • Cuando en un paciente que toma levodopa y un anticolinérgico aparece confusión mental, en primer lugar, se recomienda disminuir la dosis o retirar este último. • Ante un paciente con síndrome parkinsoniano farmacológico, si es preciso continuar con un antipsicótico, cambiar a uno atípico como risperidona, siempre que el paciente no tenga una enfermedad cardiovascular. • La EP requiere un abordaje multidisciplinar, contando con el paciente y su familia. En fases avanzadas la enfermedad es muy incapacitante y hay que preparar el entorno para ese momento. • El paciente y su familia deben recibir información de qué es la EP, su evolución y complicaciones. Habrá que contar con apoyos del servicio de rehabilitación con terapia ocupacional y de asociaciones de enfermos de Parkinson. Siempre es útil apoyarse en material escrito de apoyo. Para el TE, consultar el anexo 1. Para la EP, consultar la GPS, p. 33. Bibliografía recomendada 1. Tolosa E, Wenning G, Poewe W. The diagnosis of Parkinson’s disease. Lancet Neurol. 2006;5(1):75-86. Errores más frecuentes 2. Ruiz J, Arritibel I, Gorostidi A, Bergareche A, Martí JF. El temblor: actualidad y controversias. Rev Neurol. 2009;48(supl 1):S37-S41. • Desinteresarse por el paciente que tiembla (no preguntarle si el temblor le molesta o incapacita) a pesar de que no sea un motivo de consulta. • No contemplar la causa farmacológica como origen de un temblor. • No valorar suficientemente, sobre todo en pacientes ancianos, la indicación y la dosis de fármacos que pueden producir temblor. • No recomendar en la EP que se contacte con asociaciones de familiares de EP. 3. Lang AE. When and how should treatment be started in Parkinson’s disease? Neurology. 2009 Feb 17;72(7 suppl):1658-77. 4. Ferreira J, Sampaio C. Essential tremor. Clin Evid. 2005;(13):160821. 5. Morgan JC, Sethi KD. Drug-induced tremors. Lancet Neurol. 2005;4(12):866-76. 6. Bermejo PE. Tratamiento del temblor esencial. Med Clín (Barc). 2007;129:222-7. 7. Gironell A. Estrategias terapéuticas en el temblor esencial. Med Clín (Barc). 2007;129:632-7. 8. Nutt JG, Wooten GF. Clinical practice. Diagnosis and initial management ofParkinson’s disease. N Engl J Med. 2005;353(10):1021-7. 9. Schapira AH. Treatment options in the modern management of Parkinson disease. Arch Neurol. 2007;64(8):1083-8. 443 5.7 Otros movimientos anormales M. Prósper Sierra ¿De qué hablamos? Introducción Los trastornos del movimiento incluyen un grupo de enfermedades caracterizadas por un exceso de movimientos (anormales involuntarios o hipercinesias) o por una pobreza o lentitud de éstos (síndromes rígido-acinéticos o hipocinesias). La mayor parte de estos trastornos se debe a una disfunción de los ganglios basales. Para el estudio del temblor y del síndrome de Parkinson, consultar el tema 5.6. Conceptos iniciales • Distonía: movimiento anormal, sinuoso, repetitivo, lento, con contracción de músculos agonistas y antagonistas, que progresa hasta un pico en el cual el movimiento tiende a ser sostenido adoptando posturas anormales. También tienden a repetir su morfología (a diferencia del movimiento coreico) y suelen producir dolor. Según su localización, pueden ser focales (blefarospasmo, distonía orofacial o de Meige, cervical o tortícolis espasmódica, espasmo del escribiente, laríngea), segmentarias (craneal, axial, crural) o generalizadas. La distonía focal más frecuente en el adulto es la distonía cervical. • Tic: movimiento estereotipado, compulsivo, breve y brusco. Puede inhibirse de forma voluntaria por un breve período de tiempo. Aumenta su intensidad en situaciones de estrés, ansiedad y fatiga, y disminuye al realizar otros actos que requieren concentración y con el sueño. Pueden ser motores, fonatorios, o una combinación de ambos. • Corea (palabra griega que significa danza): movimiento involuntario abrupto, irregular, de duración breve y baja amplitud, que cambia de una zona corporal a otra sin una secuencia definida. A pesar de ser movimientos involuntarios, el sujeto tiende a incorporarlos a su repertorio de movimientos habituales, intentando disimularlos. Se acompaña de hipotonía muscular y reflejo rotuliano pendular. • Atetosis: movimiento lento y sinuoso, continuo y con imposibilidad de mantener una postura fija. Afecta sobre todo a las manos y pies. Suele presentarse asociada a corea y distonía. • Balismo: produce movimientos involuntarios de carácter violento por contracción de la musculatura proximal de una extremidad, frecuentemente de carácter unilateral (hemibalismo). • Mioclonías: movimientos rápidos, muy breves y de amplitud variable producidos por la contracción simultánea de músculos agonistas y antagonistas (mioclonías positivas) o inhibición del tono muscular (mioclonías negativas o asterixis) que se originan en el sistema nervioso central. • Síndrome de las piernas inquietas: trastorno consistente en la necesidad de mover las piernas por una sensación displacentera en éstas. Es más frecuente en las horas del sueño. Durante el día mejora al caminar. 444 • Discinesias: movimiento involuntario producido por el uso de fármacos, generalmente neurolépticos, aunque pueden ser otros grupos como la metoclopramida. Puede ser de instauración aguda o tardía: – Discinesia aguda: movimientos involuntarios, generalmente de tipo distónico o en forma de acatisia (sensación desagradable de inquietud interior con incapacidad de estar quieto), que aparece en las primeras horas o días de iniciar un fármaco. – Discinesia tardía: movimiento rápido, repetido, de tipo coreico, generalmente localizado en la zona orolinguobucal, producido por la administración prolongada de un fármaco, pudiendo aparecer meses después de suspender el tratamiento. ¿Qué lo puede ocasionar? Causas En la tabla 1 se muestran las entidades que pueden ocasionar movimientos anormales. • Enfermedad de Huntington: enfermedad hereditaria de carácter autosómico dominante que muestra una prevalencia de 5-10/100.000 habitantes. Suele iniciarse entre los 30-50 años y además de movimientos anormales diversos (coreoatetosis, distonía generalizada), cursa con demencia de tipo subcortical. • Enfermedad de Wilson: se transmite de forma autosómica recesiva. Es un trastorno del metabolismo del cobre con disminución de los niveles séricos de su proteína transportadora (ceruloplasmina) y depósito anormal de este metal a nivel cerebral, hepático y corneal (anillo de Kayser-Fleischer). Los síntomas neurológicos son el temblor y movimientos distónicos, mioclonía y coreoatetosis. Cursa con afectación hepática. Hay que sospecharlo en personas jóvenes con cualquier trastorno del movimiento (tema 14.12). • Síndrome de Gilles de la Tourette: denominada enfermedad de los tics, es de origen idiopático, se inicia alrededor de los 5 y 10 años de edad y predomina en hombres (relación hombre/mujer de 3:1). Cursa con tics motores y vocales. En ocasiones, con ecolalia y coprolalia (lenguaje obsceno). • Síndrome de las piernas inquietas: la mayoría son idiopáticos pero se pueden asociar a anemia ferropénica, fármacos, embarazo, neuropatías y enfermedad de Parkinson. Su prevalencia en la población adulta es alta (2-15 %), con mayor predominio en las mujeres (tabla 2). ¿Cómo orientar el diagnóstico? Anamnesis El diagnóstico sindrómico es fundamentalmente clínico. Debe interrogarse sobre los siguientes aspectos: • Antecedentes familiares: enfermedades hereditarias y antecedentes de movimientos anormales. • Antecedentes personales: consumo de fármacos, trau- 5.7 Otros movimientos anormales Tabla 1. Causas de movimientos anormalesa Primarias Secundarias Enfermedad de Huntington Fármacos: neurolépticos, levodopa, metoclopramida, cleboride, anfetaminas, cocaína, fenitoína, flunaricina, cinarizina, carbamazepina, gabapentina, antidepresivos tricíclicos, fluoxetina, sertralina, trazodona Enfermedad de Wilson Distonías primarias Síndrome de Guilles de la Tourette Corea senil Ictus Encefalitis Traumatismos SIDA Corea de Sydenham y corea gravídica Hipoxia neonatal y kernicterus Psicógenas a Para los síndromes parkinsonianos y el temblor, tema 5.6. Tabla 2. Criterios diagnósticos del síndrome de piernas inquietas Criterios esenciales Urgencia para mover las piernas, generalmente acompañada por sensaciones no confortables o desagradables en las piernas Las sensaciones desagradables o la urgencia para moverse comienzan o empeoran durante períodos de reposo o inactividad, como estar tumbado o sentado Las sensaciones desagradables o la urgencia para moverse mejoran total o parcialmente por movimientos como caminar, agacharse, estirarse, etc., al menos mientras dicha actividad continúa Las sensaciones desagradables o la urgencia para moverse están peor durante la tarde o la noche que durante el día, o sólo ocurre en la tarde o la noche Criterios de apoyo Respuesta positiva a tratamientos dopaminérgicos Presencia de movimientos periódicos de los miembros (durante la vigilia y el sueño) Historia familiar positiva de síndrome de piernas inquietas sugerente de herencia autosómica dominante Características asociadas Puede empezar a cualquier edad, pero la mayoría de los pacientes observados en la práctica clínica son de mediana edad o avanzada La mayoría de los pacientes vistos en las consultas tienen curso clínico progresivo, pero también se ha observado un curso estable, incluso remisiones de un mes o más Trastorno del sueño (el disconfort de la pierna y la necesidad de moverse causan insomnio) El examen neurológico suele ser normal en las formas idiopáticas y familiares En las formas no familiares puede verse a veces polineuropatía o radiculopatía asociada Pueden encontrarse niveles bajos de ferritina (< 50 µg/l) Según el International Restless Legs Syndrome Study Group (IRLSSG). Adaptada de Allen RP, et al. Sleep Med. 2003;4:101-19. matismo craneoencefálico (TCE), accidente cerebrovascular agudo (ACVA), síndrome de la inmunodeficiencia adquirida (SIDA), fiebre reumática, hábitos tóxicos (alcohol y otras drogas). • Enfermedad actual: edad de comienzo. Tipo de movimiento anormal observado. Síntomas asociados: demencia, focalidad neurológica o cirrosis. Fármacos asociados. Exploraciones complementarias Variarán en función de la enfermedad subyacente o de la orientación diagnóstica realizada. En general son del ám- bito del neurólogo, excepto las formas secundarias a fármacos. ¿Qué propuesta haremos? Tratamiento No farmacológico • En las de causa secundaria, tratar la enfermedad subyacente. No existen tratamientos curativos para las formas primarias. 445 5.7 Otros movimientos anormales Consejos prácticos • Los tics más comunes suelen ser transitorios, sin significación patológica y no requieren tratamiento. • Los fármacos son la causa más frecuente de movimientos anormales. • La presencia de un espasmo hemifacial obliga a descartar la presencia de una compresión extrínseca del facial por tumores o dilataciones vasculares. • La discinesia coreodistónica más frecuente es la inducida por el tratamiento con levodopa. Errores más frecuentes • En los pacientes ancianos, no dar importancia a los movimientos anormales. • No dosificar adecuadamente fármacos inductores de discinesias como la metoclopramida. • No derivar al paciente con distonías focales en las que el tratamiento de elección es la toxina botulínica. • Psicoterapia en pacientes con sospecha de causa psicógena y también como terapia de apoyo en los casos más traumatizantes. • Síndrome de las piernas inquietas: corrección de las causas desencadenantes, como la ferropenia y el consumo de café, alcohol o tabaco. Ejercicio físico y técnicas de relajación. • Corea: haloperidol 1-5 mg/6 h v.o. o clorpromazina 50 mg/8 h v.o. • Distonía: en las formas focales y segmentarias, inyecciones locales de toxina botulínica cada 3-4 meses. En las crónicas: biperideno 1 mg/12 h v.o. (máx.12 mg/día) o trihexifenidilo 2 mg/día v.o. (máximo 15 mg/día repartidos en tres tomas). • Síndrome de piernas inquietas: en los casos en que los síntomas no remitan y produzcan un deterioro importante de la calidad de vida, usar levodopa/carbidopa (100/25 mg al acostarse) o agonistas dopaminérgicos como ropinirol (dosis de inicio 0,25 mg/día) o pramipexol (dosis de inicio 0,088 mg/día). También pueden usarse benzodiazepinas, gabapentina o codeína. • Tics: no suelen precisar tratamiento. En las formas graves puede prescribirse haloperidol 0,5-2,5 mg/día. Derivación Deben derivarse, para su estudio y tratamiento, las formas primarias y las formas secundarias no farmacológicas. Bibliografía recomendada 1. Roa ES, Tolosa E. Trastornos del movimiento. Concepto, epidemiología y clasificación. Medicine. 2003;08:5075-80. 2. Tudurí I, Liaño H. Movimientos involuntarios: actualización diagnóstica y terapéutica. FMC. Form Med Contin Aten Prim. 2001; 08:511-22. 3. Muñoz E. Otros trastornos del movimiento: distonía, corea y tics. Medicine. 2003;08:5102-10. 4. Dressler D, Benecke R. Diagnosis and management of acute move- Normas específicas: tratamiento sintomático Muestran una eficacia variable: • Discinesia aguda: la suspensión del fármaco es fundamental. Si es preciso, por la intensidad o intolerancia, administrar: biperideno 2,5-5 mg i.m. o i.v. lento, repetir si es necesario cada 30 minutos hasta un máximo de 10 mg/24 h; o diazepam 5 mg i.m. o i.v. muy lento y luego 5 mg/8-12 h v.o. El diazepam y los betabloqueantes (propranolol en dosis inicial de 20 mg/8 h v.o.) pueden ser útiles en la acatisia inducida por neurolépticos. • Discinesia tardía: biperideno si hay distonía. Suspender el neuroléptico progresivamente o cambiarlo por otro más selectivo (p. ej., risperidona). Clonazepam 0,5-2 mg/8 h v.o. si las discinesias son incapacitantes. 446 ment disorders. J Neurol. 2005;252(11):1299-306. 5. Harris, MK. Movement Disorders. Med Clin North Am. 2009;93: 371-88. 6. Bayard M, Avonda T, Wadzinski J. Restless Legs Syndrome. Am Fam Physician. 2008;78(2):243. 7. Alonso-Navarro H, Jiménez-Jiménez FJ, Luquin MR. Burguera JA. Trastornos del movimiento (IV): otros trastornos del movimiento (temblor, mioclomas, tics, síndrome de piernas inquietas). Medicine. 2007;09:4753-63. 8. Allen RP, Picchietti D, Hening WA, Trenkwalder C, Walters AS, Montplaisi J, et al. Restless legs syndrome: diagnostic criteria, special considerations, and epidemiology. A report from the restless legs syndrome diagnosis and epidemiology workshop at the National Institutes of Health. Sleep Med. 2003;4:101-19. 17.1 5.8 Pérdida de fuerza en las extremidades F. J. Valderrama Zurián y S. García Domínguez ¿De qué hablamos? Introducción Es una alteración de la motilidad voluntaria que se manifiesta como paresia o parálisis. lor osteomuscular de la pérdida de fuerza verdadera. El cansancio, que se produce al repetir tareas, orienta a la astenia y la presencia de dolor al movimiento, a la patología musculoesquelética. También puede confundirse con la claudicación debida a la isquemia arterial (figura 1). ¿Qué tenemos que hacer? ¿Qué lo puede ocasionar? Anamnesis Causas La causa de la pérdida de fuerza es el resultado de una alteración que puede acontecer a diferentes niveles (tabla 1): • Motoneurona superior. • Motoneurona inferior. • Raíces nerviosas y nervios periféricos. • Unión neuromuscular y músculo. Diagnóstico diferencial Ante un paciente que se queja de pérdida de fuerza es necesario diferenciar, en primer lugar, la sensación de debilidad general y la limitación del movimiento debida al do- • Antecedentes familiares: dirigidos a la búsqueda de neuropatías o miopatías de carácter hereditario y de enfermedad cerebrovascular (ACV) o ictus. • Antecedentes personales: hábitos tóxicos (alcohol, cocaína, anfetaminas), fármacos (anticonceptivos, anticoagulantes, corticosteroides, estatinas), factores de riesgo cardiovascular, fibrilación auricular, infarto de miocardio, valvulopatías, ictus previo, lúes antigua, enfermedad tiroidea, diabetes, vacunación reciente. Cuantificar el consumo de alcohol y/o realizar algún test para descartar un problema crónico (tema 7.3). • Enfermedad actual Hay que profundizar en las características del síntoma guía: Tabla 1. Diagnóstico topográfico de la lesión en las paresias y parálisis Motoneurona superior Motoneurona inferior Nervio periférico Unión neuromuscular Músculo Debilidad Hemiparesia Paraparesia Monoparesia Cuadriparesia Asimétrica o segmentaria Según trayecto del nervio Asimétrica proximal Simétrica proximal Masa muscular Normal o ligera atrofia Atrofia marcada Atrofia moderada Normal Atrofia ligera Tono Aumentado Normal o disminuido Normal o disminuido Normal Normal Sensibilidad Normal o disminuida Normal Disminuida Normal Normal Reflejos Aumentados Signo de Babinsky Disminuidos Disminuidos Normales o disminuidos Normales o disminuidos Fasciculaciones Ausentes Presentes Ocasionales Ausentes Ausentes Enzimas músculo Normales Normales Normales Normales Aumentadas Causas más frecuentes ACV Neoplasia Enfermedad desmielinizante Infección ELA Espondilosis o traumatismo cervical ELA Siringomielia Poliomielitis Hernia discal Síndrome GuillainBarré PN diabética PN enólica Traumatismo Compresión PN familiares Tumores nerviosos Miastenia grave Botulismo Disfunción tiroidea Miopatía tóxica Distrofias musculares ACV: accidente cerebrovascular; ELA: esclerosis lateral amiotrófica; PN: polineuropatía. 447 448 Astenia Sospecha botulismo ITC preferente a neurología Valorar ITC a neurología Suspender Tto. en APS Fármacos tóxicos Analítica: CPK, VCM, transam, K, Na, Ca, Mg, P Trastorno hidroelectrolítico Sospecha miastenia grave Sí Tono muscular # Reflejos de estiramiento No Descartar: fármacosa, alcohol, trastornos electrolíticos, tóxicos No Siringomielia, neoplasias, ELA Ictus Neoplasia Con atrofia Hemiparesia Neuropatías miopatías Aguda ITC urgente a neurología No ACV, déficit de B12, Sd. cola caballo siringomielia, Hernia discal neoplasias Espástica crónica Sí Sd. de GuillainBarré Aguda ITC a cirugía vascular Sospecha isquemia arterial (tema 3.14) Sí Diabetes, alcohol Tto. analgésico y valorar evolución Sospecha patología musculoesquelética No Existe claudicación y ausencia de pulsos Crónica Paresia predominio distal Polimiositis, distrofias Crónica Paresia predominio proximal Paraparesia Flácida aguda ITC a traumatólogo Presentación súbita. Aguda Traslado a urgencias hospital Sí Valorar ITC a neurología Tto. en APS AINE, RHB, reposo infiltración Alteración moderada/ grave leve EMG: lesión plexural Grupo muscular de una extremidad Alteración leve Traumatismo Ictus, trauma, neoplasia EM Sin atrofia Monoparesia Tetraparesia ¿Cómo se distribuye la debilidad? No Se asocia a dolor Localizada a Fármacos más utilizados que producen debilidad muscular: amiodarona, AINE, antitiroideos, antirretrovirales, interferón, citostáticos, corticoides, estatinas, fibratos, penicilina. ACV: accidente cerebrovascular; AINE: antiinflamatorios no esteroideos; AIT: accidente isquémico transitorio; APS: atención primaria de salud; Ca: calcio; CPK: creatinofosfocinasa; ELA: esclerosis lateral amiotrófico; EM: esclerosis múltiple; EMG: electromiografía; ITC: interconsultor; K: potasio; Na: sodio; P: fósforo; RHB: rehabilitación; SGB: síndrome de Guillain-Barré; SNC: sistema nervioso central; VCM: volumen corpuscular medio. Figura 1. Algoritmo diagnóstico de la pérdida de fuerza. Traslado a urgencias hospital SGB Sí Fluctuación diaria, mejoría con reposo Enfermedad SNC: EM, AIT, cataplejia Cansancio al repetir tareas Ascendente Descendente Parálisis progresiva con hipo o arreflexia ITC preferente/ urgente a neurología Tema 1.7 Incapacidad para realizar una tarea física Generalizada Pérdida de fuerza 5.8 Pérdida de fuerza en las extremidades 5.8 Pérdida de fuerza en las extremidades – Grado de afectación funcional en las tareas de la vida diaria: qué cosas no puede hacer que antes sí hacía (p. ej., andar sin tropezar o subir las escaleras sin agarrarse a la barandilla o levantarse de una silla sin apoyo). Servirá para diferenciar la debilidad general de la verdadera pérdida de fuerza. – Localización: generalizada o localizada, miembros afectados (monoparesia, hemiparesia o tetraparesia), proximal o distal y simetría. – Forma de instauración: en el ictus, la instauración suele ser aguda. Las miopatías, distrofias, síndromes metabólicos, cuadros compresivos y la esclerosis lateral amiotrófica (ELA) tienen un comienzo insidioso. La esclerosis múltiple (EM) suele tener un comienzo agudo o subagudo y en brotes. Los procesos tumorales pueden debutar de cualquier forma. – Evolución: cuadros episódicos y fluctuantes son típicos de la EM; la fluctuación, relatada como fatigabilidad precoz, sugiere miastenia grave. El comienzo del ictus es brusco pero su evolución posterior es diferente: a) AIT (accidente isquémico transitorio): resolución completa en 24 horas. b) DNIR (déficit neurológico isquémico reversible): resolución entre 24 horas y 3 semanas. c) Déficit establecido: no mejora después de la tercera semana. d) Ictus evolutivo: el déficit progresa (después de 24 h en los de origen carotídeo y tras 72 h si es de origen vertebrobasilar). • Síntomas acompañantes: – Neurológicos: vértigo, temblor, calambres, fasciculaciones, convulsiones, hipoestesias, dolor, diplopía, afasias, afectación de esfínteres. – Síntomas generales: fiebre, cefalea, pérdida de peso, disnea. • Factores agravantes del cuadro: el mal control metabólico en la afectación diabética, tiroidea o suprarrenal agrava los síntomas; en la miastenia grave la repetición del movimiento también los aumenta (fatigabilidad). Tabla 2. Escala de valoración de la fuerza muscular 0 1 2 3 4 5 Ausencia de contracción muscular Fibrilación muscular o signos de contracción Movilidad activa eliminando la gravedad Movilidad activa que vence la gravedad Movilidad activa contra cierta resistencia Potencia muscular normal Exploración física • Fuerza muscular: evaluada por grupos musculares proximales y distales (balance muscular), utilizando maniobras de contrarresistencia u otras que evidencien la pérdida de fuerza (para la distal, caminar de puntillas o talones; para la proximal, levantarse de la posición de cuclillas o sentado). Siempre se cuantificará mediante una escala de graduación (tabla 2). • Fatigabilidad o debilidad del grupo muscular: empeoramiento con la repetición de un movimiento o actitud que precisa poco esfuerzo (p. ej., valorar la caída de los párpados superiores forzando la mirada hacia arriba durante un minuto estando la cabeza en posición neutra). • Trofismo muscular: atrofia, hipertrofia, seudohipertrofia. • Tono muscular: espasticidad, flacidez. • Alteraciones de la sensibilidad (tema 5.9). • Reflejos osteotendinosos y cutaneoplantares: buscando hiperreflexias e hiporreflexias, asimetrías y presencia de signo de Babinski (tabla 3). • Coordinación: mediante las maniobras dedo-nariz y talón-rodilla. • Actividad anormal del músculo: – Miotonías: dificultad/retraso de la relajación tras una contracción normal (se deben a trastornos bioquímicos neuromusculares). – Fasciculaciones: sacudidas espontáneas en el músculo en reposo, suelen ser signo de denervación. • Exploración del nivel de conciencia, pares craneales, signos meníngeos y fondo de ojo. Tabla 3. Principales reflejos tendinosos y cutáneos de las extremidades Reflejo Estimulación Respuesta Nivel afectado Tronco nervioso Bicipital Pliegue del codo Bíceps braquial y braquial anterior C6 (C5) Musculocutáneo Tricipital Por encima de olécranon Tríceps braquial C7 (C6-C7) Radial Estilorradial Por encima del estiloides radial Supinador largo y bíceps braquial C6 (C6-C7) Radial Cúbito-pronador Estiloides cubital Pronación de la muñeca C8 Cubital Aquíleo Tendón aquíleo Tríceps sural S1 (S2) Ciático poplíteo interno y mayor Rotuliano Tendón rotuliano Cuádriceps L4 (L3-L5) Crural Cutáneo-plantar (signo de Babinsky) Borde externo del pie Flexión dedos. Extensión y separación en abanico S1-S2 Vía piramidal 449 5.8 Pérdida de fuerza en las extremidades • Exploración general: que debe incluir respiración, presión arterial (PA), pulso, soplos cardíacos y carotídeos, pulsos periféricos. Exploraciones complementarias Iniciales • Analítica: glucemia para descartar una neuropatía diabética; hemograma para excluir poliglobulia en las trombosis, anemia que podría ser secundaria a un sangrado, fórmula leucocitaria y velocidad de sedimentación globular (VSG); creatinina por la posibilidad de una neuropatía urémica; gammaglutamiltranspeptidasa (GGT) y volumen corpuscular, en la sospecha de enolismo; potasio, para valorar la función renal y las hipopotasemias; calcio para descartar hipocalcemias. Posteriores • Analítica: vitamina B12 y ácido fólico para descartar mielopatía de origen carencial (en alcohólicos, déficit nutricional o terapias con antagonistas de folatos); hormona tirotropina (TSH) en caso de sospecha de miopatía de origen tiroideo; hemoglobina glucosilada (HBA1c); enzimas musculares si se sospecha miopatía inflamatoria (creatincinasa [CK], lactatodeshidrogenasa [LDH], transaminasa glutámico pirúvica [GOT], aldolasa); serología luética (tema 23.15) y anticuerpos antinucleares (ANA) en las miopatías autoinmunitarias (tema 23.14); estudio de trombofilia en caso de ACV recurrentes de origen no filiado (tema 23.5). • Electrocardiograma (tema 25.1): para valorar si hay arritmias que justifiquen embolismos de origen cardíaco. • Tomografía computarizada (TC) y resonancia magnética (RM) (temas 24.23 y 24.24): son las exploraciones que se deben realizar ante la sospecha de proceso expansivo cerebral, ictus, o de una enfermedad desmielinizante. Consejos prácticos • La anamnesis y exploración correctas permiten orientar el diagnóstico clínico en muchas ocasiones. • En pacientes con antecedentes de ACV que presentan pérdida de conciencia, descartar epilepsia secundaria. • Ante síntomas neurológicos resueltos, pensar en un AIT y no sólo en una debilidad subjetiva y un cuadro funcional. • No olvidar que un síncope de origen cardiogénico puede simular un AIT. • Una hipoglucemia puede manifestarse como déficit motor agudo. • Una migraña acompañada de paresia debe ser enviada al neurólogo. • No olvidar realizar una auscultación carotídea para buscar soplos. • Una analítica que valore la CK en un paciente que toma estatinas puede permitir descartar la miopatía debida a este fármaco. • En todo paciente con AIT o ACV descartar una arritmia embolígena, aunque en ese momento no esté presente. 450 Errores más frecuentes • Atribuir a un cuadro osteoarticular degenerativo una monoparesia sin plantearse un síndrome compartimental. • Considerar que un déficit resuelto espontáneamente no tiene importancia. • No valorar la anticoagulación oral en los pacientes en fibrilación auricular. • No tener en cuenta la importancia de la rehabilitación posterior al ACV. • Olvidar el hipotiroidismo como causa de pérdida de fuerza. • Radiografía de columna (temas 24.5 y 24.6): cuando la clínica indica afectación de raíces nerviosas, para descartar afectación degenerativa, hernias discales, estenosis de canal medular o lesiones tumorales óseas. • Electromiograma/neurograma (tema 25.15): ante una posible neuropatía por atrapamiento, diabética o enólica y en la ELA, y también para definir el momento evolutivo en que se encuentra. ¿Qué propuesta haremos? Tratamiento No farmacológico • Ante un ictus, pasada la fase aguda, hay que plantear las medidas de prevención secundaria abordando los factores de riesgo cardiovascular, comenzando con las modificaciones de estilo de vida (tema 20.7). • Control glucémico y abstención alcohólica para tratar la neuropatía diabética y enólica. • Se corregirá la obesidad como agravante de cuadros compresivos y osteoarticulares. • En ACV, después del alta hospitalaria, es fundamental la adaptación familiar y del domicilio a las nuevas circunstancias (tema 22.8) y la realización de la rehabilitación oportuna (tema 22.5). Farmacológico • Aportar vitamina B12 y ácido fólico, si hay déficit (tema 23.3). • En caso de sospecha de AIT o tromboembolismo cerebral, traslado urgente al hospital activando el código ictus. La prevención secundaria y terciaria tiene como fármaco de elección el ácido acetilsalicílico (AAS) 100 mg/ día, si existe riesgo de sangrado gastrointestinal asociar omeprazol 20 mg/día y si el paciente es alérgico al AAS utilizar clopidrogel 75 mg/día; el tratamiento anticoagulante debe considerarse cuando se asocie patología embolígena (como una fibrilación auricular [FA]) (tema 21.8). • Si hay espasticidad en el ACV establecido, administrar baclofeno 5 mg/8 h (máximo 50-75 mg/día). • Para los cuadros osteoarticulares: antiinflamatorios no esteroideos (AINE) y relajantes musculares. 5.8 Pérdida de fuerza en las extremidades Derivación Urgente La mayoría de los cuadros de inicio brusco y/o en evolución se derivarán a urgencias, para comprobar su origen vascular y confirmar si la causa es isquémica o hemorrágica. Preferente A neurología, neurocirugía o cirugía vascular, los cuadros repetitivos o fugaces (posible EM, AIT) y los susceptibles de anticoagulación o de cirugía tanto traumatológicos (hernia discal) como vasculares (estenosis carotídea > 70 %). ¿Se ha resuelto del todo? Complicaciones más frecuentes La rigidez y el dolor articular en uno o más miembros paréticos y las úlceras por presión son las complicaciones más frecuentes una vez en el domicilio. Todo paciente con un ACV tiene mayor riesgo de recidiva por lo que habrá que realizar prevención secundaria adecuada. actuación enérgica sobre los factores de riesgo cardiovascular. El tratamiento de las secuelas es fundamental, de ahí la importancia de la rehabilitación (tema 22.5), la prevención de úlceras de decúbito y de la rigidez, en las que la colaboración de enfermería es básica. Educación sanitaria Siempre es útil suministrar información escrita al paciente y a la familia. Consultar en Guía práctica de la salud, p. 37. Bibliografía recomendada 1. Erro-Aguirre E, Maisterra O, Gallego-Culiere J. Síndromes paraneoplásicos neurológicos. Med Clín. 2005;125:543-7. 2. Felipe J, Manuel J, Bermejo F. Ictus en el adulto joven. Med Clín (Barc). 2004;122:70-4. 3. Ricart C, Leno C, Altable M, Rebollo M. Factores de riesgo de los accidentes cerebrovasculares. Etiopatogenia del accidente cerebrovascular. Medicine. 2003;8:4911-7. 4. Perona AB, García-Muñozguren S. Protocolo diagnóstico del paciente con pérdida aguda de fuerza. Medicine. 2007;9(87): 5635-9. 5. Bilbao I. Protocolo diagnóstico de la hemiparesia y la hemiplejia. Pronóstico y seguimiento Dependerá de la enfermedad de base y de la extensión de la lesión. Después del alta hospitalaria de un ACV con secuelas importantes hay que hacer una intervención multidisciplinar (médico, enfermera y trabajador social) para valorar las necesidades sociosanitarias y de rehabilitación del paciente y de la familia (temas 19.1, 19.14, 22.5 y 22.8). Medicine. 2007;9(78):5033-5. 6. Lowenstein DH, Martin JB, Hauser SL. Estudio del paciente con enfermedades neurológicas. En: Fauci AS, Braunwald E, Kasper DL, Hauser SL, Longo DL, Jamenson JL, Loscalzo J., editores. Harrison. Principios de Medicina Interna. 17.ª ed. México: McGraw-Hill Interamericana; 2009. p. 2484-9. 7. Codina A, Cervera C. Síndrome de Guillain-Barré. Med Clín (Barc). 2002;118:142-5. 8. Pascuzzi RM. Peripheral neuropathy. Med Clin North Am. 2009; ¿Podemos hacer alguna cosa más? 93(2):317-42. 9. Hughes RA, Cornblath DR. Guillain-Barré syndrome. Lancet. 2005; 366(9497):1653-66. Actividades preventivas Dado que la causa más frecuente de pérdida aguda de fuerza en las extremidades es el ACV, se debe realizar una 451 5.9 Hormigueos (trastornos de la sensibilidad) J. V. Sorlí Guerola ¿De qué hablamos? que son percepciones aumentadas o desagradables, respectivamente, ante estímulos habituales. Introducción Epidemiología básica Los hormigueos o trastornos de la sensibilidad son motivo frecuente, y a su vez poco conocido, de consulta en Atención Primaria (AP). El síndrome de las piernas inquietas (SPI) se presenta en el 5-10 % de la población de 18-65 años, siendo más frecuente en las mujeres (2:1). La prevalencia del síndrome del túnel carpiano sin diagnosticar se estima en el 1 % de la población general y el 2-5 % en la población laboral. El síndrome de Guillain-Barré o polineuropatía desmielinizante inflamatoria aguda (PDIA) representa el 25-40 % de las polineuropatías de la edad adulta. Conceptos iniciales La sensibilidad somática se clasifica en superficial o de fibras pequeñas (térmica, táctil y dolorosa) y profunda o de fibras grandes (vibratoria, propioceptiva o artrocinética y de presión). Los trastornos de la sensibilidad pueden ser deficitarios o irritativos. Los deficitarios cursan en forma de hipoestesias que el paciente puede describir como «acorchamiento o falta de tacto». Los irritativos o productivos se manifiestan como parestesias (sensaciones anormales sin estímulo previo, descritas como «hormigueos o miembro dormido») o como hiperestesias o disestesias, ¿Qué lo puede ocasionar? Causas (tablas 1 y 2) Diagnóstico diferencial (figura 1) • Mononeuropatía (MN): es la lesión de un nervio periférico, habitualmente debida a una compresión local Tabla 1. Causas más frecuentes de los trastornos sensitivos Mononeuropatías Polineuropatías Síndrome del túnel carpiano Diabética Atrapamiento cubital Alcohólica Atrapamiento del radial Síndrome de «piernas inquietas» Meralgia parestésica (atrapamiento del nervio femorocutáneo) Urémica Síndrome del túnel tarsiano Paraneoplásica (oat-cell) Atrapamiento del nervio ciático-poplíteo externo Mieloma y otras paraproteinemias Afectación del nervio ciático mayor Déficit de vitamina B12 Atrapamiento del nervio occipital mayor Infecciosas: VIH, mononucleosis, brucelosis, borreliosis Afectación de pares craneales Farmacológicas: amiodarona, antirretrovirales, antineoplásicos, cloroquina, colchicina, etambutol, fenitoína, hidralazina, isoniazida, nitrofurantoína Tóxicas: acrilamida, arsénico, talio, disulfuro de carbono, hexacarbonos Familiares Acroparestesias nocturnas Radiculopatías Mielopatías Artropatías Compresiones traumáticas Hernias discales Compresiones tumorales Traumatismos vertebrales Síndrome siringomiélico (tumor, siringomielia o hemorragia) Cervicobraquialgias Síndrome cordón posterior (tabes, VIH/SIDA, déficit B12) Tumores meníngeos Infarto de la arteria espinal anterior Tumores del canal medular Afectación de plexos nerviosos VIH/SIDA: virus de la inmunodeficiencia humana/síndrome de la inmunodeficiencia adquirida. 452 5.9 Hormigueos (trastornos de la sensibilidad) Tabla 2. Clasificación de las neuropatías según el tipo de fibras afectadas Sensitiva de fibras pequeñas (neuropatías dolorosas y con alteración de la sensibilidad superficial) Neuropatía diabética de fibras pequeñas o inicial Amiloidosis Neuropatía por VIH y tratamiento antirretroviral Otras Sensitiva de fibras grandes (neuropatías atáxicas y con alteración de la sensibilidad profunda) Déficit de vitamina B12 Síndrome de Sjögren Neuropatía por cisplatino Intoxicación por piridoxina Ataxia de Friedreich Sensitiva de fibras pequeñas y grandes (neuropatías con alteración sensitiva superficial y profunda) Neuropatía sensitiva diabética avanzada Neuropatía paraneoplásica Neuropatías sensitivas hereditarias Otras Neuropatías de predominio motor Síndrome de Guillain-Barré (SGB) Neuropatías hereditarias Porfiria intermitente aguda Neuropatía por plomo Neuritis braquial Neuropatía diabética del plexo lumbosacro (amiotrofia diabética) Autonómicas Agudas: botulismo, porfiria, SGB, amiodarona, vincristina Crónicas: amiloide, diabetes, síndrome de Sjögren, paraneoplásica, enfermedad de Chagas VIH: virus de la inmunodeficiencia humana. o a microtraumatismos repetidos. Cursa con parestesias, hipoestesia táctil y alteraciones vasomotoras en la sudoración y la piloerección. Puede estar asociada, entre otras, a diabetes mellitus (DM), hipotiroidismo, gota, alcoholismo, insuficiencia renal, neoplasias, artritis reumatoide, virus de la inmunodeficiencia humana (VIH) y otras enfermedades debilitantes. • Mononeuritis múltiple o multineuritis: lesión de diversos nervios no contiguos. • Polineuropatía (PN): es la lesión simétrica de varios nervios periféricos. Clínicamente cursa con síntomas irritativos, hipoestesia profunda y lesión motora. En su desarrollo pueden confluir varios mecanismos. Así, en la PN alcohólica, el efecto enólico se suma al defecto nutricional, y en la PN asociada al VIH, la toxicidad de los antirretrovirales a la acción propia del virus. • Radiculopatía: la causa más frecuente es la compresión de una raíz o de un plexo nervioso. Predomina la afectación algésica frente a la táctil, con frecuente déficit motor e hiporreflexia, pero nunca alteración vasomotora. • Mielopatía: la causa más frecuente es la compresiva de origen vertebral. Aparece hipoestesia sublesional para algunos tipos de sensibilidad en función del grado de lesión y de su localización. Suele haber hiperreflexia patológica e hipoestesia contralateral. • Lesiones tronculares, talámicas y supratalámicas: la causa más frecuente es la vascular, sin descartar el posible origen tumoral. • Síntomas sensitivos transitorios: casi siempre son por compresión (mecánica externa, habitualmente posicional), por elongación o por isquemia local de un tronco nervioso. Pueden ser debidos a accidentes isquémicos transitorios (AIT), crisis parciales sensitivas, esclerosis múltiple (EM), migrañas, cuadros de hiperventilación o hipocalcemias. • Trastornos funcionales: las alteraciones de la sensibilidad son síntomas frecuentes en los trastornos de conversión y de somatización, así como en los simuladores. ¿Qué tenemos que hacer? Anamnesis • Antecedentes familiares: de DM, SPI, epilepsia, migraña y porfiria, entre otras. • Antecedentes personales: tareas habituales y actividad laboral, cuadros congénitos (estenosis del canal medular), DM, anemia, insuficiencia renal, artritis reumatoide, neoplasias, déficit nutricional de vitamina B12 y ácido fólico, tóxicos o fármacos (alcohol, isoniazida, quimioterapia antineoplásica y antirretrovirales), embarazos, hipotiroidismo, radioterapia, traumatismos o compresiones locales, sífilis, síndrome de la inmunodeficiencia humana (SIDA), ansiedad, depresión, somatizaciones; cuadros previos similares y su evolución. • Enfermedad actual: – Características del síntoma: si es deficitario o irritativo y tipo de sensibilidad afectada. En el síndrome del túnel carpiano (STC), el paciente refiere parestesias nocturnas en las manos que le despiertan y obligan a sacudirlas. El SPI se caracteriza por la aparición nocturna en las piernas de parestesias o disestesias, hormigueos, e incluso dolor o desasosiego. – Localización: el STC cursa con síntomas en 1.º, 2.º y 3.er dedos y en la mitad radial del 4.º dedo, aunque también puede afectar a toda la mano o a un único dedo, generalmente el 1.º o el 2.º. Las parestesias debidas a la afectación cubital se distribuyen por la mitad cubital del 4.º dedo y el 5.º completo. En el síndrome del túnel tarso (STT) se localizarán en la planta del pie. En la meralgia parestésica, a menudo debido a compresiones externas del nervio femorocutáneo a nivel del arco inguinal, habrá parestesias en una banda externa del muslo. En la estenosis del canal medular, disestesias en ambas piernas, y en el síndrome de la cauda equina, hipoestesia en silla de montar. En el SPI habrá alteraciones de la sensibilidad en la porción inferior de las piernas. Los síntomas funcionales, al contrario que los anteriores, no siguen una organización neurológica definida (p. ej., en la cara 453 454 Desmielinizante Axonal No Control de la enfermedad DM Insuficiencia renal Alcoholismo Sida Sí Tema 5.1 Dolor facial Cabeza Tema 11.6 STC Atrapamiento cubital Atrapamiento radial EMG ITC a neurología Tumor, hemorragia peritoneal, idiopática, diabetes de larga evolución Infiltración tumoral Costilla cervical Tracción del brazo hacia arriba Neuritis braquial idiopática Lesión por radiación Tracción brazo hacia abajo Debilidad antebrazo y mano b Tóxicos que pueden causar polineuropatía: acrilamida, arsénico, disulfuro de carbono, hexacarbonos, talio. Fármacos: isionazida, nitrofurantoína, amiodarona, hidralazina, antineoplásicos, antirretrovirales. CPE: ciático poplíteo externo; DM: diabetes mellitus; EMG: electromiograma; ITC: interconsulta; PDIC: polineuropatía desmielinizante inflamatoria crónica; SGB: síndrome de Guillain-Barré; STC: síndrome del túnel carpiano. a Tema 11.3 Atrapamiento CPE EMG Marcha equina Pierna Debilidad cintura escapular Braquial Radiculopatía Lumbar Lepra, neuropatías hereditarias, diabetes, amiloidosis Afectación fibras pequeño calibre Predominio algésico, frecuente déficit motor ITC a neurología Mielopatías Afectación haz espinotalámico Axonal: vasculitis Múltiple Extremidad Desmielinizante: PDIC superior Valorar ITC a traumatología o neurología Tema 1.7 Ciatalgia EMG EMG Meralgia parestésica Cara posterior muslo Extremidad inferior Mononeuropatía Única Sí Alteraciones vasomotoras, sudoración y piloerección Predominio hipoestesia táctil y parestesias Sensibilidad táctil conservada y termoalgésica alterada (disociación sensitiva) Zona externa de muslo ITC a neurología Enf. familiar Figura 1. Algoritmo de actuación en los trastornos de la sensibilidad. Revaluación terapéutica farmacológica Fármacosb Traslado a urgencias hospital Enf. previas PDIC No Crónica Desmielinizante EMG EMG Porfiria Parálisis motora Intoxicación arrefléxica: masivaa SGB Axonal Subaguda Aguda Polineuropatía Sí Afectación distal, progresiva, simétrica en guante o calcetín No Trastornos de la sensibilidad 5.9 Hormigueos (trastornos de la sensibilidad) 5.9 Hormigueos (trastornos de la sensibilidad) alcanza el cuero cabelludo sin respetar el ángulo mandibular y en las extremidades cesan al llegar a los pliegues anatómicos). – Evolución: el inicio brusco sugiere causa vascular, traumática o compresiva; síntomas cambiantes suelen ser funcionales pero también pueden deberse a AIT, crisis parciales, EM, migrañas, hiperventilación o hipocalcemias. En el síndrome de Guillain-Barré, suele tener una evolución rápidamente progresiva y ascendente, predominando la pérdida de fuerza y la arreflexia, siendo menores las alteraciones sensitivas, aunque a menudo presenta disestesias como hormiguillos en las extremidades y dolores en la espalda. El SPI suele aparecer de forma esporádica a partir de los 40-50 años, aumentando en frecuencia y gravedad con el tiempo. – Desencadenamiento por movimientos o posturas: el STC surge con los movimientos reiterados (p. ej., costureras); la afectación cubital con el martillo o la bicicleta; el STT, con la bipedestación y la marcha; la estenosis del canal lumbar aparece con la bipedestación, la marcha o la extensión lumbar, mejorando con el decúbito y la sedestación. – Otros síntomas acompañantes: pérdida de fuerza (tema 5.8), atrofias, quemaduras o lesiones en las manos (sugieren hipoestesias importantes), síntomas disautonómicos (impotencia coeundi, sudoración, hipotensión ortostática), vasomotores, alteración de esfínteres, cefalea. – Interferencia con tareas habituales: suele ser escasa o nula en los pacientes funcionales. El SPI suele interferir en los períodos de inactividad o descanso. Exploración física • Inspección general: caquexia, obesidad, etilismo, compresiones locales (yesos, férulas, fajas, cinturones), lesiones dérmicas, lesión herpética. • Exploración neurológica: determinar la localización y el tipo de afectación. – Sensibilidad (tablas 3 y 4): comparando hemicuerpos se dibujarán los resultados en un esquema y valorando déficit o aumentos en todos los tipos de sensibilidad. En los cuadros funcionales, el déficit es global y en la exploración artrocinética, al preguntar a los pacientes sobre la posición que se ha hecho adoptar al Tabla 3. Exploración de la sensibilidad Sensibilidad superficial o de fibras pequeñas Sensibilidad dolorosa Aguja de un solo uso Sensibilidad táctil Torunda de algodón Sensibilidad térmica Tubos de ensayo con agua caliente y fría Sensibilidad profunda o de fibras grandes Sensibilidad vibratoria Diapasón de 128 Hz Sensibilidad artrocinética Movimientos pasivos de dedos (3.º y 4.º de las manos y los pies) dedo explorado, muestran cerca del 100 % de fallos, cuando sólo por azar deberían acertar alrededor del 50 %. – Fuerza, movilidad y tropismo muscular (tema 5.8). – Reflejos osteotendinosos: investigando hiporreflexias (polineuropatías o radiculopatías) e hiperreflexias (mielopatía cervical). Se debe explorar el reflejo cutáneo plantar (el signo de Babinsky puede indicar una mielopatía). – Maniobras de provocación: a) Maniobra de Lasègue: dolor en la cara posterior o lateral del muslo al flexionar la cadera con el miembro extendido; supone una irritación de la raíz L5-S1 o L4-L5. b) Signo de Lhermitte: la flexión forzada del cuello produce dolor lancinante que baja por la espina dorsal. Aparece en EM y en cervicoartrosis, indica mielopatía. c) Los signos de Tinel (percutir el túnel carpiano) y el de Phalen (la flexión ventral de la muñeca durante 1 min) reproducen los síntomas del STC. d) Hiperventilar puede desencadenar las parestesias peribucales y en manos de una crisis de ansiedad. – Maniobras de distracción: en los trastornos funcionales, al hacerle girar sobre sí mismo o cruzar los brazos, frecuentemente provocará una equivocación respecto a qué lado es el afectado. – Valoración de la marcha, que en determinados trastornos sensitivos puede ser muy característica: atáxica sensitiva, en estepaje en la afectación del ciático poplíteo externo (CPE) (tema 5.10). – Test de Romberg (tema 5.4): es positivo si al cerrar los ojos tiende a caerse hacia diferentes lados, cuando existe un déficit de sensibilidad profunda propioceptiva. Exploraciones complementarias Iniciales • Analítica: glucosa, creatinina, gammaglutamil transpeptidasa (GGT), proteinograma, iones, hemograma y velocidad de sedimentación globular (VSG). • Radiografías de columna: en proyección anteroposterior, lateral y oblicua, buscando compromiso de espacio (temas 24.5 y 24.6). Posteriores • Analítica: hormonas tiroideas, creatincinasa (CK), lactato deshidrogenasa (LDH), proteinograma, crioglobulinas, porfirinas, estudios inmunológicos, VIH, sífilis, factor reumatoide, vitamina B12, ácido fólico y niveles de tóxicos. • Electromiograma (tema 25.15): es la prueba más sensible y específica en las MN o PN. • Tomografía computarizada y resonancia magnética (temas 24.23 y 24.24): indicadas cuando se sospecha compresión de origen vertebral (p. ej., por hernia discal o estrechez del canal medular). 455 5.9 Hormigueos (trastornos de la sensibilidad) Tabla 4. Neuropatías periféricas: síntomas y signos Afectación de fibras grandes o sensibilidad profunda Síntomas Entumecimiento Pinchazos Hormigueo Ataxia Signos Disminución de la sensibilidad propioceptiva y vibratoria Hiporreflexia Afectación de fibras pequeñas o sensibilidad superficial Afectación motora Afectación autonómica Dolor: urente, penetrante, como electricidad, punzante, terebrante, lancinante; desencadenamiento del dolor por pequeños estímulos (alodinia) Calambres Prensión débil Pie péndulo Fasciculaciones Sudoración aumentada o disminuida Ojos y boca secos Disfunción eréctil Gastroparesia, diarreas Lipotimias, síncopes Disminución de sensibilidades táctil, temperatura y pinchazo Disminución de fuerza Hiporreflexia Ortostatismo ¿Qué propuesta haremos? Tratamiento No farmacológico Normas generales • Mejorar el control glucémico. • Evitar el alcohol, el tabaco y otros tóxicos. • Retirar compresiones locales (fajas, corsés, cinturones). • Realizar actividad física aeróbica. • Buena higiene de sueño. Normas específicas • En el STC, férula antebraquial dorsal con la muñeca en posición neutra, preferiblemente por la noche durante 2-4 semanas. Revisar las condiciones laborales. • En la meralgia parestésica, evitar las prendas ajustadas y poner hielo local. • En el atrapamiento cubital, evitar la flexoextensión enérgica del codo o su apoyo continuado (en relación con posturas mantenidas en el trabajo). • El reposo puede aliviar una discoartropatía. • En los síndromes compresivos de nervios periféricos, una opción terapéutica es la infiltración local con anestésico y corticosteroides (temas 21.32 y 21.33). • En las radiculopatías cervicales, pueden estar indicadas las tracciones cervicales. • Descompresión quirúrgica de STC y STT en caso de afectación grave clínica y en el electromiograma o ante el fracaso con los tratamientos previos. Cirugía en la hernia discal y en compresiones extrínsecas. Farmacológico • En las PN: el fármaco de elección para el tratamiento de las hiperestesias y disestesias es la amitriptilina (de 25 mg/día a 150 mg/24 h) y en segundo lugar, carbamazepina (inicio 100 mg/12 h, hasta 1 g/día) o gabapentina (dosis inicial 300 mg/día, hasta 3,6 g/día en 456 tres tomas). En la PN enólica: complejo vitamínico B incluyendo tiamina (300 mg/día). En el déficit de B12, administrar esta vitamina por vía i.m. (1 mg/día i.m. durante 2 semanas y posteriormente 1 mg/mes de por vida) asociada con ácido fólico v.o. (5 mg/día). En la polineuropatía diabética (tema 14.3). • En el SPI son de elección la levodopa/carbidopa (100/25 mg al acostarse como dosis inicial) y los agonistas dopaminérgicos no ergóticos: pramipexol (inicialmente 0,125 mg por la tarde hasta 0,75 mg/día) y ropinirol (incrementando la dosis de 0,25 mg por la tarde a 2-4 mg/día). Los antiepilépticos mencionados anteriormente en las PN también son eficaces para el SPI. Ante una anemia o un déficit de hierro o ferritina (tema 23.3). • En las radiculopatías: antiinflamatorios no esteroideos (AINE) y miorrelajantes junto con reposo. • En los trastornos de ansiedad (tema 6.1): psicoterapia, ansiolíticos y antidepresivos. Derivación Urgente • Ante la sospecha de un síndrome de Guillain-Barré, patología vascular cerebral o tumoral y en síntomas de rápida evolución. Preferente u ordinaria • El STC y el STT con afectación en el EMG o fracaso del tratamiento, en la hernia discal con afectación neurológica y en la estenosis de canal medular muy sintomática o con déficit neurológico para valoración quirúrgica. • En los cuadros de presentación familiar (p. ej., hipopotasemias y neuropatías hereditarias) para consejo genético (tema 20.9). • En las crisis epilépticas parciales sensitivas, EM y migrañas con síntomas sensitivos asociados, el paciente debe ser remitido al neurólogo (temas 5.2 y 5.5). 5.9 Hormigueos (trastornos de la sensibilidad) ¿Se ha resuelto del todo? Errores más frecuentes Complicaciones Las complicaciones más frecuentes son las heridas y quemaduras en zonas distales cuando se trata de síntomas deficitarios. A menudo, las alteraciones sensitivas se asocian a trastornos motores y tróficos, fácilmente objetivables mediante pruebas neurofisiológicas. Éstas suelen indicar mayor afectación, y en los síndromes por atrapamiento y en la hernia discal, confirman la indicación quirúrgica. Pronóstico y seguimiento Dependerán de la patología subyacente. ¿Podemos hacer alguna cosa más? Actividades preventivas Dirigidas al paciente con el fin de controlar la diabetes, la obesidad y el alcoholismo, evitar las compresiones locales o los movimientos repetitivos. En el ámbito comunitario, prevención laboral, sistemas antivibratorios y evitar determinadas posturas y maniobras. • Confundir un síndrome de Guillain-Barré o una EM con un cuadro funcional. • No reconocer un síndrome de compresión cubital a nivel del codo. • Atribuir las parestesias bilaterales en las manos a una compresión cervical o a un problema circulatorio sin plantear la posibilidad de patología funcional, STC bilateral o tumor neural. • No valorar el factor tóxico: alcohol, fármacos (isoniazida, antirretrovirales, antineoplásicos). • Olvidar la hipocalcemia tras cirugía tiroidea como causa de parestesias. • No derivar una migraña común habitual que de pronto se acompaña de parestesias. Educación sanitaria En síndromes frecuentes como el del túnel carpiano y el ciático, conviene darle al paciente algún tipo de documento de apoyo (Guía práctica de la salud, p. 30 y 155). Bibliografía recomendada 1. Casademont J. Aproximación clínica al paciente con sintomatología del sistema nervioso periférico y muscular. En: Farreras P, Rozman C, editores. Medicina interna. 16.ª ed. Barcelona: Elsevier Es- Consejos prácticos • La anamnesis y la exploración neurológica minuciosa pueden clarificar muchas situaciones. En caso de síntomas transitorios, programar una visita de control. • Los trastornos funcionales no siguen una distribución neurológica definida y la exploración no es concordante con el déficit referido. • Un síntoma neurológico evolutivo difícilmente será funcional y menos si se acompaña de síntomas motores. • Por su alta prevalencia, las parestesias en las manos casi siempre corresponderán a un STC. • Las alteraciones sensitivas en el SPI predominan y empeoran al acostarse. paña; 2008. p. 1546-9. 2. López T, Pascual J. Neuropatías diabéticas. Factores de riesgo. Valoraciones pronósticas. Planificación de seguimiento. Medidas terapéuticas. Medicine. 2008;10(17):1130-7. 3. Valls J. Neuropatías adquiridas (III). Neuropatías secundarias a atrapamiento, compresión y otros agentes físicos. Medicine. 2003; 08:5397-404. 4. Erro ME, Maisterra O, Gallego J. Síndromes paraneoplásicos neurológicos. Med Clín (Barc). 2005;125(14):543-7. 5. Martínez M. Actualización en Medicina de Familia. Una revisión del síndrome de piernas inquietas. SEMERGEN. 2008;34(2):80-6. 6. Ly-Pen D, Andreu JL. Tratamiento del síndrome del túnel carpiano. Med Clín (Barc). 2005;125(15):585-9. 7. Varela AA, Blázquez F, Toribio ML. Síndrome de Guillain-Barré en la consulta de Atención Primaria. Aten Primaria. 2003;32(8):493-4. 457 5.10 Trastornos de la marcha A. Maiques Galán ¿De qué hablamos? ¿Qué lo puede ocasionar? Introducción Causas Las consultas por trastornos de la marcha, como signo fundamental de enfermedad neurológica y especialmente en el paciente menor de 65 años, son infrecuentes en Atención Primaria (AP). Sin embargo, una aproximación al problema es esencial para diagnosticar algunas causas tratables (déficit de vitamina B12 o enfermedad de Parkinson) o para una derivación adecuada a otro nivel asistencial. Observar la marcha de una persona es una de las maniobras más importantes en la exploración neurológica por la participación de diversos sistemas corporales y mentales. Un límite impreciso entre el deterioro de la marcha propio de la edad y la presencia de un trastorno patológico dificultan la valoración de este síntoma. La tabla 1 muestra las estructuras anatómicas afectadas y las causas más frecuentes de trastornos de la marcha. Diagnóstico diferencial Existen patrones peculiares de trastornos de la marcha que corresponden a la afectación de determinadas estructuras anatómicas (tabla 2) y otras alteraciones inespecíficas de difícil diagnóstico. El diagnóstico diferencial se desarrolla en la figura 1. ¿Qué tenemos que hacer? Anamnesis Definición Los trastornos de la marcha se definen como una disminución de la velocidad o alteraciones en la suavidad, simetría o sincronía de los movimientos del cuerpo. Epidemiología básica Los trastornos de la marcha constituyen un problema frecuente en el anciano, en un 15-25 % de los mayores de 60 años y hasta en un 50 % de los mayores de 85 años. Junto con los ambientales, constituyen los principales factores de riesgo de caídas (tema 20.17) y son causa de minusvalía. • Antecedentes familiares: algunos trastornos de la marcha tienen carácter hereditario y se presentan en pacientes jóvenes (ataxias hereditarias). • Antecedentes personales: es importante preguntar sobre los antecedentes de caídas (dos o más en 1 año requieren una evaluación exhaustiva); accidentes cerebrovasculares (ACV) o accidentes isquémicos transitorios (AIT) previos; episodios de vértigo; alteraciones cognitivas; temblor; cefaleas frecuentes o constantes; incontinencias de esfínteres; traumatismos craneoencefálicos (TCE) o de columna previos; consumo de fármacos (neurolépticos, antihipertensivos, ansiolíticos, Tabla 1. Trastornos de la marcha. Estructuras anatómicas afectadas, patrones específicos de marcha y causas más frecuentes Localización de la lesión Patrón de marcha Causas más frecuentes Lóbulo frontal Marcha apráxica Demencias (enfermedad de Alzheimer), enfermedad cerebrovascular, hidrocefalia a presión normal Cerebelo, vías aferentes y eferentes Marcha cerebelosa Enfermedades cerebrovasculares, neoplásicas, desmielinizantes, ataxias hereditarias (ataxia de Friedrich, ataxia telangiectásica), alcoholismo Sistema vestibular Marcha vestibular Diversas causas de vértigo periférico y central (tema 5.4) Ganglios basales Marcha parkinsoniana Enfermedad de Parkinson y diversos fármacos (neurolépticos, metroclopramida, clebopride, flunarizina, cinarizina) Vía piramidal Marcha hemipléjica Marcha espástica («en tijera») Unilateral: enfermedad cerebrovascular Bilateral: parálisis cerebral infantil y paraplejia espástica (mielopatías y demencia multiinfarto) Cordones posteriores de la médula Marcha atáxica sensitiva Déficit de vitamina B12 y sífilis Nervios periféricos Marcha equina o en «steppage» Bilateral: polineuropatía sensitivomotoras (temas 5.8 y 5.9) Unilateral: compresión del nervio ciático-poplíteo externo Musculatura proximal extremidades inferiores Marcha de «ánade» Miositis, polimiositis y miopatías hereditarias En negrita las causas más frecuentes en Atención Primaria. 458 5.10 Trastornos de la marcha Tabla 2. Patrones de trastornos de la marcha Trastorno de la marcha Características Manifestaciones acompañantes Marcha apráxica Tarda en iniciar la marcha, se detiene frecuentemente. Base de sustentación amplia, postura ligeramente flexionada; pasos pequeños, vacilantes y arrastrados Demencia; incontinencia esfinteriana (hidrocefalia a presión normal) Marcha atáxica cerebelosa Amplia base de sustentación, necesidad de apoyo, empeora con los giros. Tambaleante y con titubeos Dismetría, temblor intencional, adiadococinesia Marcha atáxica sensitiva Una amplia base de sustentación, mirada fija en el suelo y levanta mucho los pies y los deja caer bruscamente Romberg positivo (hacia cualquier lado), trastorno de la sensibilidad vibratoria Marcha vestibular o «en estrella» Tendencia sistemática a desplazarse hacia un lado Predominio del vértigo periférico o central (tema 5.4) Test de Romberg positivo (caída hacia el mismo lado) Marcha parkinsoniana o «festinante» Marcha a pequeños pasos, lentos y escaso despegamiento del suelo; flexión de caderas, rodillas y codos; inclinación del tronco hacia delante y ausencia de oscilación de los brazos; dificultad para parar; pérdida del equilibrio Amimia, rigidez, temblor de reposo (tema 5.6) Marcha espástica o en «tijera» Extensión y rigidez de ambos miembros inferiores. Las piernas se cruzan al caminar; pasos cortos y con mucho esfuerzo Paraparesia, hiperreflexia y signo de Babinski bilateral Marcha hemipléjica o en «segador» Miembro inferior en extensión y rígido. Miembro superior flexionado Hemiparesia, hiperreflexia y signo de Babinski unilateral Marcha equina o en «steppage» Flexión exagerada de las caderas y rodillas. Pie caído Arreflexia aquílea y atrofia de la musculatura anterolateral de la pierna Marcha de «ánade» Inclinación del tronco del lado opuesto al miembro que se eleva Debilidad muscular proximal y atrofia glútea Marcha anormal por déficit multisensoriales (senil) Inestabilidad y mareo al caminar y dar la vuelta. Uso frecuente del bastón y apoyo en paredes u objetos para dar la vuelta Alteraciones visuales y propioceptivas fármacos con efecto extrapiramidal) o alcohol. La evolución aguda del proceso puede orientar hacia una enfermedad vascular o infecciosa. Exploración física La exploración neurológica incidirá especialmente en las siguientes pruebas: Exploración de la marcha • Deambulación en la consulta: consiste en observar la marcha del paciente invitándolo a deambular por la consulta. Fijarse en el inicio, la base de sustentación, la longitud de los pasos, la velocidad, el braceo y el giro. • Patrones peculiares de la marcha (v. tabla 2): – Marcha apráxica: caracterizada por tardar en el inicio y detenerse frecuentemente. La demencia es la manifestación acompañante más frecuente de esta marcha. La tríada clínica de demencia, leve o mode- rada, incontinencia esfinteriana y trastornos de la marcha sugiere una hidrocefalia a presión normal. Ocurre por una afectación del lóbulo frontal. La marcha del envejecimiento normal remeda este tipo de marcha. – Marcha atáxica cerebelosa: sus características comprenden una amplia base de sustentación, la necesidad de apoyo mínimo para deambular e incremento de la sintomatología al girar («marcha de ebrio»). La presencia de la alteración de las pruebas de coordinación, dismetría, temblor intencional, adiadococinesia, rebote excesivo o nistagmo indican el origen cerebeloso del trastorno. – Marcha vestibular: la característica peculiar de este tipo de marcha es la tendencia sistemática a desplazarse hacia un mismo lado. El vértigo suele predominar sobre los trastornos de la marcha. El nistagmo, de origen central o periférico, es otra manifestación 459 460 Parálisis cerebral congénita Lesión troncoencefáfica Sí Miopatías, enf. de la unión neuromuscular Marcha en ánade Bilateral No Sífilis Trastorno funcional Astasia Abasia ITC a psiquiatría Marcha vestibular Marcha parkinsoniana Lenta, pasos cortos, encorvado, tiende a acelerarse: festinante Rigidez, acinesia temblor Temas 5.4 y 5.6 Nistagmo asimétrico, sensibilidad propioceptiva normal Al andar cae hacia un lado y en bipedestación ITC a neurología urgente u ordinaria, según forma de instauración y grado de gravedad del cuadro ACV, neoplasias, enfermedades desmielinizante enfermedades hereditarias Marcha Atáxica cerebelosa Dismetría, adiadococinesia No Marcha normal distraído. Se cae sin golpearse buscando apoyos Coordinación normal sentado o acostado Dorsalgia, lumbalgia, lumbocitalgia (Temas 11.2 y 11.3) Problemas musculoesqueléticos (Tema 11.7) ACV: accidente cerebrovascular; CPE: ciático poplíteo externo; EMG: electromiograma; ITC: interconsulta; RHB: rehabilitación; TPHA: Prueba de hemaglutinación de T. pallidum. ITC a traumatología EMG Sífilis Enf. hereditarias Def. Vit B12 Rodillas, pies o caderas Dolor de espalda, radicular o claudicación neurológica Romberg positivo, sensibilidad propioceptiva alterada Marcha atáxica sensitiva Sí No Amplía la base de sustentación Marcha antiálgica Analítica: TPHA, Vit B12 Sí Pérdida sensibilidad propioceptiva Marcha equina Considerar lesión compresiva de nervio CPE Sí Sí Debilidad dorsiflexión tobillo Caída del pie, elevación exagerada de pierna No Elevación y rotación de pelvis inclinando tronco al lado opuesto Debilidad muscular proximal al flexionar la cadera ITC a neurología Enf. médula espinal Paraparesia Marcha en tijeras: marcha del segador bilateral ¿Se acompaña de debilidad muscular? Figura 1. Algoritmo diagnóstico de los trastornos de la marcha. Tema 22.5 Tto. RHB Secuela de ACV Marcha segador ¿Presenta espasticidad? No Desequilibrio lateralizado y nistagmo Hemiparesia Sí No ¿Se acompaña de dolor? Trastorno de la marcha Tema 5.12 Hidrocefalia, hematoma subdural, neoplasia, enf. de Alzheimer Marcha apráxica Similar a Parkinson, pero dificultad para mantener la marcha. No festinante Deterioro cognitivo e incontinencia de esfínteres 5.10 Trastornos de la marcha 5.10 Trastornos de la marcha neurológica que acompaña a este trastorno. Los síntomas vegetativos o cocleares expresan un origen periférico del vértigo y la disartria, la diplopía o los déficit localizados de la fuerza muscular indican un origen central (tema 5.4). La prueba de Romberg es positiva con caída siempre hacia el mismo lado. – Marcha parkinsoniana: el paciente adopta en la enfermedad de Parkinson una postura encorvada característica. La marcha es a pequeños pasos y sin el balanceo asociado de los brazos. Los pasos se pueden hacer sucesivamente más rápidos (marcha festinante). La rigidez, la acinesia y el temblor de reposo conforman la tríada clásica de esta enfermedad (tema 5.6). – Marcha espástica: ocurre en enfermedades poco frecuentes que afectan bilateralmente a la vía piramidal y con frecuencia tienen carácter familiar. Los miembros inferiores en extensión y con rigidez, a veces con aducción (marcha en «tijera»), generan una marcha característica con pasos cortos. Las manifestaciones asociadas incluyen hiperreflexia, signo de Babinski bilateral y debilidad de los miembros inferiores. – Marcha hemipléjica («de segador»): es característica de la afectación unilateral de la vía piramidal. El miembro inferior afectado está rígido, en extensión, con lo que deambula rozando con el pie en el suelo y describiendo un semicírculo. – Marcha atáxica sensitiva: las características principales de esta marcha son una amplia base de sustentación, la mirada fija en el suelo y levanta mucho los pies y los deja caer bruscamente. La prueba de Romberg positiva con caída hacia cualquier lado, la alteración de la sensibilidad artrocinética y, sobre todo, de la vibratoria, expresan una afectación de los cordones posteriores. Puede asociarse arreflexia que sugiere afectación de los nervios periféricos o espasticidad y reflejos cutáneos plantares en extensión (signo de Babinski) por lesión de la vía piramidal. El déficit de vitamina B12 es la causa más frecuente. – Marcha equina o «en steppage»: se caracteriza por una flexión exagerada de la cadera y de la rodilla para evitar que la punta del pie roce con el suelo. Además, existe arreflexia aquílea y atrofia de la musculatura anterolateral de la pierna. Las polineuropatías sensitivomotoras constituyen las causas más frecuentes si es bilateral y la afectación del nervio ciático-poplíteo externo si es unilateral. – Marcha «de ánade»: la debilidad en la flexión de la cadera obliga a inclinar el tronco hacia el lado opuesto para elevar el miembro inferior durante la marcha, remedando a la de un pato. Coexiste, en ocasiones, debilidad de la cintura escapular. Esta marcha es característica de las miopatías. – Marcha senil: los pasos cortos y la reducción de la velocidad de la marcha son característicos de la marcha de la edad avanzada, más acentuados a partir de los 80 años, pero sin apreciarse un claro déficit neuroló- gico. Lo fundamental de este trastorno consiste en establecer el límite entre las alteraciones fisiológicas y las patológicas, ya que su intensidad está relacionada con la aparición de las caídas. El origen de esta marcha es multicausal: disminución de la fuerza muscular, deterioro de la visión (especialmente la periférica), enfermedad cerebrovascular, clínica o subclínica, y la polimedicación (ansiolíticos e hipotensores). • Pruebas para evaluar la intensidad del trastorno: – La marcha en tándem (avanzar en línea recta más de 10 pasos colocando el talón delante de los dedos del pie contrario), y mantenerse en equilibrio con un solo pie más de 10 segundos descarta un trastorno relevante de la marcha. – La prueba «up and go» (levantarse de una silla, caminar 3 metros, darse la vuelta y volver a sentarse) realizada en menos de 14 segundos descarta un trastorno importante y al contrario, si se supera dicho tiempo indica un riesgo aumentado de caídas. Exploración del equilibrio La prueba de Romberg consiste en observar la estabilidad del paciente cuando permanece en bipedestación y con los pies juntos. Se explora sucesivamente con los ojos cerrados y abiertos; si se pierde el equilibrio al cerrar los ojos, se habla de signo de Romberg positivo. Expresa una ataxia sensitiva si la pérdida del equilibrio ocurre hacia cualquier lado y una ataxia vestibular si se produce siempre hacia el mismo lado. En la ataxia cerebelosa el paciente está inestable en la posición de Romberg, tanto con los ojos abiertos como cerrados. Exploración de la coordinación Su disfunción orienta hacia un origen cerebeloso como causante de los trastornos de la marcha. Se realizan las siguientes pruebas: • Prueba dedo-nariz o talón-rodilla: se explora indicando al paciente que toque con el dedo índice la punta de la nariz o con el talón la rodilla contraria. Si existe dismetría, se producen errores en la distancia, que se acentúan cerca del objetivo o del punto de intención, de ahí el término de temblor intencional (tema 5.6). • Prueba del rebote excesivo: se pide al paciente que mantenga los brazos extendidos contra una resistencia que se retira repentinamente. • Adiadococinesia: es la incapacidad para realizar movimientos alternativos con rapidez, por ejemplo cuando se intenta golpear alternativamente las superficies dorsal y palmar de la mano. Exploración de la sensibilidad vibratoria y posicional La sensibilidad vibratoria se explora colocando un diapasón de 128 Hz sobre las eminencias óseas y la sensibilidad posicional, situando en diversas posiciones la falange distal del segundo dedo del pie para que el paciente, con los ojos cerrados, describa la posición. Su alteración indica afectación de la sensibilidad profunda. 461 5.10 Trastornos de la marcha Exploración del nistagmo La presencia de nistagmo es un signo de disfunción vestibular y sus características sirven para diferenciar un vértigo central o periférico (tema 5.4). Exploración del tono muscular • Hipertonía: – Espasticidad en «tubo de plomo» o en «navaja de muelle»: aumento de tono de los músculos antigravitatorios, indica afectación de la vía piramidal. – Rigidez en «rueda dentada»: indica afectación extrapiramidal (tema 5.6). • Hipotonía: orienta hacia una afectación cerebelosa o del sistema nervioso periférico. Exploración de la fuerza muscular y los reflejos de estiramiento muscular La presencia de déficit motores localizados, reflejos de estiramiento muscular u osteotendinosos y cutáneos plantares son pruebas básicas para confirmar la existencia y las características del déficit neurológico responsable del trastorno de la marcha (tema 5.8). Errores más frecuentes • Atribuir como posibles causas de un trastorno de la marcha a la arteriosclerosis o al envejecimiento sin realizar una valoración adecuada. • Pensar en los trastornos de la marcha como trastornos irreversibles en los que poco se puede hacer. • Derivar al neurólogo a un paciente sin un estudio preliminar. • Dilatar en el tiempo, derivando a otros niveles asistenciales con largas listas de espera, el tratamiento de un déficit de B12. de B12 (tema 23.3) y en la enfermedad de Parkinson (tema 5.6). Si se detecta un déficit de B12 el tratamiento precoz evita o reduce las secuelas neurológicas. • En ancianos, pasear 30 minutos al día es el mejor entrenamiento para mantener la movilidad, incluso si existe ya una alteración leve de la marcha. Las actividades preventivas se dirigirán a evitar las caídas y reducir la minusvalía (temas 19.6 y 19.8). Exploraciones complementarias Derivación Iniciales • Analítica de sangre: hemograma completo, iones, creatinina, glucemia, bioquímica hepática (bilirrubina, transaminasas, fosfatasas alcalinas y gammaglutamiltranspeptidasa [GGT]), hormonas tiroideas, vitamina B12, ácido fólico y serología de sífilis (tema 23.15). • Estudio electromiográfico, si se sospecha neuropatía o miopatía (tema 25.15). • Técnicas de neuroimagen: tomografía computarizada (TC) o resonancia magnética (RM) en los casos sin signos acompañantes al trastorno de la marcha o con sospecha de afectación cerebelosa, sensitiva, vestibular central o apráxica. Urgente • En los casos de instauración aguda del trastorno de la marcha, derivar a un servicio de urgencias hospitalarias. ¿Qué propuesta haremos? Tratamiento • El diagnóstico etiológico es importante para instaurar el tratamiento específico, como en los casos de déficit Ordinaria • Los pacientes ancianos con una prueba cronometrada de «up and go» mayor de 14 segundos deben ser valorados con especial cuidado para descartar un trastorno específico de la marcha y si es preciso, remitirlos a un servicio de rehabilitación y en algunos casos a neurología. • Los pacientes menores de 65 años con un trastorno de la marcha, después de una valoración adecuada, requieren valoración por neurología. ¿Se ha resuelto del todo? Complicaciones más frecuentes Una complicación frecuente de estos trastornos son las caídas repetidas. Consejos prácticos • Un paciente que debuta con un trastorno de la marcha requiere un estudio exhaustivo para determinar la causa y la intensidad, y no atribuirlo inicialmente a la edad. • Evitar una actitud fatalista ante un anciano con trastornos de la marcha, ya que existen tratamientos específicos que pueden minimizar el riesgo de caídas y evitar una minusvalía. • La detallada observación de la marcha es una de las más importantes maniobras exploratorias en neurología. 462 Pronóstico y seguimiento El pronóstico es bastante bueno en los trastornos reversibles de la marcha, como el déficit de B12, siempre que el diagnóstico se realice precozmente. En los ancianos el pronóstico depende de lo acusado de la alteración. La asistencia a un servicio de rehabilitación puede mejorar el pronóstico en muchos trastornos de la marcha (tema 22.5). Las caídas constituyen la causa más frecuente de morbimortalidad por accidente en mayores de 65 años. 5.10 Trastornos de la marcha ¿Podemos hacer alguna cosa más? Actividades preventivas La identificación de los trastornos de la marcha supone una actividad esencial para la prevención de las caídas. Educación sanitaria La educación sanitaria se centra en aconsejar paseos diarios de 30 minutos de duración y en las medidas encaminadas a evitar las caídas (Guía práctica de la salud, p. 227). Es importante informar al paciente o a sus familiares de las circunstancias ambientales que puedan provocar las caídas. Vigilar las condiciones del domicilio, que esté bien iluminado, y eliminar o modificar los accesorios, muebles y complementos (baño, escaleras, alfombras) que puedan favorecer las caídas. Bibliografía recomendada 1. Nnodim JO, Alexander NB. Assessing falls in older adults: a comprehensive fall evaluation to reduce fall risk in older adults. Geriatrics. 2005;60:24-8. 2. Beers MH, Jones TV, editors. Gait disorders [internet]. The Merck Manual of Geriatrics. Merck & Co. 2000-2006. Disponible en: http://www.merck.com/mkgr/mmg/sec2/ch21/ch21a.jsp 3. Castillo J, Comas B, Fiol F, Frontera G, García M, Gutiérrez B, et al. Guía de actualización clínica en neurología. Barcelona: semFYC ediciones; 2004. 4. Pérez JL. Manual de Patología General. Sisinio de Castro. 6.ª ed. Barcelona: Masson; 2006. 5. Ondo W. Gait and balance disorders. Med Clin North Am. 2003;87(4):793-801. 6. Factora R, Luciano M. When to consider normal pressure hydrocephalus in the patient with gait disturbance. Geriatrics. 2008; 63(2):32-7. 7. Thurman DJ, Stevens JA, Rao JK; Quality Standards Subcommittee of the American Academy of Neurology. Practice parameter: Assessing patients in a neurology practice for risk of falls (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2008;70 (6):473-9. 463 5.11 Trastornos del habla y del lenguaje I. M. Socias Buades ¿De qué hablamos? ¿Qué lo puede ocasionar? Introducción Causas Habla y lenguaje son conceptos distintos. El lenguaje engloba la comprensión, la formulación y la transmisión de ideas mediante el uso de signos convencionales, señales, sonidos y gestos, así como su ordenación secuencial según las reglas gramaticales establecidas. El habla se refiere a los aspectos relacionados con la articulación y la fonética de la expresión verbal. Los trastornos del lenguaje (afasias) reflejan una alteración cerebral adquirida del área perisilviana del hemisferio cerebral dominante (hemisferio izquierdo en el 95 % de la población). Las causas más frecuentes son el traumatismo craneoencefálico (TCE), la enfermedad cerebrovascular aguda (ECVA) y las neoplasias (tabla 1). Los trastornos del habla (disartria) pueden ser debidos a una lesión en diferentes áreas cerebrales o a causas extracerebrales (v. tabla 1). En la infancia, el retraso o trastorno del habla y del lenguaje puede ser primario cuando no hay causa que lo justifique o secundario a entidades como autismo, deficiencia auditiva, dificultades generales del desarrollo, dificultades conductuales y emocionales o deterioro neurológico. Las causas más frecuentes de disfonía son las laringitis agudas, los pólipos laríngeos y las inflamaciones crónicas de las cuerdas vocales. Clasificación Los trastornos del habla y del lenguaje pueden clasificarse en tres categorías: • Afasias: trastornos del lenguaje debidos a lesión cerebral con pérdida total o parcial de la capacidad de comprensión (afasias sensitivas) o de la producción del lenguaje escrito o hablado (afasias motoras). • Disartrias y anartrias: trastorno del habla debido a lesiones neurológicas con afectación de los músculos implicados en la emisión y articulación de los sonidos. Las funciones superiores están intactas. • Disfonías y afonías: disminución o pérdida total de la voz debido a alteraciones laríngeas o de su inervación siendo el lenguaje y la articulación de los sonidos normales (tema 2.9). Diagnóstico diferencial En la figura 1 se desarrolla el algoritmo diagnóstico en los trastornos del habla y del lenguaje. Tabla 1. Etiología de los trastornos del habla y del lenguaje Afasias Disartrias/anartrias Disfonías/afonías Enfermedad cerebrovascular Parálisis bulbar y seudobulbar Polineuritis aguda idiopática Traumatismos craneoencefálicos Neuropatía periférica de los pares craneales V, VII, IX, X, XII Enfermedades extrapiramidales Enfermedades del SNC: tumores, infecciones, degenerativas, epilepsia (síndrome de Landau-Kleffner) Enfermedad de Parkinson Corea Enfermedades degenerativas del SNC Espasmos glóticos Tabaquismo Neoplasias e infecciones laríngeas Miopatías, mioclonías Procesos intratorácicos: CAI, tumores, aneurisma aórtico Infecciones del SNC Endocrinopatías Tartamudez LES Encefalopatía postanóxica Artritis reumatoide Golpe de calor Pubertad masculina Intoxicación etílica aguda Causa psicógena Tratamiento con progesterona Trastornos del comportamiento o problemas familiares en niños CAI: crecimiento auricular izquierdo; LES: lupus eritematoso sistémico; SNC: sistema nervioso central. 464 5.11 Trastornos del habla y del lenguaje Trastorno del habla y el lenguajea Alteración en la comprensióna y/o construcción del lenguaje con nivel de conciencia normal No Alteración en la intensidad de la voz o su tonalidad No Alteración en la articulación del habla Tema 2.9 Disfonía Sí Habla espontánea mantenida con incapacidad para habla voluntaria o a la orden Disartria No No Sí Normal Repeticióna Discurso Denominacióna Alterado Fluido Comprensión Alterada Afasia de conducción Alterado Alterada Normal Alterada Afasia cortical Derivación preferente/ urgente a neurologíab Afasia transcortical Normal Lectura Alexia pura Derivación preferente/urgente No Sí Considerar lesión neurológica Alterada Sí Sí Alcohol, fármacos, drogas Traslado a urgencias hospital No Tema 5.3 No Afasia Sólo alterada la lectura Sí Parálisis facial Apraxia del habla Sí Considerar ITC a odontología, ORL Sí Causa mecánica Sordera verbal pura Afasia transcortical motora Afasia transcortical sensitiva Discurso Comprensióna Fluido Afasia cortical sensitiva Normal Afasia nominal Alterado Afasia cortical global Afasia cortical motora Causas más frecuentes: enfermedad cerebrovascular, TCE, neoplasias, enfermedades degenerativas e infecciones Forma de presentación Lenta progresiva fluctuante Súbita Derivación urgente al hospital ITC urgente o preferente a neurología Figura 1. Algoritmo diagnóstico de los trastornos del habla y del lenguaje. a Comprensión: Ej. ¿puede volar un perro? ¿nieva en verano? Repetición: repetir palabras sueltas o frases cortas. Denominación o nominación: nombrar objetos comunes. b Lesiones neurológicas que causan disartria: parálisis bulbar y seudobulbar, neuropatía periférica de los pares craneales V, IX, X, XII; enf. de Parkinson, corea, miopatías, mioclonías, enfermedades degenerativas del SNC, infecciones del SNC, encefalopatía postanóxica y golpe de calor. ITC: interconsulta; ORL: otorrinolaringólogo; SNC: sistema nervioso central; TCE: traumatismo craneoencefálico. 465 5.11 Trastornos del habla y del lenguaje ¿Qué tenemos que hacer? Anamnesis • Antecedentes personales: factores de riesgo para ECVA o antecedentes de accidentes cerebrovasculares o de TCE, esfuerzos vocales (profesionales de la voz), ambientes contaminados o hábitos tóxicos como el consumo de tabaco y alcohol. • Enfermedad actual: – Afasias: determinar la forma de presentación clínica y la evolución en el tiempo. Si la causa es una ECVA, el inicio de la sintomatología característicamente es súbito, mientras que si se trata de un tumor se produce un síndrome afásico progresivo. Si la afasia es una forma de aura migrañosa, se instaura de manera progresiva en 5-20 minutos y suele durar menos de 1 hora. La afasia nominal debe alertar sobre la posibilidad de un tumor en el hemisferio cerebral izquierdo o una enfermedad cerebral difusa. Se interrogará si ha habido alteraciones de conducta, cefalea de inicio reciente o si ésta aparece después de la afasia (posible aura migrañosa). – Disfonías: investigar la forma de comienzo y evolución. Recabar información sobre pérdida o ganancia de peso, tos, hemoptisis, disfagia, parestesias locales en el cuello o intolerancia al frío. Investigar antecedentes de enfermedades crónicas, sobre todo tiroideas y neurológicas. La afonía completa casi siempre es psicógena, sobre todo si se trata de una mujer joven. – Disartrias: interrogar sobre la forma de presentación y los síntomas asociados. En las disartrias, a diferencia de lo que ocurre en las afasias, el lenguaje no está alterado. El tartamudeo en un adulto que previamente no presentaba esta alteración debe hacer sospechar una lesión cerebral unilateral o bilateral. Puede aparecer también durante la recuperación de un síndrome afásico. La apraxia del habla es una alteración en la programación de los movimientos complejos utilizados para la producción del habla y se presenta como una alteración única de la expresión verbal y mantiene conservadas la comprensión, la escritura, la lectura y la cognición, se produce con frecuencia tras sufrir un ictus y en algunas ocasiones es difícil establecer el diagnóstico diferencial con la afasia. En la infancia, la tartamudez puede ser de origen genético, síntoma de una alteración neurológica o de causa psicosocial. Se detecta en dos picos de edad, entre los 2-4 años (aprendizaje del lenguaje) y entre los 6-8 años cuando el lenguaje se extiende a leer en voz alta, escribir y recitar en clase. Exploración física Debe realizarse una exploración física adecuada integrando la exploración del lenguaje con el resto del examen del estado mental y la exploración neurológica para orientar el diagnóstico (v. figura 1). • Afasias: en la exploración específica del lenguaje es importante conocer la lengua nativa del paciente, si éste es diestro o zurdo y su grado cultural previo, y se deben valorar los siguientes aspectos (tabla 2): – Discurso espontáneo: valorar la fluencia del lenguaje, dificultad en la articulación y contenido. El habla es fluida si la longitud media de las frases es superior a 4 palabras, habla sin interrupciones, no tiene disartria. – Capacidad de nombrar o denominar: objetos, colores, partes del cuerpo, etc. – Comprensión auditiva: invitar al paciente a ejecutar órdenes de complejidad creciente. – Capacidad de repetición de palabras y frases. – Capacidad de leer en voz alta y comprensión de la lectura. – Capacidad para escribir frases espontáneas, dictado y copiado. – Utilización de parafasias: utilizar un circunloquio o decir una palabra equivocada en lugar de otra. – Ecolalia: repetición de palabras o frases que se han pronunciado inmediatamente antes. • Disfonías: buscar signos asociados a la disfonía (afectación de pares craneales y de vías largas, adenopatías, bocio) y auscultación pulmonar para valorar problemas neumológicos. Exploraciones complementarias Iniciales • Laringoscopia indirecta (tema 25.8): en las disfonías es la prueba inicial principal para poder hacer una Tabla 2. Diagnóstico diferencial del síndrome afásico Afasia Discurso Nombrar Comprensión Repetición Lectura Escritura Otros signos Broca o motora No fluente Alterada Intacta Alterada Alterada Alterada HPD, HSD Wernicke o sensitiva Fluente, P Alterada Alterada Alterada Alterada Alterada HAD, ADSM Global Mutismo Alterada Alterada Alterada Alterada Alterada HPD, HSD, HAD Conducción Fluente Alterada Intacta Alterada Alterada Variable HPD, HSD, HAD Nominal Fluente Alterada Intacta Intacta Intacta Intacta Transcortical motora No fluente Alterada Intacta Intacta +/– Conservada +/– Conservada Transcortical sensitiva Fluente, E Alterada Alterada Intacta Alterada Alterada ADSM: ausencia de déficit sensitivomotor; E: ecolalia; HAD: hemianopsia derecha; HPD: hemiparesia derecha; HSD: hiposensibilidad derecha; P: parafasias. 466 5.11 Trastornos del habla y del lenguaje orientación diagnóstica provisional ya que permite visualizar la glotis con las cuerdas vocales y la epiglotis; especialmente indicada si se trata de una disfonía persistente o asociada a uso excesivo de la voz o existen factores de riesgo para cáncer de laringe. • Radiografía de tórax: en casos de disfonías, para descartar patología pulmonar, diafragmática o la existencia de un bocio intratorácico. • Electrocardiograma (ECG): en caso de afasias, para descartar ritmos cardíacos embolígenos. Posteriores • Tomografía computarizada (TC) craneal y/o resonancia magnética (RM): que se realizará para estudiar las causas de las afasias (enfermedad cerebrovascular [ECV], tumores y TCE). • Laringoscopia directa y estroboscopia laríngea: en el estudio de las disfonías. La estroboscopia se considera el más importante procedimiento diagnóstico en las disfonías; se graban con luz estroboscópica (luz de flash) los movimientos de las cuerdas vocales y se analiza la imagen resultante en un monitor observando las variables de la vibración producida al emitir un sonido. ¿Qué propuesta haremos? Tratamiento No farmacológico • Afasias: control de los factores de riesgo para ECV si ésta ha sido la causa. Valorar tratamiento con logopeda. No hay evidencia científica que permita apoyar o refutar la terapia del habla y del lenguaje en afasias y disartrias después de un daño cerebral no progresivo, aunque se ha objetivado un efecto psicológico positivo. La evidencia en adultos no puede extrapolarse a los niños en los que parece que la terapia del habla y del lenguaje puede ser más efectiva, aunque hacen falta investigaciones adicionales en este campo. • Disfonías: tratamiento etiológico. Evitar el tabaco y reducir al mínimo el uso de la voz. • Disartrias: tratamiento de la enfermedad subyacente. Farmacológico • Afasias: los ensayos clínicos realizados con donepezilo, bromocriptina, piracetam y anfetaminas no ofrecen resultados concluyentes para recomendar el tratamien- Consejos prácticos • Ante un paciente fumador con disfonía, valorar la realización de una radiografía de tórax para descartar una parálisis del nervio recurrente izquierdo por neoplasia pulmonar. • Estratificar el riesgo de tromboembolia arterial en pacientes con fibrilación auricular para valorar tratamiento antiagregante plaquetario o anticoagulante oral (tema 3.12). Errores más frecuentes • No instaurar tratamiento anticoagulante en fibrilación auricular paroxística o crónica, si está indicado. • No investigar hábitos tóxicos en pacientes con disfonía. • No modificar el tratamiento de la hipertensión arterial cuando no se consiguen los objetivos de control. • No dar importancia a una disfonía que dura varias semanas. • No contemplar el ictus como una urgencia médica impidiendo aplicar las medidas terapéuticas adecuadas para conseguir la reperfusión del área con isquemia cerebral (si procede código ictus). to farmacológico de la afasia orientado a recuperar los niveles de neurotransmisores, mejorar el flujo sanguíneo cerebral y la neuroplasticidad. Se precisa la realización de ensayos clínicos aleatorizados que confirmen los posibles efectos beneficiosos. Derivación • Afasias: derivación urgente al hospital, excepto en los casos de diagnóstico previo de migraña con aura. • Disfonías: si duran más de 3 semanas, derivar al otorrinolaringólogo con carácter ordinario o preferente, en función de que existan o no factores de riesgo para carcinoma de laringe, si no se ha llegado a un diagnóstico etiológico o si el tratamiento es quirúrgico. • Disartrias: una vez descartada la embriaguez aguda o una parálisis facial periférica, se realizará derivación urgente (posible ECV, tumor o TCE) o preferente a neurología. La tartamudez en el niño precisa tratamiento psicológico precoz, realizando terapia familiar, si se sospecha una causa psicosocial. La tartamudez persistente en un niño a partir de los 5 años debe remitirse al logopeda y someterse a un estudio del habla y del lenguaje. La sospecha de retraso/trastorno del lenguaje en niños debe derivarse al logopeda preferiblemente en etapa preescolar, derivar a neuropediatría en caso de retraso global, trastorno específico del lenguaje, disartria o afasias adquiridas. ¿Se ha resuelto del todo? Complicaciones más frecuentes • Afasias y disartrias de causa cerebral: pueden asociarse complicaciones producidas por la enfermedad causal como las convulsiones (tema 5.5). El síndrome depresivo reactivo es frecuente y puede precisar tratamiento farmacológico con antidepresivos y psicoterapia (tema 6.2). Se debe evaluar el apoyo que tiene el paciente de la familia, su capacidad vital y el grado de autonomía para las actividades de la vida diaria. Lo ideal es disponer de un equipo rehabilitador con experiencia en fisioterapia, terapia ocupacional, logopedia, psiquiatría, psicología y trabajo social. 467 5.11 Trastornos del habla y del lenguaje El pronóstico del síndrome afásico depende de la gravedad inicial de la afasia y del episodio de ECVA pero no de la edad, el sexo o el tipo de afasia. Las afasias producidas por ECVA pueden recuperarse en horas, días o incluso meses o, por el contrario, persistir. Casi la mitad de la recuperación de la afasia se produce durante el primer mes aunque puede continuar mejorando hasta 1 año después del episodio. Pequeños estudios realizados en afasias causadas por TCE indican que el curso clínico es similar al de la afasia postictus. La recuperación es más favorable en zurdos que en diestros. • Disfonías y otras disartrias: el pronóstico depende de la causa del proceso. Valorar la remisión al logoterapeuta. Bibliografía recomendada 1. Knibb JA, Xuereb JH, Patterson K, Hodges JR. Clinical and pathological characterization of progressive aphasia. Ann Neurol. 2006;59(1):156-65. 2. Berthier ML. Poststroke aphasia: epidemiology, pathophysiology and treatment. Drugs Aging. 2005;22(2):163-82. 3. Ansaldo AI, Arguin M, Lecours AR. Recovery from aphasia: a longitudinal study on language recovery, lateralization patterns, and attentional resources. J Clin Exp Neuropsychol. 2004;26(5):621-7. 4. Knollman-Porter K. Acquired Apraxia of Speech. Top Stroke Rehabil. 2008;15(5):484-93. 5. Gassió-Subirachs R. Trastornos del lenguaje. An Pediatr Contin. 2006;4(2):140-4. ¿Podemos hacer alguna cosa más? 6. Law J, Garrett Z, Nye C. Intervenciones de terapia del habla y el lenguaje para niños con retraso o trastorno primario del habla y el lenguaje (Revisión Cochrane traducida). En: La Biblioteca Coch- Actividades preventivas • Afasias y disartrias de causa cerebral: control de los factores de riesgo para enfermedad cerebrovascular, antiagregación plaquetaria para la prevención secundaria. Si el paciente es hipertenso, controlar que la presión arterial sea < 150/90 mmHg antes de iniciar el tratamiento antiagregante. De elección, ácido acetilsalicílico 100 mg/día y si está contraindicado, clopidogrel 75 mg/día. Consejo para dejar de fumar (tema 7.2). • Disfonías: consejo para dejar de fumar y abstenerse del consumo de alcohol. No hay evidencia para recomendar el entrenamiento vocal para prevenir trastornos de la voz en adultos con ocupaciones en las que el uso de la voz es fundamental y su alteración puede producir elevado absentismo laboral. Esta falta de evidencia no significa, necesariamente, que el entrenamiento vocal no sea efectivo pero se precisa la realización de estudios de calidad para poder ofrecer recomendaciones a estos pacientes. Educación sanitaria Consultar en Guía práctica de la salud, p. 21 y 37. rane Plus, 2008 Número 2. Oxford: Update Software Ltd. Disponible en: http://www.update-software.com. (Traducida de The Cochrane Library, 2008 Issue 2. Chichester, UK: John Wiley & Sons, Ltd.) 7. Ruotsalainen JH, Sellman J, Lehto L, Jauhiainen M, Verbeek JH. Intervenciones para la prevención de los trastornos de la voz en adultos (Revisión Cochrane traducida). En: La Biblioteca Cochrane Plus, 2008 Número 2. Oxford: Update Software Ltd. Disponible en: http://www.update-software.com. (Traducida de The Cochrane Library, 2008 Issue 2. Chichester, UK: John Wiley & Sons, Ltd.) 8. Morgan T, Vogel P. Intervención para la disartria asociada con la lesión cerebral adquirida en niños y adolescentes (Revisión Cochrane traducida). En: La Biblioteca Cochrane Plus, 2007 Número 4. Oxford: Update Software Ltd. Disponible en: http://www.updatesoftware.com. (Traducida de The Cochrane Library, 2007 Issue 4. Chichester, UK: John Wiley & Sons, Ltd.) 9. Consenso de la Sociedad Española de Medicina familiar y Comunitaria y del Grupo de Estudio de Enfermedades Cerebrovasculares de la Sociedad Española de Neurología. Manejo coordinado y prevención del ictus. Neurología. 2007;3(supl 2). Disponible en: http://www.semfyc.es/pfw_files/cma/noticias/noticia/Grupos/ Neurologia/Consenso%20SEMFYC%20final.pdf 10. Hillis AE. Aphasia: progress in the last quarter of a century. Neurology. 2007;69(2):200-13. 468 5.12 Pérdida de la memoria y demencia J. Espinàs Boquet y M. J. González Moneo ¿De qué hablamos? Introducción A pesar de los progresos efectuados en el diagnóstico y tratamiento de la demencia, la mejora de la calidad de vida de los pacientes y la de sus familias se fundamenta en una adecuada información y soporte profesional y humano. Conceptos iniciales • Deterioro cognitivo (DC): es la disminución del rendimiento de al menos una de las capacidades cognitivas (memoria, lenguaje, orientación, pensamiento abstracto, juicio, etc.). El envejecimiento fisiológico puede estar asociado a un cierto declive cognitivo. • DC ligero (DCL): es aquel que no llega a ser demencia. Su curso puede ser variable, dependiendo de la causa, pudiendo revertir, estacionarse o, lo más frecuente, evolucionar a una demencia (75 % de los casos). Normalmente presentan pérdida subjetiva de memoria confirmada por un observador, un test cognitivo confirma pérdida de al menos un área (memoria, orientación, cálculo, etc.), supone un declive de un nivel previo, dura 6 meses o más, no hay causas orgánicas, y no se afecta la vida diaria. El estado cognitivo y funcional de una persona es un continuo condicionado por aspectos genéticos, culturales, laborales y económicos, en el que no existe un límite perfectamente definido entre lo fisiológico y lo patológico. Aunque en desuso, en la práctica diaria en Atención Primaria (AP) pueden ser útiles los siguientes términos: – Alteración de la memoria asociada a la edad (AMAE): cuando el DC afecta básicamente a la memoria (DCL amnésico). – Deterioro cognitivo asociado a la edad (DECAE): cuando además hay afectación de las actividades complejas de la vida diaria. • Demencia: se caracteriza por un deterioro global de las funciones cognitivas, de carácter orgánico, adquirido y, por lo general, crónico y progresivo, sin alteración del nivel de conciencia. Para realizar el diagnóstico de la demencia, el DC debe ser de intensidad suficiente tal que interfiera en las actividades familiares, sociales u ocupacionales habituales del individuo y posteriormente las necesarias para el autocuidado y la supervivencia. En la demencia se pueden presentar también alteraciones neuroconductuales (personalidad, comportamiento) a cuya presencia se les da el mismo valor diagnóstico que a las alteraciones cognitivas. Los síntomas cognitivos aparecen siempre, pudiendo progresar de forma continuada. Los síntomas conductuales y psicológicos son múltiples aunque no siempre están presentes, y lo hacen en diferentes momentos de la enfermedad pudiendo ser limitados en el tiempo. Epidemiología básica La prevalencia de la demencia en mayores de 64 años es de aproximadamente el 5 % y la incidencia de demencia, del 1 % anual. En mayores de 80 años la prevalencia de demencia aumenta al 20-30 %. El 12-15 % de los pacientes diagnosticados de DCL desarrollan anualmente una demencia. En la demencia, la enfermedad de Alzheimer (EA) supone un 35-56 % del total, la demencia vascular, del 7-20 % y la asociación entre ambas, del 6-15 %. Por sus especiales características, también son destacables la demencia con cuerpos de Lewy y la demencia frontotemporal. Las demencias realmente reversibles son menos del 1 %. ¿Qué lo puede ocasionar? Causas La demencia es un síndrome que puede estar ocasionado por múltiples enfermedades (tabla 1). Los factores de riesgo más aceptados actualmente para la EA son: ApoE, A2macroglobulina y el receptor de ambas proteínas: LRP (low density lipoprotein-related protein), y el gen confirmado: alelo ε4 del gen de la apolipoproteína E (APOE) del cromosoma 19. Son factores protectores de deterioro cognitivo: evitar el aislamiento, el entrenamiento cognitivo, el control de la hipertensión arterial (HTA), la dieta mediterránea, evitar los psicofármacos y posiblemente, el ejercicio físico. La edad, la HTA, la enfermedad cerebrovascular, el tabaquismo, la diabetes mellitus, la cardiopatía y los antecedentes familiares de demencia son factores de riesgo para la demencia vascular y aumentan las posibilidades de aparición de EA en el deterioro cognitivo ligero. La diabetes es un factor de riesgo probable tanto para EA como para la demencia vascular. Parece adecuado abordar los factores de riesgo como objetivo prioritario. Diagnóstico diferencial En la práctica clínica, el diagnóstico diferencial más complejo de la demencia se debe realizar con la depresión. Algunos autores creen que debería sospecharse una depresión ante síntomas de tristeza con llanto, pensamientos de inutilidad o falta de esperanza. La opinión del cuidador puede ayudar a matizar estos cambios en fases iniciales e intermedias de la enfermedad. También deben considerarse las diferencias entre el DCL (AMAE, DECAE) y la demencia (tabla 2). ¿Qué tenemos que hacer? Detección Los síntomas característicos en una fase inicial se detallan en la tabla 3. No hay evidencias para recomendar la detección sistemática en personas asintomáticas. 469 5.12 Pérdida de la memoria y demencia Tabla 1. Clasificación etiológica de las demencias Demencias degenerativas Enfermedades en que la demencia es la manifestación principal: Enfermedad de Alzheimer Enfermedad por cuerpos de Lewy Demencia frontotemporal Enfermedades en las que la demencia puede formar parte del cuadro clínico: Enfermedad de Parkinson Otras: enfermedad de Huntington, degeneración corticobasal, parálisis supranuclear progresiva, degeneraciones cerebrales focales con demencia Otras enfermedades degenerativas que, excepcionalmente, pueden presentarse como demencia en la edad adulta: Leucodistrofias del adulto Demencias secundarias Vasculares Isquémicas (infarto, multiinfarto o enfermedad de pequeños vasos) Isquémico-hipóxicas Hemorrágicas (subaracnoidea, hematoma subdural crónico) Procesos expansivos: tumores cerebrales primarios, tumores metastásicos, síndromes paraneoplásicos Alteración dinámica del líquido cefalorraquídeo: hidrocefalia de presión normal Infecciosas: meningoencefalitis, neurolúes, criptococosis, complejo demencia-SIDA, abscesos cerebrales, etc. Demencia por priones: enfermedad de Creutzfeldt-Jakob y variante Endocrino-metabólicas: hipotiroidismo/hipertiroidismo, hipoparatiroidismo/hiperparatiroidismo, enfermedad de Addison, enfermedad de Cushing, encefalopatía urémica, hepática, enfermedad de Wilson, etc. Carenciales: déficit de vitamina B12, ácido fólico, niacina, etc. Tóxicas y medicamentosas: alcohólica, intoxicación por metales pesados u otros tóxicos. Litio, metotrexato y otros fármacos Traumáticas: demencia postraumática, demencia pugilística Enfermedades psiquiátricas: demencia por depresión u otras enfermedades psiquiátricas crónicas Demencias mixtas Enfermedad de Alzheimer con enfermedad cerebrovascular asociada Otras demencias combinadas (enfermedad de Alzheimer con cuerpos de Lewy) Tabla 2. Diagnóstico diferencial básico entre AMAE, DECAE y demencia Características DC leve Demencia AMAE DECAE Clínica: Alteración de la memoria Problemas en actividades complejas Otros trastornos cognitivos: atención, concentración, lenguaje, etc. Sí No No Sí Sí No o discreta Sí Sí Sí Alteración laboral y/o social significativas No No Sí > 24 艑 24 < 24 2 3 ⱖ4 Test: MEC Estadio evolutivo: GDS de Reisberg AMAE: alteración de memoria asociada a la edad; DC: deterioro cognitivo; DECAE: deterioro cognitivo asociado a la edad; GDS: escala global del deterioro de Reisberg; MEC: miniexamen cognitivo de Lobo. Anamnesis Se realizará en la consulta programada con un familiar o cuidador próximo para completar y confrontar la información obtenida del paciente (tabla 3). Se valorará: • La comunicación verbal y no verbal del paciente. • Las capacidades cognitivas: memoria, capacidad para llevar a cabo actividades complejas (planificar, organizar, secuenciar y abstraer), orientación temporoespa- 470 cial, lenguaje, instrumentales, reconocimiento o identificación de objetos. • La repercusión sobre las actividades de la vida diaria (laborales y/o sociales) que comportan los déficit cognitivos del paciente. Si efectivamente se sospecha un deterioro cognoscitivo, se interrogará además sobre los siguientes aspectos: • Antecedentes familiares: demencia, enfermedades psiquiátricas y cerebrovasculares. • Antecedentes personales: traumatismo craneal, 5.12 Pérdida de la memoria y demencia Tabla 3. Anamnesis básica ante el deterioro cognitivo Síntomas cognitivos ¿Olvida fechas y datos importantes y no los recuerda más tarde? ¿Pregunta varias veces la misma cosa? ¿Coloca las cosas en lugares inadecuados? Cuando pierde algo, ¿es incapaz de volver sobre sus pasos para buscar la pista de dónde lo ha dejado? ¿Le cuesta seguir las conversaciones? ¿Pierde el hilo de lo que va contando cuando habla y se repite constantemente? ¿Le cuesta encontrar palabras que usa normalmente? ¿Se pierde en lugares conocidos? ¿Le cuesta entender los planes futuros? Cuando se olvida del día en que estamos, ¿le es imposible recordarlo más tarde? ¿Ha perdido iniciativa? ¿Ha dejado de aprender cosas nuevas o hace siempre lo mismo (p. ej., la misma receta)? ¿Tiene problemas para elegir la ropa de cada día o para planificar la compra? Síntomas psicológicos ¿Se siente deprimido? ¿Ve o escucha cosas que los demás no ven? ¿Se siente últimamente suspicaz, temeroso, deprimido, nervioso, confuso, más irritable al cambiar su rutina? ¿En el fondo tiene la impresión de que le quieren mal? Síntomas conductuales ¿Presenta: vagabundeo, inquietud o agitación psicomotriz, desmotivación, trastornos del sueño (más frecuentemente hipersomnia), alteraciones de la conducta alimentaria, comportamientos repetitivos, agresividad, conductas sexuales inapropiadas? Síntomas funcionales ¿Tiene dificultad para realizar tareas complejas? ¿Pide ayuda para planificar actividades inusuales: cena de Navidad, las vacaciones, la declaración de la renta? ¿Tiene dificultades para realizar tareas instrumentales habituales: comida, dinero, teléfono, medicamentos, etc.? ¿Presenta desadaptación sociolaboral? neoplasias, gastrectomía, psiquiátricos, endocrinometabólicos, conductas sexuales de riesgo, consumo de alcohol, tóxicos laborales, factores de riesgo cardiovasculares, que pueden llevar a pensar en demencias secundarias. También es importante conocer el nivel educativo, cultural y las capacidades y actividades (laborales, familiares y sociales) habituales del paciente. Fármacos: anticoagulantes (por el riesgo de hemorragia cerebral) y otros que pueden producir efectos secundarios sobre la memoria, especialmente los anticolinérgicos, antidepresivos tricíclicos, benzodiazepinas, opioides y neurolépticos. • Enfermedad actual: inicio de los síntomas, progresión, conciencia de enfermedad, síntomas asociados (incontinencia de esfínteres, alteraciones de la marcha, temblores, mioclonías, cefalea reciente, alucinaciones, etc.), síntomas psicológicos y alteraciones del comportamiento. Ante un paciente con pérdida de memoria o sospe- cha de demencia, considerar las pautas de actuación que se detallan en la figura 1. Pruebas psicométricas Los tests psicométricos (tema 21.41) completan la exploración mental y funcional del paciente, y objetivan o cuantifican los déficit. Los más útiles en AP son los siguientes: • Test del informador (TIN). Versión de 17 preguntas. Es un test cognitivo-funcional que cumplimenta un familiar o persona próxima, habitualmente en su domicilio. Requiere una breve explicación. En los estadios iniciales de la enfermedad es más sensible que los test cognitivos efectuados al paciente. Una puntuación > 57 puntos sugiere DC. • Miniexamen cognoscitivo (MEC-35). Es el test básico para utilizar en personas alfabetizadas. Tiempo de administración: 10-15 minutos. Una puntuación < 24 puntos sugiere DC. • Set Test de Isaacs. De elección en personas analfabetas o con deterioro sensorial importante. Tiempo de administración: 5 minutos. Una puntuación < 27 puntos sugiere DC. • Escala de depresión geriátrica de Yesavage abreviada (tema 6.2). Pueden ser útiles ante la sospecha de un trastorno depresivo en el anciano. Exploración física General • Talla, peso, presión arterial. Piel y mucosas. • Palpación del tiroides (bocio). • Auscultación cardíaca, carotídea y pulmonar. Pulsos periféricos. • Abdomen. • Valorar: temperatura y tacto rectal (si se sospecha impactación fecal). Neurológica • Nivel de conciencia. • Pares craneales. • Fuerza, sensibilidad, reflejos osteotendinosos y signos meníngeos. • Signos extrapiramidales (temblor, mioclonías, rigidez, reflejos de actitud y postura) (tema 5.6). • Alteraciones en la marcha (tema 5.10). En los casos en que no exista deterioro cognitivo o éste sea leve y la exploración física anodina, se deberá realizar un seguimiento periódico cada 3-6 meses. Si se confirma el DC, se realizarán las exploraciones complementarias para detectar demencias secundarias o presencia de comorbilidad asociada. Exploraciones complementarias Iniciales • Analítica: hemograma, bioquímica (glucemia, función renal y hepática, sodio, potasio y calcio, colesterol, albúmina, hormona tiroestimulante [TSH] y T4L, vitamina B12). 471 5.12 Pérdida de la memoria y demencia Sospecha de deterioro cognitivo Anamnesis del paciente y de la familia Test psicométricos breves Exploración física y neurológica Deterioro cognitivo No Sí Alteración del nivel de consciencia No Sí Depresión (tema 6.2) Comorbilidad Sd. confusional (tema 5.14) No Afectación de la vida del paciente Deterioro cognitivo leve Demencia Control a los 3-6 meses No Progresión Sí Diagnóstico etiológico y evolutivo Sí Figura 1. Algoritmo diagnóstico de las demencias en Atención Primaria. Si hay sospecha clínica que lo justifique: serología luética, virus de la inmunodeficiencia humana (VIH), ácido fólico, perfil básico de orina y si procede, niveles de fármacos o tóxicos. En la práctica clínica, las pruebas analíticas descartan más frecuentemente comorbilidad que verdaderas demencias reversibles. • Tomografía computarizada (TC) craneal. No es una prueba concluyente en el diagnóstico de las demencias primarias y su objetivo es descartar causas secundarias (hematoma subdural crónico, tumores, hidrocefalia, lesiones isquémicas, etc.). Es cuestionable su realización en todos los casos de demencia. No estaría indicada en pacientes con demencia grave, con más de 3 años de evolución, de inicio progresivo, sin antecedentes de traumatismo craneal, ni presencia de signos o síntomas de focalidad neurológica. • Electrocardiograma (ECG) y radiografía de tórax. Si existen factores de riesgo cardiovasculares o arritmia. Posteriores • El resto de exploraciones son específicas, generalmente del medio hospitalario: resonancia magnética (RM) craneal, electroencefalograma (EEG), punción lumbar, 472 pruebas de neuroimagen funcional (SPECT, PET) y los marcadores genéticos. Diagnóstico, valoración del estado funcional y estadificación El diagnóstico de las demencias es clínico: • Diagnóstico sindrómico de demencia: los criterios más utilizados son los del Manual diagnóstico y estadístico de los trastornos mentales (DSM-IV-TR) (tabla 4) o los de la Clasificación Estadística Internacional de Enfermedades y otros Problemas de Salud (CIE-10). A menudo, solamente el seguimiento del paciente y una correcta valoración clínica proporcionarán el diagnóstico definitivo. • Diagnóstico etiológico: también es clínico: – Para el diagnóstico de la EA se utilizan preferentemente los criterios del DSM-IV-TR o del National Institute of Neurological and Communicative Disorders and the Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) o de la CIE-10. – Para la demencia vascular, los DSM-IV-TR o los del National Institute of Neurologic Disorders and Stroke-Association Internationale pour la Recherche et l’Enseignement en Neurosciences (NINDS-AIREN) o de la CIE-10. También puede ser de utilidad la escala de Hachinski. 5.12 Pérdida de la memoria y demencia ¿Qué propuesta haremos? Tabla 4. Criterios de demencia según el DSM IV-TR A. La presencia de los múltiples déficit cognoscitivos se manifiesta por: 1. Deterioro de la memoria (deterioro de la capacidad para aprender nueva información o recordar información aprendida previamente) 2. Una (o más) de las siguientes alteraciones cognoscitivas: a) Afasia (alteración del lenguaje) b) Apraxia (deterioro de la capacidad para llevar a cabo actividades motoras, a pesar de que la función motora está intacta) c) Agnosia (fallo en el reconocimiento o identificación de objetos, a pesar de que la función sensorial está intacta) d) Alteración de la ejecución (p. ej., planificación, organización, secuenciación y abstracción) B. Los déficit cognoscitivos en cada uno de los criterios A1 y A2 provocan un deterioro significativo de la actividad laboral o social y representan una merma importante del nivel previo de actividad C. Los déficit no aparecen exclusivamente en el transcurso de un delírium – Para la demencia con cuerpos de Lewy difusos, los criterios del grupo de Newcastle (McKeith, et al), validados en 1996. – Para las demencias frontotemporales, los criterios de Lund y Manchester adaptados o los del NINDS. • Valoración del estado funcional: para las actividades instrumentales de la vida diaria, las primeras que se alteran, se utilizan las escalas de Lawton y Brody o el cuestionario de actividad funcional de Pfeffer, de 1982. Para las actividades básicas de la vida diaria, el índice de Barthel o de Katz. Suelen estar preservadas en las fases iniciales de la demencia. • Estadificación de la EA: se utilizan conjuntamente la Global Deterioration Scale (GDS), de Reisberg (tabla 5) y la escala auxiliar Functional Assessment Stage (FAST). Tiene utilidad diagnóstica, de seguimiento, pronóstica y terapéutica, y es una herramienta importante para el diseño del plan de cuidados. Tratamiento Paciente No farmacológico Una vez establecido el diagnóstico etiológico de demencia, se efectuará una evaluación multidimensional e interdisciplinaria (trabajador social, personal de enfermería y médico de familia) para elaborar conjuntamente el Plan de Actuación Individualizado o Valoración Geriátrica Integral, que considera: • Los déficit y problemas psíquicos, físicos, sensoriales, emocionales, familiares, estructurales (del domicilio), comunitarios y sociosanitarios. • Define la relación de problemas, basados en las características y la realidad personal (estadio clínico), familiar, social y sanitaria. • Propone posibles actividades, el calendario y el personal sanitario responsable de su seguimiento, junto con el cuidador principal y la familia, procurando como objetivos prioritarios, el bienestar, la seguridad y la dignidad del paciente y su familia. Es importante que el paciente sea evaluado periódicamente (cada 3-6 meses en los estadios iniciales) por todo el equipo interdisciplinario. Normas específicas Actuaciones previas importantes: • Identificar al cuidador principal. • Escuchar e informar al paciente, que debería recibir toda la información necesaria para entender lo que le pasa y para que pueda colaborar con las actuaciones que se propongan. • Escuchar e informar al cuidador principal. • Escuchar e informar a la familia. • Valorar la realización de una visita domiciliaria inicial. El tratamiento no farmacológico se basa en: – La posibilidad de modificar las alteraciones de conducta mediante consejos o actuaciones ambientales, relacionales y del comportamiento. Tabla 5. Escala Global del Deterioro cognitivo. Versión abreviada Estadio Déficit cognitivo Correspondencia MEC Características clínicas 1 Inexistente 30-35 Normal 2 Muy ligero 25-30 DCL tipo AMAE 3 Ligero 20-27 DCL tipo DECAE 4 Moderado 16-23 Demencia inicial 5 Moderadamente grave 10-19 Demencia con déficit de AVD instrumentales 6 Grave 0-12 Demencia con déficit de AVD básicas 7 Muy grave 0 Demencia. Síndrome de afasia, apraxia, agnosia AMAE: alteración de la memoria asociada a la edad; AVD: actividades de la vida diaria; DCL: deterioro cognitivo leve; DECAE: deterioro cognitivo asociado a la edad; MEC: miniexamen cognoscitivo. 473 5.12 Pérdida de la memoria y demencia – El conjunto de actividades de estimulación cognitiva, sensorial, terapia de orientación temporoespacial, musicoterapia, canto, estimulación física, reminiscencia (recuperar recuerdos personales antiguos), validación, y en especial pueden recomendarse las terapias ocupacionales dirigidas al paciente; cada una de ellas adaptada a la fase en la que se encuentre. Podrán realizarse en el propio domicilio o en un centro de día. – Fases iniciales: debe asegurarse una información correcta y gradual. Es fundamental la adaptación del paciente en función de sus actividades habituales, relaciones, soporte y recursos sociales. La terapia ocupacional, el ejercicio físico moderado y el paseo son terapias de gran utilidad. El paciente debe salir de casa acompañado y documentado. – Fases moderadas: se valorará la retirada del carné de conducir o de sus responsabilidades profesionales, etc. Se mantendrá la terapia ocupacional, según sus intereses y capacidades. Se efectuarán las modificaciones en el domicilio para asegurar una mejor orientación (carteles, calendarios, etc.) y evitar accidentes y caídas (quitar alfombras, iluminar correctamente los pasillos y escaleras, control de los medicamentos, etc.). Los trastornos del comportamiento no son inherentes a la demencia, puesto que muchos pacientes con demencia no los presentan. Ante un comportamiento anómalo siempre debe buscarse una causa y, si es posible, modificarla. En estos casos, los consejos generales deben asegurar que no haya riesgo físico para el paciente, mantener la calma, tratar al paciente con cariño y respeto, proponer otra tarea, no discutir, ni regañarle, y reforzar/premiar las actitudes positivas. Debe fomentarse siempre la máxima autonomía e independencia posible durante todo el proceso. Es muy importante mantener una rutina y unos horarios fijos. – Fases avanzadas: aparecen los problemas de las actividades básicas de la vida diaria (puntuación 6 en la escala GDS). Es prioritario atender las necesidades de higiene, asegurar una nutrición e hidratación adecuadas, vestimenta, eliminación (urinaria y fecal), movilidad, etc. Farmacológico 1.Los inhibidores de la acetilcolinesterasa (IACE) están autorizados para la EA leve o moderada. Han demostrado mejoras en la puntuación de algunos test (ADAS-Cog, ADCS-CGIC, ADCS-ADL, MMSE, etc.) y en algún ítem de test de actividades de la vida diaria no validados y no practicables en la práctica clínica en AP, pero lamentablemente no frenan la enfermedad. Algunos autores afirman que las supuestas mejoras en la puntuación de los test utilizados o los parámetros de las actividades de la vida diaria, no alcanzan un nivel clínicamente significativo. Por ello, algunos países se plantean su retirada de la financiación pública y por ejemplo en Alemania su empleo es muy restringido. 474 No han demostrado reducir los ingresos en una institución sanitaria, los síntomas de comportamiento, psicológicos, la psicopatología del cuidador, el coste sanitario, ni la mortalidad. • Donepezilo es el fármaco sobre el que hay un mayor número de ensayos clínicos y comparativamente sus efectos secundarios son menores. Dosis de 5-10 mg/día v.o. por la noche. Los resultados de eficacia son muy escasos. El porcentaje de pacientes que mejoran con el tratamiento es del 7-20 %. Además, hay que tener precaución al administrarlo a pacientes con antecedentes de epilepsia, asma/enfermedad pulmonar obstructiva crónica (EPOC), úlcera gástrica activa, enfermedad del nódulo sinusal, bloqueos de tercer grado y en el embarazo. • Rivastigmina y galantamina tienen un perfil de seguridad menos favorable. La rivastigmina en parches de 9,5 mg/24 h ha mostrado una eficacia similar a las cápsulas. La formulación en parches produce menos náuseas y vómitos que las cápsulas; sin embargo, hay que tener en cuenta que la rivastigmina parece inducir una mayor incidencia de efectos adversos que otros fármacos de su grupo (donepezilo). Una proporción similar de pacientes abandona por los efectos adversos causados por los parches y las cápsulas. Por tanto, es un medicamento más en un grupo farmacológico que, de por sí, muestra una escasa eficiencia. En enero de 2005, la Agencia Española del Medicamento notificó que se había observado un incremento de la mortalidad en pacientes con deterioro cognitivo leve (datos preliminares de dos ensayos clínicos) por el uso de galantamina, fundamentalmente de origen cardiovascular. Deberían considerarse las implicaciones económicas, teniendo en cuenta sus modestos beneficios clínicos. Hay pruebas de que el tratamiento del DCL no es efectivo, y de que la retirada del donepezilo, comparado con placebo en condiciones doble ciego, no provoca efectos adversos. 2.Antagonista de los receptores NMDA: la memantina es un derivado de la amantadina autorizada en España para el tratamiento de los pacientes con EA en fases moderadamente graves a graves. Ningún estudio ha demostrado que el tratamiento aporte ventajas clínicas relevantes, ni un beneficio adicional de la asociación de memantina con anticolinesterásicos en pacientes con EA moderada. El único ensayo clínico publicado hasta el momento presenta limitaciones metodológicas importantes. El consejo asesor sobre el tratamiento farmacológico de la EA propone que debería suspenderse el tratamiento farmacológico en caso de empeoramiento cognitivo del paciente, reacciones adversas graves, presencia de enfermedades concomitantes que contraindiquen su uso o ausencia de respuesta a éste, a las 18 semanas de su instauración. 5.12 Pérdida de la memoria y demencia 3.Otros fármacos utilizados para el tratamiento paliativo de las demencias: nootrópicos y neuroprotectores (citicolina, piracetam), calcioantagonistas (nimodipino), estrógenos (estradiol), antioxidantes (vitamina E, selegilina) o Ginkgo biloba, NGF (nerve growth factor), dihidroergocristina; faltan estudios que demuestren su eficacia en este síndrome. Los profesionales sanitarios tienen el deber de informar al enfermo y a la familia, y muchas veces corregir la información recibida, en cuanto a las expectativas que puedan tener acerca de la eficacia del tratamiento farmacológico. En cuanto a la demencia vascular, resulta razonable el control estricto de los factores de riesgo cardiovascular y la administración diaria de 100 mg/día de ácido acetilsalicílico (AAS), aunque en pacientes con demencia vascular ni el control tensional ni la administración de AAS ha conseguido revertir o mejorar el cuadro. 4.Tratamiento farmacológico sintomático y de los trastornos psicoconductuales en las demencias (tema 5.13) únicamente se iniciará después del fracaso de las medidas no farmacológicas o por urgencia del proceso intercurrente. • Se propondrá la retirada de todos los fármacos que no sean imprescindibles. Se deben valorar siempre las interacciones farmacológicas y anotar los psicofármacos que se administren al paciente, su respuesta y si han producido algún efecto secundario. E informar a la familia que con frecuencia es preciso ensayar más de un fármaco y dosis. • Existen algunos problemas como la rigidez o la espasticidad, frecuentes en fases avanzadas, cuyo tratamiento principal es preventivo. Es útil la rehabilitación y también los ejercicios diarios, si es preciso en el domicilio del paciente. Hay que procurar retirar los neurolépticos. La mejoría puede tardar hasta 1 mes después de su retirada. Como sustitutos se puede plantear el uso de un antidepresivo sedante (fluvoxamina) o una benzodiazepina. El baclofeno no causa dependencia, aunque no es efectivo en la rigidez causada por enfermedad extrapiramidal o cerebrovascular. • Es fundamental que exista una buena accesibilidad de los profesionales sanitarios. • Conviene utilizar dosis iniciales bajas, efectuar incrementos en dosis pequeñas y usar la dosis efectiva más baja. No dejar nunca instaurados fármacos sin evaluar periódicamente (cada 4-8 semanas) su retirada. Atención del cuidador El papel del cuidador es fundamental dado que es el informador directo, el proveedor de cuidados, el organizador de la actividad en el domicilio y, posteriormente, el tutor del paciente. La atención del cuidador precisa también de un abordaje multidisciplinar. Los profesionales del centro de salud deberán establecer una buena relación y conocer sus creencias y conocimientos sobre la enfermedad, dudas, miedos, habilidades para el cuidado, sentimientos y el apoyo familiar y social previsible. Con frecuencia, antes de llegar al diagnóstico de sobrecarga o claudicación del cuidador, los profesionales del centro de salud han recibido múltiples visitas de trastornos que podrían catalogarse de tipo psicosomático. Es conveniente entregar por escrito los consejos básicos para el autocuidado (Guía práctica de la salud [GPS], p. 233). Deberá recibir también información sobre las asociaciones de familiares próximas y de las actividades grupales de las que actualmente existe amplia experiencia en AP. Familia La atención y valoración de la familia son importantes para su propio confort y para una mejor calidad de vida del paciente y del cuidador principal. Se informará de los recursos sociosanitarios para los pacientes y sus familiares (tabla 6). También, que la incapacitación es un acto civil que puede efectuarse desde un simple traspaso de poderes (más o menos amplio) ante notario en las fases iniciales de la enfermedad o bien mediante la interposición de la demanda de incapacitación con representación de abogado y procurador ante el Ministerio fiscal, normalmente en fases más avanzadas de la demencia. Comunidad El centro de salud debe conocer y mantener una relación fluida con la red de asociaciones y servicios sociosanitarios públicos y privados de la comunidad para poder dirigir al paciente, al cuidador principal y a la familia en función de las necesidades de cada fase. Informar a la comunidad del interés de disponer de un documento claro de instrucciones previas (voluntades anticipadas), para ayudar a que su cuidado sea el más ajustado a su voluntad. Derivación • Neurólogo: se aconseja consultar a aquellos pacientes menores de 65 años o cuando existan dudas razonables de que se trate de una EA: inicio brusco y/o curso fluctuante y/o manifestaciones atípicas (convulsiones, focalidad neurológica reciente, alteración precoz de la marcha, alucinaciones en fases no avanzadas, presencia de temblor, predominio de los trastornos conductuales o de la personalidad, etc.). Para realizar exploraciones complementarias o tratamientos farmacológicos que no sean accesibles al médico de familia. Cuando esté indicado el consejo genético (familiares de EA de inicio precoz –cuando hay dos o más personas en la familia con demencia iniciada antes de los 65 años– o enfermedad de Huntington). Solicitar los tratamientos farmacológicos no accesibles en Atención Primaria de Salud (APS). • Salud mental: ante el debut de un trastorno de personalidad o conducta con TC cerebral normal, a partir de la quinta década de la vida, se debería pensar en una posible demencia de tipo frontal. Una depresión resistente al tratamiento habitual. Síntomas de difícil control relacionados con la demencia (insomnio, irritabilidad o vagabundeo rebeldes al tratamiento, etc.). 475 5.12 Pérdida de la memoria y demencia Tabla 6. Recursos sociosanitarios para los pacientes y familiares Documentos legales Servicios Documento de instrucciones previas Pensión por jubilación Programas y servicios comunitarios: Servicios sanitarios: APS y especializada Servicios sociales: información y asesoría técnica Asociaciones de familiares Otras ONG y de voluntariado PNC de jubilación o de invalidez (M/D ⱖ 65 %) Grupos informativos o de autoayuda para cuidadores y familiares Declaración de minusvalía/discapacidad Servicios de atención diurna: Hogares y clubes de jubilados Centros de día, centros de noche Centros de atención nocturna ILT por enfermedad ILP Incapacitación civil Ayudas económicas (p. ej., Programa vivir en familia, para la adaptación de la vivienda) Ley de conciliación y vida laboral Ley de la dependencia Servicios domiciliarios: Servicio de ATDOM de APS Teleasistencia, telealarma, sistema inteligente de monitorización de alertas personales (SIMAP) Servicios de ayuda a domicilio Equipos de soporte sociosanitario de atención domiciliaria Servicios institucionales Servicios sanitarios: atención hospitalaria Servicios sociales institucionales: Estancias temporales Unidades de vida y sistemas de alojamiento alternativo Unidades de psicogeriatría en residencia o centros sociosanitarios APS: atención primaria de salud; ATDOM: atención domiciliaria; ILP: incapacidad laboral permanente; ILT: incapacidad laboral transitoria; M/D: minusvalía/ discapacidad; ONG: organización no gubernamental; PNC: pensión no contributiva. Consejos prácticos • No es posible abordar correctamente las demencias sin una actuación multidisciplinaria (médico de familia, enfermería y trabajador social). • La comunicación no verbal del paciente, como el girar la cabeza hacia su familiar durante la anamnesis, en actitud de pedirle ayuda, puede ser un signo de deterioro cognitivo. • Las personas mayores padecen mucha morbilidad encubierta no diagnosticada y que, si no se aborda, puede condicionar de forma importante la calidad de vida del paciente. • Los déficit de memoria pueden agravarse si coexiste depresión, problemas sensoriales y/o tratamientos farmacológicos sedativos. • La aparición de un extrapiramidalismo exagerado y/o alucinaciones en las fases iniciales de una demencia han de alertar sobre una demencia con cuerpos de Lewy difusos. • No hay diferenciación clínica precisa entre envejecimiento normal, deterioro cognitivo y demencia. • Hay que considerar la posibilidad de que el origen de una demencia sea debido a varias causas asociadas. La demencia mixta más frecuente es la degenerativa asociada a la vascular. • Los síntomas psicológicos en la demencia suelen presentarse de forma múltiple, recurrente y oscilante, con tendencia a desaparecer espontáneamente, por lo que hay que valorar de forma periódica la retirada de los medicamentos prescritos. • Las descompensaciones agudas deben siempre ser abordadas con carácter urgente, averiguar qué las ha desencadenado y no solamente intentar controlarlas con fármacos. 476 Errores más frecuentes • Creer que el deterioro cognitivo es normal en personas mayores. • Querer efectuar un diagnóstico rápido. Para un correcto diagnóstico es importante observar la evolución del paciente. Además, hay pruebas de que el tratamiento precoz con inhibidores de la acetilcolinesterasa (IACE) en estadio GDS 3 no es de utilidad. • Olvidar los antecedentes del paciente, relegándolos a un segundo lugar ante el diagnóstico de demencia y en ocasiones, condicionando la calidad de vida del paciente. • Pensar que los rasgos pesimistas del paciente son normales para su edad, o debidos a la demencia, cuando pueden encubrir una depresión grave. • Ofrecer poca o demasiada información al inicio, tras el diagnóstico, lo que dificulta que el paciente y la familia acepten y elaboren correctamente el proceso. • No identificar el cuidador principal. • Introducir medicamentos sin antes comprobar el resultado de las medidas no farmacológicas. • No revisar los medicamentos que toma el paciente como posible causa de los síntomas que describe. • Considerar que una demencia es sinónimo de derivación. 5.12 Pérdida de la memoria y demencia • Geriatría: pacientes mayores con pluripatología de difícil manejo. • Otras especialidades: rehabilitación (déficit motor o trastornos del habla), traumatología (valorar la conveniencia de emplear ortesis y mejorar la deambulación), oftalmología (déficit visuales), otorrinolaringología (déficit auditivos), odontología (falta de piezas dentarias), con el objetivo de mejorar su calidad de vida. • Trabajador social (para valoración de la ley de la dependencia, recursos sociosanitarios, etc.), asociaciones de voluntarios, etc. ¿Se ha resuelto del todo? Bibliografía recomendada 1. Grupo de Trabajo de Demencias de la Sociedad Española de Medicina Familia y Comunitaria. Demencias desde la Atención Primaria. Barcelona: semFYC; 2005. 2. González Moneo MJ. ¿Son útiles los fármacos para el Alzheimer? FMC. 2008;(15)5:332-4. 3. Guía de tests y criterios diagnósticos en Atención Primaria. semFYC. Barcelona: semFYC; 2007. 4. Dementia: A NICE-SCIE Guideline on supporting people with dementia and their carers in health and social care. National clinical practice guideline 42. National Collaborating Centre for Mental Health. Social Care Institute for Excellence (SCIE), National Institute for Health and Clinical Excellence (NICE). London: Pronóstico y seguimiento La expectativa de vida varía en función de la causa de la demencia, la edad, el momento del diagnóstico, la patología asociada y la atención recibida. La media en la EA es de 4-10 años, y menor en la demencia vascular. Aldenn Press 2007. Disponible en: http://www.nice.org.uk/ nicemedia/pdf/CG42Dementiafinal.pdf 5. Boada M. Tárraga LL, Morera A, Guitart M, Acosta C. Alzheimer: vivir cuando dos y dos ya no son cuatro. 2.ª ed. Barcelona: Vienas Ediciones; 2008. 6. Scottish Intercollegiate Guidelines Network (SIGN). Management of patients with dementia. A national clinical guideline. ¿Podemos hacer alguna cosa más? Educación sanitaria Individual Tanto del paciente como del cuidador principal y la familia. Se puede realizar en cualquier visita que efectúe algún miembro de la familia. Se aconseja también entregar hojas informativas (GPS, p. 34-36). Edinburgh (Scotland): Scottish Intercollegiate Guidelines Network (SIGN); 2006 Feb. 53 p. (SIGN publication; no. 86). [183 references]. Disponible en: http://www.sign.ac.uk/pdf/sign86.pdf 7. Grupo de Trabajo de Atención al Mayor de la Sociedad Española de Medicina de Familia y Comunitaria. Atención a las personas mayores desde la Atención Primaria. Barcelona: semFYC; 2004. 8. Selmes J, Antoine M. Vivir con la enfermedad de Alzheimer u otra demencia. Madrid: Meditor; 2003. 9. Ray WA, Chung CP, Murray KT, Hall K, Stein CM. Atypical Antipsychotic Drugs and the Risk of Sudden Cardiac Death. N Engl J Grupal Existen dos tipos de actividad grupal, las sesiones informativas dirigidas a cuidadores y familiares, y los grupos de ayuda mutua o autoayuda. Ambas pueden efectuarse y hay experiencia en AP. Los estudios más conocidos son el Proyecto Elois (Madrid) y Cuida’l (Cataluña). Med. 2009;360:225-35. 10. Golden J, Lawlor B. Treatment of dementia in the community. BMJ. 2006;333(7580):1184-5. 477 5.13 Trastornos psicoconductuales en las demencias N. Ruiz Hombrebueno ¿De qué hablamos? Introducción Los síntomas psicopatológicos y de alteración de la conducta en la demencia son un aspecto de gran importancia por la elevada prevalencia en la comunidad, su asociación a una más rápida progresión de la demencia y la alteración en la calidad de vida que genera tanto para el paciente como para el cuidador principal y su familia. Actualmente, el abordaje terapéutico pasa por un tratamiento no farmacológico que consiste en terapia conductual-ambiental y un posible tratamiento farmacológico que, consiguiendo un aceptable control de los síntomas, presente efectos secundarios asumibles. No está demostrado que la aparición de estos síntomas psicoconductuales sea un marcador de mayor mortalidad per se, pero lo cierto es que el padecimiento de estos trastornos determina una mayor morbilidad por las complicaciones a las que pueden dar lugar como, por ejemplo, las caídas. Definición En la Conferencia de Consenso de la International Psychogeriatric Association (IPA), de 1996, se decidió, reemplazar el término trastornos conductuales por el de síntomas psicológicos y conductuales de las demencias (SCPD), definidos como un conjunto de «síntomas de alteración en la percepción, en el contenido del pensamiento, el humor o el comportamiento que a menudo se presentan en pacientes con demencia». Conceptos iniciales • Agitación: es definida por Cohen-Mansfield como «la actividad verbal o motora inadecuada asociada a una sensación de tensión interna, que no parece resultado directo de las necesidades básicas del paciente (alimentación, aseo, etc.) o de un estado confusional que le afecte en ese momento». En la demencia, la agitación puede comprender un amplio abanico expresivo, bien como un síntoma más del propio síndrome, o bien como la manifestación de otro tipo de trastornos en fases más avanzadas de la demencia, o como la forma de expresar un malestar provocado por un proceso intercurrente. • Síndrome depresivo: presente en el 50% de los pacientes con demencia en estadios iniciales. Si se sospecha, se ha de tratar siempre aunque no cumpla criterios estrictos. • Insomnio: se considera que la mitad de los enfermos con enfermedad de Alzheimer (EA) tienen dificultades para conciliar el sueño y dormir adecuadamente, pero no son los únicos trastornos del sueño; también son frecuentes la hipersomnia diurna y las alteraciones conductuales añadidas al insomnio, como los paseos nocturnos y los trastornos de conducta que aparecen o se acentúan al anochecer (sundown syndrome). 478 Realmente las alteraciones conductuales nocturnas suponen un gran estrés en el entorno del paciente, de ahí la importancia de una correcta aproximación a este problema. En cuanto a los mecanismos implicados en los trastornos del sueño de los pacientes con demencia, parece ser que en la EA, y en otras demencias, se produce una alteración profunda de los mecanismos que regulan el sueño (ciclos REM-no REM), lo que se traduce en la alteración del ritmo vigilia-sueño, la fragmentación de la etapa de sueño e insomnio. Pero, además, hay que insistir, como en el caso de la agitación, en que pueden estar implicados múltiples procesos intercurrentes en los pacientes con demencia y trastornos del sueño. Estos factores pueden ser físicos, psíquicos (es muy frecuente el insomnio en la depresión y la ansiedad) o ambientales (horarios de comidas, higiene, etc.). Clasificación La clasificación de estos síntomas es compleja, pues algunos de ellos son difíciles de encuadrar. En la tabla 1 se expone una de las clasificaciones propuestas. Epidemiología La prevalencia de los síntomas neuropsiquiátricos en pacientes con demencia, según diferentes estudios, se expone en la tabla 2. La depresión está presente entre el 15 y el 57 % de los pacientes con demencia avanzada. Se considera que la mitad de los enfermos con EA tienen dificultades para conciliar el sueño y dormir adecuadamente. Los trastornos de la conducta se producen en más del 50 % de los enfermos con demencia en fases avanzadas de la enfermedad. Y el delírium puede aparecer hasta en el 85 % de los pacientes con demencia al final de la vida. Las dificultades de alimentación se desarrollan en todos los enfermos a medida que la demencia progresa, independientemente de su causa. Existe una clasificación en la que se dividen los síntomas psicológicos y los conductuales en función de la frecuencia con que aparecen, así como la repercusión o la tolerabilidad sobre el cuidador (tabla 3). La aparición de estos trastornos en las demencias puede estar relacionada con la fase evolutiva de la enfermeTabla 1. Contenido de los síntomas psicológicos y de conducta en las demencias (SCPD) Alteraciones en el pensamiento y percepción (delirios, falsos reconocimientos, ilusiones, alucinaciones) Alteraciones en la afectividad (depresión, manía, ansiedad) Trastornos en la personalidad y la conducta (apatía, agitación psicomotriz, hiperactividad) Trastornos del sueño (insomnio) Trastornos en la alimentación (anorexia, bulimia) 5.13 Trastornos psicoconductuales en las demencias Tabla 2. Prevalencia de síntomas neuropsiquiátricos en pacientes con demencia, según diferentes estudios Porcentaje (%) Síntomas Ideas delirantes 12-31 8-15 Alucinaciones Agitación 47-85 Disforia 12-62 Ansiedad 24-65 Euforia 0-18 Apatía 47-92 Desinhibición 15-40 Irritabilidad 35-54 Conducta motora aberrante 12-84 Depresión 25-80 Cambios en la personalidad 90-100 Insomnio 40-60 dad y, generalmente, son más frecuentes los síntomas psicopatológicos en las primeras fases y los síntomas conductuales en las fases más avanzadas de la enfermedad, pero siempre teniendo en cuenta que hablándose trata de unos trastornos que aparecen en el curso de un síndrome con múltiples causas. ¿Qué lo puede ocasionar? Causas Entre los factores desencadenantes de SCPD se reconocen los siguientes: yatrogénicos (tabla 4), infecciones (tracto urinario, neumonía), enfermedades (agudas o crónicas con exacerbaciones), traumatismos (fractura de cadera, hematoma subdural, etc.), impacto fecal, retención urinaria, cambios ambientales o depresión. Desde el punto de vista neurobiológico, la agitación se ha relacionado con disminución en la actividad neuroquímica en el lóbulo frontal y temporal, con alteración en el metabolismo de diferentes neurotransmisores como GABA (ácido gammaaminobutírico), histamina, serotonina y acetilcolina (tabla 5). Tabla 3. Clasificación de los síntomas psicológicos y conductuales de la demencia (SCPD), en función de su frecuencia y repercusión Grupo I. Más frecuentes y más exasperantes Ideas delirantes, alucinaciones, ánimo depresivo, insomnio, ansiedad, agresividad física, vagabundeo, inquietud Grupo II. Moderadamente frecuentes y moderadamente exasperantes Falsos reconocimientos, falta de motivación, agitación, conducta culturalmente inapropiada y desinhibición, gritos Grupo III. Menos frecuentes y controlables Llanto, lenguaje malsonante, preguntas repetitivas, seguir a otra persona Tabla 4. Fármacos con potenciales efectos adversos de tipo psiquiátrico Grupo farmacológico Fármaco Efectos AINE AAS, indometacina Delirios, alucinaciones, hipomanía, paranoia Anticonvulsivos Fenitoína Carbamazepina Psicosis Antidepresivos tricíclicos Amitriptilina Nortriptilina Trazodona Delirios, paranoia Delirio paranoide Hiperactividad, paranoia Benzodiazepinas Diazepam Triazolam Delirios Paranoia Cardiovascular Digoxina Propranolol Tiazidas Delirio, depresión, paranoia Depresión, paranoia, alucinaciones, insomnio Depresión Bloqueadores H2 Ranitidina Cimetidina Depresión Agitación, confusión, insomnio, manía Corticosteroides Prednisona Prednisolona Depresión, hipomanía Alucinaciones, paranoia Antihistamínicos Manía, depresión Hormonas tiroideas Psicosis, alucinaciones, ansiedad, delírium AAS: ácido acetilsalicílico; AINE: antiinflamatorios no esteroideos. Modificada de Vilalta Franch J. 479 5.13 Trastornos psicoconductuales en las demencias Tabla 5. Clasificación de los síntomas psicológicos y conductuales de la demencia según la vía neurotransmisora defectuosa Vía Clasificación Acetilcolinérgica Trastorno cognitivo, confusión, delirio Dopaminérgica Manía, delirio, alteraciones sensoperceptivas Acinesia, bradicinesia, bradifrenia, enlentecimiento psicomotor Noradrenérgica Depresión mayor Serotoninérgica Depresión, ansiedad, agitación, inquietud, agresividad, falta de control de los impulsos Glutaminérgica Síntomas psicóticos Diagnóstico diferencial Ante la aparición de un deterioro cognitivo con agitación, en primer lugar hay que descartar la presencia de otras entidades que pueden producirlo (fármacos, delirio, depresión, dolor, inactividad, etc.). En referencia específica a los trastornos del sueño en los pacientes con demencia, hay que considerar siempre la posibilidad de que estos trastornos sean secundarios a una depresión o que formen parte de un proceso psicótico. ¿Qué tenemos que hacer? Anamnesis • Detectar el trastorno predominante: «Regla del ABC»: A: Antecedentes o evento gatillo, B: Behavior (conducta), C: Consecuencia de dicha conducta. El enfoque general más útil es el siguiente: – Identificar el síntoma psicológico o conductual que se pretende corregir: definir claramente el problema en colaboración con el cuidador; es preferible abordar los síntomas de uno en uno. – Recabar información sobre el SCPD: frecuencia, cuándo, dónde, en presencia de quién, etc. – Identificar qué sucede antes y después de la aparición del síntoma; es decir, los desencadenantes y los resultados (puede haber más de un antecedente y de un trastorno conductual). – Precipitantes ambientales: cambios en la rutina, viajes, nuevo cuidador o dificultades en el mismo, mudanzas, muerte de un familiar, sobreestimulación, temperaturas extremas. – Precipitantes médicos: antecedentes cardiovasculares, enfermedad pulmonar obstructiva crónica (EPOC), infección, anemia, trastorno metabólico, dolor, diabetes mellitus mal controlada, desnutrición, deshidratación, retención urinaria, impactación fecal. • Se puede detectar y cuantificar la presencia de trastornos de la conducta, mediante el inventario neuropsiquiátrico de Cummings (NPI) (tabla 6). El NPI es un instrumento habitual en la evaluación de este tipo de trastornos en pacientes con demencia que consiste en una entrevista semiestructurada a un informador fiable del entorno del paciente. La existencia de trastornos conductuales y psicoló- 480 gicos en pacientes con deterioro cognitivo leve (DCL) podría ser un factor predictivo del desarrollo ulterior de demencia. El NPI puede ser una herramienta útil en la detección y evaluación de estos trastornos. Exploración física Exploración completa orientada a descartar organicidad. Presión arterial (PA), frecuencia cardíaca (insuficiencia cardíaca, fiebre), frecuencia respiratoria (infección respiratoria, insuficiencia cardíaca, tromboembolismo pulmonar), temperatura axilar (si caquexia: temperatura rectal), piel, auscultación respiratoria y cardíaca, auscultación y palpación abdominal (globo vesical), tacto rectal (fecaloma), movilización y exploración de las extremidades, el raquis y exploración neurológica. Exploraciones complementarias Iniciales • Analítica urgente: tira de orina (infección urinaria); glucemia capilar (hipoglucemia); SaO2 por pulsioximetría (hipoxemia). • Analítica: hemograma, creatinina, glucemia, iones (incluido Ca), alanino aminotransferasa (ALT); sedimento y cultivo de orina. • Según la sospecha clínica: radiografía de tórax, radiografía simple de abdomen, osteoarticular, etc. Es fundamental descartar causas físicas desencadenantes cuando un paciente empieza a presentar alguna conducta anómala. Sólo cuando todas las causas físicas son descartadas, así como las ambientales y psicológicas, se puede concluir que la alteración de la conducta está primariamente causada por la demencia. Posteriores • Según la sospecha clínica: electrocardiograma, tomografía computarizada. ¿Qué propuesta haremos? Tratamiento Paciente No farmacológico Las intervenciones no farmacológicas son, en general, de primera elección en el tratamiento de los SCPD; sobre 5.13 Trastornos psicoconductuales en las demencias Tabla 6. NPI (Inventario Neuropsiquiátrico de Cummings) Frecuencia Gravedad Delirios 01234 123 Alucinaciones 01234 123 Trastorno No valorable (marcar) Agitación 01234 123 Depresión/disforia 01234 123 Ansiedad 01234 123 Euforia/júbilo 01234 123 Apatía/indiferencia 01234 123 Desinhibición 01234 123 Irritabilidad/labilidad 01234 123 Conducta motora sin finalidad 01234 123 Total (Frecuencia × Gravedad)a Puntuación total a Trastornos neuropsiquiátricos: multiplicar frecuencia por gravedad. Significado de los valores Frecuencia: 0 = Ausente 1 = Ocasionalmente (menos de una vez por semana) 2 = A menudo (alrededor de una vez por semana) 3 = Frecuentemente (varias veces por semana, pero no a diario) 4 = Muy frecuentemente (a diario o continuamente) Gravedad: 1 = Leve (provoca poca molestia al paciente) 2 = Moderada (más molesto para el paciente, pero puede ser redirigido por el cuidador) 3 = Grave (muy molesto para el paciente, y difícil de redirigir) NOTA: el rendimiento psicométrico del Inventario Neuropsiquiátrico (Neuropsychiatric Inventory, NPI) es muy elevado para la valoración de los síntomas no cognitivos en los pacientes con demencia, y permite realizar un seguimiento de la eficacia de los tratamientos sobre esos aspectos. No se han incluido aquí las subescalas de alimentación y sueño. todo en aquellos de intensidad leve y moderada. Formarán parte del Plan de Actuación Individualizado (tema 5.12) elaborado por el equipo muldisciplinar (trabajador social, médico de familia, enfermería). Se contemplará: • Entorno ambiental: Determinados factores ambientales pueden suponer intensos estímulos sensoriales en los pacientes con demencia. Hay que tratar de crear un ambiente «no estresante», modificando simplemente algunas de estas variables ambientales: – Empleo de iluminación suave y colores tenues, «sosegantes». – Empleo de absorbentes de sonido tipo alfombras, corchos, etc. – Añadir una música relajante y adecuada. – Evitar: ruidos intensos, espejos (fuera de vestidores y aseos), timbres, cambios de habitación, etc. • Programas de higiene en el sueño: Para el tratamiento del insomnio se han propuesto una serie de recomendaciones ambientales y conductuales que tratan de reforzar el estado del ritmo circadiano (alterado en los pacientes con demencia). Se intenta mantener la noción temporal, así como las condiciones ambientales más favorables para el sueño. Las recomendaciones del Instituto Nacional de la Salud (NIH) de EE.UU., incluyen: – Mantenimiento de un horario regular al acostarse y al levantarse. – Uso del dormitorio principalmente para dormir. – Horario regular en las comidas. – Evitar el alcohol, la cafeína y la nicotina. – Reducir la ingesta de líquidos al anochecer. – Establecer un ritual para la hora de acostarse. – Minimizar la luz y el ruido al acostarse y durante la noche. • Cuidados en la alimentación: Las dificultades de alimentación se desarrollan en todos los enfermos a medida que la demencia progresa, independientemente de su causa. Las dificultades motivadas por la apraxia y la descoordinación al tragar pueden manejarse al principio con ayuda de cambios en la textura de la dieta y recomendaciones de comportamiento al cuidador encargado de las comidas, mientras que la negativa a la ingesta puede responder al uso de antidepresivos. El planteamiento de medidas extraordinarias de alimentación e hidratación cuando el paciente deja de nutrirse por la boca es una de las decisiones más difíciles, tanto para los familiares como para los profesionales sanitarios. Dos revisiones sistemáticas concluyen que la alimentación por sonda en pacientes con demencia avanzada no previene la neumonía por aspiración, las úlceras por 481 5.13 Trastornos psicoconductuales en las demencias presión o las complicaciones infecciosas, ni mejora el estado funcional, el bienestar o la supervivencia. Además, la alimentación por sonda puede causar múltiples problemas mecánicos, gastrointestinales, infecciosos y metabólicos en estos pacientes. Sin embargo, en algunos trabajos realizados en pacientes con demencia avanzada, la alimentación por gastrostomía parece mejorar la supervivencia y prevenir las complicaciones, y tiene una buena valoración por parte de los familiares. • Establecer estrategias conductuales recomendadas para la agitación y la agresividad: – Intervenir cuanto antes. – Alejar al paciente de situaciones e individuos que le resulten provocadores. – Animar a los cuidadores a emplear una voz suave y sosegada. – Abordar al paciente agitado con calma, despacio y por delante. – Usar el contacto físico con criterio. – Emplear posturas no amenazantes. Procurar estar al mismo nivel ocular que el paciente. – Distraer a la persona, ir a otra sala, cambiar de actividades, dejar la situación en suspense, etc. – Establecer un entorno tranquilo. – Evitar discusiones y no tratar de razonar con el paciente agitado. Evitar la confrontación. – Evitar la restricción física siempre que sea posible. – Dar números telefónicos de urgencia a los cuidadores y familiares, y recomendarles su uso si se sintieran amenazados. Otras terapias no farmacológicas que van cobrando auge día a día en el abordaje de los trastornos SCPD son la musicoterapia, el toque terapéutico, la orientación en la realidad, la terapia artística y del movimiento, etc. Tratamiento farmacológico Antes de iniciar un tratamiento farmacológico, en estos casos, siempre deberían plantearse algunas cuestiones como: ¿realmente está justificado para este síntoma?, ¿qué grupo de fármacos es el más adecuado?, ¿cuáles son los efectos adversos potenciales y predecibles del fármaco a utilizar?, ¿cuánto tiempo debe mantenerse el tratamiento? En cualquier caso, se iniciará únicamente después del fracaso de las medidas no farmacológicas y/o la gravedad de los síntomas. Conviene utilizar dosis iniciales bajas, efectuar los incrementos poco a poco usando la dosis efectiva más baja. Los fármacos instaurados se deben revaluar periódicamente (4-8 semanas) contemplando su retirada, si es posible. Valorar las interacciones, la respuesta y los efectos secundarios. Se advertirá a la familia de la posibilidad de frecuentes cambios en las dosis y los fármacos. Trastornos psicóticos (agresividad, agitación psicomotriz, delirios) • Neurolépticos: Los neurolépticos han sido los fármacos de elección para el tratamiento de las alteraciones conductuales en 482 la demencia. Deberían reservarse sólo para las situaciones que no responden a las estrategias no farmacológicas, y que tengan una intensidad importante, evitando en todo caso su prescripción crónica. Se pueden utilizar los clásicos o los más recientes o atípicos. Parece que el aumento de muertes de origen cardiovascular (muerte súbita por arritmias) y cerebrovascular (ACV) es similar con los neurolépticos clásicos y con los atípicos. • Neurolépticos clásicos o convencionales (haloperidol, tiaprida, clorpromazina, levomepromazina): La eficacia de todos ellos es similar aunque presentan diferencias en sus efectos secundarios. Es frecuente la aparición de diversos efectos adversos: extrapiramidales (la discinesia tardía es más frecuente en los ancianos, así como babeo, rigidez, acinesia, etc.), especialmente con el uso del haloperidol; hipotensión ortostática, efectos anticolinérgicos (sequedad de boca, estreñimiento, visión borrosa, retención urinaria, confusión, etc.), sobre todo con agentes de baja potencia tipo levomepromazina o clorpromazina. Otros efectos menos frecuentes son: síndrome neuroléptico maligno, convulsiones, agranulocitosis, ictericia, etc. – Haloperidol: continúa siendo el neuroléptico clásico más usado. Su eficacia, seguridad y manejo lo configuran como un fármaco de gran utilidad. Su margen terapéutico es relativamente amplio con dosis que oscilan entre 0,5-1,0 mg/día iniciales y 3 mg/día (dosis máxima) según los casos. Además, la presentación en solución oral y también parenteral, así como su bajo coste, facilitan su manejo. Una pauta de inicio eficaz podría ser la administración de 5-10 gotas (0,5 a 1 mg) en dosis única nocturna e ir aumentando según necesidades hasta un máximo de unas 30 gotas. La limitación de uso en los pacientes con demencia estaría determinada por la frecuente aparición de efectos extrapiramidales (rigidez, bradicinesia, discinesia, etc.). – Tiaprida: es un neuroléptico tipo benzamida. Algunos autores lo consideran como un neuroléptico atípico por su perfil de acción (antagonista selectivo de los receptores de dopamina D2). A pesar de su escasa actividad antipsicótica ha demostrado su utilidad en el tratamiento de la agitación psicomotriz en el anciano, produciendo escasa sedación y manteniendo el nivel de alerta. Los comprimidos de tiaprida son de 100 mg. También puede administrarse en solución (gotas) dosis entre 5 y 50 mg/día con un amplio margen de seguridad (1 gota = 0,5 mg). – Clorpromazina, levomepromazina: fueron hasta hace unos años neurolépticos muy usados en el tratamiento de las afecciones psíquicas de ancianos, especialmente por su poder sedativo. Sin embargo, hoy día son de segunda elección, sobre todo por sus efectos adversos de tipo anticolinérgico y las posibles consecuencias de la hipotensión ortostática en este tipo de pacientes. Las dosis iniciales de clorpromazina y levomepromazina, en caso de que se decida su empleo, serán de 5-25 mg/24 h, hasta un máximo de 5.13 Trastornos psicoconductuales en las demencias 100-300 mg/día; la levomepromazina se puede utilizar en gotas (1 gota = 1 mg). • Neurolépticos atípicos (risperidona): En los últimos años, la introducción de los neurolépticos atípicos (risperidona, olanzapina, quetiapina) ha supuesto nuevas opciones en el tratamiento de los SCPD, pero sólo la risperidona tiene aceptada esta indicación. En general, presentan una menor incidencia de efectos extrapiramidales y anticolinérgicos, una relativa eficacia y una buena tolerancia; pero se mantiene la alerta sobre un aumento de los ACV isquémicos, que probablemente sea similar al de los neurolépticos clásicos. – Risperidona: su eficacia moderada y aceptable tolerancia la han convertido en un neuroléptico más usado en el tratamiento de los SCPD. Se puede iniciar con dosis de 0,5 mg hasta un máximo recomendado de 2 mg/día. Existe la presentación en comprimidos y en solución oral. En cuanto a sus efectos adversos, la aparición de extrapiramidalismo, discinesias, sedación o agitación «paradójica» está en función de la dosis. En la demencia de cuerpos de Lewy el uso de neurolépticos debe ser muy prudente. Se han publicado estudios que demuestran una susceptibilidad grave e incluso letal con el uso de estos fármacos. De modo que, ante la sospecha clínica de que un paciente presente este tipo de demencia, habrá que limitar el uso de neurolépticos clásicos y, si es necesario, recurrir a los atípicos en dosis mínimas (0,5 mg/día de risperidona), vigilando el desarrollo de extrapiramidalismo y deterioro físico graves. • Inhibidores de la acetilcolinesterasa (IACE): Los fármacos colinérgicos centrales pueden tener efectos beneficiosos sobre los SCPD, particularmente sobre la apatía, alucinaciones e ideas delirantes, pero queda por determinar su coste-eficacia. • Antidepresivos: Los antidepresivos también se han utilizado en pacientes con demencia y agitación por su efecto beneficioso, quizá debido al supuesto déficit serotoninérgico que forma parte de la plurietiología de esta alteración conductual. Los antidepresivos citalopram y trazodona han demostrado su eficacia en algunos ensayos, pero en absoluto puede generalizarse su uso con esta indicación. Trastornos del sueño y ansiedad • Benzodiazepinas: Con precaución y después de agotar las medidas no farmacológicas, pueden emplearse las benzodiazepinas. Sus efectos adversos, como sedación, somnolencia, ataxia, amnesia y confusión, limitan su utilización en los pacientes con demencia. Además, se ha comprobado que aumentan el riesgo de caídas. En caso de recurrir al uso de estos agentes, bien como ansiolíticos o como hipnóticos, se usarán las de semivida corta o media, como lorazepam y lormetazepam durante un tiempo limitado (4-6 semanas), sus- pendiéndolas de forma gradual. El lorazepam se usa en dosis entre 0,5 y 2,0 mg/día y puede administrarse por vía sublingual para favorecer una rápida absorción. El hipnótico lormetazepam se puede emplear en dosis nocturnas de 1 a 2 mg/día. Las benzodiazepinas de semivida larga están contraindicadas por el riesgo de causar reacciones paradójicas, mayor deterioro cognitivo e insomnio de rebote. • Antidepresivos sedantes: Si se precisa un tratamiento a largo plazo, pueden ser útiles los fármacos con actividad reguladora del sueño en dosis bajas, como los antidepresivos sedantes administrados por la noche. • Clometiazol: El clometiazol es un hipnótico sedante que puede ser utilizado en segunda opción, dada su farmodinámica (absorción muy rápida y semivida muy corta). Está indicado en la agitación y el insomnio. La dosis habitual suele ser 1-2 cápsulas (192 mg cada una) administradas por la noche. También puede ser de utilidad como coadyuvante de otros fármacos para el tratamiento de la agitación durante el día (de 1 a 3 cápsulas/día, según las necesidades). • Otros fármacos: En referencia específica a los trastornos del sueño en los pacientes con demencia, cabe considerar siempre la posibilidad de que estos trastornos sean secundarios a una depresión o formen parte de un proceso psicótico; los fármacos de elección serían, en este caso, los antidepresivos o los neurolépticos. Depresión • Antidepresivos: La principal indicación será el tratamiento de la depresión, frecuente en los pacientes con demencia. – Inhibidores selectivos de la recaptación de la serotonina (ISRS) (fluoxetina, sertralina, fluvoxamina, paroxetina, citalopram): parece ser que citalopram y sertralina poseen menos interacciones que el resto, lo cual los hace más indicados en estos pacientes plurimedicados. La dosis de citalopram inicial sería, en estos casos, de 10 mg/día para aumentar hasta un máximo de 30-40 mg/día (dosis habitual 20 mg/día). El uso de sertralina se inicia con 50 mg/día hasta un máximo de 200 mg/día (dosis habitual 100 mg/día). – Paroxetina: por su efecto ansiolítico-sedante puede también utilizarse en estos casos, teniendo en cuenta el déficit de atención y la sedación que se pueden deducir de sus propios efectos. La dosis de inicio es de 10 mg/día, que pueden mantenerse o aumentar a 20 a 40 mg/día si se considera necesario. Se puede administrar por la mañana o por la noche, según el efecto sedativo en cada paciente. La fluoxetina se puede utilizar cuando se asocia inhibición pero comenzando en dosis bajas 5-10 mg/día y vigilando una posible reacción hipomaníaca. – Trazodona: posee una acción serotoninérgica responsable de su eficacia. Se usa en dosis entre 50 y 100 mg/día, 483 5.13 Trastornos psicoconductuales en las demencias administrados antes de que el paciente se acueste, dado que posee un importante efecto sedante, muy útil en muchos casos en los que los SCPD predominan por la noche. Indicado en insomnio y agitación. – Antidepresivos tricíclicos clásicos (clomipramina, amitriptilina, imipramina, nortriptilina, etc.): es preferible no utilizarlos. Se asocian con frecuentes efectos adversos, sobre todo de tipo anticolinérgico, alteración de la conducción cardíaca, hipotensión ortostática, etc. En caso de depresión asociada al síndrome parkinsoniano con demencia, los antidepresivos ISRS pueden aumentar la rigidez y el temblor, en cuyo caso sería preferible la amitriptilina. Derivación Neurología/salud mental • Para realizar exploraciones complementarias o tratamientos farmacológicos que no sean accesibles al médico de familia. • Depresión resistente al tratamiento habitual. • Síntomas de difícil control relacionados con la demencia (insomnio, irritabilidad o vagabundeo rebeldes al tratamiento, etc.). Geriatría • Pacientes mayores con pluripatología difícilmente controlable. ¿Se ha resuelto del todo? Pronóstico y seguimiento El pronóstico es muy variable. Estos trastornos pueden marcar el inicio del proceso, en ocasiones serán los síntomas guía de la enfermedad causal, y habitualmente acompañan la progresión del deterioro cognoscitivo y funcional. Iniciado el tratamiento farmacológico indicado por el servicio de neurología, se plantea el establecimiento del plan de cuidados y seguimiento desde la consulta. Consejos prácticos • La descompensación aguda es un motivo importante de preocupación y sufrimiento por parte del paciente y la familia. Debemos intervenir de urgencia y averiguar qué la ha desencadenado y no solamente intentar controlarla con fármacos. • Ante la aparición de novo de un SCPD siempre habrá que descartar una causa orgánica o farmacológica. Antes de cualquier tratamiento deberá procederse a la realización de una historia clínica, anamnesis y exploración física completas. • El tratamiento de primera elección de los SCPD es la terapia conductual-ambiental. • Iniciar el tratamiento farmacológico sintomático después del fracaso de las medidas no farmacológicas, o por urgencia del proceso intercurrente. • Se retirarán todos los fármacos que no sean imprescindibles. 484 Errores más frecuentes • En muchas ocasiones los propios enfermos, sus familiares y hasta los profesionales sanitarios consideran «normales» algunas pérdidas que podrían ser compensadas con una correcta atención. Si no se solucionan, son factores que agravan innecesariamente la calidad de vida y la conexión con su entorno de los pacientes con demencia. • Tratar farmacológicamente con neurolépticos y/o benzodiazepinas cualquier síntoma psicoconductual en las personas con demencia y no hacer primero una intervención no farmacológica. • Mantener los tratamientos con psicofármacos sin hacer evaluaciones periódicas para ver si siguen siendo necesarios. • El número de pacientes con demencia avanzada portadores de sonda nasogástrica de alimentación es muy elevado. Un estudio reciente ha puesto de manifiesto que las principales causas de la colocación de sondas en pacientes con demencia avanzada son el desconocimiento del balance beneficio/riesgo por los facultativos y las presiones familiares. Insistir en la importancia del abordaje biopsicosocial y multidisciplinar, tanto del paciente como del entorno familiar, prestando especial atención al cuidador principal. ¿Podemos hacer alguna cosa más? Actividades preventivas La prevención de las demencias y, por tanto, de los SCPD es posible. Considerando las causas y el desarrollo de éstas, cabe distinguir: • Prevención primaria: – Control de los factores de riesgo cardiovasculares (PA, tabaquismo, dislipemias y diabetes mellitus). – Hábito enólico y hábitos sexuales de riesgo (lúes y VIH). – Utilización racional de los fármacos (ansiolíticos, antipsicóticos). • Prevención secundaria: – Diagnóstico temprano de la patología cardiovascular y de la EA (test del informador y miniexamen cognitivo (MEC) de Lobo preferentemente). – Diagnóstico y tratamiento correctos del paciente: a) Farmacológico: ácido acetilsalicílico en la demencia vascular (100 mg/día). b) No farmacológico: control de los factores de riesgo cardiovasculares, sobre todo en la demencia vascular pero también en la EA. – Atención del cuidador: una correcta información y formación del cuidador pretenden reducir su estrés y la posibilidad de malos tratos pasivos (infraestimulación, abandono) o activos al enfermo y mejorar la calidad de vida de ambos. 5.13 Trastornos psicoconductuales en las demencias • Prevención terciaria: – Diagnóstico, tratamiento y/o rehabilitación de todas aquellas patologías que originen discapacidad física (artrosis, ECV, etc.), sensorial (agudeza visual, sordera), mental (depresión, ansiedad) y social (aislamiento, etc.). Educación sanitaria El abordaje no farmacológico es la base tanto de la prevención como del tratamiento de estos trastornos. Numerosos trabajos corroboran la efectividad de los distintos tipos de medidas no farmacológicas para reducir la agitación y la ansiedad de los pacientes con demencia. Es imprescindible el trabajo con el cuidador, tanto para poner en marcha medidas preventivas de estos trastornos como para aportar un protocolo de intervención que permita afrontar las alteraciones conductuales. El plan de cuidados contemplará conjuntamente al paciente, el cuidador principal y el entorno familiar, siendo necesaria la coordinación entre los miembros del Equipo de Atención Primaria (médico de familia, enfermera y trabajador social), así como entre niveles asistenciales: • Se utilizarán ayudas técnicas que favorezcan la autonomía y los cuidados del paciente: utensilios adaptados, asideros, duchas, camas articuladas, etc. • Es aconsejable mantener una rutina en las actividades diarias, puesto que ayuda al paciente a orientarse en el tiempo. • Información a la familia y el cuidador principal acerca de la enfermedad: en qué consiste, evolución y fases por las que puede pasar el paciente. • Formar respecto a la comunicación entre el paciente y el cuidador. • Enseñar al cuidador las técnicas de cuidado del paciente y la forma de actuar ante trastornos del comportamiento (quejas, vagabundeo, agitación, negación, etc.). • Ofrecer asesoramiento respecto a las decisiones que deberá tomar a lo largo de la enfermedad, conforme el enfermo vaya perdiendo capacidades. • Interconsulta con el trabajador social para obtener más información y realizar un seguimiento: información so- bre recursos sociales disponibles, ley de la dependencia, asociaciones de familiares y aspectos legales. • Apoyo y cuidado del cuidador identificando sus necesidades, atendiendo sus consultas, previniendo la sobrecarga y enseñándole a manejar las situaciones de estrés y a cuidar de sí mismo. Bibliografía recomendada 1. Riu S, Alonso A, González VM. Atención al paciente con demencia avanzada en atención primaria. FMC 2008;15(3):132-43. 2. Riu S, Rodríguez JL, Casabella B, Cubero P, Hoyos MC, Espinàs J, et al. Demencias desde la atención primaria. Grupo de trabajo sobre demencias semFYC. Barcelona: semFYC ediciones; 2005. 3. Gill SS, Bronskill SE, Normand SL, Anderson GM, Sykora K, Lam K, et al. Antipsychotic drug use and mortality in older adults with dementia. Ann Intern Med. 2007;146:775-86. 4. Wang PS, Schneeweiss S, Avorn J, et al. Risk of death in elderly users of conventional vs atypical antipsychotic medications. N Engl J Med. 2005;353:2335-41. 5. González VM. Aspectos éticos y legales en la asistencia a los pacientes con demencia avanzada y sus familiares desde atención primaria. Aten Primaria. 2006;38 Supl 1:33-4. 6. Galindo J, Olivera FJ. Agitación e insomnio en pacientes demenciados. FMC. 2000;7(7):473-82. 7. Ray WA, Chung CP, Murray KT, Hall K, Stein CM. Atypical Antipsychotic Drugs and the Risk of Sudden Cardiac Death. N Engl J Med. 2009;360:225-35. 8. Ballard C, Waite J. Efectividad de los fármacos antipsicóticos atípicos para el tratamiento de la agresividad y la psicosis en la enfermedad de Alzheimer (Revisión Cochrane traducida). En: La Biblioteca Cochrane Plus, 2008 Número 4. Oxford: Update Software Ltd. Disponible en: http://www.update-software.com. (Traducida de The Cochrane Library, 2008 Issue 3. Chichester, UK: John Wiley & Sons, Ltd.) 9. Kalapatapu RK, Schimming C. Update on neuropsychiatric symptoms of dementia: antipsychotic use. Geriatrics. 2009;64(5): 10-8. 10. Selmes J. Antoine M. Vivir con… la enfermedad de Alzheimer u otra demencia. Madrid: Meditor; 2003. 485 5.14 Estado confusional agudo S. Alcántara Espadafor ¿De qué hablamos? Introducción El estado confusional agudo o delírium es una alteración aguda de la conciencia con disminución de la capacidad para centrar, mantener o dirigir la atención. Es un trastorno de la atención, la cognición, la memoria y la percepción que también puede afectar al sueño, la actividad psicomotora y las emociones. Los ancianos son más susceptibles a este trastorno. Con frecuencia es una forma de presentación atípica de una enfermedad aguda y también puede ser un marcador temprano de enfermedad neurológica. Conceptos iniciales Existe una gran variedad de términos para referirse a esta frecuente situación clínica, como delírium, síndrome confusional agudo (SCA), encefalopatía o síndrome orgánico cerebral: • El síntoma más característico es la alteración de la conciencia (con disminución de la atención o incapacidad para responder de forma adecuada al entorno) con cambios en las funciones cognitivas, como desorientación temporal y espacial, deterioro de la memoria, alteraciones del lenguaje o alteraciones perceptivas (alucinaciones). Con frecuencia los pacientes están aletargados o somnolientos pero pueden estar hiperalertas o agitados, o alternando entre ambos estados. El comienzo es rápido, con una evolución fluctuante de horas o de días. • La disminución de la atención se manifiesta con distraibilidad. Hay alteraciones en el curso del pensamiento acompañadas de trastornos en el lenguaje, que se hace confuso. Pueden observarse dificultades para nombrar algún objeto o incluso afasia. Las alteraciones sensoperceptivas más frecuentes son las alucinaciones visuales. • Como síntoma inicial pueden aparecer: ansiedad, irritabilidad, euforia, apatía y labilidad afectiva. • Las alteraciones conductuales graves en el curso de la noche pueden manifestarse como agitación psicomotora, contrastando con la somnolencia diurna. • Pueden aparecer signos neurológicos como temblor, asterixis, marcha inestable y alteraciones en los reflejos y en el tono muscular. Epidemiología En la población general la incidencia es del 1-2 %. Pero aproximadamente entre un 10-15 % de los pacientes hospitalizados presentan un estado confusional agudo. Este porcentaje es más alto en el grupo de individuos mayores de 60 años o en pacientes sometidos a intervenciones quirúrgicas o ingresados en una unidad de cuidados intensivos (UCI). El delírium aparece hasta en el 85 % de las personas al final de su vida. La mortalidad de estos cuadros es alta y existe un riesgo vital importante. En pacientes hospitalizados que de- 486 sarrollan un delírium, la mortalidad es del 22-76 %. Se ha demostrado que hasta el 25 % de los pacientes con delírium fallecen en los 6 meses siguientes al alta hospitalaria. ¿Qué lo puede ocasionar? Causas El delírium corresponde a un síndrome cerebral orgánico que carece de etiología específica y que supone una disfunción cognoscitiva global y multicausal. Generalmente aparece en el curso de una enfermedad médica, una intervención quirúrgica, una estancia hospitalaria o por el consumo o la retirada de determinadas sustancias. Habitualmente se asocian varias causas. En ancianos las más frecuentes son la demencia, las infecciones y los fármacos. En el adulto y en el joven son el alcohol (intoxicación y privación) y el abuso de drogas y fármacos y su retirada. En la etiopatogenia de este síndrome hay que distinguir tres grupos de factores: – Las causas de base que conducen a una disfunción cerebral aguda. – Los factores predisponentes que hacen que el paciente sea vulnerable. – Los factores precipitantes. • Alteraciones causales de base: – Infecciones agudas: respiratorias, urinarias, neurológicas. – Traumatismos craneoencefálicos. – Trastornos metabólicos: anoxia, hipercapnia, hipoglucemia, isquemia. – Alteraciones del equilibrio hidroelectrolítico: hipopotasemia, hiponatremia, hipercalcemia, desnutrición, deshidratación. – Alcoholismo crónico (encefalopatía de Wernicke por déficit de tiamina). – Enfermedades hepáticas, renales, respiratorias y cardíacas. – Vasculitis, lupus eritematoso, otras colagenosis. – Estimulación neuronal excesiva: epilepsia; abstinencia a alcohol, a benzodiazepinas y otras drogas; abuso a otros tóxicos. – Déficit de tiamina, piridoxina, ácido nicotínico. • Factores predisponentes: – Pérdida neuronal (demencias y ancianidad). – Disminución del metabolismo cerebral. – Disminución de la actividad de los neurotransmisores. – Desnutrición, deshidratación, alcoholismo crónico. – Privación de sueño. – Factores ambientales, como cambios del entorno habitual del paciente. – Déficit sensoriales, como visión o audición. – Hipotermia, inmovilidad, sobreestimulación, estrés. • Factores precipitantes: – Cambios bruscos en el ambiente (hospitalización, cambio de residencia). 5.14 Estado confusional agudo – Abstinencia a sustancias: alcohol, sedantes, hipnóticos, benzodiazepinas. – Fármacos: con actividad colinérgica (propantelina, trihexifenidilo, biperideno, prociclidina, escopolamina, oxibutinina, flavoxato, antidepresivos tricíclicos, atropina). Otros fármacos como anestésicos, opiáceos, levodopa, benzodiazepinas, agonistas dopaminérgicos, antihistamínicos corticosteroides, ranitidina, amantadina, teofilina, bupropión. – Intoxicación por alcohol, anfetaminas, cannabis, cocaína, opiáceos, sedantes, hipnóticos, ansiolíticos, insecticidas organofosforados. – Impactación fecal, retención urinaria, sondaje vesical. Diagnóstico diferencial Hay que realizar el diagnóstico diferencial con otros problemas que cursen con alteraciones de la conciencia, alteraciones cognitivas, alucinaciones y alteraciones de la memoria (figura 1). • Demencia: el inicio es insidioso y la evolución, crónica. El curso es lentamente progresivo con mantenimiento de la conciencia hasta los estadios finales. Se pierde la memoria a corto plazo. Con frecuencia aparecen cuadros de delírium (episodios agudos) en la evolución de una demencia (dos tercios de los casos de delírium ocurre en personas con demencia) (tema 5.13). • Esquizofrenia y otros cuadros psicóticos: el inicio es variable, no hay deterioro en la conciencia, ni en la memoria. Son personas muy retraídas con escaso contacto social. Las alteraciones sensoperceptivas se presentan fundamentalmente como alucinaciones auditivas (tema 6.5). • Cuadros depresivos graves: el inicio es variable, con un curso lento. No hay alteración de la conciencia, ni de la memoria. Predomina el trastorno del humor, con pérdida de interés y placer en las actividades habituales. La actividad delirante es ocasional y congruente con el estado de ánimo (tema 6.2). ¿Qué tenemos que hacer? Anamnesis • Antecedentes personales: demencia, accidente cerebrovascular agudo (ACVA), cirrosis hepática, insuficiencia renal, diabetes mellitus, consumo de alcohol, enfermedades mentales (depresión, esquizofrenia, delirio paranoide), enfermedad de Parkinson, cirugía reciente. Nutrición e hidratación los días previos, fiebre, focalidad neurológica, diarrea, estreñimiento, vómitos, cambio ambiental y, sobre todo, la historia detallada del consumo de fármacos en los días precedentes. • Enfermedad actual: el diagnóstico de SCA debe estar basado en la exploración del paciente, así como en la información aportada por los familiares y acompañantes del enfermo. Hay que tener en cuenta que el curso es fluctuante a lo largo del día, por lo que no es infrecuente que se pase por alto una alteración del nivel de conciencia, que durante el día esté mejor y empeore por la noche. • Síntomas que sugieren un SCA: cambios agudos del estado mental, presencia de una enfermedad médica de base, alucinaciones visuales, nivel de conciencia fluctuante, comienzo agudo de síntomas psiquiátricos sin antecedentes de enfermedad mental, paciente descrito como confuso o desorientado, distraibilidad manifiesta. Una vez realizado el diagnóstico sindrómico hay que buscar la causa de éste (v. figura 1). Exploración física • General: presión arterial (PA), pulso, frecuencia respiratoria, estado de hidratación, temperatura. Auscultación cardiopulmonar. Auscultación y palpación abdominal (globo vesical). Tacto rectal (fecaloma). Edemas, signos de trombosis venosa profunda (TVP). • Neurológica: – Debe preguntarse al paciente su nombre, edad, el lugar actual, el día y la hora. Puede comprobarse la memoria inmediata nombrando tres objetos no relacionados y pidiendo al paciente que los repita. La memoria a largo plazo puede explorarse preguntando datos biográficos. Se puede utilizar instrumentos de evaluación como el miniexamen cognoscitivo (tema 21.41). – Recuento de dígitos en sentido directo e inverso inmediatamente después del examinador (el recuento directo de dígitos normal hace improbable el SCA). – Examen de la perseverancia: al no mantener la atención, empieza una tarea pero no la acaba. – Recitar los meses del año en sentido natural e inverso. – Prueba de fluencia de categorías (Set Test de Isaacs): el paciente sin SCA es capaz de decir 10 nombres de animales, frutas, u otra secuencia de objetos en 1 minuto (tema 21.41). – Descartar rigidez de nuca, déficit o focalidad neurológica. Exploraciones complementarias Iniciales • Glucemia capilar, tira de orina y SaO2. • Hemograma, glucosa, creatinina, ionograma (sodio, potasio, calcio), aspartato aminotransferasa (AST); alanino aminotransferasa (ALT); hormona tiroestimulante (TSH), niveles de vitamina B12 y ácido fólico, urocultivo. • Radiografía de tórax (descartar neumonía). • Electrocardiograma para descartar infarto agudo de miocardio (IAM) como enfermedad de base. Posteriores • Gasometría arterial. • Exploraciones de imagen: tomografía computarizada (TC), resonancia magnética (RM). Si hay antecedente de traumatismo craneoencefálico (TCE) o se aprecia focalidad neurológica, rigidez de nuca o fiebre de origen desconocido. 487 488 Coma (estado inconsciente) Figura 1. Algoritmo diagnóstico del estado confusional agudo. Derivación a urgencias Traumatismos Golpe de calor Intoxicaciones por gases Tema 5.11 Alteración del lenguaje No ¿Responde a estímulos externos? Anciano: alcohol Fármacos (anticolinérgicos) Causa relacionada con consumo o abstinencia de sustancias Joven: alcohol, drogas (temas 7.3 y 7.4) Tema 6.5 Tema 5.12 ¿Desencadenante inminente? Percepción: ilusiones y alucinaciones Alteración de la memoria: amnesia ¿La afectación se limita a una función? Derivación a urgencias Tema 5.12 Descartar demencia secundaria Inicio insidioso curso progresivo – Enf. metabólicas: alteración de la hidratación y alteraciones iónicas – Enf. endocrinológicas: diabetes y alteraciones tiroideas – Epilepsia – Insuficiencias orgánicas (cardíaca, pulmonar, hepática y renal) ¿Descompensación de enfermedad previa? Sd. confusional agudo ¿Predomina la afectación de la atención, con alteraciones cognoscitivas, desorientación y alteración del ritmo sueño-vigilia? Paciente con alteración de las funciones cognoscitivas (confuso) Infecciones localizadas: infección urinaria y pulmonar Sepsis Forma de inicio de enfermedad nueva Tema 5.11 Afasia de Wernicke Inicio agudo pero estable y con afectación predominante del lenguaje Enf. vascular cerebral Sd. meníngeo Sd. tumoral Datos positivos de alteración neurológica signos meníngeos, focalidad o lateralidad Temas 6.2 y 6.5 Esquizofrenia Enf. depresiva monopolar o bipolar Sí Ocurre en pacientes con enfermedad mental previa 5.14 Estado confusional agudo 5.14 Estado confusional agudo • Electroencefalograma (EEG): un EEG normal ayuda a descartar el delírium como causa del deterioro cognitivo (tema 25.16). ¿Qué propuesta haremos? Tratamiento No farmacológico Es el enfoque principal y muchas veces único. • Además de controlar los síntomas es preciso buscar posibles desencadenantes, muchas veces de origen farmacológico. Tratar la enfermedad de base (p. ej., las infecciones). • Evitar cambios de personal; debe acompañarle alguna persona conocida, acercarle objetos familiares, evitar la estimulación lumínica o acústica. Dejar una luz tenue encendida por la noche. Periódicamente, intentar resituarle en el espacio y en el tiempo. • Aportarle sus gafas o audífono. • Se debe controlar y garantizar la seguridad del paciente, sobre todo la heteroagresividad y las autolesiones. • Valorar el estado mental del paciente periódicamente. • Ofrecer información y apoyo a la familia. • Se deben revisar todos los fármacos que recibe el paciente y dejar sólo los estrictamente necesarios. En caso de abstinencia a fármacos (p. ej., benzodiazepinas), introducirlos a la menor dosis posible. • Mantener una buena hidratación del paciente. Farmacológico Evitarlo si es posible y mantenerlo sólo unos pocos días y a la menor dosis necesaria. • Neurolépticos: El más utilizado es el haloperidol, el tratamiento farmacológico inicial de elección. En adultos se recomienda comenzar con 1-2 mg/6-8 h v.o. En ancianos 0,5-1 mg/ 8-12 h v.o. La vía intramuscular mejora la rapidez y la eficacia en cuadros que supongan un riesgo vital; en este caso pueden emplearse dosis de 2-5 mg/4-6 h (el máximo efecto por esta vía se alcanza a los 20-40 min), según necesidades (tener en cuenta que hay riesgo de acumulación del fármaco). La vía intravenosa sólo debe emplearse con monitorización cardíaca. Una vez resuelto el SCA y tratado el problema de base, debe retirarse el neuroléptico en unos pocos días. No se deben administrar a pacientes con síndrome de abstinencia a Consejos prácticos • Evitar sitios con demasiados estímulos sensoriales o la ausencia de ellos. • Favorecer la orientación del paciente, recordándole el día y la hora. • Simular el entorno familiar con objetos conocidos. • Utilizar luces tenues por la noche. • Dar apoyo emocional tanto al paciente como a la familia. Errores más frecuentes • Tratar el síntoma predominante (agitación, delirio) y no estudiar la causa desencadenante (infección, impactación fecal, deshidratación, etc.). • No tener en cuenta que en ancianos, los fármacos hay que empezarlos con dosis bajas, especialmente los psicofármacos. • No prever que se produce acumulación de las dosis sucesivas de neurolépticos. • Pensar que todos los delírium en ancianos son debidos a demencia. • No tener en cuenta que muchos fármacos (opiáceos, anticolinérgicos) son desencadenantes del SCA. alcohol o benzodiazepinas, insuficiencia hepática y síndrome neuroléptico maligno. Una alternativa que valorar es la risperidona en dosis iniciales de 0,5 mg/12 h v.o. Otros fármacos empleados en esta situación son la quetiapina y la olanzapina, siempre en dosis bajas. • Benzodiazepinas: En general, no deben emplearse, excepto en el tratamiento del síndrome de abstinencia a las benzodiazepinas, en el delírium por deprivación de alcohol y en el síndrome neuroléptico maligno. Son de elección las de semivida intermedia. También se utilizan, a veces, combinadas con los neurolépticos para evitar los efectos secundarios de éstos o aumentar el umbral convulsivo. La más utilizada es lorazepam: 0,5-1 mg/repetible cada 4 h si es preciso, v.o. (tener en cuenta el efecto acumulativo). En pacientes alcohólicos con SCA, administrar tiamina 100 mg/día por vía i.m. durante 5 días. A todo alcohólico al que se le administren sueros glucosados hay que aportarle tiamina para evitar una encefalopatía de Wernicke. • Colinérgicos: La fisostigmina es un fármaco anticolinesterásico que atraviesa la barrera hematoencefálica, por lo que se emplea por vía intravenosa como coadyuvante en el tratamiento del delírium causado por anticolinérgicos (hay que solicitarlo como medicamento extranjero). • Antidepresivos: La trazodona ha sido útil, pero sólo en estudios no controlados. En caso de emplearse, se ha utilizado en dosis de 25-100 mg por la noche. De momento es un fármaco de uso restringido en estos casos. Derivación Urgente • Delírium debido a enfermedad médica no controlable en Atención Primaria (AP). • Delírium con focalidad neurológica actual. • Agitación psicomotriz con agresividad y no controlable en AP. • Delírium por abstinencia de alcohol. • SCA con disminución importante del nivel de conciencia. 489 5.14 Estado confusional agudo ¿Se ha resuelto del todo? Pronóstico y seguimiento • El delírium comporta una morbimortalidad importante si no se puede corregir la causa subyacente. La duración habitual del SCA es de 1-2 días a 2 semanas. En los ancianos puede ser prolongado y en una tercera parte no hay recuperación completa. • En pacientes hospitalizados, el SCA se asocia a estancias más prolongadas y mayor número de complicaciones, como neumonías y úlceras. En ancianos el desarrollo de un SCA empeora el pronóstico vital por la asociación de complicaciones frecuentes, como deshidratación, desnutrición, estreñimiento, caídas y alteraciones hidroelectrolíticas. • Si se decide tratar un SCA en el domicilio, es necesario un seguimiento diario con ajuste frecuente del tratamiento farmacológico. • Adaptar el horario del control de constantes al ritmo de sueño del paciente. • Corregir, en lo posible, los déficit auditivos y visuales. • Evitar la privación de sueño. • Movilización precoz después de accidentes o cirugía. • Cuidar la nutrición e hidratación. Educación sanitaria Los documentos escritos de apoyo pueden ser útiles a la familia (Guía Práctica de la salud, p 36). Bibliografía recomendada 1. Tejeiro J, Gómez, B. Guía diagnóstica y terapéutica del síndrome confusional agudo. Rev Clin Esp. 2002;202(5):280-8. 2. Inouye SK. Delirium in older persons. N Engl J Med. 2006;354 (11):1157-65. 3. Gleason OC. Delirium. Am Fam Physician. 2003;67(5):1027- ¿Podemos hacer alguna cosa más? 34. 4. Sánchez JC, Baquero M, Vílchez JJ. Síndrome confusional agudo: Actividades preventivas • En pacientes ancianos, evitar la administración de fármacos que pueden desencadenar un SCA. • La retirada de las benzodiazepinas debe ser gradual. • No someter a los ancianos a cambios ambientales drásticos. Deben tener junto a ellos objetos y personas que les sean familiares. Dejarles por la noche una luz ambiental. Reloj y calendarios visibles. Evitar, en lo posible, la sujeción mecánica. 490 manejo diagnóstico y terapéutico. Medicine. 2007;9:4969-73. 5. Formiga F, San José A, López-Soto A, Ruiz D, Urrutia A, Duaso E. Prevalencia de delírium en pacientes ingresados por procesos médicos. Med Clín (Barc). 2007;129:571-3. 6. Young J, Inouye SK. Delirium in older people. BMJ. 2007;21;334 (7598):842-6. 7. Miller MO. Evaluation and management of delirium in hospitalized older patients. Am Fam Physician. 2008;78(11):1265-70. 8. Delirium. Manual diagnóstico y estadístico de los trastornos mentales (DSM-IV-TR). Barcelona: Masson; 2002. p.156-76.