Grupo anatómico: (N) SISTEMA NERVIOSO
Anuncio
 SISTEMA NERVIOSO")
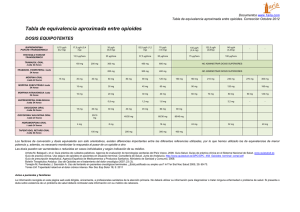
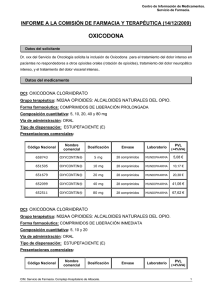
OXICODONA OXYCONTIN (Mundipharma) GRUPO TERAPÉUTICO - Grupo anatómico: (N) SISTEMA NERVIOSO - Grupo específico: N02AA. ANALGÉSICOS. OPIOIDES. Alcaloides naturales del opio INDICACIÓN AUTORIZADA Dolor severo. EL DOLOR El dolor es una experiencia sensorial y emocional de carácter desagradable que suele asociarse a una lesión real o potencial. Puede ser clasificado de diversas formas, ateniéndose a criterios variados. Según la duración, puede ser dividido en: - Agudo: Generalmente implica una señal de alarma disparada por una lesión somática o visceral, que generalmente dura lo que la lesión. - Crónico: Es un dolor que persiste, al menos, durante un mes tras la resolución de la lesión causal. En general, es un síntoma de una enfermedad que perdura y evoluciona. Según la localización, el dolor puede ser clasificado en: - Somático: Es un dolor bien localizado, circunscrito a la zona dañada y con sensaciones claras y precisas. Suele afectar a la piel, las articulaciones, los músculos, los huesos, los ligamentos, etc. - Visceral: Está difusamente localizado y normalmente suele ir acompañado de intensas reacciones vegetativas y motrices. Generalmente, está producido por lesiones en vísceras, aunque puede no tener relación con una lesión, o incluso estar referido a áreas superficiales distantes del origen. También puede clasificarse la sensación dolorosa según el origen de los estímulos álgicos, distinguiéndose entre el dolor nociceptivo o "fisiológico", que procede de los estímulos relacionados con daños somáticos o viscerales, y el dolor neuropático o "patológico", que procede del deterioro de estructuras del sistema nervioso, sin que existan lesiones en otros órganos o sistemas. Además, existen ciertos estados de hipersensibilidad que se traducen en el padecimiento del dolor. Uno de los más característicos es la hiperalgesia, que supone una respuesta exagerada a un estímulo normalmente doloroso pero de baja intensidad. Por su parte, la alodinia indica un dolor producido por un estímulo que normalmente no suele causarlo (estímulos táctiles, etc). El dolor implica la transmisión de estímulos al Sistema Nervioso Central que delatan, en general, situaciones peligrosas (“nocivas”, de ahí el término nocicepción) para el organismo. En realidad, estos estímulos “nocivos” son absolutamente indispensables para la subsistencia, ya que su ausencia impediría al cuerpo tomar ningún tipo de medida, preventiva o paliativa, frente a cualquier agresión externa o trastorno interno. Los “sensores” fisiológicos que generan tales impulsos son los nociceptores, que pueden ser definidos como terminaciones nerviosas situadas en diversos órganos y tejidos, con capacidad para discernir entre sucesos potencialmente lesivos y aquéllos de carácter inocuo, y con capacidad de enviar información (estímulos) al Sistema Nervioso Central. Los principales órganos sensoriales de los estímulos nociceptivos son los llamados nociceptores polimodales (NPM), un tipo de fibras nerviosas carentes de mielina, sensibles al dolor, al calor y a la presión. Los nociceptores generan impulsos nerviosos que se propagan en sentido aferente (desde la periferia hacia la estructuras nerviosas superiores). Esto exige la presencia de despolarizaciones de la membrana neuronal, lo que provoca potenciales de acción. Las despolarizaciones son consecuencia de las modificaciones iónicas inducidas, por diversas sustancias, como resultado del flujo iónico a través de la membrana (fundamentalmente, salida de potasio y entrada de calcio): - Neurotransmisores: serotonina (potente algógeno), acetilcolina, histamina (leve algógeno). - Metabolitos celulares: ATP, ADP, K+. - Dolor isquémico. - Prostaglandinas: sin efecto directo. Potencian los efectos algógenos de serotonina y bradicinina. - Cininas (quininas o kininas): péptidos producidos por escisión proteolítica a partir de precursores plasmáticos inactivos: bradicinina. - Otras sustancias: capsaicina (que provoca la depleción de sustancia P). Los nociceptores producen y liberan mediadores químicos de acción rápida en respuesta a la estimulación. Generalmente, se trata de aminoácidos (ácido glutámico) o pequeños péptidos (cadenas de hasta 25 aminoácidos), tales como la sustancia P, formada por 11 aminoácidos, considerado como el principal neurotransmisor nociceptivo en las fibras de tipo C. La lesión periférica (estímulo nociceptivo) activa tanto los sistemas ascendentes de trasmisión del dolor como los sistemas endógenos inhibitorios de la trasmisión nociceptiva (opiáceo, alfa-2 adrenérgico, colinérgico, etc.) situados a nivel periférico, espinal y supraespinal. La integración de la trasmisión excitatoria e inhibitoria a estos tres niveles, determina las principales características de la trasmisión y percepción del dolor. El impulso nociceptivo es transmitido por diversos tipos de conducciones nerviosas (formadas, obviamente, por neuronas). La velocidad de transmisión del impulso varía según el grado de mielinización de estas conducciones, al actuar la mielina como un auténtico aislante eléctrico. Así, se ha estimado en un valor de 6 metros por segundo (6 m/s) por cada micra (µm) de grosor de la capa de mielina. Dado que el diámetro neuronal es de 2 a 20 µm, la velocidad de conducción fisiológica de los estímulos dolorosos está comprendida entre 12 y 120 m/s. En casos especiales, el diámetro neuronal alcanza valores tan bajos como 0,2 µm, lo que implica velocidades de transmisión de apenas 0,8 m/s. Este tipo especial de neuronas juega un papel decisivo en la transmisión de los impulsos que dan origen al dolor lento, y son denominadas fibras C. Las fibras con mayor grosor en la capa de mielina (entre 1 y 6 µm) son denominadas fibras Aδ (léase A “delta”), que conducen los impulsos causantes del dolor rápido (la velocidad de transmisión es de 6 a 35 m/s). Este último tipo de fibras son las únicas activadas en los procesos en los que hay un intenso estímulo cutáneo como, por ejemplo, el que se produce por el pinchazo de un alfiler. El “viaje” de los impulsos nociceptivos es largo y complejo. Básicamente, implica un recorrido desde los nociceptores, presentes en la piel y prácticamente los restantes órganos y vísceras del cuerpo, hasta la corteza cerebral. No deja de ser una curiosidad el hecho de que el propio cerebro carezca de receptores del dolor, cuando es el órgano, por antonomasia, capaz de “traducir” los estímulos dolorosos. El impulso nociceptivo recorre los nervios periféricos, llega a las capas superficiales del asta dorsal espinal, asciende por alguna de las múltiples vías medulares, pasando por el cerebro medio, para acabar en el tálamo, desde donde se distribuye hacia la corteza cerebral. Según el tipo de fibra (lenta o rápida), el acceso al tálamo se produce por vías medulares diferentes. El complejo ventrobasal del tálamo (formado por los núcleos laterales y posteriores) recibe conexiones neuronales procedentes del sistema de conducción rápida. En estos núcleos están representados topográficamente la cara, la cabeza y el cuerpo, lo que implica un elevado grado de especialización y selectividad en la ulterior interpretación de los impulsos dolorosos a nivel de la corteza cerebral. Las neuronas que siguen el sistema ascendente múltiple de la médula transmiten los impulsos lentos (dolor lento), pasan por la formación reticular y terminan en los núcleos medial e intralaminal del tálamo. A diferencia de los otros núcleos talámicos antes indicados, estos últimos no muestran ninguna organización topográfica. Esto quiere decir que no existe, o bien no se conoce, reciprocidad entre áreas determinadas de estos núcleos y una localización orgánica específica. De los núcleos medial e intralaminal irradian fibras aferentes en dirección a la corteza, al sistema límbico (relacionado con las emociones y la memoria) y a los ganglios basales (implicados en el control de los movimientos voluntarios). Por todo esto, el tálamo aparece como el gran “discriminador” de los estímulos dolorosos que ascienden por la médula. Además de los estímulos nociceptivos aferentes (periféricos), existen vías descendentes de los centros superiores que participan en la transmisión ascendente. Este sistema de “control de apertura” o “barrera” permite modular la entrada de los impulsos aferentes. El sistema inhibitorio descendente, en el que se basa el mecanismo de control de apertura (figura 1), parte de la actividad de neuronas encefalinérgicas (capaces de producir encefalinas), de la sustancia gris presente en torno al acueducto (Area Gris Periacueductal). Estas neuronas excitan a las neuronas serotonérgicas (utilizan serotonina como neurotransmisor) de un núcleo próximo, el Núcleo del Rafe Dorsal que, a su vez, excitan a otras neuronas - también serotonérgicas - pertenecientes al Núcleo Magno del Rafe, las cuales conectan directamente con la Sustancia Gelatinosa del asta dorsal de la médula espinal, lugar donde actúan sobre neuronas encefalinérgicas, responsables últimas de la acción inhibidora sobre la aferencia de estímulos nociceptivos. Uno de los elementos esenciales del denominado “control de apertura” (barrera) es el sistema inhibitorio descendente. Gracias a él, el propio cerebro es capaz de modular, parcialmente al menos, la percepción de estímulos dolorosos. Este sistema inhibitorio descendente basa su actividad en varios tipos de neurotransmisores, entre ellos la serotonina y unas sustancias de carácter peptídico conocidas como encefalinas. Estas sustancias actúan como transmisores inhibitorios, uniéndose a receptores específicos situados en las membranas de las neuronas “encefalinérgicas”. La estimulación de tales receptores parece capaz de anular la liberación de neurotransmisores excitatorios, bloqueando así la transmisión del impulso doloroso. Estos receptores no son otros que los receptores opioides, sobre los que actúan en mayor o menor medida los analgésicos opiáceos (morfina, etc), de ahí que a las encefalinas también se las denomine como "opioides internos" o endorfinas ("morfinas endógenas"). Las encefalinas o endorfinas son péptidos de tamaño variable (desde 5 hasta 31 aminoácidos), pero con importantes puntos en común. Las secuencias de aminoácidos son muy similares y lo más importante es que el aminoácido Tirosina (Tyr) siempre es el N-terminal. Se conoce con certeza la existencia de cuatro receptores opioides: el µ (mu), el δ (delta), el κ (kappa) y el “huérfano” u ORL1 ; de ellos puede haber varios subtipos. En general, las consecuencias neuronales inmediatas de la activación de los receptores opioides consisten en una reducción de la actividad espontánea de la neurona, una menor capacidad para responder cuando es estimulada, y una menor capacidad para liberar o emitir sus neurotransmisores específicos, sean activadores o inhibidores. Queda deprimida, pues, la transmisión nerviosa. A efectos prácticos, es útil clasificar los fármacos opioides en: a) Agonistas puros: Son los fármacos que fundamentalmente interactúan con receptores µ (mu). En consecuencia producen analgesia, euforia, depresión respiratoria, miosis, náuseas y vómitos, estreñimiento, aumento de presión en vías biliares, dependencia física, y grados crecientes de sedación en función de la dosis. La intensidad de los efectos depende de la dosis; es decir, el grado de analgesia crece casi ilimitadamente con la dosis y podría alcanzar un techo antiálgico muy alto si no fuera por las limitaciones impuestas por sus efectos adversos (somnolencia y perturbación cognitiva/estupor/coma, depresión respiratoria grave, mioclonías). En la analgesia del opioide participan mecanismos críticos a nivel del asta posterior de la médula espinal, mecanismos supraespinales y cerebrales, incluidos aquéllos que participan en la tonalidad afectiva del dolor, e incluso en ocasiones mecanismos a nivel de las propias terminaciones nerviosas en donde llegan a expresarse receptores opioides. Ésta es la razón de que la analgesia opioide sea tan poderosa, si bien es importante conocer que hay dolores, fundamentalmente de carácter neuropático, que resisten la acción del opiáceo. Todos muestran potencialidad para inducir dependencia física y, en determinadas circunstancias, drogadicción. A la intensidad de la analgesia conseguida solemos llamar eficacia antiálgica; y a la cantidad de producto necesaria para conseguirla llamamos potencia, de forma que es más potente quien menos dosis requiera para conseguir la analgesia, y no quien más grado o techo de analgesia consiga. Los agonistas que tienen un techo analgésico más bajo suelen ser d e n o m i n a d o s o p i á c e o s menores, y se utilizan para dolores entre leves y moderados, unas veces solos y otras en asociación con analgésicos no opioides. En contraposición, los denominados mayores sirven para dolores entre moderados e intensos; alguno, como el tramadol, se encuentra en posición intermedia. b) Agonistas parciales: Su principal representante es la buprenorfina. Se caracterizan por presentar una menor eficacia o menor techo antiálgico, ya que su actividad intrínseca es algo inferior a la de los agonistas puros. Agonistas/antagonistas mixtos: Se caracterizan por activar preferentemente el receptor opioide κ (kappa) y por comportarse como agonista débil y antagonista débil de receptores µ (mu) . Su representante más conocido es la pentazocina. De acuerdo con estas acciones, producen analgesia también, pero de calidad algo diferente: su techo antiálgico es más bajo que el de los agonistas µ (mu) y no se acompaña del efecto euforizante que, por el contrario, puede ser disfórico. El aumento de dosis viene también limitado por la aparición de efectos psicotomiméticos. Aventajan a los agonistas puros µ (mu) en que inducen menor grado de depresión respiratoria, no producen estreñimiento ni aumentan la presión de vías biliares, y presentan menor potencialidad para crear drogadicción. Pero la disforia y la psicotomimesis han limitado mucho su utilización en la práctica. c) Antagonistas puros. Son fármacos con alta afinidad por todos los receptores opioides, pero sin actividad intrínseca. Por tanto compiten con los agonistas en su capacidad de unirse a los receptores, los desplazan de éstos, y sirven así para evitar o suprimir los efectos de los agonistas. Sus principales representantes son la naloxona y naltrexona. El dolor agudo puede ser somático (superficial, profundo) y visceral. El superficial, que asienta sobre piel y mucosas, se localiza con precisión. El dolor profundo, de músculos, huesos, articulaciones y ligamentos, es menos preciso y produce reacciones de defensa. El dolor visceral es un dolor sordo, difuso y mal localizado, cuyo punto de partida son las vísceras huecas o parenquimatosas, que generalmente es referido a un área de la superficie corporal, siendo acompañado frecuentemente por una intensa respuesta refleja motora y autonómica. Para el tratamiento del dolor agudo se utilizan tres grupos de fármacos, que actúan por distintos mecanismos: a) Los opiáceos, como la morfina, se unen predominantemente a los receptores opioides µ y, en menor proporción, a los δ y κ) y de esta forma impiden la transmisión nociceptiva. Los receptores µ se expresan en los tres niveles anatómicos donde se integra la transmisión nociceptiva (periférico, espinal y supraespinal). La unión del opiáceo al receptor induce analgesia efectiva frente a la mayor parte de estímulos dolorosos, y los agonistas opioides µ son los analgésicos más eficaces disponibles, ya que carecen de “techo” analgésico. Sin embargo, los receptores µ se encuentran ampliamente distribuidos en el organismo, y la administración sistémica de agonistas induce efectos adversos, como náuseas y vómitos, sedación, íleo, depresión respiratoria, prurito, entre otros. b) Analgésicos antiinflamatorios no esteroídicos (AINE). Los AINE convencionales inhiben de forma inespecífica las ciclooxigenasas (COX-1 y COX-2), lo que disminuye la síntesis de prostaglandinas y, como consecuencia, la inflamación periférica. Sin embargo, las PGE también participan en la transmisión de la señal nociceptiva, por lo que en la actualidad se acepta que estos fármacos tienen un mecanismo de acción tanto central como periférico. periférico. La inhibición inespecífica de las COX induce alteraciones en la coagulación y hemorragia, alteraciones gastrointestinales, renales, etc. La relevancia clínica de estos efectos adversos, cuando los AINE se utilizan por períodos de tiempo cortos (como es el caso del tratamiento del dolor agudo), no ha sido establecida de forma definitiva. Los inhibidores específicos de la COX-2 constituyen una alternativa válida para obviar algunos de los efectos indeseables mencionados. c) Analgésicos antitérmicos (A/A).Los A/A carecen de efecto antiinflamatorio y su mecanismo de acción no está completamente establecido. Aparentemente tienen una acción inhibitoria débil sobre las COX y se ha postulado que inhiben la síntesis del óxido nítrico (NO) tanto a nivel central como periférico. Carecen de efectos gastrointestinales y sobre la coagulación, aunque su uso está restringido en algunos países debido a que pueden inducir anemia aplásica. Los principales efectos indeseables que producen son sedación ligera e hipotensión. Los más utilizados en el tratamiento del dolor agudo son el metamizol (dipirona), el paracetamol y el propacetamol. d) Los anestésicos locales en el tratamiento del dolor agudo se aplican sobre o cerca de terminaciones nerviosas, nervios/troncos o bien en la médula espinal a nivel epidural o subaracnoideo. Estos fármacos impiden de forma reversible la transmisión de la conducción nerviosa (sensorial, motora y simpática). Se utilizan ampliamente en el tratamiento del dolor agudo postoperatorio, bien por infiltración/perfusión de la herida quirúrgica, bien administrados por vía espinal. En este último caso pueden inducir hipotensión, bloqueo motor, sedación y, ocasionalmente, secuelas neurológicas. Aunque disponemos de múltiples fármacos eficaces para el tratamiento del dolor agudo, todos ellos presentan efectos indeseables importantes que limitan su uso. Para prevenir la aparición de estos efectos se utilizan coadyuvantes, que son fármacos que de por sí no son analgésicos, pero que mejoran la eficacia/seguridad de los analgésicos. Como ejemplo, podemos mencionar la asociación de morfina y antieméticos, tales como el droperidol, la metoclopramida o el ondansetron. Por otra parte, la asociación de dos o más analgésicos que actúan por mecanismos diferentes permite mantener un nivel adecuado de analgesia, disminuir las dosis de cada uno de ellos y, a la vez, disminuir también los efectos indeseables. Esta estrategia constituye la base de la analgesia multimodal o balanceada, que es el tratamiento de elección en el dolor agudo postoperatorio. El dolor crónico es aquel que persiste mucho más que el tiempo normal de curación previsto, no habiéndose resuelto con los tratamientos efectuados cuando se tiene una expectativa de que esto ocurra. También se define como el dolor que dura más de entre 3 y 6 meses, a pesar de estar siendo tratado por procedimientos adecuados. Es más frecuente en la edad media de la vida (30-60 años), no distingue sexo, y no siempre hay un substrato físico que lo justifique. Los síndromes de dolor crónico afectan a un 30% de la población de los países industrializados, produciendo incapacidad total o parcial durante distintos períodos de tiempo o permanentemente, con el impacto humano y coste económico que conlleva. De ellos, el dolor de cabeza afecta al 73%, las lumbalgias al 56%, los dolores musculares al 53% y los dolores articulares al 51% de la población. En el paciente con dolor crónico es habitual la presencia de transtornos psicoafectivos que alteran el entorno familiar y laboral del paciente. Son significativas las manifestaciones depresivas en forma de alteraciones del sueño, irritabilidad, retraimiento social, desinterés por el entorno, convirtiéndose el dolor no ya en un síntoma -como en el dolor agudo-, sino en una enfermedad. Para muchos expertos sólo se debería hablar de dolor crónico cuando altera la función laboral y la actividad social, y hace frecuente la utilización de servicios sanitarios. Los diagnósticos de los pacientes con dolor crónico son diversos, pero pueden ser agrupados bajo distintas categorías: - Dolor nociceptivo, por estímulos periféricos sostenidos. Se producen alteraciones en el nociceptor y en las vías aferentes periféricas, espinales y supraespinales, persistiendo el dolor, aunque puede haber desaparecido la causa inicial. - Dolor neuropático, consecutivo a lesiones o alteraciones periféricas o centrales del sistema nervioso. - Dolor mixto, que engloba ambas etiologías (nociceptivo y neuropático: dolor neoplásico). - Dolor psicógeno o idiopático, cuando no hay ninguna causa objetiva que lo justifique. Desde el punto de vista terapéutico, el dolor crónico plantea problemas complejos, siendo la polimedicación la regla común para este tipo de pacientes. ACCIÓN Y MECANISMO Oxicodona es una analgésico opioide, con acción agonista pura sobre los receptores Mu (µ) y Kappa (κ). Además de la acción analgésica, la oxicodona, como otros agonistas puros de receptores opioides, produce efectos ansiolíticos, euforia, sensación de relajación, depresión respiratoria, estreñimiento, miosis y supresión de la tos. En términos de analgesia, la oxicodona es unas dos veces más potente que la morfina. Los agentes opiáceos, como la oxicodona, impiden la transmisión nociceptiva. Los receptores µ y κ se expresan en los tres niveles anatómicos donde se integra la transmisión nociceptiva (periférico, espinal y supraespinal). El efecto agonista sobre receptores µ y κ es responsable de la analgesia y de la aparición de náuseas y vómitos, así como de sedacción. La depresión respiratoria, el estreñimiento, la retención de orina, la hipotermia y la adición están asociadas únicamente a la estimulación de los receptores µ. Las principales diferencias entre la estimulación (agonismo) de los receptores µ y κ reside en la diuresis (inhibida por los µ y estimulada por κ) y la euforia, asociada a µ (los κ se relacionan con disforia); la dependencia física está más intensamente ligada a la acción sobre receptores µ que sobre los κ. ASPECTOS MOLECULARES DEL NUEVO FÁRMACO La oxicodona está estrechamente relacionada desde el punto de vista estructural y farmacológico con otros agonistas puros de los receptores mu (µ) opioides, como la morfina y la codeína (tiene, como ésta, eterificado el OH en C3 ). Igualmente, guarda una notable relación estructural con la naloxona, un antagonista puro de receptores opioides, de la que se diferencia únicamente en el agrupamiento ligado al átomo de nitrógeno (N), pequeño en el caso de la oxicodona (-CH3 ), lo que determina su acción agonista sobre los receptores opioides, frente al relativamente voluminoso de la naloxona (-CH2 CH=CH2 ), lo que determina su efecto antagonista, determinado por una elevada afinidad hacia el receptor, pero sin ejercer ningún efecto sobre el, lo que impide el acceso de los ligandos endógenos o de los fármacos agonistas opioides. EFICACIA CLÍNICA La eficacia y la seguridad clínicas de la oxicodona han sido adecuadamente contrastadas en estudios clínicos controlados con placebo y con comparadores activos (morfina, fentanilo, hidromorfona, etc). Los criterios primarios de eficacia utilizados en estos estudios han consistido en valoraciones subjetivas realizadas por los propios pacientes, entre las que la escala visual-analógica (VAS) que utiliza una línea que va desde el 0 (ausencia de dolor) a 10 (dolor insoportable), es la más ampliamente utilizada. También se determina en muchos estudios la cantidad de medicación analgésica adicional sobre la ensayada (medicación de rescate), utilizada para conseguir una analgesia satisfactoria. Un grupo de 30 pacientes con dolor de origen canceroso participaron en un ensayo clínico doblemente ciego y cruzado, en el que se compararon dos formas de oxicodona, una de liberación inmediada (LI) y otras de liberación controlada (LC). En una fase abierta del estudio, los pacientes fueron estabilizados mediante oxicodona LI cada 6 horas. Tras ello, los pacientes fueron aleatoriamente asignados, ya en la fase doblemente ciega, a recibir oxicodona LC (cada 12 horas) o LI (cada 6 horas), durante 3 a 7 días. Finalizados estos, se cruzó el tratamiento, durante el mismo tiempo. Tras el ajuste inicial con oxicodona LI, se redujo la intensidad del dolor desde una media de 6,0 puntos hasta 2,7. La intensidad del dolor se mantuvo estable durante la fase doblemente ciega, con niveles similares de analgesia: 2,7 con oxicodona LC y 2,8 con la forma LI. La incidencia de efectos adversos fue similar con ambos tratamientos. En otro ensayo clínico se compararon las administraciones de formas controladas de liberación de oxicodona y de morfina. El estudio comenzó con una fase abierta, durante la que se ajustó la posología para conseguir adecuadamente la analgesia en un grupo de 26 pacientes con dolor de origen oncológico, que fue seguida de dos periodos de dos semanas cada uno doblemente ciegos y cruzando el tratamiento con oxicodona o morfina, ambas en forma oral de liberación controlada (LC), permitiendo en todo momento el empleo de medicación de rescate (morfina de liberación inmediata, LI). El consumo semanal de medicación de rescate fue 1,6 veces mayor durante el empleo de morfina LC que durante la oxicodona LC. Además, durante el tratamiento con oxicodona LC, los pacientes experimentaron menos náuseas y vómitos que con morfina LC. En otro estudio multicéntrico, con distribución aleatoria, doblemente ciego y controlado con placebo, se analizó el empleo de oxicodona de liberación controlada (LC) en un conjunto de 159 pacientes con dolor intenso o moderado, debido a una neuropatía diabética. El tratamiento empezó con dosis de 10 mg de oxicodona LC o placebo cada 12 horas, incrementándose cada tres días hasta un máximo de 60 mg/12 h. El tratamiento finalizó a las seis semanas. Los pacientes tratados con oxicodona LC mostraron un valor medio de la escala visual-analógica (0-10) de 4,1 mientras que en los tratados con placebo fue de 5,3. El 80% de los tratados con oxicodona y el 68% de los tratados con placebo experimentaron efectos adversos. En una revisión retrospectiva de 12.818 pacientes terminales que emplearon morfina, oxicodona o fentanilo de liberación controlada (este último en forma de parche transdérmico), se observó que los niveles de analgesia alcanzados fueron similares entre todos ellos, al igual que la incidencia de estreñimiento. Sin embargo, los pacientes tratados con fentanilo transdérmico mostraron una mayor dificultad de comunicación con sus allegados, en comparación con las formas orales de liberación controlada con morfina u oxicodona. Los efectos adversos más frecuentemente descritos con oxicodona son las náuseas y el estreñimiento, con una frecuencia de 25-30% en la mayor parte de los estudios. ASPECTOS INNOVADORES Oxicodona es una analgésico opioide, con acción agonista pura sobre los receptores Mu (µ) y Kappa (κ). Además de la acción analgésica, la oxicodona, como otros agonistas puros de receptores opioides, produce efectos ansiolíticos, euforia, sensación de relajación, depresión respiratoria, estreñimiento, miosis y supresión de la tos. En términos de analgesia, la oxicodona es unas dos veces más potente que la morfina. La oxicodona está estrechamente relacionada desde el punto de vista estructural y farmacológico con otros agonistas puros de los receptores mu (µ) opioides, como la morfina y la codeína. La tendencia actual de considerar como inaceptable la existencia de dolor en los pacientes está provocando un resurgimiento de los analgésicos opioides. Resurgimiento que es tanto mayor en Europa, cuanto que en Estados Unidos y otros países desarrollados el uso de opioides era notablemente mayor anteriormente. La excesiva prudencia con que se han utilizado los analgésicos opioides en Europa contrasta, no obstante, con otras prácticas terapéuticas notablemente irracionales. El terror casi patológico a emplear morfina u otros analgésicos opioides “de alto techo” condenó al dolor y al sufrimiento a no pocos pacientes en las décadas pasadas. Con el agravante de que pretendía ofrecerse como prudencia lo que simplemente se trataba de ignorancia. Afortunadamente, esta situación está cambiando, gracias en parte a las formas oral de liberación controlada o retardada (aunque no es exactamente lo mismo), que están facilitando su uso, incluso controlado por el propio enfermo en determinadas condiciones. Justamente, uno de los problemas que planteaba el uso de los analgésicos opioides es que requerían una administración frecuente (cuatro a seis veces diarias), lo que resulta claramente incómodo, con el agravante adicional que la dosificación requería una regularidad elevada (around-the-clock, en expresión anglófona), con el fin de mantener un nivel constante de fármaco en el cuerpo, ayudando a prevenir la recurrencia del dolor. Las formas orales de liberación controlada han paliado, al menos en parte, estos requerimientos estrictos de regularidad y frecuencia en la administración. Junto con ellas, es preciso citar también las formas transdérmicas que, en el caso del fentanilo, pueden colocarse en el mismo nivel de eficacia en el control del dolor intenso (severo es una traducción excesivamente literal del inglés, a pesar de que se emplea en textos oficiales). La eficacia comparada de la oxicodona, en su formulación oral controlada, está en la misma línea de la conseguida con la morfina (también en esa misma formulación) o en el fentanilo transdérmico, aunque algunos estudios parecen indicar que la oxicodona permite a los pacientes utilizar algo menos de medicación analgésica de rescate. No obstante, no se trata de diferencias que parezcan tener trascendencia clínica significativa. Algo más de interés puede tener el aspecto toxocológico, dada la relativa menor incidencia de náuseas y vómitos asociadas a la oxicodona, así como el hecho de no presentar metabolitos significativamente activos (noroxicodona-oximorfona) y, lo que es más importante, resulta bastante segura en la insuficiencia renal. El hecho de que la oxicodona sea dos veces más potente que la morfina, en términos equimoleculares no tiene demasiada trascendencia, ya que ello es fácilmente compensado por la dosificación empleada, la mitad que la morfina. Al igual que esta última su techo analgésico viene determinado por sus efectos adversos, en especial la depresión respiratoria (patente solo con dosis muy elevadas). En definitiva es un fármaco OTROS FÁRMACOS SIMILARES REGISTRADOS ANTERIORMENTE EN ESPAÑA Fármaco Morfina (liberación controlada) Fentanilo (transdérmico) Especialidad MST Continus Durogesic Laboratorio Mundipharma Janssen Cilag Año 1988 1998 COSTES DIRECTOS DEL TRATAMIENTO Indicación: Dosis diarias y coste Coste diario adultos1 Coste mensual adultos (4 semanas) OXICODONA (LC) 4,60 € 128,58 € MORFINA (LC) 2,23 € 62,47 € Considerando, por ejemplo, una dosis de 40 mg/12 h de oxicodona y de 80 mg/12 h de morfina (la equivalencia entre oxicodona y morfina es de 1:2) 1 VALORACIÓN OXICODONA OXYCONTIN (Mundipharma) Grupo Terapéutico (ATC): N02AA. ANALGÉSICOS. OPIOIDES. Alcaloides naturales del opio Indicaciones autorizadas: Dolor severo. VALORACIÓN GLOBAL: INNOVACIÓN MODERADA. Aporta algunas mejoras, ♣♣ pero no implica cambios sustanciales en la terapéutica estándar. Reduce la incidencia o la frecuencia de efectos adversos de la terapia ⇑ farmacológica estándar. BIBLIOGRAFÍA • • • • • • • • • • • • • • • • • Brant JM. Opioid equianalgesic conversion: the right dose. Clin J Oncol Nurs. 2001; 5(4): 163-5. Cheville A, Chen A, Oster G, McGarry L, Narcessian E. A randomized trial of controlledrelease oxycodone during inpatient rehabilitation following unilateral total knee arthroplasty. J Bone Joint Surg Am. 2001; 83-A(4): 572-6. Chou R, Clark E, Helfand M. Comparative efficacy and safety of long-acting oral opioids for chronic non-cancer pain: a systematic review. J Pain Symptom Manage. 2003; 26(5): 1026-48. Cuéllar Rodríguez S. Analgésicos Opioides. En: Farmacología y Farmacoterapia, Módulo II. Madrid, Consejo General de Colegios Oficiales de Farmacéuticos de España 1997. Curtis GB, Johnson GH, Clark P, Taylor R, Brown J, O'Callaghan R, Shi M, Lacouture PG. Relative potency of controlled-release oxycodone and controlled-release morphine in a postoperative pain model. Eur J Clin Pharmacol. 1999; 55(6): 425-9. Flórez Beledo J. Analgésicos Opiáceos. En: Avances en Farmacología y Farmacoterapia, Módulo III. Madrid, Consejo General de Colegios Oficiales de Farmacéuticos de España 2003. Gimbel JS, Richards P, Portenoy RK. Controlled-release oxycodone for pain in diabetic neuropathy: a randomized controlled trial. Neurology. 2003; 60(6): 927-34. Ginsberg B, Sinatra RS, Adler LJ, Crews JC, Hord AH, Laurito CE, Ashburn MA. Conversion to oral controlled-release oxycodone from intravenous opioid analgesic in the postoperative setting. Pain Med. 2003; 4(1): 31-8. Hale ME, Fleischmann R, Salzman R, Wild J, Iwan T, Swanton RE, Kaiko RF, Lacouture PG. Efficacy and safety of controlled-release versus immediate-release oxycodone: randomized, double-blind evaluation in patients with chronic back pain. Clin J Pain. 1999; 15(3): 179-83. Heiskanen TE, Ruismaki PM, Seppala TA, Kalso EA. Morphine or oxycodone in cancer pain? Acta Oncol. 2000; 39(8): 941-7. Lauretti GR, Oliveira GM, Pereira NL. Comparison of sustained-release morphine with sustained-release oxycodone in advanced cancer patients. Br J Cancer. 2003; 89(11): 2027-30. MacPherson RD. New directions in pain management. Drugs Today (Barc). 2002; 38(2): 135-45. Pasero C, McCaffery M. Controlled-release oxycodone. Am J Nurs. 2004 Jan;104(1):30-2. Pearl ML, McCauley DL, Thompson J, Mahler L, Valea FA, Chalas E. A randomized controlled trial of early oral analgesia in gynecologic oncology patients undergoing intraabdominal surgery. Obstet Gynecol. 2002; 99(5 Pt 1): 704-8. Silvasti M, Tarkkila P, Tuominen M, Svartling N, Rosenberg PH. Efficacy and side effects of tramadol versus oxycodone for patient-controlled analgesia after maxillofacial surgery. Eur J Anaesthesiol. 1999; 16(12): 834-9. Stambaugh JE, Reder RF, Stambaugh MD, Stambaugh H, Davis M. Double-blind, randomized comparison of the analgesic and pharmacokinetic profiles of controlled- and immediate-release oral oxycodone in cancer pain patients. J Clin Pharmacol. 2001; 41(5): 500-6. Vallerand AH. The use of long-acting opioids in chronic pain management. Nurs Clin North Am. 2003; 38(3): 435-45. • Weschules D. Toward Evidence-Based Prescribing at End-of-Life: A Comparative Analysis of Long-Acting Morphine, Oxycodone, and Transdermal Fentanyl and Clinical Outcome Markers in the Hospice Patient. 20 th Annual Meeting of the American Academy of Pain Medicine. Poster 164, March 7 th 2004.