042
Anuncio

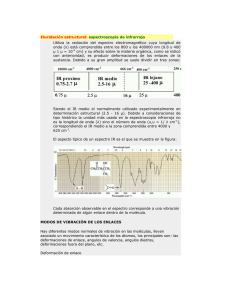
Análisis de espectroscopía UV e IR del 1,2,3,4-Tetroxano. Un estudio teórico y experimental Leiva, Laura C.1 - Romero, Jorge M.1 - Jorge, Nelly L.1 - Gómez Vara, Manuel E.1 - Castro, Eduardo A.2 1. Area de Fisicoquímica - Facultad de Cs. Exactas y Naturales y Agrimensura - UNNE. Av. Libertad 5450 - (3400) Corrientes - Argentina. E-mail: [email protected] 2. CEQUINOR - Depto. de Química - Facultad de Ciencias Exactas - UNLP. C.C. 962 - (1900) La Plata - Buenos Aires - Argentina. E-mail: [email protected] ANTECEDENTES Compuestos con grupos peróxidos juegan un papel importante en las reacciones de oxidación de compuesto orgánicos. En la mayoría de los casos, estas reacciones son producidas por la reacción con oxígeno molecular o su forma activada, como oxígeno singulete, o el anión radical superóxido[1,2]. Una comprensión de las propiedades químicas de estas especies, es esencial para la elucidación de la reacciones de oxidación de los compuestos orgánicos. Estas especies activadas juegan un rol significativo en reacciones de combustión, polimerización, metabolismo biológico, cáncer, envejeciendo, polución y en los variados aspectos de la ciencia medioambiental y tecnológica. El progreso de la química teórica, en procedimientos computacionales han contribuido a la determinación de las estructuras de éstas moléculas inestables y de las estructuras de transición en las reacciones de oxidación.[3] Estos métodos teóricos avanzados se han aplicado a la determinación de geometrías de equilibrio de intermediarios inestables y también las estructuras de transición en reacciones de la oxidación de compuestos orgánicos con varios compuestos con grupos peróxidos[4]. Recientemente, el mecanismo de un gran número de reacciones han sido elucidados a través del empleo de estas modernas técnicas de química teórica. El propósito de este trabajo es presentar los resultados teóricos y experimentales de los H espectros UV e IR y de la estructura molecular del 1,2,4,5-Tetroxano (Diperoxido de O Formaldehído, DPF, Fig. 1), con el fin de contribuir en el conocimiento de éstos compuestos O H H O que han cobrado importancia en estos momentos. O H Figura1: Estructura del 1,2,4,5-tetroxano MATERIALES Y METODOS Parte Experimental El 1,2,4,5-tetroxano se preparó por oxidación del formaldehído con peróxido de hidrógeno en presencia de ácido sulfúrico concentrado [5] y se analizó su pureza por cromatografía gaseosa en un equipo Hewlett – Packard modelo 5890, series II, usando una columna capilar HP5 (30m de longitud y 250mì de diámetro interno) con 5% de metilfenilsilicona como fase estacionaria (temperatura inicial 50°C, tiempo inicial 2min., incremento de temperatura 30°C/min., temperatura final 150°C), con un detector de ionización de llama (FID) a 300°C, inyector a 160°C y nitrógeno como gas carrier. Se prepararon soluciones del tetroxano en metanol, a las siguientes concentraciones: 1.00 x 10-3, 6.6 x 10-4, 5 x 10-4, 4.00 x 10-4, 3,08 x 10-4, 6.10 x 10-5, 1.60 x 10-5 , 1.60 x 10-6 M, para determinar el espectro de absorción UV-visible. El espectro UV-visible se realizó en el rango de 200-400 nm (celda de cuarzo, 1cm de paso óptico, con un rango de absorbancia de 0.000-2.000), en un espectrofotómetro marca Camspec modelo M330. El espectro IR fue determinado entre 400-4000 cm-1 en un espectrómetro infrarrojo Nicolet usando refractancia difusa con KBr como solvente. Detalles Computacionales Para determinar la estructura geométrica más estable se llevaron a cabo cálculos ab inito, funcional de la densidad y semiempiricos. El método semiempirico elegido fue AM1 ya que en análisis de estructuras en otros tetroxanos estudiados (3,6-difenil-1,2,4,5-tetroxano y 3,3,6,6-tetrametil-1,2,4,5-tetroxano) fue el que mejor describió su tendencia experimental [6,7]. El método ab initio Hartree Fock se realizó con la base 3-21 G y el funcional de la densidad, B3LYP, con la base 3-21G. Para el espectro UV se utilizó el software HYPERCHEM Release 6.0 para Windows [9]. El espectro electrónico teórico UV fue calculado por el método semiempírico ZINDO/S por un cálculo de single-point, con la estructura más estable obtenida del cálculo ab initio. El análisis vibracional se realizó por método ab initio, RHF usando una base 3-21G, implementada en el GAUSSIAN94.[8], con la estructura mas estable. RESULTADOS Y DISCUSION Los parámetros geométricos correspondientes a la molécula de DPF obtenidos por los tres métodos de cálculo computacionales se muestran en la Tabla 1. Los parámetros geométricos permiten asignar a una estructura completamente simétrica, contrastando con los hallazgos anteriores para los derivados. El mejor método para realizar estos estudios teóricos es RHF en base 3-21 G, por la concordancia hallada en ángulos y longitudes de enlace con los datos experimentales En la Fig.2 se muestra el espectro UV visible correspondiente al DPF en solución de metanol. Se observa que a medida que se diluye la solución los espectros mantienen el mismo máximo y todas cumplen con la Ley de Lambert-Beer. El espectro experimental dentro del rango de longitudes de onda estudiado, presenta un único pico a 203nm, y puede ser asignado al grupo peróxido O-O, a la transición π→π*. Los orbitales moleculares que intervienen en las transiciones electrónicas son el 24, 25 y 26, los cuales están localizados sobre los dos enlaces peróxidos (O1O2, O4O5) y los dos enlaces C-O. Figura 2. Espectro UV del 1,2,4,5-tetroxano en solución de metanol. En la Tabla 2 se muestran los datos calculados y correlacionados con el dato experimental. Si bien el diperóxido de formaldehído presenta diferentes confórmeros, que han sido estudiados previamente,[10] solo se trabajó con el confórmero de menor energía que es la estructura de silla, por ello la comparación entre los espectros UV teórico y experimental se realizó con respecto a la estructura de silla obtenida por calculo RHF, el espectro calculado por el método semiempírico ZINDO/S, coincide con el experimental. En la Fig. 3 se muestra el espectro IR experimental del DPF. En la Tabla 3 se muestra el espectro vibracional del 1,2,4,5-tetroxano, teórico y experimental, como así también las asignaciones a los distintos grupos presentes. Lo interesante del espectro IR es la presencia del pico de absorción del enlace peróxido O-O, característico de este tipo de compuestos. Cabe aclarar que solo se trabajo tanto teóricamente como experimentalmente con el confórmero silla. Figura 3. Espectro IR del 1,2,4,5-tetroxano. De la comparación entre los números de onda del espectro IR experimental y el obtenido por cálculo teórico vibracional utilizando RHF con una base 3-21G surge el siguiente análisis: el estiramiento simétrico y asimétrico del C-H del grupo CH2 usualmente aparece entre 2941 y 2882 cm-1 y la intensidad de estos picos es fuerte. En el DPF estos picos aparecen entre 2849 y 3272 cm-1 para el estiramiento simétrico y 3317 y 3398 cm-1 para el asimétrico. Los valores calculados fueron 3096 y 3177 para el estiramiento simétrico y 3307 y 3391 para el estiramiento asimétrico. El desplazamiento fuera del rango en el que se presentan habitualmente podría deberse a la presencia de los dos enlaces peroxídicos en el anillo. Los picos localizados a 1381 y 1431 cm-1 son asignados a la deformación simétrica y asimétrica del enlace C-H del grupo CH2. Los correspondientes valores calculados están entre 1366 y 1403 – 1492 cm-1, respectivamente. El pico a 1217 cm-1 corresponde al HCO estiramiento fuera de fase y esta correlacionado con el valor calculado 1294 cm-1. Los valores entre 1204 y 1207 cm-1 corresponden al estiramiento del enlace C-O perteneciente al anillo. No se encontró este pico en el cálculo. Las bandas localizadas entre 1169.52 y 1197 cm-1 pertenecen al estiramiento asimétrico HCO y su correspondiente valor calculado es 1130 cm-1. El estiramiento de la unión O—O se caracteriza por presentarse en el rango de 900 a 700 cm-1 [11]. Experimentalmente se obtuvieron picos entre 826 y 875 cm-1 . El valor calculado está entre 844 y 909 cm-1. Las bandas que se presentan entre 511.32 y 678 cm-1 corresponden a deformaciones en el esqueleto HCO y los valores calculados están dentro del rango 341 y 691 cm-1. CONCLUSIONES Las principales conclusiones derivadas de este estudio experimental y teórico del DPF, son las siguientes: • Absorción a 203 nm en el espectro UV puede ser asignada al grupo O-O. Hay concordancia con el valor calculado. • Los parámetros geométricos permiten asignar a una estructura completamente simétrica, contrastando con los hallazgos anteriores para los derivados. El mejor método para realizar estos estudios teóricos es RHF en base 3-21 G, por la concordancia hallada en ángulos y longitudes de enlace con los datos experimentales • En el espectro vibracional IR longitudes de onda entre 826 – 875 nm (experimental) y 844 – 908 nm (teórico) es propio del estiramiento de la unión peroxidica O-O. Tabla 1. Parámetros geométricos de la conformación silla del diperóxido de formaldehído (DPF), obtenidos de los métodos semiempírico AM1, ab intio y funcional de la densidad. 8 H 1 2 O 9 H O 6 3 O 5 H H 7 O 4 10 AM1 Longitud de Enlace (Å) O1O2 O4O5 C3O2 C3O4 C6O1 C6O5 C3H8 Angulo de Enlace (°°) O2C3O4 O1C6O5 C3O4O5 C3O1O2 C6O5O4 C6O1O2 H8C3O2 H8C3O4 H9C3O2 H9C3O4 H8C3H9 Angulo de Torsión (°°) C6-O1-O2-C3 C6-O5-O4-C3 O5-O4-C3-O2 O1-O2-C3-O4 O2-O1-C6-O5 O4-O5-C6-O1 Tabla 2. Espectro UV Experimental Valor Exp. nm 203 ∆Hf= - 0.061Ha. RHF 3-21G ∆Hf=-375.14 Ha. B3LYP 3-21G ∆Hf=-377.16 Ha. 1.300 1.300 1.440 1.440 1.440 1.440 1.110 1,467 1,467 1,435 1,435 1,435 1,435 1,074 1,529 1,529 1,458 1,458 1,458 1,458 1,091 103.24 103.24 111.14 111.14 114.14 114.14 106.78 106.77 111.71 111.71 115.73 108,95 108,95 105,19 105,18 105,18 105,18 106,70 106,70 110,24 110,23 113,83 110,63 110,60 103,22 103,19 103,21 103,19 105,64 105,61 110,43 110,44 113,91 65.63 -65.61 60.78 -60.80 -60.79 60.78 65,03 -65,01 67,66 -67,66 -67,67 67,66 64,79 -64,71 70,15 -70,19 -70,29 70,19 Calculado nm 204.6 248.5 Conformación silla MOs 24 → 26 24 → 25 Asignación π→π* π→π* Tabla 3. Espectro vibracional Experimental IR ν /cm-1 511.32 521.99 575.46 580.10 591.89 603.62 651.49 669.46 674.39 677.96 826.10 856.45 875.49 1169.52 1176.29 1188.43 1197.46 1203.90 1207.44 1217.72 1381.45 1431.91 2849.67 3272.20 3317.06 3366.43 3398.28 Calculado Conformación silla ν /cm-1 281.80 341.04 528.96 691.34 844.13 908.65 1130.45 1294.34 1366.31 1403.98 1492.55 3096.48 3177.84 3307.43 3391.19 Asignación Deformacion HCH Deformación COO Deformación OCH Deformación OCH Deformación OCH Deformación OCH Deformación OCH Deformación OCH Deformación OCH Deformación OCH Deformación OCH Deformación OCH Str. O-O Str. O-O Str. O-O Str. Asym. HCO Str. Asym. HCO Str. Asym. HCO Str. Asym. HCO Str. C-O Str. C-O Str. Out-of- phase HCO Sym Def. C-H del CH2 Asym Def. C-H del CH2 Asym Def. C-H del CH2 Str. Sym. del C-H del CH2 Str. Sym. del C-H del CH2 Str ASym. del C-H del CH2 Str ASym. del C-H del CH2 Str Asym. del C-H del CH2 AGRADECIMIENTOS Los autores agradecen a la Dra. Graciela Montiel por su valiosa asistencia técnica y al Dr. Jorge Avanza por facilitarnos el uso del espectro infrarrojo Nicolet con refractancia difusa. REFERENCIAS [1]. A. G. Davies, Organic Peroxides, Butterworths, London (1961). [2]K. Yamaguchi, K. Takada, Y. Otsuji and K. Mizuno, Organic Preoxides, Cap.1, 2-97 (1992). John Wiley &Sons Ltd. [3] K. P. Lawley, Advances in Chemical Physic, Vol. 69 I and II , Wiley, New York (1987). [4] W. J. Hehre, L. Radom, P. V. R. Schleyer and J. A. Pople, Ab initio Molecular Orbital Theory, Wiley, New York (1986). [5] Leiva L. C. A., Romero, J. M. , Jorge, N. L., M. E. Gómez Vara y L. F. R. Cafferata, Información Tecnológica, [6] N. Jorge, N. Peruchena, L. R. F. Cafferata and E. A. Castro, J. Mol. Struct. (Theochem), 1994, 433, 311. [7] N. Jorge, N. Peruchena, E. A. Castro and L. R. F. Cafferata, J. Mol. Struct. (Theochem), 1994, 309, 315. [8] M. J. Frisch, G. W. Trucks, H. B. schlegel, P. M. W. Gill, B. G. Johnson, A. Robb, J. R. Cheeseman, T. A. Keith, G. A. Petersson, J. A. Montgomery, K. Kaghavachari, M. A. Al-Laham, V. G. Kakrewski, J. V. Ortiz, J. B. Foresman, J. Cioslowski, B. B. Stefanov, A. Nanayakkara, M. Challacombe, C. Y. Peng, P. Y. Ayala, W. Chen, M. W. Wong, J. L. Andrés, E. S. Replogle, R. Gomperts, R. L. Martin, D. J. Fox, J. S. Binkley, D. J. DeFrees, J. Baker, J. P. Stewart, M. Head-Gordon, C. González and J. A. Pople, Gaussian 94, rev D3, SGI; Gaussian, Inc., Pittsburgh, PA., 1995. [9]HYPERCHEM para windows, Hypercube Inc. 1115 NW 4th Street, Gainesville, FL, USA, Release 5, 1991. [10] Leiva L. C. A. , Romero, J. M. , Jorge, N. L., M. E. Gómez Vara y E. A. Castro, trabajo en revisión. [11] G.J. Mnkoff, Proc. R. Soc. London, Ser. A224,176 (1954); H.A. Szymanski, Progree in IR Specectroscopy, Vol. 3, p. 139. Plenum Press, New York (1967); D. Lin-Vlen, N. B. Colthup, W.G. Fateley and J.G. Grasell, The Handbook of Infrared and RamnaCharacteristic Wavenumbers of Organc Molecules, p. 68. academc Press, New York (1991).