Descarga el PDF - Fundación Comparte Vida
Anuncio

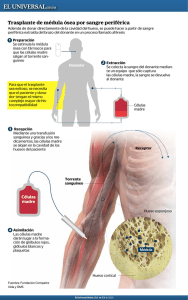
FUNDACIÓN COMPARTE VIDA, A.C. Información Médica - Anemia de Falconi Definición, características y diagnóstico La anemia de Fanconi (FA) fue descrita por primera vez por el pediatra suizo Guido Fanconi, quien en 1927 publicó sus observaciones clínicas sobre hermanos que habían heredado algunas condiciones físicas anormales y que experimentaron falla en la médula ósea. Estos niños sufrieron anemia aplásica severa. Su sistema inmunológico no pudo combatir las infecciones y se sentían crónicamente fatigados. Su cuenta de plaquetas era muy baja por lo que sufrían de hemorragias espontáneas. Las investigaciones han permitido establecer lo siguiente: - La anemia de Fanconi es uno de varios tipos de anemia que pueden ser mortales. - Ambos padres son portadores de un gen recesivo FA para que su niño nazca con esta alteración y la probabilidad de que su hijo nazca con la enfermedad es de 1 en 4 (25%). - Los pacientes con FA pueden tener una variedad de defectos físicos desde menores hasta serios, que llegan a afectar sistemas importantes del cuerpo. Otros pacientes quizá no muestren estos defectos, pero eventualmente su médula ósea fallará. - Los pacientes con FA experimentan una alta incidencia de leucemia (18-20%) y la de cáncer es mucho mayor que en la población en general. - Los cromosomas en las células de pacientes con FA se rompen y reacomodan fácilmente (esto es la base paradiagnosticar la enfermedad). La FA puede diagnosticarse cuando se manifiesta la anemia aplásica, condición en la cual la médula ósea no produce suficientes glóbulos rojos, blancos ni plaquetas. Igualmente, se pueden presentar síndromes mielodisplásticos que pueden progresar hacia leucemia. ¿La FA es lo mismo que el Síndrome de Fanconi? No. Este síndrome es un raro y es una alteración de la función renal, que ocurre principalmente en los niños. Algunos nutrientes importantes se pierden con la orina y esto ocasiona fallas en el crecimiento y alteraciones óseas, como el raquitismo. Los pacientes con FA pueden nacer con riñones anormales y sufrir problemas en su crecimiento, pero el tratamiento de la FA es muy diferente al del Síndrome de Fanconi. En la lucha contra la leucemia y otras enfermedades de la sangre o de origen genético FUNDACIÓN COMPARTE VIDA, A.C. ¿Cómo se relaciona la FA con la anemia aplásica? Los científicos dividen la anemia aplástica (AA) en dos categorías: “adquirida” y “hereditaria” (genética). Las causas de la AA adquirida pueden ser la exposición a una radiación excesiva, químicos tóxicos, pesticidas, algunos fármacos, infecciones virales y diversos agentes en el ambiente que dañan la médula ósea. En muchos casos de AA adquirida, la causa específica nunca se identifica, por lo que se le conoce como AA idiopática. La FA es una anemia heredada rara que conduce a la AA. Nadie aún ha logrado explicar por qué en los pacientes con FA les llega a fallar la médula ósea, pero se piensa que algún detonante en el medio ambiente puede contribuir a la aparición de la AA. ¿Cómo se diagnostica la FA? Esta enfermedad no ha sido, a lo largo de la historia, debidamente diagnosticada, quizá porque la aparición de sus síntomas varía desde los primeros años incluso hasta la edad adulta. Por ello es importante orientar a los médicos para que sepan identificar los síntomas y signos que indican su presencia. En general, el método más común para diagnosticar la FA consiste en tomar una pequeña muestra de sangre del paciente y combinar sus linfocitos (un tipo de glóbulos blancos) con sustancias químicas tales como el diepoxibutano (DEB) o la mitomicina C (MMC). En el laboratorio, los cromosomas que se encuentran dentro de las células con FA se romperán y reacomodarán al contacto con estos químicos, mientras que las células normales y sanas son más estables. Cabe señalar que si en una familia se está considerando el trasplante de médula ósea con un hermano, es crucial hacer la prueba del rompimiento cromosómico a cualquier donador potencial, aun si un hermano o hermana lucen sanos y normales, ya que pueden ser portadores de la enfermedad. Defectos físicos en personas con FA En la mayoría de pacientes con FA existe uno o varios defectos físicos en cualquier sistema o parte del cuerpo. No hay manera de predecir qué tipo de defectos podría tener una persona que hereda el problema; puede sospecharse la presencia de FA cuando un niño nace con un pulgar faltante, uno adicional o bien uno con forma irregular. También cuando no se le desarrolla completamente o le falta un hueso del brazo (el radio). Los principales síntomas son: Estatura baja.- Esta característica es muy común, ya que 50 % de los pacientes la padecen. Anormalidades del dedo gordo de la mano y de los brazos.-Se sospecha de FA cuando falta, está deformado o sobra un pulgar en cualquiera de las manos, o bien el En la lucha contra la leucemia y otras enfermedades de la sangre o de origen genético FUNDACIÓN COMPARTE VIDA, A.C. el radio no está bien desarrollado o no existe en el brazo. Anormalidades del esqueleto.- Aproximadamente una quinta parte de los pacientes con FA sufren de anormalidades en la cadera, costillas, malformaciones de la espina dorsal (bífida). Problemas renales.-Algunos pacientes nacen sin un riñón, aparecen unidos o bien alguno de ellos está rotado respecto a su posición normal, o su forma no es la correcta. Uno en cada cuatro pacientes tiene esta situación. Coloración de la piel.- Muchos pacientes desarrollan manchas “café con leche” (“café-au-lait” spots), que son zonas más oscuras de la piel. A veces el cuerpo entero o una gran parte de él, puede tener una apariencia de bronceado. Cabeza u ojos pequeños.- Algunos pacientes con FA pueden tener estas características, a lo que se le llama “microcefalia” y “microftalmia”, respectivamente. Retraso mental.- Algunas personas la presentan, pero no es tan común como se pensaba anteriormente. Otros tienen dificultades de aprendizaje, sin que esto signifique retraso mental. Bajo peso al nacer y dificultad de crecimiento Anormalidades del tracto intestinal.- Ciertas personas requieren cirugía del esófago, intestino o estómago. Otros muestran pobre apetito. Defectos en el corazón Algunos pacientes nacen con defectos en los tejidos que separan las cámaras del corazón. Anormalidades en los órganos sexuales.- Las mujeres pueden mostrar retraso menstrual, períodos irregulares y decremento en la fertilidad, con menopausia a menudo a los 30 años. Los hombres pueden tener órganos sexuales poco desarrollados, así como producción baja de esperma y fertilidad baja. Tumores sólidos.- Los pacientes con FA, principalmente de más de 20 años, están en riesgo de desarrollar cáncer en la cabeza, cuello y esófago. ¿Cuál es la función de la médula ósea? La parte central de los huesos está llena de un tejido esponjoso de color rojizo, llamado “médula ósea”; en él que se produce la sangre. Diariamente se producen millones de células que sostienen nuestra vida. La médula ósea alberga y es fuente de células madre, las cuales se dividen y evolucionan en células maduras: los glóbulos rojos, blancos y las plaquetas. Este proceso de formación y desarrollo de las células sanguíneas se conoce como hematopoyesis. En la lucha contra la leucemia y otras enfermedades de la sangre o de origen genético FUNDACIÓN COMPARTE VIDA, A.C. Cada tipo de células sanguíneas lleva a cabo funciones esenciales. Los glóbulos rojos (eritrocitos) llevan oxígeno desde los pulmones a todas las partes del cuerpo. Los glóbulos blancos (leucocitos) ayudan a combatir las infecciones y las enfermedades al atacar y destruir a los microorganismos. Las plaquetas (trombocitos) ayudan a cerrar las heridas y a controlar las hemorragias al formar costras en las zonas dañadas y previenen las hemorragias internas. Una persona con una cuenta baja de glóbulos rojos (anemia) presenta fatiga, dificultades para respirar y palidez. El tener pocos glóbulos blancos (leucopenia) lo hace vulnerable a infecciones y puede manifestar fiebres frecuentes; cuando un grupo de glóbulos blancos llamados neutrófilos son bajos en cantidad (neutropenia), la persona es propensa a las infecciones bacterianas. Por otra parte, una cuenta baja de plaquetas (trombocitopenia) puede manifestarse en forma de manchas rojas en la piel (petequias). Cuando Hay una cuenta baja en los tres tipos de células (glóbulos rojos, blancos y plaquetas), se tiene una condición llamada pancitopenia, igualmente conocida como anemia aplásica. ¿Cuándo ocurre la Anemia de Fanconi? Nadie puede predecir la edad en la que comienza a fallar la médula ósea en pacientes con FA. La edad promedio es a los 7 años. La mayoría de los niños experimenta los primeros síntomas de falla medular entre los 3 y 12 años. Al menos un 10 % de casos ha sido diagnosticado después de los 16 años e incluso un paciente tenía 48 años cuando se le diagnosticó la enfermedad. A otros se les identifica mediante pruebas de rompimiento cromosómico y no habían mostrado problemas de su sangre o físicos sino hasta después de los 30 años de edad. Así pues, la FA no es una enfermedad exclusiva de la niñez. ¿Cuál es el pronóstico para un paciente con FA? Nadie puede saber con exactitud cuánto tiempo vivirá un paciente con esta enfermedad, ya que es impredecible. Por los casos informados al Registro Internacional de Anemia de Fanconi, la esperanza de vida promedio es de unos 22 años; sin embargo, esta esperanza varía de un individuo a otro. Los nuevos descubrimientos científicos han hecho posible mejores tratamientos, así comolos trasplantes de médula ósea más exitosos. Cabe señalar, sin embargo, que conforme los pacientes con FA viven más años, mayor será la probabilidad de que lleguen a desarrollar tumores malignos. ¿Cuáles son los tratamientos para la Anemia de Fanconi? Existen varios tipos: a) trasplante de médula ósea, o de sangre periférica (de un familiar o de un donador no relacionado); b) trasplante de células progenitoras hematopoyéticas del cordón umbilical; c) terapia con andrógenos; d) factores de crecimiento sintéticos; y e) terapia génica. En la lucha contra la leucemia y otras enfermedades de la sangre o de origen genético FUNDACIÓN COMPARTE VIDA, A.C. Para los primeros dos tipos de tratamiento (trasplante de médula ósea, células periféricas o de cordón umbilical), se debe verificar que haya compatibilidad en el sistema genéticoHLA, (Human Leukocyte Antigens). Un hermano gemelo (univitelino) es el donador ideal pues será compatible en HLA y los demás genes. La siguiente opción es buscar entre los hermanos al donador HLA idéntico. Cuando no se tiene un grado de compatibilidad perfecto, se puede presentar una complicación posterior al trasplante, conocida como enfermedad injerto vs huésped ó EICH (GVHD ó Graft Vs. Host Disease), la cual ocurre cuando ciertos linfocitos (células T) del donador reconocen a las células del paciente como extrañas y las comienzan a atacar, lo cual puede ocasionar desde “rash” en la piel hasta falla múltiple de los órganos y eventualmente la muerte. Por ello, muchos centros hospitalarios eliminan a las células T antes del trasplante, lo que reduce notoriamente los riesgos de la GVHD. La probabilidad de encontrar un hermano idéntico es sólo de 25%. De modo que sí no lo hay, se recurre a los Registros de Donadores No Relacionados de Médula Ósea. En el caso c), terapia con andrógenos, entre un 50 y 75 % de los pacientes con FA responden a un grupo de drogas conocidas como “andrógenos”, como la oximetolona (AnadrolMR) y la oxandrolona, que son hormonas masculinas artificiales que a menudo estimulan la producción de uno o más tipos de células sanguíneas por períodos de tiempo prolongados. Ayudan a incrementar las cuentas de glóbulos rojos y plaquetas; prolongan la vida del paciente pero no son una “cura”. La mayoría, eventualmente deja de responder a los andrógenos, a veces después de varios años de uso. El consumo de estas hormonas puede causar daños severos al hígado y ocasionar efectos “masculinizantes” en quien los toma. Los factores de crecimiento hematopoyético (d) también estimulan la producción de células sanguíneas. Entre otros se encuentran las citocinas GM-CSF y G-CSF, así como la eritropoyetina y las interleucinas. La terapia génica se basa en la posibilidad de modificar los genes defectuosos que ocasionan la enfermedad, de manera que produzcan las proteínas necesarias en el cuerpo del paciente y en su sistema sanguíneo. Se han aislado cuatro de los genes de la FA (grupos A, C, F y G). Muchos laboratorios investigan los genes normales FA, las proteínas que fabrican y cóm podrían ser introducidos en las células de pacientes con Anemia de Fanconi. Las mutaciones en el gen A representan un 65% de los casos de Anemia de Fanconi. Fuente: Fanconi Anemia: A Handbook for Families. Fanconi Anemia Research Fund. Eugene, Oregon USA En la lucha contra la leucemia y otras enfermedades de la sangre o de origen genético