- Ninguna Categoria
Tecnología farmacéutica
Anuncio
TECNOLOGà A FARMACà UTICA II TEMA 1: INTRODUCCIà N Fármaco: sustancia quÃ−mica con actividad terapéutica a una dosis adecuada; sinónimo de principio activo. Forma farmacéutica = forma de dosificación: mezcla de principios activos unidas a sustancias auxiliares todo ello sometido a procesos de elaboración tecnológica (comprimidos, inyectables,...) Especialidad farmacéutica: forma farmacéutica envasada y registrada (©) Medicamento: fármaco incluido en una forma farmacéutica. Funciones de las formas farmacéuticas Las formas farmacéuticas son sistemas complejos más o menos caros a la hora de elaborarlos. ¿Por qué no administramos directamente el principio activo? - Para facilitar la manipulación y administración (se aumenta el tamaño y el volumen de principio activo) - Para incrementar la estabilidad quÃ−mica del principio activo (para evitar su degradación antes de que se produzca el efecto) - Para mejorar la aceptabilidad por parte del paciente (mejorar las propiedades organolépticas) - Para modular la liberación del principio activo - Acción retardada: liberación lenta -> podemos administrar el fármaco en perÃ−odos de tiempo más prolongados. - Para calcular la distribución del principio activo. Biodisponibilidad Concentración de fármaco en plasma efectiva para ejercer su acción. Cp CMT VÃ−a IV Cualquier otra vÃ−a CME t Caracterización de la biodisponibilidad Cmax 1 Tmax AUC CMT: concentración máxima tolerada. A partir de ella se producen efectos adversos. CME: concentración mÃ−nima efectiva. Una formulación ideal serÃ−a del siguiente tipo: Cp CMT CME t La mayor parte del tiempo la concentración se encuentra en el rango terapéutico. Criterios de diseño de una forma farmacéutica - VÃ−a de administración: la vÃ−a de elección es la vÃ−a oral (+ barata, + sencilla y + aceptada). - Dosis: según la que necesitemos utilizaremos una forma u otra. - Uniformidad de dosis. - Pureza. - Biodisponibilidad. - Aspecto. - Estabilidad. Con todos estos criterios, tengo que seleccionar un sistema en concreto y elaborar un SISTEMA TERAPà UTICO OPTIMIZADO: Más eficaz Más seguro Más fiable (fiabilidad: reproductibilidad de la dosis) La mayor parte de las formas farmacéuticas existentes no son sistemas de este tipo porque este concepto es relativamente reciente y las empresas no lo adoptan si sus compuestos son satisfactorios. Conseguir estos 3 objetivos en una sola forma farmacéutica es muy difÃ−cil y el criterio que se sigue es seleccionar un objetivo primario y los otros dos secundarios. Ej. : compuesto muy tóxico -> objetivo primario = seguridad, objetivos secundarios = razonablemente 2 efectivo y fiable. En ocasiones los objetivos son contrapuestos: Ej. : comprimidos muy blandos son poco fiables (pierden compuesto). Si para aumentar la fiabilidad aumentamos la fuerza de compresión podemos perjudicar la disolución y se rompe más fácilmente. Debemos llegar a un compromiso entre los 3 objetivos. Calidad de las formas farmacéuticas Se entiende como un conjunto de caracterÃ−sticas y procesos regidos por la calidad. En la actualidad no existe control de calidad sino aseguramiento de la calidad: - calidad de principios activos y excipientes - diseño de la forma farmacéutica optimizado - proceso de elaboración correcto y llevado a cabo de forma correcta Sólo llevando a cabo correctamente todas estas normas se puede asegurar la calidad de la forma farmacéutica. Las especificaciones de los principios activos y los excipientes se recogen en las farmacopeas (US farmacopea, farmacopea española y europea) La farmacopea española es una traducción y ordenación de la europea, y es la de obligado cumplimiento en España. Si no aparece, se ve la europea y si no, la americana (más actualizada). Forma farmacéutica: sustancia terapéutica compuesta por uno o varios principios activos y excipientes elaborados por un proceso tecnológico y con una calidad determinada, que libere el principio activo a una velocidad y en una cuantÃ−a determinada en un lugar determinado. TEMA 2: EXCIPIENTES Excipiente: sustancia sin actividad farmacológica que se añade al principio activo para: - mejorar sus propiedades - mejorar su estabilidad - facilitar el procesado - mejorar su biodisponibilidad IPEC: International Pharmaceutical Excipiens Council. Organismo internacional que estudia los excipientes y establece las caracterÃ−sticas adecuadas para cada uno de sus usos. Excipiente (1989): cualquier sustancia distinta del medicamento o del promedicamento con caracterÃ−sticas adecuadas de seguridad que se incorpora para: 3 - facilitar el procesado de la forma de dosificación (ej. Paracetamol -> comprime muy mal, por eso se le añaden excipientes para poder obtener comprimidos y lubrificantes para mejorar las propiedades de flujo) - proteger o incrementar la estabilidad tanto quÃ−mica (ej. ProteÃ−nas liofilizadas -> agente crioprotector para evitar la degradación) como la fÃ−sica (ej. Plastificantes ->evitan la rotura de la cubierta) - incrementar o mejorar la biodisponibilidad. Ej. : Aumento de tamaño de un comprimido con excipientes. Antes no ejercÃ−a efecto farmacológico porque el excipiente disminuÃ−a la absorción. Cp CMT CME t Se obtenÃ−an perfiles de biodisponibilidad de este tipo. Se utilizó otro excipiente y se obtuvieron perfiles de este tipo: Cp CMT CME t Puso de manifiesto la importancia de los excipientes en la biodisponibilidad. - facilitar la aceptabilidad por parte del paciente (ej. Jarabes -> mejora del sabor) - facilitar la identificación del producto (generalmente si se administra en distintas concentraciones, se utilizan distintos colores) - aumentar la seguridad y efectividad en el almacenamiento y uso. Las caracterÃ−sticas de calidad están especificadas en las farmacopeas. El tipo de información que dan es quÃ−mico (ej. Celulosa microcristalina ->pH, solubilidad,...) Las compañÃ−as vieron que tenÃ−an un material con las especificaciones de la farmacopea y al cambiar de lote o de proveedor, obtenÃ−an unas caracterÃ−sticas del producto diferentes y poco aceptables. Esto es asÃ− porque existen algunas diferencias que no se evalúan en las caracterÃ−sticas de la farmacopea. En un intento de recopilar toda la información acerca de excipientes, se realizó una recopilación de monografÃ−as sobre cada excipiente de uso farmacéutico (handbook). La base fue que un mismo excipiente se puede utilizar para distintos usos y para cada uno las propiedades crÃ−ticas serán diferentes y deben ser tenidas en cuenta al elaborar la forma farmacéutica. Selección de excipientes: 4 - producto aprobado por autoridades sanitarias - lote que cumple las especificaciones Hoy en dÃ−a existe una preocupación creciente por armonizar las especificaciones acerca de cada excipiente. Hay distintas especificaciones: Japón, EEUU, Europa) Por eso el IPEC ha intentado armonizar las monografÃ−as, porque un producto fabricado en Japón no puede venderse en EEUU. El primer candidato es la LACTOSA. Este organismo trabaja en la aplicación de normas ISO, acerca de la calidad. Las normas GMP (Good Manufacturing Practise) hacen referencia a como deben ser las instalaciones, métodos de trabajo,...) TEMA 3: MECANISMOS IMPLICADOS EN LA LIBERACIà N DE LOS MEDICAMENTOS A PARTIR DE FORMAS FARMACà UTICAS Tenemos el principio activo en la forma farmacéutica, se libera y se disuelve. Solo el principio activo disuelto se absorbe, llega a la circulación y ejerce efecto sistémico. Existen 2 posibilidades de administración de medicamentos: 1- formas de LIBERACIà N INMEDIATA: si queremos que el medicamento se libere lo más rápido posible. Si existen problemas en la liberación, aparecerán problemas de biodisponibilidad. 2- formas de LIBERACIà N CONTROLADA: si queremos que el medicamento se libere de modo ralentizado y que la forma de dosificación controle la liberación. La forma de dosificación permite modular la biodisponibilidad. Procesos que tienen lugar durante la liberación del principio activo: la liberación se modula por distintos procesos fÃ−sico-quÃ−micos. 1- LIBERACIà N INMEDIATA Disolución De la velocidad de disolución depende la velocidad de absorción por lo que Noyes-Whitney estableció una ecuación matemática que explicaba el proceso. Posteriormente Nerst-Brunner establecieron otra teorÃ−a, según la cual aparece una pelÃ−cula de difusión donde en la zona más cercana existe mayor concentración y en la más alejada menor concentración. Establece que: - la cantidad disuelta por unidad de tiempo es proporcional a la superficie de las partÃ−culas, la diferencia de concentraciones y a un coeficiente de difusión D que depende de las caracterÃ−sticas del producto a evaluar e inversamente proporcional al espesor de la pelÃ−cula de difusión y del volumen. DC/dt =D/h â ª S/V â ª (Cs-C) Para mantener una difusión constante mantendremos constantes los distintos parámetros. Mantener constantes estos parámetros solo puede hacerse hipotéticamente por lo que intentó explicarse la disolución suponiendo que: 5 - todas las partÃ−culas son esféricas - el procedimiento se desarrolla en condiciones SINK Ecuación de Hixon-Cronwell 3 Mo - Mt = Kd â ª t Métodos de ajuste para los perfiles de disolución de estos autores: - modificación del tamaño de partÃ−cula (s) - formación de sales o derivados más solubles del principio activo (aumenta Cs). Ej. Ciclodextrinas = carbohidratos cÃ−clicos con radical hidrófobo en el interior e hidrófilo en el exterior. Pueden formar complejos con el principio activo => derivado más soluble. - en ocasiones (pocas) se quiere disminuir la disolución. Ej. Principio activo que se degrada en el medio gástrico, intentaremos que se disuelva tras pasar el estómago mediante cubiertas poliméricas (portadores insolubles) 2- LIBERACIà N SOSTENIDA Difusión Proceso quÃ−mico más importante, controlamos asÃ− la liberación provocándola o ralentizándola. - Tenemos el principio activo disperso en una forma sólida; tenemos una sustancia que se disuelve formando canales en contacto con el medio; forman una red en 3D de canales por lo que las sustancias que forman parte del principio activo se disuelven y difunden a través del canal siempre a nivel molecular y a favor de gradiente. - También puede difundir si en vez de formarse canales se forma un gel-medio semisólido en el cual pueden difundir las partÃ−culas de principio activo previamente disuelto. Solo depende del gradiente y las propiedades del gel. - Otro ejemplo de difusión son las microcápsulas que el contacto con el medio permiten el paso a través de la membrana permeable del principio activo que se encuentra en el interior (también los comprimidos recubiertos) - Parches transdérmicos - Sistemas matriciales = geles y canales (existen parches transdérmicos matriciales) - Sistemas reservorio = microcápsulas, comprimidos recubiertos, parches transdérmicos. La ecuación general que rige el proceso de difusión es la establecida por Fick para el flujo de un principio activo. El flujo de un principio activo es proporcional al gradiente de concentraciones e inversamente proporcional a la distancia. Ley de Fick: f = flujo D = coeficiente de difusión 6 f = -D â ª dC/dX c = concentración x = distancia Sistemas reservorio Núcleo = principio activo + membrana polimérica (espesor = d) El sistema reservorio una vez contacta con el medio existe un periodo de hidratación de la membrana hasta que se alcanza un equilibrio, en ese momento podemos definir el coeficiente de reparto entre en medicamento disuelto antes y después de la membrana: Coeficiente de reparto (K) = Cm (o) / C (o) Incluyendo este coeficiente de reparto en la ecuación se llegó a la expresión: f = D â ª K â ª Î c / d K = Cm (o) / C (o) a x = 0 K = Cm (d) / C (d) a x = d Si lo expresamos en función de la superficie: dM / dt = A â ª D â ª K â ª Î c / d La velocidad de cesión del sistema reservorio es función de la superficie y espesor y del coeficiente de reparto que a su vez dependerá de las caracterÃ−sticas del polÃ−mero. Al variar éstas, variará el flujo por lo que podemos controlarlo de este modo. Si aumenta la velocidad de disolución => aumenta espesor + Ventajas: se consiguen perfiles de cesión de orden 0, la velocidad de liberación es constante. + Inconvenientes: contienen elevadas dosis de principio activo (elevado Cmax), pero si el sistema funciona bien se libera la dosis adecuada. El problema aparece si se rompe la membrana => sobredosificación (efecto Burst) Se pueden producir periodos de latencia importantes después de la administración. No existe principio activo disuelto => esto se produce porque la membrana no se hidrató, no se disuelve el principio activo y no se libera. Sistemas matriciales El frente de difusión se va desplazando hacia el interior quedando el polÃ−mero remanente. Se intentaron ajustar las cinéticas para sistemas porosos y no porosos, ya que no coinciden. Se hicieron varios estudios Perfiles de liberación: ECUACIONES DE HIGUCHI + Sistemas no porosos M = 2 â ª Cs â ª Dm â ª Co â ª t 7 M = medicamento cedido Co = [medicamento] en la matriz Cs = [medicamento] en saturación Dm = coeficiente de difusión del medicamento en la matriz La concentración de medicamento es directamente proporcional a la â t y obtenemos: M â t + Sistemas porosos M = [Da â ª Ca â ª p / T â ª (2 Co - p Ca) â ª t p = porosidad de la matriz T = tortuosidad = forma del poro (tubo o intrincado) Ca = solubilidad del medicamento en el medio Da = coeficiente de difusión en el medio La concentración de medicamento es directamente proporcional a la â t y obtenemos: M â t La matriz a tiempo 0 está completa y se va desplazando hacia el interior y va quedando el polÃ−mero remanente. M=Kâ ªâ t => sistemas porosos y no porosos + Ventajas: >fáciles de producir y baratos >útiles para formular compuestos de elevado peso molecular +Inconvenientes: >no dan perfiles de orden 0 a no ser que la liberación inicial no produzca variaciones en la matriz >dejan residuos -> matriz puede producir obstrucción intestinal 3- SISTEMAS QUE FUNCIONAN POR MECANISMO MIXTO DIFUSIà N + DISOLUCIà N 8 Normalmente se encuentran en el mercado medicamentos que utilizan un mecanismo de: Difusión + Disolución - Bioerosión: Se disuelve el polÃ−mero, no existe remanente (ventaja) Mi/Mo = 1 - (1 - Ko â ª t / Co â ª n)┿ n = 3 -> esfera n = 2 -> cilindro n = 1 -> lámina Formas de elaborar sistemas de este tipo: + Unión quÃ−mica covalente entre medicamento y polÃ−mero Ej. : absorción colónica -> la flora colónica es capaz de romper fibra que antes se creÃ−a insoluble. M - S - S - GALACTOMANANO -> vÃ−a oral Existen bacterias azorreductoras que rompen el enlace disulfuro. Por lo tanto, el colon es una zona adecuada para administrar péptidos porque allÃ− no existen enzimas que los degraden. + Utilizar polÃ−meros que se hinchan Ej. : derivados de ésteres de celulosa en contacto con un medio de disolución se hinchan y se dispersa el fármaco. Inconvenientes de la bioerosión: >es difÃ−cil controlar la cinética de cesión porque el sistema no mantiene sus propiedades fÃ−sicas. >los productos de degradación del polÃ−mero pueden ser tóxicos, por eso son adecuados carbohidratos naturales o semisintéticos. Los comprimidos bioerosionables son los más usados para conseguir cesión sostenida. - Presión osmótica Tenemos una membrana semipermeable, que se introduce en un medio de disolución en el que no existe medicamento o su concentración es muy pequeña. Tenemos el principio activo dentro de un núcleo osmótico solo o separado por una envoltura. El gradiente de concentración genera una presión osmótica que hace que el agua entre dentro de la forma farmacéutica, se expande y se disuelve el principio activo; la membrana es rÃ−gida, se realiza un poro por el que pueda salir la disolución de principio activo formada en el interior. 9 Saldrá medicamento a la misma velocidad que entre agua en el sistema. Si conseguimos modularla, también modularemos la cantidad de principio activo que sale. dV / dt = A â ª K / h â ª (Î Ï“ - Î P) => dV / dt = A â ª K / h â ª Î Ï“ flujo de entrada de agua en el depósito Aumento del área de la membrana => aumento del flujo de agua que entra Aumento de K (permeabilidad) => aumento de la velocidad Aumento de h (espesor) => disminución de la velocidad Î Ï“ (diferencia de presión osmótica) Î P (diferencia de presión hidrostática) => aumento de Ph (hidrostática) => más le cuesta entrar al agua. Î P desaparece al realizar un orificio en la membrana. Si el flujo de entrada de agua = flujo de salida de medicamento dM / dt = dV / dt â ª Cs Cs = concentración saturada de principio activo. AsÃ− podemos obtener perfiles de cesión definidos y de orden 0. Se usan para administración oral y ocular. + Ventajas: >conseguir perfiles de orden 0 >adaptables a distintos medicamentos >cesión independiente del entorno + Inconvenientes: >caros >control de calidad complejo => agujero con láser >existen antecedentes de obstrucción intestinal por acumulación del fármaco - Intercambio iónico Están constituidos por una resina de intercambio iónico a la que se une el medicamento. Cuando se administra y se encuentra en el medio con un ion competidor que tenga más afinidad por el medicamento, éste se unirá a la resina y se liberará el medicamento. Variables crÃ−ticas: + Ôrea de difusión: de ella depende la cantidad de iones que accedan al interior de la formulación 10 + Camino que tenga que seguir la difusión a través de los poros o de la resina hidratada. + Rigidez o viscosidad del sistema. Se usan sobre todo por vÃ−a oral. +Ventajas: >protegen al principio activo de la degradación enzimática. + Inconvenientes: >la cesión depende de la concentración de iones en el medio. Presenta más variabilidad interindividual. TEMA 4: CÔPSULAS Son formas sólidas que presentan una cubierta de tamaño y capacidad variable, destinadas a la vÃ−a oral. Se distinguen 2 tipos: 1- GELATINA Rà GIDA - Formulación de principios activos a dosis muy altas con malas propiedades de compresión. - Formulación conjunta de principios activos incompatibles. - Formulación de sistemas multiparticulares = gránulos de tamaño grande, pellets, por su estructura suelen dar lugar a formas de cesión sostenida. 2- GELATINA BLANDA - Inclusión de principios activos lÃ−quidos, disueltos o dispersos. El lÃ−quido debe ser de naturaleza oleosa; no puede ser acuoso porque solubilizarÃ−a la gelatina. - Principios activos con problemas de biodisponibilidad. El principio activo ya está disuelto por lo que se absorbe muy rápidamente. - Principios activos volátiles, hidrolizables u oxidables. 1- GELATINA Rà GIDA Cubiertas Se compran hechas, no las fabrican las compañÃ−as. Componentes: - Gelatina: se ha utilizado debido a sus propiedades >por su supuesta falta de toxicidad. Aunque está en entredicho debido a que es una proteÃ−na que se extrae de tendones, cartÃ−lagos,... >dispersable en fluidos biológicos => se disuelve inmediatamente salvo que añadamos algo que lo insolubilice 11 >da lugar a pelÃ−culas resistentes >los cambios de fase sólido - lÃ−quido son reversibles a baja temperatura (aproximadamente Tª corporal) >existen distintos tipos de rigidez y viscosidad variables - Plastificantes: >proporcionan elasticidad a la cubierta >glicerina, propilenglicol, goma arábiga, sorbitol - Colorantes: >solubles: azoicos y no azoicos >insolubles: óxido de titanio (opacificante), óxidos de hierro (rojo, negro y amarillo) - Agentes conservadores: la gelatina incorpora agua por lo que puede contaminarse >antifúngicos >antimicrobianos - Modificadores de la disgregación: las hacen gastrorresistentes >acetoftalato de celulosa Existen distintos tipos de cubiertas: >convencionales >gastrorresistentes >para liberación en zonas especÃ−ficas Las cápsulas están formadas por 2 partes: tapa y cuerpo. El diseño no ha variado a excepción del cierre o hendidura ahora más seguros. Se han elaborado en distintos tamaños; cada tamaño de corresponde con un volumen interior estandarizado. Los más grandes (000 y 00) se usan para el ganado, del 0 al 4 para terapéutica humana y el 5 para animales de experimentación. Contenido - Principio activo solo o junto a otros componentes (sólido pulverulento) - Granulado: el llenado es volumétrico => el producto debe tener buenas propiedades de flujo, para ello se recurre al granulado. - Puede incorporarse un granulado por una parte y un polvo por otra. - Un sólido en comprimidos para evitar el recubrimiento de comprimidos con una cubierta entérica que es más complicado. 12 - Comprimidos junto a material pulverulento o granulado para modular la velocidad de disolución. - Incorporar una cápsula de menor tamaño junto a sólido pulverulento o granulado. - Pellets de distintos tipos o pellets + sólido pulverulento. - Incorporación de un material que se funde o tixotrópico => material que al removerlo se fluidifica. Objetivos - Liberación inmediata - Liberación en zonas especÃ−ficas - Liberación gradual - Obviar las incompatibilidades Cálculos para la selección de la cubierta Para sólidos pulverulentos: el volumen que entra en cada cápsula, está en relación con su densidad aparente. Ï” aparente = masa material / V ocupa V ocupa: se mide en una probeta graduada sin apelmazarlo V ocupa = masa / Ï” ap = V cápsula Como existe un número de cápsulas definidas, se han calculado los volúmenes para todas las masas y Ï” ap posibles (normograma): Ï” ap masa / cápsula 1´8 0´7 g En el punto de corte de la recta de cápsulas y la recta con las caracterÃ−sticas de nuestro producto, se encuentra la cápsula. Si está en medio hay que elegir la mayor. Puede modularse la cantidad de principio activo que entra añadiendo excipiente para completar si elegimos una cápsula un poco mayor. Producción de cápsulas - Preparación del material a encapsular: >a pequeña escala: se coloca una cápsula abierta en cada alvéolo, añadir el polvo, enrasar, cerrar y valorar. >a escala industrial: tolva con cápsula cerrada, sistema que las orienta y las gira para que el cuerpo esté 13 hacia abajo, sistema de aspiración que quita la tapa, tolva con polvo que las rellena y cierre. Luego rodillos que las pule y abrillanta y selección de las defectuosas. 2- GELATINA BLANDA Se diferencian de las rÃ−gidas en que llevan como plastificante glicerol en elevado porcentaje y la gelatina es menos densa. La composición general es similar a la de las rÃ−gidas: colorantes, opacificante, acetoftalato de celulosa, conservantes. En el interior se encuentra un lÃ−quido inmiscible en agua: aceites vegetales, minerales y triglicéridos de cadena media y otro miscible en agua: PEG400 y PEG600. En ambos casos no volátiles. El llenado y sellado se realiza a la vez y se puede hacer de 2 modos diferentes: - Matrices rotativas cilÃ−ndricas - Inyectores concéntricos En ambos casos: 1º- dispersión del principio activo, del resto de ingredientes y de la glicerogelatina. 2º- aquÃ− aparecen las diferencias entre los 2 métodos: - Matrices rotativas cilÃ−ndricas Existen 2 tanques para la mezcla de glicerogelatina y la dispersión del medicamento. La glicerogelatina forma una lámina por un lado y otra por otro, se unen y se mete la disolución en medio antes de cerrar. - Inyectores concéntricos Mezcla de glicerogelatina en un tanque y del medicamento en otro, se unen los 2 inyectores y se forma una doble gota que cae en un colector, se centrifuga, separa, pule y abrillanta. Control de calidad Se habla de calidad a lo largo de todo el proceso aunque ahora prefiere hablarse de aseguramiento de la calidad: 1º- Control de la materia prima 2º- Control del proceso 3º- Control del producto acabado En las de gelatina rÃ−gida lo habitual es que el control de calidad de la industria que produce las cubiertas mande a la industria farmacéutica documentación especÃ−fica de la calidad de la cápsula: - Control microbiológico de las cubiertas 14 - Espesor - Dimensiones Control de calidad de los principios activos y excipientes que se van a emplear - Evaluación de caracterÃ−sticas especificadas en las farmacopeas - Evaluación de las caracterÃ−sticas crÃ−ticas de los excipientes para llevar a cabo con éxito el producto deseado Control del proceso Evaluación de los parámetros crÃ−ticos de los productos intermedios: - Propiedades de flujo de las mezclas >mediante el ángulo de reposo o control masa >por el tamaño de partÃ−cula Si tiene malas propiedades de flujo, existe mala dosificación - Contenido en humedad: si el producto queda húmedo provocaremos una mala encapsulación. Estos controles los establece cada empresa farmacéutica. - Evaluación visual del proceso de impresión (señores con pinzas y gafas de aumento. Control del producto acabado - Calidad microbiológica de formas farmacéuticas orales: >solubilización de la cápsula en un determinado volumen de lÃ−quido. >siembra en un medio de cultivo adecuado, incubación y recuento de colonias. >lÃ−mites: no más de 1000 bacterias aerobias viables por ml. no más de 100 hongos por ml. no Escherichia coli - CaracterÃ−sticas farmacotécnicas o galénicas: >uniformidad del contenido en principio activo (prácticas) >uniformidad de masa: hay que asegurarse de que la cápsula lleva la dosis adecuada (prácticas): se pesan 20 unidades individualmente, se calcula la media y la desviación estándar y se ve si el lote está entre los lÃ−mites establecidos que son 7´5 - 10 % respecto a la masa media. >disgregación: aptitud de la cápsula para disgregarse en medio lÃ−quido. 15 >disolución del principio activo: aptitud de las cápsulas para verter el principio activo de modo adecuado. Para llevar a cabo los 2 últimos puntos se utilizan: â ª Disgregadores con cubetas en las que está inserto un rotor. Lleva 6 cilindros de vidrio y en cada uno va una cápsula y sobre ellos puede ir un disco. El rotor sufre movimiento ascendente y descendente que es lo que da lugar a la disgregación del principio activo en el lÃ−quido que puede ser agua, jugo gástrico o tampón fosfato con jugo entérico. LÃ−mite = 30 min. sin discos. Esto se utiliza para cápsulas rÃ−gidas, en las gastrorresistentes la primera evaluación se hace en jugo gástrico y posteriormente en jugo entérico. Pasan el ensayo si la disgregación se produce en el jugo entérico. Las dimensiones del equipo están caracterizadas en las farmacopeas por lo que los datos obtenidos son comparables con los de otros laboratorios. â ª Disolución: proceso estandarizado y caracterizado en farmacopeas que señalan como aparatos posibles: + tipo I: baño termostatizado con 6 cubetas con un vástago donde va la cápsula a analizar. Cada cubeta lleva también medio de disolución caracterizado también en la farmacopea. + tipo II: se produce agitación con una paleta y la cápsula estará en el interior de una cubeta. Medio de disolución = HCl 0´1 N -> jugo gástrico Se coloca una cápsula por cubeta, se agita y se va produciendo la disolución. Medimos la concentración disuelta a distintos tiempos y se evalúa la cantidad de principio activo que existe en la muestra. Representamos % disuelto frente al tiempo: 100 % cant disuelta t Para una cápsula de cesión inmediata, el tiempo de disolución del 100 % será corto (aproximadamente 1 hora) Si es de cesión sostenida el tiempo será mucho más largo: Si fuese orden 0 100 % % disuelto si existen pellets 16 t + Aparatos de flujo continuo Para elaboración de cápsulas entéricas que permiten cambiar de medio de forma progresiva. Poseen uno o varios depósitos de medios de disolución, una bomba que bombea medio gástrico o entérico y lo lleva a la cámara donde está la muestra a 31º C. El medio disuelve el principio activo y luego hay un colector que nos mide la cantidad disuelta a distintos tiempos y obtenemos una curva. Se programa el aparato de tal forma que se muestre el medio al que se expone la cápsula. Ej. : la digestión dura 2 horas -> en el programa sometemos el medicamento a 2 horas de exposición al medio gástrico. Una vez ha pasado el control de calidad se tienen en cuarentena y continúa el proceso de producción con el envasado. Los lotes después de la cuarentena van al mercado, se mantiene una muestra 3 - 4 años en cuarentena para detectar cualquier fallo. TEMA 5: COMPRIMIDOS Formas sólidas que se obtienen por compresión enérgica de una mezcla de principios activos y excipientes. + Ventajas: >administración cómoda; bien aceptado por los pacientes >permiten el acondicionamiento de dosis elevadas en pequeños volúmenes (diferencia con cápsulas) >aptos para producción a escala industrial; baratos Comprimidos convencionales = liberación inmediata del principio activo. Comprimidos especiales: - Efervescentes - Liberación en zonas especÃ−ficas â ª bucales â ª sublinguales â ª entéricos â ª de liberación colónica - Cesión sostenida Compresión 17 No todos los materiales sirven para elaborar comprimidos: - Buenas propiedades de flujo: el llenado de las matrices se hace de forma volumétrica. - Buenas caracterÃ−sticas de comprimibilidad. + Propiedades de flujo >tamaño (elevado) y forma de partÃ−cula (redondeada). >propiedades superficiales (más liso -> mayor fluidez). >densidad (mayor densidad, mejor flujo). + Comportamiento frente a la compresión (comprimibilidad) >plasticidad (ej. : plastilina) >elasticidad (ej. : gominola) >tendencia a la fragmentación (ej. : cereales) No solo depende de la intensidad de la fuerza, sino también de la velocidad => el mecanismo de deformación varÃ−a. Debemos usar una mezcla de materiales que den lugar a una buena comprimibilidad. Cuando aplicamos una fuerza sobre un material, se van a reordenar las partÃ−culas: EI II III IV Log F + Efectos de la fuerza de compresión sobre la porosidad de un material pulverulento: >fase I: reordenación de las partÃ−culas >fase II: deformación elástica >fase III: deformación plástica o fragmentación particular >fase IV: deformación de la red cristalina Procedimientos de elaboración + Compresión directa 18 Elaboración a partir del principio activo solo o junto a una serie de excipientes. Ej. : ac acetilsalicÃ−lico => se le añaden excipientes para mejorar el disgregado. Etapas: >pulverizar el principio activo. Si es necesario, debe adecuarse el tamaño del principio activo a los excipientes, si no aparecerán procesos de segregación. >mezclar (mezcladoras en V o cúbicas) el principio activo con los excipientes: â ª diluyentes: destinados a incrementar el volumen de cada unidad de dosificación y hacerlo más manejable. * celulosa microcristalina (avicel®) -> vegetal * lactosa SD -> animal * fosfato dicálcico dihidratado (emcompress®) -> mineral â ª disgregantes: facilitan el desmoronamiento de los comprimidos. * carboximetilcelulosa sódica reticulada (sin reticular es aglutinante) â ª lubrificantes: favorecen el flujo del material o evitan adherencias del material a las partes metálicas de la máquina. * estearato de magnesio * talco * sÃ−lice coloidal >compresión â ª limitaciones * capacidad de incorporación de principio activo limitada según sus propiedades. Si se comprime mal y tenemos que añadir mucho excipiente -> comprimido muy grande. * segregación de componentes -> materiales con distintos tamaños. * producción de polvo -> problemas de toxicidad, posibles mezclas con otros fármacos (contaminación cruzada). * excipientes de compresión directa muy caros. + Compresión previa granulación Granulación: incrementa el tamaño de partÃ−cula de un material. - previene problemas de segregación 19 - mejora las propiedades de flujo y compresión - reduce la contaminación cruzada Puede ser granulación vÃ−a húmeda o vÃ−a seca - Granulación por vÃ−a húmeda (granulación convencional) Se mezcla el principio activo y los excipientes (diluyentes, disgregantes, adsorbentes y colorantes. En algunos casos el aglutinante se incorpora en seco en esta mezcla (CMCNa sin reticular) o también puede añadirse dispersada en el lÃ−quido de humectación. Luego se procede al malaxado hasta que alcanza unas caracterÃ−sticas adecuadas. Se hace pasar la masa humectada a través de una malla de unas dimensiones adecuadas y los gránulos deben secarse (estufa, lecho fluido) Se someten a una doble tamización para separar la fracción de interés. La parte que pasa el tamiz de menor abertura de malla son los finos. Se le incorpora el lubrificante y se vuelve a mezclar. Los gránulos ya tendrán unas propiedades de flujo adecuadas. - Granulación en lecho fluido Consiste en suspender las partÃ−culas de polvo en una corriente de aire en una columna de lecho fluido. Se provoca la dispersión de una sustancia aglutinante y al interaccionar las partÃ−culas van formándose los gránulos a medida que se va evaporando el lÃ−quido en el que está disuelto el aglutinante. Esto es muy rápido, normalmente se hace un secado del granulado (12 h. en estufa o microondas) después de la granulación convencional; en lecho fluido con la corriente ya se seca. - Granulación por vÃ−a seca El procedimiento transcurre en ausencia de lÃ−quidos (los disolventes no acuosos no son recomendables por los vapores, peligro de explosión y su eliminación para evitar contaminación). Esto supone una ventaja desde el punto de vista de la estabilidad; gran parte de las degradaciones de fármacos son debidas a hidrólisis. Se mezclan todos los componentes: principio activo + diluyente + disgregantes + aglutinantes + colorantes. Se compacta la mezcla utilizando compactadores = dispositivos que actúan de forma similar a máquinas de comprimir que dan lugar a briquetas (briqueteado). También se utilizan compactadores de rodillos que dan lugar a láminas. Se pulverizan y se obtienen fracciones homogéneas después de tamizar; se añade el lubrificante que forma una pelÃ−cula en torno a los gránulos y disminuye las fuerzas de fricción entre ellas. - Granulación en paila Se usa si algún componente cede agua (también si existe algún componente lipÃ−dico) que actúa como aglutinante y facilita que las partÃ−culas se unan. Esto, junto al giro produce la formación de gránulos aproximadamente esféricos. 20 Excipientes para compresión previa granulación >Diluyentes: â ª Almidón/lactosa (60/40%) Tienen propiedades que se complementan. El almidón es insoluble pero muy hidrófilo (se hincha) y es un disgregante eficaz. La lactosa es soluble. â ª Celulosa microcristalina: es insoluble en agua e hidrófila y disgregante. â ª Fosfato dicálcico dihidratado >Absorbentes: no siempre están presentes. Disponen la necesidad de incorporar lÃ−quidos (principio activo lÃ−quido, extracto vegetal). También se utilizan como protectores frente a la humedad de principios activos que se hidrolizan fácilmente. Los absorbentes fijan el agua. Almidón, sÃ−lice coloidal >Aglutinantes: aumentan la cohesividad. PVP, CMCNa sin reticular. >Disgregantes: se añaden para que cuando los comprimidos se utilicen puedan disgregarse y ceder el principio activo. Mecanismos: â ª Liberación de gas: efervescencia -> disgregación antes de la ingestión. Carbonato o bicarbonato + ac orgánico â ª Hinchamiento: desmoronamiento por fragmentación. Almidón â ª Disolución: lactosa es soluble y se erosiona el comprimido >Lubrificantes: forman una pelÃ−cula que disminuye las fuerzas intergranulares y promueven el flujo. También actúan como antiadherentes en el punzón de la máquina. Talco, estearato magnésico. >Correctivos organolépticos: colorantes, edulcorantes, aromatizantes. >Otros: antioxidantes, reguladores de pH (evitar inestabilidad y efectos irritantes) Comprimibilidad: aptitud de un material para dar lugar a un comprimido Compresión Un comprimido se forma aplicando a una masa de polvo que contiene una cantidad de principio activo determinada, una presión para que se agreguen y den lugar a una unidad compacta. La presión se aplica en una máquina de comprimir, en lo que se conoce como matriz => orificio en principio cilÃ−ndrico dentro del cual se desplazan los punzones, uno inferior que determina el peso según su posición y el superior que aplica la fuerza. Existen 2 tipos de máquinas de comprimir: â ª Rotativas: para producir grandes lotes, velocidad más elevada. â ª Excéntricas: para pequeña producción, laboratorios de investigación. Las 2 cuentan con 3 elementos básicos: 21 ->Matriz-punzones: determinan la forma y el anagrama o letras. Las máquinas excéntricas sólo tienen punzón superior y las rotativas, inferior y superior. ->Sistema de alimentación o tolva: hace llegar el polvo o granulado a la matriz. Puede ser fija (en las rotativas) o móvil (en las excéntricas). ->Dispositivo de expulsión: en las excéntricas, el movimiento de la tolva hace posible el desplazamiento del comprimido una vez realizado; en las rotativas existe un elemento metálico. Problemas durante la compresión 1- Excesiva variabilidad en el peso de los comprimidos: es inevitable que existan diferencian de peso, pero si suceden estas cosas la variabilidad será mayor a la inherente al proceso. Causas: >desplazamiento incorrecto del punzón inferior en la matriz. >deficiente lubrificación: produce deficientes propiedades de flujo. Si nos pasamos aparecen problemas mecánicos y de cesión. >excesivo contenido en finos: se pueden colar entre la superficie lateral de los punzones y la matriz. >holgura del punzón inferior en la matriz. >excesivo contenido en humedad del granulado: se producen adherencias al punzón. 2- Formación de crestas = que aparezca un reborde. Esto es debido a que se haya colado polvo en el espacio que queda entre los punzones y la matriz debido a holguras o defectos en los punzones. Se produce elevada friabilidad. 3- Aparición de defectos en la superficie: estrÃ−as en los laterales, irregularidades en la superficie o falta de brillo. Causas: >deficiente lubrificación. >falta de capacidad antiadherente de la superficie. Se resuelve lubrificando adecuadamente. 4- Exfoliación o capping: Normalmente se produce horizontalmente; el comprimido se rompe en dos capas. Es dependiente de la velocidad a la que tenga lugar la compresión; según ésta los materiales varÃ−an sus propiedades. Cuando aplicamos una fuerza a una masa que se encuentra en una matriz, la fuerza se transmite a las paredes laterales y a la parte inferior del punzón. Si tenemos un lÃ−quido ideal, las fuerzas se transmitirÃ−an a todos los puntos de la superficie por igual. Como tenemos partÃ−culas sólidas aparecen fricciones entre las partÃ−culas. Zonas en las que la transmisión de fuerzas es mayor 22 Esta diferencia en la aplicación de la fuerza se va a traducir en el comprimido en la aparición de zonas más densificadas que otras. La distribución de fuerzas heterogénea es tanto mayor cuanto peor lubrificado esté el comprimido. La densificación de un material es el resultado de 3 procesos que son los que tienen lugar cuando aplicamos una presión: >deformación elástica: el material recupera su forma después de aplicar la fuerza. >deformación plástica: se mantiene después de interrumpir la aplicación de la fuerza. >fragmentación En la práctica estos 3 procesos coexisten; en un momento determinado aparecen los 3 procesos en distintos puntos del comprimido porque las fuerzas no se distribuyen homogéneamente. Esto supone que se generan tensiones importantes en el comprimido y el resultado va a ser la exfoliación. Podemos intentar resolverlo: >lubrificar mejor el material a comprimir. >ajustar la velocidad de la máquina de comprimir a las caracterÃ−sticas del material. Control de calidad en la producción Se consideran 3 aspectos: >materias primas: principio activo y excipientes. >proceso de fabricación. >producto acabado. Cuando se recibe un material, se almacena en cuarentena hasta que el departamento de control de calidad da su autorización de uso, por cumplir las especificaciones de las farmacopeas y las de la empresa. El control del proceso de producción consiste en evaluar una serie de caracterÃ−sticas de productos intermedios: mezclas homogéneas, granulados adecuados,... y también la realización de determinaciones a pie de máquina (peso, dimensiones, resistencia a la rotura,...) Con estos dos controles prevenimos que un lote no cumpla las caracterÃ−sticas especificadas. Control del producto acabado: >control microbiológico: no son formas estériles a diferencia de colirios e inyectables, pero deben cumplir unos requerimientos mÃ−nimos de pureza microbiana recogidos en las farmacopeas: no más de 1000 bacterias aerobias viables por ml. no más de 100 hongos por ml. ausencia de Escherichia coli. 23 El control se realiza mediante un cultivo en un medio adecuado, incubación y recuento en placa. >caracterÃ−sticas farmacotécnicas o galénicas: â ª uniformidad de contenido en principio activo â ª uniformidad de masa -> 20 unidades. LÃ−mites 7´5-10% respecto a la masa media. â ª resistencia a la rotura evalúan resistencia mecánica. â ª pérdida de peso por friabilidad La resistencia a la rotura se mide aplicando una fuerza sobre un comprimido colocado verticalmente en durómetros. La pérdida de peso por friabilidad está relacionada con la tendencia a perder partÃ−culas por erosión. Se utiliza un tambor giratorio con un asa que produce el roce de los comprimidos; se pesan antes y después. AsÃ− se evalúa su resistencia para mantenerse Ã−ntegros en las manipulaciones posteriores. â ª ensayo de disgregación: asegura que los comprimidos en contacto con el agua se desintegran en un perÃ−odo de tiempo razonable para poder ceder el principio activo. Se establece un tiempo de disgregación máximo para los comprimidos según el tipo de principio activo. â ª ensayo de disolución: se evalúa la velocidad de disolución del principio activo a partir de la formulación (se obtiene un perfil de disolución). % disuelto t Se realiza “in vitro” en un aparato de disolución. Al realizarse en un paciente la gráfica no es superponible, pero estos ensayos nos dan una idea de cómo va a ir el proceso. Tienen un valor comparativo entre diferentes preparados. Si queremos extraer conclusiones más exactas, debe establecerse una correlación in vivo-in vitro entre los dos perfiles obtenidos con datos en las mismas condiciones. El ensayo de disgregación es anterior al de disolución. Se introdujo en el momento en que se detectaron problemas en formas sólidas similares a otras que sÃ− eran eficaces. Se comprobó que era debido a un fallo en la cesión del principio activo y se observaba que in vitro tenÃ−an un tiempo de disgregación alto, por lo que se incluyó el tiempo de disgregación como dato en las farmacopeas; pero al profundizar en las bioequivalencias, se comprobó que era preferible incluir como criterio de calidad el tiempo de disolución debido a que no siempre existe una correlación entre valores de tiempo de disgregación y velocidad de disolución. Estas diferencias eran debidas a la evolución del proceso de disgregación. En general los comprimidos se elaboran por compresión de granulados y la disgregación de un comprimido con ese origen tiene lugar en dos fases: 1º desintegración = separación de gránulos 24 2º desagregación de los gránulos para dar lugar a las pequeñas partÃ−culas que hemos aglomerado para hacer el gránulo. El ensayo de disgregación se da por concluido cuando el primer proceso de completó (desintegración). t disgregación (de comprimido a gránulo) es independiente de que los gránulos de desagreguen dando partÃ−culas más rápida o más lentamente. Cuando se administra un comprimido, la cesión del principio activo tiene lugar desde el comprimido; pueden disolverse partÃ−culas de la superficie, desde los gránulos y también desde las partÃ−culas, aumentando progresivamente la velocidad. La disolución producida por estas 3 vÃ−as da lugar a la absorción del principio activo en el tracto gastrointestinal para pasar a la circulación sistémica. La velocidad de disolución está condicionada principalmente por la velocidad a la que se produzca la desagregación. TEMA 6: COMPRIMIDOS ESPECIALES Todos los que no son de liberación inmediata y no deban ceder inmediatamente el principio activo después de la deglución. Tipos: - Efervescentes: se disuelven en agua antes de tomarlos. - Múltiples: están constituidos por granulados diferentes, en forma de capas distintas o en forma de núcleo y cubierta diferentes. - Liberación en zonas especÃ−ficas: â ª Masticables ceden el p.a. en la boca, absorción bucal â ª Bucales y sublinguales â ª Entéricos: diseñados para ceder el principio activo en el intestino y evitar la acidez para principios activos inestables en medio ácido o que se hidrolicen. â ª De liberación colónica: algunos principios activos se absorben eficazmente. Sobre todo interesantes para enfermedades locales (colitis ulcerosa, enfermedad de Chrom). Son especialmente útiles para acceder al colon proximal. Esto se consigue aprovechando las caracterÃ−sticas especÃ−ficas del medio intestinal en estas zonas, normalmente el pH. En el caso del colon lo más adecuado suele ser aprovechar la actividad metabólica de las bacterias fecales que da lugar a la presencia de enzimas no presentes en otras zonas del organismo => azorreductasas â ª De cesión sostenida: proporcionan niveles eficaces de principio activo, hacen más cómodos los regÃ−menes en tratamientos prolongados, los niveles plasmáticos se mantienen en el nivel óptimo para que sigan unas pautas muy incómodas, porque cuanto más complicada es la pauta más fácil es olvidarse del tratamiento. 25 COMPRIMIDOS EFERVESCENTES Son una variedad de comprimidos especiales que no cubren un gran espacio en la terapéutica, aunque existe una serie de razones que justifican su uso: Desde el punto de vista terapéutico: 1. Facilitan la administración porque llevan edulcorante 2. La presencia de CO2 promueve que se intensifique el riego sanguÃ−neo en el estómago y favorece la absorción 3. Resultan atractivos para el paciente Desde el punto de vista tecnológico la mezcla efervescente cumple 2 funciones: 1. Facilita que el comprimido se disgregue en contacto con el agua 2. El propio CO2 disuelto en el H2O produce un efecto atenuador de las papilas gustativas Todo esto supone que el proceso de elaboración de la mezcla sea complejo porque: - Se activa en contacto con el agua. Si existe humedad no controlada se va a inactivar. - Tiene que desarrollarse en un medio de humedad controlada => humedad relativa del 20%. - El personal que trabaja sufre la incomodidad de ambientes en condiciones de humedad relativa baja. Procedimientos de elaboración Podemos preparar dos granulados vÃ−a húmeda por separado, uno con cada componente de la mezcla efervescente, pero tenemos que asegurar la humedad relativa adecuada. Otro modo de hacerlo es utilizar otro lÃ−quido, como disolventes orgánicos que no promuevan el desprendimiento del CO2, pero tiene inconvenientes: 1. Relacionado con el personal y la posible toxicidad de los vapores de estos disolventes 2. Inflamabilidad de los disolventes. Hay que adecuar los sistemas de seguridad por riesgo de incendio 3. Posibles residuos de disolventes orgánicos en la forma de dosificación. Las farmacopeas clasifican según el riesgo y pata cada grupo fija unos lÃ−mites máximos Las alternativas a esta granulación vÃ−a húmeda son: - Granulación vÃ−a seca - Granulación en paila: utilizando ácido cÃ−trico es posible realizarla. Al calentar las moléculas de agua son suficientes para promover la granulación sin que llegue a producirse liberación de CO2 Ensayos 26 Se someten a los controles habituales y además: - Contenido en humedad < 0´1% - Tiempo de disgregación < 5 min. COMPRIMIDOS Mà LTIPLES Pueden ser multicapa o con núcleo. No se utilizan demasiado, pero cuando se usan, su función es formular conjuntamente principios activos incompatibles y conseguir perfiles de liberación modificados. Ej: paracetamol + ácido ascórbico. Se elaboran utilizando máquinas especiales y el proceso es muy complicado, suelen aparecer problemas: - El núcleo tiene que quedar perfectamente centrado - La cubierta tiene que adherirse a la superficie del núcleo y para ello es necesario establecer las caracterÃ−sticas de los granulados y una variable crÃ−tica es la porosidad. COMPRIMIDOS DE LIBERACIà N EN ZONAS ESPECà FICAS - Masticables > CaracterÃ−sticas: â ª Actúan en la boca â ª En la disgregación o disolución van cediendo el principio activo en la cavidad bucal â ª Principios activos utilizados: analgésicos, antiácidos, antisépticos > Procedimiento de elaboración: granulación por vÃ−a húmeda â ª Se utiliza manitol como diluyente, cuando se disuelve absorbe calor, el proceso de disolución es endotérmico. â ª Es importante dotarlos de caracterÃ−sticas de sabor y olor agradables. Se usan edulcorantes como la sacarina, esencias de limón, menta,... - Disolución instantánea > CaracterÃ−sticas: â ª Se disgregan y disuelven en la boca inmediatamente después de haberlos ingerido. > Procedimiento: â ª Compresión con excipientes muy solubles. â ª Liofilización. Estas formas surgieron por los inconvenientes que presentaba el deglutir formas sólidas. 27 - Bucales > CaracterÃ−sticas: â ª Se aplican sobre la mucosa gingival (entre encÃ−as y carrillo). La absorción es rápida y evita el efecto de primer paso porque evita el paso por el aparato digestivo. â ª Se utiliza para principios activos inestables en medio ácido o que sufren efecto de primer paso (progesterona). - Sublinguales > CaracterÃ−sticas: â ª Administración bajo la lengua. â ª Absorción rapidÃ−sima, también evita el efecto de primer paso. â ª El comprimido debe tener una forma lenticular, sin superficies rugosas ni aristas. â ª No está bien aceptada por los pacientes. â ª Principios activos -> vasodilatadores coronarios (nitroglicerina). - Entéricos > CaracterÃ−sticas: Responden a la necesidad de que el principio activo se libera en el intestino y no en el estómago por diferentes razones: â ª Necesidad de proteger el fármaco de las condiciones de inactivación (pH gástrico). â ª Cuando la liberación del fármaco produzca algún efecto indeseable se retrasa para atenuar un efecto irritante. > Procedimiento: Se consiguen formas entéricas utilizando un excipiente insoluble en el estómago y soluble en el medio intestinal. La diferencia entre las condiciones es el pH que tiene una serie de variaciones según el paciente, contenido del estómago,... que pueden suponer una limitación para la formulación. Se pueden utilizar cubiertas gastrorresistentes de polÃ−meros acrÃ−licos (ej: Eudragit ®) que está comercializado en distintas variedades para distintos pH (con cesión dependiente de pH). También pueden usarse cubiertas gastrorresistentes de acetoftalato de celulosa. - Colónicos > CaracterÃ−sticas: â ª Ceden el fármaco en el colon. 28 â ª Se administran: + principios activos convencionales; ej: macromoléculas que se absorben vÃ−a linfática. + principios activos para tratamiento local. > Tipos: Llevar el fármaco hasta el colon y que se libere allÃ− puede conseguirse de distintos modos: â ª Mediante el uso de un profármaco, una molécula emparentada con la del fármaco y que en algún momento se transforma en éste. Ej: ác. 5-aminosalicÃ−lico es degradado por azorreductasas que sólo existen en el colon (producidas por bacterias) para dar el fármaco. â ª Comprimidos elaborados con polÃ−meros que sólo son degradables por la flora colónica (azorreductasas, glucuronidasas). â ª Comprimidos de liberación programada en el tiempo -> se evita que inicien la liberación en el estómago con una cubierta entérica. Suele introducirse el comprimido en un compartimento con un medio de polÃ−mero hidrofÃ−lico (pulsicap). COMPRIMIDOS DE CESIà N SOSTENIDA - Objetivo > Mantienen la concentración plasmática de principio activo por encima de la concentración mÃ−nima eficaz durante un tiempo prolongado. > Permiten reducir el número de tomas y evitar el incumplimiento de las pautas terapéuticas. > El uso de la forma de cesión sostenida contribuye a atenuar los niveles plasmáticos del fármaco en la ventana terapéutica. concentración máxima tolerada concentración plasmática intervalo terapéutico concentración mÃ−nima eficaz 6 horas tiempo Las fluctuaciones en ventanas terapéuticas pequeñas, pueden ser un problema y dar lugar a efectos secundarios, con este tipo de comprimidos éstas se reducen. En caso de que se produzca un fallo, el riesgo que suponen estos comprimidos es mayor que en una forma de liberación inmediata, por lo que el control de calidad debe ser muy riguroso. 29 - Fundamento La absorción se produce como consecuencia de la sucesión de 3 etapas secuenciales: 1. Liberación a partir de la forma de dosificación 2. Paso a través de la pared intestinal 3. Arrastre por la vena porta para la distribución sistémica La etapa limitante es en la que se produce la cesión de la forma farmacéutica, por lo que la velocidad de absorción se modula con la velocidad de cesión. à nicamente cuando la liberación es la etapa limitante modificando la velocidad de cesión variaremos la velocidad y el perfil. Se diseñan para fármacos que se absorben en todo el intestino y en un tiempo razonable, la existencia de ventanas de absorción en el intestino serÃ−a una limitación. Lo que hacemos es administrar la misma cantidad de principio activo, pero disminuir la velocidad de absorción. - Aproximaciones tecnológicas > Comprimidos de múltiples unidades que controlan individualmente la cesión: Las unidades pueden ser micropartÃ−culas o de tamaño mayor hasta 1 mm. = “pellets”. La forma de actuar de estos sistemas consiste en su disgregación en el aparato digestivo y regeneración de las partÃ−culas iniciales que proporcionan el control de la liberación. Generalmente tanto los pellets como las micropartÃ−culas deben estar recubiertos para conseguir una velocidad de liberación que se ajuste a la que necesitamos. A los pellets se les aplica una cubierta polimérica; se han intentado descubrir sistemas sin cubiertas entéricas que serÃ−an útiles con principios activos poco solubles. Si la compresión de estas unidades genera muchos problemas, se administran dentro de cápsulas rÃ−gidas. > Comprimidos que retienen el principio activo, liberándolo de forma progresiva: El comprimido actúa como elemento activo en el control de la liberación, se disgrega muy lentamente. Existen diferentes tipos de sistemas dentro de este grupo: 1.- Comprimidos matriz a) Matrices inertes Su componente mayoritario es un polÃ−mero insoluble en agua que se mezcla con el principio activo y los excipientes restantes y se comprime. Obtenemos una matriz inerte porosa en la que penetra el agua disolviendo el principio activo que se cede por difusión. Inconveniente: el excipiente es insoluble, la forma del comprimido no se modifica una vez administrado y 30 puede acumularse en ciertas partes del intestino dando lugar a obstrucción. b) Matrices hidrofÃ−licas Su componente mayoritario es un excipiente hidrofÃ−lico que tiene una gran afinidad por el agua que provoca la expansión de sus cadenas cuando entra en la matriz. Se forma una capa de gel que es objeto de erosión progresiva, hasta que transcurrido un tiempo la matriz desaparece. La capa de gel controla la liberación del principio activo por difusión y el perfil es tipo raÃ−z cuadrada. c) Matrices lipÃ−dicas Están formadas por el principio activo junto a excipientes de naturaleza lipÃ−dica que se va emulsionando y erosionando progresivamente por acción de sales biliares y se va liberando el principio activo con una cinética compleja. Inconveniente: las grasas experimentan cambios importantes en su comportamiento por lo que es difÃ−cil prever la cesión. 2.- Comprimidos de resinas cambiadoras de iones Medicamento de carácter iónico. El proceso de cesión es por intercambio en el medio en el que están presentes iones por moléculas de fármaco. Inconvenientes: perfil de cesión dependiente de la fuerza iónica 3.- Comprimidos osmóticos Dispositivo con estructura de reservorio, en cuyo interior está el principio activo junto a una sustancia de naturaleza osmótica. Se comporta como una membrana semipermeable; al entrar en contacto con el agua, la sustancia activa promueve un flujo de ésta constante, ya que la concentración de esta sustancia osmótica también es constante. La entrada de agua constante, proporciona una velocidad de cesión del principio activo de orden 0, independiente de las condiciones del medio. Inconvenientes: â ª Son dispositivos no disgregables, pueden dar lugar a problemas de obstrucción. â ª Son sistemas de estructura compleja y de fabricación complicada; los costes de control de calidad son elevados (control orificio de salida) CONTROL DE CALIDAD Comprende los mismos aspectos que en los comprimidos convencionales: 31 - Control de materias primas - Control de proceso - Control de producto acabado > Calidad microbiológica > Control galénico: cesión del principio activo importante: Comprimido con cubierta entérica: Debe realizarse un ensayo para comprobar que no cede el principio activo en medio gástrico y sÃ− en las condiciones intestinales. Puede hacerse un ensayo en aparato I o II; antes se realizaba en un solo medio de disolución, pero actualmente se utiliza jugo gástrico durante un cierto tiempo en el que hay marcado un lÃ−mite superior de cesión; y posteriormente se sustituye por jugo entérico con el fin de asegurar que en el jugo gástrico la cantidad no supera el lÃ−mite superior y en el entérico no sea inferior. Comprimidos de liberación controlada: Debe realizarse un control de la velocidad de cesión en un aparato de flujo continuo que nos permite hacer una sustitución progresiva de jugo gástrico por entérico. Se producen cambios sucesivos de pH y las condiciones evolucionan lentamente. Este sistema es más parecido a las funciones fisiológicas que el cambio brusco del aparato I y II. TEMA 7: COMPRIMIDOS RECUBIERTOS OBJETIVOS: - Protección del principio activo frente a agentes atmosféricos - Enmascaramiento de sabor desagradable - Favorecimiento de la deglución del comprimido - Enmascarar posibles diferencias interlote derivadas de pequeñas diferencias en las materias primas - Facilitar la identificación por el fabricante, el farmacéutico y el paciente - Aumentar la resistencia mecánica de los comprimidos - Protección de principios activos frente a la degradación en el estómago - Reducción de efectos secundarios - Control de la cesión del principio activo TIPOS DE CUBIERTAS 32 - Cubiertas de sacarosa: son las que presentan una forma de dosificación denominada gragea. Inconveniente: funcionalidad, son permeables, higroscópicas, intercambian temperatura, humedad -> se forman fracturas. - Cubiertas peliculares poliméricas: son las más utilizadas. Están aprobadas con excipientes de caracterÃ−sticas y propiedades muy diferentes; el proceso de aplicación es más rápido y fácil que el proceso de grageado. - Cubiertas aplicadas por compresión: el material utilizado es un granulado. RECUBRIMIENTO EN PAILA Procedimiento clásico: utiliza una paila de grageado en la que se introducen los comprimidos que experimentan un desplazamiento relativo unos con respecto a los otros y al mismo tiempo se dispersa una disolución o dispersión acuosa del material formador de la cubierta y a tiempos progresivos se pasan corrientes de aire para eliminar el agua. Al evaporarse ésta, sobre los comprimidos se va formando una pelÃ−cula muy fina. - Dificultades que plantea el proceso â ª Es necesario que la disolución de recubrimiento se disperse en gotas muy pequeñas â ª Establecer una agitación uniforme para depósito homogéneo â ª Aportación de calor para evitar que se peguen los comprimidos y evaporar el disolvente â ª Eliminación de polvo y disolventes: preferiblemente utilizar agua y evitar orgánicos, por toxicidad y eliminación más fácil RECUBRIMIENTO EN PAILA CON SACAROSA ELABORACIà N DE GRAGEAS (GRAGEADO CLÔSICO) - Etapas 1. Aplicación de la pelÃ−cula de aislamiento: Partimos de comprimidos a los que queremos aplicar una cubierta (NUCLEOS) elaborados con punzones cóncavos. El objetivo de la pelÃ−cula es proteger al comprimido y a sus componentes frente a la humedad. Se realiza dispersando un polÃ−mero insoluble en agua y soluble en disolventes orgánicos. 2. Aplicación de la cubierta de aislamiento: El objetivo de la cubierta es facilitar la adhesión de las capas posteriores. Se aplican disoluciones de sacarosa que incorporan goma arábiga y gelatina, de forma sucesiva; entre las aplicaciones se añade un absorbente (talco). Finaliza cuando desaparecen los bordes de los comprimidos; desecado: 12 h. 33 3. Fase de alisado: Elimina las rugosidades; se realiza añadiendo en sucesivas fases una disolución diluida de sacarosa. 4. Coloración: Se añaden colorantes junto a una disolución saturada de sacarosa. Pueden ser colorantes hidrosolubles y colorantes insolubles o colorantes adsorbidos en soporte sólido. 5. Abrillantado: Suele llevarse a cabo en el bombo de abrillantado -> bombo cilÃ−ndrico con las paredes cubiertas de fieltro. Se incorpora parafina o cera de abejas o carnauba. 6. Impresión: Tintas no tóxicas. Es un proceso lento, por lo que si puede se sustituye por: RECUBRIMIENTO EN LECHO FLUIDO Es adecuado, casi exclusivamente, cuando el material de recubrimiento es polimérico. Se suspenden los núcleos a recubrir en una columna de lecho fluido y al mismo tiempo se dispersa el polÃ−mero que formará la cubierta. Preferiblemente se dispersará el polÃ−mero en agua. - Ventajas â ª Procedimiento muy rápido â ª Transferencia de calor muy eficaz, desecación casi instantánea. Suele ser necesario añadir plastificantes que le dan unas mejores propiedades mecánicas a los polÃ−meros, hacen más flexible la cubierta y evita la formación de grietas. Deben utilizarse lacas y colorantes insolubles en agua y en general insolubles en el disolvente que disperse el polÃ−mero porque existen movimientos muy rápidos de agua; si es soluble en agua aparece un color muy poco homogéneo. CUBIERTAS DE SACAROSA VS POLIMà RICAS Cubiertas de sacarosa Cubiertas poliméricas Comprimidos Aspecto Redondeado, pulido, brillante Incremento de peso Logotipo o ranura 30-50% No Formas convencionales y entéricas Funcionalidad Mantiene el contorno del núcleo, mate 2-3% Posible Formas convencionales, entéricas y de cesión sostenida Proceso 34 Entrenamiento del personal Adaptación GMP Etapas Complejo DifÃ−cil Múltiples Sencillo Más fácil Una CUBIERTAS GASTRORRESISTENTES O ENTà RICAS - Objetivos: â ª Prevenir la inactivación del principio activo â ª Reducir efectos secundarios â ª Evitar la disolución del principio activo en el estómago - Excipientes: â ª PolÃ−meros con grupos ácido carboxÃ−lico (acetoftalato de celulosa) CUBIERTAS PARA CESIà N SOSTENIDA - Excipientes: â ª PolÃ−meros acrÃ−licos, éteres de celulosa CONTROL DE CALIDAD - Control de materias primas: â ª Principio activo y excipientes - Control de proceso: â ª Productos intermedios = núcleos â ª Seguimiento de variables crÃ−ticas = Tª - Control de producto acabado: â ª Calidad microbiológica: requerimientos igual que comprimidos â ª Control galénico: > Control del aspecto externo: + Zonas con el núcleo descubierto: se produce por la adhesión entre unidades por secado en condiciones inadecuadas. + Fracturas en la cubierta, formación de puentes en las inscripciones o ranuras: se debe a una formulación inadecuada, incorrecto ajuste de la temperatura del aire. > Disgregación: 35 El control es caracterÃ−stico porque existen comprimidos de distintos tipos: + Cubierta gastrosoluble: ensayo en agua, si no se disgrega en el tiempo requerido, se hace en HCl 0´1 N. t => grageas = 60 min. // cubierta polimérica = 30 min. + Cubierta entérica: HCl 0´1 N y luego tampón fosfato 6-8. La integridad de la cubierta debe mantenerse 1 h. como mÃ−nimo y después de 2 h. sustituir el medio por tampón y debe disgregarse en 60 min. Se pretende garantizar la integridad de la cubierta en medio gástrico y luego su disgregación en el intestino. TEMA 8: FORMAS Là QUIDAS ORALES Clasificación: - Disoluciones: â ª Soluciones orales: disolución de un fármaco en agua y sustancias auxiliares (edulcorantes, correctores del color,...) â ª Jarabes: solución oral que incorpora una concentración elevada de azúcar (principalmente sacarosa o glucosa) â ª Elixir: solución oral donde el disolvente es una mezcla hidro-alcohólica en vez de agua únicamente. Suelen contener también azúcar o en todo caso edulcorantes sintéticos. â ª Colutorios: soluciones usadas para hacer enjuagues para el tratamiento de procesos locales en la cavidad bucal. - Suspensiones - Emulsiones VENTAJAS DE LAS FORMAS Là QUIDAS ORALES - Desde el punto de vista de la facilidad de manejo tecnológico, las formas orales lÃ−quidas son mejores que las sólidas. También lo son respecto a la dosificación, donde permiten hacer ajustes de dosis fácilmente, sobre todo en niños o en ancianos. - Fácil deglución. - Disminución de la irritación de la mucosa gástrica debido a la dispersión del fármaco en una zona más amplia de la mucosa que en el caso de formas orales sólidas. - Reducción de problemas de biodisponibilidad: evita el proceso de disolución previo a la absorción que ocurre en formas orales sólidas, ya que en las lÃ−quidas el principio activo ya está disuelto. Sin embargo el cambio de estado, después de estar muy disuelto en la forma oral lÃ−quida, al aparato digestivo, donde el ambiente no es tan adecuado, el principio activo precipita (esto ocurre en muy baja cantidad). JARABES - Funciones del azúcar: 36 â ª Conservante: Origina un medio hipertónico que impide el crecimiento de microorganismos. Sin embargo si existe una pequeña dilución del jarabe, éste se convertirá en un excelente medio de cultivo para los mismos. Si la dilución se produce en una zona determinada del jarabe, habrá una dilución parcial (en una zona determinada) donde podrá aumentar el número de microbios. â ª Viscosizante: Permite evitar la segmentación de distintos componentes del jarabe. â ª Solubilizante: La elevada concentración produce un cambio considerable en la constante dieléctrica, que puede ocasionar que distintos fármacos aumenten mucho su hidrosolubilidad. Es un ejemplo de hidrotropismo. â ª Edulcorante: Enmascara el sabor de los principios activos. - Componentes de los jarabes: â ª Agua purificada o destilada: debe cumplir los requerimientos establecidos en la farmacopea. â ª Azúcar (sacarosa): puede sustituirse por glucosa o por un viscosizante glucogénico + un edulcorante. La glucosa es al mismo tiempo viscosizante y edulcorante. Existen además viscosizantes no glucogénicos (no dan lugar a glucosa en el organismo tras su ingestión). Son especialmente útiles en diabéticos. â ª Conservantes: evitan que la contaminación que se produce al abrir un frasco del jarabe, no da lugar a la aparición de microbios o gérmenes. â ª Cosolventes: facilitan la solubilidad de sustancias poco polares modificando la constante dieléctrica (ε) â ª Correctivos organolépticos: aromatizantes y colorantes. - Elaboración de jarabes: 1.- Primero se prepara el jarabe simple (es una disolución de sacarosa que se puede preparar en frÃ−o o en caliente. â ª En frÃ−o: añadimos agua y agitamos. También podemos usar un percolador o sacarolizador para preparar el jarabe simple pero en mayor cantidad. â ª En caliente: aceleramos el proceso de disolución pero también facilitamos la hidrólisis de sacarosa = inversión parcial. Consecuencias de esta inversión: >se intensifica el sabor dulce >se reduce la viscosidad >sistema más susceptible de fermentar 37 >oscurecimiento porque se carameliza parte del azúcar 2.- Incorporación del principio activo: lo añadimos en polvo y disolvemos en el jarabe simple. Otras veces se parte de un extracto (solución o soluciones de principio activo). Se mezcla el lÃ−quido con jarabe simple en una proporción adecuada (5:95%) para que no se diluya mucho el principio activo. Disolución del azúcar en la disolución de fármaco. 3.- Clarificación: puede ser necesaria porque la disolución esté turbia y presente partÃ−culas en suspensión. Se puede hacer de dos formas: >filtración: filtros papel o filtros prensa >añadir un material insoluble o absorbente como la pasta de papel. Después filtramos. 4.- Alteraciones en los jarabes: desde el punto de vista de la estabilidad hay problemas: >posibilidad de que al cabo de un cierto tiempo se produzca un enturbiamiento o la aparición de un precipitado => puede ocurrir que la disolución de sacarosa esté sobresaturada (es un sistema metaestable que con el tiempo evoluciona la separación de dos fases) >los jarabes son susceptibles de sufrir alteraciones derivadas del crecimiento de gérmenes (básicamente hongos y levaduras capaces de crecer en medios bastante hipertónicos). Conque se produzca una dilución local del jarabe puede producirse un rápido crecimiento de gérmenes => aparecen burbujas de gas y colonias a simple vista. ¿Cómo podemos prevenir el riesgo de deterioro de un jarabe? +evitar que se produzcan diluciones locales +incorporar agentes antimicrobianos ¿Cómo se producen diluciones locales? Puede ser que acondicionemos el jarabe y lo ponemos en frascos no totalmente secos. También puede ser causada porque preparamos el jarabe en caliente y no esperamos a que enfrÃ−e (el vapor de agua condensa sobre el cuello del frasco) ELIXIRES Disoluciones hidro-alcohólicas de uno o más fármacos. Son lÃ−quidos y están edulcorados. Interés como forma de dosificación: escasa (se usan más en el mercado norteamericano) - Componentes de los elixires: â ª Agua purificada o destilada â ª Etanol (20%) â ª Cosolventes: propilenglicol, polietilenglicol, glicerina. También son viscosizantes â ª Edulcorantes 38 â ª Antioxidantes â ª Correctivos organolépticos: aromatizantes y colorantes â ª Conservantes - Preparación de elixires: La presencia de alcohol se justifica para resolver un problema de hidrosolubilidad. 1.- Disolución de componentes hidrosolubles en agua 2.- Incorporación de la sacarosa 3.- Disolución de los componentes solubles en etanol 4.- Mezclado de las disoluciones, añadiendo la acuosa sobre la oleosa 5.- Filtración en caso necesario 6.- Clarificación SUSPENSIONES ORALES Parecidas a las soluciones orales pero aquÃ− el principio activo se incorpora en forma de partÃ−culas insolubles. - Aplicaciones: â ª Si la solubilidad del fármaco no es suficientemente elevada (ej: mebendazol -> antiparasitario) â ª Ciertos fármacos en disolución saben muy mal: usamos algún derivado menos soluble que administramos en forma de suspensión (muchas veces es un profármaco) â ª Conseguimos una cesión sostenida (esperamos que el fármaco se ceda hacia la fase lÃ−quida) - Tipos de suspensiones: â ª Dispuestas para ser administradas: serán útiles cuando en fármaco no plantee problemas de estabilidad quÃ−mica. Los problemas serán de estabilidad fÃ−sica. Pueden comportarse como sedimentos aglomerados (difÃ−cilmente redispersables) o floculados (fácilmente redispersables). Los sedimentos aglomerados son tÃ−picos cuando la suspensión transcurre lentamente. Los floculados son tÃ−picos cuando la suspensión transcurre rápidamente (minutos-horas). Los floculados son más adecuados (agÃ−tese antes de usar). â ª Extemporáneas: el paciente las prepara justo antes de tomarlas. No importa que dé sedimento aglomerado porque lo tomamos antes de que sedimente. Las suspensiones extemporáneas interesan cuando el fármaco es inestable en contacto con el agua. También si es una forma de cesión sostenida. - Composición de las suspensiones: 39 Está condicionada por el hecho de que sean extemporáneas o preparadas para ser administradas. â ª Dispuestas para ser administradas: llevan un agente floculante. Se usan electrolitos (modifican el potencial Z) y coloides hidrofÃ−licos. Nos sirven para que el sistema tienda a flocular (sedimentos fácilmente redispersables). â ª Extemporáneas: llevan un agente suspensor -> reduce la velocidad de sedimentación pero cuando están presentes dan lugar a sedimentos aglomerados (no importa). EMULSIONES ORALES Formas lÃ−quidas que incorporan un medicamento lÃ−quido de naturaleza oleosa o un fármaco lipofÃ−lico disuelto en un vehÃ−culo oleoso como fase interna de una emulsión. - Aplicaciones: â ª Incorporar medicamentos oleosos o fármacos lipofÃ−licos que no pueden disolverse en agua. â ª Administración de vitaminas liposolubles. - Componentes: â ª Fase acuosa: agua, agentes conservantes, viscosizantes (aumentan la viscosidad de la fase externa y dificultan la coalescencia) y emulsificantes (disminuyen la tensión superficial). â ª Fase oleosa: antioxidantes (estabilizan los principios activos), secuestradores de iones (estabilizan los principios activos) y conservantes (protegen frente a microorganismos). CONTROL DE CALIDAD 1.- Control de materias primas: - Calidad del agua (imp): la farmacopea la estipula. 2.- Control de proceso: no tiene importancia porque es muy fácil. 3.- Control de producto acabado: - Control de calidad microbiológica: la farmacopea establece los lÃ−mites. - Control galénico: comprende: â ª Uniformidad de contenido en principio activo. â ª Uniformidad de masa. â ª Algunas soluciones orales están pensadas para ser dosificadas en gotas => control de uniformidad de contenido para asegurar que un determinado número de gotas tienen la cantidad de principio activo que interesa. - En jarabes debe controlarse: 40 â ª Densidad: relacionada con concentración de sacarosa. â ª Punto de ebullición: relacionada con concentración de sacarosa. â ª Viscosidad: determina la consistencia. Afecta a la palatabilidad del jarabe. También relacionada con cantidad de sacarosa. â ª Determinación de sacarosa: método Fehling. â ª Azúcar invertido: nos informa del procedimiento seguido en la preparación del jarabe (en caliente mayor cantidad). Puede estar relacionado con problemas de contaminación microbiana (actúan las invertasas). Método Fehling o polarimetrÃ−a. - En elixires debe controlarse: â ª Grado alcohólico: se controla midiendo la densidad y acudiendo a la picnometrÃ−a. â ª Determinación de sacarosa: método Fehling. â ª Azúcar invertido: método Fehling o polarimetrÃ−a. - En suspensiones debe controlarse: â ª Tamaño de partÃ−cula y distribución de tamaños medios (problemas de crecimiento cristalino -> interesa que la distribución sea homogénea). De estos dos factores depende la velocidad de sedimentación. â ª Propiedades reológicas: nos informan del tipo de sistema que estamos usando. - En emulsiones debe controlarse: â ª Viscosidad â ª Distribución de tamaños de gota â ª Movilidad electroforética: interesa porque nos permite hacer predicciones sobre la aglomeración de los glóbulos o de las partÃ−culas de la fase interna. A todo esto hay que añadir los controles de estabilidad del principio activo. TEMA 9: FORMAS FARMACà UTICAS DE ADMINISTRACIà N PARENTERAL - VÃ−a subcutánea (sc), intramuscular (im), intravenosa (iv), intradérmica son las más frecuentes. - VÃ−a intraarterial, intracisternal, intracardÃ−aca, intraabdominal, intraarticular, epidural son las menos frecuentes. Và A INTRACUTÔNEA O INTRADà RMICA Requiere la colocación del volumen adecuado de la forma de dosificación entre epidermis y dermis. Es para administrar volúmenes muy reducidos. Su campo de aplicación se limita a vacunas y determinados medios de diagnóstico. 41 Và A SUBCUTÔNEA Permite administrar fármacos como disoluciones, suspensiones o implantes sólidos. Se usa para administrar insulina. Las suspensiones e implantes se usan para conseguir liberación prolongada (cesión sostenida). Ej: depots. En forma de implantes se administran anticonceptivos y sobre todo agentes citostáticos que se usan básicamente en el tratamiento de cáncer de próstata. También antagonistas narcóticos (una vez implantados se produce liberación del fármaco como consecuencia de la biodegradación del polÃ−mero que lo constituye). Se usan preparados depot (microsferas de ácido poliláctico glicólico). Và A INTRAMUSCULAR Bastante frecuente. Permite la administración de suspensiones y disoluciones acuosas y oleosas. Las suspensiones acuosas son útiles cuando se requiere una cesión sostenida. Soluciones se usan cuando no se puede emplear vehÃ−culo acuoso. La inyección de suspensiones acuosas es más dolorosa que las soluciones acuosas. Suspensiones oleosas: muy dolorosas. Pueden producir problemas de enquistamientos. Se administran vÃ−a im fármacos de cualquier tipo, es alternativa a vÃ−a oral. Fuera del ámbito hospitalario no se suele usar (necesita personal especializado). Và A INTRAVENOSA Se usa para administrar fármacos de distintos tipos cuando se requiere efecto inmediato. Se usa con el objetivo de reponer lÃ−quidos y también para alimentación parenteral. VÃ−a parenteral se administran hidratos de carbono, proteÃ−nas y también lÃ−pidos como fase interna de una emulsión (controlar el tamaño de glóbulo de forma rigurosa). En vÃ−a oral, el acceso del fármaco a la circulación general implica paso a través del hÃ−gado. El proceso de absorción requiere tiempo considerable. En vÃ−as parenterales no hay efecto de 1º paso hepático. En la forma iv el paso a la circulación general es casi instantáneo. En general las formas parenterales dan lugar a una absorción completa de la totalidad de la dosis administrada. - Ventajas de la administración parenteral: â ª Respuesta rápida y reproducible â ª Adecuada para principios activos que no se pueden administrar vÃ−a oral. â ª Alimentación no enteral. - Inconvenientes de la administración parenteral: 42 â ª Sensación dolorosa, molestias. â ª Riesgos de infección o reacciones locales. â ª Administración por personal especializado. â ª Más complejas y más caras que las formas orales. La tecnologÃ−a de las formas parenterales es compleja porque deben cumplir unos requisitos muy estrictos en cuanto a contaminación por partÃ−culas. - Limpidez: es imposible que no exista ninguna partÃ−cula pero hay que asegurarse de que no contiene más partÃ−culas por unidad de dosificación de las que se marcan en los lÃ−mites. - Esterilidad: preparado parenteral (asegurarse de que está libre de contaminación biológica) - Apirogenia: ausencia de pirógenos -> producen una respuesta pirogénica: la manifestación más evidente es un aumento de temperatura corporal. Proceden de los microorganismos: + si proceden de la actividad metabólica de los microorganismos: pirógenos exógenos. + si proceden de la lisis de microorganismos (restos de la pared): endopirógenos o endotoxinas. Puede existir un sistema estéril pero contaminado por pirógenos. - IsotonÃ−a: relacionada con presión osmótica. Cuando se administra una forma parenteral lÃ−quida va a estar en contacto con las células del organismo. Es importante que presente caracterÃ−sticas osmóticas idénticas o similares a las del medio en que se encuentran las células sanguÃ−neas. Asumimos que la pared celular es una membrana semipermeable que intercambia sustancias y si existen diferencias osmóticas pueden producirse problemas. - pH: el fisiológico es aproximadamente 7´4. Es óptimo para un inyectable, aunque se toleran pH distintos (los ácidos se toleran mejor) pero producen sensación dolorosa; puede haber ciertas necrosis locales. REQUISITOS DE PREPARADOS INYECTABLES a) Esterilidad: ausencia de microorganismos viables en todas las unidades del lote. Se obtiene: - Trabajando en zonas adecuadas (limpias, blancas, donde la contaminación del ambiente está controlada). - Sometiendo las unidades del lote a un proceso de esterilización terminal (al final del proceso). à nicamente cuando existe algún problema relacionado con la estabilidad, no se lleva a cabo esta esterilización terminal (por calor húmedo). b) Ausencia de pirógenos (endotoxinas y exotoxinas): sustancias procedentes del metabolismo de los gérmenes o restos de microorganismos muertos capaces de provocar un proceso febril. Su actividad depende de la estructura del resto proteico y del resto polisacarÃ−dico, aunque la proteica es más importante. 43 - CaracterÃ−sticas quÃ−micas: â ª Solubles en agua: lo que permite que en la destilación sea posible su arrastre en corriente de vapor, a pesar de no ser volátiles. El agua se contamina como consecuencia del transporte de los pirógenos en moléculas de agua y no de su volatilización. â ª Son termorresistentes durante tiempos prolongados. Ej: destrucción de pirógenos por calor seco -> Tª = 250ºC y tiempo = 30 min. â ª Estables en disolución frente al calor; son únicamente destructibles a pH muy ácidos o básicos en calefacciones prolongadas. - Toxicidad de los pirógenos > Las endotoxinas (pared bacteriana) procedentes de G(-) se caracterizan por la especial intensidad de sus efectos. > La intensidad pirogénica (respuesta) depende de la especie animal. Tradicionalmente, la experimentación se hacÃ−a en conejos (son muy sensibles a los pirógenos). > VÃ−a de administración: â ª VÃ−a im y cutánea: respuesta menos intensa y prolongada. â ª VÃ−a iv: respuesta más intensa y prolongada Esto es por dos motivos: + Mayor biodisponibilidad. + Permite administrar mayores volúmenes. > SÃ−ntomas de la respuesta pirogénica: â ª Fiebre â ª Respiración entrecortada â ª Cianosis > Prevención de contaminación por pirógenos â ª Antiguamente: se actuaba en las etapas intermedias en el proceso de elaboración -> adsorción con C activado en la disolución y filtrado posterior (eliminamos el C activado con los pirógenos). â ª Actualmente: se intenta prevenir la contaminación + Trabajo en condiciones asépticas + Controlar la calidad pirogénica de las materias primas + Evitar contaminación del material de vidrio (calor a 250ºC, 30 min.) 44 + Evitar contaminación pirogénica del agua: usamos el agua inmediatamente después de su purificación. c) Limpidez: contaminación por partÃ−culas controlada. Comprende: - Ausencia de partÃ−culas visibles: revisión de envases uno por uno. - Ausencia de partÃ−culas no visibles a simple vista: recuento por un método basado en la dispersión de la luz. La farmacopea establece unos lÃ−mites permitidos. - Origen de las partÃ−culas: > Personal > Envases (tapones de caucho,...) > Ambiente (procedentes de los aparatos,...) > Formulación (partÃ−culas insolubles,...) - Problemas de las partÃ−culas: > Administración iv -> posible formación de granulomas o microtrombos. > Las partÃ−culas sirven de soporte para los gérmenes. La calidad se define como número de partÃ−culas/ml3 de aire. d) IsotonÃ−a: presión osmótica igual o similar, en la medida de lo posible a los tejidos tisulares. Hace que la inyección sea menos dolorosa, menos efectos necróticos. La presión será tanto mayor, cuanto mayor sea la diferencia de concentraciones entre ambas disoluciones. El inyectable debe tener la misma presión osmótica (Ï“) que los lÃ−quidos tisulares. Sin embargo existe la posibilidad de desviarse de la condición de isotonÃ−a en diferentes formas de administración. En principio una disolución isoosmótica serÃ−a lo mismo que una isotónica. Sin embargo existen determinadas sustancias que, si bien en experimentación no atraviesan una membrana semipermeable, “in vivo” atraviesan la membrana celular. Es por ello que no: DISOLUCIà N ISOTà NICA â DISOLUCIà N ISOOSMà TICA Disolución isoosmótica con los fluidos corporales: disolución con las mismas propiedades coligativas (descenso crioscópico, presión osmótica,...) que los fluidos corporales. Disolución isotónica con los fluidos corporales: medio en el que el volumen de una célula (eritrocito) no se modifica. AJUSTE DE LA ISOTONà A Presión osmótica (Ï“) Î = Râ ªTâ ªC 45 C = concentración de especies osmóticamente activas expresadas como osmolalidad. T = temperatura R = constante No es lo mismo osmolalidad (concentración de especies osmóticamente activas) y la osmolaridad. Osmolalidad del plasma: 0´302 osm/Kg Osmolaridad del plasma: 0´284 osm/l Pretenderemos que la disolución del fármaco (inyectable) tenga estos valores. Ej: disolución hipotónica -> añadimos una sustancia osmóticamente activa que aumente la presión osmótica (Ï“). Ej: ClNa. PROCEDIMIENTOS Fà SICO-QUà MICOS DE ISOTONIZACIà N - Cálculo de la concentración osmolar Cálculo de la osmolaridad debida al fármaco y al aditivo (ClNa) â ª Fármacos no electrolÃ−ticos (no se disocia) osmolaridad = molaridad â ª Fármacos electrolÃ−ticos osmolaridad = molaridad â ª i i = factor de Van´t Hoff: números de osmoles que se forman a partir de un mol de soluto. NaCl Na+ + Cl+ Inicialmente 1 mol 0 0 Final 0 1 mol 1 mol 2 moles = 2 osmoles Sin embargo la disociación nunca es completa. Ej : cada unidad de NaCl no da una disociación completa de Na+ y Cl-. El factor de Van´t Hoff nos indica esta disociación. i expresa: > Número de iones que genera una molécula > Está condicionado por el grado de disociación. Grado de disociación: fracción de mol que se disocia (α) 46 Cl2Ca Ca2+ + 2 Cl1-1 - α α 2α i = especies activas = 1 - α + 3α i = 1 + α (V-1) V = número de iones generados 0´284 osmoles/litro = g/l de pa / PM pa â ª i pa + g/l isotonizante / PM isot â ª i isot + ... osmolaridad del pa osmolaridad del isot Añadimos los gramos necesarios de isotonizante para obtener la osmolaridad igual a la del plasma sanguÃ−neo. Lo único que se desconoce son los gramos de isotonizante y es lo que hallaremos para obtener al final 0´284 osmol/litro. - Basados en el descenso crioscópico: descenso por debajo de la temperatura de congelación debido a la adición de sustancias) Descenso crioscópico (Î T) = K â ª C (osmolar) â ª i (factor de Van´t Hoff) Está directamente relacionado con la concentración osmolar (igual que la Ï“). Dos disoluciones con la misma Ï“ tienen el mismo Î T. Î T plasma = 0´52 K (constante crioscópica) = 1´86 ºKâ ªl/mol 0´52 = K â ª [g pa / PM pa â ª i pa + g isot / PM isot â ª i isot + ...] (1) (2) Si (1) no es igual a 0´52, el parámetro responsable de la igualdad será (2) (calculamos a partir de él los gramos de isotonizante). Tanto K (constante crioscópica) como i (factor de Van´t Hoff) están tabulados. Pueden aparecer juntos como Liso. Liso para un no electrolito = 1´86 Liso para un electrolito monovalente = 3´4 Liso para un electrolito divalente = 4´8 Liso = K â ª i - Concepto de equivalente en NaCl Equivalentes en NaCl de un pa (Epa) son los gramos de NaCl que tienen el mismo efecto osmótico que 1 gramo de pa. 47 NaCl 0´9% es isotónica con el plasma. 0´9 g/100ml = g pa / 100 ml â ª Epa + g isot / 100 ml â ª Eisot +... disolución isotónica con el suero sanguÃ−neo (0´9%) para NaCl Eisot = 1 Cálculo del equivalente en NaCl de un pa (Epa) Î T = Liso â ª C Î T1g pa = Î TNaCl Liso pa â ª 1g pa/l / PMpa = Liso NaCl â ª Epa/l / PMNaCl PMNaCl = 58´45 Liso NaCl = 3´4 Epa = 58´45 â ª Liso pa / 3´4 â ª PMpa Epa = 17 â ª Liso pa / PMpa PROCEDIMIENTOS BIOLà GICOS DE ISOTONIZACIà N - Cálculo del factor de Van´t Hoff: método del estudio hemolÃ−tico 1.- Preparación de dos series de tubos conteniendo glóbulos rojos • con concentración creciente de NaCl • con concentración creciente de medicamento 2.- Preparación de un tubo con Na2CO3 al 0´1% y con glóbulos rojos para producir una hemólisis total de los mismos. 3.- Centrifugamos y eliminamos el sedimento y tenemos disoluciones de Hb en mayor o menor cantidad en función de los glóbulos rojos que existÃ−an inicialmente. La relación de las absorbancias de cada tubo con respecto a la del tubo con Na2CO3 que produce el 100% de la hemólisis nos informa del porcentaje de hemólisis en cada tubo. - Razones por las que la hemólisis del NaCl es distinta del pa â ª El factor de Van´t Hoff del pa y del NaCl es distinto. â ª El PM del NaCl y del pa son distintos. Por todo esto el % de hemólisis del NaCl es distinto al del pa. Sólo son iguales cuando la concentración osmolar de pa y de NaCl es la misma. - Factor de Van´t Hoff del pa: ipa = i' + i'' + i''' / 3 48 Lo obtenemos de diferentes análisis (25, 50, 75% de hemólisis) y hacemos la media. e) pH - Valor fisiológico: 7´4 - VÃ−a intraespinal: pH = 7´0-7´6 - Restantes vÃ−as: amplio rango de tolerancia â ª Dolor e irritación â ª Estabilidad del pa y la formulación Solución de compromiso â ª Actividad farmacológica â ª Crecimiento de microorganismos f) Densidad - Importante en administración epidural de anestésicos â ª Hipobárico: densidad < 1´0059 a 37ºC â ª Hiperbárico: densidad > 1´0059 a 37ºC SISTEMAS PARA ADMINISTRACIà N PARENTERAL - Soluciones inyectables: â ª Soluciones de pequeño volumen (< 100 ml) > Principio/s activo/s > VehÃ−culo/s o disolvente/s > Sustancias auxiliares â ª Soluciones de gran volumen (> 100 ml) > Soluciones de gran volumen para uso intravenoso (fluidos intravenosos) + Aporte de necesidades básicas hidroeléctricas y de glucosa + Correctores de desequilibrios hidroelectrolÃ−ticos + Correctores de desequilibrio ácido-base + Expansores o sustitutivos del plasma + Aporte de elementos nutricionales 49 > Soluciones para irrigación > Soluciones para diálisis > Soluciones cardiopléjicas Agua para preparaciones inyectables: obtenida a partir de agua potable o purificada por destilación u ósmosis inversa. Es estéril y apirógena. - Suspensiones inyectables: Permiten administrar en forma lÃ−quida fármacos de baja hidrosolubilidad y permiten conseguir una cesión gradual y absorción prolongada del pa (vÃ−a intramuscular por ejemplo). Aspectos crÃ−ticos: â ª Forma y tamaño de partÃ−culas del fármaco que tiene una incidencia directa sobre la sensación dolorosa (más dolor que las soluciones). El tamaño influye en la velocidad de cesión y, por lo tanto de incorporación al torrente del pa. También influye sobre la jeringabilidad: facilidad con que ese inyectable se carga en una jeringa que ha de producirse a través de una aguja. Las partÃ−culas haciculares (de forma alargada) presentan peor jeringabilidad que las redondeadas. Hay que poner mucha atención en el crecimiento cristalino que consiste en el incremento de tamaño de las partÃ−culas grandes a costa de las pequeñas que se disuelven en la fase externa. â ª Propiedades reológicas: Sobre todo la viscosidad y la tixotropÃ−a (la viscosidad se reduce cuando se agita y se mantiene un tiempo para luego volver a la viscosidad inicial). Interesa que las suspensiones no sean muy viscosas y que sean tixotrópicas (menos viscosas al introducir y luego aumenta evitando la sedimentación). Tiene que cargarse con facilidad la jeringa y tiene que ser elevado el drenaje (vaciamiento del recipiente que lo contenÃ−a). â ª CaracterÃ−sticas del sedimento: Queremos sedimentos fácilmente redispersables y que no se produzca caking. Para conseguir todo esto hay que usar las técnicas apropiadas, ya vistas (agentes humectantes y estabilizantes, etc). - Emulsiones inyectables: Son similares a las suspensiones solo que aquÃ− son glóbulos inmiscibles en la fase externa. El tamaño de glóbulo suele ser de 0´25 a 0´4 μm ya que a partir de 3 μm se pueden producir embolias. Son de tamaño semejante a los quilomicrones. 50 Hay que controlar el tamaño de glóbulos y su estabilidad para que no se unan en glóbulos mayores. â ª Aplicación -> Alimentación parenteral (O/W) por vÃ−a IV: > Indicaciones: + Enfermos postoperatorios + Obstrucción del aparato digestivo + Coadyuvante (complemento) en pacientes sometidos a radio o quimioterapia > Composición: + Hidratos de carbono, grasas, aminoácidos, vitaminas, iones, electrolitos, elementos traza. > Fase acuosa: Constituida sobre todo por agua y contiene: + Glucosa, sorbitol y glicerina para isotonizar > Fase oleosa: + Aceite de sésamo, oliva o soja (naturales). Son de toxicidad baja pero pueden sufrir procesos de enranciamiento. > Emulsificantes: + Lecitina, fosfolÃ−pidos y polisorbatos. Otra aplicación es como sistemas reservorios de pa (W/O) por vÃ−a IM. Tiene que producirse el reparto entre la fase acuosa y oleosa y luego entre la oleosa y el organismo. - Sistemas coloidales: Sistemas dispersos cuya fase interna tiene un tamaño inferior a 1 μm. Se sitúan entre las suspensiones o emulsiones (dispersiones groseras) y las disoluciones (dispersión molecular) â ª Aplicaciones: > Vectorización de medicamentos: para dirigirlos a los lugares de acción óptimos. > Obtener sistemas de caracterÃ−sticas similares a las disoluciones con fármacos de baja hidrosolubilidad. â ª Tipos de sistemas coloidales de administración parenteral: 1.- Emulsiones submicroscópicas: nanoemulsiones 2.- Sistemas coloidales vesiculares: liposomas -> pequeñas vesÃ−culas lipÃ−dicas que contienen la fase acuosa en su interior. 51 3.- Sistemas coloidales particulares: > NanopartÃ−culas: partÃ−culas poliméricas de estructura y naturaleza matricial. > Nanocápsulas: partÃ−culas constituidas por una cubierta polimérica que rodea otra fase lÃ−quida (generalmente). Esa porción recubierta por la cubierta representa las partÃ−culas que aparecen en la disolución. 1.- NANOEMULSIONES: Emulsiones de tamaño de glóbulo < 1 μm. - Aplicación: â ª Administración de medicamentos y alimentos lipofÃ−licos en un vehÃ−culo acuoso. - Composición: â ª Aceites de cadena media esterificados â ª Tensioactivos â ª Principios activos, generalmente lipofÃ−licos No es perfectamente transparente pero casi - Estabilidad limitada 2.- LIPOSOMAS: Estructuras vesiculares formadas por una o varias capas lipÃ−dicas concéntricas. (a) (b) (c) • Liposomas unilaminares pequeños (SUV) -> (25-200 nm) • Liposomas unilaminares grandes (LUV) -> (200-1000 nm) • Liposomas multilaminares (MLV) -> (0´5-5 μm) Una caracterÃ−stica importante es el tamaño. De esto dependen sus aplicaciones parenterales. Cuando se administran por vÃ−a parenteral tienden a ser captados por células del sistema retÃ−culo-endotelial por lo que su permanencia en el torrente depende del tamaño. Cuanto mayor es el liposoma, más rápido son captados por el sistema retÃ−culo-endotelial. Se usarán mejor liposomas pequeños. Los liposomas convencionales debido a la captación se usan para patologÃ−as en hÃ−gado, bazo, médula ósea, etc. Esta captación es su mayor problema por lo que deberemos introducir modificaciones para que lleguen a otros lados: - Incorporación de anticuerpos monoclonales que reconozcan antÃ−genos especÃ−ficos y asÃ− se dirijan a ciertas células (tumorales por ejemplo) 52 - Fijar sobre su superficie carbohidratos por lo que se disminuye su tendencia a fijar proteÃ−nas y se prolonga la presencia en el torrente y su especificidad por los macrófagos. - Fijar cadenas poliméricas hidrofÃ−licas (restos de polietilenglicoles). El tiempo de permanencia en el torrente se prolonga mucho y se aumenta la eficiencia de fármacos como la doxorrubicina. 3.- NANOPARTà CULAS Y NANOCÔPSULAS: Son sistemas coloidales cuyo componente diferencial es un polÃ−mero (poliláctico, etc.) biocompatible y biodegradable. En las nanopartÃ−culas el principio activo puede estar disperso en el interior o adsorbido en el polÃ−mero; en las nanocápsulas igual. Aplicaciones vÃ−a parenteral: vectorización de fármacos hacia órganos y tejidos concretos (igual que los liposomas). Sobre todo se usan con agentes antitumorales. - Implantes: Formas sólidas constituidas por un polÃ−mero que se desintegra continuamente y libera el pa bajo la piel. El precedente histórico son los pellets (comprimidos pequeños constituidos únicamente por principio activo y no porosos, que se aplicaban con una jeringa especial bajo la piel). Eran principios activos poco solubles que eran usados frecuentemente en veterinaria para la administración de hormonas. Actualmente implantes poliméricos => por su bioerosión proporcionan: â ª Niveles plasmáticos estables (debido a la liberación controlada) â ª Mejor cumplimiento del régimen de dosificación. Permiten aumentar el tiempo de administración entre dosis consecutivas. â ª Mayor eficacia del tratamiento: > Niveles plasmáticos más estables > Menor riesgo de omisión en la administración del fármaco (por olvido,...) â ª Reducción del coste Inconvenientes: â ª Su colocación requiere una pequeña intervención quirúrgica. â ª Dolor e incomodidad del paciente â ª Coste del tratamiento â ª Reacción en tejidos adyacentes => sensibilidad incluso de carácter sistémico. Es por ello que para que un polÃ−mero sea aprobado deben hacerse previamente unos estudios de biocompatibilidad (toxicidad,...) del mismo y de sus productos de degradación. 53 Son pocos los polÃ−meros que según esto pueden emplearse debido: > Existen pocos productos con caracterÃ−sticas de biocompatibilidad adecuadas. > Elevado coste para la introducción de nuevos materiales (investigación). PolÃ−meros usados: siliconas (algunas), EVAC (copolÃ−mero de polietileno acetato de vinilo) â ª Potencial toxicidad debida a una liberación brusca del fármaco. â ª DifÃ−cil la interrupción del tratamiento. PREPARACIà N DE FORMAS DE DOSIFICACIà N PARENTERAL - Fabricación de inyectables tipo solución esterilizados en autoclave: El aspecto crÃ−tico es asegurar la esterilidad y la ausencia de pirógenos al final del proceso de fabricación. Ampollas -> se llenan con unas jeringas especiales y se cierran por fusión con calor. â ª Ampollas 1º Lavamos para eliminar posibles restos de vidrio es su interior y que podrÃ−an contaminar el inyectable. Para este lavado, el agua empleada debe ser adecuada y ajustada a unos estándares de pureza. 2º Secado y esterilización (horno a calor seco) â ª Componentes de la formulación (principios activos, vehÃ−culos y excipientes) 1º Disolución de los distintos componentes 2º Proceso de filtración esterilizante a 0´22 μm. Esta eficacia mayor de retención que los filtros de 0´45 μm le permite retener Pseudomonas. â ª Paso global 1º Dosificación de ampollas y cierre 2º Esterilización en autoclave (calor húmedo) 3º Acondicionamiento de ampollas - Fabricación de polvos liofilizados de uso parenteral y esterilizados por filtración: Se emplea este tipo de polvo con la finalidad de aumentar la estabilidad del pa evitando la posible degradación del mismo que ocurrirÃ−a al tenerlo durante un largo tiempo de disolución. En este caso, además del vial debe esterilizarse el tapón de caucho. Preparamos la disolución que posteriormente se filtra y se dosifica en cada vial. Realizamos la liofilización y en la propia cámara de liofilización se incorpora el tapón de caucho. 54 â ª Acondicionamiento final: no existe etapa final de esterilización por calor húmedo, debido a que no es compatible con el liofilizado. Es por ello que las etapas previas de esterilización terminal deben controlarse mucho más que en el caso anterior. - Fabricación de preparaciones inyectables tipo solución, suspensión y emulsión: Esterilización de pa, excipientes y vehÃ−culos por separado. Tampoco existe etapa final de esterilización por calor húmedo debido a que las condiciones fÃ−sicas del producto no le permiten resistir a este proceso. Además, aunque existieran gérmenes en partÃ−culas del sólido no es seguro que el calor húmedo las eliminase. Es por ello que lo mejor serÃ−a hacer una disolución de estas partÃ−culas y una filtración esterilizante. ELABORACIà N DE INYECTABLES Requiere unas zonas blancas, limpias o estériles para asegurar condiciones de esterilidad. En esa zona blanca se regula la contaminación por partÃ−culas en el ambiente y con ello los posibles gérmenes, ya que las partÃ−culas actúan como medio de transporte de los mismos. Asimismo, las zonas limpias: - Protegen al personal - Protegen el medio ambiente; evitamos contaminaciones cruzadas. - Estándares de limpieza de ambiente: Zona aséptica: zona sin gérmenes Zona limpia: zona de contaminación controlada Existen diferentes clases de limpieza de aire: â ª British Standard > Clase (1) es el nivel más alto > Clase (2) es un nivel menor â ª US Federal Standard > Clase (100) es el nivel más alto > Clase (10000) es un nivel menor Tanto el clase (1) como el (100) aseguran un mayor control, siendo útil en caso de que sea fácil la contaminación. - Cámaras de flujo laminar: Es donde se lleva a cabo la elaboración de los inyectables. En ella se crea una corriente de aire en flujo 55 laminar desde dentro hacia fuera que elimina cualquier partÃ−cula que se origina. En caso de que el riesgo de contaminación sea mayor son más útiles las cámaras de flujo laminar vertical. Ambas cabinas se sitúan en zonas limpias. â ª Estructura de una zona limpia En ella la presión del aire es ligeramente mayor a la exterior (1´5 mm de H2O) lo que permite la salida del aire. Además el aire que entra en la zona limpia pasa por un sistema de purificación que tiene dos funciones: > Eliminar las partÃ−culas del aire > Acondicionar el aire (Tª, humedad relativa) Las partÃ−culas se eliminan primero mediante una pre-filtración y posteriormente una filtración a través de filtros HEPA. El acondicionamiento del aire se realiza: > Primero deshumidificación del aire > Se le hace llegar vapor de agua con unas caracterÃ−sticas de pureza adecuadas hasta una humedad relativa 30-60% y una Tª de 18-24ºC. Si la humedad relativa es muy baja el trabajo en la zona limpia se hace incómodo. El acceso a la zona limpia es a través de un vestuario. Desde el mismo podemos llegar a dos zonas: de clase 1 o de clase 2 ambas separadas por barreras. A su vez, en la propia sala el flujo de aire que existe es continuo y se encarga de eliminar posibles partÃ−culas procedentes de los trabajadores del laboratorio. Puede existir un flujo unidireccional (o laminar; mayor control) o convencional. En general: recintos clase 2 -> flujo convencional y recintos clase 1 -> flujo laminar. CONTROL DE CALIDAD DE INYECTABLES - Control de materias primas: â ª Principios activos y excipientes de la formulación â ª VehÃ−culos. Agua para inyección â ª Envases y cierres - Control de proceso: â ª Control de ambiente, personal, equipos... 56 â ª Evaluación de parámetros crÃ−ticos de productos intermedios. - Control del producto acabado: â ª Control de la calidad microbiológica > Ensayo de esterilidad > Evaluación de pirógenos o endotoxinas bacterianas â ª Control galénico > Contenido en pa > Tonicidad > pH > Densidad > Controles especÃ−ficos: + Soluciones: * Limpidez + Suspensiones: * Distribución de tamaños de partÃ−cula * Viscosidad * CaracterÃ−sticas de cesión del pa + Emulsiones: * Distribución de tamaños de gotÃ−cula * Signo de la emulsión * Viscosidad + Sistemas coloidales: * Potencial Z * CaracterÃ−sticas de cesión del pa + Implantes: * CaracterÃ−sticas de cesión del pa 57 ENSAYO DE ESTERILIDAD Dos procedimientos: 1) Filtración de la disolución a través de una membrana estéril semipermeable. Una vez filtrado, se toma el filtro y se lleva a dos medios de cultivo diferentes para cubrir el espectro de gérmenes posibles de contaminación. Se observa si existe crecimiento de gérmenes: â ª si existe crecimiento -> se rechaza el lote â ª si no existe crecimiento bacteriano -> se acepta el lote La principal ventaja de este método es que si el preparado tiene un agente conservante, permite su eliminación y por tanto el crecimiento, en caso de que existan, de gérmenes. 2) Siembra directa en placa Preparado parenteral diluido 1/10 para lÃ−quidos o 1/100 para sólidos TEST BIOLà GICO DE DETECCIà N DE PIRà GENOS Se emplea como animal de experimentación un conejo caracterÃ−stico (blanco, de ojos rojos) debido a su mayor sensibilidad a pirógenos. El número varÃ−a de 3 a 12 al igual que las condiciones de experimentación. Se observa el aumento de la Tª corporal del conejo que se produce tras la inyección de un determinado volumen de inyectable. Primero se mide la Tª rectal y en la vena marginal de la oreja. Al final se mide la Tª rectal. Es un ensayo lÃ−mite, permite únicamente saber si existe contaminación o no pero no cuantificarla. Vale para pirógenos de cualquier tipo: - Pirógenos endógenos: proceden de la pared bacteriana - Pirógenos exógenos: proceden de la actividad metabólica. Mà TODO “IN VITRO” PARA LA DETECCIà N DE PIRà GENOS Se basa en una reacción de aglutinación que emplea como reactivo un lisado de células “amebocitos” del cangrejo de herradura Limulus polyphemus. Inicialmente se desarrolló como un ensayo lÃ−mite: Se mezcla con el reactivo la disolución a tratar y se observa si existe o no aparición de turbidez (indicarÃ−a aglutinación y contaminación pirogénica de la solución). Esta respuesta de aglutinación ha permitido el desarrollo: Existen métodos cuantitativos (colorimetrÃ−a de punto final) y métodos cinéticos (observación de la variación en el tiempo de un parámetro, ej: Abs,... y valorar a través de la pendiente de la recta que describe la cinética si existe contaminación por pirógenos). Procedimiento: 58 Muestra -> se vierte sobre una solución que contiene el lisado de amebocitos -> incubación (además de un blanco, sin lisado de células) -> comparación de turbidez, gelificación o precipitación. La intensidad de la respuesta depende de: - Concentración de pirógenos - Temperatura - pH Los tres parámetros deben estar perfectamente controlados para poder realizar la prueba. El test LAL es muy sensible y selectivo; es especÃ−fico para endotoxinas (pirógenos endógenos) procedentes de la pared de G (-). Para determinar la concentración de pirógenos necesitamos rectas patrón que se establecen por las distintas farmacopeas. Para el test LAL los patrones que se emplean son a partir de Escherichia coli. CONTROL DE LIMPIDEZ Limpidez: contaminación por partÃ−culas controlada. - Requerimientos de contaminación por partÃ−culas (Real Farmacopea Española) â ª PartÃ−culas sub-visibles: recuento por un método basado en la dispersión de la luz. â ª PartÃ−culas visibles: evaluación de todos los envases sobre fondo blanco y negro. TEMA 10: POMADAS Y LOCIONES Formas semisólidas y lÃ−quidas para la administración sobre la piel o mucosas. Además de pomadas y lociones también son de aplicación sobre la piel los parches transdérmicos que se diferencian en su estructura y en su uso para aplicar pa a nivel sistémico a través de la piel. Solo se usan para un número limitado de fármacos. La parte más externa de la piel es la epidermis que es donde suelen actuar las pomadas y lociones. También puede pasar a la dermis y presentará efectos sistémicos (poco frecuente este uso). También se pueden usar sobre glándulas sebáceas, sudorÃ−paras, etc. Las pomadas tienen más importancia que las lociones que se suelen usar sonde las pomadas no son muy adecuadas (cuero cabelludo). Las pomadas están formadas por principios activos y la base de la pomada (engloba los excipientes -> uno o más). Las bases se diferencian por su comportamiento cuando se aplican con el fármaco incorporado sobre la piel. Clasificación: 59 - Bases de hidrocarburos: Son de naturaleza lipofÃ−lica. Ej: vaselina, parafina. El rasgo más caracterÃ−stico de comportamiento es un efecto oclusivo => impide la transpiración y produce el reblandecimiento del estrato córneo (efecto emoliente). No se usan demasiado. Solo para proteger la piel. Son difÃ−ciles de eliminar ya que no son lavables. - Bases de absorción: Son sistemas más complejos. Son capaces de incorporar agua como fase interna de una emulsión. Hay 2 tipos: â ª Incorporan una solución acuosa para dar una emulsión W/O Se emplea una fase oleosa y un agente tensioactivo y al producirse los exudados se incorporan como fase interna de la emulsión. â ª Son emulsiones W/O capaces de incorporar soluciones acuosas SerÃ−a una crema lipófila u oleosa. - Bases “eliminables con agua”: Son emulsiones O/W. Son cremas hidrofÃ−licas. - Bases solubles en agua: Mezclas de polietilenglicoles de alto y bajo PM. Son de consistencia ligera. EXCIPIENTES No tienen efecto terapéutico, pero sÃ− sobre la liberación del pa y sobre la absorción a través de la piel. - Componentes lipofÃ−licos: â ª Hidrocarburos: vaselina filante, aceite de parafina y plastibase. â ª Ceras: parafina sólida. â ª Aceites vegetales: cacahuete, almendras, sésamo, oliva,... â ª Siliconas: dimeticonas, dimetilpolisiloxanos. - Componentes hidrofÃ−licos: â ª Agua. â ª Alcoholes y glicoles: etanol, alcohol isopropÃ−lico, etc. Aumentan la solubilidad de algunos pa. Se mezclan con los componentes lipofÃ−licos para aumentar la fluidez de la pomada. 60 - Agentes tensioactivos: Son agentes solubilizantes, humectantes, emulsificantes,... - Agentes espesantes: Aumentan la viscosidad de las fases acuosas. Los más usados son polÃ−meros. - Formadores de matrices estructuradas: Forman parte de la fase oleosa para estructurarla y aumentar la consistencia. Son alcoholes de cadena larga, esteroles,... - Agentes humectantes: Disminuyen el ángulo de contacto con el agua y facilitan el contacto con el pa. Son glicerina, propilenglicol, sorbitol,... - Conservantes: Sirven para evitar el crecimiento de microorganismos ya que las bases son caldos de cultivo. Son ácidos, alcoholes, ésteres, mercuriales,... - Antioxidantes: Protegen a los componentes de los procesos oxidativos. Son: â ª Verdaderos: tocoferoles. â ª Agentes reductores: vitamina C y bisulfito sódico. - Agentes secuestrantes: Forman complejos con los iones del medio que puedan intervenir en procesos degradativos como catalizadores. - Tampones y otros: Los tampones estabilizan el pH. FORMULACIà N DE LAS POMADAS - Elección de los componentes: â ª Parámetros a tener en cuenta. > Propiedades del pa: Ej: estabilidad. Si es hidrolizable no podremos usa agua en la pomada. Si es oxidable trataremos de prevenir un proceso de oxidación. > Velocidad de cesión: los efectos que se buscan en las pomadas se producen en los estratos más externos de la piel, aunque puede interesar la penetración algo más profunda. Esto se consigue incorporando determinados disolventes (DMSO) y tensioactivos. 61 > Efecto de la propia base de la pomada: influye en la penetración. Algunas bases tienen un efecto oclusivo (no dejan evaporar el agua y se produce efecto emoliente) por lo que el papel de barrera del estrato córneo disminuye y la pomada penetra mejor. > Atractivo cosmético y facilidad de aplicación y eliminación: importante desde el punto de vista comercial. > Localización del área a tratar: lo más crÃ−tico es si es una zona sin pelo o con pelo. En el segundo caso se prefieren cremas más fluidas y lociones. > Esterilidad y contaminación microbiana: debe tener los requerimientos mÃ−nimos estipulados en este campo en la farmacopea. En algunos casos han de ser estériles (para el ojo o sobre heridas abiertas). > Estabilidad fÃ−sica: es un aspecto fundamental porque muchas pomadas son sistemas fÃ−sico-quÃ−micos complejos -> emulsiones, etc por lo que las posibilidades de alteraciones fÃ−sico-quÃ−micas son considerables. Además los componentes lipÃ−dicos son fácilmente oxidables (hay que poner antioxidantes). > Otras propiedades: la propia pomada cumple por sÃ− misma (depende de la base) otras propiedades -> efecto lubricante, emoliente, etc. Son efectos indirectos que tienen cierta relevancia. - Preparación de una pomada: Depende de las caracterÃ−sticas de la base y de la naturaleza del principio activo (sólido, lÃ−quido, que se quiera disolver, etc.). De esto depende el que una pomada sea una suspensión, emulsión, solución, etc. â ª Si el pa se incorpora a la base en forma de polvo: > Pomada solución: si es soluble en ella > Pomada suspensión: si no es soluble en ella â ª Si el pa se incorpora a la base disuelto: > Pomadas lipofÃ−licas (W/O): pomadas emulsión de fase externa oleosa (W/O). > Pomadas hidrofÃ−licas (O/W): pomadas emulsión de fase externa hidrofÃ−lica (O/W). La lipófila es más oclusiva e incorpora menos agua. EQUIPAMIENTO - A nivel de oficina de farmacia: Se mezclan los componentes en un mortero. Dentro de los componentes lipófilos se suele incorporar alguno sólida a temperatura ambiente. Esto lleva a tener que hacer una fusión antes de la mezcla. Hay que fundir los componentes de la fase oleosa y calentar la acuosa a la misma temperatura y se mezclan. La fusión: â ª Facilita el malaxado 62 â ª Permite la disolución de pa poco solubles â ª Excipientes con puntos de fusión muy diferentes â ª Facilita la formación de una emulsión - A nivel industrial: Se usan mezcladoras de elevado tamaño que están constituidas por un elemento batidor y un recipiente de doble pared entre las que pasa un lÃ−quido termostatizado. También se necesita un proceso de homogeneización con un homogeneizador de válvula o un molino coloidal. CONTROL DE CALIDAD - Control de materias primas: igual que cualquier otra forma de dosificación. - Control de proceso: no tiene demasiado peso. - Control de producto acabado: â ª Control de la calidad microbiológica. â ª Control galénico > CaracterÃ−sticas organolépticas (homogeneidad) Comprende el olor y el aspecto externo que se caracteriza por una observación macroscópica. Luego se hace una observación microscópica y dentro de esto es importante una caracterización del tamaño de las partÃ−culas sólidas si las hay (de esto depende la textura de la pomada) > Consistencia De ella depende la extensibilidad. La consistencia se caracteriza con un penetrómetro que es un sistema que consiste en medir la resistencia a la penetración de un elemento metálico cónico estándar. Para la caracterización de la consistencia también se puede usar la reometrÃ−a (para medir viscosidad, etc.). > pH Interesa si la pomada incorpora una fase acuosa. Interesa por dos razones: influye sobre la estabilidad quÃ−mica y porque afecta al pH fisiológico de la piel que afecta al crecimiento de determinados gérmenes patógenos. > Esterilidad > Cesión in vitro Tiene valor en el control de calidad para ver la homogeneidad en lotes sucesivos. Se hace por una diálisis a través de una membrana impermeable. 63 Los resultados no son extrapolables a su comportamiento in vivo. La determinación de la difusión en células de difusión con el uso de capas de piel permite obtener información más fisiológica de la penetración en la piel aunque no es totalmente extrapolable. > Actividad y biodisponibilidad Ensayos en animales de experimentación y (si es posible) en humanos. > Estudios de estabilidad (conservación) Envejecimiento en situaciones controladas (estudios acelerados). Además también hay estudios a largo plazo. LOCIONES Difieren de las pomadas en que son lÃ−quidos. Son disoluciones, emulsiones y suspensiones. Se componen de uno o varios pa, agua o solventes. También pueden tener promotores de la absorción (DMSO). Las lociones son preparados lÃ−quidos que se aplican sin fricción sobre la piel. Su mayor aplicación es sobre el cuero cabelludo. TEMA 11: SISTEMAS TRANSDà RMICOS Formas de administración sobre la piel para conseguir un efecto sistémico, es decir, que se absorba, alcance la circulación general y produzca su efecto. Dentro de éstas, diferenciamos: - Algunas pomadas de nitroglicerina para tratamiento de la angina de pecho. - Parches de nicotina, nitroglicerina, estradiol (estados postmenopáusicos), clonidina, escopolamina (mareo). VENTAJAS DE LA ADMINISTRACIà N TRANSDà RMICA - Buena aceptación por parte del paciente. - Son autoadministrables (no requieren personal especializado). - Permiten mejor cumplimiento del régimen posológico (1 cada dÃ−a). - Fácil interrupción de la administración. Si existe efecto adverso, retiramos el parche. - El medicamento no sufre efecto de primer paso hepático. - Optimización de los perfiles concentración plasmática-tiempo. Concentración Plasmática Concentración Tóxica 64 Formulación transdérmica ideal Concentración mÃ−nima eficaz Dosis I Dosis II Dosis III Tiempo La concentración plasmática/t oral es diferente de la transdérmica. à sta última es un perfil optimizado sin fluctuaciones dentro de la ventana terapéutica en un tiempo largo. Al no existir fluctuaciones aparecen menos efectos secundarios y menor ineficacia terapéutica porque siempre será menor que la dosis tóxica y mayor que la concentración mÃ−nima eficaz. INCONVENIENTES DE LA ADMINISTRACIà N TRANSDà RMICA - No todos los medicamentos son capaces de atravesar la piel que en el organismo cumple una función protectora contra pérdida de lÃ−quidos, radiaciones, cambios de temperatura. Solo son buenos para principios activos con propiedades fÃ−sicas, quÃ−micas y fÃ−sico-quÃ−micas determinadas (tamaño molecular bajo, coeficiente de reparto adecuado y coeficiente de difusión adecuado). Son también buenos candidatos los principios activos farmacológicamente muy activos. A pequeñas dosis tienen elevado efecto (principios activos que actúan a dosis menores de 5 mg). - Provocan irritación cutánea o sensibilización inmunológica. Importante añadir excipientes o promotores de la absorción que son sustancias usadas para disminuir la resistencia de la piel al paso de medicamentos lo que supone una agresión. - Sólo cuando exista una necesidad clÃ−nica justificada se usarán las formas transdérmicas que son caras. Ej: nitroglicerina sublingual en ataques de angina de pecho. â ª Tiempo de vida media muy corto â ª Efecto de primer paso 90% â ª Existe elevado número de pacientes De ahÃ− que existan parches transdérmicos. ABSORCIà N A TRAVà S DE LA PIEL - Piel: â ª Hipodermis: tejido subcutáneo altamente irrigado. â ª Dermis: también irrigada. Zona hidrofÃ−lica con elevado número de terminaciones nerviosas. â ª Epidermis: 65 > Lámina basal > Estrato germinativo > Estrato córneo: células muertas, sin núcleo, rellenas de queratina y rodeadas de una matriz lipÃ−dica. Matriz muy cerrada (estructura de ladrillos y cemento) que desprende las células muertas superficiales; es la verdadera barrera. â ª Atravesando dermis y epidermis encontramos las glándulas sudorÃ−paras y folÃ−culos pilosos que en su base están irrigados. El medicamento tendrá que atravesar todas estas capas. Và AS DE PENETRACIà N - Transfolicular: a través de los folÃ−culos pilosos. Difunde hacia la base del folÃ−culo a un medio lipÃ−dico (coeficiente de difusión a tener en cuenta). La irrigación del folÃ−culo piloso está en la base. Luego pasa del medio lipÃ−dico al acuoso (coeficiente de reparto) REPARTO - DIFUSIà N - REPARTO - Transcelular: a través de los queratocitos. REPARTO - DIFUSIà N - REPARTO - DIFUSIà N - Intercelular: entre las células del estrato córneo. REPARTO - DIFUSIà N - REPARTO - DIFUSIà N Con estos sistemas se persigue una cesión sostenida. La cesión puede estar controlada por una membrana (sistema reservorio) o por medio de difusión (sistema matricial). TIPOS DE PARCHES TRANSDà RMICOS - Tipo reservorio: núcleo separado por una membrana que controla la liberación. Liberación y difusión del pa en función de las caracterÃ−sticas de ésta. Estructura: â ª Cubierta protectora externa (parte exterior del parche) â ª Reservorio de principio activo (sólido o semisólido) â ª Membrana de control de liberación â ª Adhesivo â ª Lámina protectora interna (se quita para poner el parche) - Tipo matricial: núcleo en contacto con la piel. Liberación del pa de acuerdo con una cinética 66 determinada. Primer tipo de estructura: â ª Cubierta protectora externa â ª Adhesivo â ª Núcleo de principio activo sin propiedades adhesivas â ª Lámina protectora interna Segundo tipo de estructura: â ª Cubierta protectora externa â ª Matriz polimérica con propiedades adhesivas + principio activo â ª Lámina protectora interna Ester tipo de sistemas está diseñado para atravesar la piel por lo que tenemos que disminuir la resistencia de la piel, del estrato córneo. Para ello incorporamos unos excipientes denominados promotores de la absorción. A estos excipientes se les exige una serie de caracterÃ−sticas: â ª No tóxicos â ª No irritantes â ª No alergénicos â ª Deben disminuir la resistencia del estrato córneo solo de fuera a dentro porque si no se perderÃ−an sustancias del interior del organismo. â ª Ejercerán una acción de disminución de la resistencia reversible. â ª Serán fÃ−sica y quÃ−micamente compatibles con el principio activo y excipientes de la formulación. â ª Buen disolvente del principio activo. â ª Incoloro, inodoro e insÃ−pido. Ejemplo de excipientes: alcoholes, alquilsulfóxidos, ácido oleico, ácido caprÃ−lico, azona (único diseñado como promotor), urea, ácido salicÃ−lico, laurocepram. Mà TODOS PARA INCREMENTAR LA PERMEABILIDAD => Utilización de PROMOTORES DE LA ABSORCIà N - Actúan sobre la ruta apolar: es decir, sobre la transfolicular e intercelular (la transcelular es polar porque atraviesa la queratina que es proteica). Fluidifican los lÃ−pidos, los disuelven (con lo que también varÃ−an el coeficiente de reparto) y forman micelas. 67 - Actúan sobre la ruta polar: alteran las proteÃ−nas, la ruta hidrofÃ−lica y el coeficiente de reparto. Ambos mecanismos son posibles en una sola sustancia: Ej: dimetilsulfóxido: el inconveniente es que no sólo actúa en una dirección y no es reversible totalmente. Si no es asÃ− se emplean mezclas de los de ruta polar y apolar: Ej: azona (apolar) + dioles (polares) => Aplicación de corriente eléctrica Una corriente produce una variación en la permeabilidad de la piel. Una de baja intensidad un tiempo prolongado se conoce como IONTOFORESIS. Una corriente de elevada intensidad se llama ELECTROPORACIà N. => Aplicación de ultrasonidos Se llama SONOFORESIS. Se usa en las ecografÃ−as. => Otras técnicas (en experimentación) Eliminar un trozo de capa córnea con láser FABRICACIà N DE PARCHES TRANSDà RMICOS - Preparación del medicamento - Mezclado con el resto de componentes - Laminado y secado - Disposición de todas las láminas que hacen falta - Cortado y embalaje CONTROL DE CALIDAD DE PARCHES TRANSDà RMICOS - Control de material primas - Control del proceso â ª Presión aplicada para la formación de las láminas â ª Presión aplicada para el ensamblaje de todas las láminas del dispositivo - Control del producto acabado â ª Controles fÃ−sicos: ver si los parches salen con las dimensiones adecuadas, aspecto, ensamblado, etc â ª Control del principio activo 68 > Evaluar la cantidad de pa de una muestra aleatoria extraÃ−da de cada lote. â ª Determina el perfil de liberación â ª Ensayo de permeabilidad TEMA 12: FORMAS DE ADMINISTRACIà N OCULAR Son las que se aplican sobre el ojo (tópicas) para el tratamiento de patologÃ−as oculares. Se pretende un efecto local. El ojo es una zona de difÃ−cil acceso sistémico. Para conseguir los mismos efectos por vÃ−a sistémica que ocular se necesitarÃ−an dosis elevadas. - Formas tópicas utilizadas para el tratamiento de patologÃ−as o alteraciones oculares. â ª Se reducen los efectos secundarios frente a la vÃ−a sistémica â ª Mayor rapidez en el principio de la acción â ª Dosis eficaz más reducida - Principios activos usados: midriáticos, mióticos, antibióticos, anestésicos locales, antiinflamatorios. - Sistemas: â ª Colirios: soluciones o suspensiones de naturaleza acuosa u oleosa. Son los más usados aunque presentan ciertos inconvenientes: > Pérdida precorneal de colirio por parpadeo > Absorción no productiva > Tiempo de permanencia en el saco precorneal del pa es pequeño porque además del parpadeo existe drenaje nasofarÃ−ngeo. > Alta impermeabilidad del epitelio corneal Algunos inconvenientes se han suavizado modificando las caracterÃ−sticas de los colirios. â ª Pomadas oftálmicas â ª Formas de liberación controlada REQUISITOS DE LOS COLIRIOS 1) Esterilidad: La contaminación por microorganismos puede provocar ceguera. Los microorganismos más usuales son S. Aureus y Pseudomonas aeruginosa (más peligroso). Se usan agentes conservantes que garanticen la esterilidad en el tiempo de viabilidad del colirio (cloruro de benzalconio, clorhexidina, etc) 69 Se requiere que la fabricación sea en zonas limpias o cabinas de flujo laminar. 2) Limpidez (excepto algunos colirios suspensión): Ausencia de partÃ−culas en suspensión 3) Isotonicidad: El ojo tiene la misma que el suero fisiológico aunque admite mayor rango. Admite mejor los hipo que los hipertónicos salvo el ojo enfermo al que hay que poner isotónico. 4) pH adecuado: El pH del fluido lacrimal es de 7´4 pero soporta hasta 11´6. Los ajustes de pH se hacen por medio de tampones. La selección del pH se hace en base a una serie de factores: â ª Factores fisiológicos â ª Estabilidad del pa â ª pH de máxima efectividad â ª pH de máxima solubilidad del pa Esto se debe a que la biodisponibilidad ocular normalmente es muy baja. 5) Viscosidad: Aumentos de ésta pretenden disminuir la pérdida precorneal de los colirios. Para ello se incluyen agentes viscosizantes como alcohol polivinÃ−lico, metilcelulosa (no se puede autoclavar), carbopol, etc. En colirios oleosos no se usan viscosizantes sino tensioactivos para facilitar el contacto con la superficie del ojo. 6) Correcto acondicionamiento: Se hace en envases parecidos a los de inyectables y con especial cuidado en los multidosis. ELABORACIà N DE COLIRIOS - Materias primas: â ª Principios activos de adecuada pureza y, en colirios suspensión, adecuado tamaño de partÃ−cula. â ª VehÃ−culos: acuosos y oleosos del mismo tipo que en inyectables. â ª Coadyuvantes: isotonizantes, sistemas tampón, conservantes, antioxidantes y humectantes. - Procedimiento: Es sencillo pero caro. Las instalaciones necesarias son del mismo tipo que en inyectables. 70 â ª Disolución o suspensión que se hace por agitación â ª Filtración clarificante para evitar partÃ−culas en suspensión (excepto en suspensiones) y luego se esteriliza en autoclave. Para los pa que no se pueden meter en autoclave o formulaciones con elementos no autoclavables se hace una filtración clarificante y esterilizante con filtros de tamaño de poro más pequeño. - Colirios suspensión: Se recurre cuando el pa es poco soluble o se quiere tener cierta cesión sostenida. Siempre que se puede se evita porque ocasiona ciertos inconvenientes: sedimentación, formación de aglomerados. También requiere una agitación vigorosa justo antes de usar para instilar la misma dosis siempre. - Acondicionamiento: â ª Envases unidosis o multidosis: últimamente se tiende a los unidosis. â ª Envases fabricados con materiales inertes > Polietileno de baja densidad â ª Especificaciones similares a las de los preparados inyectables. - Conservación: â ª PerÃ−odo de validez de 4 semanas (envases multidosis). POMADAS OFTÔLMICAS - Preparados semisólidos, estériles para la aplicación sobre la conjuntiva ocular: Debido a sus propiedades reológicas tienen efectos más prolongados que los colirios y éstas propiedades deben ser adecuadas para ser extensibles por simple parpadeo. Hay 2 tipos: â ª Pomadas grasas â ª Pomadas hidrosolubles o hidrogeles - Componentes habituales: â ª Vaselina filante y parafina lÃ−quida â ª Lanolina â ª Excipientes hidrofÃ−licos: > PolÃ−meros hidrofÃ−licos, PEGs y agentes emulsificantes â ª Antioxidantes, estabilizantes y conservantes - Elaboración: 71 â ª Principios activos lipofÃ−licos: la base es grasa (se disuelven directamente en ella) â ª Principios activos hidrofÃ−licos se disuelven previamente en un pequeño volumen de agua y después se incorporan a la base. Si el pa es poco soluble se pueden añadir solubilizantes. â ª Los principios activos que no puedan ir por los métodos anteriores se pulverizan finamente y se dispersan en la pomada. - Acondicionamiento: â ª Tubos de plástico o metálicos provistos de una cánula para su aplicación directa sobre el ojo. Los metálicos se recubren con material aislante porque pueden tener iones. SISTEMAS DE LIBERACIà N CONTROLADA La biodisponibilidad ocular es muy baja por pérdida precorneal elevada y penetración de pa muy baja. SerÃ−a adecuado, en algunos casos, conseguir liberación sostenida sobre todo en pa con vida media muy baja o muchos efectos secundarios. - Aproximaciones: â ª Incrementar la penetración y disminuir la pérdida precorneal. > Empleo de agentes viscosizantes, promedicamentos o promotores de la absorción en colirios suspensión y pomadas oftálmicas. Usar pomadas en vez de colirios. > Empleo de sustancias que formen suspensiones coloidales (nanopartÃ−culas y liposomas). > Empleo de polÃ−meros bioadhesivos: sustancias que interaccionan con las mucosas de manera que la formulación quede pegada a la conjuntiva y libera de forma continua el medicamento. â ª Sistemas de liberación continua y controlada de medicamentos. > Dispositivos sólidos: + Ocusert (USA): dispositivo tipo reservorio rodeado por una membrana que controla la liberación. Esto se inserta en el saco conjuntival. Se desarrolló para la administración de pilocarpina. + SODI (Ruso): dispositivo tipo matriz polimérica (mezcla de polÃ−meros) con el pa disperso en ésta. â ª Lentillas que ceden pa (en investigación): lentillas formadas por una capa polimérica que haga las funciones de lentilla y libere pa de forma prolongada. CONTROL DE CALIDAD - Control de materias primas y envases El control de envases en multidosis es importante ya que éste es el que instila la dosis en el ojo y hay que calibrarlo. - Control de proceso 72 - Control de producto acabado Hay que controlar: contenido en pa, pH, viscosidad, descenso crioscópico, capacidad de tampón, limpidez (salvo colirios suspensión), tamaño de partÃ−cula (en colirios suspensión y pomadas). Todas las preparaciones oftálmicas deben satisfacer el ensayo de esterilidad que es el mismo que en inyectables. El ensayo de tamaño de partÃ−culas se tiene que hacer por microscopÃ−a en un área correspondiente a 10 μg de pa en estado sólido. Recuento de partÃ−culas: se establecen los lÃ−mites en partÃ−culas superiores a 25 μm. - Menos de 20 partÃ−culas > 25 μm. - Menos de 2 partÃ−culas > 50 μm. • Ninguna > 90 μm. TEMA 13: FORMAS DE ADMINISTRACIà N PULMONAR 1.- SISTEMAS PRESURIZADOS a) INHALADORES DOSIFICADORES PRESURIZADOS (MDIs) Es un campo en estudio debido a que las enfermedades respiratorias han avanzado mucho. Tradicionalmente se usa para patologÃ−as pulmonares como asma bronquial o fibrosis quÃ−stica. Ahora también se piensa en la vÃ−a pulmonar como alternativa para administración sistémica de medicamentos con problemas por otras vÃ−as: degradación vÃ−a oral, efecto de primer paso importante. Se usan los sistemas de inhalación = aerosoles. La deposición de principio activo en el tracto respiratorio depende de: - Propiedades fÃ−sico-quÃ−micas del principio activo - Propiedades de la formulación: la más crÃ−tica es el tamaño de partÃ−cula. - Sistema de administración - Estado del paciente, capacidad pulmonar TAMAà O DE PARTà CULA O GOTà CULA DE AEROSOL Aerosol: partÃ−culas de sólido o lÃ−quido (fase interna) en gas (fase externa). Hasta hace poco la valoración se hacÃ−a en función del tamaño, no se tenÃ−a en cuenta la densidad. Para tener en cuenta la densidad se ideó un parámetro (diámetro aerodinámico) que estandariza los tamaños teniendo en cuenta la densidad. Se define como diámetro de una esfera de densidad 1 que sedimenta a la misma velocidad que las 73 partÃ−culas del aerosol. Da = dp â ª â Ï”/Ï”a TIPOS DE AEROSOLES Son los envases aerosol tradicionales (matamoscas, uso tópico, etc) y los primeros tratamientos del asma. Cuando hablamos de aerosol nos referimos a un envase-aerosol, es decir envase completo conteniendo medicamento (que no es un aerosol) con un gas comprimido o licuado perfectamente cerrado que cuando se presiona y libera el contenido, el sistema fÃ−sico-quÃ−mico que se forma es un aerosol. En el interior no hay un aerosol nunca. Dentro hay dos elementos: â ª Propulsor: gas que hace que salga la formulación que puede estar comprimido o licuado: > Gases comprimidos: los habituales son no tóxicos como el N2, CO2. En el interior hay un lÃ−quido (disolución, suspensión, emulsión [no para vÃ−a pulmonar]) y un gas a sobrepresión. Cada vez que se presiona, una parte del lÃ−quido sale al exterior y queda más sitio dentro y el gas se expande. Cuando se acaba el lÃ−quido el gas ocupa todo el recipiente de manera que la presión disminuye. Para que salga todo el lÃ−quido, la presión debe ser muy elevada y este es uno de los mayores inconvenientes de este sistema. La presión baja y la dosificación en difÃ−cil. > Gases licuados: se hace a presión elevada o temperatura baja Dentro del envase está el gas licuado, el pa y el gas ocupando el espacio que queda libre. Cuando descargamos, disminuimos la cantidad de lÃ−quido, el gas se expande y disminuye la presión por debajo de la Pinicial y esto hace que se evapore lÃ−quido que está en equilibrio. Este sistema permite una descarga completa y dosificación adecuada. Hasta hace poco se usaban los CFCs. Estos compuestos tienen muchas ventajas: + baja toxicidad + sabor y olor agradable + pureza elevada + compatibles con los envases + P de vapor adecuada + posibilidad de usar mezclas + no inflamables + baratos Inconvenientes: 74 + P de vapor dependiente de la temperatura + efecto sobre la capa de ozono: forman radicales libres que rompen la molécula de ozono Los sistemas farmacéuticos fueron de los últimos en dejar los CFCs por no encontrar otros. Luego se llegó a la utilización de los hidrofluoroalcanos (HFAs). El problema es que no son buenos disolventes del pa al contrario que los CFCs. No dañan la capa de ozono pero no tienen las mismas aplicaciones que los CFCs. El problema de solubilidad se solventa disolviendo el pa en un poco de alcohol etÃ−lico. El problema es que el tamaño de las gotÃ−culas de aerosol va a ser mayor. â ª Concentrado: conjunto de elementos restantes (pa, vehÃ−culo, disolventes, etc). En función de esto, los aerosoles se pueden dividir en dos grupos: Bifásicos: llevan dos fases o Trifásicos: llevan tres fases. Si el gas es comprimido, el lÃ−quido es una mezcla que incluye pa, vehÃ−culos, excipientes. El lÃ−quido puede ser una disolución del pa o una suspensión en un vehÃ−culo o una emulsión que contenga el pa en una de sus fases. Si es una disolución es bifásico, si es suspensión o emulsión será trifásico. Si es gas licuado: el lÃ−quido puede ser una disolución del pa en el gas (bifásico), una suspensión o una emulsión (muy raro) en el gas (trifásico). DESCARGAS Si descargamos un sistema de gas comprimido con una disolución de pa, la descarga es de aerosol niebla (el gas empuja al lÃ−quido que sale en forma de gotÃ−culas -> matamoscas). Si es una suspensión, también es un aerosol niebla. Si es una emulsión -> espuma. Un sistema de gas licuado con pa disuelto -> la salida será de aerosol humo (salen gotÃ−culas pero el gas en contacto con el exterior se evapora y quedan las partÃ−culas). Con suspensión -> aerosol humo. Con alcohol para disolver -> aerosol niebla. Los de gas licuado suelen ser los de inhalación. ELEMENTOS MECÔNICOS - Recipientes Tienen que ser ligeros, resistentes a la rotura, compactos y quÃ−micamente inertes. La mayor parte de aerosoles que no son para inhalación son de hojalata. Para vÃ−a pulmonar se usan los de aluminio. También se usa acero inoxidable o vidrio (envases de cosmética) que suelen estar recubiertos de plástico para que, en caso de rotura, el usuario no se dañe. 75 - Válvulas Pueden ser de flujo continuo o dosificadoras (sale sólo una dosis). Estas válvulas dan nombre a los aerosoles de gas licuado (MDI -> Metered Dose Inhaler´s). - Adaptadores y espaciadores El envase también está adaptado a la boca mediante una pieza de plástico (adaptador) que son cilÃ−ndricos y acodados para que el aerosol se emplee de forma invertida. También hay un espaciador que es una pieza de plástico que puede ser cilÃ−ndrica o romboidal para evitar el problema de la falta de coordinación entre la pulsación y la inhalación. SISTEMAS PRESURIZADOS: LLENADO - Llenado por presión: â ª Se llenan los envases dosificando por gravimetrÃ−a â ª Se elimina el aire del envase añadiendo unas gotas de gas licuado â ª Se fija la válvula y a través de esta válvula se inyecta el propulsor licuado dentro del envase que se controla con un manómetro que mide la presión dentro del envase. â ª Se comprueba el cierre de cada envase. Se colocan los envases en un baño a 55 ºC y ver si hay burbujeo. Se desechan los malos y a los otros se les coloca el pulsador. - Llenado por enfriamiento: â ª Se dosifica el concentrado por gravimetrÃ−a â ª Se dosifica el propulsor que será el gas licuado con una temperatura de -40 ºC y se dosifica por gravimetrÃ−a. â ª La evaporación del propulsor elimina el aire que quedaba â ª Se pone la válvula viendo la presión con un manómetro â ª Se comprueba el cierre en un baño a 55 ºC â ª Colocamos el pulsador y tenemos el aerosol Una ventaja de este llenado es que existe una elevada precisión en la dosificación del propulsor porque se realiza por pesado, pero los costes de las instalaciones son muy elevados; las pérdidas de propulsor en el momento del llenado pueden ser importantes. También la temperatura tan baja del propulsor puede inducir cambios en el principio activo. Dependiendo del pa usaremos un sistema u otro. b) INHALADORES ACCIONADOS POR INSPIRACIà N 76 También son presurizados pero la válvula se acciona al inspirar. AsÃ− evitaremos el problema de la falta de coordinación. Se llama autohaler. Usan de propelente los HFAs. El problema es que requiere un flujo inspiratorio sustancial. 2.- SISTEMAS NO PRESURIZADOS Son los nebulizadores e inhaladores de polvo seco (DPIs -> Dry Powder Inhaler´s). - Nebulizadores: son equipos que generan en el ambiente un sistema aerosol (agua con eucalipto). Son para gente inconsciente, bebés, enfermos que no pueden usar otros sistemas. Son sistemas de elevado tamaño. â ª Ultrasónico: utiliza ultrasonidos que en contacto con el medicamento dan lugar a un producto nebulizado. â ª Airjet: tiene una entrada de gas que pasa por un lÃ−quido y sale el producto nebulizado por efecto Bernuilli. Los dos son aerosoles niebla. - Inhaladores de polvo seco: Dispositivos para inhalación de pequeñas partÃ−culas sólidas. â ª Sistemas monodosis: llevan una cápsula con la dosis que se rompe y cae el pa a la cámara dosificadora y al inspirar se inhala ya que tiene entrada de aire por el otro lado. El problema es que cada dosis hay que abrir y meter otra. â ª Sistemas multidosis: tiene muchas dosis dentro. Se gira una rosca y tenemos otra dosis. - Aspectos crÃ−ticos Están relacionados con las caracterÃ−sticas de las partÃ−culas. Hay pocos sistemas con las caracterÃ−sticas adecuadas para poder dispensar por efecto de la inhalación. La capacidad de penetración de una partÃ−cula depende del diámetro medio y la densidad, es decir, del diámetro aerodinámico. El material debe presentar unas propiedades de flujo adecuadas. Se consigue usando mezclas de materiales de caracterÃ−sticas óptimas como la lactosa (excipiente soluble) que da lugar a mezclas poco cohesivas. También podemos modificar la superficie de las partÃ−culas. También podemos usar metodologÃ−a basada en fluidos supercrÃ−ticos (C02 licuado) para obtener pa poco cohesivos y de pequeño tamaño. 3.- SISTEMAS PRESURIZADOS (MDIs) VS SISTEMAS NO PRESURIZADOS (DPIs) - Ventajas de MDIs: â ª Más baratos â ª No dependen de las caracterÃ−sticas del paciente (capacidad inhalatoria). 77 - Ventajas de DPIs: â ª Evitan el problema de coordinación entre inhalación y pulsación â ª No contaminan â ª Menor deposición en la zona orofarÃ−ngea. Llega a zonas más profundas del sistema respiratorio. En los presurizados, la velocidad de las gotÃ−culas es mayor choca contra la cavidad orofarÃ−ngea. 4.- CONTROL DE CALIDAD EN SISTEMAS PRESURIZADOS Y NO PRESURIZADOS - Control de materias primas - Control de componentes: â ª Recipientes â ª Válvulas â ª Control de concentrados - Control en proceso: â ª En los presurizados será control del concentrado â ª En los no presurizados será control del material pulverulento - Control de producto acabado: â ª Ensayo de estanqueidad (visto en llenado de presurizados) â ª Descarga (aerosol presurizado) > Tamaño de partÃ−cula/gotÃ−cula (diámetro aerodinámico) Se usan métodos de separación inercial que consiste en una torre de fraccionamiento con distintos platos. Se descarga en la parte superior y se hace vacÃ−o en la inferior para que salga. Las partÃ−culas grandes sedimentan contra el plato y las más pequeñas continúan el recorrido. AsÃ− se separan por tamaño. Para la observación se usa difracción láser (para gotÃ−culas) que mide la sombra de la partÃ−cula y a partir de ahÃ− se saca el tamaño. Para partÃ−culas se usa la microscopÃ−a para medir el diámetro. También se usa el método Coulter (para aerosol humo, no para niebla). > Forma y densidad (por microscopÃ−a) â ª También hay que evaluar el principio activo en cada descarga: > Para presurizados dando una dosis > Para los de polvo se saca una dosis del aparato 78 TEMA 14: ADMINISTRACIà N NASAL Y à TICA 1.- Và A NASAL Es una vÃ−a de las que están recibiendo más atención para el uso sistémico. Esto está muy ligado a la vÃ−a pulmonar. Era empleada para patologÃ−as de las vÃ−as aéreas superiores pero el hecho de que los medicamentos por esta vÃ−a no sufrÃ−an efecto de primer paso y la actividad enzimática sea muy baja, la convierten en una alternativa para la administración sistémica. ANATOMà A DE LA NARIZ Tres regiones fundamentales: - VestÃ−bulo nasal: acondiciona el aire en temperatura y humedad y retiene las partÃ−culas. - Región respiratoria: donde se absorben los pa. Tiene elevada vascularización. - Región olfatoria: destinada a captar los olores. La superficie total es de 160 cm2 aproximadamente. FISIOLOGà A DE LA NARIZ - Clearance mucociliar: mecanismo de defensa del tracto respiratorio. Es el encargado de retener sustancias extrañas o de conducirlas al estómago en caso de que pasen la zona. - Barrera enzimática: impide la entrada a moléculas grandes. SISTEMAS DE ADMINISTRACIà N NASAL - Gotas nasales: soluciones de aplicación local para aliviar los sÃ−ntomas de la rinitis o el catarro común. - Aerosoles nasales: â ª Sistemas de tipo dosificadores presurizados â ª Sistemas en polvo, tipo turbuhaler - Pomadas FORMULACIà N - Factores a considerar â ª Tamaño y peso molecular: hasta 1000 daltons se absorben hasta un 85%, por encima de este tamaño la absorción disminuye. â ª pH y osmolaridad del preparado: afecta a la ionización del medicamento (no ionizado se absorbe mejor). Desviaciones de la osmolaridad afectan a la fisiologÃ−a de las células. 79 â ª Solubilidad y velocidad de disolución: sobre todo en los administrados en polvo seco. El tamaño y la morfologÃ−a condicionan la zona de impactación de las partÃ−culas. - Lugar de administración â ª Tipo de preparado: depende de si son aerosoles, pomadas, gotas. Los aerosoles se depositan en la parte anterior de la nariz y las gotas se dispersan en el interior. La biodisponibilidad de las gotas es menor y por ello se han hecho aproximaciones para aumentar la biodisponibilidad. APROXIMACIONES - Aumento del tiempo de residencia: se aumenta el tiempo de residencia de la formulación en el lugar de absorción. Se hace aumentando la viscosidad, usando bioadhesión. - Aumento de absorción: se hace por medio de promotores (disminuyen la resistencia al paso de sustancias). Problema: presentan ciliotoxicidad. - Modificación de las propiedades fÃ−sico-quÃ−micas del pa: se hace para aumentar la biodisponibilidad. Se usan las ciclodextrinas (polisacáridos cÃ−clicos con estructura tronco-cónica). El interior es hidrofóbico y el exterior hidrofÃ−lico. Forman complejos de inclusión englobando una parte de la molécula de pa. El complejo tiene mejores propiedades de solubilidad porque es una entidad quÃ−mica nueva. Tiene que pasar todos los ensayos previos (toxicidad, etc). 2.- ADMINISTRACIà N à TICA Se usa para tratamiento de patologÃ−as locales del oÃ−do externo y medio: - Acumulación de cera - Dermatitis del oÃ−do externo - Procesos infecciosos de la zona externa y media ANATOMà A Y FISIOLOGà A DEL Oà DO - Tres regiones principales: â ª Canal auditivo externo â ª OÃ−do medio (tÃ−mpano y cadena de huesecillos) â ª OÃ−do interno (clóquea) - Transmisión de los sonidos: â ª Conducto auditivo externo â ª Membrana timpánica (vibra) â ª Cadena de huesecillos: lleva la vibración a la clóquea 80 â ª Clóquea: transformación en impulso nervioso â ª Cerebro: percepción auditiva MECANISMOS DE PROTECCIà N â ª Forma externa tÃ−pica en “S” â ª pH ácido (microorganismos patógenos crecen a pH alcalino) â ª Capa de queratina â ª TÃ−mpano: membrana continua que protege y mantiene estéril el oÃ−do medio y el interno â ª Cera, tapón de cerumen y una serie de secreciones de glándulas PREPARADOS à TICOS â ª LÃ−quidos, semisólidos o incluso polvos â ª Preparados de lavado para eliminar tapones de cera Si el tÃ−mpano está intacto, la formulación nunca llegará a los fluidos corporales. Se persigue una acción local. Se administran antisépticos, antifúngicos, anestésicos locales, antibióticos solos o con corticoides (acción local). FORMULACIà N - Excipientes (caracterÃ−sticas) â ª Garantizar y mantener el contacto directo entre el pa y la superficie del conducto auditivo. â ª Deben facilitar la difusión del pa en los tejidos. - VehÃ−culos más usados: agua, glicerina, propilenglicol y aceites. - Espesantes y humectantes: para garantizar el contacto (humectantes) y que no se eliminen rápido. Ej: metilcelulosa. La glicerina y el propilenglicol también son buenos solubilizantes y facilitan la eliminación de exudados. ACONDICIONAMIENTO - Frascos cuentagotas (10 ml) son los más frecuentes y son multidosis. Se exige la presencia de un conservante para evitar contaminación. Se usan frascos de vidrio o de plástico flexible. - Aplicación directa desde el envase - Aplicación mediante torundas: existe riesgo de contaminación 81 - Aplicación mediante aerosoles: â ª Pulsador y dosificador adaptado al oÃ−do â ª La presión sobre el tÃ−mpano no debe ser elevada CONTROLES No se indican ensayos especiales. Debe tratarse de formulaciones estériles, sin conservantes e isotónicas y deberán superar los ensayos de esterilidad (esto es para uso en oÃ−do con tÃ−mpano dañado). TEMA 15: FORMAS DE ADMINISTRACIà N RECTAL INDICACIONES â ª Principios activos que se absorben mal en el intestino delgado. â ª Principios activos inestables frente a enzimas proteolÃ−ticos. â ª Principios activos que producen irritación en estómago e intestino delgado. â ª Principios activos que requieren una dosis mayor de 500 mg (son difÃ−ciles de comprimir). No se utiliza mucho la vÃ−a rectal. VENTAJAS â ª Alternativa frente a la vÃ−a oral, para ciertos pa y determinados grupos de población (ancianos y niños). â ª Tratamiento de patologÃ−as en órganos de difÃ−cil acceso (ojo, piel, oÃ−do, la mucosa bucal). â ª Efecto local o para enfermedades del colon o del recto (tratamiento de colitis ulcerosa, hemorroides, etc) ANÔTOMO-FISIOLOGà A DEL RECTO â ª Irrigado por las venas hemorroidales (inferiores, medias y superiores). Las inferiores y medias no van a dar al hÃ−gado (no efecto de primer paso). Determinados medicamentos (propranolol, etc) por vÃ−a rectal tienen una biodisponibilidad mucho mayor que por vÃ−a oral. â ª Elevada circulación linfática: contribuye al aumento de la absorción. No es una zona diseñada para la absorción (pocas vellosidades y microvellosidades) pero la superficie de absorción y la permeabilidad son suficientes para el uso de algunos medicamentos. â ª Poco movimiento: puede ser una ventaja porque se mantienen los gradientes de concentración, lo cual puede favorecer la absorción. â ª El volumen de lÃ−quido del colon descendente y del recto es bajo (2-3 ml). Esto hace que las concentraciones de fármaco puedan ser elevadas y favorece la absorción. 82 â ª El inconveniente es que las uniones entre las células son más estrechas que en otras zonas del intestino (no todas las células pasan -> sobre todo pasan las pequeñas). FORMAS DE ADMINISTRACIà N RECTAL Los supositorios son la forma principal. Son preparados sólidos cuyo peso es variable dependiendo de a quien se pretenda administrar. Tienen una forma adecuada. En la composición se incluye uno o más pa y un excipiente base que puede ser de tipo graso o hidrosoluble. EXCIPIENTES DE SUPOSITORIOS CaracterÃ−sticas del excipiente ideal: â ª No debe tener efecto terapéutico â ª Inercia quÃ−mica : no debe interaccionar con otras sustancias, sobre todo con el principio activo. â ª Resistencia mecánica suficiente para superar la resistencia del esfÃ−nter anal. â ª Adecuado coeficiente de contracción para facilitar la salida del molde una vez formado. â ª Punto de fusión próximo a la temperatura corporal para liberar el principio activo. â ª Capacidad de absorción de agua buena para poder llegar a formar una emulsión y ayudar a absorberse. â ª Estable y bien tolerado por la mucosa rectal. â ª Puntos de ablandamiento y fusión próximos: esto es importante cuando el pa no es soluble en el excipiente y se pretende evitar la precipitación. TIPOS DE BASES - Excipientes grasos o lipofÃ−licos (los más usados) â ª Manteca de cacao: se usó mucho pero ahora se ve reemplazado. > se enrancia con relativa facilidad > baja capacidad de absorción de agua > intervalo de reblandecimiento estrecho > transformaciones alotrópicas: se transforma en otras variedades con propiedades fÃ−sicas diferentes. â ª Aceites hidrogenados (se usan poco): son productos céreos que son mezcla de aceites naturales con distintos grados de hidrogenación. > difÃ−cil control de la hidrogenación > no permite incorporar disoluciones acuosas 83 â ª Excipientes semisintéticos (más usados) > tienen composición constante > punto de fusión bien definido > sin problemas de polimorfismo (no varÃ−a el punto de fusión) > Ã−ndice de contracción adecuado > son estables y baratos Los más útiles son los ADEPS (ácidos grasos saturados de entre 10-18 átomos de C) - Excipientes autoemulsionables Son una variedad de los anteriores con más capacidad de absorber agua. Una vez fundidos pueden dar lugar a emulsiones en el recto lo que facilita la absorción. - Excipientes hidrosolubles (no se usan mucho) Se incluyen los PEG. No plantean problemas de conservación pero producen irritación y problemas de incompatibilidad con algunos pa y tienen efecto laxante por razones osmóticas. - Excipientes hidrodispersables o mucilaginosos (no interesan) Son muy higroscópicos y tienen efecto osmótico elevado. A veces se incorporan conservantes, endurecedores, correctores de la viscosidad o productos que den lugar a tixotropÃ−a. PREPARACIà N DE SUPOSITORIOS La cantidad de principio activo la podemos conocer fácilmente. Para elaborar 100 supositorios de 0´5 g de pa -> 100 â ª 0´5 = 50 g. El problema es saber la cantidad de excipiente. Para ello se usa el factor de desplazamiento (g de excipiente desplazados por 1 g de pa). La dosificación de pa se realiza por volumetrÃ−a y está condicionada por el volumen de los alveolos del molde (tamaño del molde). Queremos saber la cantidad de excipiente para rellenar el molde que contiene una cantidad de pa. Si la densidad (Ï”) del pa fuese igual a la del excipiente, conociendo la cantidad de excipiente necesaria para llenar esos alveolos no habrÃ−a problema. Normalmente las densidades son distintas. Es necesario conocer el factor de desplazamiento para poder calcular la cantidad de excipiente que contendrá el supositorio. El factor (f) depende del excipiente. El volumen del pa = Vm = Pm / Ï”m 84 Ve = Pe / Ï”e igualamos -> Pm / Ï”m = Pe / Ï”e -> Pe = Pm⠪ϔe / Ï”m Si Pm = 1 -> Pe = Ï”e / Ï”m = f Gramos de excipiente desplazado: excipiente principio activo Excipiente puro El volumen de pa es igual al volumen que tuve que sacar de excipiente. Cálculo del excipiente necesario para un supositorio con una dosis determinada: Excipiente desplazado (Pe) = dosis â ª f Cantidad de excipiente para un supositorio de excipiente puro = P Un supositorio de cualquier dosis P' = P - (dosis â ª f) PREPARACIà N DE PA Y EXCIPIENTE - Principio activo soluble en el excipiente: no son necesarias precauciones, se funden y se mezclan. - P.a. insoluble en el excipiente y muy soluble en agua: hay que tomar precauciones para evitar que el principio activo se acumule en la punta. Se disuelve el pa en un pequeño volumen de agua y luego se emulsifica en el excipiente fundido. PREPARACIà N DE LA MASA A escala industrial la mezcla se hace en reactores de acero inoxidable que tienen unos mezcladores de alta velocidad a temperatura adecuada. Se pueden usar homogeneizadores o molinos coloidales (sobre todo en supositorios emulsión). Es importante una agitación constante para facilitar que esté homogéneamente distribuido. FORMACIà N DE SUPOSITORIOS POR FUSIà N â ª Fundir el excipiente, incorporar el pa y mezclar â ª Se vierte en los alveolos donde solidifica â ª Cuando se enfrÃ−a el supositorio se contrae el excipiente (esto facilita el desmoldeo) â ª Llenado en exceso de los alveolos para evitar supositorios defectuosos debido a la contracción. El exceso se saca luego con cuchilla. TIPOS DE SUPOSITORIOS 85 - Supositorios suspensión Se elaboran a partir de una suspensión (pa no soluble en excipiente fundido). Se hace una suspensión homogénea (el pa distribuido uniformemente y no se produce acumulación en la punta). La velocidad de sedimentación será baja. Se vierte la masa sobre los alveolos a una temperatura ligeramente mayor que la de fusión para que solidifique rápido y no sedimente. También se pueden usar moldes refrigerados. - Supositorios emulsión Se emulsiona el excipiente con un pequeño volumen de agua, que contiene el pa, con agitación intensa. Se mezclan ambas fases a igual temperatura. No lleva tensioactivos. - Supositorios solución El pa es soluble en el excipiente fundido. Es importante saber la solubilidad del pa en el excipiente para que no precipite. PRODUCCIà N POR FUSIà N A ESCALA INDUSTRIAL â ª Vertido de la masa (Tª adecuada), desde una tolva a los alveolos situados en una platina giratoria. â ª Retirada de la masa sólida en exceso mediante una cuchilla caliente. â ª Abertura de la platina molde para dejar libres los supositorios. â ª Uso de los envases (celulosa, cloruro de polivinilo, polietileno, etc) como moldes. Hoy se tiende a usar el envase final como molde. FORMACIà N DE SUPOSITORIOS POR PRESIà N â ª Se usa sólo para pa termolábiles (no se puede fundir el excipiente). â ª Mezcla homogénea del excipiente pulverizado y el pa (inconveniente principal). â ª Se introducen en prensas que terminan en una pieza que actúa como molde. â ª No se pueden obtener supositorios emulsión. ENSAYOS DE CONTROL - Sobre el excipiente â ª Controles fÃ−sicos > color > punto de fusión > punto de solidificación 86 > viscosidad > dureza (penetrómetro) > densidad > contracción volumétrica â ª Controles quÃ−micos (ensayos para determinar la pureza de sustancias de naturaleza grasa) > Ã−ndice de refracción > Ã−ndice de peróxidos > Ã−ndice de acidez â ª Ensayos biológicos > ensayo de tolerancia en mucosa rectal - Sobre los supositorios â ª Apariencia > forma adecuada y aspecto homogéneo > superficie uniforme, lisa y brillante > parte basal plana â ª Homogeneidad de contenido y distribución del pa > cuantificación del contenido en pa (10% del contenido teórico) > determinación en diferentes fracciones de la relación peso pa/peso supositorio â ª Uniformidad de peso: puede haber burbujas de aire en la masa. Puede variar un 5% â ª Control de dureza: peso máximo que soporta un supositorio colocado de forma vertical 1 h â ª Ensayo de disgregación > cilindro de vidrio o plástico, cerrado por placas perforadas (orificios de 4 mm) > baño a 36-37 ºC, giros cada diez minutos tdisg â ¥ 30 min con excipientes hidrosolubles tdisg â ¥ 60 min con excipientes grasos â ª Velocidad de cesión del pa 87 > difusión a través de una membrana, en general de celofán > parámetros crÃ−ticos: Tª, velocidad de cesión, área de contacto â ª Ensayos fisiológicos (dependen de la naturaleza del excipiente y del pa) > determinación del pa en sangre u orina o de la respuesta > ensayo de tolerancia OTRAS FORMAS RECTALES - Soluciones o dispersiones rectales (enemas) â ª Se usan poco (incómodas y mal aceptadas) â ª Se administran agentes de contraste y para limpiar â ª Absorción mayor que a partir de supositorios, por eso se buscaron los: - Microenemas (poco volumen) â ª Aplicación sistémica - Geles, espumas y pomadas â ª Mejor aceptados por mejor retención â ª Tratamientos locales o intestinales de localización distal. â ª Se aplican con cánula para tratamiento de hemorroides y otras inflamaciones Controles correspondientes a cada forma farmacéutica. TEMA 16: FORMAS DE ADMINISTRACIà N VAGINAL E INTRAUTERINA Similares a las rectales. Muy usada. PECULIARIDADES DE LA Và A VAGINAL â ª Conducto de 8-12 cm desde el cuello del útero hasta la vulva â ª Mucosa con pliegues y cavidades â ª No vierten glándulas a excepción de un trasudado seroso (tipo látex) que contiene un bacilo Gram (+) -> Bacilo de Döderlein. Es una defensa del aparato genital femenino y es responsable del pH ácido. FORMAS VAGINALES - à vulos â ª Formas semisólidas (aplicación de espermicidas, anestésicos locales) o sólidas (estrógenos o 88 progestágenos). Los sólidos son parecidos a los supositorios (salvo en tamaño). Se elaboran por fusión y compresión. El peso es entre 10-15 g (elaboración igual que supositorios). Son mal aceptados por ser muy grandes y generan mucho lÃ−quido (incómodo). â ª Excipientes Igual que supositorios. El más usado es una mezcla. - Comprimidos vaginales Más recientes. Tecnológicamente parecidos a los de vÃ−a oral. Se disuelven antes de usar (enema vaginal). Peso entre 0´5-3 g. De forma clásica o alargada. â ª Problemas de fabricación Los tiempos de disgregación tienen que ser muy cortos aunque estén en contacto con poco lÃ−quido. Esto permite entrar en contacto con todas las cavidades y pliegues de la vagina. Otro requisito es que no altere el pH de la zona. Se usan para infecciones locales o para efecto sistémico con hormonas y derivados. Presentan mayor biodisponibilidad que la vÃ−a oral. Esto es porque la zona es muy irrigada y no sufre efecto de primer paso. â ª Excipientes (clásicos de comprimidos) > diluyentes que se disgreguen rápido -> lactosa; se transforma en ácido láctico que ayuda a mantener pH. > aglutinantes y lubrificantes > agentes tensioactivos para favorecer la interacción entre los componentes de la forma farmacéutica y el agua de la zona. â ª Controles Igual que los otros tipos de comprimidos Và A INTRAUTERINA Son los dispositivos intrauterinos (DIUs) Inicialmente eran todos en forma de T; ahora también con forma de 7. Funcionaban como efecto barrera impidiendo la implantación del embrión en el endometrio. Mecanismo de acción poco claro. â ª DIUs recientes Son pequeños y elaborados con plástico y en el interior llevan un reservorio de medicamento. Pueden 89 llevar metales anticonceptivos (Cu) u hormonas esteroideas. El control de la liberación lo realiza una membrana. Libera una cantidad diaria de medicamento y no se produce el embarazo. TEMA 17: CORRECTIVOS Se agrupan los aditivos de las formas farmacéuticas. Se usan para conseguir un mejor aspecto, sabor y olor y asÃ− mejorar la aceptación por parte de los pacientes. COLORANTES - Justificación del coloreado: â ª Formas farmacéuticas de color consistente y apariencia agradable (aumenta la confianza y la aceptación). â ª Incremento de la eficacia del tratamiento. En los tranquilizantes el color azul aumenta la eficacia. â ª Identificación de las especialidades. â ª Prevenir la degradación del pa. Es el caso de los colorantes que opacifican la cápsula. - CaracterÃ−sticas del colorante ideal: â ª Inocuo y sin actividad farmacológica. â ª Composición quÃ−mica definida (los naturales no la tienen). â ª Potente y estable. â ª Compatible y sin capacidad de interferir en la valoración y ensayos generales de la forma farmacéutica. â ª Sin olor y sabor desagradables. â ª Fácil de obtener y barato. - Clasificación: â ª Colorantes orgánicos: > Hidrosolubles: se usan para formas orales lÃ−quidas. Si se adsorben sobre un soporte de alúmina hidratada forman: > Lacas: insolubles. Se usan para colorear superficies. â ª Colorantes inorgánicos o pigmentos: > Insolubles en agua > Estables frente a la luz (normalmente opacos) > Cumplen bien las distintas regulaciones de los paÃ−ses. Se usan para comercializar en todo el mundo 90 > Gama de colores limitada > Son normalmente sales de Fe, Ti y Ca > Se usan para formas sólidas y de uso externo â ª Colorantes naturales: > Son inestables frente a la luz > Poder de coloración reducido y más caros para lograr que su composición sea constante > Muy bien aceptados - Criterios de selección: â ª Forma de dosificación: > Colorantes solubles (0´0005-0´001%) -> granulados, cápsulas de gelatina, formas lÃ−quidas. > Lacas (0´1%) -> recubrimiento de comprimidos â ª Estabilidad quÃ−mica del preparado: formas lÃ−quidas más inestables que las sólidas. â ª Facilidad de incorporación al sistema. Para recubrir hay que usar un colorante insoluble. â ª Aptitud estética: color, tipo de cubierta. â ª Otras propiedades organolépticas: sabor, aroma. SABORIZANTES Y AROMATIZANTES - Justificación de su uso: â ª Formas orales: corrección de caracterÃ−sticas olfativas y gustativas. â ª Formas de uso externo: corrección de caracterÃ−sticas olfativas. - Principios generales de corrección del sabor y olor: â ª Aceptación influida por la edad (niños dulce y ancianos amargo). â ª Umbral de sabor diferente de cada persona. â ª Sabores básicos: dulce, amargo, ácido y salado. â ª Sensaciones gustativas secundarias: fresco, picante, astringente. - Corrección del sabor: â ª Se usan aromatizantes y saborizantes 91 > Uno o la combinación de varios > Corrección de las caracterÃ−sticas olfatogustativas: aromatizante + saborizante > Corrección de las caracterÃ−sticas organogustativas: un saborizante Ej: para enmascarar sabor amargo se usa un correctivo con sabor persistente (cacao, melocotón), con un ligero efecto anestésico. También sabores antagonistas: cÃ−tricos o aromatizantes con relación al sabor amargo (monda de naranja). Para sabor salado: edulcorante. Para sabor ácido: cÃ−tricos. â ª Métodos especÃ−ficos: > Modificación fÃ−sica y/o quÃ−mica del pa + modificación del tamaño de partÃ−cula + formación de un derivado menos soluble + utilización de granulado o comprimidos efervescentes (cierto efecto anestésico del CO2 - Edulcorantes: â ª Sustancias que comunican sabor de dulzor y pueden ser naturales o sintéticas. â ª Pertenecen al grupo de los saborizantes. â ª Clasificación: > De origen natural (azúcares) + sacarosa, glucosa, fructosa > Otros + poder edulcorante bajo (se compara con la sacarosa): sorbitol, manitol, glicerina, etc + poder edulcorante medio y alto: glicirrina, sacarina, etc. Dejan una sabor amargo metálico en la boca después de su degustación. Por esto cuando se usan, se mezclan uno de poder edulcorante alto y otro bajo para disminuir el sabor residual. Los edulcorantes son excipientes de declaración obligatoria. - Aromatizantes Son procedentes de frutas, aceites esenciales, resinas vegetales. También los hay de sÃ−ntesis (menos usados). 92 - Criterios de selección: â ª VÃ−a de administración: en el caso de aromatizantes. Para tópicos usan unos y para oral otros. â ª Compatibilidad con el resto de la formulación. â ª Grupo de población: dependiendo de la cultura, los sabores y los olores son aceptados o no. â ª Paneles de degustación y análisis sensorial. TEMA 19: TIPOS DE ENVASES Las formas farmacéuticas tienen que envasarse y acondicionarse - Requerimientos de materiales: â ª Proteger el producto frente a agentes externos (luz, humedad, etc) â ª Adecuado para la producción industrial â ª CaracterÃ−sticas estéticas aceptables â ª Inerte frente al contenido â ª Aprovisionamiento, manipulación y almacenaje fáciles â ª Precio reducido VIDRIO No es un compuesto de composición quÃ−mica definida y propiedades quÃ−micas y fÃ−sicas modulables. - Composición y estructura: Producto inorgánico de fusión, enfriado a un estado rÃ−gido sin cristalización. Se forma una estructura vÃ−trea -> retÃ−culo vÃ−treo formado por un tetraedro. O Si Se forma una red (Si O4)n al que se pueden añadir sustituyendo al Si -> B, P, As, etc. Se puede añadir Ca, Na que deforman la red. También pueden añadirse otros para vidrios especiales -> Al, Fe, Mn. El Fe y el Mn dan color. Para los envases farmacéuticos se usan 4 tipos de vidrio especificados en la farmacopea: â ª Tipo I -> es el más puro. Se llama Borosilicatado (vidrio neutro). Es el de mayor resistencia mecánica (shock térmico). También tiene una elevada resistencia hidrolÃ−tica (resiste ácidos y bases fuertes). 93 Son los más caros. Se pueden usar para cualquier tipo de preparado. â ª Tipo II -> se llama Sódico-cálcico tratado. El tratamiento consiste en calentar el vidrio a una temperatura elevada en presencia de SO2 + O2 y se genera una pelÃ−cula de sulfato sódico. Punto de fusión más bajo y es un vidrio más barato. No es reutilizable (la pelÃ−cula es arrastrada con los lavados). No vale para soluciones orales de pH > 7. â ª Tipo III -> Sódico-cálcico sin tratar. Se usa para polvos y soluciones no acuosas de uso parenteral. Resistencia mecánica e hidrólitica media. â ª NP (no parenteral) -> vidrio sódico-cálcico para formas orales y de uso tópico. - Tipos de envases â ª Ampollas: cerradas con calor (vidrio tipo I) â ª Viales: cierre de elastómero y metálico (vidrio tipo I) â ª Recipientes de mayor volumen - Ensayos â ª Dimensiones del recipiente â ª Resistencia mecánica > Resistencia a presión interna > Resistencia al shock térmico â ª Ensayos de transmisión de luz y color â ª Resistencia al ataque hidrolÃ−tico > Recipientes Ã−ntegros (se ve si el recipiente aguanta) > Se pulveriza el vidrio del envase y se hace el ensayo PLÔSTICO Son polÃ−meros derivados del petróleo, orgánicos, de elevado peso molecular y fácilmente moldeables. Son más resistentes que el vidrio (resistencia mecánica). 94 Desventajas: â ª Estabilidad: ceden componentes â ª Absorción del producto - Tipos de plástico â ª Termoplásticos (más flexibles) > PVC > Poliestireno > Polietileno de diferentes densidades â ª Termoendurecidos (más duros) - Aditivos â ª Plastificantes: favorecen el moldeado â ª Estabilizantes y antioxidantes â ª Catalizadores â ª Pigmentos - Aplicaciones â ª Blister y botes de pastillas convencionales (formas sólidas) â ª Parenterales de elevado volumen (polietileno de baja densidad -> permite velocidad de goteo constante, el vidrio no) â ª Formas tópicas: tubos de crema - Controles Dependiendo del tipo de plástico o de envase se hacen unos u otros: â ª Aspecto â ª pH â ª Residuo seco â ª Permeabilidad > Al vapor de agua: repercute en el tiempo de validez > Al CO2 y al O2: para proteger al pa de la oxidación 95 > A las radiaciones â ª Toxicidad ELEMENTOS DE CIERRE. ELASTà MEROS - Requerimientos â ª Cierre hermético â ª Impermeable a lÃ−quidos y gases â ª No puede ceder ni tomar sustancias de la formulación - Tipos de caucho: â ª Caucho natural (poliisopreno) > Vulcanizado: no libera partÃ−culas, vale para resellado. Al atravesarlo con una aguja no se fragmenta ni desprende pedazos (a esto se le llama coring). Su envejecimiento es más rápido, presenta absorción de algunos compuestos. â ª Cauchos sintéticos Derivados del caucho natural con mayor estabilidad quÃ−mica, más resistente a procesos de esterilización, envejecimiento, absorción y permeabilidad. â ª Mezclas de caucho natural (semisintético) Se suman las ventajas de ambos - Aditivos: â ª Agentes vulcanizantes (azufre, aceleradores, retardadores y activadores de la vulcanización) â ª Antioxidantes â ª Plastificantes, etc - Controles â ª Evaluar la capacidad de sorción de diferentes componentes (normalmente son los conservantes). Esto disminuye la capacidad de conservar el medicamento. â ª Hinchamiento por absorción de agua u otros componentes. Se meten los tapones en el medio que queramos y vemos si existe incremento de peso. â ª Permeabilidad al vapor de agua. Puede afectar a la estabilidad. â ª Desprendimiento de partÃ−culas (coring). Sobre todo en administración parenteral. 96 â ª Compatibilidad biológica. En algunos casos los elastómeros producen alteraciones en hematÃ−es que pueden ser muy peligrosas. Los elastómeros se sujetan al envase de vidrio mediante cápsulas de Al. Se usan para: â ª Poros para formas semisólidas â ª Envase de aerosoles â ª Complejos laminares: láminas de AL + barniz termosoldable Los estudios de las formas farmacéuticas se deben hacer con el medicamento en el envase porque ahÃ− es donde van a soportar ciertas condiciones. TEMA 20: ASEGURAMIENTO DE LA CALIDAD Todas las formas farmacéuticas han de ser de calidad (grado de adecuación de un producto a lo que el consumidor busca). Un medicamento de calidad es aquella forma farmacéutica envasada que se adecúa a las necesidades del paciente. Este concepto de calidad objetiva se puede dividir en dos grupos: - Calidad de diseño: tiene que cumplir: â ª Criterios tecnológicos â ª Criterios biofarmacéuticos â ª Criterios de estabilidad â ª Adecuación a las necesidades del paciente â ª Poca complejidad (la menor posible) â ª Menor coste posible - Calidad de conformidad: forma farmacéutica bien diseñada pero mal fabricada no se adecúa al diseño realizado. Su calidad final es la suma de las dos. En el pasado se hablaba del control de calidad (del producto acabado). Hoy en dÃ−a la calidad no se entiende como un control sino como un dominio no sólo del producto final sino de todo el proceso de fabricación. Hoy se habla de aseguramiento de la calidad y “control de calidad” se refiere al control de materias primas, control intermedio y final. El aseguramiento de la calidad comprende: â ª Good Manufacturing Practise (GMP) 97 â ª Validación de procesos: obtención de pruebas documentales demostrativas de que un método analÃ−tico o cualquier otro proceso son suficientemente fiables como para producir los resultados esperados. â ª Control estadÃ−stico de la calidad: se emplea para el control de materias primas, productos intermedios y finales. GMP - Organización del personal: â ª Independencia entre fabricación y control. El personal de fabricación está bajo supervisión del de control de calidad. â ª Asignación de responsabilidades por escrito. Ej: quien tiene que pesar los compuestos, etc. - Locales: â ª Almacenamiento â ª Fabricación â ª Maquinaria - Higiene: â ª Personal â ª General: al cambiar de producto de fabricación se tiene que hacer un lavado de maquinaria, suelos, paredes, etc. Existen protocolos de lavado. - Documentación: todo tiene que ir documentado. â ª Instrucciones para cualquier tipo de trabajo realizado en la empresa â ª Fórmula patrón â ª Método patrón para cada procedimiento de trabajo de la empresa (PNT -> procedimientos normalizados de trabajo) â ª GuÃ−a de fabricación: documento que relata todos los pasos necesarios para la fabricación de una forma farmacéutica â ª Archivo de guÃ−as de fabricación y muestras de referencia (hasta un año después a la caducidad) - Fabricación: â ª Materias primas â ª Procedimientos de trabajo â ª Productos intermedios 98 â ª Fabricación por terceros DEPARTAMENTO DE CONTROL DE CALIDAD Se ocupan de todos los aspectos relacionados con el control de calidad. Son los organizadores del proceso. â ª Establecen las especificaciones y métodos analÃ−ticos â ª Procedimientos de muestreo para hacer los controles â ª Velar por el cumplimiento de GMP â ª Evaluar si las guÃ−a de fabricación están diseñadas de forma correcta â ª Decisiones (aceptar o rechazar) sobre materias primas, productos intermedios o acabados â ª Llevar a cabo estudios de estabilidad (supervisando el departamento de desarrollo) â ª Organizar sistemas de autoinspección CONTROL ESTADà STICO DE CALIDAD Para materias primas, productos intermedios o acabados. â ª Técnicas de control en proceso: se usa para detectar problemas de fabricación y ser capaces de parar la producción si hay unidades defectuosas. 104 80 99
 0
0
Anuncio
Documentos relacionados




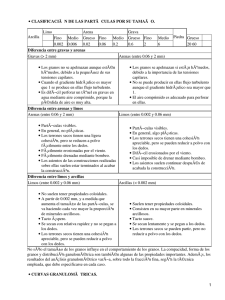
Descargar
Anuncio
Añadir este documento a la recogida (s)
Puede agregar este documento a su colección de estudio (s)
Iniciar sesión Disponible sólo para usuarios autorizadosAñadir a este documento guardado
Puede agregar este documento a su lista guardada
Iniciar sesión Disponible sólo para usuarios autorizados