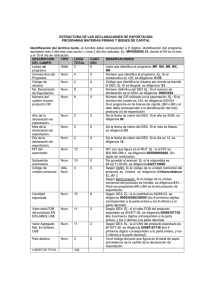
dispositivo medico implantable de biocompatibilidad y
Anuncio

19 OFICINA ESPAÑOLA DE PATENTES Y MARCAS 11 Número de publicación: 2 239 948 51 Int. Cl. : A61F 2/02 7 A61F 2/24 A61F 2/06 A61F 2/04 A61F 2/01 A61M 5/142 ESPAÑA 12 TRADUCCIÓN DE PATENTE EUROPEA T3 86 Número de solicitud europea: 00101782 .1 86 Fecha de presentación : 28.01.2000 87 Número de publicación de la solicitud: 1023879 87 Fecha de publicación de la solicitud: 02.08.2000 54 Título: Dispositivo médico implantable de biocompatibilidad y bioestabilidad mejoradas. 30 Prioridad: 29.01.1999 US 117837 P 29.04.1999 US 301842 45 Fecha de publicación de la mención BOPI: 16.10.2005 73 Titular/es: Medtronic, Inc. 710 Medtronic Parkway Minneapolis, Minnesota 55432-5604, US 72 Inventor/es: Fernandes, Brian C.A.; Donovan, Maura G.; Sparer, Randall V.; Casas-Bejar, Jesus W. y Torrianni, Mark W. 45 Fecha de la publicación del folleto de la patente: 74 Agente: Ungría López, Javier ES 2 239 948 T3 16.10.2005 Aviso: En el plazo de nueve meses a contar desde la fecha de publicación en el Boletín europeo de patentes, de la mención de concesión de la patente europea, cualquier persona podrá oponerse ante la Oficina Europea de Patentes a la patente concedida. La oposición deberá formularse por escrito y estar motivada; sólo se considerará como formulada una vez que se haya realizado el pago de la tasa de oposición (art. 99.1 del Convenio sobre concesión de Patentes Europeas). Venta de fascículos: Oficina Española de Patentes y Marcas. C/Panamá, 1 – 28036 Madrid ES 2 239 948 T3 DESCRIPCIÓN Dispositivo médico implantable de biocompatibilidad y bioestabilidad mejoradas. 5 10 15 20 Esta invención se refiere en general a dispositivos médicos implantables, específicamente bombas de infusión implantables con biocompatibilidad y bioestabilidad mejoradas. La enfermedad de la válvula cardiaca se divide en dos categorías principales: adquirida o congénita. En cualquier caso, la válvula y/o el aparato subvalvular está dañado. El daño de estas estructuras puede hacer que las válvulas sean incompetentes o estenóticas. Las válvulas incompetentes sufren cambios degenerativos que dan lugar a la ampliación del anillo de la válvula. La ampliación del anillo separa las lengüetas. Cuando las lengüetas se separan más, las lengüetas no se juntan apropiadamente, produciendo coaptación inadecuada de las lengüetas. Esta incapacidad de cerrarse correctamente da lugar a flujo sanguíneo impropio a través del corazón y eventualmente requiere corrección quirúrgica por reparación o sustitución de la válvula. La reparación de la válvula, también denominada anulopastia valvular, implica reparar el anillo de válvula reforzando la estructura con un dispositivo o cinta en forma de aro hecho de materiales de tela. Mediante el apropiado dimensionamiento e instalación del dispositivo, denominado aro de anulopastia, el cirujano puede restaurar el anillo de la válvula a su circunferencia no dilatada original. Esta restauración anular pondrá de nuevo las lengüetas en alineación correcta y restablecerá la competencia de la válvula. Las válvulas estenóticas son válvulas en las que las lengüetas han perdido su capacidad de moverse libremente. Además de permitir el flujo regurgitante a través de la válvula, las válvulas estenóticas tienen una disminución en el área del orificio de válvula. La disminución del área de orificio crea grandes gradientes de presión transvalvular. Este trastorno obliga al corazón a trabajar más. El resultado es la ampliación de los ventrículos. Los procedimientos quirúrgicos para corregir tales patologías requieren la sustitución de la válvula. 25 Actualmente hay dos tipos de válvulas protésicas que se pueden usar para sustituir las válvulas que fallan: 1) mecánicas y 2) bioprotésicas. 30 35 40 Las válvulas mecánicas son válvulas hechas de materiales no biológicos, y constan de una carcasa de válvula, un oclusor de flujo, y un aro de cosido. Las válvulas protésicas se componen de alojamiento (denominado un stent), un oclusor de flujo (generalmente tejido de pericardio o raíz aórtica de fuentes animales que ha sido conservado químicamente) y aro de cosido. Como los aros de anulopastia, los aros de cosido de las válvulas mecánicas y bioprotésicas se componen de material de tela de poliéster construido en una configuración entretejida o tejida. La configuración de la tela permite la infiltración celular a los intersticios del tejido permitiendo que la prótesis “agarre”. El proceso de “agarre” crea una superficie biológica que no es trombogénica y hace de una barrera a escapes transvalvulares. Históricamente, se eligió poliéster como el material de tela a causa de la respuesta de curación que provocaba. El poliéster también se elegía por su inertidad química y resistencia a la degradación enzimática. Sin embargo, el implante de estos materiales no resorbibles altera permanentemente el microentorno del tejido donde está implantado. Al tiempo del implante, el tejido del anillo de válvula experimenta trauma por extracción de lengüetas, disección del tejido dañado y/o mineralizado, manipulación, operaciones de unión y sutura. El trauma da lugar a la generación de una herida, implicando la curación de la herida una respuesta inflamatoria. La naturaleza y extensión de la respuesta inflamatoria depende del tamaño del fondo de la herida así como de la naturaleza del material implantado en la herida. El resultado puede ser curación incompleta o inadecuada. 45 50 55 60 65 En sus términos más simples, la respuesta inflamatoria se divide en dos fases, una fase aguda y una fase crónica. Los detalles completos de los eventos de cada fase han sido bien documentados por Shankar y otros (Capítulo 5, “Inflammation and Biomaterials” en Implantation Biology, Host Response and Biomedical Devices, RalpH S. Greco, Ed, CRC Press, Inc, Boca Raton, FL, 1994, 68-80). Si la respuesta inflamatoria aguda no se corrige totalmente, puede progresar a la fase crónica. Las secuelas de eventos en la respuesta inflamatoria crónica pueden dar lugar a serias complicaciones de la función de la válvula. Junto con la deposición de células inflamatorias y la acumulación de elementos celulares y proteínicos sanguíneos, se produce formación de una envuelta fibrosa (de origen en el huésped) denominada panus. Este tejido fibroso se desarrolla como una extensión del tejido que cura el aro de cosido. En el caso de válvulas bioprotésicas de tejido, si la respuesta inflamatoria no es resuelve, el panus resultante continúa creciendo, saliendo el tejido fibroso, al avanzar, a las lengüetas, produciendo estenosis y/o incompetencia. Además de producir estenosis que inhibe la función de las lengüetas, se ha demostrado que el excesivo crecimiento de tejido produce retracción de las lengüetas en las válvulas pericárdicas, dando lugar a regurgitación clínicamente significativa. El estancamiento o la acumulación de sangre es otro resultado de las lengüetas en peligro. La progresión de la disfunción de la válvula en bioprótesis depende del paciente, y está relacionada con lo agresiva que sea la respuesta inflamatoria del paciente para el implante. El progreso de la curación para receptores de válvulas mecánicas es similar al observado con las bioprotésicas, pero pueden tener consecuencias aún más graves. El crecimiento de tejido sobre la válvula puede obstruir el oclusor produciendo fallo catastrófico de la válvula. Así, las válvulas de sustitución, tanto bioprotésicas como mecánicas, suelen tener inconvenientes que pueden hacer necesaria su extracción. Sin embargo, a causa del crecimiento exuberante del pannus a y sobre el aro de cosido de estas válvulas, la extracción de las válvulas requiere disección extensa, haciendo aún más difíciles las operaciones siguientes. Es sabido que los dispositivos médicos que contienen polímeros incluyen agentes terapéuticos para administración al tejido circundante. Por ejemplo, se han diseñado stents con recubrimientos o películas poliméricos que incorporan 2 ES 2 239 948 T3 5 10 15 20 una amplia variedad de agentes terapéuticos, tal como agentes antiinflamatorios, agentes antitrombogénicos, y agentes antiproliferativos, para una amplia variedad de efectos. Se han incorporado compuestos antimicrobianos a porciones poliméricas de dispositivos médicos para liberación sostenida al tejido circundante para mejorar la resistencia a la infección. Los cables eléctricos médicos han incorporado esteroides a o en la punta del electrodo conductor, para reducir la impedancia de la fuente y disminuir los umbrales de estimulación máximos y crónicos. Sin embargo, hasta la fecha los agentes antiinflamatorios no se consideran útiles para mejorar la biocompatibilidad y/o bioestabilidad de los biomateriales utilizados en dispositivos médicos implantables, especialmente los que es posible que deban ser extraídos. El tejido de poliéster usado típicamente para fabricar aros de cosido de válvulas protésicas cardiacas, así como los aros de anulopastia y cubiertas de stent, es trombogénico e inflamatorio por naturaleza. Es sabido que activa las cascadas de complemento y coagulación, y ha demostrado una mayor propensión a la deposición de plaquetas y la formación de trombos antes de la curación e incorporación de tejido. Se ha postulado que la tromborreactividad del material del aro de cosido es la principal culpable de las altas incidencias de fenómenos relacionados con trombos observados en el primer período postoperatorio (T. Orszulak y otros, 8th Annu. Meeting Eur. Assoc. Cardiacthoracic Surg. 1994:98; P. Perier y otros, en E Bodner y otros, Eds, Biologic and Bioprosthetic Valves, York Medical Books, 1986: 511-520). Se han realizado numerosos intentos de tratar el poliéster al objeto de mejorar su función in vivo. La mayor parte se han centrado en la reducción de la tromborreactividad del poliéster con la esperanza de que se acelere la respuesta curativa, evitando por ello el contacto adicional con elementos sanguíneos. Los ejemplos incluyen: impregnación con albúmina, recubrimiento de carbono, precoagulación del tejido de poliéster, siembra de células, unión de péptidos sintéticos, unión de factor de crecimiento de fibroblastos básicos (bFGF), recubrimiento con fibrina conteniendo bFGF y heparina, y recubrimiento de poliuretano. 25 Idealmente, los implantes médicos deberán curar bien, sin excesivo crecimiento de tejido, y permitir el establecimiento de una suave transición neoíntima entre el huésped y la prótesis. La reducción de la respuesta inflamatoria a materiales porosos tal como tejidos de poliéster sería un paso importante hacia la completa curación del implante quirúrgico sin las consecuencias a largo plazo de estenosis, regurgitación o fallo catastrófico. 30 Muchas de las listas siguientes de patentes y documentos no patentes describen información relacionada con dispositivos médicos conteniendo agentes antiinflamatorios, en particular esteroides. Otros en las listas siguientes se refieren a respuestas inflamatorias a tejidos de poliéster tal como DacronTM ; otros se refieren en general a biomateriales y mecanismos de respuesta humana. 35 (Tabla pasa a página siguiente) 40 45 50 55 60 65 3 ES 2 239 948 T3 TABLA 1a Patentes 5 10 15 20 25 30 35 40 45 50 55 60 65 4 ES 2 239 948 T3 TABLA 1b Documentos no patentes 5 Ackerman y otros, “Purification of Human Monocytes in Microexudate-Coated Surfaces”, J. Immunol., 120, 13721374.(1978). Alderson y otros, “A Simple Method of Lymphocyte Purification of Human Peripheral Blood”, J. Immunol. Methods, 11, 297-301 (1976). 10 Anderson, “Mechanisms of Inflammation and Infection with Implanted Devices”, Cardiovasc. Pathol., 2, 335-415 (1993). Anderson, “Inflammatory Response in Implants”, TASAIO, XXXIV, 101 (1988). 15 20 Boyum y otros, “Density-Dependent Separation of White Blood Cells”, Blood Separation and Plasma Fractionation, 217-239, (eds) James Harris, Wiley-Liss, Inc. (1991). Bonfield y otros, “Cytokine and Growth Factor Production by Monocytes/Macrophages on Protein Preadsorbed Polymers”, J. Biom. Mat. Res., 26, 837-850 (1992). Brais y otros, “Acceleration of Tissue Ingrowth on Materials Implanted in the Heart”, The Annals of Thoracic Surgery, 21, 221-229 (1976). 25 Cardona y otros, “TNF and IL-1 Generation by Human Monocytes in Response to Biomaterials”, J. Biom. Mat. Res., 26, 851-859 (1992). Casas-Bejar y otros, “In vitro Macrophage-Mediated Oxidation and Stress Cracking in a Polyetherurethane”, Transactions of the Fifth World Biomaterials Congress, Toronto, Canadá, pg. 609 (1996). 30 35 Cenni y otros, “Platelet and coagulation factor variations induced in vitro by polyethylene terephthalate (Dacron®) coated with pyrolytic carbon”, Biomaterials, 16, 973-976 (1995). French y otros, “Rifampicin Antibiotic Impregnation of the St. Jude Medical Mechanical Valve Sewing Ring: A Weapon Against Endocarditis”, The Journal of Thoracic and Cardiovascular Surgery, 112, 248-252 (1996). Fujimoto y otros, “Ozone-Induced Graft Polymerization onto Polymer Surface”, J. Polym. Chem. 31, 1035-1043 (1993). 40 Kadoba y otros, “Experimental comparison of albumin-sealed and gelatin-sealed knitted Dacron conduits”, The Journal of Thoracic and Cardiovascular Surgery, 103, 1059-1067 (1992). Kao y otros, “Role of Interteukin-4 in Foreign-Body Giant Cell Formation on a Poly(etherurethane urea) in vivo”, J. Biomed. Mat.. Res., 29, 1267-1275 (1995). 45 50 Karck y otros, “Pretreatment of prosthetic valve sewing-ring with the antibiotic/fibrin sealant compound as prophylactic tool against prosthetic valve endocarditis”, Eut. J. Cardio-thorac Surg., 4, 142-146 (1990). Leake y otros, “Comparative Study of the thromboresistance of Dacron® combined with various polyurethanes”, Biomaterials, 10, 441-444 (1989). Marchant y otros, “In vivo Biocompatibility Studies. I. The Cage Implant System and a Biodegradable Hydrogel”, J. Biomed. Mat.. Res., 17, 301-325 (1983). 55 Miller y otros, “Generation of L1-like activity in response to biomedical polymer implantss: A comparation of in vitro and in vivo methods”, J. of Biomed. Mat.. Res., 23, 1007-1026 (1989). Miller y otros, “Human Monocyte/Macrophage Activation and Interleukin-1 Generation by Biomedical Polymers”, J. Blom. Mat.. Res., 22, 713-731 (1988). 60 Mond y otros, “The Steroid-Eluting Electrode: A 10-Year Experience”, PACE, 12, 1016-1020 (1996). Onuki y otros, “Accelerated Endothelialization Model for the Study of Dacron Graft Healing”, Annals of Vascular Surgery, 11, 141-148 (1997). 65 Orszulak y otros, “The risk of stroke in the early postoperative period following mitral valve replacement”, European Journal of Cardiothorasic Surgery, 9, 615-620 (1995). 5 ES 2 239 948 T3 Perier y otros, “Comparison of thromboembolic and anticoagulant-related complications after aortic valve replacement using Starr Edwards, BjorkShiley and porcine valve prostheses”, Bodnar E. Yacoub M (eds.) Biologic and Bioprosthetic Valves: York Medical Books, 511-520 (1986). 5 Rubin y otros, “Preincubation of Dacron grafts with recombinant tissue factor pathway inhibidor decreases their trombogenicity in vivo”, J. Vasc. Surg., 24, 865-870 (1996). Schubert y otros, “Oxidative Biodegradation Mechanisms of Biaxially Strained Poly(etherurethane urea) Elastomers”, J. Biomed. Mat.. Res., 29, 337-347 (1995). 10 Schwartz y otros, “Local anticoagulation of prosthetic heart valves”, Circulation, 43 (Supl. III), 85-89 (1973). Shanbhag y otros, “Macrophage/Particle Interactions: Effects of Size, Composition and Surface Area”, J. Biomed. Mater. Res. 28, 81-90 (1994). 15 Shankar y otros, “Chapter 5: Inflammation and Biomaterials”, Implantation Biology, Host Response and Biomedical Devices, (eds) RalpH S. Greco, CRC Press, Inc., Boca Raton, FL 67-80 (1994). Stokes y otros, “Polyurethane Elastomer Bioestability”, J. of Biomaterials Applications, 9, 321-354 (1995). 20 Takahashi, “Adsorption of Basic Fibroblast Growth Factor on Dacron Vascular Prosthesis and its Biological Efficacy”, Artif Organs, 21, 1041-1046 (1997). 25 Tweden y otros, “Accelerated Healing of Cardiovascular Textiles Promoted by a RGD Peptide”, J. Heart Valve Dis. 4 (Supl. I), S90-97 (1995). Van Der Lei y otros, “Improved healing of small-caliber polytetrafluoroethylene prostheses by induction of a clot layer: a review of experimental studies in rats”, International Angiology, 10, 202-208 (1991). 30 Wilkerson y otros, “Biomaterials Used in Peripheral Vascular Surgery” (eds) Ralph S. Greco, CRC Press Inc., Implantation Biology: The Host Responses and Biomedical Devices, 179-190 (1994). Zhao y otros, “Cellular Interactions and Biomaterials: In vivo Cracking of Pre-stressed Pellethane 2363-80A”, J. Biom. Mat.. Res., 24, 621-637 (1990). 35 Zhao y otros, “Glass Wool-H2 O2 /CoCl2 Test System for In Vitro Evaluation of Biodegradative Stress Cracking in Polyurethane Elastomers”, J. Biomed.Mat. Res., 29, 467-475 (1995). 40 Un dispositivo médico implantable que tiene una capa de agente antiinflamatorio dispuesta sobre la superficie y una capa de poliéster colocada encima de dicha primera capa se describe en WO-A-98 36 784. 45 La presente invención se dirige a mejorar la biocompatibilidad y/o bioestabilidad de bombas de infusión implantables. Para hacerlo, la presente invención no implica modificar las químicas de los materiales constituyentes de los dispositivos, sino que más bien implica usar agentes antiinflamatorios como moduladores de respuesta biológica para controlar la respuesta del huésped a los materiales constituyentes. 50 55 60 El término “bioestable” se usa en la presente memoria con referencia a la estabilidad química y física de un material durante el implante en tejido vivo. Más específicamente, se refiere a la resistencia a los fenómenos degradativos a los que está expuesto el material durante la respuesta aguda y crónica del huésped (por ejemplo, inflamación). En el contexto de la presente invención, mejorar la bioestabilidad de un material no implica cambiar la química del material; sino que más bien se centra en disminuir la respuesta celular al material. Así, en el sentido en que se usa aquí, bioestabilidad se refiere a los efectos de células y tejidos en materiales. El término “biocompatible” se usa en la presente memoria con referencia al grado de respuesta del huésped provocada por un material después del implante. Típicamente, ésta se evalúa evaluando el fenómeno inflamatorio, en particular en tejidos circundantes. Menos inflamación o perturbación biológica sugiere mejor biocompatibilidad y viceversa. Así, en el sentido en que se usa aquí, la biocompatibilidad se refiere a los efectos de materiales en células y tejidos. Varias realizaciones de la presente invención pretenden cumplir uno o varios de los objetos siguientes: mejorar la biocompatibilidad del material; mejorar la bioestabilidad del material; reducir la inflamación aguda; reducir la inflamación crónica; y reducir la formación de tejido fibroso (por ejemplo, reducida encapsulación o pannus del tejido). Específicamente, la presente invención pretende minimizar la respuesta inflamatoria del huésped a una bomba de infusión implantada. 65 La presente invención tiene varias ventajas sobre los dispositivos implantables de la técnica anterior incluyendo curación controlada, mínimo crecimiento del pannus, función sostenida de las lengüetas, menor riesgo de infección, menor riesgo de formación de trombo y embolización, y mayor durabilidad de la válvula. La reducción de la respuesta 6 ES 2 239 948 T3 5 10 inflamatoria aguda a dispositivos implantables tiene un efecto beneficioso en la respuesta inflamatoria crónica. Si la respuesta inflamatoria postquirúrgica es capaz de resolverse en la fase aguda, se puede producir verdadera curación del implante quirúrgico sin las consecuencias a largo plazo de estenosis, regurgitación o fallo catastrófico. El control de la respuesta de curación evita o reduce los efectos nocivos de formación de pannus en la función y durabilidad de la válvula. Los dispositivos médicos implantables de la invención incluyen bombas de medicamento rellenables para administración prolongada de medicamento a largo plazo, tal como el sistema de infusión implantable SynchroMed® (Medtronic, Inc, Minneapolis, MN). Un dispositivo implantable de la invención tiene una superficie de contacto con tejido corporal o fluido (una “sobrecapa”) incluyendo un tejido que puede tomar la forma de una bolsa de tal manera que el tejido esté en contacto con tejido corporal o fluidos tal como sangre. Un tejido es por definición poroso, y puede estar tejido o entretejido. Un tejido posee una superficie áspera o texturada, lo que es ventajoso porque una superficie texturada facilita la respuesta curativa. Se prefiere en particular un tejido de poliéster bioestable para uso en la presente invención. 15 20 25 30 35 40 45 El dispositivo implantable incluye además, bajo la sobrecapa de tejido, una porción de cuerpo que sirve como un soporte físico. La porción de cuerpo imparte resistencia mecánica al dispositivo y proporciona una estructura o armazón que da forma y configuración al dispositivo. La porción de cuerpo no se limita a un material particular constituyente, y puede ser porosa o no porosa, biodegradable o bioestable, flexible o inflexible, como indique el uso previsto. Puede incluir, por ejemplo, uno o varios de un polímero, un metal, una aleación de metal, un tejido corporal (por ejemplo, pericardio, fascia lata, duramadre, mucosa intestinal), una esponja de colágeno o gel, o mezclas o geles de péptidos sintéticos. La porción de cuerpo del dispositivo puede tomar la forma de una capa inferior, estructura, o inserto, por ejemplo. Se deberá entender que la sobrecapa de tejido y la porción de cuerpo del dispositivo pueden, aunque no tienen que, estar en contacto físico directo entre sí; en otros términos, puede haber, pero no tiene que haber, un espacio entre toda o una porción de la sobrecapa de tejido y toda o una porción de la porción de cuerpo del dispositivo. El contacto físico entre la sobrecapa y la porción de cuerpo, si se produce, puede ser incidental, o puede ser intencional, como donde la sobrecapa de tejido se adhiere a la porción de cuerpo. Además, aunque la sobrecapa de tejido rodea típicamente completamente, encierra, o abarca la porción de cuerpo del dispositivo implantable, se ha de entender que la invención incluye no obstante dispositivos implantables donde solamente una porción de la porción de cuerpo del dispositivo es recubierta por la sobrecapa de tejido. Por ejemplo, se podría adherir una lámina de tejido a una superficie seleccionada de la porción de cuerpo del dispositivo. El material constituyente de la porción de cuerpo del dispositivo implantable está en contacto íntimo con un agente terapéutico, tal como un agente antiinflamatorio esteroidal o no esteroidal, que controla, reduce o atenúa la respuesta inflamatoria del cuerpo al dispositivo médico implantado, y que es capaz de eluirse a través de la sobrecapa de tejido. El agente antiinflamatorio es preferiblemente, un glucocorticosteroide, tal como dexametasona, su derivado o su sal. El agente terapéutico se puede recubrir sobre, o impregnar o embeber en, o unir covalentemente a, la porción de cuerpo del dispositivo, por ejemplo. Alternativamente, el agente terapéutico se puede colocar en un núcleo líquido de la porción de cuerpo, de tal manera que se pueda difundir a través de una porción de cuerpo de metal poroso o polímero o administrarse mediante canales formados en una porción de cuerpo por lo demás no porosa. Preferiblemente, la porción de cuerpo del dispositivo contiene un polímero bioestable en contacto íntimo con un agente antiinflamatorio. Además o alternativamente, la porción de cuerpo del dispositivo puede incluir uno u otros varios agentes terapéuticos, incluyendo agentes antiinfección tales como medicamentos antimicrobianos y agentes bacteriostáticos tales como plata. Por ejemplo, la endocarditis de válvula protésica, que sigue siendo una complicación peligrosa posterior a la sustitución cardiaca, se puede tratar prospectivamente incorporando un antibiótico al inserto del aro de cosido de la válvula cardiaca protésica. La presente invención proporciona una bomba de infusión implantable incluyendo: 50 una bomba incluyendo un espacio interior para contención de un líquido; un catéter de administración para administración del líquido a un paciente; y una bolsa de poliéster rodeando la bomba; 55 incluyendo además dicha bomba un material constituyente en contacto íntimo con un agente antiinflamatorio, siendo dicho agente antiinflamatorio capaz de ser liberado de la bomba y de eluirse a través de la bolsa de poliéster. 60 65 Por consiguiente, uno o varios objetos de la presente invención se logran con un dispositivo médico implantable que tiene una o varias de las características siguientes: (a) una sobrecapa de tejido mediante la que se puede eluir un agente terapéutico; (b) una sobrecapa de tejido que sirve de unos medios para fijar el dispositivo al cuerpo del paciente, y funciona como una superficie que promueve el crecimiento de tejido; (c) una porción de cuerpo en contacto íntimo con un agente terapéutico liberable; (d) uno o varios agentes terapéuticos recubiertos sobre o unidos a la porción de cuerpo del dispositivo debajo de la sobrecapa de tejido, de tal manera que el (los) agente(s) terapéutico(s) sean capaces de eluirse mediante la sobrecapa de tejido; (e) una porción de cuerpo conteniendo un depósito de medicamento encapsulado conteniendo uno o varios agentes terapéuticos capaces de eluirse de la porción de cuerpo mediante la sobrecapa de tejido; y (f) un agente antiinflamatorio, tal como dexametasona, capaz de eluirse mediante la sobrecapa de tejido para mediar una respuesta de curación sobre-exuberante al dispositivo implantado. 7 ES 2 239 948 T3 5 10 Un aspecto importante de la presente invención es que la respuesta inflamatoria del cuerpo a la sobrecapa de tejido o el encapsulamiento del dispositivo implantable se controla no modificando la sobrecapa de tejido propiamente dicho, sino modificando la porción de cuerpo del dispositivo que está rodeada, cubierta o encapsulada por la sobrecapa de tejido. Así, no se ponen en peligro propiedades deseables de la sobrecapa de tejido (por ejemplo, su porosidad, flexibilidad, textura, suturabilidad, resistencia a la tracción, durabilidad, esterizabilidad y biocompatibilidad). Es significativo que se puede utilizar dispositivos médicos implantables de la invención para modular la encapsulación de tejido, la formación de pannus, y la degradación de polímeros cuando se implantan en un paciente. Así, la presente invención se puede usar para modular la encapsulación de tejido, la formación de pannus, o la degradación de un cable eléctrico médico o catéter residente implantando las válvulas protésicas cardiacas (tanto válvulas mecánicas como válvulas de tejido), aros de anulopastia para reparación de válvulas, injertos vasculares, aros de cosido, stents, hilos médicos y catéteres, y marcapasos descritos anteriormente. Un método de hacer un dispositivo médico implantable incluye: 15 incorporar un agente terapéutico en un inserto anular incluyendo un material constituyente, de tal manera que el agente terapéutico esté en contacto íntimo con el material constituyente; encerrar el inserto anular en una envuelta de tejido. 20 Ahora se describirán realizaciones preferidas, a modo de ejemplo solamente, con referencia a los dibujos anexos. 25 30 La figura 1 es una vista esquemática simplificada de dispositivos médicos implantados significativos: (a) un marcapasos en una bolsa de poliéster y (b) válvulas cardiacas mecánicas y bioprotésicas con aros de cosido de tejido de poliéster; SVC, vena cava superior; RA, aurícula derecha; TV, válvula tricúspide; IVC, vena cava inferior; RV, ventrículo derecho; LV, ventrículo izquierdo; PV, vena pulmonar; LA, aurícula izquierda, PA, arteria pulmonar; Ao, aorta. La figura 2 muestra una válvula cardiaca bioprotésica: (a) vista en planta, (b) sección a lo largo de la línea 4-4 de (a); y (c) vista superior de la válvula. La figura 3 es muestra una válvula cardiaca mecánica: (a) válvula; (b) válvula sin aro de cosido; y (c) vista superior de la válvula. 35 La figura 4 es una vista en planta de un aro de anulopastia. La figura 5 es una vista en sección a lo largo de la línea 2-2 de la figura 4. 40 45 50 La figura 6 es un gráfico que muestra la formación de hidroperóxido in vitro en: medio de cultivo estándar (no células) conteniendo especímenes de polieteruretano (preimpregnados en acetona “AS”); especímenes de polieteruretano (AS) almacenados en oscuridad en condiciones ambientales; y medio de cultivo estándar con Mo/M∅s de conejo conteniendo especímenes de polieteruretano (AS). La figura 7 es un gráfico que muestra la formación de hidroperóxido in vitro en: medio de cultivo estándar con Mo/M∅s humano conteniendo especímenes de polieteruretano (con y sin impregnación en acetona); medio de cultivo estándar con linfocitos humanos conteniendo especímenes de polieteruretano (AS); y medio de cultivo estándar con Mo/M∅s humano conteniendo especímenes de polieteruretano (AS) más dexametasona fosfato sódico a 0,024 µg/ml (+) y 240 µg/ml (+++). La figura 8 es un diagrama de barras de la concentración de hidroperóxido en especímenes de polímero con y sin dexametasona después de un paso de tratamiento de macrofago de 40 días. La figura 9 muestra un gráfico de la cantidad de elución de dexametasona por área superficial de material (cm2 ) durante un período de 32 días. 55 La figura 10 es un diagrama de barras mostrando gráficamente la fisuración general por esfuerzo ambiental en explantes a las 6 semanas y 10 semanas. Los datos se resumieron usando la puntuación más alta (más severa) de daño superficial observado en las muestras de bioestabilidad explantadas. Observación con microscopio óptico a 70X de ampliación total. 60 La figura 11 es una representación gráfica del recuento celular comparativo total en exudado de jaula en respuesta a diferentes materiales PU: 1D = 1% dexametasona en poliuretano; 20D = 20% dexametasona en poliuretano; A = control; y EC = jaula vacía. 65 La figura 12 es una representación gráfica de la elución in vitro de dexametasona de cables recubiertos con dexametasona. Se utilizaron cargas “baja” (1% DEX/PU) y “alta” (5% DEX/PU). Los porcentajes de elución de la carga de dexametasona teórica total se determinaron en PBS a 37ºC. 8 ES 2 239 948 T3 La figura 13 es una representación gráfica de la elución in vitro de dexametasona de cables recubiertos con dexametasona por área superficial en función del tiempo. Se utilizaron cargas “bajas” (1% DEX/PU) y “altas” (5% DEX/PU). La elución se realizó en PBS a 37ºC. 5 10 La figura 14 es representación gráfica de la elución in vitro de dexametasona de cables recubiertos con dexametasona después 90 días de implante in vivo. Se utilizaron cargas “bajas” (1% DEX/PU) y “altas” (5% DEX/PU). Los porcentajes de elución de la carga de dexametasona teórica total se determinaron en PBS a 37ºC. La figura 15 muestra perfiles de elución in vitro para insertos de silicona cargados con DEX que tienen diferentes porcentajes de peso de DEX. La figura 16 muestra (a) una comparación de los perfiles de elución de insertos cargados con DEX e insertos de DEX/DMP; y (b) una descomposición de la elución de DEX y DMP de insertos de DEX/DMP. 15 La figura 17 muestra los efectos supresivos de dexametasona eluida de insertos cargados con DEX en la producción de las citoquinas inflamatorias (a) factor de necrosis tumoral α. (TNF- α) y (b) interleuquina 1α (IL-1α). La figura 18 muestra el efecto de una encapsulación de poliéster en elución de dexametasona de una muestra de silicona (a) 1% DEX; (b) 2,5% DEX; (c) 1% DEX/DMP. 20 La figura 19 muestra la densidad de glóbulos blancos en función deltiempo para ratas tratadas con muestras de Si/DEX y ratas de control. 25 La figura 20 muestra secciones histológicas teñidas con hemotoxilina y eosina de muestras de silicona explantadas después de 21 días; (a) muestra de control (Si sin DEX) a una ampliación original de 100X (1 = superficie interior del tejido de Dacron&auml;, el lado que estaba en contacto con el inserto de silicona; 2 = superficie exterior del DacronTM ; * = fibras individuas de DacronTM ); (b) imagen 500X del control teñido; las cabezas de flecha apuntan a la presencia de macrófagos solos y multinucleados alrededor de las fibras; (c) imagen 100X de una muestra conteniendo DEX; y (d) imagen 200X de una muestra conteniendo DEX. 30 35 La presente invención se dirige a mejorar la biocompatibilidad y/o bioestabilidad de materiales de poliéster modulando el comportamiento celular implicado en mecanismos biológicos de defensa, tal como fagocitosis y mecanismos enzimáticos y oxidativos, y la regulación por incremento de señales de citoquina que amplifica la respuesta inflamatoria. Esto no implica modificar la química de los polímeros, sino que implica usar moduladores de respuesta biológica para “proteger” los polímeros. Se ha descubierto de forma significativa que la elución de tales moduladores de respuesta biológica en la interface entre el polímero y el tejido circundante (tejidos sólidos o líquidos, por ejemplo, sangre), modula el comportamiento de células en dicha interface. Como resultado, el polímero está expuesto a menos agentes dañinos producidos por células, tal como especies oxígeno reactivas. En esencia, los mecanismos de defensa de células en respuesta a materiales extraños son regulados hacia abajo por la presente invención. 40 45 50 55 60 65 Para ello, una realización de la presente invención proporciona un dispositivo médico implantable que tiene una porción de cuerpo incluyendo un material constituyente en contacto íntimo con un agente terapéutico, tal como un agente antiinflamatorio, estando solapada la porción de cuerpo por un tejido poroso. El agente antiinflamatorio se eluye de la porción de cuerpo del dispositivo, a través del tejido, y es eficaz al modular el comportamiento de células en contacto con el dispositivo implantado. Es significativo que el agente antiinflamatorio modere algunas actividades celulares en el lugar del implante que produce inflamación, por ejemplo. Tal actividad celular incluye regulación por incremento de recorridos de señalización específicos que amplifican la respuesta inflamatoria, dando lugar a crecimiento exuberante de tejido, aumento de la actividad de la metaloproteasa, y ráfaga oxidativa. Crecimiento exuberante de tejido se refiere a la formación de tejido fibroso como resultado de la proliferación celular y deposición de componentes extracelulares, incluyendo colágeno, elastina, y fibronectina. El resultado es la encapsulación del implante y/o la formación de pannus, que puede ser nocivo porque el crecimiento exuberante de tejido puede disminuir el área de orificio de la válvula protésica e invadir las lengüetas de válvula, inhibiendo la libertad de movimiento que da lugar a estenosis de la válvula. Ráfaga oxidativa se refiere a la capacidad de los fagocitos de consumir oxígeno y producir especies oxígeno reactivas tales como radicales hidroxilo, superóxido, peróxido de hidrógeno, y otros óxidos y peróxidos reactivos. Tiende a producir degradación del polímero del que se hace el implante. Algunos polímeros son más resistentes a la degradación oxidativa que otros; incluyen fluoroelastómeros tales como poli(tetrafluoroetileno) (PTFE), poli(tetrafluoroetileno-hexafluoropropileno) (FEP), poli(tetrafluoroetileno-perfluoro-(poliviniléter)) (PFA), poli (etil-tetrafluoretileno) (ETFE); y otros polímeros tales como cloruro de polivinilo (PVC), poliimidas, polisulfonas, poliolefinas tales como polipropileno, polietileno y copolímeros de etileno-propileno, y siliconas. La hidrólisis de materiales poliméricos también es una preocupación. El agente antiinflamatorio está localizado preferiblemente en la superficie de la porción de cuerpo del dispositivo, desde la que pasa después a través de la sobrecapa de tejido. Alternativamente, se puede eluir de un lugar remoto dentro del dispositivo médico, a condición de que, después de la elución, pase a través de la sobrecapa de tejido. Se considera que la liberación inicial del agente antiinflamatorio en el lugar de implante reduce la propagación asociada con células de la señal inflamatoria. Se cree que la liberación sostenida mantiene un nivel bajo de activación y diferenciación de células que entran en contacto con la superficie en contacto con tejido. 9 ES 2 239 948 T3 5 10 15 20 25 La porción de cuerpo del dispositivo médico de la invención incluye preferiblemente aproximadamente 0,01 por ciento en peso (% en peso) a aproximadamente 10,0% en peso de agente terapéutico, más preferiblemente de aproximadamente 0,1% en peso a aproximadamente 5% en peso de agente terapéutico. Naturalmente, la cantidad de agente terapéutico cargado en el dispositivo depende de la eficacia del agente, la finalidad para la que está siendo administrado, la edad y el estado del paciente, y la duración prevista del tratamiento. Cae dentro de los conocimientos de los expertos en la técnica relevante determinar las dosis terapéuticas efectivas. Opcionalmente, la porción de cuerpo del dispositivo médico incluye un agente inerte que facilita la detección no invasiva del dispositivo implantado. Por ejemplo, se puede usar sulfato de bario y óxido de platino para permitir la detección del dispositivo por rayos X o fluoroscopia. Alternativamente, la superficie de la porción de cuerpo del dispositivo se puede hacer ecogénica (véase, por ejemplo, Bosley, Patente de Estados Unidos número 5.289.831; Bosley y otros, Patente de Estados Unidos 5.201.314; Bosley y otros, Patente de Estados Unidos número 5.081.997; y Rammler, Patente de Estados Unidos Número 5.327.891) para mejorar la formación de imágenes por ultrasonido. Por ejemplo, se puede incorporar un material ecogénico, tal como óxido de zinc, óxido de hierro, o óxido de titanio, a la porción de cuerpo del dispositivo para hacerla visible por ultrasonido. Se debe incluir una cantidad suficiente de un polímero bioestable para impartir a la porción de cuerpo del dispositivo las características deseadas, tal como resistencia a la tracción y flexibilidad durométrica. Por ejemplo, donde se utiliza silicona, la porción de cuerpo del dispositivo incluye preferiblemente de aproximadamente 43% en peso a aproximadamente 57% en peso de silicona. En una realización especialmente preferida, la porción de cuerpo del dispositivo médico incluye de aproximadamente 50% en peso a aproximadamente 55% en peso de silicona de calidad médica, de aproximadamente 50% en peso a aproximadamente 45% en peso de sulfato de bario, y de aproximadamente 0,1% en peso a aproximadamente 1,5% en peso de agente terapéutico, preferiblemente dexametasona. La invención se describe aquí con referencia especial a bombas de infusión implantables. No obstante, se ha de entender que los principios de la invención son aplicables en general a cualquier dispositivo médico implantable, incluyendo además, por ejemplo, stents, hilos médicos, catéteres, y marcapasos, que pueden incluir una sobrecapa de tejido de cualquier tipo, incluyendo por ejemplo una vaina, una envuelta, una capa, o un recubrimiento, de tal manera que la sobrecapa de tejido esté en contacto con tejido corporal o fluidos tales como sangre. 30 35 40 45 50 55 60 65 Por ejemplo, la figura 1(a) es una vista esquemática simplificada de un dispositivo médico implantado 200. El cable de estimulación cardiaca y detección 218 está unido a un recinto herméticamente cerrado 214 e implantado cerca del corazón humano 316. El dispositivo médico implantable 200 está encerrado en una bolsa de poliéster 201; y el cuerpo (212, 214) del dispositivo médico implantable 200 contiene un fármaco antiinflamatorio eluible tal como dexametasona. En caso de que el dispositivo médico implantado 200 sea un marcapasos incluye al menos uno o ambos cables de estimulación cardiaca y detección 216 y 218. Los cables de estimulación cardiaca y detección 216 y 218 detectan señales eléctricas concomitantes a la despolarización y repolarización del corazón 316, y proporcionan pulsos de estimulación cardiaca para producir la despolarización del tejido cardíaco cerca de sus extremos distales. El dispositivo médico implantable 200 puede ser un marcapasos cardíaco implantable tal como los descritos en la Patente de Estados Unidos número 5.158.078 de Bennett y otros, la Patente de Estados Unidos número 5.312.453 de Shelton y otros, o la Patente de Estados Unidos número 5.144.949 de Olson. El dispositivo médico implantable 200 también puede ser un PCD (Marcapasos-Cardioversor-Desfibrilador) correspondiente a cualquiera de los varios PCDs implantables comercializados, con la sustitución del módulo conector de cables de estimulación cardiaca y detección 212 por el conjunto de bloque conector de otro modo presente. Los principios de presente invención se pueden llevar a la práctica en unión con PCDs tal como los descritos en la Patente de Estados Unidos número 5.545.186 de Olson y otros, la Patente de Estados Unidos número 5.354.316 de Keimel, la Patente de Estados Unidos número 5.314.430 de Bardy, la Patente de Estados Unidos número 5.131.388 de Pless o la Patente de Estados Unidos número 4.821.723 de Baker y otros. Los dispositivos se pueden emplear directamente en unión con la presente invención, y muy preferiblemente se llevan a la práctica de tal manera que los alimentadores que interconectan la circuitería a sus bloques conectores estén situados para permitir el acceso fácil entre los alimentadores y los conectores eléctricos dispuestos dentro de los agujeros de módulo conector o de cabecera 212. Alternativamente, el dispositivo médico implantable 200 puede ser un estimulador nervioso o muscular implantable tal como el descrito en la Patente de Estados Unidos número 5.199.428 de Obel y otros, la Patente de Estados Unidos número 5.207.218 de Carpentier y otros o la patente de Estados Unidos número 5.330.507 de Schwartz, o un dispositivo supervisor implantable tal como el descrito en la Patente de Estados Unidos número 5.331.966 concedida a Bennet y otros. Se cree que los principios de la presente invención encuentran amplia aplicación a cualquier forma de dispositivo eléctrico implantable para uso en unión con cables eléctricos, y se considera que es especialmente ventajoso en los contextos donde se emplean y desean múltiples cables eléctricos médicos. En general, el recinto herméticamente cerrado 214 incluye una pila electroquímica tal como una batería de litio, circuitería que controla las operaciones del dispositivo y registra episodios EGM arrítmicos, y una antena de transceptor de telemetría y circuito que recibe órdenes de telemetría de enlace descendente y transmite datos almacenados en un enlace ascendente de telemetría al programador externo. La circuitería y memoria se pueden implementar en lógica discreta o un sistema basado en microordenador con conversión A/D de los valores de amplitud EGM muestreados. No se considera que las características electrónicas concretas y las operaciones del dispositivo médico implantable sean de significado absoluto con respecto a la puesta en práctica de la presente invención. Un sistema operativo ejem10 ES 2 239 948 T3 plar se describe en la Solicitud de Patente de Estados Unidos, en tramitación, del mismo cesionario, número de serie 08/678.219, presentada el 11 de julio de 1996, para “Dispositivo implantable mínimamente invasivo para supervisar eventos fisiológicos”. 5 10 15 La figura 1(b) es una vista esquemática simplificada de válvulas cardiacas mecánicas y bioprotésicas implantadas. El aparato de válvula bioprotésica 30 se representa aquí sustituyendo a la válvula aórtica, y la válvula mecánica 40 se representa aquí sustituyendo a la válvula mitral. La figura 2 es una ilustración más detallada de una válvula cardiaca bioprotésica. La válvula bioprotésica 30 contiene tres lengüetas de tejido 26, que funcionan conjuntamente como un oclusor de flujo. La carcasa de válvula 32 se compone de una estructura polimérica cubierta de tejido que tiene tres largueros 28 a los que están unidas las lengüetas 26. Un aro de cosido 20 está unido circunferencialmente, mediante sutura, a la base de la carcasa de válvula 32. El aro de cosido 20 se fabrica de una envuelta o malla en forma de tela 24 que forma un lumen 21 conteniendo un bastidor de soporte polimérico anular 22, denominado aquí un inserto polimérico. La envuelta 24 es típicamente de tela de poliéster, tal como DacronTM , y se hace doblando una lámina de tela alrededor del inserto polimérico 22 y cosiendo los extremos plegados. La combinación del inserto polimérico 22 y la envuelta 24 da lugar a un aro de cosido 20 que es completamente flexible pero esencialmente inextensible. El inserto polimérico 22 se hace típicamente de caucho de silicona flexible radioopaco, que permite comprobar la presencia del dispositivo después de terminar la cirugía de implante. 20 La figura 3 es una ilustración más detallada de una válvula cardiaca mecánica. La válvula mecánica 40 incluye una carcasa de válvula anular metálica 42 conteniendo un larguero metálico central 44 a lo largo del que se mueve el disco oclusor de flujo 46. La envuelta en forma de tela 24 encierra el inserto 43 (representado como una línea en la figura 3 (c)) para formar el aro de cosido 48 que está unido a la carcasa de válvula 42. 25 30 35 40 45 50 Los insertos de aros de cosido para válvulas bioprotésicas o mecánicas se fabrican típicamente de uno o varios de un polímero, preferiblemente silicona; un metal, preferiblemente titanio o tántalo; una aleación de metal, preferiblemente aleaciones de titanio, aleaciones de cobalto cromo, aleaciones de níquel cromo, o aceros inoxidables; o sus combinaciones. Se utilizan para conformación y/o soporte estructural del dispositivo implantado, rodeado por o encerrado en un material tipo tela que se puede suturar. El inserto puede ser plegable o rígido, pueden variar sus dimensiones físicas dependiendo del dispositivo, y está encerrado comúnmente con tejido de poliéster. Las figuras 4 y 5 ilustran un aro convencional de anulopastia (un aro Duran) que realiza la presente invención. El aro 10 tiene un lumen 11 conteniendo un núcleo interior generalmente rectangular 12 de caucho de silicona radioopaco que es completamente flexible radialmente. El núcleo 12 está encerrado completamente por una envuelta 14 de tela de poliéster, tal como DacronTM . La envuelta 14 se hace doblando una hoja de tela alrededor del núcleo 12 y cosiendo los extremos plegados en 13. La combinación del núcleo 12 y la envuelta 14 da lugar a un aro que es completamente flexible pero esencialmente inextensible. Esta propiedad permite que el aro de anulopastia o cinta, cuando esté implantado en el corazón, evite que el anillo de válvula se distienda, sin impedir considerablemente el movimiento natural del anillo. El aro 10 tiene tres marcadores trigonales 15 cosidos a intervalos de 120º para ayudar al cirujano en la colocación de suturas. El dispositivo médico implantable de la invención es un dispositivo implantable de infusión de medicamento, tal como la bomba SynchroMedTM (Medtronic Inc, Minneapolis, MN), que está encerrado en una bolsa de poliéster. Dicha bomba se diseña para administración de los medicamentos, tal como baclofen o morfina, directamente al fluido de la médula espinal, inyectándolos al espacio intratecal. La bomba se implanta típicamente quirúrgicamente justo debajo de la piel del abdomen, e incluye un disco metálico redondo de aproximadamente 2,5 cm de grosor y aproximadamente 7,5 cm de diámetro. El medicamento se inyecta mediante un catéter de diámetro pequeño que se introduce en el fluido espinal que rodea la médula espinal. Según la presente invención, la bomba está encerrada en una bolsa de poliéster, y un agente terapéutico, preferiblemente un agente antiinflamatorio, se reviste o adhiere a la superficie del disco metálico, de tal manera que el agente terapéutico sea capaz de eluirse a través de la bolsa. Se espera que un dispositivo implantable de infusión de medicamento según la invención que tiene una porción de cuerpo incluyendo un agente antiinflamatorio eluible, sea mucho más fácil de explantar, cuando disminuya la respuesta inflamatoria crónica responsable de la formación de tejido fibroso. 55 Sobrecapa de tejido 60 65 La “sobrecapa” porosa del dispositivo médico implantable se fabrica de un tejido o tela tejida que promueve el crecimiento de tejido. El tejido es un tejido o tela tejida de fibras de poliéster. Se prefiere un material en forma de tela para dispositivos que se implantan usando técnicas de sutura. Ejemplos de fibras poliméricas que se pueden entretejer o tejer a un tejido poroso incluyen polímeros naturales tales como colágeno, seda, quitina, celulosa y polímeros sintéticos tales como poliésteres, poliamidas, poliuretanos, polipropilenos, tereftalatos de polietileno (PETs), poli(tetrafluoroetileno)s (PTFEs), polietilenos, poli(alcohol vinílico)s, poliacrilonitrilos, poli(ácidos glicólicos), poli(ácidos lácticos), polidimetilsiloxanos, aramidas, y celulosas regeneradas. Preferiblemente, el material polimérico poroso es un tejido o tela tejida de fibras PET. También se puede utilizar una tela hecha de fibras de PTFE expandido, que se hacen usando un proceso de calentamiento y estiramiento mecánico (D. Willkerson y otros, “Biomaterals Used in Peripheral Vascular Surgery”, R. Greco, Ed, Implantation Biology: The Host Responses and Biomedical Devices, 179-190, CRC Press 11 ES 2 239 948 T3 Inc, (1994)); un tejido hecho de fibras de PTFE expandido tiene mejores características de manipulación y exhibe menos deshilachado en las líneas de sutura que los tejidos de PTFE convencionales. 5 La sobrecapa de tejido poroso tiene forma de una bolsa. Preferiblemente, la sobrecapa de tejido tiene una resistencia a la tracción de fuerte a bastante alta, es flexible, posee una superficie áspera de inducción de neoíntima, es fácilmente penetrable a las suturas, y es resistente al rasgado a la penetración de agujas y suturas, y es biocompatible. Porción de cuerpo 10 15 20 25 30 35 40 45 En el sentido en que se usa aquí, el término “porción de cuerpo” de un dispositivo médico implantable se refiere a la porción del dispositivo médico implantable que se cubre, encierra o solapa por la sobrecapa de tejido. En otros términos, el dispositivo incluye opcionalmente otras estructuras o porciones que no están cubiertas por tejido. La porción de cuerpo de un dispositivo médico implantable de la invención se puede fabricar de cualquier material o materiales deseados constituyentes, sin limitación. Preferiblemente, el material constituyente de la porción de cuerpo del dispositivo es biocompatible. La elección del material constituyente dependerá de la estructura y función previstas del dispositivo. En realizaciones del dispositivo destinado a uso a largo plazo, tal como stents, válvulas cardiacas, aros de anulopastia, y marcapasos, se prefiere que la porción de cuerpo del dispositivo se forme a partir de un material bioestable, tal como un metal o polímero bioestable. Preferiblemente, el material que forma la porción de cuerpo del dispositivo no está destinado a crecimiento de tejido, en contraposición a encerrar el tejido. El dispositivo implantable de la invención está destinado típicamente a estar en contacto con tejidos o fluidos corporales durante períodos prolongados de tiempo (por ejemplo, días, meses, años). Los ejemplos de polímeros bioestables adecuados para ser utilizados como materiales para formar la porción de cuerpo del dispositivo incluyen poliuretanos, tales como poliéter uretano, siliconas; poliamidas, tal como nylon-66; poliimidas; policarbonatos; poliéteres; poliésteres, tal como tereftalato de polietileno; polivinil aromáticos, tal como poliestirenos; politetrafluoroetilenos y otras clases de fluoropolímeros tal como poli (etileno-cloro-trifluoroetileno), poli(etileno-tetrafluoroetileno), poli(cloro-trifluoroetileno), copolímeros de etileno-propileno fluorados, copolímeros de perfluoroalkoxi y fluoroelastómeros; poliolefinas, tal como polietilenos, polipropilenos, poliisoprenos, y copolímeros de etileno-alfa olefina; polímeros acrílicos y copolímeros; polímeros y copolímeros de haluro de vinilo, tal como cloruro de polivinilo; polivinil éteres, tal como polivinil metilo éter; polivinil ésteres, tal como acetato de polivinilo; polivinil cetonas; haluros de polivinilidino, tal como fluoruro de polivinilideno y cloruro de polivinilideno; poliacrilonitrilo; así como copolímeros de monómeros de vinilo entre sí y olefinas, tales como copolímeros de etileno-metacrilato de metilo, copolímeros de acrilonitrilo-estireno, resinas de acrilonitrilo butadieno estireno (ABS), polisulfonas, polieterimidas, polieteretercetonas, poliarilcetonas, resinas epoxi, polímeros cristalinos líquidos, sulfuros de polifenileno, óxido de polifenileno, poliamidaimidas, poliacetales, policetonas, poliarilatos, copolímeros de etileno-acetato de vinilo, y mezclas de los anteriores. Los poliuretanos y siliconas, o sus combinaciones, son sustratos poliméricos actualmente preferidos en el contexto de esta invención. Preferiblemente, la porción de cuerpo de un dispositivo implantable de la invención se fabrica de silicona, poliuretano, o su combinación. No obstante, deberá entenderse que cae dentro del alcance de la invención incluir dispositivos que tienen porciones de cuerpo fabricadas de materiales biodegradables o resorbibles en lugar o además de materiales bioestables, según requiera una aplicación especial, tal como los destinados a tratamiento o terapia a corto plazo. Los polímeros biodegradables adecuados para uso en la porción de cuerpo del dispositivo incluyen poli(ácidos lácticos), poli(ácidos glicólicos), poli(ácidos láctico-co-glicólicos), polianhídridos, poli(ortoésteres), poli(hidroxibutiratos), poli(hidroxivaleratos), polidioxanonas, polifosfazona, policaprolactonas, poliaminoácidos, y colágeno. Cuando se utiliza polímero biodegradable para contener el agente terapéutico, no es necesario que el agente terapéutico se eluya del polímero; el agente terapéutico puede, en cambio, liberarse del polímero como consecuencia de la biodegradación. Carga de medicamento de la porción de cuerpo 50 55 60 65 El material constituyente de la porción de cuerpo de un dispositivo médico implantable de la invención está en contacto íntimo con uno o varios agentes terapéuticos (también denominados aquí simplemente “medicamentos”), y la porción de cuerpo se fabrica de tal manera que el agente terapéutico se pueda eluir alejándose de su superficie y en último término a través de la sobrecapa de tejido poroso. El agente terapéutico se puede incorporar a la porción de cuerpo del dispositivo médico implantable de varias formas. Por ejemplo, el agente terapéutico puede ser injertado covalentemente en una superficie de la porción de cuerpo del dispositivo, solo o con un polímero de injerto superficial. Alternativamente, se puede recubrir sobre la superficie de la porción de cuerpo del dispositivo solo o mezclado con un polímero de recubrimiento. También se puede mezclar con un adhesivo, tal como un adhesivo de silicona, y aplicar a la superficie de la porción de cuerpo del dispositivo, después de lo que la sobrecapa de tejido se puede adherir, si se desea, a la porción de cuerpo del dispositivo. Se puede mezclar físicamente con un polímero de la porción de cuerpo del dispositivo como en una solución sólido-sólido. Se puede impregnar en el polímero hinchando el polímero en una solución del disolvente apropiado, o embeber en una porción de cuerpo metálica porosa. Un polímero conteniendo un agente terapéutico se puede extruir, moldear o recubrir en otro material (por ejemplo, metal), injertarse sobre otro material, embeberse dentro o mezclarse con otro material, adsorberse en otro material, etc, para formar la porción de cuerpo del dispositivo. Cae dentro del alcance de la presente invención cualquier medio por el que el agente terapéutico se pueda incorporar al dispositivo médico de tal manera que esté en contacto íntimo con un material constituyente de la porción de cuerpo del dispositivo. 12 ES 2 239 948 T3 En una realización, un polímero de la porción de cuerpo del dispositivo y un agente terapéutico se mezclan íntimamente mezclando o usando un solvente en el que ambos son solubles (por ejemplo, xileno para silicona y dexametasona fosfato). Esta mezcla se puede formar posteriormente en la forma deseada e incorporar al dispositivo médico o recubrirse sobre una estructura subyacente del dispositivo médico. 5 10 15 20 25 30 35 40 Alternativamente, se mezclan íntimamente un polímero de recubrimiento, que puede ser o no el mismo polímero que forma un polímero primario de una porción de cuerpo del dispositivo, y un agente terapéutico, mezclando o usando un solvente en el que ambos son solubles, y recubierto sobre la porción de cuerpo del dispositivo. Los polímeros de recubrimiento son preferiblemente cualquiera de los polímeros bioestables enumerados anteriormente, a condición de que sean capaces de unirse (química o físicamente) al polímero de la porción de cuerpo del dispositivo. Alternativamente, sin embargo, pueden ser cualquiera de una amplia variedad de polímeros bioabsorbibles, a condición de que sean capaces de unirse (química o físicamente) al polímero de la porción de cuerpo del dispositivo. Los ejemplos de polímeros bioabsorbibles adecuados incluyen poli(ácido L-láctico), policaprolactona, poli(lactida-co-glicólido), poli (hidroxibutirato), poli(hidroxibutirato-co-valerato), y otros, como se describe en la Patente de Estados Unidos número 5.679.400 (Tuch). Además, se puede usar un recubrimiento en la superficie de la porción de cuerpo del dispositivo, por ejemplo, para controlar las tasas de liberación de medicamento de la porción de cuerpo del dispositivo. Otra realización incluye hinchar el polímero de una porción de cuerpo del dispositivo con un disolvente apropiado y dejar que el agente antiinflamatorio impregne el polímero. Por ejemplo, para poliuretano se puede usar tetrahidrofurano, N-metil-2-pirrolidona, y/o cloroformo. En otra realización se injerta covalentemente un agente terapéutico sobre el polímero de una porción de cuerpo del dispositivo. Esto se puede hacer con o sin un polímero de injerto superficial. El injerto superficial se puede iniciar por descarga en corona, irradiación UV, y radiación ionizante. Alternativamente, se puede usar el método de ion cérico, descrito anteriormente en la Patente de Estados Unidos número 5.229.172 (Cahalan y otros) para iniciar el injerto superficial. Por consiguiente, un polímero conteniendo medicamento se puede extruir, moldear o recubrir sobre otro material (por ejemplo, metal), injertar sobre otro material, embeber dentro o mezclarse con otro material, adsorberse en otro material, etc, para formar la porción de cuerpo del dispositivo. Se deberá observar que aquí, cuando se utiliza un polímero de recubrimiento secundario, polímero de injerto superficial, o análogos, la porción de cuerpo del dispositivo se define de manera que incluya este polímero secundario así como el polímero primario que forma la estructura básica del dispositivo médico (por ejemplo, el inserto de aro de cosido o el injerto vascular). Igualmente, la porción de cuerpo del dispositivo implantable puede ser una estructura compuesta en capas. A efectos de la invención, es importante solamente que la porción de cuerpo del dispositivo, de la forma que se modele, contenga un agente terapéutico, y el agente terapéutico sea capaz de eluirse, difundirse o liberarse de otro modo de la porción de cuerpo del dispositivo, para después eluirse a través de la sobrecapa de tejido poroso del dispositivo. Sin embargo, a pesar de lo anterior, se carga preferiblemente a granel un agente terapéutico en la porción de cuerpo del dispositivo, tal como un inserto de silicona, al tiempo de la fabricación. La eficacia del medicamento cargado en la respuesta inflamatoria del huésped depende de la velocidad de elución del medicamento del inserto y a través del tejido poroso. El perfil de liberación de medicamento de un dispositivo médico implantable de la invención se extiende preferiblemente durante aproximadamente 30 días al menos. Agentes terapéuticos 45 50 55 60 65 Los agentes antiinflamatorios adecuados para uso en la presente invención incluyen compuestos tanto esteroidales como no esteroidales. Preferiblemente, los agentes antiinflamatorios esteroidales incluyen glucocorticoides, sales, y sus derivados. Los ejemplos de tales esteroides incluyen cortisol, cortisona, fludrocortisona, prednisona, prednisolona, 6α-metilprednisolona, triamcinolona, betametasona, dexametasona, beclometasona, aclometasona, amcinonida, clebetasol, clocortolona. Se prefieren en particular dexametasona (9α-fluoro-11(β,17α21-trihidroxi-16α-metilpregna1,4-dieno-3,20-diona), sus derivados, y sus sales. Dexametasona fosfato sódico y dexametasona acetato son sales adecuadas y dexametasona-21-ortofosfato y su sal disódica son derivados adecuados. Los medicamentos antiinflamatorios no esteroidales incluyen tiomalato de oro, tiosulfato de oro, auranofina, D-penicilamina, e inhibidores Cox-2 tales como rofecoxib (VioxxTM , Dupont Merck) y celecoxib (CelebrexTM , Pfizer). El agente antiinflamatorio se puede usar en cualquier cantidad que produzca la respuesta deseada sin efectos perjudiciales, tal como efectos citotóxicos o la supresión de la respuesta inmune. Típicamente, se utiliza en una cantidad o dosis apropiada durante la duración deseada e intensidad del efecto antiinflamatorio. En último término, esto viene dictado por el tipo de dispositivo al que se aplique esta invención. Sin embargo, se cree en general que se puede usar menos de aproximadamente 1 mg de un agente antiinflamatorio por centímetro cuadrado de área superficial de una superficie conteniendo polímero para producir los resultados ventajosos descritos en la presente memoria. Otros agentes terapéuticos incluyen agentes antibacterianos o antimicrobianos, agentes anticoagulantes, agentes antitrombóticos, agentes antiplaquetas, agentes antimitóticos, antisépticos, antioxidantes, agentes antimetabolito, agentes antiproliferativos, agentes anticalcificación, agentes antitrombogénicos, agentes quelantes, enzimas, catalizadores, hormonas, factores de crecimiento, lectinas, vitaminas, anticuerpos, antígenos, ácidos nucleicos tales como DNA y RNA, proteínas o péptidos, polisacáridos, colorantes, compuestos radiactivos, o cualquier combinación de los mismos. Un agente terapéutico preferido es heparina. Preferiblemente, la heparina se incluye en una cantidad eficaz para evitar o limitar la trombosis. Se puede incorporar heparina a la porción de cuerpo del dispositivo por recubrimiento, unión covalente, o cualquiera de varias técnicas conocidas para incorporar heparina a un dispositivo médico. En una realización, se une 13 ES 2 239 948 T3 covalentemente heparina a la porción de cuerpo de un dispositivo que también contiene un agente antiinflamatorio eluible. 5 10 15 20 Los agentes antimicrobianos incluyen aminoglicósidos, tales como gentamicina, canamicina, neomicina, paromomicina, estreptomicina y tobramicina, ansamicinas tales como rifamicina y rifampina, cefalosporinas tales como cefalexina, cefaloridina, cefalotina, cefazolina, cefapirina, cefradina, y cefaloglicina, macrólidos tales como erotromicina, tilosina, oleandomicina y espiramicina, penicilinas tales como penicilina G, penicilina V, feneticilina, meticilina, oxacilina, cloxacilina, dicloxacilina, floxacilina, nafcillina, ampicilina, amoxicilina, y carbenicilina, sulfonamidas, cloranfenicoles, tetraciclinas tales como tetraciclina, oxitetraciclina, clortetraciclina, metaciclina, demeclociclina, rolitetraciclina, doxiciclina, y minociclina, polipéptidos tales como bacitracina, polimixinas, tirotricina y vancomicina, y otros incluyendo trimetoprim-sulfamethoxazol, lincomicina, clindamicina y espectinomicina. Los agentes antimicrobianos especialmente preferidos incluyen rifampicina y gentamicina. Los agentes antisépticos incluyen plata, clorohexidina, irgasan, yodo y compuestos de amonio cuaternario tales como cloruro de benzalconio. Otros ejemplos de agentes terapéuticos incluyen medicamentos citostáticos tales como ametopterina, sulfato de vincristina, un sulfato de vinblastina; agentes inmunosupresores tales como ciclosporina A y azatioprina, moléculas antiadhesión celular (anti-CAMs), tal como lactosa-1-fosfato y anticuerpos antiintegrina, antioxidantes tales como ácido ascórbico y αtocoferol, y otros tales como pentoxifilina y citocalasina B. Las dosis y los protocolos de administración estándar se pueden hallar, por ejemplo, en el Merck Index, 12ª Ed. (Merck Research Laboratories, Merck & Co, Inc, Whitehouse Station, NJ, (1996)), Physician’s Desk Reference, 52ª Ed. (Medical Economics Company, Inc, Montrale, NJ, (1998)), o Manual of Medical Therapeutics/Department of Medicine, Washington University School of Medicine, 28ª Ed. (G. A. Ewald y otros, Eds, Little, Brown Press, Boston (1995)). Ejemplos 25 Los ejemplos siguientes tienen efectos ilustrativos solamente. Todos los porcentajes son en peso a no ser que se especifique lo contrario. Ejemplo 1 30 Oxidación biológica in vivo y fisuración por esfuerzo ambiental en polieteruretano A. Materiales y métodos 1. Aislamiento de células 35 40 45 En estos experimentos se utilizó sangre humana y de conejo como fuentes de las células. La sangre se coaguló con 2 unidades/ml heparina sódica (Upjohn Co, Kalamazoo, MI). Se aislaron células mononucleares (linfocitos, monocitos) dentro de 15 minutos por un procedimiento monoetápico de centrifugación de gradiente de densidad usando Isopaque1077 (una solución de gradiente de densidad) según un método de Boyum modificado (Boyum y otros, Blood Separation and Plasma Fractionation, 217-239, Wiley-Liss, Inc. (1991)). Las células mononucleares se recogieron y lavaron dos veces con solución salina equilibrada Hanks fría (HBSS) sin Ca2+ y Mg2+ para minimizar el agregado celular. Las células se resuspendieron después en medio estándar (RPMI-1640, 10% suero bovino fetal, 0,2M L-glutamina, 10 UI/ml penicilina-G, y 0,1 mg/ml estreptomicina). La suspensión de células se sembró en varios frascos de plástico para cultivo de tejidos e incubó en presencia de 5% CO2 a 37ºC durante 1 hora (Ackerman y otros, J. Immunol, 20, 1372-1374 (1978)). Después de esta incubación, se rasparon suavemente (monocitos) adherentes de la superficie y resuspendieron en medio estándar. Las células (linfocitos) no adherentes contenidas en el supernadante se recuperaron en tubos estériles, y las células no adherentes restantes se lavaron con HBSS fría. Los frascos de cultivo se lavaron tres veces con HBSS fría, y las células adherentes restantes (monocitos) se rasparon suavemente de la superficie y resuspendieron en medio estándar. Ambos tipos de células se resuspendieron a una densidad de 3 x 106/mI. 50 2. Materiales de prueba 55 Se cortaron discos poliméricos, de 6 mm de diámetro, 0,12 ± 0,008 mm de grosor, de hojas de polieteruretano (PEU) usando agujas de biopsia (Prestwick Line, S.M.S. Inc, Columbia, MD). Se impregnó un grupo de discos poliméricos en acetona (AS) durante 1 hora para extraer antioxidantes poliméricos, y secaron a temperatura ambiente durante 4 horas. El otro grupo se utilizó sin pretratamiento (no-AS). A continuación se montaron especímenes de polímero en la parte inferior de las cavidades de placas de cultivo celular de 96 microcavidades en condiciones estériles. 3. Tratamientos poliméricos in vitro 60 Se realizó un tratamiento bietápico in vitro a 37ºC para imitar el entorno in vivo y facilitar la biodegradación de las hojas de PEU. 65 Tratamiento de macrófagos. Los especímenes de película de PEU (AS y no AS) en las placas de microcavidades se cubrieron con macrófago derivado de monocito humano o de conejo recién aislado (Mo/M∅s), o linfocitos humanos (3 x 105 células por cavidad) y cultivaron en un medio estándar (RPMI-1640, 10% suero bovino fetal, 0,2M Lglutamina, 10 UI/ml penicilina-G y 0,1 mg/ml estreptomicina). Se realizó un tratamiento de macrófagos de 49 días en condiciones estándar (es decir, presencia de 5% CO2 , y 95% humedad a 37ºC). Otras variaciones experimentales 14 ES 2 239 948 T3 5 10 incluían añadir dexametasona fosfato sódico (DSP) a concentraciones de 0,024 µg/ml y 240 µ/ml al medio de cultivo en las microcavidades. También se estudiaron dos condiciones en blanco. En una, el medio de cultivo se colocó solamente en las microcavidades. La otra se preparó sin medio de cultivo y almacenó en la oscuridad. Todos los especímenes de polímero se incubaron durante el tiempo del primer tratamiento. Se extrajeron muestras por triplicado después de varios períodos de tiempo para determinación de hidroperóxido. Después de una incubación de 49 días, se prepararon especímenes por triplicado para el segundo paso del protocolo de tratamiento de muestras. Tratamiento con FeCl2 . Después del tratamiento durante 49 días con macrófagos, los especímenes se plegaron por la mitad y fijaron en esta posición termosellando los dos extremos opuestos en tal forma que hubiese un área de esfuerzo incrementado en la región central de los especímenes. Este diseño permitió una caracterización de estados poliméricos no deformados y moderadamente deformados. Los especímenes sometidos a esfuerzo se incubaron en 5 mm FeCl2 a 37ºC durante 10 días. La evaluación al microscopio óptico (OM) de las muestras se realizó durante el tratamiento. Se tomaron muestras triplicadas de cada condición después de 10 días de tratamiento para evaluación al microscopio electrónico de exploración (SEM) de la superficie del polímero. 15 4. Yodometría 20 Especímenes poliméricos tomados en varios períodos de tiempo durante el paso de tratamiento de macrófagos se sonicaron durante 15 minutos en agua destilada, enjuagaron tres veces, y secaron a 25ºC durante 4 horas. Se realizó determinación de hidroperóxido (ROOH) usando un ensayo yodométrico como describen Fujimoto y otros, J. Polym. Chem, 31, 1035-1043 (1993). Este método se basa en la reactividad del grupo hidroperóxido, que oxida yoduro a yodo. El complejo triyoduro resultante (I3 − ) se midió espectrofotométricamente a 360 nm λ con un espectrofotómetro Beckman DU-8 (Beckman Instruments, Irvine, CA). Este método mide la concentración total (superficie más volumen) de hidroperóxido en el polímero. 25 5. Morfología celular y análisis superficial 30 35 40 Las películas poliméricas (0,12 mm de grosor) eran suficientemente finas y transparentes para permitir la visualización de células en sus superficies durante el cultivo celular usando microscopía óptica (OM) con un microscopio de luz Olympus BX40. Para evaluación de la morfología celular SEM, se tomaron especímenes después de 21 días de cultivo celular de macrófagos. Se prepararon para evaluación SEM colocándolos en una solución de fijación fría conteniendo “PLASMA-LYTE” A (solución isotónica de Baxter Scientific, IL) y 1,5% glutaraldehído. A continuación se almacenaron a 4ºC durante 48 horas. Las muestras se sacaron posteriormente del fijador de glutaraldehído, enjuagaron en “PLASMA-LYTE” A tres veces durante 15 minutos cada una. A continuación, se post-fijaron con fijador de Palade (tetróxido de osmio en solución a 4%, Polysciences, Warrington, PA) durante 2 horas. Después de la post-fijación, las muestras se enjuagaron en “PLASMA-LYTE” A tres veces durante 10 minutos cada una, y después deshidrataron lentamente usando concentraciones crecientes de etanol. Finalmente se secaron al punto crítico usando CO2 . También se evaluaron las superficies poliméricas usando SEM después de los 10 días de tratamiento con FeCl2 . Todos los especímenes SEM se montaron y recubrieron por deposición con oro-paladio durante 2 minutos a 10 mA (100 Angstroms de grosor del recubrimiento), usando un Humme IV Sputterer Coater (Anatech, Alexandria. VA). La observación a diferentes ampliaciones se hizo con un microscopio electrónico de exploración Stereoscan 360 (Cambridge Instruments). B. Resultados 45 1. Morfología de monocapas Mo/M∅ 50 55 60 65 Los cambios morfológicos de la monocapa de células durante el paso de tratamiento de macrófagos en las diferentes superficies se estudiaron usando análisis OM y SEM. Usando análisis OM, en las primeras fases de cultivo, las células en el medio estándar comenzaron a aumentar su tamaño, que continuó creciendo con el tiempo. Las monocapas Mo/M∅ en el medio estándar mostraron varias formas, morfologías, y grados de difusión citoplásmica. Los cambios morfológicos que se produjeron entre 0 y 33 días en estas células eran tamaño extensivo - incrementado, difusión citoplásmica, formas inusuales asumidas con 60 µm de diámetro a lo largo del eje más grande. Se observó una disminución del número de células con el tiempo en el medio de cultivo estándar. Las monocapas Mo/M∅ cultivadas con DSP no mostraron aumento de tamaño de célula; sin embargo, se observó que unas pocas células desarrollaron morfología parecida a las cultivadas con medio estándar. El análisis SEM de células cultivadas 21 días (medio estándar) mostró un alto grado de unión de células y difusión de Mo/M∅s en PEU. Las células eran generalmente semiesféricas con un núcleo central y extensos rizos de membrana indicando activación celular. Las dimensiones de las células variaron entre 25 µm y 60 µm dependiendo del grado y excentricidad de la difusión. En contraposición, Mo/M∅s cultivadas en presencia de 0,024 µg/ml DSP mostraron un menor grado de difusión celular. Estas últimas células también mostraron numerosos procesos citoplásmicos (prolongaciones de membrana). Los núcleos de estas células tendían a ser bastante semiesféricos, aunque las superficies de las células, que frecuentemente incluían salientes, adoptaron varias formas. Estas células eran de tamaño altamente variable, pero eran generalmente inferiores a 35 mm a lo largo de su eje más grande. Otras condiciones de prueba -Mo/M∅s humanos cultivadas en presencia de 240 µg/ml DSP y linfocitos humanos cultivados en medio estándar en el que se observó una monocapa viable de células bajo OM- no mostraron células en la superficie del polímero cuando se evaluaron con SEM. 15 ES 2 239 948 T3 2. Evaluación de hidroperóxido polimérico 5 10 15 Las figuras 6 (conejo) y 7 (humano) muestran la concentración de hidroperóxido en los especímenes de polímero tratados bajo las diferentes condiciones descritas anteriormente. Estas condiciones incluían: (1) medio de cultivo estándar solamente (sin células); (2) espécimen de polímero almacenado en la oscuridad en condiciones ambientales (sin medio de cultivo); (3) Mo/M∅s humanos y de conejo en medio de cultivo estándar; (4) linfocitos humanos en medio de cultivo estándar; y (5) Mo/M∅s humanos en medio de cultivo estándar más DSP a 0,024 y 240 µg/ml. Los datos muestran una concentración incrementada de hidroperóxido en función del tiempo de cultivo y la presencia de Mo/M∅s. Este efecto se marcó en especímenes AS (especímenes de polímero impregnados en acetona antes del tratamiento) cultivados con Mo/M∅s de ambas fuentes (conejo o humano) en medio estándar. En contraposición, los especímenes AS cultivados con linfocitos o Mo/M∅s en presencia de DSP mostraron concentraciones de hidroperóxido considerablemente más bajas. Esto era comparable a niveles de concentración de hidroperóxido en especímenes incubados en medio de cultivo solamente y los almacenados en la oscuridad en condiciones ambientales. Igualmente, especímenes de polímero no AS (no impregnados en acetona antes del tratamiento) cultivados con Mo/M∅s mostraron la menor cantidad de hidroperóxidos. 3. Análisis superficial (SEM) 20 25 El examen SEM reveló picadura y fisuración sustanciales en las muestras AS PEU expuestas a Mo/M∅s, con el área sometida a esfuerzo (plegada) como la región superficial más afectada. En esta región se habían desarrollado fisuras de hasta 20 µm de ancho. Las fisuras se iniciaron primero en agujeros adoptando una estructura fibrillar y más tarde se propagaron perpendiculares a la dirección de deformación aplicada producida por el plegado. En contraposición, los especímenes cultivados con linfocitos o DSP no mostraron daño significativo. Las muestras AS PEU expuestas a Mo/M∅s seguido de FeCl2 mostraron daño más extensivo. En contraposición, las muestras no AS PEU expuestas a Mo/M∅s seguido de FeCl2 no mostraron daño superficial apreciable. Las muestras AS PEU expuestas a Mo/M∅s más DSP seguido de FeCl2 mostraron solamente agujeros muy ocasionales. C. Conclusión 30 35 Este estudio indica que los macrófagos están implicados en la oxidación de polieteruretano, induciendo probablemente formación de hidroperóxido en la estructura polimérica. Bajo la influencia de esfuerzo o deformación, los polímeros con suficientes hidroperóxidos se degradaron en presencia de iones Fe2+ de una manera que se asemeja mucho a la fisuración por esfuerzo observada in vivo. Igualmente, se demostró una reducción de la formación de hidroperóxido y ningún desarrollo posterior de ESC en PEU cultivadas en macrófagos en presencia de DSP. Ejemplo 2 40 Modulación in vitro de fenotipo de macrófago en polímero cargado con dexametasona y su efecto en la estabilidad del polímero en un sistema de macrófago humano/Fe/Esfuerzo A. Materiales y métodos 1. Línea celular de prueba; macrófagos derivados de monocito humano (Mo/M∅) 45 El método in vitro usado se describe en el Ejemplo 1. Se utilizó sangre venosa humana como la fuente de células, que se aislaron como se describe en el Ejemplo 1. 2. Materiales de prueba 50 55 60 65 “PELLETHAN E” 80A cargado con dexametasona (DEX/Pe80AS). Para preparar estos materiales, antes de la extrusión, se extrajo “PELLETHANE” 80A (Pe 80A, comercializado por Dow Chemical, Midland, MI) durante 24 horas en un extractor Soxhlett usando acetona. La finalidad de este proceso era la extracción de antioxidante del polímero. Después de la extracción, el material se secó bajo vacío a 50ºC durante 4 días. BP/EP micronizado de dexametasona USP (Lote 78AFT, Upjohn Co.) se secó al vacío durante la noche a 40ºC. Para preparar materiales con diferentes concentraciones de dexametasona (DEX), se varió la relación de medicamento a polímero para lograr niveles de carga de medicamento (p/p) de 0,1% y 1%. La extrusión de películas de 0,02 pulgada (0,5 mm) se obtuvo a formulaciones de 0,1% DEX/Pe80A y 1% DEX/Pe80A). Control “PELLETHANE” 80A (Pe80A). Usando el mismo polímero “PELLETHANE” 80A (extraída la acetona), se extruyeron películas de 0,5 mm (0,02 pulgada) sin DEX. Las condiciones de extrusión con y sin dexametasona eran similares y eran las recomendadas por el fabricante. Se cortaron discos poliméricos, de 6 mm de diámetro, de la prueba de Pe80A y hojas de película de control usando agujas de biopsia. Después se montaron especímenes de polímero (n = 16 por condición) en la parte inferior de las cavidades de placas de cultivo celular de 96 microcavidades en condiciones estériles. 16 ES 2 239 948 T3 3. Tratamientos poliméricos in vitro Se realizó un tratamiento bietápico in vitro a 37ºC, sustancialmente como se describe en el Ejemplo 1. 5 10 Tratamiento de macrófagos. Los especímenes de película de PEU (prueba y controles) en las placas de microcavidades se cubrieron con una monocapa de macrófago derivado de monocito humano recién aislado (hMo/M∅s) a una densidad de 3 x 105 células por cavidad y cultivaron en un medio estándar (RPMI-1640, 10% suero bovino fetal, 0,2M L-glutamina, 10 UI/ml penicilina-G y 0,1 mg/ml estreptomicina). Se realizó un tratamiento de macrófagos durante 40 días en condiciones estándar (es decir, 5% CO2 , 95% humedad, 37ºC). Se añadieron hMo/M∅s recién aislados a las cavidades una vez por semana. Inmediatamente antes del último refresco celular, todas las cavidades se enjuagaron enérgicamente con medio de cultivo para liberar y quitar todos los componentes y restos celulares, después de lo que se aplica una monocapa fresca de macrófago. Después de este tratamiento de macrófagos durante 40 días, se extrajeron muestras de polímero por triplicado para la determinación de hidroperóxido y por quintuplicado para el segundo paso del tratamiento. 15 Tramiento de FeCl2 . Después del tratamiento de macrófagos durante 40 días, se prepararon y trataron especímenes como se describe en el Ejemplo 1. 4. Yodometría 20 Especímenes poliméricos tomados después del paso de tratamiento de macrofago de 40 días se sonicaron durante 15 minutos en agua destilada, enjuagaron tres veces, y secaron a 25ºC durante 4 horas. Se realizó determinación de hidroperóxido (ROOH) usando un ensayo yodométrico como se describe en el Ejemplo 1. 25 30 35 40 5. Morfología celular y análisis superficial La observación OM de cultivadas células se realizó durante el paso de tratamiento de macrófagos. Igualmente, las superficies poliméricas sometidas a esfuerzo se evaluaron usando SEM después de los 10 días de tratamiento con FeCl2 . Los especímenes para evaluación SEM se enjuagaron en agua destilada y secaron a temperatura ambiente. Todos los especímenes SEM se montaron y recubrieron por deposición con oro-paladio durante 2 minutos a 10 mA (=100 Angstoms de grosor del recubrimiento), usando una Humme IV Sputterer Coater (Anatech, Alexandria, VA). La observación a diferentes ampliaciones se hizo con un microscopio electrónico de exploración “STEREOSCAN” 360 (Cambridge Instruments). 6. Cinética de la elución de DEX de materiales de prueba DEX/PEU El perfil de liberación de DEX de 0,1% DEX/Pe80A y 1% DEX/Pe80A se determinó en vitro a 37ºC en PBS. Cada uno de los materiales se procesó por triplicado. El procedimiento implicaba la inmersión de cuatro discos de 15 mm de diámetro (0,3659 ± 0,02 g) en 15 ml de tampón fosfato (Producto número p-4417, Sigma Chemical Co, St. Louis, OM). Los grosores medios de los discos eran 0,47 ± 0,06 mm. En un período de 32 días en varios puntos en el tiempo, se extrajeron 800 µl de solución tampón para análisis y sustituyeron por solución tampón fresca para mantener constante el volumen de elución. Las alícuotas se almacenaron en frío (4ºC) hasta el análisis por HPLC. 7. Análisis HPCL 45 50 Se analizó DEX usando cromatografía de fase invertida y detección visible por UV. Se eligieron para ello una columna de octadecilsilano (Producto número 07125, Tosohaas Bioseparations Specialists, Montgomeryville, PA) y fase móvil que consta de metanol y tampón fosfato (100 mm, pH 5,6). Además, el caudal (1,0 ml/minuto) y el uso de longitud de onda de detección, zonas pico y la autointegración permanecieron constantes durante todos los experimentos. A partir de estos datos se calculó un perfil de elución acumulativo y una elución diaria de DEX. 8. Análisis de citoquina 55 60 Para evaluar la expresión in vitro de IL-1α e IL-8, se incubaron monocitos primarios humanos con varias concentraciones de DEX (2,5, 0,25, y 0,025 µg/ml) y metotrexato (50, 5, y 0,5 µg/ml). Con estos agentes se observó una mayor velocidad de inhibición de IL-1 e IL-8, teniendo DEX los niveles de inhibición más altos. La inhibición parecía ser dependiente de la dosis y del tiempo de incubación. Estos resultados soportan además la capacidad antiinflamatoria y los efectos de estos agentes en macrófagos humanos. B. Resultados 1. Morfología de monocapas Mo/M∅ 65 Se estudiaron los cambios morfológicos de la monocapa de células durante el paso de tratamiento de macrófagos en las diferentes superficies. Una observación OM 100X mediante una película de control de Pe80A mostró uniforme distribución de células. Los cambios morfológicos que se produjeron entre 1 y 40 días en estas células eran extensos y mostraron ser diferentes para cada condición material. Monocapas Mo/M∅ humanos en las superficies de prueba (DEX/Pe80AS) y en las superficies de control (Pe80A) a 3 días de cultura evidenciaron pocas o nulas diferencias. 17 ES 2 239 948 T3 5 En un análisis posterior, 20 días, se observaron diferencias apreciables entre las monocapas en las diferentes superficies. Aunque se observaron una proporción mucho mayor de macrófagos con mayor tamaño y altos grados de difusión citoplásmica en material de control Pe80A, se observó que los Mo/M∅s cultivados en DEX/Pe80AS tenían forma redondeada con diámetros más cortos y con menos densidad. La evaluación a 40 días del tratamiento polimérico mostró el mismo fenotipo celular visto a 20 días, aunque un efecto más marcado, o células con un máximo de 60 mm de diámetro a lo largo de su eje más grande en controles y hasta 20 µm en materiales de prueba. 2. Evaluación de hidroperóxido polimérico 10 La figura 8 muestra la concentración de hidroperóxido en los especímenes de polímero después del paso de tratamiento de macrofago de 40 días. La formación de ROOH en Pe80A siguió un efecto dependiente de DEX. Se observó una concentración de ROOH considerablemente más baja en especímenes DEX/Pe80A. Así, después de 40 días de tratamiento hMo/M∅, 0,5 ± 0,1 y 0,9 ± 0,04 mol ROOH/g de polímero eran contenido en DEX/Pe80AS (0,1% y 1% p/p, respectivamente). En contraposición, el material de control contenía 1,4 ± 0,02 mol ROOH/g de polímero. 15 3. Análisis superficial (OM y SEM) 20 25 30 Durante el tratamiento con FeCl2 se realizó una observación OM diaria de especímenes de polímero sometidos a esfuerzo a 40X de ampliación total en un Olympus SZH 10 Research Stereo Microscope (Olympus Optical Co. LTD). Eran evidentes cambios superficiales observables comenzando a los 4 días de incubación en FeCl2 a 37ºC. A los 6 días eran visibles agujeros y fisuras bien desarrollados en las zonas de mayor esfuerzo en todas las muestras de material de control Pe80A. En las mismas condiciones de tratamiento, ambos especímenes DEX/Pe80A, 0,1% y 1%, mostraron una superficie brillante sin daño evidente. Para expandir el daño en las muestras de prueba y para inducir daño en materiales de prueba, el tratamiento con FeCl2 se prolongó hasta 10 días, después de lo que las muestras se analizaron bajo SEM. El examen SEM reveló picadura y fisuración sustanciales en las muestras de control de Pe80A, con el área sometida a esfuerzo (plegada) como la región superficial más afectada. En esta región se habían desarrollado fisuras de hasta 70 µm de ancho. Las fisuras se iniciaron primero en agujeros adoptando una estructura fibrilar y propagaron más tarde perpendiculares a la dirección de deformación aplicada producida por el plegado. En contraposición, ninguno de los especímenes de DEX/Pe80A mostró daño; más bien, se observó una superficie lisa en ambos especímenes conteniendo DEX. 40 En un intento por obtener datos semicuantitativos de esta evaluación, se adoptó un sistema de clasificación X/Y experimental. En un sistema X/Y, que evalúa la profundidad de las fisuras (X) y la extensión de la superficie afectada por daño (Y) de fisuración por esfuerzo ambiental (ESC), el producto se utiliza para comparar las diferentes condiciones de la prueba. Los resultados pueden oscilar de 0 a 25, indicando el más bajo el menor daño. La Tabla 2 muestra la clasificación de la evaluación de bioestabilidad de especímenes después de un tratamiento durante 40 días con Mo/M∅s humanos y 10 días con FeCl2 . La clasificación final en este método de puntuación experimental se expresa como la media del producto de los valores X e Y. 45 (Tabla pasa a página siguiente) 35 50 55 60 65 18 ES 2 239 948 T3 TABLA 2 Evaluación de bioestabilidad in vitro de películas de DEX/Pe80A 5 10 15 20 25 30 n = 5. Clasificación final expresada como media. Observación a 70-100X. Clasificación experimental = X/Y, X cuantifica la profundidad de las fisuras y Y cuantifica la extensión de cobertura superficial sometida a esfuerzo. X = 0 (sin cambios); 1 (cambio pero sin fisuras, zonas congeladas); 2 (agujeros); 3 (fisuras hasta la mitad a través de la pared de la película); 4 (fisuras confluentes); 5 (fisuras 100% a través de la pared del tubo, fallo). Y = 0 (sin cambios); 1 (en <20% de las superficie); 2 (en >20 y <40% de la superficie); 3 (en >40 y < 60% de la superficie); 4 (en >60 y <80% de la superficie); 5 (en >80% de la superficie). 4. Perfil de la elución DEX de DEX/Pe80AS 35 40 La figura 9 muestra la cantidad de elución DEX por área superficial de material (cm2 ) durante un período de 32 días. Independiente de la carga de DEX en el material, se observó una ráfaga inicial de liberación de DEX el día 1. Como se sospechaba, la cantidad de liberación de DEX era directamente dependiente de la concentración total de DEX en el polímero. El primer día se eluyó 1,6 ± 0,2 y 19,5 ± 0,4 µg de DEX por cm2 de material (0,1% y 1% DEX/Pe80AS, respectivamente). Esta liberación disminuyó bruscamente después. Del día 5 al día 32 había un nivel de elución lentamente decreciente. Después de esta disminución gradual, se registró una liberación de 0,02 ± 0,01 y 0,06 ± 0,03 mg/día/cm2 el día 32. C. Conclusión 45 50 55 Este sistema biológico in vitro ha demostrado ser una herramienta efectiva para estudiar la degradación del polímero. El uso de componentes que están presentes y disponibles en el cuerpo durante las respuestas del huésped (es decir, Mo/M∅s, Fe, esfuerzo) lo convierten en un método bastante realista de replicar la degradación de ESC. Estas observaciones sugieren que los iones Fe+2 aceleran la descomposición de hidroperóxido, dando lugar a un polímero degradado, y que la modulación hacia abajo de la capacidad de los macrófagos de generar especies oxígeno reactivas mediante una liberación controlada de DEX evita los pasos iniciales que conducen a la degradación del polímero. La eficacia de este acercamiento se demostró en este estudio por la reducción de formación de hidroperóxido y nulo daño de ESC siguiente en poliuretanos (Pe80A) cargados con DEX y tratados en el sistema Mo/M∅/Fe/esfuerzo. Ejemplo 3 Bioestabilidad in vivo de recubrimientos de dexametasona/polímero en un modelo de prueba acelerado A. Materiales y métodos 60 65 1. Configuración de la muestra de bioestabilidad Cada muestra de bioestabilidad constaba de una pieza de tubo de prueba recubierto o tubo de control deformado a 400% de elongación. Se utilizaron mandriles de polisulfona para soportar el tubo deformado. Se utilizó una sutura de 2-0 Ticron para sostener la deformación de las muestras de tubo en los mandriles. Los torones de material de implante constaban de cinco muestras hechas específicamente para condiciones de prueba o control. A cada conejo se le implantaron en el tejido subcutáneo de su espalda cuatro torones de 5 muestras. Cada torón se identificó por una perla de vidrio unida cuyo color se codificó para reflejar la condición de recubrimiento/control. Los torones de 19 ES 2 239 948 T3 material de implante medían aproximadamente 0,3 cm de diámetro y 7,0 cm de longitud. Se implantó un total de 120 muestras de 6 condiciones en 6 conejos, 20 por animal y 5 de cada condición. 2. Recubrimientos de prueba 5 10 Se prepararon varias formulaciones de DEX/Pe80A con diversa concentración de DEX. En base a las concentraciones de DEX (p/p) las soluciones eran 0,1% DEX/Pe80A, 1% DEX/Pe80A, 5% DEX/Pe80A y Pe80A (p/o DEX). Las soluciones se prepararon a concentración de 5% de sólidos en THF y se utilizaron para recubrimiento por inmersión de tubo de “PELLETHANE” 2363 80A (Pe80A, Dow Chemical Co, Midland, MI), c/c (proceso de extrusión frío/frío), 0,070 pulgada DI x 0,080 pulgada DE. Para controles negativos se utilizó tubo Pe 2363 80A, h/h (proceso caliente/caliente), 0,070 pulgada DI x 0,080 pulgada DE. Las secciones de los tubos de Pe80A frío/frío se recubrieron con las diferentes preparaciones de DEX/Pe80A con 1 o más inmersiones como sigue: 15 20 25 30 Tubo de Pe80A c/c recubierto (1 inmersión) con 0,1% DEX/Pe80A -dando lugar a aproximadamente 2,4 µg/cm2 DEX inicialmente y aproximadamente 0,6 µg/cm2 DEX después de 400% de elongación (denominado aquí 1/0,1 DEX/Pe80A); Tubo de Pe80A c/c recubierto (1 inmersión) con 1% DEX/Pe80A -dando lugar a aproximadamente 22 µg/cm2 DEX inicialmente y aproximadamente 5,4 µg/cm2 DEX después de 400% de elongación (denominado aquí 1/1 DEX/Pe80A); Tubo de Pe80A c/c recubierto (1 inmersión) con 5% DEX/Pe80A -dando lugar a aproximadamente 120 µg/cm2 DEX inicialmente y aproximadamente 30 µg/cm2 DEX después de 400% de elongación (denominado aquí 1/5DEX/ Pe80A); y Tubo Pe80A c/c recubierto (4 inmersiones) con 5% de DEX/Pe80A -dando lugar a aproximadamente 373 µg/cm2 DEX inicialmente y aproximadamente 93 µ/cm2 DEX después de 400% de elongación (denominado aquí 4/5DEX/ Pe80A). Todas las muestras se esterilizaron con un ciclo de óxido de etileno como es conocido en la materia. 3. Recubrimientos de control 35 40 Para controles positivos se utilizó tubo de Pe 2363 80A recubierto con Pe80A (1 inmersión), c/c, 1,77 mm (0,070 pulgada) DI x 2,03 mm (0,080 pulgada) DE. Para controles negativos, se utilizó tubo Pe 2363 80A no recubierto, h/h, 1,77 mm (0,070 pulgada) x 2,03 mm (0,080 pulgada) DE. El esfuerzo de las muestras de bioestabilidad en esta condición se liberó (S.R.) a 150ºC durante 15 minutos. Todas las muestras se prepararon a 400% de deformación. Los controles se esterilizaron con óxido de etileno. 4. Animales de prueba 45 50 Se utilizaron seis (6) conejos New Zealand White adultos macho y hembra sanos. Todas las muestras de bioestabilidad de prueba y control se implantaron bajo anestesia general. En cada animal se implantó un total de 20 muestras de bioestabilidad. Debido al efecto cruzado potencial de la dexametasona, a dos animales se les implantaron controles y a cuatro animales muestras conteniendo DEX. Las muestras individuales se montaron en torones, con cinco muestras por torón. Cada torón tenía una perla de vidrio de color para identificar cada condición experimental. Se implantaron en el tejido subcutáneo en los lomos de los conejos. Se implantaron dos torones en el lado izquierdo de la espina paralela a la línea dorsal media. Se implantaron dos torones en el lado derecho de la espina paralela a la línea dorsal media. La eutanasia y explantación de las muestras se realizaron en dos puntos de tiempo, 6 y 10 semanas (10 muestras por condición y por punto de tiempo). 5. Modelo acelerado de prueba de bioestabilidad 55 Se utilizó un modelo acelerado de bioestabilidad in vivo. Se prepararon secciones de tubos de prueba y control a 400% de elongación. El control negativo (Pe80A h/h) se sometió a tratamiento de relajación de tensiones a 150ºC durante 15 minutos. Los torones de muestra se implantaron después de un ciclo de esterilización por óxido de etileno. 60 65 6. Análisis de muestras Las muestras se explantaron a la terminación de los conejos. No se observó macroscópicamente ninguna respuesta de tejido anormal en los lugares de implante. Las muestras se desbridaron de tejido y enjuagaron en agua destilada. Después de secarlas, las muestras se examinaron por microscopía óptica a 70X sin preparación adicional de la muestra. Para análisis, las muestras se clasificaron por fisuración por esfuerzo ambiental de forma parecida a la descrita en el Ejemplo 2 (Tabla 2). Cada clasificación individual era ligeramente diferente, sin embargo, los rangos de valores para X y Y eran similares (X = 0 (sin cambios) a 5 (100% de fisuras a través de la pared del tubo, fallo) y Y = 0 (sin cambios) a 5 (sobre >80% de superficie). 20 ES 2 239 948 T3 B. Resultados 5 Al final de las semanas 6 y 10 después del implante, se sacrificaron 3 animales por punto de tiempo y se explantaron las muestras. Las muestras explantadas se desbridaron de tejido y secaron para evaluación por microscopía óptica (OM). También se evaluaron muestras representativas por microscopia electrónica de exploración (SEM) (las muestras se secaron, montaron y recubrieron por deposición con oro paladio como se describe en el Ejemplo 1). Bajo OM, las muestras se inspeccionaron para ver si contenían defectos e imperfecciones. Los resultados generales mostraron lo siguiente: 10 1. Control positivo (peor caso), Pe80A (NoDEX). A las 6 semanas, 4 muestras mostraron fallo ESC (puntuación 5/1), con fisuras poco profundas y casi fallo observado en 3 muestras. A las 10 semanas, se produjo fallo ESC en todas las muestras menos 2. 15 20 2. Recubrimiento de prueba 1/0,1 DEX/Pe80A (0,6 µg DEX/cm2 ). A las 6 semanas, 6 muestras mostraron fallo ESC, y cuatro no mostraron cambios. A las 10 semanas, 6 muestras fallaron y 3 no mostraron cambios ESC. 3. Recubrimiento de prueba 1/1 DEX/Pe80A (5,4 ± 0,71 µg DEX/cm2 ). A las 6 semanas, 4 muestras mostraron fallo, 1 casi fallo, y 5 muestras no presentaron cambios ESC. A las 10 semanas, todas las muestras a excepción de una presentaron fallo ESC. 4. Recubrimiento de prueba 1/5DEX/Pe80A (30 ± 0,6 µg DEX/cm2 ). A las 6 semanas, no se encontraron muestras con fallo. A las 10 semanas, 6 muestras mostraron fallo ESC. 25 30 5. Recubrimiento de prueba 4/5DEX/Pe80A (93,1 µg DEX/cm2 ). A las 6 semanas, ninguna de las 10 muestras presentó fallo ESC. A las 10 semanas, 4 muestras mostraron fallo ESC. Las 6 muestras restantes no presentaban ESC. 6. Control negativo (mejor caso), Pe80A h/h S.R. (tratamiento de relajación de tensiones). A las 6 semanas, no se halló ESC en 8 muestras, mientras que 2 muestras mostraron cambios mínimos y fisuras poco profundas. A las 10 semanas, 4 de 10 muestras presentaban ESC poco profundo. Las 6 muestras restantes no presentaban ESC. En algunos de los especímenes recubiertos con 1 inmersión, se observaron zonas ovales de recubrimiento defectuoso. Este defecto parecía estar correlacionado con daño ESC (fisuras y fisuras poco profundas) en el área. 35 40 El mecanismo protector parece ser efectivo mientras una cantidad adecuada de DEX está presente en el recubrimiento. Esto queda evidenciado por la clara dependencia de dosis DEX de los resultados. La figura 10, que ilustra un resumen de la clasificación más alta en términos de clasificación ESC por espécimen y por punto de tiempo, muestra gráficamente que aunque el recubrimiento con 30 µg/cm2 DEX (1/5D EX/Pe80A) era efectivo al evitar daño superficial hasta 6 semanas, a las 10 semanas se observó un daño amplio, parecido a la condición de control positivo. En contraposición, los recubrimientos conteniendo 93,1 µg/cm2 DEX (4/5DEX/Pe80A) funcionaron mejor que el control positivo en ambos puntos de tiempo. C. Conclusión 45 Este estudio muestra que la dexametasona tiene un efecto protector en la biodegradación de polímeros y evita el desarrollo de fisuración por esfuerzo ambiental en poliuretano susceptible a oxidación. Ejemplo 4 50 Dispositivos antiinflamatorios: Estudios in vivo A. Materiales y métodos 1. Animales de prueba 55 Los animales usados para el implante eran ratas hembra Sprague Dawley de tres meses de edad y 250-300 g de peso corporal adquiridas de Charles River Laboratories, Wilmington, MA. 2. Sistema de prueba de jaula 60 65 La malla metálica de la que estaban hechas las jaulas era de acero inoxidable tipo 304 con un tamaño de malla de 24, un diámetro del hilo de 0,254 mm, e intersticios de 0,8 mm x 0,8 mm (Cleveland Wire Cloth and Manufacturing Co, Cleveland, OH). Las dimensiones de las jaulas eran aproximadamente 3,5 cm de largo y 1,0 cm de diámetro. Cada jaula contenía una pieza del material de control y prueba de interés. Se utilizaron jaulas vacías como controles de prueba. Estas jaulas se empaquetaron y esterilizaron con óxido de etileno como es conocido en la materia. 21 ES 2 239 948 T3 3. Materiales de prueba 5 10 15 20 25 Poliuretano cargado con dexametasona. Un poliuretano alifático segmentado descrito en la Patente de Estados Unidos número 4.873.308 (Coury y otros) sin aditivos se cargó con dexametasona base libre micronizada USP (DEX, Upjohn Co.) usando un proceso de cosolvación. Se disolvió la cantidad apropiada de DEX en tetrahidrofurano (sin hidroxitolueno butilado), Aldrich Chemical Co, Milwaukee, WI), seguido del polímero. Las soluciones contenían 14% sólidos y 1% y 20% DEX. La solución se vertió en bandejas de “TEFLON” de 9,5 cm x 9,5 cm. La película conteniendo 20% DEX se secó en un congelador a -17ºC durante 4 días y después en un horno al vacío a 50ºC y 1,01 bar (-30 pulgadas Hg) durante 2 días. La película conteniendo 1% DEX y la película de control (sin DEX) se secaron en condiciones ambientales durante 1 día, a 50ºC durante 4 días, y después a 50ºC y 1,01 bar (-30 pulgadas Hg) durante 3 días. La película de 20% secada tenía un grosor de 0,7 mm, y la película de 1% y la película de control tenían grosores del orden de 0,44 mm a 0,62 mm. Se prepararon especímenes de 24,97 ± 0,04 mg (control), 24,98 ± 0,05 mg (ID), y 25,01 ± 0,06 mg (20D) de peso, colocaron en jaulas, y esterilizaron con óxido de etileno. 4. Procedimiento de implante Se implantó una jaula subcutáneamente en cada uno de los lados derecho e izquierdo de animales de prueba anestesiados. A efectos de implante, las 33 ratas se dividieron en 2 grupos. En el primer grupo, se implantaron 15 animales. En el segundo grupo se implantaron 18 animales. Se realizó una incisión de 1,0 cm a 1,5 cm en la piel a aproximadamente 2 cm por encima de la cola y a lo largo de la línea media. Se realizó una cavidad en el espacio subcutáneo justo por debajo del omoplato derecho o izquierdo usando disección roma. A continuación se introdujo un espécimen de jaula mediante la incisión y colocó al nivel del panículo carnoso, con la costura colocada contra el músculo subyacente. Se implantó otro espécimen de jaula en el otro lado de la rata de la misma manera. La incisión cutánea se cerró con clips (Fisher Scientific, Pittsburgh, PA). La herida cerrada se roció después suavemente con solución de Betadina. 5. Análisis del exudado 30 35 Se aspiró exudado con jeringas de las jaulas los días 4, 7, 14 y 21 después del implante. Para evitar la interferencia con la respuesta inflamatoria del cuerpo, se recogió no más de 0,3 ml de exudado de cada jaula en cada período de tiempo. Los recuentos de células total y diferencial los realizó personal sin información acerca de la identificación del exudado usando técnicas estándar. Después de la toma de muestras de exudado el día 21, las ratas se sacrificaron por asfixia con dióxido de carbono. 6. Recuento celular total Para examinar la presencia de infección, se cultivó una alícuota de cada muestra de exudado en placas de agar sangre de oveja 5%. Inmediatamente después de sacar el exudado los días 4, 7, 14 y 21 después del implante, el recuento celular total para cada exudado se determinó por recuento con hemocitómetro. 40 7. Recuento celular diferencial 45 50 Se transfirió una alícuota del exudado conteniendo aproximadamente 15000 células sanguíneas blancas (leucocitos) a un tubo de prueba con 300 ml RPMI-1640. Alícuotas (200 µl) de la suspensión de células se colocaron sobre microportaobjetos de vidrio limpio usando una citocentrífuga (Shandon Inc, Pittsburgh, PA). Estos microportaobjetos se tiñeron con tinción “DIFF-QUICK” (Baxter Scientific, McGraw, IL) según las recomendaciones del fabricante y usaron para un recuento celular cuantitativo diferencial. Polimorfonucleares (PMNs), macrófagos derivados de monocito (Mo/M∅s) y linfocitos eran los tipos de células que se contaron durante este análisis. 8. Análisis de jaulas 55 Después de la extracción de exudado a los 21 días, se extrajeron las jaulas implantadas de los animales sacrificados y evaluaron inmediatamente macroscópicamente. El borde superior de la jaula se cortó con un par de tijeras a lo largo de la costura superficial interior. Se examinaron y describieron jaulas intactas y abiertas. Después del análisis, las jaulas se sumergieron en jarras de formalina a 1%. 60 Para evaluar la cantidad de tejido fibroso en las jaulas explantadas, las jaulas se secaron a 60ºC durante 72 horas y se registró su peso en seco. Después de la digestión de tejido por inmersión de la jaula en 6N KOH durante 2 horas a 80ºC, se registró de nuevo el peso de cada jaula de acero inoxidable. Se calculó el peso de tejido seco (tejido seco/(peso total de la jaula - peso del acero inoxidable de la jaula) por jaula. 9. Análisis de la superficie del material 65 Se recuperaron especímenes poliméricos con pinzas donde fue posible, enjuagaron en “PLASMA-LYTE” A (Baxter Scientific, McGraw Park, IL), y colocaron sobre un microportaobjetos. Los especímenes se cortaron después en dos piezas con una cuchilla. Una parte se colocó en un fijador frío conteniendo “PLASMA-LYTE” A y 1,5% glutaraldehído y almacenó a 4ºC. La otra pieza se colocó en un fijador de alcohol y después tiñó con tinción “DIFFQUICK”. 22 ES 2 239 948 T3 5 Las películas poliméricas (0,6 mm de grosor) eran suficientemente finas y transparentes para permitir la visualización de leucocitos adheridos teñidos usando microscopía óptica (OM) con un microscopio de luz Olympus BX40. Los especímenes de polímero teñidos se caracterizaron inicialmente en ambos lados, que eran muy similares, con numerosos leucocitos adheridos a cada superficie. Cada célula unida a la superficie del sustrato se contó diferencialmente a 45X. Cada célula gigante cuerpo extraño (FBGC) se contó como una célula; aunque también se registró el número de núcleos contenidos dentro de cada FBGC. Para evaluación SEM, se extrajeron los especímenes del fijador de glutaraldehído, enjuagaron en “PLASMALYTE” A tres veces durante 15 minutos cada uno y prepararon como se describe en el Ejemplo 1. 10 10. Análisis estadístico 15 Los datos se presentan como la media ± DE. Para recuentos celulares totales se utilizó la prueba de Student no pareada a 95% de nivel de confianza (p < 0,05) para comparar las medias de grupo. Se compararon materiales de prueba 1D (1% DEX) y 20D (20% DEX) con la película de control PU hecha usando THF (A) y una jaula vacía (EC). B. Resultados 1. Análisis de exudado 20 25 30 35 40 45 Las densidades de leucocitos en las muestras de exudado tomadas de los materiales diferenciales los días 4, 7, 14 y 21 después del implante se visualizan gráficamente en la figura 11. Una disminución gradual en la densidad de células después de 4 días era evidente en todas las condiciones de la prueba. Los materiales conteniendo DEX (1D y 20D) produjeron claramente menores cantidades de células durante todo el tiempo de implante. Este efecto era estadísticamente significativo los días 14 y 21 para 1D y el día 7 para 20D. A los 4 días, 1D provocó 90% y 20D solamente provocó 40% del número de células provocado en el material de control (A). A los 21 días, los exudados de 1D contenían solamente 13,9% del número de células observado en exudados del poliuretano de control. Por desgracia, el análisis para material 20D se interrumpió a los 7 días a causa de infección. La comparación de los materiales de control mostró que la elección del solvente (THF y NMP) utilizado en la preparación de la película PU produjo un efecto en los resultados. Todos los tipos de células en el exudado, que incluían PMNs, macrófagos, y linfocitos, disminuyó con el tiempo después del implante. El día 4, los leucocitos estaban dominados por los polimorfonucleares (PMNs) y macrófagos (Mos). Sin embargo, en puntos temporales posteriores había una disminución rápida del porcentaje de PMNs, reflejando el establecimiento de una respuesta inflamatoria crónica. Aunque la concentración de los tres tipos de células leucocitos en el exudado disminuyó con el tiempo, la disminución considerable de PMNs proporcionó aumentos porcentuales observados para los dos tipos de células mononucleares. Solamente macrófagos y FBGCs estaban presentes en superficies del material a los 21 días, aunque se observaron macrófagos, linfocitos y PMNs de forma característica en exudados. El recuento total de células de exudado para materiales de prueba que contienen 1% DEX (1D) y 20% DEX (20D) provocó recuentos celulares más bajos de materiales de control A y EC, evidenciando que tienen considerablemente menos potencial de provocar inflamación. Este efecto subsistió durante todo el estudio para 1D. Los bajos números PMN observados a los 14 días (aproximadamente 5 células/µl de exudado) y los 21 días (aproximadamente 0,5 célula/µl de exudado) en los exudados 1D, sugiere que había poca o nula influencia de PMNs recién reclutados en el lugar inflamatorio. En otros términos, prevaleció un estado inflamatorio crónico suave después de haber concluido la fase aguda. En contraposición, todavía había PMNs los días 14 y 21 en exudados de los controles A y EC, lo que es indicativo de que la dexametasona puede acelerar el proceso que conduce a una respuesta de plena curación de la herida. 2. Análisis de la superficie del material 50 55 Se observaron diferencias macroscópicas drásticas entre jaulas conteniendo DEX/PU y otras jaulas. La formación de cápsulas fibrosas era considerablemente más baja en jaulas 1D (40,6 ± 10,6 mg de tejido seco por jaula) que en jaulas del control A (218,1 ± 72 mg de tejido seco por jaula) o jaulas vacías (207,9 ± 70,7 mg de tejido seco por jaula). Esto muestra la efectividad de la dexametasona en reducir la producción de colágeno en los tejidos que rodean un implante. El análisis superficial de materiales después de 21 días del implante mostró adherencia de células de la línea de macrófagos. Bajo microscopia óptica a 45X, la mayor parte de células adherentes se identificó fácilmente como FBGCs, aunque algunas de las células observadas mostraban morfología macrofágica clásica. 60 65 En las superficies había diferentes densidades de leucocitos adherentes. La mayor parte de las superficies evidenció una distribución de células más o menos aleatoria; sin embargo, había zonas de alta densidad de población de células, zonas de células dispersas y agregados de células adicionales, y zonas de muy pocas células. Los leucocitos adherentes evidenciaron diversas morfologías y grados de difusión citoplásmica. Algunas células habían asumido formas inusuales, y algunas exhibían un deterioro de la membrana celular, dando lugar a supresión considerable de la arquitectura celular. El día 21 había residuos celulares en todas las superficies, a excepción de en el material 1D. Puesto que no se hizo análisis superficial en puntos de tiempo anteriores (por ejemplo, 4, 7, 14 días), no se exploró la progresión en el proceso de distribución/adhesión de células. 23 ES 2 239 948 T3 Las superficies teñidas de material 1D evidenciaron una mayor relación de macrófagos a FBGC en sus superficies. En estas superficies se observaron varios macrófagos y solamente FBGCs dispersos. En contraposición, un número considerable de FBGCs y solamente macrófagos ocasionales estaban presentes en el material de control A (película de poliuretano hecha con THF y sin dexametasona). 5 C. Conclusión Este estudio muestra que el poliuretano cargado con dexametasona es efectivo para reducir la inflamación en respuesta a implante de biomaterial. 10 Ejemplo 5 Evaluación in vivo de cables de estimulación cardiaca transvenosos recubiertos con dexametasona 15 A. Materiales, métodos y resultados de cables de estimulación cardiaca tratados con DEX 1. Preparación de prototipos de cables 20 25 30 35 40 45 Se diseñó un conjunto de experimentos para comprobar la viabilidad de recubrir cables de estimulación cardiaca con formulaciones de PU cargadas con DEX. Un poliuretano alifático segmentado descrito en la Patente de Estados Unidos número 4.873.308 (Coury y otros) se cargó con DEX mediante cosolvación en THF como se describe en el Ejemplo 4. La relación de medicamento a polímero se varió para lograr niveles de carga de medicamento en solución de 1% y 5%. Primero se disolvió en THF la cantidad apropiada de medicamento. Después se añadió el polímero y dejó que se disolviese en la solución. A la terminación, las soluciones eran 11% sólidos (p/p). Bajo una campana de flujo laminar filtrado, se pesaron cables transvenosos de estimulación cardiaca Modelos números 4023 y 4523 (Medtronic Inc, Minneapolis, MN) y después sumergieron en las soluciones de DEX/PUITHF. Para variar la carga de DEX en los dispositivos, las soluciones contenían 0%, 1%, y 5% de DEX (p/p), a 11% pt/pt sólidos totales. Un control incluía solamente un recubrimiento de PU (11% sólidos). Un dispositivo de inmersión estaba configurado para controlar la velocidad de inmersión de los cables en la solución. Antes del recubrimiento, las puntas y dientes de electrodo se protegieron con una pieza de tubo de polipropileno y parapelícula. Para facilitar la inmersión de los cables en la solución de DEX/polímero, se unió distalmente a cada cable un inmersor de disparo redondo de 6,5 g recubierto con silicona (Water Gremlin Co, White Bear Lake, MN). Los cables se bajaron después a la solución de recubrimiento a una profundidad de 15 cm (cuerpo del cuerpo) a una velocidad de 1,9 cm por segundo y después se elevaron inmediatamente de la solución. Entre las inmersiones los cables recubiertos se dejaron al menos aproximadamente 4 horas en un horno de aire forzado (80ºC) y después se secaron al vacío (-30 pulgadas Hg) durante al menos aproximadamente 24 horas. El peso total de los recubrimientos en los dispositivos incrementó con cada inmersión adicional, mostrando una buena linealidad de peso a inmersión. Para obtener un rango variado de carga total de DEX en estos dispositivos, el recubrimiento se consideró terminado después de 2 inmersiones para los cables de control recubiertos con PU, 3 inmersiones para cables recubiertos con DFEX/PU a 1%, y 4 inmersiones para cables recubiertos con DEX/PU a 5%. Después de la inmersión, los dispositivos se liberaron de su protección de electrodo/dientes y cortaron bajo microscopio. El contenido de DEX final se determinó pesando cada cable. El recubrimiento por inmersión de cables en las soluciones de DEX/PU dio lugar a la deposición de una capa polimérica homogónea en la superficie corporal. En base al contenido de DEX por cable, se prepararon tres condiciones. La carga de DEX final se expone en la Tabla 3. Los dispositivos recubiertos se empaquetaron y esterilizaron en óxido de etileno antes de su uso para estudios de elución o para implante canino. 50 TABLA 3 Carga de DEX en cables recubiertos (15 cm) 55 60 65 24 ES 2 239 948 T3 2. Cinética de la elución de DEX in vitro de cables de estimulación cardiaca recubiertos con DEX 5 10 15 20 25 30 El perfil in vitro de liberación de DEX de las dos condiciones de cables conteniendo DEX, 1% DEX/PU y 5% DEX/PU (carga de DEX “baja” y “alta” respectivamente) se determinó mediante experimentos de elución realizados a 37ºC en PBS. Para estos análisis se utilizó la porción recubierta (15 cm) de dos cables de cada condición. Después de la separación de la porción de electrodo/dientes del cuerpo de cable (para quitar la dexametasona del electrodo), los cuerpos de cable recubiertos se sumergieron en PBS a 37ºC y los eluatos se analizaron usando HPLC como se describe en el Ejemplo 1 en diferentes puntos de tiempo dentro de un período de 24 días. La figura 12 muestra la elución de DEX acumulada con el tiempo, expresada como el porcentaje de la carga total de DEX por cable. Ambas condiciones de cable evidenciaron perfiles similares de elución de DEX. Después de una elución acelerada que duró hasta 10 días, se observó que la elución se ralentizaba. A los 24 días, 15,5% y 18,7% de la carga teórica total de DEX se eluyó de los cables recubiertos con DEX “baja” (1% DEX) y “alta” (5% DEX), respectivamente. La figura 13 muestra la cantidad de elución de DEX por área superficial de material (cm2 ) por día durante un período de 24 días. En una ráfaga inicial de liberación de medicamento, se liberó 2,7 ± 0,4 µg y 30,8 ± 1,9 µg de DEX por cm2 de cables recubiertos con las condiciones de DEX “bajas” y “altas”, respectivamente. La liberación de DEX declinó después bruscamente. Desde el día 4 al día 24 había una disminución gradual de liberación de DEX. El día 24 de este experimento, se liberó 0,07 ± 0,09 µg y 0,7 ± 0,1 µg de DEX por cm2 para las condiciones de DEX “bajas” y “altas”, respectivamente. Aunque las velocidades de elución in vitro pueden ser considerablemente diferentes de las velocidades de elución in vivo, estos estudios eran útiles al comprobar y validar las cargas de DEX y los perfiles de elución de dispositivos recubiertos con DEX. Este perfil de liberación y la velocidad de elución (datos no representados) eran similares a los obtenidos con materiales usados en el estudio de jaulas in vivo en el Ejemplo 4. Variando la concentración de DEX en las soluciones de recubrimiento, el porcentaje de sólidos en las soluciones de recubrimiento, o el número de inmersiones, se logró un rango biológico útil de carga de DEX. Esto demuestra la viabilidad de obtener recubrimientos que exhiben cargas y perfiles de liberación de DEX deseados para aplicaciones de dispositivos específicos. Naturalmente, el recubrimiento por inmersión no puede ser el único método de aplicar esta tecnología a cables y otros dispositivos. Es posible que se pueda usar extrusión y/o coextrusión de materiales DEX/PU. B. Materiales, métodos y resultados de la evaluación in vivo de cables de estimulación cardiaca recubiertos con DEX 1. Animales de prueba 35 Los animales usados para el implante eran caninos de sexo aleatorio y de 25 kg de peso corporal. 2. Implante de cables 40 45 Se implantaron en caninos tres condiciones de cables de estimulación cardiaca recubiertos. Como se representa en la Tabla 4, se implantaron en 6 caninos dos condiciones de cable recubierto con DEX/PU (carga de DEX “baja” y “alta”) y una condición de cable recubierto con PU (control). Para este estudio se implantaron por perro 3 cables (2 ventriculares y 1 atrial) de las condiciones de tratamiento de prueba o control. Se adoptó un modelo de 3 cables por perro para incrementar la cantidad de equipo físico dentro de las cámaras intracardiacas. El número de animales y especímenes por material/condición se exponen en la Tabla 4. TABLA 4 Distribución experimental de animales y cable recubierto/condiciones 50 55 60 65 Los cables ventriculares se implantaron mediante toracotomía derecha intercostal tercera a través del tronco costocervico-vertebral (CCTV). Los cables atriales se implantaron mediante venotomía yugular derecha. Con la ayuda de fluoroscopia se colocó un cable ventricular en el vértice RV y el otro cable ventricular se colocó en la pared posterior 25 ES 2 239 948 T3 5 RV al menos 1 cm del cable apical. Umbrales de menos de 1,0 V a 0,5 ms verificaron la adecuada colocación de los cables ventriculares y atriales usando el Patient System Analyzer Modelo 5311 (Medtronic Inc, Minneapolis, MN). Después de fijar cables en los vasos, los extremos de conexión del cable se tunelaron a la pared torácida derecha y coronaron con tapones IS-1. Las colocaciones de los cables se documentaron además con análisis lateral y dorsoventral por rayos X. En general, las actividades quirúrgica y postquirúrgica se desarrollaron sin complicaciones. Debido a la naturaleza de este estudio, no se administraron medicaciones esteroides a ningún canino. 3. Evaluación de parámetros sistémicos 10 15 20 Se evaluó la influencia reguladora de esteroides circulantes durante la etapa de implante in vivo. Se ha referido depresión severa de los niveles circulantes de monocitos usando hidrocortisona (0,6 mg/g de peso corporal, s.c.). Los esteroides también pueden disminuir el nivel circulante de linfocitos T en ratones, ratas y humanos. Igualmente, los esteroides, en particular a dosis inmunosupresoras, reducen la resistencia a la infección bacteriana. Las infecciones, cuando están presentes, pueden ser detectadas por los cambios de la distribución diferencial de las células sanguíneas o por cultivo bacteriano positivo. Para evaluar los cambios sistémicos que podrían ser atribuibles a liberación de DEX (es decir, corticoesteroide excesivo, infección, etc) de los dispositivos tratados, se realizaron análisis de sangre y hemogramas las semanas 1, 2, 4, 8 y 12 después del implante. Al menos uno de estos análisis se realizó en el preoperatorio. Los resultados demostraron que las cantidades de linfocitos estaban dentro de un rango normal o ligeramente elevado. En resumen, un hallazgo consistente o progresivo de linfopenia y/o eosinopenia no se observó en ningún perro en el estudio. 4. Evaluación patológica macroscópica intracardiaca 25 A las 13 semanas (3 meses) del implante del cable, los animales fueron heparinizados, examinados a rayos X y sacrificados siguiendo un procedimiento estándar. La necropsia la realizó un patólogo desconocedor de las diferentes condiciones del estudio. El énfasis especial se centró en el compartimiento intracardiaco para evaluar las relaciones cable-tejido, la encapsulación de los dispositivos, su extensión, grosor, etc. 30 Después de la eutanasia, el corazón se diseccionó, abrió y extrajo con cuidado. Las cavidades cardiacas derechas se abrieron mediante una incisión longitudinal para exponer los cables implantados. Se tomaron fotografías de ampliación baja y alta antes y después de abrir el corazón y después de la extracción del corazón. Después del análisis completo y la descripción de las conclusiones, los corazones se colocaron en formalina tamponada a 10%. 35 Con diferencias mínimas entre los perros, los extremos próximos de los tres cables estaban rodeados por tejido fibroso en el subcutis sobre el tórax derecho. En general, dos cables (ventriculares) entraron en el tórax directamente a través de la pared torácica derecha y después al sistema venoso mediante el CCTV derecho en un lugar de venotomía. Un cable (atrial) avanzó anteriormente a través del subcutis sobre la escápula derecha al cuello ventral derecho, donde entró en la vena yugular mediante un lugar de venotomía. Las envueltas subcutáneas eran finas y estrechamente apuestas a los cables. 40 45 50 55 60 65 Perro receptor de cables sin DEX. Se hallaron dos focos de engrosamiento endocardial multifocal amarillo blando (12 x 4 y 0,5 mm de diámetro respectivamente) detrás del tubérculo intravenoso. Cable atrial. Una envuelta de tejido translúcida corta (<1 cm) rodeaba el cable inmediatamente distal a un lugar de venotomía yugular seguro. Se verificó que su lugar de implante estaba dentro del apéndice auricular derecho (RAA) en el que se observó una envuelta de tejido uniforme, lisa y brillante (8 mm de largo). El resto del cable del cuerpo estaba libre de tejido adherente desde el nivel del CCTV a su lugar de implante en el apéndice auricular derecho (RAA). Cables ventriculares. Se observó una envuelta de tejido (4 mm de largo) cubriendo los dos cables inmediatamente distal al lugar de venotomía CCTV. Esta envuelta de tejido se complicaba distalmente por la presencia de un trombo antemortem excéntrico (5 mm de largo). Dentro de la vena cava anterior (AVC), aurícula derecha (RA), y ventrículo derecho (RV), ambos cables estaban predominantemente libres de tejido adherente. Sin embargo, dentro de la AVC, ambos cables estaban en una envuelta de tejido común, que a su vez estaba unida a la pared luminal de la parte superior de la AVC. Este tejido corto tenía características lisas translúcidas y uniformes, y cubría 7 mm y 9 mm de los dos cables, respectivamente. El cable que se implantó más en la pared RV se adhirió a la pared libre del RV por una unión lateral. Esta envuelta de tejido con un músculo trabecular y características de tejido liso translúcido brillante cubría 12 mm del cable. Perro receptor de cables sin DEX. Atrial. El lugar de implante estaba fijo y situado en la pared libre del RAA. Ventricular. Inmediatamente distales al CCTV, ambos cables están en una envuelta de tejido común que se bifurca ligeramente en su extremo distal. Esta envuelta de tejido se extendía distalmente desde el lugar de venotomía CCTV y cubría 1,2 cm del cable apical y 1,1 cm del cable de pared. El cable apical se adhirió al aparato de válvula tricúspide en una distancia de aproximadamente 1,5 cm. El extremo más distal del cable apical no era visible. El lugar de implante para el cable apical era seguro. La porción caudal de la lengüeta parietal de la válvula tricúspide tiene sus márgentes engrosados por tejido amarill blando. Perro receptor de cables recubiertos con 1% DEX/PU. Cable atrial. En la unión de la AVC y la cresta de la RAA, había un engrosamiento endocardial blando prominente. El cable estaba implantado fijamente en la pared libre de la RAA. Dentro de la AVC dorsal, había un engrosamiento endocardial nodular firme amarillo multifocal. Cada nódulo (2-3 mm de diámetro) estaba distribuido en un área aproximadamente 1,5 cm x 1,0 cm. Cables ventriculares. El lugar 26 ES 2 239 948 T3 5 10 15 20 25 30 35 40 45 50 de venotomía CCTV era seguro. Ambos cables, inmediatamente distales al lugar de venotomía CCTV, estaban en una envuelta de tejido común con un trombo suave en ella. Los cables apical y de pared salían del CCTV y estaban libres de adhesión de tejido o material hasta llegar a la válvula tricúspide, y presentaban una adhesión a la lengüeta parietal de 5 mm de largo. En este lugar de adhesión, el margen de la válvula estaba engrosado por un tejido nodular firme liso brillante. Distales a la válvula tricúspide, los cables estaban libres de tejido adherente o material hasta que llegaban a la RVA y el tabique interventricular, en el que mostraban una fijación firme. Cable RVW. Este cable está libre de toda adherencia de tejido u otro material hasta que llega a su lugar de implante en el vértice RV. El otro cable (pared RV) tiene, inmediatamente al lugar de venotomía CCTV, dos nódulos de tejido o material pálido adherente, cada uno de < 1 mm de diámetro y una envuelta de tejido muy transparente en una longitud de 3 mm. Aproximadamente a 7 cm de distancia del lugar de venotomía CCTV se observaron dos segmentos de envueltas de tejido (5 mm y 2 mm de largo respectivamente); ninguna envuelta estaba adherida a tejidos cardiacos adyacentes. Este cable pasaba por la válvula tricúspide en su comisura caudal; no se observaron adhesiones. Se observó engrosamiento endocardial amarillo blando focal de 6 x 3 mm en la pared libre RV. Perro receptor de cables recubiertos con 1% DEX/PU. Atrial. Inmediatamente distal al lugar de venotomía yugular, el cable estaba dentro de una envuelta de tejido lisa brillante de 1,5 cm de largo y de grosor variable. Esta envuelta de tejido se complicaba distalmente por un trombo antemortem organizado que tenía 0,5 cm de largo. Este cable estaba implantado fijamente en el RAA. Se observaron múltiples engrosamientos endocardiales blandos, lisos y brillantes en la parte superior de la AVC, en la superficie anterior del tubérculo intravenoso y a la unión de la RAA con la RA y la AVC. El endocardio RA junto al origen de la RAA estaba engrosado por un tejido opaco pálido. Ventricular. Inmediatamente distal al lugar de venotomía CCVT, ambos cables estaban en una envuelta fibrosa común de 1,0 cm de largo que se complicaba distalmente con un trombo organizado de 1,0 cm de largo. Se observó que un cable pasaba a la boca del seno coronario y a la vena cardiaca media. Se abrió la vena cardiaca media, y se consideró que el cable estaba envuelto en los 6 cm distales del cable. El otro cable pasaba de la aurícula derecha al RV, donde estaba implantado fijamente cerca del RVA. Este cable pasaba por la válvula tricúspide cerca de la comisura caudal; quedaba libre de adhesiones al aparato de válvula. El extremo distal de este cable estaba metido dentro del músculo trabecular. Las lengüetas de la válvula tricúspide tenían un engrosamiento blando, liso y brillante en zonas apuestas al cable RV. Perro receptor de cables recubiertos con 5% DEX/PU. Dentro del mediastino anterior, situado en la entrada torácica y adherida al primer nervio derecho, se halló una esponja de gasa encapsulada. Cable atrial. Se verificó su implante en la pared libre de la RAA cerca del origen de la RAA. El cable estaba libre de tejido o material adherente. La punta del electrodo era visible desde la superficie epicardial a través del epicardio, pero no era evidente ninguna perforación. Cables ventriculares. Inmediatamente distal al lugar de venotomía CCTV, el cable apical estaba encerrado en una envuelta de tejido 7 mm de largo. Distal a ésta, el cable estaba libre de adherencia de tejido u otro material hasta que llegaba a su lugar de implante en el vértice RV. El otro cable (pared RV), inmediatamente distal al lugar de venotomía CCTV, tenía dos nódulos de tejido o material pálido adherente, cada uno de < 1 mm de diámetro y una envuelta de tejido muy transparente de 3 mm. Aproximadamente 7 cm distales al lugar de venotomía CCTV, se observaron dos segmentos de envueltas de tejido (5 mm y 2 mm de largo, respectivamente); ninguna envuelta estaba adherida a tejidos cardiacos adyacentes. Este cable pasaba por la válvula tricúspide en su comisura caudal. No se observaron adhesiones. Perro receptor de cables recubiertos con 5% DEX/PU. Cable atrial. El cable que entraba en el sistema venoso por la vena yugular tenía su extremo distal dentro de la AVC (desplazado). Una envuelta de tejido (1 cm de largo) estaba presente alrededor del cuerpo de cable inmediatamente distal al lugar de venotomía yugular. Se observó que este cable era móvil dentro de su ligadura de venotomía. Dentro de la RA, la pared septal, y alrededor del origen RAA, había engrosamientos opacos del endocardio. No se observó ningún otro tejido adherente en el resto del cable de cuerpo. Cables ventriculares. Inmediatamente distal al lugar de venotomía CCTV se observó una envuelta de tejido fina transparente de 3 mm de largo, cubriendo un cable. El otro cable tenía una envuelta de tejido más translúcida de 8 mm de largo, que se complicaba distalmente por la presencia de un trombo excéntrico organizado en parte. Este último cable estaba adherido a la pared de la AVC. Distales a estas envueltas de tejido, ambos cables estaban libres de tejido o material adherente dentro de la AVC o RA. Ambos cables pasaban por el aparato tricúspide y quedaban libres de adhesiones. Un cable que pasaba más anteriormente en el RV no estaba implantado en su extremo distal sino que se podía mover libremente. El cable que pasaba más caudal en el RVA estaba implantado fijamente y tenía una envuelta de 3 mm en su lugar de implante. 55 5. Histología de tejidos intracardiacos asociados con cable 60 65 En general, se observaron envueltas ocasionales de tejido durante la evaluación patológica macroscópica. Los tejidos asociados con cable, situados en la porción de cable desde al menos 1 cm distal del lugar de venotomía a al menos 1 cm próximo al lugar de fijación de electrodo, fueron evaluados al microscopio por un patólogo. Se procesó para histología una envuelta de tejido por condición, y se tiñó una sección transversal con hematoxilina y eosina. El patólogo se mantuvo ciego a la condición de tratamiento relacionada con cada espécimen. Aunque los resultados de la evaluación de estos tres especímenes no pueden ser concluyentes (n = 1), ilustran importantes conclusiones que pueden ser relevantes para los diferentes tratamientos superficiales en el estudio. Sigue un breve resumen de las características inflamatorias y el ranking de estas muestras. El tejido menos inflamado es el primero: 27 ES 2 239 948 T3 Espécimen de perro receptor de cables recubiertos con 5% DEX/PU. Envuelta más fina de tejido, sin zona interior de trombo parcialmente organizado, escasa inflamación compuesta de macrófagos y pocos neutrófilos. 5 Espécimen de perro receptor de cables recubiertos con 1% DEX/PU. Envuelta de tejido moderadamente gruesa, zona interior de trombo parcialmente organizado, ligeramente más inflamación compuesta de macrófagos. Espécimen de perro receptor de cables sin DEX. Envuelta de tejido moderadamente gruesa, zona interior de trombo parcialmente organizado, moderada inflamación compuesta de linfocitos, células de plasma, macrófagos, eosinófilos, y pocos neutrófilos. 10 6. Evaluación de órganos 15 20 25 30 Se evaluó la influencia reguladora de esteroides circulantes durante la etapa de implante in vivo. El control de realimentación negativa de esteroides en el eje neuroendocrino y en la pituitaria anterior es bien aceptado. La prolongada supresión de la liberación de ACTH por esteroides está asociada con cambios degenerativos del hipotálamo y de la pituitaria anterior. Estos procesos dan lugar a cambios histopatológicos de las glándulas adrenales. Para estos análisis se recogieron de cada animal secciones de tejido de las glándulas adrenales. Otros tejidos para evaluación incluían hígado, bazo, riñón y pulmones. Las observaciones brutas de los órganos fueron documentadas por el patólogo. Estos especímenes de tejido se procesaron para estudios histológicos rutinarios. En resumen, los resultados no mostraron grandes anomalías en los órganos evaluados. En las cortezas adrenales de todos los perros, las células de la zona fasciculata tenían el citoplasma espumoso o vacuolado típico de células activas secretoras de esteroides. No había evidencia microscópica de linfopenia, que podría resultar de la excesiva administración de esteroides. La evaluación microscópica de otros órganos reveló tejidos solamente con pequeñas anomalías, ninguna de las cuales era atribuible a recubrimiento de corticosteroides en los cuerpos de cable. No se observó una linfopenia y/o eosinopenia consistente o progresiva en ningún perro en el estudio. Las conclusiones brutas incluían a) la presencia de un cuerpo consistente con una esponja de gasa hallada dentro de la cavidad torácica de uno de los perros y b) hinchamiento carpal subcutáneo bilateral de tamaño moderado en otro de los perros. En general, se halló que todos los órganos examinados estaban dentro de límites normales. 7. Estudios de elución de DEX de cables explantados 35 40 Los cables recuperados inmediatamente después de la evaluación patológica se sometieron a elución in vitro en PBS a 37ºC. Los experimentos de elución in vitro se han descrito anteriormente (“Cinética de elución de DEX in vitro de cables de estimulación cardiaca recubiertos con DEX”); se realizaron análisis en eluatos a 1 y 5 días de la elución, y la elución de DEX se calculó en términos de porcentaje del cargas iniciales de DEX en cada cable (figura 14). La liberación de DEX acumulada de 5 días (en PBS) de condiciones “bajas” y “altas” de cables explantados era 2,6% y 3,9% de la carga total de DEX, respectivamente. Esto indica que durante el período de implante in vivo, hasta 90 días, todavía había DEX para elución. C. Conclusión 45 50 El recubrimiento, como una modalidad para aplicar la tecnología a dispositivos, ha sido útil en la preparación de prototipos de cables de estimulación cardiaca y para demostrar la viabilidad de este concepto. Sin embargo, es posible que la extrusión y/o coextrusión de materiales DEX/PU pueda ser favorable para uso a gran escala y la fabricación de dispositivos biomédicos de DEX. En general, el estudio in vivo para evaluar el rendimiento biológico de cables de estimulación cardiaca de DEX no mostró complicaciones. Los resultados no demostraron toxicidad sistémica relacionada con DEX durante los 3 meses de implante, como evidencian los parámetros hematológicos o la histología de los órganos deseados. En la porción intracardiaca de los cables (porción tratada con DEX), se observó encapsulación de tejido mínima o nula asociada en condiciones de control y recubrimiento con DEX. La encapsulación de tejido observada se caracterizó como una reacción típica a poliuretano, como se evaluó macroscópicamente. 55 60 65 La histología microscópica de las envueltas de tejido ocasionales (intracardiacas) asociadas con cable (1 por condición) mostró varios grados de inflamación, cuya intensidad estaba inversamente relacionada con la presencia y la dosis de DEX en las superficies del dispositivo de prueba. Estas conclusiones inflamatorias diferenciales en envueltas de tejido asociadas con cable pueden sugerir una activa modulación hacia abajo de la funcionalidad celular en la interface atribuible a una liberación localizada de DEX. No se halló evidencia de infección sistémica ni histológica en los caninos implantados con dispositivos recubiertos con DEX (n = 4) o con dispositivos de control (n = 2). Después de 3 meses de implante in vivo, los cables recubiertos con DEX explantados mostraron una elución detectable de DEX, con una elución acumulada de 2,6% y 3,9% de la DEX total a 5 días de condiciones “bajas” y “altas” de tratamiento con DEX, respectivamente. Esto indica la presencia de DEX en la matriz polimérica durante el período in vivo, proponiendo una elución activa de DEX en la interface célula- biomaterial. La información sobre liberación de DEX de cables explantados sugirió que todavía había una liberación sostenida de DEX después de 3 meses de implante in vivo. 28 ES 2 239 948 T3 Ejemplo 6 Perfil de elución in vitro para insertos de silicona y con dexametasona 5 10 15 Materiales. Los insertos de silicona estándar para aros de cosido y aros de anulopastia son de aproximadamente 52,5 por ciento en peso de Silicona de Calidad Médica y aproximadamente 47,5 por ciento en peso de sulfato de bario (para impartir radiopacidad). El porcentaje de sulfato de bario puede ser de sólo aproximadamente 40% aunque todavía permite la radiodetección del aro. Durante la mezcla del inserto cargado con medicamento, se añadió una forma de dexametasona (DEX) no soluble en agua a la mezcla, y se extrajo una cantidad correspondiente de sulfato de bario. Por ejemplo, un inserto cargado con dexametasona a 1% contenía Si 52,5%, BaSO4 46,5%, DEX 1%. Se prepararon insertos conteniendo 5%, 2,5%, 1% y 0,5% DEX. También se preparó un inserto de silicona conteniendo una forma de dexametasona soluble en agua (DMP) y DEX. Este inserto contenía un total de 1% dexametasona, en una relación de 3:1 de DEX a DMP. Métodos. La liberación cumulativa de DEX de las muestras cargadas con medicamento se determinó en 71 días. Las muestras se sumergieron completamente en salina fosfato tamponada (PBS) y colocaron en una incubadora a 37ºC. En varios puntos de tiempo se extrajeron alícuotas de PBS y sustituyeron por solución tampón fresca para mantener constante el volumen de elución. El contenido de DEX de las alícuotas extraídas se analizó por cromatografía de líquido de alto rendimiento (HPLC). 20 25 30 35 Resultados. La figura 15 muestra perfiles de elución para insertos cargados con DEX. Aspectos notables de estos perfiles son la respuesta dependiente de dosis y la liberación lenta, siendo ésta última una característica que augura una aplicación a largo plazo. El día 71 el inserto cargado con 5% había liberado aproximadamente 5% de su cantidad teóricamente cargada, el inserto cargado con 2,5% había liberado aproximadamente 7,5% de su cantidad teóricamente cargada, y el inserto cargado con 0,5% había liberado aproximadamente 20% de la cantidad cargada. Dado que la no resolución de la curación durante la etapa de inflamación aguda (que se produce 4-7 días después del implante) puede conducir a eventos prolongados y crónicos severos in vivo, se realizó un esfuerzo por incrementar la cantidad de dexametasona liberada en las primeras fases en el perfil. Se decidió incorporar DMP, una forma de dexametasona soluble en agua, a la mezcla. DMP eluye rápidamente y proporciona una ráfaga de medicamento en las primeras ocho horas. La figura 16 compara perfiles de elución de insertos cargados con DEX a 1% con insertos cargados con DEX/DMP a 1% (relación 3:1). Como se ve en la figura 16(a), la combinación (DEX/DMP) presentó un perfil más alto, observándose una ráfaga inicial el día 1. Ésta era atribuible a la DMP soluble en agua presente en la mezcla. El día 29 se había liberado aproximadamente 8% de la DEX y aproximadamente 13% de la mezcla de medicamento. La figura 16(b) descompone la curva DEX/DMP de la figura 16(a) en sus componentes individuales (es decir, DEX y DMP). Esto tenía principalmente la finalidad de determinar qué cantidad de los dos componentes sale en los diferentes puntos de tiempo. El día 29 se había liberado aproximadamente 9% de la DEX y aproximadamente 25% de DMP. Ejemplo 7 40 45 50 55 60 65 Estudios de bioactividad para insertos de silicona con dexametasona La actividad de dexametasona se evalúa típicamente evaluando el efecto del medicamento en citoquinas inflamatorias liberadas, a saber interleuquina 1α (IL-1 α) y factor de necrosis tumoral a (TNF- α). La dexametasona activa suprime la producción de citoquina. Materiales y métodos. Se prepararon insertos de silicona conteniendo 5%, 2,5%, y 0,5% dexametasona soluble en agua (DEX) como se describe en el Ejemplo 6. Además, también se prepararon insertos de silicona conteniendo 1% DEX, 1% DMP, y una mezcla de DEX/DMP (50:50) a 1%. Células sanguíneas blancas humanas recién aisladas, suspendidas en medios RPM 11640 y complementadas con 10% FBS, penicilina/estreptomicina y L-glutamina, se sembraron en tubos de polipropileno estériles (2.000.000 células/tubo) conteniendo las muestras a evaluar. Los tamaños de las muestras eran aproximadamente 1 cm x 1 cm y la solución de células añadida a cada tubo era 1 ml. Para imitar una relación de cuerpos extraños las células sanguíneas blancas se activaron con el lipopolisacárido endotoxina bacteriano (LPS) a 10 µg/ml. Después de incubación durante 24 horas a 37ºC se extrajeron las muestras, y el contenido de las cavidades se centrifugó a 3000 rpm durante 5 minutos para aglomerar las células no unidas. Los supernadantes se recogieron y almacenaron a -85ºC hasta que se usaron. El contenido de TNF-α e IL-1α en los extractos se analizó usando ELISAs (QuantikineTM Human TNF-α Immunoassay and Human IL-1α Immunoassay) de R&D Systems (Minneapolis, MN). Resultados. La cuantificación de marcadores de citoquina es un método tradicional para evaluar la inflamación. Se analizaron en los medios de células IL-1α y TNF-α, dos potentes citoquinas que son liberadas por macrófagos activados y de las que se sabe que, mediante acciones de autocrina, paracrina y endocrina, desempeñan funciones multifuncionales en los procesos inflamatorio e inmune. Como se representa en la figura 17, la elución de DEX de los insertos suprimió la liberación de TNF-α e IL-1α en forma dependiente de la dosis, mostrando la muestra cargada con 5% el mayor efecto inhibidor. La figura 17 también muestra las diferencias entre 1% DEX, 1% DMP, y una mezcla de DEX/DMP (50:50) a 1%. Se observó que DMP, al ser soluble en agua, se eluía mucho más rápidamente y por lo tanto tenía el mayor efecto a las 24 horas. DEX, al no ser soluble en agua, se eluía lentamente y el efecto no era muy pronunciado a las 24 horas. La mezcla tenía, como era comprensible, un efecto intermedio. 29 ES 2 239 948 T3 Ejemplo 8 Perfil de elución in vitro para insertos de silicona y dexametasona encerrados en poliéster 5 Preparación de muestras. Se prepararon muestras cargadas con DEX como se describe en el Ejemplo 6. Se comprobaron y compararon las configuraciones siguientes: a) silicona cargada con DEX 1% con y sin encapsulación de poliéster 10 b) silicona cargada con DEX 2,5% con y sin encapsulación de poliéster c) una silicona cargada con DEX 1%/DMP (50:50) con y sin poliéster silicona 15 Se cortaron muestras de silicona cargadas con medicamento de dimensiones aproximadamente similares (1,5 cm x 1,5 cm). Piezas rectangulares de tejido de poliéster, que medían aproximadamente 3 cm x 1,5 cm, se conformaron en bolsas doblando las piezas de tejido por la mitad e introduciendo las muestras de silicona entre los pliegues. Se cosieron los extremos libres del tejido junto con suturas de poliéster trenzado 4-0 TI-CRON. La elución de medicamento era la descrita en el Ejemplo 6, durante un período de tiempo de 34 días. 20 Resultados. La figura 18 muestra que las bolsas de poliéster eran permeables a la dexametasona. Parecía haber un retardo ligero, aunque insignificante, del medicamento eluido con el material encapsulado en comparación con el material desnudo. 25 Ejemplo 9 Estudio del implante de jaulas en ratas para insertos de silicona y dexametasona encerrados en poliéster 30 35 40 45 La biocompatibilidad in vivo de silicona cargada con DEX se averiguó usando el sistema de implante de jaula, un método ampliamente referido en la literatura (R. Marchant y otros, J. Biomed. Mat.. Res, 17:301-325 (1983). Jaulas cilíndricas de acero inoxidable, conteniendo las muestras de prueba, se implantaron subcutáneamente en ratas. Para simular un aro de costura/anuloplastia, las muestras de prueba de Si/DEX se introdujeron dentro de bolsas de poliéster DacronTM . Con mínima interferencia mecánica de los tejidos circundantes, el modelo de jaula proporciona un “entorno inflamatorio estándar” en el que la biocompatibilidad del material puede ser estudiada en términos de respuesta celular e interacciones célula-material. En este sistema, los componentes humorales y celulares son comprobados dinámicamente en diferentes puntos de tiempo sin sacrificar el animal. Materiales. Se obtuvieron dexametasona base libre micronizada USP (EDP# 221900) y dexametasona fosfato sódico USP (EDP# 221940) de Upjohn (Kalamazoo, MI); se obtuvo hilo de poliéster TI-CRON # 4 de Davis and Geck (Canadá) y se adquirió malla metálica de acero inoxidable 304 (tamaño de malla =24, diam.= 0,254 mm, intersticios = 0,8 x 0,8 mm) de Cleveland Wire Cloth and Manufacturing Co. (Cleveland, OH). Formulación de silicona/medicamento. Se seleccionaron para este estudio dos formas diferentes de dexametasona. Se utilizó DEX, una forma de dexametasona no soluble en agua, y DMP, una forma de dexametasona soluble en agua, en una mezcla de 75% DEX:25% DMP. Los medicamentos (DEX y DEX/DMP) se mezclaron con silicona de calidad médica (Si) (Tabla I). Estas formulaciones de Si/medicamento también contenían sulfato de bario, un componente que se añade normalmente para impartir radioopacidad al producto final. TABLA I 50 55 1 2 3 Conc DEX. (%) Peso DEX Peso silicona Peso sulfato de bario Peso total 1% 1% (75DEX: 25DMP) 0% 0,2 gms DEX 0,15 gms DEX + 0,05 gms DMP 0,00 gms 10,5 gms 10,5 gms 9,3 gms 9,3 gms 20 gms 20 gms 10,5 gms 9,5 gms 20 gms 60 65 Preparación de muestras. Se cortaron muestras de dimensiones aproximadamente similares (0,25 cm x 2,5 cm) y pesos (67-71 mg) de las placas de silicona fabricadas. A una carga de medicamento de 1% las cantidades teóricas de medicamento de DEX en las muestras reflejaban un rango de aproximadamente 0,67-0,71 mg DEX. Piezas rectangulares de tejido de poliéster, que medían aproximadamente 1 cm x 2,75 cm, se conformaron en bolsas o almohadas. Esto se realizó doblando las piezas de tejido por la mitad e introduciendo las muestras de silicona entre los pliegues. Los tres extremos libres de las bolsas se cosieron a mano con hilo de poliéster TI-CRON # 4. Se utilizó un total de 5 nudos para mantener la muestra dentro de la bolsa. La muestra perteneciente al grupo de tratamiento E (véase la Tabla II) se encapsuló dentro de poliéster que se heparinizó usando tecnología de fotoenlace. Todas las operaciones 30 ES 2 239 948 T3 de manipulación, dimensionamiento, corte y pesaje se realizaron de la forma más limpia posible en una campana de cultivo de tejido Clase II. Las muestras de silicona se limpiaron con aire forzado antes de colocarlas en las bolsas. 5 Sistema de prueba de jaulas. Se fabricaron jaulas cilíndricas, que medían aproximadamente 3,5 cm de longitud y 1,0 cm de diámetro, de mallas metálicas de acero inoxidable. Las jaulas se sometieron después a un ciclo prolongado de lavado con detergente y limpieza. En cada jaula limpiada se colocó una bolsa conteniendo el control (silicona sin medicamento), o el material de prueba de interés (silicona con medicamento). También se utilizaron jaulas vacías como controles de prueba. Los conjuntos de jaula /material se metieron después en bolsas triples y sometieron a un ciclo de esterilización ETO. 10 Animales de prueba. Los animales usados para el implante eran ratas Sprague Dawley hembra de cuatro meses de 250-300 g adquiridas de Charles River Laboratories, Wilmington, MA. Se pusieron en cuarentena a la llegada. Cada animal se numeró con un tatuaje antes de la cirugía. Este estudio se realizó según las normas reconocidas de cuidado esbozadas en la “Guide for the Care and Use of Laboratory Animals” (NIH 1985). 15 20 25 Procedimiento de implante. Brevemente, se hicieron dos incisiones de 1,0 a 1,5-cm en la piel en el lado de la línea media y aproximadamente 2 cm encima de la cola. Se realizó una cavidad en el espacio subcutáneo justo por debajo del omoplato derecho e izquierdo usando disección roma. A continuación se introdujo un conjunto de jaula mediante la incisión y colocó al nivel del panniculus carnosus, con la costura colocada contra el músculo subyacente. El segundo montaje se implantó en el otro lado de la rata de la misma manera. La incisión cutánea se cerró con clips y la herida cerrada se roció después suavemente con solución de Betadina. El número de animales y especímenes por condición, así como sus IDs designadas, se exponen en la Tabla II. Se implantó un total de 30 muestras (2 por rata) en 15 ratas. TABLA II 30 35 40 45 50 55 60 65 A&B = controles; B es el control auténtico para C, D, y E. Análisis del exudado. Se aspiró exudado con jeringas de las jaulas los días 4, 7, 14, y 21 después del implante. Para evitar la interferencia con la respuesta inflamatoria del cuerpo, se recogió no más de 0,3 ml de exudado de cada jaula en cada período de tiempo. Para analizar la presencia de infección, se cultivó una gota de cada muestra de exudado extraída en placas de agar sangre de oveja 5%. El recuento total de leucocitos se determinó usando un hemacitómetro. Para determinación diferencial de leucocitos, se transfirieron exudados conteniendo aproximadamente 30.000 células sanguíneas blancas (leucocitos) a tubos de muestra con 300 µl RPMI-1640. Alícuotas (200 µl) de la suspensión de células se colocaron sobre vidrio limpio usando una citocentrífuga. Estos microportaobjetos se tiñeron con Diff-Quik Stain y cuantificaron diferencialmente para leucocitos polimorfonucleares (PMNs), monocito-macrófagos (Mo/M∅s), y linfocitos. Después del muestreo de exudado durante 21 días, las ratas se sacrificaron por asfixia con dióxido de carbono. Análisis de jaulas. Después de la extracción de exudado durante 21 días, las jaulas implantadas se extrajeron de los animales sacrificados y fueron evaluadas inmediatamente macroscópicamente por un patólogo. El borde superior de la jaula se cortó con un par de tijeras a lo largo de la costura superficial interior. Se examinaron y describieron jaulas intactas y abiertas. Después del análisis, las jaulas se sumergieron en formalina a 10%. Para evaluar la cantidad de tejido fibroso en las jaulas explantadas, primero se sacaron las bolsas de poliéster de las jaulas. Las jaulas se secaron después a 60ºC durante 48 horas y se registraron sus pesos en seco. El tejido se quitó después por digestión en 6N KOH durante 2 horas a 80ºC, y de nuevo se registró el peso de cada jaula sin tejido. La diferencia entre estos dos pesos proporcionó una indicación de la cantidad de tejido seco fibroso asociado con cada jaula. Histología. Una muestra de cada grupo B, C, D, y E, después de la fijación con formalina, se lavó en salina fosfato tamponada, deshidrató mediante lavados con etanol graduado y procesaron para embibición en parafina. Se cortaron 31 ES 2 239 948 T3 secciones transversales de cuatro micras de grosor y tiñeron con tinción tricromo Masson o colorantes de hematoxilina y eosina. 5 10 15 20 Análisis estadístico. Todos los datos se expresan como media (± error medio estándar). Para recuentos celulares totales se utilizó el método Dunnett a un nivel de confianza de 95% (p < 0,05) para comparar medias de grupo. Las comparaciones de los diferentes subgrupos de material eran las siguientes: materiales de prueba (C, D, E) frente a materiales de control (B). El día 14 se extrajo un punto alto de datos de los grupos B y C para el análisis estadístico. Igualmente, el día 21, se extrajo un punto alto de datos de todos los grupos. Resultados. Las densidades de glóbulos blancos que se determinaron en los exudados extraídos los días 4, 7, 14, y 21 después del implante se visualizan en la Tabla III y la figura 19. En general, se observó una disminución gradual de los recuentos después de 4 días con todos los grupos de prueba. Los materiales conteniendo DEX (C, D, E) generaron cantidades que eran considerablemente menores que ambos controles (A, B) en todos los puntos de tiempo. Una comparación directa de los recuentos mediados con DEX con el del grupo de control B, el auténtico control para este estudio, reveló una reducción estadísticamente considerable en todos los puntos de tiempo. Agrupando los valores de todos los materiales conteniendo DEX (C, D, E) y comparándolos con el control B calculamos reducciones porcentuales de las densidades de leucocitos en el orden de 88,5% para el día 4, 82,5% para el día 7, 85% para el día 14, y 97,4% para el día 21. TABLA III Densidad de células sanguíneas blancas (células/µl) 25 30 35 40 45 50 55 60 65 Parecía no haber diferencias obvias entre el grupo C, que contenía la combinación de medicamento (DEX/DMP), y el grupo D, que contenía el medicamento único (DEX). Aparentemente, en el primer punto de tiempo de 4 días, ya había suficiente DEX presente para efectuar una reducción de los recuentos de células inflamatorias. La DEX, por lo tanto, parecía haber enmascarado el efecto de DMP, y que para observar un efecto DMP, habría que considerar un punto de tiempo muy anterior (tal vez el día 1). En el caso de los grupos D y E, en los que la variación entre los dos grupos de tratamiento era el recubrimiento de heparina aplicado al tejido de poliéster, los recuentos en el grupo E, aunque no considerablemente diferentes de los del grupo D, eran consistentemente más bajos en todos los puntos de tiempo. Un examen macroscópico de las jaulas después de la explantación resaltó además las diferencias entre Si/DEX y los controles. Las jaulas no conteniendo DEX se marcaron por considerable crecimiento de tejido (grupos A y B). Éste era especialmente pronunciado en las jaulas que contenían la bolsa de poliéster (grupo B), corroborando de nuevo la naturaleza inflamatoria del material. En general, las jaulas de grupo B estaban ampliamente cubiertas con tejido crecido engrosado translúcido a opaco que también se marcó con rojez granular multifocal. Se halló que el tejido estaba cubierto con tejido fino y tenía coágulos de sangre multifocales en la superficie. En estas jaulas aproximadamente 530% de la malla era visible. En fuerte contraposición, las jaulas que contenían las muestras cargadas con medicamento (grupos C, D, E), en general, parecían estar considerablemente libres del crecimiento de tejido, con aproximadamente 90-95% de la malla claramente visible. El material de poliéster estaba cubierto con una capa transparente fina de tejido. De un total de dieciocho muestras conteniendo DEX solamente una estaba infectaba, y la infección no estaba relacionada probablemente con medicamento como ya era evidente el día 4, cuando se examinó el exudado. Además, la infección permaneció muy localizada durante los 21 días del estudio y no afectó a la segunda jaula en el mismo animal. Se cuantificó y comparó la cantidad de tejido seco fibroso presente en las jaulas. La formación de cápsulas fibrosas 32 ES 2 239 948 T3 5 10 15 en las jaulas cargadas con medicamento (C = 45 ± 20,63 mg, D = 22,5 ± 11,66 mg, E = 13,33 ± 7,63 mg) se consideró considerablemente menor que las jaulas de control B (307 ± 74,81 mg) y las jaulas vacías (249,5 ± 31,3 mg). De los tres grupos conteniendo medicamento, las jaulas con las bolsas heparinizadas en el grupo E registraron el peso de cápsulas fibrosas más bajo. Este hallazgo parecía ser consistente con las menores densidades de leucocitos observadas en la Tabla III. Las bolsas de DacronTM de las jaulas se analizaron histológicamente para determinar la naturaleza y extensión del crecimiento de tejido. Como era el caso de las jaulas de control (Grupo B), las bolsas de Dacron de las jaulas de control también se caracterizaron por extenso crecimiento celular y de tejido (figura 20 (a)). Era evidente una persistente respuesta inflamatoria, principalmente en forma de macrófagos y unos pocos PMNs. También eran visibles células gigantes de cuerpos extraños multinucleadas que se forman alrededor de las fibras individuales del tejido de poliéster (figura 20 (b)). Además, la muestra mostró un alto grado de vascularidad en y alrededor de la muestra. El tejido observado en la superficie exterior del Dacron estaba suelto y orientado aleatoriamente, una respuesta que es consistente con la observada en implantes porosos. La superficie interior del Dacron, por otra parte, reveló tejido fibroso más denso, más organizado, altamente orientado, que se extendía paralelo al plano de la muestra de silicona. Esta formación es característica de una reacción del huésped a implantes no porosos. En contraposición a los controles, las bolsas de DacronTM que rodeaban las muestras de silicona conteniendo DEX indicaron una ausencia notable del crecimiento de tejido (figuras 20 (c) y (d)). En casi todos los casos se halló una capa muy fina de tejido fibroso, escasamente poblada con macrófagos redondeados, rodeando la superficie exterior del tejido. 20 25 Las realizaciones específicas anteriores son ilustrativas de la práctica de la invención. La presente invención también tiene utilidad, por ejemplo, en cirugía reconstructiva, tal como implantes de mama, implantes de pantorrilla, reconstrucción facial y análogos. Es fácilmente evidente a los expertos en la materia que la presente invención, que se refiere a un dispositivo médico incluyendo un material biocompatible de elución de medicamento solapado con un tejido que promueve el crecimiento de tejido, se puede llevar a la práctica en una amplia variedad de aplicaciones tecnológicas de tejido. 30 35 40 45 50 55 60 65 33 ES 2 239 948 T3 REIVINDICACIONES 1. Una bomba de infusión implantable incluyendo: 5 una bomba incluyendo un espacio interior para contención de un líquido; un catéter de administración para administración del líquido a un paciente; y 10 una bolsa de poliéster rodeando la bomba; incluyendo además dicha bomba un material constituyente en contacto íntimo con un agente antiinflamatorio, siendo capaz dicho agente antiinflamatorio de ser liberado de la bomba y eluido a través de la bolsa de poliéster. 15 2. La bomba de infusión implantable de la reivindicación 1, donde el agente antiinflamatorio está revestido o adherido a la superficie de la bomba. 3. La bomba de infusión implantable de la reivindicación 2, donde el agente antiinflamatorio es dexametasona, su derivado, o su sal. 20 25 30 35 40 45 50 55 60 65 34 ES 2 239 948 T3 35 ES 2 239 948 T3 36 ES 2 239 948 T3 37 ES 2 239 948 T3 38 ES 2 239 948 T3 39 ES 2 239 948 T3 40 ES 2 239 948 T3 41 ES 2 239 948 T3 42 ES 2 239 948 T3 43 ES 2 239 948 T3 44 ES 2 239 948 T3 45 ES 2 239 948 T3 46 ES 2 239 948 T3 47 ES 2 239 948 T3 48 ES 2 239 948 T3 49