- Ninguna Categoria
ALQUENOS I
Anuncio
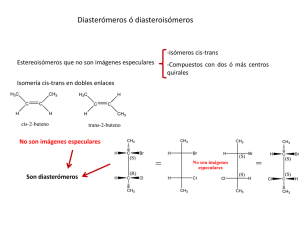
ALQUENOS I Estructura y Enlace en el Etano: El Enlace Pi El enlace Pi del etano es relativamente débil ¿ Cuanto contribuyen los enlaces σ y π a la fuerza total del doble enlace?. Sabemos que los enlaces se forman por solapamiento de orbitales y que su fortaleza relativa depende de la efectividad de dicho solapamiento. Así, debemos esperar que el solapamiento en un enlace σ sea considerablemente mejor que en un enlace π, puesto que los orbitales sp2 se solapan sobre el eje intranuclear. Esta situación se ilustra en los diagramas de energía siguientes: En la región de enlace carbono-carbono deben entrar dos electrones más. Cada átomo p no hibridizado. El orbital 2p p consta de dos de carbono contiene todavía un orbital 2p lóbulos y a cada uno se le da un signo que representa el signo algebraico de la función de onda en las diferentes regiones. Los signos de la función de onda (+, -) no representan cargas. Indican que la función de onda de un orbital 2p tiene valor cero en el átomo de carbono. A esto se le denomina un nodo. Los nodos son puntos que marcan un cambio de signo de la función de onda. Para que los dos orbitales p se recubran eficazmente, deben estar orientados paralelamente entre sí y perpendicularmente a la estructura del enlace sigma, y además el signo de la función de onda tiene que coincidir. Para que esto ocurra, la estructura de los enlaces sigma tiene que ser coplanar y los seis núcleos atómicos implicados en el d bl enlace doble l ti tienen que estar t en ell mismo i plano. l Si esto t ocurre, los l d dos orbitales bit l paralelos p están lo suficientemente cerca para solaparse en posición lateral y se Pueden combinar de dos maneras a) Cuando se recubren los lóbulos del mismo signo se forma un orbital molecular enlazante pi. b) Si los signos de la función de onda no coinciden se genera un orbital molecular antienlazante pi*. En el estado fundamental de un alqueno, los dos electrones que forman el enlace pi entre los átomos de carbono están en el orbital molecular enlazante pi. El solapamiento de los orbitales p es menos eficaz que el solapamiento frontal por el que se forman los orbitales sigma. g Por consiguiente g un enlace pi p es más débil que un enlace sigma. La longitud del enlace C-H es menor en el etileno que en el etano por dos razones: Primera, Primera el enlace sigma del etileno está formado por el solapamiento de dos orbitales sp2 del carbono (33.3% de carácter s), mientras que el enlace sigma en el etano está formado por el solapamiento de dos orbitales sp3 (25% de carácter s). Segunda, el solapamiento de los orbitales p que forman el enlace pi aproxima a los dos átomos de carbono. Isomería cis-trans. La energía de disociación del doble enlace C=C es aproximadamente de 146 kcal/mol y la energía de disociación de un enlace simple C-C C C es de 83 kcal/mol. kcal/mol Por tanto, tanto la energía de disociación del enlace pi debe ser de 63 kcal/mol. Los extremos de la molécula de etileno no pueden torcerse entre sí, porque para ello se debería romper el enlace pi. pi A diferencia de lo que ocurre en los enlaces simples, simples en los enlaces dobles C=C no hay libre rotación. Este es el origen de la isomería cis-trans. Por ejemplo, hay dos alquenos que responden al nombre de 2-buteno: el cis-2-buteno y el trans 2 buteno: trans-2-buteno: Estabilidades relativas de los alquenos. Las energías relativas de los alquenos se pueden comparar midiendo el calor de hidrogenación que es el calor que se libera (ΔH hidrogenación, (ΔH°)) durante la hidrogenación catalítica. catalítica La reacción se lleva a cabo tratando el alqueno en una atmósfera de hidrógeno en presencia de un catalizador metálico. El alqueno se reduce a un alcano. La hidrogenación es ligeramente exotérmica y se desprenden aproximadamente entre 20 y 30 kcal por mol de hidrógeno consumido. Por ejemplo, para el 1-buteno y el (E)-2buteno se desprenden 30.3 kcal/mol y 27.6 kcal/mol respectivamente. La diferencia de estabilidad entre el 1-buteno y el (E)-2-buteno es la diferencia entre sus calores de hidrogenación, por tanto el (E)-2-buteno es 2.7 kcal/mol más estable que el 1-buteno. Esta diferencia de estabilidad es típica p entre un alqueno q monosustituido ((1buteno) y uno trans-disustituido [(E)-2-buteno]. La hidrogenación del 3-metil-1-buteno desprende 30.3 kcal/mol y la del 2-metil-2buteno 26.9 26 9 kcal/mol. kcal/mol Por tanto, tanto el alqueno trisustituido es más estable en 3.4 3 4 kcal/mol que el monosustituido. En los casos que se acaban de comentar se comparan los calores de hidrogenación de alquenos que dan el mismo alcano en la hidrogenación. En la práctica, los calores de hidrogenación se pueden emplear para comparar la estabilidad relativa de alquenos diferentes siempre que se hidrogenen para dar alcanos de energías similares. La conclusión más importante que se desprende de los calores de hidrogenación de los alquenos es que: L dobles Los d bl enlaces l más á estables t bl son aquellos ll que tienen ti ell mayor número ú de d grupos alquilo como sustituyentes. El factor que explica el aumento de la estabilidad de los alquenos con el aumento de la sustitución es fundamentalmente el efecto estérico. En un alcano los grupos alquilo están separados por el ángulo tetrahédrico de enlace de aproximadamente 109.5°. Un doble enlace aumenta esta separación a 120°. En general, los grupos alquilo están más separados en el doble enlace más sustituido y la compresión estérica que experimenta la molécula es menor. Los calores de hidrogenación de los isómeros cis y trans muestran que los isómeros trans son, por lo general, más estables que los cis. La mayor estabilidad termodinámica de los alquenos trans se debe también a su menor compresión estérica, puesto que en los isómeros trans los sustituyentes alquilo están más separados que en los cis . Propiedades Físicas de los Alquenos. Los puntos de ebullición de los alquenos son muy similares a los de los correspondientes di t alcanos. l A í eteno, Así, t propeno y los l butenos b t son gases a temperatura t t ambiente. Los puntos de fusión, sin embargo, dependen, en parte, del empaquetamiento de las moléculas en las celdas cristalinas, lo que está en función de sus formas moleculares. moleculares El doble enlace en los cis-alquenos cis alquenos disustituidos, disustituidos impone una forma de U doblada a la molécula, que dificulta el empaquetamiento y reduce el punto de fusión, usualmente por debajo del que muestra el correspondiente alcano ó el transalqueno . Un doble enlace cis es el responsable de los puntos de fusión, fusión por debajo de la temperatura ambiente de los aceites vegetales. El grupo funcional también afecta a otras propiedades físicas de los alquenos, incluyendo la polaridad y la acidez. Dependiendo de su estructura, los alquenos pueden mostrar crácter dipolar débil. Ello es debido a que los enlaces entre los grupos alquilo y un carbono alquenílico están polarizados en la dirección del átomo con hibridación sp2, porque el grado de caráter s en un orbital híbrido sp2 es más grande que en uno sp3. Los electrones en orbitales con mayor caráter s, se mantienen más próximos ó i all núcleo ú l que aquellos ll en orbitales bit l poseyendo d más á carácter p. Éste efecto hace al carbono sp2 relativamente atrayente de los electrones ( aunque mucho menos que átomos fuertemente electronegativos tales como el O ó el Cl) y crea un dipolo débil a lo largo del enlace sustituyente-carbono alquenílico. En alquenos cis-disustituidos, los dos dipolos individuales se combinan para dar un dipolo molecular neto. Esos dipolos son opuestos en alquenos trans-disustituidos y tienden a cancelarse el uno al otro. Los alquenos cis-disustituidos, más polares, presentan puntos de ebullición ligeramente más altos que los de sus correspondientes isómeros trans. Las diferencias en puntos de ebullición se hacen mayores cuando los dipolos de enlace son mayores, es el caso, p. ej. para los dos isómeros del 1,2-dichloroetano, en los cuales la elevada electronega tividad del àtomo de Cl, hace que los dipolos se inviertan. Otra consecuencia del carácter electrón-atrayente del carbono sp2 es la mayor acidez del hidrógeno alquenílico. Mientras que el etano tiene un pKa aproximado a 50, el eteno es algo más ácido con un pKa de 44. Incluso así, el eteno es una fuente de protones muy débil, comparado con otros compuestos tales como los ácidos carboxílicos ó los alcoholes. Así, p. ej. El etenil litio (vinil litio), no se prepara, generalmente, por deprotonación por metalación del cloruro de vinilo. directa del eteno ( con hidruro de Litio ) , sino p ¿ Cual sería el producto de la reacción de vinil litio con acetona?. Preparación de Alquenos a partir de haluros y sulfonatos de alquilo : (un recordatorio de de las eliminaciones E2) La regioselectividad de las reacciones E2 depende de la base. Ya discutimos como los haloalcanos (ó los sulfonatos de alquilo) pueden, en presencia de bases fuertes,, sufrir eliminación de HX con formación simultánea de un doble enlace carbono-carbono. Con muchos sustratos, la remociòn de un hidrógeno puede tener lugar desde más de un átomo de carbono en la molécula dando lugar a dobles enlaces isómeros constitucionales. En tales casos, ¿ podemos controlar la regioselectividad de la reacción?. La respuesta es sí, en limitada extensión. Un ejemplo simple es la eliminación de bromuro de hidrógeno en 2-bromo-2metilbutano con etóxido de sodio en etanol caliente, que da principalmente 2-metil-2-buteno, pero también algo de 2-metil-1buteno. En nuestro ejemplo ,el producto mayoritario contienen un doble enlace trisustituido y es, así, termodinamicamente más estable que el producto minoritario. En efecto, muchas eliminaciones son regioselectivas g en esta forma, con el p producto más estable termodinámicamente predominando. Este resultado puede explicarse por análisis del estado de transición de la reacción (Fig abajo). La eliminación de HBr procede a través de ataque de la base sobre uno de los hidrógenos vecinos situado anti a el grupo saliente. En el estado de transición hay una ruptura parcial del enlace C–H, formación parcial del doble enlace C–C y apertura parcial del enlace C–Br. El estado de transición que lleva al 2-metil-2-buteno está ligeramente más estabilizado que el que da lugar al 2metil-1-buteno. El producto mas estable se forma f más á rapidamente porque la estructura del estado de transición de la reacción se parece más a la de los productos. Reacciones de eliminación de éste tipo, que dan lugar al alqueno más sustituido tit id se dice di que siguen i l regla la l de d Saytzev: S t El doble d bl enlace l se forma f preferentemente entre el carbono que contiene el grupo saliente y el àtomo de carbono adyacente más sustituido que porta un hidrógeno. Cuando se emplea una base mas impedida estericamente, se obtiene una distribución de productos diferente; en ese caso el producto mayoritario de la eliminación es el alqueno terminal, terminal termodinamicamente menos favorecido. favorecido Para ver porqué el alqueno terminal es ahora el favorecido, examinemos de nuevo el g secundario ( desde C3 en el bromuro estado de transición. Remoción de un hidrógeno de partida) es más dificil estéricamente que abstraer uno de los hidrógenos del metilo, mas expuesto. Cuando se usa como base ter-butóxido, muy voluminoso, la energía del estado de transición que da lugar al producto más estable incrementa, por interferencia estérica, en relación a la energía del que da lugar al isómero menos sustituido (ver Fig. en pg. siguiente); así, el isómero menos sustituido es ahora el producto mayoritario. Una reacción E2 que genera el alqueno isómero menos favorecido se dice que sigue la regla de Hofmann, en honor al químico que investigó una serie de eliminaciones que proceden con éste particular mode de regioselectividad. Las Reacciones E2 favorecen, en general al alqueno trans frente al cis Dependiendo de la estructura del sustrato alquilo, alquilo la reacción E2 puede dar lugar a mezclas de alquenos cis-trans, en algunos casos con selectividad. Por ejemplo, tratamiento de 2-bromopentano con etóxido sódico produce 51% de trans-penteno y sólo 18% de cis cis-penteno, penteno, siendo el resto del producto el regioisómero terminal. El resultado de ésta y de racciones relacionadas parece estar controlado, de nuevo y en alguna extensión, por las estabilidades termodinámicas relativas de los productos, siendo el doble enlace trans,, más estable,, el q que se forma p preferentemente. Desafortunadamente y desde el p punto de vista sintético,, completa p selectividad trans es rara en las reacciones E2. Más adelante veremos algunos métodos alternativos de preparar cis y trans alquenos estereoquimicamente puros. Algunos procesos E2 son Estereoespecíficos. Recuerda que el estado de transición preferido de la eliminación coloca el protón a ser removido y al grupo saliente, en posición geométrica relativa anti el uno con respecto al otro Así, otro. Así antes de que ocurra reacción de eliminación E2, E2 debe producirse una rotación en torno al enlace, de modo que se alcance tal conformación anti. Èste hecho tiene consecuencias adicionales cuando la reacción puede dar lugar a los estereoisómeros Z ó E. E Por ejemplo, ejemplo la reacción E2 de los dos diastereómeros de 2 2-bromo-3-metilpentano bromo 3 metilpentano para dar 3-metil-2-penteno es estereoespecífica. Ambos, el isómero (R,R) y su enantiómero (S,S) dan exclusivamente el isómero (E) del alqueno. Inversamente, sus diastereómeros (R,S) (R S) y (S,R) (S R) dan únicamente el alqueno (Z) (ver Fig, Fig pg. pg siguiente). siguiente) La reacción es así estereoespecífica: Un diastereómero (y su imagen especular) del haloalcano, producen únicamente un estereoisómero del alqueno, mientras que el otro diastereómero ( y su imagen g especular) p ) dan lugar g exclusivamente al alqueno q de configuración opuesta. Preparación de Alquenos por deshidratación de Alcoholes Ya vimos que el tratamiento de alcoholes con un àcido mineral, a elevada temperatura, resulta en la formación de un alqueno por pérdida de agua, agua un proceso llamado deshidratación, que puede proceder por un mecanismo E1 ó E2. El modo usual de deshidratar un alcohol ,es calentarlo en presencia de ácido sulfúrico ó fosfórico a temperatura relativamente alta (120 (120-170ºC). 170 C). La facilidad de eliminación de agua en los alcoholes, incrementa a medida que aumenta el grado de sustitución del carbono que porta el grupo hidroxi. Los alcoholes 2º y 3º se deshidratan a través del mecanismo de eliminación unimolecular E1 ya discutido. Protonación del oxígeno, debilmente básico, del grupo hidróxi forma un ión alcoxonio q que contiene ahora agua, g un p potencial buen g grupo p saliente; la pérdida de agua da lugar, entonces, a los respectivos carbocationes secundarios ó terciarios , cuya deprotonación produce el alqueno. La reacción está sometida, así, a todas las reacciones secundarias que son capaces de sufrir los carbocationes, particularmente a través de reagrupamiento por desplazamiento de hidrógeno ó alquilo. Tipicamente, la deshidratación unimolecular de alcoholes en presencia de ácidos minerales da el alqueno ó mezcla de alquenos mas favorecidos termodinamicamente . Así siempre que ello sea posible, Así, posible se genera el alqueno mas sustituido; si hay elección los alquenos trans-sustituidos predominan sobre los isómeros cis. Por ejemplo, la deshidratación catalizada por ácido del 2-butanol da una mezcla en equilibrio de butenos consistente en 74% de trans-2-buteno, trans-2-buteno 23% de del isómero cis y sólo 3% de Tratamiento de alcoholes primarios con ácidos minerales a elevada temperatura, tambien da lugar a la formación de alquenos.
 0
0
Anuncio
Documentos relacionados






Descargar
Anuncio
Añadir este documento a la recogida (s)
Puede agregar este documento a su colección de estudio (s)
Iniciar sesión Disponible sólo para usuarios autorizadosAñadir a este documento guardado
Puede agregar este documento a su lista guardada
Iniciar sesión Disponible sólo para usuarios autorizados