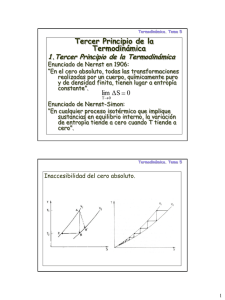
Apunte sobre termodinámica, parte II
Anuncio

Taller de Enseñanza de Física – UNLP en su XXVII aniversario Termodinámica: Parte II Segundo Principio, Potenciales y Aplicaciones Tabla de Contenidos 9- Irreversibilidad: Segundo Principio de la Termodinámica....................................................................2 9.1 Concepto de proceso espontáneo...................................................................................................2 9.2 Segundo Principio de la Termodinámica........................................................................................3 9.3 Ejemplo de predicción no trivial: motores y heladeras..................................................................4 10- Potenciales termodinámicos................................................................................................................7 10.1- El plano T-S.................................................................................................................................7 10.2- La energía interna como potencial: el plano V-S........................................................................9 10.3- Potenciales termodinámicos y transformaciones de Legendre.................................................10 10.4- El plano P-S y la Entalpía (H)...................................................................................................11 10.5- El plano T-V: Función de Helmholtz (F)...................................................................................12 10.6- El plano T-P: Función de Gibbs (G)..........................................................................................13 10.7- El plano U-V y la entropía........................................................................................................14 10.8- Relaciones de Maxwell.............................................................................................................14 10.9- Los potenciales y el Segundo Principio....................................................................................16 Mínimo de la energía mecánica......................................................................................................16 Máximo de la Entropía...................................................................................................................17 Mínimo de la Función de Gibbs.....................................................................................................18 10.10- Ejemplo de uso: H y G en química.........................................................................................19 10.11- Ejemplo de uso: “Motores Químicos” (pilas, fotosíntesis, etc)..............................................21 11- Termodinámica más allá del equilibrio..............................................................................................22 11.2- Equilibrio local..........................................................................................................................23 11.3- Creación de entropía.................................................................................................................23 12- Conexiones con lo microscópico.......................................................................................................29 12.1- ¿Qué quiere decir “microscópico”?..........................................................................................29 12.2- Las “moléculas” durante el siglo XIX......................................................................................30 12.3- La misteriosa constante R.........................................................................................................31 12.4- Las ideas de Boltzman..............................................................................................................32 12.5- Disparen sobre Boltzman..........................................................................................................33 12.6- Tour guiado por la Mecánica Estadística..................................................................................34 12.7- ¿Cómo sé si necesito de la mecánica estadística para resolver un problema?..........................35 13- La Termodinámica Realmente Existente...........................................................................................35 13.1- Fenómenos térmicos.................................................................................................................36 Dilatación térmica...........................................................................................................................36 “Transferencia de calor”, calor específico......................................................................................38 13.2- Calorimetría..............................................................................................................................39 13.3- Primer Principio en varios dialectos.........................................................................................40 1 Convenciones de signos..................................................................................................................40 13.4- Fenómenos de Transporte.........................................................................................................41 Conducción.....................................................................................................................................42 Convección.....................................................................................................................................43 Radiación........................................................................................................................................43 13.5- Exergía......................................................................................................................................44 13.6- La “energía” en la nutrición......................................................................................................45 13.7- Termodinámica en Ecología......................................................................................................46 El flujo de energía en la ecología ortodoxa....................................................................................46 La Ecología de Odum, Margalef y Jacques Cousteau....................................................................47 14- Desmantelando tabúes: ¿Cómo se usa la Termodinámica en Ciencias Naturales?...........................49 9- Irreversibilidad: Segundo Principio de la Termodinámica (esta es una de las secciones centrales del apunte) 9.1 Concepto de proceso espontáneo Empecemos observando que hicimos un poquito de trampa. En los problemas 1 y 2 sobre modelización de interacciones térmicas (secciones 8.1 y 8.2) asumimos que la temperatura final de los sistemas es homogenea, pero nada en la teoría obliga a que eso sea así. En lo que al primer principio se refiere, nada impide imaginar que un pedazo del agua de un vaso se vuelve un cubito de hielo mientras el resto del agua se calienta, siempre que en el proceso se conserve la energía. Es cierto que para este sistema es bastante claro que el estado final es uno en el que la temperatura es uniforme, pero sería deseable que una teoría sobre interacciones térmicas incluyera eso. Pero hay razones más prácticas para buscar un principio que permita hacer ese tipo de predicciones: no siempre la dirección en que un proceso es espontaneo resulta tan clara como en el caso del cubito en el vaso de agua. Piensen por ejemplo en las reacciones químicas o en la estabilidad de estructuras cristalinas de minerales. Ese principio es el Segundo Principio de la Termodinámica. Esencialmente el segundo principio lo que hace es permitir predecir qué procesos ocurren en la naturaleza, y cuáles no. A los primeros se les llama “espontaneos”, y a los otros, “no espontáneos”. Esos nombres pueden llevar a equívocos, porque la idea de espontaneidad del lenguaje coloquial es muy distinta. Lo opuesto a “espontáneo” en lenguaje coloquial es “forzado”. Si un sospechoso declara de motu propio ante el juez su declaración es “espontánea”, pero si va a buscarlo la policía y lo lleva a la fuerza con el juez la declaración no es “espontánea” y el tipo está en el horno. Pero desde el punto de vista de la termodinámica los dos son procesos que pueden ocurrir (y de hecho ambos ocurren bastante seguido) así que si modelizamos termodinámicamente al sospechoso y al sistema judicial, suponiendo que tal cosa fuera posible, los dos son “procesos espontáneos”. 2 Pongamos un ejemplo más físico. Si una pava de agua caliente se deja en la mesa, casi todo el mundo estaría de acuerdo en afirmar que el agua “se enfría espontáneamente”. En cambio, si encendemos la hornalla y ponemos una pava de agua a temperatura ambiente, uno se siente tentado a decir que el agua “no se calienta espontáneamente”, sino porque el fuego “la fuerza a calentarse”. Esa es una forma coloquial de pensar, pero si nos atenemos a nuestra definición termodinámica de “proceso espontáneo” claramente el agua se calienta espontáneamente. ¡De lo contrario sería imposible calentar una pava de agua con una hornalla! En termodinámica los procesos espontáneos son los que realmente ocurren, y los no espontáneos los que no pueden ocurrir. En el caso de la pava de agua sobre la hornalla encendida, lo que ocurre espontáneamente es que los gases calientes que rodean la pava se enfrían un poco, y que el agua se calienta. Un ejemplo de proceso no espontáneo sería que el agua de la pava se enfríe y los gases de la llama se calienten todavía más; por más que la energía se conserve, el proceso es imposible. Es entonces importante reconocer que para saber qué procesos de un sistema son espontáneos es imprescindible establecer el entorno. No son los mismos los procesos espontáneos del agua dentro de una pava si la pava está sobre la mesada, en una hornalla encendida o en un freezer. Justamente, la peculiaridad del segundo principio es que no se puede prescindir de las transformaciones del entorno para formularlo. Si no se comprende esto es imposible darle sentido al enunciado del segundo principio. 9.2 Segundo Principio de la Termodinámica Analicemos el caso de un cuerpo caliente en contacto térmico con uno frío. Ya sabemos que el proceso espontaneo es el que lleva a que las dos temperaturas se igualan. Los dos bloques de la figura de la izquierda están aislados del resto del Universo, pero en contacto térmico entre sí. Supongamos que TA es mayor que TB. El calor que B le hace a A (Q A) es menos el calor que A le hace a B (Q B). De la definición de calor, sabemos entonces que T A dS A=−T B dS B (34) El cambio de entropía del Universo es dS U =dS A +dS B , así que usando (34) tendremos: dS U =dS A – TA T dS A= 1− A dS A (35) TB TB Si asumimos que la dirección espontanea es la que lleva a que la temperatura de A disminuya y la de B aumente, B debe hacer sobre A un calor negativo, por lo que dS A será negativa. Por otra parte, T A / T B es mayor que 1, de modo que el coeficiente que acompaña a dS A en (35) es negativo. Por lo tanto, dS U es positivo. Si consideramos A como el objeto de estudio, y B como su entorno, vemos que la entropía del objeto de estudio disminuye, pero la entropía del entorno aumenta más de lo que disminuye la del objeto. Lo relevante es que al Universo (objeto más entorno) la entropía le aumenta. Supongamos que el objeto es 3 la pava caliente y el entorno la mesada. La entropía de la pava disminuye, y la de la mesada aumenta. Pero como la temperatura de la pava es mayor que la de la mesada, y como los calores son iguales y opuestos, la entropía de la mesada aumenta más de lo que disminuye la entropía de la pava. La entropía del Universo aumenta, por lo que el proceso espontáneo es aquel en el que la pava se enfría. Ahora analicemos el caso en que el objeto es la pava fría, el entorno es la hornalla encendida. La entropía de la pava aumenta, la entropía de los gases calientes de la llama disminuye un poco, pero como estos gases están a una temperatura mucho más alta que la pava, la entropía de la pava aumenta más que lo que disminuye la entropía de los gases calientes, de modo que de nuevo el proceso espontáneo es aquel en el que la pava se calienta. Todo este análisis para “predecir” lo que ya sabemos que ocurrirá suena a usar cañonazos para matar hormigas sobre las que alguien ya había usado insecticida. En realidad estamos mostrando casos en los que es facil ver que la entropía del Universo aumenta, para justificar postular, como principio, que eso es lo que ocurre siempre. Aunque parezca mentira, es un postulado que, usado con ingenio, puede llevar a predecir cosas que están lejos de ser obvias. Lo que dice el Segundo Principio de la Termodinámica es que sólo pueden ocurrir procesos en los que la entropía del Universo aumenta: Segundo Principio de la Termodinámica: Sólo pueden ocurrir procesos en los que la entropía del Universo aumenta. A los procesos que pueden ocurrir se les llama espontaneos. ΔS U 0 para procesos espontaneos Algunas aclaraciones: • Insistimos: no podemos decidir si un proceso es espontaneo o no sin estudiar lo que le ocurre al entorno. Por eso en el segundo principio se habla del aumento de la entropía del Universo. • El “Universo” no es la totalidad del cosmos. Es el sistema de estudio más una parte del entorno lo bastante grande como para considerar al total como cerrado (basta con que el sistema de estudio no interaccione con nada fuera de esa parte del Entorno. • En principio uno puede pensar en procesos ideales para los que la entropía del Universo no aumenta. Esto ocurre si y sólo si el proceso es cuasiestático y el sistema de estudio es tal que las partes mecánicas sufren un trabajo nulo de las fuerzas no conservativas. En este caso, el proceso inverso tampoco cambia la entropía del Universo, y por eso se los llama procesos reversibles. En la práctica ningún proceso es realmente reversible porque ningún proceso es cuasiestático. • Un corolario de esto es que todos los procesos espontaneos son irreversibles. Esto no significa que un cierto objeto de estudio no pueda volver a estar en un estado que ya hubiera estado. Lo que no puede hacerse es devolver al objeto y al entorno a un estado en el que ya hayan estado. 9.3 Ejemplo de predicción no trivial: motores y heladeras Esta subsección sirve para mostrar con un ejemplo simple por qué el segundo principio de la 4 Termodinámica es poderoso. Es un ejemplo algo típico de libro de texto, pero por desgracia es más ingenieril que de interés para las ciencias naturales. Se recomienda su lectura a quien quiera hacer la conexión entre el tratamiento que damos a la termodinámica en este curso y las exposiciones más convencionales. Hay muchos, muchísimos ejemplos de predicciones no triviales hechas a partir del segundo principio. Muchas son sumamente técnicas y sería complejo trabajarlas tan pronto. Pero hay una aplicación histórica que de hecho es la que motivó el desarrollo de la Termodinámica, hija de la revolución industrial: los límites a la eficiencia de las máquinas. Una gran cantidad de inventores intentó por décadas (en internet puede verse que algunos siguen intentando) hacer máquinas “de movimiento perpetuo”, es decir máquinas que permitan producir sin ponerles nafta, ni enchufarlas, ni latiguear mulas para que las muevan. Algunas ideas son muy ingeniosas y lleva un esfuerzo grande descubrir en qué fallan. Lo interesante es que todas esas máquinas deben fallar si es que los principios de la termodinámica son válidos, y hoy por hoy la evidencia a su favor es abrumadora. Las máquinas que pretenden funcionar creando energía de la nada son las que se conocen como “móviles perpetuos de la primera especie”, y están prohibidas por el Primer Principio (en su versión completa). Los que resultaron más desafiantes son los que respetan el primer principio, que reciben el nombre de “móviles perpetuos de la segunda especie”. Por ejemplo, un motor de barco que funcione con calor que haga el agua de mar. Vamos a hacer un modelo de máquina muy simple que ilustra cuál es el problema con este tipo de máquinas. La máquina en sí será un sistema con partes mecánicas y con sustancias modelizables termodinámicamente, no importa cómo. La única suposición que vamos a hacer es que en todo momento se le puede asignar una energía mecánica y una energía interna, y que opera cíclicamente (o sea, que vuelve a pasar una y otra vez por el mismo estado). El entorno puede hacer sobre el sistema calor y trabajo. Supongamos que el entorno está a una temperatura T, y tratemos de hacer una máquina que aproveche el calor que pueda hacerle el mar para mover un barco. Consideremos un ciclo, de modo que el cambio de energía interna del motor valga cero. Si queremos que el motor haga un trabajo positivo sobre el entorno (léase: el barco) el entorno hará sobre el motor un trabajo negativo. Aplicando el Primer Principio al motor a lo largo de un ciclo: W entorno+Q entorno=0 en un ciclo (36) También debido a que el motor opera en un ciclo, el cambio de entropía del motor en el ciclo es nulo, por lo que el cambio de entropía del Universo es simplemente el del entorno. Ahora bien, si queremos que el motor funcione como tal, el trabajo del entorno es negativo por lo que el calor es positivo. Entonces el calor del sistema sobre el entorno (en nuestro ejemplo, el calor del motor sobre el mar) es negativo (el mar “se enfría”), de modo que el cambio de entropía del entorno es negativo. Por lo dicho, el cambio de entropía del Universo es negativo: este proceso no es espontaneo. ¡Por lo tanto este motor no puede funcionar! Vean que a la inversa no hay problemas: el entorno puede hacer un trabajo positivo sobre el “motor” de manera que la entropía del entorno aumente. Un buen ejemplo de este tipo de “motor” es un calentador eléctrico. Lo interesante acá es que nos hemos independizado de todo detalle de cómo está hecho el motor. Esto es lo fuerte de la termodinámica, y es la razon que hace que la modelización macroscópica resulte tan 5 poderosa. Los detalles microscópicos pueden ser extremadamente complicados, pero hay muchas cosas que se pueden concluír con independencia de esa complejidad. Entonces necesitamos complicar un poco nuestro modelo de motor termodinámico para hacerlo funcionar. Una buena idea puede ser que la interacción térmica ocurra con dos “focos” a temperaturas diferentes. Por ejemplo, en un barco el motor puede estar en contacto con una caldera (que sería el “foco caliente”) y con el mar (“foco frío”). En un ciclo tendremos entonces W entorno+Q caliente+Q frío =0 sobre un ciclo (37) El foco caliente hace un calor positivo sobre el motor, y el motor hace un trabajo positivo sobre el entorno y un calor positivo sobre el foco frío. Una forma de leer la expresión (37) es que por cada joule que hace el foco caliente una parte la hace el motor como trabajo y el resto “se pierde” como calor sobre el foco frío. Esta forma de pensarlo motiva la siguiente definición de “eficiencia” de una máquina termodinámica que opera cíclicamente: Eficiencia= W entorno Q caliente (38) La “eficiencia” así definida es simplemente la fracción del calor hecho por el foco caliente que el motor logra hacer como trabajo. Es facil calcular la eficiencia de un motor reversible operando entre estos dos focos. El calor que el foco caliente hace sobre el motor es Q caliente=−T calientedṢ caliente (39) donde dS caliente es el cambio de entropía en el foco caliente, mientras que el calor que el foco frío hace sobre el motor es Q frío =−T frío dS frío (40) (Los signos “-” se deben a que no hablamos del calor sobre los focos sino sobre el motor). Como la entropía del motor no cambia (estamos analizando un ciclo) el cambio de la entropía del Universo es la suma de los cambios de ambis focos; por el Segundo Principio, si el motor es reversible tendremos dS caliente+dS frío =0 (41) Usando (37), (39), (40) y (41), el trabajo que entorno hace sobre el motor reversible hace será: W entorno=−Q caliente−Q frío =−T caliente+T frío dS caliente (42) Reemplazando (42) y (39) en (38) obtenemos finalmente para la eficiencia de una máquina reversible: Eficiencia de un motor reversible = 1 – T frío T caliente (43) Vean que, no importa cómo se haga el motor, si es reversible la eficiencia depende sólo de las temperaturas de los focos. El motor sería perfectamente eficiente si el foco frío estuviera a temperatura absoluta cero, y tendría eficiencia nula si el foco caliente y el frío estuvieran a la misma temperatura. Vean que, como el motor es reversible, se lo puede usar al reves, haciendo trabajo negativo contra el entorno, calor negativo sobre el foco frío y calor positivo sobre el caliente. Una pregunta interesante, por no decir molesta, es por qué nos tomamos tanto trabajo en calcular la 6 eficiencia de un motor que sabemos es imposible de construír. ¿No será posible construír una máquina irreversible que sea más eficiente que esta? Bueno, esta es la parte más divertida del análisis. Supongamos que tenemos una máquina más eficiente que la reversible; entonces podríamos usar parte del trabajo producido por esa máquina para, con la máquina reversible andando al revés, hacer el calor contrario al que hizo la primera máquina. El resultado sería una máquina de movimiento perpetuo de la segunda especie: el trabajo que hace la máquina es igual al calor neto que hace el foco caliente sobre ella, y el calor neto sobre el foco frío es cero. Pero ya vimos que esa máquina viola el Segundo Principio, o sea que si admitimos que el Segundo Principio es válido no puede haber máquinas más eficiente que una reversible: Eficiencia de un motor real < 1 – T frío T caliente (44) Como hemos mencionado, este es el origen histórico del segundo principio. Por eso, muchos libros arrancan por acá: postulan como segundo principio Segundo Principio de la Termodinámica (versión habitual en libros de texto) No puede hacerse una máquina térmica que opere cíclicamente sobre un foco a una temperatura, haga un trabajo positivo contra el entorno y no haga ningún otro proceso sobre el entorno. Una vez establecido esto, se calcula la eficiencia de un motor reversible (usando un ciclo concreto 1, porque la entropía en este enfoque aún no está construída) y se demuestra, tal como lo hicimos acá, que ninguna máquina térmica puede ser más eficiente que esta. Recién entonces se define la función S como la integral de 1/T respecto del calor hecho reversiblemente, y se demuestra que es una función de estado. Por eso en la mayoría de los libros de texto las máquinas y su eficiencia ocupan un lugar tan destacado en el formalismo, así como los procesos en los gases ideales. 10- Potenciales termodinámicos 10.1- El plano T-S Hasta ahora hemos representado los estados de equilibrio (interno) de un fluído como puntos de un diagrama P-V, dejando fijo el número de moles n. Después hemos escrito el resto de las variables de estado termodinámico como funciones sobre estas variables: T(P,V,n); U(P,V,n); S(P,V,n); etc. Una pregunta que uno puede hacerse (si está muy aburrido) es: ¿Por qué usar la presión y el volumen como las variables independientes? Por qué no “despejar” algunas de las funciones de estado y escribir cosas del estilo P(T,S,n); U(V,T,n), etc? La verdad es que con cierta restricción, uno siempre puede poner 1 Para calcular la eficiencia de una máquina reversible se usa como “motor” una cantidad fija de gas ideal que opera isotérmicamente sobre los focos y adiabáticamente para ir de una isoterma a la otra. El calor específico es lo primero que se define, de manera que el cálculo puede hacerse sin apelar al concepto de entropía. Este ciclo es conocido en los libros como “ciclo de Carnot”. 7 todas las variables en términos de las tres 2 variables que a uno se le ocurra. La cierta restricción es simple: como algunas funciones de estado son extensivas, las tres que elijamos como variables independientes deben incluír por lo menos una extensiva. Quien no estuviese tan aburrido para plantearse semejante pregunta, probablemente se preguntará (muy sensatamente) qué tiene de interesante tomarse ese trabajo. Bueno, lo cierto es que, dependiendo del problema, puede ser mucho más cómodo trabajar con un conjunto de variables que con otro. Vamos a poner un ejemplo simple de sistema alternativo de variables independientes y jugar un poco con él. Después vamos a tener en cuenta varios conjuntos interesantes de variables independientes en la subsección siguiente. Supongamos que en vez de presión, volumen y número de moles, como hasta ahora, usamos como conjunto de variables independientes la entropía, la temperatura y el número de moles. Si fijamos el número de moles, los estados de equilibrio (interno) del fluído se representan como puntos en el plano T-S. En el plano T-S las isotermas son simplemente líneas horizontales y las adiabáticas son líneas verticales. En cambio, las isobaras y las isocoras (triviales en el plano P-V) son curvas complejas que dependen del modelo. Para graficarlas necesitaríamos la ecuación de estado y algún calor específico. En la figura de arriba mostramos varias curvas isotérmicas (líneas punteadas) y adiabáticas (líneas cortadas) tanto en el plano P-V como en el plano T-S para cierto fluído hipotético. Recuerden que para conocer las curvas isotérmicas (curvas de nivel de T(P,V,n)) debe conocerse la ecuación de estado, mientras que para conocer las curvas adiabáticas (curvas de nivel de S(P,V,n)) deben conocerse los calores específicos. En el diagrama P-V se muestra cierto proceso que va del estado A al estado B. En el plano T-S hemos dibujado las mismas isotermas y las mismas adiábatas que en el otro diagrama. Entonces, observando los estados sobre el proceso que va de A a B relativamente a las curvas isotérmicas y adiabáticas, se ve que el mismo proceso, para esa sustancia, en el plano T-S corresponde aproximadamente al mostrado a la derecha. Es un ejercicio interesante verificarlo, así como hacer isocoras e isobaras aproximadas para esta sustancia en el plano T-S. Algo interesante de ver es que en el plano T-S el calor durante un proceso cuasiestático es el área bajo la curva (por la definición de calor) independientemente de la sustancia de la que se trate, mientras que para calcular el trabajo en ese proceso necesitaremos la ecuación de estado y el calor específico. Los 2 Tres variables para el caso de un fluído. Otros modelos (por ejemplo medios elásticos) necesitan de más variables independientes. 8 cambios de variable permiten simplificar una parte de la descripción, casi siempre complicando otra. 10.2- La energía interna como potencial: el plano V-S Elijamos como variables independientes el volumen V, la entropía S y el número de moles n. Podemos entonces escribir la energía interna como función de la entropía, el volumen y el número de moles, U(V,S,n). Ahora supongamos que variamos infinitesimalmente las distintas variables y queremos ver cómo cambia U: dU= ∂U ∂V S,n dV+ ∂∂US dS+ V,n ∂U∂n V,S dn (45) Ahora bien, usando el primer principio: dU=−PdV+TdS+μdn (46) Comparando (45) con (46) tenemos: P=− ∂∂VU S,n T= μ= ∂U ∂S V,n ∂∂Un (47) V,S Es decir que si U es escrita como función de el volumen, la entropía y el número de moles, tres cantidades importantes de la teoría pueden obtenerse derivando U. Por eso se dice que, en este caso, U es un “potencial” (recuerden que calculando derivadas de la energía potencial puede calcularse la correspondiente fuerza conservativa). La notación extravagante en (47) merece comentarios. Las dos variables que se ponen como subíndice afuera del paréntesis son las cantidades que permanecen constantes al derivar. Por ejemplo, la primer expresión dice que la presión es menos la derivada de U respecto de V, a S y n constantes. La aclaración es relevante en termodinámica: si uno dijera simplemente que la presión es menos la derivada de U respecto de V, sin aclaraciones, se estaría siendo ambiguo. Supongamos por ejemplo que expresamos U como función de P, V y n. Entonces, la derivada de esa función respecto de V ya no es -P (si intentan hacer algo parecido a la expresión (46) lo van a ver). La razón práctica para expresarlo de esta manera en lugar de simplemente aclarar que (47) es válido únicamente cuando U es escrita en función de S, V y n es que casi nunca uno tiene una función explícita. Los razonamientos son formales, y en un punto uno usa el hecho de que S es constante, por ejemplo. Esta forma de trabajar resulta muy poderosa. Siguiendo esta línea uno encuentra relaciones entre 9 variables medibles que uno no hubiera sospechado como interdependientes. De esa manera se logra una gran cantidad de predicciones a partir de una mínima cantidad de mediciones, siguiendo razonamientos completamente independientes de los detalles microscópicos del sistema estudiado. 10.3- Potenciales termodinámicos y transformaciones de Legendre Esta subsección es sobre la herramienta matemática detrás de la construcción de los potenciales termodinámicos aparte de U. Su lectura puede omitirse sin pérdida de continuidad. La forma de trabajo con potenciales en termodinámica resulta tan poderosa que uno se pregunta si no hay una manera sistemática de generarlos. Por ejemplo: ya sabemos que U escrita como función de V, S y n funciona como potencial para P, T y μ. No podrá encontrarse otra función, en otras variables, que funcione como potencial para, por ejemplo, V? Estamos pidiendo una nueva función que nos permita obtener la misma información que la nueva U, pero que resulte más útil que U en otros contextos. Supongamos que tenemos la función f(x,y), que funciona como potencial de u y v; es decir: u= v= ∂∂ xf y ∂∂ fy x (48) (49) Ahora queremos hallar una función g que sea un potencial para la variable x. Si hacemos g u,y =f u,y –ux u,y (50) esa es exactamente la función que necesitábamos, es decir un potencial para x: ∂∂ gu = ∂∂ uf –x u,y –u ∂∂ xu y y y (51) y como por regla de la cadena ∂f ∂f ∂x ∂f ∂y = (52) ∂ u y ∂ x y ∂u y ∂ y y ∂ u y Usando (48) y el hecho de que la derivada de y a y constante es cero, (52) se escribe: ∂f ∂x =u (53) ∂u y ∂u y Reemplazando (53) en (51) se obtiene finalmente ∂∂ gu =x y o sea, g es un potencial para x. 10 (54) Sintetizando: Si se tiene una función f (de x y otras variables) que es potencial para la variable u, la función g=f −ux es un potencial para x si se la escribe como función de u. La función g se denomina la transformada de Legendre de la función f. Observemos: • • La función f y su transformada g tienen “la misma información”, en el sentido de que ambas permiten conocer tanto x como u, o bien por ser la variable independiente o bien derivando. En un sistema termodinámico el “potencial” es parte de la modelización del sistema. Por ejemplo, si la función es la energía potencial U en términos del volumen y la entropía, entonces dado un valor del volumen y la entropía (un estado) podemos conocer la presión del sistema para ese estado. Si armamos una transformada de Legendre de U que sea un potencial para V, nuevamente esa función será una caracterización termodinámica del sistema tan buena como U. 10.4- El plano P-S y la Entalpía (H) Ya vimos cómo U funciona como potencial cuando se la escribe como función de el volumen, la entropía y el número de moles. Veamos ahora un potencial muy popular, que puede obtenerse a partir de la energía interna para que sea un potencial del volumen. La forma en que se hace eso es la llamada transformada de Legendre, que en esta subsección vamos a abordar como es habitual en los libros de texto. Ante insatisfacciones o deseos de profundizar, puede acudirse a la subsección anterior. Definamos sin anestesia la función H: H=U+PV (DEFINICIÓN DE ENTALPÍA) Si U se escribe como función de S, V y n, puede obtenerse P como función de S, V y n (derivando U) y entonces H es función de S, V y n. Pero al igual que toda función de estado, H puede ponerse en función de las tres variables independientes de nuestra preferencia. Hagamos ahora un cambio infinitesimal de H: dH=dU+PdV+VdP (55) Usando el primer principio, dU=− PdV+TdS+μdn , con lo que (55) queda: dH=VdP+TdS+μdn 11 (56) Supongamos ahora que escribimos H como función de la presión, la entropía y el número de moles. dH es entonces: dH= ∂∂ HP dP+ S,n ∂∂HS dS+ P,n ∂∂Hn P,S dn (57) Comparando (56) y (57) vemos que ∂∂ HP ∂H T= ∂S ∂H μ= ∂n V= S,n P,n (58) P,S Cuando H es expresado como función de la presión, la entropía y el número de moles, funciona como potencial para el volumen, la temperatura y el potencial químico. Hay dos preguntas que siempre se hace quien se topa por primera vez con la entalpía: “qué es” y “para qué sirve”. La primer pregunta no es realmente legítima. Uno tiende a sentirse más tranquilo con algo que se llama “energía” porque siente que le puede atribuir un significado, mientras que la palabra “entalpía” es intimidante. Pero en el fondo las dos son igual de “raras” e “inventadas”. La energía interna es una función de estado y la entalpía otra. Más aún, la entalpía es la energía interna más presión por volumen, y eso es todo. La segunda pregunta es la realmente relevante y la que le da sentido a la entalpía. En principio uno podría decir simplemente que sirve para poder expresar (58), pero hay más. Si se fijan en la expresión para dH van a ver que en procesos a presión constante y número de moles constante el cambio de la entalpía es igual al calor sobre el sistema, si este se hace cuasiestáticamente. Como casi toda la tarea del químico (y muchos otros procesos) se hace a presión atmosférica, los cambios de entalpía resultan particularmente útiles y fáciles de medir. La entalpía funciona como potencial en el plano P-S. Es decir, al considerársela como función de P, S y n. H P,S,n 10.5- El plano T-V: Función de Helmholtz (F) De un modo similar puede pensarse en escribir un potencial en las variables V, T y n, para que funcionen como potenciales de P, S y µ. Eso puede hacerse definiendo el potencial de Helmholtz (a veces se lo llama “energía libre de Helmholtz”) F: F=U −TS (DEFINICIÓN DE LA FUNCIÓN DE HELMHOLTZ) Procediendo análogamente a como hicimos con la entalpía llegamos a: 12 dF=− PdV − SdT+μdn (59) por lo que si pensamos F como función del volumen, la entropía y el número de moles llegaremos a: ∂∂VF ∂F S=− ∂T ∂F μ= ∂n P=− T,n V,n (60) T,V A volumen y temperatura fijos la variación de F es igual al trabajo químico cuasiestático sobre el sistema. En cuanto a la pregunta de qué “es” F nos remitimos a la pregunta análoga sobre la entalpía. 10.6- El plano T-P: Función de Gibbs (G) Análogamente puede pasarse al plano T-P definiendo la función de Gibbs (o el potencial de Gibbs, o la “energía libre” de Gibbs) G así: G=U+PV–TS (DEFINICIÓN DE LA FUNCIÓN DE GIBBS) Procediendo análogamente a como hicimos con H y F llegamos a: dG=VdP−SdT+μdn (61) por lo que si pensamos G como función de la presión, la entropía y el número de moles llegaremos a: ∂∂ GP ∂G S=− ∂T ∂F μ= ∂n V= T,n P,n (62) P,V A presión y temperatura fijos la variación de G es igual al trabajo químico cuasiestático sobre el sistema. Esto es interesante porque presión constante (atmosférica) y temperatura constante (ambiente) son condiciones habituales para muchos sistemas de interes. Junto con la entalpía es una de las funciones más usadas. En cuanto a la pregunta de qué “es” G nos remitimos nuevamente a la pregunta análoga sobre la entalpía. 13 10.7- El plano U-V y la entropía Usando el Primer Principio puede pasarse al plano U-V usando la función Entropía. Para eso despejamos dS: dS= 1 P μ dU+ dV − dn T T T (FORMULACIÓN ENTRÓPICA DEL PRIMER PRINCIPIO) Acá no estamos definiendo un potencial nuevo por medio de una transforamada de Legendre, sino aprovechando el primer principio para escribir como potencial una que ya conocíamos (la entropía). Si ahora consideramos a la entropía en función de U, V y n (S(U,V,n)) y razonando análogamente como para el resto de los potenciales, obtendremos las relaciones: Procediendo análogamente a como hicimos con H y F llegamos a: ∂U∂ S ∂V∂ S ∂∂ Sn 1 = T P = T μ − T V,n U,n (63) U,V 10.8- Relaciones de Maxwell Tenemos ahora a nuestra disposición montones de relaciones entre variables involucrando derivadas. Por otra parte, rara vez uno scribe explícitamente los potenciales termodinámicos como funciones de las variables de estado, salvo para los modelos más sencillos. De hecho, lo habitual es usar tablas de cambio de entalpía por mol y de “energía libre” de Gibbs por mol (sobre todo en química) así que alguno puede estarse preguntando qué tiene que ver todo este barullo de derivadas con el uso habitual de U, G, S y H. Un ejemplo concreto lo damos más abajo: el uso de G para decidir si cierta reacción es o no espontanea. En esta subsección vamos a aclarar un poco lo que dijimos al empezar el estudio de potenciales termodinámicos: uno raramente deriva explícitamente funciones, solo hace cálculos formales que le permiten conectar entre sí cantidades medibles en principio independientes. Este tipo de relaciones (llamadas “de Maxwell” en honor a quien las explotó por primera vez) aprovecha el hecho de que ciertas cantidades son a su vez derivada de otras, y usando propiedades generales de las derivadas va estableciendo las conexiones. Acá va un ejemplo: ∂T∂V = ∂V∂ ∂U∂ S S,n ∂ ∂ ∂ ∂U ∂P U S,V,n = =− ∂V ∂ S ∂ S ∂V ∂S S,n V,n V,n S,n (EJEMPLO DE RELACIÓN DE MAXWELL) = 14 V,n De la misma manera se pueden demostrar muchísimas identidades, simplemente reorganizando la forma de escribir las cosas y usando las propiedades de las derivadas parciales. Como prácticamente todas las cantidades de interés pueden escribirse como derivadas de otras, pueden establecerse relaciones no triviales. Mostremos un ejemplo concreto: la relación entre calores específicos en un fluído genérico (ejemplo tomado del “Modern Thermodynamics” de Kondepudi y Prigogine, reproducido sin autorización del editor). Vamos a relacionar entre sí cuatro parámetros medibles de un fluído: Calor específico a volumen constante (definido en la sección 8.1). De su definición, observando que a volumen constante el trabajo es nulo, y usando el Primer Principio, obtenemos: dU V,n =nC V dT V,n por lo que CV = 1 ∂U n ∂T V,n ( C V EXPRESADO COMO DERIVADA) Calor específico a presión constante (definido en la sección 8.1): De su definición, y recordando que a presión constante el calor es el cambio de entalpía, resulta: dH P,n =nC P dT P,n por lo que C P= 1 ∂H n ∂T P.n ( C P EXPRESADO COMO DERIVADA) Coeficiente de expansión a presión constante: Si a un material (sometido a presión constante, como por ejemplo la atmosférica) se le aumenta su temperatura en una cantidad (pequeña) dT, su volumen cambiará en dV. Ese cambio de volumen es proporcional tanto al volumen de material como al cambio de temperatura: dV P,n =αV dT P,n (64) donde α es el coeficiente de proporcionalidad. Por lo tanto: α= 1 ∂V V ∂T P,n (DEFINICIÓN DEL COEFICIENTE DE DILATACIÓN TÉRMICA) Compresibilidad a temperatura constante: Si a un material (sometido a temperatura constante, por ejemplo la ambiente) se lo somete a un cambio (pequeño) de presión dP, su volumen cambiará en dV. Ese cambio de volumen es proporcional tanto al volumen del material como al cambio de presión: dV T,n=−κ T V dP T,n (65) El signo “-” se debe a que queremos definir a κ T positivo, mientras que un aumento en la presión provoca una disminución en el volumen. De (65) se deduce: 15 κ T =− 1 ∂V V ∂P T,n (DEFINICIÓN DEL COEFICIENTE DE COMPRESIBILIDAD) Observen que los cuatro parámetros son cantidades medibles independientemente. Nos las hemos ingeniado para expresarlos como derivadas de variables de estado. Entonces, usando “relaciones de Maxwell” al estilo del ejemplo de arriba puede demostrarse (el cálculo es engorroso y está en el libro citado) que: 2 C P –C V = Tα V κT n (66) Los detalles de la cuenta no nos interesa. De hecho se necesita mucha práctica y desenvoltura en termodinámica para usar este tipo de relaciones. Lo que sí nos interesa señalar es que uno usa las propiedades sin en ningún momento derivar realmente alguna función concreta, y llega a relaciones entre cantidades medibles al estilo de (66) que uno jamás hubiera adivinado: los calores específicos, la compresibilidad y la dilatación térmica de un fluído están interrelacionados en cualquier fluído, sin importar su composición química ni ningún otro detalle. Basta el primer principio de la termodinámica para demostrarlo. 10.9- Los potenciales y el Segundo Principio Observen que en todo nuestro trabajo sobre potenciales hasta ahora solo hemos usado el primer principio. Varias conclusiones interesantes se alcanzan cuando el segundo entra en juego. En general, bajo distintas condiciones de las fronteras el estado de equilibrio será un extremo (mínimo o máximo) de algún potencial termodinámico. Mínimo de la energía mecánica Si una pelotita rueda por un paisaje escarpado (ver figura más adelante), la experiencia cotidiana nos indica que esta terminará quieta, en el fondo de algún valle. Muchas veces se afirma en libros que lo que ocurre es que “el sistema alcanza el estado de mínima energía”. Veamos si la termodinámica permite acomodar este tipo de fenómenos y justificar semejante expresión. Antes que nada notemos que la expresión “el sistema alcanza el estado de mínima energía” es demasiado ambigua, y en ciertos contextos puede ser completamente falsa. Lo que vamos a hacer a continuación es mostrar que, si la energía interna del sistema de estudio (en este caso, la pelotita) es constante, el sistema de estudio no hace trabajo (ni mecánico ni termodinámico) sobre el entorno y tanto el sistema de estudio como el entorno permanecen a una temperatura homogenea, entonces la energía mecánica del sistema de estudio alcanza un mínimo. En el caso de la pelotita, esta alcanzará su mínima energía cuando su energía cinética sea nula (pelotita quieta) y cuando la energía potencial gravitatoria sea mínima (en el fondo de un valle). 16 Veamos: si las fuerzas no conservativas hicieran trabajo nulo, el sistema no iría a un estado de mínima energía mecánica: la energía mecánica se conservaría. Claro que es una situación extremadamente idealizada: un mínimo trabajo de fuerzas no conservativas es esperable. En dinámica no hay restricción para los posibles valores de ese trabajo, pero usando el primer principio con las condiciones enunciadas (sólo pueden cambiar la energía mecánica del sistema de estudio y la energía interna del entorno) el trabajo de las fuerzas no conservativas termina siendo igual a menos la variación de energía interna: ΔU ent=−ΔE M (conservación de la energía) ΔE M =LFNC (teorema del trabajo y la energía) Por lo tanto: ΔU=− L FNC (67) Como no hay trabajo sobre el entorno, el cambio de U en (67) es igual al calor, que es igual a TΔS ent . La entropía del sistema de estudio no cambia, por lo que el cambio de la entropía del entorno es la del Universo; por el segundo principio esa cantidad es positiva. Entonces: L FNC =−Q=−TΔS<0 (68) Vemos entonces que los principios de la Termodinámica usados bajo estas condiciones implican que la energía mecánica sólo puede disminuir. El estado de equilibrio es el de mínima energía potencial. Algo interesante que ilustramos en la figura es que si bien hay un estado de equilibrio “absoluto”, puede también haber mínimos relativos que también son estables, como el “valle alto” a la izquierda del mínimo. A veces, dependiendo de los detalles de cómo evoluciona el sistema hacia el equilibrio, se alcanza un estado “metaestable” que, sin ser el equilibrio teórico, es estable. Algo similar ocurre para otros sistemas con otros potenciales (ver más abajo). Máximo de la Entropía Un extremo que surge inmediatamente del segundo principio es el de la entropía: si el sistema estudiado está térmicamente aislado, entonces el cambio de entropía del sistema será igual al cambio del Universo. Por lo tanto el estado de equilibrio será el máximo valor posible para la entropía. Los posibles valores de la Entropía estarán dados por las condiciones del problema. Supongamos, por ejemplo, que tenemos dos gases separados por un tabique rígido y adiabático. En esas condiciones cada uno de esos dos gases puede estar en un estado de equilibrio a temperaturas y presiones diferentes. Cada gas tiene una función entropía, y la entropía total es la suma de ambas. Si en un momento permitimos al tabique moverse libremente, pero mantenemos la aislación térmica, el gas a mayor presión se va a expandir y el gas a menor presión se va a contraer, pero no habrá interacciones térmicas 17 entre ellos. Uno espera que entonces el estado de equilibrio sea uno en el que las presiones se igualen, pero no necesariamente las temperaturas. Si en vez de permitir que el tabique se mueva, pero retiramos la aislación térmica, esperamos que el gas más caliente se enfríe y el más frío se caliente hasta que las temperaturas se equilibren, pero no necesariamente las presiones. Finalmente, si se retira tanto la aislación térmica como la rigidez, el sistema evolucionará a un estado de equilibrio tanto térmico como de presiones. ¿Cómo puede hallarse ese estado de equilibrio usando el segundo principio? Bueno, uno puede plantear la función entropía sujeta a los vínculos del problema. Entonces, dado que el sistema está térmicamente aislado, el estado de equilibrio será el correspondiente a la máxima entropía compatible con esos vínculos. De ese cálculo surgirá no solo que se igualan las presiones, las temperaturas o ambas, sino en qué valores concretos. Si se parte de una situación alejada del equilibrio (por ejemplo, dentro del recipiente hay un gradiente de presión), siempre que se parta de una situación de equilibrio local (ver la sección sobre termodinámica más allá del equilibrio) se puede usar exactamente el mismo principio. Se necesita sin embargo una fuerte sofisticación matemática, porque ahora en vez de dos subsistemas de equilibrio aparecen infinitos (cada elemento de volumen), pero aunque los cálculos sean mucho más complejos el principio es exactamente el mismo. Mínimo de la Función de Gibbs Vamos a analizar un caso de gran importancia práctica: el caso en que el entorno del sistema de estudio puede considerarse a temperatura y presión constantes. Como ya hemos dicho en varias oportunidades ese es un caso relevante porque es la situación habitual en el laboratorio y en la vida (temperatura ambiente y presión atmosférica). En este caso el sistema de estudio puede hacer tanto trabajo como calor, así que la energía y la entropía del entorno pueden cambiar. Observemos que la función de Gibbs del sistema de estudio es: G sist =U sist +PV sist –TS=H sist −TS sist (69) siendo T la temperatura tanto del sistema de estudio como del entorno. Si la temperatura es constante, el cambio de G será: ΔG sist =ΔH sist –TΔS sist (70) Si queremos conocer la condición para que el proceso sea espontaneo debemos estudiar, aparte del cambio de entropía del sistema, el del entorno. A temperatura constante, el cambio de entropía del entorno por la temperatura es igual al calor cuasiestático que el sistema hace sobre el entorno. Como el proceso ocurre a presión constante, el cambio de entalpía del sistema es igual al calor cuasiestático que el entorno hace sobre el sistema. El proceso real seguramente no es cuasiestático, pero en la medida que estudiemos cambios de estado no importa qué procesos usemos, solo los estados inicial y final, así que lo mejor es usar un proceso que sepamos calcular. Entonces, el cambio de entropía del entorno es: ΔS ent = Q ent T =− Q sist T 18 =− ΔH sist T (71) de donde ΔH sist =−TΔS ent (72) Reemplazando (72) en (70): ΔG sist =−T ΔS ent +ΔS sist (73) Usando el segundo principio en (73) obtenemos finalmente: ΔG sist 0 PARA PROCESOS ESPONTANEOS A P y T CONSTANTES Y TRABAJO QUÍMICO NULO Esto significa que para sistemas cuyo entorno no hace trabajo químico y se encuentra a presión y temperatura constantes la función de Gibbs alcanza un mínimo. A veces el mínimo alcanzado no es el mínimo absoluto sino uno local, como en el ejemplo de la pelotita al comienzo de esta sección. Eso es lo que se llama un estado metaestable. Un buen ejemplo de ese fenómeno es el agua subenfriada: a veces puede ponerse agua en el freezer que sigue líquida por debajo de cero grados. Si eso ocurre, al agarrar la botella y darle un golpe se observa una formación súbita de cristales de hielo. El golpe obligó al sistema a pasar al auténtico mínimo de G. 10.10- Ejemplo de uso: H y G en química. Vamos a repasar ahora el uso de la entalpía y la función de Gibbs en el estudio de las reacciones químicas. Empecemos por aclarar qué son las “tablas de entalpía molar” y de “energía libre molar” que se usan en química. Lo que se hace es asignar arbitrariamente entalpía cero y función de Gibbs cero a las sustancias formadas únicamente por un elemento. Esto puede hacerse porque lo único relevante son los cambios. Así, por ejemplo, al oxígeno gaseoso y al hidrógeno gaseoso en condiciones de laboratorio se les asigna Hm=0, Gm=0 (el subíndice “m” indica “molar”, es decir por cada mol). Para calcular la entalpía de un mol de agua se mide el calor cuasiestático necesario para hacer reaccionar el oxígeno y el hidrógeno que forman un mol de agua a presión y temperatura constantes. Por eso a esa cantidad se le llama “calor de formación” o “entalpía de formación”. Lo más habitual es que las reacciones no puedan realmente hacerse a presión y temperatura constantes, por lo que se debe usar algún otro procedimiento, relacionando una serie de medidas con el cambio de entalpía usando la teoría termodinámica (por ejemplo, por medio de relaciones de Maxwell). Lo mismo puede hacerse para calcular funciones molares de Gibbs, y así podemos atribuir a cada sustancia un valor de H y de G. Supongamos ahora que nos dan una ecuación química balanceada correspondiente a cierta reacción, como por ejemplo: 2 CO+O 2 2CO 2 (74) ¿Cómo podemos calcular el cambio de H o de G en esta reacción? Por empezar, veamos que la entalpía de los reactivos es (recuerden que al oxígeno le atribuimos entalpía cero): 19 mientras que la de los productos será 2H m,CO (75) 2H m,CO (76) 2 (los factores “2” en (75) y (76) se deben a que estamos considerando dos moles de sustancia). Para calcular, entonces, el cambio de entalpía, imaginemos que primero disociamos los reactivos en sus componentes (el cambio de entalpía es entonces −H m por cada mol de reactivo) y luego recombinamos esos elementos en los productos (el cambio en ese paso será H m por cada mol de producto). El cambio entonces al ir de los reactivos a los productos es la suma de ambos cambios, o sea la entalpía de formación de los productos menos la entalpía de formación de los reactivos. Del mismo modo con cualquier función de estado M. Si tenemos una ecuación balanceada n R1 R1 +n R2 R2 n P1 P 1 +nP2 P 2 . .. (77) el cambio de la función M será ΔM=n P1 M m,P +n P2 M m,P −n R1 M m,R −n R2 M m,R −.. . (78) 1 2 1 2 La tabla de formación suele estar dada a cierta temperatura, presión y fase. Si los reactivos o productos están en otra situación, deberá incluírse en el cálculo la variación necesaria para llevar cada sustancia a las condiciones tabuladas. Pero para no meternos con sutilezas que distraigan de la esencia, vamos a considerar solamente reacciones que ocurren para reactivos y productos en la situación registrada en tablas. De acuerdo a lo que sabemos, a presión constante el cambio de entalpía del sistema de estudio es igual al calor que le hace el entorno, y por lo tanto es igual a menos el calor que el sistema hace sobre el entorno. Por eso, si en esas condiciones una reacción es tal que el cambio de entalpía es negativo, el sistema hará calor positivo contra el entorno (sentiremos que el tubo de ensayo se calienta). Por esa razon tales reacciones se llaman “exotérmicas”. Si el cambio de entalpía es positivo, la reacción se llama “endotérmica”3. Otro uso importante de los potenciales de estado en química está vinculado a la predicción del caracter espontaneo o no de la reacción estudiada. En la subsección anterior mostramos que en un sistema a temperatura constante los procesos espontaneos son aquellos para los que la “energía libre” de Gibbs del sistema disminuye. Esto crea la falsa ilusión de que uno está pudiendo prescindir del entorno en la decisión de si el proceso es o no espontaneo. Es falsa justamente porque este criterio es válido sólo si el entorno se mantiene en ciertas condiciones (presión y temperatura constantes). Si repasan la forma en que se llegó a que G disminuye verán que se usa el hecho de que la entropía del sistema más la del entorno aumentan. Esto lleva también a malentendidos respecto del significado de espontaneidad (vean la subsección 9.1). A veces una reacción para la que G aumenta puede ocurrir (por ejemplo calentando el tubo de ensayo, sometiendo a presión al sistema, etc). Esto no significa que ocurre una reacción “no espontanea” (cosa que por definición no puede pasar) sino que el criterio de disminución de G ya no es un buen criterio de espontaneidad, porque el entorno ha cambiado y no es más a T o P constante. 3 En un modelo en el que el calor se considera una sustancia esto se lee como que si la entalpía crece el entorno tiene que “darle” calor al sistema, mientras que si la entalpía disminuye es porque parte del calor va del sistema al entorno. Este lenguaje es habitual en los textos de química. 20 10.11- Ejemplo de uso: “Motores Químicos” (pilas, fotosíntesis, etc) Vamos a hablar ahora de una aplicación relevante para estudiar sistemas vivos (¡Al fin!). Recordemos que cuando usamos en química el criterio con el cambio de G estamos pensando en una reacción “de tubo de ensayo”, en el que el objeto de estudio es toda la mezcla dentro del tubo. Si nuestro objeto de estudio hubiera sido una sola sustancia hubiera habido trabajo químico sobre él, pero sobre el contenido del tubo el entorno no hace trabajo químico. Esto no ocurre siempre así: para muchos sistemas interesantes el entorno hace un trabajo químico sobre el sistema mientras la reacción progresa. Un ejemplo paradigmático son las pilas y las baterías: mientras ocurre una reacción de óxido-reducción la batería hace trabajo positivo contra el entorno. Durante un proceso de fotosíntesis, la radiación solar hace un trabajo químico imprescindible sobre el sistema (agua y dióxido yendo a hidratos de carbono). Esta es la razón histórica por la que a la función de Gibbs se le dio el nombre de “energía libre”: el cambio de G se interpretó como “el máximo trabajo químico que un sistema puede hacer sobre el entorno a presión y temperatura constante”. Veamos qué significa esto. Para verlo, recordemos que el cambio de G a T constante es: ΔG sist =ΔH sist +TΔS sist +W quim (79) Repitiendo los razonamientos ondujeron a (73) obtenemos: ΔG sist =−T ΔS ent +ΔS sist +W quim (80) Entonces se cumple: TΔS U =W quim− ΔG sist (81) Usando el segundo principio tenemos entonces: W quim−ΔG sist 0 PARA PROCESOS ESPONTANEOS A P y T CONSTANTES Este es probablemente el resultado más importante de la termodinámica en cuanto a sus implicaciones para sistemas vivos. Vean: si queremos que una reacción química en la que el cambio de G es positivo sea espontanea necesitamos un trabajo químico del entorno sobre el sistema más grande que el cambio de G. Por ejemplo, si queremos transformar dióxido de carbono y agua en glucosa y oxígeno (sistema de estudio) necesitaremos hacer un enorme trabajo químico por mol. Esto es precisamente lo que ocurre en la fotosíntesis: la radiación solar hace ese trabajo. Después un animal (o la propia planta) puede “quemar” esa glucosa con el oxígeno produciendo dióxido de carbono y agua; si lo hace con suficiente cuidado puede hacerlo de modo que ese sistema haga sobre el entorno un trabajo positivo (el entorno hace sobre el sistema un trabajo negativo). Ese trabajo químico puede “usarse” para hacer el trabajo químico para sintetizar todas las proteínas y el ATP que hacen funcionar nuestros cuerpos. 21 11- Termodinámica más allá del equilibrio 11.1- ¿Qué es la termodinámica de no equilibrio? Como en la práctica nada está en equilibrio, una respuesta facilista es que “todo” es termodinámica de no equilibrio. De tan simplista, esa respuesta es perfectamente falsa. La verdad es que varios aspectos vinculados a procesos de no equilibrio entre estados de equilibrio (aproximado) pueden estudiarse con termodinámica de equilibrio (estados de equilibrio y procesos cuasiestáticos), mientras que muchos fenómenos son demasiado complejos para ser abordados por la termodinámica. En concreto, la “termodinámica de no equilibrio” es un marco teórico aún en construcción 4 en el que se asume la validez de los dos principios pero en el que los “estados” ya no son de equilibrio interno. El aspecto realmente complejo es la caracterización de esos estados y la construcción de funciones de estado, que ya no pueden ser funciones de un número pequeño de variables. La única condición para seguir considerándosela “termodinámica” es que todas las variables involucradas sigan siendo macroscópicas: la temperatura, la densidad de entropía o un gradiente en un “punto” son cantidades que involucran porciones de materia que aún pueden considerarse contínuas (un milímetro cúbico de aire contiene unas 1019 moléculas). Entonces tenemos, como variables en la termodinámica de no equilibrio, todas las viejas variables intensivas (presión, temperatura, potencial químico, calor específico...) más versiones “locales” de las extensivas (energía interna por unidad de volumen, entropía por unidad de volumen, etc), todas ellas funciones de la posición y del tiempo. Y aparte aparecen variables nuevas, que en la termodinámica de equilibrio no hubieran tenido cabida, como “conductividades térmicas”, “coeficientes de difusión” y otras que vinculan entre sí distintos gradientes. Y aparece una cantidad fundamental, vinculada directamente con el segundo principio: la producción de entropía por unidad de volumen y de tiempo. Algunos problemas que no pueden estudiarse con termodinámica de equilibrio son: • Estados estacionarios en sistemas abiertos. Por ejemplo: el perfil de temperaturas en una barra cuyos extremos se mantienen a temperaturas diferentes. En estas situaciones uno no va de un estado inicial de equilibrio a otro final de equilibrio sino que busca un cierto estado de noequilibrio. • Detalles de la dinámica al pasar de un estado de equilibrio a otro. Por ejemplo, en la expansión libre de un gas uno puede estudiar el cambio de U o de S con termodinámica de equilibrio (porque es el mismo que para procesos cuasiestáticos) pero no hay manera de saber cuánto tarda ni cómo son los gradientes en los instantes intermedios. Estos últimos son “estados de no equilibrio”. Volvamos a nuestro ejemplo de la fotosíntesis: usando termodinámica de equilibrio sabemos que el trabajo químico que la planta debe hacer sobre el sistema de estudio (el agua y el dióxido de carbono que pasan a ser carbohidratos) es mayor que el cambio de G para pasar de dióxido de carbono y agua a carbohidratos. ¿Cuánto mayor? Si queremos detalles tendremos que estudiar un objeto bastante más complicado que las meras sustancias y modelizarlo más allá del equilibrio. Tal vez 4 “Un marco teórico en construcción” es lo mismo que decir que no hay un marco teórico, sino un montón de ideas más o menos desordenadas y la esperanza de que algún día se va a poder construír ese marco. 22 termodinámicamente, tal vez microscópicamente, tal vez lo que salga. Lo cierto es que el problema es entonces mucho más complejo. 11.2- Equilibrio local Este es el punto donde queda claro por qué el estudio de los estados de equilibrio es tan importante en la termodinámica: muchos sistemas que no están en equilibrio pueden pensarse como sistemas inhomogeneos en los que cada elemento de volumen es un estado de equilibrio interno. Esto implica que: • cada elemento de volumen tiene una temperatura, una presión y una densidad bien definidos y medibles en todo instante de tiempo • La entropía de cada elemento de volumen es función de esas variables, la misma función que para los estados de equilibrio. Esto resulta ser una excelente aproximación en muchísimas situaciones, porque es mucho más rápida la llegada al equilibrio en escalas pequeñas que grandes. ¿Qué sucede cuando no hay ni siquiera equilibrio local? Sobre esto no hay acuerdo claro. Hay quienes piensan que no tiene sentido asignar una entropía a estados que no sean de equilibrio. Según esta visión, los casos en que no hay equilibrio local quedan fuera del alcance de la termodinámica. Otros generalizan la definición de entropía, haciéndola depender también de gradientes. El hecho es que no existe una teoría termodinámica sólida más allá del equilibrio local (aunque en rigor más acá tampoco es del todo sólida). Por suerte, muchos problemas interesantes (incluso algunos muy alejados del equilibrio global) pueden ser atacados satisfactoriamente bajo la hipótesis de equilibrio local. 11.3- Creación de entropía Un ejemplo Supongamos que ponemos una barra, digamos, metálica, uno de cuyos extremos está en contacto con un sistema a temperatura T1, y el otro está en contacto con un sistema a temperatura menor T2. Supongamos que esos sistemas son enormes y pueden mantenerse a esas temperaturas cuando pasa el tiempo. Después de un rato, uno espera que se alcance una situación estacionaria, en la que se desarrolla un gradiente de temperaturas a lo largo de la barra que no cambia en el tiempo. Observemos que la situación estacionaria no es de equilibrio: hay un proceso de calor negativo sobre el sistema a temperatura T1 (cuya energía interna va disminuyendo) y uno de calor positivo sobre el sistema a temperatura T2 (cuya energía interna va aumentando). Suponiendo que cada elemento de volumen sobre la barra es un estado de equilibrio interno (o sea, hay equilibrio local) y como la situación es estacionaria, la barra no cambia de volumen (el trabajo termodinámico sobre la barra es nulo) y su energía interna permanece constante, el primer principio implica que el calor que el 23 sistema “caliente” le hace a la barra por segundo es igual al calor por segundo que la barra le hace al sistema “frío” (a modo de ejercicio, verifíquenlo). Esto se puede interpretar como se hace habitualmente en los libros como un “flujo de calor” atravesando la barra, y es por eso que se usa la expresión “conducción del calor”5. Veamos qué pasa si intentamos hacer algo similar con la entropía: igual que antes, debido a que la situación es estacionaria y de equilibrio local, la entropía total de la barra no cambia en el tiempo. En el extremo caliente de la barra “entra” por segundo una cantidad de entropía que es el calor por segundo q dividido la temperatura “caliente”; en el extremo frío, “sale” de la barra una entropía por segundo igual q q a q dividido la temperatura menor. Pero como T 2 <T 1 resulta T T . En síntesis: por el lado frío 2 1 “sale” más entropía que la que “entra” por el lado caliente, pero la entropía “dentro” de la barra permanece constante. Eso, que parece tan extraño, significa simplemente que la entropía no se conserva, y muestra los problemas en los que se puede caer por porfiar en pensar en las funciones de estado extensivas como fluídos. De hecho lo que ocurre es casi una obviedad: estamos hablando de un proceso espontaneo, de modo que la entropía del Universo debe estar aumentando. Lo que pasa es que antes comparábamos “fotos” (estados de equilibrio inicial y final) mientras que ahora el sistema permanece en un estado estacionario, así que debe estar “produciendo” entropía todo el tiempo. Veamos con más detalle lo que está pasando en cada punto de la barra: en esencia hay creación de entropía porque hay un gradiente de temperatura. Por cualquier sección de la barra en cada segundo la parte derecha hace sobre la izquierda un calor q, y la izquierda sobre la derecha un calor -q. Introduzcamos un sistema de coordenadas horizontal sobre la barra, llamémosle x. La sección ubicada en x está a temperatura T(x); la parte derecha tiene un aumento de la entropía de q/T(x) cada segundo. Consideremos una rodaja de espesor Δx , la entropía creada por segundo por esa rodaja en el entorno será ΔS ent =− q q T x T x+Δx (82) En este caso, dado que no hay trabajo, podemos pensar a q como un flujo de energía interna por segundo a traves de la sección. Supongamos que la sección tiene área A. Si llamamos ju al flujo de energía interna por unidad de tiempo y de area, q=j u AΔt , (82) puede reescribirse como: ΔS ent =j u AΔtΔ T1 (83) Como además 1 T 1 d 1 Δ = Δx ~ Δx T Δx dx T Δ podemos reescribir (83) como 5 d 1 dx T ΔS ent =j u AΔxΔt (84) (85) Las “leyes de la conducción del calor” las estableció Fourier hacia 1820, pleno auge de la teoría del calórico. 24 y como AΔx=δV es el volumen (infinitesimal) de la rodaja, (85) puede expresarse como: ΔS ent d 1 =j δVΔT u dx T (86) La cantidad sobre el miembro izquierdo es la producción de entropía del entorno por unidad de tiempo y de volumen, a la que se le suele llamar σ σ=j u d 1 dx T (PRODUCCIÓN DE ENTROPÍA POR FLUJO DE ENERGÍA) Observen que σ no es el cambio de entropía del elemento de volumen (en situación estacionaria esa entropía no varía) sino la producción de entropía en el entorno debida a ese volumen. Una cosa remarcable es que ese entorno no es necesariamente el entorno inmediato. Para la barra analizada, por ejemplo, las dos rodajas adyacentes a la estudiada también se encuentran en estados estacionarios, de modo que sus entropías tampoco varían, y sí están “produciendo entropía en el entorno”. En este caso las entropías varían para los focos caliente y frío entre los que está la barra, que no pueden estar en un estado estacionario. Es interesante observar que no puede suponerse que todo el Universo está en estado estacionario: la entropía de alguna parte está aumentando. Si aplicamos el segundo principio de la termodinámica a cada elemento de volumen, y si llamamos s a la “densidad de entropía” (entropía por unidad de volumen) a lo que llegamos es a que en cada punto se debe tener (¡verifiquen!) σ+ ∂s 0 EN PROCESOS ESPONTANEOS ∂t En procesos estacionarios la densidad de entropía no cambia con el tiempo, con lo que σ es positiva, y por lo tanto la energía interna fluye en la dirección del gradiente de 1/T. Vamos a ver a continuación que este esquema se generaliza: la producción de entropía de un elemento de volumen en su entorno es una suma de productos entre flujos de variables extensivas (“flujos”) y gradientes de variables intensivas (“fuerzas”). La interpretación habitual es que el gradiente es una “fuerza”6 que provoca el “flujo” de la cantidad extensiva correspondiente. Fuerzas y Flujos Consideremos el primer principio en su versión entrópica (sección 10.7) dS= 6 1 P μ dU+ dV − dn T T T (87) Acá “fuerza” no se usa en el sentido newtoniano. De hecho usa una concepción del sentido común que es antinewtoniana: que el “movimiento” (de la energía, en este caso) está causada por la “fuerza”. El uso es metafórico y no es contradictorio con las leyes de Newton porque la energía interna no es una partícula con masa. 25 Supongamos que nuestro sistema de estudio está en un estado de equilibrio local (pero no de equilibrio global). Tambien, para simplificar, supongamos que la presión, la temperatura y el potencial químico varían en una única dirección, a lo largo de la que definimos un eje x. Analicemos la producción de entropía en un disco de área A y espesor Δx como se muestra en la figura (el área es perpendicular al eje x, es decir, a todos los gradientes). Supongamos que hay un flujo ju de energía interna por unidad de tiempo y de área. Entonces ΔU derecha=j u AΔt (88) Si ju es positivo entonces aumenta U a la derecha (y disminuye en la misma cantidad a la izquierda) del área A, y a la inversa si ju es negativo. Entonces, la producción de entropía debida al primer término en (87) en tiempo Δt será: ΔS ent =j u AΔt − 1 1 . .. T x T x+Δx (89) (el signo menos en el primer miembro de (89) se debe a que corresponde al cambio de U a la izquierda del disco). Si el espesor del disco es muy pequeño podemos hacer lo mismo que hicimos en el ejemplo de la barra más arriba, y la cantidad entre paréntesis es la derivada de 1/T respecto de x por el espesor Δx . La producción de entropía resulta entonces: ΔS ent =j u AΔxΔt ∂ 1 . .. ∂x T (90) análogamente a lo concluído para la barra sometida a un gradiente de temperatura. Veamos ahora la variación de entropía en el elemento de volumen debido a la variación de energía interna, de acuerdo a (87). Por empezar, el cambio de U es el cambio de la densidad de energía interna u por el volumen por el tiempo (infinitesimal) transcurrido: ΔU sist = ∂u AΔxΔt ∂t de modo que ΔS sist =AΔxΔt (91) ∂u 1 .. . (92) ∂t T Como la suma de la energía mecánica y la interna se conservan, existe una relación entre la densidad de energía y su flujo que tiene una expresión matemática al estilo de la ecuación de continuidad que estudiamos para fluídos. Mencionamos esa relación, pero no vamos a entrar en los detalles matemáticos. Sí debemos decir que en estos casos (variable extensiva conservada localmente) modelizar la variable como sustancia resulta muy conveniente. Lo interesante es que a partir de (87) tenemos una expresión para la variación de entropía del elemento de volumen como de la entropía producida por ese elemento de volumen en su entorno, y que esta última resulta del producto entre un flujo y un gradiente. 26 Veamos los otros dos términos en (87). El segundo es el referido a cambios de volumen. Ese término contribuye al cambio de la entropía del elemento de volumen si cambia su densidad. Pero hay una contribución a la entropía del entorno si por ese elemento hay un caudal a lo largo de un gradiente de P/T. Veamos: el cambio de volumen a la derecha del área A será el caudal por el tiempo infinitesimal transcurrido, y a su vez el caudal es la componente de la velocidad normal al área por esa área. Si la velocidad es normal al área entonces el caudal es la rapidez por el área; en ese caso el cambio de entropía en el entorno será: ΔS ent =+vAΔt − P x P x+Δx .. . (93) T x T x+Δx Como antes, al hacer tender a cero el espesor de la rodaja la cantidad entre paréntesis pasa a ser el gradiente (ahora de P/T) por el espesor del disco: ΔS ent =+vAΔxΔt ∂ P .. . ∂x T (94) Algo similar ocurre con el tercer término. Sumando los tres, resulta que la entropía producida por nuestro elemento de volumen en el entorno en un tiempo infinitesimal Δt será: ΔS ent =AΔxΔt j u ∂ 1 ∂ P ∂ μ +v –j n ∂x T ∂x T ∂x T (95) Si pasamos dividiendo el área por el espesor por el transcurso de tiempo al lado izquierdo de (95), obtendremos la producción de entropía por unidad de volumen y de tiempo en el entorno de ese volumen: σ=ju ∂ 1 ∂ P ∂ μ +v –j ∂x T ∂x T n∂x T (96) Si la variación de las cantidades intensivas ocurriese no solo en el eje x, la expresión (95) tendría la forma más sofisticada que aparece en la bibliografía, pero cuyo significado es esencialmente el mismo: 1 P − = j u . ∇ v .∇ jn .∇ T T T (97) Recordemos que σ es la entropía por unidad de tiempo que un punto produce en su entorno. Aparte, la densidad de entropía del propio punto puede estar cambiando, de modo que (recordamos) la expresión del segundo principio será: σ+ ∂s 0 EN PROCESOS ESPONTANEOS ∂t (98) Veamos un ejemplo bastante transitado: la biósfera terrestre. Si consideramos un proceso corto en 27 comparación con los tiempos evolutivos podemos suponer que la biósfera se encuentra, más o menos, en un estado estacionario: la densidad de entropía no varía. En ese caso es σ el que debe ser estrictamente positivo. Entonces el primer término en (97) señala que debe haber un flujo de energía interna de las zonas cálidas a las zonas frías. El segundo término indica que el viento y las corrientes oceánicas son una fuente de generación de entropía también. ¿Cuál es el entorno en este caso? El interior de la Tierra y el espacio exterior. En algunos textos de ecología puede leerse que el Sol aporta “energía de baja entropía” y que la Tierra emite al espacio, a través de los polos, “energía de alta entropía”, y que ese flujo de energía es el que hace “funcionar” a los ecosistemas. Esas frases algo vagas (y que retomaremos en la sección 13) adquieren su sentido en las expresiones (97) y (98). Conviene no extrapolar esa forma de hablar demasiado lejos de esas expresiones. 11.4- El régimen lineal y las relaciones de Onsager Si bien a partir de las relaciones (97) y (98) se pueden hacer varias inferencias cualitativas sobre lo que ocurre en sistemas fuera del equilibrio, no se puede entrar en mayores detalles sin conocer los flujos. Es razonable que para muchos sistemas de interés, si el desequilibrio no es muy grande, los flujos sean proporcionales a las fuerzas. Este es el llamado “régimen lineal” y abarca los fenómenos habitualmente llamados “de transporte”. Estudiemos el caso de un fluído como en la subsección anterior. En el régimen lineal: 1 ∇ j u=1 ∇ 1 T 1 ∇ v =3 ∇ 2 T 1 ∇ j n=5 ∇ 6 T P 2 ∇ T P 4 ∇ T P 3 ∇ T T T T (99) donde los coeficientes α representan la proporcionalidad entre el flujo y su fuerza conjugada, y los coeficientes γ las contribuciones “cruzadas”. Esos números dependen de la sustancia y del estado en que esta se encuentra, y deben ser medidos (o bien predichos a partir de una modelización microscópica). Se puede vincular las expresiones en (99) con leyes fenomenológicas. Por ejemplo la llamada “conducción del calor”: la Ley de Fourier (caloricista) establecía que el “flujo de calor” es proporcional T donde κ es la “conductividad de calor” (para más j q=−κ ∇ a menos el gradiente de temperatura: detalles vean en la sección siguiente). Desarrollando la expresión (99) para la corriente de energía interna: γ γ α γ P γ μ j u=− 2 1 2 2 2 ∇ T 1∇ P 2 ∇ μ T T T T T (100) Comparando (100) con la Ley de Fourier se ve que la cantidad entre paréntesis es la conductividad térmica. El segundo término representa posibles flujos de energía debidos a un gradiente de presión, y el tercero uno debido a un gradiente en la concentración. 28 Lo cierto es que se ha detectado que las contribuciones cruzadas (los términos con γ ) suelen ser iguales dentro del error experimental. Por ejemplo, en las expresiones (99), γ 1 =γ 3 . No hay una explicación de esta observación a partir de los dos principios de la Termodinámica. Lars Onsager, alrededor de 1940, mostró que si los sistemas se modelizan microscópicamente y se usa la reversibilidad subyaciente en el sistema microscópico, esos coeficientes “cruzados” son iguales. Como no puede demostrarse estas relaciones desde una modelización termodinámica, puede considerárselo como un principio más; de hecho, en algunos textos aparece como el “cuarto principio de la Termodinámica”7. 11.5- Fenomenología vs marco teórico En las subsecciones anteriores hemos expuesto los elementos más firmes y consensuados de un marco teórico en construcción. La termodinámica fuera del equilibrio no es propiamente una teoría, sino más bien una colección de teoremas, algunos procedentes de la termodinámica de equilibrio, otros de las modelizaciones microscópicas, más otra colección de conjeturas sin tanto consenso (la más notable, probablemente, sea el “principio de mínima producción de entropía” de Prigoyine). Lo cierto es que a la hora de la verdad, y excepto algunos argumentos en ecología, el formalismo que hemos expuesto acá se usa muy poco. Ante situaciones concretas de no equilibrio se recurre a leyes fenomenológicas, bien establecidas en el sentido de que dan respuestas sólidas a gran cantidad de fenómenos prácticos (vean la subsección “fenómenos de transporte” en la sección 13). Por supuesto, eso no significa que estudiar termodinámica de no equilibrio sea inutil: cuando los problemas son más complejos y no están en el espectro de lo atacable por las leyes fenomenológicas, ese conocimiento puede ser la única guía disponible a una solución. Pero es importante tener claro en mente que mucho de lo expuesto en la bibliografía como “aplicaciones de la termodinámica” no está en el marco teórico de la termodinámica contemporánea , y las distintas herramientas pueden entrar en contradicción mutua o severos problemas si se las aplica demasiado lejos de sus dominios. 12- Conexiones con lo microscópico 12.1- ¿Qué quiere decir “microscópico”? “Microscópico” no necesariamente significa “extremadamente chico”. Se habla de un nivel “micro” cada vez que un sistema puede estudiarse también a un nivel “macro”, que es el resultante del comportamiento colectivo de los constituyentes del nivel “micro”. Por ejemplo, la microeconomía estudia las decisiones de individuos en situaciones económicas, mientras que la macroeconomía estudia el comportamiento de las cuentas nacionales, como el PBI y la cantidad de moneda circulante. Un bosque a nivel micro es cada uno de sus árboles y animales en interacción, mientras que su nivel macro es el ecosistema. 7 El “Tercer Principio”, también inspirado en modelizaciones microscópicas, afirma que la entropía de una sustancia pura es cero a temperatura absoluta cero. Esto no es verdad para un gas ideal, pero no se conoce ningún gas que no se haga líquido a temperatura lo bastante baja. 29 Si lo que estudiamos es la materia según sus propiedades físicas, el nivel macro es lo que hicimos en este curso: un auto completo puede ser estudiado por sus coordenadas en cinemática, sus coordenadas y su masa en dinámica, como cuerpo rígido o como sistema elástico, y de muchas otras maneras. Lo cierto es que modelizamos colectivamente porciones de materia sin preocuparnos por preguntas del estilo “de qué está hecho”. El nivel micro, en cambio, consiste en alguna especulación sobre los detalles internos del sistema. Estas especulaciones han cambiado mucho en los últimos tres siglos. Newton, por ejemplo, imaginaba que los gases estaban formados por “partículas” idénticas, estáticas y que se repelían. Boltzman imaginaba también partículas idénticas y diminutas, pero en vez de repeliéndose, viajando a grandes velocidades y chocando desordenadamente entre sí y con las fronteras del recipiente. La imagen actual de la materia a nivel microscópico es que está formada por objetos complejos (que llamamos moléculas) que interaccionan entre sí y con radiación a traves de una teoría conocida como Física Cuántica. Hay a su vez un marco teórico con el que se relacionan ambos niveles: la Mecánica Estadística. Esta es una teoría sutil y compleja, que no reduce lo macroscópico a lo microscópico pero sí logra hacer corresponder propiedades del modelo microscópico (como la masa o la “forma” de las moléculas) y propiedades macroscópicas (como calores específicos o compresibilidades). Sin embargo, muchas de las concepciones “térmicas” que aparecen en libros de texto (muchas de las cuales se usan actualmente en varios contextos científicos) bajo el título genérico de “termodinámica” son las que fueron apareciendo tentativamente durante el siglo XIX, cuando ni la Termodinámica ni la Mecánica Estadística estaban formuladas. Muchas son incompatibles entre sí porque corresponden a teorías “competidoras” en su época, y todas tienen un rango de aplicación mucho más acotado que la termodinámica o la mecánica estadística contemporaneas. En esas ideas tentativas las propiedades macroscópicas aparecen entrecruzadas con modelizaciones microscópicas altamente especulativas: la teoría atómica de Dalton es de 1802, el número de Avogadro se midió por primera vez alrededor de 1905, y la mecánica cuántica se desarrolló entre 1905 y 1930. ¡Todo es extremadamente nuevo! De modo que durante el siglo XIX la física térmica era un cuerpo desordenado y confuso de ideas. Vamos a repasar muy someramente algunas de las ideas “históricas” que siguen teniendo vigencia en los libros de texto, y finalmente vamos a hacer un “tour” por la física estadística contemporanea. 12.2- Las “moléculas” durante el siglo XIX Durante el siglo XVIII se desarrollaron las ideas “macroscópicas” de la química: elemento, sustancia y reacciones químicas. Pero no fue hasta 1802 que apareció la noción química de “átomo” y “molécula”: John Dalton logró explicar varios fenómenos (sintetizados en las llamadas “leyes estequiométricas”) haciendo la suposición de que las sustancias están formadas por unidades mínimas (las “moléculas”) formadas por la unión de unidades indestructibles correspondientes a cada elemento (los “átomos”), de modo que las reacciones químicas consistirían en la desintegración de las viejas moléculas y la formación de nuevas, siempre preservando la cantidad de cada tipo de “átomo”. La idea era de una utilidad indudable en química, pero muchos se mantuvieron escépticos, a lo largo del siglo XIX, sobre la posibilidad de que las moléculas fuesen objetos físicos (es decir objetos que se mueven, interaccionan y a los que se pudieran aplicar las leyes de Newton). Durante el siglo XX se concluyó que no lo eran, pero para entonces los átomos y las moléculas eran tan importantes que se hizo a la inversa: se reformuló la física para que, bajo las nuevas leyes (la “mecánica cuántica”) las moléculas resultasen estudiables físicamente. Lo cierto es que en el interín, mientras las moléculas eran entes 30 sobre todo químicos, algunos osados les atribuyeron propiedades físicas como posición, velocidad o energía mecánica. Esos eran los osados. En el marco de la química, en cambio, a las moléculas se les atribuía una masa (aunque se le llamaba “peso molecular”) y se desarrolló la idea de “mol” como una cierta cantidad de moléculas. Primero se definió el “número de Avogadro” A como la cantidad de moléculas de hidrógeno presentes en dos gramos de gas hidrógeno. Aunque A no se midió hasta el siglo XX, no importaba: estudiando las reacciones químicas se podía medir cuánto pesaba un “mol” de cualquier sustancia. Por lo tanto, se podía decir, para una masa de sustancia, cuántas moléculas había, expresadas como múltiplos del número A. Y aparecieron las sorpresas. 12.3- La misteriosa constante R Durante los siglos XVII y XVIII se estudió el comportamiento de los gases que llevarían a la formulación de la ecuación de estado de los gases ideales, que podríamos expresar como PV=CmT donde m es la masa de gas estudiada, y C una constante que depende del gas. Ahora bien, la masa de gas puede expresarse como el número de moles por la masa de un solo mol de gas (la llamada “masa molar” M) la expresión anterior puede escribirse como PV= CM nT Y acá aparece la primer sorpresa: si bien C y M son distintos para cada gas, el producto CM es básicamente el mismo para todos los gases: la consabida constante R. Y la sorpresa no se termina: si se expresa el calor específico a volumen constante por mol (en vez de por unidad de masa) para algunos 3 5 6 gases C V = 2 R , para otros C V = 2 R y para algunos otros C V = 2 R= 3R . Hasta acá uno podría pensar que hay un misterio sobre los gases, pero resulta que si uno expresa el calor específico de varios sólidos (metales, diamante y otros cristales) por mol en vez de por unidad de masa el calor específico resulta ser, con buena aproximación, 3R. Para colmo, todas estas “leyes” son aproximadas. Parece casi un capricho que los gases tengan un calor específico de “aproximadamente” un semientero de R. Y para algunas sustancias (el almidón, por ejemplo) la falla es estrepitosa. Y como si todo esto fuera poco, para todas las sustancias todas estas aproximaciones fallan por completo si la temperatura baja lo suficiente. Este era el rompecabezas que enfrentaban químicos, físicos e ingenieros interesados en cuestiones térmicas durante el siglo XIX. Lo que parecía lógico era suponer que algo ocurría molécula por molécula. Por ejemplo, si se supone que en un sólido cáda molécula “absorbe” una cantidad de calor (pensado como sustancia, que era la teoría dominante en la primer mitad del siglo XIX) por grado puede admitirse que todos los sólidos tengan el mismo calor específico molar, pero el hecho de que sea un múltiplo semientero de R sigue siendo un misterio bajo esta mirada. 31 Por un tiempo, la idea de que el calor es una sustancia que es atraída por las moléculas y se repele a sí mismo gozó de gran popularidad a pesar de no poder explicar esos extraños coeficientes semienteros porque en cambio explicaba muchas cosas interesantes. A modo de ejemplo: explicaba la expansión térmica (cuanto más calor, más repulsión) o el calor latente considerando que la diferencia entre fases era la cantidad de calor por molécula. Pero hacia la segunda mitad del siglo XIX el calórico cayó en desgracia y le llegó una oportunidad a la mecánica. 12.4- Las ideas de Boltzman El calórico tenía un problema serio: no podía explicar el calor “producido” durante una fricción en la que la estructura de los materiales no se modificara. Pero, aunque parezca sorprendente, tuvo su golpe de gracia cuando la teoría ondulatoria de la luz pudo ponerse en términos de campos electromagnéticos. Por entonces se creía firmemente que la luz y el calor eran manifestaciones de un mismo fenómeno (aún hoy circula la idea popular de que el calor es “radiación infraroja”), y al caer la idea sustancialista para la luz simplemente se dejó de creer en el calórico. Por entonces se desempolvó una idea antigua de Leibnitz, quien para justificar el hecho de que su “vis viva” (la actual energía cinética) no se conservaba postuló que lo que ocurría era que el movimiento que dejaba de observarse macroscópicamente pasaba a ser oscilaciones microscópicas inobservables del material. En tiempos de Leibniz no se podía avanzar, pero en el siglo XIX estaban las moléculas, listas para cumplir las leyes de Newton y tener energía cinética. No dejaba de ser una especulación osada, pero ahora, gracias a los avances de la química, se podían hacer algunos cálculos. A esta alternativa al calórico se la llamó “teoría mecánica del calor”, y es en esta corriente en la que se desarrolló la idea de “entropía”. Ellos fueron también los primeros en hablar de “orden” y “desorden”: según ellos, lo que comunmente se llamaba “energía mecánica” correspondía a “movimiento ordenado” (todos los átomos en la misma dirección), mientras que la energía interna correspondería a “movimiento caótico”. Aunque esta idea era bastante vaga, cuando empezó a quedar claro el rol de la entropía en la teoría lo lógico es que este grupo buscara vincular de algún modo a esta con la idea de desorden. Pero el más osado de todos ellos fue un tal Ludwig Boltzman (Austria, 1844-1906), quien estaba convencido de que podía aplicar la teoría de Newton a las moléculas de un gas y deducir de allí las propiedades termodinámicas de este. Esta idea reduccionista suele darse por hecho en no pocos textos. La idea fundamental era involucrar la estadística: Boltzman se preguntaba cómo debía distribuirse la energía total (que ahora era la suma de las energías mecánicas de las “moléculas”. En un caso extremo, un solo átomo tiene toda la energía y los demás están quietos: un solo microestado. Si la energía empezaba a “repartirse” había más microestados correspondientes al mismo macroestado (energía total U). Boltzman partió de la suposición de que el estado de equilibrio correspondía al que “repartía” la energía en un mayor número de microestados. Fue entonces cuando concibió la fórmula que figura en su tumba: S = k log W, donde k = R/A (hoy conocida como “constante de Boltzman”) y W es el número de microestados compatibles con el macroestado. Esto último, el “número de microestados”, es una cantidad terriblemente ambigua. Hace falta toda una serie de reglas adicionales para darle sentido. Pero lo cierto es que todo este marco (conocido como “teoría cinética”) era muy exitoso para dar cuenta de las propiedades de los gases. 32 De todos los triunfos de la teoría cinética, probablemente el más convincente fue el que permitió explicar que los calores específicos sean múltiplos semienteros de R: de acuerdo a la prescripción de que el número de estados sea máximo Boltzman fue capaz de deducir que la energía se “reparte” en partes iguales para todos los “grados de libertad” de las “moléculas” (resultado conocido como “equipartición de la energía”). La energía por grado de libertad de cada molécula resultaba de kT/2. Así, razonaba Boltzman, la energía de moléculas monoatómicas, que podían pensarse como puntos, era en promedio 3kT/2 (porque la molécula puede moverse en cualquiera de los tres ejes). Una molécula diatómica puede girar sobre dos ejes además de moverse como un todo a lo largo de tres ejes: cinco grados de libertad. Entonces su energía promedio será de 5kT/2. Y así sucesivamente. Los átomos en sólidos cristalinos tendrían tres grados “cinéticos” y tres grados de libertad “potenciales” debido a la interacción elástica entre ellos. Energía promedio: 3R. Consideren ahora un mol de cualquiera de estas sustancias y vean en cuánto cambia su energía al cambiar la temperatura a volumen constante, y tendrán el resultado. Todo esto estaba muy bien, pero (le criticaban) nada en las leyes de Newton permitía deducir la regla de que la energía se repartía en el mayor número de estados. Aunque Boltzman suponía que era un resultado “lógico”, se embarcó en el proyecto de demostrar que aplicando las leyes de Newton a un número enorme de “moléculas” podía demostrarse su hipótesis. La tarea no fue sencilla, Boltzman tuvo que generar un conjunto de ideas audaces y complejas, hasta que finalmente anunció lo que llamó el “teorema H”. Según Boltzman, podía construírse una función H (función del microestado del sistema) tal que, al evolucionar el sistema de acuerdo a las leyes de Newton, nunca puede crecer. Identificó esa función como el opuesto de la entropía. Así, según Boltzman, un montón de partículas newtonianas interactuando con fuerzas conservativas terminaba comportándose irreversiblemente, dando lugar al comportamiento termodinámico. 12.5- Disparen sobre Boltzman Si las ideas de Boltzman habían sido resistidas antes del teorema H, a partir de entonces las críticas fueron despiadadas. Pero antes de empezar a comentar esas críticas, veamos una objeción que entonces no existió o no fue muy tenida en cuenta: algo huele mal con la equipartición de la energía. Por ejemplo ¿Por qué las moléculas diatómicas no podrían girar sobre su eje? ¿Por qué no podrían vibrar? Cualquier grado de libertad, por rígido que sea, en principio debería acaparar la misma energía que los demás modos. ¿Cómo sabe uno cuándo dejar de incorporar modos? Para colmo, si uno aumenta lo bastante la temperatura el calor específico aumenta, pasando rápidamente, por ejemplo, de 5/2R a 3R. Como si los grados de libertad más rígidos se fueran “despertando” a medida que la temperatura aumenta. Desde el punto de vista de la Física clásica eso no tiene ni pies ni cabeza. Pero lo que más revuelo armó era la idea de que podía hacerse un sistema irreversible (termodinámica de un gas) juntando muchísimos sistemas que interactúan reversiblemente (“moléculas” cumpliendo con las leyes de Newton a través de fuerzas conservativas). Para que todo funcione, las “moléculas” deben interactuar a traves de fuerzas conservativas, o si no su energía mecánica no se conservaría. Y eso es fundamental para “explicar” el primer principio a partir de la física microscópica. Ahora bien, ¿Qué pasa si tomamos un sistema cuyo H no ha parado de disminuir, e invertimos todas las velocidades de cada una de las “moléculas”? De acuerdo a las leyes de Newton el sistema simplemente irá marcha 33 atrás hasta el estado inicial. ¡Y en ese proceso H tendrá que aumentar! Ese es el núcleo de la crítica de Loschmidt (hoy conocida como la “paradoja de Loschmidt”): algo tiene que estar mal en la demostración del teorema H. Hoy se sabe exactamente dónde está el error de Boltzman: en un punto, sin explicitarlo, Boltzman supuso que las probabilidades de lo que ocurriera en la interacción entre dos “moléculas” es independiente de la historia previa. Eso, por supuesto, no puede ser verdad en un sistema reversible. Esta hipótesis adicional se conoce hoy como hipótesis de caos molecular, y sin ella es imposible conectar la microfísica con la termodinámica. 12.6- Tour guiado por la Mecánica Estadística Hoy en día existe una teoría sólida que permite conectar modelos microscópicos con las propiedades termodinámicas de un sistema: la Mecánica Estadística. Pero, recalcamos, eso no significa que la termodinámica queda reducida a lo microscópico: en un punto se requiere de una hipótesis al estilo del caos molecular para terminar de establecer la conección. Por empezar, debemos aclarar que es muy limitado lo que puede hacerse con física newtoniana. Durante el siglo XX se desarrolló un nuevo marco teórico que pudiera usarse para describir átomos y moléculas, y fue una teoría extraordinariamente exitosa: la Mecánica Cuántica. Y también extremadamente antiintuitiva, muy diferente a la física newtoniana en muchos sentidos. Pero, curiosamente, la Mecánica Estadística encaja mucho mejor con la física cuántica que con la newtoniana. Y además da excelentes resultados: por ejemplo da perfecta cuenta del fenómeno del “despertado” de grados de libertad en los calores específicos que mencionamos en la subsección anterior. El primer paso consiste en imaginar todos los posibles microestados de nuestro sistema. Esto es lo que se conoce como “ensemble”. Cualquier cantidad que deseemos (la energía, por ejemplo) será un promedio de la correspondiente cantidad de los microestados, pesada por una probabilidad para ese microestado. Esta asignación de probabilidades es la que juega un rol similar a la hipótesis de caos molecular: es imposible justificarla a partir de las propiedades microscópicas del sistema. Hay tres tipos de ensemble: el “microcanónico”, el “canónico” y el “grancanónico”. En el primero se supone que el sistema está perfectamente aislado (la energía total es siempre la misma) y se hace la suposición de que cada microestado es igualmente probable. La entropía se define como el logaritmo del número de microestados. Entonces, usando esa función como un potencial, se obtienen la presión, la temperatura o cualquier otra variable de interés. En el ensemble canónico se supone que se mantiene el número de partículas, pero en lugar de mantener la energía lo que se hace es tener al sistema en contacto con uno mucho mayor a temperatura T. Considerando al “universo” (sistema de estudio más sistema a temperatura T) como un ensemble microcanónico, uno llega a demostrar que la probabilidad E /kT de cada microestado es proporcional a e , donde E i es la energía del estado correspondiente de acuerdo al modelo microscópico empleado. La cantidad central a calcular es la “función de partición”, que es la suma sobre todos los microestados de ese factor. Todas las cantidades termodinámicas resultan de manipulaciones adecuadas de esa función. Finalmente, en el grancanónico se supone que el sistema se mantiene a temperatura y potencial químico constantes, y se hacen cuentas distintas pero i 34 similarmente esotéricas. La mecánica estadística es extremadamente técnica. Se requiere de una teoría microscópica en el marco de la física cuántica y una fuerte dosis de cálculo matemático. Solo despues de todo eso, como una descripción, se habla de cosas como “desorden” o “información”. Si uno trata de llegar a conclusiones sobre la entropía con una interpretación personal de lo que sea el “desorden” en nuestro sistema, sin usar mecánica estadística, probablemente llegue a conclusiones sin sentido. 12.7- ¿Cómo sé si necesito de la mecánica estadística para resolver un problema? No pueden darse reglas de aplicación mecánica para decidir cuándo debe usarse física macroscópica o cuándo es imprescindible acudir a lo microscópico. Pero sí pueden darse algunos criterios útiles. Por empezar, si las variables relevantes del problema son todas macroscópicas es casi seguro que el problema puede resolverse a nivel macroscópico. En cambio, si el problema consiste en estudiar los efectos de usar cierto tipo de molécula en lugar de otro para cierto proceso, casi sin duda se necesita una descripción microscópica. Supongamos que uno hizo algún tipo de modelización microscópica en que las “moléculas” son esferas rígidas de radio R, y llega a calcular un calor latente (por decir algo) que queda como función de ese radio. En ese caso el modelo es fuertemente dependiente de lo microscópico. En cambio, si el resultado al que uno llega es totalmente independiente de las características de la molécula, no está claro si ese resultado depende o no de lo microscópico que uno supuso. En ese caso conviene tratar de resolver el problema macroscópicamente. Eso a veces puede ser complejo o sutil (un ejemplo son las “relaciones de Maxwell”). Otras veces uno puede estudiar ciertos fenómenos que uno mide macroscópicamente y encontrar que se dan regularidades que uno no puede explicar macroscópicamente. Uno podría entonces tratar de correlacionar las cantidades estudiadas con algún dato micro (un ejemplo es el de los calores específicos en relación al número de moléculas). Si uno encuentra ese tipo de correlaciones es muy probable que para entenderlas uno necesite una descripción microscópica. Si no aparecen correlaciones así, puede ser simplemente que no hemos dado con el razonamiento macroscópico adecuado, o bien con la variable microscópica relevante. En definitiva, sólo la práctica dota a uno de la capacidad de decidir si el problema que uno tiene entre manos es resoluble macroscópicamente o si se requieren dosis de física estadística. 13- La Termodinámica Realmente Existente8 8 Varios teóricos de orientación marxista han llamado “socialismo realmente existente” a las experiencias socialistas concretas del siglo XX (Unión Soviética, China, Cuba, Vietnam, etc) por oposición con el socialismo teórico de Marx. 35 Hasta acá hemos intentado dar una visión coherente de la termodinámica. O sea, hemos armado un formalismo, hemos construído un marco teórico y nos hemos preocupado por expresar todos los conceptos y los métodos en ese marco, y hemos explicitado cada vez que nos salimos de ese marco. La idea de proceder así es poder conectar entre sí las ideas termodinámicas, y hacer contacto con las herramientas conceptuales y de procedimiento del resto del curso (objeto de estudio, entorno, fronteras, interacciones, estados, procesos). Pero ese objetivo tiene un costo: algunas de las herramientas y mucho del lenguaje habitualmente usados quedan afuera de ese formalismo, y a veces queda la sensación de que se ha aprendido un dialecto exótico mientras que los demás hablan inglés. El propósito de esta sección es mitigar esa sensación y proveer un puente con los usos corrientes de las herramientas termodinámicas que aparecen tanto en textos introductorios como en aplicaciones. Cuando las desviaciones de la ortodoxia termodinámica sean graves vamos a rezongar un poco. 13.1- Fenómenos térmicos Hay ciertas leyes fenomenológicas que se presentan en la bibliografía como contenidos en sí: la dilatación térmica, el cambio de temperatura “por transferencia de calor”, la compresibilidad, etc. Dilatación térmica El asfalto de la calle se expande con el aumento de la temperatura. Esa es la razon por la que muchas veces se asfalte por bloques separados por juntas de brea. También es la razon por la que a veces en verano el asfalto se levanta. En realidad casi todas las sustancias se expanden al aumentar su temperatura (un contraejemplo notable es el agua entre cero y cuatro grados centígrados). Los termómetros de mercurio funcionan así: cuanto mayor la temperatura, más volumen ocupa el mercurio y entonces indica más alto en la escala del termómetro. Para ser concretos, tomemos un bloque de un material cualquiera como el de la figura. Si el cambio de temperatura es pequeño, puede hacerse la aproximación de que el cambio de tamaño de un objeto es proporcional a ese cambio: ΔL ∝ ΔT (101) Va a ocurrir análogamente con h, e o cualquier otra dimensión lineal (por ejemplo, una diagonal). Pero el cambio de tamaño también debe ser proporcional al propio tamaño: si una sisterna de acero de 40 metros de largo que se calienta cincuenta grados se agranda en cinco centímetros, no es razonable que con el mismo cambio de temperatura un prendedor de acero de dos centímetros se agrande cinco. Lo lógico es que el aumento de tamaño sea un cierto porcentaje del tamaño del propio objeto, o sea que el cambio del tamaño es proporcional al tamaño mismo: ΔL ∝ L 36 (102) Las expresiones (101) y (102) pueden sintetizarse en la siguiente expresión: ΔL=αLΔT EXPANSIÓN TÉRMICA LINEAL (103) donde α es el coeficiente de expansión lineal, que depende del material. Por supuesto, para el resto de las dimensiones lineales tendremos expresiones similares: Δh=αhΔT Δe=αeΔT Veamos lo que ocurre con un área. Razonando análogamente a como hicimos para llegar a (103) obtenemos ΔA=βAΔT EXPANSIÓN TÉRMICA SUPERFICIAL (104) Pero como las superficies están relacionadas con las dimensiones lineales, β no puede ser independiente de α . Veamos como ejemplo el área frontal del bloque cuya superficie es e.h: ΔA= e+Δe h+Δh −eh (105) Desarrollando (105) y usando (103) para expresar los cambios de e y h tendremos: ΔA= 2 αAΔT+ αAΔT 2 (106) Como se supone que el cambio es chico en comparación con el área, el término cuadrático en (106) puede despreciarse, y comparando el otro término con (104) llegamos a que β= 2α RELACIÓN ENTRE LOS COEFICIENTES LINEAL Y SUPERFICIAL (107) Se puede razonar idénticamente con la expansión de volumen: ΔV=γVΔT EXPANSIÓN TÉRMICA VOLUMÉTRICA (108) con γ= 3α RELACIÓN ENTRE LOS COEFICIENTES LINEAL Y VOLUMÉTRICA (109) Este coeficiente de expansión es el mismo del que hablamos en la subsección 10.8 (Relaciones de Maxwell), solo que allí le llamamos α en lugar de γ , como es habitual en termodinámica (la notación usada en esta subsección es habitual en textos introductorios). Como vimos en la subsección 10.8 ya mencionada, el coeficiente de dilatación volumétrica entra en razonamientos termodinámicos sofisticados en los que se lo puede relacionar con otros coeficientes. Las dilataciones lineal y superficial pueden entrar si la modelización termodinámica no es de fluído (por ejemplo, en un medio elástico). Pero sin insertarlas en razonamientos termodinámicos las expresiones (103), (104) y (108) tienen aplicaciones prácticas directas, por ejemplo si queremos saber qué distancia tenemos que dejar entre rieles en invierno para que en verano no se arruinen, o cuánto podemos llenar un tanque de petroleo a la madrugada de manera que no se vuelque al mediodía. 37 “Transferencia de calor”, calor específico Un problema típico de texto introductorio sobre “termodinámica” puede decir algo así como: ¿Cuánta energía debe transferirse a 10 gramos de agua para elevar su temperatura en 15 grados? O ¿Cuánto calor debe...? En algún lado habrá una tabla o se dejará el dato de que el calor específico del agua es de una caloría por gramo y grado centígrado, y que una caloría son 4,2 joules. En este apunte hay más que suficiente teoría para resolver estos problemas, el inconveniente es el lenguaje. El calor específico suele estar definido como la “cantidad de energía en forma de calor que hay que entregarle a un gramo de sustancia para elevar su temperatura en un grado centígrado”. Esa definición es problemática por varias razones. Por un lado, parece implicar que la temperatura cambia si y sólo si hay un proceso de calor, lo cual como vimos no es cierto. Por otro lado considera al calor como una sustancia que se le “agrega” al sistema (y por lo tanto, una vez que se le “agrega”, queda “en” el sistema). Nosotros sabemos que el calor que el entorno hace sobre el sistema depende del proceso (y por eso, cuando definimos calores específicos, los definimos para ciertos procesos). Suele estar implícito que se trata de calores específicos a presión constante, porque es el caso habitual en el laboratorio. Finalmente sólo tiene en cuenta sistemas cuyo calor específico no depende de la temperatura. Dado todo esto, se escribe algo como Q=CmΔT (110) que es, desde luego, idéntica a la expresión que obtendríamos si usáramos lo visto en la sección 8.1 para una situación en la que el calor específico C es constante. Lo que cambia es la interpretación: mientras que en el lenguaje de la sección 8.1 (110) sería “el proceso de calor que el entorno hace sobre el sistema para llevarlo de la temperatura inicial a la temperatura final a presión constante”, en el lenguaje habitual de los textos introductorios se trata de “el calor (o la energía, o la energía en forma de calor) que hay que transferirle al sistema para elevar su temperatura de la temperatura inicial a la final”. En esa forma de decirlo desaparece el entorno (la presión constante) y el calor Q parece ser el único ente capaz de llevar al sistema del estado inicial al estado final. Esa forma de leer la expresión (110) tiene, por supuesto, una raíz histórica. En los tiempos del calórico se consideraba que la ocurrencia de cambios de temperatura era la forma en que se manifestaba el flujo de calórico, a menos que pasara algo más (como un cambio en la composición o del estado de agregación). La caloría se definió como la cantidad de calórico necesaria para elevar la temperatura del agua de 14,5 grados centígrados a 15,5 grados centígrados, y en base a esta definición se midieron los calores específicos (a presión constante) de las demás sustancias. Esta concepción es la que se usa en calorimetría, que es la subsección siguiente. Consideraciones análogas pueden hacerse respecto del “calor latente” (en este lenguaje sería la diferencia de calor almacenado en cada estado de agregación) y del “calor de reacción” (el cambio de entalpía en una reacción química, que en este lenguaje se interpreta como la diferencia de calor almacenado entre reactivos y productos). 38 13.2- Calorimetría A pesar de que la teoría del calórico se considera obsoleta desde alrededor de 1850, mucho del lenguaje generado entonces y de las estructuras de pensamiento permanecen. Parte del problema es que las técnicas de laboratorio con las que se miden procesos de calor son exactamente las mismas que en tiempos del calórico, aunque la interpretación sea muy distinta. El término calorimetría se refiere a los fenómenos térmicos empleados para medir procesos de calor. El problema 1 (subsección 8.1) y el problema 2 (subsección 8.2) son problemas típicos de “calorimetría”. La idea es suponer un sistema aislado (en el que no puede entrar ni salir “calor”) en el que partes inicialmente están a temperaturas distintas y terminan teniendo la misma al alcanzar el equilibrio. El planteo es que, dado que el “calor” se queda en el sistema, el “calor” que “entrega” una parte caliente lo “recibe” la parte fría. La inspiración caloricista de esta forma de pensar es evidente. La razón de semejante nombre se debe a un artefacto, el llamado “calorímetro”, que es lo que se usa para medir procesos de calor desde el siglo XVIII. El calorímetro es un tacho con un tacho más chico adentro. El tacho de afuera tiene una cantidad de agua conocida (medida previamente) y está aislado del exterior por una frontera lo más adiabática posible. En el tacho interior se coloca el sistema de estudio. La frontera entre éste y el agua es impermeable y diatérmica. En el agua se coloca un termómetro, que registra las temperaturas inicial y final de esta. Entonces se hace el proceso a considerar sobre el objeto de estudio. Por definición, el calor específico del agua es de una caloría por gramo por grado centígrado, de manera que sabiendo que el calor que el sistema le hace al agua es menos el calor que el agua le hace al sistema, usando (110) y la diferencia de temperatura medida se puede determinar el calor que ocurrió sobre el sistema de estudio. En “lenguaje calórico” diríamos: “el calor que le entregó el sistema de estudio al agua elevó su temperatura, así podemos medir en cuánto aumentó la energía del agua”. Debe decirse que el lenguaje de la teoría del calórico es tan cómodo de usar para calorimetría que es dificil criticar a quienes se apegan al uso. El problema no es en realidad el uso de ese lenguaje sino su naturalización, el pasar a sentir como “evidente” el caracter sustancial del calor. Esos son los riesgos de empezar una exposición de la teoría termodinámica a partir de la calorimetría. Fue también un calorímetro el que permitió establecer la “equivalencia mecánica del calor”, la famosa equivalencia entre 1 caloría y 4,2 joules. Lo hizo, por supuesto, Joule. A la izquierda se muestra un esquema del calorímetro de Joule: un tanque con una cantidad conocida de agua, en su interior una paleta que puede ser accionada por el movimiento de una pesa que baja. Suponiendo que la masa y la fricción en las poleas es despreciable, la diferencia de energía mecánica de la pesa desde que sale hasta que está por tocar la mesa es igual al trabajo que sobre ella hace el agua. Usando el primer principio en su versión completa vemos que el cambio de energía mecánica de la pesa (antes de que la mesa accione sobre la pesa) es igual a menos el 39 cambio de energía interna del agua. Como no hay trabajo termodinámico sobre el agua, debe actuar un calor (este análisis es similar al que hicimos como ejemplo ilustrativo del primer principio en la sección 7). Actúa calor a presión constante, de modo que puede usarse (110). En efecto, si este experimento se hace con todo cuidado, se nota que la temperatura del agua aumenta. Comparando las calorías calculadas mediante (110) con el cambio de energía mecánica de la pesa puede determinarse, como hizo Joule, que 1 cal = 4,2 J. Veamos cómo se describe este experimento habitualmente en la bibliografía. Muchas veces se dice “Joule demostró que el calor y la energía se pueden interconvertir, y que por lo tanto el calor es una forma de energía”. Es probable que Joule haya usado un lenguaje así: corría 1850 (la termodinámica contemporanea alcanzó su forma actual recién hacia 1900) y la teoría dominante era todavía la teoría del calórico. Entonces, con el calor pensado como una sustancia, lo que parecía ocurrir era que la fricción “producía” calor. El calor era una sustancia que se podía crear. Y se lo podía crear a expensas de “destruír” trabajo. La energía era concebida todavía, mecanicistamente, como “capacidad de producir trabajo”, de modo que el calor era una forma de energía porque al entrar en una máquina térmica una parte de él podía “convertirse” en trabajo. Esta misma concepción, pero ya incorporando una mirada del segundo principio, está detrás de otra expresión habitual: “la energía mecánica de la pesa se disipa en forma de calor”. Esto tiene en mente la idea de que la energía, una vez “convertida” en calor, no puede ser completamente “convertida” en energía mecánica, porque para “reconvertirla” una parte debe “entregarse” a un sistema a menor temperatura (vean la sección 9.3). Todavía hay otra expresión habitual en los libros, que hace uso de una concepción microscópica del agua: “la energía ordenada de la pesa pasa a ser energía desordenada de las moléculas del agua”. Esta expresión es propia de la “teoría mecánica del calor” (~1870, vean la sección 12.4). Los calorímetros son artefactos de gran popularidad entre los físicos, asociados desde siempre a la medición de “energías” en el imaginario de la comunidad. Tal es así que, en los aceleradores de partículas como el LHC, ciertos detectores sensibles a la energía total de las partículas que colisionan, a los que la temperatura no les varía en absoluto, son conocidos como “calorímetros”. Tal vez por eso se insiste tanto con problemas y teorías vinculados a la calorimetría. 13.3- Primer Principio en varios dialectos Convenciones de signos En este apunte nos hemos apegado al uso de considerar el cambio de estado del sistema de estudio causado por las acciones del entorno sobre este. Si el entorno hace sobre el sistema un trabajo o un calor positivos, la energía interna del sistema aumenta. Pero en el origen fueron las máquinas térmicas, y las máquinas térmicas actúan sobre el entorno. O mejor dicho, el interés de quien las estudia es lo que la máquina hace sobre el entorno. El trabajo que el sistema de estudio (máquina o no) hace sobre el entorno es menos el trabajo que el entorno hace sobre él. Así, si Q es el calor que el entorno hace sobre el sistema de estudio (o, en lenguaje sustancialista, el “calor neto que entra al sistema”) y W es el trabajo que el sistema hace sobre el entorno, la primera ley será: ΔU=Q–W (PRIMER PRINCIPIO EN FORMATO REVOLUCIÓN INDUSTRIAL) A veces se escribe el primer principio de un modo aún más desfachatadamente ingenieril, pensando ya 40 en una máquina de Carnot, y en vez del calor neto se especifica el “calor recibido” (por una fuente caliente) y el “calor entregado” (a una fuente fría) ΔU=Q recibido −Qentregado –W (PRIMER PRINCIPIO EN FORMATO DESFACHATADAMENTE INGENIERIL) Huelga decir que el lenguaje sustancialista usado aquí, a imagen y semejanza de varios manuales de física, no es adecuada para la visión contemporanea de la termodinámica. En particular, la forma “desfachatadamente ingenieril” está lista para describir, una vez establecido que el motor es cíclico y por lo tanto ΔU= 0 , que “una parte del calor que entra en la máquina se transforma en trabajo, y el resto se entrega al foco frío”. Esta forma de hacer contabilidad del calor y del trabajo es la más cómoda para el interés ingenieril de calcular el rendimiento de la máquina: qué porcentaje del calor “entregado” “se convierte en” trabajo. En la subsección 9.3 se detallan estos manejes. Deltas de más Una expresión que suele aparecer con frecuencia para el primer principio es: ΔU=ΔQ+ΔW (PRIMER PRINCIPIO CON DEMASIADAS DELTAS) Esta forma de escribir el primer principio pone en pie de igualdad a la energía interna, el calor y el trabajo. Cuando uno lee la forma en que se describe o usa el primer principio en los contextos en que se lo expresa así se ve que las tres cantidades son concebidas como sustancias que se “interconvierten”. En el fondo lo que queda en evidencia es la incomprensión del significado de la delta en expresiones termodinámicas. En la figura mostramos dos posibles caminos para ir del estado 1 (cuya energía interna es U1) y el estado 2 (cuya energía interna es U2). La energía interna depende sólo del estado, por eso es una función de estado. En este ejemplo ΔU significa simplemente U 2 –U 1 , el cambio de la función de estado. El trabajo para ir entre ambos estados, en cambio, es muy diferente: de la definición de trabajo como la integral de P(V) respecto de V se ve que el trabajo para ir de 1 a 2 es mucho menor (en valor absoluto) por el proceso 1 que por el proceso 2, porque es el área bajo la curva. Si el cambio de U es el mismo pero el trabajo es muy distinto, por el primer principio, el trabajo es muy distinto. Entonces no puede decirse algo como que el trabajo es “el trabajo en 2 menos el trabajo en 1”, no es la diferencia de una función de estado, y lo mismo puede decirse para el calor. Por eso las deltas, adecuadas para funciones de estado, no lo son para procesos. Por supuesto que en cualquier situación en que se busque establecer algo así como un balance de energía el primer principio “con demasiadas deltas” funciona perfectamente, porque en un balance puede pensarse en términos de sustancia. Pero en cualquier razonamiento donde sea importante distinguir procesos de estados esa forma de pensar resultará insuficiente. 13.4- Fenómenos de Transporte 41 Al cerrar la sección 11 sobre termodinámica de no equilibrio prometimos una introducción fenomenológica a las situaciones de no equilibrio habitualmente encontradas en aplicaciones. Aquí está, esta es. Vamos a hablar de las famosas tres maneras en que “se propaga el calor”: conducción, convección y radiación. Conducción La “ley de Fourier” de la conducción del calor es de la época de la teoría del calórico, por lo que trata al calor como una sustancia que se propaga por los materiales. Si podemos despreciar el posible trabajo sobre cada parte de ese material la aproximación es excelente, porque entonces por el primer principio el calor total sobre cada elemento de volumen es igual al cambio de energía interna de ese elemento. El calor por unidad de tiempo puede pensarse entonces como un flujo de energía interna. La Ley de Fourier afirma que en presencia de un gradiente de temperatura se desarrolla una corriente “de calor” que es proporcional a menos el gradiente, es decir que “va” de zonas calientes a las frías: T j q=− ∇ LEY DE “CONDUCCIÓN DEL CALOR” (111) En términos caloricistas (que es el tratamiento habitual en libros de texto y en su aplicación) j q es el “flujo de calor” por unidad de área y de tiempo. La constante κ depende del material y se denomina “conductividad térmica” de este. En términos modernos hablaríamos más bien de un flujo de energía interna como en la subsección 11.4, o bien en términos de procesos de calor por unidad de área y de tiempo en la frontera de cada elemento de volumen si queremos usar la forma de pensar de la sección 6.2. Pero es cierto que en este caso particular una visión sustancialista del calor es particularmente cómoda, siempre que el material no se esté deformando o transformando químicamente. En cualquier caso (111) no puede deducirse de los principios de la termodinámica y debe considerársela una ley fenomenológica, válida para muchos materiales siempre que el gradiente no sea muy elevado. Consideremos el ejemplo de la barra sometida a dos temperaturas constantes de la sección 11.3 (vean la figura allí). Usemos el lenguaje caloricista (en la sección 11.3 ya usamos para este problema el lenguaje de la termodinámica contemporanea). De acuerdo con (111), por cada sección de la barra “pasa” por unidad de tiempo una cantidad de calor dQ dT =− Aκ dt dx (112) Si la barra tiene largo L, la integral definida de la derivada de T respecto de x a lo largo de la barra es la diferencia de temperatura. En estado estacionario el “flujo de calor” es constante a lo largo de la barra (no se acumula energía interna en el camino). Por lo tanto, integrando respecto de x a lo largo de toda la barra a ambos lados de (112) obtenemos: L es decir: dQ =− AκΔT dt dQ Aκ =− ΔT dt L 42 (113) La forma en que se interpreta(ba) (113) es la siguiente: el “flujo de calor” a lo largo de una barra de sección constante en estado estacionario es proporcional a menos la diferencia de temperatura. La constante de proporcionalidad es una especie de “conductancia térmica”, su inversa es la “resistencia térmica” (la resistencia que la barra ofrece al “flujo de calor”). Esa resistencia es proporcional al largo de la barra (cuanto más larga la barra, más “le cuesta pasar al calor”) e inversamente proporcional al área transversal y a la conductividad térmica. El caso (113) es el uso más sencillo de (111). En realidad, (111) junto con (110) permiten escribir una ecuación diferencial conocida como “ecuación del calor”, que es el modelo formal de una serie de fenómenos conocidos como “difusión”. La “ecuación del calor” es de gran importancia práctica: permite averiguar el perfil de temperaturas como función del tiempo de cualquier configuración, si uno conoce la condición inicial, el calor específico y la conductividad del material. También tiene un gran valor histórico porque fue una vía importante hacia el testeo de modelizaciones microscópicas en teoría cinética como la descripta en la sección 12. Convección Cuando se habla de “transmisión de calor por convección”, en rigor se habla de movimiento de materia. La conductividad térmica del agua o del aire es muy baja, pero como el agua y el aire calientes son menos densos que los fríos, por lo que un elemento de volumen de aire caliente rodeado de aire frío sufrirá una fuerza del aire circundante mayor al que le hace la Tierra (estamos usando el teorema de Arquímedes) y como consecuencia subirá. Pongamos como ejemplo agua calentándose en una olla. El fuego hace un proceso de calor sobre el fondo de la olla, y este a su vez lo hace sobre la capa inferior de agua. Si dependiese de la conductividad térmica, el calentamiento del resto del agua sería muy lento. En vez de eso, ocurre que esas capas inferiores se hacen menos densas y suben, arrastrando hacia abajo al agua más fría. Esa agua se calienta y se reinicia el ciclo. Así empiezan a producirse “corrientes convectivas” que mezclan el agua y aceleran el calentamiento. Similarmente, si se enfría el aire encima de agua tibia, la capa de más arriba se enfriará y bajará, haciendo subir el agua tibia que a su vez se enfría y mantiene las corrientes. Es interesante mencionar un caso muy curioso e importante: el agua entre 4 grados centígrados y cero se expande en lugar de contraerse al enfriarse. Eso significa que un lago en un invierno muy frío se enfriará por convección hasta los cuatro grados, y a partir de ese momento se estabilizará y cesarán las corrientes convectivas. El agua más fría de arriba será tambien menos densa y permanecerá así, hasta eventualmente congelarse. Así, aunque en la superficie haya una temperatura bajo cero por meses, sólo se congelará la superficie y abajo podrán seguir subsistiendo plantas y peces hasta la primavera. Radiación La llamada “propagación del calor por radiación” probablemente sea de las tres la que genera más equívocos. Supongamos que tenemos una lámpara de escritorio. Si ponemos la mano encima a cierta distancia podemos sentir el aire caliente subiendo de las que hablamos antes (las corrientes convectivas), pero si ponemos la mano por delante sentimos “el calor”. Más aún, lo sentiríamos aunque no hubiese aire entre la lámpara y nuestra mano. 43 La decisión de encomillar “el calor” que sentimos no es arbitraria. Existe la tentación de suponer que hay un “calor” que viaja de la lámpara a nuestra mano, entidad a la que se le dio el nombre de “radiación”. La luz también es “radiación” (tambien viaja por el espacio vacío desde los objetos luminosos) y por bastante tiempo se supuso que la luz y el “calor” eran manifestaciones del mismo fenómeno. Aún hoy se oye popularmente la idea de que el calor son “rayos infrarrojos” (por ejemplo es habitual oír de los aparatos de visión nocturna infrarroja que “detectan el calor”). Esta visión está superada desde hace mucho tiempo. De acuerdo a las mejores teorías de las que disponemos, lo que viaja de la lámpara, o del Sol, hasta nosotros no es “calor” sino “radiación electromagnética”, un ente dinámico que no puede ser entendido ni desde la mecánica ni desde la termodinámica, pero que sí ha recibido una descripción satisfactoria como oscilaciones de campos eléctricos y magnéticos. Al igual que un sistema material, la “radiación” dentro de un recinto puede tener una temperatura, una entropía y una energía interna, e interactuar con los objetos materiales. En la visión más moderna que tenemos, un objeto a cierta temperatura irradia radiación electromagnética. Si el objeto está realmente caliente (más de 2000 K) parte de esa radiación será luminosa, como pasa con un hierro candente. Si el entorno está a una temperatura inferior, el objeto caliente emitirá radiación con una potencia que va como la diferencia de temperaturas a la cuarta. Cuando la radiación alcanza un sistema material, si esta radiación es más que la que emite el objeto alcanzado, hará un proceso de calor y el sistema se “calienta”. El proceso por el que un objeto caliente emite radiación puede entenderse como que sobre el objeto actúa un calor negativo, “enfriándolo”, y sobre la radiación uno positivo que aumenta su energía interna y su entropía, o sea aumentando la cantidad de radiación. El ejemplo más importante en ciencias naturales de calentamiento por radiación es lo que ocurre con el Sol. Del Sol nos llegan radiaciones que interactúan con el aire, las piedras, el agua y las plantas. En esa interacción hacen calor y trabajo (la radiación hace trabajo sobre las hojas, y eso permite que la fotosíntesis sea un proceso espontaneo, hace trabajo sobre paneles solares permitiendo generar electricidad, etc). No nos podemos extender en el manejo detallado de la radiación, que nos saca de los marcos que hemos visto en el curso, de modo que dejamos el tema en este punto. Lo bueno es que una vez que han hecho calor o trabajo sobre objetos que sabemos modelar, a partir de ese punto podemos limitarnos a hacer la termodinámica de siempre. 13.5- Exergía Cuando uno ya tiene bastante con la energía, la entropía, la entalpía y la energía libre, en muchos textos (sobre todo en ingeniería y ecología) apabullan con un término más: la “exergía”. Por lo general se trata de textos en los que se sigue concibiendo a la energía como “la capacidad de hacer trabajo”, pero la energía interna no puede usarse toda para hacer trabajo: las máquinas térmicas tienen límites de eficiencia. Entonces se dicen cosas extrañas: se dice que no toda la energía interna es del todo buena, cuanto más entropía tenga peor, porque será menos la energía “utilizable”. La “exergía” se define entonces como “la energía utilizable”, y uno se queda con la sensación de, a lo sumo, haber entendido a medias. ¿Qué quiere decir “la energía utilizable”? En ese contexto se habla, en realidad, del máximo trabajo que el sistema de estudio puede hacer (por aquella concepción de la energía como capacidad de hacer trabajo). Pero acá surge una dificultad: el trabajo que un sistema puede hacer sobre el entorno no 44 depende solo del estado del sistema: depende del estado inicial, del estado final y del entorno. Por ejemplo, si uno dispone en el entorno de un subsistema a temperatura absoluta cero, podría usar la totalidad de la energía interna del sistema de estudio para hacer trabajo (vean subsección 9.3). Entonces, la definición de exergía debería ser: La exergía de un sistema de estudio que se transforma entre dos estados sometido a ciertas condiciones del entorno es el máximo trabajo que el sistema puede hacer sobre el entorno en esas condiciones al transformarse. Así, la exergía no es una cierta función de estado, sino una cantidad que depende del problema. En muchos problemas prácticos la condición del entorno es temperatura y presión fijas. Entonces, recordando lo que vimos en la sección 10.11, el máximo trabajo que un sistema puede hacer a presión y temperatura constante es el cambio de la función de Gibbs (alias “diferencia de energía libre”) durante el proceso considerado. Por ejemplo, la exergía de un litro de nafta, si vamos a usar la nafta en un motor, quemándola, y abiertos a la atmósfera, será el cambio de energía libre entre un litro de nafta y el producto de la combustión (esencialmente dióxido de carbono y agua). Este es el sentido que se le da en ecología. 13.6- La “energía” en la nutrición Es un lugar común hablar de la “energía” de los alimentos, pensar en ellos como un combustible que al ser consumido su energía pasa a nuestros músculos, listos para la acción. Y a diferencia de otros usos de la palabra “energía” en este caso está cuantificada: todos hemos oído hablar de las “calorías” que tiene un chocolate o una galletita. Extrañamente, en nutrición las “calorías” engordan. Alguno podrá sublevarse contra la idea de que estemos por poner esto en duda. Después de todo, en este mismo apunte hemos hablado de trabajo vinculado a la fotosíntesis y a la respiración y lo hemos vinculado con cambios de “energía libre” (función de Gibbs, vean la subsección 10.11). Es cierto, no vamos a negar que los alimentos se transforman en nuestro interior, y que al hacerlo hacen trabajo sobre nuestro cuerpo, y que ese trabajo nos permite sintetizar proteínas, mover músculos y otras funciones fisiológicas. Lo que vamos a hacer en esta subsección es matizar hasta qué punto las “calorías” de un alimento tiene que ver con todo eso. Empecemos por explicitar qué significa que un churrasco tenga una determinada cantidad de “calorías”. Para determinar cuántas calorías por gramo “tiene” un alimento lo que se hace es pesar cierta cantidad de este (un carré de cerdo, digamos). Una vez pesado se lo seca (se le quita la humedad haciéndole pasar aire tibio, pero tratando de no degradar las sustancias orgánicas que lo componen. Y, finalmente, se lo coloca en un calorímetro (vean subsección 13.2), se lo prende fuego (sí, como leen) y se mide el cambio de temperatura del agua del calorímetro. Como se conoce la cantidad de agua y su calor específico, se puede calcular cuántas calorías hizo el alimento sobre el agua. Esa es la cantidad que figura como “valor nutricional” en calorías por gramo en las cajas de los alimentos. El procedimiento, por supuesto, no es completamente arbitrario. El argumento para proceder así es que a nivel celular el organismo “quema” el alimento para poder hacer el trabajo que hace funcionar a las células. Pero hay varios problemas. Por empezar, el trabajo máximo que puede obtenerse a partir de la 45 combustión del alimento es la diferencia en la función de Gibbs, y lo que se obtiene con el calorímetro es, en el mejor de los casos, una diferencia de entalpía. Aún peor, no todo el alimento se “quema”: las proteínas se usan como tales en la constitución de estructuras celulares, y sólo en caso de hambre extremo se queman. Parte de las grasas suelen almacenarse como reserva para eventualmente quemarlas y el resto no se asimila, y las fibras directamente no se asimilan porque el cuerpo humano no puede digerirlas. Sólo los hidratos de carbono se queman prioritariamente. Por eso, los dietólogos que algunas décadas atrás se basaban exclusivamente en las calorías han ido incorporando otros parámetros (como la “calidad de las proteínas”) para caracterizar los alimentos. Entonces, las famosas “calorías” de un alimento no son una medida completamente fiable de la energía (ni la exergía) disponible por un organismo que lo come. ¿Por qué es tan difundido su uso entonces? Esencialmente porque es facil de medir. Con paciencia puede medirse el cambio de la función de Gibbs para diversas sustancias inorgánicas, pero hacer eso para un pedazo de bacalao es prohibitivamente costoso. En cambio, secarlo y quemarlo es muy sencillo. Además, aunque no sea la “exergía real” del alimento, resulta un indicador no tan desatinado. En definitiva, es facil de medir y resulta razonablemente útil. Pero siempre conviene recordar que se trata de un indicador, que cuando uno habla de la “eficiencia en la acumulación de energía” de la vaca al producir, a partir de pasto, carne y grasa, uno no está haciendo cálculos termodinámicos sino más bien un balance contable. 13.7- Termodinámica en Ecología Nos dejamos para el final de la sección los usos más polémicos de conceptos termodinámicos. Esta va a ser, probablemente, la subsección con más rezongos. Debe decirse, sin embargo, que los ecosistemas son extremadamente complejos y están extremadamente fuera del equilibrio. La necesidad tiene cara de hereje: los problemas complejos suelen requerir abordajes osados, y alguna que otra tropelía puede llegar a ser considerada “collateral damage”. Con todo, aunque no aboguemos por un uso inmaculado de las ideas termodinámicas, siempre es bueno recordar que el norte de toda ciencia es la coherencia y que es deseable avanzar con el formalismo sobre las partes más oscuras y peor comprendidas. Antes que nada debemos aclarar que hay dos enfoques de la ecología. Una visión ortodoxa centrada en la interrelación entre las poblaciones y de estas con los factores abióticos, que en rigor no usa termodinámica pero como habla de flujo de energía le vamos a dedicar un par de párrafos, y una visión más holística, más romántica, más Jaques Cousteau, más Gaia, que despliega una frondosa terminología termodinámica e incluso arriesga principios como el de máxima producción de entropía, máxima “acumulación de información” o máximo flujo de energía a los que vamos a dedicar buena parte de nuestra, ejem, energía. El flujo de energía en la ecología ortodoxa En 1942 un tal Lindemann publicó un paper que inició el estudio del “flujo de energía” en ecosistemas. En ecología “ortodoxa” lo central son las interdependencias entre poblaciones, y central en esto son las “redes tróficas” (el grafo orientado que indica qué especies comen cada especie). Lindemann trata de establecer qué pasa con la “energía” a medida que los bichos se comen entre sí, y cuando decimos “energía” en este contexto estamos hablando de nutrición, como en la subsección anterior. Es decir, en las “calorías”. Algo así como “las vacas de la Pampa Húmeda comen tantas toneladas de pasto (biomasa) que les aporta una energía de tantas calorías. Con eso generan tal biomasa de vacas por 46 tantas y tantas calorías” (que siempre son menos que las aportadas por el pasto comido). Se diría, en paralelo con el discurso en los libros introductorios de física térmica, que el resto de la “energía” se disipa en forma de bosta. Humoradas aparte, el enfoque de Lindemann no pretende ser termodinámico. La inspiración de la teoría es más bien la macroeconomía (el estudio de los balances contables de los países), como se desprende de las terminologías utilizadas. Por ejemplo: la fijación total de energía por fotosíntesis es la productividad bruta primaria PBP (compárese con el PBI), la diferencia entre la PBP y la respiración autótrofa (la respiración de la propia planta, que en esta visión sería el “costo” en la producción de biomasa vegetal) es la productividad neta primaria PNP, y así siguiendo. La idea es rastrear la producción de biomasa y su “energía” con vistas a la producción de alimentos. Debe comprenderse todo esto en su contexto: el mundo venía de una debacle económica sin precedentes y estaba embarcada en la mayor guerra de la historia (por lo menos hasta ahora). Por otra parte, las nuevas teorías económicas de Keynes (que impulsaron una comprensión de las cuentas nacionales, que antes no se seguían9) eran enormemente exitosas y populares: EEUU crecía a todo vapor. La perspectiva hacia fines de la guerra y la posguerra era que el dominio de la ciencia y la comprensión de la economía podían terminar con el hambre y evitarían otra guerra como la recién vivida. Durante 15 o 20 años esta expectativa parecía comprobarse con los hechos: una transformación sin precedentes de la estructura de la economía de los países centrales elevó enormemente la productividad de los campos, en parte gracias a estudios como el de Lindemann, y se vivieron años de una prosperidad sin precedentes (ni postcedentes, debe decirse). Por aquellos años de reconfiguración de la arquitectura financiera internacional se llegó a sugerir reemplazar al oro por la “energía” como reserva de valor internacional. Lo cierto es que el flujo agregado de “energía” en los ecosistemas tiene las mismas limitaciones que la idea correspondiente en nutrición (vean sección 13.6). La Ecología de Odum, Margalef y Jacques Cousteau Hacia mediados de los años '50 algunos ecologistas empezaron a usar ideas termodinámicas en ecología. A partir de los años '70, cuando el sueño de prosperidad eterna de la posguerra se desvanecía y empezó a ser claro que el mundo es pequeño y arruinable, estas ideas cobraron una gran popularidad. Odum señala en una entrevista que el factor crucial en ese cambio cultural fue, probablemente, la llegada de las primeras imágenes de la Tierra vista desde lejos, en especial aquella tomada desde la Luna. Un mundo fragil como ese no parecía el reino del crecimiento sin límites. Tiene mucha lógica que una teoría que habla de la irreversibilidad, del aumento de la entropía, pareciese una herramienta imprescindible para entender esa capita verde y azul suspendida en medio de la nada. El uso de la termodinámica ha llevado a muchos malos entendidos, desde la afirmación de que el segundo principio no vale para sistemas biológicos hasta la afirmación de que la biodiversidad y la evolución “se explican” con termodinámica. En realidad lo que se hace al usar termodinámica en ecosistemas es verificar las condiciones que los dos principios imponen. La vida en este planeta no ocurre gracias a la termodinámica sino más bien a pesar de ella. Es decir que la termodinámica no prohibe la evolución o la biodiversidad, pero tampoco la explica. 9 El primer país que midió su PBI fue la Unión Soviética hacia los años '20. En países capitalistas esas mediciones se empezaron a hacer a mediados de los '30. 47 El equívoco respecto a la supuesta no validez del segundo principio se debe a que la biósfera es un sistema abierto, mantenido fuera del equilibrio merced a una “corriente de energía”. Es habitual ver en textos de termodinámica ecológica decir que “la Tierra recibe del Sol energía de baja entropía y reemite al espacio energía de alta entropía. Esa corriente de energía que va del ecuador a los polos es la que necesita la biósfera para sostener los procesos irreversibles que ocurren en ella”. Es facil mistificar a partir de este tipo de frases que parecen salidas de la película Avatar. No es en absoluto un disparate, pero conviene entenderla en su contexto y no tratar de hacer extrapolaciones. El flujo de energía al que alude esa frase tiene muy poco que ver con el flujo de “energía” en las cadenas tróficas estudiadas por Lindemann. Tampoco con un espíritu estilo Gaia (o llegado el caso, tipo “Espíritu Santo”) moviendo al mundo. Es una corriente de energía interna en el sentido de la sección 11.3: hay gradientes de temperatura, por lo tanto hay “producción de entropía”. Parte de esa corriente tiene que ver con las cadenas tróficas (pero no con las calorías de los alimentos sino más bien con los trabajos originados en los procesos de fotosíntesis y respiración) pero también con las corrientes oceánicas y con el “ciclo del agua”. La evolución de menor a mayor complejidad es probablemente la que genera los mayores equívocos. Se han sugerido una diversidad de principios “termodinámicos” para dar cuenta de este aspecto intrigante. Pero es un error suponer que se trata de “explicaciones termodinámicas”. No es que se parta de los principios de la termodinámica y se demuestren cosas como el principio de máximo flujo de energía, o de máxima generación de entropía, o de máxima “acumulación de información”. Todos esos “principios” son propuestas de leyes propias de los ecosistemas, todas son altamente especulativas y ninguna tiene gran consenso. De hecho no está claro que la “tendencia a la complejidad” pueda describirse desde la termodinámica, porque esa complejidad alcanza los niveles microscópicos (moléculas de ADN o de proteínas) y mesoscópicas (células y tejidos). Es decir, hay complejidad en todas y cada una de las escalas, de modo que los seres vivos no están ni siquiera localmente en equilibrio (vean la sección 11.1). Respecto del uso del concepto de “información” en ecología hay que rezongar enérgicamente. Hay un concepto de “información” en mecánica estadística, bastante abstracto y ni siquiera totalmente consensuado. Como explicamos en la sección 12.6 es central en el formalismo la idea de “ensemble” y una asignación de probabilidades a cada microestado. En el equilibrio todos los microestados tienen la misma probabilidad, por lo tanto si conocemos el macroestado y sabemos que es de equilibrio nuestra “información” sobre el microestado es mínima. Pero fuera del equilibrio algunos estados son “más probables” que otros, entonces tenemos más “información” del posible microestado. Hay un cálculo que se puede hacer en mecánica estadística de la probabilidad de que cierto estado de no equilibrio ocurra como fluctuación de un estado de equilibrio (aunque no está claro qué significa en la práctica esa probabilidad). Eso permite hacer un cálculo de la “información” contenida en un sistema termodinámico. Pero la palabra “información” debe entenderse en ese sentido restringido, no es la cantidad de megabytes que pueden almacenarse como si se tratase de un pendrive. No es información como la que se lee de los diarios o la que lleva el Voyager en su famoso disco de oro. En termodinámica, a partir de cálculos de “exergía” (en este caso, diferencia del potencial de Gibbs entre las sustancias orgánicas y sus contrapartes inorgánicas si toda la biomasa se quemara) puede hacerse un cálculo de “información” en el sentido de la mecánica estadística. Algunos autores identifican esa “información” con la información genética del ecosistema. Este es un típico error por multiplicidad de significados, como cuando se piensa que al mezclar la baraja se aumenta su entropía porque “se desordena”, o como pensar que la fuerza de las convicciones de un candidato se mide en newtons. 48 14- Desmantelando tabúes: ¿Cómo se usa la Termodinámica en Ciencias Naturales? Las subsecciones de esta sección son preguntas que consideramos, en buena medida, ilegítimas. Usar un concepto determinado no está ni bien ni mal, es solo más o menos adecuado a los problemas que queremos resolver, porque ningún concepto de la Física refiere a la realidad: sólo son modelos para tratar de interpretar esa realidad. Deliberadamente las hemos escrito en el formato en que se oyen por ahí los tabúes del TEF, cosas como “En el taller no se puede decir que el calor es energía”. Varias de las palabras que aparecen en este apunte aparecen en otros contextos bajo la forma de “aplicaciones de la Termodinámica”, aunque muchas veces designando variaciones sutiles (o no tanto) de los correspondientes conceptos. La divergencia no es en una sola dirección, corresponde a varias ideas (incompatibles entre sí) que han ido apareciendo a lo largo de la historia de la Termodinámica. ¿Qué hacemos con lo aprendido en el TEF a la hora de enfrentarnos con esos dialectos de la Termodinámica? ¿Está “mal” decir que el calor entra o sale de los cuerpos? La primer regla de oro de la física aplicada es: si un conjunto de procedimientos, con su lenguaje, se usa para resolver cierto tipo de problemas y funciona, entonces está “bien” (léase: es adecuado). Si en ese contexto el calor está modelizado como algo que entra y sale, que fluye, o lo que sea, entonces usen esa terminología y esos procedimientos sin culpa, siempre que estén atacando el tipo de problemas para los cuales se supone que esas ideas funcionan. Ese formalismo es una modelización en el mismo grado que lo que vimos en este curso. Ahora bien: si el problema que quieren atacar está por fuera de lo que tradicionalmente se resuelve usando esos métodos... DUDEN. La termodinámica macroscópica, tal como la vimos en este apunte, funciona tan bien que no se conocen ejemplos de casos en que sus principios fallen. Se ha aplicado esta versión a sistemas tan diversos como seres vivos, estrellas o agujeros negros. Y mucha gente muy ingeniosa ha tratado de encontrarle problemas o contradicciones sin éxito. Por lo tanto, los dos principios tal como los hemos visto acá son un puerto seguro ante la duda. Como regla general, la idea de que el calor “fluye” funciona siempre que consideremos un subconjunto de procesos que, operando cíclicamente, den calor nulo. Por ejemplo, si nos restringimos a presión constante (lo que suele no estar mal si estudiamos procesos abiertos a la atmósfera) o a volumen constante (no hay trabajo). Las razones para esto están en el último comentario sobre el Primer Principio, donde explicamos por qué el calor no puede considerarse función de estado. ¿Está “mal” decir que el calor es una forma de energía? Si el contexto es la termodinámica contemporanea sí, está mal, en la medida que el calor es un proceso. Está tan “mal” como decir que el trabajo es energía. Pero en cualquier contexto en que el “calor” sea considerado una función de estado tiene perfecto sentido pensar a la energía interna como “el calor” más alguna otra cosa. Remitimos entonces a la pregunta anterior. Al final ¿Qué “es” el “calor”? 49 El calor es una abstracción humana. Bah, en realidad varias, porque en distintos contextos (aunque todos ellos sean del ámbito de la física) la palabra “calor” puede ser usado de maneras diferentes. En otras palabras, el calor es lo que hagamos de él, igual que el amor o la belleza. Alguno podrá sentir “evidente” que el amor es una abstracción, que no existe como tal, mientras que es “evidente” que el calor es real. Es la evidencia del sentido común, la misma que lleva tantas veces a meter la pata. En definitiva ¿Cuál es la evidencia que uno tiene de su existencia? Sólo el efecto de interacciones sobre porciones de materia, a veces nosotros mismos. Si tocamos la pava caliente nos quemamos, si nos patean nos duele. Si es “evidente” que nos quemamos porque “el calor pasa de la pava a nuestra mano” ¿Por qué no convencernos de que el pie del otro tiene “pateadura” que nos es transmitida durante la patada? ¿Está “mal” considerar que “la energía fluye”? Para considerar que la “energía” pasa de un lugar a otro atravesando fronteras se necesita que ese ente al que llamemos “energía” cumpla con dos cosas: • • Esa “energía” tiene que ser conservada, de manera que lo que “aparezca” en algún lado “se vaya” de algún otro. Cada porción de energía debe ser pensable como residente en un sitio, o de otro modo no podría “mudarse”. La primer condición no parece tan alocada a primera vista: el primer principio dice algo no muy diferente. Solo que el primer principio es muy específico: es la suma de la energía mecánica más la energía interna lo que se conserva. En muchos contextos en los que la “energía” fluye eso a lo que se denomina “energía” suele no ser esa suma; a veces no es ni siquiera una función de estado desde el punto de la termodinámica contemporanea (ejemplo: el “calor”) y por eso usamos comillas para nombrar a la “energía” en este apartado. Seguramente el marco en cuestión es tal que esa cantidad pueda ser considerada una cantidad conservada. Por lo tanto, la validez de ese modelo estará acotada a ciertos problemas, y nuevamente habrá que ser cuidadoso cada vez que se aplique en contextos novedosos. La segunda condición puede parecer una obviedad a quien piense en la “energía” como en una cosa sustancial. Recordemos que cualquier “energía” en cualquier marco teórico es siempre una cantidad abstracta inventada por el hombre, así que nada es obvio sobre su naturaleza. Pongamos un ejemplo: hay una energía potencial asociada a la atracción de la Tierra (la “Energía Potencial Gravitatoria”). ¿Dónde “está” esa energía? ¿En el objeto? ¿En la Tierra? Desparramada entre ellos? La verdad es que al construír la idea de “Energía Potencial Gravitatoria” no hizo falta asignarle una “posición”, de modo que en principio no la tiene. Puede ser conveniente asignarle una en algunos contextos, en otros puede no tener sentido o dar lugar a problemas o inconsistencias. Por lo tanto, un modelo sustancial para la “energía” puede ser útil, inútil o contraproducente, según el caso, pero nunca necesario. ¿Es “mejor” la termodinámica actual que los modelos en los que el calor y la energía 50 “fluyen”? No necesariamente. No puede decidirse si un marco teórico es “mejor” o “peor” que otro sin tener en cuenta el tipo de problemas que se quiere resolver. Para muchos problemas (como el estudio del gradiente de temperaturas en un sistema a presión constante) el uso de la termodinámica completa puede ser demasiado engorroso. Si uno tiene un modelo que permite resolver el problema que uno tiene entre manos y es mucho más simple que la termodinámica contemporanea, ese modelo es mejor para resolverlo, en el mismo sentido en que un destornillador es mejor que un cuchillo Tramontina para aflojar un tornillo, y vice versa para comer un asado. La termodinámica contemporanea es superior a cualquier otro marco sobre procesos térmicos siempre que estemos en duda sobre la validez de ese marco para resolver nuestro problema. En caso de duda, cualquier cosa que pueda fundamentarse seriamente con los principios de la termodinámica es esencialmente segura. ¿Está “mal” usar átomos, moléculas u otros detalles microscópicos para hacer razonamientos en termodinámica? Este es un punto delicado. Lo que obtengamos aplicando rigurosamente termodinámica macroscópica es muy sólido, pero a veces puede resultar insuficiente. A veces la información microscópica puede completar información relevante que no puede obtenerse de otro modo. Por otra parte, la mecánica estadística rigurosa es mucho más compleja que la termodinámica macroscópica, y la física microscópica usada con poco rigor lleva facilmente a razonamientos equivocados. Nuestro consejo, por lo tanto, es usar termodinámica macroscópica toda vez que sea posible, y si se necesita en algún punto usar física microscópica ser cuidadoso. Ese cuidado puede consistir simplemente en usar procedimientos estandar sólo para resolver problemas estandar y dudar sistemáticamente de cualquier uso de esas herramientas en contextos novedosos. En ese último caso lo mejor es pedir asesoramiento. ¿Está “mal” mezclar lo microscópico con lo macroscópico? Es imposible hacer termodinámica a partir de lo microscópico sin, en algún punto, hacer “trampa” y usar lo macroscópico. Eso significa que encontrar conceptos micro y macro en un mismo contexto no necesariamente es incorrecto. Pero que ambos niveles aparezcan no significa que estén “mezclados”. “Mezclarlos” significa hacer cosas como atribuir propiedades macroscópicas a lo microscópico (por ejemplo, decir que como el agua es transparente las moléculas de agua son transparentes). “Mezclar” es lo que mata, eso sí está decididamente mal. De todas maneras a veces se hacen engendros útiles. Nuestro consejo, en ese caso, es que consideren cualquier conclusión que obtengan de tales engendros como conjeturas provisorias, casi como si viniesen del sentido común, y que busquen modos mejores de chequearlos. ¿Está “mal” decir que la temperatura es la energía por molécula? Hablamos algo de esto al exponer las ideas de Boltzman. Recuerden que la base para esta caracterización es la llamada “equipartición de la energía”, y que este reparto se da no entre las moléculas sino entre los distintos grados de libertad de estas. Contar grados de libertad no es sencillo: los modos que se deben incluír y los que no dependen tanto de la temperatura como de la dinámica de la molécula (cálculo en el marco de la mecánica cuántica). Hay que tener mucha suerte para contar 51 correctamente esos grados de libertad mirando una representación esquemática de una molécula. Encima uno puede estar justo a una temperatura a la que se está “despertando” un grado de libertad. La cosa empeora si las moléculas son enormes y complejas como las de una proteína o un polímero, que tiene tantos modos de vibración que a cualquier temperatura se están “despertando” varios. En concreto: en un contexto donde tengamos ciertas garantías de cuáles son los modos de vibración (por ejemplo, porque alguien más ya lo calculó) puede ser útil usar esta idea de que la temperatura es “la energía por grado de libertad”. Sin esas garantías lo mejor es no pensarlo así. En particular, este criterio no es adecuado como definición de temperatura. Al final ¿Qué tienen que ver la entropía, el desorden y la información? La entropía de la termodinámica no tiene absolutamente nada que ver con desorden. ¿Cómo se “desordena” un gas? Acá va un típico ejemplo de falacia: un gas agitado, en el que cada parte va a velocidades diferentes, está más “desordenado” que un gas perfectamente uniforme, por lo tanto tiene más entropía. ¡NO! De hecho, si el gas agitado se lo deja evolucionar en ausencia de acciones del entorno, va a evolucionar a un estado de homogeneidad, y por el segundo principio (como el sistema está aislado la entropía del entorno no cambia) su entropía tuvo que aumentar. Otro: ordenando un mazo de cartas por palo y de menor a mayor le bajamos la entropía. Un disparate termodinámico: las variables termodinámicas de una carta no pueden depender del símbolo impreso en ella, La idea de “desorden” como sinónimo de “entropía” sólo tiene sentido en un contexto muy acotado, como metáfora para describir ciertos cálculos en teoría cinética. Con la “información” la cosa es aún más resbaladiza. La asociación viene de un problema tecnológico: cómo separar señal de ruido en una transmisión. Se supone que algo en equilibrio térmico no puede contener “información” (para transmitir información se necesita algo como pulsos, algo fuera del equilibrio). Así, hoy en día se usa la palabra “entropía” en criptografía, lo cual no quiere decir que los mensajes cifrados tengan temperatura o presión. Moraleja: tomen todas estas asociaciones como metáforas, y tómenlas con pinzas. Y sobre todo, no las usen como base para un razonamiento físico, porque lo más probable es que concluyan disparates. ¿Está “mal” no justificar rigurosamente todos los modelos y todas las herramientas que uno usa? No, no está “mal”. La rigurosidad es una herramienta metodológica, y como toda herramienta hay que usarla con juicio. El celo excesivo en el rigor puede resultar paralizante, mientras que el exceso de laxitud puede llevarnos a cometer errores groseros. Buscar el punto justo de rigor que permite avanzar con riesgo mínimo en cada paso de la propia investigación tiene algo (o mucho) de arte, y en parte tiene que ver con el perfil, el estilo y los gustos de cada quien como investigador. En esta materia hemos expuesto los temas con todo el rigor posible, de manera que ustedes ya saben que existe ese rigor, y pueden apelar a él cada vez que lo necesiten. 52