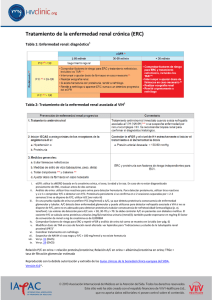
3 ERA UNIDAD RENAL:SINDROME NEFROTICO PROTEINURIA: DE 3.5 G POE 1.73 M2 EN 24 HRS HIPOALBUMINEMIA, HIPERCOLESTEROLEMIA, LIPIDURIA HIPERLIPIDEMIA , EDEMA Es el resultado del infcrmento de la permeabilidad glomerular, esto quiere decir que el paciente presentara proteinuria e hipoalbuminea y se acompaña de edema, este sindrome incluye un factor de mal pronostico en la evolucion de la lesion renal, y este se asocia a complicaciones sistemicas, que podemos prevenir o reconocer cuando aparescan Etiología del síndrome nefrótico: enfermedades glom erulares secundarias ENFERMEDADES SISTÉMICAS Lupus eritematoso sistémico* Síndrome de Goodpasture Lipodistrofia parcial Dermatomiositis Púrpura de Schönlein-Henoch Síndrome de Sjögren Enfermedad mixta del tejido conectivo Vasculitis sistémicas Sarcoidosis Artritis reumatoide Crioglobulinemia mixta esencial Glomerulonefritis fibrilar ENFERMEDADES METABÓLICAS Y HEREDOFAMILIARES Diabetes mellitus* Amiloidosis* Enfermedad de Fabry Síndrome de Alport Síndrome nefrótico congénito Hipotiroidismo Drepanocitosis Enfermedad de Graves-Basedow Déficit de a1-antitripsina ENFERMEDADES INFECCIOSAS Bacterianas (GN postestreptocócicas, endocarditis infecciosa, nefritis de shunt, sífilis, tuberculosis, pielonefritis crónica) Víricas (hepatitis B y C, inmunodeficiencia humana [HIV], citomegalovirus, Epstein-Barr) Otras (paludismo, toxoplasmosis, filariasis, tripanosomiasis) *Las más frecuentes. GN: glomerulonefritis; HTA: hipertensión arterial. FÁRMACOS AINE* Captopril* Mercurio* Sales de oro* Penicilamina* Heroína Interferón a Warfarina Contrastes yodados Probenecid Rifampicina Litio Clorpropamida NEOPLASIAS Tumores sólidos (carcinomas y sarcomas)* Linfomas y leucemias OTROS Preeclampsia HTA vasculorrenal unilateral Nefropatía crónica del injerto renal* Nefropatía por reflujo Nefroangiosclerosis Obesidad mórbida FISIOPATOLOGIA Fisiopatologicamente el síndrome nefrótico se desencadena tras la alteración de la barrera de filtración glomerular, por una lesion generada antes en los podocitos,dejando par las proteinas que condiciona la pérdida de proteínas por la orina conocida como proteinuria y como consecuencia, la hipoalbuminemia y el resto de las alteraciones del síndrome nefrótico. PROTEINURIA : puede producirse por una pérdida de la electronegatividad de la barrera de filtración o por una desestructuración que condiciona un aumento del tamaño de los poros. Pues la proteinuria es altamente selectiva (se pierde sobre todo albúmina y otras proteínas negativas, mientras quedan retenidas las de mayor peso molecular, como la IgG HIPOPROTEINEMIA : se genera cuando la proteinuria y el catabolismo tubular renal de la albúmina filtrada superan la tasa de síntesis hepática. EDEMA : Existen dos mecanismos 1-por hipovolemia, la presión oncótica plasmática desencadena la aparición de edema, hipovolemia y la activación del sistema renina-angiotensina que lleva a un aumento de la reabsorción renal de sodio. 2-Se da por la liberación de angiotensina II y oxidantes tras el PRUEBAS DE LABORATORIO DE BASE Suero: glucosa, creatinina, urea, iones, proteínas totales, albúmina, colesterol LDL y HDL y triglicéridos Orina: proteinuria de 24 h (al menos dos determinaciones; en el seguimiento puede utilizarse el cociente proteínas/creatinina en muestra de orina reciente), iones Filtrado glomerular estimado por fórmulas. Puede considerarse aclaramiento de creatinina Sedimento urinario Fisiopatologia To empieza por que se genera una lesion renal , como consecuencia del deposito de inmunocomplejos en el interior de y liberacion de mediadores inflamatoriosl capilar glomerular, activacion del complemento, el deposito glomerular de C3 y de IgG, se da por una disminucion de las concentraciones plasmaticas de C3, del proactivador c3 y de la properdina. Normalmente vamos haber concentracion del C4 , que nos indicaran la activacion del complemento. Donde abra liberacion den citocinas que amplifican la reaccion inmunologica como el TNF alfa y interleucinas 1 y 6,como consecuencia de la inflamacion glomerular se produce disminucion en la excresion renal de agua, sodio y la hipervolemia, la disminucion del filyrado glomerular genera una disminucion de la creatinina y la permeabilidad de la membrana basal glomerular ocasiona hematuria y proteinuria, por una disminucion de la actividad de la de la renina, aldosterona y vasopresina, así como un aumento del péptido natriurético atrial, tras la supresion de la actividad del sistema renina plasmatica, van actuar los inhibidores de la enzima de conversion de la angiotensina, para generar un aumento del filtrado glomerular y esto nos indica que hay un daño en la actividad intrarenal de la angiotensina 2 y pues las prostanglandinas E, F y la calicreina van a estar disminuidas en la orina FALLA RENAL AGUDA Incremento ede creatinina:2,4 y urea 150 Analitica, examen de orina, examen de proteinas en 24 horas, Ecografia renal para ver si hay lesiones Insuficiencia renal cro Falla renal cronica reagudizada d. Tbc Sindrome donde se evidenciara un descenso de la tasa de filtracion glomerular, por un aumento de la concentración plasmática de BUN y de creatinina,, tambien puede acompañarse de un descenso de la diuresis. Asimismo puede tener lugar en riñones con función basal normal o en riñones con insuficiencia crónica previa Tabla 90-1 Cla ESTADIO AKIN sificación funcional de la IRA según la AKIN y RIFLE CRITERIOS DE CREATININA CRITERIOS DE DIURESIS 1 creat. ≥ 0,3 mg/dL o hasta ≥ 150%-200% < 0,5 mL/kg/h durante 6 h 2 creat. creat. hasta > 200%-300% < 0,5 mL/kg/h durante 12 h 3 creat. hasta > 300% o creat. ≥ 4 mg/dL con un aumento agudo ≥ 0,5 mg/dL < 0,3 mL/kg/h durante 24 h o anuria durante 12 h ESTADIO RIFLE CRITERIOS DE CREATININA CRITERIOS DE DIURESIS R (risk) creat. 1,5 veces o FG > 25% < 0,5 mL/kg /h durante 6 h I (injury) creat. 2 veces o FG > 50% < 0,5 mL/kg/h durante 12 h creat. 3 veces o FG > 75% o < 0,3 mL/kg/h durante 24 h o Fisiopatologia Fisiopatologicamente la IRA, tiene mecanismos que desencaden dicha patologia por una necrosis tubular aguda izquemica o toxica , pues esta se basa debido a 2 allteraciones debido a la hipoxia y depleción de ATP: la lesión endotelial-vascular y la lesión tubular, ambas van a contribuir al descenso de la tasa de filtracion glomerular, pues las causas que descencadenn esta patologia son debido a una serie de procesos como un tono vasomotor aumentado con reducción del flujo sanguíneo renal, lesión del epitelio tubular con necrosis y apoptosis frecuente y desprendimiento de las células tubulares de la membrana basal que conduce a obstrucción de la luz tubular más distal por cilindros compuestos por células tubulares desprendidas, restos necróticos ensamblados todos en proteína de TammHorsfall, retrodifusión del FG desde la luz tubular a los vasos por la pérdida de la barrera epitelial y, en la reperfusión, inflamación y lesión de estrés oxidativo mediado por la infiltración de leucocitos. Hay que tener encuenta que la necrosis tubular aguda se divide en 4 fases: 1-iniciación: lugar la agresión renal que conduce a la IRA y a la cascada de fenómenos microvasculares y tubulares que interaccionan y contribuyen al desarrollo y al mantenimiento de la IRA. 2-extensión: 1-3 días de la agresión al riñón las lesiones microvasculares y la inflamación inciden sobre la función renal. 3-mantenimiento: se prolonga a semanas, tiene lugar la regeneración del tejido renal. 4-recuperación: restablecimiento total o parcial de la función renal debido a los procesos de rediferenciación y repolarización de las células tubulares. PRUEBAS DE LABORATORIO Falla renal cronica La enfermedad renal crónica (ERC) se define como la presencia persis- tente durante > 3 meses de alteraciones estructurales o funcionales del riñón que tienen implicaciones para la salud y que se manifiestan por: a) indicadores de lesión renal, como alteraciones en estudios de laboratorio en sangre u orina (p. ej., elevación de la creatinina sérica, proteinuria o hematuria glomerular), en estudios de imagen (p. ej., riñón poliquístico) o en una biopsia (p. ej., glomerulopatía crónica), independientemente de que se acompañen o no de una disminución del filtrado glomerular (FG), y b) un FG menor de 60 mL/min por 1,73 m2 de superficie corporal, independientemente de que se acom- pañe o no de otros indicadores de lesión renal. FISIOPATOLOGIA Fisiopatologicamnete la enfermedad renal cronica, va estar acompañada por una disfuncion progresiva de de la funcion renal, lo que genera: 1) alteraciones del equilibrio hidroelectrolítico y acidobásico, 2) acumulación de solutos orgánicos que normalmente son excretados por el riñón, y 3) alteraciones en la producción y metabolismo de ciertas hormonas, como la eritropoyetina y la vitamina D, cuando la funcion renal disminuye se activan una serie de mecanismos de compensacion, es por eso que muchos pueden estar asintomaticos, a pesar de haber perdido un gran porcentaje de masa renal, dentro de los mecanismo tenemos a la hiperfiltracion glomerular, en donde las nefronas no dañadas por la lesion inicial se vuelven hiperfuncionantes, asimismo esta permite mantener un balance de liquidos y electrolitos hasta una fase avansada de la ERC, induciendo una glomerulosclerosis, que contribuye a aumentar el daño en las nefronas y cuando estan nefronas llegan a un nivel critico , ya los mecanismo de compensacion no seran suficientes, y se va a generar alteraciones bioquímicas y clínicas del síndrome urémico. Hay que tener encuenta que la hiperfiltración en las nefronas es un aumento de la presión hidrostática a nivel de los capilares glomerulares (hipertensión glomerular), generada tras latransferencia de presión sistémica a los glomérulos o tambien de cambios hemodinámicos locales, como incremento del flujo plasmático secundario a vasodilatación de la arteriola aferente. Vamos haber que se va adar hipertension capilar glomerular, la cual daña los capilares glomerulares y va a distender las celulas mesagiales, generando aumeto de la síntesis de citocinas que inducen proliferación y fibrogénesis, pues estos cambios en el paciente se manifiestan como hipertrofia glomerular y finalmente glomerulosclerosis que favorece la hiperfiltración en las nefronas aún sanas, con lo que finalmente progresa la ERC terminal Otros factores que secundariamente pueden contribuir a la progresión de la lesión renal incluyen: •Proteinuria. Contribuye debido al efecto tóxico de algunas proteínas sobre las células y matriz mesangiales, y las células tubulares, así como por inducción de la síntesis de moléculas proinflamatorias y citocinas. •Hipertensión arterial. Además de ser un factor de riesgo cardiovas- cular favorece la progresión de la ERC al aumentar la presión capilar glomerular y, por tanto, los fenómenos de hiperfiltración y glomerulosclerosis; además, las alteraciones vasculares propias de la hipertensión arterial (hiperplasia e hialinosis arteriolar) causan disminución del flujo plasmático renal y del FG, tanto más acusado cuanto mayor es el incremento de las cifras de PA y su duración. Lesiones tubulointersticiales. La dilatación tubular y la fibrosis inters- ticial ocurren en prácticamente todos los casos de ERC independientemente de la causa y suelen ser un mejor índice pronóstico de la reducción del FG que las mismas lesiones glomerulares. Aunque el mecanismo no se conoce del todo, varios factores pueden contribuir a su desarrollo, como la participación del intersticio en el proceso inflamatorio que acompaña a las glomerulonefritis, el depósito en el intersticio de fosfato cálcico y amonio (este último se acumula en las nefronas hiperfuncionantes que excretan una canti- dad mayor de ácido y puede causar lesión al activar el complemento por la vía alternativa), las alteraciones (activación y rarefacción) del endotelio de los capilares peritubulares debidas a isquemia y el posible efecto proinflamatorio de las proteínas y el hierro libre en contacto con los túbulos. También contribuye el efecto fibrogénico de la angiotensina II y la aldosterona. • Hiperlipemia. Puede contribuir a la lesión renal al activar la proli- feración de las células mesangiales (las cuales tienen receptores para LDL), fibronectina (que es un componente de la matriz mesangial), factores quimotácticos de los macrófagos y especies reactivas de oxígeno. Sin embargo, la evidencia clínica es insuficiente para confirmar el efecto del tratamiento de la hiperlipemia en la pro- gresión de la ERC, así como el objetivo terapéutico más apropiado. • Tabaquismo. Puede contribuir a la progresión de la ERC por varios mecanismos, como hiperfiltración glomerular, disfunción endote- lial e incremento de la proteinuria. Sin embargo, no hay estudios prospectivos que demuestren el efecto de la suspensión del tabaco en la progresión de la ERC. • Factores genéticos. Ciertos polimorfismos del gen de la apolipo- proteína E (apoE) se asocian con un mayor riesgo de aterosclerosis y progresión de la ERC. • Otros factores. Aunque la anemia, la hiperuricemia, la acidosis y las alteraciones del metabolismo mineral pueden contribuir a la progresión, la evidencia es todavía insuficiente para recomendar su corrección únicamente con el fin de retrasar la progresión de la ERC. DIABETES MELLITUS Puedo definir a la diabetes mellitus, como un desorden metabolico, que se debe a causas multiples, desde una destruccion inmunitaria de las celulas b, hasta una deficiencia en la accion de la insulina en sus tejidos diana, el cual se va a carcaterizar por la hiperglucemia y esta se va asosiar a alteraciones en el metabolismo de hidratos de carbono, proteinas y grasas, las cuales se han generado debido a una consecuencia de defectos en la secresion o accion de la insulina. Tambien hay que tener en cuenta que la hiperglucemia cronica que es propia de la DM ,se asoca con disfunciones, lesiones o fracasos que se da en diversos organos, en especial en (vasos sanguineos, nervios,riñones,corazon,ojos). Dentro de sus signos o sintomas, veremos que el paciente puede presentar, polifagia,sed, poliuria, perdida de peso, ETIOLOGIA 1.DIABETES DE TIPO 1 (destrucción de la célula b que usualmente tiende a la deficiencia absoluta de insulina) A. Autoinmunitaria B. Idiopática 2.DIABETES DE TIPO 2 (desdeunpredominioderesistenciaala insulina con relativa deficiencia a un predominio de defecto secretor con resistencia a la insulina) FACTORES DE RIESGO DE LA DIABETES TIPO 2 -Indice de mas corporal (IMC)mayor a 25 o al percentil 85 - Perimetro de la cintura >80 cm en mujeres y >90 cm en mujeres indican un exceso de grasa visceral - Antecente familiar de diabetes en primer o segundo grado. - Procedencia rural con urbanizacion reciente. - Antecendente obstetrico de diabetes gestacional o hijos con peso mayor de 4 kg al nacimiento - Enfermedad izquemica coronaria o vascular de origen ateroesclerotico. - Hipertension arterial. - Trigliceridos ≥ 150 mg/dl. - Colesterol HDL <40mg/dl. - Masocrosomia - Sedentarismo - Enfermedades asociadas(deterioro cognitivo, deficit de audicion, esquizofrenia, apnea, canceres y esteatosis hepatica) - Sindrome de ovario poliquistico. PRUEBAS DE DIAGNOSTICO: -prueba de tolerancia a la glucosa oral -La prueba de hemoglobina glicosilada DIAGNOSTICO DIFERENCIAL: -Cetoacidosis ENFERMEDAD CARDIOVASCULAR Dislipemia Hiperglucemia Hipertensión arterial PIE DIABÉTICO Neuropatía motora proximal Enfermedad renal) fisiopatologia La diabetes mellitus tipo 2, se desarrolla tras una respuesta inadecuada de celulas b pancretica, resistentes a la insulina, que esta asociada principalmente a la obesidad,sedentarismo y envejecimiento. Tras la acumulacion excesiva de grasa en nuestro paciente, va a producir una disfuncion y agotamiento de la capacidad de almacenamiento del tejido adiposo. Hay que tener encuenta que hay grasa que no se puede almacenar en el tejido adiposo, lo cual va a roducir en este organo un efecto inflamatorio, donde habra infiltracion de macrofagos, que liberan citocinas proinflamatorias que contribuyen al desarrollo de la resistencia de insulina en el tejido adiposo y organos perifericos. Tras la incapacidad de almacenar grasa en el tejido adiposo, , hara que la almacene de manera ectopica en musculo, celulas b, higado, y pues estos claramente no estan diseñados para almacenar este tipo de lipidos, lo que conlleva a producir una lipotoxicidad que contribuye a la resistencia de insulina y a la disfuncion mitocondrial. La Resistencia de insulina disminuye la utilizacion de glucosa a nivel del musculo y en el higado incremeta la produccion hepatica de glucosa, las celulas b se ven afectadas tambien por la lipotoxicidad e inflamacion del tejido adiposo, musculo, higado y celulas b. Las manifestaciones clinicas de este sindrome metabolico, dependen del grado de vulnerabilidad genetica en las celulas b, tambien hay genes relacionados con el desarrollo de diabetes mellitus tipo 2 como son dKAL1, CdKN2A, CdKN2B, HHEX, IGF2BP2, WFS1, MTNR1B, IRS1, SLC30A8, PPARg, KCNJ11 y TCF7L2, que se asocian con el inico temprano de la diabetes, pues estan relacionados con la secresion de la insulina y pues la hiperglucemia presente en la diuabetes mellitus tipo 2 se va a dar como resultado de una falla en la secresion y sensibilidad insulinica, que va adepender de la interacciom entre factores aambientles y los genomas que confiere suceptibilidad genetica Para hablar de DM 2 es útil diferenciarla en dos grandes grupos: a) DM 2 asociada a obesidad, y b) DM 2 no asociada a obesidad. INSUFICIENCIA RENAL AGUDA, INJURIA RENAL AGUDA, FRACASO RENAL AGUDO