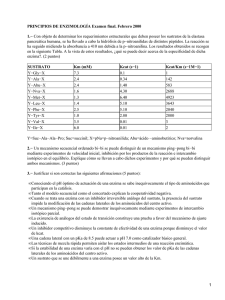
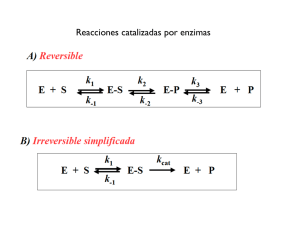
Texto para el curso
Anuncio

Material de apoyo para el módulo de CINÉTICA RÁPIDA.
Rogelio Rodríguez Sotres
2015-04-21
Curso: Cinética Enzimática Avanzada
Modalidad: Intensiva
Coordinador: Dr. Ismaél Bustos Jaimes
Programa de Maestría y Doctorado en Ciencias Bioquímicas
UNIVERSIDAD NACIONAL AUTÓNOMA DE MÉXICO
Profesor del módulo: Rogelio Rodríguez Sotres
Semestre: 2015-2 (Febrero-mayo, 2015).
0.1
Abreviaturas
vi , rapidez instantánea.
v0 , rapidez inicial.
t1/2 , vida media.
SS , Estado estacionario.
v, rapidez de reacción.
ode, ecuación diferencia ordinaria.
1
Diferencias conceptuales entre las condiciones de estado
estacionario y las condiciones de estado preesatacionario.
Los procesos biológicos ocurren a diferentes escalas de tiempo. Comúnmente consideramos que
numerosas reacciones químicas ocurren exclusivamente en los seres vivos, porque su ocurrencia es
muy lenta en ausencia de catalizadores - a veces - casi imperceptible.
Por el contrario, la ocurrencia de mucha reacciones enzimáticas es demasiado rápida in vivo.
Así, con el propósito de estudiarlas y conocer el efecto de diversas condiciones sobre la rapidez de la
reacción (reactantes, productos, cofactores, catalizador, pH, temperatura, etc.), las condiciones in
vitro se manipulan para hacerlas más lentas y seguirlas con instrumentos convencionales, tales como
un cronómetro y un colorímetro. Además, para que la medición sea posible, el régimen de flujo
de la reacción catalizada se limita al rango en que la cantidad del reactante consumido o producto
acumulado cae dentro del rango de sensibilidad del método de medición. El régimen de flujo más
comúnmente empleado y que cumple con estas condiciones se denomina estado estacionario (SS ).
Sin embargo, Mediante el uso de instrumentos especializados, se pueden auscultar eventos que
ocurren en fracciones de segundo, antes de que se establezca el
SS. Estos eventos pueden brindar información muy valiosa acerca de la catálisis.
1
1.1
Ejemplo 1.
Para ejemplificar lo anterior, consideremos un caso simple:, la desintegración radiactiva de un
material.
Suponga un material radiactivo cuya emisión se detecta con un contador Geiger. Para conocer
la cantidad de radiaciones emitidas por unidad de tiempo, el contador nos permite medir las cuentas
por minuto o CP M y, mediante la calibración de la eficiencia del contador, es posible calcular las
desintegraciones por minuto (DP M ). Dicha medición es una medida en condiciones de
SS.
Lo anterior nos dice si el material es peligroso o no, pues mide la dosis de radiación que recibiría
un sujeto expuesto al material, pero dice poco de la naturaleza de dicho material.
Si ahora registramos la variación en la emisión respecto del tiempo, es decir, hago esa misma
determinación varias veces en los siguientes 150 días, tengo el resultado que se ilustra en la figura
1.
La teoría de la desintegración radiactiva nos dice que los átomos radiactivos son inestables,
pero se descomponen de modo aleatorio. En un número muy grande de átomos radiactivos la
probabilidad de observar una desintegración es proporcional a la cantidad de material radiactivo.
es decir:
dpm = kC (Ecn. 1)
En donde C es la cantidad de átomos radiactivos y k una constante de proporcionalidad. Cada
desintegración significa que perdimos un átomo radiactivo, es decir, en un intervalo de tiempo
suficientemente pequeño (dt) el material radiactivo perdido (−dC) se reduce de manera proporcional
a su concentración:
(Ecn. 2)
− dC
dt = kC
La ecuación 2 es lo que se conoce como una ecuación diferencial ordinaria (ode ), porque
contiene la variables y sus diferenciales. Resolver una ecuación diferencial requiere expresar la
variable dependiente (C) como una función de la variable independiente (t) que cumpla con lo que
expresa la ode. En este ejemplo, la variable t, sólo aparece como diferencial y se puede separar de
la Variable C que aparece en ambas formas. Entonces las diferenciales se pueden eliminar mediante
la operación inversa, es decir la integración.
RC
Rt
− C0 dC
C = k t=0 dt,
que resulta en:
ln C = C0 − kt (Ecn. 3)
podemos ajustar la curva de la figura 1 a la ecuación 3 y deducir que la vida media del material
(t1/2 = lnk2 = 17.17 ± 0.23 días). Las vidas medias son invariantes características de cada material.
De aquí, podemos deducir qué, para este ejemplo, el radionúclido era 32P y dar la cantidad de
átomos de 32P presentes al día cero (quizá antes de recibir acceso al material).
1.2
Recapitulemos:
1. La medición de la cantidad de radiactividad es una medida de rapidez instantánea (vi ) de
descomposición, el tiempo que toma la medida es un instante, si se compara con la vida media
del material (días).
2. La vi sólo depende del isótopo que se tiene y de su cantidad en la muestra.
3. La vi varía de modo definido en función del tiempo y se puede modelar con una ecuación
diferencial, que define a la vida media, la cual es una firma del elemento.
2
Figura 1. Cuentas radiactivas por minuto emitidas por un material en función del tiempo transcurrido (días). La línea muestra el ajuste a una curva de decaimiento exponencial con un valor
−1
inicial de 25017 ± 53 DP M y una constante de decaimiento 0.0403481 ± 0.0003735 días . Además,
la estabilidad del isótopo es inversamente proporcional a la t1/2 y a la energía de las partículas
emitidas.
3
De modo análogo, si estudiamos la rapidez de una reacción química en SS , la concentración de
los diversos intermediarios de la enzima ya es constante. Podemos saber quizá que intermediario
es al más abundante y como es que el balance de las especies en el SS responde a cambios en la
temperatura, concentración de reactante, etc. Pero las vidas medias de los intermediarios serán
desconocidas y no sabremos nada de su estabilidad relativa.
Por el contrario, si podemos saber la vida media de un intermediario, podremos inferir sobre su
estabilidad, su energía y podemos hacer propuestas acerca de su naturaleza.
A nivel macroscópico, los principios que gobiernan el comportamiento de los sistemas en equilibrio no son distintos de los que gobiernan las situaciones en SS o en pre-SS . Sin embargo, estudiar
un sistema en equilibrio es más sencillo que estudiar un SS , y estudiar el pre-SS se complica aún
mas.
La diferencia entre estudiar un sistema en SS y estudiarlo en otros regímenes de flujo está en
la manera en los métodos de medida y la elección de las condiciones experimentales. Tal elección
determina también los instrumentos a usar y la manera en que analizaremos nuestros datos.
2
Conceptos básicos: Equilibrio, estado estacionario, ciclos
límite y estados transitorios.
Todo proceso natural obedece a las leyes de la termodinámica. Al aplicarlas al caso especial de las
transformaciones químicas se pueden derivar ciertos principios básicos que son centrales en cinética
química.
1. La energía se conserva. Por lo tanto, los valores de energía de las partes, mídanse como se
midan, suman siempre la energía del todo. Multiplicar energías equivaldría a crear energía y
dividir dos energías equivale a desaparecer energía.
2. En las transformaciones químicas, la masa también se conserva. Es decir, la suma de la masa
de todas las especies presentes en el sistema no cambia con el tiempo.
3. Una consecuencia de la segunda ley de la termodinámica es que todos los sistemas tienden
al equilibrio. Tal tendencia, sin embargo, no nos dice cual es el camino que los sistemas
siguen para llegar a dicho equilibrio. En la mayoría de las transformaciones químicas, el proceso de aproximación al equilibrio ocurre transitando por un cierto número de intermediarios
definidos. Cada intermediario aparece como resultado de la transformación del intermediario
anterior. Por ende, el sistema sigue una trayectoria con secuencia definida, aunque puede
estar ramificada.
4. En el equilibrio, los sistemas obedecen a un principio termodinámico postulado por Le Chatelier, (Henri-Louis Le Chatelier; 1850 - 1936) que al ser aplicado a sistemas químicos da lugar
a la llamada ley de acción de masas y se resume así:
“un proceso químico ocurre con un rapidez proporcional a la concentración de las
especies que participan en dicha transformación.”
5. Este principio se aplica a un sistema en equilibrio. Sólo puede aplicarse a las transformaciones
parciales dentro de la trayectoria hacia el equilibrio, si conocemos cuales especies individuales
participan en cada transformación parcial y el orden en que aparecen. A esta última secuencia
se le conoce como el mecanismo cinético de la reacción.
4
Según lo anterior se pueden modelar un mecanismo cinético si plantemos una propuesta correcta
para los intermediarios. Nuestro modelo debe coincidir ocn los datos experimentales de la aparición
y/o desaparición de uno o más intermediarios. La concordancia es evidencia indicativa, pero no es
garantía) de que el mecanismo sea correcto.
En un modelo cinético la concentración de cada intermediario se describe mediante una función
del tiempo. Dicha función depende de los intermediarios previos y de los que se generan después.
Consideremos pues un sistema genérico:
R → n1 I1 → n2 I2 ... → mP
La concentración de cada especies puede describirse como una función del tiempo:
CR = f0 (t); CIi = fi (t)...; CP = fp (t)
Es decir, para n intermediarios habrán n funciones del tiempo. Además, la masa se conserva,
por lo que la enésima función puede obtenerse por diferencia entre la masa total y un conjunto de
n − 1 funciones cualesquiera.
CP = m(MT OT AL − CR +
1
1
1
C I1 −
CI2 ... +
CI
)
n1
n2
nm−1 m−1
En su tránsito hacia el equilibrio pueden el régimen de flujo de un sistema cambia y su modelado
se facilita o se complica. A saber, los regímenes de flujo pueden ser:
1. Caóticos: Son aquellos en que la función que describe la concentración presenta cambios
que no siguen ningún patrón regular apreciable. Usualmente los sistemas químicos bajo
condiciones controladas no siguen este tipo de regímenes, pero los sistemas complejos de
múltiples reacciones pueden mostrar comportamientos que son cuasicaóticos.
2. Ciclo límite o cuasiperiódicos: Son aquellos en que el sistema presenta oscilaciones regulares
y los intermediarios aumentan y disminuyen su concentración entre valores definidos, con
intervalos de tiempo (periodos) definidos. Generalmente, tardan fracciones de segundo en
establecerse y se agotan después de un tiempo. Mientras duran, es posible observar que la
aparición de las especies finales (productos) ocurre con una rapidez constante (véase figura
2).
3. Decaimiento o crecimiento exponencial, o cuasiexponencial: Ocurren cuando el aumento o la
disminución en la concentración de una especie es proporcional a la concentración de dicha
especie, o de alguna otra especie relacionada.
4. Estado estacionario (SS ): Son aquellos en los que las especies intermediarias se mantienen
casi constantes en función del tiempo. Su duración es corta, ya que persisten mientras la concentración de las especies iniciales no cambia de manera importante y los flujos se mantienen
razonablemente constantes (ver figuras 3 y 4).
La cinética de pre-SS considera los procesos que ocurren antes del establecimiento de un SS .
En el caso de las reacciones enzimáticas, los procesos suelen seguir regímenes del tipo (3), aunque
se han reportado algunos casos en los que se observan comportamientos del tipo (2).
El modelado de los regímenes de tipo (3) es posible empleando métodos analíticos o métodos
numéricos. Los métodos analíticos se pueden emplear si el sistema de ode puede resolverse, lo que
solo es posible para unos pocos casos. Una vez establecido un modelo válido, este puede usarse para
5
Figura 2. Un ejemplo de un ciclo límite es el flujo glucolítico en la levadura S. cereviciae incubada con concentraciones muy elevadas de glucosa y en presencia de cianuro, mismo que puede
monitorearse mediante la absorbencia de NADH en las células completas (Danø et al., 1999).
calcular el valor de los parámetros ajustables que mejor describen el comportamiento observado.
Para tal propópisto se recurre a los métodos estadísticos de regresión no lineal (NRL ). Cuando el
sistema de ode no puede resolverse analíticamente, es posible combinar los métodos numéricos de
integración de las ode con los métodos de NRL para estimar los parámetros del sistema.
Dado que el modelo se refiere a las diferentes especies del sistema, dichos parámetros indican
propiedades de los intermediarios como estabilidad, cambios en la energía libre asociados a su
aparición, etc. Todos estos datos pueden proporcionar gran información acerca de los actores
participantes, particularmente, acerca de la enzima en estudio.
3
Ejemplos que ilustran el poder de esta estrategia.
3.1
Ejemplo 1: Un sistema en equilibrio no equivale a reposo absoluto.
Supongamos una proteína P se une a una substancia L y forma PL
k1
P +L
k2
PL
Esquema 1.
Entonces, suponiendo que la reacción ocurre con un mecanismo simple, la rapidez con que P se
transforma en PL será:
− dP
dt = k1 [P ]EQ [L]EQ
y, de igual manera, al alacanzar el equilibrio, PL se estará convirtiendo de regreso en P según:
L
− dP
dt = k2 [PL ]EQ .
El equilibrio implica que la rapidez de formación de PL iguala a la rapidez de disociación:
dPL
− dP
y,
dt = − dt
por lo tanto:
k1 [P ]EQ [L]EQ = k2 [PL ]EQ
Así, si las concentraciones no son cero, tenemos:
[PL ]EQ
= kk21 = KF (Ecn. 4)
[P ]EQ [L]
EQ
6
A) Equilibrio
Nivel de agua
constante
Ejemplo físico: Dos tanques conectados por un tubo
Liberación de
energía
P
Energía
potencial
acumulada
P=0
Situación de
equilibrio
El volumen de agua es diferente,
pero las presiones son iguales.
Ejemplo químico: Unión del oxígeno a la Mioglobina.
En el equilibrio el potencial químico se
iguala (es decir G=0).
Mb
+
O2
MbO2
EL equilibrio puede describirse numéricamente
mediante una constante de equilibrio.
K equilibrio=
[ MbO 2 ]eq
[ Mb ]eq⋅[ O 2 ]eq
B) Estado estacionario.
P1
Nivel de agua
aparentemente
constante
P2
Estado
estacionario
(SS)
P2
P1
Ejemplo físico: Imaginemos una orificio en la base del tanque pequeño del
sistema anterior.
Más tarde, P1 se reduce y P2 aumenta, la
fuga en el orificio iguala al flujo entre tanques.
Inicio: el agua fluye rápidamente
hacia el segundo tanque, pero sale
lentamente por el orifício.
Ejemplo químico: Catálisis enzimática. La velocidad con la que E.S es formado, por la reacción
entre E y S, iguala a la velocidad con la que E..S se disocia
o cataliza la conversión de S a P.
E
+
KM
S
E·S
kCAT
E
+
productos
K M=
[ E ]ss [ S ] ss
[ E.S ] ss
La constante KM no es una constante de equilibrio, se denomina constante de Michaelis
Figura 3. Comparación entre sistemas físicos y químicos en equilibrio (A), o en SS (B).
7
E
+
KM
E·S
S
kCAT
productos
+
E
Figura 4. Comportamiento clásico de un sistema enzimático simple en su aproximación al régimen
de SS que conocemos como rapidez inicial ( v0 ).
El equilibrio es un proceso terminal que alcanza un sistema si su energía libre ha sido agotada. El
sistema permanece inmutable de modo indefinida y la concentración de las especies ya no cambiará.
Pero,
¿Es el equilibrio un estado en el que las moléculas se encuentran en reposo?
¡Claro que no!
veamos:
3.1.1
Ejercicio resuelto 1. Tasa de conversión en el equilibrio.
Consideremos el sistema anterior (Esquema 1) iniciando con una concentración de 1 × 10−9 M de
proteína total y 1 × 10−5 M total del ligando, siendo:
k1 = 100 M −1 s−1 y k2 = 0.1 s−1 ,
en estas condiciones:
M −1 s−1
KF = kk12 = 1000.1
= 1 × 103 M −1
s−1
y, cuando se alcanza el equilibrio podemos hacer las siguientes consideraciones:
Sea xEQ la concentración del complejo proteína-ligando en equilibrio ( [PL ]EQ ). Entonces, la
estequiometría de reacción indica que:
[P ]EQ = (1 × 10−9 M − xeq ) y
[L]EQ = (1 × 10−5 M − xeq )
y substituyendo en la Ecn. 4 tendremos:
xeq
−1
KF = (1×10−9 M −xeq )(1×10
,
−5 M −x ) = 1000 M
eq
es decir:
1000 M −1 x2eq − (1 + 1 × 10−2 + 1 × 10−6 )xeq + 1 × 10−11 M = 0,
de donde:
8
√
(1+1×10−2 +1×10−6 )±
(1+1×10−2 +1×10−6 )2 −4×10−8
M = 9.9 × 10−12 M ,
xEQ =
2000
por lo que:
[P ]EQ = (1 × 10−9 − 9.9 × 10−12 ) M = 9.901 × 10−10 M y,
[L]eq = (1 × 10−5 − 9.9 × 10−12 ) M = 9.999 × 10−6 M .
L]
−1
De lo anterior, se sigue: d[P
)(9.9 × 10−12 M ) = 9.9 × 10−13 M/s.
dt = k2 [PL ]EQ = (0.1s
Si consideramos una cubeta de 1 ml y tomamos en cuenta el número de Avogadro (6.022 ×
1023 moléculas mol−1 ) tendremos:
d[PL ]
= (9.9 × 10−13 mmol ml−1 s−1 )(6.022 × 1020 moléculas mmol−1 )
dt
= 5.962 × 109 moléculas/ s
Es decir, aún en el equilibrio y a concentraciones tan bajas como las que se suele manejar en los
laboratorios, la tasa de reacción es muy elevada, sólo que ocurre en ambos sentidos por igual.
3.1.2
Ejercicio no resuelto 1.
Calcule la tasa de interconversión usando ahora la expresión equivalente
− dP
dt = k1 [P ]EQ [L]EQ
y compruebe que el flujo en ambas direcciones es el mismo.
3.2
Ejemplo 2: Tiempo en que se requiere para que se alcance el equilibrio en el sistema del ejemplo anterior.
Para modelar la trayectoria de aproximación al equilibrio, requerimos considerar la velocidad de
ambas reacciones (asociación como la de disociación) pero mientras no se ha establecido el equilibrio.
La tasa neta de acumulación de PL dependerá de la diferencia entre las tazas de formación y de
descomposición. si “x” es la cantidad de PL formado al tiempo t, tenemos:
k2
dx
2
(Ecn 5).
dt = k1 (P0 − x)(L0 − x) − k2 x = k1 P0 L0 − k1 (P0 + L0 + k1 )x + k1 x
Este caso sencillo, da un ecuación diferencial no tan sencilla, que es de primer orden, pero
no homogénea (“x” aparece sin derivar) y de segundo grado (está elevada al cuadrado). Existen
métodos para resolver esta ecuación y los casos frecuentes que tienen solución analítica ya han sido
descritos (Consultar la tabla 2 de la referencia: Kuby, SA. A study of Enzymes. Vol. I Chap. 10
pp. 377-415).
Pero en lugar de tomar la solución completa, aprovechemos el sistema para aprender a simplificar
problemas complejos mediante la manipulación de la condiciones experimentales.
En nuestro caso, la concentración de proteína total es cuatro órdenes de magnitud inferior a la
de ligando. Por lo tanto, la cantidad de complejo formado (x) será despreciable en comparación
con la de ligando libre. Es válido asumir entonces que:
(x L0 ) ⇒ ([L] = L0 − x ≈ L0 ) ,
con lo que, el mecanismo del esquema 1 se simplifica para dar el esquema 2.
y la ode 5 se simplifica a la ecuación 6.
AP P
dx
k1 P0 − (AP P k1 + k2 )x (Ecn. 6)
dt = k1 (P0 − x)[L0 ] − k2 x =
9
APP
P
k1= k
1
L
k2
PL
Esquema 2
3.2.1
Ejercicio no resuelto 2.
Resuelva la ode anterior (Ecn. 6) por el método de separación de variables ilustrado en la solución
de la ecuación 2 (pag. 2, confronte su resultado con la solución indicada en seguida, Ecn. 7)
AP P
k1 +k2 )t
x = ([P ]0 − [P ]EQ )(1 − e−(
) (Ecn. 7),
en la que:
[P ]EQ = AP P kk21 +k2 [P ]0
3.3
Recapitulando:
1. Antes de atacar un sistema elimine las complejidades innecesarias (aquellas que no tienen
efecto apreciable en el comportamiento del sistema bajo las condiciones de estudio). Esto
facilita el modelo y el relacionar el modelo con el experimento. En este caso:
La solución (Ecn. 7) indica que x se aproxima a su concentración de equilibrio
exponencialmente, ([P ]0 − [P ]EQ ) alcanzándola cuando la exponencial vale cero,
es decir a tiempo infinito.
2. La tasa con la que el sistema se aproxima al equilibrio sigue una exponencial decreciente, lo
que quiere decir que se acerca muy rápido al inicio, pero se va frenando conforme más cerca
está del equilibrio.
Como consecuencia de los dos puntos anteriores: tendríamos que suponer que nada llega jamás
al equilibrio, si bien puede aproximarse mucho.
Esto es causado por las funciones de variables continuas empleadas, siendo que los sistemas
moleculares son discretos, i.e.:
Las soluciones presentadas aproximan bien el comportamiento del sistema mientras se
refieran a predicciones que dependan de muchas moléculas, pero fallan al tener unas
pocas decenas de moléculas, cuando la fracción de molécula en el resultado ya no
resulta despreciable. Una fracción de molécula es un sinsentido.
3.3.1
Ejercicio resuelto 2. Tiempo requerido para que la concentración de PL remanente sea 99% de la concentración al equilibrio.
Llamemos t99 al tiempo en el que:
x = 0.99 [PL ]EQ = 0.99 ([P ]0 − [P ]EQ ).
De la ecuación 7 :
x = ([P ]0 − [P ]EQ )(1 − e−(k1 L+k2 )t99 ).
Substituyendo los valores dados en el ejercicio resuelto 1 tendremos:
10
0.99 ([P ]0 − [P ]EQ ) = ([P ]0 − [P ]EQ )(1 − e−(k1 L+k2 )t99 ),
por lo que:
= 45.6 s
e−(k1 L+k2 )t99 = (1 − 0.99) = 0.01 y t99 = − kln(0.01)
1 L+k2
Para fines prácticos, aún es esta reacción, que podemos considerar lenta,
¡se requiere menos de un minuto para que se establezca el equilibrio!
Note que si aumentamos la concentración de proteína, esto no tendrá un efecto apreciable en
el tiempo en el que se alcance el equilibrio, en tanto no se viole nuestra aproximación de que la
concentración de ligando inicial se mantenga casi constante durante la aproximación al equilibrio
(es decir, mientras no pongamos proteína a un nivel cercano al de ligando).
Una particularidad de este ejemplo, que sorprende, es que la concentración de proteína no
determina el tiempo en el que se alcanza el equilibrio. Además, la concentración de ligando tampoco
tiene un efecto notable, en tanto [L] se inferior a 10 mM, ya que 100 M −1 s−1 [L] k2 (ver los
datos en el ejemplo 1). Es decir, a pesar de que numéricamente, la constante de rapidez para la
disociación del complejo parece pequeña y se antoja poco relevante - lo que determina la rapidez
de aproximación al equilibrio es la rapidez de la ruptura del complejo - ¡no la rapidez con la que se
forma!
Esta es una situación común en muchos sistemas dinámicos, los pasos que determinan la dinámico
del proceso global son a menudo pasos que intervienen en el proceso que ocurre en la dirección
opuesta a aquella en la que se observa un flujo neto, es decir “los frenos pueden ser más significativos
que el empuje”.
Note también que, en una alícuota de 1 nl de la disolución anterior, tendríamos:
0.99 [PL ]EQ = 0.99 × 9.9 × 10−12 nmol nl−1 × (6.022 × 1014 moléculas nmol−1 )
= 5902.2 moléculas ml−1
Es decir aún con tan poco proteína y en un volumen muy pequeño tenemos muchas moléculas
y la diferencia entre 5902 y 5902.2 moléculas es despreciable. En otras palabras el uso de funciones
continuas se justifica aún en situaciones nanoscópicas.
3.3.2
Ejercicio no resuelto 3.
Estime el tiempo adicional que tendré que esperar para pasar de 99% de la concentración de PLEQ
para llegar a 99.99% de su concentración de equilibrio.
3.4
Ejemplo 3. Aceleración de una reacción enzimática en su camino
hacia el SS .
En el sistema anterior, consideramos un proceso físico de asociación de proteína y ligando. Es de
notar, que las ecuaciones que describen la cinética de estos procesos no difieren de las que describen
la cinética de transformaciones químicas. Consideremos la forma más sencilla de reacción química
catalizada enzimáticamente, un único reactante, un sólo producto y sólo dos formas de la enzima
(Esquema 3).
Consideramos que este proceso no está en equilibrio y se refiere a una trasformación química,
pero para modelarlo recurrimos a las mismas herramientas que antes, es decir, la rapidez de la
reacción química se mide como:
11
k1
E
S
k2
kCAT
E·S
E·P
P
E
Esquema 3
= kCAT [E.S] (Ecn. 8)
y la rapidez de formación y desaparición del complejo está dada por:
d[E·S]
dt = k1 [E][S] − (k2 + kCAT )[E ·S] = k1 (E0 − [E ·S])[S] − (k2 + kCAT )[E ·S],
que se simplifica a:
d[E·S]
dt = k1 E0 [S] − (k1 [S] + k2 + kCAT )[E ·S] (Ecn.9).
Asumimos nuevamente que [S] [E] y que durante el tiempo del experimento [P ] [S]. La
[S] consumida en formar E · S + P es despreciable y se puede considerar que [S]≈[S]0 y es casi
constante. Ahora, derivando la ecuación 8, obtenemos la Ecn. 10.
d2 [P ]
= kCAT d[E·S]
(Ecn.10)
dt
dt2
entonces, multiplicando la ecuación 9 por kCAT e igualando, generamos la Ecn. 11.
d2 [P ]
= kCAT d[E·S]
dt = kCAT k1 E0 [S] − (k1 [S] + k2 + kCAT ) kCAT [E · S] (Ecn.11).
dt2
Substituyendo 8 en 11 tendremos:
d2 [P ]
= kCAT k1 E0 [S] − (k1 [S] + k2 + kCAT ) dP
dt
dt2
y que se ordena para dar:
d2 [P ]
+ (k1 [S] + k2 + kCAT ) dP
dt − kCAT k1 E0 [S] = 0 (Ecn.12).
dt2
Esta ecuación diferencial ya no se puede resolver con los métodos que empleamos para resolver
la ecuación 3 (pag. 2), pero observemos que [P] sólo aparece como derivadas (es decir, se trata de
una ode homogénea), por lo que se puede simplicficar con un truco sencillo (substituir la variable).
Sea
]
ν = d[P
dt ,
dP
dt
2
d [P ]
por lo tanto: dν
dt = dt2 de donde se tiene:
dν
dt + (k1 [S] + k2 + kCAT )ν − kCAT k1 E0 [S] = 0 (Ecn.13).
Ahora si podemos separar las variables y ordenarla, es decir:
dν
= dt
−(k1 [S] + k2 + kCAT )ν + kCAT k1 E0 [S]
−([S] +
dν
= k1 dt
])ν + kCAT E0 [S]
CAT
[ k2 +k
k1
(Ecn. 14).
Si ahora definimos:
CAT
KM = k2 +k
(Ecn.15)
k1
y
VM AX = kCAT [E0 ] (Ecn.16),
y se substituyen 15 y 19 en 14 y como a t = 0 , [E · S] = 0 y ν0 = 0 nos queda la siguiente
integral definida:
12
ν
Z
0
dν
= k1
−([S] + KM )ν + VM AX [S]
Z
t
dt
,
t=0
que se resuelve para dar:
−(
1
) [ln(−([S] + KM )v + VM AX [S]) − ln(VM AX [S])] = k1 t
[S] + KM
o bien:
ln
VM AX [S]
+VM AX [S] − ([S] + KM )ν
,
= k1 ([S] + KM )t
(17)
,
que es la solución en su forma logarítmica, en su forma exponencial tendremos:
VM AX [S] = ek1 ([S]+KM )t VM AX [S] − ek1 ([S]+KM )t ([S] + KM )ν
es decir:
(18)
,
d[P ]
VM AX [S] =ν=
1 − e−k1 ([S]+KM )t
dt
[S] + KM
(Ecn. 19).
¡Sorpresa! - La ecuación de Michaelis y Menten multiplicada por un factor exponencial. Si
ahora, si queremos determinar como varía la concentración de producto en función del tiempo,
basta con integrar esta función.
Suena a una integral complicada, pero como [S] ≈ [S]0 desde el planteamiento inicial, podemos
simplemente separar las variables e integrar, considerando que [P ] = 0 a t = 0.
Z
0
P
Z t
([S] + KM )
d[P ] =
1 − e−k1 ([S]+KM )t dt
VM AX [S]
0
e−k1 ([S]+KM )t −1
VM AX [S]
t+
[P ] =
([S] + KM )
k1 ([S] + KM )
(Ecn. 20) ,
que equivale a:
VM AX [S]
[P ] =
([S] + KM )
k1 ([S] + KM )t + e−k1 ([S]+KM )t −1
k1 ([S] + KM )
Para analizar el comportamiento de esta ecuación consideremos primero que la reacción se encuentra
en los primeros momentos; así, la función exponencial se puede simplificar considerando sólo los
n
2
primeros tres términos de la expansión de Taylor ( ex = 1 + x + x2 ! + ... xn !), es decir, el numerador
de la expresión anterior dentro del paréntesis se convierte en:
k 2 ([S] + KM )2 t2
−k1 ([S] + KM )t + 1 − k1 ([S] + KM )t + (1/2)k12 ([S] + KM )2 t2 − 1 = 1
2
y toda la expresión se simplifica a:
13
VM AX [S]
k1 kCAT E0 [S] 2
(k1 ) =
t
(21)
2
2
Una gráfica de [P] vs. t2 es aproximadamente lineal y tiene pendiente (1/2)k1 kCAT E0 S0 .
Ahora, si kCAT y KM han sido determinadas de los datos de SS , E0 es conocida y aceptando la
ecuación 15 como una definición correcta de KM , entonces es posible calcular todas las constantes.
A tiempos largos, la ecuación 20 puede simplificarse considerando que el término de decaimiento
exponencial se hace despreciable, con
lo que la ecuación se reduce a:
VM AX [S]
1
t
−
(Ecn. 22).
[P ] = ([S]+K
k1 ([S]+KM )
M)
En estas condiciones se alcanza el SS , el cual es ahora descrito por esta recta cuya pendiente
es:
[P ] =
VM AX [S]
([S]+KM )
y que intercepta
a la ordenada está del lado negativo del eje en:
−VM AX [S]
k1 ([S]+KM )2 .
Siahora extrapolamos
la recta a la abscisa ([P ] = 0), nos da:
k1 ([S]+KM )t−1
= 0.
k1 ([S]+KM )2
Despejando el tiempo, al que llamaremos periodo de retardo ( τ ) o fase “lag”, tenemos:
1
.
τ = k1 ([S]+K
M)
De nuevo, si se conoce KM , o si la concentración de sustrato es saturante ([S] + KM ) ' [S], se
puede determinar el valor de k1 .
Este caso puede no ser muy realista, porque se asume que la reacción es completamente irreversible y que en el proceso catalítico sólo interviene un complejo E ·S (lo cual no siempre es una
aproximación válida). Sin embargo, ilustra el poder de la medición de la reacción en condiciones
previas al establecimiento del SS y muestra que la cinética de pre-SS nos puede decir que tan
rápido se asocian la enzima y el sustrato, que tan rápido se disocia el complejo y EL VALOR
VERDADERO DE LA CONSTANTE DE DISOCIACIÓN DEL COMPLEJO E·S.
Este enfoque fue empleado por Gutfreund para determinar las constantes de la reacción de
hidrólisis de esteres sintéticos de aminoácido catalizada por la quimitripsina. El siguiente ejemplo
es una adaptación de sus resultados.
[S] ( µM )
3.4.1
δP/δt (µ mol min−1) ± SE.
3.5
1.14
0.10
6.0
1.69
0.02
11.0
2.34
0.08
20.0
2.97
0.14
35.0
3.42
0.33
60.0
3.91
0.23
Ejercicio no resuelto 4.
La quimotripsina cataliza la hidrólisis de ésteres de aminoácido como la acetil-tirosina (AcY). lo
reacción puede seguirse por el cambio en la fluorescencia de TYR cuando se desacetila. Se realizaron
14
las siguientes mediciones con 0.3µ moldeenzima, en cubetas de 3ml y variando la concentración del
sustrato (según se indica arriba), bajo condiciones de SS .
Luego se siguió la reacción en el pre-SS mezclando dos disoluciones (par partes iguales) una con
20µM de enzima y otra con 100µM de sustrato en un equipo de flujo detenido (sección siguiente).
Los resultados se muestran en seguida.
tiempo (ms)
[P ](µM )
tiempo (ms)
[P ](µM )
tiempo (ms)
[P ](µM )
0.0
0.000
6.2
0.362
14.1
1.200
0.9
0.012
6.5
0.387
14.6
1.261
1.0
0.014
6.7
0.406
15.0
1.312
1.2
0.021
7.0
0.432
15.5
1.364
1.5
0.030
7.3
0.459
16.0
1.427
1.8
0.040
7.5
0.487
16.6
1.490
2.0
0.051
7.8
0.509
17.0
1.543
2.3
0.063
8.1
0.538
17.5
1.607
2.5
0.078
8.3
0.561
18.0
1.661
2.8
0.091
8.5
0.584
18.5
1.725
3.0
0.105
8.7
0.607
19.0
1.779
3.2
0.121
9.0
0.640
19.5
1.834
3.5
0.141
9.3
0.664
20.0
1.900
3.8
0.156
9.5
0.689
21.0
2.021
4.0
0.175
9.7
0.714
22.1
2.142
4.2
0.192
10.0
0.748
23.1
2.264
4.5
0.214
10.5
0.800
23.9
2.365
4.8
0.232
11.0
0.854
25.0
2.499
5.0
0.252
11.5
0.909
26.1
2.622
5.2
0.272
12.0
0.965
27.0
2.735
5.5
0.293
12.5
1.023
28.1
2.858
5.7
0.315
13.0
1.081
29.1
2.982
6.1
0.344
13.5
1.140
30.0
3.091
Asumiendo que esta enzima sigue el modelo del Esquema 3 (Pag. 12) determine el valor de las
constantes k1 , k2 y kCAT .
15
Figura 5. Diseño de un equipo clásico de flujo detenido. En el esquema se muestran los componentes
básicos, incluyendo dos diferentes diseños de celdas y un mezclador de 4 vías.
4
Métodos para cuantificar los tiempos de relajación. “Stopped
Flow” “Quenched Flow” y “Equilibrium Jump”.
Los primeros experimentos de cinética rápida se reportaron por Hartridge y Roghton en 1923, con
base en un instrumento diseñado por Reschig (1905), quién estudió reacciones en fase gaseosa.
Es esos experimentos, Hartridge y Roghton mezclaron una disolución de hemoglobina con otra de
monóxido de carbono y la mezcla se hizo fluir por un tubo delgado que tenía una serie de termopares
(sensores) a los largo de tubo. La reacción de la hemoglobina con el monóxido desprende calor y
los autores siguieron la reacción por el calor desprendido.
En este sistema, a flujo constante, se obtiene una disolución con una edad fija, después del
mezclado, en cada punto del tubo. Cada termopar detecta el estado de la reacción a un tiempo
fijo después del mezclado. La concepción del diseño es simple y el equipo requerido no es muy
sofisticado en resolución y tiempo de respuesta. La información es certera y repetida, pues la
disolución en cada termopar es continuamente reemplazada por otra disolución igual fresca. Sin
embargo se requieren cantidades enormes de proteína, que difícilmente pueden obtenerse cuando
no se trata de la hemoglobina equina.
En 1940, Chance realizó un experimento semejante, pero detuvo el flujo abruptamente en la celda
de un detector. Si el flujo se detiene cuando la disolución ha envejecido apenas unos milisegundos,
se puede seguir el devenir de la reacción en los segundos siguientes, observando la celda con algún
espectroscopio (espectrofotómetro o espectrofluorómetro, comúnmente).
El diseño clásico de un equipo de flujo detenido se muestra en la figura 5. En ella se muestran la
jeringas de reactantes que contienen dos disoluciones distintas, con los componentes de la reacción
16
!"#$%"&'()#*"+,%"&'('-'#*'.'
/)+0(123451
!
67%'.8(9:))7%'&"8#':"8
"
#
Figura 6. A) Diseño de un equipo clásico de flujo extinto (QUENCHED FLOW). En el esquema se
muestran los componentes esenciales, pero en este equipo el tiempo de reacción está fijado por la
longitud del trayecto ente los mezcladores y la velocidad del flujo. B) se muestra una fotografía de
un equipo comercial. C) Equipo básico de flujo extinto pulsado. En este equipo luego de un pulso
en el impulsor A, un sistema adicional de válvulas puede retener el líquido en el tubo de incubación
por el periodo deseado, en tanto que un pulso del impulsor B desplaza el líquido de la incubadora
al segundo mezclador para detener la reacción.
separados de manera que la reacción no se inicia, hasta después del mezclado. La mezcla ocurre en
un dispositivo mezclador que provoca un régimen de flujo hidrodinámico turbulento en la conjunción
de las tuberías.
En un equipo de flujo rápido típico (Fig 5), la disolución se conduce hacia una cámara de observación y se recoge finalmente en una jeringa receptora. Las jeringas de reactantes son empujadas
simultáneamente, generalmente por un dispositivo automatizado. El émbolo de la jeringa receptora
está también acoplado a un transductor que enciende el registro al ser empujado hasta su posición
final. Los detectores estaban inicialmente acoplados a un osciloscopio para permitir un registro
de lo ocurrido en la celda en los primeros 10 a 100 ms de la reacción. En los equipos modernos
dispositivos electrónicos computarizados colectan datos en formato digital. Sin embargo, se trata de
equipos costosos debido a que los procesadores de la señal deber ser capaces de registrar y procesar
los datos con gran rapidez y la sincronía entre la mecánica y la electrónica debe ser muy precisa.
Para algunas reacciones que no es posible seguir en los espectroscopios, se han diseñado equipos
que se conocen como de flujo extinto (“quenched flow”). un esquema básico de ellos se muestra
en la figura 6. Los equipos de flujo extinto difieren de los de flujo detenido en que tienen un
asegunda cámara de mezclado que permite combinar la reacción con una disolución que la detiene,
generalmente con ácido u otra substancia que ejerza su efecto de manera casi inmediata, esto en
comparación con la rapidez del proceso en estudio.
Se requieren cantidades grandes de proteína y para seguir la reacción en el tiempo hay que
recurrir a trucos como pulsar o variar el flujo, lo que generalmente se realiza con un sistema de
válvulas controladas por computadora. Esto generalmente se traduce en experimentos más largos,
con buen gasto de proteína y se obtiene una cantidad abundante de muestra por analizar.
El tiempo que transcurre para que el líquido llegue desde la cámara de mezclado a la celda se
llama “tiempo muerto” del equipo. Aunque puede reducirse acortando la longitud de la tubería, en la
;<=>6?/=@A@(<A!=B<AC(A;DE<BFA(@6(FGH=!B
9I0(3JKLJ
17
práctica hay un límite a esta distancia, ya que los fenómenos de transporte en la disoluciones durante
el mezclado impiden que los reactantes formen una mezcla homogénea en tiempos demasiado cortos.
A pesar de los enormes adelantos tecnológicos, los tiempos muertos de los equipos de flujo
detenido no han logrado reducirse gran cosa desde que se fabricaron los primeros equipos. Recientemente, equipos que emplean los adelantos en la tecnología de materiales para fabricar tubería
muy delgada, con muy poca fricción y cámaras de mezclado óptimas, con diseños asistidos por
computadora han logrado reducir los tiempos muertos de 2 o 3 ms a tan sólo 0.5 ms. Como se ve,
el avance no ha sido espectacular.
Cuando se desea estudiar reacciones en tiempos aún más corto se ha recurrido a la técnica de
perturbación del equilibrio. Esta técnica tiene la ventaja de que no depende de la difusión para
el mezclado, porque la reacción se mezcla y se deja llegar a equilibrio dentro de la cámara de
observación. Entonces, se emplea un cambio en las condiciones del medio que modifique la posición
del equilibrio y se detecta como es que las especies se redistribuyen para satisfacer la condición de
equilibrio. Estos estudios reciben el nombre de “salto del equilibrio” (EQUILIBRIUM JUMP).
Dos tipos equipos han sido los más empleados:
I) los que modifican la temperatura, lo cual puede hacerse de manera muy eficiente en tiempos
tan cortos como 0.1 ms, sobretodo si el cambio es de unos pocos grados. Aunque su diseño puede
considerarse tan sencillo como el de un espectrofotómetro con control de temperatura por efecto
Peltier, la necesidad de transmitir el calor en tiempos muy cortos y permitir su flujo hacia el cuerpo
de la disolución en periodos igualmente cortos, hace de este tipo de instrumentos equipos muy
costosos. Recientemente el uso de luz láser (Fig. 7) permite calentar específicamente el solvente
(usando un láser de Oxidos de Ytrio y Aluminio envenenados con Neodimio, conocido como NdYAG, que emite luz infrarroja de 1064 nm) en tiempos aún más cortos. En teoría el límite inferior
de resolución de esta estrategia puede ser llevado al orden de 10 ps.
II) Los que modifican la presión, cuyos efectos en la disolución se transmiten de manera casi
instantánea, especialmente en solvente poco compresibles como el agua. Sin embargo, para que
se observen cambios importantes en las constantes de equilibrio se requiere aplicar cambios de
presión de varios cientos o, incluso, miles de atmósferas, lo que no es resistido por cualquier equipo,
especialmente en la cámara de observación, que tiene que permitir el registro de los eventos que
ocurren en su interior. Frecuentemente en estos equipos la celda de observación es un conductímetro
y el diseño general del equipo se realiza en acero inoxidable, excepto por el disco o membrana de
ruptura que es de Nylan u otros materiales poliméricos resistentes a la deformación.
Finalmente, algunas reacciones pueden estudiarse mediante “saltos” del equilibrio que pueden
provocarse con luz o con electricidad mediante efectos fotoquímicos o electroquímicos. Aunque
estas estrategias están limitadas a reacciones en las que el proceso químico lo permite, su aplicación
puede ser muy informativa, dado que se puede tener un control muy estricto del inicio del proceso.
Un aspecto muy importante de esta estrategia es el tipo de registro espectroscópico que se
ha de realizar. Se han empleado muy comúnmente espectrofotómetros y fluorímetros en el rango
UV/visible. Pero más recientemente se ha introducido el uso de espectrofotometría infrarroja (IR),
resonancia paramagnética del electrón (EPR), fluorescencia de energía transmitida por resonancia
(FRET) y resonancia magnética nuclear (NMR). En algunos casos, ha sido posible el introducir
marcaje isotópico en sitios específicos del ligando o de la proteína. Esta técnica es de utilidad si
se combina con la espectrofotometría IR, ya que el espectro diferencial entre la mezcla sin y con
el marcaje isotópico proporciona la señal de aquellos átomos que están covalentemente unidos al
isótopo.
18
B
A
Figura 7. A) Diseño esquemático de un espectroscopio de salto de temperatura basado en
calentamiento por medio de luz láser. B) Emisión a 340 nm en respuesta a un salto de
temperatura de 5 a 25 oC de una disolución con 20 µM de triosa fosfato isomerasa (TIM).
(a) apo-TIM; (b) TIM con 5 mM gliceraldehído-3-fosfato. La línea roja es el registro de la
señal y la verde el ajuste de los datos. La señal transitoria entre 14 y 50 ns no tiene aún
explicación satisfactoria (tomado de Callender y Dyer, 2002).
5
Escalas de tiempo, resolución de los espectroscopios y
límites de la técnica.
Para dar idea de la capacidad de resolución de esta técnica consideraremos lo siguiente:
1. Los diferentes métodos tienen diferente capacidad para mostrar información dependiendo de
la rapidez con la que es posible “disparar” el fenómeno. Las técnicas que implican mezclado
(stopped flow or quenched flow) tienen un límite técnico 0.1 ms, aunque en la práctica los
mejores equipos actuales usualmente llegan a los 0.5 ms. Las técnicas de salto del equilibrio
son mucho más eficientes y permiten registrar los fenómenos en tiempos de hasta 0.05 s.
Algunos los equipos modernos utilizan un láser "sintonizado" para calentar el solvente (agua)
y permiten llevar la resolución a límites 10 veces inferiores.
2. Sin embargo, para el control y el registor de datos, la tecnología moderna se apoya principalmente en los dispositivos electrónicos. Tales dispositivos dependen en general de un circuito
básico llamado oscilador sinusoidal que genera corriente alterna de diversas frecuencias. Las
frecuencias determinan en gran medida el tamaño de los fenómenos que se pueden codificar
o inspeccionar con tal señal. En general, aquellos fenómenos cuya frecuencia es menor que la
frecuencia del oscilador se pierden. Nuestros dispositivos actuales trabajan en frecuencias de
0.1 a 100 MHz (millones de oscilaciones por segundo). Esto quiere decir que se puede sondear
bien cosas que ocurren en rangos de µs o menos. Desafortunadamente, entre mayor se la
frecuencia de la onda, mayor su tendencia a viajar por el aire fuera del conductor (circuito),
lo que genera pérdida dde sensibilidad y ruido. Por lo mismo, las señales observadas deben
tener una intensidad apreciable para que puedan separarse del ruido. Este problema ha sido
obviado en parte gracias a la conversión digital. Los mejores circuitos disponibles en la actualidad manejan conversiones en el orden de 96 MHz. Lo que significa que para analizar una
19
C
A
B
Figura 8. Diseño clásico de un equipo de salto de presión A) esquema general B) diagrama
de la bomba de presión para detección con conductividad. C) diagrama de la celda de
conductividad y su montaje (tomado de Takahashi y Alberti)
curva con una duración de 1 µs podemos procesar unos 96 puntos de la señal. Si vemos la
curva de la figura 9, observaremos que para analizar una curva de este tipo se requiere de 3
o 4 puntos para definir la región lineal, pero se requieren10 o más puntos para definir bien la
parte curva.
3. En la reacciones químicas catalizada por enzimas los fenómenos generalmente ocurren en
disoluciones acuosas. Por ello los fenómenos de segundo orden no superan un valor de 4 ×
109 M −1 s−1 , que el la rapidez de difusión. Para determinar el valor de una constante de
segundo orden de 1 × 109 M −1 s−1 , si el menor valor de τ que podemos medir es de digamos
1m s y necesitamos una concentración de sustrato de 1µ M , pero con una concentración de
sustrato de ese nivel, la cantidad de producto acumulado sería 20 veces menor a la que se
muestra en la figura 7, lo que significaría que aún por fluorescencia sería casi imposible detectar
la señal del producto acumulado.
4. La espectroscopía empleada también requiere consideraciones. Con la fluorescencia la excitación lumínica emite fluorescencia en tiempos que pueden exceder un milisegundo, por lo
que es posible que no se mida la fluorescencia de SS y entonces habría que resolver también los
tiempos de relajación debidos a la acumulación o pérdida de la forma excitada del fluoróforo.
5. Con las constantes de primer orden el problema es que no dependen de la concentración, lo
que significa que son inversamente proporcionales a los tiempos de relajación y, por lo tanto,
valores superiores a 1 × 104 s−1 escapan a auscultación directa por técnicas de flujo detenido,
o bien 1 × 106 s−1 para las técnicas de salto del equilibrio.
6. Por último, cuando un intermediario sufre dos isomerizaciones consecutivas, sus tiempos de
relajación aparecen sumados en las ecuaciones, es decir, lo que veremos es la suma de ambos
20
Figura 9. Datos del problema 4 ( pag. 14) en forma gráfica. La línea verde es resultado del ajuste
por regresión no lineal a la parte final de la curva, la línea azul es una parábola de la forma y = x2
ajustada al inicio de la curva. Ambas líneas fueron extendidas más allá de la región en que fueron
ajustadas.
tiempos de relajación, si uno de ellos es muy lento con respecto al otro, su valor dominará en
el proceso y sólo podremos detectar uno de ambos intermediarios.
6
Solución analítica de mecanismos cinéticos comunes y sus
implicaciones.
En la primera parte de este escrito tratamos de un caso relativamente sencillo, que seguramente
no puede ser aplicado más que a unos pocos casos, ya que supone que la formación de producto
se da en un sólo paso y la liberación del producto ocurre sin la presencia de formas intermediarias
del complejo enzima producto. Si se asume que la concentración de sustrato no es limitante y que
permanece constante en los experimentos, es posible obtener una solución analítica para el sistema
completo considerando la reacción reversible y varias formas intermediarias. Consideremos por
ejemplo el esquema 4.
Por supuesto, no es difícil imaginar que la concentración de producto en función del tiempo
tendrá una forma compleja.
Para empezar podemos ver que se trata de un sistema que generará 4 ecuaciones diferenciales
independientes, una por cada especie enzimática menos una, más la ecuación de balance de masa
para la enzima. La formación de producto en función del tiempo dependerá de la conversión de ES
en EP, la cual puede además revertirse, es decir:
]
f
r
− d[P
dt = kCAT [E ∗ S] − kCAT [E ∗ P ] (Ecn 23)
Pero para resolver la ecuación 23 en términos de la concentración de sustrato, producto, enzima
21
E
k1
S
k2
k2
E·S
k4
i
kCAT
f
i
E*S
r
kCAT
i
E·P
i
k6
k8
E*P
k4
k3
P
E
Esquema 4
total y los valores de las constantes de rapidez, necesitamos saber como varía la concentración de
E*S y E*P en función del tiempo, lo que implica conocer también las demás formas, ya que todas
ellas están interconectadas por los eventos químicos.
Matemáticamente se puede demostrar que un sistema de ecuaciones diferenciales lineales de
primer orden con 5 ecuaciones independientes (incluyendo el balance de masa de la enzima) y 5
funciones desconocidas (una describiendo la variación en la concentración de cada especie como
función del tiempo) tiene al menos una solución completa.
La manera más evidente de obtener tales soluciones es usar las funciones exponenciales, ya
que esta función, al ser derivada da lugar a si misma, lo que significa que puede eliminarse de las
ecuaciones igualadas a cero (por factorización) y se resuelve entonces una ecuación algebraica con
los coeficientes, que permite determinar los valores de todos los coeficientes, el resultados será de
la forma:
[P ](t) = A0 + f (t) + A1 et/τ1 +A2 et/τ2 +A3 et/τ2 +A4 et/τ4 +A5 et/τ5
en donde f (t) es usualmente una función algebraica de t, A0 , A1 , etc. son las amplitudes de los
componentes exponenciales y τ1 , τ2 , etc. son los llamados tiempos de relajación de los componentes
exponenciales., que son funciones de las constantes de rapidez. Los tiempos de relajación son
funciones algebraicas racionales de las constates de velocidad, la concentración de sustrato y la
concentración de enzima total.
En la actualidad, es incluso posible ajustar las curvas experimentales a los datos directamente a
las ecuaciones diferenciales, ya que hay programas capaces de resolver el sistema en forma numérica
y luego aproximar los valores de las constantes mediante ajuste por iteraciones sucesivas. Si bien,
tales algoritmos son complejos, requieren numerosos controles para solventar las complicaciones
numéricas de hacer cálculos repetidamente y son, por ello, costosos en tiempo de cálculo. Afortunadamente, la aproximación puede realizarse en la computadoras de escritorio modernas en tiempo
bastante razonables, pero uno tiene que realizar experimentos con diversas condiciones iniciales contrastantes, con el fin de asegurar que el modelo es consistente con los datos en todas ellas. Además,
existen programas que manejan las ecuaciones en forma simbólica y pueden ayudar a obtener la
solución de un sistema en forma completa.
En cualquier caso, como se podría esperar, hay un pelo en la sopa, ya que las sumas de funciones
exponenciales tienen formas que siguen pareciéndose a una función exponencial y para determinar
el número de componentes exponenciales que explican un sistema, se requiere que los valores de
los tiempos de relajación ( τ ) y las amplitudes se encuentren bien separadas (es decir, serán de
magnitudes razonablemente distintas). En la práctica, resulta muy difícil separar razonablemente
más de 3 componentes a partir de un sólo conjunto de datos experimentales.
Un análisis completo de un sistema como el mostrado en el esquema 4 es posible si se asume
que las concentraciones de los ligandos S y P , así como otros ligandos intermedios que puedan
aparecer en el mecanismo, se pueden mantener en niveles cuasiconstantes. Es decir, que existen
22
valores promedio L̄i promedio que describen el comportamiento del sistema de manera razonable,
bajo las condiciones experimentales en estudio. Tal análisis ha sido descrito en detalle por Hammes
y Schimmel (1966; 1967).
Estos esquemas ilustran que se pueden analizar secuencias de reacciones consecutivas y resolver
la aparición y desaparición de los intermediarios, para determinar las constantes del sistema.
Ejemplos de la aplicación de este análisis hay muchos, menciono tres y en los dos primeros los
resultados pudieron ajustarse a modelo planteado en esquema 5 y cuya solución estaría dada por
las Ecn. 24 y 25.
CA (t) = CA (0)e−k12 t (Ecn.24)
12
(e−k12 t − ek23 t ) (Ecn.25)
CB (t) = CA (0) k12k+k
23
1. La emisión de luz por la aequorina reportada por Hastings et al., (1969).
2. El reporte por Halford, Johnson & Grinsted (1979), usando la enzima de Resitricción EcoRI.
En dicho trabajo, se aprovecha que la enzima realiza el corte en cada hebra en momentos
distintos y usando un plásmido superenrrollado se pueden separa el DNA superenrrollado
(sustrato) del DNA circular relajado (cortado en una hebra, pero no abierto) y el DNA
abierto (cortado en ambas hebras).
3. La aplicación este tipo de análisis para el estudio de la enzima piranosa-2-oxidasa por Prongjit
y colaboradores (2009). En dicho estudio los autores aseguran haber resuelto hasta 5 componentes exponenciales independientes, lo cual es muy poco común.
7
Integración numérica de ecuaciones diferenciales.
Vamos ahora a usar octave para resolver numéricamente un sistema de ecuaciones que simula el
comportamiento completo de una enzima monosustrato, pero sin hacer ninguna consideración.
Esta simulación debería ser capaz de predecir el comportamiento de la catálisis en el pre-SS
, en el SS e incluso al alcanzarse el equilibrio.
El software que emplearemos se denomina octave y es el equivalente abierto de MatLab™.
El script necesario es el que se incluye en seguida y está disponible en línea para que eviten
tener que teclearlo. Las líneas que inician con # son comentarios y no se ejecutan
# 1. THESE ARE COMMENTS, CHANGE THEM AT YOUR CONVINIENCE
# SYSTEM DEFINITION:
#
kf1*[S]
kfc
kf2
# E <==========> Es <===========> Ep <=========> E
#
kr1
krc
kr2*[P]
# So initial substrate concentration
# Et total enzyme concentration
#- THE LINES BELOW DEFINE TEH VALUES OF THE CONSTANTS
# AND CONCENTRATIONS
#- THE MUST BE WRITTEN TWICE, ONCE HERE
# AND ONCE INSIDE THE FUNCTION
#rate constants kvel= [kfc=120, krc=10, kf1=1100, kr1=700, kf2=300, kr2=115];
23
# Co=[Substrate, Enzyme], initial total concentrations
#------------ begins section that needs to be copied into the function ----kv=[120, 10, 1100, 700, 300, 115];
Co=[0.747 0.000010];
#------------ end of section that needs to be copied into the function -----# Mxtm=MAXIMUM TIME OF THE SIMULATION,
# Finess is the number of points to integrate and plot
# the more points the smoother the ploted line.
Mxtm=0.025;
Finess=500;
# pltflg are FLAGS TO INDICATE WHICH SPECIES
# YOU WANT IN THE PLOT, 1= plot, 0= don’t plot.
pltFlg = [1,1,1,0,1];
# sclFact this number is used to multiply the final values, so all number can
# be seen in the plot.
# the first sclFact is the factor aaplied to the x axis, which is by default in s.
# but if you want the number in ms use 1E3 as sclFact.
# i.e. if you want to plot the [S] in mM and the [E.S] in nM
# you need to multiply the [E.S] by 1E6
# so the values do not appear compressed on the X axis a flat line.
sclFact = [1E3 1E6 1E6 1E6 1 1E6];
# plotnames is the titles to put in the x (first label) and in the Legend (the rest).
plotnames=char(’time (s)’,’[E] ’,’[Es] ’,’[Ep] ’,’[S]’,’[P]’);
# the next labels indicate the file names for the output.
data_filename=’progress_MM_1.txt’;
plot_filename=’progress_MM_1.pdf’;
# your results will be saved to files unscaled,
# so units are uniform in numeric data.
plotfile_format=’-dpdf’;
function xdot = mm (x, t)
# in this function x is a vector with concentrations of
# E Es P
# xdot would be derivatives:
# dE/dt, dEs/dt and dP/dt, respectively.
# due to the inflexible fortran syntax of lsode
# declaring the variables as global would not work
# so after changing them above in the main script
# copy them here
#
1
2
3
4
5
6
#rate constants kv= [kfc=120, krc=10, kf1=1100, kr1=700, kf2=300, kr2=115];
#---copy from above in here (2 lines)
kv=[120, 10, 1100, 700, 300, 115];
Co=[0.747 0.000010];
S=Co(1)-x(3);
Ep=Co(2)-x(1)-x(2);
xdot(1)=kv(4)*x(2)+kv(5)*Ep-(kv(3)*S+kv(6)*x(3))*x(1);
24
xdot(2)=kv(3)*S*x(1)+kv(2)*Ep-(kv(4)+kv(1))*x(2);
xdot(3)=kv(5)*Ep-kv(6)*x(3)*x(1);
endfunction
# - END OF DEFINITIONS, THE REST MAY BE LEFT
# UNCHANGED FOR MOST CASES.
# make a set of color choices (black, red, green, blue, magenta, cyan)
colrlst=["k", "r", "g", "b", "m", "c" ];
# set initial conditions vector and time points vector for interpolation
x0=[ Co(2), 0, 0];
t=linspace(0, Mxtm, Finess)’;
# lsode does the job, this is actually the core of this script
x=lsode("mm",x0 ,t );
# calculate the remaning concetrations such as substrate and
# Ep order everything in
# a matriz matching plotnames
z=[ t x(:,1) x(:,2) (Co(2).-x(:,1).-x(:,2)) (Co(1).-x(:,3)) x(:,3) ];
# save unscaled data to a file
save(data_filename,’z’) ;
# scale data for the plot
z=z.*sclFact;
# create the plot
grph=figure;
figure(grph), hold on;
leyendas="";
for i = 1:length(pltFlg)
if ( pltFlg(i) )
leyendas=char(leyendas,sprintf("%sx%.1e",plotnames(i+1,:),sclFact(i+1)));
plot(z(:,1),z(:,i+1),colrlst(mod(i-1,length(colrlst))+1));
endif
endfor
xlabel(sprintf("%s x %.1e",plotnames(i+1,:), sclFact(1)));
legend(leyendas(2:end,:));
ylabel(’Concentration (mM)’);
legend(’boxoff’);
legend(’left’);
legend("location","north");
refresh;
# save the plot to a file;
25
print(plot_filename, plotfile_format);
figure(grph), hold off;
k12
A
k23
B
C
Esquema 5
8
Manipulación de la condiciones para reducir la complejidad
del problema.
Los ejemplos anteriores y la sección previa nos hacen ver que la elección de las condiciones experimentales es crucial para reducir la complejidad del problema.
Por ejemplo, consideremos el esquema 4 (Pag. 22). Si la concentración de sustrato no sólo es
constante, sino además saturante, podemos considerar que la formación de complejo E ·S es muy
rápida y su disociación en E + S prácticamente no ocurre. Esto puede hacer que el paso de E ·S a
E ∗S y los posteriores sean mucho más lentos y determinen la cinética, por lo que la contribución
del primer paso de asociación se puede “esconder” a los ojos del espectroscopio.
Usualmente, la presencia de producto es también muy baja y la reversión de la reacción (determinado por k3 P ) no es observable, lo que simplifica bastante la forma de las ecuaciones y su
análisis.
Por otro lado, se podría añadir una cierta [P ], provisto que enmascare la medida del que se está
formando. También, si existe alguna señal espectroscópica de la proteína que sea característica de
alguna de las formas intermediarias (vgr. fluorescencia al unir algún ligando). En este caso, se
enlentece el paso S → P , con lo que este paso podría las curvas y darnos señales fáciles de analizar.
Finalmente, es posible modificar la rapidez de los cambios conformacionales de la proteína
alterando la temperatura, el pH, la viscosidad del medio o la presión. Con lo cual, si logramos que
alguno de los pasos sea afectado de manera particular, podemos hacerlo suficientemente lento para
estudiarlo sin interferencia apreciable de los demás eventos.
Desde luego no existe una receta única, cada sistema tiene sus peculiaridades propias y es
necesario conocerlas bien, a fin de explotarlas cabalmente. Es decir, cada sistema requiere una
buena cantidad de trabajo de caracterización previa para que su análisis pueda realizarse con éxito.
Veamos un caso más, reconsideremos el ejemplo 3, pero ahora hagamos que la cantidad de
enzima y sustrato sean tales que la formación de producto casi se agote luego de un ciclo catalítico.
Sensu estricto deberíamos resolver nuevamente el modelo, porque se había asumido que [S] ≈ [S]0 ≈
constante, lo que ya no es válido en la nueva situación. Sin embargo, en aras de la simplicidad
consideremos que existe una “concentración de sustrato promedio ( [S̄])” tal que los valores de las
constantes serán también promedio y que las desviaciones causadas por las variaciones de [S] afectan
el valor de las constantes, dentro de la incertidumbre del error experimental, pero no la forma de
las curvas.
26
Consideremos que:
1. la cantidad de enzima es igual o superior a la de sustrato, por lo que la formación del complejo
E ·S será bimolecular y mucho más rápida que otros eventos.
2. la enzima es de tal forma que su complejo E·S se convierte en un intermediario I que genera
una señal espectroscópica semejante a la del producto (para una deshidrogenasa, equivale a
decir que el complejo E ·N ADH absorbe casi igual al N ADH soluble);
3. la reacción no se revierte con las cantidades de producto que se generan durante el experimento, es decir la termodinámica de la reacción dificulta su reversibilidad.
El modelo puede escribirse a hora como sigue:
E
kf
S
kr
P
kL
I
E
Esquema 6
Para simplificar el esquema y la solución haremos un truco introducido originalmente por Cleland
y usado de manera amplia por Fersht, que consiste en abreviar el mecanismo escribiendo constantes
de velocidad aparentes que reemplazan a las verdaderas (net rate constants), pero que reducen cada
paso a un sólo evento pseudoirreversible. como se muestra en el esquema 7.
E
a
I
b
P
E
Esquema 7
Para estas constantes a y b, consideramos que el tránsito de E a E + P pasando por I es igual a
la probabilidad de que E pase a I ( kf [S̄]) multiplicada por la probabilidad de que I pase a E + P
L
en lugar de romperse de nuevo ( kLk+k
) , es decir:
r
kL
a = kL +kr kf [S̄] = (kCAT /KM )[S̄] (Ecn. 26),
en tanto que el paso de I a E + P estará determinado únicamente por
b = kL = AP P kCAT
(Ecn.27),
entonces, la formación de intermediario I estará determinada por:
d[P ]
d[I]
dt = a[E] − b[I], la liberación de producto por dt = b[I] y tenemos [E]0 = [E] + [I].
Substituyendo [E] en la primera expresión se obtiene:
d[I]
(Ecn.28),
dt = a[E]0 − (a + b)[I]
Que de nuevo puede resolverse por separación de variables, ya que [E]0 es constante.
27
Usemos esta ecuación para aprovechar la herramienta maxima, que nos permite obtener soluciones simbólicas. La figura 10 presenta el trabajo de obtención de la solución en este software, en
donde la solución (%o9) se puede simplificar
a:
a
1 − e−(a+b)t
(Ecn.29),
[I](t) = [E]0 a+b
ahora, como:
d[P ]
ab
−(a+b)t
,
dt = b[I] = [E]0 a+b 1 − e
obtenemos una ecuación diferencial integrable fácilmente de la que se obteine [P ](t).
ab
ab
t − [E]0 (a+b)
1 − e−(a+b)t (Ecn.30).
[P ](t) = [E]0 a+b
2
La ecuación 30 es de hecho equivalente a la ecuación 20 (Pag. 13), lo que se puede ver reemplazando a y b por sus valores originales. Recordando que el intermediario y la señal espectroscópica
del producto no son distinguibles, tendremos que la señal total F(t) = [I](t) + [P](t) (de Ecn. 29+
Ecn. 30) :
ab
a
ab
t − [E]0 (a+b)
1 − e−(a+b)t + [E]0 a+b
1 − e−(a+b)t , o bien,
[P ](t) + [I](t) = [E]0 a+b
2
a
b
ab
t + [E]0 a+b
[F ](t) = [E]0 a+b
1 − e−(a+b)t 1 − a+b
ab
a2
[F ](t) = [E]0 a+b
t + [E]0 (a+b)
1 − e−(a+b)t (Ecn. 31)
2
Esta expresión tiene un término lineal en t y un término exponencial decreciente. A tiempos
muy cercanos a cero, el término exponencial se acerca a cero. Por lo tanto, esta expresión describe
un comportamiento inicial de una recta que parte del origen y representa la acumulación inicial
ab
del complejo [I], es decir [F ](t) = [E]0 a+b
t. La recta adquiere curvatura conforme el término
exponencial alcanza un valor detectable y, cuando t es grande y el término exponencial se aproxima
ab
a2
a cero (ver figura 11, pag 30), nos queda ahora una recta dada por [F ](t) = [E]0 a+b
t + [E]0 (a+b)
2 .El
2
a
valor del corte con el eje “Y” de esta segunda fase es iY = [E]0 (a+b)
2 , que como se puede ver, es
proporcional a [E]0 .
Aquí podemos tener dos situaciones:
[S̄]2
(kCAT [S̄]/KM )2
a2
i) [S̄] es relativamente saturante, (a+b)
2 = (k
2 = ([S̄]+K )2 ≈ 1 ,
CAT [S̄]/KM +kCAT )
M
lo que significa iY = [E]0 . En este caso deben usarse [E] muy elevadas, ya que [S̄] ≈ [E0 ].
ii) si [S̄] no es saturante, la gráfica
√ iY vs. [S̄] sigue una curva de tipo sigmoidal, pero que puede
convertirse a una recta tomando iY como función de [S̄].
8.0.2
Ejercicio no resuelto 5.
√
Derive la forma de la función iY vs. [S̄], demuestre que es una línea recta y determine la forma
algebraica de su pendiente y su intersección con las ordenadas en dicho regráfico.
28
(%i1) assume(nra>0,nrb>0,E0>0);
(%i2) declare(E0,constant); declare(nra,constant);declare(nrb, constant);
(%o1) [nra > 0, nrb > 0, E0 > 0] | (%o2) done |(%o3) done |(%o4) done
(%i5) mmodeq: ’diff(In, t)= nra*E0 - (nra+nrb)*In;
dIn
(%o5)
--- = nra E0 - (nrb + nra) In
dt
(%i6) gsolmmodeq: ode2(mmodeq, In, t);
- (- nrb - nra) t
nra E0 %e
(- nrb - nra) t
(%o6)In = (-------------------------- + %c) %e
nrb + nra
(%i9) icsolmmodeq: expand(ic1(gsolmmodeq, t=0, In=0));
(- nrb t - nra t)
nra E0
nra E0 %e
(%o9)
In = -------- - ---------------------------nrb + nra
nrb + nra
(%i10) velmmodeq: ’diff(P,t)= nrb*rhs(icsolmmodeq);
- nrb t - nra t
dP
nra E0
nra E0 %e
(%o10)
-- = nrb (--------- - ------------------------)
dt
nrb + nra
nrb + nra
(%i11) solvelmmodeq: ode2(velmmodeq, P,t);
(- nrb - nra) t
nra nrb E0 %e
nra nrb E0 t - ----------------------------- nrb - nra
(%o11)
P = -------------------------------------------- + %c
nrb + nra
(%i12) icsolvelmmodeq: expand(ic1(solvelmmodeq, t=0, P=0));
- nrb t - nra t
2
nra nrb E0 %e
nra nrb E0 t
(%o12) P = ---------------------------- + ----------------------2
2
2
2
nrb + 2 nra nrb + nra
nrb + 2 nra nrb + nra
2
nra nrb E0 t
nra nrb E0
+ ------------------------------------------------2
2
2
2
nrb + 2 nra nrb + nra
nrb + 2 nra nrb + nra
Figura 10. Solución de la Ecn. 28 en el programa maxima. El resultado (%o9) es equivalente
a la Ecn. 29, en tanto (%o12) es equivalente a la Ecn. 30.
29
Figura 11. Resultado de un experimento de cinética de la explosión, que permite ver la variación
en el intercepto a la ordenada (respecto de la enzima total) con la concentración de sustrato empleada. Note que a concentraciones elevadas de sustrato este valor se aproxima a 1. La presente
estrategia se ha empleado en numerosos artículos para titular el total de sitios activos realmente
funcionales de una enzima.
Un aspecto interesante que surge de la comparación entre el análisis presentado en el ejemplo
del apartado 3 (pag 12) y este último es el hecho de que la constantes respectivas k2 y kr no son
necesariamente equivalentes. Ello debido a que en un caso se está midiendo la liberación neta del
producto del sitio activo de la enzima, en tanto que en el segundo la medición monitorea tanto al
producto liberado como aquel que se encuentra atrapado en los complejos intermediarios EP (que
pueden ser uno o más, pero que se ven juntos). Es decir, nuestro análisis parte de una simplificación,
30
al aplicarla los valores de algunas constantes son aparentes y pueden incluir pasos adicionales. Dadas
la condiciones experimentales y la estrategia de monitoreo de la reacción, dichos pasos no pueden
resolverse y quedan ocultos.
Un mecanismo que contiene en realidad los pasos mínimos para la catálisis por una hidrolasa
como la mayoría de las proteasas, carbohidrolasas, fosfatasas y lipasas, asumiendo que la hidrólisis
es esencialmente irreversible en medio acuoso diluido, debería ser:
E
k1
S
k2
E·S
kCAT
E·PQ
k4
k3
EQ
.
P
k6
k5
Q
E
Esquema 8
En el que se asume que los productos se liberan ordenadamente (lo cual es cierto para las serina
proteasas y las lipasas, pero no necesariamente para otras hidrolasas). Note entonces que en este
último análisis, la concentración de intermediario que se mide es [E.PQ]. En cambio, [E] + [E.Q] ,
son vistas como “enzima libre” y [E.S] es una especie “invisible” cuyo impacto en la cinética queda
oculto, dadas las condiciones experimentales y cuya influencia está enmascarada en los valores de
algunas constantes. Es decir algunos parámentros estimados son valores aparentes, que dependen
de otras variables y que serán afectados por un cambio en las condiciones del experimento.
En cambio, en el primer análisis (ejemplo 3, apartado 3, pag 12)) los valores de k1 y k2 deben
ser reales, pero obs kCAT es un valor aparente que involucra tanto a kCAT , k4 y k6 , quizá con alguna
influencia menor de k3 [P ] y k5 [Q], en la medida en que su acumulación no resulta tan despreciable
como usualmente se asume.
Nuevamente, para conocer en detalle un mecanismo cinético es necesario recurrir a una variedad
de condiciones experimentales que permitan realizar un análisis completo de los diversos pasos de
la reacción.
8.0.3
Ejercicio no resuelto 6.
En el siguiente experimento se midió la actividad de la quimotripsina con p-nitrofenilacetato, cuya
señal se detecta desde la ruptura del enlace ester en el sitio activo de la enzima, antes de la liberación
del segundo producto (que permanece unido covalentemente a la proteína), misma que es el paso
lento de la reacción y determina la rapidez neta de la catálisis.
Asuma que los parámetros cinéticos ( KM y VM AX ) determinados en el ejercicio 4 (Pag. 14)
son válidos para este sustrato (lo que no implica que las constantes de velocidad también los sean).
Para estas medidas, la cantidad de sustrato empleada fue cercana a la concentración de proteína
(1:1 o 3:1), variando entre 10 y 90 µM (0.21 a 1.89 mg/ml). Otras condiciones experimentales
fueron iguales a las del problema 1. Los resultados obtenidos fueron los siguientes:
[S]0
[E]0 =1
[S]0
[E]0 =3
[S]0 =10
30 (µM)
50 (µM)
90 (µM)
30 (µM)
60 (µM)
150 (µM)
270 (µM)
t
t
t
t
∆A/ε t
∆A/ε t
∆A/ε t
∆A/ε
∆A/ε t
∆A/ε
∆A/ε
∆A/ε
(ms) (µM) (ms) (µM) (ms) (µM) (ms) (µM) (ms) (µM) (ms) (µM) (ms) (µM) (ms) (µM)
31
2.6
0.89
1.7
4.68
1.3
9.38
1.9
34.29
2
1.93
1.1
8.65
1.1
21.44
1.1
55.23
4.4
1.44
3
7.70
2.4
15.70
2.9
43.95
3.5
3.14
2.2
14.60
2.4
34.31
1.9
71.09
6
1.86
4.3
9.86
3.6
20.12
3.9
50.68
4.9
4.03
3.3
18.60
3.1
38.31
2.9
79.89
7.3
2.19
5.5
11.55
4.7
23.38
4.8
55.65
6.1
4.72
4.4
21.42
4.6
43.43
3.9
84.55
8.6
2.48
6.6
12.89
5.7
25.94
5.8
59.46
7.3
5.29
5.4
23.46
5.4
45.11
5
87.14
9.8
2.73
7.7
14.02
6.7
27.99
6.8
62.50
8.4
5.77
6.5
25.02
6.1
46.39
6.1
88.86
10.9
2.96
8.7
14.97
7.8
29.69
7.8
65.04
9.5
6.18
7.6
26.22
7.7
48.29
7.2
90.11
12
3.16
9.7
15.81
8.7
31.13
8.8
67.19 10.6
6.55
8.6
27.18
8.5
48.99
8.4
91.32
13
3.35
10.7 16.55
9.7
32.41
9.8
68.99 11.6
6.87
9.7
27.97
9.2
49.63
9.5
92.52
14.1
3.52
11.7 17.21 10.7 33.51 10.8 70.60 12.6
7.16
10.8 28.67
10
50.23 10.6 93.73
15
3.68
12.7 17.82 11.7 34.50 11.7 72.04 13.6
7.43
11.9 29.26 11.6 51.33 11.8 94.96
16
3.82
13.7 18.36 12.7 35.39 12.7 73.30 14.6
7.67
13
29.83 12.4 51.83 12.9 96.27
17
3.96
14.6 18.86 13.6 36.20 13.7 74.45 15.6
7.90
14
30.33 13.2 52.38 14.1 97.51
17.9
4.10
15.6 19.34 14.6 36.94 14.7 75.54 16.6
8.11
15.1 30.82
18.8
4.22
16.5 19.77 15.5 37.63 15.7 76.53 17.5
8.31
16.2 31.27 15.6 53.85 16.3 100.2
19.7
4.34
17.5 20.18 16.5 38.27 16.7 77.43 18.5
8.49
17.3 31.71 16.4 54.37 17.5 101.5
20.6
4.46
18.4 20.56 17.5 38.87 17.7 78.28 19.4
8.67
18.4 32.15 17.2 54.86 18.6 102.9
21.5
4.57
19.3 20.91 18.4 39.43 18.7 79.08 20.4
8.84
19.5 32.58
22.4
4.67
20.3 21.27 19.4 39.96 19.7 79.78 21.3
9.00
20.6 32.99 19.6 56.41 20.9 105.6
23.2
4.77
21.2 21.60 20.4 40.44 20.8 80.49 22.3
9.15
21.7 33.41 20.4 56.94
24.1
4.87
22.1 21.91 21.3 40.92 21.8 81.16 23.2
9.30
22.8 33.81 21.2 57.43 23.1 108.4
25
4.96
23.1 22.21 22.3 41.39 22.9 81.77 24.1
9.44
23.9 34.24
25.8
5.05
9.58
24.9 34.65 23.6 59.02 25.4 111.1
26.6
5.14
24.9 22.76 24.3 42.21
27.5
5.22
25.9 23.03 25.3 42.59 26.1 83.41 26.9
9.85
27.1 35.48 25.2 60.06 27.7 113.8
28.3
5.31
26.8 23.28 26.3 42.97 27.1 83.84 27.9
9.98
28.2 35.90
29.2
5.39
28.7 23.75 28.3 43.66 28.2 84.32 28.8 10.10 29.3 36.31 28.4 62.13
30
5.47
24
30
22.49 23.3 41.79 23.9 82.31 25.1
24.06
30
44.20
25
30
82.89
84.95
26
30
9.72
10.26
26
30
14
18
22
52.87 15.2 98.87
55.40 19.7 104.2
35.07 24.4 59.53 26.5 112.4
36.57
26
30
60.57 28.8 115.2
63.19
El problema del mezclado.
Como se puede deducir de la discusión anterior, a pesar de que se ha realizado buena dosis de
investigación en el tema (véase por ejemplo el artículo Wong et al., 2004, Sensors and Actuators
B 100:359–379), uno de las limitaciones a las que no se les ha logrado superar es la reducción de
32
107.0
57.95 24.3 109.7
¿cuántos sitios activos hay por molécula de quimotripsina?
¿Cuánto vale la constante de liberación de producto i.e. la tasa de catálisis neta?
¿cómo se compara este valor con la kCAT obtenida en el primer experimento?
9
22
30
116.5
30
116.6
los tiempos de mezclado. Los mejores resultados se aproximan a los 0.1 ms, pero esto depende
mucho de aspectos como la viscosidad de medio, que, en el caso de las proteínas, se eleva de manera
importante. Sin embargo, en lo que se ha avanzado substancialmente es en lo referente al volumen de
la muestra requerido para cada lectura y en el filtrado electrónico de los datos que permite mejorar
la sensibilidad a través de elevar la relación señal/ruido. Se ha logrado también avances importantes
en lo referente a la versatilidad de los equipos de “quench-flow”, ya que el control computarizado de
la instrumentación permite la manipulación de los tiempos de incubación de las mezclas previas a
la extinción, ya sea variando la rapidez del flujo, o bien recurriendo a flujos pulsados con un control
muy preciso de la demora entre el mezclado de la enzima con el sustrato y su posterior mezclado
con la disolución de extinción.
Por otro lado, la reducción en el volumen de la muestra también trae asociado una limitante
que, de nuevo, tiene que ver con el mezclado. El mezclado es más eficiente cuando el flujo se vuelve
turbulento, pero la turbulencia del flujo es directamente proporcional a la velocidad, inversamente
proporcional a la viscosidad y directamente proporcional a los que se conoce como la longitud
característica del flujo, la cual es una función directa del diámetro de la tubería (para tuberías de
sección circular).
Esto significa que al disminuir el volumen de muestra es necesario reducir el diámetro de las
tuberías y esto lleva a aumentar los flujos. En consecuencia, los experimentos requieren presión,
pero la presión genera cambios en los valores de las constantes de rapidez, lo que también puede
complicar el análisis de los experimentos.
Una alternativa a estas complicaciones es el uso de los métodos de relajación. En estos métodos
la limitante es que la reacción debe ser susceptible de alcanzar una situación de equilibrio, lo cual
no siempre es posible, pero en muchos casos si lo es.
Consideremos la reacción:
k1
P +L
k2
PL
Esquema 9
El sistema parte de un condición de equilibrio hacia la condición final nueva en la que [P ]EQi =
p0 , [PL ]EQ = c0 y [L]EQi = l0 . Después de un intervalo de tiempo t después del cambio de condiciones
el c0 se encuentra a una distancia del equilibrio que llamaremos x.
Así [P ] = p = p0 + x, [L] = l = l0 + x y [PL ] = c = c0 − x.
El cambio en el sistema puede describirse como:
que puede reordenarse en:
d(c0 − x)
= k1 (p0 + x)(l0 + x) − k2 (c0 − x)
dt
dc0
dx
−
= k1 p0 l0 − k2 c0 + k1 (p0 + l0 )x + k2 x + k1 x2
dt
dt
33
Pero cuando se alcance la nueva situación de equilibrio tendrá que verificarse que:
dc0
0 0
0
dt = k1 p l − k2 c = 0, por lo que la ecuación de arriba se reduce a:
−dx
= k1 (p0 + l0 )x + k2 x + k1 x2
dt
Dado que la diferencia entre ambos equilibrios no es usualmente muy grande x, es usualmente lo
bastante pequeña para despreciar el término de segundo grado x2 .
La ecuación anterior se integra entonces para dar:
0
0
x = (p − p0 ) ek1 (p +l )+k2
Podemos entonces ver que la relajación del sistema hacia el nuevo equilibrio es un decaimiento
exponencial simple con un tiempo de relajación:
1
0
0
τ = k1 (p + l ) + k2 .
Una gráfica del tiempo de relajación ( τ ) obtenido a diferentes concentraciones de ligando y
proteína contra la concentración de ligando más proteína libre al equilibrio ( p0 + l0 ) dará una recta
cuya pendiente es igual a la constante de segundo orden y cuyo intercepto a la ordenada es el valor
de la constante de primer orden.
Evidentemente los mecanismos en los que hay más de dos especies de proteína en equilibrio
involucran múltiples tiempos de relajación, lo cuales están usualmente acoplados. Por ejemplo en
un esquema como el que sigue:
k1
E
S
k2
k2
i
E·S
k4
i
E*S
Esquema 10
El análisis del experimento rinde el valor de los dos tiempos de relajación que se recuperan
acoplados y que pueden resolverse ajustando la señal a sumas de exponenciales (Naraghi, 1997), de
la forma:
SOBS (t) =
n
X
k=1
ak
τk
τ
1+
e−t/τk −
e−t/τ
τ − τk
τ − τk
en la que τ es el tiempo de relajación del equipo y del detector combinados (convolucionados).
El producto y como la suma de los tiempos de relajación estarían dados por las expresiones
siguientes:
i
i
1
1
τ1 + τ2 = k2 + k1 (eEQ + sEQ ) + k2 + k4
1 1
)( ) = k2 i k4 + k1 (i k2 + k2 )(eEQ + sEQ )
τ1 τ2
en las que las eEQ y sEQ son las concentraciones de enzima y sustrato libres al equilibrio (final).
De estos valores el posible recalcular los valores de las constantes, en la mayoría de los casos.
Un ejemplo de este tipo de análisis se muestra en la figura 7 (pag. 19).
(
34
10
Información susceptible y refractaria a la auscultación
mediante estas metodologías.
Después de toda este análisis, cabe aún preguntarse que es lo que puedo averiguar con estas
metodología sobre una enzima (u otro catalizador) y que es lo que está fuera del alcance de estos métodos.
Para contestar a esta pregunta consideremos lo siguiente:
En estos métodos las determinaciones consisten en cuantificar la presencia de alguna especie
química detectable. Sin embargo, hay especies químicas cuya vida media es muy corta y no presentan señales espectroscópicas únicas que permitan diferenciarlas de otros componentes en la mezcla.
En muchos casos, puede tratarse de estados de transición o confórmeros desconocidos por no haberse
aislado, i.e. sus propiedades no nos son familiares y, por ello, no siempre es posible asociarlos a
alguna señal detectable.
A pesar de que la electrónica y la espectroscopia se han sofisticado substancialmente, las especies
de vida media muy corta suelen encontrarse en cantidades muy pequeñas, por lo que su baja
concentración puede impedir el alcanzar certidumbre cuando se intenta medirla, aún si conocemos
alguna propiedad que nos permita su detección.
Los métodos aquí estudiados muestran fenómenos en función del tiempo, pero en términos
generales, nuestros modelos asumen que los diferentes estados son en sí especies químicas definidas.
En formulaciones alternativas (como la teoría de la fluctuaciones y la teoría de las tasas de reacción)
se plantea que cada especie química es en si un conjunto confórmeros semejantes (ensambles), pero
no idénticos; o bien, que un confórmero puede atravesar por una infinidad de microestados siguiendo
un paisaje de casi infinitas trayectorias, algunas de los cuales son de menor energía por un estrecho
margen (figura 12). Así, las “barreras energéticas” que parecen existir y que representamos como
constantes de velocidad dependientes de la temperatura y la presión, son en sí una consecuencia
de un ensamble en el que sólo algunos de los individuos pueden llegar “al otro lado del camino” y
todos los demás estados deben esperar a que el azar los lleve a esos estados “privilegiados” capaces
de transitar hacia el siguiente intermediario (que a su vez es otro ensamble), o alternativamente,
sólo algunos confórmeros “aciertan” a seguir la trayectoria que finalmente conduce al siguiente
intermediario, sin desviarse en el camino. Estas formulaciones son difíciles de concebir con moléculas
pequeñas, dadas los escasos grados de libertad que se presentan cuando existes sólo unos cuantos
átomos, pero son particularmente atractivas con macromoléculas, especialmente, dado lo que hemos
aprendido de la macromoléculas con el análisis conformacional que permite la espectroscopia de
resonancia magnética nuclear.
El comportamiento predicho de las macromoléculas en tales modelos no es tan diferente al
propuesto por la teoría clásica, excepto por detalles sutiles de difícil auscultación, lo que explica
que no exista todavía un consenso en la literatura acerca del modelo que mejor describe a la
naturaleza.
Dado que las metodología estudiadas en este apartado se basan en la inspección de los intermediarios a través de técnicas que tienen a promediar las propiedades de los posibles confórmeros,
estas metodologías no permiten establecer cual de estos modelos es más cercano a la realidad ya
que todos ellos pueden ajustarse para explicar las observaciones experimentales de la cinética de
estado pre-estacionario.
A pesar de lo anterior, la combinación de las técnicas fisicoquímicas y analíticas modernas puede
darnos información sobre la termodinámica, la química y los fenómenos cuánticos asociados a los
fenómenos químicos observados. Toda esta información en conjunto podría en el futuro ayudarnos
35
estado de
alta energía
múltiples estados
de energía
intermedia
Multiples veredas
Un camino definido con tiempos de tránsito
de un estado a otro
diversos
Multiples trajectorias,
pero sólo una efectiva
Figura 12. Dos tipos de modelos para explicar la barrera energética para la conversión de una
especie química en otra. El modelo clásico de Eyring propone un intermediario con configuración
poco favorable requiere de elevada energía en forma transitoria. En otros, la teoría de las tasas de
reacción y las teorías de las fluctuaciones proponen que las moléculas no atraviesan por estado realmente, pero si pueden quedar temporalmente “atrapadas” en confórmeros que no pueden convertirse
directamente en productos, o bien transitar por tiempos largos entre estados de energía intermedia
sin “encontrar la ruta” hacia los productos. En ambos casos, de la población de moléculas presente,
sólo una fracción está en posibilidad real de completar la reacción, ya sea por que tiene la energía
suficiente, o porque alcanzó el confórmero o trayectoria efectiva.
a resolver estas interrogantes.
Adicionalmente, el análisis de la cinética de estado pre-estacionarios es excelente para detectar intermediarios y medir el curso temporal de reacciones fisiológicas en las que las respuestas
de la célula depende de respuestas transitorias controladas por la rapidez relativa de reacciones
alternativas que compiten por la misma posa de reactantes.
A pesar del poder de estas técnicas existen consideraciones importantes que uno debe hacer
para planear un experimento informativo:
a) ¿Las condiciones experimentales son posibles?
c) ¿El modelo matemático de la situación a inspeccionar tiene soluciones expresas? o bien,
¿Contamos con métodos numéricos que integren el modelo de manera confiable?
36
b) Los métodos numéricos y el poder de las computadoras disponibles permiten extraer la
información buscada en forma fidedigna.
11
Ejemplos de la literatura que ilustran el poder de esta
estrategia.
Shore & Gutfreund (1970) Estudiaron la reacción catalizada por la alcohol deshidrogeneas hepática
(LDH). En este estudio existe la ventaja de que tanto la proteína como el nucleótido poseen señales
fluorescentes características que varían a lo largo de la reacción. Además, es posible seguir la
liberación de protones mediante el uso de indicadores de pH y se puede usar alcohol deuterado para
determinar la relevancia de la transferencia del hidruro en la reacción general.
Ellos observaron que el alcohol deuterado era oxidado con una tasa de reacción mucho más lenta
y que la liberación de protones precedía ligeramente a la reducción del nucléotido en el sitio activo
de la enzima, por lo que propusieron un mecanismo como el que sigue:
NAD+
EH+
NADH
E
NADH
EH+
etOH
EH+
etO
Esquema 11
Travers et al. (1979) encontraron que por debajo de 4oC hay un cambio dramático en la energía
de activación de la reacción catalizada por la Creatina cinasa (CK).
Trabajando a estas temperaturas pudieron subdividir el transcurso inicial de la reacción catañlizada por la CK en una fase lag, una explosión y un SS . En esta enzima el complejo ternario
(E.ATP.Creatina) está en equilibrio rápido con las especies libres y aparece un intermediario que
no contiene cretina fosfato, pero es diferente al complejo ternario.
ATP
E
E
ATP
E
Cr
k1
k2
EX
k3
k4
ADP
E
k5
E
CrP
Esquema 12
Un éxito notable de este trabajo fue el haber podido determinar el valor de las cinco constantes
de rapidez involucradas en los pasos entre el complejo ternario y la regeneración de la enzima libre.
Otra enzima estudiada extensamente con estas técnicas es la miosin-ATPsa. Numerosos trabajos
realizados con esta enzima allanaron el camino de la interpretación de la información estructural de
esta proteína en relación con su función en la contracción muscular, todo ello, apesar de que en esa
37
época (70s y 80s) no se contaba con suficiente información cristalográfica de esa proteína (Greeves,
1991).
Estos métodos has sido empleados también para analizar el comportamiento de reacciones
acopladas del tipo R −Eb→ I −Ea→ P. Cuyo entendimiento facilita la interpretación de diversas vía metabólicas en la célula, pero también es importante en el diseño de ensayos enzimáticas
acoplados, procesos industriales y otras situaciones de índole práctico.
12
Referencias:
Ballew R M, Sabelko J, Reiner C, y Gruebele M, (1996) A single-sweep, nanosecond time resolution
laser temperature-jump apparatus Rev. Sci. Instrum. 67(10):3694-3699
Callender R, Dyer R B (2002) Probing protein dynamics using temperature jump relaxation
spectroscopy. Current Opinion in Structural Biology 12(5):628-633.
Danø S, Sørensen PG, & Hynne F (1999) Nature 402(6759):320-2.
Greeves (1991) Biochem. J. 274:1-14.
Halford S E, Johnson N P, and Grinstead J (1969). Biochem. J. 179: 353-365
Hammes G G, Schimmel R P (1966) J. Phys. Chem. 70(7):2319-2324
Hammes G G, Schimmel R P (1967) J. Phys. Chem 71(4):917-923
Hastings J W Mitchell G, Mattingly P H, Blinks J R van Leeuwen M (1969) Nature 222:10471050.
Naraghi M (1997) T-jump study of calcium binding kinetics of calcium chelators. Cell Calcium
22(4), 255-268
Prongjit M, Sucharitakul J, Wongnate T, Haltrich D, Chaiyen P (2009) Biochemistry 48(19):41704180
Sadqi M,Lapidus L J, Muñoz V (2003) How fast is protein hydrophobic collapse? Proc Natl
Acad Sci U S A. 14 100(21):12117-12122.
Shore & Gutfreund (1970) Biochem. 9, 4655-4659
Travers, Barman y Bertrnad (1979) Eur. J. of Biochem. 100:149-155
38