QUÉ ES LA MITOCONDRIA? Las mitocondrias son orgánulos
Anuncio

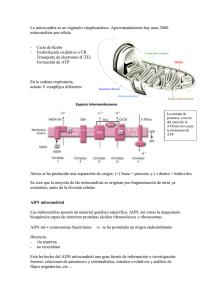
QUÉ ES LA MITOCONDRIA? Las mitocondrias son orgánulos celulares encargados de suministrar la mayor parte de la energía necesaria para la actividad celular (respiración celular). Actúan, por lo tanto, como centrales energéticas de la célula y sintetizan ATP a expensas de los carburantes metabólicos (glucosa, ácidos grasos y aminoácidos). En las mitocondrias se consume la glucosa y otras sustancias energéticas del organismo en un proceso de respiración celular que requiere oxígeno y en el que se genera energía. Para que la energía se pueda aprovechar al máximo y no se pierda en forma de calor, se almacena en un compuesto, el ATP. El ATP permite transportar y utilizar la energía generada dentro la mitocondria, en los tejidos, órganos y sistemas que la requieran para que puedan realizar su función eficazmente. Cuando existe un defecto en la producción de energía las reacciones metabólicas que la requieren no funcionan eficazmente, ni tampoco lo hacen los órganos y sistemas de nuestro organismo, especialmente aquéllos que necesitan más energía para su función (cerebro y sistema nervioso en general, músculo, hígado, riñón). El niño puede ya nacer con problemas, ya que la energía es necesaria para todos los procesos vitales. No obstante, las enfermedades mitocondriales pueden manifestarse a cualquier edad, en cualquier órgano o tejido que requiera energía, aún cuando los síntomas predominantes son neuromusculares. 1. Fármacos con riesgo de toxicidad para la mitocondria Antibióticos: Tetraciclinas, ciprofloxacino, aminoglicosidos (en pacientes con la mutación A1555G). Antivirales: Azydothymidina (AZT). Fialuridine y drogas que deplecionan al ADNmt en pacientes con depleción del mismo. Antiepilépticos: Especialmente el valporato sódico, que inhibe la fosforilación oxidativa y afecta a la oxidación de los ácidos grasos. También se deben evitar los barbitúricos y las hidantoinas. Anestésicos: Se debe evitar la administración de etomidato y thiopental en el síndrome de Kearns–Sayre. En los pacientes con enfermedad mitocondrial se debe tener en cuenta la gran sensibilidad al atracurium y rocuronium. Algunos autores han descrito complicaciones con la utilización de Fentanilo y Thiopental. Los pacientes con afectación mitocondrial presentan mayor riesgo de fallo respiratorio en el postoperatorio quirúrgico debido en parte a la mayor producción de citoquinas y la consiguiente formación de óxido nítrico que en grandes cantidades afecta la producción de energía. Se aconseja siempre que sea posible la utilización de anestesia epidural. También se debería evitar la hipoxia crónica, que pueden provocar anestesias irreversibles. EL GEN TK2 La miopatía con depleción de ADN mitocondrial corresponde al tipo 2 de los síndromes de depleción de ADN mitocondrial (MTDPS2). Este proceso se caracteriza por el desarrollo de debilidad muscular, generalmente de comienzo en la infancia, y está relacionado con la depleción del ADN mitocondrial (mtDNA) en los músculos esqueléticos. La causa de este síndrome son las mutaciones en el gen TK2, situado en el brazo largo del cromosoma 16 (16q22-q23.1). Este gen codifica la timidina-kinasa 2 mitocondrial, una deoxiribonucleósido kinasa que fosforila la timidina, deoxicitidina y deoxiuridina, dando lugar a los correspondientes nucleósidos requeridos en las mitocondrias para la síntesis del ADN mitocondrial. Se han descrito varias mutaciones en este gen: I22M (I95M), H90N (H163N), T77M (T150M), I181N (I254N), entre otras. Este proceso es de carácter autosómico recesivo, por lo que se requiere que ambas copias del gen, la procedente del padre y la procedente de la madre, estén afectadas para que ocurra la manifestación. EL TRATAMIENTO CON NUCLEÓTIDOS: UNA PUERTA A LA ESPERANZA Hace más de 10 años, y por iniciativa de la Fundación para el Discapacitado Muscular Nacional con sede en Madrid (www.fundismun.org), se inició en el Columbia University Medical Center en Nueva York, EEUU, una investigación sobre la miopatía mitocondrial por déficit de timidin kinasa 2 o tk2. Descubrieron que dicho déficit se debía a una mutación en el gen tk2 del cromosoma 16, y utilizando ratones con la misma mutación , comprobaron que la administración oral de dTMP (deoxitimidina monofosfato) y dCMP (deocitidinamonofosfato) triplicaba la supervivencia de los ratones y mejoraba notablemente los síntomas. Al existir tan pocos casos en el mundo y tratarse de una situación de riesgo vital para los niños que la padecían, se aprobó su uso en humanos directamente, ya que no produce efectos secundarios. Actualmente, los pacientes que están tomando este tratamiento con nucleótidos han mejorado objetivamente, abriendo una puerta a lo que podría ser la curación de dicha enfermedad. El equipo de investigación del Columbia Medical Center, al mando del Dr. Michio Hirano, y en permanente comunicación con investigadores españoles, sigue trabajando en este proyecto para perfeccionar el tratamiento.