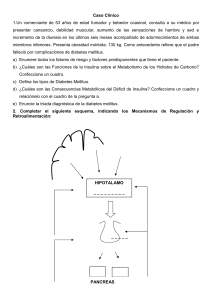
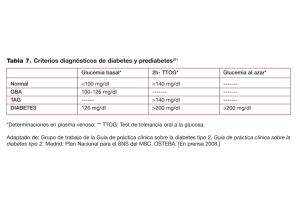
18 a EDICIÓN Medical Knowledge Self-Assessment Program Programa de autoaprendizaje de conocimientos médicos Endocrinología y Metabolismo ® Sumario Trastornos del metabolismo de la glucosa Diabetes mellitus . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1 Cribado de la diabetes mellitus . . . . . . . . . . . . . . . . . . . . 1 Criterios diagnósticos de la diabetes mellitus . . . . . . . . 1 Clasificación de la diabetes mellitus . . . . . . . . . . . . . . . . 1 Manejo de la diabetes mellitus . . . . . . . . . . . . . . . . . . . . 8 Hiperglucemia inducida por fármacos . . . . . . . . . . . . . . . . 18 Manejo hospitalario de la hiperglucemia . . . . . . . . . . . . . . 18 Pacientes con diabetes mellitus hospitalizados . . . . . 20 Pacientes sin diabetes mellitus hospitalizados . . . . . . 20 Complicaciones agudas de la diabetes mellitus . . . . . . . . . 20 Cetoacidosis diabética/síndrome hiperosmolar hiperglucémico . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20 Complicaciones crónicas de la diabetes mellitus . . . . . . . 23 Morbilidad cardiovascular . . . . . . . . . . . . . . . . . . . . . . 23 Retinopatía diabética . . . . . . . . . . . . . . . . . . . . . . . . . . . 23 Nefropatía diabética . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25 Neuropatía diabética . . . . . . . . . . . . . . . . . . . . . . . . . . . 25 Úlceras del pie diabético . . . . . . . . . . . . . . . . . . . . . . . . 26 Hipoglucemia. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26 Hipoglucemia en pacientes con diabetes mellitus . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26 Hipoglucemia en pacientes sin diabetes mellitus . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 27 Hipoglucemia inadvertida . . . . . . . . . . . . . . . . . . . . . . . 28 Trastornos de la hipófisis Anatomía y fisiología del hipotálamo y la hipófisis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29 Alteraciones de la hipófisis . . . . . . . . . . . . . . . . . . . . . . . . . . 30 Masas hipofisarias detectadas de manera incidental . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30 Silla turca vacía . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30 Otras alteraciones . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31 Efectos de masa de los tumores hipofisarios . . . . . . . . 32 Evaluación de los tumores hipofisarios . . . . . . . . . . . . 32 Tratamiento de los tumores hipofisarios clínicamente no funcionales . . . . . . . . . . . . . . . . . . . . . 33 Déficit hormonal hipofisario. . . . . . . . . . . . . . . . . . . . . . . . . 34 Panhipopituitarismo . . . . . . . . . . . . . . . . . . . . . . . . . . . 34 Déficit de corticotropina (déficit secundario de cortisol) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 34 Déficit de tirotropina . . . . . . . . . . . . . . . . . . . . . . . . . . . 35 Déficit de gonadotropinas . . . . . . . . . . . . . . . . . . . . . . . 35 Déficit de hormona del crecimiento . . . . . . . . . . . . . . 36 Diabetes insípida central . . . . . . . . . . . . . . . . . . . . . . . . 36 Exceso de hormonas hipofisarias. . . . . . . . . . . . . . . . . . . . . 36 Hiperprolactinemia y prolactinoma . . . . . . . . . . . . . . 36 Acromegalia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 38 Tumores secretores de tirotropina . . . . . . . . . . . . . . . . 39 Secreción excesiva de hormona antidiurética . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 39 Exceso de corticotropina de origen hipofisario (enfermedad de Cushing) . . . . . . . . . . . . . . . . . . . . . . . 39 Trastornos de las glándulas suprarrenales Anatomía y fisiología suprarrenal. . . . . . . . . . . . . . . . . . . . 40 Exceso de hormonas suprarrenales . . . . . . . . . . . . . . . . . . 41 Exceso de cortisol (síndrome de Cushing debido a masa suprarrenal) . . . . . . . . . . . . . . . . . . . . . . . . . . . . 41 Hiperaldosteronismo primario . . . . . . . . . . . . . . . . . . . 42 Feocromocitoma y paraganglioma . . . . . . . . . . . . . . . . 44 Tumores suprarrenales productores de andrógenos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 47 Déficit de hormonas suprarrenales . . . . . . . . . . . . . . . . . . . 47 Insuficiencia suprarrenal primaria . . . . . . . . . . . . . . . 47 Función suprarrenal durante la enfermedad crítica . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 50 Masa suprarrenal . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 50 Masas suprarrenales descubiertas de manera incidental . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 50 Carcinoma de corteza suprarrenal . . . . . . . . . . . . . . . . 52 Trastornos de la glándula tiroides Anatomía y fisiología de la tiroides . . . . . . . . . . . . . . . . . . . 52 Exploración de la tiroides . . . . . . . . . . . . . . . . . . . . . . . . . . . 53 Trastornos estructurales de la glándula tiroides . . . . . . . . 53 Nódulos tiroideos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 53 Bocio . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 55 Cáncer de tiroides . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 56 Evaluación de la función tiroidea. . . . . . . . . . . . . . . . . . . . . 58 Trastornos de la función tiroidea . . . . . . . . . . . . . . . . . . . . . 58 Exceso de hormonas tiroideas (hipertiroidismo y tirotoxicosis) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 58 Déficit de hormonas tiroideas . . . . . . . . . . . . . . . . . . . . 62 Disfunción tiroidea inducida por fármacos . . . . . . . . 64 Función y disfunción tiroidea en el embarazo. . . . . . . . . . 65 Síndrome de enfermedad no tiroidea (síndrome del enfermo eutiroideo). . . . . . . . . . . . . . . . . . . . . . . . . . . . . 66 Emergencias tiroideas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 66 Crisis tirotóxica (tormenta tiroidea) . . . . . . . . . . . . . . . 66 Coma mixedematoso . . . . . . . . . . . . . . . . . . . . . . . . . . . 67 Trastornos de la reproducción Fisiología de la reproducción femenina . . . . . . . . . . . . . . . 68 Amenorrea. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .68 Cuadro clínico . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 68 Evaluación de la amenorrea . . . . . . . . . . . . . . . . . . . . . 69 Tratamiento de la amenorrea . . . . . . . . . . . . . . . . . . . . 71 Síndromes de hiperandrogenismo . . . . . . . . . . . . . . . . . . . 71 Hirsutismo y síndrome del ovario poliquístico . . . . . 71 Infertilidad femenina. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 72 Fisiología de la reproducción masculina. . . . . . . . . . . . . . . 73 Hipogonadismo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 73 Causas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 73 Cuadro clínico . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 74 Evaluación . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 74 Manejo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 74 Abuso de esteroides anabólicos en los varones. . . . . . . . . . 74 Cambios de la testosterona en el envejecimiento . . . . . . . 75 Infertilidad masculina. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 77 Ginecomastia. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 77 Manejo de la terapia hormonal transgénero. . . . . . . . . . 77 Trastornos del calcio y los huesos Homeostasis del calcio y fisiología ósea . . . . . . . . . . . . . . . 78 Hipercalcemia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 79 Manifestaciones clínicas de la hipercalcemia . . . . . . . 79 Causas y diagnóstico de la hipercalcemia . . . . . . . . . . 80 Manejo de la hipercalcemia . . . . . . . . . . . . . . . . . . . . . . 83 Hipocalcemia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 83 Manifestaciones clínicas de la hipocalcemia . . . . . . . 83 Causas y diagnóstico de la hipocalcemia . . . . . . . . . . . 84 Manejo de la hipocalcemia . . . . . . . . . . . . . . . . . . . . . . 84 Enfermedad ósea metabólica . . . . . . . . . . . . . . . . . . . . . . . . 85 Masa ósea baja y osteoporosis . . . . . . . . . . . . . . . . . . . . 85 Déficit de vitamina D . . . . . . . . . . . . . . . . . . . . . . . . . . . 90 Enfermedad ósea de Paget . . . . . . . . . . . . . . . . . . . . . . . 90 Bibliografía. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 91 Endocrinología y Metabolismo Trastornos del metabolismo de la glucosa La hiperglucemia tiene su origen en un metabolismo anómalo de los hidratos de carbono secundario a déficit de insulina, resistencia periférica a la acción de la insulina o una combinación de ambos. La hiperglucemia que supera el intervalo normal de los niveles de glucosa pero no cumple los criterios diagnósticos de diabetes mellitus se define como prediabetes, condición que aumenta el riesgo de aparición de diabetes. Diabetes mellitus Cribado de la diabetes mellitus El cribado de la diabetes mellitus tipo 2 en la población adulta general está indicado porque: 1) la diabetes tipo 2 suele ir precedida por un período prolongado de hiperglucemia asintomática en el cual pueden producirse daños microvasculares y macrovasculares; 2) se ha demostrado que las intervenciones en el estilo de vida y el tratamiento farmacológico tienen la capacidad de retrasar o evitar la aparición de diabetes tipo 2 en las personas con prediabetes, y 3) un control glucémico y un manejo intensivo inicial de la hiperlipemia y la hipertensión pueden prevenir o reducir la progresión de la enfermedad cardiovascular (ECV) microvascular y macrovascular. La American Diabetes Association (ADA) y el U.S. Preventive Services Task Force (USPSTF) incluyen la edad, el índice de masa corporal (IMC), la raza/etnia y otros factores de riesgo como parte de los criterios de las recomendaciones de cribado para la diabetes tipo 2 (Tabla 1). Estos factores de riesgo están asociados a un riesgo elevado de aparición de diabetes. No se recomienda el cribado de la diabetes tipo 1. Las pruebas de detección de anticuerpos en una persona de alto riesgo con un familiar con diabetes tipo 1 deben realizarse en el contexto de un ensayo clínico (www.diabetestrialnet.org). Criterios diagnósticos de la diabetes mellitus La diabetes mellitus puede diagnosticarse con un resultado anómalo en uno de estos tres parámetros: hemoglobina A1c, glucemia en ayunas o basal (GB) o glucemia a las dos horas (G2h) después de una prueba tolerancia a la glucosa oral (PTGO) con 75 g de hidratos de carbono (Tabla 2). Los resultados anómalos en personas asintomáticas deben confirmarse con la repetición de la prueba. Un único valor glucémico aleatorio ≥200 mg/dl (11,1 mmol/l) en el contexto de una hiperglucemia sintomática es diagnóstico de diabetes. Los resultados de las pruebas de diabetes difieren en función de la prueba realizada, GB, G2h o hemoglobina A1c. La G2h tiene una mayor sensibilidad para el diagnóstico de diabetes que la GB o la hemoglobina A1c. Es necesario tener en cuenta las ventajas y los inconvenientes de las pruebas a la hora de determinar la mejor opción de cribado para un paciente (Tabla 3). PUNTOS CLAVE • La diabetes mellitus puede diagnosticarse con un resultado anómalo en uno de los siguientes parámetros en las pruebas de cribado: hemoglobina A1c, glucemia basal o glucemia a las dos horas después de una prueba de tolerancia a la glucosa oral con 75 g de hidratos de carbono. • Los resultados anómalos en personas asintomáticas deben confirmarse con la repetición de la prueba. Clasificación de la diabetes mellitus La anomalía insulínica subyacente, ya sea déficit de insulina absoluto o relativo, resistencia periférica a la insulina o ambos a la vez, es importante para clasificar el tipo de diabetes mellitus y tiene implicaciones en las opciones de tratamiento (Tabla 4). Déficit de insulina Diabetes mellitus tipo 1 La diabetes mellitus tipo 1 se caracteriza por un estado de déficit de insulina secundario a la destrucción de células beta del páncreas productoras de insulina. La destrucción puede ser secundaria a autoinmunidad, idiopática o adquirida. Diabetes mellitus inmunomediada La diabetes tipo 1 inmunomediada (tipo 1A) representa la causa subyacente en entre el 5% y el 10% de las personas con diabetes de nuevo diagnóstico. El mecanismo de la destrucción de las células beta es multifactorial y se debe probablemente a factores ambientales en personas con susceptibilidad genética. Determinados alelos del antígeno leucocitario humano (HLA) muestran una estrecha relación con la diabetes tipo 1 inmunomediada. Típicamente, en el diagnóstico hay uno o varios autoanticuerpos dirigidos a las dianas siguientes: ácido glutámico descarboxilasa (GAD65), tirosina fosfatasas IA-2 e IA-2β, células de los islotes pancreáticos, insulina y transportador de zinc (Zn T-8). Debido a la disponibilidad de ensayos altamente automáticos, se recomienda utilizar los autoanticuerpos contra GAD65 y contra IA-2 para el cribado inicial. Los 1 Trastornos del metabolismo de la glucosa Tabla 1. Guías para el cribado de la diabetes mellitus tipo 2 en adultos asintomáticos Criterios para el cribado ADA (2018)a USPSTF (2015) Cribar a los adultos con sobrepeso (IMC ≥25 o ≥23 en norteamericanos de origen asiático) con al menos un factor de riesgo adicional: Cribar a los adultos de entre 40 y 70 años de edad que tengan sobrepeso u obesidad como parte de una evaluación del riesgo de enfermedad cardiovascular. Familiar de primer grado con diabetes Raza/etnia de alto riesgo (negros, hispanos/latinos, nativos americanos, asiáticos, nativos de Hawai/islas del Pacífico) Otros factores de riesgo: Alto porcentaje de grasa abdominal Antecedentes de diabetes mellitus gestacional Hiperlipemia Antecedentes de enfermedad cardiovascular Hipertensión Inactividad física Inactividad física Hipertensión (≥140/90 mmHg o con tratamiento antihipertensivo) Tabaquismo Colesterol-HDL <35 mg/dl (0,90 mmol/l) y/o triglicéridos >250 mg/dl (2,82 mmol/l) Síndrome del ovario poliquístico Hemoglobina A1c ≥5,7% (39 mmol/mol), TAG o GBA en pruebas anteriores Otros trastornos asociados a resistencia a la insulina (obesidad grave, acantosis pigmentaria [nigricans]) Criterios adicionales para el cribado Todos los adultos ≥45 años de edad — Consideraciones adicionales sobre el cribado Considerar el cribado de los pacientes que toman medicamentos de los que se sabe aumentan el riesgo de diabetes, como glucocorticoides, diuréticos tiazídicos, medicamentos contra el VIH y antipsicóticos atípicos La diabetes puede darse en pacientes más jóvenes o con un IMC inferior. Considerar el cribado en un momento anterior en presencia de uno de los factores de riesgo siguientes: Antecedentes familiares de diabetes Antecedentes de diabetes gestacional Síndrome del ovario poliquístico Raza/etnia de alto riesgo (negros, hispanos/latinos, norteamericanos de origen asiático, nativos americanos/ de Alaska, nativos de Hawai/islas del Pacífico) Intervalos de cribado aSe Repetir el cribado cada 3 años si los resultados son normales. Se recomienda realizar las pruebas anualmente en caso de diagnóstico de prediabetes (hemoglobina A1c entre 5,7% y 6,4%, TAG, GBA) Los datos que respaldan unos intervalos óptimos de cribado son limitados. Puede ser razonable repetir el cribado cada tres años hallará un instrumento de cribado opcional del riesgo de diabetes de la ADA en www.diabetes.org/are-you-at-risk/diabetes-risk-test/. Consultado el 16 de mayo de 2018. ADA: American Diabetes Association; ECV: enfermedad cardiovascular; GBA: glucemia basal alterada; HDL: lipoproteínas de alta densidad; IMC: índice de masa corporal; TAG: tolerancia alterada a la glucosa; USPSTF: U.S. Preventive Services Task Force; VIH: virus de la inmunodeficiencia humana. Recomendaciones de la American Diabetes Association. 2. Classification and diagnosis of diabetes: standards of medical care in diabetes-2018. Diabetes Care. 2018;41:S1 3-S27. [PMID:29222373] Recomendaciones de Siu AL; US Preventive Services Task Force. Screening for abnormal blood glucose and type 2 diabetes mellitus: U.S. Preventive Services Task Force Recommendation Statement. Ann Intern Med. 2015;163:861-8. [PMID: 26501513] doi:10. 7326/M15-2345 autoanticuerpos contra GAD65 tienen una prevalencia elevada (70%) en el momento del diagnóstico y pueden seguir siendo detectables durante años. La diabetes tipo 1 inmunomediada tiene una presentación variable que va desde la hiperglucemia moderada hasta una cetoacidosis diabética (CAD) potencialmente mortal. En el momento del diagnóstico, aproximadamente el 90% de las células beta funcionales han sido destruidas. Iniciar la insuli2 na en el momento del diagnóstico puede reducir la toxicidad asociada a la hiperglucemia extrema, ya que permite a las células beta recuperar cierta capacidad de producir insulina. Aunque este período, llamado de «luna de miel», puede durar de semanas a años, el uso de insulina debe proseguir para disminuir el estrés impuesto a las células beta funcionales restantes y prolongar su vida. El déficit de insulina requiere el uso de tratamiento con insulina durante toda la vida. Trastornos del metabolismo de la glucosa Tabla 2. Criterios diagnósticos de la diabetes mellitusa Prueba Intervalo normal Riesgo elevado de diabetes (prediabetes) Diabetes Glucemia aleatoria — — Síntomas hiperglucémicos más una glucemia aleatoria ≥200 mg/dl (11,1 mmol/l) <100 mg/dl (5,6 mmol/l) 100-125 mg/dl (5,6-6,9 mmol/l) ≥126 mg/dl (7,0 mmol/l) Glucemia a las 2 horas en una PTGO <140 mg/dl (7,8 mmol/l) 140-199 mg/dl (7,8-11,0 mmol/l) ≥200 mg/dl (11,1 mmol/l) Hemoglobina A1cd,e <5,7% (39 mmol/mol) 5,7%-6,4% (39-46 mmol/mol) ≥6,5% (48 mmol/mol) Glucemia basalb c a En ausencia de síntomas hiperglucémicos, los resultados anómalos de la glucemia basal, la PTGO o la hemoglobina A 1c deben confirmarse repitiendo la misma prueba un día separado. Si dos pruebas diferentes arrojan resultados discordantes, la American Diabetes Association recomienda repetir la prueba con el resultado anómalo. bAyuno durante un mínimo de ocho horas. c Una PTGO implica el consumo de una carga de 75 g de glucosa disuelta en agua. dLa American Diabetes Association recomienda un ensayo de hemoglobina A 1c acreditado por el National Glycohemoglobin Standardization Program (NGSP) que esté estandarizado según el ensayo de Control and Complication Trial (DCCT). eLas directrices del Veterans Affairs/Department of Defense recomiendan la confirmación de la diabetes sobre la base de un valor elevado de hemoglobina A de entre 6,5% y 1c 6,9% con una glucemia basal elevada ≥126 mg/dl (7,0 mmol/l) debido a la existencia de pruebas sólidas de diferencias raciales entre el control glucémico y los valores de hemoglobina A1c para el diagnóstico y el tratamiento. PTGO: prueba tolerancia a la glucosa oral (PTGO). Datos de la American Diabetes Association. 2. Classification and diagnosis of diabetes: Standards of Medical Care in Diabetes—2018. Diabetes Care. 2018;41 (Suppl. 1):S13-S27. [PMID:29222373] Datos del U.S. Department of Veterans Affairs/U.S. Department of Defense. VA/DoD Clinical Practice Guidelines for the management of diabetes mellitus in primary care. 2017. www.healthquality.va.gov/guidelines/cd/diabetes. Consultado el 16 de mayo de 2018. Los pacientes con diabetes tipo 1 inmunomediada presentan también un mayor riesgo de otros trastornos autoinmunes, entre los que se encuentran la enfermedad celíaca, trastornos tiroideos, el vitiligo y la insuficiencia primaria autoinmune de las glándulas suprarrenales. La diabetes autoinmune latente del adulto (LADA, por sus siglas en inglés) se caracteriza por el desarrollo de anticuerpos que provocan la destrucción de células beta y, en última instancia, déficit de insulina. Normalmente, las personas con LADA no necesitan insulina en el primer momento y muchas veces son clasificadas erróneamente como afectadas de diabetes tipo 2. Con el paso de los meses y años posteriores al diagnóstico se produce una lenta progresión hacia la dependencia de la insulina en el contexto de autoanticuerpos positivos. PUNTO CLAVE • Los autoanticuerpos contra la ácido glutámico descarboxilasa (GAD65) y la tirosina fosfatasa IA-2 muestran una estrecha relación con la diabetes tipo 1 inmunomediada y deben medirse en el diagnóstico inicial para determinar la etiología. tipo 2, y la enfermedad es más frecuente en los pacientes asiáticos y afroamericanos, sobre todo de ascendencia africana subsahariana. Diabetes mellitus tipo 1 adquirida La destrucción de células beta puede tener su origen en enfermedades que afectan al páncreas o en el efecto de fármacos o infecciones (véase la Tabla 4). Esto puede dar lugar a la alteración de la producción o secreción de insulina, con la aparición posterior de diabetes tipo 1. Resistencia a la insulina La resistencia a la insulina se caracteriza por el uso ineficaz de la insulina por parte de las células periféricas para utilizar la glucosa y los ácidos grasos. Los niveles de glucemia se mantienen dentro del intervalo normal mientras las células beta puedan incrementar la producción de insulina. La hiperglucemia se debe a un déficit relativo de insulina que tiene lugar cuando el páncreas deja de ser capaz de producir suficiente insulina. La obesidad aumenta el riesgo de resistencia a la insulina, que también forma parte del síndrome metabólico y predispone a la aparición de diabetes tipo 2. Diabetes mellitus tipo 1 idiopática Síndrome metabólico La diabetes tipo 1 idiopática (tipo 1B) se caracteriza por un déficit variable de insulina con origen en la destrucción de células beta sin presencia de autoanticuerpos. Las personas con diabetes tipo 1 idiopática pueden experimentar CAD episódica. Por lo general, las personas con diabetes idiopática tienen antecedentes familiares importantes de diabetes El síndrome metabólico comprende una constelación de factores de riesgo de aparición de diabetes tipo 2 y ECV que incluye obesidad abdominal, metabolismo de la glucosa alterado, hiperlipemia e hipertensión. Varias organizaciones definen el síndrome metabólico de maneras diferentes (Tabla 5). La Endocrine Society recomienda cribar a los pacientes con fac3 Trastornos del metabolismo de la glucosa Tabla 3. Comparación de las pruebas de cribado de la diabetes mellitus Prueba Ventajas Inconvenientes Hemoglobina A1c Cómoda: no requiere ayuno y no hay restricciones en cuanto al momento de la obtención Menor sensibilidad diagnóstica que la GB o la G2h No está influida por la enfermedad, el estrés, etc. Mide la glucemia durante las 8-12 semanas previas Aumentos o descensos erróneos del resultado de la hemoglobina A1c secundarios a factores que afectan a la supervivencia de los eritrocitosa: Anemia ferropénica Hemorragia/hemólisis Variabilidad biológica mínima Enfermedad renal La muestra de sangre se mantiene estable Enfermedad hepática Ensayo estandarizado Exactitud de la prueba controlada La medición está correlacionada con los resultados microvasculares y macrovasculares Embarazo Variantes de la hemoglobina en personas de ascendencia africana, del Sudeste Asiático y mediterráneab Valor más alto en los negros que en las personas blancas no hispanasc Influencia de algunas variantes de la glucosa-6-fosfato deshidrogenasad No está disponible en algunas regiones del mundo Cara Glucemia basal (en ayunas) No es cara Amplia disponibilidad Ensayo automático Inconveniente: ayuno obligatorio ≥8 horas y restricciones en cuanto al momento de la obtención Influida por la enfermedad y el estrés Mide un único punto temporal Alta variabilidad biológica en el mismo paciente Muestra de sangre no estable después de la obtención Variación diurna Las complicaciones de la diabetes no están tan ligadas a la GB como a la hemoglobina A1c El origen de la muestra (sangre capilar, venosa o arterial) altera la medición Estandarización de los ensayos incompleta G2h en una PTGO Muy sensible para detectar el riesgo de aparición de diabetes Detecta las anomalías iniciales del metabolismo de la glucosa Inconvenientes parecidos a los de la prueba de GB Preparación prolongada del paciente Riesgo de hipoglucemia a las 4-6 horas en las personas normales Mala reproducibilidad Cara aEn caso de recambio eritrocítico alterado, deben usarse pruebas de glucemia en lugar de hemoglobina A 1c para el cribado de la diabetes. bAlgunos métodos empleados para determinar la hemoglobina A1c pueden medir con exactitud la hemoglobina A1c en las personas que son heterocigotas para HbS, HbE, HbC, HbD y HbF elevada. En las personas que son homocigotas para HbS, HbC o HbSC, debe usarse la glucemia en lugar de la hemoglobina A 1c para fines diagnósticos. Las personas de raza negra que son heterocigotas para HbS pueden tener una hemoglobina A 1c el 0,3% inferior que las personas sin el rasgo en cualquier nivel de glucemia media. cLa hemoglobina A es más alta en la personas de raza negra que en los individuos blancos no hispanos en el contexto de valores parecidos de la GB y la glucosa pospandrial. 1c A pesar de esto, el riesgo de complicaciones asociadas a A1c es similar en las personas de raza negra y blanca no hispanas. dExiste una relación entre una hemoglobina A inferior y los varones hemicigotos y las mujeres homocigotas con la glucosa-6-fosfato deshidrogenasa G202A ligada 1c al cromosoma X en el 0,8% y el 0,7%, respectivamente. G2h: glucosa prandial a las 2 horas; GB: glucemia basal; PTGO: prueba tolerancia a la glucosa oral. Datos de la American Diabetes Association. 2. Classification and diagnosis of diabetes: Standards of Medical Care in Diabetes—2018. Diabetes Care. 2018;41 :S13-S27. [PMID:29222373] Datos de la American Diabetes Association. 6. Glycemic targets: Standards of Medical Care in Diabetes—2018. Diabetes Care. 2018;41:S55-S64. [PMID: 29222377] Datos de Sacks DB. A1C versus glucose testing: a comparison. Diabetes Care. 2011;34:518-23. [PMID: 21270207] Datos de NGSP: Harmonizing Hemoglobin A1c Testing web site. www.ngsp.org. Consultado en junio de 2018. Datos del National Institute of Diabetes and Digestive and Kidney Diseases. Comparing tests for diabetes and prediabetes. Mar. 2014. NIH Publication No. 14-7850. www. diabetes.niddk.nih.gov. Consultado en junio de 2018. 4 Trastornos del metabolismo de la glucosa Tabla 4. Clasificación de la diabetes mellitus Déficit de insulinaa Autoinmune (tipo 1A) Diabetes tipo 1 LADA Formas infrecuentes: síndrome de la «persona rígida», anticuerpos contra los receptores de insulina Idiopática (tipo 1B) (seronegativa) Adquirida Enfermedades del páncreas exocrino: pancreatitis, traumatismo/pancreatectomía, neoplasia, fibrosis quística, hemocromatosis, pancreatopatía fibrocalculosa Medicamentosa: Vacor (veneno para ratas), pentamidina intravenosa Infecciones: rubéola congénita, enterovirus Resistencia a la insulina Diabetes tipo 2b Con tendencia a la cetosisc Otros tipos o tipos infrecuentes Defectos genéticos del funcionamiento de las células beta (incluyendo seis síndromes MODY distintos) Defectos genéticos de la acción de la insulina Endocrinopatías: Acromegalia, síndrome de Cushing, glucagonoma, feocromocitoma, hipertiroidismod Somatostatinoma, aldosteronomad Relacionados con fármacos: Glucocorticoides, tiazidas, betabloqueates, diazoxida, tacrolimus, ciclosporina, niacina, inhibidores de la proteasa del VIH, antipsicóticos atípicos (clozapina, olanzapina)e Síndromes genéticos: Síndrome de Downf Síndrome de Wolfram (DIDMOAD)g Síndromes de Klinefelter, Turner y Prader-Willi; distrofia miotónicad aNormalmente de insulina. bResistencia la destrucción de las células beta produce déficit absoluto a la insulina con déficit relativo progresivo de insulina. cMás frecuente en las personas de raza no blanca que presentan cetoacidosis diabética pero dejan de depender de la insulina con el tiempo. dAcción alterada de la insulina. eSecreción alterada de insulina, acción alterada de la insulina o metabolismo alterado de la glucosa hepática. fDéficit gDéficit de insulina autoinmune. de insulina. DIDMOAD: diabetes insípida, diabetes mellitus, atrofia óptica y ceguera; LADA: diabetes autoinmune latente del adulto; MODY: diabetes del adulto de inicio juvenil, VIH: virus de la inmunodeficiencia humana. Datos de la American Diabetes Association. 2. Classification and diagnosis of diabetes: Standards of Medical Care—2018. Diabetes Care. 2018;41:S13-S27. [PMID:29222373] tores de riesgo de síndrome metabólico cada tres años para evaluar la glucemia basal, los lípidos en ayunas, la presión arterial y la circunferencia de la cintura. En los pacientes con síndrome metabólico se recomienda calcular el riesgo cardiovascular a 10 años, ya sea mediante la puntuación del riesgo de Framingham o utilizando la calculadora de riesgo del American College of Cardiology (ACC)/American Heart Association (AHA). Diabetes mellitus tipo 2 La mayoría de los casos de diabetes (del 90% al 95%) cumplen los criterios de diabetes tipo 2. La diabetes tipo 2 se define por una hiperglucemia que cursa con resistencia a la insulina y/o déficit relativo de insulina. El grado de disfunción de las células beta determina el grado de hiperglucemia, que puede empeorar con el tiempo a medida que desciende la producción de insulina. La patogénesis de la diabetes tipo 2 es multifactorial, con influencias de factores tanto genéticos como ambientales. La diabetes tipo 2 suele estar presente en los familiares de primer grado de las personas de alto riesgo y de los diagnosticados de diabetes tipo 2. Además, algunos grupos étnicos presentan un mayor riesgo, incluyendo hispanos/latinos, afroamericanos, nativos americanos y norteamericanos de origen asiático. Otros factores de riesgo de diabetes son el envejecimiento y la actividad física reducida. Clásicamente, la diabetes tipo 2 se presenta en adultos, aunque hay una incidencia creciente en niños y adolescentes a medida que aumenta la tasa de sobrepeso/obesidad en estas poblaciones. La diabetes tipo 2 tiene un inicio gradual, y la mayoría de las personas afectadas permanecen asintomáticas durante varios años. En el momento del diagnóstico, estos pacientes pueden presentar ya ECV microvascular y/o macrovascular. Aunque las células beta no producen suficiente insulina para superar la resistencia a la insulina y mantener la normoglucemia, en la diabetes tipo 2 la producción de insulina basta para inhibir la lipólisis y evitar la CAD. En la diabetes tipo 2, puede producirse CAD raramente, en el contexto de estrés extremo o enfermedad. La aparición de diabetes tipo 2 en las personas de alto riesgo puede retrasarse o evitarse con modificaciones del estilo de vida (dieta, ejercicio), intervenciones farmacológicas o cirugía metabólica. El objetivo de estas intervenciones es la pérdida de peso y la reducción de la resistencia a la insulina. En el Diabetes Prevention Program (DPP), las modificaciones del estilo de vida redujeron la incidencia de diabetes tipo 2 en personas con prediabetes en el 58%. Por ello, la ADA recomienda los objetivos del DPP de una pérdida de peso del 7% a lo largo de seis meses y al menos 150 min/semana de ejercicio de intensidad moderada para reducir el riesgo de aparición de diabetes. Se recomienda una dieta rica en grasas monoinsaturadas, cereales integrales, verduras, frutas enteras y frutos secos. Hay varias intervenciones farmacológicas con eficacia demostrada para reducir el riesgo de diabetes (Tabla 6). Es necesario tener en cuenta los datos de la seguridad, el coste y la durabilidad a largo plazo de cada intervención en cada pa5 Trastornos del metabolismo de la glucosa Tabla 5. Criterios para la definición del síndrome metabólico Criterios de calificación NCEP ATP III 2005 (cumple al menos tres de los cinco criterios) International Diabetes Federation (2006) (obesidad central imprescindible y al menos dos de los cuatro criterios restantes) Circunferencia de la cintura Varones ≥102 cm Personas de origen europeo Mujeres ≥88 cm Varones ≥94 cm Mujeres ≥80 cm Personas del sudeste asiático Varones ≥90 cma Mujeres ≥80 cm Chinos Varones ≥90 cma Mujeres ≥80 cm Japoneses Varones ≥90 cma Mujeres ≥80 cm Originarios de América del Sur/Central Varones ≥90 cma Mujeres ≥80 cm Subsaharianos Varones ≥94 cm Mujeres ≥80 cm Originarios del Mediterráneo Oriental y Oriente Medio Varones ≥94 cm Mujeres ≥80 cm TG basales ≥150 mg/dl (1,7 mmol/l) o ≥150 mg/dl (1,7 mmol/l) o Tratamiento farmacológico para TG elevados Tratamiento farmacológico para TG elevados Colesterol-HDL Varones <40 mg/dl (1,0 mmol/l) Varones <40 mg/dl (1,0 mmol/l) Mujeres <50 mg/dl (1,3 mmol/l) o Mujeres <50 mg/dl (1,3 mmol/l) o Tratamiento farmacológico para el colesterol-HDL bajo Tratamiento farmacológico para el colesterol-HDL bajo Presión arterial Glucemia basal (en ayunas) aLa Sistólica ≥130 mmHg Sistólica ≥130 mmHg Diastólica ≥85 mmHg o Diastólica ≥85 mmHg o Tratamiento farmacológico para la hipertensión Tratamiento farmacológico para la hipertensión Glucemia ≥100 mg/dl o Glucemia ≥100 mg/dl o Tratamiento farmacológico para la glucemia elevada Tratamiento farmacológico para la glucemia elevada circunferencia de la cintura es de 90 cm según la International Diabetes Foundation y de 88 cm según el NCEP ATP III. HDL: lipoproteínas de alta densidad; NCEP ACP III: National Cholesterol Education Program - Adult Treatment Panel III; TG: triglicéridos. Datos de Alberti KG, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, et al; International Diabetes Federation Task Force on Epidemiology and Prevention. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation. 2009;120:1640-5. [PMID: 19805654] Datos de la International Diabetes Federation. The IDF consensus worldwide definition of the metabolic syndrome, 2006. www.idf.org/our-activities/advocacy-awareness/ resources-and-tools/60:idfconsensus-worldwide-definitionof-the-metabolic-syndrome.html. Consultado en junio de 2018. ciente concreto. La metformina redujo la incidencia de diabetes en el 31% en comparación con placebo en el DPP. Además, hay datos de la seguridad a largo plazo de la metformina. La ADA y la American Association of Clinical Endocrinologists (AACE) recomiendan utilizar metformina en un primer 6 momento para prevenir el riesgo de diabetes en las personas con prediabetes, sobre todo en quienes presentan valores crecientes de hemoglobina A1c a pesar de las modificaciones del estilo de vida o que tienen menos de 60 años de edad, obesidad o antecedentes de diabetes gestacional. Trastornos del metabolismo de la glucosa Tabla 6. Estrategias para prevenir o retrasar el inicio de la diabetes mellitus tipo 2 Intervención Eficacia Dieta y ejercicioa Se ha observado que retrasa el inicio de la diabetes un máximo de 10-20 años Deshabituación tabáquica Moderadamente eficaz mientras no provoque aumento de peso, aunque está siempre recomendado Cirugía bariátrica Eficaz si se utiliza en personas con obesidad mórbida (IMC >40) a Metformina Se ha observado que retrasa el inicio de la diabetes un máximo de 10 años Inhibidores de la lipasa (orlistat) Se ha observado que retrasan el inicio de la diabetes un máximo de cuatro años Inhibidores de la α-glucosidasa (acarbosa, voglibosa) Se ha observado que retrasan el inicio de la diabetes un máximo de tres años Tiazolidindionas (troglitazona, rosiglitazona, pioglitazona) Se ha observado que retrasan el inicio de la diabetes un máximo de tres años Agonistas de los receptores del GLP-1 (exenatida, liraglutida) Pérdida de peso significativa y mejoras del control glucémico en personas de alto riesgo en estudios a corto plazo Insulina y secretagogos de insulina (sulfonilureas, meglitinidas) Ineficaces Inhibidores de la ECA y bloqueadores de los receptores de angiotensina Ineficaces Estrógeno-progestágenos Efecto solo moderado aPreferible. ECA: enzima de conversión de la angiotensina; GLP-1: péptido similar al glucagón tipo 1; IMC: índice de masa corporal. Datos de la American Diabetes Association. 5. Prevention or delay of type 2 diabetes: Standards of Medical Care in Diabetes—2018. Diabetes Care. 2018;41:S51-S54. [PMID: 29222376] Datos de Garber AJ, Abrahamson MJ, Barzilay JI, Blonde L, Bloomgarden ZT, Bush MA, et al. Consensus statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the comprehensive type 2 diabetes management algorithm—2018 Executive Summary. Endocr Pract. 2018;24:91-120. [PMID: 29368965] • La American Diabetes Association (ADA) y la American Association of Clinical Endocrinologists (AACE) recomiendan utilizar metformina en un primer momento para prevenir el riesgo de diabetes en las personas con prediabetes, sobre todo en quienes presentan valores crecientes de hemoglobina A1c a pesar de las modificaciones del estilo de vida o que tienen menos de 60 años de edad, obesidad o antecedentes de diabetes gestacional. Diabetes mellitus con tendencia a la cetosis El concepto «diabetes con tendencia a la cetosis» integra varios síndromes glucémicos también denominados diabetes tipo 2 propensa a la cetosis, «diabetes Flatbush», diabetes tipo 1B o diabetes atípica. Estos síndromes se manifiestan con CAD episódica resultante del déficit de insulina, pero cursa con períodos variables de dependencia e independencia de la insulina. En las personas con diabetes con tendencia a la cetosis, es necesaria una terapia con insulina para tratar la CAD hasta que esta se haya resuelto y las células beta dejen de estar deterioradas por la toxicidad de la glucosa, si es posible, y puedan producir cantidades suficientes de insulina para suprimir la lipólisis. Dada la variabilidad de la evolución clínica que muestra la diabetes con tendencia a la cetosis, prima la incertidumbre acerca de la necesidad de regímenes de tratamiento con insulina a corto y largo plazo. Por lo tanto, se han elaborado cuatro sistemas de clasificación para que sirvan de orientación a la hora de predecir la duración del tratamiento. Un estudio longitudinal demostró que el sistema Aβ tenía una mayor exactitud para predecir la reserva de células beta y la dependencia de la insulina 12 meses después del episodio inicial de CAD en comparación con los demás sistemas de clasificación, con una sensibilidad del 99% y una especificidad del 96%. En el sistema Aβ, el estado de los anticuerpos (A) y el funcionamiento de las células beta (β) constituyen los factores principales que determinan si una persona necesitará insulina a largo plazo. Los datos longitudinales de las cohortes con diabetes con tendencia a la cetosis indican que las personas sin reserva de células beta, independientemente del estado de los anticuerpos (A+β– y A–β–), tienen más probabilidades de presentar un mal control glucémico y desarrollar dependencia de la insulina tras la aparición de CAD que las personas con preservación del funcionamiento de las células beta. Diabetes mellitus gestacional PUNTOS CLAVE • Según el Diabetes Prevention Program (DPP), las modificaciones del estilo de vida, incluyendo la pérdida de peso, una alimentación saludable y el ejercicio, redujeron la incidencia de diabetes tipo 2 en personas con prediabetes en el 58%. El aumento de la resistencia a la insulina durante el segundo y el tercer trimestres de embarazo es un fenómeno fisiológico normal provocado por las hormonas placentarias. Debido al funcionamiento alterado de las células beta, la producción de insulina es insuficiente para superar la resistencia a la insulina, con la aparición consiguiente de hiperglucemia. La diabetes gestacional se define como la presencia de hiperglucemia durante el segundo trimestre o el tercer trimes7 Trastornos del metabolismo de la glucosa tre en mujeres sin diagnóstico de diabetes tipo 1 o tipo 2 antes del embarazo. Los factores de riesgo incluyen edad superior a 25 años, sobrepeso/obesidad, antecedentes familiares de diabetes tipo 2 y grupo racial/étnico de alto riesgo (negros, hispanos/latinoamericanos, originarios del sudeste asiático o Asia oriental, nativos de las islas del Pacífico y nativos americanos). Los resultados adversos maternos y neonatales relacionados con la diabetes gestacional aumentan a medida que empeora la hiperglucemia. Las complicaciones incluyen macrosomía, complicaciones en el período de dilatación y el expulsivo, preeclampsia, defectos fetales, hipoglucemia neonatal, aborto espontáneo y muerte fetal intrauterina. Dada la prevalencia creciente de la diabetes tipo 2 no diagnosticada en la población general, la ADA recomienda un cribado estándar en todas las embarazadas con factores de riesgo de diabetes en la primera visita prenatal. Las mujeres con hiperglucemia detectada en el primer trimestre se clasifican como afectadas de diabetes tipo 2 y no de diabetes gestacional. En todas las demás embarazadas sin diagnóstico previo de diabetes, debe realizarse un cribado de la diabetes gestacional entre las semanas de gestación 24 y 28. El método de cribado recomendado varía en los diferentes grupos de expertos. La PTGO «en un paso» consiste en medir la glucemia basal (en ayunas) y una y dos horas después de una sobrecarga oral de 75 g de glucosa. Un valor anómalo por encima del valor de corte es diagnóstico de diabetes gestacional. La PTGO «en dos pasos» consiste en una medición de la glucemia inicial una hora después de una PTGO de 50 g. Si la glucemia es anómala, se inicia el segundo paso. La glucosa se mide en el momento basal (en ayunas) y una, dos y tres horas después de una sobrecarga oral de 100 g de glucosa. Dos valores anómalos de la glucemia después de la sobrecarga de 100 g son diagnósticos de diabetes gestacional. En la mayoría de las mujeres con diabetes gestacional la glucosa se normaliza después del embarazo, aunque persiste un riesgo elevado de aparición de diabetes gestacional recidivante y diabetes tipo 2. La ADA recomienda una PTGO de 75 g entre cuatro y 12 semanas después del parto para confirmar la resolución de la hiperglucemia. Si la primera prueba en el posparto es normal, deberán realizarse pruebas de cribado cada 1-3 años con una PTGO de 75 g, hemoglobina A1c o glucemia basal durante toda la vida. los síntomas tiene lugar antes de los 25 años de edad, y normalmente hay importantes antecedentes familiares de diabetes atípica en pacientes no obesos. La producción excesiva de hormonas asociada a varias endocrinopatías también puede alterar la secreción o la acción de la insulina y provocar hiperglucemia (véase la Tabla 4). Manejo de la diabetes mellitus La mejor manera de manejar eficazmente la diabetes consiste en una estrategia centrada en el paciente en la que los pacientes y sus cuidadores establezcan objetivos y planes de tratamiento individualizados compatibles con las preferencias de los pacientes, su estilo de vida, las comorbilidades y la seguridad. Además, el manejo debe incorporar la educación del paciente, la automonitorización de la glucemia, modificaciones del estilo de vida y tratamientos farmacológicos. PUNTO CLAVE • El manejo eficaz de la diabetes requiere una estrategia centrada en el paciente que individualice los objetivos y los planes de tratamiento, teniendo en cuenta las características particulares y las preferencias del paciente. Educación del paciente Los programas de educación y apoyo para el autocontrol de la diabetes (DSMES, por sus siglas en inglés) ofrecen los conocimientos y habilidades que permiten a los pacientes asumir los cuidados personales relacionados con la diabetes y elaborar estrategias eficaces de resolución de problemas. La ADA recomienda tener en cuenta la derivación a DSMES en diferentes períodos críticos de la atención: en el momento del diagnóstico, anualmente para reevaluar las necesidades durante las transiciones asistenciales y cuando los cambios del estado de salud afecten a las habilidades de autocontrol. Se ha demostrado que los programas de DSMES mejoran los resultados, como la hemoglobina A1c y la calidad de vida, y también reducen los costes, ya que permiten a los pacientes reducir la utilización de la atención aguda y de las unidades de hospitalización para el manejo de la diabetes. Automonitorización de la glucemia Tipos infrecuentes de diabetes mellitus Los defectos genéticos que provocan un deterioro en la secreción o la acción de la insulina constituyen formas infrecuentes de diabetes mellitus (véase la Tabla 4). La diabetes del adulto de inicio juvenil (MODY, por sus siglas en inglés) se describe como un defecto monogénico autosómico dominante en diferentes loci cromosómicos que da lugar a seis subtipos definidos por el gen concreto afectado. Aunque la acción de la insulina sigue siendo normal en la MODY, la sensibilidad a la glucosa y la secreción de insulina están afectadas. No hay autoanticuerpos. Las personas con MODY presentan una evolución clínica muchas veces atípica de diabetes tipo 1 o tipo 2. El inicio de 8 La automonitorización de la glucemia (AMG) está recomendada en los pacientes con regímenes intensivos de insulina (regímenes de múltiples dosis de insulina o tratamiento con bomba de insulina). Las pautas específicas para la AMG son individualizadas; las mediciones pueden realizarse antes de las comidas, a la hora de acostarse, antes y después del ejercicio y antes de manejar maquinaria. La AMG puede utilizarse para detectar y corregir la hipoglucemia. El control puede ser informativo cuando los valores de glucemia preprandial se sitúan dentro del intervalo ideal, pero en este sentido la hemoglobina A1c es superior. La medición de los valores de glucemia posprandial puede identificar una hiperglucemia no detectada. Trastornos del metabolismo de la glucosa La AMG puede considerarse en los pacientes motivados que siguen regímenes no intensivos de insulina; sin embargo, en estos pacientes no se ha determinado la frecuencia óptima de las pruebas. Por lo general, la hemoglobina A1c está correlacionada con el nivel medio de la glucemia de tres meses en los pacientes sin hemoglobinopatías o recambio eritrocítico elevado; por lo tanto, es posible medir la eficacia del tratamiento combinando la AMG y los datos de la hemoglobina A1c (Tabla 7 y Tabla 8). Otra opción es un sistema de monitorización continua de la glucosa (SMCG), que puede advertir al usuario de las tendencias actuales y retrospectivas de la hipoglucemia y la hiperglucemia. Además, la Food and Drug Administration (FDA) ha aprobado un SMCG para la administración y el control de la insulina en tiempo real. Los objetivos del empleo de un SMCG son mejorar el cuidado de la diabetes reduciendo la hemoglobina A1c y evitar la hipoglucemia, algo fundamental para las personas con hipoglucemias inadvertidas. La ADA recomienda el uso de SMCG en adultos (≥18 años) con diabetes tipo 1 que no cumplen los objetivos glucémicos. La Endocrine Society recomienda el uso de un SMCG en pacientes con diabetes tipo 1 y hemoglobina A1c elevada o el nivel objetivo de A1c cuando se usa diariamente, ya que los datos demuestran que el control glucémico mejora con el uso prolongado del SMCG. En el futuro, puede que el SMCG esté indicado también para los pacientes con diabetes tipo 2 que siguen regímenes intensivos de insulina. PUNTO CLAVE • Se recomienda la automonitorización de la glucemia o el uso de un sistema de monitorización continua de la glucosa en los pacientes con regímenes intensivos de insulina (regímenes de múltiples dosis de insulina o tratamiento con bomba de insulina). Vacunas recomendadas y cribado Las personas con diabetes deben recibir las vacunas adecuadas para su edad que recomienden las directrices del Advisory Committee on Immunization Practices. Además, los pacientes con diabetes deben recibir la vacuna antigripal anualmente, la vacuna antineumocócica polisacárida (PPVS23) y la serie de vacunas contra la hepatitis B. El calendario de vacunación recomendado por los Centers for Disease Control and Prevention (CDC) puede consultarse en: https://www.cdc.gov/ vaccines/schedules/hcp/imz/adult.html. Estrategias no farmacológicas para el manejo de la diabetes Los cambios en el estilo de vida son esenciales para el manejo a largo plazo de la diabetes y la prevención de complicaciones cardiovasculares. Aunque las medidas deben ser individualizadas, la dieta y la actividad física constituyen elementos básicos para los pacientes con diabetes tipo 1 y tipo 2. El tratamiento nutricional con un dietista proporciona educación individualizada específica sobre la diabetes para fomentar opciones de alimentación saludables y lograr los objetivos glucémicos y el control de peso, y también se ha asociado a reducciones de la hemoglobina A1c en pacientes con diabetes tipo 1 y tipo 2. La ADA no recomienda una dieta específica; sin embargo, en los pacientes con sobrepeso y obesidad y diabetes tipo 2 se recomienda un objetivo de pérdida de peso de al menos el 5%, ya que se ha demostrado que mejora el control glucémico. Las recomendaciones relativas a la actividad física son las mismas que las del programa DPP: actividad aeróbica de intensidad moderada o alta durante 150 minutos/semana, actividad aeróbica de intensidad alta durante 75 minutos/semana o una combinación de ambas pautas. Se ha demostrado que esto disminuye el nivel de hemoglobina A1c, reduce el peso, mejora la sensación de bienestar y mejora los factores de riesgo de CAD. Se recomienda realizar ejercicios de resistencia dos o tres veces por semana. Los ancianos con diabetes deben realizar ejercicios de flexibilidad y equilibrio dos o tres veces por semana, si es posible. Las conductas sedentarias prolongadas deben interrumpirse a intervalos de 30 minutos con actividades ligeras o poniéndose de pie. Los medicamentos para perder peso o la cirugía metabólica son opciones alternativas que deben tenerse en cuenta si el tratamiento nutricional y la actividad física fracasan (véase el módulo «Medicina interna general» de MKSAP 18). La cirugía metabólica debe tenerse en cuenta en las personas obesas con diabetes tipo 2. Después de la operación pueden producirse pérdidas de peso significativas y mejoras del control glucémico, incluida la remisión de la diabetes. Otros factores que deben tenerse en cuenta y abordarse en los pacientes con diabetes mellitus son la ansiedad, la depresión y el sufrimiento provocado por la diabetes. En el momento del diagnóstico de diabetes y de manera periódica deben llevarse a cabo pruebas de cribado de problemas psicosociales y trastornos conductuales. Estos trastornos pueden tener consecuencias adversas en el control glucémico, tanto directamente como debido a las dificultades que suponen para el cumplimiento de los planes terapéuticos por parte del paciente. Tratamiento farmacológico El tratamiento farmacológico debe ser individualizado y tener en cuenta la edad de la persona, su estado de salud y peso, la fisiopatología de su hiperglucemia, los riesgos/beneficios específicos de un posible agente terapéutico, el coste de la medicación y el estilo de vida y los objetivos personales del tratamiento del paciente. Por lo general, los objetivos de hemoglobina A1c no son estrictos en los pacientes con trastornos concomitantes significativos, ECV macrovascular, esperanza de vida reducida, diabetes de larga evolución, recursos o apoyo social limitados, nivel bajo de alfabetización/matemáticas, falta de adherencia terapéutica y riesgo elevado de complicaciones hipoglucémicas. La mayoría de las guías de práctica clínica, incluidas las de la ADA, recomiendan basar los um9 Trastornos del metabolismo de la glucosa Tabla 7. Objetivos glucémicos ambulatorios recomendados por la American Diabetes Association para los adultos con diabetes mellitus Estado de salud Características de los pacientes Hemoglobina A1ca Glucemia capilar preprandial Glucemia capilar posprandial (1-2 horas después de comer)c Sanos Primeros estadios de evolución de la enfermedad <7,0% 80-130 mg/dl (4,4-7,2 mmol/l) <180 mg/dlc (10,0 mmol/l) Pocas enfermedades concurrentes <6,5% en determinados pacientesb Glucemia capilar a la hora de acostarse Preconcepción Preferencias del paciente Esperanza de vida >10 años Problemas de salud complejos Comorbilidades significativas, incluidas la aterosclerosis avanzada o las complicaciones microvasculares <8,0% Mayor tiempo de evolución de la diabetes con dificultades para lograr los objetivos glucémicos a pesar de un manejo correcto Hipoglucemia frecuente Hipoglucemia inadvertida Esperanza de vida <10 años Ancianos Sanos <7,5% 90-130 mg/dl (5,0-7,2 mmol/l) 90-150 mg/dl (5,0-8,3 mmol/l) <8,0% 90-150 mg/dl (5,0-8,3 mmol/l) 100-180 mg/dl (5,6-10,0 mmol/l) <8,5% 100-180 mg/dl (5,6-10,0 mmol/l) 110-200 mg/dl (6,1-11,1 mmol/l) Pocas comorbilidades Esperanza de vida larga Ausencia de alteraciones cognitivas o del funcionamiento Complejos/intermedios Varias comorbilidades Riesgo de hipoglucemia Riesgo de caídas Deterioro de múltiples AVD instrumentales Deterioro cognitivo leve o moderado Muy complejos/mala salud Comorbilidades crónicas con enfermedad terminal Ingreso en centros de asistencia a largo plazo Deterioro cognitivo moderado o grave Dependencia en múltiples AVD Esperanza de vida limitada (Continúa en página siguiente) 10 Trastornos del metabolismo de la glucosa Tabla 7. Objetivos glucémicos ambulatorios recomendados por la American Diabetes Association para los adultos con diabetes mellitus (continuación) Estado de salud Características de los pacientes Hemoglobina A1ca Glucemia capilar preprandial Glucemia capilar posprandial (1-2 horas después de comer)c Embarazadasd Diabetes tipo 1 preexistente, diabetes tipo 2 preexistente o diabetes gestacional 6,0%-6,5% sin hipoglucemia gravee Basal (en ayunas) ≤95 mg/dl (5,3 mmol/l) 1 hora después de comer ≤140 mg/dl (7,8 mmol/l) (<6,0% puede ser óptimo a medida que progresa el embarazo) Glucemia capilar a la hora de acostarse o 2 horas después de comer ≤120 mg/dl (6,7 mmol/l) a Recomendado si es posible alcanzar el objetivo sin hipoglucemia recurrente grave. En caso de presencia de hipoglucemia recurrente grave, no se recomienda ningún objetivo de hemoglobina A1c, ya que debe tener prioridad modificar el régimen antidiabético del paciente para resolver la hipoglucemia recurrente grave. Una vez resuelta la hipoglucemia recurrente grave, puede escogerse una hemoglobina A1c objetivo y tomar las decisiones de tratamiento otra vez en función de ese objetivo individualizado sin hipoglucemias frecuentes. La hemoglobina A1c debe determinarse en el diagnóstico, y a intervalos de tres meses en adelante, ya que se producen cambios en las modificaciones del estilo de vida y/o los tratamientos farmacológicos. Las mediciones de la hemoglobina A 1c pueden reducirse a cada seis meses una vez logrados los objetivos glucémicos. bPuede considerarse en los pacientes con un diagnóstico inicial de diabetes mellitus, sin enfermedades cardiovasculares significativas, una esperanza de vida larga o manejados con modificaciones del estilo de vida o metformina. cCuando la hemoglobina A no es la ideal a pesar de la consecución de los objetivos de glucemia preprandial, hay que abordar los valores de la glucemia posprandial. Los valores 1c de la glucemia posprandial tienen un mayor impacto en los valores de A 1c cercanos a 7%. dEn las embarazadas se recomienda el control de la glucemia preprandial y posprandial. eLas mediciones de la glucemia preprandial y posprandial deben representar el método primario de evaluación del control glucémico, ya que los valores de hemoglobina A 1c descienden a medida que aumenta el recambio de eritrocitos asociado al embarazo. AVD: actividades de la vida diaria. Recomendaciones de la American Diabetes Association. 6. Glycemic targets: Standards of Medical Care in Diabetes—2018. Diabetes Care. 2018;41 (Suppl. 1):S55-S64. [PMID: 29222377] Recomendaciones de la American Diabetes Association. 11. Older adults: Standards of Medical Care in Diabetes—2018. Diabetes Care. 2018;41 (Suppl. 1):S119-S125. [PMID: 29222382] Recomendaciones de la American Diabetes Association. 13. Management of diabetes in pregnancy: Standards of Medical Care in Diabetes—2018. Diabetes Care. 2018;41:S137-S143. [PMID: 29222384] CONT. brales ideales de hemoglobina A1c en el estado de salud del paciente (véase la Tabla 7). En cambio, las guías del Veterans Affairs/Department of Defense (VA/DoD) de Estados Unidos para el manejo de la diabetes tipo 2 recomiendan un intervalo objetivo de hemoglobina A1c en lugar de un umbral objetivo. Las directrices VA/DoD intentan evitar la intensificación de la farmacoterapia basada únicamente en cambios marginales de la hemoglobina A1c causados por las características conocidas del paciente y las limitaciones del laboratorio que podrían provocar más daños que beneficios en las personas con enfermedades concurrentes importantes, complicaciones microvasculares o edad avanzada. El American College of Physicians (ACP) recomienda un nivel de hemoglobina A1c de entre 7% y 8% en la mayoría de los pacientes con diabetes tipo 2, y los clínicos deben plantearse reducir la intensidad de la farmacoterapia en los pacientes que logren unos niveles de hemoglobina A1c inferiores a 6,5%. El fundamento de estos objetivos se basa en la evidencia que demuestra colectivamente que tratar hasta objetivos inferiores a 7%, en comparación con objetivos en torno a 8%, no redujo la muerte ni los eventos macrovasculares durante 5-10 años de tratamiento, sino que dio lugar a daños importantes. Los objetivos más estrictos pueden ser adecuados en los pacientes con una esperanza de vida larga (>15 años) que Tabla 8. Comparación del valor de la hemoglobina A1c y el valor estimado de glucemia Hemoglobina A1c Nivel de glucemia medio estimado Nivel de glucemia medio estimado % mg/dl (IC del 95%) mmol/l (IC del 95%) 6 126 (100-152) 7,0 (5,5-8,5) 7 154 (123-185) 8,6 (6,8-10,3) 8 183 (147-217) 10,2 (8,1-12,1) 9 212 (170-249) 11,8 (9,4-13,9) 10 240 (193-282) 13,4 (10,7-15,7) 11 269 (217-314) 14,9 (12,0-17,5) 12 298 (240-347) 16,5 (13,3-19,3) IC: intervalo de confianza. Datos de la American Diabetes Association. 6. Glycemic targets: Standards of Medical Care in Diabetes—2018. Diabetes Care. 2018;41:S55-S64. [PMID:29222377] estén interesados en un control glucémico más intensivo con el tratamiento farmacológico a pesar del riesgo de daños, entre los que se incluyen, entre otros, hipoglucemia, carga para el paciente y costes farmacológicos. Además, el ACP reco11 Trastornos del metabolismo de la glucosa CONT. mienda no establecer un nivel de hemoglobina A1c objetivo en los pacientes con una esperanza de vida inferior a 10 años debido a edad avanzada (80 años o más), internamiento en una residencia geriátrica o trastornos médicos crónicos, ya que en esta población los daños superan los beneficios. Varios estudios de referencia ofrecen orientación sobre los objetivos glucémicos y la reducción del riesgo de ECV. En comparación con un control estándar, el control glucémico intensivo reduce de manera significativa la incidencia y progresión de las complicaciones microvasculares en los pacientes con diabetes tipo 1 y tipo 2, como demuestran el Diabetes Control and Complications Trial (DCCT) y el UK Prospective Diabetes Study (UKPDS). El seguimiento a largo plazo puso de manifiesto reducciones constantes en las complicaciones microvasculares a pesar de la convergencia del control glucémico en los diferentes grupos del estudio. Los estudios Action to Control Cardiovascular Risk in Diabetes (ACCORD), Action in Diabetes and Vascular Disease: Preterax y Diamicron MR Controlled Evaluation (ADVANCE) y Veterans Affairs Diabetes Trial (VADT) reforzaron aún más la relación entre la reducción de las complicaciones microvasculares y un control glucémico estricto, pero también pusieron de relieve que los pacientes y profesionales deben sopesar los riesgos/beneficios de un régimen intensivo con la posible morbimortalidad en poblaciones concretas. La evaluación del seguimiento a largo plazo de los participantes de los grupos de terapia insulínica intensiva de los ensayos DCCT y UKPDS que se encontraban en los primeros estadios de la enfermedad demostró una reducción significativa de ECV y mortalidad. En cambio, los estudios ACCORD, ADVANCE y VADT evaluaron el control glucémico estricto en ancianos con diabetes tipo 2 más avanzada y ECV preexistente o factores de riesgo de ECV. La ECV no disminuyó de manera significativa en los ensayos ACCORD y ADVANCE. El ensayo VADT reveló una reducción significativa de los eventos cardiovasculares, pero sin cambios en la mortalidad cardiovascular o global. Recientemente, el estudio EMPA-REG Outcome, un ensayo aleatorizado controlado (EAC), puso de manifiesto que, en los pacientes con ECV establecida, la empagliflozina, un inhibidor del contransportador de sodio y glucosa 2 (SGLT2), redujo el criterio de valoración compuesto (muerte cardiovascular, infarto de miocardio no mortal o ictus no mortal); la reducción se debió sobre todo al descenso significativo del riesgo relativo de las tasas de muerte cardiovascular del 38%. Asimismo, se observó una reducción significativa de la mortalidad por cualquier causa del 32% y de hospitalización por insuficiencia cardíaca del 35%. Como consecuencia de este ensayo, la FDA aprobó la empagliflozina para la reducción de la muerte cardiovascular en los adultos con diabetes tipo 2 y ECV. Otro inhibidor del SGLT2, la canagliflozina, demostró también una reducción de los eventos cardiovasculares, pero no de la muerte cardiovascular, en los pacientes con diabetes tipo 2 y alto riesgo de enfermedad cardiovascular en comparación con placebo en el programa CANVAS (Canagliflozin Cardiovascular Assessment Study). 12 El EAC Liraglutide Effect and Action in Diabetes: Evaluation of Cardiovascular Outcome Results (LEADER) incluyó a sujetos en riesgo de ECV y puso de manifiesto que la liraglutida, un análogo del péptido similar al glucagón tipo 1 (GLP-1), redujo el criterio de valoración principal compuesto (muerte cardiovascular, infarto de miocardio no mortal o ictus no mortal) el 13% en comparación con placebo (reducción del riesgo relativo). Además, la liraglutida produjo una reducción significativa de la muerte cardiovascular (22%) y la mortalidad por cualquier causa (15%) en comparación con placebo. Sobre la base de los datos de LEADER, la FDA aprobó la liraglutida para la reducción de los eventos cardiovasculares mayores y la muerte cardiovascular en los adultos con diabetes tipo 2 y ECV. PUNTO CLAVE • El tratamiento farmacológico debe ser individualizado y tener en cuenta la edad de la persona, su estado de salud y peso y la fisiopatología de su hiperglucemia, los riesgos/beneficios específicos de un posible agente terapéutico, el coste de la medicación y el estilo de vida y los objetivos personales del tratamiento del paciente. Tratamiento para la diabetes mellitus tipo 1 Debido a la destrucción de células beta y el subsecuente déficit de insulina, las personas con diabetes mellitus tipo 1 necesitan tratamiento con insulina de por vida. Idealmente, debe prescribirse un régimen de insulina intensivo que incluya múltiples dosis diarias de insulina (MDI) que imiten la acción fisiológica del páncreas. El régimen de insulina debe incluir cobertura basal para mantener el control glucémico en ayunas y entre las comidas, cobertura prandial y aportes complementarios de insulina para corregir la hiperglucemia. Esto puede conseguirse con inyecciones subcutáneas de insulina, preparaciones inhaladas de insulina o infusión continua de insulina subcutánea (ICIS) con bomba de insulina. En los pacientes con diabetes tipo 1, la dosis diaria total de insulina inicial oscila entre 0,4 y 1,0 U/kg/día. Normalmente, la insulina basal comprende en torno al 50% de la dosis diaria total de insulina, mientras que la insulina prandial supone el 50% restante. Las formulaciones de insulina disponibles y sus perfiles de actividad se resumen en la Tabla 9. El momento y el modo de administración de la insulina prandial varían en función de las necesidades o preferencias del paciente y de sus hábitos alimentarios. La administración prandial de MDI puede llevarse a cabo con dosis fijas o a partir del recuento de hidratos de carbono o el recuento de hidratos de carbono modificado. En general, una unidad de insulina cubre entre 10 y 20 g de hidratos de carbono consumidos. Cuando no es posible contar con exactitud los gramos de hidratos de carbono consumidos, puede emplearse un método de recuento de hidratos de carbono modificado. Con este método, es posible ajustar las dosis de insulina regular o análogo en el 50 % según la porción de alimento consumida. Por ejemplo, la dosis para toda la comida sería la siguiente: pe- AMAV Trastornos del metabolismo de la glucosa Tabla 9. Propiedades farmacocinéticas de los productos de insulinaa Tipo de insulina Inicio Máximo (pico) Duración Lispro, aspart, glulisina 5-15 min 45-90 min 2-4 h Insulina inhalada 5-15 min 50 min 2-3 h 5-15 min 45-90 min 2-4 h 0,5 h 2-5 h 4-8 h 1-3 h 4-10 h 10-18 h 0,5 h 2-5 h 13-24 h 1-2 h Ningunob Glargina 2-3 h b Ninguno 20-24+ h Degludec 1-3 h Ninguno 24-42 h Glargina (300 U/ml) 6h Ninguno 24-36 h Degludec (200 U/ml) 1-3 h Ninguno 24-42 h 70% NPH/30% regular 0,5-1 h 2-10 h 10-18 h 75% NPL/25% lispro 10-20 min 1-6 h 10-18 h 50% NPL/50% lispro 10-20 min 1-6 h 10-18 h 70% NPA/30% aspart 10-20 min 1-6 h 10-18 h 70% degludec/ 30% aspart 10-30 min 0,5-2 h 24+ h Análogos de acción rápida Análogo concentrado de acción rápida Lispro (200 U/ml) Acción breve Regular humana Acción intermedia Insulina NPH Regular humana concentrada Regular humana U-500 (500 U/ml) Análogos basales de acción prolongada Detemir 12-24 hc Análogo basal concentrado (acción ultraprolongada) Insulinas premezcladasd aLa evolución temporal de cada insulina varía de manera significativa entre las personas y en una misma persona en días diferentes. Por lo tanto, los períodos de tiempo mencionados deben considerarse únicamente pautas generales. bTanto la insulina detemir como la insulina glargina pueden producir un efecto máximo en algunas personas, sobre todo a dosis elevadas. cLa duración de la acción de la insulina detemir varía en función de la dosis administrada. dLas insulinas premezcladas que contienen una mayor proporción de insulina de acción rápida o breve tienden a mostrar máximos más grandes en un momento anterior que las mezclas que contienen proporciones menores de insulina de acción rápida y breve. NPA: aspart protamina neutra; NPH: Hagedorn protamina neutra; NPL: lispro protamina neutra queña (50%), normal (100%), grande (150%). Además, las MDI incorporan aportes complementarios de insulina para corregir la hiperglucemia. Un método frecuente para calcular la dosis correctora de insulina consiste en administrar una dosis adicional de insulina regular o análogo en el momento de la medición previa a la comida por cada valor de glucemia de 50 mg/dl (2,8 mmol/l) por encima del valor ideal de glucemia en las personas sensibles a la insulina y de una unidad por cada 25 mg/dl (1,4 mmol/l) en las personas con resistencia a la insulina. El aporte de insulina puede administrarse con la insulina prandial en una inyección. Por ejemplo, si la glucemia objetivo es de 120 mg/dl (6,7 mmol/l) y la glucemia actual de 270 mg/dl (15,0 mmol/l) en una persona con diabetes tipo 1, se administrarán otras tres unidades de insulina con la insulina prandial. Las formulaciones de insulina premezclada combinan insulina basal de acción intermedia o prolongada e insulina de acción rápida o breve en concentraciones fijas. Por lo general, estas formulaciones se administran dos veces al día y deben tenerse en cuenta en quienes no pueden o no quieren practicarse inyecciones diarias de insulina más frecuentes. Las formulaciones premezcladas pueden aumentar las oscilaciones glucémicas, incluida la hipoglucemia, porque constituyen un régimen no fisiológico. La insulina inhalada es una formulación de acción rápida para administración prandial. La disponibilidad de la insulina inhalada en ampollas con dosis predeterminadas de insulina (4, 8 y 12 unidades) limita la flexibilidad de la administración. Es necesario evaluar y controlar la función pulmonar en el momento inicial, porque puede descender con el uso de insulina inhalada. La ICIS permite la administración continua de insulina basal y utiliza una calculadora de bolos programada para lograr los objetivos glucémicos individuales con el fin de calcular las dosis prandiales y los bolos correctores. La Endocrine Society recomienda la ICIS sobre la MDI en todos los adultos con diabetes tipo 1 que no han alcanzado un nivel de hemoglobina A1c objetivo y en quienes lo han logrado pero presentan una gran variabilidad glucémica, hipoglucemias graves o hipoglucemias inadvertidas. Otras consideraciones incluyen la necesidad de flexibilidad en la administración de insulina, la hiperglucemia matinal («fenómeno del alba»), un estilo de vida activo o las preferencias del paciente. Hay sistemas de ICIS que reducen o detienen la administración de insulina si los niveles de glucemia descienden por debajo del valor umbral especificado en el sistema de ICIS y aumentan la insulina administrada si los niveles de glucemia están por encima de un valor umbral. La administración de insulina se reinicia o se aumenta/reduce al nivel basal cuando deja de alcanzarse el umbral. El uso de insulina conlleva riesgo de hipoglucemias y aumento de peso. El riesgo de hipoglucemias es menor con los análogos de insulina que con la insulina regular debido que tienen una duración de acción menor. La hipoglucemia provocada por el efecto acumulativo de la insulina se produce cuando se administra insulina con demasiada frecuencia y 13 CONT. Trastornos del metabolismo de la glucosa CONT. esta se solapa con la duración de la acción de una inyección de insulina anterior. Puede evitarse dejando que transcurran al menos tres o cuatro horas entre las inyecciones secuenciales de análogos de insulina. La pramlintida, un análogo de la amilina, es un tratamiento complementario cuyo uso está aprobado con insulina en la diabetes tipo 1. La pramlintida puede dar lugar a un mejor control glucémico, necesidad de dosis menores de insulina y pérdida de peso gracias al retraso del vaciamiento gástrico, el aumento de la saciedad y la disminución de la secreción de glucagón. PUNTOS CLAVE • Las personas con diabetes mellitus tipo 1 necesitan tratamiento con insulina de por vida. • La Endocrine Society recomienda la infusión continua de insulina subcutánea (ICIS) sobre el régimen de múltiples dosis de insulina (MDI) en todos los adultos con diabetes tipo 1 que no han alcanzado el nivel de hemoglobina A1c objetivo y en quienes lo han logrado pero presentan una gran variabilidad glucémica, hipoglucemias graves o hipoglucemias inadvertidas. Tratamiento para la diabetes mellitus tipo 2 Cuando disminuye el funcionamiento de las células beta, muchas veces es necesario combinar los tratamientos farmacológicos con modificaciones del estilo de vida para lograr el control glucémico. Las opciones terapéuticas pueden ser una monoterapia o una combinación de fármacos orales con fármacos inyectables (Tabla 10). La ADA recomienda iniciar la monoterapia si el nivel de hemoglobina A1c es inferior a 8% en el momento del diagnóstico. La metformina es el fármaco oral de primera línea recomendado para la diabetes tipo 2 de diagnóstico reciente, debido a su eficacia conocida y al bajo riesgo de hipoglucemia. Los efectos secundarios gastrointestinales de la metformina son frecuentes y pueden reducirse con un ajuste lento y progresivo de las dosis, la administración de alimento y/o el uso de formulaciones de liberación extendida. La acidosis láctica es un riesgo posible pero infrecuente asociado al uso de metformina. La insuficiencia cardíaca que requiere tratamiento farmacológico y la disfunción hepática pueden aumentar el riesgo. Para evitar una posible acidosis láctica con disfunción renal, se recomienda una tasa de filtración glomerular estimada (TFGe) superior a 45 ml/min/1,73 m2 para iniciar la metformina. Los clínicos deben evaluar los riesgos y beneficios de proseguir con el tratamiento en los pacientes cuya TFGe desciende a menos de 45 ml/min/1,73 m2 durante el tratamiento. La metformina está contraindicada con TFGe inferiores a 30 ml/min/1,73 m2. Si se administra un medio de contraste yodado con una TFGe de entre 30 y 60 ml/min/1,73 m2, es necesario interrumpir la metformina hasta que la función renal permanezca estable durante 48 horas. También hay que interrumpir la metformina en situaciones que puedan provocar deshidratación, como los vómitos o la diarrea. Hasta el 30% de los pacientes 14 que reciben metformina experimentan una reducción de la absorción intestinal de vitamina B12, mientras que entre el 5% y el 10% sufren déficit de vitamina B12. Puede estar justificado realizar controles periódicos, sobre todo en caso de anemia o neuropatía periférica. El control glucémico debe evaluarse cada tres meses con ajustes del tratamiento hasta alcanzar el objetivo glucémico, y cada seis meses en adelante. Cuando no se alcanzan los objetivos glucémicos con metformina y modificaciones del estilo de vida, hay pocos datos comparativos de la eficacia que sirvan de orientación para añadir otros fármacos; por lo tanto, muchas guías están basadas en opiniones de expertos. Si el nivel de hemoglobina A1c es ≥9% en el momento del diagnóstico o después de tres meses de tratamiento con metformina, la ADA recomienda pasar al tratamiento doble, definido como la combinación de metformina con otro agente terapéutico (véase la Tabla 9 y la Tabla 10). En las personas con ECV, para el tratamiento doble deben considerarse los agentes terapéuticos que han demostrado reducir los eventos adversos cardiovasculares mayores (canagliflozina, empagliflozina y liraglutida) y la mortalidad cardiovascular (empagliflozina y liraglutida). La AACE y el American College of Endocrinology (ACE) recomiendan iniciar la metformina si el nivel de hemoglobina A1c es inferior a 7,5% en el diagnóstico. Debe iniciarse un tratamiento doble si el nivel de hemo­ globina A1c es ≥7,5% en el diagnóstico o después de tres meses de monoterapia. La ADA y la AACE/ACE recomiendan pasar a un tratamiento triple si el tratamiento doble no logra alcanzar los objetivos glucémicos al cabo de tres meses. Si los objetivos siguen sin cumplirse, hay que pasar del tratamiento triple a un tratamiento combinado inyectable. Este consiste en continuar con la metformina e iniciar un agonista de los receptores de GLP-1 o insulina basal, si no se ha prescrito aún, iniciar insulina basal con un tratamiento agonista de los receptores de GLP-1 de base o iniciar un agonista de los receptores de GLP-1 o insulina prandial con insulina basal optimizada. Los algoritmos de la ADA y la AACE/ACE sirven de guía para iniciar y administrar regímenes de insulina basal y prandial (Tabla 11). La ADA recomienda un tratamiento inyectable combinado inicial en el contexto de hiperglucemia sintomática (polidipsia, poliuria), hemoglobina A1c ≥10% o un nivel de glucemia ≥300 mg/dl (16,6 mmol/l). La AACE/ACE recomienda iniciar el tratamiento con insulina con otros fármacos si la hemoglobina A1c inicial está por encima de 9% en una persona sintomática. Si la hiperglucemia persiste tras optimizar la dosis de insulina basal, debe añadirse insulina prandial antes de la comida más importante. Para la hiperglucemia persistente debe emplearse un régimen con insulina basal en bolo, con insulina prandial antes de dos o más comidas. Los análogos de insulina basal de acción ultraprolongada pueden tener ventajas respecto a los análogos de insulina basal de acción prolongada debido a la duración de su acción (>24 horas), a la administración de insulina sin picos y a la menor variabilidad de la acción entre las personas y en una Trastornos del metabolismo de la glucosa Tabla 10. Agentes farmacológicos utilizados para reducir los niveles de glucemia en la diabetes mellitus tipo 2a,b Clase Mecanismo de acción Efecto en el peso Inconvenientes Estudios a largo plazo de los resultados definitivos Insulina Reduce la producción de glucosa hepática Aumento Hipoglucemia, aumento de peso, requiere entrenamiento, formas inyectables, toxicidad pulmonar con insulina inhalada Descenso de los eventos microvasculares (UKPDS)c,d Aumenta la captación de glucosa periférica Inhibe la cetogénesis Sulfonilureas (tolbutamida, clorpropamida, glipizida, glibenclamida, gliclazida, glimepirida) Estimulan la secreción de insulina Aumento Hipoglucemia (sobre todo en los fármacos con semividas prolongadas o en poblaciones de edad avanzada); aumento de peso; reducción de la glucemia sin durabilidad Descenso de los eventos microvasculares (UKPDS)c; posible aumento de los eventos de ECV Biguanidas (metformina) Reducen la producción de glucosa hepática Neutro Diarrea y molestias abdominales, déficit de vitamina B12, acidosis láctica (infrecuente) Descenso de los eventos de ECV (UKPDS)d Aumentan la captación insulinomediada de glucosa en los músculos Contraindicadas con insuficiencia hepática, renal o cardíaca progresiva Inhibidores de la α-glucosidasa (acarbosa, miglitol) Inhiben la absorción de polisacáridos Neutro Flatulencia, molestias abdominales Posible descenso de los eventos de ECV en la prediabetes (STOP-NIDDM)e Tiazolidindionas (rosiglitazona, pioglitazona) Aumentan la captación periférica de glucosa Aumento Retención de líquidos, insuficiencia cardíaca, edema, fracturas, posible aumento del riesgo de cáncer vesical con pioglitazona Posible descenso de los eventos de ECV con pioglitazona (PROactive)f Meglitinidas (repaglinida, nateglinida) Estimulan la liberación de insulina Aumento Hipoglucemia, aumento de peso, administración frecuente Ninguno Amilina mimética (pramlintida) Ralentiza el vaciamiento gástrico Disminución Náuseas, vómitos, agrava la gastroparesia, riesgo elevado de hipoglucemia con el uso concomitante de insulina, requiere entrenamiento, inyectable, administración frecuente Ninguno Disminución Hipoglucemia cuando se usan en combinación con sulfonilureas, náuseas, vómitos, diarrea, agravan la gastroparesia, frecuencia cardíaca elevada, posible pancreatitis, los estudios con animales demuestran hiperplasia de células C y tumores tiroideos medulares, requieren entrenamiento, inyectable Descenso de los eventos de ECV y la mortalidad en las personas de alto riesgo con diabetes tipo 2 con liraglutida (LEADER)g Neutro Hipoglucemia cuando se usan en combinación con sulfonilurea, riesgo elevado de infecciones, posible riesgo elevado de pancreatitis, reacciones dermatológicas, requieren ajustes de la dosis en caso de declive de la función renal excepto con la linagliptina Aumento de las hospitalizaciones por insuficiencia cardíaca (saxagliptina [SAVOR-TIMI 53])h Inhibe la secreción de glucagón Aumenta la saciedad Agonistas de los receptores de GLP-1 (exenatida, exenatida de liberación extendida, liraglutida, albiglutida, lixisenatida, dulaglutida, semaglutida) Aumento dependiente de la glucosa en la secreción de insulina Ralentizan el vaciamiento gástrico Inhibición dependiente de la glucosa de la secreción de glucagón Aumentan la saciedad Inhibidores de la DPP-4 (sitagliptina, saxagliptina, linagliptina, alogliptina) Aumento dependiente de la glucosa en la secreción de insulina Inhibición dependiente de la glucosa de la secreción de glucagón (Continúa en página siguiente) 15 Trastornos del metabolismo de la glucosa Tabla 10. Agentes farmacológicos utilizados para reducir los niveles de glucemia en la diabetes mellitus tipo 2a,b (continuación) Clase Mecanismo de acción Efecto en el peso Inconvenientes Estudios a largo plazo de los resultados definitivos Inhibidores del SGLT2 (canagliflozina, dapagliflozina, empagliflozina, ertugliflozina) Aumentan la excreción renal de glucosa Disminución Hipoglucemia con secretagogos de insulina, deshidratación/hipotensión, daño renal agudo (canagliflozina, dapagliflozina), reacciones de hipersensibilidad, aumento de las infecciones por cándidas y de las infecciones urinarias, CAD «normoglucémica», posible aumento de las amputaciones (canagliflozina), hiperpotasemia (canagliflozina), fracturas (canagliflozina), cáncer vesical (dapagliflozina) Descenso de los eventos de ECV y la mortalidad en las personas de alto riesgo con diabetes tipo 2 con empagliflozina (EMPA-REG OUTCOME)i Secuestradores de ácidos biliares (colesevelam) No se sabe del todo: Descenso de la aparición o el empeoramiento de nefropatía en las personas de alto riesgo de ECV con diabetes tipo 2 (EMPA-REG OUTCOME)i Descensos de los eventos de ECV en las personas de alto riesgo con diabetes tipo 2 con canagliflozina (Programa CANVAS)j Neutro Estreñimiento, dispepsia, triglicéridos elevados, posible interferencia en la absorción de otros medicamentos Ninguno Neutro Náuseas, ortostatismo, fatiga Posible descenso de los eventos de ECV (Cycloset Safety Trial)k Posible descenso de la producción de glucosa hepática Posible aumento de los niveles de incretina Agonistas de la dopamina 2 (bromocriptina de liberación rápida) Aumenta la sensibilidad a la insulina Altera el metabolismo mediante el hipotálamo aDatos de la American Diabetes Association. 8. Pharmacologic approaches to glycemic treatment: Standards of Medical Care in Diabetes—2018. Diabetes Care. 2018;419 Suppl. 1):S73-S85. [PMID: 29222379] b Recomendaciones de Garber AJ, Abrahamson MJ, Barzilay JI, Blonde L, Bloomgarden ZT, Bush MA, et al; American Association of Clinical Endocrinologists (AACE). Consensus Statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the Comprehensive Type 2 Diabetes Management Algorithm-2018 Executive Summary. Endocr Pract. 2018;24:91-120. [PMID: 29368965] cDatos de Intensive blood-glucose control with sulfonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998;352:837-53. [PMID: 9742976] dDatos de Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998;352:854-65. [PMID: 9742977] e Datos de Chiasson JL, Josse RG, Gomis R, Hanefeld M, Karasik A, Laakso M; STOP-NIDDM Trial Research Group. Acarbose treatment and the risk of cardiovascular disease and hypertension in patients with impaired glucose tolerance: the STOP-NIDDM trial. JAMA. 2003;290:486-94. [PMID: 12876091] fDatos de Dormandy JA, Charbonnel B, Eckland DJ, Erdmann E, Massi-Benedetti M, Moules IK, et al; PROactive Investigators. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet. 2005;366:1 279-89. [PMID: 16214598] g Datos de Marso SP, Daniels GH, Brown-Frandsen K, Kristensen P, Mann JF, Nauck MA, et al; LEADER Steering Committee. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2016;375:311-22. [PMID: 27295427] h Datos de Scirica BM, Bhatt DL, Braunwald E, Steg PG, Davidson J, Hirshberg B, et al; SAVOR-TIMI 53 Steering Committee and Investigators. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med. 2013;369:1317-26. [PMID: 23992601] i Datos de Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, et al; EMPA-REG OUTCOME investigators. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373:2117-28. [PMID: 26378978] jDatos de Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, et al; CANVAS Program Collaborative Group. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. 2017;377:644-657. [PMID: 28605608] kDatos de Gaziano JM, Cincotta AH, O’Connor CM, Ezrokhi M, Rutty D, Ma ZJ, et al. Randomized clinical trial of quick-release bromocriptine among patients with type 2 diabetes on overall safety and cardiovascular outcomes. Diabetes Care. 2010;33:1503-8. [PMID: 20332352] CAD: cetoacidosis diabética; DPP-4: dipeptidil peptidasa 4; ECV: enfermedad cardiovascular; GLP-1: péptido similar al glucagón tipo 1; SGLT2: contransportador de sodio y glucosa 2. CONT. misma persona. El perfil farmacodinámico puede reducir la hipoglucemia en los pacientes de alto riesgo, mejorar las fluctuaciones glucémicas y permitir la flexibilidad en la administración más allá de los períodos de 24 horas. 16 En los pacientes con diabetes tipo 2 que siguen sin alcanzar la glucemia ideal a pesar de la adherencia a la monitorización la glucemia y de múltiples modalidades de tratamiento, puede considerarse la ICIS. Trastornos del metabolismo de la glucosa Tabla 11. Comparación de los algoritmos de administración de insulina de la ADA y la AACE/ACE Inicio o modificación de la insulina ADA AACE/ACE Insulina basal Dosis inicial: Dosis inicial: 10 U/día o 0,1-0,2 U/kg/día Si A1c <8%: 0,1-0,2 U/kg/día Si A1c >8%: 0,2-0,3 U/kg/día Ajuste de la dosis de insulina basal Para hiperglucemia: Para hiperglucemia: Aumentar la dosis 1-2 veces/semana hasta alcanzar el objetivo glucémico Aumentar la dosis cada 2-3 días hasta alcanzar el objetivo glucémico Aumentar la dosis en: Aumentar la dosis en: 10%-15% o 2-4 U 2Uo 20% si la GB >180 mg/dl (10 mmol/l) 10% si la GB es 140-180 mg/dl (7,8-9,9 mmol/l) 1 U si la GB es 110-139 mg/dl (6,1-7,7 mmol/l) Para hipoglucemia: Para hipoglucemia: Reducir la dosis en: 10%-20% o 4 U Reducir la dosis en: 10%-20% si la GB <70 mg/dl (3,9 mmol/l) 20%-40% si la GB <40 mg/dl (2,2 mmol/l) Insulina prandial más insulina basal (1 comida) Iniciar insulina prandial en la comida más grande Iniciar insulina prandial en la comida más grande Dosis inicial: Dosis inicial: 4 U, 10% de la dosis basal o 0,1 U/kg 5 U o 10% de la dosis basal Para evitar la hipoglucemia, considerar la reducción de la dosis basal en la misma cantidad si A1c <8% Régimen de insulina bolo-basal (≥2 comidas) Dosis inicial de insulina prandial: Dosis inicial de insulina prandial: 4 U, 10% de la dosis basal o 0,1 U/kg/comida 0,3-0,5 U/kg = DDT Para evitar la hipoglucemia, considerar la reducción de la dosis basal en la misma cantidad si A1c <8% 50% de la DDT = insulina basal 50% de la DDT = insulina prandial Cada dosis a la hora de comer = 1/3 dosis de insulina prandial Ajuste de la dosis de insulina prandial Para hiperglucemia: Para hiperglucemia: Aumentar la dosis 1-2 días/semana hasta alcanzar el objetivo glucémico Aumentar la dosis cada 2-3 días hasta alcanzar el objetivo glucémico Aumentar la dosis en: Aumentar la dosis en: 10%-15% o 1-2 U 10% o 1-2 U si la glucemia >140 mg/dl (7,8 mmol/l) 2 horas después de la comida o en la comida siguiente Si la hiperglucemia persiste en otras comidas, añadir dosis de insulina adicionales en el momento de la comida (bolo basal) Si la hiperglucemia persiste en otras comidas, añadir dosis de insulina adicionales en el momento de la comida (bolo basal) Para hipoglucemia: Para hipoglucemia: Reducir la dosis en: Reducir la DDT (basal y/o prandial) en: 10%-20% o 2-4 U 10%-20% si la glucemia <70 mg/dl (3,9 mmol/l) 20%-40% si la glucemia <40 mg/dl (2,2 mmol/l) Insulina premezclada 2× al día Dosis inicial: Análogo de insulina premezclada 3× al día Añadir dosis adicional de insulina en el almuerzo Dosis basal actual administrada en el desayuno y la cena distribuida a las 2/3 h y 13/15 h o 1/2 h y 13/14 h (Continúa en página siguiente) 17 Trastornos del metabolismo de la glucosa Tabla 11. Comparación de los algoritmos de administración de insulina de la ADA y la AACE/ACE (continuación) Inicio o modificación de la insulina ADA Ajuste de la dosis de insulina premezclada Para hiperglucemia: AACE/ACE Aumentar la dosis 1-2 veces/semana hasta alcanzar el objetivo glucémico Aumentar la dosis en: 10%-15% o 1-2 U Para hipoglucemia: Reducir la dosis en: 10%-20% o 2-4 U AACE: American Association of Clinical Endocrinologists; ACE: American College of Endocrinology; ADA: American Diabetes Association; DDT: dosis diaria total; GB: glucemia basal. Datos de la American Diabetes Association. 8. Pharmacologic approaches to glycemic treatment: Standards of Medical Care in Diabetes—2018. Diabetes Care. 2018;40 (Suppl. 1): S73-S85. [PMID: 2922379] Datos de Garber AJ, Abrahamson MJ, Barzilay JI, Blonde L, Bloomgarden ZT, Bush MA, et al; American Association of Clinical Endocrinologists (AACE). Consensus statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the comprehensive type 2 diabetes management algorithm—2018 Executive Summary. Endocr Pract. 201 8:24:91 -1 20. [PMID: 29368965] PUNTOS CLAVE • La metformina es el fármaco oral de primera línea recomendado para la diabetes tipo 2 de diagnóstico reciente, debido a su eficacia conocida y al bajo riesgo de hipoglucemia. • El control glucémico debe evaluarse cada tres meses, con ajustes posteriores de los agentes terapéuticos hasta alcanzar el objetivo glucémico y cada seis meses en adelante. Tratamiento para la diabetes mellitus gestacional En las pacientes con diabetes gestacional, cuando las intervenciones en el estilo de vida no logran los objetivos glucémicos debe prescribirse un tratamiento farmacológico para mejorar los resultados perinatales. La insulina es el tratamiento recomendado. Si bien puede considerarse el uso de un tratamiento con metformina o una sulfonilurea, ambas atraviesan la placenta y no hay datos de la seguridad largo plazo de su uso durante el embarazo. Además, el tratamiento con sulfonilureas se ha asociado a tasas más altas de macrosomía e hipoglucemia neonatal. Hiperglucemia inducida por fármacos Varios fármacos pueden provocar hiperglucemia mediante múltiples mecanismos: aumento de la producción de glucosa hepática, alteración de la acción de la insulina o reducción de la secreción de insulina (Tabla 12). Si bien la hiperglucemia producida por tratamientos farmacológicos temporales puede resolverse tras su suspensión, muchos de estos fármacos se utilizan de manera indefinida para trastornos médicos crónicos. Las personas en riesgo de hiperglucemia y de aparición de diabetes inducida por fármacos deben realizarse controles periódicos. 18 Manejo hospitalario de la hiperglucemia Un control glucémico hospitalario estricto (80-110 mg/dl [4,4-6,1 mmol/l]) no siempre está asociado a una mejora de los resultados y puede aumentar la mortalidad. En consecuencia, los objetivos glucémicos hospitalarios actuales intentan evitar las complicaciones de la hipoglucemia y la hiperglucemia graves, tales como alteraciones electrolíticas y deshidratación. En caso de valores persistentes por encima de 140 mg/dl (7,8 mmol/l), es necesario realizar modificaciones en la dieta. Si la hiperglucemia persiste, hay que iniciar un tratamiento. Los cambios del estado clínico pueden aumentar el riesgo de eventos adversos asociados a tratamientos no insulínicos. Por lo tanto, en la mayoría de los pacientes se prefiere la insulina para el manejo hospitalario de la hiperglucemia ≥180 mg/dl (10,0 mmol/l), con ajustes para mantener un nivel de glucemia de entre 140 y 180 mg/dl (7,8-10,0 mmol/l). Según la ADA y la AACE, si se evita la hipoglucemia pueden ser razonables unos valores de glucemia inferiores a 140 mg/dl (7,8 mmol/l) en determinados pacientes en estado no crítico. En cambio, el American College of Physicians (ACP) no recomienda valores de glucemia inferiores a 140 mg/dl (7,8 mmol/l), debido al mayor riesgo de hipoglucemia. Varios factores pueden provocar hipoglucemia hospitalaria: estado mental alterado, ayuno (previsto o imprevisto), enfermedad, desajuste de la hora de la insulina y las comidas, escasa ingesta oral y cambios en los tratamientos inductores de hiperglucemia. PUNTO CLAVE • Un control glucémico hospitalario estricto (80-110 mg/dl [4,4-6,1 mmol/l]) no siempre está asociado a una mejora de los resultados y puede aumentar la mortalidad. AMAV Trastornos del metabolismo de la glucosa Tabla 12. Hiperglucemia inducida por fármacos Categoría del fármaco Fármaco Mecanismo de la hiperglucemia Glucocorticoides Todos los glucocorticoides sistémicos Reducción de la producción de insulina Aumento de la resistencia periférica a la insulina Aumento de la producción de glucosa hepática Inmunodepresores Inhibidores de la calcineurina: Reducción de la producción y la liberación de insulina Sirolimus Tacrolimus Ciclosporina Antirretrovirales Medicamentos cardiovasculares Inhibidores de la proteasa Aumento de la resistencia a la insulina periférica ITINN Daño pancreático mediante pancreatitis inducida por fármacos (didanosina) Niacina Aumento de la producción de glucosa hepática Estatinas Alteración de la función de las células beta pancreáticas Aumento de la resistencia periférica Betabloqueantes: Reducción de la liberación de insulina Atenolol Aumento de la resistencia a la insulina periférica Metoprolol El carvedilol (bloqueador α) tiene un efecto neutro en la glucosa Propranolol Tiazidas: Reducción de la secreción de insulina secundaria a hipopotasemia Hidroclorotiazida Aumento de la resistencia a la insulina Clortalidona Clorotiazida Indapamida Vasopresores Reducción de la secreción de insulina Adrenalina Aumento de la glucogenólisis Noradrenalina Aumento de la producción de glucosa hepática Estimulación del glucagón y el cortisol Medicamentos hormonales Anticonceptivos orales: Metabolismo anómalo de la glucosa hepática Estrógeno-progestágenos combinados Aumento de la resistencia a la insulina periférica Solo progestágenos Reducción del riesgo de hiperglucemia con pastillas de dosis bajas con ≤35 µg de etinilestradiol Progestágeno: Aumento de la resistencia a la insulina periférica Megestrol acetato Antipsicóticos atípicos (segunda generación) Hormona del crecimiento Aumento de la resistencia a la insulina periférica Clozapina Incierto Olanzapina Posible aumento de la resistencia a la insulina periférica Ziprasidona Quetiapina Risperidona Iloperidona Paliperidona Antibióticos Moxifloxacina Alteración de la secreción de insulina Gatifloxacina ITINN: inhibidores de la transcriptasa inversa no nucleósidos. Datos de Fathallah N, Slim R, Larif S, Hmouda H, Ben Salem C. Drug-induced hyperglycaemia and diabetes. Drug Saf. 2015;38:1153-68. [PMID: 26370106] Datos de Thomas Z, Bandali F, McCowen K, Malhotra A. Drug-induced endocrine disorders in the intensive care unit. Crit Care Med. 2010;38:S219-30. [PMID: 20502175] 19 Trastornos del metabolismo de la glucosa Pacientes con diabetes mellitus hospitalizados En los pacientes críticos con diabetes mellitus tipo 1 y tipo 2, se recomienda un tratamiento con insulina intravenosa. Los ajustes de la dosis de insulina intravenosa deben basarse en un algoritmo validado que incorpore controles en la cama del paciente cada una o dos horas. En los pacientes no críticos, es adecuada la insulina subcutánea. Las personas con diabetes tipo 1 necesitan un tratamiento continuo con insulina. Debe proporcionarse insulina basal para evitar la aparición de CAD. Las personas con diabetes tipo 2 y valores de glucemia ≥180 mg/dl (10,0 mmol/l) también deben recibir tratamiento con insulina. Si el paciente come, el régimen de insulina ideal consiste en un régimen bolo-basal con cobertura prandial y bolos correctores para la hiperglucemia previa a las comidas. Antes del consumo de la comida deben realizarse mediciones en la cama del paciente e inyecciones de insulina prandial. Puede que sea adecuada la administración de insulina posprandial para ajustar la dosis en caso de ingesta oral reducida en algunas personas o en quienes presentan vaciamiento gástrico retardado. Las mediciones en la cama durante la noche están justificadas si hay inquietud por la hipoglucemia no detectada; de lo contrario, hay que evitar las comprobaciones nocturnas de la glucemia para no interrumpir el sueño ni aumentar el riesgo de un efecto acumulativo de la insulina. El uso exclusivo de insulina correctora («escala móvil de insulina») no está recomendado, porque se trata de una estrategia reactiva no fisiológica que provoca grandes fluctuaciones de la glucemia. Continuar con un tratamiento ambulatorio con ICIS puede ser adecuado para los pacientes con estado mental normal capaces de manejar el dispositivo bajo la supervisión de profesionales sanitarios que conozcan bien esta tecnología. Si un paciente hospitalizado no es capaz de manejar con seguridad el tratamiento con ICIS, este debe suspenderse y sustituirse por un régimen de insulina subcutánea o insulina intravenosa. La continuación de los fármacos orales o inyectables no insulínicos no se recomienda cuando los pacientes están ingresados, debido a las posibles alteraciones hemodinámicas y/o nutricionales que pueden producirse. Para manejar la glucemia debe iniciarse un tratamiento con insulina. Cuando se aproxima el alta de un paciente con estado nutricional y hemodinámica estables, puede considerarse reiniciar estos fármacos si la función orgánica ha vuelto a la situación basal. PUNTOS CLAVE • Los pacientes críticos con diabetes mellitus tipo 1 y tipo 2 necesitan tratamiento con insulina intravenosa con una posología basada en un algoritmo validado que incorpore controles en la cama del paciente cada una o dos horas. 20 • Los pacientes no críticos con diabetes tipo 1 necesitan insulina basal además del tratamiento con insulina prandial; las personas con diabetes tipo 2 y valores de glucemia ≥180 mg/dl (10,0 mmol/l) también deben recibir tratamiento con insulina. • El uso exclusivo de insulina correctora («escala móvil de insulina») no está recomendado, porque se trata de una estrategia reactiva no fisiológica que provoca grandes fluctuaciones de la glucemia. Pacientes sin diabetes mellitus hospitalizados El estrés asociado a la enfermedad aguda, la nutrición enteral/parenteral y los medicamentos inductores de hiperglucemia en el marco hospitalario pueden provocar anomalías de la glucemia en las personas sin diabetes. El manejo de la hiperglucemia debe seguir las mismas pautas que en los pacientes diabéticos hospitalizados. Es importante saber que la hiperglucemia hospitalaria puede darse en el contexto de una diabetes previamente indetectada. Una medición hospitalaria de la hemoglobina A1c ≥6,5% es indicativa de anomalías de la glucemia previas a la hospitalización, y estos pacientes necesitan planificar el alta para el manejo de una diabetes de nuevo diagnóstico. Complicaciones agudas de la diabetes mellitus Cetoacidosis diabética/síndrome hiperosmolar hiperglucémico La cetoacidosis diabética (CAD) y el síndrome hiperosmolar hiperglucémico (SHH) se producen en caso de hiperglucemia extrema y deben tratarse pronto y de manera agresiva para evitar las consecuencias potencialmente mortales de la deshidratación y las alteraciones electrolíticas. La hiperglucemia grave es consecuencia de unos niveles de insulina insuficientes junto con el aumento de las hormonas contrarreguladoras. Esto altera la utilización eficiente de la glucosa y, subsecuentemente, provoca glucogenólisis y gluconeogénesis para la producción de combustible. La CAD típicamente se presenta en personas con diabetes tipo 1 menores de 65 años de edad. Se trata de un estado de déficit absoluto de insulina que da lugar a lipólisis sin inhibición. Los ácidos grasos experimentan oxidación, con la producción posterior de cuerpos cetónicos y el desarrollo de acidosis metabólica. El SHH por lo común se presenta en personas con diabetes tipo 2 que tienen más de 65 años de edad. Está asociado a una tasa de mortalidad superior a la de la CAD. Se caracteriza por un déficit parcial de insulina capaz de inhibir la lipólisis y evitar la producción de cuerpos cetónicos, pero incapaz de corregir la hiperglucemia o prevenir la deshidratación y las alteraciones electrolíticas posteriores. Los pacientes jóvenes con diabetes tipo 1 presentan una tasa de filtración glomerular más alta que permite un mayor nivel de AMAV Trastornos del metabolismo de la glucosa hidratación y las anomalías electrolíticas, el trastorno puede progresar a letargo, obnubilación y muerte. La evaluación inicial incluye la medición de los niveles séricos de glucosa, electrolitos séricos, cetonas en suero, nitrógeno ureico en sangre y creatinina sérica, osmolalidad plasmática, hemograma completo, gasometría arterial, análisis de orina y cetonuria. También debe revisarse un electrocardiograma. En caso de presunta infección después de la anamnesis y la exploración, pueden obtenerse cultivos (sangre, esputo, orina) y una radiografía de tórax. La CAD y el SHH cursan con múltiples anomalías analíticas. En la CAD hay acidosis metabólica con gap aniónico secundario a la producción de ácido acetoacético y β-hidroxibutirato. Aunque algunos pacientes con SHH pueden tener elevado el gap aniónico, normalmente con niveles de glucemia superiores a 400-600 mg/dl (22,2-33,3 mmol/l), no experimentan una cetoacidosis significativa como la que se observa en la CAD. En la CAD se produce una reducción de moderada a grave de los niveles de bicarbonato sérico, pero estos pueden ser normales o estar ligeramente reducidos (<20 mEq/l [20 mmol/l]) en 28 26 Hiperglucemia hiperosmolar no cetósica Intervalo normal 24 22 20 Bicarbonato sérico CONT. glucosuria que quienes tienen diabetes tipo 2. En consecuencia, el SHH está asociado a una hiperglucemia más extrema que la CAD (Figura 1). Entre los factores que provocan la aparición de CAD o SHH se incluyen infección, infarto de miocardio, incumplimiento accidental o deliberado del tratamiento antidiabético, estrés, traumatismo y confusión de medicamentos (antipsicóticos atípicos, glucocorticoides e inhibidores del SGLT2). La CAD o el SHH pueden ser la presentación inicial en una persona con diabetes sin diagnosticar. La CAD y el SHH pueden manifestarse con una gran cantidad de síntomas y niveles de glucemia entre normales y muy altos. Por lo general, los síntomas de CAD se dan en las 24 horas posteriores al inicio, mientras que los síntomas de SHH pueden no aparecer durante varios días. Los síntomas de CAD y SHH pueden incluir dolor abdominal, náuseas, diarrea, poliuria, polidipsia, vómitos, pérdida de peso o disnea. La glucosuria extrema provoca una diuresis osmótica y una depleción de volumen grave, que pueden agravarse por las pérdidas gastrointestinales de volumen y electrolitos. Sin un tratamiento agresivo temprano para la hiperglucemia, la des- 18 16 14 12 10 8 6 4 CAD clásica CAD normoglucémica Espectro 2 0 100 5,6 200 11,1 300 16,7 400 22,2 500 27,8 600 33,3 700 38,9 800 44,4 900 50,0 1.000 55,5 Glucosa plasmática (mg/dl [mmol/l]) F i g u r a 1 . Espectro de la descompensación metabólica que tiene lugar en la cetoacidosis diabética. CAD: cetoacidosis diabética. 21 Trastornos del metabolismo de la glucosa CONT. el SHH. Por lo general, el pH sérico es >7,3 en el SHH. En la CAD y el SHH puede haber seudohiponatremia, con hiperglucemia extrema y desplazamientos osmóticos del agua del compartimento intracelular al extracelular. Un nivel de sodio normal o elevado indica deshidratación grave. En comparación con la diabetes tipo 1, el SHH suele cursar con osmolalidad elevada, con frecuencia >320 mOsm/kg de H2O, secundaria a hiperglucemia más grave y a pérdida de agua por la diuresis osmótica. Los niveles de potasio sérico pueden estar elevados por causa de los desplazamientos del espacio intracelular al extracelular producidos por la cetoacidosis y por la ausencia de insulina suficiente. Los niveles de potasio sérico normales o bajos indican que las reservas del organismo se han agotado y requieren un aporte complementario antes del tratamiento con insulina para evitar las arritmias cardíacas. El estrés puede provocar una leucocitosis leve, pero unos niveles más altos pueden ser indicativos de una causa infecciosa de la CAD o el SHH. El tratamiento de la CAD y el SHH requiere una estrategia múltiple (Tabla 13). Es necesaria una hidratación intravenosa para recuperar el volumen. Los déficits de electrolitos, como el potasio, deben ser subsanados. La hiperglucemia debe corregirse, preferentemente con insulina intravenosa y mediciones de la glucosa cada hora que puedan servir de guía en los ajustes de la dosis. Durante la hidratación y el tratamiento con insulina hace falta realizar mediciones frecuentes de los electrolitos que sirvan de orientación para revertir su Tabla 13. disminución. Dada la gran complejidad del tratamiento necesario, el manejo de la mayoría de los pacientes con CAD o SHH tiene lugar en la unidad de cuidados intensivos (UCI). Deben tratarse los otros trastornos que contribuyeran a la aparición de la CAD o el SHH, como la infección PUNTOS CLAVE • La cetoacidosis diabética y el síndrome hiperosmolar hiperglucémico se producen en caso de hiperglucemia extrema y deben tratarse pronto y de manera agresiva para evitar las consecuencias potencialmente mortales de la deshidratación y las alteraciones electrolíticas. • Entre los factores que provocan la aparición de cetoacidosis diabética y síndrome hiperosmolar hiperglucémico se incluyen infección, infarto de miocardio, incumplimiento accidental o deliberado del tratamiento antidiabético, estrés, traumatismo y confusión de medicamentos (inhibidores del SGLT2, antipsicóticos atípicos y glucocorticoides). • El tratamiento de la cetoacidosis diabética y el síndrome hiperosmolar hiperglucémico requiere corrección de la hiperglucemia, hidratación intravenosa, repleción de electrolitos y tratamiento las presuntas infecciones. Manejo de la crisis hiperglucémica: cetoacidosis diabética y síndrome hiperosmolar hiperglucémico Líquidos Insulina (regular) Potasio Corrección de la acidosis Evaluar el estado de volumen, administrar solución salina IV al 0,9% a 1 l/h inicialmente a todos los pacientes y continuar en caso de hipovolemia grave. Cambiar a solución salina al 0,45% a 250-500 ml/hora si el nivel del sodio sérico corregido pasa a ser normal o alto Administrar insulina regular, 0,1 U/kg, en forma de bolo IV seguido de 0,1 U/kg/h en infusión IV Comprobar que la función renal es adecuada, con una diuresis suficiente (aproximadamente 50 ml/h) Si el nivel de glucosa plasmática no desciende el 10% en la primera hora, administrar un bolo adicional de 0,14 U/kg y reanudar la velocidad de infusión anterior Si el potasio sérico es <3,3 mEq/l (3,3 mmol/l), no iniciar la insulina, sino administrar cloruro potásico IV, 20-30 mEq/h, mediante un catéter central hasta que el nivel de potasio sérico sea >3,3 mEq/l (3,3 mmol/l) Si el pH es <6,9, considerar la administración de bicarbonato sódico, 100 mmol en 400 ml de agua, y cloruro potásico, 20 mEq, mediante infusión durante 2 horas Cuando el nivel de glucosa plasmática llegue a 200 mg/dl (11,1 mmol/l) en los pacientes con CAD o a 300 mg/dl (16,7 mmol/l) en el SHH en el marco de insulina IV continua, cambiar a dextrosa al 5% con solución salina normal al 0,45% a 150-250 ml/h para mantener la glucemia y evitar la hipoglucemia Cuando el nivel de glucosa plasmática llegue a 200 mg/dl (11,1 mmol/l) en la CAD o a 300 mg/dl (16,7 mmol/l) en el SHH, reducir a 0,02-0,05 U/kg/h y mantener el nivel de glucosa plasmática entre 150 y 200 mg/dl (8,3-11,1 mmol/l) hasta que resuelva la acidosis con gap aniónico en la CAD En el SHH la glucosa plasmática debe mantenerse entre 250 y 300 mg/dl hasta que el paciente esté alerta y se resuelva el estado hiperosmolar Si el pH es de 6,9 o superior, no administrar bicarbonato sódico A continuación, añadir 20-30 mEq de cloruro potásico a cada litro de líquido IV para mantener el nivel de potasio sérico en el intervalo de entre 4,0 y 5,0 mEq/l (4,0-5,0 mmol/l) Si el nivel de potasio sérico es >5,2 mEq/l (5,2 mmol/l), no administrar cloruro potásico, sino iniciar la administración de insulina y líquidos IV y comprobar el nivel de potasio sérico cada 2 horas CAD: cetoacidosis diabética; IV: intravenoso; SHH: síndrome hiperosmolar hiperglucémico. Recomendaciones de Kitabchi AE, Umpierrez GE, Miles JM, Fisher JN. Hyperglycemic crises in adult patients with diabetes. Diabetes Care. 2009;32:1335-43. [PMID: 19564476] 22 Trastornos del metabolismo de la glucosa Complicaciones crónicas de la diabetes mellitus Morbilidad cardiovascular La ECV constituye una causa importante de morbimortalidad en las personas con diabetes mellitus. La diabetes es un factor de riesgo independiente de ECV. Otros factores de riesgo significativos de ECV son la hipertensión, la dislipemia, el tabaquismo, los antecedentes familiares y la albuminuria. Para reducir la morbimortalidad, se recomienda el manejo simultáneo de los factores de riesgo de ECV. Las pautas en cuanto a los intervalos de cribado de los factores de riesgo se presentan en la Tabla 14. La hipertensión contribuye a la aparición de complicaciones macrovasculares y microvasculares. La ADA define la hipertensión como una presión arterial ≥140/90 mmHg de manera constante. Mencionando la inquietud por el aumento de las complicaciones del tratamiento con una presión arterial objetivo <130/80 mmHg, la ADA establece un objetivo del tratamiento por debajo de 140/90 mmHg en la mayoría de las personas. Las personas con riesgo alto de ECV pueden buscar una presión arterial objetivo inferior, si es posible hacerlo de manera segura. En cambio, las guías de la AACE/ACE y el ACC/AHA y nueve organizaciones más defienden un objetivo terapéutico por debajo de 130/80 mmHg para la mayoría de los pacientes con diabetes. Las estrategias terapéuticas recomendadas por la ADA incluyen modificaciones del estilo de vida (para una presión arterial >120/80 mmHg) y tratamientos farmacológicos (para una presión arterial >140/90 mmHg). Los regímenes antihipertensivos iniciales recomendados son inhibidores de la enzima de conversión de la angiotensina (ECA), bloqueadores de los receptores de angiotensina (BRA), bloqueadores de los canales de calcio dihidropiridínicos y diuréticos tiazídicos. Con frecuencia hacen falta varios fármacos para lograr la presión arterial objetivo. Las comorbilidades subyacentes deben determinar la elección de los agentes terapéuticos, como el uso de un inhibidor de la ECA o un BRA en presencia de microalbuminuria. El manejo de los lípidos en la diabetes muchas veces requiere una combinación de modificaciones del estilo de vida y agentes farmacológicos. La calculadora de riesgo del ACC/AHA puede determinar el riesgo de enfermedad cardiovascular ateroesclerótica (ECVA) a 10 años y servir de orientación para el manejo terapéutico. El tratamiento farmacológico inicial recomendado para todas las personas diabéticas que reúnan los requisitos es la terapia con estatinas (véase el módulo «Medicina interna general» de MKSAP 18). La ADA recomienda un tratamiento antiagregante plaquetario con ácido acetilsalicílico (75-162 mg/día) para la prevención secundaria en las personas con diabetes y ECVA. El tratamiento con ácido acetilsalicílico para la prevención primaria de la ECVA en las personas con diabetes tipo 1 y tipo 2 puede no ser beneficioso para todo el mundo, por lo que puede considerarse un tratamiento con ácido acetilsalicílico (75-162 mg/día) en las personas de 50 años de edad y mayores con al menos un factor de riesgo de ECVA adicional. La ADA no recomienda el tratamiento con ácido acetilsalicílico para las personas menores de 50 años y bajo riesgo de ECVA. PUNTOS CLAVE • La American Diabetes Association (ADA) recomienda una presión arterial ≤140/90 mmHg como objetivo terapéutico en las personas en alto riesgo de ECV si es posible alcanzarla de manera segura; la American Association of Clinical Endocrinologists/American College of Endocrinology (AACE/ACE), el American College of Cardiology/American Heart Association (ACC/AHA) y nueve organizaciones más recomiendan un objetivo <130/80 mmHg para la mayoría de los pacientes con diabetes. • El manejo de los lípidos en la diabetes requiere muchas veces una combinación de modificaciones del estilo de vida y agentes farmacológicos; el tratamiento farmacológico inicial recomendado para todas las personas con diabetes que reúnan los requisitos es la terapia con estatinas. AMAV • La ADA no recomienda el tratamiento con ácido acetilsalicílico para las personas menores de 50 años y bajo riesgo de enfermedad cardiovascular ateroesclerótica. Retinopatía diabética La retinopatía constituye la principal causa de ceguera evitable en las personas con diabetes de entre 20 y 74 años de edad en los países desarrollados. Los factores de riesgo de retinopatía incluyen el tiempo de evolución de la diabetes, el grado de hiperglucemia, la hipertensión, la albuminuria y la dislipemia. Las alteraciones de la retinopatía diabética se clasifican como no proliferativas (tienen lugar dentro de la retina) o proliferativas (se producen en el vítreo o la superficie interna de la retina). Los signos de retinopatía no proliferativa pueden incluir microaneurismas, hemorragias puntiformes o en manchas, exudados duros (depósito de lípidos), exudados blandos o manchas algodonosas (fibras nerviosas superficiales isquémicas), hemorragia venosa y anomalías microvasculares intrarretinianas. La neovascularización debida a la isquemia crónica es característica de la retinopatía proliferativa, que puede provocar hemorragia intraocular, desprendimiento de retina y pérdida de visión. El edema macular puede darse con retinopatía proliferativa y no proliferativa. Se han elaborado directrices de cribado para una detección temprana de las anomalías asintomáticas que permita efectuar intervenciones terapéuticas con el fin de prevenir la pérdida de visión (véase la Tabla 14). El control óptimo de la presión arterial, la glucemia y los parámetros lipídicos puede prevenir y retrasar la progresión de la retinopatía. La fotocoagulación panretiniana con láser 23 Trastornos del metabolismo de la glucosa Tabla 14. Recomendaciones para el cribado de las complicaciones crónicas de la diabetes mellitus Complicación Situación clínica Cuándo iniciar el cribado Frecuencia del cribado Prueba de cribado preferible Retinopatía Diabetes tipo 1 5 años después del diagnóstico Anualmentea Examen oftalmoscópico exhaustivo con dilataciónb Diabetes tipo 2 En el diagnóstico Anualmentea Examen oftalmoscópico exhaustivo con dilataciónb En embarazadas con cualquier tipo de diabetes Primer trimestre Cada trimestre y luego controles estrictos durante 1 año después del parto Examen oftalmoscópico exhaustivo con dilataciónb En mujeres con cualquier tipo de diabetes que tengan previsto quedarse embarazadas Durante la planificación previa a la concepción Igual que en las recomendaciones para las embarazadas una vez lograda la concepción Examen oftalmoscópico exhaustivo con dilataciónb Diabetes tipo 1 5 años después del diagnóstico Anualmentec Cociente albúmina/creatinina en micción aislada aleatoria, TFGe Diabetes tipo 2 En el diagnóstico Anualmentec Cociente albúmina/creatinina en micción aislada aleatoria, TFGe Diabetes tipo 1 5 años después del diagnóstico Anualmente Evaluación cutánea, evaluación de las deformidades del pie, evaluación del pulso de la extremidad inferior, evaluación neurológica (monofilamento de 10 g más diapasón de 128 Hz, reflejos de los tobillos, pinchazo o temperatura) Diabetes tipo 2 En el diagnóstico Anualmente Evaluación cutánea, evaluación de las deformidades del pie, evaluación del pulso de la extremidad inferior, evaluación neurológica (monofilamento de 10 g más diapasón de 128 Hz, reflejos de los tobillos, pinchazo o temperatura) Hipertensión En el diagnóstico Cada visita Medición de la presión arterial Dislipemia En el diagnóstico y antes de iniciar la terapia con estatinas Anualmentee Perfil lipídico Nefropatía Neuropatía (polineuropatía distal simétrica)d Enfermedad cardiovascular aEs razonable efectuar el cribado cada 1-2 años en ausencia de retinopatía diabética y más de una vez al año en caso de retinopatía diabética avanzada o de progresión rápida. bLa fotografía retiniana constituye un posible método alternativo para detectar retinopatía diabética que puede mejorar el acceso a la atención y reducir los costes. La fotografía retiniana con interpretación por oftalmólogos especialistas puede detectar la mayoría de la retinopatía diabética clínicamente significativa. cLas guías de la American Diabetes Association afirman que es razonable evaluar la progresión de la enfermedad y la respuesta a las intervenciones terapéuticas con controles continuos de la excreción urinaria de albúmina y creatinina. dAunque la diabetes suele provocar neuropatía periférica, en el proceso de cribado deben tenerse en cuenta otros diagnósticos diferenciales como el déficit de vitamina B 12, el alcoholismo, el hipotiroidismo, la enfermedad renal, el cáncer y las quimioterapias, la vasculitis y las neuropatías hereditarias. eCribado anual o periódico para controlar la respuesta terapéutica tras el inicio de la terapia con estatinas. Si el paciente no toma estatinas, el cribado puede llevarse a cabo cada cinco años. TFGe: tasa de filtración glomerular estimada. Recomendaciones de la American Diabetes Association. 9. Cardiovascular disease and risk management: Standards of Medical Care in Diabetes—2018. Diabetes Care. 2018;41 (Suppl. 1):S86-S104. [PMID: 29222380] Recomendaciones de la American Diabetes Association. 10. Microvascular complications and foot care: Standards of Medical Care in Diabetes—2018. Diabetes Care. 2018;41 (Suppl. 1):S105-S118. [PMID: 29222381] Recomendaciones de Garber AJ, Abrahamson MJ, Barzilay JI, Blonde L, Bloomgarden ZT, Bush MA, et al; American Association of Clinical Endocrinologists (AACE). Consensus Statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the comprehensive type 2 diabetes management algorithm—2018 executive summary. Endocr Pract. 2018;24:91-120. [PMID: 29368965] permite tratar la retinopatía diabética proliferativa de alto riesgo y la retinopatía no proliferativa grave. Además, las inyecciones intravítreas de antifactor de crecimiento vascular endotelial (anti-VEGF) para reducir la pérdida de visión aso24 ciada a la retinopatía proliferativa no son inferiores a la fotocoagulación panretiniana con láser. La retinopatía puede surgir o acelerarse durante el embarazo o con las mejoras glucémicas rápidas y necesitar fotocoagulación con láser para re- Trastornos del metabolismo de la glucosa ducir el riesgo de pérdida de visión. El edema macular se trata idealmente con inyecciones intravítreas de anti-VEGF para mejorar la pérdida de visión. Las inyecciones de anti-VEGF deben efectuarse mensualmente durante al menos 12 meses para seguirse de inyecciones intermitentes con el fin de prevenir el edema macular recidivante. PUNTOS CLAVE • Los pacientes con diabetes tipo 2 deben someterse a un examen oftalmoscópico en el momento del diagnóstico. • El control óptimo de la glucemia, la presión arterial y los parámetros lipídicos puede prevenir y retrasar la progresión de la retinopatía. • La fotocoagulación panretiniana con láser o las inyecciones intravítreas de antifactor de crecimiento vascular endotelial pueden tratar la retinopatía diabética proliferativa de alto riesgo y la retinopatía no proliferativa grave. Nefropatía diabética La nefropatía diabética es la principal causa de enfermedad renal terminal (ERT). Habitualmente, la diabetes está presente 5-10 años antes de la aparición de nefropatía. Las personas con un familiar de primer grado con ERT debido a nefropatía diabética presentan un mayor riesgo de experimentar progresión a ERT. Se recomienda la medición de la TFGe y el cribado de la presencia de microalbuminuria para la detección temprana de enfermedad renal (véase la Tabla 14). La excreción urinaria de albúmina puede determinarse a partir de una recogida de orina aleatoria en forma de cociente de albúmina/ creatinina (CAC). Un nivel elevado de CAC (≥30 mg/g de crea­ tinina) debe confirmarse mediante múltiples mediciones a lo largo de entre tres y seis meses, porque la variabilidad biológica, la enfermedad, la hiperglucemia, la insuficiencia cardíaca, la hipertensión, el ejercicio y la menstruación pueden provocar elevaciones temporales. Las mediciones anuales de la TFGe y el CAC pueden detectar la progresión de la nefropatía y servir de orientación a la hora de tomar decisiones terapéuticas. En caso de empeoramiento de la función renal, pueden ser necesarias mediciones más frecuentes. Una TFGe <30 ml/min/1,73 m2 justifica la derivación a un nefrólogo. La hiperglucemia y la hipertensión no controladas son factores de riesgo de nefropatía diabética, por lo que se recomienda un tratamiento para lograr los objetivos de glucemia y presión arterial. La ADA recomienda un inhibidor de la ECA o un BRA como tratamiento de primera línea para ralentizar la progresión de la nefropatía y prevenir la ECV en las personas con diabetes, hipertensión, TFGe reducida (<60 ml/min/1,73 m2) y CAC elevado (≥300 mg/g de creatinina), salvo en las embarazadas. También se recomienda un inhibidor de la ECA o un BRA para el tratamiento de un CAC elevado de entre 30 y 299 mg/g de creatinina en las personas con hipertensión, salvo las embarazadas. No se recomienda el tratamiento con un inhibidor de la ECA o un BRA en los pacientes con diabetes que tengan una presión arterial normal, un CAC <30 mg/g de creatinina y una TFGe >60 ml/min/1,73 m2. PUNTOS CLAVE • Se recomienda la medición de la tasa de filtración glomerular estimada y el cribado de la presencia de microalbuminuria para la detección temprana de enfermedad renal. • La American Diabetes Association recomienda un inhibidor de la ECA o un bloqueador de los receptores de angiotensina como tratamiento de primera línea para ralentizar la progresión de la nefropatía y prevenir la enfermedad cardiovascular en las personas con diabetes, hipertensión, tasa de filtración glomerular estimada reducida (<60 ml/min/1,73 m2) y cociente albúmina/creatinina en orina elevado (≥300 mg/g de creatinina), salvo en las embarazadas. AMAV • No se recomienda el tratamiento con un inhibidor de la ECA o un bloqueador de los receptores de angiotensina en los pacientes con diabetes que tengan una presión arterial normal, un cociente albúmina/creatinina en orina inferior a 30 mg/g de creatinina y una tasa de filtración glomerular estimada superior a 60 ml/min/1,73 m2. Neuropatía diabética La neuropatía diabética cursa con daños en los nervios o las raíces nerviosas provocados por la hiperglucemia. Los síntomas dependen del nervio o los nervios afectados y pueden ser de naturaleza focal o difusa. La neuropatía puede darse a nivel periférico y/o afectar al sistema nervioso autónomo. El control glucémico puede prevenir la neuropatía periférica y la neuropatía autonómica cardíaca en las personas con diabetes tipo 1 y retrasar la progresión de la neuropatía en la diabetes tipo 2. Normalmente, la neuropatía periférica diabética (polineuropatía distal simétrica) tiene una presentación ascendente con una distribución en manos y pies (en «calcetines y guantes»). Puede cursar con daños en las fibras nerviosas tanto pequeñas como grandes. Los síntomas del daño de las fibras nerviosas pequeñas incluyen dolor, quemazón y cosquilleo. Las anomalías de las fibras nerviosas pequeñas pueden detectarse en la exploración mediante la evaluación de la sensibilidad al pinchazo y la temperatura. Las alteraciones sensitivas de la posición, la vibración y el tacto ligero son indicativas de daños en las fibras nerviosas grandes y suponen un mayor riesgo de úlceras en el pie. La evaluación de los daños de las fibras nerviosas grandes puede efectuarse mediante la valoración de los reflejos del tobillo y con un diapasón de 128 Hz y un monofilamento de 10 g. Dado que la neuropatía periférica puede ser asintomática, es necesario realizar un 25 Trastornos del metabolismo de la glucosa CONT. cribado para una detección precoz que evite la pérdida de la extremidad (véase la Tabla 14). La neuropatía autonómica puede afectar a uno o varios órganos, con síntomas que varían en función del órgano afectado. Los síntomas pueden incluir hipoglucemia inadvertida, gastroparesia, estreñimiento, diarrea, disfunción eréctil y disfunción vesical. Los síntomas de disfunción autonómica cardíaca pueden ser hipotensión ortostática, taquicardia sinusal en reposo e intolerancia al ejercicio. La neuropatía autonómica cardíaca constituye un factor de riesgo independiente de muerte súbita. El objetivo del tratamiento de la neuropatía diabética es el control sintomático. Como tratamiento inicial se recomiendan dos medicamentos aprobados por la FDA, la pregabalina o la duloxetina. Hay otros fármacos que pueden ofrecer alivio sintomático, pero no están aprobados por la FDA; se trata de los antidepresivos tricíclicos, la venlafaxina, la carbamacepina, la capsaicina y la gabapentina. El tratamiento primario de la hipotensión ortostática es no farmacológico e incluye dieta, uso de medias compresivas y cambios lentos de posición. Es necesario suspender los medicamentos que causan o empeoran las alteraciones ortostáticas y añadir otros fármacos (fludrocortisona, midrodina o droxidopa) para los síntomas refractarios. Las comidas frecuentes bajas en grasas y fibra pueden mejorar los síntomas de gastroparesia. La metoclopramida es el único fármaco procinético aprobado por la FDA para el tratamiento de la gastroparesia. Dado el riesgo de efectos secundarios, entre ellos distonía, debe realizarse una evaluación atenta de los riesgos y beneficios antes de la prescripción (véase el módulo «Gastroenterología y Hepatología» de MKSAP 18). La amiotrofia diabética es un trastorno infrecuente que afecta al plexo lumbosacro y puede ser secundario a infarto o vasculopatía inmunitaria. Tiene una presentación aguda y está asociada a dolor asimétrico intenso o debilidad proximal en una pierna, con atrofia muscular progresiva asociada. Puede producirse una remisión parcial a lo largo de muchos meses. El tratamiento es sintomático. Los pacientes con diabetes pueden experimentar mononeuropatías y síndromes de compresión nerviosa (síndrome del túnel carpiano, parálisis peroneal). Con frecuencia las mononeuropatías se resuelven sin intervención alguna en el plazo de unos meses. Los síndromes de compresión pueden responder al manejo conservador o requerir cirugía para conseguir el alivio sintomático. PUNTOS CLAVE • La neuropatía periférica diabética (polineuropatía distal simétrica) se manifiesta con una distribución en «calcetines y guantes» y cursa con daños en las fibras nerviosas tanto pequeñas como grandes; los síntomas incluyen dolor, quemazón y cosquilleo. • El control glucémico puede retrasar la progresión de la neuropatía en la diabetes tipo 2. 26 Úlceras del pie diabético Las úlceras de la extremidad inferior y las amputaciones tienen asociada una morbimortalidad significativa (véase el módulo «Enfermedades infecciosas» de MKSAP 18). Es necesario examinar las extremidades inferiores en cada visita y realizar una exploración exhaustiva del piel, que incluya una prueba con monofilamento de 10 g, al menos una vez al año. Los factores de riesgo de úlcera incluyen: hiperglucemia, enfermedad arterial periférica, antecedentes de úlcera en el pie o amputación, deformidad en el pie, neuropatía periférica, visión alterada, tabaquismo y nefropatía diabética. En las personas con pulsos pedios ausentes o síntomas inquietantes de claudicación es necesario llevar a cabo una evaluación vascular. En la atención de las personas de alto riesgo deben intervenir podólogos especialistas. Hay que educar a los pacientes sobre la importancia de las inspecciones diarias de los pies y del uso de calzado adecuado. Hipoglucemia La hipoglucemia se define como un valor de glucemia inferior a 70 mg/dl (3,9 mmol/l). Los valores de glucemia por debajo de 54 mg/dl (3,0 mmol/l) son graves y clínicamente significativos. Se considera hipoglucemia grave cualquier valor glucémico en el cual una persona necesite ayuda externa para corregir la glucemia. La respuesta fisiológica normal a la aparición de hipoglucemia implica la aparición de síntomas hiperadrenérgicos (sudor, temblores, ansiedad, taquicardia). Posteriormente, el organismo libera hormonas contrarreguladoras (glucagón, adrenalina, noradrenalina, cortisol y hormona del crecimiento) para corregir la hipoglucemia. Los signos neuroglucopénicos (estado mental alterado, disartria, confusión) están asociados a hipoglucemia grave. Si la hipoglucemia grave no se corrige con rapidez, puede producirse obnubilación, convulsiones y muerte. Hipoglucemia en pacientes con diabetes mellitus En muchas personas, la hipoglucemia puede convertirse en un obstáculo para lograr los objetivos glucémicos. La hipoglucemia recidivante grave está asociada a déficits cognitivos adquiridos y puede provocar demencia. Es necesario ajustar los tratamientos para eliminar las hipoglucemias, así como personalizar los objetivos glucémicos para que su consecución sea posible de manera segura. Varios factores contribuyen a la aparición de hipoglucemia, incluyendo los desajustes entre el consumo de comida y la administración de insulina, el aumento del ejercicio físico, la pérdida de peso, el empeoramiento del deterioro renal, las anomalías de la motilidad y la absorción gastrointestinal y las sobredosis accidentales/intencionadas de insulina. Los ancianos también presentan un riesgo elevado de hipoglucemia. Trastornos del metabolismo de la glucosa CONT. La hipoglucemia puede darse también con el uso de antidiabéticos debido a una posología incorrecta, a las interacciones entre fármacos y a las enfermedades intercurrentes que alteran el metabolismo o la excreción de los fármacos. El tratamiento para la hipoglucemia en una persona alerta incluye el consumo de entre 15 y 20 g de hidratos de carbono de absorción rápida seguido de una medición de la glucemia 15 a 20 minutos después. Si la glucemia no ha mejorado, debe repetirse el tratamiento con 15 g de hidratos de carbono. Una vez normalizada la glucemia, es necesario consumir una comida o tentempié (>70 mg/dl [3,9 mmol/l]) para evitar la hipoglucemia recidivante. A las personas en riesgo de experimentar hipoglucemias clínicamente significativas (<54 mg/dl [3,0 mmol/l]) hay que facilitarles glucagón, para que puedan administrarlo por vía intramuscular los allegados si la persona no puede consumir hidratos de carbono de manera segura para corregir la hipoglucemia. La hipoglucemia relativa se caracteriza por la presencia de síntomas de hipoglucemia en el contexto de valores de glucosa en plasma >70 mg/dl (3,9 mmol/l). Esto puede ocurrir ante un descenso grande y rápido de la glucemia o en caso de normalización rápida de la glucemia con la intensificación del tratamiento en una persona con valores glucémicos prolongados por encima de 200 mg/ml (11,1 mmol/l). La hipoglucemia relativa puede prevenirse evitando las grandes oscilaciones glucémicas y con la corrección lenta de la hiperglucemia de larga evolución hasta el valor objetivo para que el período de ajuste sea más prolongado. micos obtenidos en la cama del paciente no son fiables en este contexto. La hipoglucemia en personas sin diabetes puede atribuirse a las causas siguientes: fármacos, alcohol, enfermedad, disfunción orgánica (renal o hepática), déficits hormonales (insuficiencia suprarrenal), malnutrición e insulinoma o no insulinoma pancreatógeno (hipoglucemia hiperinsulinémica endógena que no tiene su origen en un insulinoma). Aunque puede haber solapamiento en la presentación clínica, la hipoglucemia suele darse en ayunas o en estado posprandial. Durante un episodio hipoglucémico en el que se ha demostrado la tríada de Whipple, deben obtenerse cultivos diagnósticos de sangre y orina. Si no hay testigos de un episodio espontáneo, es necesario aplicar medidas para recrear las circunstancias que normalmente provocarían la hipoglucemia (ayuno o ingesta de una comida típica que produzca un episodio en ese paciente concreto). Solo deben llevarse a cabo estudios de imagen para la localización del tumor tras la confirmación de hiperinsulinismo endógeno a partir de los estudios diagnósticos de sangre y orina. PUNTO CLAVE AMAV • Solo deben llevarse a cabo estudios de imagen para la localización tumoral tras la confirmación de hiperinsulinismo endógeno a partir de los estudios diagnósticos de sangre y orina. Hipoglucemia en ayunas PUNTOS CLAVE • La hipoglucemia se define como un valor glucémico inferior a 70 mg/dl (3,9 mmol/l) y la hipoglucemia grave, por un valor inferior a 54 mg/dl (3,0 mmol/l). • El tratamiento inicial de la hipoglucemia consiste en el consumo oral de hidratos de carbono o en la administración de glucagón con el objetivo de elevar la glucemia a más de 70 mg/dl (3,9 mmol/l). • La hipoglucemia relativa puede prevenirse evitando las grandes oscilaciones glucémicas y con la corrección lenta de la hiperglucemia de larga evolución hasta el valor objetivo para que el período de ajuste sea más prolongado. Hipoglucemia en pacientes sin diabetes mellitus La hipoglucemia sin diabetes concomitante es infrecuente y debe dar lugar a una evaluación en mayor profundidad. Es necesario realizar una evaluación de la hipoglucemia si se cumplen los criterios de la tríada de Whipple: síntomas neuroglucopénicos, hipoglucemia ≤55 mg/dl (3,1 mmol/l) y resolución de los síntomas con el consumo de glucosa. La determinación analítica de la glucosa debe confirmar la presencia de hipoglucemia verdadera, ya que los valores glucé- Si normalmente la hipoglucemia se produce durante el ayuno, debe realizarse un ayuno prolongado de hasta 72 horas. Se extraen cinco muestras de sangre simultáneas cada seis horas para determinar: glucosa, péptido C, insulina, proinsulina y β-hidroxibutirato. Al inicio del ayuno debe hacerse también un cribado de anticuerpos contra la insulina y de hipoglucemiantes orales. Si la medición de la glucemia es inferior a 60 mg/dl (3,3 mmol/l), es necesario incrementar la recogida de muestras de sangre hasta cada una o dos horas. Las pruebas finalizan cuando se cumple uno de los parámetros siguientes: glucosa plasmática de 45 mg/dl (2,5 mmol/l) o inferior con neuroglucopenia o glucosa plasmática inferior a 55 mg/dl (3,1 mmol/l) en caso de tríada de Whipple demostrada anteriormente. Los valores glucémicos obtenidos en la cama del paciente y los síntomas hiperadrenérgicos no deben utilizarse para determinar el final del ayuno. Si no se ha cumplido ninguno de los criterios anteriores, deben volver a obtenerse muestras de sangre al término del período de 72 horas. La interpretación de los resultados de las pruebas diagnósticas se muestra en la Tabla 15. Para reducir el coste del procedimiento, la muestra para glucosa en plasma debe enviarse al laboratorio lo antes posible y, en caso de ser inferior a 60 mg/dl (3,3 mmol/l), deberán enviarse las otras cuatro muestras de sangre. 27 Trastornos del metabolismo de la glucosa Tabla 15. Diagnóstico diferencial de la hipoglucemia basal espontáneaa en una persona sin diabetes mellitus Diagnóstico Insulina en suero Péptido C en plasmab Proinsulina en plasmab β-hidroxibutirato en plasmac Anticuerpos antiinsulina en suero Metabolitos de las sulfonilureas o meglitinidas en orina o sangre Insulinomad,e ↑ ↑ ↑ ↓ Negativos Negativos Uso subrepticio de sulfonilureas o meglitinidas ↑ ↑ ↑ ↓ Negativos Positivos Uso subrepticio de insulina ↑ ↓ ↓ ↓ Negativos Negativos Hipoglucemia autoinmune por insulina ↑ ↑ ↑ ↓ Positivos Negativos IGF-IIf ↓ ↓ ↓ ↓ Negativos Negativos aHipoglucemia sintomática, glucemia basal de 55 mg/dl (3,1 mmol/l) o inferior y alivio sintomático inmediato con la corrección de la hipoglucemia (tríada de Whipple). bEl péptido C y la proinsulina son indicativos de producción de insulina. cEl β-hidroxibutirato se inhibirá en presencia de insulina, pero será elevado con una hipoglucemia no mediada por la insulina. d Es necesario obtener muestras de sangre al término de las pruebas y administrar glucagón tras las mediciones seriadas de la glucosa a lo largo de 30 minutos. La insulina inhibe la glucogenólisis y preserva las reservas de glucógeno. En el contexto de un insulinoma, la glucosa aumentará en respuesta al glucagón debido a la utilización de las reservas de glucógeno. eTambién pueden observarse resultados similares con el síndrome de hipoglucemia pancreatógena sin insulinoma o la hipoglucemia posterior a una derivación gástrica. fLos tumores pueden producir factor de crecimiento insulínico II (IGF-II) o sus precursores, que pueden inducir hipoglucemia mediante la estimulación de los receptores de insulina, con el consiguiente aumento del uso de glucosa. Datos de Cryer PE, Axelrod L, Grossman AB, Heller SR, Montori VM, Seaquist ER, et al; Endocrine Society. Evaluation and management of adult hypoglycemic disorders: an Endocrine Society Clinical Practice Guideline. Clin Endocrinol Metab. 2009;94:709-28. [PMID: 19088155] PUNTO CLAVE AMAV • Para la hipoglucemia en ayunas, se extraen cinco muestras de sangre simultáneas cada seis horas para determinar: glucosa, péptido C, insulina, proinsulina y β-hidroxibutirato; para reducir el coste del procedimiento, la muestra para glucosa en plasma debe enviarse al laboratorio lo antes posible y, en caso de ser inferior a 60 mg/dl (3,3 mmol/l), deberán enviarse las otras cuatro muestras de sangre. Hipoglucemia posprandial Normalmente, la hipoglucemia posprandial se produce en las cinco horas posteriores a la última comida. Con frecuencia la causa es una alteración de las estructuras anatómicas gastrointestinales, como puede ocurrir tras una derivación gástrica en Y de Roux. Muchas veces tiene su origen en comidas consistentes en hidratos de carbono simples (tortitas, sirope, zumos). Para determinar la causa, debe llevarse a cabo una prueba de comida mixta con los tipos de alimentos que suelen provocar hipoglucemia. Las pruebas analíticas basales, incluyendo determinación de los niveles séricos de glucosa, péptido C, insulina y proinsulina, deben obtenerse antes de la ingestión de la comida. Estas pruebas se repetirán cada 30 minutos durante cinco horas. En caso de aparición de neuroglucopenia, es necesario repetir las pruebas antes de la administración de hidratos de carbono. Para reducir el coste del procedimiento, la mues28 tra de sangre para la determinación de glucosa en plasma debe enviarse al laboratorio lo antes posible, remitiendo el resto en caso de que sea inferior a 60 mg/dl (3,3 mmol/l). También deberán realizarse pruebas de detección de anticuerpos contra la insulina e hipoglucemiantes orales en caso de aparición de hipoglucemia sintomática. La interpretación de los resultados es similar a la de los obtenidos para la hipoglucemia en ayunas (véase la Tabla 15). Por lo general, el tratamiento consiste en pequeñas comidas mixtas fijas con una cantidad equilibrada de proteínas, grasas e hidratos de carbono. PUNTO CLAVE • En la hipoglucemia posprandial, deben obtenerse pruebas analíticas basales que incluyan determinaciones séricas de glucosa, péptido C, insulina y proinsulina; para reducir el coste del procedimiento, la muestra para glucosa en plasma debe enviarse al laboratorio lo antes posible y, en caso de ser inferior a 60 mg/dl (3,3 mmol/l), deberán enviarse las otras tres muestras de sangre. Hipoglucemia inadvertida La hipoglucemia inadvertida se caracteriza por la liberación insuficiente de hormonas contrarreguladoras y por una respuesta autonómica inadecuada a la hipoglucemia. Los episodios anteriores de hipoglucemia aumentan el riesgo de experi- AMAV Trastornos de la hipófisis CONT. mentar una hipoglucemia inadvertida. El tratamiento consiste en la relajación de los objetivos glucémicos y en la modificación de los tratamientos para la diabetes inductores de hipoglucemia con el fin de evitar la hipoglucemia continua. Evitar la hipoglucemia durante varias semanas puede revertir la hipoglucemia inadvertida en algunas personas y dar lugar a la reaparición de síntomas adrenérgicos con niveles glucémicos inferiores a 70 mg/dl (3,9 mmol/l). Un sistema de monitorización continua de la glucosa puede ser beneficioso en las personas adecuadas para permitir la detección precoz de una hipoglucemia grave inminente y una intervención temprana. Trastornos de la hipófisis Anatomía y fisiología del hipotálamo y la hipófisis La hipófisis está situada en la silla turca, posteriormente al seno esfenoidal. Por encima de la hipófisis está situado el quiasma óptico, y a los lados se encuentran las arterias carótidas (Figura 2). La glándula está compuesta por la hipófisis anterior (adenohipófisis), formada por tejido glandular, y la hipófisis posterior (neurohipófisis), que proviene del tejido neural. Una rica red vascular portal conecta el hipotálamo con la hipófisis anterior, mientras que la hipófisis posterior está formada por terminaciones nerviosas proyectadas por las neuronas de los núcleos supraóptico y paraventricular del hipotálamo. Tanto la red portal como las neuronas hipotalámicas viajan desde el hipotálamo hasta la hipófisis a través del tallo hipofisario. La irrigación de la hipófisis depende de las arterias hipofisarias, ramas de las carótidas, y el drenaje venoso tiene lugar a través de los senos petrosos hasta la vena yugular. Las hormonas estimuladoras e inhibidoras, secretadas a la sangre portal por el hipotálamo, regulan la hipófisis anterior, mientras que las hormonas de la hipófisis posterior se sintetizan en los núcleos hipotalámicos y se desplazan por las neuronas para ser liberadas por la hipófisis posterior. La hipófisis anterior secreta seis hormonas hipofisarias, que suelen denominarse por sus siglas en inglés: hormona luteinizante (LH), hormona foliculoestimulante (FSH), corticotropina u hormona adrenocorticotropa (ACTH), prolactina (PRL), tirotropina u hormona estimulante de la tiroides (TSH) y hormona del crecimiento (GH). La hormona liberadora de gonadotropina (GnRH) es liberada por el hipotálamo en pulsos y, a su vez, controla la liberación de LH y FSH. La LH y la FSH regulan la reproducción masculina y femenina, incluyendo la estimulación de las gónadas para producir testosterona y estrógeno, así como la estimulación de los folículos ováricos y la espermatogénesis (véase «Trastornos reproductivos»). La corticoliberina u hormona liberadora de corticotropina (CHR), producida en el hipotálamo, estimula en la hipófisis la producción de ACTH, que, a su vez, estimula la producción de cortisol en las glándulas suprarrenales. La prolactina se sintetiza en las células lactotropas, y su síntesis y secreción es inhibida por la dopamina producida en el hipotálamo, que atraviesa el tallo hipofisario a través de la circulación portal. La TSH se libera en respuesta a la estimulación de la hormona liberadora de tirotropina (TRH) producida en el hipotálamo. La TSH se une a los receptores de la glándula tiroides, lo que da lugar a la síntesis y la secreción de hormonas tiroideas. La liberación de GH está regulada por dos hormonas hipotalámicas, la hormona liberadora de hormona del crecimiento (GHRH), que estimula la liberación de GH, y la somatostatina, que inhibe la liberación de GH. La hipófisis posterior secreta oxitocina, necesaria para el parto y la lactancia, y hormona antidiurética (ADH), que mantiene el equilibrio hídrico. F i g u r a 2 . Resonancia magnética (RM) frontal (izquierda) y RM sagital (derecha) que muestran la hipófisis (flecha gruesa), el tallo hipofisario (flecha delgada), el quiasma óptico (punta de flecha), el seno esfenoidal (estrella) y la arteria carótida (flecha curva). 29 Trastornos de la hipófisis Tabla 16. Pruebas iniciales para el déficit y el exceso de hormonas hipofisarias Exceso de hormona hipofisaria Hormona hipofisaria Hormona periférica Prueba(s) inicial(es) ACTH Cortisol Cortisol libre en orina de 24 horas (×2) O cortisol salival nocturno (×2) O prueba de la dexametasona en dosis bajas durante la noche ADH ADH Sodio sérico simultáneo, osmolalidad sérica, sodio en orina y osmolalidad urinaria GH IGF-1 IGF-1 TSH Tiroxina, triyodotironina TSH, tiroxina libre (o total) PRL Prolactina Prolactina en suero Déficit de hormona hipofisaria Hormona hipofisaria Hormona periférica Prueba(s) inicial(es) Prueba de confirmacióna ACTH Cortisol ACTH simultánea, cortisol Prueba de estimulación con ACTH ADH ADH Sodio sérico simultáneo, osmolalidad urinaria y sérica Prueba de privación de agua LH y FSHb Testosterona o estradiol LH simultánea, FSH, testosterona total (hombres), estradiol (mujeres) TSH Tiroxina, triyodotironina TSH simultánea, tiroxina libre (o total) GH IGF-1 IGF-1 Prueba de la GHRH-arginina Prueba de tolerancia a la insulina aVéase la Tabla 18 para hallar más información sobre las pruebas de confirmación para la disfunción hipofisaria. b No se recomienda el cribado sistemático de déficits si no hay signos específicos de déficit tales como amenorrea, ginecomastia o impotencia. ACTH: corticotropina; ADH: hormona antidiurética; FSH: hormona foliculoestimulante; GH: hormona del crecimiento; GHRH: hormona liberadora de hormona del crecimiento; IGF-1: factor de crecimiento insulínico tipo 1; LH: hormona luteinizante; PRL: prolactina; TSH: tirotropina. En la Tabla 16 se enumeran las hormonas hipofisarias y la evaluación inicial de la sospecha de exceso o déficit de cada hormona hipofisaria. Alteraciones de la hipófisis Masas hipofisarias detectadas de manera incidental Cuando se descubre incidentalmente una lesión hipofisaria en una prueba de imagen obtenida por un motivo sin relación alguna, la lesión se denomina «incidentaloma hipofisario». Las lesiones hipofisarias de pequeño tamaño detectadas de este modo son bastante frecuentes. En los pacientes que se someten a una resonancia magnética (RM) por motivos ajenos a la hipófisis, se observan microadenomas en el 10-38% de los casos, mientras que en el 0,2% se detectan macroadenomas. La mayoría de los incidentalomas hipofisarios son adenomas hipofisarios no funcionales benignos; sin embargo, en un pequeño porcentaje pueden ser quistes de la bolsa de Rathke, craneofaringiomas o meningiomas. En los pacientes con antecedentes de cáncer, debe considerarse la posibilidad de que se trate de una enfermedad metastásica. Los adenomas hipofisarios que miden ≥1 cm son macroadenomas; 30 los que miden >1 cm son microadenomas (véase «Evaluación de los tumores hipofisarios»). PUNTO CLAVE • La mayoría de las masas hipofisarias detectadas de manera incidental son adenomas hipofisarios no funcionales benignos; sin embargo, en un pequeño porcentaje pueden ser quistes de la bolsa de Rathke, craneofaringiomas o meningiomas. Silla turca vacía La silla turca vacía también suele ser un hallazgo incidental en las pruebas de imagen obtenidas por motivos no relacionados con la hipófisis. No se trata tanto de un trastorno médico como de un hallazgo radiológico. Este término se usa cuando la silla turca está hipertrofiada y el tejido hipofisario no la llena del todo. La glándula no puede visualizarse o es extremadamente pequeña. La silla turca vacía primaria es consecuencia de una hernia del espacio subaracnoideo en la silla turca que comprime la hipófisis normal. La silla turca vacía primara tiene su origen en la incompetencia del diafragma selar, una presión intracraneal elevada o alteraciones volumétricas de la hipófisis (como puede suceder en el embarazo, especial- Trastornos de la hipófisis mente en las mujeres multíparas). La silla turca vacía puede deberse a un infarto de un tumor hipofisario o a otras causas, entre las que pueden incluir infección, enfermedad autoinmune, traumatismo o radioterapia. Por lo general, los pacientes en los que se detecta la silla turca vacía tienen una función hipofisaria normal, porque la glándula está presente, pero se encuentra recubriendo la silla turca agrandada como la cáscara de una naranja. Todos los pacientes con silla turca vacía deben someterse a evaluaciones clínicas para detectar signos y síntomas de déficits hipofisarios. La hiperprolactinemia, la alteración hipofisaria más frecuente en caso de silla turca vacía, puede tratarse con una terapia con agonistas de la dopamina en caso necesario. En los pacientes asintomáticos debe realizarse un cribado con medición del nivel de cortisol a las 8:00 horas, así como una medición de la TSH y la tiroxina (T4). En caso de signos o síntomas de déficit, es necesario llevar a cabo otras pruebas dirigidas a los ejes hipofisarios. Es improbable que los pacientes sin anomalías iniciales experimenten alteraciones hormonales o radiológicas. No obstante, dado el riesgo teórico de progresión, se recomienda que los pacientes asintomáticos con silla turca vacía repitan la evaluación endocrina, radiológica y oftalmoscópica al cabo de 24 o 36 meses. En caso de ausencia de progresión, las evaluaciones posteriores pueden limitarse a quienes las necesiten clínicamente. Tabla 17. PUNTOS CLAVE • La silla turca vacía se diagnostica cuando la silla turca está hipertrofiada y el tejido hipofisario no la llena del todo; todos los pacientes con silla turca vacía deben someterse a una evaluación clínica para detectar signos y síntomas de déficits hipofisarios. Otras alteraciones Otros procesos patológicos también pueden afectar a la hipófisis, como pueden ser las enfermedades autoinmunes, las infecciones, las infecciones infiltrativas, la enfermedad metastásica o el infarto (Tabla 17). Alteraciones inducidas por fármacos Hay varios fármacos que pueden afectar a la función de la hipófisis. Toda hormona administrada exógenamente tiene efectos negativos en las células normales de la hipófisis. El estrógeno o la testosterona exógenos inhibirán las gonadotropinas, la LH y la FSH, mientras que el exceso de hormona tiroidea exógena inhibirá la TSH. De igual modo, las dosis fisiológicas y suprafisiológicas de glucocorticoides inhibirán la ACTH. Los opiáceos ejercen varios efectos. Destaca sobre todo el uso crónico de opiáceos, que inhibe la función gonadotrófica, provoca hipogonadismo hipogonadotrófico y constituye una causa cada vez más conocida de déficit de ACTH. Otras alteraciones de la hipófisis Patología Causa Efecto de masa Anomalías hormonales Contexto clínico Autoinmune Posible La más frecuente es el déficit de ACTH Embarazo, posparto Metástasis de tumor maligno en la hipófisis Disfunción del nervio óptico en el 24% La diabetes insípida es la más frecuente Puede cursar con parálisis de los pares craneales y cefaleas También puede cursar con déficits de la hipófisis anterior Pacientes con los siguientes cánceres malignos: mama (el más frecuente), pulmón, linfoma, células renales Tumor hipofisario maligno A menudo la presentación inicial es un macroadenoma En la mayoría de los casos hay cierta hipersecreción hipofisaria y no un déficit Pacientes con adenoma hipofisario con invasión significativa del seno cavernoso o diseminación supraselar Sarcoidosis Infiltración hipofisaria Posible Combinación de anomalías de la hipófisis anterior y posterior Presente hasta en el 10% de los pacientes con sarcoidosis Hemocromatosis Depósito de hierro No Déficit de gonadotropina Mutación del gen HFE Histiocitosis de células de Langerhans Depósito de células de Langerhans en el interior de la hipófisis No Diabetes insípida Rara vez se da en adultos Inflamación Hipofisitis linfocítica Cáncer Metástasis hipofisaria Carcinoma hipofisario (raro) 0,1% de todos los tumores hipofisarios Infiltrativas Menos probablemente déficits de la hipófisis anterior ACTH: corticotropina. 31 Trastornos de la hipófisis Una clase de fármacos relativamente nueva, la de los anticuerpos bloqueadores de puntos de control inmunitario, se ha asociado a alteraciones hipofisarias relacionadas con hipofisitis. Estos fármacos, entre los que se incluyen el nivolumab, el ipilimumab, el tremelimumab y el pembrolizumab, se utilizan para el tratamiento del melanoma metastásico, el carcinoma de células renales, el cáncer de pulmón no microcítico y los cánceres de cabeza y cuello. La hipofisitis se da en entre el 0,5% y el 5% de los pacientes y suele manifestarse con cefalea y fatiga. Normalmente, la evaluación endocrina revela insuficiencia suprarrenal secundaria (déficit de ACTH) e hipotiroidismo secundario (déficit de TSH), así como niveles bajos de LH, GH y prolactina. Las pruebas de imagen ponen de manifiesto potenciación y/o hipertrofia de la hipófisis con engrosamiento del tallo hipofisario. La diabetes insípida es infrecuente. El tratamiento consiste en la reposición de los déficits hormonales junto con la administración de glucocorticoides en dosis altas para tratar el proceso inflamatorio. A pesar de la resolución de la inflamación, es frecuente que persistan los déficits hormonales. Efectos de masa de los tumores hipofisarios En la mayoría de los casos, los efectos de masa de los tumores hipofisarios incluyen compresión de la hipófisis, que produce déficits hormonales, o compresión del quiasma óptico, que en la mayoría de los casos da lugar a hemianopsia bitemporal; también son posibles otros patrones de pérdida visual. La compresión del tallo hipofisario puede provocar hiperprolactinemia. Las cefaleas pueden ser síntoma de tumor hipofisario, pero no están bien correlacionadas con el tamaño del tumor. La cefalea sola no constituye una indicación para la cirugía. Los déficits hipofisarios relacionadas con la compresión de la glándula pueden ir del déficit de una hormona aislada, en la mayoría de los casos la gonadotropina, al panhipopituitarismo (déficit de todas las hormonas de la hipófisis anterior). De igual modo, los tumores hipofisarios pueden tener efectos variables en la compresión de las estructuras circundantes. Un tumor hipofisario que crece o se expande con rapidez debido a apoplejía hipofisaria (hemorragia súbita o infarto de un adenoma hipofisario) y comprime el quiasma óptico puede dar lugar a hemianopsia bitemporal completa o incluso ceguera. La apoplejía hipofisaria puede provocar incluso parálisis de los pares craneales III, IV y VI, mientras que un tumor hipofisario de crecimiento lento que contacta con el quiasma óptico puede producir una pérdida de visión periférica mínima o inexistente. Todos los pacientes con tumores hipofisarios que tocan o comprimen el quiasma óptico deben someterse a una evaluación oftalmológica (preferentemente por un neurooftalmólogo). Una anomalía en la exploración visual constituye una indicación para cirugía, a menos que el tumor sea un prolactinoma. Los tumores hipofisarios pueden invadir el seno cavernoso, pero rara vez provocan un efecto de masa en el tejido cerebral ni un estrechamiento de la carótida dentro del seno cavernoso. 32 PUNTO CLAVE • En la mayoría de los casos, los efectos de masa de los tumores hipofisarios incluyen compresión de la hipófisis, que produce déficits hormonales, o compresión del quiasma óptico, que en la mayoría de los casos da lugar a hemianopsia bitemporal; la compresión del tallo puede provocar hiperprolactinemia. Evaluación de los tumores hipofisarios En los pacientes con un tumor hipofisario en la tomografía computarizada (TC), debe obtenerse una RM hipofisaria especial con y sin contraste con cortes dinámicos de la silla turca. Todo tumor que toque o comprima el quiasma óptico requiere una exploración formal del campo visual. Es necesario descartar hipersecreción hipofisaria mediante la medición de la prolactina y el factor de crecimiento insulínico tipo 1 (IGF-1). En los pacientes sin signos o síntomas de exceso de cortisol no hace falta una evaluación para detectar enfermedad de Cushing. Los tumores hipofisarios también pueden provocar hipopituitarismo. Se recomienda un cribado del hipopituitarismo en todos los tumores hipofisarios, independientemente de los síntomas, con medición de FSH, LH, cortisol, TSH, T4 libre y, adicionalmente, testosterona total en los varones. En las mujeres premenopáusicas es posible evaluar el hipogonadismo hipogonadotrófico mediante el historial menstrual. Los antecedentes de oligomenorrea o amenorrea serían indicativos de hipogonadismo hipogonadotrófico y requieren más pruebas hormonales, mientras que los antecedentes de menstruaciones normales descartarían esencialmente el hipogonadismo hipogonadotrófico. Un resultado anómalo basal puede dar lugar a la realización de otras pruebas estimuladoras para confirmar el hipopituitarismo (Tabla 18); se recomienda la derivación a un endocrinólogo. Si un paciente no necesita una intervención quirúrgica por efecto de masa o hipersecreción hipofisaria, debe repetirse la evaluación hormonal y mediante pruebas de imagen de la hipófisis en el plazo de seis meses en caso de macroadenoma y posteriormente cada año si no hay cambios. Los microadenomas deben reevaluarse con pruebas de imagen en el plazo de un año y posteriormente cada uno o dos años. En los microadenomas, si las pruebas iniciales son normales y no ha habido cambios clínicos ni en la RM de hipófisis, no es necesario repetir la evaluación de la función hipofisaria. PUNTOS CLAVE • Es necesario descartar hipersecreción hipofisaria mediante la medición de la prolactina y el factor de crecimiento insulínico tipo 1; en los pacientes sin signos ni síntomas de exceso de cortisol no hace falta una evaluación para detectar enfermedad de Cushing. (Continúa en página siguiente) AMAV Trastornos de la hipófisis Tabla 18. Pruebas dinámicas para la disfunción hipofisaria Indicación Prueba Técnica Interpretación Déficit de ACTH (cortisol) Prueba de estimulación con ACTH Medir el nivel de cortisol sérico basal Un nivel de cortisol sérico >18 µg/dl (496,8 nmol/l) en cualquier medición indica una respuesta normal Administrar 250 µg de ACTH sintética IM o IV Medir los niveles de cortisol a los 30 y 60 minutos Déficit de ADH (DI) Prueba de privación de agua, seguida de prueba de desmopresina, si está indicado El paciente vacía la vejiga y se mide el peso basal Interpretación de la prueba de privación de agua: Medir el volumen de orina y la osmolalidad cada hora Una osmolalidad urinaria >750-800 mOsm/kg H2O es una repuesta normal a la privación de agua e indica que la producción de ADH y el efecto periférico están intactos Medir el sodio sérico, la osmolalidad y el peso cada 2 horas La prueba se detiene en cualquiera de las circunstancias siguientes: Osmolalidad urinaria/osmolalidad plasmática >2 El paciente ha perdido >3% del peso corporal Una osmolalidad sérica >295 mOsm/kg H2O y/o un sodio sérico >145 mEq/l (145 mmol/l) con una orina indebidamente diluida (osmolalidad urinaria/osmolalidad plasmática <2) es diagnóstica de DI La osmolalidad urinaria permanece estable durante 2-3 h, mientras que la osmolalidad sérica aumenta Osmolalidad plasmática >295 mOsm/kg H2O Sodio sérico >145 mEq/l (145 mmol/l) Prueba de desmopresina: Si osmolalidad urinaria final <600 mOsm/kg, osmolalidad sérica >295 mOsm/kg H2O o sodio sérico >145 mEq/l (145 mmol/l): administrar 2 µg de desmopresina por vía subcutánea Medir la osmolalidad urinaria y el volumen de orina cada hora durante 4 horas después de la desmopresina. Exceso de hormona del crecimiento (acromegalia) Prueba de tolerancia a la glucosa Prueba de tolerancia a la glucosa oral con 75 g glucosa. Medir la glucosa y la GH a los 0, 30, 60, 90, 120 y 150 minutos Interpretación de la prueba de desmopresina: Una osmolalidad urinaria >800 mOsm/kg después de la desmopresina es compatible con DI central completa La ausencia de elevación de la osmolalidad urinaria (sigue <300 mOsm/kg) es diagnóstica de DI nefrogénica completa Una osmolalidad urinaria entre 300 y 800 mOsm es compatible con DI parcial Una GH <0,2 ng/ml (0,2 µg/l) es una respuesta normal. Un nadir de GH ≥1,0 ng/ml (1,0 µg/l) (o ≥0,3 ng/ml [0,3 µg/l] en un ensayo ultrasensible) es diagnóstico de acromegalia ACTH: corticotropina; ADH: hormona antidiurética; DI: diabetes insípida; GH: hormona del crecimiento. P untos clave (continuación) AMAV • Los antecedentes de oligomenorrea o amenorrea serían indicativos de hipogonadismo hipogonadotrófico y requieren más pruebas hormonales, mientras que los antecedentes de menstruaciones normales descartarían esencialmente el hipogonadismo hipogonadotrófico. Tratamiento de los tumores hipofisarios clínicamente no funcionales Los pacientes con un tumor hipofisario no funcional deben ser derivados a una evaluación neuroquirúrgica en presencia de cualquiera de estos signos o síntomas: déficits visuales relacionados con el tumor, una lesión que toque o comprima el quiasma o los nervios ópticos en la RM de hipófisis o apoplejía hipofisaria con alteración visual. Además, debe considerarse la cirugía en los tumores de crecimiento clínicamente significativo, como en el caso de un crecimiento hacia el quiasma óptico, y en los pacientes con nueva pérdida de la función endocrina. En las mujeres con un macroadenoma cercano al quiasma óptico que tengan previsto quedarse embarazadas puede ser beneficiosa la descompresión quirúrgica del tumor hipofisario, dado el riesgo de crecimiento durante el embarazo. Los microadenomas rara vez aumentan de tamaño durante el em33 Trastornos de la hipófisis barazo. La estrategia quirúrgica más frecuente para estos tumores es el abordaje transesfenoidal a través de los orificios nasales o la boca. En ocasiones, es necesaria una craneotomía para tumores muy grandes. La mayor parte de los macroadenomas no funcionales mostrarán una inmunocitoquímica compatible con adenoma gonadotrópico y son clínicamente «silentes» (sin hipersecreción de gonadotropinas funcionales). Tabla 19. Causas de hipopituitarismo Neoplásicas Adenoma hipofisario Meningioma Craneofaringioma Cáncer metastásico PUNTOS CLAVE Linfoma • Los pacientes con un tumor hipofisario no funcional deben ser derivados a una evaluación neuroquirúrgica si hay déficits visuales relacionados con el tumor, si una lesión toca o comprime el quiasma o los nervios ópticos en la RM de hipófisis o ante una apoplejía hipofisaria con alteración visual. Enfermedad infiltrativa • Debe considerarse la cirugía en los tumores de crecimiento clínicamente significativo, como en el caso de un crecimiento que podría afectar a la visión, y en los pacientes con nueva pérdida de la función endocrina. Sarcoidosis Hemocromatosis Histiocitosis de células de Langerhans Inflamación Hipofisitis linfocítica Iatrogénicas Cirugía Radioterapia Déficit hormonal hipofisario El hipopituitarismo se define como el déficit de una o varias hormonas hipofisarias. Puede producirse como consecuencia de la compresión de las células de la hipófisis normal por parte de un tumor o ser una complicación de la cirugía o la radioterapia craneal. Las células somatotropas y las gonadotropas parecen ser las más sensibles al daño, por cuyo motivo el déficit de GH, además de los de LH y FSH, constituyen los déficits hipofisarios más habituales. El déficit de ACTH y el de TSH son menos frecuentes, pero más graves. La apoplejía hipofisaria y el síndrome de Sheehan (infarto de hipófisis asociado a hemorragia puerperal) pueden provocar hipopituitarismo potencialmente mortal debido a déficit de ACTH. En la Tabla 19 se presenta la lista completa de las causas de hipopituitarismo. Panhipopituitarismo El panhipopituitarismo se presenta cuando un paciente tiene una producción insuficiente de todas las hormonas de la hipófisis anterior, por lo general debido a un tumor de gran tamaño o a las complicaciones de una cirugía de hipófisis. Los pacientes con panhipopituitarismo requieren la reposición diaria de tiroxina y cortisol. La reposición de esteroides sexuales y GH es individualizada y se basa en la situación clínica y en la evaluación de los riesgos y beneficios del tratamiento. Los pacientes con panhipopituitarismo deben llevar consigo una tarjeta de alerta médica, porque el déficit de glucocorticoides puede ser potencialmente mortal. Déficit de corticotropina (déficit secundario de cortisol) La causa más frecuente de déficit de ACTH es iatrogénica y radica en la administración de glucocorticoides exógenos y en 34 Déficits congénitos Vasculares Infarto de hipófisis Apoplejía hipofisaria Silla turca vacía Enfermedad hipotalámica Lesión cerebral traumática Medicamentos Opiáceos Inhibidores de puntos de control inmunitario (nivolumab, ipilimumab, tremelimumab, pembrolizumab) la inhibición de la producción de ACTH. Los glucocorticoides orales, los inyectables (intraarticulares, intramusculares) e incluso, ocasionalmente, los tópicos pueden inhibir la ACTH. Los glucocorticoides atenúan la recuperación de la producción de ACTH endógena, pero rara vez provocan directamente la inhibición de la producción de ACTH. Los pacientes con insuficiencia suprarrenal iatrogénica tienen intactos los sistemas de la renina y la aldosterona y presentan un menor riesgo de hipotensión y crisis suprarrenal. En caso de prescripción de glucocorticoides a dosis suprafisiológicas durante tres semanas o más, es necesario disminuir la dosis de manera progresiva para que el eje hipófiso-suprarrenal pueda recuperarse. Una vez que la dosis de glucocorticoides esté próxima a la fisiológica (equivalente a 15-20 mg de hidrocortisona), deben sustituirse por hidrocortisona. A continuación, la dosis puede reducirse en 5 mg cada una o dos semanas, según se tolere. La administración solo por la mañana puede facilitar la recuperación del eje suprarrenal. Trastornos de la hipófisis Una vez que se utilice hidrocortisona fisiológica solo por la mañana, pueden realizarse pruebas para comprobar que el eje suprarrenal se ha recuperado. Un nivel de cortisol a las 8:00 horas superior a 10 µg/dl (276 nmol/l) después de la retirada de los glucocorticoides durante 24 horas es indicativo de recuperación del eje hipófiso-suprarrenal. Esta debe confirmarse con una prueba de estimulación con ACTH. A pesar de la recuperación del eje hipófiso-suprarrenal, puede que los pacientes tarden más tiempo en ser capaces de responder al estrés y que durante un año necesiten dosis de glucocorticoides para las situaciones de estrés o los días de malestar en caso de enfermedad. El déficit de ACTH puede darse también en el contexto de daño hipofisario. Los síntomas de déficit secundario de cortisol pueden ser fatiga, malestar general, pérdida de peso, náuseas, vómitos, hipoglucemia asintomática, mareo e hiponatremia. Dado que solo está afectada la producción de cortisol (la producción de mineralocorticoides está intacta), los pacientes no experimentan hiperpotasemia y tienen menos probabilidades de sufrir hipotensión. Además, los pacientes con insuficiencia suprarrenal secundaria no presentan hiperpigmentación. No obstante, los pacientes con insuficiencia suprarrenal secundaria necesitan reposición de glucocorticoides y dosis de estrés durante la enfermedad. Los niveles matinales de cortisol <3 µg/dl (82,8 nmol/l) son diagnósticos de déficit de cortisol; por el contrario, un nivel de cortisol matinal >15 µg/dl (414 nmol/l) probablemente lo descarta. Los pacientes con niveles de cortisol de entre 3 y 15 µg/dl (82,8-414 nmol/l) deben someterse a la prueba de estimulación con ACTH (véase la Tabla 18). Un nivel máximo de cortisol ≥18 µg/dl (496,8 nmol/l) a 0, 30 o 60 minutos descarta el déficit de cortisol. Una vez diagnosticada, la insuficiencia suprarrenal secundaria debe tratarse con hidrocortisona a entre 15 y 20 mg en dos dosis divididas, por ejemplo, 10-15 mg por la mañana y 5 mg por la tarde. En situación de emergencia, como puede ser una apoplejía hipofisaria, debe administrarse una dosis intravenosa inmediata de hidrocortisona de 100 mg. En el contexto de estrés fisiológico o enfermedad aguda, es necesario ajustar la dosis de glucocorticoides. La administración de entre dos y tres veces la dosis basal de reposición de cortisol durante dos o tres días suele ser suficiente para las enfermedades menores o moderadas, incluidas las cirugías menores o moderadas. En los pacientes con estrés fisiológico, incluyendo cirugía mayor o parto activo, deben administrarse 100 mg de hidrocortisona mediante inyección intravenosa, seguidos de la infusión continua de 200 mg cada 24 horas o de 50 mg en inyección intravenosa cada seis horas (véase «Trastornos de las glándulas suprarrenales»). PUNTOS CLAVE AMAV • La causa más frecuente de déficit de corticotropina es iatrogénico y radica en la administración de glucocorticoides exógenos y en la inhibición de la producción de ACTH. • En caso de prescripción de glucocorticoides a dosis suprafisiológicas durante tres semanas o más, es necesario disminuir la dosis de manera progresiva para que el eje hipófiso-suprarrenal pueda recuperarse. • La dosis de glucocorticoides debe ajustarse en el contexto de estrés fisiológico o enfermedad aguda. Déficit de tirotropina El déficit de TSH provoca la incapacidad de la glándula tiroides para producir T4. El resultado es una producción insuficiente de T4 con una TSH baja o inadecuadamente normal. Los síntomas clínicos de hipotiroidismo secundario son los mismos que los que se observan en el hipotiroidismo primario. El tratamiento consiste en la administración de diaria de levotiroxina. La TSH no puede utilizarse para controlar el tratamiento y no debe medirse. Una dosificación basada en el nivel de TSH puede provocar infradosificación. Para controlar la adecuación de la dosis debe tomarse como parámetro la T4 libre, que debe mantenerse en la mitad media o superior del intervalo normal. Aunque para que la TSH refleje con exactitud el estado hormonal tiroideo en el hipotiroidismo primario hacen falta de seis a ocho semanas, en el hipotiroidismo secundario los niveles de T4 libre pueden comprobarse cada dos o tres semanas después de un cambio de dosis para evaluar su adecuación. PUNTO CLAVE • El tratamiento del déficit de tirotropina consiste en la administración diaria de levotiroxina; para controlar la adecuación de la dosis solo puede utilizarse la tiroxina libre, que debe mantenerse en la mitad media o superior del intervalo normal. Déficit de gonadotropinas El déficit de gonadotropinas puede deberse a enfermedad hipofisaria o a un déficit de hormona liberadora de gonadotropinas (GnRH) como el que se observa en el síndrome de Kallmann y la amenorrea hipotalámica. Algunos fármacos, entre los que se encuentran los opiáceos, también pueden inhibir la GnRH. El déficit de gonadotropinas, LH y FSH, da lugar a déficit de hormonas sexuales masculinas y femeninas. La combinación de un nivel bajo o inadecuadamente normal de LH y FSH junto con esteroides sexuales bajos se denomina hipogonadismo «central» o «hipogonadotrófico». Por lo general, en las personas sin contraindicaciones que no desean tener hijos, el tratamiento del hipogonadismo hipogonadotrófico puede consistir en la reposición de los esteroides sexuales; en los varones se utiliza un tratamiento con testosterona, mientras que en las mujeres se recurre a un tratamiento combinado con estrógeno y progesterona. Si bien las píldoras anticonceptivas orales pueden ser más aceptables para este fin en las mujeres jóvenes, en algunos casos pueden ser preferibles otras formas de estrógeno y progesterona (como los parches de estradiol y los ciclos de progesterona oral). En 35 Trastornos de la hipófisis los varones y mujeres que desean tener hijos, es necesario reponer las gonadotropinas, porque la testosterona y el estrógeno exógenos inhiben la espermatogénesis en los varones y la ovulación en las mujeres, respectivamente. Déficit de hormona del crecimiento La GH es necesaria para un crecimiento lineal. El déficit de GH en los niños da lugar a una estatura reducida. Los síntomas de déficit de GH en los adultos son más sutiles e incluyen fatiga, pérdida de masa muscular y aumento del cociente tejido graso/masa magra. Mientras que el déficit de GH aislado puede darse en niños, el déficit de GH aislado idiopático en adultos es bastante raro. La evaluación de un posible déficit de GH aislado de inicio en la etapa adulta solo debe considerarse en los pacientes con antecedentes de enfermedad hipotalámica o hipofisaria, cirugía o radioterapia en esas regiones, traumatismo craneal u otros déficits de hormonas hipofisarias. Dada la naturaleza pulsátil de la GH, su medición directa no puede interpretarse, por lo que el déficit de GH debe evaluarse mediante la medición del IGF-1. Un nivel de IGF-1 por debajo del intervalo normal para el sexo y la edad es altamente indicativo de déficit de GH, mientras que un nivel normal de IGF-1 no descarta del todo el déficit de GH cuando la sospecha pretest es alta. Las pruebas de provocación, como la prueba de tolerancia a la insulina o la prueba de la GnRH-arginina, pueden llevarse a cabo en interconsulta con un endocrinólogo para establecer el diagnóstico de déficit de GH en el adulto. Los beneficios del tratamiento en las personas con déficit de GH incluyen la mejora de la capacidad de ejercicio, la composición corporal y la densidad ósea. La decisión de iniciar la reposición de GH debe ser individualizada. En caso de cáncer o en pacientes con un tumor hipofisario no tratado, está contraindicada, debido al potencial de estimulación del crecimiento tumoral. Además, en las personas con diabetes mellitus debe actuarse con precaución, ya que puede empeorar la hiperglucemia. Cuando el tratamiento está indicado en adultos, la GH puede remplazarse por una inyección diaria de dosis bajas ajustadas según el nivel normal de IGF-1 y la evaluación clínica. PUNTO CLAVE AMAV • El déficit de hormona del crecimiento aislado idiopático en el adulto es infrecuente; solo debe evaluarse en los adultos con antecedentes de enfermedad hipotalámica o hipofisaria, cirugía o radioterapia en esas regiones, traumatismo craneal u otros déficits de hormonas hipofisarias. frecuente en los pacientes con el mecanismo de la sed intacto y acceso libre al agua, esta situación puede ser grave si los pacientes no pueden beber cuando tienen sed. Una osmolalidad urinaria inadecuadamente baja en el contexto de osmolalidad sérica elevada e hipernatremia en un paciente con poliuria (>50 ml/kg/24 horas en ausencia de glucosuria) es diagnóstica de DI. En caso de duda diagnóstica, puede realizarse una prueba de privación de agua (véase la Tabla 18). La DI se trata con la administración de desmopresina (DDAVP) por vía intranasal, oral o subcutánea. La biodisponibilidad de la DDAVP oral es mucho menor y las dosis son considerablemente más altas que con las vías intranasal y subcutánea. Aun así, las preparaciones orales pueden ser preferibles en determinadas circunstancias, ya que la absorción intranasal de DDAVP puede cambiar en caso de alteraciones de la mucosa nasal. Normalmente, las dosis se administran una vez por la noche, para evitar la nicturia y garantizar un sueño ininterrumpido, o dos veces al día, si los síntomas interfieren en el funcionamiento diario. Es necesario actuar con precaución para evitar una reposición excesiva, ya que esta puede provocar hiponatremia, hiperhidratación hipotónica y sobrecarga de volumen. PUNTOS CLAVE • Una osmolalidad urinaria inadecuadamente baja en el contexto de osmolalidad sérica elevada e hipernatremia en un paciente con poliuria (>50 ml/kg/24 horas en ausencia de glucosuria) es diagnóstica de diabetes insípida. • La diabetes insípida se trata con la administración de desmopresina (DDAVP) por vía intranasal, oral o subcutánea; es necesario actuar con precaución para evitar una reposición excesiva, ya que esta puede provocar hiponatremia, hiperhidratación hipotónica y sobrecarga de volumen. Exceso de hormonas hipofisarias Los tumores hipofisarios se consideran funcionales cuando secretan hormonas hipofisarias en exceso. Los tumores hipofisarios más frecuentes son los prolactinomas. Aunque los tumores hipofisarios productores de ACTH o GH son menos frecuentes, es importante reconocerlos, debido a sus consecuencias clínicas. Los adenomas secretores de TSH son una causa muy poco frecuente de hipertiroidismo. Los tumores hipofisarios rara vez secretan más de una hormona en exceso al mismo tiempo. La cosecreción se da sobre todo con la GH y la prolactina. Diabetes insípida central Hiperprolactinemia y prolactinoma La incapacidad de la hipófisis posterior de producir una cantidad suficiente de ADH provoca diabetes insípida (DI) central. La ausencia de ADH (ID completa) y la ADH reducida (DI parcial) impiden la reabsorción de agua en los riñones y dan lugar a poliuria y polidipsia. Aunque la hipernatremia es in- La causa más frecuente de hiperprolactinemia es fisiológica y está relacionada con el embarazo y la lactancia. Otras causas no patológicas de hiperprolactinemia leve son estrés fisiológico, coito, sueño y estimulación de los pezones. En la Tabla 20 se presenta una lista exhaustiva de las causas de hiperprolac- 36 Trastornos de la hipófisis Tabla 20. Causas de hiperprolactinemia Fisiológicas Medicación Otras Coito Antipsicóticos Ejercicio Antipsicóticos típicos Traumatismo de la pared torácica Lactancia Clorpromazina Estimulación de los pezones Flufenazina Enfermedad renal crónica Haloperidol Cirrosis Proclorperazina Cocaína Antipsicóticos atípicos Síndrome de la silla turca vacía Embarazo Sueño Estrés Amisulprida Olanzapina (raras veces) Paliperidona Risperidona Herpes zóster Síndrome del ovario poliquístico Ziprasidona (raras veces) Prolactinoma ISRS Hipotiroidismo grave Citalopram Escitalopram Fluoxetina Convulsiones Compresión del tallo hipofisario Paroxetina Sertralina Antihipertensivos Metildopa Verapamilo Otros Cimetidina Domperidona Estrógeno Metoclopramida Opiáceos ISRS: inhibidor selectivo de la recaptación de serotonina. tinemia. Los síntomas de hiperprolactinemia incluyen amenorrea y a veces galactorrea, en las mujeres premenopáusicas. En los varones suele manifestarse más tarde con síntomas de efecto de masa o hipogonadismo, como disminución de la libido o problemas de erección; con menor frecuencia experimentan ginecomastia y sensibilidad mamaria. La causa más frecuente de hiperprolactinemia patológica no tumoral son los medicamentos. De los pacientes que toman antipsicóticos típicos (véase la Tabla 20), entre el 40% y el 90% experimentarán hiperprolactinemia causada por el efecto antagonista de la dopamina de estos medicamentos. Aunque en la mayoría de los casos la hiperprolactinemia inducida por medicamentos da lugar a niveles de prolactina de entre 25 y 100 ng/ml (25-100 µg/l), fármacos como la metoclopramida, la risperidona y las fenotiazinas pueden provocar niveles de prolactina superiores a 200 ng/ml (200 µg/l). Confirmar que la hiperprolactinemia está relacionada con medicamentos puede ser complicado. Cuando sea posible, el medicamento responsable debe retirarse durante tres días para saber si los niveles de prolactina vuelven a la normalidad. Los fármacos psicotrópicos solo pueden suspenderse en interconsulta con el psiquiatra del paciente. Si no es posible retirar el medicamento y la elevación de la prolactina no puede relacionarse con el momento del inicio del fármaco, deberá obtenerse una RM de hipófisis para descartar un prolactinoma. Idealmente, la hiperprolactinemia inducida por antipsicóticos se trata en interconsulta con el psiquiatra del paciente cambiando a un fármaco con menos probabilidades de provocar hiperprolactinemia. Aunque la hiperprolactinemia asintomática relacionada con medicamentos no requiere tratamiento, los pacientes con hipogonadismo deben recibir estrógeno o testosterona para preservar la masa ósea. El tratamiento de la hiperprolactinemia inducida por medicamentos con un agonista de la dopamina (cabergolina o bromocriptina) es motivo de controversia, ya que puede agravar la psicosis. En todos los pacientes con hiperprolactinemia de origen desconocido está indicada la obtención de una RM. Seguidamente, la evaluación y las decisiones terapéuticas se basan en el nivel de prolactina y los hallazgos de la RM. Un nivel de prolactina por encima de 500 ng/ml (500 µg/l) es diagnóstico de macroprolactinoma. Aunque los niveles superiores a 250 ng/ml (250 µg/l) son indicativos de macroprolactinoma, hay algunos medicamentos que pueden elevar la prolactina hasta ese nivel. Por lo general, los niveles de prolactina están correlacionados con el tamaño del tumor. Por lo tanto, cuando hay un macroadenoma con un nivel de prolactina inferior a 100 ng/ml (100 µg/l) debe sospecharse que la causa de la hiperprolactinemia es la compresión del tallo hipofisario por un tumor no funcional y no el prolactinoma. Los pacientes con microadenomas asintomáticos no requieren tratamiento. Las mujeres con hipogonadismo relacionado con un microadenoma pueden ser tratadas con un anticonceptivo oral combinado si no desean tener hijos o con un agonista de la dopamina en caso contrario. Las mujeres posmenopáusicas con microadenomas no requieren tratamiento. En los pacientes con macroadenomas, se recomienda un tratamiento con agonistas de la dopamina para reducir el nivel de prolactina, reducir el tamaño del tumor y recuperar la función gonadal. El fármaco de elección es la cabergolina, debido a que tiene una mayor eficacia para reducir la prolactina y reducir el tumor que la bromocriptina. Además, la cabergolina se administra dos veces por semana, mientras que la bromocriptina se debe tomarse entre una y tres veces al día. Puede llevarse a cabo un control de la prolactina de dos a cuatro semanas después del tratamiento y posteriormente cada tres o cuatro meses una vez esté estable. La RM por microadenoma debe repetirse en el plazo de un año. Si tanto el tumor como la prolactina permanecen estables en el seguimiento a un año, no hacen falta más pruebas de imagen. En caso de macroadenoma, las pruebas de imagen deben repetirse tres meses después 37 Trastornos de la hipófisis del tratamiento médico y cada 6-12 meses una vez que se confirme que está estable. Si el nivel de prolactina aumenta a pesar del tratamiento, deben repetirse las pruebas de imagen. La cirugía no es el tratamiento de primera línea, porque hasta el 50% de los prolactinomas recidivan después de la resección. La cirugía solo debe plantearse en los prolactinomas de pacientes sintomáticos que no toleran el tratamiento con agonistas de la dopamina o cuyos tumores no encogen o incluso crecen durante el mismo. PUNTOS CLAVE AMAV • La causa más frecuente de hiperprolactinemia es fisiológica y está relacionada con el embarazo y la lactancia. AMAV • La causa más frecuente de hiperprolactinemia patológica no tumoral son los medicamentos. AMAV • Los pacientes con microadenomas asintomáticos no requieren tratamiento; en los pacientes con macroadenomas, se recomienda un tratamiento con agonistas de la dopamina para reducir el nivel de prolactina, reducir el tamaño del tumor y recuperar la función gonadal. AMAV • La cirugía no es el tratamiento de primera línea, porque hasta el 50% de los prolactinomas recidivan después de la resección. es fácil de reconocer en niños, las manifestaciones del exceso de GH son más sutiles en los adultos y con frecuencia pasan inadvertidas durante muchos años. En la Tabla 21 se hallará una lista de las manifestaciones clínicas de la acromegalia. En caso de presunta acromegalia debe realizarse un cribado mediante la obtención del nivel de IGF-1. En los pacientes con un nivel elevado, debe llevarse a cabo una PTGO para confirmar el diagnóstico (véase la Tabla 18). Un nivel por encima de 1 ng/ml (1 µg/l) confirma el diagnóstico de acromegalia. Una vez demostrada la existencia de un exceso de GH, debe obtenerse una RM de la hipófisis. La resección transesfenoidal (RTE) del tumor secretor de GH constituye el puntal del tratamiento. Los pacientes que no logran la remisión con la cirugía pueden recibir tratamiento médico y/o radioterapia estereotáctica. Los medicamentos de elección son los análogos de la somatostatina, ya que dan lugar a la reducción del tamaño tumoral, así como a un descenso de los niveles de GH. El pegvisomant, un antagonista de los receptores de GH, puede usarse en combinación con un análogo de la somatostatina en caso necesario; a veces puede usarse también la cabergolina. En algunos casos se utiliza la radioterapia estereotáctica (cuchillo gamma). Una vez lograda la remisión, se realiza un seguimiento anual mediante RM y medición de los niveles de IGF-1. Los pacientes con acromegalia pueden presentar una mortalidad elevada debido a enfermedad cardíaca, apnea del Prolactinomas y embarazo Debido a la hiperplasia lactotropa del embarazo, el crecimiento de los prolactinomas es motivo de inquietud en el embarazo. Dada la improbabilidad de que los microadenomas crezcan durante el embarazo, el tratamiento con agonistas de la dopamina debe suspenderse cuando se descubre un embarazo. No obstante, las pacientes con macroadenomas sin tratamiento quirúrgico o radioterapia anterior presentan un riesgo significativo de crecimiento del tumor. En estas pacientes puede ser necesaria una citorreducción tumoral quirúrgica antes del embarazo o un tratamiento con agonistas de la dopamina durante el mismo. La bromocriptina es el fármaco de elección durante el embarazo. Las pacientes con macroadenomas deben hacerse controles con exámenes del campo visual cada trimestre, mientras que las que presentan microadenomas pueden ser objeto de controles clínicos. Las cefaleas o alteraciones del campo visual deben dar pie a una RM de la hipófisis sin contraste. Acromegalia En el 95% de los pacientes, la acromegalia está causada por la secreción excesiva de GH con origen en un tumor hipofisario. En menos del 5% de los pacientes con exceso de GH, la causa de la acromegalia es un tumor secretor de hormona liberadora de hormona del crecimiento (GHRH) o un tumor neuroendocrino. Cuando hay tumores hipofisarios secretores de GH en niños antes de la pubertad, el resultado es el aumento del crecimiento longitudinal, que da lugar a gigantismo. Si bien el gigantismo 38 Tabla 21. Manifestaciones de la acromegalia Signos y síntomas clínicos Prognatismo Macroglosia Dientes muy separados (normalmente el primer signo de exceso de hormona del crecimiento) Nariz ancha Manos y pies hipertrofiados e hinchados Sudoración excesiva Acrocordón Dolor articular Cefalea Asociaciones patológicas Apnea del sueño Hipertensión Resistencia a la insulina Miocardiopatía hipertrófica Pólipos colónicos y cáncer de colon Nódulos tiroideos y cáncer de tiroides Enfermedad valvular cardíaca Artropatía Síndrome del túnel carpiano Trastornos de la hipófisis sueño y cáncer, pero el riesgo vuelve al nivel basal cuando el IGF-1 se mantiene dentro del intervalo normal. Un cribado adecuado y el tratamiento de las enfermedades concurrentes son tan importantes como el manejo del nivel de IGF-1. PUNTOS CLAVE • En caso de presunta acromegalia debe hacerse un cribado mediante la obtención del nivel de factor de crecimiento insulínico tipo 1; en los pacientes con un nivel elevado, debe llevarse a cabo una prueba de tolerancia a la glucosa oral para confirmar el diagnóstico. • La resección transesfenoidal del tumor secretor de hormona del crecimiento constituye el puntal del tratamiento; no obstante, los análogos de la somatostatina reducen el tamaño del tumor, así como los niveles de hormona del crecimiento, en quienes no logran la remisión con cirugía. Tumores secretores de tirotropina Los tumores hipofisarios secretores de TSH son extremadamente infrecuentes. Los signos y síntomas de adenoma secretor de TSH son los que se observan en el hipertiroidismo, aunque la evaluación analítica revela niveles elevados de T4 y triyodotironina (T3) con un nivel de TSH inadecuadamente normal o elevado. Una vez descartadas otras causas de las anomalías analíticas (interferencia en el ensayo tiroideo, resistencia a la hormona tiroidea o hipertiroxinemia disalbuminémica familiar), debe obtenerse una RM de hipófisis. El tratamiento de elección es la RTE del tumor productor de TSH. El tratamiento médico con análogos de la somatostatina puede usarse para controlar el hipertiroidismo antes de la cirugía y después de esta en los pacientes que no logren la remisión. me de Cushing endógeno) es el término que se utiliza para describir el hipercortisolismo con origen en la secreción excesiva de ACTH por un tumor hipofisario. Los síntomas y signos del síndrome de Cushing se presentan en la Tabla 22. Para diagnosticar el síndrome de Cushing, primero hay que demostrar la presencia de hipercortisolismo (véase «Trastornos de las glándulas suprarrenales»). La medición de la ACTH determina si es dependiente o independiente de ACTH. Una vez demostrado el diagnóstico de síndrome de Cushing dependiente de ACTH, debe obtenerse una RM de hipófisis de confirmación. Si no se observa ningún tumor hipofisario o si este tiene un tamaño <6 mm, se lleva a cabo una prueba de inhibición con 8 g de dexametasona para evaluar la presencia de un tumor productor de ACTH ectópico (en la mayoría de los casos carcinoma de pulmón, páncreas o timo), que tiene una gran resistencia a la inhibición con dexametasona. A menudo se recomienda la obtención de muestras de los senos petrosos inferiores antes de la RTE para confirmar que el exceso de ACTH tiene origen hipofisario, debido a la baja sensibilidad y especificidad de la prueba de inhibición con dexametasona con dosis altas. El tratamiento de elección es la RTE del adenoma hipofisario. Por lo general, la remisión se define como un valor de cortisol sérico matinal <5 µg/dl (138 nmol/l) en el plazo de sie- Tabla 22. Síntomas y signos de síndrome de Cushing Síntomas Depresión Fatiga Aumento rápido de peso Disminución de la libido Secreción excesiva de hormona antidiurética Anomalías menstruales El síndrome de secreción inadecuada de hormona antidiurética (SIADH) provoca retención de agua, con la hiponatremia resultante, a menudo grave. Pueden dar lugar a una liberación excesiva de ADH trastornos del sistema nervioso central (SNC) (traumatismo, ictus, metástasis cerebrales, infección), fármacos, enfermedades pulmonares y la cirugía hipofisaria (entre tres y siete días después de la operación). El SIADH es un diagnóstico de exclusión. El tratamiento consiste en la corrección de la patología subyacente, restricción de líquidos, antagonistas de los receptores de vasopresina y solución salina hipertónica en los casos de hiponatremia grave. Si se considera el uso de solución salina hipertónica, se recomienda una interconsulta con un endocrinólogo o un nefrólogo (véase el módulo «Nefrología» de MKSAP 18). Signos Exceso de corticotropina de origen hipofisario (enfermedad de Cushing) El síndrome de Cushing es un término que se utiliza para describir el hipercortisolismo independientemente de su causa; la enfermedad de Cushing (la causa más frecuente de síndro- Estrías (sobre todo si son de un rojo purpúreo y >1 cm de ancho)a Formación de hematomas con facilidada Congestión faciala Debilidad proximal (miopatía proximal) Obesidad abdominal Desgarros cutáneos (secundarios a adelgazamiento de la epidermis) Acné Hirsutismo Almohadilla de grasa dorsocervical (joroba de búfalo)a Almohadilla de grasa supraclaviculara Hipopotasemia Hipertensión Diabetes aLos signos más diferenciadores del síndrome de Cushing de la población general. 39 Trastornos de las glándulas suprarrenales Tabla 23. Manejo farmacológico para el tratamiento del síndrome de Cushing Inhibidores de la esteroidogénesis (inhiben la síntesis de colesterol) Dirigidos a la hipófisis (inhiben la secreción de ACTH) Antagonista de los receptores de glucocorticoides (inhibe la acción del cortisol) Ketoconazol Pasireotida Mifepristona Metirapona Cabergolina Mitotano Etomidato ACTH: corticotropina. te días desde la cirugía. Los pacientes requieren la reposición de glucocorticoides después de la operación hasta que las células corticotropas se recuperan de la prolongada inhibición del cortisol. La recuperación puede llevar un máximo de un año, y en ocasiones no hay recuperación, en cuyo caso el paciente necesitará tratamiento de reposición con cortisol durante toda la vida. Si no se consigue la remisión después de la operación, puede que haga falta recurrir a radioterapia o un tratamiento médico (Tabla 23). En raras ocasiones, en los pacientes que no responden a ninguno de los demás tratamientos hace falta una suprarrenalectomía bilateral; estos pacientes necesitarán reposición de glucocorticoides y mineralocorticoides durante toda la vida para la insuficiencia suprarrenal primaria adquirida. Además, debido a la estimulación desmesurada de la producción de ACTH, existe el riesgo de crecimiento del tumor hipofisario después de la suprarrenalectomía (síndrome de Nelson). Los pacientes con enfermedad de Cushing deben ser objeto de seguimiento con pruebas de imagen y bioquímicas (medición del cortisol libre en orina o del cortisol salival nocturno) anualmente durante varios años, y con menor frecuencia después. El primer signo bioquímico de recidiva suele ser los niveles elevados de cortisol salival nocturno. Las recidivas se manejan con la repetición de la RTE, la radioterapia y/o el tratamiento médico. PUNTOS CLAVE • Para diagnosticar el síndrome de Cushing primero hay que demostrar la presencia de hipercortisolismo; una vez confirmada la enfermedad de Cushing dependiente de corticotropina, debe obtenerse una RM de hipófisis. • El tratamiento de elección para la enfermedad de Cushing dependiente de corticotropina es la resección transesfenoidal del adenoma hipofisario, seguida de la reposición de glucocorticoides hasta que las células corticotropas normales se recuperan de la inhibición del colesterol. 40 Trastornos de las glándulas suprarrenales Anatomía y fisiología suprarrenal Aunque cada glándula suprarrenal se considera un órgano único, consta de dos regiones con funciones diferentes: la corteza externa y la médula interna. La corteza secreta hormonas que se clasifican en mineralocorticoides (aldosterona), glucocorticoides (cortisol) y andrógenos (deshidroepiandrosterona [DHEA]), mientras que la médula secreta catecolaminas. La corteza suprarrenal está compuesta de tres zonas: la zona glomerular (externa), la zona fascicular (media) y la zona reticular (interna). La zona glomerular se encarga de la producción de aldosterona, que está regulada por el sistema renina-angiotensina y favorece la reabsorción de sodio y la excreción de potasio a través de los túbulos distales del riñón. La expansión resultante del volumen extracelular aumenta la presión arterial. Además, la aldosterona ejerce efectos inflamatorios y fibróticos directos en otros órganos que son independientes de sus efectos en la presión arterial. Los principales estímulos para la secreción de aldosterona son la hipotensión, la hipovolemia y la hiperpotasemia. La producción de cortisol en la zona fascicular está regulada por la liberación de ACTH de la hipófisis. El cortisol muestra un ritmo diurno diferenciado que se caracteriza por niveles máximos al despertar que descienden hasta niveles muy bajos a la hora de acostarse. A este ritmo diurno se sobreimponen pequeñas oscilaciones de la secreción durante la vigilia. La mayoría del cortisol circula por la sangre unido a la proteína fijadora de cortisol, y solo una pequeña fracción circula en forma de hormona libre biológicamente activa. El cortisol es fundamental para la respuesta adaptativa del organismo al estrés fisiológico, y sus niveles aumentan en respuesta al estrés psicológico, así como a la enfermedad física. Las acciones del cortisol son diversas e incluyen efectos inmunitarios, vasculares, antiinflamatorios y metabólicos. La DHEA, producida en la zona reticular, y su sulfato, el DHEAS, son andrógenos suprarrenales débiles que ejercen sus efectos mediante la conversión periférica a testosterona. En las mujeres, las glándulas suprarrenales representan un factor contribuyente importante a los niveles de andrógenos circulantes. En los varones, la contribución suprarrenal al efecto andrógeno es irrelevante. La médula suprarrenal secreta las catecolaminas noradrenalina y adrenalina en respuesta a la hipotensión, la hipoglucemia, el miedo, la ansiedad, la enfermedad aguda y otras causas de estrés psicológico y físico. Las catecolaminas interactúan con los receptores adrenérgicos α y β para aumentar la frecuencia cardíaca y la presión arterial, relajar el músculo liso, dilatar los bronquiolos e incrementar el ritmo metabólico. Una pequeña fracción de la noradrenalina y la adrenalina se excreta en la orina en forma de hormona libre; el resto se Trastornos de las glándulas suprarrenales degrada en el hígado a metadrenalina y normetadrenalina antes de ser excretado en la orina. Exceso de hormonas suprarrenales Exceso de cortisol (síndrome de Cushing debido a masa suprarrenal) Los adenomas suprarrenales secretores de cortisol y, en raras ocasiones, los carcinomas que también lo producen representan el 20% de las causas endógenas de síndrome de Cushing. La secreción excesiva de cortisol de estos tumores inhibe la producción de ACTH de la hipófisis, lo que da lugar a una forma de síndrome de Cushing clasificada como independiente de la ACTH. El síndrome de Cushing dependiente de ACTH es más frecuente y tiene su origen sobre todo en los adenomas hipofisarios (véase «Trastornos de la hipófisis»). Aunque muchos de los síntomas y signos del síndrome de Cushing son frecuentes en la población general, algunos, incluyendo las almohadillas de grasa supraclavicular, la debilidad muscular proximal, la congestión facial y las estrías violáceas anchas (Figura 3), se consideran más diferenciadores (véase «Trastornos de la hipófisis»). El diagnóstico del síndrome de Cushing es complicado, porque los pacientes presentan un espectro que va desde enfermedad leve con manifestaciones sutiles hasta enfermedad grave potencialmente mortal. Además, también puede darse una hipercortisolemia por estrés psicológico y debido a enfermedad física en ausencia de síndrome de Cushing. En los estados de estrés grave, como depresión mayor, ansiedad, psicosis, diabetes mellitus mal controlada y obesidad visceral grave, puede producirse un estado seudo-Cushing en el que coexiste la hipercortisolemia con manifestaciones clínicas inespecíficas de síndrome de Cushing. No se recomienda una evaluación para detectar síndrome de Cushing en pacientes sin signos específicos de este. La evaluación del síndrome de Cushing implica: 1) pruebas iniciales seguidas de pruebas de confirmación del síndro- F i g u r a 3 . Estrías violáceas anchas observadas en el abdomen de un paciente con síndrome de Cushing. Las estrías con un ancho >1 cm tienen una alta especificidad para el hipercortisolismo. me de Cushing; 2) determinación de si el síndrome de Cushing es independiente o dependiente de la ACTH, y 3) localización del origen de la ACTH en la enfermedad dependiente de la ACTH o confirmación de la presencia de una (o varias) masas suprarrenales en la enfermedad independiente de la ACTH. Es imprescindible confirmar el síndrome de Cushing bioquímico con seguridad antes de buscar su origen, ya que un diagnóstico erróneo puede dar lugar a pruebas y tratamientos innecesarios. Para diagnosticar el síndrome de Cushing hay que combinar y repetir pruebas (Figura 4). La determinación del cortisol sérico matinal o aleatorio no es fiable debido a la superposición de los niveles de cortisol sérico en los pacientes normales, en quienes tienen el síndrome de Cushing y en quienes presentan hipercortisolismo leve/estado seudo-Cushing en ausencia de síndrome de Cushing. Además, los niveles de cortisol no son fiables cuando las proteínas fijadoras están afectadas por el estrógeno oral, la enfermedad aguda o los estados que cursan con niveles bajos de proteínas. Las pruebas iniciales del síndrome de Cushing tienen una exactitud diagnóstica similar e incluyen la medición del cortisol libre en orina de 24 horas, mediciones seriadas de cortisol salival nocturno y la prueba de inhibición con 1 mg de dexametasona por la noche. Cuando el grado de sospecha de síndrome de Cushing es bajo, una única prueba, si es negativa, indica que el síndrome de Cushing es improbable. En caso de un mayor índice de sospecha de síndrome de Cushing, se recomiendan dos pruebas iniciales diferentes. El cortisol libre en orina y el cortisol salival nocturno representan la fracción de cortisol libre en suero y evitan las dificultades en la interpretación relacionadas con las alteraciones de la proteínas fijadora de cortisol. La falsa elevación del cortisol libre en orina puede deberse a hipercortisolemia no relacionada con síndrome de Cushing/estado seudo-Cushing o a presencia de una poliuria significativa (>5 l/día). Los resultados falsos negativos solo pueden darse en caso de enfermedad renal avanzada o en pacientes con tasas variables de secreción de cortisol. La muestra para la determinación del cortisol salival nocturno es recogida por el paciente en su domicilio entre las 23:00 horas y la medianoche en un mínimo de dos noches diferentes. Esta prueba evalúa el ritmo diurno normal de cortisol, que se pierde en el síndrome de Cushing, por lo que el nivel de cortisol no será tan bajo como lo previsto. La prueba no se recomienda en pacientes con trabajo por turnos con un patrón de sueño desigual. El consumo reciente de cigarrillos o la contaminación de la muestra por glucocorticoides tópicos puede elevar falsamente los resultados. La prueba de inhibición con 1 mg (dosis baja) de dexametasona depende del principio de que la secreción de cortisol autonómica no está sujeta a una inhibición en respuesta a los glucocorticoides exógenos. La dexametasona se toma a las 23:00 horas y el cortisol total en suero se mide a las 8:00 horas de la mañana siguiente. Un nivel de cortisol posterior a la dexametasona >5 µg/dl (138 nmol/l) se considera una prueba 41 Trastornos de las glándulas suprarrenales Sospecha clínica de SC Realizar pruebas iniciales: — CLO de 24 ha — PID con un IMG — Cortisol salival Na Descartar el uso de glucocorticoides exógenos Anormal: descartar hipercortisolismo fisiológico Hipercortisolismo fisiológico descartado Anormal: SC confirmado Sospecha de hipercortisolismo fisiológico Otras pruebas: repetir la prueba anormal; realizar otras pruebas positiva. Se ha defendido que un valor de corte del cortisol inferior >1,8 µg/dl (49,7 nmol/l) mejora la sensibilidad de la prueba, pero a costa de una menor especificidad. Los resultados falsos positivos pueden darse con el uso concomitante de medicamentos (carbamacepina, fenitoína y pioglitazona) que inducen enzimas hepáticas CYP3A4 y aceleran el metabolismo de la dexametasona. La medición simultánea de la dexametasona sérica puede confirmar el cumplimiento del paciente o la alteración del metabolismo de la dexametasona. Hay muchos factores que pueden elevar los niveles de cortisol en ausencia de síndrome de Cushing, por lo que la interpretación de la prueba debe incorporar la probabilidad pretest de síndrome de Cushing. Un nivel de cortisol libre en orina que supere tres veces el intervalo normal superior en el contexto de manifestaciones clínicas de síndrome de Cushing se considera diagnóstico del trastorno, mientras que una prueba positiva en el marco de un bajo grado de sospecha de síndrome de Cushing no confirma el diagnóstico. Si las pruebas iniciales son positivas, la confirmación y la evaluación posterior deben incluir la consulta a un endocrinólogo. Una vez establecido el diagnóstico de síndrome e Cushing, el paso siguiente consiste en la medición de la ACTH; si está inhibida (<5 pg/ml [1,1 pmol/l]), lo que apunta a que el síndrome de Cushing tiene una causa independiente de la ACTH, está indicada una TC o RM suprarrenal específica. Si las glándulas suprarrenales parecen normales en las pruebas de imagen, debe cuestionarse el diagnóstico de síndrome de Cushing. La resección quirúrgica constituye el tratamiento definitivo para los tumores suprarrenales secretores de cortisol tanto benignos como malignos. Tras la suprarrenalectomía, los pacientes necesitan un tratamiento diario con hidrocortisona para recuperarse de la prolongada inhibición de la ACTH provocada por el hipercortisolismo. La recuperación de la función fascicular suprarre42 Normal SC improbable Normal F i g u r a 4 . Algoritmo para confirmar o descartar el diagnóstico de síndrome de Cushing. CLO: cortisol libre en orina; cortisol salival N: cortisol salival nocturno; PID: prueba de inhibición con dexametasona; SC: síndrome de Cushing. aDebe obtenerse en un mínimo de dos ocasiones. nal puede tardar un año o más, en función de la gravedad del síndrome de Cushing (véase «Trastornos de la hipófisis»). PUNTOS CLAVE • La evaluación del síndrome de Cushing implica: 1) pruebas iniciales seguidas de pruebas de confirmación del síndrome de Cushing; 2) determinación de si el síndrome de Cushing es independiente o dependiente de la corticotropina (ACTH), y 3) localización del origen de la ACTH en la enfermedad dependiente de la ACTH o confirmación de la presencia de una (o varias) masas suprarrenales en la enfermedad independiente de la ACTH. • Las pruebas iniciales del síndrome de Cushing tienen una exactitud diagnóstica similar e incluyen la medición del cortisol libre en orina de 24 horas, determinaciones seriadas de cortisol salival nocturno y la prueba de inhibición con 1 mg de dexametasona por la noche. • No se recomienda una evaluación para detectar síndrome de Cushing en pacientes sin signos específicos de síndrome de Cushing. Hiperaldosteronismo primario El hiperaldosteronismo primario es una causa frecuente de hipertensión secundaria. Tradicionalmente, para su diagnóstico se consideraba como una condición bioquímica indispensable la presencia de hipopotasemia, pero ahora se sabe que más del 60% de los pacientes tienen valores de potasio normales. Dado que muchas veces la hipertensión es el único signo de hiperaldosteronismo primario, muchas veces este trastorno no se diagnostica. Es importante identificar a los pacientes con hiperaldosteronismo primario, porque la aldosterona tiene efec- AMAV Trastornos de las glándulas suprarrenales tos perjudiciales en el sistema cardiovascular y el tratamiento previene la progresión y, en ocasiones, puede revertir las alteraciones. Se ha observado una morbimortalidad cardiovascular más alta en los pacientes con hiperaldosteronismo primario en comparación con quienes tienen hipertensión primaria con un control similar de la presión arterial. El posible impacto en la salud del hiperaldosteronismo primario no tratado y la importancia de identificarlo se reflejan en las guías actualizadas sobre las pruebas para detectar casos de hiperaldosteronismo primario (Tabla 24). El hiperaldosteronismo primario está causado por una hiperplasia de las glándulas suprarrenales (hiperaldosteronismo idiopático) en dos terceras partes de los casos y por un adenoma productor de aldosterona (APA) unilateral en un tercio de los casos. El diagnóstico del hiperaldosteronismo primario requiere la realización de una detección de caso escalonada, así como de pruebas de confirmación y localización. La prueba de detección de caso más fiable consiste en el cálculo del cociente aldosterona/renina en plasma (CAR) mediante la determinación de la concentración de aldostero- Tabla 24. Indicaciones para detectar los casos de hiperaldosteronismo primario y feocromocitoma na en plasma y la actividad de la renina plasmática (o la concentración de renina directa) en una muestra obtenida en sedestación a media mañana. En los pacientes que toman un inhibidor de la ECA o un bloqueador de los receptores de angiotensina, la renina debería estar elevada, por cuyo motivo en estos pacientes una prueba inicial sencilla es la medición de la actividad de la renina plasmática. Si la actividad de la renina plasmática está inhibida, hay una alta probabilidad de hiperaldosteronismo primario y debe obtenerse el CAR; de lo contrario, se descarta un estado de hiperaldosteronismo. Los antagonistas de los receptores de mineralocorticoides (espironolactona y eplerenona) y la amilorida en dosis altas pueden interferir de manera significativa en la interpretación del CAR y deben suspenderse seis semanas antes de la evaluación. No es necesario suspender los otros antihipertensivos, pero, dado que algunos pueden tener efectos menores en los niveles de aldosterona y/o renina (Tabla 25), los resultados del CAR deben interpretarse teniendo en cuenta dichos efectos. La hidralazina, un bloqueador selectivo de los receptores adrenérgicos α, y el verapamilo de liberación lenta ejercen efectos mínimos sobre la secreción de aldosterona y renina y, cuando sea factible, pueden reemplazar otros fármacos en caso de CAR dudoso. Un CAR superior a 20 con una concentración Hiperaldosteronismo primario Hipertensión no tratada con PA >150/100 mmHg Hipertensión (>140/90 mmHg) con un tratamiento farmacológico triple Hipertensión y masa suprarrenal descubierta de manera incidental Tabla 25. Efecto de los medicamentos de prescripción habitual en las mediciones de la actividad de la renina plasmática y la concentración de aldosterona en plasma ARP CAP CAP/ ARP Agonista de los receptores α2-adrenérgicos ↓↓ ↓ ↑ ↓↓ ↓ ↑ Feocromocitoma Bloqueadores de los receptores β-adrenérgicos Crisis tipo adrenérgico (cefalea, sudoración y taquicardia) con o sin hipertensión Inhibidor directo de la renina ↓ ↓ ↑ Masa suprarrenal descubierta de manera incidental con o sin hipertensión AINE ↓↓ ↓ ↑ Inhibidor de la ECA/BRA ↑↑ ↓ ↓ Hipertensión con una edad de inicio <20 años BCC dihidropiridínicos ↑ ↓ ↓ Miocardiopatía idiopática Diuréticoa ↑↑ ↑ ↓ Episodio hipertensivo inducido por anestesia, cirugía o angiografía Antagonista de los receptores de mineralocorticoides ↑↑ ↑ ↓ ↑ ↓ Hipertensión asociada a hipopotasemia espontánea o inducida por diuréticos Hipertensión en el contexto de un familiar de primer grado con hiperaldosteronismo primario Efecto en los resultados de las pruebas Clase de medicamentos Falsos positivos Hipertensión en el contexto de antecedentes familiares de hipertensión de inicio a una edad <40 años Hipertensión (>140/90 mmHg) con un tratamiento farmacológico triple Paraganglioma Síndromes familiares que predisponen al feocromocitoma: VHL, NF-1 y NEM 2 Antecedentes familiares de feocromocitoma o paraganglioma NEM 2: neoplasia endocrina múltiple tipo 2; NF-1: neurofibromatosis tipo 1; PA: presión arterial: VHL: Von Hippel-Lindau. Falsos negativos ISRS a Diuréticos ahorradores de potasio (amilorida) y diuréticos causantes de pérdida de potasio (hidroclorotiazida). ARP: actividad de la renina plasmática; BCC: bloqueador de los canales de calcio; BRA: bloqueador de los receptores de angiotensina; CAP: concentración de aldosterona en plasma; ISRS: inhibidor selectivo de la recaptación de serotonina. 43 Trastornos de las glándulas suprarrenales de aldosterona en plasma de al menos 15 ng/dl (414 pmol/l) se considera un resultado positivo, y en tal caso los pacientes deben ser derivados a un endocrinólogo, que podrá llevar a cabo otras pruebas para confirmar que la secreción de aldosterona es inadecuada en un estado de repleción de sal. El estudio de localización básico para el hiperaldosteronismo primario es una TC suprarrenal específica. Los hallazgos pueden incluir glándulas suprarrenales normales o adenoma(s)/hiperplasia unilateral o bilateral. En una tercera parte de los pacientes, es posible que la TC no identifique la causa del hiperaldosteronismo primario porque muchos APA son demasiado pequeños para ser visibles o porque hay una masa suprarrenal causal que no está relacionada con el hiperaldosteronismo primario. En consecuencia, la mayoría de los pacientes con hiperaldosteronismo primario confirmado deben someterse a obtención de muestras de la vena suprarrenal para confirmar el origen del hiperaldosteronismo. El tratamiento médico con un antagonista de los receptores de aldosterona (espironolactona o eplerenona) constituye el tratamiento de elección para el hiperaldosteronismo primario debido a un hiperaldosteronismo idiopático o cuando los pacientes con APA no son candidatos, o no desean someterse, a cirugía. Muchas veces se prefiere la espironolactona a la eplerenona porque es menos cara y tiene más potencia. Sin embargo, los pacientes que toman espironolactona tienen más probabilidades de experimentar efectos secundarios dependientes de la dosis como son ginecomastia y disfunción eréctil en los varones e irregularidades menstruales en las mujeres. La hiperpotasemia se resuelve casi invariablemente con el tratamiento, aunque el control de la presión arterial puede requerir fármacos adicionales. No hay estudios que demuestren claramente la superioridad de la suprarrenalectomía con respecto al tratamiento médico para los APA, pero la cirugía puede ser más coste-efectiva a largo plazo. La suprarrenalectomía laparoscópica es eficaz para la enfermedad unilateral y reduce la aldosterona en plasma y el riesgo elevado de enfermedad cardiovascular consiguiente. La hipertensión mejora en la mayoría de los pacientes y se cura en alrededor del 40% de los casos. La hipertensión persistente posterior a suprarrenalectomía puede deberse a las alteraciones vasculares provocadas por la hipertensión crónica o a una hipertensión primaria coexistente. Los pacientes tienen más probabilidades de lograr la resolución de la hipertensión sin tomaban menos de tres antihipertensivos antes de la operación, así como si tienen uno o pocos familiares de primer grado con hipertensión. Es necesario controlar el potasio sérico semanalmente durante el primer mes posterior a la operación, y hay que indicar a los pacientes que sigan una dieta alta en sal debido al riesgo de hiperpotasemia por la reducción transitoria de la producción de aldosterona en la glándula suprarrenal restante ocasionada por la inhibición crónica del sistema renina-angiotensina durante el período de hiperaldosteronismo. En los pacientes que experimentan hiperpotasemia es necesaria la reposición de mineralocorticoides a corto plazo. 44 PUNTOS CLAVE • La prueba de detección de caso más fiable para el hiperaldosteronismo primario consiste en el cálculo del cociente aldosterona/renina en plasma mediante la determinación de la concentración de aldosterona en plasma y la actividad de la renina plasmática (o la concentración de renina directa) en una muestra obtenida en sedestación a media mañana; si la actividad de la renina plasmática está inhibida, haya una alta probabilidad de hiperaldosteronismo primario. • En los pacientes que toman un inhibidor de la ECA o un bloqueador de los receptores de angiotensina, la renina debería estar elevada, por cuyo motivo en estos pacientes una prueba inicial sencilla es la medición de la actividad de la renina plasmática; si la actividad de la renina plasmática está inhibida, hay una alta probabilidad de hiperaldosteronismo primario y debe obtenerse el cociente aldosterona/renina en plasma; de lo contrario, se descarta un estado de hiperaldosteronismo. • La suprarrenalectomía laparoscópica para la enfermedad unilateral da lugar a la reducción de la aldosterona en plasma; el tratamiento médico con un antagonista de los receptores de aldosterona (espironolactona o eplerenona) constituye el tratamiento de elección para el hiperaldosteronismo primario derivado de un hiperaldosteronismo idiopático o cuando los pacientes con un adenoma productor de aldosterona no son candidatos, o no desean someterse, a cirugía. Feocromocitoma y paraganglioma Los feocromocitomas y paragangliomas son tumores secretores de catecolaminas que tienen su origen de células cromafines de la médula suprarrenal (80%) y los ganglios simpáticos extrasuprarrenales (en su mayor parte abdominales), respectivamente. Los tumores también pueden surgir de los ganglios parasimpáticos de la cabeza y el cuello, pero estos rara vez secretan catecolaminas. Al menos la tercera parte de los feocromocitomas/paragangliomas están asociados a mutaciones en la línea germinal. Los feocromocitomas pueden darse en síndromes familiares, incluyendo la neoplasia endocrina múltiple tipo 2, el síndrome de Von Hippel-Lindau y la neurofibromatosis tipo 2 (Tabla 26). Por lo tanto, todos los pacientes con tumores secretores de catecolaminas deberían recibir asesoramiento genético. La hipertensión asociada a feocromocitoma/paraganglioma puede mostrar un patrón prolongado, con o sin paroxismos, o manifestarse solo de forma paroxística. Algunos pacientes (del 10% al 15%) permanecen normotensos. La tríada clásica de palpitaciones, cefalea y diaforesis se observa en menos del 50% de los pacientes con feocromocitoma. Puede haber múltiples síntomas relacionados con el exceso de cateco- AMAV Trastornos de las glándulas suprarrenales Tabla 26. Síndromes de neoplasia endocrina múltiple Tipo Mutación Característica más frecuente Características asociadas 1 MEN1 (la herencia de un alelo mutado con mutación somática en otro alelo provoca neoplasia) Adenoma paratiroideo (a menudo múltiple) Tumores de las células de los islotes pancreáticos y entéricos (el gastrinoma y el insulinoma son los más frecuentes) Adenoma hipofisario Otras (tumores carcinoides, adenoma corticosuprarrenal) 2A RET (exón 11, codón 634a) 2B a) Carcinoma medular de tiroides Feocromocitoma (a menudo multifocal) Hiperplasia paratiroidea RET (exón 16, codón 918 Carcinoma medular de tiroides Feocromocitoma (a menudo multifocal) Neuroma mucoso Ganglioneuroma gastrointestinal Hábito corporal marfanoide aMutación observada más frecuente. laminas, entre los que se incluyen el dolor abdominal, la palidez cutánea, la visión borrosa o la poliuria. En raras ocasiones, los pacientes pueden presentar infarto de miocardio, miocardiopatía o ictus. Las indicaciones para las pruebas de detección de feocromocitoma se presentan en la Tabla 24. Las pruebas iniciales para detectar un feocromocitoma incluyen la medición de la metadrenalina libre en plasma obtenida en decúbito supino o los niveles de metadrenalina fraccionada en orina de 24 horas y catecolaminas. La elevación de las catecolaminas puede observarse en pacientes sometidos a estrés psicológico o físico. Los medicamentos pueden afectar a los resultados (Tabla 27) y deben suspenderse al menos dos semanas antes de las pruebas. El diagnóstico exacto puede confundirse también debido al hecho de que los pacientes con o sin hipertensión pueden sufrir crisis tipo adrenérgico en ausencia de tumor secretor de catecolaminas. La interpretación de los resultados de las pruebas debe tener en cuenta el grado de elevación de la metadrenalina y no si el resultado es normal o anormal. Las elevaciones leves pueden requerir la repetición de la prueba. Los niveles más de cuatro veces por encima del límite superior de la normalidad, en ausencia de estrés o enfermedad agudos, son compatibles de tumor secretor de catecolaminas. La metadrenalina libre en plasma tiene una gran sensibilidad (entre el 96% y el 100%); la especificidad se sitúa entre el 85% y el 89%. La metadrenalina fraccionada en orina y las catecolaminas tienen una mayor especificidad (98%) y una alta sensibilidad (de hasta el 97%). Ninguna prueba es superior a las otras, por lo que los clínicos pueden usar una estimación de la probabilidad pretest para seleccionar la prueba inicial. En caso de un índice elevado de sospecha, se escoge la metadrenalina libre en plasma, y cuando la sospecha es baja, la metadrenalina fraccionada en orina y las catecolaminas pueden ser una mejor opción. Para evitar el diagnóstico erróneo de una masa suprarrenal no funcional detectada de manera incidental como feo- Tabla 27. Sustancias asociadas a falsos positivos en las pruebas bioquímicas para detectar feocromocitoma Clase farmacológica Sustancia/medicamento Analgésicos Paracetamol Antieméticos Proclorperazina Antihipertensivos Fenoxibenzaminaa Medicamentos psiquiátricos Antipsicóticos Buspirona Inhibidores de la monoaminooxidasa Inhibidores de la recaptación de serotonina y noradrenalina (IRSN) Antidepresivos tricíclicosa Estimulantes Anfetaminas Metilfenidato Cocaína Cafeína Otros fármacos Levodopa Descongestionantes (seudoefedrina) Reserpina Síndrome de abstinencia Clonidina Alcohol Drogas a El que tiene más probabilidades de causar resultados falsos positivos. cromocitoma, la búsqueda de un tumor debe iniciarse cuando los resultados analíticos respaldan un diagnóstico bioquímico de feocromocitoma/paraganglioma. Es difícil determinar la relevancia clínica de unos niveles de metadrenalina significativamente elevados en pacientes hospitalizados. La modalidad de imagen de elección es una TC abdominal y pélvica potenciada con contraste, ya que el 85% de los tumores 45 Trastornos de las glándulas suprarrenales añade una vez logrado el bloqueo α-adrenérgico para manejar la taquicardia refleja, pero nunca debe iniciarse antes de un bloqueo α-adrenérgico adecuado, porque una vasoconstricción α-adrenérgica sin oposición puede provocar una crisis hipertensiva. En caso de feocromocitomas de gran tamaño con una elevada tasa de secreción de hormonas, se añaden otros fármacos al régimen de tratamiento, como un bloqueador de los canales de calcio y/o metirosina. Los bloqueadores de los canales de calcio pueden usarse también en los pacientes que experimentan hipotensión significativa con dosis pequeñas de un bloqueador α-adrenérgico. Los bloqueadores selectivos de los receptores α-1 como la doxazosina pueden usarse como alternativa para la fenoxibenzamina si la disponibilidad o la ausencia de cobertura de la última suponen un problema. Después de la operación, los pacientes pueden presentar hipotensión significativa, y la mayoría necesitan soporte con líquidos y vasopresores al menos brevemente en el período postoperatorio. Los pacientes con feocromocitoma pueden tener una glucemia basal alterada o diabetes tipo 2 relacionada con la resistencia a la insulina con origen en el exceso de catecolaminas. Esta puede mejorar o revertirse tras la suprarrenalectomía. Aproximadamente el 83% de los feocromocitomas/paragangliomas son benignos. Los hallazgos patológicos no predicen qué tumores se volverán malignos y producirán metástasis. Dado que las metástasis pueden aparecer décadas después del diagnóstico inicial, la mayoría de los pacientes deben someterse a un cribado bioquímico anual a largo plazo, normalmente con la determinación de metadrenalina libre en plasma. secretores de catecolaminas son intrasuprarrenales (y el 95% están alojados en el abdomen o la pelvis). Los signos clásicos de los feocromocitomas en las pruebas de imagen se presentan en la Tabla 28. El tamaño medio de los feocromocitomas sintomáticos en el diagnóstico es de 4 cm. Si la TC es negativa, el primer paso consiste en reconsiderar el diagnóstico; sin embargo, en caso de un alto grado de sospecha de tumor secretor de catecolaminas, el paso siguiente es una prueba con yodo-123 (123I)-metayodobencilguanidina Esta prueba puede estar indicada también en pacientes con feocromocitomas de tamaño muy grande (>10 cm), para detectar enfermedad metastásica, o paragangliomas, para detectar múltiples tumores. La tomografía por emisión de positrones (PET) con fludeoxiglucosa es más sensible para la detección de enfermedad metastásica, pero por lo general su uso se reserva a los pacientes con tumores malignos demostrados. El tratamiento definitivo para el feocromocitoma/paraganglioma consiste en la resección quirúrgica. El bloqueo preoperatorio de los receptores β-adrenérgicos con fenoxibenzamina durante 10-14 días antes de la cirugía es esencial para prevenir las crisis hipertensivas durante la misma. La dosis se incrementa de manera progresiva hasta alcanzar una presión arterial ≤130/80 mmHg o una frecuencia cardíaca de entre 60 y 70/min en sedestación y una presión sistólica de ≥90 mmHg con una frecuencia cardíaca entre 70 y 80/min en bipedestación. Los efectos secundarios incluyen mareo, congestión nasal y fatiga. Para facilitar el aumento escalonado de la dosis y mitigar los efectos de la contracción del volumen del bloqueo α-adrenérgico, se indica a los pacientes que liberalicen la ingestión de sal y líquidos. El bloqueo β-adrenérgico se Tabla 28. Características clásicas de las masas suprarrenales en las pruebas de imagen Masa suprarrenal Generales TC Intensidad de la señal de RMa Adenoma suprarrenal Diámetro <4 cm Densidad <10 UH Potenciación homogéneab Lavado de contraste >50% (10 min) Isointenso en las imágenes potenciadas en T2 Márgenes redondeados y claros Carcinoma de corteza suprarrenal Normalmente >4 cm Densidad >10 UH Potenciación heterogénea Lavado de contraste <50% (10 min) Hiperintenso en las imágenes potenciadas en T2 Márgenes irregulares Calcificaciones, necrosis Feocromocitoma Densidad >10 UH Tamaño variable b Potenciación heterogénea , áreas quísticas Lavado de contraste <50% (10 min) Hiperintenso en las imágenes potenciadas en T2 Márgenes redondeados y claros Puede ser bilateral Metástasis aIntensidad Márgenes variables Densidad >10 UH Puede ser bilateral Lavado de contraste <50% (10 min) Hiperintenso en las imágenes potenciadas en T2 de la señal en comparación con el hígado. bPotenciación tras la administración de contraste intravenoso. RM: resonancia magnética; TC: tomografía computarizada; UH: unidades Hounsfield (medida de radiodensidad en comparación con el agua). 46 Trastornos de las glándulas suprarrenales PUNTOS CLAVE • Las pruebas iniciales para detectar un feocromocitoma incluyen la medición de los niveles de metadrenalina libre en plasma o los niveles de metadrenalina fraccionada y catecolaminas en orina de 24 horas; algunos medicamentos pueden afectar a los resultados y deben suspenderse al menos dos semanas antes de las pruebas. • Para evitar el diagnóstico erróneo de una masa suprarrenal no funcional detectada de manera incidental como feocromocitoma, la búsqueda de un tumor debe iniciarse cuando el diagnóstico bioquímico de feocromocitoma/paraganglioma está claro en los resultados analíticos. AMAV • El tratamiento definitivo del feocromocitoma/paraganglioma es la resección quirúrgica; un bloqueo α-adrenérgico preoperatorio con fenoxibenzamina es esencial para prevenir las crisis hipertensivas durante la cirugía. Los bloqueadores selectivos de los receptores α-1 como la doxazosina pueden usarse como alternativa para la fenoxibenzamina si la disponibilidad o la ausencia de cobertura de la última suponen un problema. Tumores suprarrenales productores de andrógenos Los tumores suprarrenales productores de andrógenos son infrecuentes y provocan irregularidades menstruales y virilización en las mujeres, incluyendo hirsutismo, voz profunda, aumento Tabla 29. de la masa muscular, aumento de la libido y clitoromegalia. Los tumores secretan DHEA/DHEAS y androstenodiona, que posteriormente se convierten en testosterona en los tejidos periféricos. Los tumores de las glándulas suprarrenales secretores de DHEAS son fácilmente visibles en la TC, y rara vez hace falta la obtención de muestras venosas suprarrenales para localizar el tumor. Aproximadamente el 50% de los tumores son benignos, y el tratamiento de elección es la resección. Déficit de hormonas suprarrenales Insuficiencia suprarrenal primaria Causas y cuadro clínico La insuficiencia suprarrenal primaria es un trastorno potencialmente mortal que suele manifestarse con un inicio sintomático gradual que dificulta el diagnóstico (Tabla 29). También puede manifestarse con una crisis suprarrenal, muchas veces desencadenada por una enfermedad aguda o por el inicio de un tratamiento de reposición de hormona tiroidea en un paciente con insuficiencia suprarrenal crónica inadvertida. Aunque la hiperpigmentación cutánea por estimulación de los melanocitos por los elevados niveles de ACTH se considera un signo distintivo de la insuficiencia suprarrenal primaria, no está presente en el 5% de los pacientes aproximadamente. La causa más frecuente de insuficiencia suprarrenal primaria es la destrucción autoinmune de todas las capas de la corteza suprarrenal, que da lugar a déficit progresivo de mineralocorticoides, glucocorticoides y andrógenos suprarrena- Manifestaciones clínicas y analíticas de la insuficiencia suprarrenal primaria Déficit hormonal Síntomas Signos Signos analíticos Cortisol Fatiga Hiperpigmentaciónb (pliegues palmares, superficies extensoras, mucosa bucal) ↓ cortisol sérico Debilidad Febrícula Descenso de la PA ↑ ACTH en plasma ↓ sodio séricoc ↓ glucosa plasmáticad Pérdida de peso Anorexia Náuseas/vómitos Dolor abdominal Artralgia Mialgia Aldosterona Antojo de sal Ortostatismo ↑ ARP Mareo Hipotensión ↓ sodio sérico Disminución de la libidoa Vello axilar o púbico reducidoa ↓ DHEAS sérico ↑ potasio sérico DHEAS aSolo bSe cEl mujeres. da exclusivamente en la insuficiencia suprarrenal primaria. cortisol inhibe la secreción de hormona antidiurética (ADH), por lo que la hipocortisolemia dará lugar a un aumento de la secreción de ADH y a hiponatremia. dInfrecuente en adultos. ACTH: corticotropina; ARP: actividad de la renina plasmática; DHEAS: sulfato de deshidroepiandrosterona; PA: presión arterial. 47 Trastornos de las glándulas suprarrenales les. La mayoría de los pacientes tienen anticuerpos contra la 21-hidroxilasa positivos, y en torno al 50% experimentarán otro trastorno endocrino autoinmune en la vida (hipotiroidismo primario, insuficiencia ovárica primaria, enfermedad celíaca, hipoparatiroidismo o diabetes mellitus tipo 1). La insuficiencia suprarrenal primaria puede estar causada también por trastornos infiltrativos como infección (tuberculosis, infecciones micóticas), sarcoidosis y linfoma, que dan lugar a hipertrofia bilateral de las glándulas suprarrenales. La enfermedad metastásica que afecta a las glándulas suprarrenales, en la mayoría de los casos debido a cáncer de pulmón, carcinoma de células renales y melanoma, rara vez provoca insuficiencia suprarrenal, aun cuando afecte a las dos glándulas suprarrenales. La hemorragia suprarrenal bilateral puede manifestarse en forma de insuficiencia suprarrenal aguda y debe tenerse en cuenta si se produce una hipotensión imprevista. Los factores de riesgo de hemorragia suprarrenal bilateral incluyen déficit de proteína C, anticoagulación, coagulopatía intravascular diseminada y septicemia. Diagnóstico En la Figura 5 se presenta un algoritmo para el diagnóstico de la insuficiencia suprarrenal. La evaluación inicial incluye la medición del cortisol total sérico matinal y los niveles de ACTH. La insuficiencia suprarrenal primaria se confirma con la combinación de niveles de cortisol sérico bajos y niveles de ACTH sérica elevados. En la Figura 5 se mencionan las consideraciones importantes que deben tenerse en cuenta a la hora de interpretar los resultados y que a menudo exigen la derivación a un endocrinólogo. Otras pruebas pueden incluir la determinación de anticuerpos contra la 21-hidroxilasa; aproximadamente en el 90% de los casos de suprarrenalitis autoinmune se observan anticuerpos contra la 21-hidroxilasa positivos. Si son negativos, debe obtenerse una TC de las glándulas suprarrenales. Tratamiento Para tratar la insuficiencia suprarrenal primaria es necesario un tratamiento tanto con glucocorticoides como con mineralocorticoides. El glucocorticoide de elección es la hidrocortisona, a una pauta de dos o tres veces al día. El cumplimiento de múltiples dosis diarias puede ser complicado, por lo que una posible alternativa es el uso de prednisona (Tabla 30). El principio de la reposición consiste en administrar una dosis alta por la mañana y evitar la reposición por la noche. A pesar de este intento por imitar la variación diurna, los pacientes con insuficiencia suprarrenal primaria refieren una disminución de la calidad de vida relacionada con la salud. Es imprescindible evitar una reposición excesiva de glucocorticoides para prevenir el síndrome de Cushing iatrogénico, con el riesgo que comporta de obesidad, diabetes mellitus tipo 2, hipertensión, hiperlipemia, pérdida ósea y enfermedad cardiovascular. Algunos pacientes «se sienten mejor» con una reposición a dosis «más altas» que las fisiológicas, pero la administración de dosis suprafisiológicas conlleva más riesgos que beneficios. La reposición de mineralocorticoides se logra con la administra- Manifestaciones clínicas indicativas de insuficiencia suprarrenal Medir el cortisol a las 8:00 horas Descartar las condiciones que alteran las globulinas fijadoras de transporte (estrógenos orales, estados de proteínas bajas) Conocer el uso reciente o actual de glucocorticoides (orales, inhalados tópicos, intraarticulares) Cortisol >15 μg/dl Cortisol 3-15 μg/dl Cortisol <3 μg/dl Prueba de estimulación con ACTH Administrar 250 μg de ACTH Medir el cortisol a los 0, 30 y 60 minutos Descarta insuficiencia suprarrenal Cortisol máximo >18 μg/dl Cortisol máximo ≤18 μg/dl Presencia de insuficiencia suprarrenal Medir la ACTH Baja o inadecuadamente normal Alta Insuficiencia suprarrenal secundaria Insuficiencia suprarrenal primaria 48 F i g u r a 5 . Algoritmo para el diagnóstico de la insuficiencia suprarrenal. ACTH: corticotropina. Trastornos de las glándulas suprarrenales Tabla 30. Equivalencia de las dosis y potencias relativas de los glucocorticoides orales sintéticos habituales Glucocorticoide sintético Dosis equivalente (mg) Semivida biológica (horas) Potencia antiinflamatoria relativaa Potencia mineralocorticoide relativab Hidrocortisona 20 8-12 1 1/125 Prednisolona/prednisona 5 18-36 4 1/150 Metilprednisolona 4 18-36 5 0 Dexametasona 0,75 36-54 25-50 0 a Potencia antiinflamatoria en relación con la hidrocortisona. b Potencia mineralocorticoide en relación con la fludrocortisona. Tabla 31. Tratamiento médico crónico de la insuficiencia suprarrenal primaria Medicamento Dosis basal Consideraciones Glucocorticoidesa «Reglas de los días malos»: Hidrocortisona Normalmente 15-25 mg/día, divididos en 2-3 dosis a lo largo del día Prednisona Prednisona 5 mg una vez al día El paciente hace el seguimiento en casa En caso de estrés fisiológico menor (infección respiratoria alta, fiebre, cirugía menor bajo anestesia local): Administración de la dosis: Ajustar en función de la respuesta clínica con el objetivo de que desaparezcan los signos y síntomas de déficit o exceso de cortisol (aumentar la dosis si persisten los síntomas de déficit de cortisol; reducirla en presencia de signos y síntomas de SC) 2-3 veces la dosis basal durante 2-3 días Dosis de estrés: Los profesionales sanitarios hacen el seguimiento mientras el paciente está en el hospital En caso de estrés fisiológico moderado (cirugía menor o moderada con anestesia general): Hidrocortisona 25-75 mg/día por vía oral o IV durante 1-2 días En caso de estrés fisiológico mayor (cirugía mayor, traumatismo, enfermedad crítica o parto): Hidrocortisona 100 mg IV. seguidos de 50 mg cada 6 horas IV.; reducción rápida de la dosis y cambio a régimen oral en función del estado clínico Mineralocorticoides Fludrocortisona 0,05-0,2 mg una vez al día por la mañana Administración de la dosis: La fludrocortisona no es necesaria si la dosis de hidrocortisona es >40 mg/día Ajustar hasta: 1. PA normal 2. Na, K en suero normales Andrógenos suprarrenales DHEA 25-50 mg una vez al día Considerar DHEA en las mujeres con estado anímico o sensación de bienestar alterados una vez optimizada la reposición de glucocorticoides aLos glucocorticoides de acción breve son preferibles a los fármacos de acción más prolongada debido al menor riesgo de exceso de glucocorticoides. Las preparaciones de acción más prolongada tienen la ventaja de su administración una vez al día. DHEA: deshidroepiandrosterona; IV: intravenoso; K: potasio; Na: sodio; PA: presión arterial; SC: síndrome de Cushing. ción diaria de fludrocortisona. La reposición de DHEAS es controvertida en las mujeres debido a la ausencia de datos sólidos de sus beneficios y a causa de la inquietud por la seguridad y la calidad de las preparaciones en Estados Unidos, que son suplementos y no están reguladas como fármacos. En caso de enfermedad, los pacientes no pueden incrementar adecuadamente el cortisol y, por lo tanto, enseñar las reglas de los «días malos» es fundamental para prevenir las crisis suprarrenales (Tabla 31). En los estados de estrés fisiológico menor, como infección respiratoria, fiebre o cirugía menor bajo 49 Trastornos de las glándulas suprarrenales CONT. anestesia local, los pacientes deben duplicar o triplicar la dosis base de glucocorticoides durante dos o tres días. Durante los episodios de estrés fisiológico moderado o mayor son necesarias dosis más altas de glucocorticoides. Los pacientes que presentan una crisis suprarrenal deben recibir reanimación con líquidos y una dosis inicial inmediata de hidrocortisona intravenosa (100 mg), seguida de hidrocortisona intravenosa (100 mg) cada ocho horas durante las 24 horas siguientes, tras las cuales la dosis dependerá del estado clínico. Los pacientes con insuficiencia suprarrenal e hipotiroidismo concomitantes no tratados deben recibir siempre tratamiento de reposición de glucocorticoides en primer lugar para evitar que la reposición de hormonas tiroideas desencadene una crisis suprarrenal. Asimismo, debe aconsejarse a los pacientes que lleven consigo una tarjeta de alerta médica en todo momento. En caso de enfermedad, no es necesario incrementar la dosis de mineralocorticoides. El término «fatiga suprarrenal» es utilizado por algunos profesionales que practican medicina alternativa en referencia a un grupo de síntomas que supuestamente se producen en pacientes que experimentan estrés emocional o físico crónico y que, según dicen, están provocados por el hipercortisolismo y el hipocortisolismo simultáneos. No hay evidencias científicas que respalden la existencia de dicho trastorno. Los pacientes pueden hacerse pruebas de cortisol salival, pero la interpretación de los resultados no suele ser fiable. A algunos pacientes con supuesta «fatiga suprarrenal» se les administra un tratamiento con hidrocortisona o extracto de glándulas suprarrenales de origen animal que puede contener glucocorticoides activos, lo que provoca la inhibición exógena de la producción de ACTH y síndrome de Cushing iatrogénico. La suspensión repentina de estos productos puede causar insuficiencia suprarrenal aguda. En los pacientes con «fatiga suprarrenal» es necesario reducir con cuidado todo tratamiento glucocorticoide y explorar otras posibles causas de los síntomas. PUNTOS CLAVE • La causa más frecuente de insuficiencia suprarrenal primaria es la destrucción autoinmune de todas las capas de la corteza suprarrenal, que da lugar a déficit progresivo de mineralocorticoides, glucocorticoides y andrógenos suprarrenales. • Para tratar la insuficiencia suprarrenal primaria es necesario un tratamiento tanto con glucocorticoides como con mineralocorticoides. • En caso de enfermedad, los pacientes no pueden incrementar adecuadamente el cortisol y, por lo tanto, enseñar las reglas de los «días malos» sobre administración de glucocorticoides es fundamental para prevenir las crisis suprarrenales. Función suprarrenal durante la enfermedad crítica Durante los momentos de estrés fisiológico, el eje hipotálamo-hipófiso-suprarrenal recibe estímulos para producir mayores niveles de cortisol. En algunos pacientes, se cree que el 50 aumento de la secreción de cortisol es subóptima y se denomina «insuficiencia suprarrenal relativa». No obstante, hay debate sobre si esta entidad es una verdadera enfermedad. La globulina fijadora de cortisol y la albúmina descienden en la enfermedad crítica, lo que reduce el cortisol total medido. A pesar de la capacidad de medir el nivel de cortisol libre, el cortisol libre calculado y el cortisol total estimulado por la ACTH en los pacientes en estado crítico, no hay consenso en cuanto al conjunto de criterios diagnósticos de la insuficiencia suprarrenal. Hasta la fecha, los estudios no revelan mejoras en la supervivencia de los pacientes con insuficiencia suprarrenal relativa tratados con glucocorticoides en dosis altas. Sin embargo, es posible mejorar la recuperación del choque, por cuyo motivo en la actualidad se recomienda administrar hidrocortisona a dosis de estrés a los pacientes con choque resistente al tratamiento estándar con líquidos y vasopresores. Masa suprarrenal Masas suprarrenales descubiertas de manera incidental Un incidentaloma suprarrenal se define como una masa suprarrenal de con un diámetro >1 cm que se detecta en una prueba de imagen realizada para fines distintos de presunta enfermedad suprarrenal. La prevalencia del incidentaloma suprarrenal aumenta con la edad y se estima que es aproximadamente del 10% en las personas ≥70 años de edad. La mayoría de las lesiones son adenomas no funcionales benignos, y alrededor de entre el 10% y el 15% secretan hormonas en exceso. Otras causas son metástasis (la probabilidad aumenta en caso de cáncer primario conocido), mielolipoma, quistes y carcinoma corticosuprarrenal. El hallazgo casual de una masa suprarrenal plantea dos interrogantes principales: 1) si secreta hormonas en exceso (aldosterona, cortisol o catecolaminas) y 2) si es benigna o maligna. Los pacientes con hipertensión o hipopotasemia deben hacerse pruebas para detectar hiperaldosteronismo primario. Es necesario llevar a cabo pruebas bioquímicas para detectar feocromocitoma, como la determinación de la metadrenalina total en orina de 24 horas, en todos los pacientes, incluso en ausencia de los síntomas clásicos o hipertensión. Además, hay que evaluar a todos los pacientes para detectar un síndrome de Cushing subclínico, trastorno que se caracteriza por una secreción de cortisol independiente de la ACTH que puede provocar los efectos metabólicos (hiperglucemia e hipertensión) y óseos (osteoporosis) del hipercortisolismo, pero no las manifestaciones clínicas más específicas del síndrome de Cushing, como almohadilla de grasa supraclavicular, estrías violáceas anchas, congestión facial y debilidad muscular proximal. La prueba inicial del síndrome de Cushing subclínico consiste en una prueba de inhibición con 1 mg de dexametasona por la noche; un nivel de cortisol superior a 5 µg/dl (138 nmol/l) se considera un resultado positivo. Tras un resultado positivo, son necesarias más pruebas para confirmar la autonomía del cortisol; estas pueden incluir la determinación de la ACTH (inhibida), el DHEAS (bajo), el cortisol Trastornos de las glándulas suprarrenales infección. Hay que descartar los feocromocitomas antes de la biopsia, a fin de evitar la posibilidad de una crisis hipertensiva. No debe llevarse a cabo una biopsia en caso de sospecha de carcinoma corticosuprarrenal primario porque, sin una revisión de la muestra completa, la histopatología no puede diferenciarlo con seguridad de un adenoma benigno y puede producirse una diseminación tumoral. Si se sospecha lo anterior, el diagnóstico debe establecerse mediante suprarrenalectomía. En la Figura 6 se presenta un algoritmo para el manejo de los incidentalomas suprarrenales, incluyendo el control de las lesiones que no requieren suprarrenalectomía. libre en orina de 24 horas y una prueba de inhibición con 8 mg de dexametasona por la noche; en este punto se recomienda la derivación a un endocrinólogo. La decisión de proceder a una suprarrenalectomía debe tener en cuenta los riesgos y beneficios del tratamiento. Los estudios realizados hasta la fecha no son lo suficientemente sólidos para revelar una mejora postoperatoria clara en resultados clínicamente importantes, pero apuntan a una mejora de las mediciones de la glucemia, los lípidos, la presión arterial y la densidad ósea. Los resultados de las pruebas de imagen pueden ayudar a diferenciar entre una masa suprarrenal benigna y una maligna (véase la Tabla 28). La mayoría de los carcinomas corticosuprarrenales miden más de 4 cm en el momento en que son descubiertos. No obstante, aproximadamente el 75% de las masas suprarrenales benignas se encuentran también dentro de este intervalo de tamaño. En torno al 66% de los adenomas benignos son ricos en lípidos y muestran una baja atenuación (<10 unidades Hounsfield [UH]) en la TC. Además, los adenomas benignos muestran un lavado rápido (>50%) del material de contraste en comparación con las lesiones no benignas. La biopsia suprarrenal tiene una función limitada en la evaluación de los incidentalomas y se reserva para las lesiones con sospecha de metástasis o de un proceso infiltrativo, como un linfoma o una Masa suprarrenal descubierta de manera incidental Fenotipo en las pruebas de imagen Evaluación hormonal Seguimientoa • Repetir TC o RM a los 6-12 meses y una vez al año posteriormente durante 1-2 años PUNTOS CLAVE • Todos los pacientes con un incidentaloma suprarrenal deben ser evaluados para detectar un posible feocromocitoma; quienes presenten hipertensión o hiperpotasemia deben ser evaluados también para detectar hiperaldosteronismo primario, y todos los pacientes deben hacerse pruebas de síndrome de Cushing subclínico. • Los resultados de las pruebas de imagen pueden ayudar a diferenciar entre una masa suprarrenal benigna y maligna. Fenotipo en las pruebas de imagenb A favor de una naturaleza benigna: • Tamaño <4 cm • Densidad <10 UHc • Lavado de contraste >60 (10 min)c Sospechoso: • Tamaño ≥4 cm • Densidad >10 UHc • Lavado de contraste ≤60 (10 min)c • Quién: Todos los pacientes • Prueba: PIDB hormonasd Aldosterona: • Quién: HTN o ↓ K+ • Prueba: ARP/CAP Catecolaminas: Andrógenos: • Prueba: Metadrenalinas en plasma u orina, catecolaminas en orinac • Prueba: DREAS, testosterona, androstenodionac • Quién: Todos los pacientes Pruebas de imagen sospechosas Crecimiento >1 cm/año Pruebas para detectar exceso de Cortisol: Indicaciones para la suprarrenalectomía Tumores funcionales • Feocromocitoma • Tumor suprarrenal productor de aldosterona • SC subclínico con complicacionesg • Quién: En caso de sospechaf F i g u r a 6 . Algoritmo para la evaluación diagnóstica inicial y el seguimiento de una masa suprarrenal descubierta de manera casual. ARP: actividad de la renina plasmática; CAP: concentración de aldosterona en plasma; DHEAS: sulfato de deshidroepiandrosterona sulfato; HTN: hipertensión; K: potasio; PIDB: prueba de inhibición con dexametasona en dosis bajas (1 mg); RM: resonancia magnética; SC: síndrome de Cushing; TC: tomografía computarizada; UH: unidades Hounsfield. aEn las masas suprarrenales que no cumplen los criterios para la cirugía en el diagnóstico está indicada la repetición de las pruebas de imagen. b Para hallar más hallazgos de la TC y la RM, consultar la Tabla 28. Si la imagen es sospechosa en un paciente con cáncer conocido, debe considerarse la realización de una biopsia para confirmar la presencia de metástasis suprarrenal tras realizar el cribado de feocromocitoma. c Hallazgos de la TC. dNormalmente las pruebas de cribado positivas requieren nuevas evaluaciones bioquímicas para confirmar el diagnóstico (véase el texto). eMedir las metadrenalinas en plasma si la apariencia radiológica es clásica de feocromocitoma; de lo contrario, medir las metadrenalinas en orina de 24 horas y las catecolaminas. fLa evaluación hormonal de un tumor suprarrenal productor de andrógenos solo está indicada en caso de sospecha clínica sobre la base de la presencia de hirsutismo, virilización o irregularidades menstruales en las mujeres. gLa suprarrenalectomía se considera para los casos confirmados de SC subclínico asociado a inicio reciente de diabetes, hipertensión, obesidad o baja masa ósea. 51 Trastornos de la glándula tiroides Carcinoma de corteza suprarrenal El carcinoma de corteza suprarrenal (CCS) es un tumor agresivo infrecuente que suele secretar cortisol y/o andrógenos en exceso. Los pacientes pueden presentar un síndrome de Cushing de inicio rápido, con o sin virilización, y/o síntomas del efecto de masa tales como aumento de la circunferencia abdominal y edema de las extremidades inferiores. El 50% de los CCS secretan cortisol; el 20% secretan varias hormonas, entre las que se incluyen el cortisol y los precursores de la aldosterona, y menos del 10% secretan aldosterona. Algunos CCS se descubren de forma incidental como una masa suprarrenal indeterminada o sospechosa de CCS (véase la Tabla 28). Las lesiones suelen ser de gran tamaño (>4 cm) y la enfermedad por lo general se encuentra en un estadio avanzado en el momento del diagnóstico. En caso de enfermedad localizada, el tratamiento de primera línea consiste en la resección abierta. La cirugía citorreductora, la radioterapia y/o la quimioterapia pueden ser también opciones paliativas en el CCS avanzado. El mitotano es un fármaco adrenolítico que se usa habitualmente como tratamiento adyuvante y del que se ha observado que está asociado a una supervivencia sin recidiva más prolongada. El mitotano provoca insuficiencia suprarrenal primaria y requiere un tratamiento de reposición de glucocorticoides diario, a menudo en dosis suprafisiológicas, debido a que acelera el metabolismo de los glucocorticoides. Algunos pacientes experimentan también déficit de aldosterona, que se manifiesta con hiponatremia, hiperpotasemia, niveles elevados de renina e hipotensión ortostática, y necesitan reposición de fludrocortisona. Las tasas de supervivencia a los cinco años se sitúan entre el 62% y el 82% en los pacientes con enfermedad confinada en la glándula suprarrenal y en el 13% en caso de tumores asociados a metástasis distantes. PUNTOS CLAVE • El carcinoma de corteza suprarrenal es un tumor agresivo infrecuente que suele secretar cortisol y/o andrógenos en exceso, así como precursores hormonales causantes de hipertensión y síndrome de Cushing; los pacientes pueden presentar un síndrome de Cushing de inicio rápido, con o sin virilización, y/o síntomas del efecto de masa tales como aumento de la circunferencia abdominal y edema de las extremidades inferiores. • En caso de enfermedad localizada, el tratamiento de primera línea consiste en la resección abierta; la cirugía citorreductora, la radioterapia y/o la quimioterapia pueden ser también opciones paliativas en el carcinoma de corteza suprarrenal avanzado. • El mitotano es un fármaco adrenolítico que se usa habitualmente como tratamiento adyuvante y que está asociado a una supervivencia libre de recidiva más prolongada. 52 Trastornos de la glándula tiroides Anatomía y fisiología de la tiroides La glándula tiroides es el órgano exclusivamente endocrino de mayor tamaño. La ubicación anatómica habitual del istmo es justo delante del segundo y el cuarto anillos traqueales. Las glándulas paratiroides suelen estar situadas detrás de las partes superior y media de cada lóbulo tiroideo. Los nervios laríngeos recurrentes, que inervan la mayoría de los músculos intrínsecos de la laringe, transcurren por detrás de la glándula tiroides. La patología tiroidea puede provocar síntomas progresivos como disnea, tos, disfagia y alteraciones vocales debido a la estrecha proximidad de la tiroides y la tráquea, el esófago y los nervios laríngeos recurrentes. La glándula tiroides contiene células foliculares tiroideas y células parafoliculares (células C). La calcitonina, producida por las células parafoliculares, inhibe la resorción ósea; sin embargo, desempeña una función menor en la fisiología ósea. Las células foliculares producen las hormonas tiroideas, la tiroxina (T4) y la triyodotironina (T3). La síntesis y la secreción de las hormonas tiroideas están estrictamente reguladas por el eje hipotálamo-hipófiso-tiroideo. La hormona liberadora de tirotropina (TRH) del hipotálamo provoca la liberación pulsátil de TSH de las células tirotropas de la hipófisis anterior. La TSH, mediante la activación del receptor de TSH, estimula el crecimiento de las células tiroideas, el metabolismo del yodo y la síntesis y secreción de hormonas tiroideas. La T4 y la T3 ejercen una retroalimentación negativa en el hipotálamo y la hipófisis para moderar la síntesis de nuevas hormonas. El yodo constituye un micronutriente alimentario esencial y un componente estructural fundamental de la T4 y la T3. La glándula tiroides es la única fuente de T4, mientras que aproximadamente el 80% de la T3 tiene su origen en la eliminación de una molécula de yodo de la T4 a través de la actividad de la desyodinasa en los tejidos periféricos. Esto tiene lugar principalmente en el hígado y el riñón. La mayoría de la T4 (99,96%) y la T3 (99,6%) está ligada a proteínas en el suero. Aproximadamente el 70% de la T4 y la T3 está ligado a la globulina fijadora de tiroxina. La albúmina, la transtirretina y las lipoproteínas transportan una proporción menor. Solo la diminuta porción de T4 y T3 libres es biológicamente activa. Mientras que la T4 actúa mayormente como prohormona, la T3 se une con una gran afinidad a los receptores nucleares de las hormonas tiroideas que afectan a la transcripción génica en los tejidos diana y median sus efectos fisiológicos. Tiene efectos inotrópicos y cronotrópicos positivos en el corazón y potencia la sensibilidad adrenérgica del miocardio. Además, aumenta la tasa de relajación diastólica miocárdica, incrementa el volumen intravascular y reduce la resistencia vascular periférica. Aumenta la motilidad gastrointestinal y el recambio óseo y regula la generación cardíaca y el gasto de energía. Trastornos de la glándula tiroides Exploración de la tiroides La glándula tiroides está situada en el cuello, a mitad de camino entre la escotadura esternal y el cartílago tiroides. Está unida a la tráquea por la parte de atrás y se eleva con la deglución. La exploración incluye la visualización y la palpación mientras el paciente ingiere líquido. El examinador puede palparla desde detrás del paciente colocando la mano en forma de circunferencia para centrarse en la palpación o con el examinador delante del paciente, lo que le permite ver la tiroides durante la palpación. La estrategia anterior es preferible con los cuellos de gran diámetro. Trastornos estructurales de la glándula tiroides Nódulos tiroideos Los nódulos tiroideos son lesiones estructurales diferenciadas, distintas del parénquima glandular de base en la ecografía. En la mayoría de los casos, se detectan de manera incidental en estudios de imagen realizados por otros motivos. La prevalencia de los nódulos palpados en la exploración se sitúa en el 5% en las mujeres y en el 1% en los varones. Se detectan en la ecografía en el 40% de la población estadounidense y son más frecuentes a medida que aumenta la edad. Los nódulos tiroideos pueden deberse a varios procesos patológicos, que van desde quistes benignos y nódulos inflamatorios hasta cánceres como el cáncer de tiroides primario, el linfoma o las lesiones metastásicas. Las lesiones no tiroideas, como pueden ser los adenomas paratiroideos, también pueden manifestarse en forma de nódulos. Las neoplasias tiroideas primarias tienen origen clonal e incluyen los adenomas foliculares y el cáncer de tiroides. La evaluación inicial de un nódulo tiroideo empieza con la medición de la TSH sérica (Figura 7). La inhibición de la TSH puede indicar la presencia de nódulos funcionales autónomos o «calientes», que representan entre el 5% y el 10% de los nódulos tiroideos palpables. Los nódulos autónomos pueden provocar hipertiroidismo y están asociados a un riesgo muy bajo de cáncer. No requieren biopsia por aspiración con aguja fina (BAAF). Los pacientes con nódulos tiroideos e inhibición de la TSH se evalúan con gammagrafía tiroidea. Se administra un isótopo radiactivo, preferentemente yodo-123 (123I), se calcula el porcentaje captado por la tiroides (captación de yodo radiactivo [CYR]) y se obtiene una imagen (escáner de tiroides). Los nódulos calientes concentran yodo radiactivo en mayor grado que el tejido tiroideo normal. En los pacientes con TSH normal o elevada se obtiene una ecografía de tiroides/cuello para confirmar la presencia de nódulos tiroideos. El manejo de los nódulos tiroideos no funcionales viene determinado por los resultados de la ecografía y la presencia de síntomas. Las guías de la American Thyroid Association de 2015 clasifican los nódulos tiroideos en cinco patrones ecográficos basados en la ecogenicidad, en si son sólidos, quísticos o ambos y en las características cancerosas (Tabla 32). Los nódulos hiperecoicos son más brillantes, los nódulos isoecoicos son igual de brillantes y los nódulos hipoecoicos son más oscuros que el parénquima de base. La ecografía puede determinar el tamaño del nódulo. La BAAF no está recomendada para nódulos subcentimétricos, a menos que cursen con síntomas, linfadenopatía patológica o diseminación extratiroidea. Cuando sea posible, la BAAF de nódulos tiroideos debe llevarse a cabo con guía ecográfica, debido a su mayor exactitud en comparación con la biopsia por palpación. Normalmente, la citopatología tiroidea se interpreta y clasifica en función de los criterios elaborados en el Thyroid Fine Needle Aspiration State of the Science Conference del National Cancer Institute (Congreso de Bethesda). El sistema de clasificación de Bethesda se resume en la Tabla 33. La citología de la BAAF tiroidea puede ser no diagnóstica en hasta el 5-10% de las muestras. En torno al 60-70% de los nódulos biopsiados Anamnesis, exploración física TSH TSH alta o normal Escáner tiroideo T4 libre, T3 total Ecografía ≥1 cm Evaluar para BAAF con guía ecográficaa TSH baja <1 cm Repetir ecografía en 6-24 meses Funcional «Caliente» No funcional «Frío/templado» No BAAF Rx hipertiroidismo si está indicado Evaluar para BAAF con guía ecográficaa F i g u r a 7 . Evaluación inicial de un nódulo tiroideo. Hay umbrales de tamaño para la BAAF basados en la apariencia ecográfica. En una lesión menos sospechosa, puede no ser necesaria la BAAF hasta que tenga más de 2 cm; en los nódulos sospechosos, si tienen de más de 1 cm. BAAF: biopsia por aspiración con aguja fina; T3: triyodotironina; T4: tiroxina; TSH: tirotropina. aLa necesidad de BAAF con guía ecográfica depende de los factores de riesgo clínicos de cáncer de tiroides, del tamaño del nódulo y de la apariencia ecográfica. 53 Trastornos de la glándula tiroides Tabla 32. Resumen de las directrices de la American Thyroid Association del 2015: patrones ecográficos y recomendaciones para la biopsia por aspiración con aguja fina Patrón ecográfico Imagen representativa Descripción Riesgo estimado de cáncer Umbral del tamaño para biopsia por aspiración con aguja fina Benigno Quiste puro (anecoico sin flujo sanguíneo interno) <1% Biopsia por aspiración con aguja fina no recomendada Sospecha muy baja Algunos nódulos quísticos y sólidos mixtos (nódulos espongiformes)a <3% 2 cmc Sospecha baja Nódulos sólidos isoecoicos/ hiperecoicos; algunos nódulos quísticos y sólidos mixtos 5%-10% 1,5 cm Sospecha intermedia Nódulos sólidos hipoecoicos 10%-20% 1 cm Sospecha alta Nódulos sólidos hipoecoicos con una o varias características sospechosasb >70%-90% 1 cm a Los espacios microquísticos que ocupan más del 50% del volumen del nódulo están altamente correlacionados con una citología benigna. bMicrocalfcificaciones, forma más alta que ancha en la proyección transversal, márgenes irregulares, diseminación extratiroidea o ganglios linfáticos patológicos (la imagen muestra un nódulo sólido hipoecoico con márgenes irregulares). cLa observación clínica constituye una alternativa aceptable. Reproducido y adaptado de: Haugen Bryan R, Alexander Erik K, Bible Keith C, Doherty Gerard M, Mandel Susan J, Nikiforov Yuri E, Pacini Furio, Randolph Gregory W, Sawka Anna M, Schlumberger Martin, Schuff Kathryn G, Sherman Steven I, Sosa Julie Ann, Steward David L, Tuttle R. Michael, and Wartofsky Leonard. Thyroid. Ene 2016. Avance de la edición impresa http://doi.org/10.1089/thy.2015.0020 Publicado en Volume: 26 Issue 1: 12 de enero de 2016; avance en Internet antes de la edición: 14 de octubre de 2015. tienen una citología benigna, el 20% son indeterminados y entre el 5% y el 10% muestran signos de malignidad. El manejo de los nódulos tiroideos de citología indeterminada (Bethesda III-IV) puede ser complicado y se recomienda la derivación a un endocrinólogo. Esta indicado un control clínico de todos los nódulos tiroideos que incluya la determinación de la TSH sérica, dado que las glándulas tiroides estructuralmente anómalas conllevan un riesgo elevado de disfunción tiroidea (véase «Trastornos de la función tiroidea»). El patrón ecográfico de la American Thyroid Asso54 ciation y el contexto clínico sirven de orientación a la hora de decidir el momento de inicio del seguimiento ecográfico de los nódulos benignos y los no evaluados con BAAF. Debe obtenerse una ecografía repetida en el plazo de entre 6 y 12 meses de todos los nódulos sospechosos, al cabo de entre 12 y 24 meses con los nódulos con un índice de sospecha intermedio y bajo y a los 24 meses o más en caso de nódulos con un índice de sospecha muy bajo. La BAAF repetida está indicada para todos los nódulos con un índice de sospecha muy alto, los nódulos con hallazgos ecográficos nuevos preocupantes y Trastornos de la glándula tiroides Tabla 33. Diagnósticos de nódulos tiroideos obtenidos mediante biopsia por aspiración con aguja fina y riesgo de cáncer Diagnóstico con biopsia por aspiración con aguja fina Riesgo de cáncera Manejo Benigno 0%-3% Repetir ecografía en 6-24 mesesb Atipia de significación incierta/lesión folicular de significación incierta 10%-30% Repetir BAAF en 3 mesesc Presunta neoplasia folicular 25%-40% Lobectomíab Presunto cáncer 50%-70% Lobectomía o tiroidectomía total Maligno 97%-99% Lobectomía o tiroidectomía total No diagnóstico 5%-10% Repetir BAAF En caso de dos BAAF no diagnósticas, cirugía aEl riesgo de cáncer por diagnóstico citológico incluye la neoplasia tiroidea folicular no invasiva con características nucleares de tipo papilar (noninvasive follicular thyroid neoplasm with papillary-like nuclear features [NIFTP]), una lesión «precancerosa». Si se cuenta como benigna, el riesgo de cáncer se reduce en todas las categorías Bethesda, exceptuando las lecturas no diagnósticas y benignas. Datos de Cibas ES, Ali SZ. The 2017 Bethesda System for reporting thyroid cytopathology. Thyroid. 2017 Nov;27(11):1 341 -1346. doi:10.1089/thy.201 7.0500. PubMed PMID: 29091573. bSi corresponde al patrón de «sospecha alta» de la American Thyroid Association, repetir la ecografía y la BAAF en 6-12 meses. aLas estrategias terapéuticas complementarias incluyen el análisis genético molecular del nódulo y el uso selectivo de gammagrafía tiroidea (cuando el nivel de hormona estimulante de la tiroides en suero es bajo o normal). BAAF: biopsia por aspiración con aguja fina. los nódulos con un índice de sospecha intermedio o bajo que experimenten un aumento de tamaño significativo, definido como un aumento del 20% en al menos dos dimensiones o un aumento del volumen del nódulo superior al 50%. No se recomienda repetir la BAAF en caso de nódulos con dos biopsias negativas. Bocio El término «bocio» denota una glándula tiroides hipertrofiada. El bocio puede darse en el contexto de función tiroidea normal, hipotiroidismo o hipertiroidismo. La causa más frecuente en todo el mundo es el bocio endémico causado por un déficit de yodo grave. En los pacientes que presentan bocio, es necesario preguntar por el consumo de yodo, la velocidad del cambio de tamaño y los factores de riesgo de cáncer de tiroides (véase «Cáncer de tiroides»). La anamnesis debe centrarse en los síntomas indicativos de exceso o déficit de hormona tiroidea y de compresión. Los síntomas compresivos incluyen disnea, tos, disfagia y alteraciones vocales y son evidentes en entre el 10% y el 20% de los pacientes con bocio, la mayoría de los cuales presentan también una tiromegalia clínicamente evidente. En la exploración, es necesario evaluar el desvío traqueal y observar el tamaño, la simetría y la consistencia de la tiroides y la presencia de nódulos. Hay que evaluar una posible obstrucción venosa haciendo que el paciente eleve los brazos por encima de la cabeza. El hallazgo de dilatación venosa yugular, congestión facial y rubor indican posible obstrucción de la salida torácica con retorno venoso reducido (signo de Pemberton) (Figura 8). El nivel de TSH debe evaluarse en los pacientes con bocio. Si es bajo, es necesario determinar la T4 y la T3 total y llevar a cabo una gammagrafía tiroidea. Si es normal o alto, en los pacientes con factores de riesgo de cáncer de tiroides, nódulos tiroideos palpables, asimetría glandular, bocios grandes, patrón de crecimiento rápido o síntomas compresivos está indicada una ecografía de tiroides/cuello. En los pacientes con signos o síntomas indicativos de compresión hay que llevar a cabo las pruebas adicionales que se indican más adelante, y es posible que sea necesaria una cirugía para el manejo sintomático. El tratamiento de los trastornos hipotiroideos e hipertiroideos en el contexto de bocio se describe a continuación. PUNTOS CLAVE • La evaluación inicial de un nódulo tiroideo empieza con la determinación de la tirotropina en suero. • Los pacientes con inhibición de la tirotropina se evalúan con gammagrafía tiroidea; los pacientes con hormona estimulante de la tiroides normal o elevada se evalúan con ecografía. • Los pacientes con nódulos ≥1 cm deben evaluarse con biopsia por aspiración con aguja fina. AMAV • La biopsia por aspiración con aguja fina no está recomendada para nódulos subcentimétricos, a menos que cursen con síntomas, linfadenopatía patológica o diseminación extratiroidea. F i g u r a 8 . El signo de Pemberton. Cabeza y cuello con los brazos abajo (izquierda) y los brazos elevados (derecha). 55 Trastornos de la glándula tiroides PUNTOS CLAVE • La causa más frecuente de bocio en todo el mundo es el déficit de yodo grave. • En los pacientes que presentan bocio es necesario preguntar por el consumo de yodo y la velocidad del cambio de tamaño, evaluar los signos y síntomas de compresión y valorar las manifestaciones clínicas de exceso o déficit de hormonas tiroideas. • En los pacientes con bocio debe evaluarse la tirotropina en suero; si es baja, deben medirse la tiroxina libre (T4) y la triyodotironina (T3) total y realizar una gammagrafía tiroidea; si la T4 y la T3 son normales o altas, está indicada una ecografía de tiroides/cuello en presencia de factores de riesgo de cáncer de tiroides, nódulos tiroideos palpables, asimetría glandular, bocios grandes, patrón de crecimiento rápido o síntomas compresivos. Bocio multinodular El bocio multinodular es la causa de bocio más frecuente en las personas de edad avanzada de Estados Unidos. La evaluación consiste en la determinación de la TSH en suero; cuando no está inhibida, debe realizarse una ecografía de tiroides/cuello y evaluar los nódulos diferenciados, como se ha comentado con anterioridad (véase «Nódulos tiroideos»). La frecuencia del cáncer de tiroides en los pacientes con bocio multinodular es parecida a la de los nódulos tiroideos solitarios. Los signos y síntomas de compresión o presunta diseminación subesternal requieren pruebas adicionales. La TC o la RM de cuello y tórax (en caso de presunto bocio subesternal) pueden definir las relaciones anatómicas y detectar estrechamientos traqueales. Es necesario evitar la administración de medios de contraste yodados en la medida de lo posible para no provocar un hipertiroidismo inducido por yodo (fenómeno de Jod-Basedow). En los pacientes con síntomas de compresión de las vías respiratorias o en caso de luz traqueal <1 cm de diámetro en la TC o la RM, está indicado un estudio espirométrico con curva de flujo-volumen (véase el módulo «Neumología y Cuidados intensivos» de MKSAP 18). Una endoscopia o un estudio de la deglución pueden evaluar la compresión extrínseca del esófago en los pacientes con disfagia cervical. Para confirmar clínicamente una presunta parálisis de las cuerdas vocales, está indicada una interconsulta con un otorrinolaringólogo. La cirugía está indicada en los casos de compresión significativa o sospecha de cáncer. más, el bocio difuso puede darse en pacientes eutiroideos en ausencia de procesos inflamatorios o neoplásicos predisponentes. La predisposición genética, la insuficiencia de yodo y el tabaquismo son factores contribuyentes. En los pacientes eutiroideos con bocio difuso está indicada una ecografía de tiroides/cuello, también recomendada en los pacientes con enfermedad de Graves o tiroiditis de Hashimoto en caso de asimetría de la glándula tiroides o nódulos en la exploración. Como se ha dicho anteriormente, en presencia de signos o síntomas de compresión está indicado realizar pruebas adicionales. En el contexto de compresión significativa, se considera una cirugía tiroidea. Cáncer de tiroides El cáncer de tiroides se diagnostica en 13,9 por cada 100.000 personas al año en Estados Unidos. La incidencia del cáncer de tiroides ha aumentado en las cuatro últimas décadas y en la actualidad representa el quinto cáncer más frecuente en las mujeres. Gran parte de este cambio puede atribuirse al aumento del diagnóstico de los pequeños cánceres no invasivos, que inicialmente se detectaban de manera incidental en las pruebas de imagen obtenidas por otros motivos (ecografías Doppler de la carótida, TC de cuello/tórax, PET). Las tasas de mortalidad han permanecido estables, con una tasa de supervivencia a cinco años del 98,1%. El carcinoma papilar tiroideo y el carcinoma folicular tiroideo, denominados conjuntamente cáncer de tiroides diferenciado, representan la mayoría de los diagnósticos de cáncer de tiroides en Estados Unidos. El carcinoma papilar tiroideo se disemina con frecuencia a los ganglios linfáticos cervicales, pero está asociado a un riesgo bajo de metástasis a distancia; aunque las metástasis en los ganglios linfáticos son infrecuentes en el carcinoma folicular tiroideo, pueden observarse metástasis en pulmón, huesos y otras localizaciones. Los tipos y la frecuencia del cáncer de tiroides se presentan en la Figura 9. 1 1 3 10 Papilar Folicular Medular Anaplásico Bocio difuso La causa más frecuente de bocio difuso es la enfermedad tiroidea autoinmune asociada a disfunción de la tiroides (tiroiditis de Hashimoto y enfermedad de Graves). Los trastornos infiltrativos, como la tiroiditis de Riedel (relacionada con la IgG4), constituyen causas infrecuentes de bocio difuso. Ade56 85 Otros (linfoma tiroideo y metástasis de otros cánceres) F i g u r a 9 . Frecuencia relativa de los tipos de cáncer de tiroides. Datos de Hundahl SA, Fleming ID, Fremgen AM, Menck HR. A National Cancer Data Base report on 53,856 cases of thyroid carcinoma treated in the U.S., 1985-1995. Cancer. 1998 Dec 15;83(12):2638-48. [PMID: 9874472] Trastornos de la glándula tiroides Muchas veces el cáncer de tiroides se detecta de manera incidental; sin embargo, puede ser detectado en una exploración del cuello. Los factores de riesgo de cáncer de tiroides incluyen antecedentes personales de exposición a radiación ionizante, con una mayor prevalencia de carcinoma papilar tiroideo en las personas expuestas a radiación ionizante (>10 rads) y un riesgo especialmente elevado tras la exposición en la infancia, como en el caso de accidentes nucleares (Chernóbil), y antecedentes personales o familiares de cáncer tiroideo. Otros factores de riesgo de cáncer de tiroides son las edades extremas (menos de 30 o más de 60 años) y el sexo masculino. Los hallazgos indicativos de cáncer incluyen crecimiento nodular rápido, un nódulo fijo duro, disfagia, parálisis de las cuerdas vocales (ronquera) y linfadenopatía cervical. El diagnóstico se confirma mediante biopsia por aspiración con aguja fina. La cirugía es el puntal del tratamiento del cáncer de tiroides. Tanto la tiroidectomía total como la hemitiroidectomía son aceptables para los cánceres de tiroides diferenciados unilaterales que miden 1-4 cm, siempre que no haya presunta diseminación locorregional. De lo contrario, está indicada la tiroidectomía total. Los riesgos específicos de la cirugía de tiroides son hipocalcemia como consecuencia de la extirpación o la desvascularización de las glándulas paratiroides y dificultad para respirar o alteraciones vocales por daño en los nervios laríngeos recurrentes. Dado el menor riesgo de complicaciones postoperatorias, es preferible la derivación a un cirujano que tenga un gran volumen de operaciones de tiroides (>90 casos al año). Debe considerarse el uso postoperatorio de yodo radiactivo (131I) con el objetivo doble de la ablación de los restos tiroideos y la administración de un tratamiento adyuvante a los pacientes con cáncer de tiroides diferenciado y un riesgo intermedio o alto de recidiva, como en los casos de diseminación extratiroidea, invasión linfovascular, histología poco diferenciada/más agresiva o enfermedad metastásica. Para favorecer la captación del 131I por parte de las células foliculares tiroideas es necesario estimular la TSH, ya sea mediante la retirada de la reposición hormonal tiroidea (levotiroxina) o bien con la administración de TSH humana recombinante (rhTSH). Tras el tratamiento con 131I, los pacientes se someten a una gammagrafía de rastreo corporal total para detectar las regiones de captación de 131I correspondientes a enfermedad metastásica. Se prevé captación en el lecho tiroideo después de la cirugía, pero no en otros sitios. El tratamiento con 131I se utiliza también para tratar las recidivas del cáncer de tiroides no aptas para resección quirúrgica. Después del tratamiento inicial contra el cáncer, se controlan los títulos de tiroglobulina (Tg) sérica, un marcador sensible para la detección de enfermedad persistente o recidivante, y de anticuerpos antitiroglobulina (AcTg). Cuando hay AcTg, los niveles de Tg no pueden interpretarse, porque los anticuerpos pueden disminuir falsamente la determinación de Tg. En este caso, el nivel de AcTg hace las veces de indicador indirecto. Un descenso del título de AcTg con el tiem- po está correlacionado con un pronóstico favorable, mientras que un título en aumento apunta a enfermedad persistente o recidivante. En los controles rutinarios del cáncer de tiroides se obtienen ecografías del cuello de manera habitual, normalmente entre 6 y 12 meses después del tratamiento anticanceroso inicial. En los pacientes con un riesgo elevado de recidiva de la enfermedad, puede llevarse a cabo un rastreo corporal total con yodo radiactivo (123I o 131I) con determinación de Tg estimulada con TSH. En caso de sospecha de cáncer de tiroides residual o recidivante, como en los casos en que la Tg sérica permanece elevada de manera persistente o aumenta con el tiempo pero no se detecta nada en la radiografía de cuello ni en el rastreo corporal total con yodo radiactivo, las pruebas de imagen complementarias, incluyendo la TC, la RM, la gammagrafía ósea o la PET/TC, pueden ser útiles para localizar la enfermedad. El tratamiento del cáncer de tiroides diferenciado de riesgo intermedio o alto consiste en la inhibición de la TSH con levotiroxina diaria. Las células foliculares tiroideas responden a la TSH, al igual que la mayoría de los cánceres de tiroides bien diferenciados. A fin de reducir la recidiva del cáncer, se administra una dosis suficiente de levotiroxina para inhibir la TSH sérica por debajo de la normalidad con un valor objetivo personalizado. En los pacientes con un riesgo bajo de cáncer de tiroides, puede buscarse un nivel de la TSH sérica ≤2, pero situado en el límite inferior del intervalo de referencia o por encima de este. Es correcto que los internistas se encarguen del control y el ajuste de la dosis en conjunción con el endocrinólogo. El cáncer de tiroides metastásico se maneja con vigilancia activa, cirugía adicional o tratamiento con 131I, seguido de radioterapia de haz externo y/o quimioterapia (inhibidores de la tirosincinasa). El cáncer de tiroides anaplásico es un cáncer tiroideo infrecuente pero agresivo que puede presentarse en pacientes con cáncer de tiroides diferenciado preexistente o de nueva aparición. Tiene un pronóstico desalentador, con una mediana de la supervivencia de cinco meses. El cáncer de tiroides anaplásico se manifiesta con una masa en el cuello que crece con rapidez y puede ser irresecable en el momento del diagnóstico. El tratamiento es paliativo en la mayoría de los casos y consiste en cirugía, radioterapia de haz externo y quimioterapia. El cáncer medular de tiroides tiene su origen en las células parafoliculares. En el cáncer medular de tiroides familiar y la neoplasia endocrina múltiple (NEM) 2A y 2B hay mutaciones del oncogén de la línea germinal RET. Teniendo en cuenta su asociación con el feocromocitoma, es necesario descartar NEM con análisis genéticos antes de la cirugía. El cáncer medular de tiroides se trata con tiroidectomía total y disección de los ganglios linfáticos de la zona central del cuello. La levotiroxina está indicada para tratar el hipotiroidismo postoperatorio en los pacientes con cáncer medular de tiroides con un nivel de TSH en suero objetivo dentro del intervalo de referencia. La calcitonina sérica, los niveles de antígeno 57 Trastornos de la glándula tiroides carcinoembrionario sérico y la ecografía del cuello forman parte de la vigilancia rutinaria del cáncer. El cáncer papilar de tiroides de bajo riesgo, confinado a la glándula tiroides, que se ha resecado por completo, no ha metastatizado y no demuestra características patológicas agresivas (invasión linfovascular o variante de células altas) no requiere más tratamiento. El riesgo de muerte relacionada con la enfermedad es inferior al 1%, y el riesgo de recidiva de enfermedad estructural de los microcarcinomas papilares unifocales de bajo riesgo se sitúa entre el 1% y el 2%. Los pacientes que se someten a lobectomía o tiroidectomía obtienen resultados excelentes similares. PUNTOS CLAVE • Muchas veces el cáncer de tiroides se detecta de manera incidental, pero puede detectarse cuando se aprecia un nódulo tiroideo o un ganglio linfático anómalo en el cuello en la exploración. • La cirugía constituye el puntal del tratamiento del cáncer de tiroides; tanto la hemitiroidectomía como la tiroidectomía total son aceptables para los cánceres de tiroides diferenciados unilaterales que miden 1-4 cm, siempre que no haya presunta diseminación locorregional, mientras que la tiroidectomía total es preferible en todos los demás casos. • Además de la cirugía, los pacientes con cáncer de tiroides diferenciado y riesgo de recidiva intermedio o alto reciben tratamiento con yodo radiactivo (131I) e inhibición de la tirotropina con levotiroxina. Evaluación de la función tiroidea La medición de la TSH sérica es la prueba inicial más sensible y recomendada para evaluar la función tiroidea. Se utiliza para determinar si el paciente presenta eutiroidismo, hipotiroidismo o hipertiroidismo. Si la TSH está inhibida, es necesario evaluar la T4 libre y la T3 total para detectar hipertiroidismo manifiesto o subclínico, y si la TSH está elevada, es necesario evaluar la T4 libre para detectar hipotiroidismo manifiesto o subclínico. La determinación de la TSH sérica sola es suficiente excepto en determinadas circunstancias, como un presunto hipotiroidismo central, en cuyo caso también está indicada la determinación de la T4 libre (véase «Trastornos de la hipófisis»). Hay diversos ensayos que permiten evaluar y determinar con exactitud las concentraciones de T4 total y libre y de T3 total en la mayoría de los pacientes con disfunción tiroidea manifiesta. Los ensayos para la T3 libre disponibles en el mercado son menos fiables. Pueden producirse alteraciones de la globulina fijadora de tiroxina y otras proteínas fijadoras de transporte en ciertos estados fisiológicos (embarazo), en determinados estados patológicos (síndrome nefrótico) y como consecuencia del uso de medicamentos (tratamiento con estrógenos orales). Los niveles de T4 total y T3 variarán en fun58 ción del aumento o descenso de las proteínas fijadoras y no reflejan el funcionamiento tiroideo real. Normalmente, la T4 libre, que corresponde a la fracción de T4 no ligada proteínas en suero, se determina utilizando ensayos inmunométricos que están disponibles de manera generalizada. Estas pruebas son exactas en la mayoría de los contextos clínicos, incluyendo los pacientes con alteraciones leves de las proteínas fijadoras; sin embargo, pueden ser inexactas en caso de alteraciones más significativas (hipertiroxinemia disalbuminémica familiar). La medición de la T4 mediante diálisis de equilibrio tiene una gran exactitud, pero es cara, su disponibilidad es limitada y rara vez es necesaria. Es necesario aconsejar a los pacientes que toman más de 5-10 mg/día de biotina que suspendan su consumo durante dos días antes de la evaluación analítica de la función tiroidea. La biotina es una vitamina hidrosoluble habitualmente presente en suplementos alimentarios de venta sin prescripción médica. Se ha observado que los niveles altos de biotina circulante interfieren en los análisis que utilizan la estreptavidina-biotina como sistema de inmovilización. La biotina arroja resultados falsamente elevados en los inmunoanálisis competitivos utilizados para medir moléculas pequeñas (T4 libre, T3 libre, T4 total y T3 total) y da lugar a resultados falsamente bajos en los ensayos tipo sándwich empleados para medir moléculas grandes (TSH). Se recomienda la medición de la T3 en tres contextos: 1) en la evaluación de la tirotoxicosis para identificar la toxicosis por T3 aislada, 2) para evaluar la gravedad del hipertiroidismo y la respuesta al tratamiento y, 3) potencialmente, para diferenciar el hipertiroidismo de la tiroiditis destructiva. En la toxicosis por T3, el cociente de T3/T4 suele ser >20 debido a la secreción preferencial de T3. Varios fármacos pueden afectar a la función y la reposición tiroideas (Tabla 34). La medición de la T3 en el contexto de hipotiroidismo no es necesaria ni está recomendada; los niveles se mantienen normales a menos que el hipotiroidismo sea grave. En el hipotiroidismo, se elevará la TSH en un primer momento y posteriormente se producirán anomalías en el nivel de T4. No hay indicaciones clínicas para la evaluación de los valores de T3 inversa, por lo que esta no se recomienda. PUNTO CLAVE • La medición de la triyodotironina en el contexto de hipotiroidismo no es necesaria ni está recomendada; los niveles se mantienen normales a menos que el hipotiroidismo sea grave. Trastornos de la función tiroidea Exceso de hormonas tiroideas (hipertiroidismo y tirotoxicosis) El término «tirotoxicosis» describe la exposición de los tejidos a niveles altos de hormonas tiroideas circulantes (T4 y/o T3) por cualquier causa. El hipertiroidismo corresponde a la tirotoxicosis causada por la producción endógena excesiva de hor- AMAV Trastornos de la glándula tiroides Tabla 34. Medicamentos que afectan a la función o reposición tiroideas Mecanismo de acción Fármacos Comentarios Disminución de la absorción o la circulación enterohepática de la levotiroxina Calcio Se recomienda que la administración de levotiroxina esté separada de estos medicamentos en varias horas Inhibidores de la bomba de protones Hierro Colestiramina Hidróxido de aluminio Aceite de soja Sucralfato Psyllium Aumento del metabolismo de la levotiroxina Fenitoína Las dosis más altas de levotiroxina pueden requerir el mantenimiento de la TSH en el intervalo normal Carbama Cepina Rifampicina Fenobarbital Sertralina Tiroiditis Amiodarona Puede causar hipotiroidismo o hipertiroidismo Litio Interferón alfa Interleucina 2 Inhibidores de la tirosincinasa Sunitinib Inhibidor de puntos de control inmunitario Nivolumab, pembrolizumab Aparición nueva de anticuerpos antitiroideos Interferón alfa Puede desarrollarse tiroiditis de Hashimoto, enfermedad de Graves o tiroiditis indolora Inhibición de la síntesis o liberación de TSH Glucocorticoides Provoca inhibición de la TSH; es necesario volver a comprobar la TSH 6-8 semanas después de la interrupción de estos medicamentos para ver si ha vuelto a la normalidad Dopamina Dobutamina Octreotida Aumento de la globulina fijadora de tiroxina Estrógeno Tamoxifeno Falsa elevación de los niveles de T3 total y T4 total; la T3 libre y la T4 libre reflejan con mayor exactitud los niveles hormonales Metadona Disminución de la globulina fijadora de tiroxina Tratamiento androgénico Glucocorticoides Falsa disminución de los niveles de T3 total y T4 total; la T3 libre y la T4 libre reflejan con mayor exactitud los niveles hormonales Niacina T3: triyodotironina; T4: tiroxina; TSH: tirotropina. monas tiroideas. La prevalencia global del hipertiroidismo en Estados Unidos se sitúa en el 1,3%. En el hipertiroidismo primario, la disfunción tiene origen en la glándula tiroides. La secreción elevada de TSH es una causa secundaria infrecuente de hipertiroidismo. Cuadro clínico y diagnóstico En la Tabla 35 se presentan los signos y síntomas del exceso de hormonas tiroideas. Los pacientes de edad avanzada con hipertiroidismo pueden estar apáticos en lugar de presentar los síntomas clásicos. En la tirotoxicosis por cualquier causa pue- de observarse retracción palpebral, que se debe al aumento del tono adrenérgico. El diagnóstico de hipertiroidismo está basado en las pruebas bioquímicas que demuestran un nivel bajo de TSH y concentraciones elevadas de T4 libre y/o T3 total. La gammagrafía tiroidea con captación de yodo radiactivo (CYR) puede comprobar la causa. La CYR es alta (>30%) o inadecuadamente normal en el hipertiroidismo y baja (<10%) en otras causas de tirotoxicosis. Pueden realizarse otras pruebas cuando el diagnóstico clínico es incierto, en caso de que la CYR no esté disponible o no sea fiable (pacientes que toman amiodarona o litio o han 59 Trastornos de la glándula tiroides Tabla 35. Manifestaciones clínicas de tirotoxicosis y déficit de hormonas tiroideasa Signo o síntoma Tirotoxicosis Déficit de hormonas tiroideas Generales Fatiga, pérdida de pesob, intolerancia al calor Fatiga, aumento de peso, intolerancia al frío Neuropsiquiátricos Disminución de la concentración, ansiedad, irritabilidad, insomnio Disminución de la concentración, depresión, retraso psicomotor, hipersomnolencia Hiperreflexia, temblor, retracción palpebral Relajación tardía de los RTP Cardiovasculares Palpitaciones, taquicardia, hipertensión sistólica, insuficiencia cardíaca por alto gasto Bradicardia, hipertensión diastólica Gastrointestinales Hiperfagia, aumento de la frecuencia de las deposiciones, heces sueltas, diarrea Estreñimiento Genitourinarios Alteraciones menstruales (oligomenorrea, amenorrea) Alteraciones menstruales (menorragia) Musculoesquéleticos Debilidad muscular Mialgia, artralgia Cutáneos Pérdida de cabello, aumento de la producción de sebo/acné; edema periorbital Pérdida de cabello, piel seca, uñas frágiles, edema periorbital, truncación lateral de las cejas, alteraciones cutáneas mixedematosas aEl bocio puede estar presente en la tirotoxicosis o en el déficit de hormonas tiroideas. Véase el texto para hallar las manifestaciones físicas características de la enfermedad de Graves. bEl hipertiroidismo subclínico puede cursar con un aumento leve de peso (inhibición de la hormona estimulante de la tiroides sin elevación de la T o la T ) debido 4 3 a la estimulación del apetito. RTP: reflejos tendinosos profundos. estado expuestos recientemente a medio de contraste yodado) o cuando la gammagrafía está contraindicada (embarazo y lactancia). Si se sospecha la presencia de enfermedad de Graves pero el diagnóstico clínico sigue siendo dudoso, las pruebas incluyen la determinación de inmunoglobulina estimuladora de la tiroides (TSI) o la de anticuerpos contra los receptores de tirotropina (AcTR), así como la ecografía tiroidea para evaluar los patrones de vascularidad. PUNTOS CLAVE • El diagnóstico de hipertiroidismo está basado en las pruebas bioquímicas que demuestran nivel bajo de tirotropina y concentraciones elevadas de tiroxina libre y/o triyodotironina total. • La gammagrafía tiroidea con determinación de la captación de yodo radiactivo puede verificar la causa del hipertiroidismo. Causas Las causas de tirotoxicosis se enumeran en la Tabla 36. La enfermedad de Graves, el bocio multinodular tóxico y el adenoma tóxico constituyen las causas más frecuentes de hipertiroidismo. Enfermedad de Graves La enfermedad de Graves es una enfermedad multiorgánica que puede afectar a la tiroides, los músculos oculares y la piel. Causa el 80% del hipertiroidismo en las regiones con yodo suficiente. Se trata de un trastorno tiroideo autoinmune que afecta de manera predominante a mujeres, con una incidencia máxima en los pacientes de entre 30 y 60 años de edad. 60 Además, la enfermedad de Graves es más frecuente en pacientes con otros trastornos autoinmunes o con antecedentes familiares de autoinmunidad tiroidea. Los linfocitos T se sensibilizan a los antígenos tiroideos y estimulan a los linfocitos B a producir anticuerpos contra el receptor de TSH (TSI o AcTR). La tiroides muestra una hipertrofia difusa, puede presentar un soplo y tiene una textura firme y lisa en la exploración; puede haber linfadenopatía cervical. A menudo cursa con hipertensión sistólica, taquicardia, hiperreflexia y piel húmeda y caliente. La oftalmopatía de Graves afecta al 25% de los pacientes. El consumo de tabaco es un factor de riesgo. Las manifestaciones clínicas incluyen edema periorbital, quemosis (edema conjuntival), proptosis ocular (protrusión del globo ocular), diplopía (debida a paresia oculomotora) y pérdida de visión. La oftalmopatía de Graves no responde al tratamiento del hipertiroidismo y muchas veces requiere glucocorticoides o cirugía. El mixedema pretibial es una dermopatía infiltrativa infrecuente de la enfermedad de Graves que afecta a entre el 2% y el 3% de los pacientes; se trata de un edema sin fóvea indurado, con apariencia de piel de naranja, normalmente en las crestas tibiales (véase el módulo «Dermatología» de MKSAP 18). PUNTOS CLAVE • En la enfermedad de Graves, la tiroides muestra una hipertrofia difusa, puede presentar un soplo y tiene una textura firme y lisa en la exploración; puede haber linfadenopatía cervical. • La oftalmopatía de Graves no responde al tratamiento del hipertiroidismo y muchas veces requiere glucocorticoides o cirugía. Trastornos de la glándula tiroides Tabla 36. Causas de tirotoxicosis Tabla 37. Causas de tiroiditis destructiva Trastorno Comentarios Trastorno Comentarios Enfermedad de Graves Frecuente; activación del TSHR activada por AcTR Tiroiditis indolora (silente) Bocio multinodular tóxico Frecuente; tejido tiroideo de funcionamiento autónomo Se observa con enfermedad tiroidea autoinmune subyacente (tiroiditis de Hashimoto) Tiroiditis puerperal Adenoma tóxico Frecuente; tejido tiroideo de funcionamiento autónomo Tiroiditis indolora que se produce en el posparto; hipotiroidismo permanente en el 20% Tiroiditis (aguda, subaguda, indolora) Frecuente; inflamación tiroidea que produce la liberación de las hormonas tiroideas almacenadas Tiroiditis inducida por medicamentos Tiroiditis indolora; amiodarona, litio, interferón alfa, interleucina 2, inhibidores de la tirosincinasa, inhibidores de puntos de control inmunitario Inducida por medicamentos Frecuente; amiodarona, litio, interferón alfa, interleucina 2, inhibidores de la tirosincinasa, inhibidores de puntos de control inmunitario Tiroiditis subaguda (de Quervain o granulomatosa subaguda) Tiroiditis dolorosa; sigue a una infección vírica de las vías respiratorias altas Tirotoxicosis facticia Frecuente; administración de hormona tiroidea exógena; ingestión de productos cárnicos porcinos o bovinos contaminados Infecciosa (supurativa) Tiroiditis dolorosa; infección por especies de Staphylococcus o Streptococcus que normalmente se observa en pacientes inmunodeprimidos Mediada por hCG (embarazo, enfermedad trofoblástica, tumor de células germinales) Frecuente en el embarazo, las otras formas son infrecuentes; unión indiscriminada de la hCG al TSHR debido a la subunidad alfa común de la TSH y la hCG Tiroiditis inducida por radiación Tiroiditis dolorosa; se da después de un tratamiento con yodo radiactivo o radioterapia de haz externo en el cuello Estruma ovárico Infrecuente; tejido tiroideo de funcionamiento autónomo en un teratoma ovárico que representa >50% del tumor Metástasis del cáncer folicular de tiroides Infrecuente; metástasis de carcinoma folicular tiroideo de funcionamiento autónomo Adenoma tirotropo Infrecuente; adenoma hipofisario secretor de TSH AcTR: anticuerpos contra los receptores de tirotropina; hCG: gonadotropina coriónica humana; TSH: tirotropina; TSHR: receptor de tirotropina. Adenoma tóxico y bocio multinodular Por lo general, el adenoma tóxico y el bocio multinodular afectan a ancianos, ya que la prevalencia de los nódulos tiroideos aumenta con la edad. Los nódulos tiroideos sintetizan y secretan hormonas tiroideas independientemente de la estimulación de la TSH. La exposición a medios de contraste yodados puede provocar la conversión de adenomas no tóxicos a adenomas tóxicos, como ocurre con las TC con contraste y los cateterismos cardíacos. Manejo En la mayoría de los pacientes tirotóxicos, los betabloqueantes (atenolol, metoprolol, propranolol) son beneficiosos para reducir los síntomas adrenérgicos con rapidez. El propranolol reduce la conversión periférica de T4 a T3, pero no es cardioselectivo y requiere una administración dos o tres veces al día. Son preferibles el atenolol y el metoprolol, porque su administración una vez al día incrementa el cumplimiento del tratamiento por parte del paciente y su naturaleza cardioselectiva reduce los efectos secundarios en el sistema nervioso central. Hay tres modalidades terapéuticas para el hipertiroidismo: tioamidas (metamizol y propiltiouracilo [PTU]), terapia ablativa con yodo radiactivo (131I) y tiroidectomía. La elección del tratamiento se basa en la causa del hipertiroidismo y en las preferencias del paciente. En los pacientes ≥65 años de edad que tienen enfermedad cardiovascular prevalente o múltiples comorbilidades, se recomienda el uso de metimazol a corto plazo para normalizar la función tiroidea antes del tratamiento con 131I o una tiroidectomía. Está recomendada la derivación a un endocrinólogo. Tiroiditis destructiva La tirotoxicosis observada en la tiroiditis destructiva se produce como consecuencia de la liberación no regulada de hormona tiroidea preformada de los folículos tiroideos dañados por la inflamación. Las causas se enumeran en la Tabla 37. Normalmente, la tiroiditis tiene tres fases: tirotóxica, hipotiroidea y regreso al eutiroidismo. Las dos primeras fases pueden durar hasta tres meses cada una. Una persona tiene un mayor riesgo de sufrir otros episodios de tiroiditis una vez resuelta la tiroiditis inicial. Enfermedad de Graves Los fármacos antitiroideos (tionamidas) se utilizan con frecuencia en el tratamiento inicial del hipertiroidismo de Graves, porque hasta el 50% de los casos experimentarán una remisión espontánea del hipertiroidismo en el plazo de 24 meses, sobre todo si el bocio es de tamaño pequeño y solo se necesitan dosis pequeñas de tionamida para lograr el eutiroidismo. Cuando los niveles de AcTR siguen elevados en el momento 61 Trastornos de la glándula tiroides de la suspensión del fármaco, es probable que se produzca una recidiva del hipertiroidismo. Si el hipertiroidismo de Graves recidiva, se recomienda un tratamiento definitivo. La agranulocitosis y la disfunción hepática son dos efectos secundarios infrecuentes pero graves de las tionamidas. Antes del tratamiento, debe obtenerse un hemograma completo basal con fórmula diferencial y un perfil hepático. En el contexto de fiebre o faringitis, hay que sospechar la presencia de agranulocitosis y evaluar la cifra de neutrófilos. La función hepática debe evaluarse en cualquier paciente con síntomas o signos de disfunción hepática (ictericia). El fármaco antitiroideo de elección es el metimazol, excepto en el primer trimestre de embarazo, ya que el PTU se ha asociado a hepatonecrosis mortal. El objetivo de la terapia ablativa con 131I en la enfermedad de Graves es lograr que el paciente sea hipotiroideo. Las mujeres que reciben tratamiento con 131I deben evitar el embarazo durante 6-12 meses después del tratamiento. En los pacientes con oftalmopatía de Graves, se observa un incremento agudo de los títulos de autoanticuerpos tiroideos tras el tratamiento con yodo radiactivo que puede agravar los síntomas oculares. El pretratamiento de la oftalmopatía de Graves o la elección de otros tratamientos depende de la gravedad de la enfermedad ocular. La tiroidectomía es especialmente adecuada en el hipertiroidismo de Graves cuando hay un bocio de gran tamaño y síntomas compresivos, oftalmopatía de Graves moderada o grave y/o cáncer de tiroides coexistente o hiperparatiroidismo primario. Otras causas El tratamiento de primera línea para un adenoma tóxico o un bocio multinodular tóxico consiste en el uso de 131I o la cirugía tiroidea. La elección está determinada por las preferencias del paciente, la presencia de síntomas compresivos y el acceso a un cirujano con un gran volumen de intervenciones tiroideas. La tiroiditis destructiva se maneja con espera y betabloqueantes para controlar los síntomas adrenérgicos y antiinflamatorios no esteroideos (AINE), seguidos de un tratamiento con glucocorticoides en dosis altas para el control del dolor en la tiroiditis dolorosa. PUNTOS CLAVE • En la mayoría de los pacientes tirotóxicos, los betabloqueantes son beneficiosos para reducir los síntomas adrenérgicos. • Las tres modalidades de tratamiento para el hipertiroidismo son las tionamidas (metimazol y propiltiouracilo), la terapia ablativa con yodo radiactivo (131I) y la tiroidectomía; la elección del tratamiento se basa en la causa del hipertiroidismo y en las preferencias del paciente. • Los fármacos antitiroideos (tionamidas) están asociados a una tasa de remisión espontánea de hasta el 50% en el plazo de 24 meses; la agranulocitosis y la disfunción hepática son dos efectos secundarios infrecuentes pero graves. 62 Hipertiroidismo subclínico El diagnóstico del hipertiroidismo subclínico se basa en la inhibición de la TSH sérica con niveles normales de T4 y T3. El hipertiroidismo subclínico afecta al 0,7% de la población estadounidense. Aproximadamente entre el 0,5% y el 7% progresan a hipertiroidismo manifiesto al año y entre el 5% y el 12% revierten a función tiroidea normal. La causa más frecuente es el bocio multinodular tóxico. El hipertiroidismo subclínico se ha asociado a un riesgo elevado de fibrilación auricular y complicaciones cardiovasculares. Un gran estudio de cohortes prospectivo reciente demostró mayores tasas de fractura de cadera con hipertiroidismo subclínico (7% en el hipertiroidismo subclínico frente a 4,5% en los pacientes eutiroideos); sin embargo, no se sabe si el tratamiento reduce el riesgo de fractura. En más del 25% de los pacientes con hipertiroidismo subclínico, la TSH se normalizará en el plazo de seis semanas. Por lo tanto, es razonable observar y volver a comprobar la función tiroidea antes de iniciar un tratamiento, a menos que haya un riesgo alto de contraindicaciones, como en el caso de los pacientes con enfermedad cardíaca. El riesgo de complicaciones cardiovasculares y óseas observado es mayor con un nivel de TSH sérica <0,1 µU/ml (0,1 mU/l). El tratamiento del hipertiroidismo subclínico está recomendado en los pacientes con niveles de TSH sérica <0,1 µU/ml (0,1 mU/l) y con síntomas, factores de riesgo cardíacos, enfermedad cardíaca u osteoporosis, así como en las mujeres posmenopáusicas que no tomen tratamiento con estrógenos o bifosfonatos. PUNTOS CLAVE • El hipertiroidismo subclínico se diagnostica sobre la base de la inhibición de la tirotropina en suero, pero con niveles normales de tiroxina y triyodotironina. • El tratamiento del hipertiroidismo subclínico está recomendado en los pacientes con niveles de tirotropina en suero inferiores a 0,1 µU/ml (0,1 mU/l) y síntomas, factores de riesgo cardíacos, enfermedad cardíaca u osteoporosis, así como en las mujeres posmenopáusicas que no tomen tratamiento con estrógenos o bifosfonatos. Déficit de hormonas tiroideas El déficit de hormonas tiroideas afecta a más de 10 millones de estadounidenses. Es 10 veces más frecuente en las mujeres que en los varones. Cuadro clínico y diagnóstico Los signos y síntomas de déficit de hormonas tiroideas se enumeran en la Tabla 35. El déficit de hormonas tiroideas está asociado también a anomalías analíticas como anemia, niveles elevados de colesterol ligado a lipoproteínas de baja densidad (LDL) e hiponatremia. El diagnóstico de hipotiroidismo primario se obtiene con la medición de la TSH sérica y, en caso de resultado elevado, Trastornos de la glándula tiroides con la determinación de la T4 libre, que puede añadirse en la mayoría de los laboratorios. La TSH sérica está elevada en el hipotiroidismo tanto manifiesto como subclínico, pero la T4 libre es normal en el hipotiroidismo subclínico y baja en el hipotiroidismo manifiesto. La mayor parte de los pacientes con tiroiditis de Hashimoto presentan autoanticuerpos tiroideos (anticuerpos contra la peroxidasa tiroidea [TPO]); no obstante, la medición del título de anticuerpos contra la TPO no es necesaria a menos que el diagnóstico sea dudoso. El diagnóstico del hipotiroidismo se describe en otro lugar (véase «Trastornos de la hipófisis»). PUNTOS CLAVE • El diagnóstico de hipotiroidismo primario se obtiene con la medición la tirotropina en suero y, en caso de resultado elevado, con la medición posterior de la tiroxina libre. • La tirotropina en suero está elevada en el hipotiroidismo tanto manifiesto como subclínico, pero la tiroxina libre es normal en el hipotiroidismo subclínico y baja en el hipotiroidismo manifiesto. AMAV • La mayor parte de los pacientes con tiroiditis de Hashimoto presentan autoanticuerpos tiroideos (anticuerpos contra la peroxidasa tiroidea [TPO]); no obstante, la medición del título de anticuerpos contra la TPO no es necesaria a menos que el diagnóstico sea dudoso. Causas Las causas de hipotiroidismo se enumeran en la Tabla 38. La causa más frecuente en Estados Unidos es la insuficiencia autoinmune de la glándula tiroides (debido a tiroiditis de Hashi­ moto), mientras que el déficit de yodo, que afecta a 2.000 millones de personas en todo el mundo, es la causa más frecuente a nivel global. El déficit de yodo es infrecuente en Estados Unidos gracias a las iniciativas para enriquecer los alimentos (sal yodada). El hipotiroidismo central también es infrecuente. Hipotiroidismo primario La tiroiditis de Hashimoto (tiroiditis linfocítica crónica) es un trastorno tiroideo autoinmune que se caracteriza por la infiltración difusa de linfocitos y células plasmáticas en la glándula tiroides con atrofia folicular y posterior formación de cicatrices. Es más frecuente en pacientes con otros trastornos autoinmunes o con antecedentes familiares de autoinmunidad tiroidea. El bocio difuso se observa sobre todo en pacientes jóvenes. La mayoría de los pacientes (90%) presentan anticuerpos contra la TPO, y el riesgo de experimentar hipotiroidismo es cuatro veces mayor en los pacientes eutiroideos con anticuerpos contra la TPO. El hipotiroidismo se da en todos los pacientes tras tiroidectomía y en el 20% de los pacientes tras lobectomía tiroidea. El hipotiroidismo postablativo surge tras el tratamiento con Tabla 38. Causas de déficit de hormonas tiroideas Trastorno Comentarios Tiroiditis de Hashimoto Trastorno tiroideo autoinmune asociado a anticuerpos anti-TPO Después de tiroidectomía Tratamiento para enfermedad de Graves, bocio, nódulos tiroideos o cáncer de tiroides Después de terapia con yodo radiactivo Tratamiento para enfermedad de Graves o adenoma tóxico/bocio multinodular Radioterapia de haz externo en el cuello Tratamiento para linfoma de Hodgkin y cánceres de cabeza y cuello Tiroiditis (aguda, subaguda, supurativa) Normalmente hipotiroidismo transitorio previo a la recuperación de un estado eutiroideo Hipotiroidismo central Déficit de TSH por enfermedad hipotalámica o hipofisaria; no debe usarse la TSH para evaluar la adecuación de la dosis de reposición; para determinar la dosis debe usarse el nivel de T4 Hipotiroidismo congénito Cribado neonatal universal en los Estados Unidos, donde la incidencia es de 1/3.500 Déficit de yodo Frecuente en todos los países en desarrollo con déficit grave de yodo Inducida por fármacos Amiodarona, litio, interferón alfa, interleucina 2, yodo, tionamidas (metimazol), etionamida, inhibidores de la tirosincinasa (sunitinib), inhibidores de puntos de control inmunitario (ipilimumab) Anti-TPO: anti-peroxidasa tiroidea; TSH: tirotropina estimulante de la tiroides; T4: tiroxina. 131I en el 90% de los pacientes con enfermedad de Graves en el año posterior a la terapia con yodo radiactivo, así como en el 60% de los pacientes con bocio multinodular tóxico, aunque en el último caso el inicio del hipotiroidismo puede postergarse muchos años. La radioterapia de haz externo en el cuello también puede provocar hipotiroidismo, como ocurre en el tratamiento del linfoma de Hodgkin y los cánceres de cabeza y cuello. El hipotiroidismo inducido por fármacos es otra causa de hipotiroidismo primario (véase la Tabla 38). PUNTOS CLAVE • La causa más frecuente de hipotiroidismo primario es la insuficiencia autoinmune de la glándula tiroides debido a tiroiditis de Hashimoto, normalmente asociada a anticuerpos contra la peroxidasa tiroidea. • Además de la tiroiditis de Hashimoto, las causas frecuentes de hipotiroidismo incluyen cirugía tiroidea e hipotiroidismo postablativo posterior a terapia con 131I para el tratamiento de la enfermedad de Graves o el bocio multinodular tóxico. 63 Trastornos de la glándula tiroides Hipotiroidismo subclínico El hipotiroidismo subclínico suele ser asintomático y se diagnostica con un nivel de TSH en suero por encima del límite superior del intervalo de referencia y un nivel normal de T4 libre. Afecta a entre el 5% y el 10% de la población general. Es necesario descartar una elevación transitoria de la TSH sérica repitiendo la medición en seis u ocho semanas. La tasa de progresión de hipotiroidismo subclínico a hipotiroidismo manifiesto se sitúa entre el 2% y el 4% anual, mientras que una tercera parte de los pacientes revierten a función tiroidea normal de manera espontánea. El intervalo normal de la TSH aumenta con la edad, y un nivel de TSH de hasta 10 µU/ml (10 mU/l) se encuentra dentro del intervalo normal en las personas ≥80 años. El hipotiroidismo subclínico con TSH >10 µU/ml (10 mU/l) puede ser un factor de riesgo de enfermedad coronaria e insuficiencia cardíaca. No hay evidencias de que tratar el hipotiroidismo subclínico mejore la calidad de vida, la función cognitiva, la presión arterial o el peso, pero, en los pacientes con colesterol-LDL elevado, la normalización de la TSH reducirá el colesterol-LDL. Manejo El tratamiento de elección para el déficit de hormonas tiroideas consiste en la reposición hormonal con levotiroxina. Los objetivos del tratamiento son normalizar la TSH sérica (en el hipotiroidismo primario) o la libre T4 (en el hipotiroidismo central) y resolver los signos y síntomas de hipotiroidismo. Empezar con una dosis de reposición completa (1,6 µg/kg de peso corporal magro) es adecuado en la mayoría de los pacientes con hipotiroidismo, exceptuando a los ancianos y a los pacientes con enfermedad cardiovascular, en quienes se recomiendan dosis iniciales más bajas (25-50 µg/día). Es necesario evaluar la adecuación del tratamiento con un nivel de TSH sérica repetido al menos seis semanas después del inicio del tratamiento o de una modificación de la dosis con el objetivo de lograr una TSH normal. Los compuestos que contienen T3 no están recomendados para el tratamiento del hipotiroidismo debido a su breve semivida, que da lugar a picos en los niveles de T3. Los estudios no han logrado demostrar que la T3 sola o en combinación con T4 tenga beneficios claros en el tratamiento del hipotiroidismo. Hay que tratar el hipotiroidismo subclínico con un valor de la TSH en suero por encima de 10 µU/ml (10 mU/l). El tratamiento inicial consiste en entre 25 y 50 µg de levotiroxina al día. El tratamiento del hipotiroidismo subclínico leve (TSH >5-10 µU/ml [5-10 mU/l]) no tiene beneficios claros y puede ser perjudicial. En más de una tercera parte de los pacientes mayores de 65 años se observa sobretratamiento, que puede aumentar el riesgo de arritmia y pérdida ósea. La levotiroxina oral se absorbe en el yeyuno y el íleon. Idealmente, debe tomarse con el estómago vacío (60 minutos antes del desayuno o el café de la mañana). Si al paciente le cuesta tomársela por la mañana, puede hacerlo antes 64 de acostarse. Los pacientes jóvenes con dificultades con el cumplimiento de la pauta diaria pueden tomarse las dosis omitidas al día siguiente. La absorción de una dosis administrada por vía oral se sitúa entre el 70% y el 80% en las condiciones óptimas de ayuno. Los trastornos gastrointestinales (como la enfermedad celíaca) pueden influir en la absorción y dar lugar a requisitos de dosis de levotiroxina más altos de los esperados. Los medicamentos también pueden interferir en la absorción o el metabolismo de la levotiroxina (véase la Tabla 34). PUNTOS CLAVE • La levotiroxina es el tratamiento de elección del déficit de hormonas tiroideas; debe tomarse con el estómago vacío 60 minutos antes del desayuno o el café de la mañana. • Los compuestos que contienen triyodotironina no están recomendados para el tratamiento del hipotiroidismo debido a su breve semivida, que da lugar a picos en los niveles de triyodotironina. AMAV • El sobretratamiento del hipotiroidismo subclínico es frecuente, sobre todo en pacientes mayores de 65 años de edad, y debe evitarse. AMAV Disfunción tiroidea inducida por fármacos Los numerosos medicamentos que pueden afectar en la función tiroidea se presentan en la Tabla 34. La amiodarona es un antiarrítmico con un alto contenido en yodo (37%) y una semivida prolongada, de 60 días aproximadamente. En torno al 25% de los pacientes tratados con amiodarona sufren disfunción tiroidea, que puede manifestarse en forma de hipotiroidismo, hipertiroidismo, tiroiditis indolora o bocio. El hipotiroidismo, presente en el 20% de los pacientes afectados, suele observarse en el contexto de una tiroiditis de Hashimoto. Todos los pacientes pueden experimentar una elevación transitoria de los niveles de TSH en los primeros meses de tratamiento con amiodarona debido al efecto de Wolff-Chaikoff (descenso temporal de la producción tiroidea debido a la carga de yodo), pero la mayoría recuperan una función tiroidea normal. La tirotoxicosis afecta al 5% de los pacientes tratados con amiodarona. La tirotoxicosis inducida por amiodarona tipo 1 (hipertiroidismo) se da en pacientes con enfermedad de Graves o nódulos tiroideos. Se trata de una forma de hipertiroidismo inducido por el yodo (fenómeno de Jod-Basedow). El tratamiento consiste en la administración de tionamidas, normalmente metimazol. La tirotoxicosis inducida por amiodarona tipo 2 (tiroiditis destructiva) es más frecuente y se produce en pacientes sin enfermedad tiroidea subyacente. Normalmente se resuelve de manera espontánea, pero a veces requiere tratamiento con glucocorticoides en dosis moderadas o altas. La decisión de suspender la amiodarona depende del estado cardíaco del paciente y del tipo de tirotoxicosis (tipo 1 o 2). Trastornos de la glándula tiroides PUNTO CLAVE • En torno al 25% de los pacientes que toman amiodarona sufren disfunción tiroidea, que puede manifestarse en forma de hipotiroidismo, hipertiroidismo, tiroiditis indolora o bocio. TGB T4 total Función y disfunción tiroidea en el embarazo Las hormonas tiroideas son esenciales para un desarrollo fetal normal. El tamaño de la glándula tiroides materna puede aumentar hasta el 40% durante el embarazo. La producción de T4 y T3 aumenta hasta el 50% para compensar la mayor producción de globulina fijadora de tiroxina asociada al aumento de estrógenos relacionado con el embarazo. Las necesidades de yodo también se incrementan hasta el 50%. Por lo tanto, hay que aconsejar a las mujeres embarazadas y en período de lactancia que complementen el consumo de yodo de la alimentación con un aporte suplementario oral diario que contenga 150 µg de yodo y que está incluido en algunos, aunque no en todos, los complejos vitamínicos prenatales con o sin prescripción médica. No se recomienda el cribado universal de la TSH en las embarazadas. Sin embargo, este debe realizarse en las que presenten un riesgo elevado de disfunción tiroidea, entre las que se incluyen las mujeres con una edad >30 años, las que tengan hipotiroidismo conocido o antecedentes familiares importantes de disfunción tiroidea, las que hayan recibido radioterapia previa en la cabeza o el cuello, las que se hayan sometido a cirugía previa en el cuello, las que presenten TPO, TSI o AcTR positivos o las que sufran otros trastornos autoinmunes. Las alteraciones de las pruebas de la función tiroidea se describen en la Figura 10. La gonadotropina coriónica humana (hCG) placentaria estimula la secreción de hormonas tiroideas y, en consecuencia, la TSH puede estar ligeramente inhibida. A finales del primer trimestre (semanas 7-12), el límite inferior del intervalo de referencia de la TSH desciende en 0,4 µU/ml (0,4 mU/l) y el límite superior lo hace en 0,5 µU/ml (0,5 mU/l). La TSH en suero vuelve al intervalo de referencia de manera gradual en el segundo y el tercer trimestres. Las concentraciones medidas de T4 total experimentan un aumento lineal durante el embarazo. Después de la semana 16, el límite superior del intervalo de referencia de la T4 puede calcularse multiplicando el límite superior sin embarazo por 1,5. La determinación de la T4 libre mediante inmunoanálisis análogos indirectos es inexacta en el embarazo, a menos que se apliquen métodos e intervalos de referencia específicos para el trimestre. Para el manejo de la tirotoxicosis durante el embarazo está indicada una interconsulta con un endocrinólogo. La tirotoxicosis gestacional por niveles elevados de hCG es la causa más frecuente de inhibición transitoria de la TSH. Si la T4 total o libre en suero permanece dentro del intervalo de refe- TSH T4 libre hCG Semana 10 20 30 40 F i g u r a 1 0 . Alteraciones de las pruebas de función tiroidea en el embarazo. hCG: gonadotropina coriónica humana; T4: tiroxina; TGB: globulina fijadora de tiroxina; TSH: tiroxina. rencia específica para el trimestre, no es necesario ningún tratamiento. Las mujeres con hipertiroidismo moderado o grave en las primeras etapas del embarazo deben recibir tratamiento con PTU, debido a que sus posibles efectos teratógenos son menos graves que los del metimazol. Después del primer trimestre, las mujeres pueden cambiar a metimazol. Para evitar el hipotiroidismo fetal, es necesario controlar estrictamente la función tiroidea y mantener la T4 total o libre en suero dentro del intervalo de referencia específico del trimestre o justo por encima. La enfermedad de Graves afecta al 0,2% de las embarazadas y puede confirmarse con las manifestaciones físicas clásicas o los niveles elevados de TSI o AcTR. Los embarazos en mujeres con enfermedad de Graves se consideran de alto riesgo y deben ser objeto de seguimiento por parte de especialistas maternofetales durante toda la gestación. El hipotiroidismo en el embarazo está asociado a aumentos de abortos espontáneos, partos prematuros y recién nacidos de bajo peso, así como a disminuciones de la función neurocognitiva del niño. El tratamiento de elección es la levotiroxina. En las mujeres con hipotiroidismo preexistente, puede aumentarse empíricamente la dosis levotiroxina en el 30% cuando se confirme el embarazo. En las embarazadas con anticuerpos anti-TPO positivos que no han recibido tratamiento anteriormente, la levotiroxina se inicia si el nivel de TSH es ≥2,5 µU/ml (2,5 mU/l). En las embarazadas con TPO negativa, el tratamiento está indicado cuando la TSH esté 65 Trastornos de la glándula tiroides por encima del intervalo de referencia específico para el embarazo. La TSH debe medirse cada cuatro semanas en la primera mitad del embarazo y en torno a las 30 semanas en todas las mujeres hipotiroideas y en las que están en riesgo de hipotiroidismo (anticuerpos positivos o antecedentes de hemitiroidectomía o tratamiento con 131I). En las mujeres hipotiroideas tratadas debe buscarse un nivel de TSH <2,5 µU/ml (2,5 mU/l) tanto antes de la concepción como durante el embarazo. La detección de nódulos tiroideos en las embarazadas debe evaluarse como en los pacientes sin embarazo. El momento de obtención de la BAAF, ya sea durante el embarazo o después, está determinado por la probabilidad de cáncer y las preferencias de la paciente. Para el manejo del cáncer de tiroides detectado durante el embarazo está indicada una interconsulta con un endocrinólogo. Las embarazadas con antecedentes de cáncer de tiroides deben ser manejadas como si no estuvieran embarazadas. PUNTOS CLAVE • En las mujeres con hipotiroidismo preexistente, puede aumentarse empíricamente la dosis levotiroxina en el 30% cuando se confirme el embarazo. • En las embarazadas con anticuerpos contra la peroxidasa tiroidea (TPO) positivos que no han recibido tratamiento anteriormente, la levotiroxina se inicia si el nivel de hormona estimulante de la tiroides es ≥2,5 µU/ml (2,5 mU/l); en las embarazadas con TPO negativa, el tratamiento está indicado cuando el nivel de hormona estimulante de la tiroides esté por encima del intervalo de referencia específico para el embarazo. Síndrome de enfermedad no tiroidea (síndrome del enfermo eutiroideo) El síndrome de enfermedad no tiroidea (SENT) suele producirse en pacientes que están hospitalizados en situación crítica. Hasta el 75% de los pacientes hospitalizados presentan anomalías en las pruebas de función tiroidea. En la enfermedad no tiroidea está inhibida la hormona liberadora de tirotropina, lo cual normalmente da lugar a una TSH inhibida pero detectable. Una TSH indetectable no es compatible con SENT. En raras ocasiones, la TSH puede estar ligeramente elevada en el SENT, aunque un nivel de TSH ≥20 µU/ml (20 mU/l) no es compatible con SENT. La T4 suele ser normal, pero, debido a la menor actividad de la desyodinasa en el metabolismo de la T4, la T3 desciende y la T3 inversa aumenta (inactiva biológicamente). La globulina fijadora de tiroxina baja en caso de enfermedad, lo que reduce los niveles totales de T4 y T3. El SENT puede interpretarse como una respuesta adaptativa a la enfermedad sistémica y la restricción de macronutrientes. 66 Debido a la ausencia de beneficio clínico, no se recomienda tratar el SENT. En general, la función tiroidea no debe evaluarse en los pacientes hospitalizados a menos que haya una fuerte sospecha clínica de disfunción tiroidea. En caso de diagnóstico de SENT, es necesario volver a comprobar la TSH unas seis semanas después de que el paciente se haya recuperado de la enfermedad no tiroidea para evaluar la vuelta a la normalidad. PUNTOS CLAVE • Debido a la ausencia de beneficio clínico, no se recomienda tratar el síndrome de enfermedad no tiroidea. AMAV • La función tiroidea no debe evaluarse en los pacientes hospitalizados a menos que haya una fuerte sospecha clínica de disfunción tiroidea. AMAV Emergencias tiroideas Crisis tirotóxica (tormenta tiroidea) La crisis tirotóxica, también conocida como tormenta tiroidea, es un trastorno infrecuente con una elevada mortalidad (de hasta el 30%) que se caracteriza por una tirotoxicosis grave (TSH inhibida, T4 libre y/o T3 total elevadas) y una descompensación hemodinámica sistémica (choque). Las concentraciones de hormonas tiroideas en suero no diferencian la crisis hipertiroidea de la tirotoxicosis grave; lo que determina el diagnóstico de crisis hipertiroidea es la presencia de choque. La presentación clínica suele ser posterior a suspensión de un tratamiento con fármacos antitiroideos, enfermedad sistémica, parto, cirugía o traumatismo. Los pacientes con enfermedad de Graves tienen un riesgo más alto. Las manifestaciones clínicas incluyen fiebre alta, taquicardia, estado mental alterado y disfunción cardíaca y hepática. Un sistema de puntuación, como la escala de Burch y Wartofsky (Tabla 39), puede respaldar el diagnóstico, pero la crisis hipertiroidea se diagnostica clínicamente. El manejo consiste en atención a nivel de UCI, tratamiento de la enfermedad desencadenante, tratamiento dirigido a la tirotoxicosis y medidas sintomáticas. La tirotoxicosis se trata con betabloqueantes intravenosos (infusión de esmolol), tionamida (normalmente PTU, pasando a metimazol cuando el paciente esté más estable), glucocorticoides intravenosos en dosis altas y yoduro potásico. El yodo debe administrarse más de una hora después de los fármacos antitiroideos, para evitar proporcionar sustrato a la glándula. El tratamiento con glucocorticoides es un potente inhibidor de la conversión periférica de T4 a T3. Es posible usar secuestradores de ácidos biliares para reducir los niveles de T4 y T3, sobre todo en los pacientes que no pueden tomar tionamidas. En los pacientes con una respuesta escasa al tratamiento médico puede recurrirse a la plasmaféresis o a una tiroidectomía urgente. En los pacientes que sobreviven a una crisis hipertiroidea está indicado un tratamiento definitivo con tiroidectomía o terapia con 131I. Trastornos de la glándula tiroides Tabla 39. Escala de Burch y Wartofsky para confirmar el diagnóstico de crisis tirotóxica (tormenta tiroidea)a Criterios Puntos Disfunción de la termorregulación Temperatura en °C 37,2-37,7 5 37,8-38,2 10 38,3-38,8 15 38,9-39,3 20 39,4-39,9 25 >40 30 Cardiovasculares PUNTOS CLAVE • La crisis tirotóxica o tormenta toroides es un trastorno infrecuente con una elevada mortalidad (de hasta el 30%) que se caracteriza por tirotoxicosis grave y descompensación hemodinámica sistémica. • La crisis tirotóxica suele darse con la suspensión de un tratamiento con fármacos antitiroideos, enfermedad sistémica, parto, cirugía o traumatismo. • Para manejar la crisis tirotóxica se emplea atención a nivel de UCI, tratamiento de cualquier enfermedad desencadenante y uso de betabloqueantes intravenosos, tionamida, glucocorticoides intravenosos en dosis altas y yoduro de potasio. Taquicardia (lpm) 100-109 5 110-119 10 120-129 15 130-139 20 >140 25 Fibrilación auricular Ausente 0 Presente 10 Insuficiencia cardíaca congestiva Ausente 0 Leve 5 Moderada 10 Grave 20 Disfunción gastrointestinal-hepática Ausente 0 Moderada (diarrea, dolor abdominal, náuseas, vómitos) 10 Grave (ictericia) 20 Alteración del sistema nervioso central Ausente 0 Leve (agitación) 10 Moderada (delirio, psicosis, letargo extremo) 20 Grave (convulsiones, coma) 30 Antecedentes desencadenantes Ausentes 0 Presentes 10 Puntuaciones totalizadas <25 Crisis tirotóxica improbable 25-45 Crisis tirotóxica inminente >45 Crisis tirotóxica probable a La crisis tirotóxica se diagnostica clínicamente en presencia de deterioro hemodinámico. De Burch HB, Wartofsky L. Life-threatening thyrotoxicosis: thyroid storm. Endocrinol Metab Clin North Am. 1993 Jun;22(2):263-77. Review. PMID: 8325286. Coma mixedematoso El coma mixedematoso es una presentación potencialmente mortal del hipotiroidismo grave con deterioro hemodinámico que afecta a 0,22 personas por millón al año. La mortalidad es elevada (de hasta el 40%), y requiere atención a nivel de UCI. Los factores de riesgo de coma mixedematoso son sexo femenino, edad avanzada, exposición al frío o un acontecimiento desencadenante en pacientes con hipotiroidismo no diagnosticado, como infarto de miocardio, septicemia, traumatismo o ictus. Las alteraciones del estado mental, que oscilan entre letargo y coma y psicosis, junto con la hipotermia (temperatura inferior a 34,4 °C), son las manifestaciones clínicas más habituales. También son frecuentes la bradicardia, la hipotensión o la frecuencia respiratoria reducida con hipoxia e hipercapnia resultantes. Es fundamental una exploración atenta del cuello para detectar una cicatriz de tiroidectomía. En el coma mixedematoso, el nivel de T4 libre es bajo. La TSH suele estar elevada, pero sin una T4 claramente baja es improbable que haya coma mixedematoso, independientemente de lo alta que sea la TSH. Otras alteraciones metabólicas incluyen hiponatremia e hipoglucemia. Es necesario analizar el cortisol con las pruebas analíticas iniciales para evaluar el déficit concomitante de cortisol. Las medidas sintomáticas agresivas incluyen administración de líquidos, vasopresores en caso necesario, soporte respiratorio y calentamiento pasivo en lugar de activo para evitar la vasodilatación, que puede empeorar la hipotensión. Para tratar la posible insuficiencia suprarrenal concomitante, se administran dosis de estrés de glucocorticoides (100 mg de hidrocortisona intravenosa cada ocho horas) de manera empírica antes de iniciar las hormonas tiroideas. Si un nivel de cortisol aleatorio está por encima de 18 mg/dl (496,8 mmol/l), puede suspenderse la hidrocortisona. Para una reposición de hormonas tiroideas hay que tener en cuenta la necesidad de normalizar el nivel con rapidez y el riesgo de un evento cardíaco mortal causado por la administración de hormonas tiroideas. El tratamiento inicial consiste en la administración intravenosa de levotiroxina, con una dosis de carga de entre 200 y 400 µg seguida de una dosis oral diaria de 1,6 µg/kg. La 67 Trastornos de la reproducción CONT. dosis deberá reducirse al 75% si se administra por vía intravenosa. En caso de edad avanzada y/o enfermedad cardíaca, se recomiendan dosis menores de levotiroxina. Los objetivos del tratamiento son mejorar el estado mental, los parámetros metabólicos y la función cardiopulmonar. Cuando el paciente se encuentra estable, el objetivo es pasar a levotiroxina oral. – – GnRH LH FSH Tecas foliculares Células de la capa granulosa Androstenediona Estradiol – PUNTOS CLAVE • El coma mixedematoso es una presentación potencialmente mortal del hipotiroidismo grave con deterioro hemodinámico; se da sobre todo con el solapamiento de una enfermedad sistémica y un hipotiroidismo no diagnosticado anteriormente. • Además de medidas sintomáticas agresivas, se administran dosis de estrés de glucocorticoides (100 mg de hidrocortisona intravenosa cada ocho horas) de manera empírica antes de iniciar las hormonas tiroideas para tratar la posible insuficiencia suprarrenal concomitante. Inhibina B Ovulación F i g u r a 1 1 . Eje de la reproducción femenina Los pulsos de GnRH llevan a la producción de LH y FSH. La LH actúa en las tecas foliculares para estimular la producción de andrógenos (principalmente androstenodiona). La androstenodiona se metaboliza a estradiol en las células de la capa granulosa. La FSH actúa en las células de la capa granulosa para potenciar la maduración folicular. Las células de la capa granulosa producen inhibina B como regulador de la respuesta a la producción de FSH. FSH: hormona foliculoestimulante; GnRH: hormona liberadora de gonadotropinas; LH: hormona luteinizante; (rodeado con un círculo): retroalimentación negativa. Trastornos de la reproducción Amenorrea Fisiología de la reproducción femenina La amenorrea, la ausencia de menstruación, puede ser intermitente o permanente. Puede deberse a trastornos hipotalámicos, hipofisarios, ováricos, uterinos o del tracto de salida. Las acciones coordinadas del hipotálamo, la hipófisis y los ovarios (eje hipotálamo-hipófiso-ovárico) originan los ciclos ovulatorios en las mujeres. La liberación pulsátil de hormona liberadora de gonadotropinas (GnRH) lleva a las células de la hipófisis anterior a secretar hormona foliculoestimulante (FSH) y hormona luteinizante (LH) (Figura 11). La FSH regula la producción de estradiol y el crecimiento de los folículos en la fase folicular del ciclo menstrual. Una elevación repentina de los niveles de LH provoca la liberación de un óvulo en mitad del ciclo, lo que señala el inicio de la fase lútea, con una duración constante de 14 días. Si no se implanta un embrión fecundado, la disminución de los niveles de estrógenos o progesterona se sigue de la descamación endometrial. La menstruación tiene lugar cada 25-35 días; los ciclos menstruales de menos de 25 días o de más de 35 días en mujeres menores de 40 años son probablemente anovulatorios y dan lugar a un sangrado uterino anormal o a oligomenorrea. Antes de la pubertad, los ovarios están quiescentes debido a la inmadurez del hipotálamo. Después de la menopausia, cesa toda la función reproductiva y la mayor parte de la función endocrina de los ovarios. PUNTO CLAVE • En las mujeres menores de 40 años, los ciclos menstruales de menos de 25 días o de más de 35 días son probablemente anovulatorios. 68 Cuadro clínico Amenorrea primaria La amenorrea primaria se define como la ausencia de menstruación a los 15 años de edad en presencia de crecimiento y caracteres sexuales secundarios normales. En la mayoría de los casos, la amenorrea primaria está causada por una anomalía genética (50%) o anatómica (15%). La mayor parte de las causas de amenorrea secundaria pueden manifestarse también como amenorrea primaria. La causa más frecuente de amenorrea primaria es una disgenesia gonadal, sobre todo con síndrome de Turner (45,X0). El síndrome de Turner se debe a la pérdida de parte o de la totalidad de un cromosoma X. Se da en 1/2.500 nacimientos femeninos vivos. Está asociado a estatura corta e insuficiencia ovárica primaria (IOP); la amenorrea primaria se da en aproximadamente el 90% de las pacientes con síndrome de Turner. Las anomalías anatómicas causantes de amenorrea primaria comprenden himen intacto, tabique vaginal transverso y agenesia vaginal. La agenesia vaginal (también conocida como agenesia mülleriana o síndrome de Mayer-Rokitansky-Kuster-Hauser) es la segunda causa más frecuente de amenorrea primaria, con una incidencia de 1/5.000 nacimientos femeninos vivos. Las mujeres con agenesia vaginal tienen cariotipo y funcionamiento ovárico normales y, por lo Trastornos de la reproducción tanto, genitales externos y caracteres sexuales secundarios normales. Amenorrea secundaria La amenorrea secundaria se define como la ausencia de menstruación durante más de tres meses en las mujeres que anteriormente tenían ciclos menstruales regulares o durante seis meses en las mujeres con menstruaciones irregulares. En las mujeres con oligomenorrea, que se define como menos de nueve ciclos menstruales al año o ciclos de más de 35 días de duración, la evaluación es la misma que en la amenorrea secundaria. La amenorrea hipotalámica funcional (AHF) está causada por una alteración del eje hipotálamo-hipófiso-ovárico y constituye la causa más frecuente de amenorrea secundaria después del embarazo. La alteración de la liberación pulsátil de GnRH hipotalámica puede deberse al estrés, a una pérdida de peso o al ejercicio. En muchos casos, están presentes los tres factores. La AHF es un diagnóstico de exclusión; es necesario obtener la anamnesis, la exploración física, pruebas bioquímicas y, cuando corresponda, pruebas de imagen para descartar otras causas de amenorrea secundaria, entre ellas tumores intracraneales, trastornos infiltrativos o destructivos como la hipofisitis linfocítica, histiocitosis X, sarcoidosis, síndrome de Sheehan y enfermedad sistémica aguda o crónica. Las mujeres premenopáusicas con hiperprolactinemia presentan oligomenorrea o amenorrea con mayor frecuencia que galactorrea. La hiperprolactinemia representa entre el 10% y el 20% de la amenorrea no mediada por el embarazo. La disfunción menstrual en la hiperprolactinemia se debe a la inhibición de la GnRH. La disfunción menstrual es frecuente en las mujeres con trastornos tiroideos; si bien las hemorragias abundantes son típicas en el hipotiroidismo, también puede darse una amenorrea secundaria. Los trastornos hiperandrogénicos están asociados a amenorrea; de ellos, el síndrome del ovario poliquístico (SOP) es, con diferencia, la causa hiperandrogénica más frecuente. La IOP espontánea puede diagnosticarse en las mujeres menores de 40 años con disfunción menstrual en asociación a dos niveles de FSH sérica dentro del intervalo menopáusico. La IOP afecta a 1/100 mujeres. Además de alteraciones menstruales, las mujeres afectadas pueden experimentar síntomas relacionados con el déficit de estrógenos, como síntomas vasomotores, alteraciones del sueño y dispareunia relacionada con la sequedad vaginal. La mayoría de los casos son esporádicos, pero un familiar de primer grado con IOP apunta a una etiología familiar, mientras que los antecedentes personales de trastornos autoinmunes pueden ser indicativos de un síndrome poliglandular autoinmune. Las mujeres con IOP tienen un riesgo elevado de sufrir insuficiencia suprarrenal autoinmune. Las adherencias intrauterinas son la única causa uterina de amenorrea secundaria. La amenorrea tiene su origen en la formación de tejido cicatricial en el interior de la cavidad uterina que impide la acumulación y el desprendimiento de las células endometriales. Las adherencias se forman después de una instrumentación uterina y en la mayoría de los casos están asociadas a legrado uterino por complicaciones del embarazo (síndrome de Asherman). PUNTOS CLAVE • La amenorrea primaria se define como la ausencia de menstruación a los 15 años de edad en presencia de crecimiento y caracteres sexuales secundarios normales; en la mayoría de los casos se debe a una anomalía genética (50%) o anatómica (15%). • La amenorrea secundaria se define como la ausencia de menstruación durante más de tres meses en las mujeres que anteriormente tenían ciclos menstruales regulares o durante seis meses en las mujeres con menstruaciones irregulares. Evaluación de la amenorrea El primer paso de la evaluación de la amenorrea consiste en una anamnesis y una exploración física exhaustivas. Son datos importantes el uso de medicamentos y la exposición a drogas, los cambios de peso, los antecedentes de ejercicio, los factores estresantes psicosociales y los antecedentes familiares relacionados con la menarquia. Los síntomas pueden incluir cefalea o alteraciones visuales indicativas de patología hipofisaria, síntomas de exceso o déficit de hormonas tiroideas, galactorrea indicativa de hiperprolactinemia o síntomas vasomotores asociados a déficit de estrógenos. La exploración física deberá incluir un examen pélvico para evaluar posibles anomalías en la vagina, el cuello uterino y el útero. Para confirmar que el útero es normal pueden hacer falta pruebas de imagen. La exploración física deberá incluir también una evaluación para detectar manifestaciones del síndrome de Turner, como una línea de implantación capilar baja, cuello alado, tórax ancho y pezones muy separados. En las pacientes con amenorrea primaria o secundaria, es importante medir la estatura, el peso y el IMC. Un IMC bajo (<18,5 kg/m2) puede indicar AHF debido a un trastorno alimentario, ejercicio excesivo o enfermedad sistémica. Un IMC alto (≥30 kg/m2) es frecuente en las mujeres con SOP; otros hallazgos de la exploración física indicativos de este síndrome son el acné y el hirsutismo. El hipercortisolismo está asociado a acné e hirsutismo, así como a distribución anómala de almohadillas de grasa, obesidad centrípeta, congestión facial, debilidad de los músculos proximales y estrías violáceas anchas (>1 cm). El vitiligo u otros signos de enfermedad autoinmune aumentan la probabilidad de IOP autoinmune. La exploración mamaria debe incluir la evaluación de galactorrea por presión. La atrofia vulvovaginal indica déficit de estrógenos. Tras descartar el embarazo, las pruebas analíticas iniciales en la amenorrea tanto primaria como secundaria deberán incluir la determinación de los niveles de FSH, TSH, T4 libre y prolactina. Los resultados analíticos determinarán los pasos siguientes (Figura 12). 69 Trastornos de la reproducción Amenorrea Obtener hCG sérica Positiva Negativa Embarazo Obtener FSH, TSH, T4 libre, prolactina Prolactina elevada TSH anormal FSH elevada FSH baja o normal Repetir prolactina Revisar medicamentos Obtener RM de hipófisis Evaluar y tratar la disfunción tiroidea Obtener cariotipo Considerar IOP Proceder con prueba de retirada de progesterona No hay sangrado con retirada de progesterona En caso de cefaleas o pérdida de visión temporal, obtener RM craneal Considerar AHF en caso de trastorno alimentario, ejercicio excesivo o estrés Sangrado con retirada de progesterona Si hay antecedentes de procedimientos ginecológiccos, considerar histeroscopia para detectar adherencias uterinas Considerar hiperandrogenismo; obtener testosterona, DHEAS y prueba con 17-hidroxiprogesterona F i g u r a 1 2 . Algoritmo para evaluar la amenorrea. AHF: amenorrea hipotalámica funcional; DHEAS: sulfato de deshidroepiandrosterona; FSH: hormona foliculoestimulante; hCG: gonadotropina coriónica humana; IOP: insuficiencia ovárica primaria; RM: resonancia magnética; T4: tiroxina; TSH: tirotropina. Si el nivel de TSH es anormal, es necesario evaluar y manejar la disfunción tiroidea (véase «Trastornos de la glándula tiroides»). Si el nivel de prolactina está elevado, es necesario repetir las pruebas de prolactina para confirmar el diagnóstico. Dado que muchos fármacos pueden provocar hiperprolactinemia, es esencial una revisión atenta de los medicamentos. También hacen falta pruebas de función renal y hepática (véase «Trastornos de la hipófisis»). Si el nivel de FSH está elevado, la prueba deberá repetirse al cabo de un mes, con análisis simultáneos del estradiol sérico. Si el nivel de FSH está elevado en las pruebas repetidas y el nivel de estradiol es bajo, está indicado un análisis del cariotipo para detectar síndrome de Turner. La IOP y la menopausia también provocan niveles altos de FSH. En las mujeres con niveles de FSH normales o bajos, los pasos siguientes están determinados por la anamnesis y los resultados de la exploración física. Las pruebas posteriores del estado de los estrógenos pueden depender de una prueba de retirada de progesterona. Si se confirma que el estado estro70 génico es normal (hemorragia en la semana posterior a la interrupción de la progesterona), debe considerarse la presencia de hiperandrogenismo. Las pruebas analíticas para detectar hiperandrogenismo incluyen la determinación de los niveles de testosterona total y globulina fijadora de hormonas sexuales (SHBG). El SOP es la causa más frecuente de disfunción menstrual en las mujeres con hiperandrogenismo; sin embargo, antes de diagnosticar SOP hay que descartar otros trastornos hiperandrogénicos. En ausencia de sangrado, es muy probable que haya un estado estrogénico debido a hipogonadismo hipotalámico. Los estudios de imagen que pueden utilizarse en la evaluación de la amenorrea incluyen la RM de la región selar (para evaluar la integridad estructural de la hipófisis), la ecografía pélvica (para evaluar las anomalías anatómicas del útero, la vagina y los ovarios), la histerosalpingografía y la histeroscopia (para detectar obstrucciones del tracto de salida uterino). La elección del estudio de imagen, así como la necesidad de obtener pruebas de imagen, se basan en los resultados de las pruebas bioquímicas previas que indican la causa de la amenorrea. Trastornos de la reproducción PUNTO CLAVE • Tras descartar el embarazo, las pruebas analíticas iniciales en la amenorrea tanto primaria como secundaria deberán incluir la determinación de los niveles de hormona foliculoestimulante, tirotropina, tiroxina libre y prolactina. Tratamiento de la amenorrea Casi todas las mujeres con síndrome de Turner necesitarán tratamiento estrogénico exógeno con un progestágeno cíclico para prevenir la hiperplasia endometrial. El tratamiento con estrógeno-progestágeno continúa hasta los 51 años de edad, la edad media de la menopausia. El tratamiento para la AHF incluye patrones alimentarios menos restrictivos, aumento de peso o reducción del ejercicio intenso para recuperar la menstruación. Además, es importante tratar los trastornos asociados a la AHF, incluyendo una masa ósea baja, trastornos alimentarios, ansiedad y otros trastornos del estado de ánimo. La amenorrea debida a hiperprolactinemia con un adenoma lactotropo se maneja con tratamiento con agonistas de la dopamina si la paciente desea tener hijos o con estrógeno/progestágeno si no tiene este plan, para prevenir la pérdida ósea. Si la amenorrea inducida por hiperprolactinemia está relacionada con medicamentos que no pueden interrumpirse (como los antipsicóticos), está indicada una terapia con estrógeno/progestágeno. El tratamiento de la IOP incluye terapia con estrógeno/progestágeno a hasta los 51 años de edad aproximadamente. El apoyo psicosocial es importante debido a las mayores puntuaciones en las escalas de depresión, ansiedad y afecto negativo de las pacientes con IOP; también está indicado consultar a un especialista para hablar de las opciones para tener hijos. El tratamiento de la agenesia vaginal consiste en dilatación vaginal no quirúrgica; si el tratamiento no quirúrgico fracasa, pueden considerarse las opciones quirúrgicas. Síndromes de hiperandrogenismo Hirsutismo y síndrome del ovario poliquístico En la mayoría de los casos, las concentraciones elevadas de andrógenos en mujeres se manifiestan con hirsutismo, y pue- Tabla 40. den cursar también con acné, alopecia androgénica y/o virilización. El hirsutismo es la presencia de pelo terminal excesivo en una distribución masculina; afecta a alrededor del 10% de las mujeres. La virilización (voz más profunda, clitoromegalia, calvicie de patrón masculino, acné grave) solo se da en los casos graves de hiperandrogenismo y despierta inquietud por la posible de presencia de una hipertecosis ovárica o un tumor ovárico o suprarrenal productor de andrógenos. El inicio del hirsutismo en una mujer de más de 30 años de edad también apunta a la presencia de un tumor productor de andrógenos. Aunque los tumores ováricos secretores de andrógenos son infrecuentes, deben tenerse en cuenta en las pacientes con hirsutismo abrupto de progresión rápida o hiperandrogenismo grave, así como en las mujeres con hiperandrogenemia notable (testosterona total >150 ng/dl [5,2 nmol/l]). Las mujeres con hirsutismo crónico y ciclos menstruales cada 25-35 días tienen muy probablemente hirsutismo idiopático o SOP. Entre el 15% y el 40% de las mujeres con hiperandrogenismo y menstruación cada 21-35 días presentan disfunción ovulatoria. El SOP es la causa más frecuente de hirsutismo y explica el 95% de los casos. El SOP es un trastorno caracterizado por hiperandrogenismo y disfunción ovulatoria. Afecta a entre el 6% y el 10% de las mujeres y constituye la causa más frecuente de infertilidad anovulatoria. Está asociado a pulsos rápidos de GnRH, exceso de LH y secreción insuficiente de FSH, que dan lugar a una producción ovárica de andrógenos excesiva y a disfunción ovárica. Cursa con resistencia a la insulina. Los niveles elevados de insulina en el SOP potencian en mayor medida la producción ovárica y suprarrenal de andrógenos, así como el aumento de la biodisponibilidad de andrógenos relacionada con la reducción de la SHBG. El SOP está asociado a una mayor incidencia de síndrome metabólico, prediabetes, diabetes mellitus tipo 2, hipercolesterolemia y obesidad. Hay diversos criterios diagnósticos para el SOP (Tabla 40). Es importante recordar que el SOP es un diagnóstico de exclusión; es necesario tener en cuenta otras causas de oligoovulación/anovulación, entre las que se incluyen la disfunción tiroidea, la hiperplasia suprarrenal congénita no clásica, la hiperprolactinemia y los tumores secretores de andrógenos. Criterios diagnósticos del síndrome del ovario poliquístico Criterios consensuados de los NIH de 1990 (todos son necesarios) Criterios de Rotterdam de 2003 (hacen falta dos o tres) Exceso de andrógenos y criterios de la Polycystic Ovary Syndrome Society (todos son necesarios) Irregularidades menstruales debido a oligoovulación/anovulación Oligoovulación/anovulación Signos clínicos y/o bioquímicos de hiperandrogenismo Signos clínicos y/o bioquímicos de hiperandrogenismo Signos clínicos y/o bioquímicos de hiperandrogenismo Disfunción ovárica, oligoovulación/anovulación y/u ovarios poliquísticos en la ecografía Exclusión de otros trastornos Ovarios poliquísticos en la ecografía Exclusión de exceso de otros andrógenos o de trastornos ovulatorios 71 Trastornos de la reproducción PUNTOS CLAVE • El síndrome del ovario poliquístico es un trastorno caracterizado por hiperandrogenismo y disfunción ovulatoria que afecta a entre el 6% y el 10% de las mujeres. • El síndrome del ovario poliquístico es un diagnóstico de exclusión; es necesario tener en cuenta otras causas de oligoovulación/anovulación, entre las que se incluyen la disfunción tiroidea, la hiperplasia suprarrenal congénita no clásica, la hiperprolactinemia y los tumores secretores de andrógenos. Evaluación del hiperandrogenismo La anamnesis y la exploración física deben incluir los detalles del inicio del hirsutismo y otros síntomas/signos de hiperandrogenismo, antecedentes menstruales, antecedentes familiares de hiperandrogenismo, signos de resistencia a la insulina (obesidad, acantosis pigmentaria, acrocordones), distribución del crecimiento del pelo terminal y pérdida de cabello. La exposición a testosterona exógena (tópica, oral o inyectada) debe evaluarse como posible causa del hiperandrogenismo y la virilización. En las mujeres con hirsutismo debe medirse la testosterona total con SHBG, así como la 17-hidroxiprogesterona matinal para detectar hiperplasia suprarrenal congénita. También está indicada una evaluación analítica para detectar oligomenorrea o amenorrea (hCG, prolactina, FSH, TSH, tiroxina libre). En los casos de inicio reciente de hirsutismo y/o virilización de rápida progresión, es necesaria una medición de DHEAS en suero. Unos niveles notablemente elevados de DHEAS y/o testosterona no son compatibles con SOP. En las pacientes con niveles de testosterona total >200 ng/dl (6,9 nmol/l) o valores de DHEAS >7,0 µg/ml (18,9 µmol/l) hay que obtener pruebas de imagen para detectar un tumor suprarrenal (TC o RM suprarrenal) u ovárico (ecografía transvaginal). Manejo del hiperandrogenismo En las mujeres con hirsutismo idiopático puede ser adecuada la eliminación mecánica del vello (roscado, depilatorios, electrólisis, láser) por motivos estéticos. El tratamiento farmacológico de primera línea para el hirsutismo consiste en anticonceptivos hormonales combinados (estrógeno y progestágenos) orales; estos fármacos inhiben la secreción de gonadotropina y la producción de andrógenos ováricos, además de aumentar los niveles de SHBG. Para una mejor respuesta estética puede añadirse una terapia antiandrogénica (espironolactona); debido a sus efectos teratógenos en los fetos de sexo masculino, es obligatoria una anticoncepción concomitante con este tratamiento. La eflornitina tópica también está aprobada para el tratamiento del crecimiento de vello no deseado. En el SOP, la pérdida de peso es una intervención de primera línea en las pacientes con IMC ≥25 kg/m2. Una pérdida 72 de peso prolongada de entre el 5% y el 10% mejora los niveles de andrógenos, la función menstrual y posiblemente la fertilidad. Los anticonceptivos orales constituyen el tratamiento farmacológico de primera línea para el hirsutismo y la disfunción menstrual, a menos que se desee tener hijos. Si los anticonceptivos orales tienen una eficacia cosmética subóptima, al cabo de seis meses se añade un fármaco antiandrogénico. En el caso de que la paciente desee tener hijos, puede usarse clomifeno citrato o letrozol para corregir la oligoovulación/anovulación. La metformina reduce la hiperinsulinemia y los niveles de andrógenos, pero tiene efectos mínimos en el hirsutismo y la ovulación. En las pacientes con SOP es necesario hacer un cribado de prediabetes/diabetes mellitus, hipercolesterolemia, obesidad, hipertensión y apnea obstructiva del sueño, debido al mayor riesgo de estos trastornos. Cuando la tolerancia alterada a la glucosa, la prediabetes o la diabetes mellitus tipo 2 no responden adecuadamente a la modificación del estilo de vida, está indicado el uso de metformina. PUNTOS CLAVE • Los anticonceptivos orales constituyen el tratamiento farmacológico de primera línea para el hirsutismo y la disfunción menstrual; para una mejor respuesta estética, puede añadirse un fármaco antiandrogénico. • En las pacientes con síndrome del ovario poliquístico es necesario hacer un cribado de prediabetes/diabetes mellitus, hipercolesterolemia, obesidad, hipertensión y apnea obstructiva del sueño. Infertilidad femenina Conviene evaluar la infertilidad al cabo de un año de relaciones sexuales sin protección, una media de dos veces por semana, en las mujeres <35 años, y al cabo de seis meses en las mujeres ≥35 años. Normalmente, el responsable del tratamiento de la infertilidad es un endocrinólogo especialista en reproducción. Es necesario evaluar simultáneamente a los dos miembros de la pareja; a menudo intervienen varios factores (véase «Infertilidad masculina»). La anamnesis y los hallazgos de la exploración física pueden apuntar a la causa de la infertilidad. Los factores clave comprenden los antecedentes menstruales (para determinar el estado ovulatorio) y las pruebas para detectar disfunción tiroidea, galactorrea, hirsutismo, dolor pélvico, dismenorrea y dispareunia. Deben explorarse los antecedentes de embarazos anteriores, tratamiento anticanceroso, trastorno por consumo de sustancias, infecciones de transmisión sexual, enfermedad inflamatoria pélvica y los procedimientos ginecológicos. La frecuencia del coito es una información importante. La exploración física debe incluir el IMC y una evaluación para detectar signos de hiperandrogenismo, déficit de estrógenos, hiperprolactinemia y disfunción tiroidea. El primer paso de la evaluación es la valoración de la función ovulatoria. Las mujeres con menstruaciones cada 28 días Trastornos de la reproducción aproximadamente y síntomas de molimina (sensibilidad mamaria, plenitud abdominal, dolor ovulatorio) son probablemente ovulatorias. En las mujeres sin ciclo menstrual, se evalúa el estado ovulatorio con una medición del nivel de progesterona en suero en mitad de la fase lútea (obtenido una semana antes de la fecha prevista de la menstruación); un nivel de progesterona >3 ng/ml (9,5 nmol/l) demuestra una ovulación reciente. Si se sospecha que los ciclos son anovulatorios, la evaluación inicial consiste en la determinación de la prolactina, la TSH y la FSH, con pruebas posteriores para detectar SOP. La histerosalpingografía se utiliza para detectar oclusión tubárica y para evaluar la cavidad uterina. En caso de sospecha de endometriosis o adherencias pélvicas, puede recurrirse a una laparoscopia exploratoria. Si no se detectan anomalías, se ofrecerán tratamientos de fertilidad dirigidos por un endocrinólogo especialista en reproducción, que posiblemente incluyan estimulación ovárica con clomifeno citrato o letrozol, inseminación intrauterina y fecundación in vitro, que puede ofrecerse a las mujeres ≥40 años como tratamiento de primera línea. PUNTOS CLAVE • Conviene evaluar la infertilidad al cabo de un año de relaciones sexuales sin protección en las mujeres menores de 35 años y al cabo de seis meses en las mujeres de 35 años o más. AMAV - - GnRH LH FSH Células de Leydig Células de Sertoli Testosterona Espermatogénesis - Inhibina B Estradiol Dihidrotestosterona F i g u r a 1 3 . Eje de la reproducción masculina. Los pulsos de GnRH provocan pulsos de LH y FSH. La FSH actúa en las células de Sertoli, que facilitan la maduración de los espermatozoides y producen inhibina B, el principal regulador negativo de la producción basal de FSH. Las células de Leydig producen testosterona, que retroalimenta la inhibición de GnRH y la liberación de LH. Parte de la testosterona se convierte irreversiblemente en dihidrotestosterona o estradiol, que son más potentes que la testosterona para inhibir la GnRH y la LH. FSH: hormona foliculoestimulante; GnRH: hormona liberadora de gonadotropinas; LH: hormona luteinizante; (rodeado con un círculo): retroalimentación negativa. • Es necesario evaluar simultáneamente a los dos miembros de la pareja; a menudo intervienen varios factores. Hipogonadismo • El primer paso de la evaluación de la infertilidad femenina es la valoración de la función ovulatoria. El hipogonadismo masculino es un síndrome clínico que tiene su origen en la incapacidad de los testículos de producir niveles fisiológicos de testosterona y un número normal de espermatozoides debido a una alteración del eje hipotálamo-hipófiso-testicular. El hipogonadismo primario está causado por anomalías testiculares. Las causas frecuentes de hipogonadismo primario adquirido en adultos incluyen la orquitis por parotiditis, las secuelas de la radioterapia, los fármacos o toxinas antineoplásicos, los traumatismos o torsiones testiculares y las enfermedades sistémicas agudas y crónicas. El síndrome de Klinefelter (47,XXY) constituye la causa congénita más frecuente de hipogonadismo primario y está asociado a gran estatura, testículos pequeños, retraso del desarrollo y problemas de socialización. El hipogonadismo secundario es reflejo de un déficit hipotalámico (GnRH) y/o hipofisario (LH/FSH). Tiene causas congénitas infrecuentes, como el síndrome de Kallmann, que está asociado a anosmia. Las causas frecuentes de hipogonadismo hipogonadotrófico adquirido son hiperprolactinemia, medicamentos, enfermedad crítica, trastornos del sueño no tratados, obesidad, enfermedad hepática y renal, alcoholismo, consumo de marihuana y trastornos de la alimentación. Son causas infrecuentes tumores, traumatismos, talasemias y enfermedades infiltrativas que provocan la alteración de la producción de gonadotropina (como la sarcoidosis y la hemocromatosis). Fisiología de la reproducción masculina Los testículos contienen dos unidades anatómicas: los túbulos espematogénicos, compuestos de células germinales y células de Sertoli, y el intersticio, que contiene células de Leydig. Los tres esteroides de importancia fundamental en la reproducción masculina son la testosterona, la dihidrotestosterona y el estradiol. La secreción pulsátil de GnRH por el hipotálamo provoca la secreción pulsátil de LH y por las células gonadotropas de la hipófisis anterior, La LH regula la síntesis de testosterona en las células de Leydig en un patrón diurno; la secreción de LH está regulada por la retroalimentación negativa de la testosterona y el estradiol. La FSH regula la espermatogénesis en las células de Sertoli. La inhibina B es un importante inhibidor peptídico de la secreción de FSH hipofisaria (Figura 13). El eje hipotálamo-hipófiso-testicular es sensible a los factores estresantes, entre los que se incluyen la enfermedad aguda y crónica, el ayuno y el ejercicio intenso, todos los cuales pueden reducir los niveles de testosterona. Causas 73 Trastornos de la reproducción PUNTO CLAVE • El hipogonadismo primario está causado por anomalías testiculares; el hipogonadismo secundario es reflejo de disfunción hipotalámica y/o hipofisaria. Cuadro clínico Los síntomas específicos de hipogonadismo en el varón adulto incluyen disminución de las erecciones matinales y espontáneas, disminución de la libido, mastodinia, ginecomastia, menor necesidad de afeitarse y/o reducción del vello axilar y genital. Los sofocos, la reducción de la masa ósea y las fracturas por traumatismos leves están asociados a déficit de testosterona profundo y/o de larga evolución. Son síntomas inespecíficos las disminuciones del estado anímico, la energía, la concentración, la fuerza y volumen muscular y la resistencia, así como sueño de mala calidad y poca memoria. La infertilidad es más probable con el hipogonadismo primario que con el hipogonadismo secundario. Los varones que experimentan hipogonadismo antes de la pubertad tienen testículos y falo pequeños y carecen de caracteres sexuales secundarios masculinos normales. Si se inicia después de la pubertad, puede cursar con cierta regresión de los caracteres sexuales secundarios. La disminución del tamaño de los testículos y/o el falo y la aparición de ginecomastia en adultos se deben probablemente a una causa primaria. Evaluación No se recomienda realizar un cribado a los varones con síntomas inespecíficos de hipogonadismo. En los varones con signos y síntomas específicos, está indicado medir el nivel de testosterona total a las 8:00 horas. Si el nivel de testosterona es bajo, se realiza una segunda medición del nivel de testosterona a las 8:00 horas. El diagnóstico se emite con dos mediciones bajas de testosterona en suero. En los varones obesos conviene determinar la testosterona libre, porque la obesidad reduce la SHBG y arroja una medición del nivel de testosterona total falsamente bajo. En caso de resultado bajo de la medición de testosterona, está indicado medir la LH sérica. Un nivel alto de LH es reflejo de hipogonadismo primario, y las pruebas siguientes deben buscar la identificación de la causa. Un nivel de LH bajo o normal con una testosterona baja simultánea es reflejo de hipogonadismo secundario. Medicamentos como los análogos de la GnRH (tratamiento prostático), los esteroides gonadales (como el uso de esteroides anabólicos o megestrol para la estimulación del apetito), los glucocorticoides en dosis altas y los opiáceos utilizados de manera crónica pueden inhibir las gonadotropinas y provocar hipogonadismo secundario. Otras pruebas incluyen la medición de la prolactina sérica y el cribado de hemocromatosis. La evaluación de otros posibles déficits de hormonas hipofisarias está indicada en presencia de signos y síntomas. Es necesario realizar una RM de hipófisis específica si hay hiperprolactinemia, si se detectan otras anomalías de las hormonas hipofisa74 rias, si el nivel de testosterona es <150 mg/dl (5,2 nmol/l) o si hay signos o síntomas de efecto de masa (Figura 14). PUNTOS CLAVE • No se recomienda realizar un cribado a los varones con síntomas inespecíficos de hipogonadismo. AMAV • El hipogonadismo masculino se diagnostica con dos mediciones de la testosterona total en suero a las 8:00 horas. Manejo En los varones con hipogonadismo demostrado bioquímicamente, es posible iniciar un tratamiento con testosterona una vez determinada la etiología. Hay disponibles varias preparaciones de reposición de testosterona (Tabla 41). El objetivo consiste en reponer la testosterona para que el valor de testosterona total medido se sitúe en el intervalo medio o normal. Los beneficios clínicos del tratamiento con testosterona incluyen aumento de la libido, la masa muscular magra, la masa muscular sin grasa, la densidad ósea y los caracteres sexuales secundarios. Los posibles efectos adversos son acné, efecto en el tejido prostático, apnea obstructiva del sueño, trombofilia y eritrocitosis (Tabla 42). La terapia con testosterona solo está indicada para el tratamiento del déficit de testosterona; no se utiliza para las alteraciones de la espermatogénsis y, en realidad, la altera mediante la inhibición de la secreción de FSH hipofisaria. Es necesario asesorar a los pacientes sobre la disminución de la fertilidad que conlleva el tratamiento con testosterona exógena. PUNTOS CLAVE • Una vez determinada la etiología del hipogonadismo, debe iniciarse un tratamiento con testosterona con el objetivo de lograr una medición de la testosterona total situada en el intervalo medio o normal. • La terapia con testosterona solo está indicada para el tratamiento del déficit de testosterona; no se utiliza para las alteraciones de la espermatogénesis. Abuso de esteroides anabólicos en los varones El abuso de esteroides anabólicos constituye un grave problema de salud pública que va más allá del deportista profesional. La prevalencia del consumo de esteroides en los varones se sitúa cerca del 7%. Con frecuencia los esteroides se adquieren a través de Internet y los patrones de administración varían. El estado de ánimo voluble, una masa muscular excesiva y unos testículos pequeños pueden indicar abuso de esteroides anabólicos. Los efectos secundarios reproductivos del abuso de esteroides anabólicos incluyen ginecomastia (debido a conversión periférica de testosterona a estradiol), atrofia testicular, disminución de la espermatogénesis y la fertilidad e hipogonadismo hipogonadotrófico iatrogénico, que AMAV Trastornos de la reproducción Presunto hipogonadismo Cribado: testosterona total en suero (×2) Testosterona baja Testosterona normal Testosterona biodisponible/libre y/o SHBG en los pacientes adecuados Seguimiento Testosterona baja confirmada: comprobar niveles de FSH y LH Nivel de FSH o LH elevado Nivel de FSH o LH bajo/normal Hipogonadismo primario Hipogonadismo secundario: comprobar nivel de PRL, estudios de hierro Nivel de PRL elevado, otros déficits hipofisarios Signos/síntomas de efecto de masa, nivel de testosterona <150 ng/dl (5,52 nmol/l) RM de hipófisis Saturación de transferrina y nivel de ferritina elevados Hemocromatosis F i g u r a 1 4 . Algoritmo para evaluar el hipogonadismo masculino. FSH: hormona foliculoestimulante; LH: hormona luteinizante; PRL: prolactina; RM: resonancia magnética; SHBG: globulina fijadora de hormonas sexuales; ×2: dos mediciones separadas. puede ser permanente. Las evidencia analíticas indicativas de abuso de esteroides anabólicos incluyen hematocrito elevado, nivel de LH indetectable o bajo, nivel bajo de SHBG y nivel bajo de testosterona total con elevación de precursores de la testosterona como la androstenodiona. PUNTOS CLAVE • El estado de ánimo voluble, una masa muscular excesiva y unos testículos pequeños pueden indicar abuso de esteroides anabólicos. • Los efectos secundarios del abuso de esteroides anabólicos incluyen ginecomastia, atrofia testicular, disminución de la espermatogénesis y la fertilidad e hipogonadismo hipogonadotrófico iatrogénico, que puede ser permanente. Cambios de la testosterona en el envejecimiento Con el envejecimiento, descienden los niveles de testosterona y testosterona libre en los varones y aumenta la SHBG, lo que da lugar a una disfunción testicular e hipotálamo-hipofisaria. Aunque los niveles de testosterona disminuyen del 1% al 2% anual, la mayoría de los varones no experimentan hipogonadismo. La producción de semen no se altera de manera significativa con la edad. Las consecuencias de la «andropausia» no se conocen del todo, pero los efectos adversos pueden incluir un impacto negativo en la función sexual, la masa muscular, la eritropoyesis y la salud ósea. La Endocrine Society respalda el tratamiento de los varones de edad avanzada con déficit de testosterona confirmado bioquímicamente. El objetivo consiste en la reposición de tes75 Trastornos de la reproducción Tabla 41. Terapia de reposición de testosterona recomendada Vía de administración Preparacióna Patrón de administración típico Calendario de control inicial Ventajas; inconvenientes Inyección intramuscular Cipionato de testosterona 100-200 mg cada 2 semanas Testosterona en el punto medio entre inyecciones Bajo coste; fluctuación del nivel de testosterona Inyección intramuscular Enantato de testosterona 100-200 mg cada 2 semanas Testosterona en el punto medio entre inyecciones Bajo coste; fluctuación del nivel de testosterona Parche transdérmico Parche transdérmico de testosterona de 24 horas 2-6 mg/día Testosterona matinal ~14 días después del inicio del tratamiento Niveles estables; exantema cutáneo/mala adherencia a la piel Gel transdérmico AndroGel 1% 50-100 mg diarios Testosterona matinal ~14 días después del inicio del tratamiento Niveles estables; posible transferencia de la piel a otros Gel transdérmico AndroGel 1,62% 20,25-81 mg diarios Testosterona matinal 14-28 días después del inicio del tratamiento Niveles estables; posible transferencia de la piel a otros Gel transdérmico Fortesta 10-70 mg diarios 2 horas después de la aplicación ~14 días después del inicio del tratamiento Niveles estables; posible transferencia de la piel a otros Gel transdérmico Testim 50-100 mg diarios Testosterona matinal ~14 días después del inicio del tratamiento Niveles estables; posible transferencia de la piel a otros Gel transdérmico Vogelxo 50-100 mg diarios Testosterona matinal ~14 días después del inicio del tratamiento, antes de la aplicación Niveles estables; posible transferencia de la piel a otros Solución transdérmica Axiron 30-120 mg diarios 2-8 horas después de la aplicación ~14 días después del inicio del tratamiento Niveles estables; posible transferencia de la piel a otros Implantes subcutáneos Implantes de pellets de testosterona 150-450 mg cada 3-6 meses Medir al término del intervalo de administración Administración infrecuente/ Necesidad de incisión para la inserción; riesgo de hipogonadismo sintomático recidivante debido a la gran variabilidad de la duración de la acción aNombres comerciales en Estados Unidos. Tabla 42. Directrices clínicas de la Endocrine Society para el control de los efectos adversos de la terapia de reposición de testosterona Parámetro Calendario de cribado recomendado Alertas Hematocrito Valor obtenido en el momento basal y a los 3 y 6 meses después del inicio del tratamiento, seguidos de mediciones anuales Valor >54% Nivel de PSA En los pacientes >40 años de edad con un valor basal >0,6 µg/ml (0,6 ng/l), TR y nivel de PSA (determinados 3 y 6 meses después del inicio del tratamiento, seguidos de cribados regulares) Aumento >1,4 µg/ml (1,4 ng/l) en 1 año o >0,4 µg/ml (0,4 ng/l) al cabo de 6 meses de uso; resultados anómalos del TR; puntuación de síntomas prostáticos de la AUA/IPSS >19 AUA: American Urological Association; IPSS: International Prostate Symptom Score; PSA: antígeno prostático específico; TR: tacto rectal. Datos de Bhasin S, Cunningham GR, Hayes FJ, Matsumoto AM, Snyder PJ, Swerdloff RS, et al; Task Force, Endocrine Society. Testosterone therapy in men with androgen deficiency syndromes: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2010;95:2536-59. [PMID: 20525905] tosterona hasta un intervalo bajo-normal. Antes de iniciar el tratamiento, algunos expertos recomiendan compartir la toma decisiones con el paciente y comentar las dudas en cuanto a los daños y beneficios del tratamiento con testosterona. Esta estrategia es motivo de controversia. No se ha demostrado que el tratamiento con testosterona sea beneficioso en los varones sin evidencias bioquímicas de déficit, y los estudios han puesto de manifiesto que el uso de tratamientos con testosterona conlleva un mayor riesgo de en76 fermedad y muerte cardiovascular, tromboembolismo venoso y cáncer de próstata. PUNTOS CLAVE • Los cambios de la testosterona asociados al envejecimiento en los varones no provocan hipogonadismo sintomático en la mayoría de ellos. (Continúa en página siguiente) Manejo de la terapia hormonal transgénero P untos clave (continuación) AMAV • No se ha demostrado que el tratamiento con testosterona sea beneficioso en los varones sin evidencias bioquímicas de déficit, y los estudios han puesto de manifiesto que el uso de tratamientos con testosterona conlleva un mayor riesgo de enfermedad y muerte cardiovascular, tromboembolismo venoso y cáncer de próstata. Infertilidad masculina En las parejas con infertilidad, es necesario realizar una evaluación de la infertilidad masculina al mismo tiempo que la femenina. Una anamnesis exhaustiva deberá centrarse en las posibles causas de infertilidad: historial del desarrollo, enfermedad crónica, infección, cirugía, exposición a fármacos/drogas y ambiental, antecedentes sexuales y fertilidad previa. La exploración física deberá centrarse en las evidencias de déficit de andrógenos, con una exploración atenta de los genitales externos. Si la exploración testicular es normal, debe considerarse la derivación a un urólogo. El análisis del semen constituye la evaluación analítica inicial; debe recogerse después de dos o tres días de abstinencia sexual, pero no más, para evitar la disminución de la motilidad espermática. Si el análisis del semen es anómalo, debe repetirse un mínimo de dos semanas después y, en caso de que los resultados sean anómalos, se recomienda la derivación a un endocrinólogo especialista en reproducción. PUNTO CLAVE • El análisis del semen constituye la evaluación analítica inicial de la infertilidad masculina; si los resultados son anómalos, la prueba debe repetirse un mínimo de dos semanas después, con derivación a un endocrinólogo especialista en reproducción en caso de resultados nuevamente anómalos. Ginecomastia La ginecomastia, una proliferación benigna de tejido glandular mamario producida por un aumento de la acción de los estrógenos en relación con los andrógenos, se da en entre una y dos terceras partes de los varones de edad avanzada. Es necesaria una anamnesis exhaustiva y una revisión atenta de los medicamentos que se están tomando. Los fármacos antiandrogénicos, como la espironolactona, la cimetidina y los inhibidores de la proteasa, están claramente asociados a ginecomastia. Otras causas identificadas son trastornos por consumo de sustancias, malnutrición, cirrosis, hipogonadismo, tumores testiculares de células germinales, hipertiroidismo y enfermedad renal crónica. En la exploración física, la ginecomastia se manifiesta en forma de masa subaerolar gomosa y concéntrica. Normalmente es bilateral y puede ser sensible en los primeros estadios del desarrollo. Las masas mamarias unilaterales, no sensibles y/o fijas deben dar pie a una evaluación para cáncer mama con una mamografía. La seudoginecomastia se caracteriza por el aumento de la grasa subaerolar sin hipertrofia glandular. En un varón que presenta ginecomastia dolorosa sin que identifique una causa clara en la anamnesis y la exploración física, debe obtenerse una medición de los niveles de hCG, LH, testosterona total matinal y estradiol. El tratamiento de una causa específica de ginecomastia durante la fase proliferativa activa puede provocar su regresión. Si la ginecomastia es de larga evolución, la regresión (espontánea o con tratamiento médico) es improbable, debido a las alteraciones fibróticas. En este contexto, la mejor manera de lograr una mejora estética puede ser la derivación a cirugía plástica. PUNTO CLAVE • El tratamiento de una causa específica de ginecomastia durante la fase proliferativa activa puede provocar su regresión; si la ginecomastia es de larga evolución, la regresión es improbable, debido a las alteraciones fibróticas. Manejo de la terapia hormonal transgénero La medicina transgénero corresponde a la atención a las personas cuya identidad de género difiere del sexo asignado en el nacimiento. La incongruencia de género es la incongruencia persistente entre la identidad de género y la anatomía sexual externa en el nacimiento que no surge de un trastorno de confusión mental; la disforia de género es el malestar que provoca la incongruencia entre la identidad de género de una persona y su anatomía sexual externa en el nacimiento. Un varón transgénero es una persona con identidad sexual masculina y sexo femenino asignado en el nacimiento; una mujer transgénero es una persona con identidad sexual femenina y sexo masculino asignado en el nacimiento. Las personas transgénero pueden evitar la atención sanitaria debido a las interacciones discriminatorias o irrespetuosas en relaciones previas con el personal sanitario. Proporcionar un entorno seguro es fundamental para garantizar el establecimiento y mantenimiento de la atención primaria y de afirmación de sexo en las personas transgénero. Es importante que los profesionales comprendan la terminología básica utilizada por la comunidad trans, que varía en función de la región. La atención psicológica y médica debe procurarse en un entorno que evite los prejuicios; con este fin, es imprescindible que la señalización del entorno y la terminología sean adecuadas y que el personal tenga la debida formación (Standards of Care de la World Professional Association for Transgender Health [WPATH]). También es importante obtener la información exacta de la identidad de género; muchas organizaciones utilizan un método en «dos etapas» para recabar 77 Trastornos del calcio y los huesos estos datos: 1) la identidad de género y 2) el sexo mencionado en el certificado de nacimiento original, evitando así la invisibilidad del estado transgénero. Antes de la exploración física, es necesario obtener la anamnesis para conocer las alteraciones anatómicas de la persona asociadas a la terapia hormonal para la afirmación de género (gender-affirming hormone therapy [GAHT]) y la intervención quirúrgica. Los caracteres sexuales secundarios se manifiestan en un amplio espectro del desarrollo en los pacientes transgénero. Los profesionales deben ofrecer un cribado del cáncer y un mantenimiento de la salud adecuados en función de la anatomía de la persona. La GAHT es la intervención médica más frecuente que solicitan las personas transgénero y no requiere atención especializada. Pueden prescribirla profesionales de atención primaria, ginecólogos y endocrinólogos. El tratamiento incluye medicamentos para el hirsutismo (espironolactona), anticoncepción (estradiol/progestágeno), sangrado uterino anormal (estradiol/progestágeno), menopausia (estradiol/progestágeno), déficit de testosterona (testosterona) e hiperplasia prostática benigna (inhibidores de la 5-α reductasa). La GAHT debe estar centrada en el paciente y personalizarse según los objetivos de este. Antes de iniciar el tratamiento, es esencial comentar los riesgos/beneficios asociados al tratamiento y obtener el consentimiento informado. Los criterios que deben tenerse en cuenta antes de iniciar la GAHT incluyen disforia de género persistente y demostrada, capacidad de tomar una decisión plenamente informada, mayoría de edad en un país determinado y, si los hay, tener controlados los trastornos médicos o psicológicos significativos. La GAHT limita la fertilidad, por lo que es necesario comentar las opciones de reproducción con los pacientes antes de iniciarla. La Endocrine Society dispone de guías de práctica clínica para la GAHT. Con la GAHT, la mayoría de los cambios tienen lugar en el transcurso de dos años, pero el calendario exacto es muy variable. La terapia hormonal feminizante suele consistir en estradiol combinado con un bloqueador androgénico. Los objetivos son el desarrollo de las mamas, la redistribución de la grasa y la reducción de la masa corporal, el vello corporal, la función eréctil, el recuento de espermatozoides y el tamaño testicular. La terapia estrogénica aumenta el riesgo de trombosis venosa profunda (TVP) y, en menor grado según los resultados de estudios de cohortes, ictus isquémico e infarto de miocardio; las contraindicaciones de la terapia estrogénica incluyen los antecedentes de TVP, las neoplasias sensibles a los estrógenos y la enfermedad hepática terminal. Dado el mayor riesgo de TVP, es necesario alentar al paciente a dejar de fumar antes del inicio de la terapia androgénica. La terapia antiandrogénica, como la espironolactona, reduce los caracteres sexuales secundarios masculinos y minimiza la dosis de estrógenos necesaria, por lo que reduce los riesgos asociados al tratamiento con estrógenos exógenos en dosis altas. Durante el primer año hay que controlar los niveles de testosterona y estradiol para determinar si la respuesta al tratamiento es adecuada. 78 El tratamiento con hormonas masculinizantes consiste en el uso de testosterona tópica o inyectada con el objetivo de lograr el cese de la menstruación, el crecimiento de vello facial, la profundización de la voz, la redistribución de la grasa, el aumento de la masa muscular y el vello corporal y el crecimiento clitoriano. Las contraindicaciones del tratamiento con testosterona incluyen embarazo, enfermedad coronaria inestable y policitemia. Durante el primer año deben controlarse los niveles de testosterona y estradiol para determinar si la respuesta al tratamiento es adecuada. También hay que controlar la hemoglobina. La cirugía de confirmación de género suele ser la última intervención en las personas transgénero. Muchas personas transgénero no solicitan la cirugía, pero esta es fundamental para aliviar la disforia de género en otras. En las mujeres transgénero, las intervenciones quirúrgicas pueden incluir mamoplastia de aumento, cirugía genital (penectomía, orquiectomía, vaginoplastia, clitoroplastia, vulvoplastia) y cirugía no genital y no mamaria (feminización facial, cirugía de la voz, reducción del cartílago tiroides). En los varones transgénero, las intervenciones quirúrgicas pueden incluir mastectomía, histerectomía con ovariectomía, faloplastia, vaginectomía, escrotoplastia e implantación de prótesis peneanas y/o testiculares. Antes de someterse a una cirugía de reasignación de género irreversible hay que cumplir unos criterios estrictos. PUNTOS CLAVE • La terapia hormonal para la afirmación de género es la intervención médica más frecuente que solicitan las personas transgénero y no requiere atención especializada; los criterios para recibir terapia hormonal para la afirmación de género incluyen disforia de género persistente y demostrada, capacidad de tomar una decisión plenamente informada, mayoría de edad en un país determinado y, si los hay, tener controlados los trastornos médicos o psicológicos significativos. • El cribado y la medicina preventiva en los pacientes transgénero deberá basarse en la anatomía de la persona. Trastornos del calcio y los huesos Homeostasis del calcio y fisiología ósea La regulación del nivel de calcio sérico es compleja y depende de las acciones de la vitamina D y la hormona paratiroidea (PTH). El efecto primario de la vitamina D es potenciar la absorción del calcio en el tubo intestinal, mientras que los efectos de la PTH están mediados principalmente por la regulación de la retención y la excreción de calcio en el riñón (Figura 15). La medición de los niveles de calcio depende de la cantidad que esté Trastornos del calcio y los huesos 1,25 (OH)2 vitamina D Alimentación 1.000 mg Intestino Absorción 300 mg Excreción 100 mg Heces 800 mg Formación 200 mg Calcio en sangre Hueso Reabsorción 200 mg Filtrado 9.000 mg Riñón Reabsorbido 880 mg Orina 200 mg PTH F i g u r a 1 5 . El flujo neutro del calcio entre el hueso y la sangre en adultos está coordinado por la hormona paratiroidea (PTH). Aunque la mayor parte del calcio filtrado en la orina se reabsorbe independientemente de la PTH, la PTH aumenta en mayor medida la retención de calcio de la orina. La PTH incrementa de manera indirecta la absorción de calcio en el intestino aumentando la producción de 1,25 (OH)2 vitamina D. Los dos efectos de la PTH adquieren mayor relevancia con la menor ingesta de calcio y a medida que descienden los niveles de este. Las cantidades de calcio que se presentan aquí ilustran la contribución relativa de cada órgano a la homeostasis del calcio en un adulto sano. unida a albúmina, que puede estar afectada por la nutrición y el estado ácido-base. La hipoalbuminemia por cualquier causa, como cirrosis o caquexia relacionada con un cáncer, provocará niveles bajos de calcio. Cuando la concentración de calcio es baja, es necesario realizar una medición del calcio ionizado o calcular el calcio total corregido para evaluar con exactitud los niveles de calcio. El nivel de vitamina D está determinado por la producción en la piel en respuesta a la luz solar y por la ingesta ya sea de los alimentos o de aportes complementarios. Aunque la vitamina D2 (ergocalciferol) y la vitamina D3 (colecalciferol) están disponibles en forma de suplementos, la última puede ser más eficaz debido a la mayor potencia, la semivida más prolongada y el hecho de que es idéntica a la que se forma con la exposición a la luz ultravioleta. La activación de las vitaminas D2 y D3 requiere la hidroxilación en un primer momento por parte del hígado y posteriormente por el riñón, lo que da lugar a la forma activa de la vitamina D, la 1,25-dihidroxivitamina D, el calcitriol. La 25-hidroxivitamina D es la forma en que se la vitamina D se almacena en el organismo, y la medición de la 25-hidroxivitamina D constituye la prueba más exacta para evaluar las reservas vitamínicas. La respuesta inicial a un descenso del calcio sérico es un aumento de la secreción de PTH, que reduce la excreción de calcio renal y aumenta la resorción ósea para elevar el nivel de calcio sérico. Además, la PTH induce una mayor conversión renal de 25-hidroxivitamina D al metabolito activo 1,25-dihidroxivitamina D, lo cual mejora la eficiencia de la absorción intestinal de calcio. La movilización continua del calcio del hueso mediada por la PTH a lo largo de meses o años en respuesta a un equilibrio cálcico negativo crónico puede provocar enfermedad ósea metabólica. En cambio, los huesos, el intestino y el metabolismo de la vitamina D no contri- buyen de manera significativa a la corrección de la hipercalcemia. Por el contrario, el aumento de la carga filtrada y la inhibición de la secreción de PTH dan lugar a una importante excreción de calcio por medio de los riñones, siempre que el volumen circulante efectivo sea suficiente. Además de su función en el metabolismo mineral, el esqueleto adulto constituye un depósito de calcio y da soporte estructural a la movilidad, la fijación muscular y la protección de los órganos vitales. La remodelación ósea permite la adaptación y la reparación continua del esqueleto. Los osteocitos coordinan la remodelación ósea, que se inicia con la resorción osteoclástica y sigue con la formación ósea osteoblástica y la mineralización de una matriz de colágeno/proteína, mucho más lentas. El esqueleto completo se remodela cada 10 años aproximadamente. PUNTOS CLAVE • El efecto primario de la vitamina D es potenciar la absorción del calcio en el tubo intestinal, mientras que los efectos de la hormona paratiroidea están mediados principalmente por la regulación de la retención y excreción de calcio en el riñón. • La medición de la 25-hidroxivitamina D constituye la prueba más adecuada para detectar déficit de vitamina D. Hipercalcemia Manifestaciones clínicas de la hipercalcemia La hipercalcemia se diagnostica cuando el nivel de calcio supera los niveles normales, normalmente 10,5 mg/dl (2,6 mmol/l). Es frecuente la detección incidental de hipercalcemia asintomática en análisis de sangre de rutina o de cribado. 79 Trastornos del calcio y los huesos tituye un primer paso fundamental para diagnosticar la causa y clasificar la hipercalcemia en mediada por PTH o no mediada por PTH. La determinación del calcio ionizado no es útil cuando el nivel de albúmina sérica es normal o cuando no hay trastornos ácido-base agudos. Todos los pacientes con hipercalcemia deben ser objeto de una anamnesis y una exploración física exhaustivas, así como de una revisión atenta de todos los medicamentos que tomen, incluidos los suplementos. Los síntomas clásicos de hipercalcemia incluyen poliuria, polidipsia y nicturia. Otros síntomas pueden ser anorexia, náuseas, dolor abdominal, estreñimiento y alteraciones del estado mental. A niveles más altos, los pacientes pueden experimentar embotamiento. Los síntomas no muestran una relación lineal con los niveles séricos de calcio o PTH. La hipercalcemia y la hipercalciuria graves pueden provocar depleción de volumen y daño renal agudo, nefrolitiasis o nefrocalcinosis. Las manifestaciones óseas son reflejo de la causa subyacente de la hipercalcemia. El hiperparatiroidismo primario puede manifestarse con osteoporosis con fracturas por fragilidad y baja densidad ósea. El hiperparatiroidismo grave por carcinoma paratiroideo o el hiperparatiroidismo secundario debido a enfermedad renal pueden cursar con dolor óseo y osteítis fibrosa quística (un diagnóstico radiográfico). Muchas veces la hipercalcemia asociada a lesiones óseas líticas es consecuencia de mieloma múltiple o cáncer de mama. PUNTO CLAVE • Las pruebas diagnósticas iniciales de hipercalcemia requieren la medición simultánea del calcio y la hormona paratiroidea (PTH) en suero, que permite clasificar la enfermedad en relacionada con la PTH o no relacionada con la PTH. Medicamentos causantes de hipercalcemia Los diuréticos tiazídicos pueden provocar hipercalcemia leve, sobre todo en el marco de un hiperparatiroidismo primario leve no detectado anteriormente. La hipercalcemia asociada a tratamiento con litio tiene su origen en la secreción alterada de PTH y puede presentarse años después del inicio del tratamiento. Si es posible, la interrupción del medicamento y la repetición de la medición de los niveles de calcio representan un primer paso lógico en el manejo. El hecho de que el calcio vuelva a la normalidad es indicativo del papel causal del medicamento. PUNTO CLAVE • Los síntomas de hipercalcemia son variables, pero pueden incluir poliuria, polidipsia, nicturia, anorexia, náuseas, dolor abdominal, estreñimiento y alteraciones del estado mental, y pueden estar asociados a daño renal agudo, nefrolitiasis, nefrocalcinosis y alteraciones óseas. Causas y diagnóstico de la hipercalcemia Los indicios de la causa subyacente de la hipercalcemia se basan en la gravedad, el carácter agudo de la enfermedad y factores del paciente, incluyendo las comorbilidades. La hipercalcemia se clasifica en leve (<12 mg/dl [3 mmol/l]), moderada (12-14 mg/dl [3-3,5 mmol/l]) o grave (>14 mg/dl [3,5 mmol/l]). Cuando la hipercalcemia se descubre de manera incidental, está indicado repetir la medición. Si se confirma la hipercalcemia, la medición simultánea del calcio y la PTH en suero cons- Hipercalcemia mediada por la hormona paratiroidea La secreción de la PTH desciende de manera repentina en respuesta a la elevación de la concentración de calcio en suero. Por lo tanto, un nivel de PTH elevado o inadecuadamente normal (por lo general, en la mitad superior del intervalo de referencia) en un paciente con hipercalcemia es diagnóstico de hipercalcemia mediada por la PTH (Figura 16). El manejo 200 180 PTH intacta (pg/dl) 160 140 120 100 Hiperparatiroidismo secundario 80 Hiperparatiroidismo primario 60 40 Normal 20 0 Hipoparatiroidismo 6 7 8 Hipercalcemia o cáncer 9 10 11 Calcio sérico (mg/dl) 80 12 13 14 15 F i g u r a 1 6 . Relación del calcio y la hormona paratiroidea (PTH) en condiciones normales y en varias enfermedades. Trastornos del calcio y los huesos de los pacientes con nivel elevado de PTH pero niveles normales de calcio y vitamina D (hiperparatiroidismo primario normocalcémico) puede ser similar al de quienes tienen hiperparatiroidismo primario asintomático. Hiperparatiroidismo primario El hiperparatiroidismo primario suele deberse a un adenoma paratiroideo solitario. Las mujeres se ven afectadas con mayor frecuencia que los varones, con una incidencia máxima en la séptima década de vida. La hipercalcemia suele ser leve (dentro de 1 mg/ml [0,25 mmol/l] del límite superior de la normalidad) y puede ser normal de manera intermitente. La hipercalciuria está presente hasta en el 30% de los pacientes. Dado que la PTH aumenta la excreción de fosfato renal, las concentraciones de fósforo sérico bajas o bajas-normales respaldan el diagnóstico. La evaluación de la densidad mineral ósea (DMO) con absorciometría de rayos X de energía dual (DEXA) debe incluir el brazo no dominante, que puede estar especialmente afectado en los pacientes con hiperparatiroidismo. Aunque las pruebas de imagen paratiroideas con sestamibi o ecografía de cuello pueden localizar un adenoma, la presencia de un adenoma no influye en la decisión de proceder con la cirugía en el contexto de un hiperparatiroidismo primario. Las pruebas de imagen pueden ser útiles para el cirujano a la hora de planificar la intervención quirúrgica. En ausencia de antecedentes de nefrolitiasis por calcio, puede estar indicado obtener pruebas de imagen renales para descartar cálculos ocultos si su hallazgo sería causa de modificación del tratamiento. El consumo de calcio con la alimentación debe ser de 1.000 mg/día aproximadamente para evitar nuevos aumentos de la excreción de calcio en la orina y de la secreción de PTH. Es importante medir la 25-hidroxivitamina D y corregir con precaución el déficit de vitamina D. La recuperación se recomienda en los pacientes cuyos niveles estén situados por debajo de 30 ng/dl (75 nmol/l), prestando atención especial a la excreción de calcio en la orina y al calcio sérico una vez que los valores de vitamina D superen los 30 ng/dl (75 nmol/l). Los cambios en los criterios de valoración específicos durante el control que llevan a recomendar la cirugía paratiroidea se presentan en la Tabla 43. Si la efectúa un cirujano experimentado con técnicas mínimamente invasivas, la cirugía tiene una tasa de curación del 95% y una tasa de complicaciones inferior al 1%. La corrección preoperatoria del déficit de vitamina D es importante para prevenir la hipocalcemia postoperatoria, que es consecuencia de un hipoparatiroidismo relativo y de la producción reducida de 1,25-dihidroxivitamina D mediada por PTH que culminan en un flujo rápido de calcio en el hueso (síndrome del hueso hambriento). Los pacientes con hiperparatiroidismo primario leve suelen necesitar un aporte complementario de calcio durante un máximo de una semana después de paratiroidectomía hasta que el tejido paratiroideo residual normaliza las concentraciones de calcio sérico. La reevaluación de la DMO un año después de la paratiroidectomía puede revelar una mejoría de la DMO, sobre todo en la columna. Tabla 43. el control Indicaciones para la cirugía paratiroidea durante Evaluación Indicacióna Calcio sérico (> límite superior de la normalidad) >1 mg/dl (>0,25 mmol/l) Ósea Puntuación T <–2,5 en columna lumbar, cadera total, cuello femoral o radio 1/3 distal; o reducción significativa de la DMOa Fractura vertebral según radiografía, TC, RM o EFV Renal ACr <60 ml/min Desarrollo clínico de un cálculo renal o con pruebas de imagen (radiografía, ecografía o TC) aUn cambio significativo se define como una reducción que es mayor que el cambio menos significativo según la definición de la International Society for Clinical Densitometry. ACr: aclaramiento de creatinina; EFV: evaluación de fracturas vertebrales; RM: resonancia magnética; TC: tomografía computarizada. De Bilezikian JP, Brandi ML, Eastell R, Silverberg SJ, Udelsman R, Marcocci C, et al. Guidelines for the Management of Asymptomatic Primary Hyperparathyroidism: Summary Statement from the Fourth International Workshop. J Clin Endocrinol Metab. 2014;99:3561-9. [PMID: 25162665] Aproximadamente uno de cada tres pacientes con hiperparatiroidismo primario asintomático que en un primer momento aplazan la cirugía presentará indicaciones para esta durante 10-15 años de observación. En los pacientes que no se consideraron aptos o que decidieron no someterse a cirugía, la evaluación debe incluir la medición anual del calcio sérico, la creatinina y la tasa de filtración glomerular (TFG). En caso de presuntos cálculos renales, debe plantearse la obtención de pruebas de imagen y la recogida de orina de 24 horas para determinar el perfil bioquímico de los cálculos. La DMO debe obtenerse cada dos años y, si el paciente presenta una pérdida significativa de estatura o dolor de espalda en el contexto de una DMO normal, debe considerarse la obtención de pruebas de imagen. PUNTO CLAVE • Es importante medir la 25-hidroxivitamina D y corregir con precaución el déficit de vitamina D en los pacientes con hiperparatiroidismo primario. Carcinoma paratiroideo El carcinoma paratiroideo es muy infrecuente, pero puede manifestarse con síntomas de hipercalcemia grave, con niveles séricos >14 mg/dl (3,5 mmol/l) y concentraciones de PTH notablemente elevadas. Las pruebas de imagen no son útiles, y la aspiración con aguja fina no está recomendada debido a la posibilidad de siembra del tumor. El tratamiento primario consiste en la resección quirúrgica. Lamentablemente, el 50% de los pacientes pueden presentar enfermedad residual o recidivante. La hipercalcemia grave en el carcinoma paratiroi81 Trastornos del calcio y los huesos deo que no es apto para cirugía puede tratarse de manera crónica con cinacalcet. Las opciones farmacológicas son limitadas, y es muy probable que los pacientes mueran por complicaciones de la hipercalcemia. Hiperparatiroidismo terciario En los pacientes con enfermedad renal terminal, se produce una hiperplasia multiglandular debida al estímulo crónico de la PTH (una secuela del hiperparatiroidismo secundario de larga evolución) que tiene su origen en la hipocalcemia y la hiperfosfatemia mal controladas. En algunos casos de hiperparatiroidismo secundario, el calcio sérico puede normalizarse si la hiperplasia y la secreción de PTH asociada son firmes. La estimulación crónica de las glándulas paratiroides puede provocar la producción autónoma de PTH por parte de las cuatro glándulas, que da lugar a hipercalcemia. En la mayoría de los casos, el hiperparatiroidismo terciario se presenta tras trasplante renal. Aunque históricamente se ha tratado con paratiroidectomía multiglandular total, en la mayoría de los pacientes la hipercalcemia puede resolverse con un tratamiento con paricalcitol o cinacalcet. Causas genéticas de la hipercalcemia Hipercalcemia hipocalciúrica familiar La hipercalcemia hipocalciúrica familiar (HHF) es un trastorno autosómico dominante y representa el tipo más frecuente de hipercalcemia familiar. Los pacientes están asintomáticos. Las glándulas paratiroides y el riñón detectan las concentraciones de calcio en suero a través del receptor sensible al calcio (CaSR). En la HHF, una mutación de pérdida de actividad del gen del CaSR hace que las glándulas paratiroides perciban las concentraciones séricas como bajas, lo que da lugar al aumento de la secreción de PTH y a un nivel de calcio sérico elevado. Al mismo tiempo, en el riñón el CaSR mutado incrementa la absorción renal de calcio, lo que provoca hipocalciuria paradójica en el contexto de hipercalcemia. Aunque estos pacientes parecen tener hiperparatiroidismo primario, la HHF es un trastorno benigno que no se trata con paratiroidectomía. La hipercalcemia no se resolverá con la cirugía. Los pacientes no sufren secuelas de la hipercalcemia como cálculos u osteoporosis. Los signos indicativos de HHF son: hipercalcemia leve desde la infancia; baja excreción de calcio en la orina de 24 horas, sobre todo si el cociente de calcio y aclaramiento de la creatinina está por debajo de 0,01, y/o antecedentes familiares de paratiroidectomía sin resolución de la hipercalcemia. En caso de ambigüedad clínica, el diagnóstico puede confirmarse mediante análisis genéticos del CaSR. PUNTO CLAVE • La distinción entre hiperparatiroidismo primario e hipercalcemia hipocalciúrica familiar puede hacerse con una recogida de orina de 24 orina para determinar el calcio y la creatinina, que demostrará la cantidad de excreción renal de calcio y permitirá evaluar el cociente de calcio y aclaramiento de la creatinina. 82 Síndrome de neoplasia endocrina múltiple El primer signo de síndrome de neoplasia endocrina múltiple (NEM) en adolescentes y jóvenes puede ser el hiperparatiroidismo primario. El hiperparatiroidismo primario está asociado a los síndromes NEM1 y NEM2A. Si los antecedentes familiares revelan hiperparatiroidismo primario, tumor hipofisario, síndrome de Zollinger-Ellison, muerte prematura por neoplasia pancreática, feocromocitoma o cáncer medular de tiroides, la NEM es más probable y debe considerarse un cribado. A diferencia del hiperparatiroidismo primario esporádico, los síndromes de NEM cursan con recidiva del hiperparatiroidismo debido a la hiperplasia continua del tejido paratiroideo restante tras la paratiroidectomía. La NEM1 está asociada a mutación del gen inhibidor tumoral MEN1, y la NEM2 está asociada a una mutación del gen RET. El manejo debe estar a cargo de un endocrinólogo o hacerse en conjunción con un endocrinólogo. Hipercalcemia no mediada por la hormona paratiroidea El diagnóstico diferencial de la hipercalcemia con PTH inhibida es amplio. En los pacientes con hipercalcemia grave, la anamnesis, los síntomas y los hallazgos pueden apuntar a la causa subyacente. El tratamiento debe iniciarse sin demora mientras se espera a los resultados de las pruebas analíticas. En los estados hipercalcémicos independientes de la PTH, la hipercalciuria puede ser grave y preceder a la hipercalcemia. Normalmente, la PTH es indetectable, aunque puede ser muy baja (<20 pg/ml [20 ng/l]) en caso de hipercalcemia leve. Hipercalcemia asociada a cáncer La causa más frecuente de hipercalcemia no mediada por hormonas paratiroideas es el cáncer; por lo general se trata de una hipercalcemia grave (>14 mg/dl [3,5 mmol/l]). A menudo es consecuencia de la proteína relacionada con la PTH (PTHrP) producida por el tumor, que provoca una importante resorción ósea. El carcinoma de células renales, el cáncer de mama y los cánceres epiteliales están asociados a hipercalcemia relacionada con la PTHrP. En contadas ocasiones, la osteólisis mediada localmente producida por metástasis óseas extensas, normalmente en el mieloma múltiple y en el cáncer de mama, puede causar la salida del calcio del hueso y dar lugar a hipercalcemia significativa. Para más información, véase el módulo «Hematología y Oncología» de MKSAP 18. Hipercalcemia dependiente de la vitamina D La hipercalcemia dependiente de la vitamina D está asociada a niveles de fósforo sérico normales o altos, porque la vitamina D potencia la absorción intestinal de fósforo y la inhibición de la secreción de PTH reduce la excreción de fósforo a través del riñón. En el tejido granulomatoso asociado a infección fúngica, tuberculosis, sarcoidosis y linfoma puede producirse una conversión no regulada de 25-hidroxivitamina D a 1,25-dihidroxivitamina D, lo que da lugar a una mayor absorción in- Trastornos del calcio y los huesos testinal de calcio. Estos trastornos están asociados a un nivel de 1,25-dihidroxivitamina D inadecuadamente normal o notablemente elevado y a inhibición de la PTH. La disminución del calcio sérico y urinario después de la ingesta de calcio y vitamina D está restringido; un descenso rápido del calcio después de un tratamiento glucocorticoide (que inhibe la hidroxilación de la 25-hidroxivitamina D) es compatible con estos trastornos. La intoxicación por vitamina D por el consumo prolongado de vitamina D en dosis altas (normalmente >50.000 unidades diarias en pacientes con condiciones de malabsorción) y el aumento de las reservas de grasa produce hipercalciuria prolongada, nefrolitiasis, función renal alterada y niveles elevados de 25-hidroxivitamina D. Otras causas El consumo de grandes cantidades de calcio, normalmente por el uso de antiácidos (p. ej., carbonato de calcio), sobre todo en caso de enfermedad renal crónica concurrente, provoca el síndrome de leche y alcalinos. La reposición de glucocorticoides y mineralocorticoides y el aumento del volumen resuelven la hipercalcemia leve que a veces está asociada a crisis addisonianas. En ocasiones, la tirotoxicosis grave produce hipercalcemia o hipercalciuria al aumentar la resorción ósea. La inmovilización prolongada aguda, como la que se observa en las lesiones de la médula espinal, puede provocar un gran eflujo de calcio de los huesos a través de la remodelación ósea desajustada que supone la menor actividad osteoblástica a pesar del aumento de la actividad osteoclástica. Los pacientes con hiperparatiroidismo primario o metástasis óseas tienen predisposición a sufrir hipercalcemia debido a la inmovilización, igual que los pacientes jóvenes en los que la remodelación ósea elevada es normal. Manejo de la hipercalcemia El manejo de la hipercalcemia depende de su gravedad. En los casos leves (<12 mg/dl [3 mmol/l]), el tratamiento del trastorno subyacente (p. ej., paratiroidectomía en el hiperparatiroidismo primario) es suficiente. En los pacientes con daño renal agudo, alteraciones del estado mental o niveles de calcio por encima de 12 mg/dl (3 mmol/l), puede ser necesaria la hospitalización. El tratamiento inicial de la hipercalcemia grave es la hidratación agresiva para recuperar la pérdida de volumen y aumentar la excreción renal de calcio. Los diuréticos de asa no están recomendados, a menos que haya insuficiencia renal o insuficiencia cardíaca, en cuyo caso el aumento de volumen debe ser anterior a su administración para evitar la hipotensión y la aparición de nuevos daños renales. En los pacientes con síntomas agudos puede emplearse la calcitonina subcutánea; sin embargo, el efecto del fármaco se difumina al cabo de 48 horas. El manejo a largo plazo de la hipercalcemia puede requerir tratamiento con bifosfonatos intravenosos para prevenir la movilización del calcio de los huesos, pero hace falta que la función renal sea adecuada. Los glucocorticoides y la restricción de la ingesta de calcio y vitamina D son especialmente beneficiosos en la hipercalcemia dependiente de la vitamina D. La hemodiálisis se reserva para el tratamiento de la hipercalcemia grave en los pacientes oligúricos. PUNTO CLAVE • El tratamiento inicial de la hipercalcemia moderada o grave es la hidratación agresiva para recuperar la pérdida de volumen y aumentar la excreción renal de calcio; los diuréticos de asa no están recomendados, a menos que haya insuficiencia renal o insuficiencia cardíaca, en cuyo caso el aumento de volumen debe ser anterior a la administración de diuréticos de asa para evitar la hipotensión y nuevos daños renales. Hipocalcemia Manifestaciones clínicas de la hipocalcemia Los signos y síntomas de la hipocalcemia son reflejo de su gravedad y agudeza. Los trastornos hipocalcémicos en pacientes ambulatorios suelen detectarse en análisis de cribado y son leves, con un nivel de calcio en suero de entre 7,5 y 8,9 mg/dl (1,9-2,2 mmol/l). También puede detectarse durante la evaluación por fracturas traumáticas de baja intensidad o baja masa ósea. La mayoría de los pacientes serán asintomáticos o referirán parestesias intermitentes en las manos y los pies o hipoestesia peribucal. La hipocalcemia debida a hipoparatiroidismo crónico puede cursar también con formación de cataratas, calcificación de los núcleos basales, papiledema e hipoplasia del esmalte dental. Los pacientes con hipocalcemia grave pueden presentar síntomas y signos neuromusculares. El espasmo carpopedal con postura característica de la mano (flexión en las articulaciones metacarpofalángicas y extensión en las articulaciones interfalángicas) puede ser espontáneo o provocado por isquemia transitoria de la extremidad distal durante la medición de la presión arterial (signo de Trousseau). La hiperirritabilidad y espasmos musculares de los nervios faciales pueden demostrarse mediante percusión en el nervio facial justo por delante de la oreja (signo de Chvostek). Cabe destacar que el laringoespasmo, las convulsiones, la disfunción miocárdica y la prolongación del intervalo QT causantes de muerte súbita cardíaca debido a hipocalcemia grave (<7,5 mg/dl [1,9 mmol/l]) pueden producirse sin parestesias o calambres musculares prodrómicos. PUNTO CLAVE • El laringoespasmo, las convulsiones, la disfunción miocárdica y la prolongación del intervalo QT causantes de muerte súbita cardíaca debido a hipocalcemia grave (<7,5 mg/dl [1,9 mmol/l]) pueden producirse sin parestesia o calambres musculares prodrómicos. 83 Trastornos del calcio y los huesos Causas y diagnóstico de la hipocalcemia La hipocalcemia debe confirmarse con una segunda medición, que requiere la evaluación y corrección de las concentraciones de albúmina sérica. La medición del calcio ionizado está indicada en el contexto de estado ácido-base fluctuante. El paso siguiente consiste en la medición simultánea del calcio, el fósforo, la creatinina y la PTH en suero. En caso de hipocalcemia, la PTH debería estar elevada (véase la Figura 16). Hipoparatiroidismo En la mayoría de los casos, el hipoparatiroidismo está causado por daño inadvertido durante una cirugía de cuello anterior (tiroidectomía, paratiroidectomía) o una cirugía para tratar la hiperplasia de las glándulas paratiroides; en ambos casos, se manifiesta en las horas posteriores a la intervención quirúrgica. En función del grado del daño o la resección, el hipoparatiroidismo quirúrgico puede durar días o semanas. El hipoparatiroidismo permanente puede ser parcial o completo; el último está asociado a niveles séricos de PTH indetectables y a una mayor prevalencia de hiperfosfatemia. El primero cursa con niveles inadecuadamente normales de PTH e hipocalcemia concurrente. Otras causas de hipocalcemia debida a secreción insuficiente de PTH incluyen trastornos infiltrativos (hemocromatosis o enfermedad de Wilson), radiación, autoinmunidad y trastornos congénitos (como el síndrome de deleción de 22q11.2). La hipocalcemia crónica con PTH inadecuadamente normal que se da dentro de una familia puede representar una mutación activadora del gen CaSR. La hipomagnesemia, observada en el contexto de malnutrición, alcoholismo y uso de diuréticos de asa y tratamiento crónico con inhibidores de la bomba de protones, provoca hipofunción paratiroidea funcional reversible y debe descartarse antes de atribuir un nivel de PTH bajo o inadecuadamente normal a hipoparatiroidismo. La resistencia a la PTH (seudohipoparatiroidismo) es una causa genética infrecuente de hipocalcemia. PUNTO CLAVE • La hipomagnesemia provoca hipofunción paratiroidea funcional reversible y debe descartarse antes de atribuir un nivel de hormona paratiroidea bajo o inadecuadamente normal a hipoparatiroidismo. Otras causas de hipocalcemia Puede sospecharse malnutrición y/o malabsorción de vitamina D o calcio sobre la base de los antecedentes clínicos (cirugía bariátrica, enfermedad celíaca); la confirmará un nivel bajo de 25-hidroxivitamina D en suero o una baja excreción de calcio en orina de 24 horas (un indicador indirecto de la ingesta y absorción de calcio). La causa más frecuente de hipocalcemia adquirida es la insuficiencia renal crónica debida a la producción alterada de 1,25-dihidroxivitamina D e hiperfosfatemia. La hipercalciuria es a menudo idiopática, pero también puede tener su origen en el uso crónico de diuréti84 cos de asa. La rabdomiólisis y el síndrome de lisis tumoral aumentan el fósforo sérico y la unión del fosfato cálcico en el espacio vascular, lo que da lugar a un nivel bajo de calcio ionizado. El síndrome del hueso hambriento (flujo rápido de calcio hacia el hueso después de paratiroidectomía por hiperparatiroidismo primario grave) y las metástasis osteoblásticas extendidas (cáncer de próstata, cáncer de mama) pueden provocar hipocalcemia, igual que la saponificación del calcio (y el magnesio) en la grasa necrótica en la pancreatitis aguda. Los fármacos antirresortivos potentes, como los bifosfonatos intravenosos y el denosumab, pueden provocar hipocalcemia grave y prolongada al alterar el eflujo fisiológico de calcio de los huesos en los pacientes con déficit de vitamina D. Por lo tanto, es importante evaluar los niveles de vitamina D y corregir el déficit antes de iniciar el tratamiento con un fármaco antirresortivo. Manejo de la hipocalcemia Dado que la hipocalcemia puede tener complicaciones neuromusculares graves en ausencia de tetania muscular prodrómica, la hipocalcemia grave (<7,5 mg/dl [1,9 mmol/l]) requiere tratamiento urgente con calcio intravenoso. Es preferible la administración mediante acceso intravenoso central con control electrocardiográfico. Otra posibilidad es administrar 20 µg de teriparatida dos veces al día, que elimina rápidamente los síntomas hipocalcémicos en el hipoparatiroidismo posquirúrgico agudo (indicación no recogida en la ficha técnica). El aporte complementario de entre 1.000 y 4.000 UI/día de vitamina D y carbonato de calcio o citrato de calcio oral a dosis de entre 1 y 3 g/día puede normalizar o tratar adecuadamente la hipocalcemia leve o crónica. El calcitriol es necesario en el contexto de hipoparatiroidismo con PTH indetectable o insuficiencia renal, porque la activación de la 1,25-dihidroxivitamina D requiere PTH y una función renal suficiente. En el hipoparatiroidismo crónico, los objetivos del tratamiento consisten en eliminar los síntomas a la vez que se evitan las complicaciones del tratamiento. Un objetivo razonable para la mayoría de los pacientes es una concentración de calcio sérico dentro del intervalo de referencia o justo por debajo sin hipercalciuria. Es imprescindible controlar la excreción de calcio matinal, ya que muchas veces la hipercalciuria limita el tratamiento. También es necesario corregir la hipomagnesemia coexistente. Habitualmente se utilizan diuréticos tiazídicos, porque reducen la excreción de calcio en la orina. El tratamiento inicial de la hiperfosfatemia consiste en reducir el fósforo de la alimentación, pero en ocasiones requiere la adición de aglutinantes del fosfato orales cuando el fósforo sérico supera el intervalo normal. Hay disponible PTH humana recombinante para los pacientes que no cumplen los objetivos terapéuticos con el tratamiento con calcitriol y calcio solamente. Trastornos del calcio y los huesos PUNTO CLAVE • Un objetivo razonable para la mayoría de los pacientes con hipoparatiroidismo es una concentración de calcio sérico dentro del intervalo de referencia o justo por debajo sin hipercalciuria. Enfermedad ósea metabólica Masa ósea baja y osteoporosis La masa ósea, el contenido de minerales y tanto la macroarquitectura como la microarquitectura determinan la resistencia de los huesos. La densidad mineral ósea (DMO) es reflejo de la masa ósea y del contenido de minerales y, en los ancianos, predice el deterioro de la microarquitectura. Esta relación y los datos epidemiológicos sustentan el uso de la determinación de la DMO, mediante DEXA, para diagnosticar una masa ósea baja y afinar la evaluación del riesgo de fracturas en los ancianos. Las fracturas por fragilidad (las que tienen lugar con traumatismos mínimos, equivalentes o inferiores a una caída desde la altura en bipedestación) después de los 50 años indi- Tabla 44. can baja resistencia ósea y definen la osteoporosis clínica independientemente de la DMO. Por definición, las fracturas de cráneo, pies y mano no pueden ser fracturas por fragilidad. Fisiopatología Una masa ósea baja en adultos puede representar mala formación ósea, pérdida ósea o ambas. Los factores que pueden afectar a la formación de masa ósea incluyen trastornos genéticos, factores del estilo de vida y mala salud, sobre todo en la segunda década de vida. La pérdida neta de masa ósea puede darse en adultos cuando el remodelado óseo osteoclástico es más rápido que la formación ósea osteoblástica. La lista de factores de riesgo de masa ósea baja y osteoporosis es extensa y se presenta en la Tabla 44 y la Tabla 45. Sin embargo, algunos pacientes presentan osteoporosis provocada por causas secundarias. Las pruebas para detectar las causas secundarias se resumen en la Tabla 46. Cribado de osteoporosis Las guías actuales recomiendan realizar un cribado a las mujeres posmenopáusicas de riesgo medio a partir de los 65 años Factores de riesgo de densidad ósea baja y osteoporosis Estilo de vida/modificables No modificables Medicamentos/suplementos Consumo de alcohol Raza/grupo étnico Anticonvulsivos Inmovilización Edad Tratamiento antirretroviral (tenofovir) IMC <17 kg/m2 Sexo Inhibidores de la aromatasa Bajo consumo de calcio Familiar de primer grado con DMO baja Inhibidores de la calcineurina Tabaquismo Genéticas Depo-medroxiprogesterona Déficit de vitamina D Fibrosis quística Pérdida de peso Hipofosfatasia Caídas recurrentes Síndrome de Ehlers-Danlos Glucocorticoides (≥5 mg/día de prednisona o equivalente durante ≥3 meses) Heparina Agonistas de la GnRH Osteogénesis imperfecta Inhibidores de la bomba de protones Tiazolidindionas Litio Terapia de privación androgénica DMO: densidad mineral ósea; GnRH: hormona liberadora de gonadotropinas; IMC: índice de masa corporal. Tabla 45. Trastornos y comorbilidades asociados a un riesgo elevado de masa ósea baja y osteoporosis Endocrinos Gastrointestinales Hematológicos Reumatológicas Neurológicos Otros Anorexia nerviosa Cirugía bariátrica Amiloidosis Enfermedad celíaca Esclerosis múltiple Diabetes mellitus Enfermedad inflamatoria intestinal Leucemia y linfoma Espondilitis anquilosante Infección por VIH/sida Síndrome de Cushing Artritis reumatoide Distrofia muscular Hiperparatiroidismo Hipogonadismo Tirotoxicosis Malabsorción biliar primaria Síndrome de Turner Gammapatías monoclonales Mieloma múltiple Amiloidosis Lupus sistémico Lesión de médula espinal con parálisis Enfermedad pulmonar obstructiva crónica Enfermedad renal terminal Hipercalciuria idiopática VIH: virus de la inmunodeficiencia humana. 85 Trastornos del calcio y los huesos Tabla 46. Estudios diagnósticos para evaluar las causas secundarias de osteoporosis Análisis de sangre Hemograma completo Calcio, fósforo y magnesio Pruebas de función renal Pruebas de función hepática Tirotropina. 25-hidroxivitamina D Testosterona total (varones jóvenes) Triptasa Análisis de orina Calcio en orina de 24 horas Nivel de cortisol libre en orina La extensión de las pruebas debe depender de la sospecha clínica y de la gravedad de la osteoporosis. de edad. Las recomendaciones de las guías varían en cuanto al cribado sistemático de la osteoporosis en los varones. El American College of Rheumatology recomienda realizar una determinación de la DMO en los seis meses posteriores al inicio de un tratamiento glucocorticoide a largo plazo en los adultos ≥40 años de edad y en los adultos <40 años con factores de riesgo de osteoporosis o antecedentes de fracturas por fragilidad. Los pacientes con factores de riesgo de masa ósea baja u osteoporosis, fracturas de fragilidad de fémur, vértebras (Figura 17), pelvis, húmero o radio, pérdida de estatura ≥4 cm o cifosis deben someterse a una determinación de la DMO antes de lo que indican las recomendaciones de cribado estándar. La medición de la DMO también puede estar indicada si el riesgo de fractura es elevado según los resultados de herramientas de evaluación del riesgo como: Simple Calculated Osteoporosis Risk Estimate (SCORE), Osteoporosis Self-Assessment Tool (OST), Osteoporosis Risk Assessment Instrument (ORAI) y Fracture Risk Assessment Tool (FRAX). En la Tabla 47 se presentan las recomendaciones para realizar determinaciones de la DMO y pruebas de imagen de las vértebras. En las mujeres jóvenes, el cribado puede estar indicado en presencia de uno o varios factores de riesgo de osteoporosis. En las mujeres premenopáusicas sin factores de riesgo, la evaluación de la DMO para determinar el riesgo de fractura no está aconsejada ni validada. Sin embargo, si las pruebas se llevan a cabo en una persona por lo demás sana, en general los resultados inferiores a las medias de la edad o el sexo (puntuación Z <0) no requieren más pruebas ni controles seriados. PUNTO CLAVE • Las guías actuales recomiendan realizar un cribado a las mujeres posmenopáusicas de riesgo medio a partir de los 65 años de edad; las recomendaciones de cribado para los varones varían según la organización. 86 F i g u r a 1 7 . Fracturas vertebrales por compresión asintomáticas detectadas mediante radiografía de columna en un paciente con pérdida de estatura y cifosis. En la imagen se muestra una depresión en el platillo vertebral sin pérdida de altura vertebral en las vértebras lumbares superiores (flechas vacías) y enclavamiento grave y pérdida de altura de varios cuerpos vertebrales mesotorácicos (flechas llenas). Diagnóstico En las mujeres posmenopáusicas y los varones mayores de 50 años de edad, el diagnóstico de osteopenia se determina mediante la DMO (puntuación T de entre –1 y –2,5) solamente, mientras que la osteoporosis puede diagnosticarse clínicamente sobre la base de la presencia de fracturas por fragilidad, fractura de cadera o fractura por compresión vertebral o mediante DEXA. Si no hay causas secundarias de DMO baja o de una fractura por fragilidad, la osteoporosis se diagnostica cuando la puntuación T del cuello femoral, la cadera total, el radio no dominante o la columna lumbar compuesta (dos o más vértebras diagnósticas consecutivas) es ≤–2,5, según la definición de la Organización Mundial de la Salud (OMS). Si no es posible medir de manera exacta la DMO de la cadera o la columna, puede usarse la DEXA del tercio distal del radio. En las mujeres premenopáusicas y los varones menores de 50 años, la osteoporosis puede diagnosticarse ante una fractura por fragilidad o una masa ósea baja en la DEXA, de- Trastornos del calcio y los huesos Tabla 47. Recomendaciones para medición de la densidad mineral ósea y pruebas de imagen vertebrales Pruebas de medición de la densidad mineral óseaa Mujeres ≥65 años de edad En los varones, no hay evidencias suficientes para evaluar el riesgo/beneficio del cribado de la osteoporosisb Mujeres posmenopáusicas y varones de entre 50 y 69 años, en función del perfil de riesgo-beneficio Personas que han sufrido una fractura, para determinar la gravedad de la enfermedad AMAV • La TC cuantitativa ofrece una puntuación T equivalente a la de la DEXA, no se ve obstaculizada por las alteraciones degenerativas de la columna lumbar y es altamente sensible para la detección de fracturas vertebrales por compresión, pero su coste y la exposición a la radiación son mayores y no está recomendada para el cribado de la osteoporosis. Evaluación de las causas secundarias de masa ósea baja Mujeres de 65-69 años y varones de 75-79 años si la puntuación T de la columna, la cadera total o el cuello femoral es ≤–1,5 Las causas más frecuentes de masa ósea baja son factores no modificables, pero hay muchas otras (véase la Tabla 44 y la Tabla 45). Una masa ósea baja puede ser la presentación de trastornos como hipercalciuria idiopática o enfermedad celíaca. Es necesario obtener una anamnesis y una exploración física exhaustivas en todos los pacientes, así como otras pruebas basadas en los hallazgos (véase la Tabla 46). Aunque la determinación de la DMO puede no ser necesaria en todos los pacientes para emitir el diagnóstico de osteoporosis, puede ser útil para establecer si hacen falta más pruebas y como orientación para el tratamiento. En las mujeres posmenopáusicas de 50-64 años y los varones de 50-69 años con los factores de riesgo siguientes: Osteomalacia Signos radiográficos indicativos de osteoporosis o deformidad vertebral Tratamiento glucocorticoide durante más de 3 meses Hiperparatiroidismo primario Pruebas de imagen vertebralesc Mujeres ≥70 años y varones ≥80 años si la puntuación T de la columna, la cadera total o el cuello femoral es ≤–1,0 Fracturas por pequeños traumatismos Pérdida de estatura histórica ≥4cm Pérdida de estatura ≥2 cm Tratamiento con glucocorticoides a largo plazo reciente o en curso a Las mediciones de la densidad mineral ósea deben llevarse a cabo en centros con absorciometría de rayos X de energía dual (DEXA) que utilicen medidas aceptadas de garantía de la calidad. bLa National Osteoporosis Foundation recomienda el cribado en los varones ≥70 años, y la Endocrine Society recomienda el cribado en los varones ≥70 años y los varones de 50-69 años que presenten factores de riesgo como bajo peso corporal, fractura anterior en la etapa adulta o tabaquismo. cLas pruebas de imagen vertebrales deben repetirse cuando se advierta una nueva pérdida de estatura o se refiera un dolor de espalda nuevo. finida como una puntuación Z <–2, que indica «masa ósea baja para la edad». La evaluación de la DMO mediante ecografía calcánea puede usarse para detectar osteoporosis, pero los resultados anómalos deben confirmarse mediante DEXA. La TC cuantitativa ofrece una puntuación T equivalente a la de la DEXA, no se ve obstaculizada por las alteraciones degenerativas de la columna lumbar y es altamente sensible para la detección de fracturas vertebrales por compresión, pero su coste y la exposición a la radiación son mayores y no está recomendada para el cribado de la osteoporosis. PUNTOS CLAVE • La osteoporosis puede diagnosticarse clínicamente sobre la base de la presencia de fracturas por fragilidad, fractura de cadera, fractura vertebral por compresión o una medición de la densidad mineral ósea ≤–2,5. La osteomalacia está causada sobre todo por el déficit grave y prolongado de vitamina D, que tiene su origen en concentraciones insuficientes de calcio y/o fosfato en el hueso, que, a su vez, impiden la mineralización de la matriz ósea recién formada. A diferencia de la osteoporosis, la osteomalacia no da lugar a pérdida permanente de estructura ósea. Hace falta un período de entre meses y años de mineralización alterada para que la resistencia ósea se vea afectada. Los síntomas de osteomalacia comprenden dolor óseo difuso, sensibilidad ósea a la palpación (meseta tibial y esternón) y debilidad muscular proximal; sin embargo, la osteomalacia inicial puede manifestarse solamente con una masa ósea baja en la DEXA y ser indistinguible de la osteoporosis sin otras pruebas. La osteomalacia se diferencia de la osteoporosis por una DMO muy baja (puntuación Z ≤–2), niveles de calcio y fósforo bajos o bajos-normales, un nivel muy bajo de 25-hidroxivitamina D (<10 ng/ml [25 nmol/l]) y la presencia de hiperparatiroidismo secundario. Un nivel elevado de fosfatasa alcalina sérica es especialmente indicativo de osteomalacia, aunque puede observarse una elevación leve en caso de fractura osteoporótica reciente. En la osteomalacia grave, las radiografías pueden mostrar una distribución inusual de las fracturas (Figura 18). Cuando el dolor óseo no se explica con los hallazgos radiográficos convencionales en el contexto de presunta osteomalacia, las pruebas de imagen con gammagrafía ósea o RM pueden revelar fracturas. El objetivo del tratamiento es optimizar las condiciones de la mineralización ósea mediante el uso de suplementos para normalizar las concentraciones de 25-hidroxivitamina D sérica (>30 ng/ml [75 nmol/l]), calcio y fósforo. La respuesta ósea al tratamiento se refleja en la normalización gradual del nivel de fosfatasa alcalina y el alivio de los síntomas. La reso87 Trastornos del calcio y los huesos na D de al menos 30 ng/ml (75 nmol/l), un aporte complementario de entre 1.000 y 2.000 UI/día de vitamina D puede ser apropiado en el contexto de la atención de la osteoporosis. Manejo farmacológico F i g u r a 1 8 . Las seudofracturas se muestran en forma de áreas radiotransparentes perpendiculares a la superficie ósea y suelen ser bilaterales y/o simétricas. Son lugares frecuentes las ramas púbicas (que se muestran aquí), el aspecto medial del fémur proximal, el peroné proximal y los metatarsos. lución de la osteomalacia puede demorar 12 meses; las determinaciones de la DMO posteriores revelarán aumentos significativos y/o normalización de la DMO. Otras causas La masa ósea baja y las fracturas por fragilidad en adultos jóvenes y de mediana edad pueden tener su origen en trastornos genéticos (véase la Tabla 45). Los pacientes con osteogénesis imperfecta (tipo 1) pueden presentar antecedentes de fracturas en la infancia cuya frecuencia disminuye en la etapa adulta. La hipermovilidad apunta a síndrome de Ehlers-Danlos y otros síndromes relacionados. En los pacientes de mediana edad con fracturas y niveles de fosfatasa alcalina muy por debajo del intervalo de referencia debe sospecharse la presencia de hipofosfatasia. Manejo no farmacológico El ejercicio que implica carga, resistencia y equilibrio es importante para la salud ósea y puede reducir el riesgo de fractura en cualquier edad, pero especialmente en los pacientes mayores de 65 años. Aunque el efecto mensurable del tratamiento puede ser pequeño, en todos los ensayos farmacoterapéuticos de la osteoporosis se administraron aportes complementarios de calcio y vitamina D; la National Academy of Medicine recomienda la ingesta de entre 1.000 y 1.200 mg/día de calcio, idealmente de fuentes alimentarias. En los pacientes con dietas con un contenido de calcio insuficiente puede utilizarse un aporte complementario de calcio, pero este no debe recomendarse independientemente de una evaluación y la intervención alimentarias. Dado que numerosos ensayos clínicos de la osteoporosis, incluidos los que probaban farmacoterapias, intentaron alcanzar niveles de 25-hidroxivitami88 El objetivo del tratamiento es reducir el riesgo de fracturas en los pacientes con osteoporosis o en quienes presentan un riesgo elevado de fractura (prevención primaria). La U.S. National Osteoporosis Foundation recomienda el tratamiento farmacológico en los pacientes con fracturas de cadera o columna relacionadas con la osteoporosis, en quienes tienen una puntuación T de DMO ≤2,5 y en los que tienen una puntuación T de DMO de entre –1 y –2,5 con un riesgo a 10 años de fractura de cadera ≥3% o un riesgo de fractura mayor relacionada con la osteoporosis ≥20%, según la estimación de la FRAX. Algunos estudios indican que en otras poblaciones en riesgo es necesario considerar diferentes umbrales para el inicio del tratamiento. El American College of Rheumatology recomienda el tratamiento de la osteoporosis inducida por glucocorticoides en función de la edad, el sexo y el riesgo de fractura. La farmacoterapia puede usarse también para prevenir la pérdida de DMO en las mujeres posmenopáusicas en riesgo de osteoporosis y para prevenir o tratar la osteoporosis inducida por glucocorticoides. Aunque puede plantearse una terapia estrogénica para el manejo de los síntomas vasomotores en las mujeres posmenopáusicas, esta ya no considera el tratamiento de primera línea para la prevención de la osteoporosis. Recientemente la FDA ha aprobado una píldora combinada, que incluye bazedoxifeno y estrógenos conjugados, para la prevención de la osteoporosis posmenopáusica. Los fármacos aprobados por la FDA para el tratamiento de la osteoporosis y las opciones para la prevención se presentan en la Tabla 48. Bifosfonatos Los bifosfonatos orales, el alendronato y el risedronato, son fármacos antirresortivos y en general se utilizan como tratamiento de primera línea en las mujeres posmenopáusicas y los varones mayores de 50 años de edad. Se ha demostrado que reducen el riesgo de fracturas de columna, de cadera y no vertebrales. Otro bifosfonato, el ibandronato, solo ha mostrado eficacia en la reducción de las fracturas vertebrales. En la osteoporosis inducida por glucocorticoides con riesgo de fractura moderado o alto, los bifosfonatos orales se recomiendan como tratamiento de primera línea en los varones adultos y en las mujeres de cualquier edad. La administración de ácido zoledrónico intravenoso una vez al año representa una opción para los pacientes que experimentan síntomas gastrointestinales o tienen dificultades para tomarse la medicación según las instrucciones. Después de la administración de la primera dosis puede producirse una reacción de respuesta de fase aguda, con pirexia y mialgias, en uno de cada tres pacientes, aunque normalmente no se da con las dosis posteriores. Los bifosfonatos están contraindicados en los pacientes con función renal reducida (TFG <35 ml/min/1,73 m2) y no deben admi- Trastornos del calcio y los huesos Tabla 48. Medicamentos aprobados por la Food and Drug Administration (FDA) para el tratamiento y la prevención de la osteoporosis y sitios esqueléticos con prevención de fracturas demostrada cuando se usan para tratar la osteoporosis Prevención OPM Prevención de fracturas documentada OIG Recidivante Cadera Vertebral No vertebral √ √ √ √ √ √ √ √ Bifosfonatos Alendronato √ Risedronato √ Ibandronato √ Ácido zoledrónico (IV) √ √ √ √ √ Denosumab Raloxifeno √ √ √ √ √ √ √ √ Anabólicos Abaloparatida √ √ Teriparatida √ √ IV: intravenoso; OIG: osteoporosis inducida por glucocorticoides; OPM: osteoporosis posmenopáusica. nistrarse hasta que se hayan tratado el déficit de vitamina D y la hipocalcemia, si las hay. Los efectos secundarios infrecuentes de los fármacos antirresortivos son osteonecrosis de mandíbula y fracturas atípicas de fémur. La osteonecrosis de mandíbula puede darse en cualquier momento del tratamiento; en cambio, el riesgo de fractura atípica del fémur parece aumentar con la duración del tratamiento. En la mayoría de los pacientes, los beneficios de la reducción de las fracturas de osteoporosis superan en mucho el riesgo de estos efectos secundarios infrecuentes. En las mujeres que no presentan riesgo alto de fracturas, puede considerarse un descanso farmacológico al cabo de tres años de tratamiento con bifosfonatos, en el caso de su administración intravenosa, o de cinco años, cuando sea oral. En las mujeres posmenopáusicas de alto riesgo debido a una puntuación T ≤–3,5, fractura osteoporótica previa o fractura durante el tratamiento, hay que considerar la continuación del tratamiento durante un máximo de 10 años (tratamiento oral) o seis años (tratamiento intravenoso). Si bien no hay datos que sirvan de orientación en cuanto a la duración del descanso farmacológico, los factores que deben tenerse en cuenta incluyen el bifosfonato utilizado y si la DMO se mantiene en la DEXA. PUNTOS CLAVE • Por lo general, los bifosfonatos son el tratamiento de primera línea para la osteoporosis; solo el alendronato y el risedronato reducen el riesgo de fractura de columna, cadera y no vertebral. • En las mujeres posmenopáusicas que no presentan riesgo alto de fracturas puede considerarse un descanso farmacológico al cabo de tres años de tratamiento con bifosfonatos, en el caso de su administración intravenosa, o de cinco años, cuando sea oral. Inhibidores del ligando del receptor activador del factor nuclear κB (RANK) El denosumab es un anticuerpo monoclonal que inhibe la activación de los osteoclastos. Administrado por vía subcutánea dos veces al año, el denosumab inhibe la resorción ósea, aumenta la densidad ósea y reduce la incidencia de fracturas osteoporóticas en varones y mujeres. Los efectos del denosumab no se mantienen con la interrupción del tratamiento. El denosumab puede ser preferible en los pacientes con enfermedad renal crónica en estadio 4 y en quienes son intolerantes al tratamiento con bifosfonatos o no responden del todo al mismo. Los efectos adversos incluyen hipocalcemia, sobre todo en los pacientes de edad avanzada con déficit de vitamina D, y una mayor tasa de celulitis y bronquitis. También se ha notificado osteonecrosis de mandíbula relacionada con el medicamento y fractura atípica de fémur con el uso de denosumab. Fármacos anabólicos La teriparatida, rhPTH (1-34), está aprobada para uso en mujeres posmenopáusicas y en varones o mujeres con osteoporosis inducida por glucocorticoides que presentan un riesgo elevado de fractura osteoporótica. También se utiliza para mejorar la masa ósea en los varones con osteoporosis primaria o relacionada con hipogonadismo y riesgo alto de fractura. La abaloparatida, rhPTHrP (1-34), está aprobada para el tratamiento de las mujeres posmenopáusicas con osteoporosis y riesgo alto de fractura. Ambos fármacos estimulan la formación de hueso y deben inyectarse diariamente por vía subcutánea. El tratamiento debe limitarse a dos años. La mejora de la DMO es especialmente evidente en la columna. Para evitar la pérdida del hueso recién formado, es necesario iniciar un tratamiento secuencial con un fármaco antirresortivo en el plazo de un mes desde la finalización del ciclo de tratamiento anabólico. 89 Trastornos del calcio y los huesos Seguimiento de los pacientes con masa ósea baja Las mediciones seriadas sistemáticas con DEXA de la DMO no están indicadas para el seguimiento de los pacientes de bajo riesgo que no presentan osteoporosis. Las pruebas posteriores de la DMO dependen de la DMO basal. Puede ser razonable repetirlas en 15 años si la puntuación T de la cadera es normal (>–1), mientras que si la puntuación T se sitúa entre –2 y –2,4 puede considerarse repetirlas a los dos años. El motivo principal para repetir las mediciones de la DMO en los pacientes que toman fármacos antirresortivos es detectar el fracaso del tratamiento. La disminución de la DMO, evidente con un descenso porcentual estadísticamente significativo en g/cm2 del hueso (puntuaciones T que no descienden en las DEXA posteriores) o una fractura durante el tratamiento, lleva a pensar en una causa secundaria no detectada, incumplimiento del tratamiento o una respuesta insuficiente que requiere evaluación. El American College of Physicians de­ saconseja controlar la DMO durante el tratamiento, porque los datos de varios estudios pusieron de manifiesto que las mujeres con tratamiento antirresortivo sufrían menos fracturas aun cuando no se incrementara la DMO. En cambio, el seguimiento debe incluir la revisión de la indicación del tratamiento, el control del cumplimiento del tratamiento y el refuerzo de las medidas del estilo de vida para prevenir las fracturas, minimizar la pérdida ósea y evitar la fragilidad. Normalmente, el descanso del tratamiento antirresortivo implica la medición de la DMO para establecer una medición basal y repetirla al cabo de dos o tres años. Aunque esta estrategia sería altamente informativa para las decisiones terapéuticas posteriores, no se ha validado. Déficit de vitamina D La prueba más adecuada para evaluar la adecuación de la vitamina D es la medición de la 25-hidroxivitamina D en suero, que refleja la vitamina D alimentaria y derivada de la piel. Para prevenir la enfermedad ósea metabólica en las poblaciones por lo demás sanas, se recomiendan al menos 20 ng/ml (50 nmol/l), nivel que puede alcanzarse con una ingesta de entre 600 y 800 UI/día. El cribado sistemático de déficit de vitamina D no está recomendado en las poblaciones sanas; sin embargo, conviene hacer pruebas de detección de déficit en los grupos de alto riesgo o en los pacientes que presenten masa ósea baja, fracturas, hipocalcemia o hiperparatiroidismo. En los pacientes con trastornos óseos, paratiroideos o del calcio, elevar los niveles sanguíneos de manera uniforme a más de 30 ng/dl (75 nmol/l) puede requerir entre 1.000 y 2.000 U/día. Los pacientes con malabsorción, falta crónica de exposición al sol, IMC >40 kg/m2, enfermedad hepática avanzada y que reciben medicamentos que interfieren en el metabolismo de la vitamina D, como la fenitoína y el fenobarbital, pueden necesitar de 2.000 a 4.000 U/día para recuperar y mantener las reservas de vitamina D, guiándose en los niveles de 25-hidroxivitamina D obtenidos tres meses después del inicio del tratamiento. 90 Las dosis de carga de 50.000 U/día de vitamina D2 (ergocalciferol) o vitamina D3 (colecalciferol) una vez por semana durante ocho semanas son adecuadas en los casos de déficit grave, sobre todo en el contexto de malabsorción. Los candidatos para las dosis de carga incluyen: 25-hidroxivitamina D indetectable, hipocalcemia y osteomalacia debido a déficit de vitamina D. Para mantener un nivel suficiente tras una dosis de carga, se recomienda un tratamiento de mantenimiento de entre 1.000 y 4.000 U/día. Los efectos autocrinos y paracrinos en la mayoría de los tejidos, así como los extensos datos observacionales que asocian los niveles de 25-hidroxivitamina D a resultados de la salud ósea, han despertado un interés público generalizado y han dado pie a una gran cantidad de publicaciones científicas sobre la vitamina D. Sin embargo, los estudios de intervención no han demostrado de manera convincente que el aporte complementario de vitamina D sean beneficioso para la mayoría de los resultados patológicos, lo que permite suponer que los niveles bajos de 25-hidroxivitamina D pueden ser un marcador, y no una causa, de mala salud. PUNTO CLAVE • El cribado sistemático de déficit de vitamina D no está recomendado en las poblaciones sanas; sin embargo, conviene hacer pruebas de detección de déficit en los grupos de alto riesgo o en los pacientes que presenten baja masa ósea, fracturas, hipocalcemia o hiperparatiroidismo. Enfermedad ósea de Paget Aunque la enfermedad ósea de Paget puede manifestarse con síntomas localizados, en la mayoría de los casos se diagnostica en pacientes de edad avanzada asintomáticos que presentan niveles elevados de fosfatasa alcalina o hallazgos radiográficos casuales. Los huesos afectados habitualmente son el cráneo, la columna, el sacro, la pelvis, el fémur y la tibia. El lugar esquelético y el grado de afectación, incluyendo la tasa de recambio óseo, determinan el cuadro clínico. La afectación de los huesos de carga de la extremidad inferior puede provocar dolor óseo, deformidad o fractura. La afectación de un hueso próximo a una articulación puede contribuir a la aparición de enfermedad articular degenerativa (Figura 19). La expansión del hueso pagético de la columna superior o la base del cráneo puede provocar compresión de la médula espinal o los pares craneales. El diagnóstico se basa en los hallazgos radiográficos de engrosamiento del hueso cortical, marcas trabeculares engrosadas y distorsión y expansión del hueso afectado. Rara vez es necesaria una biopsia. La fosfatasa alcalina en suero, un marcador de un recambio óseo elevado, suele estar alta, aunque puede no ser así en algunos pacientes con enfermedad de larga evolución que se ha vuelto metabólicamente inactiva. La fosfatasa alcalina puede estar elevada en otros trastornos, entre los que se incluyen el déficit de vitamina D, otras enferme- AMAV Bibliografía PUNTOS CLAVE • Aunque la enfermedad ósea de Paget puede manifestarse con síntomas localizados, en la mayoría de los casos se diagnostica en pacientes de edad avanzada asintomáticos que presentan niveles elevados de fosfatasa alcalina o hallazgos radiográficos incidentales. • Las indicaciones para el tratamiento de la enfermedad de Paget incluyen dolor óseo; riesgo de fractura y deformidad progresiva que podrían poner en peligro el hueso, la articulación o la función neurológica, y concentraciones elevadas de fosfatasa alcalina. • Con frecuencia una dosis única de ácido zoledrónico por vía intravenosa logrará el objetivo terapéutico de reducir el dolor y normalizar los niveles de fosfatasa alcalina en los pacientes con enfermedad ósea de Paget. Bibliografía F i g u r a 1 9 . El engrosamiento de las estructuras trabeculares (flecha vacía) sin deformidad cortical es diagnóstico de la enfermedad ósea de Paget en la tibia izquierda proximal. También se muestra una artrosis avanzada en la rodilla izquierda adyacente (flecha llena). dades óseas metabólicas (como la osteomalacia) o en caso de fractura reciente. Por lo tanto, en todos los pacientes con presunta enfermedad ósea de Paget debe realizarse una evaluación del calcio sérico y la 25-hidroxivitamina D, así como una gammagrafía ósea de todo el cuerpo. Si la gammagrafía revela otros sitios esqueléticos con presunta enfermedad ósea de Paget, hay que hacer una radiografía de los mismos para continuar la evaluación. El manejo de la enfermedad de Paget se basa en los síntomas, la ubicación de la afectación y la actividad de la enfermedad. Las indicaciones para el tratamiento incluyen: dolor óseo; riesgo de fractura y deformidad progresiva que podrían poner en peligro el hueso, la articulación o la función neurológica, y concentraciones elevadas de fosfatasa alcalina. Los bifosfonatos, especialmente en forma de una dosis única de 5 mg de ácido zoledrónico por vía intravenosa, suelen alcanzar el objetivo del tratamiento de reducir el dolor y normalizar la fosfatasa alcalina durante un máximo de cinco años. También pueden usarse bifosfonatos orales, pero la duración del tratamiento es variable. Es necesario controlar anualmente los niveles de fosfatasa alcalina. Si los niveles anteriormente normalizados de fosfatasa alcalina superan los niveles normales, está indicado repetir el tratamiento. En caso de aumento espectacular de los niveles de fosfatasa alcalina o aceleración de los síntomas, que apunta a la infrecuente transformación sarcomatosa del hueso pagético, es necesario obtener pruebas de imagen. Trastornos del metabolismo de la glucosa American Diabetes Association. Standards of medical care in diabetes—2018. 2018; 41(Suppl 1): S1-S159. Bergenstal RM, Klonoff DC, Garg SK, Bode BW, Meredith M, Slover RH, et al; ASPIRE In-Home Study Group. Threshold-based insulin-pump interruption for reduction of hypoglycemia. N Engl J Med. 2013;369:224-32. [PMID: 23789889] doi:10.1056/NEJMoa1303576 Cryer PE, Axelrod L, Grossman AB, et al. Evaluation and management of adult hypoglycemic disorders: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metabl. 2009; 94:709-28. [PMID: 19088155] Duckworth W, Abraira C, Moritz T, Reda D, Emanuele N, Reaven PD, et al; VADT Investigators. Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med. 2009;360:129-39. [PMID: 19092145] doi:10.1056/NEJMoa0808431 Garber AJ, Abrahamson MJ, Barzilay JI, et al. Consensus statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the comprehensive type 2 diabetes management algorithm-2018 executive summary. Endoc Pract. 2018; 24:91-120. [PMID: 29368965] Hayward RA, Reaven PD, Wiitala WL, Bahn GD, Reda DJ, Ge L, et al; VADT investigators. Follow-up of glycemic control and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2015;372:2197-206. [PMID: 26039600] doi:10.1056/NEJMoa1414266 Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med. 2008;359:1577-89. [PMID: 18784090] doi:10.1056/NEJMoa0806470 Ismail-Beigi F, Craven T, Banerji MA, Basile J, Calles J, Cohen RM, et al; ACCORD trial group. Effect of intensive treatment of hyperglycaemia on microvascular outcomes in type 2 diabetes: an analysis of the ACCORD randomised trial. Lancet. 2010;376:419-30. [PMID: 20594588] doi:10.1016/ S0140-6736(10)60576-4 Knowler WC, Barrett-Connor E, Fowler SE, Hamman RF, Lachin JM, Walker EA, et al; Diabetes Prevention Program Research Group. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346:393-403. [PMID: 11832527] Maldonado M, Hampe CS, Gaur LK, et al. Ketosis-prone diabetes: dissection of a heterogeneous syndrome using an immunogenetic and β-cell functional classification, prospective analysis, and clinical outcomes. J Clin Endocrinol Metab 2003;88:5090-98. [PMID: 14602731] Marso SP, Daniels GH, Brown-Frandsen K, et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2016; 375:311-322. [PMID: 27295427] Nathan DM, Cleary PA, Backlund JY, Genuth SM, Lachin JM, Orchard TJ, et al; Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Study Research Group. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med. 2005;353:2643-53. [PMID: 16371630] 91 Bibliografía Nathan DM, Zinman B, Cleary PA, Backlund JY, Genuth S, Miller R, et al; Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Research Group. Modern-day clinical course of type 1 diabetes mellitus after 30 years’ duration: the diabetes control and complications trial/epidemiology of diabetes interventions and complications and Pittsburgh epidemiology of diabetes complications experience (1983-2005). Arch Intern Med. 2009;169:1307-16. [PMID: 19636033] doi:10.1001/archinternmed.2009.193 Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, et al; Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. 2017;377:644-57. [PMID: 28605608] Patel A, MacMahon S, Chalmers J, Neal B, Billot L, Woodward M, et al; ADVANCE Collaborative Group. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med. 2008;358:2560-72. [PMID: 18539916] doi:10.1056/NEJMoa0802987 Peters AL, Ahmann AJ, Battelino T, Evert A, Hirsch IB, Murad MH, et al. Diabetes technology-continuous subcutaneous insulin infusion therapy and continuous glucose monitoring in adults: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2016;101:3922-3937. [PMID: 27588440] Qaseem A, Wilt TJ, Kansagara D, Horwitch C, Barry MJ, Forciea MA; Clinical Guidelines Committee of the American College of Physicians. Hemoglobin A1c targets for glycemic control with pharmacologic therapy for nonpregnant adults with type 2 diabetes mellitus: a guidance statement update from the American College of Physicians. Ann Intern Med. 2018 Mar 6. doi:10.7326/M17-0939. [Epub ahead of print] PubMed PMID: 29507945. Trastornos de las glándulas suprarrenales Bornstein SR, Allolio B, Arlt W, Barthel A, Don-Wauchope A, Hammer GD, et al. Diagnosis and treatment of primary adrenal insufficiency: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2016;101:364-89. [PMID: 26760044] doi:10.1210/jc.2015-1710 Fassnacht M, Arlt W, Bancos I, Dralle H, Newell-Price J, Sahdev A, et al. Management of adrenal incidentalomas: European Society of Endocrinology Clinical Practice Guideline in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol. 2016;175:G1-G34. [PMID: 27390021] doi:10.1530/EJE-16-0467 Funder JW, Carey RM, Mantero F, Murad MH, Reincke M, Shibata H, et al. The management of primary aldosteronism: case detection, diagnosis, and treatment: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2016;101:1889-916. [PMID: 26934393] doi:10.1210/jc.2015-4061 Lacroix A, Feelders RA, Stratakis CA, Nieman LK. Cushing’s syndrome. Lancet. 2015 Aug 29;386(9996):913-27. doi:10.1016/S0140-6736(14)61375-1. Epub 2015 May 21. Review. PubMed PMID: 26004339. Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK, Murad MH, et al; Endocrine Society. Pheochromocytoma and paraganglioma: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2014;99:1915-42. [PMID: 24893135] doi:10.1210/jc.2014-1498 Libé R. Adrenocortical carcinoma (ACC): diagnosis, prognosis, and treatment. Front Cell Dev Biol. 2015;3:45. [PMID: 26191527] doi:10.3389/fcell.2015.00045 Siu AL; U.S. Preventive Services Task Force. Screening for abnormal blood glucose and type 2 diabetes mellitus: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2015;163:861-868. [PMID: 26501513] Nieman LK, Biller BM, Findling JW, Newell-Price J, Savage MO, Stewart PM, et al. The diagnosis of Cushing’s syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2008;93:1526-40. [PMID: 18334580] doi:10.1210/jc.2008-0125 U.S. Department of Veterans Affairs/U.S. Department of Defense. VA/DoD Clinical Practice Guidelines for the management of diabetes mellitus in primary care. 2017. www.healthquality.va.gov/guidelines/cd/diabetes. Accessed May 16, 2018. Rossi GP, Cesari M, Cuspidi C, Maiolino G, Cicala MV, Bisogni V, et al. Long-term control of arterial hypertension and regression of left ventricular hypertrophy with treatment of primary aldosteronism. Hypertension. 2013;62:62-9. [PMID: 23648698] doi:10.1161/HYPERTENSIONAHA.113.01316 Whelton PK, Carey RM, Aronow WS, Casey DE Jr, Collins KJ, Dennison Himmelfarb C, et al; 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/ NMA/PCNA guidelines for the prevention, detection, evaluation, and management of high blood pressure in adults: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. 2017; doi:10.1016/j.jacc.2017.11.006. Trastornos de la glándula tiroides Zinman B, Wanner C, Lachin JM, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015; 373: 2117-2128. [PMID: 26378978] Burch HB, Cooper DS. Management of Graves disease: a review. JAMA. 2015; 314:2544-54. [PMID: 26670972] doi:10.1001/jama.2015.16535 Trastornos de la hipófisis Corsello SM, Barnabei A, Marchetti P, De Vecchis L, Salvatori R, Torino F. Endocrine side effects induced by immune checkpoint inhibitors. J Clin Endocrinol Metab. 2013;98:1361-75. [PMID: 23471977] doi:10.1210/jc.2012-4075 Fleseriu M, Hashim IA, Karavitaki N, Melmed S, Murad MH, Salvatori R, et al. Hormonal replacement in hypopituitarism in adults: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2016;101:3888-3921. [PMID: 27736313] Freda PU, Beckers AM, Katznelson L, Molitch ME, Montori VM, Post KD, et al; Endocrine Society. Pituitary incidentaloma: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2011;96:894-904. [PMID: 21474686] doi:10.1210/jc.2010-1048 Guielman M, Basavilbaso, NG, Vitale M et al. Primary empty sella (PES). Pituitary. 2013;16:270-274. [PMID: 22875743] Katznelson L, Laws ER Jr, Melmed S, Molitch ME, Murad MH, Utz A, et al; Endocrine Society. Acromegaly: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2014;99:3933-51. [PMID: 25356808] doi:10.1210/jc.2014-2700 Melmed S, Casanueva FF, Hoffman AR, Kleinberg DL, Montori VM, Schlechte JA, et al; Endocrine Society. Diagnosis and treatment of hyperprolactinemia: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2011;96:273-88. [PMID: 21296991] doi:10.1210/jc.2010-1692 Molitch ME, Clemmons DR, Malozowski S, Merriam GR, Vance ML; Endocrine Society. Evaluation and treatment of adult growth hormone deficiency: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2011;96:1587-609. [PMID: 21602453] doi:10.1210/jc.2011-0179 Nieman LK, Biller BM, Findling JW, Murad MH, Newell-Price J, Savage MO, et al; Endocrine Society. Treatment of Cushing’s syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2015;100: 2807-31. [PMID: 26222757] doi:10.1210/jc.2015-1818 Nieman LK, Biller BM, Findling JW, Newell-Price J, Savage MO, Stewart PM, et al. The diagnosis of Cushing’s syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2008;93:1526-40. [PMID: 18334580] doi:10.1210/jc.2008-0125 92 Alexander EK, Pearce EN, Brent GA, Brown RS, Chen H, Dosiou C, et al. 2017 Guidelines of the American Thyroid Association for the diagnosis and management of thyroid disease during pregnancy and the postpartum. Thyroid. 2017;27:315-389. [PMID: 28056690] doi:10.1089/thy.2016.0457 Cohen-Lehman J, Dahl P, Danzi S, Klein I. Effects of amiodarone therapy on thyroid function. Nat Rev Endocrinol. 2010;6:34-41. [PMID: 19935743] doi:10.1038/nrendo.2009.225 Cooper DS, Biondi B. Subclinical thyroid disease. Lancet. 2012;379:1142-54. [PMID: 22273398] doi:10.1016/S0140-6736(11)60276-6 De Leo S, Lee SY, Braverman LE. Hyperthyroidism. Lancet. 2016;388:906-18. [PMID: 27038492] doi:10.1016/S0140-6736(16)00278-6 Demers LM, Spencer CA. Laboratory medicine practice guidelines: laboratory support for the diagnosis and monitoring of thyroid disease. Clin Endocrinol (Oxf). 2003;58:138-40. [PMID: 12580927]. Fliers E, Bianco AC, Langouche L, Boelen A. Thyroid function in critically ill patients. Lancet Diabetes Endocrinol. 2015;3:816-25. [PMID: 26071885] doi:10.1016/S2213-8587(15)00225-9 Haugen BR, Alexander EK, Bible KC, Doherty GM, Mandel SJ, Nikiforov YE, et al. 2015 American Thyroid Association management guidelines for adult patients with thyroid nodules and differentiated thyroid cancer: the American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid. 2016;26:1-133. [PMID: 26462967] doi:10.1089/thy.2015.0020 Hennessey JV. The emergence of levothyroxine as a treatment for hypothyroidism. Endocrine. 2017;55:6-18. [PMID: 27981511] doi:10.1007/s12020016-1199-8 Kundra P, Burman KD. The effect of medications on thyroid function tests. Med Clin North Am. 2012;96:283-95. [PMID: 22443976] doi:10.1016/j.mcna. 2012.02.001 Ross DS, Burch HB, Cooper DS, Greenlee MC, Laurberg P, Maia AL, et al. 2016 American Thyroid Association guidelines for diagnosis and management of hyperthyroidism and other causes of thyrotoxicosis. Thyroid. 2016;26: 1343-1421. [PMID: 27521067] Trastornos de la reproducción Bhasin S, Brito JP, Cunningham GR, Hayes FJ, Hodis HN, Matsumoto AM, Snyder PJ, Swerdloff RS, Wu FC, Yialamas MA. Testosterone therapy in men with hypogonadism: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2018 May 1;103(5):1715-1744. [PMID: 29562364] doi:10.1210/jc.2018-00229. Bibliografía Bhasin S, Cunningham GR, Hayes FJ, Matsumoto AM, Snyder PJ, Swerdloff RS, et al; Task Force, Endocrine Society. Testosterone therapy in men with androgen deficiency syndromes: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2010;95:2536-59. [PMID: 20525905] doi:10.1210/jc.2009-2354 Bondy CA; Turner Syndrome Study Group. Care of girls and women with Turner syndrome: a guideline of the Turner Syndrome Study Group. J Clin Endocrinol Metab. 2007;92:10-25. [PMID: 17047017] Brennan MJ. The effect of opioid therapy on endocrine function. Am J Med. 2013;126:S12-8. [PMID: 23414717] doi:10.1016/j.amjmed.2012.12.001 Fourman LT, Fazeli PK. Neuroendocrine causes of amenorrhea—an update. J Clin Endocrinol Metab. 2015;100:812-24. [PMID: 25581597] doi:10.1210/jc. 2014-3344 Gordon CM. Clinical practice. Functional hypothalamic amenorrhea. N Engl J Med. 2010;363:365-71. [PMID: 20660404] doi:10.1056/NEJMcp0912024 Legro RS, Arslanian SA, Ehrmann DA, Hoeger KM, Murad MH, Pasquali R, et al; Endocrine Society. Diagnosis and treatment of polycystic ovary syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2013;98:4565-92. [PMID: 24151290] doi:10.1210/jc.2013-2350 Lindsay TJ, Vitrikas KR. Evaluation and treatment of infertility. Am Fam Physician. 2015;91:308-14. [PMID: 25822387] Pope HG Jr, Wood RI, Rogol A, Nyberg F, Bowers L, Bhasin S. Adverse health consequences of performance-enhancing drugs: an Endocrine Society scientific statement. Endocr Rev. 2014;35:341-75. [PMID: 24423981] doi:10.1210/ er.2013-1058 Manejo de la terapia hormonal transgénero American Psychological Association. Guidelines for psychological practice with transgender and gender nonconforming people. Am Psychol. 2015 Dec;70(9):832-64. doi:10.1037/a0039906. PubMed PMID: 26653312. www. apa.org/practice/guidelines/transgender.pdf. Accessed May 15, 2018. Center for Excellence for Transgender Health website. www.transhealth.ucsf. edu/trans?page=guidelines-home. Accessed May 15, 2018. Coleman E, Bockting W, Botzer M, et al. Standards of care for the health of transsexual, transgender, and gender-nonconforming people, version 7. Int J Transgend. 2012;13:165. Getahun D, Nash R, Flanders WD, Baird TC, Becerra-Culqui TA, Cromwell L, et al. Cross-sex hormones and acute cardiovascular events in transgender persons: a cohort study. Ann Intern Med. 2018 Jul 10. doi:10.7326/M17-2785. [Epub ahead of print] PubMed PMID: 29987313. Hembree WC, Cohen-Kettenis P, Delemarre-van de Waal HA, Gooren LJ, Meyer WJ 3rd, Spack NP, et al; Endocrine Society. Endocrine treatment of transsexual persons: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2009;94:3132-54. [PMID: 19509099] doi:10.1210/ jc.2009-0345 Wylie K, Knudson G, Khan SI, Bonierbale M, Watanyusakul S, Baral S. Serving transgender people: clinical care considerations and service delivery models in transgender health. Lancet. 2016;388:401-11. [PMID: 27323926] doi:10.1016/S0140-6736(16)00682-6 Trastornos del calcio y los huesos Adler RA, El-Hajj Fuleihan G, Bauer DC, Camacho PM, Clarke BL, Clines GA, et al. Managing osteoporosis in patients on long-term bisphosphonate treatment: report of a task force of the American Society for Bone and Mineral Research. J Bone Miner Res. 2016;31:1910. [PMID: 27759931] doi:10.1002/jbmr.2918 Bilezikian JP, Brandi ML, Eastell R, Silverberg SJ, Udelsman R, Marcocci C, et al. Guidelines for the management of asymptomatic primary hyperparathyroidism: summary statement from the Fourth International Workshop. J Clin Endocrinol Metab. 2014;99:3561-9. [PMID: 25162665] doi:10.1210/jc.2014-1413 Black DM, Rosen CJ. Postmenopausal osteoporosis [Letter]. N Engl J Med. 2016;374:2096-7. [PMID: 27223157] doi:10.1056/NEJMc1602599 Brandi ML, Bilezikian JP, Shoback D, Bouillon R, Clarke BL, Thakker RV, et al. Management of hypoparathyroidism: summary statement and guidelines. J Clin Endocrinol Metab. 2016;101:2273-83. [PMID: 26943719] doi:10.1210/jc.2015-3907 Buckley L, Guyatt G, Fink HA, Cannon M, Grossman J, Hansen KE, et al. 2017 American College of Rheumatology Guideline for the Prevention and Treatment of Glucocorticoid-Induced Osteoporosis. Arthritis & Rheumatology. 2017;69(8):1521-37.[PMID: 28585373] doi:10.1002/art.40137 Camacho PM, Petak SM, Binkley N, Clarke BL, Harris ST, Hurley DL, et al. American Association of Clinical Endocrinologist and American College of Endocrinology Clinical Practice Guidelines for the Diagnosis and Treatment of Postmenopausal Osteoporosis - 2016. Endocrine Practice. 2016; 22(9):1111-8. [PMID 27643923] doi:10.4158/EP161435.ESGL Cosman F, de Beur SJ, LeBoff MS, Lewiecki EM, Tanner B, Randall S, et al; National Osteoporosis Foundation. Clinician’s guide to prevention and treatment of osteoporosis. Osteoporos Int. 2014;25:2359-81. [PMID: 25182228] doi:10.1007/s00198-014-2794-2 Manson JE, Brannon PM, Rosen CJ, Taylor CL. Vitamin D deficiency - is there really a pandemic? N Engl J Med. 2016;375:1817-1820. [PMID: 27959647] Singer FR, Bone HG 3rd, Hosking DJ, Lyles KW, Murad MH, Reid IR, et al; Endocrine Society. Paget’s disease of bone: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2014;99:4408-22. [PMID: 25406796] doi:10.1210/jc.2014-2910 93