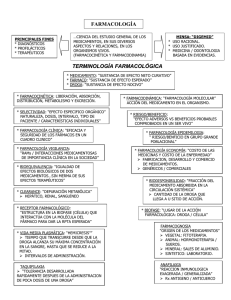
FARMACOLOGIA TEMA 1: INTRODUCCION A LA FARMACOLOGIA La Farmacología se impartirá durante el tercer año de la carrera y consta de dos asignaturas , que son: Farmacología general comprende los contenidos y habilidades que son esenciales para abordar la Farmacología Clínica y favorece que el estudiante desarrolle la competencia de analizar de manera crítica la información sobre medicamentos. Farmacología clínica que estudia los medicamentos en relación con el proceso salud enfermedad y favorece que el estudiante realice con bases científicas un uso racional de los medicamentos, constituye el sustento científico de la terapéutica que se imparte en la Clínica. La Farmacología como ciencia biomédica se encarga del estudio de los fármacos o medicamentos y sus acciones sobre el organismo. La asignatura Farmacología General comprende los contenidos y habilidades que son esenciales para abordar la Farmacología Clínica y favorece el desarrollo de la competencia de analizar de manera crítica la información sobre medicamentos; emplea precedentes como: Morfofisiología y Morfofisiopatología humana, pues éstas aportan elementos necesarios para poder comprender el mecanismo y las acciones de los medicamentos en el organismo, se relaciona estrechamente con la Clínica y constituye el sustento científico de la terapéutica que se imparte en todas las asignaturas clínicas. La farmacología es una ciencia joven y sus raíces están en la llamada materia médica, el papiro de Ebers (1500 a.n.e.) contiene referencias de unas 7000 sustancias, muchas de las cuales muestran con claridad su origen mágico. Se considera que Hipócrates y la medicina griega elevaron a la farmacología del rango empírico y mágico en el que se hallaba a otro que cabe denominar científico y técnico. A pesar de esto el hallazgo de fármacos siguió siendo azaroso y empírico y no fue hasta bien entrado el siglo XVIII que se realizaron los primeros ensayos de validación experimental de fármacos. Claude Bernard y Francois Magendie influyeron notablemente en el desarrollo de esta ciencia y proporcionaron los medios para descubrir lo que exactamente hacen los fármacos en el organismo. El desarrollo en el campo de la química permitió un rápido ascenso en la farmacología. El pasado siglo XX ha sido considerado, por muchos, el siglo de la farmacología, donde pudiéramos mencionar la introducción de la morfina como analgésico, la aspirina como antipirético y los antimicrobianos. Para profundizar en la historia de esta especialidad a lo largo de los siglos La farmacología es la ciencia biológica que estudia las propiedades de los fármacos y sus acciones sobre el organismo. Es importante conocerla ya que en su relación profesional con el paciente el médico debe resolver dos problemas fundamentales: hacer el diagnóstico y establecer la mejor terapéutica, es la Farmacología la ciencia que otorga al médico las bases necesarias efectuar una terapéutica racional. Aunque el campo terapéutico es amplio, la terapéutica farmacológica es la de mayor importancia y extensión en medicina humana. Por esto el médico debe ineludiblemente conocer la Farmacología. En los últimos años ha ocurrido una verdadera “explosión” de fármacos, paradójicamente dicha profusión de drogas ha ocasionado grandes problemas de salud, llamados por algunos autores, “patología farmacológica”. Esta patología se origina principalmente en el desconocimiento de los aspectos fundamentales de la farmacología de las drogas utilizadas y su consecuencia, la aplicación de una terapéutica irracional. La severidad de la “enfermedad por medicamentos” adquiere cada día mayor trascendencia requiriéndose acciones de farmacovigilancia, investigación epidemiológica y difusión sanitaria para disminuir sus efectos. Fármaco: Sustancia utilizada para el tratamiento, prevención, curación o diagnóstico de una enfermedad, también se le llama fármaco al principio activo del medicamento. Medicamento: Sustancia medicinal y sus asociaciones o combinaciones destinadas al uso humano o animal. Especialidad farmacéutica: El medicamento de composición e información definidas, de forma farmacéutica y dosificación determinadas, preparado para su uso medicinal inmediato, dispuesto y acondicionado para su dispensación al público, con denominación, embalaje, envase y etiquetado uniformes al que la Administración del Estado otorgue autorización sanitaria e inscriba en el Registro de Especialidades Farmacéuticas. Por último se ha definido a los medicamentos genéricos como la especialidad con la misma forma farmacéutica e igual composición cualitativa y cuantitativa en sustancias medicinales que otra especialidad de referencia (patentado). La Farmacología abarca diversos campos entres las que se encuentran: Farmacognosia que estudia el origen y características de las drogas o fármacos en su estado natural. Farmacocinética: Comprende el estudio del paso de las drogas a través del organismo, es decir los procesos de absorción, distribución, metabolismo y excreción. Farmacodinamia: Esta rama se ocupa principalmente del mecanismo de acción de las drogas y los efectos bioquímicos, fisiológicos o directamente farmacológicos que desarrollan los medicamentos. Farmacogenética: Es la rama que se ocupa del estudio de factores genéticos relacionados con la respuesta individual a las drogas o fármacos. Farmacovigilancia: tiene como objetivo la identificación y valoración cuantitativa del riesgo que representa el uso agudo y crónico de los medicamentos en la población o en subgrupos específicos de ella. Toxicología: Es una disciplina muy amplia que estudia el origen, investigación, diagnóstico y tratamiento de las intoxicaciones. Farmacoepidemiología: Ciencia que estudia el impacto de los medicamentos en poblaciones humanas, utilizando métodos epidemiológicos. El conocimiento de los lugares de unión de los fármacos y los mecanismos por los que la asociación entre una molécula de medicamento y su punto de unión producen una respuesta fisiológica constituye el principal impulso para la investigación en farmacología. La mayoría de los fármacos ejercen sus efectos uniéndose primero a proteínas aunque existen excepciones como antimicrobianos, antitutumorales que interactúan directamente con el ADN; los bifosfonatos usados en el tratamiento de la osteoporosis; los agentes quelantes o los antiácidos actúa por antagonismo químico. Las proteínas que sirven de diana para la acción farmacológica son: 1. receptores como los beta adrenérgicos 2 (beta 2) del músculo bronquial sobre los que actúa el salbutamol o los receptores opiodes a los que se une la morfina para conseguir la analgesia; 2. canales iónicos como los de calcio que pueden ser bloqueados por la nifedipina; 3. enzimas como la ciclooxigenasa que es inhibida por el ácido acetil salicílico y se produce el efecto analgésico, antipirético y antiinflamatorio de este medicamento y 4. moléculas transportadoras como la bomba de Na+ – K+ (célula cardíaca) a la que se unen los digitálicos como la digoxina. Para demostrar el efecto farmacológico, la eficacia y seguridad de un nuevo medicamento, requiere de varios años y tiene un costo elevado, ya que para su introducción en la práctica clínica habitual internacionalmente. debe cubrir una serie de requisitos establecidos Tras la caracterización físico-química de un nuevo compuesto de origen natural, sintética o semisintético, debe cumplirse una ruta crítica para su evaluación que incluye estudios preclínicos (farmacológicos y toxicológicos) que transcurren en el laboratorio y se realizan en animales de experimentación o sistemas biológicos disponibles. Los estudios de farmacología clínica, ensayos clínicos, se realizan en condiciones controladas para evaluar la eficacia y seguridad del nuevo compuesto en el hombre. Luego de la aprobación, por las agencias regulatorias para la comercialización del medicamento continúan los estudios postcomercialización o de farmacovigilancia. El tamizaje farmacológico y el bioensayo son los diseños fundamentales de la farmacología preclínica. El tamizaje es la búsqueda de una o varias acciones presentes en un compuesto determinado, puede ser dirigido si se trata de una acción farmacológica determinada en uno o varios compuestos y a ciegas cuando se buscan múltiples actividades biológicas que pudieran estar presentes en uno o varios compuestos. El bioensayo es un método cualitativo que permite valorar el efecto y la potencia de una sustancia. Los estudios toxicológicos preclínicos se diseñan para determinar los efectos tóxicos del compuesto en animales y pueden ser agudos que se realizan con el objetivo de determinar mortalidad a corto plazo, subagudos y crónicos para la búsqueda de efectos tóxicos acumulativos. Actualmente se ha establecido como requisito evaluar la reproducción, el potencial carcinogénico y mutagénico. Estas evaluaciones son conocidas como estudios especiales. Un ensayo clínico es cualquier investigación en sujetos humanos dirigida a verificar los efectos farmacológicos, farmacocinéticas e identificar las reacciones adversas del producto de investigación con el objetivo de determinar su eficacia y seguridad. Los pilares básicos sobre los cuales se debe desarrollar un ensayo clínico controlado y que permiten garantizar la validez de los resultados, son cuatro: Comparación concurrente que se logra al evaluar la eficacia de un producto en dos grupos de pacientes lo más parecido posible, excepto en la intervención que están recibiendo, es decir un grupo de estudio y un grupo control. Asignación aleatoria que se cumple cuando el paciente a incluir en el estudio tiene la misma posibilidad de estar en cualquiera de los grupos, el Enmascaramiento que permite disminuir la influencia de la subjetividad de investigadores y pacientes, el más utilizado es a doble ciegas donde ni el paciente ni el investigador conocen el tratamiento que recibe y la objetividad de la observación que se cumple a través de la utilización de variables de medición lo más objetiva posibles como sería emplear escalas validadas de medición del dolor para evaluar la eficacia de un analgésico en lugar de la percepción del paciente sobre el alivio del dolor. El desarrollo clínico de un producto consta de 4 fases. La fase I es la primera administración del producto en el hombre y generalmente se realiza en voluntarios sanos aunque en ocasiones se utilizan enfermos, como en el cáncer o el SIDA, su objetivo es verificar si el medicamento es tolerado satisfactoriamente. La fase II se realiza de manera general en pacientes y tiene como objetivo principal comenzar a evaluar la eficacia. La fase III incluye un mayor número de pacientes, es la última fase de evaluación de un medicamento (confirmación) antes de su comercialización y su objetivo es determinar la relación beneficio/riesgo en comparación con medicamentos de eficacia conocida. Al finalizar esta etapa se presentan los resultados en la autoridad sanitaria y si el producto es aceptado y registrado, se inicia su comercialización. La fase IV comienza después del registro y en la misma se desarrollan estudios de farmacovigilancia dirigidos a conocer mejor el perfil de seguridad del fármaco en las condiciones de la práctica clínica habitual. TEMA 2: FARMACOCINETICA El médico debe conocer los procesos a los que están sometidos los fármacos en el organismo que determinan la velocidad de inicio de acción de un fármaco, la intensidad de esta acción y la duración de su efecto. Estos procesos son: la absorción que es el paso del medicamento desde su sitio de administración hacia el plasma, la distribución como proceso mediante el cual el fármaco puede dejar de forma reversible el torrente sanguíneo para distribuirse en los líquidos intersticiales e intracelulares. En tercer lugar en órganos como el hígado, riñón u otros el agente puede transformarse en una sustancia más polar, más hidrosoluble, es el metabolismo, por último ocurre la excreción cuando el fármaco y sus metabolitos se eliminan del organismo a través de diferentes vías como la orina, heces o la bilis. En todos estos procesos el fármaco está atravesando membranas, movimiento conocido como biotransporte y que se realiza por diferentes mecanismos. La absorción de los medicamentos, independientemente de la vía de administración, se ve afectada por diferentes factores como la velocidad de disolución que determina que una forma líquida se absorba más rápido que una forma sólida. A mayor concentración del fármaco la absorción será mayor. El pH del medio influye en la absorción de medicamentos como las sales de hierro que lo hacen mejor cuando este es ácido. La mayor vascularización del sitio de absorción determina una rápida absorción del medicamento, como sucede con la vía sublingual. A mayor superficie mayor absorción, como sucede en los alvéolos y en la mucosa del intestino delgado. Por último la asociación con fármacos que modifiquen el pH, la circulación en el sitio de absorción o la motilidad del tracto gastrointestinal puede también afectar la absorción. La vía oral de administración de los medicamentos es la que con mayor frecuencia utilizamos, por lo que consideramos importante exponer los factores que condicionan la absorción por esta vía, por ejemplo: algunos medicamentos como la metoclopramida pueden acelerar el vaciamiento gástrico e influir en la absorción, enfermedades como el hipotiroidismo o fármacos pueden endentecer el tránsito intestinal y afectarla también. En la insuficiencia cardíaca se reduce el flujo sanguíneo esplácnico y se afecta la absorción. La velocidad de disolución depende del tamaño de la partícula y de la forma farmacéutica y como señalamos previamente este factor condiciona la absorción. Características fisicoquímicas como la polaridad, la naturaleza proteica o el pH también influyen en la absorción oral como sucede con los aminoglucósidos o las tetraciclinas. Se conoce como biodisponibilidad a la fracción del fármaco administrado que logra acceder a la circulación sistémica, la misma puede ser menor si se afecta la absorción o si ocurre el efecto del primer paso que se produce cuando los medicamentos sufren reacciones de inactivación inicial a su paso por la mucosa intestinal y el hígado, de esta forma se reduce la cantidad de medicamento que llega activo a la circulación general. Las vías de administración son las diferentes formas de aplicación de un medicamento en el organismo. Para la selección de las mismas es necesario tener en cuenta el estado del paciente, el efecto terapéutico deseado y las características físico-químicas del medicamento. La vía entérica incluye a: la vía oral que es la más común, consiste en la administración a través de la boca, la absorción ocurre en el estómago y en el duodeno que es con frecuencia el sitio de mayor capacidad de absorción, el efecto no aparece rápidamente y algunos medicamentos pueden causar irritación gástrica; la vía sublingual consiste en la administración del medicamento debajo de la lengua, se trata de una zona muy vascularizada que permite la rápida absorción de medicamentos liposolubles, además se evita el efecto del primer paso, sin embargo solo un reducido número de medicamentos puede administrarse por esta vía; por último la vía rectal que se puede utilizar para ejercer una acción local, sistémica o para provocar la evacuación del colón. Es una zona de rica vascularización, se evita parcialmente el efecto del primer paso pero puede resultar molesta para algunos pacientes y la absorción es irregular e incompleta La vía parenteral consiste en la inyección de medicamentos en los tejidos o líquidos corporales, incluye a la vía endovenosa en la que el medicamento se introduce directamente en la circulación y su efecto aparece de forma inmediata, lo que la convierte en la elección en las urgencias. Cuando la inyección se realiza en el tejido muscular se ha utilizado la vía intramuscular que permite la administración de sustancias oleosas y volúmenes de 1 a 10 ml, aunque los volúmenes superiores a 5 ml pueden provocar dolor por distensión. Es muy utilizada también la vía subcutánea que consiste en la administración del medicamento en el tejido celular subcutáneo que como es una zona poco vascularizada la velocidad de absorción es menor que para la intramuscular, permite la administración de suspensiones y micorscristales que forman pequeños depósitos a partir de los cuales el medicamento se libera gradualmente. Clasificadas dentro de la vía parenteral también se utilizan la intradérmica, intraarterial, intratecal, epidural, intraarticular e intraperitoneal, La vía inhalatoria es la administración de un fármaco en forma de gas, polvo o líquido vehiculizado por el aire inspirado con el fin de obtener un efecto local en el árbol bronquial como el de los broncodilatadores o sistémico después de su absorción como sucede con las gases anestésicos. Los efectos aparecen rápidamente y permite la autoadministración pero el alivio rápido de los síntomas puede inducir al abuso. La administración de principios activos por la piel para ejercer una acción sistémica es la vía percutánea, para la cual se han diseñado sistemas transdérmicos que garantizan el aporte percutáneo del principio activo a una velocidad programada o durante un período de tiempo determinado. Evita el efecto del primer paso y permite obtener un mejor cumplimiento terapéutico, pero solo es útil en pacientes crónicos y para medicamentos liposolubles y de pequeña masa molecular. La vía tópica consiste en la administración directa de los medicamentos a través de la piel y mucosas de orificios naturales, excepto los del tubo digestivo. A diferencia de la percutánea el objetivo es obtener un efecto local después de la administración. Pueden aplicarse altas concentraciones del fármaco en la lesión y es una técnica sencilla pero no siempre se alcanzan las concentraciones requeridas en las capas más profundas de la piel siendo necesaria la utilización simultánea de otra vía para lograr el efecto sistémico. Las formas farmacéuticas son las formas en las que se puede administrar un medicamento en el organismo, pueden ser sólidas, líquidas y gaseosas y guardan correspondencia con la vía de administración. Para la administración oral los medicamentos pueden presentarse en forma de jarabe, elixir, suspensiones o gotas, en el caso de la fitoterapia se pueden utilizar, para esta vía, formas específicas como la infusión, decocción y las tinturas, en cuanto a las formas sólidas se destacan los comprimidos (liberación retardada, grageas, cubierta entérica o efervescentes), las cápsulas (duras, blandas, cubierta entérica o liberación modificada), los granulados y los polvos. Para la administración de medicamentos liposolubles por vía sublingual se pueden emplear gotas, comprimidos y aerosoles. Los supositorios constituyen la forma farmacéutica más conocida para la vía rectal, en la que también se utilizan las pomadas, soluciones y dispersiones rectales. En la administración parenteral se utilizan formulaciones estériles para inyectarse o implantarse en el organismo y pueden ser: preparaciones inyectables en las cuales el principio activo ya se encuentra disuelto, emulsionado o suspendido (Vitaminas, Dipirona); preparaciones para diluir antes de inyectarse o administrase en infusión (Aminofilina); las preparaciones inyectables para infusión como la Dextrosa 5 % o la Solución salina al 0.9 %; polvo para preparaciones extemporáneas a las que se les debe añadir un volumen de líquido determinado (Hidrocortisona, Penicilina G) y los implantes o pellets que se colocan por vía subcutánea para garantizar la liberación gradual del principio activo (Depósito de contraceptivos hormonales). En la vía inhalatoria se emplean aerosoles y nebulizaciones pero el acceso al lugar de acción depende del tamaño de la partícula (por encima de 20ц se deposita en la orofaringe y las vías respiratorias altas y por debajo de 1ц no se deposita) y de la técnica empleada (inhaladores o nebulizadores). Los sistemas transdérmicos utilizados en la vía percutánea son los parches y la iotoforesis, ambos permiten la liberación continúa del fármaco desde el sitio de administración. Diversas son las formas farmacéuticas que se emplean para administrar los medicamentos por la vía tópica, entre las que se encuentran los colirios que son soluciones isotónicas para ser instiladas en los ojos, las gotas óticas y nasales, lociones para aplicar sobre la piel sin friccionar, linimentos y tinturas, también se emplean formas sólidas como las cremas, pomadas y pastas que debes distinguir pues tienen indicaciones diferentes según las características de la lesión, por último para la administración de medicamentos a través de la vagina se emplean los óvulos y tabletas vaginales La distribución es el proceso mediante el cual el fármaco puede dejar de forma reversible el torrente sanguíneo para distribuirse en los líquidos intersticiales e intracelulares. Los fármacos pueden hallarse en la circulación en forma libre, que es la farmacológicamente activa y unidos a proteínas plasmáticas, ambas se encuentran en equilibrio. Los medicamentos tienen diferente afinidad por las proteínas plasmáticas, así tenemos agentes como el nitroprusiato de sodio (utilizado en la emergencia hipertensiva) que no se une a éstas, en cambio la digitoxina (útil en la Insuficiencia cardiaca) se une extensamente y la penicilina G lo hace más débilmente. Un fármaco puede ser desplazado de su sitio de unión a las proteínas plasmáticas por otro que posea mayor afinidad, aumenta, entonces la forma libre del desplazado y secundariamente sus efectos en el organismo. El volumen de distribución se concibe como el volumen de fluido al cual accede un fármaco teniendo en cuenta la dosis y la concentración plasmática en un momento determinado. El valor óptimo depende de los objetivos que se pretendan alcanzar. El valor del agua intercambiable en el organismo es de 0.6 L/kg y se toma como referencia. Un Vd como el de la Claritromicina (antimicrobiano) de 2.6 L/kg significa que la concentración del mismo en los tejidos será alta y baja en el plasma lo que se corresponde con los fármacos liposolubles. La cetirizina es un antihistamínico que posee un Vd de 0.4 L/kg que significa que su concentración en los tejidos es baja y alta en el plasma, característico de los fármacos hidrosolubles. Cuando el Vd se superpone al valor del volumen plasmático 0.1 L/Kg como sucede con el piroxicam lo podemos interpretar como un fármaco que se une de manera considerable a las proteínas plasmáticas y su transferencia a los tejidos está restringida. Mediante el metabolismo los fármacos se transforman en sustancias más polares y más hidrosolubles para poder ser eliminadas del organismo, este ocurre fundamentalmente en el hígado a través de reacciones de Fase I como la oxidación, reducción e hidrólisis que pueden aumentar, disminuir o dejar sin cambios la actividad farmacológica de una sustancia y reacciones de Fase II como la conjugación. Durante las reacciones de fase I el medicamento se hace más hidrosoluble pero no siempre excretable o inactivo, debido a esto muchos fármacos pasan después a la fase II como el ejemplo de el ácido acetil salicílico que se conjuga con el ácido glucurónido para poder ser eliminado. La biotransformación o metabolismo de los medicamentos varía con la edad, en los recién nacidos la capacidad metabolizadota está disminuida por inmadurez enzimática, mientras que en el anciano la biotransformación es más lenta por disminución del flujo sanguíneo hepático. Las velocidades con las cuales las personas metabolizan son variables y esta variabilidad tiene una fuerte base genética, de esta manera existen acetiladores rápidos y lentos para fármacos como la isoniacida (antituberculoso), estos últimos son más propensos a padecer reacciones adversas por incremento de las concentraciones plasmáticas del fármaco. El incremento de la actividad enzimática se conoce como inducción, un fármaco puede inducir su propio metabolismo como lo hace el fenobarbital (anticonvulsivante) o influenciar el metabolismo de otros. La inhibición enzimática se traduce en una disminución del metabolismo, por ejemplo la eritomicina (antimicrobiano) inhibe el metabolismo de la carbamacepina (anticonvulsivante) con el consiguiente aumento de su concentración plasmática y la aparición de reacciones adversas. La excreción es el proceso mediante el cual un fármaco o un metabolito se elimina del organismo sin que se modifique más su estructura química. Los medicamentos son eliminados del organismo a través de la eliminación hepática y de la excreción renal, esta última es considerada la principal vía, pero existen otras vías como la bilis, la saliva, el sudor, las lágrimas, la leche materna, etc. Los mecanismos de la excreción renal son 3: filtración glomerular, secreción activa y reabsorción tubular pasiva. El conocimiento de las vías de excreción puede ser importante para tratar enfermedades como la sepsis biliar con medicamentos que se eliminen preferentemente por la bilis, la sepsis urinaria con medicamentos que se eliminen de forma activa por vía renal o realizar ajustes de dosis en caso de insuficiencia hepática o renal. El tiempo de vida media es el tiempo que tarda la concentración del fármaco en caer a la mitad de su valor inicial, es directamente proporcional al volumen de distribución e inversamente proporcional al aclaramiento del fármaco; la vida media es útil para calcular el tiempo que permanece el fármaco en el organismo y de esta forma establecer la frecuencia de su administración. Los fármacos eliminados a la luz intestinal en forma activa a través de la bilis pueden reabsorberse pasivamente en el intestino a favor de un gradiente de concentración. Estos procesos dan origen a una circulación enterohepática , lo que retrasa la caída de las concentraciones plasmáticas y prolonga la duración del efecto. TEMA 3: FARMACOLOGIA AUTONOMICA DE LOS RECEPTORES Y LA NEUROTRANSMISION Desde el punto de vista anatómico el sistema nervioso se divide en sistema nervioso central y sistema nervioso periférico, constituido por neuronas que entran o salen del sistema nervioso central. La porción eferente del SNP se divide en dos porciones funcionales, el sistema nervioso somático y el sistema nervioso autónomo (SNA). Este último consta de 2 grandes divisiones morfológicas, simpática y parasimpática. La mayoría de los órganos reciben inervación simpática y parasimpática, cuya activación suele ocasionar efectos contrarios. Desde el punto de vista farmacológico presenta gran interés la porción eferente del SNA, porque los fármacos que modifican sus funciones, actúan sobre este. El sistema nervioso simpático tiene los cuerpos celulares en las astas laterales de los segmentos torácicos y lumbares desde D1 hasta L3, las fibras preganglionares se dirigen en busca de la neurona posganglionar ubicada en el ganglio correspondiente, que se encuentra alejado del órgano efector y de donde se origina la fibra posganglionar que se encaminan hacia los diferentes sitios donde actúa el sistema simpático. La noradrenalina es el neurotransmisor de la mayoría de las fibras simpáticas posganglionares que interactúan con receptores alfa y beta localizados en los órganos efectores. En el sistema nervioso parasimpático las neuronas provienen de los pares craneales III, VII, IX y X y los segmentos S2, S3, S4 de la médula sacra. A diferencia del sistema simpático, posee generalmente largas neuronas preganglionares, los ganglios están próximos o dentro del mismo órgano efector y las fibras posganglionares son cortas. La acetilcolina es el neurotransmisor de las fibras pre y posganlionares que interactúan con los receptores muscarínicos y nicotínicos en los órganos efectores. Varias investigaciones han permitido alcanzar a comprender todos los pasos involucrados en la neurotransmisión autonómica, en general estos pasos son similares para el simpático y el parasimpático y lo que varía son los neurotransmisores. Así tenemos: Biosíntesis del neurotransmisor, almacenamiento, liberación, acción en receptores específicos e inactivación. Su conocimiento nos permitirá comprender la utilidad terapéutica de varios medicamentos que modifican las funciones del SNA. La síntesis del neurotransmisor en el sistema simpático comienza con la hidroxilación del aminoácido tirosina catalizada por la tirosina hidroxilasa, se considera el paso limitante de la síntesis, esta enzima es bastante selectiva a diferencia de las otras que intervienen en este proceso. L-dopa obtenida es transformada en dopamina por acción de la enzima dopa descarboxilasa, reacción que también ocurre en el citoplasma de la neurona adrenérgica, en el próximo paso la enzima dopamina beta hidroxilasa convierte a la dopamina en noradrenalina en el interior de las vesículas sinápticas y finalmente esta última puede metilarse para formar adrenalina por acción de la feniletanoamino-Nmetiltransferasa. Las catecolaminas sintetizadas se almacenan en vesículas formando complejo con el ATP y con proteínas denominadas cromograninas. La liberación al espacio sináptico se lleva a cabo a través de un proceso de exocitosis dependiente de calcio. La despolarización ocasiona apertura de los canales de Ca voltaje sensibles y la entrada de Ca por estos da lugar a la fusión de la membrana de la vesícula con la membrana celular, se forma un poro de fusión y se libera todo el contenido vesicular. Pueden liberarse también por un proceso que es independiente de Ca, que consiste en el desplazamiento de sus lugares de depósito por las aminas simpaticomiméticas de acción indirecta y que se conoce como liberación inducida por drogas. Una vez liberados, la acción en sus receptores debe ser breve, precisa y localizada, lo que se garantiza por los mecanismos de inactivación: recaptación e inactivación enzimática, que son complementarios y nunca excluyentes. La recaptación I o intraneuronal es un transporte activo dependiente de sodio, la recaptación II o extraneuronal tiene menor afinidad por la noradrenalina, pero tiene más capacidad, ambos pueden ser bloqueados por fármacos. La inactivación enzimática se debe a la acción de las enzimas MAO y COMT, la primera está presente sobre todo en neuronas noradrenérgicas, mientras que la COMT se encuentra en tejido neuronal y no neuronal Como señalamos al inicio el SNA es de gran interés desde el punto de vista farmacológico, muchas sustancias modifican sus funciones y son de utilidad terapéutica como la L-dopa, medicamento empleado en el tratamiento de la enfermedad de Parkinson, la que se manifiesta por movimientos involuntarios y trastornos del tono muscular motivados por un déficit de dopamina en las vías nigroestriatales, sin embargo no es posible administrar dopamina porque no atraviesa la barrera hematoencefálica, este medicamento es capaz de atravesar esta barrera, transformarse en dopamina por acción de la dopa descarboxilasa, de esta forma eleva sus niveles y permite controlar los síntomas descritos. La alfametildopa, considerada el antihipertensivo de elección para la tratar la HTA gestacional, puede ser captada por la neurona adrenérgica donde constituirá un sustrato de la enzima dopa descarboxilasa (poco específica) que la transforma en alfametildopamina, más tarde por acción de la dopamina beta hidroxilasa es convertida en alfametilnoradrenalina y actúa como un falso neurotransmisor a nivel de receptores alfa 2 possinápticos inhibitorios de la liberación de noradrenalina en el SNC, como consecuencia disminuye la frecuencia cardiaca y la tensión arterial. Otros medicamentos pueden tener acciones en otros pasos de la neurotransmisión autonómica, es el caso de: Reserpina que bloquea el transporte de noradrenalina a la vesícula de almacenamiento, hace que las terminaciones nerviosas agoten sus depósitos de noradrenanalina, disminuye la actividad del sistema nervioso simpático y se reduce la tensión arterial, por lo que durante muchos años se ha utilizado como antihipertensivo sin embargo su uso ha disminuido considerablemente motivado por los efectos adversos que posee relacionados con la depleción que provoca de otros neurotransmisores como dopamina y serotonina. La anfetamina es una amina simpaticomimética usada por su acción estimulante del SNC consecuencia del efecto estimulante de la liberación de noradrenalina de la terminación nerviosa en ausencia de despolarización. Las acciones sobre el rendimiento físico y el estado de animo han provocado su abuso en numerosas circunstancias. La teofilina es capaz de bloquear la recaptación II extraneuronal de catecolaminas, lo que puede provocar un incremento de noradrenalina en los receptores beta 2 del músculo liso bronquial lo que puede contribuir al efecto antiasmático de este fármaco. La selegilina o deprenil es un inhibidor selectivo de la isoenzima MAO-B, disminuye la degradación enzimática de dopamina en SNC y es de gran utilidad junto a la L-Dopa en el tratamiento de la enfermedad de Parkinson. La acetilcolina es sintetizada en la terminación nerviosa colinérgica a partir de la colina, requiere de acetilación para la que se utiliza acetil CoA y la presencia de la enzima colinoacetilasa. Se almacena en vesículas sinápticas a concentraciones elevadas, se libera por exocitosis mediada por Ca al igual que el neurotransmisor en el sistema simpático, actúa sobre receptores muscarínicos y nicotínicos y es inactivada por acción de las enzimas acetilcolinesterasa y pseudocolinesterasa, la primera es relativamente específica para la acetilcolina y responsable de la hidrólisis rápida en las sinapsis colinérgicas, por su parte la pseudocolinesterasa es relativamente poco selectiva y se encuentra en el plasma y muchos tejidos. Los anticolinesterásicos son aquellos fármacos capaces de inhibir a la colinesterasa y favorecer la transmisión colinérgica, han demostrado ser eficaces en enfermedades como el glaucoma donde logra reducir la tensión intraocular, mejorando los síntomas y signos de la misma. El íleo paralítico, al aumentar las concentraciones de acetil colina en receptores muscarínicos del músculo liso intestinal, provoca un incremento de la actividad peristáltica y alivio de la distensión abdominal. En la miastenia grave, trastorno autoinmune caracterizado por un defecto en la transmisión neuromuscular y que se manifiesta por debilidad muscular y fatiga, producen un aumento significativo de acetil colina, se estimula la transmisión sináptica y mejoran los síntomas de la enfermedad. Recordemos que uno de los mecanismos fundamentales por los que pueden actuar los fármacos en el organismo es a través de la interacción con un receptor. Se han propuesto varios conceptos para definir un receptor, pero se acepta que son moléculas con las que un fármaco debe interactuar selectivamente, para generar una modificación constante y específica de la función celular. Los receptores son generalmente de naturaleza proteica aunque otras macromoléculas como los ácidos nucleicos pueden comportarse como receptores de fármacos, sucede con los receptores para medicamentos antitumorales y antimicrobianos Los receptores desencadenan muchos tipos de efectos celulares y existen también tipos muy diferentes de conexión entre la ocupación del receptor y la aparición de la respuesta. Basados en la naturaleza de esta conexión y en la estructura molecular, podemos distinguir cuatro tipos de receptores o familias. Los canales iónicos controlados por ligandos que se conocen también como receptores ionotrópicos, en los que el acoplamiento es directo, los efectos aparecen en milisegundos y el receptor nicotínico es un ejemplo. Los receptores acoplados a proteínas G o metabotrópicos, son la familia más numerosa y comprenden receptores de muchas hormonas y transmisores lentos como el receptor de acetil colina. Los receptores ligados a quinasas constituyen un grupo extenso y heterogéneo que responden a mediadores proteicos, contienen un dominio intracelular de tipo enzimático con actividad de proteína quinasa o guanilato ciclasa e incluyen receptores de insulina, citoquinas y factores de crecimiento, sus efectos aparecen en horas. Los receptores nucleares regulan la transcripción génica, el acoplamiento es a través del ADN, engloba los receptores de hormonas esteroideas, tiroidea, Vitamina D y sus efectos pueden tardar horas. Para que un fármaco produzca un efecto biológico necesita reunir dos propiedades fundamentales: La capacidad que posee el fármaco de unirse al receptor y formar el complejo fármaco-receptor se conoce como afinidad y la capacidad que tienen los fármacos, de una vez unidos al receptor, generar un estímulo y desencadenar la respuesta o efecto farmacológico es la actividad intrínseca. Los fármacos que poseen afinidad y actividad intrínseca son los agonistas, mientras que aquellos que tiene afinidad pero carecen de actividad intrínseca son los antagonistas, debido a que bloquean los efectos normalmente inducidos por el agonista. Los agonistas dan lugar al inicio de una respuesta, mientras que los antagonistas simplemente ocupan el receptor. El antagonismo es la interferencia de la acción de una sustancia química mediante la acción de otra y tiene una importancia extraordinaria en el campo de la farmacología. El antagonismo químico se produce como resultado de una reacción química entre dos sustancias, lo que origina la pérdida el efecto farmacológico o tóxico de la sustancia química, se conoce también como antagonismo por neutralización y es el que se produce cuando utilizamos sulfato de protamina (con carga electropositiva) en el tratamiento de las hemorragias graves causadas por el anticoagulante heparina (con carga electronegativa). El antagonismo fisiológico se produce cuando dos agonistas actuando sobre poblaciones de receptores diferentes ocasionan efectos opuestos, sucede cuando sustancias como la noradrenalina (agonista 1) provoca relajación del músculo liso bronquial al actuar en receptores beta 2 mientras que la acetil colina (agonista 2) causa broncoconstricción al actuar en receptores muscarínicos M3. El antagonismo farmacológico es el más importante desde el punto de vista clínico y puede ser competitivo cuando dos agentes compiten por el mismo sitio receptor y no competitivo cuando el antagonista no actúa en el receptor, sino en otro sitio diferente que forma parte del mecanismo de transducción de señales, no puede ser contrarrestado con el aumento de la concentración del agonista y es el tipo de antagonismo que se produce cuando fármacos como la nifedipina bloquean la entrada de calcio a través de los canales responsables de la entrada lenta de esta ion e impiden su participación en la contracción del músculo liso. El antagonismo farmacológico competitivo puede ser reversible cuando se puede lograr desplazar el antagonista con el aumento de la concentración del agonista e irreversible debido a la formación de enlaces covalentes que hacen estable el complejo fármaco–receptor y no puede lograrse el desplazamiento del antagonista aumentando las concentraciones del agonista. Las aplicaciones terapéuticas de agonistas y antagonistas adrenérgicos y colinérgicos son el mejor ejemplo del antagonismo farmacológico competitivo reversible, el bloqueo irreversible de receptores alfa 1 y 2 por la fenoxibenzamina de utilidad en el tratamiento del feocromocitoma es un ejemplo del farmacológico competitivo irreversible. Los receptores farmacológicos pueden ser: Adrenérgicos, colinérgicos, purinergivos, de hormonas, de autacoides, de opoides y de neurotransmisores centrales. Por su importancia estudiaremos los receptores adrenérgicos y colinérgicos. Los receptores adrenérgicos pueden ser: Múltiples ejemplos ponen de manifiesto la importancia clínica de la teoría de receptores, así tenemos: el salbutamol es un agonista selectivo de receptores beta 2 del músculo liso bronquial que causa broncodilatación y mejora la disnea en pacientes asmáticos. La pilocarpina es un agonista de receptores muscarínicos M3 que produce contracción del esfínter de la pupila y de los músculos ciliares, de esta forma reduce la resistencia al drenaje del humor acuoso y la tensión intraocular que se encuentra elevada en los pacientes con glaucoma. El atenolol es un antagonista de receptores beta 1 del músculo cardíaco, el bloqueo de este receptor se traduce en una disminución de la fuerza de contracción y la frecuencia cardiaca de gran utilidad en la hipertensión arterial. El bromuro de ipratropio es un antagonista de receptores muscarínicos M3 en músculo liso bronquial que provoca broncodilatación y mejora los síntomas secundarios a la obstrucción bronquial en pacientes con bronquitis crónica. TEMA 4: MEDIADORES QUIMICOS Y SU RELACION CON DIFERENTES SUSTANCIAS MEDICAMENTOSAS Los principales mediadores químicos son: histamina, eicosanoides como protaglandinas, tromboxanos y leucotrienos, serotonina, angiotensina, quininas, péptido natriurético auricular, citoquinas, interleuquinas, interferones y óxido nítrico. La histamina se sintetiza a partir del aminoácido histidina, se encuentra en casi todos los tejidos, se almacena asociada a la heparina en el interior de los mastocitos y básofilos, especialmente en los pulmones, en el estómago y en neuronas histaminérgicas del cerebro. Dentro de las acciones más importantes se encuentran: contracción del músculo liso bronquial e intestinal, relajación en vasos sanguíneos, estimulación cardiaca, mediador importante de reacciones alérgicas e inflamatoria inmediatas y estimulo de la secreción gástrica, además de ser un neurotransmisor en algunas áreas del cerebro. El uso clínico de la histamina es discutido en la actualidad y solo se usa ocasionalmente como medio diagnóstico, en cambio sus antagonistas son de gran utilidad. Los antagonistas de los receptores H1 pueden ser de primera generación como la difenhidramina, meclicina, dimenhidrinato (gravinol) y de segunda generación como la terfenadina, loratadina, cetiricina, estos últimos no atraviesan la barrera hematoencefálica, no provocan sedación y la duración del efecto farmacológico es más prolongada lo que permite su administración una vez al día. Los antagonistas de receptores H2 más utilizados son la cimetidina, ranitidina y famotidina. Los fármacos antagonistas de la histamina son de gran utilidad en múltiples afecciones como, las reacciones alérgicas en la que se emplean los antihistamínicos H1 como la difenhidramina, en la úlcera péptica donde están indicados los antagonistas H2 (cimetidina). El uso de los antihistamínicos H1 de primera generación se acompaña con frecuencia de sedación y somnolencia relacionada con su acción a nivel de receptores H1 en el SNC. Los eicosanoides son derivados importantes de los fosfolípidos de la membrana, se detectan en fluidos biológicos y actúan como moduladores y mediadores de procesos fisiológicos y patológicos del organismo. Por acción de la fosfolipasa A2 se obtiene el ácido araquidónico y a partir de este por acción de la lipoxigenasa se forman leucotrienos, lipoxinas y hepoxilinas. Por otra vía en la que participa la enzima ciclooxigenasa se forman prostaciclina, prostaglandinas y tromboxano El término prostanoide abarca a prostaglandinas y tromboxanos. La prostaciclina o prostaglandina I2 procede fundamentalmente del endotelio vascular y tiene como efectos principales la vasodilatación e inhibición de la agregación plaquetaria. El tromboxano procede fundamentalmente de las plaquetas y sus principales efectos consisten en agregación plaquetaria y vasoconstricción. Las prostaglandinas tienen acciones diversas, así tenemos que las PGE2 es la que predomina en las respuestas inflamatorias y es un mediador de la fiebre, también produce contracción del músculo liso bronquial y del aparato digestivo, inhibe la secreción ácida gástrica, aumenta la secreción mucosa gástrica y libera neurotransmisores autónomos, al igual que la F2 alfa provoca contracción uterina y de forma similar a la D2 que se deriva de los mastocitos causa vasodilatación e inhibición de la agregación plaquetaria. El leucotrieno LTB4 es un importante mediador de todos los tipos de inflamación que al actuar sobre receptores específicos provoca adherencia, quimiotaxis y activación de los polimorfos y monocitos, estimula la proliferación y la síntesis de citoquinas en macrófagos y linfocitos. Los cistenil leucotrienos (C4, D4 E4), especialmente importantes en el asma, provocan contracción del músculo liso bronquial, vasodilatación y constricción coronaria. Varios son los fármacos relacionados con los eicosanoides, incluye inhibidores de la síntesis, análogos y antagonistas a nivel de sus receptores. A continuación te presentamos un grupo de fármacos que pueden afectar la biosíntesis de estos mediadores químicos como: los glucocorticoides (Prednisona, hidrocortisona) que inducen la síntesis de una proteína llamada lipocortina que inhibe a la fosfolipasa A2, los AINEs (ASA, Ibuprofeno, Piroxicam) que inhiben a la ciclooxigenasa responsable de la formación de las PGs y el zileutón que inhibe a la 5-lipoxigenasa encargada de la formación de los leucotrienos. A partir del conocimiento de las acciones de las PGs se han desarrollado varios análogos: Dinoprostona, misoprostol y gemeprost de la PGE2, carboprost y latanoprost de la PGF2 alfa, alprostadil de la PGE1 y epoprostenol de la PGI2. El zafirlukast y el montelukast se comportan como antagonistas de los receptores CIST-LT1 para leucotrienos cisteínicos (LTC4, D4, E4). Múltiples ejemplos ponen de manifiesto la importancia clínica de los eicosanoides y los fármacos relacionados, por ejemplo: La dinoprostona es un análogo de la PGE2 que actúa a nivel de sus receptores facilitando la contracción uterina y dilatación del cuello uterino por lo que se pueden utilizar para iniciar y estimular el trabajo de parto a término y postérmino. El ibuprofeno es un AINE que inhibe a la COX y por tanto la síntesis de PGs mediadores importantes de la inflamción, fiebre y dolor lo que explica la utilidad de este fármaco como analgésico, antipirético y antiinflamatorio. El misoprostol es un análogo de la PGE2 relacionada con la disminución de la secreción ácido péptica y aumento de la secreción mucosa gástrica indicado en la profilaxis de la úlceras por AINEs. El montelukast es un antagonista del receptor Cist-LT1 de los leucotrienos C4, D4, E4, el bloqueo de este receptor se traduce en la relajación del músculo liso bronquial, lo que los hace especialmente útiles en el asma bronquial. La serotonina o 5 hidroxitriptamina (5-HT) se sintetiza a partir del aminoácido triptófano y más del 90 % de esta se encuentra en las células enterocromafines, existe cierta cantidad en las células nerviosas del plexo mientérico, plaquetas y sistema nervioso central. En la periferia la serotonina participa en la peristalsis, el vómito, la agregación plaquetaria. Es un neurotransmisor del SNC y a este nivel interviene en el control del sueño, el estado ánimo y apetito, entre oras como la temperatura corporal, las alucinaciones, la percepción del dolor y el vómito. Estas acciones se producen al interactuar la serotonina con receptores específicos (5-HT). Se utilizan en terapéutica agonistas de estos receptores como: sumatriptán y naratriptán que actúan en los receptores 5-HT1B, 1D, de ellos el naratriptán ofrece las mayores ventajas desde el punto de vista farmacocinético. Se comportan como agonistas del 5-HT4 en tubo digestivo la cisaprida y la metoclopramida, ellos aumentan motilidad y secreciones intestinales. Son de importancia antagonistas de los receptores de la serotonina como: la ciproheptadina y el Ketotifeno que son antagonistas no selectivos de los receptores 5-HT2, estos también muestran afinidad por receptores alfa adrenérgicos e histaminérgicos. A nivel del receptor 5-HT 3 se comporta como un antagonista el ondansetron y otros del mismo grupo como granisetrón y tropisetrón. La clozapina debe parte de su perfil terapéutico al antagonismo de receptores 5-HT 2A, 2C y 7. De gran utilidad son también los inhibidores de la recaptación de Serotonina como la fluoxetina, sertralina, fluvoxamina, etc. Después de conocer las acciones de la Serotonina y los fármacos relacionados podemos comprender la importancia clínica de este mediador químico, por ejemplo: El sumatriptán es un agonista de receptores 5-HT 1B, 1D localizados en el cerebro, y ganglios basales que median analgesia y vasoconstricción, lo que permite utilizarlo en el ataque agudo de la migraña, condición común que afecta hasta un 15 % de las personas y en cuya génesis esta involucrada la serotonina. El ondansetrón es un antagonista selectivo de receptores 5-HT3 en neuronas entéricas donde bloquea la emésis y su uso está aprobado en la prevención de náuseas y vómitos inducidos por la radioterapia y quimioterapia antineoplásica. Los bloqueadores de la recaptación de serotonina como la fluoxetina han demostrado ser útiles en el tratamiento de la depresión ya que la inhibición de la recaptación de 5-HT en la terminal nerviosa determina niveles más altos de serotonina en el espacio sináptico y en consecuencia la modificación del estado de ánimo. La ruta del sistema renina-angiotensina se inicia con la renina, enzima proteolítica que actúa sobre el angiotensinógeno, generando un metabolito inactivo que es la angiotensina 1, esta constituye el sustrato de la enzima convertidora de angiotensina (ECA) que da lugar a la angiotensina II, principal efector del sistema. La ECA no es una enzima específica y también se comporta como una quinasa que degrada e inactiva a la bradiquinina. Dentro de las principales acciones de la angiotensina II, mediadas por los receptores AT1, tenemos: estimulación generalizada del sistema nervioso simpático, contracción de células musculares lisas, el crecimiento de células musculares lisas y fibroblastos. A nivel renal, reduce flujo sanguíneo y aumenta la reabsorción tubular de sodio y en las suprarrenales aumenta la síntesis y liberación de aldosterona y la liberación de catecolaminas. Los fármacos relacionados con la angiotensina pertenecen a dos grandes grupos, el primero los Inhibidores de la ECA, conocidos como IECA, al que pertenecen fármacos como el captopril, enalapril y fosinopril. El segundo grupo es el de los antagonistas de los receptores AT1 de la angiotensina II conocidos como ARAII al que pertenece el losartán. Tanto los IECA como los ARAII son fármacos muy útiles en el tratamiento de enfermedades cardiovasculares, debido al bloqueo farmacológico del sistema reninaangiotensina. Los inhibidores de la enzima convertidora de angiotensina han sido los que han demostrado mayor utilidad clínica, dentro de todos los fármacos que actúan bloqueando el sistema renina-angiotensina, por ejemplo se ha probado la eficacia del captopril en el tratamiento de la hipertensión arterial y la insuficiencia cardíaca, al reducir la resistencia vascular periférica, la tensión arterial, facilitar la excreción renal de sodio, retrasan el inicio o reaparición de insuficiencia cardiaca y aumentan la supervivencia de estos pacientes. Los ARAII como el losartán inhiben la actividad biológica de la angiotensina II, independientemente de su ruta metabólica de formación, tiene indicaciones terapéuticas similares a los IECA y la ventaja que representa su uso consiste en la menor frecuencia de efectos adversos, por lo que resultan una alternativa de tratamiento para los pacientes en los que el uso de los IECA está contraindicado. TEMA 5: FARMACOVIGILANCIA, PRESCRIPCION Y USO RACIONAL DE MEDICAMENTOS El concepto de reacción adversa podemos definirlo como cualquier respuesta nociva, indeseable que se presenta cuando se administran dosis normales de un medicamento en el hombre para la profilaxis, tratamiento o diagnostico de una enfermedad o con el objetivo de modificar una función fisiológica como en la planificación familiar. La clasificación de las reacciones indeseables teniendo en cuenta el mecanismo de producción es la siguiente: las de tipo A son reacciones relacionadas con la dosis, predecibles, esperadas, debidas a la forma farmacéutica, alteraciones farmacocinéticas o causas farmacodinámicas, por ejemplo la somnolencia por antihistamínicos H1; las de tipo B no tienen relación con la dosis, no se pueden predecir, son más graves, raras y el ejemplo más significativo son las reacciones de hipersensibilidad o idiosincráticas, las tipo C se relacionan con la administración de tratamientos prolongados en enfermedades crónicas, como la hipertrofia gingival de la difenilhidantoína en el tratamiento de la epilepsia y las D aparecen varios meses o años después de haber retirado el medicamento como el cáncer de vagina de células claras que apareció en muchachas de 16 años cuyas madres habían consumido durante el periodo de gestación dietilestilbestrol. Si tenemos en cuenta la intensidad se clasifican en: las letales son reacciones que como su nombre indica provocan la muerte del paciente y generalmente no son frecuentes, las graves pueden dejar secuelas, provocar ingresos hospitalarios o prolongarlo en el caso de pacientes hospitalizados, las moderadas son reacciones que no ponen en peligro la vida del paciente pero requieren tratamiento médico mientras que las leves no requieren tratamiento y desaparecen según el fármaco se vaya eliminando de nuestro organismo como la cefalea por nifedipina. Se muestran los diferentes tipos de reacciones adversas según sus características y se clasifican en reacciones de hipersensibilidad, idiosincrasia, efecto colateral, tóxico, efecto paradójico, de rebote, taquifilaxia, tolerancia, resistencia, intolerancia, efecto teratógeno, farmacodependencia, reacción de Herxheimer y la reacción por la interacción fármaco infección viral como el síndrome de Reye en niños con infecciones virales que se le administra acido acetilsalicílico para la bajar la fiebre. Se muestra la diferencia entre la hipersensibilidad y la idiosincrasia, en la primera existen contactos previos con el medicamento que desencadena la reacción o con similares del mismo grupo y tiene una base inmunológica, es decir se produce en virtud de una reacción antígeno-anticuerpo, el edema angionéurotico o la urticaria por fármacos como los AINEs, son ejemplos de esta reacción adversa. Mientras que la idiosincrasia aparece cuando el paciente se pone en contacto por primera vez con el fármaco y es una reacción genéticamente determinada, generalmente relacionada con deficiencias enzimáticas como sucede en la metahemoglobinemia por sulfas y nitritos o la neuropatía periférica por isoniacida en acetiladores lentos. El efecto colateral depende del mecanismo de acción del medicamento por lo que aparece casi siempre que se administra el fármaco y no pone en peligro la vida del paciente, la somnolencia por antihistamínicos H1, la bradicardia por atenolol o la tos por captopril son ejemplos de efecto colateral. El efecto tóxico depende de la dosis, el tiempo de exposición, ciertos estados patológicos y la susceptibilidad del enfermo, se produce por el medicamento o sus metabolitos y puede poner en peligro la vida del paciente o dejar secuelas, nefrotoxicidad por aminoglucósidos o gingivitis hipertrófica por difenilhidantoína, son ejemplos de esta reacción. El efecto teratógeno aparece por la administración de medicamentos a la mujer embarazada siendo el periodo más crítico las primeras 8 semanas de la gestación aunque el peligro persiste a todo lo largo del embarazo, el fenómeno de rebote es una reacción común en la que se produce una reactivación del cuadro clínico inicial cuando algunos fármacos se suspenden bruscamente como las convulsiones por la suspensión brusca de antiepilépticos, la farmacodependencia es un trastorno conductual, en el cual como resultado de los efectos biológicos de una determinada sustancia, una persona tiene disminuido el control sobre su consumo, es uno de los inconvenientes en el uso de las drogas que alteran el estado de animo y la afectividad, por su acción sobre el SNC, el paciente se habitúa y no puede dejar de usarla, como ocurre con el alcohol, la cocaína, el LSD, el tabaco etc. Cuando dos o más medicamentos se administran simultáneamente a un paciente pueden aparecer fenómenos de sinergismo o antagonismo, o puede provocar alteraciones en la farmacocinética con el consiguiente aumento o disminución del efecto son las interacciones medicamentosa. Estas pueden ser de carácter físico – químico cuando se mezclan los fármacos para ser administrados y estos precipitan, cambian de coloración o se inactivan como la infusión de Fenitoína en Dextrosa al 5%, las interacciones farmacocinéticas provoca cambios en la absorción, distribución, metabolismo o excreción del fármaco y en la farmacodinamia la interacción ocurre entre agonista y antagonistas en los receptores específicos o sistemas fisiológicos como el simpático y parasimpático que tienen muchos efectos opuestos en nuestro organismo. Podemos definir a la Farmacovigilancia como el conjunto de actividades destinadas a identificar y valorar los efectos del uso agudo y crónico de los tratamientos farmacológicos en poblaciones o subgrupos de estas y cuyo objetivo fundamental es identificar, definir, cuantificar y prevenir las reacciones por medicamentos, para poder generar alertas e informar a la comunidad científica y a la población cualquier aspecto relacionado con la seguridad de un fármaco y poder tomar medidas relacionadas con cambios de dosis o retiro del mercado de un medicamento que provoca reacciones adversas por encima de lo aceptado para poder ser comercializado. Los métodos más utilizados por la farmacovigilancia para detectar reacciones adversas son el reporte de casos, el reporte espontáneo de sospecha de reacción adversa que es el más importante en la detección precoz de reacciones y para lo cual debe salir preparado el médico integral comunitario, los estudios de caso-control, los de cohorte, los ensayos clínicos, los reportes de morbi-mortalidad y la supervisión intensiva de pacientes hospitalizados. Para seleccionar un fármaco el médico debe basarse en los criterios de eficacia, seguridad, conveniencia y costo, en ese orden. La eficacia y seguridad de un medicamento deben ser demostradas durante las diferentes fases del ensayo clínico hasta su registro en la fase III, poniendo de manifiesto la relación beneficio/riesgo que el médico debe tener siempre en cuenta al prescribir un medicamento, la conveniencia esta dada por lo que sea mas cómodo para el paciente, la frecuencia de administración, la vía de administración, etc; por último el costo es algo que debe tenerse en cuenta para afectar lo menos posible la economía del paciente y del sistema de salud. El uso irracional de medicamentos implica no utilizar el fármaco de elección a la dosis correcta y por el tiempo establecido, en esto pueden contribuir varios factores como: la formación inadecuada en farmacología clínica, la falta de educación continuada, la presión ejercida por el paciente, dudas en el diagnóstico, las consultas numerosas o la interpretación personal de la literatura científica, es importante destacar que la experiencia personal de un médico no debe prevalecer por encima de las evidencias científicas pues en ocasiones la experiencia personal puede ser la repetición de los errores que se pueden cometer a diario. La prescripción irracional puede ser: incorrecta cuando se prescribe el medicamento inadecuado como los antimicrobianos en infecciones virales, excesiva cuando se utilizan altas dosis o se prescriben fármacos por largos periodos de tiempo como los ansiolíticos o los antidepresivos, la submedicación , cuando se utilizan bajas dosis o no se prescriben los fármacos necesarios, como la tardanza en prescribir analgésicos en el dolor crónico, o múltiple cuando se prescriben innecesariamente dosis fijas de un medicamento, por ejemplo muchas veces se comercializan varios antiparasitarios en un solo producto y la mayoría de las veces el paciente no tiene un poli parasitismo intestinal. La prescripción de fármacos en el embarazo puede traer como consecuencia efectos teratogénicos a los que se expone el feto cuando se administran medicamentos a la madre fundamentalmente durante el primer trimestre, efectos sobre el desarrollo fetal que pueden producirse durante todo el embarazo y efectos secundarios que se producen sobre todo en el último trimestre. Durante el embarazo se producen cambios fisiológicos en la madre que alteran la farmacocinética y farmacodinamia de los medicamentos, estos cambios se producen de manera gradual se acentúan en el tercer trimestre y vuelven a la normalidad pocas semanas después del parto, produciendo cambios en la absorción, distribución, metabolismo y excreción de algunos medicamentos como las beta lactámicos, aminoglucósidos, propranolol, diazepan, salbutamol, teofilina, las alteraciones en la unión al receptor son menos conocidas y se ha descrito una disminución del efecto de la heparina. También se pueden ver alteraciones en la dinámica del parto con el uso de algunos medicamentos como los AINEs que si se utilizan durante las últimas semanas de la gestación disminuyen las contracciones uterinas, estos fármacos han sido clasificados por la FDA como categoría D en el embarazo por lo que su uso siempre debe ser valorado. La Farmacoepidemiología es considerada una de las ramas más jóvenes de la Farmacología y se ocupa de estudiar el impacto del uso de los medicamentos en la población con el objetivo de alcanzar el uso racional de los medicamentos, es decir el fármaco de elección, a dosis terapéutica y por el menor tiempo posible. Surge aparejado al desarrollo creciente de la industria farmacéutica durante el siglo XX y las tragedias que produjeron el consumo de fármacos sin los estudios necesarios como las muertes por un jarabe de sulfas en los EE. UU o las malformaciones de la talidomida que marcaron pautas en la farmacovigilancia, además por la diferencia entre la eficacia y efectividad de un medicamento, debemos señalar que la eficacia es la capacidad que tiene el fármaco de producir un resultado beneficioso en las condiciones controladas del ensayo clínico, mientras que la efectividad se ve en la práctica médica habitual sin restricciones de uso, por lo general la efectividad de un medicamento siempre será menor que la eficacia. Por último veremos los estudios que utiliza la farmacoepidemiología para demostrar la causalidad entre la aparición de un efecto y la causa que lo provoca y que se han expuesto en forma ascendente de menor a mayor especificidad. Así tenemos el reporte del caso clínico, la serie de casos, los estudios transversales, el caso-control, el estudio de cohorte, el ensayo clínico como patrón de oro de la causalidad y los metanálisis que se hacen a partir de ensayos clínicos con metodología similar para aunar criterios con relación a un fármaco o a una estrategia terapéutica.