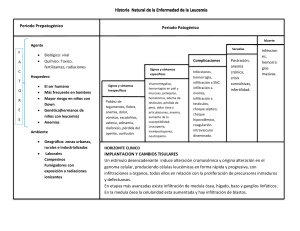
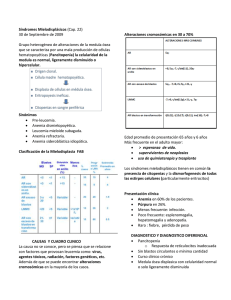
Insuficiencia renal aguda Caso clínico MUJER, 28 años MC= Vómitos y diarrea. Fiebre y oliguria EF PA 80/60 P. 120/m o Mucosas y piel secas. o Dolor abdominal difuso. o RHA aumentados Lab= Hto 45 GB 12.300 o N 85 % PCR 24 o Na 135; K 3,6; Clor 108. o O: D 1.025 Uremia 75 Creat: 1,6 Hipovolemia Leucocitosis Neutrofilia Gastroenteritis infecciosa PRERRENAL Hidratarle y expandir su volumen HOMBRE, 72 años MC= Nauseas. Dolor en hipogastrio Anuria de 12 horas EF PA 180/90 P. 120/m o Mucosas y piel normales. o Dolor en hipogastrio. Lab= Hto 38 GB 8.500 o N 85 % PCR <6 o Na 145; K 5,0; Clor 108. O: D 1.020 Uremia 100 Creat: 1,8 Hiperplasia prostática que provoco una retención aguda de orina ↑ de la capsula de Bowman ↑ de los elementos nitrógenos POSTRRENAL No hay daño estructural MUJER, 45 años Sometida a aborto provocado por empírica Ingresa confusa, febril, con secreción purulenta por vagina, no orina desde 24 horas atrás. EF: PA 70/50 P: 140/m Filiforme. FR: 28/m o Hipotérmica o Deshidratación severa o Dolor abdominal difuso con signos de irritación peritoneal. o Anemia severa o LAB. Hgb6,5 GB 24.000 N 90 % PH 7,33 Na 145 K 6,8 Urea 220 Creatinina 5,6 Cuadro séptico Hipotensión severa Hipoperfusión, toxicidad, daño isquémico por anemia NTA. IRA RENAL grave Definición Fracaso brusco e importante de las funciones del riñón Incapacidad de realizar funciones de: o Eliminación de productos de desecho, que se evalúa con la urea y la creatinina. o Mantener constante el medio interno. Eliminacion de radicales ácidos. o Endócrinas Incapacidad renal para o Manejar el agua y los electrolitos. o Acumulación de productos derivados del metabolismo de las proteínas: Lleva a la Uremia y Oliguria Cuadro grave 5 % de hospitalizaciones 30 % de ingresos a UTI con IRA 25 % de hospitalizados desarrollan IRA La disminución o suspensión súbita en la filtración glomerular acompañada de la retención de desechos nitrogenados y alteraciones en el equilibrio hídrico, ácido base y metabólico, con grado variable en el volumen urinario. IRA. Clasificación IRA prerrenal o funcional IRA renal o parenquimatosa IRA postrrenal u obstructiva IRA. Síntomas Náuseas y vómitos Malestar HTA, por la retención e electroliticos Frote, derrame y taponamiento pericárdicos Arritmias Hemorragias Encefalopatía Oliguria IRA Prerrenal Llamada “funcional” ya que no existe lesión anatómica ni bioquímica Consecuencia de la caída de la presión sanguínea < 60 mmHg Múltiples causas IRA Prerrenal. Etiología 1. Hipovolemia o Hemorragia o Deshidratación. Tercer espacio 2. Bajo gasto cardiaco o Taponamiento o IAM. Arritmias graves 3. Vasodilatación sistémica o Shock séptico 4. Vasoconstricción renal o Preeclampsia. Síndrome Hepatorrenal. 5. Alteración de la autorregulación del flujo renal o AINES, IECA, ARA II. La autoregulacion intrínseca de la arteriola aferente y eferente que se ve severamente afectado por el uso de antiinflamatorios o inhibidores de la enzima convertidora de los antagonistas del receptor ARAII, que puede también provocar hipoperfusión glomerular y caída de la producción de orina. Estas drogas cuando hay creatinina de 2,5- 2,6, esta droga se suspenden, porque pueden empeorar la situación. 1. Hipovolemia disminución “contenido” 2. Bajo GC FE del VI insuficiente (postcarga) Precarga disminuida Disminución PA 3. Vasodilatación: aumento del “continente” Disminución presión intraglomerular Cese Filtración 4. Vasoconstricción renal Preferente arteriola aferente Disminución flujo glomerular Caída presión hidrostática Cese Filtración Fracaso Renal Fracaso renal 5. Pérdida autorregulación ↓ PA: apertura aferente. Inhibida por AINES + AII cierre eferente. Inhibida IECA. ARA II IRA Prerrenal. Clínica Común para todas las etiologías Hipotensión arterial El riñón está intacto, está sano, entonces por todos los medios trata de retener líquido, retener agua. Presión venosa disminuida, excepto en los secundarios a bajo GC Oliguria con orinas concentradas Disparidad en los incrementos de Urea y Creatinina >20:1 Hiperpotasemia Osmolaridad de la orina ↑ FeNa(%): <1% Densidad alta IRA Parenquimatosa Lesión anatómica del parénquima renal Túbulo-Intersticio. Glomérulos. Vasos Casi todas las enfermedades renales la pueden producir La lesión clásica es la NTA (Necrosis Tubular Aguda) IRA RENAL Necrosis tubular aguda o Toxinas, Antb, contrastes, AINES, RM, MM, hemólisis o Isquemia la más frecuente Glomerulonefritis aguda o Complejos inmunitarios o Pauciinmunitaria (ANCA +) o Antimembrana basal glomerular Nefritis intersticial aguda o Fármacos o Infecciones o Inmunitarias IRA Parenquimatosa. NTA. Etiología 1. Isquémica o Todas las causas del prerrenal si perduran 2. Tóxica Endógena: Hb; MHb; Bilirrubina… Exógenas o Yatrógenas: Aminoglucósidos, cefalosporinas.. o Industriales: etilenglicol, Pb, As… 3. Inmunoalérgica o Medicamentosas o Infecciosas: virus Hanta, leptospirosis,… Patogenia Anuria < 50 ml/día Oliguria < 400 ml/día Puede presentarse: Sin oliguria Con oliguria Inicial u oligúrica (7-10días) o Oliguria y retención nitrogenada progresiva Mantenimiento o fase diurética o Diuresis osmótica y retención nitrogenada constante. Recuperación o Aumento progresivo de FG y descenso de la retención de productos nitrogenados. IRA Parenquimatosa. NTA. Clínica Fase inicial: Dura entre horas a varios días, se limita hasta el momento que aparece la uremia Fase de uremia: Hay oliguria, aumento de nitrogenados y alteraciones metabólicas. Duración aproximada: 10 a 20 días Fase de recuperación: Comienza cuando se recupera paulatinamente la diuresis y disminuyen los nitrogenados en sangre Fases Fase de instauración Fase de oligoanuria Fase poliúrica ineficaz Fase de restauración Se presenta con: ↑ de las concentraciones de urea Sobrecarga circulatoria ↑ de la creatinina Insuficiencia cardiaca o 1 – 1,5 mg/100 ml al día Edema pulmonar Anorexia HTA Nauseas Edema cerebral Vómitos Aumento del potasio extracelular Alteraciones del estado de conciencia Arritmias Trastornos hematológicos Muerte Oliguria Se observa: Macroscópicamente aumentados de tamaño La unión cortico-medular se observa congestiva y pálida Glomérulos normales Túbulos congestivos Edema intersticial Osmolaridad de la orina disminuida hiperpotasemico FeNa: >1% Índice IRA prerrenal IRA Renal Osm orina >500 mOs <350 mOs Urea u / Urea p >10 <5 Cr u / Cr p >40 <20 Na+ orina <10 mEq >30 mEq EF de Na+ (1) <1% >1% IRA Postrrenal. Obstructiva Obstrucción de los dos uréteres o uretra Riñón único funcionante La obstrucción contrarresta la presión de filtración Disminución del Flujo Sanguíneo Renal y lesión de las células tubulares con edema e infiltrado intersticial Si persiste en el tiempo puede evolucionar a NTA Anuria Na alto Osmolaridad urinaria baja NECROSIS CORTICAL ANURIA Diagnóstico diferencial Anamnesis o Antecedentes de nefropatía, uropatía… o Indagar tóxicos, medicamentos…… Exploración física o Estado de hidratación o Estado cardiovascular…. Ecografía o Por el tamaño renal diferencia un cuadro agudo de crónico a excepción de la amiloidosis o Valorar presencia de hidronefrosis: confirma obstrucción, su ausencia no la descarta Gammagrafía o Detecta procesos vasculares (obstrucción arteria renal) TRATAMIENTO de la IRA Controlar las fases oligúricas volviéndolas no oligúricas Mantener los niveles de volumen adecuados Controlar la hiperpotasemia Controlar la acidosis metabólica Pronóstico Siempre es malo. Mortalidad del 40 al 50% Causas de mortalidad: o Septicemia o Alteraciones cardiacas o Insuficiencia respiratoria o Hemorragias o Hiperpotasemia Tumores renales Adenocarcinoma renal (también conocido como carcinoma de células renales, gran simulador, tumor del internista) Corresponde a la gran mayoría de canceres de origen renal 2-3% de todos los cánceres A nivel urológico ocupa el tercer lugar después del cáncer de próstata y de vejiga Es el 90% de tumores malignos del riñón Más frecuentes en hombres que en mujeres (aproximadamente el doble) Mayor frecuencia entre los 50 y 70 años de edad. Factores de riesgo: tabaquismo, HTA, DM, Sx metabólico, sedentarismo, sobrepeso u obesidad, enfermedad renal previa, factores hormonales (más en mujeres que nulíparas), hábitos dietéticos, consumo moderado o escaso de alcohol (se relaciona de forma inversa con el cáncer renal), etc. Hallazgos incidentales: en aumento, generalmente en estadios tempranos cuando aún no presenta síntomas. Se lo divide en tres formas: 1. Sintomático: triada clásica (masa abdominal, hematuria (60%) y dolor) síntomas constitucionales característicos de las neoplasias como pérdida de peso, sudoración nocturna, fiebre, astenia, anorexia, etc. (la triada clásica se encuentra en la mayoría de las veces ausente) 2. Asintomático: más del 80% se detecta con la ecografía pues posee buena sensibilidad. 3. De hallazgo Incidental: sobrevida media más del doble que poseen la triada clásica. Clínica: hematuria, dolor en flanco, masa abdominal, hematoma perirenal, edema en MI, varicocele, si hay metástasis (dolor óseo, tos persistente, adenopatías cervicales, etc) Diagnóstico: Buena anamnesis (muy importante, antes que nada) Diagnóstico por imagen: Ecografía (diferencia quistes), TAC (estadiaje clínico, gold standard cuando hay sospecha), RNM en casos seleccionados con resultados comparables a la TAC. Análisis de sangre: anemia, aumento de fosfatasa alcalina, aumento de calcio (señal de que la enfermedad está avanzada). Pruebas de función renal: urea y creatinina, alteraciones del filtrado Clasificación de TNM: tumor, nódulos, metástasis (importante para el tto y diagnóstico) Limitado solo al parénquima: T1 (< 7 cm); T2 (>7cm) Localmente avanzado: T3a (grasa perineal), T3b (trombo tumoral vena renal-cava infra diafragmática) T3c (trombo tumoral en cava supra diafragmática) Afectación de órganos vecinos: T4 (cuando se atraviesa la fascia de Gerota) N0: sin evidencia de afectación ganglionar N1: un ganglio afectado N2: más de un ganglio M0: sin evidencia de metástasis M1: metástasis Tipos histológicos: Adenocarcinoma típico Carcinoma papilar: menor estadio y pronóstico, familiar y IRC Carcinoma cromófobo: origen túbulos colectores-medular, buen pronóstico Carcinoma de conductos colectores: medular, papilar/tubular, pronóstico malo Carcinoma medular: variante agresiva Ca colectores, jóvenes, negros, sickle cell Carcinoma sarcomatoide: tumores > 10 cm, células fusiformes, mal pronóstico, spv 8 años. Gradación histológica del adenocarcinoma renal: G1, G2, G3, G4 Pronóstico: factores clínicos: bajo peso, anemia, aumento de VSG, hipercalcemia, aumento de FA Estadio patológico: T1-2 (70% a 5 años), N1-2 (30% a 5 años) y M1 (0% a 5 años) Tipo histológico: grado nuclear Diagnóstico diferencial con Oncocitoma renal: 5-7% de los tumores renales Tumor sólido benigno más común. La clínica y radiología idéntica Histología: presencia de oncocitos (células grandes con citoplasma eosinófilo), no hay mitosis, se encuentra en el túbulo distal. Se debe a la pérdida y/o la translocación del cromosoma 11q13 Diagnóstico diferencial con adenocarcinoma cromófobo variante eosinófilo Posee buen pronóstico, excepcionalmente hace metástasis. El tratamiento suele ser la nefrectomía. Angiomiolipoma: Hamartoma renal, pero pos sus características histológicas es una neoplasia. Tumor benigno Poco frecuente Se puede diagnosticar con ECO, TAC (por su gran componente graso) Tiene tres componentes: proliferación de vasos sanguíneos, musculo liso y tejido adiposo) Más frecuente en mujeres Aparece en la pubertad Asociada a la esclerosis tuberculosa (enfermedad bourneville-pringle) Adenoma sebáceo en cara Manifestaciones neurológicas: convulsiones, retraso mental, etc. La principal complicación es el sangrado (hemorragia retroperitoneal masiva) Son tumores renales múltiples y bilaterales Tumor de wilms Neoplasia relativamente rara Pero en niños 5% de todos los tumores del tracto urinario Habitualmente es esporádico (no 11p13) Signos y síntomas: masa abdominal palpable (75%) indoloro, dolor abdominal (28%), fiebre (22%), náuseas, vómitos, hematuria (18%), hipertensión (26%), etc. Diagnóstico: masa palpable, TAC, RMN, ECO Pronóstico bueno en niños Diagnóstico alrededor de los 3 años (75% antes de los 5 años) y (90% antes de los 7 años) Igual incidencia entre sexos Hallazgos raros: aniridia (1%), hemihipertrofia (2%) Confirmación radiológica: ECO y TAC Estadiaje NWTS: ESTADIO 1 Tumor limitado a riñón ESTADIO 2 Se extiende más allá del riñón pero es resecado, aun puede extirparse quirúrgicamente. ESTADIO 3 Tumor residual en abdomen ganglios +, afectación peritoneal, margen + o irresecable ESTADIO 4 Metástasis a distancia, pulmón, hígado, hueso, cerebro, etc. ESTADIO 5 Bilateral Repaso de Fisiología de los GR - - - - - - - - - Se encargan del transporte del oxig a los tejidos y de retirar el dióxido de carbono hacia los pulmones para el intercambio gaseoso. Función secundaria liberación de oxido nítrico para vasodilatación de los capilares. Como es la célula? Bicóncava, sin núcleo, no tienen tp organelas como mitocondrias pq si tuvieran esto consumirían oxig metabólicamente. Son células muy básicas, exclusivamente diseñadas para el transporte de oxigeno. Tiempo de vida de 120 días. Hemoglobina es la estructura que capta el oxígeno y el anhídrido carbónico para el transporte, estructura metida dentro del GR formada por varias moléculas proteicas unidas a moléculas de hierro, que deben estar presentes para q se produzca esta unión con el oxigeno Los GR sufren el proceso de destrucción en el bazo, tb en el hígado, periferia, capilares. La membrana de los GR son flexibles y se van adaptando a los capilares, esta capacidad se va perdiendo al pasar los días, y al llegar a los 120 dias ya pierden esa flexibilidad y se rompen y se destruyen y la hemoglobina sufre un proceso metabólico de reciclado y el hierro tb, para formar nuevos GR y tb la bilirrubina. Proceso de transformación de Hb a bilirrubina, bilis q se produce en el hígado y se almacena en la vesícula, para digestión de grasas sirve esto. La fabrica de producción de todas esta series de células está en la medula ósea, sobre todo de huesos largos. Para producirse los GR se necesita materia prima, ej el reciclado de GR, vit b12, acido fólico y Fe. El Fe una parte se recicla de GR y el resto viene de nuestra alimentación. Una de las causas de anemias es el déficit de hierro, las anemias ferropénicas se puede dar por múltiples factores (falla en la dieta, problemas metabólicos de Fe, problemas de absorción, déficit de factor intrínseco, alterac genéticas que dificultan el transporte de Fe, problema de entrada del Fe transportado por las proteínas a la medula ósea). Los GR se destruyen por un proceso fisiológico, pero tb pueden aparecer procesos fisiopatológicos, por ej: el bazo esta muy agrandado y empiezan a destruirse más precozmente los globulos rojos, cuerpos extraños en el torrente de circulatorio como una protesis valvular aortica, estas antes rompían y producían hemolisis. En las TVP, se suele poner una sombrilla en la vena cava para evitar migración de coágulos y eso causaba hemolisis. En vascular periférica se utilizan prótesis muy a menudo, y producen hemolisis por contacto con la sangre. Proceso MECANICO. Virus que pueden producir hemolisis, mas bien los ANTICUERPOS. (la superficie membranosa de los GR están llenos de glicoproteínas, lq da la tipificación, factor, etc), estos antígenos q están en la sup de membrana a veces son reconocidos por anticuerpos q se producen contra virus o bacterias En ciertas circunstancias patológicas autoinmunes, sin virus o bacterias, se pueden producir anticuerpos q pueden destruir GR… se produce ANEMIA. Entre las sustancias que estimulan la transformación de eritrocitos, y reticulocitos. Esta la eritropoyetina, que es una hormona que se produce en el riñon. En ptes renales crónicos hay déficit de eritropoyetina: anemia Causas de anemia: sangrados, deficiencia de hierro, deficiencia de eritropoyetina, déficit de ac fólico, def de vit b12 En ptes con anemia, tres procesos: - Déficit de producción: no se producen los GR Aumento de la destrucción de los GR y procesos hemolíticos Perdidas, sangrados, agudos o crónicos: cortes, lesión, ulcera grande, divertículos sangrantes del intestino son agudas. Perdidas crónicas por ej en la menstruación, hay mujeres que tienen un sangrado mayor que puede llevar a anemia crónica, por gastritis, ulceras. ANEMIAS Los criterios clásicos de la OMS se establecen en límites de 13 g/dl para varones y 12 g/dl para mujeres Hablando de Hb: Grados de anemia normal, medio, moderada y severa - Severa: 6,5 y 8 g/dl de Hb Moderada: 8 -9,5 A quién se le hace una transfusión? A partir de 7,5 g/dl de Hb, salvo q el pte tenga comorbilidades, como cardiopatía isquémica, antecedente de infarto, ptes renales crónicos, insuf cardiaca, en estos ptes en torno a 10 g/dl Hemoglobina menor de: - 11 g/dl en niños menores de 6 años 12 g/dl en niños de 6-14 años 12 g/dl en mujeres 11 g/dl en mujeres 13 g/dl en hombres Ojo: Esto tiene variaciones, personas q viven en grandes alturas tienen mayor concentración de Hb en la sangre, pq la posibilidad de oxigeno es más baja entonces se producen mas GR y Hb Trastornos que pueden dar falsa concentración de hemoglobina Aumento del volumen plasmático (hemodilución) 1- Embarazo 2- Anemias carenciales Si a un pte le ponemos un litro de suero, la sangre se hemodiluye y las concentraciones de Hb bajan Disminución del volumen plasmático (hemoconcentración) 1- Deshidratación 2- Síndromes diarreicos Los niveles de Hb y hematocrito mas aumentados Disminución del volumen plasmático y volemia eritrocitaria 1- Hemorragia aguda 2- Neoplasias Mecanismos de adaptación de la anemia - Estimulo eritropoyetico, se da en las alturas, el riñón produce más cantidades de eritropoyetina y estimula a la medula ósea. Algunos deportistas hacen esto para tener mas condición. - - Mejor aprovechamiento de la Hb disponible : curva de asociación entre oxig y Hb, eso aumenta este porcentaje de saturación de Hb, cuando hay déficit de algún motivo, hay mayor captación de oxg por la hb Adaptación del sistema cardiocirculatorio: en anemias hay déficit de oxig y si hay déficit de transporte, el corazón empieza a bombear mas rápido, taquicardia. Clasificación -Primero los ya mencionados (produc, sangrado..) - Anemias agudas por sangrado o perdidas agudas o hemolisis agudas, y anemias crónicas Clasif morfológica: Anemia - Normocítica: VCM 80-100 fl Macrocítica: VCM mayor de 100 fl Microcítica: VCM menor de 80 fl Y de acuerdo a la coloración: hipercrómicos (raro), normocrómicos o hipocrómicos. Lo mas frecuente en anemia ferropenica, q gralmente son crónicas, se tiene anemia microcitica tamaño pequeño e hipocrómicas (coloración pálida). Por qué el déficit de hierro habla de proceso crónico? Pq nuestras reservas de hierro, sobre todo al nivel del hígado son muy altas, entonces para tener deficiencia debe pasar semana o meses para consumir todas esas reservas, solo luego se da la producción disminuida y estas características de GR. Proceso lento no rápido. Se debe considerar primero q nada restituir esas reservas de hierro, administrarse de forma prolongada, oral o endovenosa Otra forma de evaluación: Por el recuento de reticulocitos De proeritrocitos, que están en la med ósea y son cél nucleadas, van transformándose en reticulocitos, primera sin núcleo, y precede al eritrocito. Estos reticulocitos llegan a salir a la sangre (1% - 2,5% en sangre periférica en frotis normal) Recuento de reticulocitos para saber si es perdida de sangre o prod de glóbulos rojos en medula ósea: - Reticulocitos bajos: producción de GR, la medula ósea tiene el problema. Reticulocitos altos: hemolisis, destrucción o hemorragia. Medula reacciona y produce GR y saca a la sangre periférica GR muy jóvenes, reticulocitos igual o mayor a 2,5%. De 10 a 15%. Obs: Hipoproliferativa: - - Medula ósea infiltrada por leucemia: GB empiezan a invadir toda la medula ósea y no le da espacio a las plaquetas y GR: hiproplif de eritrocitos (esto se puede dar por GB aumentados o plaquetas aumentadas por hiperplaquetosis) Infiltración o fibrosis de la medo sea: mielofibrosis, general disminución de las tres líneas celulares: anemias normociticas normocromicas con eritrocitos bajos Diagnóstico : Fácil, se le nota a un pte que esta anémico. - - ANAMNESIS Y EXPLORACIÓN FÍSICA En adultos jóvenes la forma más frecuente es la que se presenta en mujeres como consecuencia de la menstruación y es anemia ferropénica (porque con el sangrado se va perdiendo Fe y las reservas de Fe se van consumiendo y aparece la anemia ferropénica) De hecho las mujeres tienen niveles de Hb menor q el varón: 12 g/dl Manifestaciones clínicas 1- Cardiovasculares y respiratorios - - - - Pueden extenderse desde disnea de esfuerzo, taquicardia (compensatorios, taquicardia es reacción del corazón para tratar de bombear más rápido la sangre con menor concentración de Hb, y el pte con baja Hb se cansa más rápido) Hipotensión postural (al levantarse bruscamente, baja la presión mas de 10mmHg en menos de 6min) infarto del miocardio (por eso un pte con antecedentes de cardioaptia isquémica o diabético, es conveniente mantener niveles de Hb de 10 o mas) Claudicación intermitente (los ptes con ateroesclerosis vasculares periféricas, con arterias de los MI dañadas por procesos de ateroesclerosis o obstructivos, al caminar tienen un dolor intenso en la pantorrilla, camina dos tres cuadras y el pte debe quedarse un tiempo a descansar hasta q se recupera y vuelve a caminar, “miradores de vidrieras”), edemas, soplos sistólicos, incluso cuadros sincopales (sobretodo en personas con anemias agudas, perdida muy brusca de Hb, ptes propensos a producir perdida del estado de conciencia sobretodo en cambios e posición brusca, disminución del flujo sanguíneo cerebral. En ptes que hacen anemias crónicas, el cuerpo va compensando, por ej un pte que dps de hemorragia tuvo un descenso de Hb importante, va a tener taquicardia, pero un pte con perdica crónica, renal crónico por ej, déficit de eritropoyetina maneja sicfras bajas de Hb, pero no tiene taquicardia, de 70 a 75 lat/min) Una dilatación cardiaca está casi siempre presente en pacientes politransfundidos 2- Neurológicos: - Cefaleas, acúfenos, vértigo, mareo, pérdida de concentración, astenia, menor tolerancia al frío Síntomas neurológicos que son más específicos de la anemia por deficiencia de Vit B12 los cuales comienzan con parestesias en dedos de manos y pies, juntos con alteraciones en la sensibilidad vibratoria y propioceptiva, progresando si no se trata a ataxia espástica (dificultades para mantenerse parado, sobre todo la coordinación de los movimientos), por desmielinización de los cordones laterales y dorsales de la médula espinal. 3 - Cutáneos, mucosas y faneras - Es típica la palidez de piel y mucosas La piel y mucosas tienen un alto requerimiento de hierro debido al alto recambio y crecimiento Glositis la cual se caracteriza por una lengua enrojecida, lisa, brillante y dolorosa debido al adelgazamiento del epitelio Rágades (estomatitis angular): pequeñas llagas en la comisura de los labios Atrofia gástrica También piel seca, uñas frágiles, y caída de cabello Manifestaciones específicas: 1. 2. Deficiencia de acido fólico: lengua cuadriculada y con superficie lisa Deficiencia de hierro: llagas en la comisura labial 4-Gastrointestinales: - - Anorexia, náuseas, estreñimiento o diarrea La atrofia gástrica estará también presente en la anemia perniciosa En caso de coexistir la glositis, úlcera o inflamación de la boca, disfagia y déficit de hierro estaremos ante el llamado Sx de Plummer Vinson - En ptes con talasemia (destrucción congénita de los glóbulos rojos, alteración de la Hb) una hepatoesplenomegalia, por hematopoyesis extramedular (La hematopoyesis cuando hay problemas de anemias crónicas, trata de producirse en el bazo y en el hígado) Las anemias hemolíticas inmunes y no inmunes (por causas mecánicas) pueden presentar litiasis vesicular por cálculos de bilirrubinato cálcico Obs: En las alturas la digestión es mucho más lenta y difícil, es lo mismo que estar con anemia 5-Genitourinarios: - Puede presentar amenorrea. Pérdida de la libido e impotencia Otros: - Un síntoma peculiar y típico de la deficiencia de hierro severa es la pica, coiloniquia (uñas tienen una forma invertida, convexa, en forma de cuchara y son frágiles) También en caso de anemia ferropénica veremos escleras azules Ante anormalidades óseas pensar en talasemia como resultado de expansión e hipertrofia medular Dolor en anemia de células falciformes Obs: Talasemia tb causa Hb dismorfa o amorfa: se destruyen precozmente los GR (típico en zonas del mediterraneo) Eritrocitos: VN: 4500000-5700000 Ojo con este esquema de VCM: siempre sale dice el profe Obs: Anemia de enf crónica: trastornos oncológicos reumatológicos ANEMIA FERROPENICA Definición: estado en el cual el contenido de hierro en el cuerpo es menor de lo normal Causas: Enfermedad celiaca: una de las manifestaciones es la ANEMIA. Si hay un pte que tiene anemia, problemas dermatológicos, caída del cabello: hacer prueba de enf celiaca Colestriramina: para tto de las dislipidemias Hipermenorrea: perdidas de sangre con la menstruación prolongada, en úteros miomatosos o en úteros que tienen engrosamiento endometrial: puede ser cáncer de endometrio Hemoglobinuria paroxística nocturna: en traumatismos, un ej puede ser en grandes maratones y una persona que usa un calzado deportivo inapropiado. Manifestaciones clínicas - Palidez Escleróticas azules Irritabilidad Anorexia Taquicardia Dilatación cardiaca Soplos sistólicos Déficit neurológicos e intelectuales Estomatisis: en cualquier lugar de la mucosa oral pero sobre todo en las comisuras labiales Hallazgos de laboratorio - Eritrocitos microcíticos- hipocrómicos. VCM menor de 80 fl y por consiguiente disminución de HCM Disminución de la concentración de hierro sérico: Todo el panel de hierro se debe pedir, hierro, ferritina, transferrina, porcentaje de saturación de la transferrina Aumento de la capacidad de fijación del hierro Ferritina sérica baja. Menor de 10 Ug/lt en muejres y menor de 20 en hombres Disminución del hierro en médula ósea Frotis de sangre: GR bien pálidos y decolorados Aspirado de medula ósea: GR color más apagado Tto (no sale): Tratamiento oral: - Sulfato ferroso: 325 mg v. o 3 v/d durante 6 meses Efectos colaterales: estreñimiento, cólicos, diarrea, náuseas - El gluconato y fumarato ferroso son alternativas bien toleradas Tratamiento parenteral: - Útil en ptes con escasa absorción, requerimientos muy altos, intolerancia a los preparados orales. Preparados existentes: hierro dextrano, gluconato de sodio férrico ANEMIA MEGALOBLÁSTICA Definición: Desórdenes causados por la alteración en la síntesis de DNA, caraterizado por la presencia de células megaloblásticas La más común es la DEFICIENCIA DE FOLATO Y LA DEFICIENCIA DE CIANOCOBALAMINA Clasificación: Obs: Ingesta disminuida: puede ser tb por aumento de requerimiento en embarazo Causada por drogas: Obs: Zidovudina para tto de VIH Deficiencia de ácido fólico. Manifestaciones clínicas: - Malestar general Glositis Pérdida de peso Diarrea Hipocalcemia, osteomalacia y osteroporosis pueden ocurrir Def de ac fólico. Hallazgos específicos: - Historia y lab que demuestran deficiencia de folato. VCM: MAYOR A 100 Ausencia de las manifestaciones neurológicas de déficit de cobalamina Como discernir si es por déficit de folato o de VitB12?: cuando hay def de vitb12 aparecen síntomas neurológicos como la ataxia o de hormigueo, parestesia. - Respuesta completa al tto con folato Exámenes de laboratorio: - Todas las líneas se afectan Eritrocitos aumentados en tamaño y forma Reticulocitos disminuidos VCM: 100 a 150 fl Tratamiento (no sale): 12- Deficiencia de cobalamina Cianocobalamina parenteral o hidroxocobalamina 1 a 5 mg 1mg 1M diario por 2 semanas, luego cada semana hasta que el Ht sea normal Luego 1 mg/m de por vida Deficiencia de ac fólico 1 mg/d vo hasta corregir la deficiencia - En ptes con sx de malabsorción se pueden requerir hasta 5mg/d ANEMIAS HEMOLITICAS - Anomalías de membrana: GR tienen otras formas de las que deberían tener Por defecto en la forma de los glóbulos rojos: esferocitosis, eliptosis, anomalías de membrana. Por defecto estructural o metabolico del glóbulo rojo (enzimas y hemoglobina). Secundarias a alteraciones del medio que rodea los hematíes. Autoinmunes Eritroblastosis fetal Causas mecánicas Agente tóxicos Hemoglobinopatías Defectos de la Hb (talasemia) Hemolisis microangiopatica: por lesiones vasculares, capilares Algunas de las causas de AH son: Infecciones, ciertos medicamentos, trastornos autoinmunitarios y trastornos hereditarios. Síntomas - Escalofríos Fatiga Palidez Dificultad respiraroria Frecuencia cardiaca rápida Ictericia: un aumento de la destrucción de GR puede producir un aumento de bilirrubina, que se manifiesta como ictericia Agrandamiento del bazo Laboratorio - Niveles elevados de bilirrubina indirecta Haptoglobulina sérica baja Hemoglobina en la orina Hemosiderina en la orina Aumento del urobilinógeno urinario y fecal Conteo de reticulocitos absoluto elevado Conteo de glóbulos rojos y hemoglobina bajo Tratamiento (no sale) Hay que tener cuidado con las transfusiones pq en algunas casos es como echar leña al fuego - El tto depende del tipo y la causa de la anemia hemolítica. Se puede usar ácido fólico, suplementos de hierro y corticoesteroides. Es posible que en casos de emergencia se necesiten transfusiones sanguíneas Complicaciones Las complicaciones varían de acuerdo con el tipo específico de anemia hemolítica. La anemia severa puede producir colapso cardiovascular y agravar una cardiopatía, una enfermedad pulmonar o una enfermedad cerebrovascular preexistentes LEUCEMIAS Enfermedad neoplásica progresiva del sistema hematopoyético caracterizado por la proliferación no regulada de las células progenitoras. • • Puede ir a otros tejidos como ganglios, bazo, igado mas comun, entre otros. Origen en MO. Clasificación Leucemias Agudas (LA) • • Leucemia Mieloblastica Aguda (LMA) Leucemia Linfoblastica Aguda (LLA) Leucemias Crónicas (LC) • • Leucemia Mieloide Crónica (LMC) Leucemia Linfática Crónica (LLC) Leucemias agudas implican mayor gravedad y peor pronóstico. LEUCEMIA MIELOIDE AGUDA Son un grupo heterogéneo de enfermedades que caracterizan por se la infiltración de la sangre, la medula ósea y otros tejidos, por células neoplásicas del sistema hematopoyético. Cel mieloblásticas - progranulocitos Epidemiologia • • • • En Estados Unidos en el año del 2006 hubo 16,430 casos nuevos de leucemia mieloide. Se estima una incidencia anual de de 3.5 casos por 100,000 habitantes. Mayor en varones que en mujeres (4.3 contra 2.9) La edad promedio al momento del diagnóstico es de 67 años. Etiología Herencia, radiación, exposiciones químicas y otras, fármacos, insecticidas y pesticidas. NO evidencia Viral. • Una tomografía equivale a 400 placas rayos X! Manejan los hematólogos. Primer grupo: anomalías genéticas. Segundo: con displasias Tercero: relacionados con tratamientos, fármacos. Cuarto: sin clasificación especial. Presentación Clínica (<3 meses) Síntomas • • • • • • • • • Fatiga Debilidad Hiporexia Pérdida de peso Fiebre (primero en 10%) Sangrado Diaforesis Dolor óseo Cefalea Signos • • • • • • • • Fiebre Hepatomegalia Esplenomegalia Adenopatías Hemostasia anormal (5%) Hipersensibilidad a la palpación del esternón Hemorragia Meningitis Leucémica Hallazgos Hematológicos Anemia normocítica normocrómica (intensa) Recuento leucocitario: 15, 000/µL (mediana). Globulos blancos inmaduros que no sirven. • • < 5000/µL (25 – 40 % de los casos) > 100000/µL (20 % de los casos) Menos del 5% de personas carecen de células leucémicas detectables. Neutropenias severas en el 30% a 40 % de los pacientes . Fiebre por infecciones. Plaquetas: • • < 100000/µL (75 % de los pacientes) < 25000/µL (25 % de los pacientes) Diagnostico Alteraciones importantes en la citología hemática. • Presencia de blastos 20-30% con granulación Auer. citoplásmica y cuerpos de Estudio de la medula ósea. • Citoquímicas. • Biología molecular. • Citofluorometría. Tinción citoquímica de mieloperoxidasa. Sudán negro. Esterasas inespecíficas. Morfología Normal Figura 1. Frotis de médula ósea normal en que están representadas todas las series hematopoyéticas normales. CARACTERISTICAS CELULARES PUEDEN SALIR EN EL EXAMEN! Se encuentra en el frotis de SP y MO. Figura 2. Frotis de médula ósea de una leucemia mieloblástica aguda tipo M-6 (eritroide). Se observan eritroblastos inmaduros con hiperbasofilia y vacuolización citoplasmática. Figura 3. Frotis de sangre periférica de una leucemia mieloblástica aguda tipo M-7 (megacarioblástica). Se observa una célula blástica de estirpe megacarioblástica, núcleo desnudo de micromegacariocito y dismorfia plaquetaria. Blastos de la leucemia aguda mieloide con mutación en el gen de la nucleolosmina. Inclusión cristaloide rectangular en el interior de una célula blástica (LAM4). Leucemia aguda promielocítica o LAM3. Obsérvese la presencia de astillas en los promielocitos atípicos. Tratamiento—Importante tratamiento precoz es muy agresiva, para mejor pronostico. El tratamiento de un paciente con diagnostico de LMA suele dividirse en 2 fases: 1. La Inducción. 2. Posterior a la remisión. □El objetivo inicial es inducir pronto una remisión completa. Quimioterapia ideal es con una combinación de 2 o 3 de los de los siguientes medicamentos: • • • • • Citarabina 100 – 200 mg/mt2 c/dia x / 7dias Daunorrubicina 45 mg/mt2 x 3 dias Arabinósido de citosina(ARA-C) Etopósido. Tioguanina o 6-mercaptopurina. Tratar Neutropenia febril. (Ceftazidima 1gr/c 8 h) Agregar Anfo B si persiste por mas de 7 dias. Primer ciclo se logra la remisión del 60-75% de los casos. • • ARA C por 7 días. Daunorrubicina. Por tres días. Segundo ciclo. Tratamiento de apoyo • • • Transfusiones de plaquetas para mantener el recuento entre > 10000 y 20000/µL Transfundir los eritrocitos necesarios para mantener la cifra de hemoglobina > 8 g/Dl Aseo extremo. • Aislamiento relativo y alimentos que no sean riesgosos. Complicaciones infecciosas de LMA Las complicaciones infecciosas siguen siendo la principal causa de morbilidad y mortalidad durante la quimioterapia de inducción y posremisión de la LMA. □ Nistatina o el Cotrimazol por vía oral para evitar candidosis local. □ Personas con virus del Herpes Simple: Aciclovir. □ La administración empírica precoz de antibióticos principalmente las quinolonas. LEUCEMIA LINFOIDE AGUDA □ □ □ □ □ PREDOMINA EN VARONES . ESPECIALMENTE ENTRE LOS 3-5 AÑOS. 5,400 CASOS POR AÑO. 75% DE TODAS LAS LEUCEMIAS DE LA INFANCIA. SE DIAGNOSTICAN 240,000 EN EL MUNDO DE LEUCEMIA AGUDA EN NIÑOS CADA AÑO. LLA en adultos • • • a los 50 años. Sub aguda. El FSP es casi suficiente para establecer el diagnostico. LAL1: niños, 80% a 85% LAL2: adultos 60-70%. Niños 15% LAL3: menos 5% adultos y niños. Diagnostico • • • • • • • Hx . clínica. • Biometría hemática. Examen físico. • FSP. Aspirado de médula ósea. Se requiere de un 20% de linfoblastos en MO. Para establecer el diagnostico. Dolor articular • Sangrado Fatiga • Adenopatías Fiebre • Hepatomegalia Perdida de pesos • Esplenomegalia Cuadro clínico SINTOMA LAL(%) LAM(%) FATIGA 92 22 FIEBRE 71 75 INFECCION 17 31 HEMORRAGIA 51 22 DOLOR OSEO 79 18 SIGNO LAL(%) LAM(%) ESPLENOMEGALIA 83 61 HEPATOMEGALIA 74 55 ADENOMEGALIA 76 55 DOLOR OSEO 69 65 HEMORRAGIA 50 46 INFILTRACION SNC 80 38 Célula blástica linfoide en una Célula linfoide inmadura en una LAL3. Obsérvese la intensa leucemia linfática crónica en basofilia y la presencia de transformación vacuolas citoplasmáticas Tratamiento El tratamiento se divide en 4 etapas: I.Inducción a la remisión. II.Profilaxis al SNC. III.Intensificación pos inducción. IV.Mantenimiento o terapia continua de erradicación.(2 años) • • Supervivencia del 60 – 65 % de los casos. TMO 30% en pacientes con recaídas. Medicamentos 1. VINCRISTINA. 5. MITOXANTRONA. 2. PREDNISONA. 6. CICLOSFOSFAMIDA. 3. DOXORRUBICINA. 7. ASPARAGINASA. 4. DAUNOMICINA. 8. 6MERCAPTOPURINA. 9. METROTEXATO. 10. ETOPÓSIDO. LEUCEMIA MIELOBLASTICA CRONICA • • • • • 1.5 CASOS POR 100,000 PERSONAS ALAÑO. 15-20% DE TODAS LAS LEUCEMIAS. MEDIANA DE EDAD A LOS 50 AÑOS. PICO DE INCIDENCIA 30-40AÑOS LA INCEDENCIAAJUSTADAS A EDADES ES MAYOR EN VARONES QUE EN MUJERES:-2.0 EN VARONES.-1.2 EN MUJERES CML Se confirma cuando se descubre una expansión clonal de citoblastos hematopoyéticos portador de una translocación reciproca de material citogenético entre los cromosomas t(9 y 22.) CROMOSOMA FILADELFIA Fases de la LMC 1) Fase crónica. 2 – 4 años. 2) Fase acelerada. Anemia que no guarda relación con actividad de enfermedad o tratamiento. 3) Fase blástica o fase terminal. Cuadro clínico de LMA, la mitad de los casos se convierte en LMA Manifestaciones clínicas • • • Insidioso. Asintomáticos. Fatiga. • • Malestar general. Perdida de peso. • • • Esplenomegalia. Prurito. Rubefacción. Manifestaciones hematológica • • • • • • • • GB. <50,000 - >200,000por micro litro. Predominio en bandas y formas maduras. Plaquetas cifras de un millón por micro litros. Anemia N-N. Descenso de la fosfatasa alcalina de leucocitaria. La función fagocítica es normal. < 5% de células blásticas. ≥20% de células blásticas en fase terminal. MO: Hipercelular con hiperplasia de granulocitos. FSP Las células mieloide muestra todos los estadios de diferenciación: Mielocitos. Eosinófilos. Basófilos. Eosinófilo que presenta un núcleo en anillo en un síndrome mielodisplásico. Basófilo desgranulado en un síndrome mieloproliferativo crónico. Leucemia mieloide crónica en crisis blástica. En la imagen obsérvese la presencia de un blásto y de una marcada trombocitosis. Tratamiento • • • TERAPEUTICA CURATIVA. ( TRASPLANTE ALOGENETICO.) NUEVO TRATAMIENTO CON ORIENTACION ESPECIFICA. • • • • Interferón alfa. Quimioterapia. Hidroxiurea. Busulfan. (IMATINIB 400 mg/dia) Tx. de la crisis de blástos. suelen ser ineficaz, incluso con imatinib. mediana de supervivencia global fue de 6.6 meses. LEUCEMIA LINFOCITICA CRONICA Síndromes Linfoproliferativos Crónicos con expresión hemoperiferica: constituyen una serie de enfermedades que tienen en común la existencia de una proliferación clonal de células linfoides B o T maduras en sangre periférica. Epidemiologia • • • • • Leucemia mas frecuente en adultos occidentales. En EEUU. se reportan 17,000 casos nuevos al año. Edad media 65 años Incidencia en <65 años 1,2/100.000 Incidencia en >65 años 19,7/100.000 Leucemias pueden ir a linfomas. B Y T son primariamente leucemias. Laboratorio ü Leucocitosis entre 20.000 y 150.000 glóbulos Blancos, con 75% de linfocitos o superiores. ü La hipogammaglobulinemia. ü El 5-10% de los pacientes presentan gammapatía monoclonal. Disparo de producción de globulinas monoclonales. Aspirado de medula Ósea □ Aspirado de MO. Se observa una infiltración por linfocitos de apariencia madura mas del 30%, pequeños, con núcleos redondos y cromatina condensada en uno de los 4:nodular, intersticial, mixto y difuso. □ CITOFLUOROMETRIA. Los linfocitos de la LLC expresan sobre su superficie los antígenos CD19,20, 21,24 Y 5. PRONOSTICO RAI PRONOSTICO BINET Tratamiento • FLUDARABINA. • CICLOFOSFAMIDA. • • • CLORAMBUCILO. PREDNISONA. QUIMIOTERAPIA COMBINADA. • • VINCRISTINA. ADRIBLASTINA. Leucemia linfática crónica B en la que las células linfoides muestran una marcada granulación maru acosta Linfomas Definición: • Padecimiento maligno del sistema linfático, aferente caracterizado por el crecimiento progresivo e indoloro de los ganglios linfáticos. Tiene su origen en los ganglios linfáticos y sus estructuras: - Vasos linfáticos aferentes: entran al ganglio - Vasos linfáticos eferentes: sale - Nódulos y su corteza - Hílum (centro) - Cápsula A partir de estas estructuras, pueden empezar a proliferar los linfocitos y generar los diferentes tipos de linfomas. • • Los linfomas son tipos de cáncer que comienzan con la transformación maligna de un linfocito en el sistema linfático, principalmente a nivel de los ganglios. El linfoma de Hodgkin y todos los linfomas no Hodgkin son consecuencia de una lesión en el ADN de un linfocito. Linfoma de Hodgkin Epidemiología • Sexo masculino <10 años • En adolescentes la incidencia entre mujeres y hombres es igual • En sistema inmune bajo o deprimido adquirido o congénito • Estudios realizados sugieren que la enfermedad de Hodgkin está relacionada con: o Herpes virus 6 o CMV o Virus de Epstein Barr (VEB) • La proteína latente de membrana del VEB ha sido encontrada Se cree que el primer pico se da entre los 15 – 20 años y después cae, pero en la edad adulta (60-75 años) vuelve a aumentar. El pronóstico de la enfermedad de Hodgkin depende esencialmente delà tipo histológico. TEMA DE EXAMEN Patología • La célula de Reed Sternberg es una célula grande de 15-45um de diámetro con núcleos múltiples o multilobulados. • No producen Ig ni expresan BCR, CD19, CD20 • Ausencia o disminución de genes OCT-2, BOB-1 • Resiste a la apoptosis Subtipos histológicos • El sistema de clasificación de R y E define 4 subtipos histológicos: o Predominio linfocitarioà mejor pronóstico o Celularidad mixta maru acosta o o Depleción linfocitaria Esclerosis nodular (++) Citocinas producidas por la enfermedad de Hodgkin Citocina IL-1 IL-2 IL-4 IL-5 IL-6 FNT-α FNT-β GM.C5F TGF-B Características biológicas Linfoproliferación, fiebre, sudoración nocturna, inmunodeficiencia, fibrosis Similar a IL-1 Infiltración eosinofílica Linfoproliferación Pérdida de peso Infiltración leucocitaria y eosinófila Mieloproliferación Fibrosis Presentación clínica: Linfadenopatía: • • Usualmente los pacientes presentan adenopatías supraclaviculares o cervical indolora. Los nódulos linfoides afectados son firmes • Puede haber una masa mediastinal que provoca tos o síntomas de compresión bronquial o traqueal. Síntomas sistémicos: Puede incluir fatiga, anorexia y pérdida de peso Fiebre inexplicable, T°> 38°C, sudoración toda la noche Pérdida de peso inexplicable con pérdida del apetito Prurito o dolor • • • • La afectación ganglionar más frecuente en etapas tempranas esà ganglio cervical. TEMA DE EXAMEN Diagnóstico: • • • • Historia clínica Debe realizarle radiografía de tórax por la frecuencia de aparición de masas ganglionares. Se prefiere biopsia por extirpación que biopsia por aguja Una vez establecido el diagnóstico debe determinarse la extensión de la enfermedad y eso nos marca el pronóstico y la sobrevida del paciente. Sistema de clasificación de Ann Arbor para la enfermedad de Hodgkin Estadio I Estadio II Estadio III Estadio IV Afectación de una única región ganglionar o de un solo órgano o lugar extralinfático Afectación de 2 o más regiones ganglionares en el mismo lado del diafragma o afectación localizada de un solo órgano o sitio extralinfático y de 1 o más regiones ganglionares en el mismo lado del diafragma Afectación de regiones ganglionares a ambos lados del diafragma, que puede acompañarse de 1 órgano o lugar extralinfático Afectación difusa o diseminada de uno o más órganos o tejidos extralinfáticos, con o sin afectación ganglionar asociada maru acosta ZONAS RICAS EN GANGLIOS Diagnóstico diferencial: • • • • • Micobacterias atípicas Toxoplasmosis Linfoma no Hodgkin Mononucleosis infecciosa Carcinoma nasofaríngeo Tratamiento: Quimioterapias cada 28 días, depende mucho del pronóstico y del estadio Linfoma no Hodgkin • Linfomas no-Hodgkin son tumores malignos originados del tejido linfoide, principalmente de los nódulos. Origen o causa: o Translocación cromosómica o Infecciones retrovirales o Factores ambientales o Estados de inmunodeficiencia o Inflamación crónica Fisiopatología Linfomas se dividen en 2 grandes de neoplasias: Hodgkin y no-Hodgkin Aproximadamente el 85% de los tumores malignos son NHL La edad media de diagnóstico se presenta en la 6ta década de vida NHL incluye varios subtipos clinicopatologicos con diferentes epidemiologias, etiologías, morfología, inmunofenotipo, genética y respuesta terapéutica LNH representan una progresiva expansión clonal de células B, células T o natural killers nacientes del acumulo de lesiones afectando los proto-oncogenes o los genes supresores de tumores Estos oncogenes pueden ser activados por translocaciones cromosomales, o el locus supresor de tumores puede ser inactivado por eliminación o mutación cromosomal Además, el genoma de varios subtipos de linfoma puede ser alterado con la introducción de genes exógenos por varios virus oncogéneticos Alrededor del 85% corresponde a células B y un 15% - células T y NK. Un porcentaje muy pequeño proviene de macrófagos Los tumores se caracterizan por su nivel de diferenciación, el tamaño de la célula de origen, la velocidad de proliferación, y el patrón histológico de crecimiento. Todos estos datos tipifican el tipo exacto del linfoma no Hodgkin, el pronóstico y el tto. Tumores que crecen de una manera nodular tienden a ser más benignos que los que crecen en un patrón difuso Los linfomas de células pequeñas son menos agresivos que los de células grandes que son mediano y alto grado Etiología Traslocaciones cromosómicas La t (18,19) (q32; q31) translocación es la anormalidad cromosomal más frecuente asociada con LNH. Esta translocación ocurre en 85% de los linfomas foliculares y en 28% de los LNH de alto grado o o o T (11,14) (q13, q32) Translocaciones en 8q24 T (2,5) (p23, q35) Resultan en la eliminación del inhibidor de apoptosis de oncogenes, en una sobreexpresión de reguladores de ciclos celulares, expresión de proteínas aberrantes encontradas en la mayoría de linfomas de células grandes Infecciones Algunos virus están implicados en la patogénesis del LNH, probablemente por su habilidad de inducir estimulación crónica antigénica y desregulación de citosinas, que lleva a una estimulación no controlada de células B y T, proliferación y linfomagénesis El virus de Epstein-Barr está asociado a linfoma de Burkitt, linfoma sinonasal, Hodgkin, entre otros Los virus tipo 1 de leucemias de células T, causan una infección latente mediante la transcripción inversa de células T activadas o Este virus es endémico en ciertas áreas de Japón y las Islas canarias y aproximadamente 5% de los portadores desarrollan un linfoma Virus de hepatitis C está asociado al desarrollo de expansiones de células B y ciertos subtipos de LNH Sarcoma de Kaposi está asociado a producir linfomas en pacientes con VIH, el VIH por si solo sin asociarse a este puede causar linfomas Helicobacter pylori está asociado al desarrollo de linfomas gastrointestinales, particularmente linfomas MALT. (Por eso es importante tratar la gastritis) Factores ambientales Radiación (TAC, Rx, etc.), quimioterapia, y químicos (pesticidas, herbicidas, solventes, polvos, pinturas para el cabello, y químicos orgánicos) Se ve con mayor incidencia: linfomas no Hodgkin y leucemias Estados de inmunodeficiencia Estados congénitos y adquiridos de inmunodeficiencia, están asociados a alta incidencia de LNH de alta agresividad Enfermedad celiaca están asociadas a un alto riesgo de linfomas malignos. En los ptes con dietas controladas no hay riesgo porque se los considera curados, pero en los que siguen consumiendo gluten es peligroso. Inflamación crónica La inflamación crónica observada en pacientes con enfermedades autoinmunes como síndrome de Sjögren o tiroiditis de Hashimoto que promueve el desarrollo de MALT y predispone al paciente a subsecuentes malignidades linfoideas Entre el 23-56% de pacientes con tiroiditis de Hashimoto tiene linfomas primarios tiroideos Epidemiología La ACS estima que hay 65,540 casos nuevos de LNH cada año La incidencia a partir del año 1970 ha aumentado el doble sin poderse explicar exactamente por qué LNH es la neoplasia hematopoyética más prevalente, representando aproximadamente el 4% de los diagnósticos de cáncer y teniendo el 7mo lugar de más frecuente entre todos los cánceres LNH es 5 veces más frecuente que Hodgkin Incidencia entre razas: personas caucásicas tienen un mayor riesgo que afroamericanos y asiáticos LNH es ligeramente más frecuente en hombres que en mujeres siendo la relación 1,4:1 La edad media para presentar la enfermedad es mayor a 50 años, excepto el linfoblástico de alto grado y linfomas pequeños no hendidos que ocurren en niños y jóvenes adultos Linfomas de bajo grado son extremadamente raros en niños Manifestaciones clínicas Existen pacientes/sintomas A y B: A: Síntomas generales como fiebre, fatiga, sudoración, mal estar nocturno (no se ven adenomegalias, masas, etc. O sea, no hay señal física de la enfermedad) B: síntomas A + señal física de la enfermedad o o o o o o o o Fiebre sin causa aparente Sudor nocturno Cansancio constante Pérdida de peso sin causa aparente Anorexia Piel pruriginosa Petequias Adenopatía Examen físico Linfomas de bajo grado pueden producir adenopatía periférica, esplenomegalia y hepatomegalia o Esplenomegalia se observa en 40% de los pacientes Linfoma de mediano y alto grado pueden presentar: o Esplenomegalia, linfadenopatia, hepatomegalia, masa testicular, grandes masas abdominales y lesiones en piel (mucosis) Complicaciones Citopenias (anemia, neutropenia, trombocitopenia) secundaria al infiltrado de la medula, además se puede observar anemia hemolítica autoinmune en algunas variantes de LNH Sangrados secundarios a trombocitopenia o CID Infecciones secundarias a neutropenia Problemas cardiacos secundarios a efusión del pericardio o arritmias por metástasis cardiaca(tiene que estar muy avanzado el proceso para llegar a esto) Compresión de la medula por metástasis vertebral y fracturas vertebrales Problemas neurológicos por linfoma en SNC o meningitis linfomatosa (esto no es raro) • • Obstrucción, perforación y sangrado del tracto GI Dolor y leucocitosis si el paciente está en una fase de leucemia Consideraciones diagnósticas Diagnostico patológico se debe hacer con biopsia Pueden existir otras enfermedades que tengan manifestaciones clínicas similares como: o Tumores sólidos malignos: metástasis a nódulos linfoideos secundarios a carcinoma, melanoma o sarcoma, linfoma de Hodgkin; otras malignidades hematológicas como: sarcoma granulocítico o enfermedad multicéntrica de Castleman Laboratorio Citometría/hemograma: en las etapas tempranas se pueden tener valores normales o o o Anemia por hemolisis autoinmune o por enfermedad crónica Pancitopenia secundaria al infiltrado de medula ósea (cuando las 3 series están disminuidas, bicitopenia es cuando 2 de las 3 están disminuidas) Linfocitosis y trombocitosis Elevada LDH indica mal pronóstico y correlaciona incremento de tamaño Pruebas hepáticas Inflamación crónica Hipercalcemia en pacientes con linfoma-leucemia de células T Test Coombs positivo HIV (es obligatorio hacer este) Biopsia: es más para dx o o Aspiración de medula ósea sirve más para dar un estadio que para dar diagnóstico Biopsia de los sitios extranodales representan entre 30-35% de pacientes con LNH siendo el sitio más frecuente GI Punción lumbar: o Para LNH agresivos y difusos, linfomas relacionados con HIV o signos/síntomas de SNC Etapas Etapa I Etapa II LNH involucra un solo nódulo linfático o está localizado en una sola zona región extralinfática Involucra 2 o más regiones de nódulos linfáticos del mismo lado del diafragma o está localizada en un solo órgano extralinfoideo Involucra nódulos linfáticos en ambos lados del diafragma que puede ser acompañado por involucramiento de un órgano extra linfático o sitio, bazo, o ambos Compromiso de un órgano extral infático aislado en ausencia de compromiso de un ganglio linfático regional adyacente, pero junto con enfermedad en sitio(s) distante(s) Etapa III Etapa IV Nomenclatura: N: ganglios linfáticos S: bazo H: hígado P: pleura L: pulmón O: hueso M: médula ósea D: piel Tratamiento LNH adultos en crecimiento en estadio I y contiguo en estadío II o o o Radioterapia Rituximab con quimioterapia o sin esta → ac monoclonal, es nuevo Conducta expectante LNH en adultos en crecimiento lento no contiguo en estadios II, III y IV o o o o o o Conducta expectante para ptes asintomáticos Rituximab Análogos de nucleósidos de purina Alquilantes (con o sin esteroides) Tiuxetano de ibritumomab marcado con itrio-90 y tostitumomab marcado con yodo 131 Mantenimiento con rituximab Opciones de tto de LNH recidivante en adultos o o o o o Quimioterapia Rituximab Lenalidomida Ac monoclonales anto CD20 radiomarcadores Radioterapia paliativa Pronóstico La sobrevida a 5 años va ser de 63% dependiendo de los siguientes factores: o o o o Histología, etapa y tamaño del tumor (I, II, III, IV) Edad del paciente (60) Niveles de LDH (normal vs elevado) Presencia de enfermedades extranodales (0 vs 1+) Con este modelo, la sobrevida y un estado libre de relapso a 5 años son: o o o 0-1 factores: 75% 2-3 factores: 50% 4-5 factores: 25% Mal pronóstico Pacientes con inmunodeficiencias Relación entre remisión completa y tiempo de respuesta tiene un pronóstico significativo. Pacientes que no alcanzan RC para su tercer ciclo de quimio tienen peor pronóstico Pacientes con linfomas de células T o NK tienen peor pronóstico que aquellos con linfomas de células B Anormalidades en los cromosomas 1,7,17 tiene peor pronostico Aproximadamente 70% de pacientes con LNH de mediano y alto grado recaen o nunca responden al tratamiento inicial. La mayoría de las recurrencias son dentro de los primeros 2 años de acabar tratamiento Entre 5-10% de los pacientes que tienen relapso o LNH resistente siguen vivos dentro de los siguientes 2 años