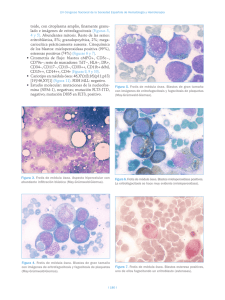
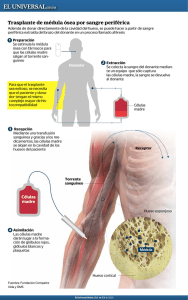
UNIDAD 5. 2. 2 Tiempo de protrombina Consiste en obtener sangre y colocarla en tubos con citrato u oxalato de sodio como anticoagulante, el cual secuestra (quela) el calcio. Esta sangre se centrifuga y se separan los glóbulos rojos del plasma. Se coloca una cantidad precisa de plasma (100 μl) en un tubo a 37°C, se añade una fuerte cantidad (200 μl) de una solución de tromboplastina tisular y calcio. Entre 10 y 12 segundos se producirá un coágulo. El tiempo transcurrido se compara al que se obtiene en un “fondo común” normal (mezcla de al menos 30 plasmas normales), y a la relación entre ambos tiempos se denomina proporción. La tromboplastina y el calcio permitirán que se active directamente el factor VII, el cual activado transformará el factor X en Xa, actuando la tromboplastina tisular como cofactor. Una vez que se ha activado el factor X, éste activará al factor II. El factor II activado o trombina en medio líquido se desprende de la capa de fosfolípidos y actúa sobre el fibrinógeno convirtiéndolo en fibrina, transformando la solución en un gel, el coágulo. El tiempo de protrombina se encontrará prolongado cuando hay una de las siguientes circunstancias: Disminución de alguno de los factores que actúan en todo el proceso. Cuando la concentración de los factores VII, V, X, II o fibrinógeno (por debajo de 100 mg/dl) esté disminuida. Cuando alguno de los factores anteriores, a pesar de encontrarse en concentraciones normales, no funcione correctamente. Cuando hay alguna sustancia que bloquee la acción de alguno o varios de estos factores, como sucede con los anticuerpos dirigidos a alguno de ellos (p. ej., anti-factor II en casos de anticoagulante lúpico). UNIDAD IV. Introducción a mecanismos hemostáticos 2 .3 Tiempo de tromboplastina parcial activado (TTPA) Esta prueba se utiliza para descartar anomalías en la vía intrínseca de la coagulación . Se realiza añadiendo al plasma fosfolípidos, como la cefalina, y caolín (de ahí la antigua denominación de “tiempo de cefalina-caolín”) e iones calcio, tras lo cual se anota el tiempo que tarda en establecerse el coágulo . Se denomina tiempo de tromboplastina parcial activada (TTPA) porque una superficie activadora inicia la activación del plasma y los fosfolípidos añaden una actividad de tromboplastina parcial equivalente al componente lipídico de las plaquetas. La normalidad de esta prueba sugiere la ausencia de anomalías de los factores XII, XI, X, IX, VIII, V y II. El TTPA se emplea para detectar la deficiencia de factores en la vía intrínseca (es muy sensible a los factores VIII y IX, por lo que es de utilidad para descartar hemofilias A y B), para realizar el cribado de anticoagulante lúpico y la monitorización de la anticoagulación con heparina. 2 .4 Tiempo de trombina El tiempo de trombina (TT) se calcula añadiendo trombina diluida al plasma del paciente. Al no añadirse iones calcio, la reacción se debe de manera exclusiva a la presencia de la trombina añadida . El alargamiento de esta prueba sugiere: • Aumento de la actividad antitrombínica del plasma; por ejemplo, presencia de heparina. • Presencia de productos de degradación del fibrinógeno (PDF) que interfieren en la polimerización de la fibrina. • Hipofibrinogenemia, cuando los niveles de fibrinógeno son inferiores a 80 mg/dl. • Disfibrinogenemias. • Presencia de inhibidores directos de trombina (dabigatrán). UNIDAD IV. Introducción a mecanismos hemostáticos 2 .5 Cifra paquetería Actualmente el recuento de plaquetas se realiza en aparatos automatizados, y las cifras normales oscilan entre 150 y 400 × 10^/l. A la hora de interpretar los recuentos, es necesario descartar las pseudotrombocitopenias, cifras falsamente disminuidas de plaquetas a consecuencia de su agrupación o de la existencia de plaquetas de tamaños muy grandes o pequeños que el contador incluye dentro de otros grupos celulares. Problemas asociados a las plaquetas Existen tres tipos de problemas asociados a las plaquetas: Trombocitopenia Trombocitosis Trombocitopatía Además, la pseudotrombocitopenia puede ser causada por aglutinación plaquetaria dependiente del anticoagulante ácido etilendiaminotetracético (EDTA), aglutininas frías plaquetarias o satelitismo plaquetario a neutrófilos o monocitos; por ello, en estos casos, es conveniente realizar el hemograma con heparina y/o citrato como anticoagulantes y revisar el frotis de sangre periférica para confirmar su existencia. La morfología plaquetar se determina en un frotis de sangre periférica mediante tinciones ordinarias . Además del número, puede valorarse el tamaño de las plaquetas, que está aumentado en pacientes con la enfermedad de Bernard-Soulier UNIDAD IV. Introducción a mecanismos hemostáticos 3 . 1 Trombocitopatias 3 . Trastornos plaquetarios Las manifestaciones clínicas de los trastornos del sangrado pueden dividirse en dos amplios grupos mal definidos: 1) sangrado superficial (como petequias, epistaxis o sangrado gingival), que suele asociarse con un defecto plaquetario o un trastorno vascular 2) sangrado de los tejidos profundos (como hematomas o hemartrosis), que por lo general se asocia con deficiencias plasmáticas de los factores de la coagulación. Los trastornos plaquetarios pueden afectar la cantidad de plaquetas, su correcto funcionamiento, o ambas. Un trastorno plaquetario afecta la coagulación normal de la sangre. Las enfermedades ocasionadas por la alteración de la función de las plaquetas se denominan trombocitopatía. Hay formas congénitas y formas adquiridas. Trombocitopatía congénita Síndrome de Bernard-Soulier. Es una enfermedad hereditaria, autosómica recesiva, en la que la adhesividad plaquetaria está anormalmente disminuida y las plaquetas no se agregan con ristocetina a pesar de que el factor VIII plasmático sea normal. El defecto básico se debe a la ausencia de un receptor proteínico específico, la glicoproteína I, para el factor VIII en la superficie plaquetaria. Estos pacientes agregan normalmente con el resto de los reactivos utilizados en el estudio de agregación plaquetaria, y se puede encontrar trombocitopenia con macroplaquetas en la sangre periférica. UNIDAD IV. Introducción a mecanismos hemostáticos 3 . 1 Trombocitopatias Trombastenia. También conocida como la enfermedad de Glanssman, es un defecto de la agregación plaquetaria. Se hereda de manera autosómica recesiva y la consanguinidad es un dato más o menos constante en la historia familiar. Clínicamente, se manifiesta por hemorragia mucocutánea con epistaxis, gingivorragia, petequias, metrorragia y sobre todo en el posoperatorio o después de extracciones dentales. El defecto principal se encuentra en las glicoproteínas Iib y IIIa de la membrana plaquetaria que están ausentes. Estas glicoproteínas son las encargadas de la expresión de los antígenos plaquetarios y los receptores para el fibrinógeno, lo cual podría explicar la alteración en la retracción del coágulo y la ausencia de la agregación inducida por todos los agentes agregantes . El diagnóstico de laboratorio resulta más o menos fácil, ya que la falta de agregación con ADP y trombina es prácticamente única en esta enfermedad; existen además alteraciones en el tiempo de sangrado y la retracción del coágulo. Los hallazgos más importantes en la trombastenia son: Los hallazgos más importantes en la trombastenia son: Morfología y recuento de plaquetas normales, ausencia de coágulos en el frotis. Tiempo de sangrado prolongado. Prueba del torniquete normal o prolongada. Retracción del coágulo anormal. Ausencia de la agregación con ADP, colágeno, adrenalina, trombina y una agregación escasa o imagen de disagregación con ristocetina. Disponibilidad del factor 3 plaquetario disminuida. TP y TTP normales. UNIDAD IV. Introducción a mecanismos hemostáticos 3 . 1 Trombocitopatias Desórdenes Granulares: Estos desórdenes incluyen a los gránulos alfa y densos. Dentro de los primeros encontramos al síndrome de plaquetas grises y síndrome de Quebec. El síndrome de plaquetas grises se caracteriza por un déficit de gránulos alfas con salidas de estas proteínas al plasma y medula ósea. Muchas de estas son factores de crecimiento capaces de inducir mielofibrosis. Se presentan con sangrado mucocutáneo espontáneo, por trauma o cirugía asociado a esplenomegalia secundario a mielofibrosis. Presentan macrotrombocitopenia, tiempo de sangría prolongado, agregación inducida por trombina y colágeno deficiente. El síndrome de Québec es raro, autosómico dominante (AD), que se caracteriza por ausencia del factor V en los gránulos alfa. Las manifestaciones son sangrados tardíos en cirugías, epistaxis, hemartrosis y hematuria. El síndrome de Hermansky-Pudlak, el síndrome de ChediakHigashi se encuentran englobados dentro de los síndromes de gránulos densos. Existen alteración en la acumulación granular de serotonina, ADP, ATP, y calcio. Son AR en general, pueden ser mixtos y en estos casos es AD. Se asocian a albinismo óculocutáneo, tirosinasa positivo y predisposición a infecciones. Trombocitopatía adquirida Hay enfermedades que producen por sí trombocitopatía, al recubrirse las plaquetas con moléculas anormales (como sucede con la uremia) o con las hepatopatías, en donde se producen fragmentos de fibrinógeno que se adhieren a las plaquetas e impiden su función. La causa más frecuente de trombocitopatía adquirida la constituye la ingesta de ácido acetilsalicílico que inhibe por acetilación de manera irreversible las enzimas de la vía del ácido araquidónico, en particular la ciclooxigenasa, alterando el funcionamiento plaquetario. A continuation se muestra una tabla con los farmacos que causan disfunción plaquetaria: UNIDAD IV. Introducción a mecanismos hemostáticos 3 . 2 Purpuras trombocitopenicas Si la trombocitopenia persiste más de seis meses o bien hay Púrpura trombocitopénica (inmune) recidivas pasado este tiempo, es probable que se trate del primer Trombocitopenia inmune aguda o posinfecciosa. Se caracteriza brote de una púrpura trombocitopénica idiopática (PTI). por la aparición, sobre todo en niños, de un cuadro purpúrico, Púrpura trombocitopénica idiopática diseminado por el organismo, más manifiesto en zonas declives. Es una enfermedad autoinmune caracterizada por la presencia Este cuadro normalmente va precedido por un proceso infeccioso de autoanticuerpos dirigidos contra las plaquetas que ocasionan agudo, sobre todo de las vías respiratorias altas, del tipo catarral una destrucción acelerada de las mismas en el sistema de origen viral. En las pruebas de detección de coagulación se reticuloendotelial. Es un padecimiento crónico, más frecuente de detecta una cifra de plaquetas profundamente disminuida, con un adultos jóvenes y niños, aunque puede verse en ancianos. Suele tiempo de protrombina y de tromboplastina parcial normales. En aparecer de dos formas: exámenes complementarios se detectan anticuerpos Aparición aguda. El enfermo, sin ninguna otra enfermedad antiplaquetarios en el suero de estos enfermos, y sus plaquetas acompañante ni alteración previa, nota la brusca aparición de un están recubiertas de una mayor cantidad de inmunoglobulinas. En cuadro hemorrágico caracterizado por púrpura petequial y ocasiones, la infección precedente puede que no sea detectada. equimosis espontáneas, hematomas al mínimo traumatismo, Ello ha llevado a una gran confusión, ya que algunos autores le gingivorragia y, en la mujer, metrorragia. En los estudios de denominan PTI aguda o del niño, considerando que se trata de laboratorio se observa una trombocitopenia notoria. La una púrpura trombocitopénica idiopática.El tratamiento consiste trombocitopenia puede corregirse bajo tratamiento y ceder o en administrar prednisona a dosis de 1 a 2 mg/kg de peso/día persistir. A este brote agudo le pueden seguir otros varios durante tres a cuatro semanas, disminuyendo progresivamente la (recidivas). Cuando la recidiva se presenta durante más de seis dosis en la cuarta semana; también pueden administrarse meses se le considera como púrpura trombocitopénica inmunoglobulinas por vía intravenosa a dosis altas. Esta idiopática. No se acompaña de fiebre, en ocasiones de astenia y trombocitopenia se resuelve antes de un mes en más del 80% de las lesiones son las de un síndrome trombocitopénicolos enfermos, aun sin tratamiento; después puede que no recidive trombocitopático. jamás o bien que sí haya varias recidivas en los primeros seis UNIDAD IV. Introducción a mecanismos hemostáticos meses. 3 . 2 Purpuras trombocitopenicas Forma asintomática. Con los contadores electrónicos, es posible que, con motivo de una revisión de empresa, un estudio preoperatorio para una intervención banal o por la realización de un análisis en otras circunstancias, se observe una trombocitopenia moderada. En la historia clínica se puede o no detectar la existencia de hematomas que no se relacionan con la intensidad del traumatismo. En el laboratorio, sólo se detecta trombocitopenia, sin anemia o si ésta existe es circunstancial y ocasionada por otras causas (ferropenia en mujeres). Esta trombocitopenia muchas veces se había detectado años atrás sin darle importancia, y al observarse una cifra inferior a 100 000 plaquetas/mm3 se acude a valoración. En la mayoría de los casos, no hay antecedentes familiares de hemorragia y no se advierte en la familia trombocitopenia. Esta forma de trombocitopenia se acompaña de aumento del volumen de las plaquetas, algunas casi del tamaño de linfocitos. Estos enfermos pasan años sin presentar hemorragia; con frecuencia, jamás tendrán hemorragia, aunque su recuento de plaquetas persistirá disminuido. Al marcarse las plaquetas con un isótopo radiactivo y reinyectarse, la vida media puede encontrarse ligeramente acortada o ser normal, pero con mucha frecuencia se aprecia un aumento de secuestro de las plaquetas por el bazo. Cuando se estudia por medio del laboratorio, sólo destaca una trombocitopenia de intensidad variable, que se acompaña de un aumento del tamaño de las plaquetas. Estas últimas circulan con una carga superior de inmunoglobulinas adheridas a su superficie, y a la vez se pueden detectar anticuerpos dirigidos contra ellas en el plasma o pegados a su superficie. Estos anticuerpos pueden ser de tipo IgG o IgM. En la médula ósea sólo se observa un moderado a notorio aumento de megacariocitos, sin alteraciones morfológicas. También se puede apreciar un incremento moderado de linfocitos. Desde el punto de vista del laboratorio, los hallazgos característicos en la PTI son: • Recuento plaquetario bajo. • Tiempo de sangrado prolongado. • Retracción del coágulo anormal, prueba del torniquete anormal. • TP y TTP normales. Sólo deben tratarse los brotes agudos cuando producen manifestaciones hemorrágicas. El tratamiento a dosis terapéuticas sólo debe sostenerse mientras haya sintomatología. UNIDAD IV. Introducción a mecanismos hemostáticos 3 . 2 Purpuras trombocitopenicas Otros medicamentos. El rituximab (Rituxan, Truxima) ayuda a Tratamiento : aumentar el recuento de plaquetas al reducir la respuesta del Corticoesteroides.: prednisona a la dosis de 1 a 2 mg/kg/ día sistema inmunitario que las está dañando. Pero este durante un mínimo de cuatro semanas. Durante la quinta semana, medicamento también puede reducir la eficacia de las vacunas, se disminuye de manera progresiva la dosis hasta su total que pueden ser necesarias si más adelante eliges la cirugía para supresión, si se ha alcanzado la desaparición de la sintomatología. extirpar el bazo Puede suceder que cesen los síntomas, pero la cifra de plaquetas Cirugía: Si tu afección es grave o persiste a pesar del tratamiento permanezca por debajo de 20 000 plaquetas/mm3. En este caso se farmacológico inicial, el médico puede sugerirte una cirugía para recomienda que la dosis de prednisona no se suprima totalmente, extirparte el bazo. Esto elimina rápidamente la fuente principal y se deje al enfermo con una dosis de 15 a 5 mg en días alternos de destrucción de plaquetas en el cuerpo y mejora el conteo de (enfermos denominados corticodependientes). Cuando el plaquetas, aunque no funciona para todos. Vivir sin bazo en individuo está asintomático con una cifra de plaquetas superior a forma permanente aumenta la susceptibilidad a las infecciones. 40 000/mm3 , puede suprimirse por completo el tratamiento. Inmunoglobulina. Si los corticosteroides no ayudan, el médico puede darte una inyección de inmunoglobulina( Ig IV) 1g / kg/ dia por 1 o 2 dias y despues 0.4 g/ kg/ dia por 5 dias . Este medicamento también se puede usar si tienes una hemorragia grave o si necesitas aumentar rápidamente tu recuento sanguíneo antes de la cirugía. El efecto suele desaparecer en un par de semanas. Medicamentos que estimulan la producción de plaquetas. Los medicamentos como el romiplostim (Nplate) y el eltrombopag (Promacta) ayudan a la médula ósea a producir más plaquetas. Este tipo de medicamentos puede aumentar el riesgo de presentar coágulos sanguíneos. UNIDAD IV. Introducción a mecanismos hemostáticos 3. 3 Purpuras vasculares Vasculares hereditatios Telangiectasia hemorrágica hereditaria (síndrome de Rendu Weber-Osler) El modo de herencia de la telangiectasia hemorrágica hereditaria (T H H ) es autosómico dominante. El defecto vascular de este trastorno se caracteriza por vasos sanguíneos de paredes delgadas con endotelio discontinuo, músculo liso inadecuado y cantidad insuficiente o falta de elastina en la estroma circundante. Las telangiectasias (vasos sanguíneos dilatados, superficiales, que crean lesiones rojas, pequeñas, focales) se producen en todo el cuerpo, pero son más evidentes debajo de la lengua; en la cara, los labios, la lengua, la conjuntiva, la mucosa nasal, los dedos de las manos y los pies, y en el tronco. Las lesiones se tornan blancas cuando se aplica presión. El trastorno suele manifestarse en la pubertad y progresa durante toda la vida. Las telangiectasias son frágiles y propensas a romperse. La epistaxis es un hallazgo casi universal y los síntomas casi siempre empeoran con la edad. La edad en la que comienza la epistaxis se correlaciona con la gravedad del trastorno. Mientras la cavidad oral, el tracto gastrointestinal y el aparato urogenital son sitios comunes de sangrado, éste puede producirse en casi todos los órganos.Las características de laboratorio se relacionan con la intensidad de la tendencia hemorrágica. El tiempo de sangría suele ser normal y el resultado de la pruea del torniquete puede ser normal o mostrar un incremento de la fragilidad capilar. El diagnóstico de THH se basa en las lesiones características de la piel o las mucosas, antecedentes de hemorragias recurrentes y una historia familiar de afección similar. Los pacientes con THH se encuentran sorprendentemente bien a pesar de la falta del tratamiento específico y la gravedad de sus manifestaciones hemorrágicas. Síndrome de Ehlers-Danlos Es un trastorno del tejido conjuntivo causado por anomalías en los genes que codifican diferentes subtipos de colágeno. En su mayoría tienen una herencia autosómica dominante. La tendencia hemorrágica parece ser debida a la adhesión defectuosa de las plaquetas al colágeno subendotelial, que es anómalo. El cuadro clínico se caracteriza por: hiperelasticidad cutánea, laxitud ligamentosa y articular, piel atrófica como “papel de fumar” y hemorragias dérmicas, preferentemente hematomas. En el subtipo 4, debido a un fallo en el colágeno de tipo III, está afectado el árbol arterial y los pacientes padecen hemorragias graves que se asocian a una alta morbimortalidad, en parte debida a la ausencia de tratamiento, que suele limitarse a la ligadura del vaso. Otros trastornos congénitos El síndrome de Marfan, el seudoxantoma elástico y la osteogénesis imperfecta son otros desórdenes del colágeno de la pared vascular que pueden cursar con púrpura cutánea u otras manifestaciones hemorrágicas generalmente leves. UNIDAD IV. Introducción a mecanismos hemostáticos 3. 3 Purpuras vasculares Adquiridas Púrpura alérgica (púrpura de Henock-Schónlein) El término púrpura alérgica o púrpura anafilactoide por lo general se aplica a un grupo de púrpuras no trombocitopénicas caracterizado por manifestaciones en apariencia alérgicas, como erupción cutánea y edema. La púrpura alérgica se asoció con algunos alimentos, fármacos, frío, picaduras de insectos y vacunas. El término púrpura de Henoch-Schónlein se aplica cuando el cuadro se acompaña por artralgia transitoria, nefritis, dolor abdominal y las lesiones cutáneas purpúreas con frecuencia se confunden con la erupción hemorrágica de la PTI. La evidencia general implica lesiones vasculares autoinmunes, pero la fisiopatología del trastorno aún es confusa. La evidencia preliminar indica que la vasculitis es mediada por complejos inmunes que contienen anticuerpos IgA. Se sugirió que la púrpura alérgica puede representar autoinmunidad contra los componentes de la pared de los vasos sanguíneos.La púrpura de Henoch-Schónlein es una enfermedad que afecta sobre todo a los niños entie los 3 y los 7 años. El comienzo es súbito, a ntenudo después de una infección del tracto respiratorio superior. El microorganismo puede dañar el revestimiento endotelial de los vasos sanguíneos, que produce vasculitis. Se hicieron intentos para implicar un agente infeccioso específico, en particular estreptococo (3-hemolítico). Los síntomas de presentación son malestar general, cefalea, fiebre y erupción. El retraso en la aparición de la erupción cutánea plantea un problema en el diagnóstico diferencial. Las lesiones cutáneas son urticarianas y en forma gradual se tornan rosadas, luego rojas y por último hemorrágicas. La aparición de las lesiones puede ser muy rápida, acompañada por prurito. Las lesiones se describieron como “púrpura palpable”, al contrario de las lesiones perfectamente planas de la trombocitopenia y de la mayor parte de las formas de púrpura vascular. Estas lesiones son más frecuentes en los pies, los codos, las rodillas, las nalgas y el tórax. Por último se observa una erupción roja pardusca. También puede haber petequias. A medida que la enfermedad progresa, suele haber dolor abdominal, poliartralgias, cefaleas y enfermedad renal. En el 60% de los pacientes presentan lesiones renales durante la segunda a la tercera semanas de la evolución del trastorno. Son frecuentes la proteinuria y la hematuria.El recuento de plaquetas es normal. Las pruebas de hemostasia, como el tiempo de sangría, la prueba del torniquete y las pruebas de coagulación de la sangre, suelen ser normales en los pacientes con púrpura alérgica. La anemia no está presente a menos que las hemorragias sean graves. UNIDAD IV. Introducción a mecanismos hemostáticos 3. 3 Purpuras vasculares El recuento de leucocitos y la velocidad de eritrosedimentación Las lesiones se limitan a las superficies extensoras de los están elevados. La enfermedad debe diferenciarse de otras formas antebrazos y el dorso de las manos, y solo en ocasiones se de púrpura no trombocitopénica. En el diagnóstico diferencial presentan en la cara y el cuello. Con excepción del aumento de ciertas enfermedades infecciosas asociadas con púrpura también la fragilidad capilar, los resultados de las pruebas de laboratorio deben considerarse. A veces pueden estar implicados fármacos o son normales y no hay otras manifestaciones de sangrado. sustancias químicas. En los niños la duración promedio del ataque inicial es de alrededor de 4 semanas. Las recidivas son frecuentes, Púrpuras vasculares inducidas por fármacos por lo general después de un período de bienestar aparente. La púrpura asociada con vasculitis inducida por fármacos se Excepto en el caso de pacientes en quienes se desarrolla la produce en presencia de plaquetas con funcionalidad adecuada. enfermedad renal crónica, el pronóstico suele ser bueno. En Hay una amplia variedad de fármacos que causan púrpura ocasiones, se produce la muerte por insuficiencia renal. El vascular, com o aspirina, cumadina, barbitúricos, diuréticos, tratamiento es sobre todo sintomático, dado que hasta el presente digoxina, metildopa y diversos antibióticos. Las sulfonamidas y no hay una terapéutica eficaz. Los corticosteroides algunas veces los yoduros fueron los implicados con mayor frecuencia. Las fueron útiles para aliviar los síntomas. La mayoría de los pacientes lesiones varían de unas pocas petequias a erupciones se recupera sin tratamiento. petequiales masivas y generalizadas. Entre los mecanismos se Púrpura senil incluyen el desarrollo de anticuerpos contra las paredes de los Este trastorno afecta con mayor frecuencia a los hombres ancianos vasos sanguíneos, el desarrollo de complejos inmunes y que a las mujeres y se debe a la falta de sostén del colágeno para cambios en la permeabilidad de la pared de los vasos. Tan los vasos sanguíneos de pequeño calible y la pérdida de la grasa rápido como se reconoce el trastorno, el fármaco ofensor debe subcutánea y las fibras elásticas. La cantidad de casos aumenta con suspenderse. No se necesita otro tratamiento. la edad. Las manchas oscuras son planas, de cerca de 1 a 10 mm de diámetro, no se blanquean con la vitropresión y se resuelven lentamente, a menudo dejan una coloración castaña en la piel (manchas de la edad). UNIDAD IV. Introducción a mecanismos hemostáticos 4 . Coagulopatias afectan a un solo factor de la coagulación, y según la magnitud de afectación pueden aparecer en la infancia, en la adolescencia, o hasta en la madurez En ocasiones sólo se pueden detectar por un análisis de laboratorio. Coagulopatías adquiridas Coagulopatías congénitas Las alteraciones de la fase plasmática de la hemostasia o coagulación propiamente dicha se denominan coagulopatías. Éstas pueden ser congénitas o adquiridas. afectan a varios factores de la coagulación en forma simultánea, y además también pueden alterar la fase celular de la hemostasia (trombocitopatía o trombocitopenia). 4 .1 Enfermedad de Von Willebrand Se debe a que no se produce el factor von Willebrand o éste es defectuoso. En cualquier caso, las plaquetas no se adhieren al colágeno del subendotelio y se produce un cuadro hemorrágico, típico de la alteración de la hemostasia primaria, pero dado que el factor von Willebrand transporta las moléculas de factor VIII, su ausencia o alteración provoca un cuadro de deficiencia de factor VIII, una hemofilia A, que puede variar de moderada a grave, dependiendo del grado de alteración del factor von Willebrand.Hay formas heterocigotas (frecuentes) que producen trastornos moderados y homocigotas o heterocigotas dobles (raras) con hemorragias intensas. La enfermedad de Von Willebrand se clasifica en 3 tipos: Tipo 1: deficiencia cuantitativa de FvW, que es la forma más común y es un trastorno autosómico dominante Tipo 2: alteración cualitativa de la síntesis de FvW que puede deberse a diversas anomalías genéticas y es un trastorno autosómico dominante UNIDAD IV. Introducción a mecanismos hemostáticos Tipo 3: trastorno autosómico recesivo raro en el que los homocigotos no tienen FvW detectable 4 .1 Enfermedad de Von Willebrand Aunque la enfermedad de von Willebrand, al igual que la hemofilia A, es un trastorno hereditario que puede causar deficiencia de factor VIII, la deficiencia del factor VIII en la enfermedad de von Willebrand suele ser solo moderada. Signos y síntomas Las manifestaciones hemorrágicas son de leves a moderadas y consisten en propensión a presentar hematomas, hemorragia mucosa, hemorragia por pequeños cortes en la piel que puede detenerse y comenzar al cabo de unas horas, a veces hipermenorrea y hemorragia anormal después de procedimientos quirúrgicos (p. ej., extracción dentaria, amigdalectomía). Las plaquetas funcionan lo suficientemente bien para que rara vez aparezcan petequias o púrpura. Diagnóstico oAntígeno total de VWF en plasma oPrueba de función de VWF oNivel de factor VIII en plasma oTTP Se sospecha enfermedad de Von Willebrand en pacientes con sangrado de causa desconocida, en particular en aquellos con antecedentes de una diátesis hemorrágica similar. Los estudios de coagulación revelan recuento plaquetario normal, INR normal y, a veces, TTP ligeramente prolongado. La prueba del tiempo de sangría es poco fiable y ya no se hace. El diagnóstico requiere determinar el antígeno de FvW plasmático total, la función del FvW determinada por la capacidad del plasma para mantener la aglutinación de plaquetas normales por medio de ristocetina (actividad del cofactor de ristocetina) y la concentración plasmática de factor VIII. Los estímulos (como el embarazo y la inflamación) que aumentan transitoriamente las concentraciones de FvW pueden causar resultados falsos negativos en la EvW leve; a veces, deben repetirse los estudios. A. En el tipo 1 de EvW, los resultados son concordantes; es decir, se observa una depresión equivalente del antígeno de FvW, la función del FvW y la concentración plasmática de factor VIII. El grado de depresión varía de alrededor del 15 al 60% de lo normal y determina la gravedad de una hemorragia anormal del paciente. Las concentraciones de antígeno de FvW pueden ser de tan solo el 40% de lo normal en individuos sanos con grupo sanguíneo 0. UNIDAD IV. Introducción a mecanismos hemostáticos 4 . 1 Enfermedad de Von Willebrand La desmopresina es ineficaz en otras variantes de EVW B. Se sospechan variantes tipo 2 si los resultados son tipo 2 y en el tipo 3, y en el tipo 2A puede exacerbar la discordantes; es decir, el antígeno de FvW es más alto que lo esperable para el grado de alteración de la actividad del trombocitopenia. Para garantizar la respuesta adecuada al cofactor de ristocetinaEl antígeno de FvW es más alto que el fármaco, los médicos administran a los pacientes una dosis previsto porque, en el tipo 2, el defecto del FvW es cualitativo de prueba y miden la respuesta del antígeno de FvW. La (pérdida de multímeros del FvW de alto peso molecular) no desmopresina en dosis de 0,3 mcg/kg administrados en 50 cuantitativo. El diagnóstico se confirma demostrando un mL de solución salina al 0,9% por vía IV en 15-30 minutos descenso de la concentración de multímeros grandes de FvW puede permitir que los pacientes sean sometidos a en la electroforesis en gel de agarosa. Se reconocen 4 procedimientos menores (p. ej., extracción dentaria, variantes tipo 2 diferentes, que se distinguen por alteraciones cirugía menor) sin necesidad de tratamiento de reemplazo. funcionales de la molécula de FvW. C. Los pacientes con EvW tipo 3 no tienen FvW detectable y Si se precisa un producto de reemplazo, la desmopresina presentan marcada deficiencia de factor VIII puede reducir la dosis requerida. Tratamiento de la enfermedad de Von Willebrand Una dosis de desmopresina es eficaz durante alrededor de •Desmopresina 4 a 6 horas. Deben transcurrir aproximadamente 48 horas •Reemplazo de FvW cuando es necesario para que se acumulen los nuevos depósitos de FvW de Los pacientes con enfermedad de Von Willebrand se tratan solo si manera que una segunda inyección de desmopresina presentan hemorragia activa o van a ser sometidos a un pueda ser tan eficaz como la dosis inicial. Para muchos procedimiento invasivo (p. ej., cirugía, extracción dentaria). En los pacientes con enfermedad de von Willebrand tipo 1 y en algunas pacientes, la desmopresina intra-nasal puede ser tan eficaz variantes del tipo 2, puede ser útil la desmopresina, un análogo de como el tratamiento IV y con frecuencia es útil para la vasopresina (hormona antidiurética) que estimula la liberación prevenir el sangrado durante procedimientos quirúrgicos de FvW al plasma y puede aumentar las concentraciones de factor menores. VIII. UNIDAD IV. Introducción a mecanismos hemostáticos 4 . 1 Enfermedad de Von Willebrand Para los pacientes con variantes de la enfermedad de von Willebrand tipo 2 que no responde al DDAVP, aquellos con enfermedad de von Willebrand tipo 3 o pacientes con enfermedad de von Willebrand tipo 1 sometidos a procedimientos invasivos más extensos, el tratamiento consiste en reemplazo del FvW por infusión de concentrados de factor VIII de pureza intermedia, que contiene componentes del FvW. Estos concentrados están viralmente inactivados y, por lo tanto, no transmiten infección por HIV ni hepatitis. Como no causan infecciones transmitidas por transfusión, se prefieren estos concentrados al crioprecipitado utilizado antes. Los concentrados de factor VIII de alta pureza se preparan por cromatografía de inmunoafinidad, no contienen FvW y no deben utilizarse. Para las mujeres con sangrado menstrual abundante debido a la enfermedad de von Willebrand, un breve período de tratamiento con ácido tranexámico por vía oral o desmopresina intranasal puede disminuir el sangrado 4 . 2 Hemofilias En estas enfermedades, los factores de la coagulación son los que se encuentran anormales. Esta anormalidad puede ser en la cantidad de proteína circulante o en la función de la misma; pueden ser hereditarias o adquiridas, de un solo factor o múltiples. Hemofilia A es una enfermedad que se hereda ligada al cromosoma X, caracterizada por la disminución de la actividad procoagulante del factor VIII. Su incidencia es de 1 caso por cada 5.000- 10.000 varones, siendo el trastorno de la coagulación más común. El 100% de las hijas de los varones hemofílicos son portadoras de la enfermedad, mientras que la padecen el 50% de los hijos varones de las mujeres portadoras . El gen que codifica el factor VIII situado en el cromosoma X es grande (consta de 26 exones y 3 dominios estructurales), y las anomalías más frecuentes encontradas en la hemofilia A son las mutaciones en los diferentes exones (7, 14, 22, 26, etc.) y las inversiones de material genético (por ejemplo, la inversión del intrón 22 o el intrón 1). La hemofilia A puede ser secundaria a un defecto cuantitativo en la síntesis del factor VIII o a un defecto cualitativo de esta proteína. UNIDAD IV. Introducción a mecanismos hemostáticos 4 . 2 Hemofilias La hemofilia A puede ser secundaria a un defecto cuantitativo en la síntesis del factor VIII o a un defecto cualitativo de esta proteína. En el 90% de los casos existe una disminución, tanto de los niveles de actividad procoagulante (VIII: C) como antigénica (VIII: Ag), mientras que en el 10% restante la actividad antigénica es superior a la procoagulante, lo que sugiere la existencia de una proteína anómala. La frecuencia y la intensidad de las manifestaciones hemorrágicas generalmente guardan correlación con los niveles de factor VIII . Diagnóstico viene marcado por la historia clínica y el alargamiento del TTPA . En el diagnóstico diferencial debe descartarse la posibilidad de que se trate de una EvW, por lo que siempre hay que cuantificar esta proteína mediante enzimoinmunoanálisis. La existencia de historia hemorrágica solo en varones y una prolongación exclusiva del TTPA, con pruebas de hemostasia primaria normal, orienta a la existencia de hemofilia. Sin embargo, existe una forma de EvW, autosómica recesiva, que puede confundirse con una hemofilia (EvW de tipo 2) por manifestarse selectivamente como una deficiencia del factor VIII. Actualmente, el diagnóstico de portadoras de hemofilia se basa en el análisis de los fragmentos de restricción polimórficos conseguidos tras la digestión del ácido desoxirribonucleico (ADN) (métodos indirectos), o empleando métodos directos (investigación de la mutación familiar conocida). Estos métodos sirven también para el diagnóstico prenatal. Tratamiento Depende de la gravedad de la enfermedad y de la circunstancia clínica. UNIDAD IV. Introducción a mecanismos hemostáticos 4 . 2 Hemofilias Deficiencia del factor IX (hemofilia B) Las opciones terapéuticas disponibles para la hemofilia A son las La hemofilia B es también una enfermedad hereditaria de siguientes: carácter recesivo ligada al sexo. Es similar a la hemofilia A en • Concentrados de factor VIII recombinante. Algunos pacientes cuanto al modo de transmisión y a sus manifestaciones clínicas, desarrollan inhibidores, lo que puede ser un problema de este pero su incidencia es menor (1/30.000 varones). El trastorno tratamiento. puede ser debido a una disminución de los niveles antigénicos • Concentrados plasmáticos de factor VIII del factor IX o, en un tercio de los casos, a la existencia de una • Crioprecipitados: actualmente no se usan en el tratamiento de las proteína inactiva. La tipificación del defecto exige no solo la coagulopatías congénitas. Cada unidad de crioprecipitado obtenida determinación de la actividad coagulante del factor IX sino de un único donante contiene más de 80 U de factor VIII (una también su cuantificación inmunológica. unidad es la cantidad de factor VIII que existe en 1 ml de plasma La utilización de sondas de ADN facilita la identificación de normal). portadores y el diagnóstico prenatal de la enfermedad. Las Además de los concentrados de factor VIII, existen otros fármacos manifestaciones clínicas de la hemofilia B son indistinguibles de adyuvantes, muy útiles en el tratamiento de esta enfermedad: las descritas para la A. La administración de DDAVP no tiene • 1-desamino-8-D-arginina-vasopresina (DDAVP) o desmopresina: ningún valor. es vasoconstrictor sintético, análogo de la hormona antidiurética vasopresina, que estimula la liberación de factor VIII por el Tratamiento de elección de la hemofilia B endotelio.La DDAVP, en dosis de 0,3 µg/kg por vía intravenosa, es se basa en la administración de factor IX recombinante o de capaz de duplicar o triplicar la concentración del factor VIII, concentrados de factor IX plasmático previamente inactivados manteniendo estos niveles unas 12 horas. para virus. Cada unidad de factor IX eleva el nivel plasmático un • Antifibrinolíticos: como el ácido tranexámico, que impiden la 1%. disolución del coágulo una vez formado. Por este motivo están La vida media del factor IX es de 18-24 horas, por lo que las contraindicados en los pacientes con hematuria. El ácido dosis de mantenimiento se administran una vez al día. tranexámico se suele administrar a dosis de 10 mg/kg cada 6-8 horas por vía intravenosa. UNIDAD IV. Introducción a mecanismos hemostáticos 4 . 2 Hemofilias El uso de 40-60 UI/kg de peso es suficiente para alcanzar niveles hemostáticos terapéuticos. En caso de hemofilias B graves, deben administrarse 50 UI/ kg de factor IX dos veces por semana en programas de profilaxis para prevenir el sangrado espontáneo. La prevalencia de inhibidores contra el factor IX en pacientes con hemofilia B es inferior al 5%. Deficiencia del factor XI Es un trastorno que se hereda con carácter autosómico recesivo, siendo muy frecuente entre los judíos askenazíes procedentes del este de Europa. Es el tercer trastorno hemorrágico en orden de frecuencia. La deficiencia del factor XI se asocia generalmente a niveles disminuidos de la proteína y rara vez a la presencia de una proteína inactiva. Los episodios hemorrágicos no suelen ser graves y suelen producirse tras intervenciones quirúrgicas o extracciones dentarias. El tratamiento de este trastorno consiste en la administración de plasma fresco y tratamiento antifibrinolítico, si bien en la actualidad se dispone de concentrados de factor XI (Hemoleven®). UNIDAD IV. Introducción a mecanismos hemostáticos 5. Trombofilias Un estado de hipercoagulabilidad o trombofilia se define como aquella situación anormal de la sangre circulante en la que se requiere, en comparación con el estado normal, un menor estímulo para provocar la aparición de trombosis. Clasificacion: Clasificacion Trombofilia primaria • Hereditaria: – Déficit de antitrombina – Déficit de proteína C – Déficit de proteína S – Factor V Leiden – Mutación G20210A de la protrombina • Adquirida: – Anticuerpos antifosfolípido • Origen mixto: – Hiperhomocisteinemia – Niveles plasmáticos elevados de factores VIII, IX y XI – Resistencia a la proteína C activada no asociada a factor V Leiden Trombofilia secundaria: cáncer, embarazo/puerperio, procesos médicos agudos, cirugías, etc. Trombofilia primaria Deficiencia de antitrombina La antitrombina (AT) es una alfa2-globulina de síntesis hepática que ejerce una importante función como regulador fisiológico de la formación de fibrina mediante la inactivación de los factores XIIa, XIa, IXa, Xa y IIa y plasmina . Dicha función inhibitoria se ve especialmente potenciada en presencia de proteoglicanos de la pared vascular y de heparina. La prevalencia de la deficiencia congénita de AT en la población general es baja, oscilando en diversos estudios entre el 0,02% y el 0,4%, mientras que se aproxima al 1% en pacientes no seleccionados con ETEV. La transmisión de la deficiencia de AT es generalmente autosómica dominante y la mayoría de los afectados son heterocigotos, con niveles de AT entre el 40% y el 70%, siendo muy raros los casos homocigotos. La existencia de una deficiencia de AT multiplica hasta por 50 veces el riesgo de ETEV frente a los individuos sin esta deficiencia, aunque la expresividad clínica es heterogénea en función del defecto molecular. UNIDAD IV. Introducción a mecanismos hemostáticos 5. Trombofilias Se distinguen dos tipos de deficiencia de AT. El tipo 1 se caracteriza por un descenso tanto de la actividad funcional como de los niveles antigénicos, como consecuencia de una disminución en la síntesis de la proteína, y se asocia a un mayor riesgo trombótico. El tipo 2, menos frecuente, se caracteriza por una reducción en la actividad funcional con normalidad de los niveles antigénicos. El tipo 2 se subdivide a su vez en tres subtipos en función de la localización del defecto: lugar de unión de la heparina, sitio reactivo o ambos. La deficiencia de AT puede condicionar cierto grado de resistencia a la heparina, por lo que en algunas situaciones clínicas como el parto y procedimientos quirúrgicos, la administración de concentrados de AT (30-50 U/kg) podría ser beneficiosa. Deficiencia de proteína C La prevalencia del déficit de PC en la población general oscila entre 1:200 y 1:700, y en pacientes con trombosis venosa no seleccionados es del 3%. El déficit de PC se asocia a una elevación del riesgo de trombosis de entre 2 y 6 veces. La incidencia de homocigosidad es de 1 por cada 500.000 individuos. En este último caso, se desarrolla un cuadro grave a las pocas horas del nacimiento, conocido como púrpura neonatal fulminante, que cursa con trombosis en la microcirculación y coagulación intravascular diseminada. El tratamiento urgente con concentrados de PC o plasma es crítico tras el diagnóstico. Desde el punto de vista fenotípico, se pueden distinguir dos tipos de deficiencia de PC: el tipo 1, caracterizado por un descenso tanto en la actividad funcional como en los niveles antigénicos, y el tipo 2, en el que la baja actividad funcional contrasta con niveles antigénicos normales, como expresión de la existencia de una molécula anormal. La proteína C (PC) es una glicoproteína dependiente de la vitamina K de síntesis hepática. La PC es activada por el complejo trombinatrombomodulina en la superficie endotelial. La PC activada (PCa) degrada proteolíticamente los factores Va y VIIIa, y requiere la presencia de la proteína S (PS) como cofactor. UNIDAD IV. Introducción a mecanismos hemostáticos 5. Trombofilias Deficiencia de proteína S La deficiencia de PC se transmite generalmente de modo autosómico dominante, si bien algunos estudios en homocigotos sugieren que algunos defectos pueden transmitirse con un patrón autosómico recesivo. Un dato relevante en estos pacientes es la posibilidad de desarrollar episodios de necrosis cutánea al comenzar la terapia anticoagulante con dicumarínicos, debido a una mayor disminución de los niveles de PC Fenómenos de necrosis dérmica en un paciente con deficiencia de proteína C. La PS es una glicoproteína dependiente de la vitamina K, cuya función es actuar como cofactor de la PC en la degradación de los factores Va y VIIIa. La PS circula en forma libre en un 40%, y en el resto lo hace unida a la fracción C4b delcomplemento, la cual es inactiva como cofactor de la PC. La prevalencia de la deficiencia de PS en la población general no se conoce con exactitud, mientras que en pacientes con trombosis no seleccionados es del 1-2%. La evidencia sobre el riesgo trombótico inherente a la deficiencia de PS parece menor que para la PC, si bien este riesgo no está claramente cuantificado. El déficit de PS es considerado como un defecto hereditario con un patrón autosómico dominante. Se distinguen tres tipos distintos de déficit de PS: el tipo 1, caracterizado por una disminución de la concentración antigénica de PS total y libre y de la actividad funcional En estos casos debe interrumpirse el tratamiento anticoagulante oral, administrar vitamina K e iniciar la anticoagulación con heparina. La clínica y las recomendaciones terapéuticas son superponibles a las definidas previamente para los estados de trombofilia UNIDAD IV. Introducción a mecanismos hemostáticos el tipo 2, con niveles antigénicos normales de PS total y libre, y caracterizado por una reducción de la actividad funcional el tipo 3, caracterizado por niveles normales de PS antigénica total y una disminución tanto de la PS libre como de la actividad funcional 5. Trombofilias Resistencia a la proteína C activada/ factor V Leiden Mediante el estudio de los inhibidores naturales de la coagulación (AT, PC, PS) es posible encontrar un factor de riesgo genético en un número reducido (aproximadamente el 5-6%) de pacientes con ETEV. En 1993 se describió el fenómeno de la resistencia a la PCa (RPCa) estudiando el plasma de pacientes con historia de ETEV. En algunos casos, la prolongación del tiempo de tromboplastina parcial activada (TTPA) tras la adición de PCa era menor que en sujetos sin trombosis. Un año más tarde se demostró que la alteración responsable de más del 90% de los casos de RPCa es una sustitución del aminoácido 506 de la molécula del factor V (glutamina en lugar de arginina). Esta sustitución es consecuencia de una mutación puntual (guanina por adenina) en el nucleótido 1691 del gen del factor V, mutación que fue denominada factor V Leiden. El factor V Leiden es la causa más frecuente de trombofilia hereditaria en la raza caucásica. La PCa inactiva al factor V activado mediante una proteólisis ordenada. La sustitución de arginina por glutamina en la posición 506 del factor V condiciona una resistencia a la acción proteolítica de la PCa. La transmisión del factor V Leiden es de carácter autosómico dominante y su prevalencia varía considerablemente en función de la raza: es frecuente en la caucásica y prácticamente ausente en la negra africana o en Extremo Oriente. La presencia del factor V Leiden aumenta el riesgo de padecer trombosis venosa de tres a cinco veces en portadores heterocigotos, y es mucho mayor en el caso de portadores homocigotos de la mutación. Como se ha comentado anteriormente, resulta especialmente evidente en el caso del factor V Leiden la importancia de las interacciones gen-gen y gen-ambiente. Aunque la causa más frecuente de RPCa es la presencia del factor V Leiden, en ocasiones es posible encontrarnos ante un paciente con RPCa en ausencia de dicha mutación. Esta RPCa no causada por el factor V Leiden puede ser de origen genético o adquirido. Entre las causas adquiridas de RPCa, las mejor conocidas son el embarazo, el consumo de anticonceptivos orales o algunos tumores, como por ejemplo el mieloma múltiple Mutación G20210A de la protrombina Esta mutación, consiste en una sustitución (guanina por adenina) del nucleótido 20210, que se encuentra en la región 3’ no traducida del gen de la protrombina. La variante 20210A se ha asociado a la presencia de mayores niveles de protrombina en plasma, que pueden ser responsables de un aumento del riesgo trombótico. UNIDAD IV. Introducción a mecanismos hemostáticos 5. Trombofilias Los portadores heterocigotos de la mutación tienen un riesgo de padecer un episodio de enfermedad tromboembólica venosa tres veces mayor que los no portadores. El diagnóstico del factor V Leiden y la mutación 20210A de la protrombina se realiza mediante técnicas de reacción en cadena de la polimerasa (PCR), que permiten saber si un paciente es portador de dichas mutaciones en pocas horas. Hiperhomocisteinemia La homocisteína es un producto intermedio del metabolismo de la metionina, e incrementos leves-moderados en sus niveles plasmáticos constituyen un factor de riesgo trombótico (tanto en el territorio venoso como en el arterial). El mecanismo por el que la hiperhomocisteinemia contribuye al riesgo trombótico no ha sido del todo aclarado, aunque se ha descrito una acción citotóxica sobre el endotelio vascular. Es posible encontrar niveles plasmáticos moderadamente elevados de homocisteína en aproximadamente el 5% de la población general. Dichos niveles están influenciados por factores tanto ambientales como genéticos. Dentro de los ambientales el más importante es la deficiencia de folatos y vitaminas B6 o B12 (cofactores en el metabolismo de la homocisteína). Entre los factores genéticos destaca un polimorfismo genético en una enzima que participa en el metabolismo de la metionina: la metilentetrahidrofolato reductasa (MTHFR). Una mutación puntual (citosina por timina) en el nucleótido 677 del gen de la MTHFR condiciona una termolabilidad de la enzima, de forma que a 37 °C su actividad es un 50% menor que la de la variante normal. Aunque la variante C677T MTHFR puede relacionarse con niveles de homocisteína elevados de manera ligera o moderada, no existe relación causal entre C677T y trombosis. Niveles plasmáticos de los factores de la coagulación Los niveles elevados de factor VIII coagulante constituyen un factor de riesgo de ETEV y de recurrencia trombótica. El principal determinante de los niveles plasmáticos de factor VIII es el grupo sanguíneo (más elevados en personas con grupo distinto al O). Sin embargo, la agregación familiar persiste después de ajustar el efecto del grupo sanguíneo, por lo que deben existir otros factores genéticos, aún no identificados, implicados en la regulación de la concentración plasmática del factor VIII que expliquen la tendencia familiar a presentar niveles plasmáticos elevados de dicho factor. UNIDAD IV. Introducción a mecanismos hemostáticos 5. Trombofilias Por otra parte, también los niveles elevados de otros factores de la coagulación como el fibrinógeno, los factores IX o XI, parecen asociarse a un aumento del riesgo de ETEV, si bien los resultados en diversos estudios no han sido uniformes. Curiosamente, también se ha descrito cierto incremento de la incidencia de episodios tromboembólicos en pacientes con deficiencia leve del factor XII, así como en algunos casos de disfibrinogenemia. Anticuerpos antifosfolípido Los anticuerpos antifosfolípido (AAF) están dirigidos frente al complejo formado por fosfolípidos aniónicos y determinadas proteínas. Los de mayor interés clínico son los anticuerpos anticardiolipina, anti-beta2-glicoproteína y el anticoagulante lúpico. A diferencia de las alteraciones anteriores, de carácter hereditario, los AAF constituyen una causa de trombofilia primaria adquirida. El síndrome antifosfolípido (SAF) se caracteriza por la asociación de episodios trombóticos (tanto arteriales como venosos), abortos de repetición, trombopenia y presencia de AAF. Se distingue entre SAF primario y secundario; este último cuando se asocia a otras enfermedades, la más frecuente el lupus eritematoso sistémico. La presencia de AAF (particularmente en el caso de anticoagulante lúpico y en pacientes “triple positivos”), independientemente de la existencia o no de enfermedad asociada, supone un aumento del riesgo de trombosis. Algunos estudios han sugerido que este riesgo llega a multiplicarse hasta nueve veces. En nuestro país, aproximadamente el 5% de los pacientes diagnosticados de trombosis venosa tienen AAF, mientras que solo están presentes en el 1-2% de la población sana. Desde el punto de vista terapéutico, clásicamente se ha venido recomendando tratamiento anticoagulante con carácter indefinido en aquellos pacientes con trombosis venosa o arterial. En el caso de ausencia de trombosis pero positividad de AAF a títulos moderados o altos, existe controversia sobre el riesgo-beneficio del tratamiento anticoagulante o antiagregante. UNIDAD IV. Introducción a mecanismos hemostáticos UNIDAD III. NEOPLASIAS HEMATOLÓGICAS 1. Leucemias agudas y crónicas 2. Síndromes mielodisplasicos 3. Síndromes mieloproliferativos crónicos 4. sxxxx 5. Trasplante de medula ósea 1. Leucemias agudas y crónicas Las leucemias conforman un grupo heterogéneo de neoplasias clonales que surgen de la transformación maligna de las células hematopoyéticas. Su característica común es el acúmulo de las células malignas anormales en la médula ósea y en la sangre, lo que provoca fallo medular (anemia, neutropenia y trombopenia) e infiltración de órganos (hígado, bazo, ganglios linfáticos, meninges, cerebro, testículos o piel). CLASIFICACIÓN Leucemias agudas Leucemias cronicas Las leucemias pueden dividirse en enfermedades agudas y crónicas sobre la base de sus signos y síntomas de presentación, y el tipo celular involucrado, si bien la discriminación entre ellas es menos clara debido al desarrollo de una gama amplia de tratamientos y su eficacia. Signos y síntomas Las leucemias agudas se caracterizan por el comienzo abrupto de signos clínicos (p. ej., infección, hemorragia y palidez) y síntomas (p. ej., fatiga, debilidad, dolor óseo y articular), y la muerte se produce en el transcurso de meses si no se instituye el tratamiento. Las leucemias agudas afectan a niños y adultos. La clasificación de FAB requiere que los blastos (células inmaduras de origen mieloide o linfoide) constituyan el 30% de todas las células nucleadas de la médula ósea. La clasificación propuesta por la Organización Mundial de la Salud modificó el porcentaje de blastos a 20%, ya sea en sangre periférica o médula ósea. En términos globales, los recuentos de leucocitos periféricos pueden estar aumentados, disminuidos o dentro de los límites de referencia, si bien en los casos típicos están aumentados. La anemia normocítica y la neutropenia también son características distintivas de las leucemias agudas. La trombocitopenia por lo general se encuentra en pacientes con leucemia aguda. Las leucemias crónicas se caracterizan por el comienzo insidioso de los signos (palidez, aumento de tamaño del bazo, el hígado o ambos, pérdida de peso, etc.) y síntomas (debilidad, fatiga, depresión, etc.), y la muerte por lo general se produce años después del diagnóstico. UNIDAD III. Neoplasias hematológicas 1. Leucemias agudas y crónicas Las leucemias crónicas se producen en los adultos, aunque hay formas juveniles de leucemia mieloide trónica Los recuentos leucociurio pueden estar disminuidos, aumentados o dentro de los límites de referencia, si bien por lo general son más elevados que los observados en la leucemia aguda. La médula ósea está infiltrada por un número aumentado de células maduras o con contrapartes reconocibles de maduración normal . A diferencia de las leucemias agudas, en los pacientes con leucemia crónica en un comienzo se observan recuentos de plaquetas normales o aumentados que en la evolución posterior de la enfermedad desarrollan trombocitopenia evidente. En las leucemias crónicas a menudo se presenta anemia. En la siguiente tabla se comparan las manifestaciones clínicas y de laboratorio de las leucemias agudas y crónicas. UNIDAD III. Neoplasias hematológicas Presentación Leucemia aguda Leucemia crónica Comienzo Abrupto Insidioso Muerte Meses Años Edad Todas Adultos Recuento Leucocitario Elevado, normal o bajo Elevado Aspecto de las células Blastos (inmaduras) Maduras Neutropenia Presente Ausente Anemia Presente Presente Raquetas Bajas Normal o aumentadas 1. Leucemias agudas y crónicas Leucemias agudas Leucemia linfoblástica aguda La leucemia linfoblástica aguda (LLA) es sobre todo una enfermedad de la niñez; la mayoría de los cosos se produce entre los 2 y los 10 años. Si bien es rara en los adultos, el segundo pico en la incidencia se produce en los pacientes ancianos El tratamiento de la LLA de la niñez es un triunfo de la hematología moderna enfermedad siempre fatal antes de la década de 1070, la LLA tiene "buen pronóstico“ tiene una tasa de remisión completa del 90% y se cura en el 60% de los pacientes .Lamentablemente, los adultos con esta enfermedad tienen una evolución más tórpida: un 68 al 91% de tasa de remisión completa y 25 al 41 % de tasa de curación. Solo la mitad de los pacientes con LLA presenta leucocitosis y puede no tener linloblastos circulantes. Suele haber neutrupenia, trombocitopenia y anemia. Debido a esta pancitopenia los pacientes muestran sintomas de fatiga (consecuencia de la anemia), fiebre (por la neutropenía y la infección) y hemorragia (resultado de la trombocitopenia). A menudo se acompañan de aumento de tamaño de los ganglios linfáticos y en el S a 10% de los pacientes se observa masa mediastinica. Puede hallarse esplenomegalia, hepatomegalia. El dolor óseo a menudo es resultado de la infiltración de celulas leucemicas en la capa que recubre al hueso . Tipos de tratamiento El éxito en el tratamiento de las leucemias linfoblásticas se evalúa de acuerde con la edad. Los fármacos utilizados en el tratamiento de la LLA del adulto son daunorrubicina,vincristina y prednisona La daunorrubicina con frecuencia ocasiona complicaciones cardiacas, de modo que para el seguimiento se necesitarán estudios de las enzimas cardiacas. La vincristina ataca la formación de microtubulos es toxica para el sistema nervioso e interfiere con la acción de las plaquetas. El trasplante de medula ósea o recolección de células madre periférica es la ultima línea de tratamiento para LLA. Leucemia mieloide aguda La LMA es la leucemia más común en los niños menores de 1 año. Es rara en niños mayores y adolescentes. Si no se trata, esta leucemia es rápidamente fatal. Hay 8 tipos de LMA . Se explican a continuacion: UNIDAD III. Neoplasias hematológicas 1. Leucemias agudas y crónicas Leucemia aguda mieloide mínimamente diferenciada (LAM0) La LAM mínimamente diferenciada o LAM0 tiene una gran dificultad diagnóstica desde el punto de vista morfológico, dado que los blastos presentan rasgos morfológicos linfoides y mieloides12. Constituye únicamente el 5% de las LAM en el adulto y tiene un mal pronóstico. Miembros del grupo FAB, utilizando citoquímica ultraestructural y anticuerpos monoclonales, demostraron que algunos casos con una cifra inferior al 3% de blastos mieloperoxidasa (MPO) positivos clasificados como leucemias agudas linfoides (LAL) eran en realidad LAM con signos mínimos de maduración. En el estudio inmunofenotípico al menos un marcador mieloide (MPO citoplasmática, CD13 o CD33) es positivo en los blastos. Los marcadores linfoides son negativos (CD3, CD22, CD79a). Los blastos son de tamaño mediano, con elevada relación núcleo-citoplasmática (N/C), contorno nuclear redondeado, núcleo de cromatina laxa e inmadura con presencia de uno o varios nucleolos prominentes . Los blastos pueden presentar una fina granulación azurófila, o algún bastón de Auer visible en el citoplasma, y una cifra superior al 3% de las células blásticas son MPO positivas. La fosfatasa ácida y la β-glucuronidasa muestran una positividad difusa. La reacción del ácido peryódico de Schiff (PAS) es positiva, débil y difusa. En el estudio inmunofenotípico se demuestra que los blastos expresan antígenos mieloides (CD33 y CD13) y pueden expresar también el antígeno CD34 Leucemia aguda mieloide sin maduración (LAM1) En este subtipo suele observarse un monomorfismo celular, con presencia en sangre periférica (SP) de blastos mieloides (>3%) en ausencia de otras células en estadíos posteriores al mieloblasto. UNIDAD III. Neoplasias hematológicas 1. Leucemias agudas y crónicas Leucemia aguda mieloide con maduración (LAM2) Constituye alrededor del 30% de todos los casos de LAM y muestra células en estadios madurativos posteriores al mieloblasto (promielocitos, mielocitos y neutrófilos) en un porcentaje superior al 10%. El tamaño de los blastos en la LAM2 es de pequeño a mediano, con una elevada relación N/C y un perfil nuclear redondeado, que a veces adopta una posición cuadrangular respecto al citoplasma. El núcleo muestra una cromatina laxa e inmadura, con uno o varios nucleolos visibles. El citoplasma es basófilo y puede contener un esbozo de granulación primaria azurófila, u ocasionalmente algún bastón de Auer. Los blastos de la LAM2 son positivos para la MPO y el Negro Sudán B, y expresan los antígenos CD34, HLA-DR, CD13 y CD15. Pueden expresar otros antígenos, tales como CD117, CD34 y HLA-DR. Una tercera parte de las LAM2 se asocian a t (8;21). En este subtipo se observan blastos de tamaño pequeño junto a otros de mayor tamaño, núcleo de perfil más irregular y citoplasma moderadamente amplio, que puede contener una granulación muy marcada. Otras alteraciones son deleciones o translocaciones a nivel del cromosoma 12, y la t(6;9). Una asociación menos frecuente es la t(8;16)(p11;p13), caracterizada por la presencia de eritrofagocitosis, positividad para la MPO y esterasas inespecíficas, negatividad para los antígenos CD34 y CD117, con positividad para el CD56 y reordenamiento de los genes MOZ/CBP. El tratamiento de inducción en la leucemia MI y M2 por lo general es efectivo, lo que genera un retorno suficiente del proceso medular normal para un trasplante autólogo de médula ósea Los fármacos utilizados con frecuencia son daunorrubicina y arabinósido de citosina. Una vez más, es necesario el control cardíaco. Leucemia aguda promielocítica (LAM3) Suele acompañarse de una cifra baja de leucocitos en SP, lo que dificulta su diagnóstico. Las células que proliferan muestran una morfología muy característica y se denominan promielocitos atípicos (hipergranulares). Puede cursar con accidentes hemorrágicos muy graves por coagulación intravascular diseminada. Los promielocitos atípicos presentan una granulación intensamente azurófila y muy abundante. UNIDAD III. Neoplasias hematológicas 1. Leucemias agudas y crónicas El núcleo suele ser de aspecto monocitoide (reniforme) y con un perfil bilobulado (en hachazo) con la presencia de una hendidura amplia, o bien de perfil irregular . El citoplasma es poco basófilo debido al elevado contenido de granulación azurófila. Algunos de los promielocitos atípicos contienen además inclusiones citoplasmáticas cristalinas alargadas o astillas, específicas de este tipo de leucemia, que suelen disponerse en cúmulos y que difieren de los bastones de Auer por la detección de una subestructura tubular cuando se estudian mediante microscopía electrónica de transmisión. Los promielocitos atípicos son muy positivos para la MPO y Negro Sudán B. La reacción del PAS muestra positividad difusa y las fosfatasas ácidas son intensamente positivas. Son HLA-DR y CD34 negativos y son positivos para los anticuerpos monoclonales CD13 y CD33. La alteración citogenética característica de la LAM3 es la t(15;17), que provoca la fusión del oncogen PML(promyelocytic leukemic gen) con el gen del receptor del ácido retinoico (RARα), con el resultado de la formación del tránscrito PML-RARα. Para detectar la t(15;17) se utilizan técnicas citogenéticas, de hibridación in situ y de biología molecular. Estas últimas identifican específicamente el tránscrito PML-RARα en todos los casos de LAM3 que responden al tratamiento con ATRA (ácido trans-retinoico). El ATRA induce la maduración de las células leucémicas (a promielocitos, mielocitos y neutrófilos) Leucemia aguda mielomonocítica (LAM4) Tiene un componente granulocítico y otro monocítico, en proporciones variables y con diversos grados de maduración. Los blastos monocíticos son de gran tamaño, moderada relación N/C y basofilia variable. El núcleo puede ser redondeado, arriñonado o de forma irregular. Los promielocitos atípicos son muy positivos para la MPO y Negro Sudán B. La reacción del PAS muestra positividad difusa y las fosfatasas ácidas son intensamente positivas. Son HLA-DR y CD34 negativos y son positivos para los anticuerpos monoclonales CD13 y CD33. UNIDAD III. Neoplasias hematológicas 1. Leucemias agudas y crónicas El núcleo suele ser de aspecto monocitoide (reniforme) y con un perfil bilobulado (en hachazo) con la presencia de una hendidura amplia, o bien de perfil irregular . El citoplasma es poco basófilo debido al elevado contenido de granulación azurófila. Algunos de los promielocitos atípicos contienen además inclusiones citoplasmáticas cristalinas alargadas o astillas, específicas de este tipo de leucemia, que suelen disponerse en cúmulos y que difieren de los bastones de Auer por la detección de una subestructura tubular cuando se estudian mediante microscopía electrónica de transmisión. Leucemia aguda monocítica (LAM5) Constituye alrededor de un 15% del total de LAM. Las células leucémicas son de estirpe monocítica (monoblastos y promonocitos). La LAM5 incluye 2 subtipos: LAM5a o leucemia aguda monoblástica, en la que predominan los monoblastos. LAM5b o leucemia aguda monocítica, en la que junto a los monoblastos se observa una elevada proporción de promonocitos y monocitos. El subtipo LAM5a constituye alrededor de un 5–8% de las LAM. Los elementos blásticos son de gran tamaño, con un núcleo de perfil redondeado de cromatina laxa e inmadura (1–3 nucleolos), y un citoplasma moderadamente amplio e intensamente basófilo. En el citoplasma es posible observar algún bastón de Auer y/o prolongaciones o mamelones UNIDAD III. Neoplasias hematológicas En la LAM5b (3–6% de las LAM) los promonocitos presentan un núcleo de perfil redondeado o arriñonado, y un citoplasma menos basófilo , con mayor contenido de granulación que los monoblastos y con la presencia de alguna vacuola. La observación de eritrofagocitosis junto a blastos monocíticos sugiere la existencia de una t (8;16). 1. Leucemias agudas y crónicas Leucemias agudas eritroides (LAM6) Ha sido definida por la clasificación FAB como una proliferación de elementos eritroides displásicos, junto a una proliferación de elementos blásticos de origen mieloide. Se ha categorizado en dos subtipos: 1.La eritroleucemia (LAM6a), con una proliferación blástica mixta mieloide y eritroide. 2.La LAM6 variante o leucemia eritroide pura (LAM6b) según la clasificación de la OMS. Eritroleucemia La eritroleucemia o LAM6a muestra una proliferación leucémica mixta de las series granulocítica y eritroblástica. Constituye únicamente un 5–6% del total de casos de LAM, y puede ser secundaria a un síndrome mielodisplásico previo. Para su diagnóstico se requiere que, en médula ósea, los precursores eritroides sean de un 50% o más de la totalidad celular y los mieloblastos un 30% de la celularidad no eritroide (20% según la clasificación de la OMS). En la eritroleucemia la morfología eritrocitaria de sangre periférica está muy alterada, con presencia de esquistocitos, «hematíes pinzados» o en forma de seta, hematíes espiculados del tipo equinocitos y acantocitos LAM6 variante (leucemia eritroide pura según la clasificación de la OMS)En la LAM6 variante, más de un 80% de la celularidad de médula ósea está constituida por elementos eritroides, siendo el componente mieloide inferior al 3%. La leucemia eritroide pura se asocia a alteraciones importantes de la morfología eritrocitaria en SP, tales como macrocitosis, punteado basófilo, cuerpos de Howell-Jolly o anillos de Cabot. En la eritroleucemia o LAM6a los blastos presentan antígenos mieloides (CD13, CD33, CD15) y, en la leucemia eritroide pura o LAM6b expresan antígenos específicos de la serie eritroide (Glicoforina A o Glicoforina C), y son negativos para los antígenos mieloides. Las anomalías cromosómicas se sitúan frecuentemente en los cromosomas 5 y 7. Leucemia aguda megacarioblástica (LAM7) Representa un 3–5% de las LAM. Los blastos muestran un aspecto morfológico muy inmaduro, y son muy polimórficos. El núcleo es excéntrico, de cromatina laxa y reticulada y con 1–3 nucleolos prominentes. UNIDAD III. Neoplasias hematológicas 1. Leucemias agudas y crónicas El citoplasma es basófilo, agranular y muestra un aspecto muy similar a las plaquetas circulantes con presencia de mamelones o seudópodos . Se observan micromegacariocitos y fragmentos megacarioblásticos en SP, así como una gran dismorfia plaquetaria (plaquetas gigantes y algunas con marcada desgranulación). Los blastos muestran positividad para CD61 (glicoproteína IIIa), CD41 (glicoproteína IIb/IIIa) y CD42 (glicoproteína Ib). La t(1;22)(p13;q13) es frecuente en niños menores de 1 año. Leucemias cronicas Hay dos tipos principales de leucemia Iinfoide crónica , la linfocitica crónica y la de células pilosas . leucemia linfocítica crónica Las células B CD5 + sufren una transformación maligna. Las células B se activan continuamente mediante la adquisición de mutaciones que conducen a la linfocitosis monoclonal de células B (LMB). La acumulación adicional de anormalidades genéticas y la posterior transformación oncogénica de las células B monoclonales produce LLC. Los linfocitos inicialmente se acumulan en la médula ósea y luego se extienden a los ganglios linfáticos y otros tejidos linfoides, lo que eventualmente induce esplenomegalia, hepatomegalia y síntomas sistémicos como fatiga, fiebre, sudores nocturnos, saciedad temprana y pérdida de peso involuntaria. UNIDAD III. Neoplasias hematológicas 1. Leucemias agudas y crónicas A medida que progresa la LLC, la hematopoyesis anormal causa anemia, neutropenia, trombocitopenia y menor producción de inmunoglobulinas. La hipogammaglobulinemia puede desarrollarse en hasta dos tercios de los pacientes, lo que aumenta el riesgo de complicaciones infecciosas. Los pacientes tienen una mayor susceptibilidad a las anemias hemolíticas autoinmunes (con una prueba de antiglobulina directa positiva) y a la trombocitopenia autoinmune. La LLC puede evolucionar a leucemia prolinfocítica de células B y puede transformarse en un linfoma no Hodgkin de mayor grado. Aproximadamente del 2 al 10% de los casos de LLC se convierten en linfoma difuso de células B grandes (llamada transformación de Richter). Signos y síntomas A menudo, los pacientes son asintomáticos al principio, con comienzo insidioso de síntomas inespecíficos (p. ej., cansancio, anorexia, descenso de peso, fiebre y/o sudoración nocturna), que pueden instar a una evaluación urgente. Más del 50% de los pacientes tienen adenopatías. La adenopatía puede ser localizada (los más frecuentemente involucrados son los ganglios linfáticos cervicales y los supraclaviculares) o generalizada. La esplenomegalia y la hepatomegalia son menos comunes que las adenopatías. La afectación cutánea (leucemia cutis) es rara. Diagnóstico •Hemograma completo y frotis periférico •Citometría de flujo de la sangre periférica •Inmunofenotipificación . El diagnóstico de leucemia linfocítica crónica se sospecha por primera vez cuando se encuentra una linfocitosis periférica absoluta > 5000/mcL. La citometría de flujo e sangre periférica puede confirmar la clonalidad de las células B circulantes. Los linfocitos circulantes deben expresar las cadenas ligeras CD5, CD19, CD20, CD23 y kappa o lambda. En los pacientes con un recuento absoluto de linfocitos < 5000/mcL pero con evidencia de clonalidad se diagnostica linfocitosis de células B monoclonales. Alrededor del 1 al 2% de los casos de linfocitosis de células B monoclonales progresa a LLC en un año. No se necesita aspirado ni biopsia de médula ósea para el diagnóstico de la LLC. Sin embargo, si se efectúa biopsia, la médula a menudo muestra > 30% de linfocitos. Otros hallazgos en el momento del diagnóstico pueden incluir hipogammaglobulinemia (< 15% de los casos), aumento de lactato deshidrogenasa (LDH), el ácido úrico y las enzimas hepáticas y, rara vez, hipercalcemia. UNIDAD III. Neoplasias hematológicas 1. Leucemias agudas y crónicas Leucemia de células pilosas Los estudios citogenéticos y moleculares realizados a partir de sangre periférica en el momento del diagnóstico ayudan a La leucemia de células pilosas es un trastorno maligno raro que conforma el 2% de todas las lucemias Por lo general afecta a determinar el pronóstico. La clasificación utiliza los sistemas de estadificación de Rai o Binet. pacientes adultos el promedio de edad es de 50 años tiene un comienzo insidioso caracterizado por debilidad y letargo. En el Ninguno de los sistemas predice efectivamente la progresión 80% de los pacientes se halla esplenomegalia. La pancitopenia temprana de la enfermedad. Las imágenes de rutina no se es característica de este trastorno y el aspirado de médula ósea recomiendan para la estadificación inicial. no produce material como resultado de su infiltración y la de los Tratamiento Quimioinmunoterapia, agentes biológicos y a veces radioterapia órganos reticuloendoteliales por las células pilosas. Éstas tienen Tratamiento de sosténLa leucemia linfocítica crónica se considera núcleos reniformes a ovalados con cromatina finamente incurable con el tratamiento de referencia actual; la terapia está granular y citoplasma de color gris fino con proyecciones dirigida a la mejoría de los síntomas. Por lo tanto, el tratamiento se (filipodia), quedan el aspecto "piloso". Estas células se colorean con fosfatasa ácida que no se destruye por la preincubación con mantiene hasta que ocurre uno de las siguientes: tartrato (denominada coloración con fosfatasa ácida resistente •Síntomas atribuidos a la LLC •Linfocitosis progresiva con un aumento ≥ 50% durante un período al tartrato=TRAP). Dado que la falta de material es frecuente cuando éste se procura obtener de médula ósea, la leucemia de de 2 meses células pilosas se estudia más a menudo con biopsias de medula •Tiempo de duplicación de linfocitos de menos de 6 meses Los síntomas que indican la necesidad de tratamiento en pacientes ósea en las que las células pilosas están muy espaciadas y tienen núcleos ovalados o dentados y centrales, lo que confiere un con LLC incluyen Síntomas constitucionales (fiebre, sudoración nocturna, fatiga aspecto de "huevo frito". Desde el punto de vista inmune, la célula pilosa es del tipo B, con un estado de maduración medio o extrema, pérdida de peso) tardio, con Slg e inmunoglobulina citoplásmica, así como Hepatomegalia significativa, esplenomegalia o linfadenopatías antigeno PCA-1 pero no PC-1. Infecciones recurrentes UNIDAD III. Neoplasias hematológicas Anemia y/o trombocitopenia sintomática 1. Leucemias agudas y crónicas A semejanza de lo que sucede con pacientes con otras leucemias crónicas, los que presentan leucemia de células pilosas pueden vivir durante años con la enfermedad; la supervivencia promedio es de 7 años. Otras leucemias crónicas de origen linfoide son la leucemia prolinfocitica, la macroglobulinemia de Waklenström y la leucemia de células linfosarcomatosas. La leucemia prolinfocitica es una leucemia de células B de novo caracterizada por células que se asemejan a las prolinfocitoides de la LLC. Desde el punto de vista clínico, el nivel de leucocitos muy elevado (superior a 100 × 10`/ l ) hay esplenomegalia importante y casos raros de linfadenopatia. La macroglobulinemia de Waldenström se caracteriza por la presencia de linfocitos plasmacitoides con inmunoglobulina M (IgM) de superficie e intracitoplasmática. La secreción de IgM genera una proteína monoclonal con síndrome de hiperviscosidad. La leucemia de células linfosarcomatosas es la fase leucémica de un linfoma maligno, por lo general un linfoma lintocitico de células clivadas pequenas". Las leucemias crónicas originadas en las células T son raras. Menos del 5% de los pacientes con LLC tiene un fenotipo de celulas T. La LLC de células T es más agresiva que la de cêlulas B, y a menudo infiltra la plel. El sincrome de Sezary, o linfoma de células T cutáneo, se produce en pacientes de mediana edad que tienen erupciones cutáneas o dermatitis (una enfermedad denominada micosis fungoide). y se caracteriza por la presencia de células de Sézary que Invaden la dermis. Éstas son linfocitos T maduros que expresan CD4. Más tarde en la evolución de la enfermedad cutânea, estas células pueden circular e infiltrar ganglios linfáticos y otras órganos. Las células de Sézary tienen núcleos cerebriformes, cromatina gruesa y nucléolos discretos. El sindrome de leucemia y linfoma de células T del adulto es un trastorno de células T agresivo, caracterizado por lesiones óseas liticas, leucocitosis (recuento de leucocitos superior a 50 x 10/L), hipercalcemia e infiltración cutánea e infección por virus de la leucemia v linfoma de células T humano 1 (HTLV-1), Las células son cerebriformes pero con un tamaño mas variable que el observado en la LLC células T o síndrome de Sézary . UNIDAD III. Neoplasias hematológicas 1. Leucemias agudas y crónicas Leucemia mieloide crónica La LMC se considera un trastorno mieloproliferativo crónico. Se caracteriza por panmielosis con predominio del componente mieloide en la médula ósea, sangre periférica y otros órganos. La LMC puede presentarse a cualquier edad pero es más común después de los 45 años. Los sintomas iniciales son pérdida de peso y fatiga (como en la mayoría las leucemias crónicas). Suele haber esplenomegalia importante como resultado de la infiltración mieloide. Esta esplenomegalia puede causar dolor abdominal en el hipocondrio izquierdo o malestar gástrico. Hay anemia, leucocitosis marcada (50-500 x 10/L o mayor), trombocitosis, eosinofilia, basofilia y un espectro de maduración de granulocitos con predominio de mielocitos en sangre periférica. Los mieloblastos constituyen menos del 10% de los leucocitos circulantes. En ocasiones se observan eritrocitos nucleados; de hecho, cuando la sangre periférica se asemeja a una muestra de aspirado de médula ósea, el diagnóstico habitual es LMC , La granularidad de las células puede ser bastante variable, oscila de muy destacadas, casi similar a la granulación tóxica, a microglanular. Puede haber células seucdo-Pelger-Huët, Suele ser difícil diferenciar entre basófilos y promielocitos anormales. La fosfatasa alcalina leucocitaria (FAL) por lo general tiene un valor próximo a cero y el cromosoma Filadelfia (Ph)', una traslocación del brazo largo del cromosoma 22 al 9, t(9:22), está presente en casi todos los casos de LMC cuando se utilizan sondas moleculares sensibles, como fluorescencia en hibridación in situ (FISH) y métodos de reacción en cadena de la polimerasa.Los pacientes con LMC por lo general sufren una fase crónica con una duración promedio de 3 años. Este estadio puede prolongarse por el uso del interferón alfa . Luego se produce una fase acelerada, que se caracteriza por manifestaciones clínicas que indican empeoramiento y que responde menos al tratamiento. Aún no se comprenden bien los mecanismos que favorecen el crecimiento de las células positivas para el cromosoma Filadelfia (Ph) por encima de las células normales en la LMC, pero podrían deberse en parte a la muerte celular programada alterada (apoptosis) y a la supervivencia más prolongada de los clones de LMC. La expresión B el 2, F as y B ox,-y la actividad de caspasa-3 no se correlacionan con la progresión de la enfermedad en la LMC, pero un nivel elevado de Fase asocia con una afección intrínsecamente peor en el momento del diagnóstico, mientras que la actividad elevada de caspasa-3 se asocia con un pronóstico mejor. uniones y otras anomalías nucleares. UNIDAD III. Neoplasias hematológicas 1. Leucemias agudas y crónicas La expresión B el 2, F as y B ox,-y la actividad de caspasa-3 no se correlacionan con la progresión de la enfermedad en la LMC, pero un nivel elevado de Fase asocia con una afección intrínsecamente peor en el momento del diagnóstico, mientras que la actividad elevada de caspasa-3 se asocia con un pronóstico mejor. Los pacientes con LMC por lo general sufren una fase crónica con una duración promedio de 3 años. Este estadio puede prolongarse por el uso del interferón alfa . Luego se produce una fase acelerada, que se caracteriza por manifestaciones clínicas que indican empeoramiento y que responde menos al tratamiento. Aún no se comprenden bien los mecanismos que favorecen el crecimiento de las células positivas para el cromosoma Filadelfia (Ph) por encima de las células normales en la LMC, pero podrían deberse en parte a la muerte celular programada alterada (apoptosis) y a la supervivencia más prolongada de los clones de LMC. La expresión B el 2, F as y B ox,-y la actividad de caspasa-3 no se correlacionan con la progresión de la enfermedad en la LMC, pero un nivel elevado de Fase asocia con una afección intrínsecamente peor en el momento del diagnóstico, mientras que la actividad elevada de caspasa-3 se asocia con un pronóstico mejor. Hace poco tiempo se agregaron otros indicadores pronósticos: el recuento absoluto de linfocitos (favorable es < 30.000/gL), el tiempo para duplicar la cantidad de linfocitos (> 1 año), el nivel de hemoglobina (> 13 g/dL), el recuento plaquetario (> 150.000/pL), la (^-microglobulina (< 2,0 mg/L) y el patrón histológico de médula ósea (no difuso), entre otros.Alrededor de un tercio de los pacientes padece la “crisis blástica”, en la que su enfermedad se asemeja a la leucemia aguda; la proporción de blastos excede el 30% de células sanguíneas y de médula ósea. Un tercio de los pacientes que experimentan la crisis blástica tiene LLA y dos tercios, leucemia mieloide aguda con crisis blástica. En el tratamiento se intentan mantener o volver a los valores de la fase inicial o crónica. Se demostró que los trasplantes de médula ósea o de células madre tienen cierta eficacia. UNIDAD III. Neoplasias hematológicas 2 . Síndromes mielodisplasicos Con este término y su forma abreviada “mielodisplasia” se describe a un grupo heterogéneo de alteraciones de la médula ósea que se caracteriza por una mala producción de células hematopoyéticas y se traduce por lo general en pancitopenia. A diferencia de la anemia aplásica, la médula ósea es celular; es decir, la celularidad de la médula es normal o ligeramente disminuida, e incluso puede ser hipercelular. En el pasado se conocía a este padecimiento con el término de anemia refractaria, ya que el tratamiento con vitamínicos o hierro era inútil y sólo se brindaba tratamiento de sostén con transfusiones de sangre y sus fracciones. Con el tiempo, muchos de los pacientes desarrollan leucemia, en especial aguda y mieloblástica, lo cual provocó que por un tiempo se denominara a este grupo de enfermedades como “estados preleucémicos”. Anemia refractaria (pancitopenia, médula sin aumento de blastos o de hierro). Anemia sideroblástica (igual a la anterior, pero con aumento de hierro en la médula ósea y eritroblastos con hierro granuloso en citoplasma en forma de anillo: sideroblastos). Anemia refractaria con exceso de blastos (igual que la primera, pero con menos de 5% de blastos en sangre periférica y entre 5 y 20% de blastos en la médula ósea). Leucemia mielomonocítica crónica (igual a la anterior, pero con monocitosis absoluta: un aumento de monocitos inmaduros en sangre periférica > 1000/μl). Anemia refractaria con exceso de blastos en "transformación" (más de 5% de blastos circulantes en sangre periférica y más de 20% de blastos en médula ósea, sin sobrepasar 30%). Clasificación de la mielodisplasia En 1976 se reunió por primera vez el Grupo Cooperativo FrancoLa clasificación se ha modificado de manera reciente por la Americano-Británico (FAB) con el fi n de establecer los criterios Organización Mundial de la Salud, la cual recomienda excluir a los diagnósticos aplicables a los entonces llamados síndromes tipos 4 y 5 por considerarse leucemias y no síndromes preleucémicos o anemia refractaria con exceso de blastos. El mielodisplásicos. A continuación se describe esta clasificación. grupo FAB se reunió de nuevo en 1982 y agregó tres subtipos adicionales para dividir a este grupo de padecimientos en cinco UNIDAD III. Neoplasias hematológicas grupos: 2 . Síndromes mielodisplasicos Anemia refractaria 5 a 10%. Anemia refractaria con sideroblastos en anillo 10 a 15%. Citopenia refractaria con displasia multilínea 24%. Anemia refractaria con displasia multilínea y sidero- Anemia refractaria con exceso de blastos tipo I (1 a 5% blastos en anillo 15%. de blastos) y tipo II (6 a 19% de blastos) 40%. Se considera que los factores que producen anemia en los pacientes son la falta de diferenciación celular, la apoptosis aumentada y las alteraciones clonales malignas. Estos factores varían en cada grupo de mielodisplasia. Se ha demostrado que hay un defecto en la autoinmunidad en algunos pacientes, al igual que sucede en los casos de anemia aplásica. La enfermedad puede observarse a cualquier edad; sin embargo, es rara en la infancia y en adultos jóvenes. Predomina en adultos de ambos géneros y mayores de 50 años. Aumenta gradualmente con el paso de los años. Los signos y síntomas de presentación están relacionados con las citopenias periféricas, por lo que son inespecíficos. Un buen número de pacientes se encuentra asintomático cuando se establece el diagnóstico de manera incidental en una biometría hemática sistemática. Otros se presentan con datos similares a los de cualquier anemia crónica: fatiga, debilidad, palidez, cefalea, mareos, intolerancia al ejercicio y, en casos graves, angina. Los síntomas se instalan de manera lenta y gradual en el transcurso de meses. En ocasiones se observan casos que simulan aplasia medular: debilidad, púrpura, fiebre. El bazo puede ser palpable en los tipos 3, 4 y 5. En las variantes con mayor número de blastos, el cuadro clínico es muy similar al que se observa en la leucemia aguda. Existe un grupo denominado síndrome 5q por tener esta alteración cromosómica y, finalmente, el grupo no clasificable. Su frecuencia no está definida todavía. Causas y cuadro clínico La causa no se conoce y quizá se relacione con factores muy similares a aquellos que pueden provocar la leucemia: virus, agentes tóxicos, radiación, factores genéticos, etcétera. Es notable que se puedan encontrar alteraciones cromosómicas en la mayoría de los casos. Aunque se desconoce su incidencia, la mediana de la edad de presentación en el adulto es de 65 años, y de seis años en el niño. Los síndromes mielodisplásicos tienen en común dos cosas: 1) la presencia de citopenias 2) la dismorfogénesis de todas las estirpes celulares, UNIDAD III. Neoplasias hematológicas particularmente de los eritrocitos. 2 . Síndromes mielodisplasicos Diagnóstico y diagnóstico diferencial El diagnóstico se establece en un paciente que presenta, por lo general, pancitopenia moderada, sin blastos circulantes o con Los neutrófilos presentan anomalías notables, que incluyen núcleos mínima cantidad de ellos, con un curso clínico crónico y una bilobulados y un citoplasma hipogranuloso; se aprecian médula ósea displásica con celularidad normal o sólo ligeramente macroplaquetas o microplaquetas. disminuida. Las alteraciones hematológicas que incluyen una o La enfermedad puede confundirse con la anemia aplásica; sin más estirpes celulares y que se acompañan de características de embargo, la celularidad normal o aumentada de la médula ósea es displasia en la morfología celular constituyen los rasgos distintivos la regla en la mielodisplasia, en tanto que en la aplasia siempre de la mielodisplasia. La anemia casi siempre está presente y se existe hipocelularidad grave que puede demostrarse mediante la relaciona con una respuesta de reticulocitos inadecuadamente biopsia correspondiente. En algunos casos se puede presentar una baja que refleja el daño medular. En el examen del frotis de la mielodisplasia con ligera hipoplasia, la cual resulta difícil de sangre periférica se aprecia la anormalidad eritrocítica distinguir de la anemia aplásica leve o moderada. Hay cambios característica de este grupo de trastornos: la ovalomacrocitosis, cromosómicos hasta en el 75% de los casos, principalmente que consiste en la presencia de glóbulos rojos muy grandes, que Afectando los cromosomas 5, 7 y 8; de éstos, el más común es el han perdido su morfología normal para adoptar una forma oval 5/5q (eliminación) seguido de monosomía -7/7q. El primero de ellos, que el observador experimentado advierte de manera fácil; que constituye el clásico síndrome 5q-, es común en mujeres de además se observa poiquilocitosis, anisocitosis, anisocromía y edad avanzada, con anemia microcítica y trombocitosis, sin punteado basófilo.En casos graves puede advertirse la presencia deficiencia de hierro; tiene buen pronóstico. La monosomía 7, por de eliptocitos, eritrocitos en forma de lágrima (llamados también el contrario, implica un mal pronóstico.La leucemia aguda se dacriocitos), esquistocitos, estomatocitos o acantocitos. En la distingue porque el número de blastos en la médula ósea es mayor tinción de hierro de la médula ósea, casi siempre se puede de 20% y el cuadro clínico es de aparición relativamente rápida. corroborar la presencia de eritroblastos con más de cinco gránulos Además, la leucemia aguda es común en pacientes más jóvenes. de hierro alrededor del núcleo, que se llaman sideroblastos en anillo. UNIDAD III. Neoplasias hematológicas 2 . Síndromes mielodisplasicos La anemia megaloblástica puede remedar a la mielodisplasia; sin embargo, los cambios de gigantismo celular son muy notables y hay aumento de la enzima deshidrogenasa láctica en el suero de casi 100% de los casos. En la mielodisplasia, esto no ocurre, a menos que haya una franca transformación leucémica. Los tratamientos con vitamínicos y hierro suelen resultar inútiles en la mielodisplasia, ya que no existe deficiencia de estos elementos. En resumen, se hace el diagnóstico en un paciente con citopenia, sin causa lógica manifiesta, sin respuesta al tratamiento previo y se confirma con un estudio de la médula ósea. Se debe tener en mente descartar la insuficiencia renal, el carcinoma oculto y el hipotiroidismo en estos pacientes. Tratamiento y pronóstico Recientemente se ha establecido un índice pronóstico internacional basado en las características citogenéticas de este grupo de padecimientos. En este índice, los pacientes son asignados a tres categorías de riesgo: bueno, intermedio y malo, con una mediana de supervivencia de más de 24, 18 y menos de 12 meses, respectivamente. Estas categorías concuerdan con los tres porcentajes de blastos en la médula ósea en los que se basa la clasificación de los síndromes mielodisplásicos. Además, se han propuesto cuatro categorías según alteraciones cromosómicas, número de citopenias y el porcentaje de blastos presente. Los pacientes de mejor pronóstico tienen sólo síntomas de anemia y menos de 5% de blastos. Si sólo existe displasia pura de la serie eritroide la sobrevida puede llegar a los 10 años, mientras que si existe anemia refractaria con exceso de blastos este periodo generalmente es menor de seis meses. El tratamiento suele ser decepcionante en muchos de los pacientes, a quienes se les mantiene con medidas de sostén, por ejemplo, transfusiones de glóbulos rojos; cuando hay trombocitopenia importante se les transfunden plaquetas. En ocasiones, la piridoxina puede ser útil en pacientes con el tipo 1 o 2 de mielodisplasia. Los andrógenos como la oximetolona, la mesterolona o el danazol pueden mejorar al paciente y mantenerlo libre de transfusiones. Éstos son útiles en los tipos 1 y 2 de las mielodisplasias. En la leucemia mielomonocítica crónica, y cuando la mielodisplasia se presenta con exceso de blastos (tipos 3, 4 y 5), la quimioterapia con dosis bajas de citarabina (ARA-C) o etopósido puede mejorar a los pacientes y en ocasiones prolongar su supervivencia. UNIDAD III. Neoplasias hematológicas 2 . Síndromes mielodisplasicos Cuando la transformación es hacia una leucemia aguda, como ocurre en el tipo 5, también se puede utilizar quimioterapia ordinaria intensiva, como sería el uso de citarabina a razón de 200 mg/m2 /día durante siete días, y daunomicina en 45 mg/m2 /día durante tres días. Recientemente se han visto respuestas alentadoras con medicamentos con capacidad hipometilante, como la dacitabina o la azacitidina; sin embargo, no ofrecen mejoría sostenida y prolongada y su costo es muy alto. Al parecer, estos medicamentos funcionan en todas las variantes de mielodisplasia. En fecha reciente se ha observado que un grupo pequeño de enfermos puede mejorar con factores estimulantes de colonias de granulocitos y de granulocitos-macrófagos. Algunos pueden mejorar con inmunosupresores, como la globulina antitimocito, o bien la ciclosporina. La eritropoyetina incrementa en ocasiones la hemoglobina de estos individuos y evita las transfusiones. Por último, hasta la fecha, la mejor alternativa de tratamiento en los síndromes mielodisplásicos la constituye el trasplante de progenitores hematopoyéticos. Este tratamiento en la actualidad se ha simplificado, ya que se puede utilizar una modalidad poco dinámica que consiste en reducir la intensidad de la quimioterapia de preparación pretrasplante; esto reduce la toxicidad y mortalidad relacionadas con el procedimiento y permite considerar esta opción de tratamiento en aquellos que cuenten con un donador HLA idéntico. Los ancianos, aquellos con citopenias graves y los que presentan un aumento en el número de blastos tienen el peor pronóstico. Los tipos 1 y 2 suelen tener mejor supervivencia y en ocasiones sobrepasan los cinco años. Hay un subgrupo especial de pacientes con síndrome mielodisplásico al que pertenecen mujeres con la variedad tipo 1 (anemia refractaria) y trombocitosis, además de la eliminación parcial de 5q (eliminación cromosómica); ésta es la forma más indolente de todos los síndromes mielodisplásicos, y la que muestra una menor tendencia a su evolución hacia una leucemia mieloblástica aguda, por lo que tienen una evolución estable y relativamente benigna, con buena respuesta a los andrógenos y a la eritropoyetina. La lenalidomida parece ser útil en este grupo de pacientes. Aquellos que cuentan con un donador HLA idéntico y que se someten a un trasplante alógenico hematopoyético pueden obtener la curación definitiva. UNIDAD III. Neoplasias hematológicas 3 . Síndromes mieloproliferativos crónicos Los síndromes mieloproliferativos crónicos se caracterizan por la proliferación crónica de una o más de las tres estirpes celulares de la sangre o la de las células del estroma medular en diferente proporción. La policitemia vera, la trombocitosis esencial, la mielofibrosis idiopática y la leucemia granulocítica crónica se conocen como síndromes mieloproliferativos crónicos. En estas enfermedades, en las que hay una transformación clonal de la célula madre pluripotencial, hay una sobreproducción de una o más de las líneas mieloides sin la presencia de un estímulo definido. Estas enfermedades comparten varias características clínicas y de laboratorio entre ellas. Policitemia vera Como otros síndromes mieloproliferativos, la policitemia vera (PV), o policitemia rubra vera, es resultado de la proliferación anormal de una célula madre pluripotencial que da lugar a hematopoyesis clonal de glóbulos rojos, blancos y plaquetas, entre los que predomina la hiperplasia eritroide. En cultivos in vitro de progenitoras hematopoyéticas de pacientes con PV se ha demostrado tanto hipersensibilidad de estas células progenitoras eritroides a la acción de la eritropoyetina como capacidad de formar colonias independientes del estímulo de ésta. Cuadro clínico Los pacientes que sufren PV presentan síntomas secundarios al aumento en el volumen sanguíneo: cefalea, astenia, mareo, alteraciones visuales, disnea, prurito, diaforesis, pérdida de peso; también tienen dolor epigástrico, una complicación común que resulta del aumento de la secreción gástrica debido a las cifras altas de histamina causadas por la basofilia existente. La eritromelalgia (dolor ardoroso en pies y manos, acompañado de eritema, palidez o cianosis) se observa con frecuencia y se considera que es secundario a complicaciones trombóticas microvasculares. El prurito en la piel, en particular después de bañarse, puede ser muy grave. Los pacientes suelen desarrollar gota y el cuadro clínico característico a causa del aumento en los niveles de ácido úrico originado por el acelerado recambio celular. Los pacientes suelen ser delgados, lo que es un reflejo del estado hipermetabólico vinculado con hematopoyesis aumentada. Durante la exploración física se puede encontrar aspecto pletórico, inyección conjuntival y cianosis en piel y mucosas; 66% de los pacientes presenta esplenomegalia e hipertensión arterial; la mitad también tiene hepatomegalia. UNIDAD III. Neoplasias hematológicas 3 . Síndromes mieloproliferativos crónicos Son comunes las manifestaciones por trastornos de la hemostasia. Las complicaciones trombóticas son la principal causa de morbilidad y mortalidad: 66% de ellas son arteriales. Las complicaciones hemorrágicas se presentan en 30 a 40% de los pacientes y son consecuencia de trastornos funcionales de las plaquetas. Datos de laboratorio La biometría hemática muestra un aumento en la concentración de hemoglobina (suele variar entre 18 y 24 g/dl) y del hematócrito (superior a 60% en la mitad de los pacientes); la masa de glóbulos rojos se halla incrementada a más de 36 ml/kg en el varón y más de 32 ml/kg en la mujer. Como consecuencia de este aumento, la viscosidad sanguínea puede ser cinco a ocho veces mayor que lo normal. Existe leucocitosis moderada, con cifras de 11 000 a 25 000/μl, con aumento en el porcentaje de bandas, basófilos y eosinófi los pero sin mieloblastos. En 75% de los casos hay trombocitosis con recuento de plaquetas superior a 450 000/μl; se habla de trombocitemiacuando el recuento excede el millón por microlitro (10% de los pacientes). Para el diagnóstico, no se requiere aspirado ni biopsia de la médula ósea. De hecho, el aspirado brinda muy poca información; sin embargo, los datos que orientan el diagnóstico en caso de realizarse son: hipercelularidad, aumento de los megacariocitos (lo que diferencia a la policitemia vera de otro tipo de policitemia) y ausencia de sideroblastos en la tinción de hierro de Peris o azul de Prusia (evidencia del consumo excesivo del hierro almacenado para la síntesis aumentada de hemoglobina); puede observarse incremento en la reticulina y en casos avanzados fi brosis, que llega a convertirse hasta en una tercera parte de los casos en verdadera mielofibrosis. Otras pruebas importantes son la tinción citoquímica de la enzima fosfatasa alcalina de los leucocitos, que se encuentra elevada, en tanto que en la leucemia granulocítica crónica suele estar disminuida o ausente. En 40% de los pacientes, la vitamina B12 sérica está aumentada (> 900 ng/L); en 70% aumenta la capacidad de saturación de la transcobalamina l, con hiperuricemia (en 30% de los individuos) y el incremento de la deshidrogenasa láctica observada en los estados caracterizados por un alto recambio celular. Las cifras de eritropoyetina suelen ser normales o reducidas. En presencia de trombocitosis, puede encontrarse hiperpotasiemia espuria. UNIDAD III. Neoplasias hematológicas 3 . Síndromes mieloproliferativos crónicos En 15 a 20% de los enfermos, hay alteraciones citogenéticas al momento del diagnóstico; las más frecuentes son: +8, +9 y eliminación 20. La mutación V617F del gen JAK-2 se encuentra presente con mucha frecuencia en los síndromes mieloproliferativos crónicos, y en particular en esta enfermedad se observa hasta en 95% de los casos. Esta mutación parece estar implicada en la patogenia. Tratamiento El paciente que no se trata muere antes de dos años, por lo general debido a algún fenómeno trombótico o hemorrágico. En 60% de los casos, el tratamiento adecuado permite una supervivencia de 10 años. Hay diferentes opciones de tratamiento que implican tipos de complicaciones particulares; sin embargo, la única potencialmente curativa es el trasplante de células hematopoyéticas. Flebotomía:Reduce el volumen sanguíneo y de la masa eritrocítica a cifras normales. Su objetivo es suprimir la eritropoyesis mediante la inducción de defi ciencia de hierro. En un inicio se practica a intervalos de dos a tres días; la meta es reducir la masa de glóbulos rojos y mantener el hematócrito por debajo de 45%. Sin embargo, este procedimiento aumenta las posibilidades de mielofibrosis y complicaciones trombóticas, por lo que no está indicado en sujetos que sufren un estado hipercoagulable o con antecedentes de accidente cerebrovascular, infarto de miocardio o trombosis venosa profunda. UNIDAD III. Neoplasias hematológicas 3 . Síndromes mieloproliferativos crónicos Quimioterapia: La hidroxiurea es el mielosupresor más utilizado en el tratamiento de los síndromes mieloproliferativos; es un agente que inhibe la síntesis de DNA y que no aumenta el riesgo de inducir leucemia secundaria. El busulfán es un alquilante que se utiliza sobre todo en individuos de edad avanzada y con el cual se puede lograr mielosupresión duradera a dosis bajas (2 a 4 mg/kg). Uno de sus principales efectos secundarios es fibrosis pulmonar. Otras opciones de tratamiento son interferón α, que puede suprimir la proliferación de progenitores hematopoyéticos y la anagrelida, una quinazolona oral que afecta la maduración de los megacariocitos y es útil para controlar la trombocitosis. Terapéutica antitrombótica Se utiliza para esto el ácido acetilsalicílico a baja dosis (80 a 100 mg/día) y se recomienda en pacientes con antecedente de trombosis o enfermedad cardiovascular en adición al tratamiento citorreductor. En aquellos en que dicho medicamento no sea suficiente, puede utilizarse el clopidogrel o la ticlopidina. Cuadro clínico En muchos de los casos, el diagnóstico es un hallazgo fortuito durante la realización de una biometría hemática; sin embargo, lo más frecuente en pacientes que refieren síntomas son las manifestaciones hemorrágicas (sobre todo del tubo digestivo y la mucosa nasal) o trombóticas (en la mayoría de los casos, trombosis arterial). Estas manifestaciones trombóticas pueden conducir a datos clínicos de ataque isquémico transitorio o incluso infarto cerebral, isquemia coronaria, claudicación intermitente y eritromelalgia (enrojecimiento y dolor intenso, manifestada como ardor y dolor en las extremidades, sobre todo en las plantas de los pies). Cuando se presenta en mujeres en edad reproductiva puede causar abortos espontáneos recurrentes y retraso del crecimiento fetal. El hallazgo más común durante la exploración física es esplenomegalia grado I (hasta en 40% de los individuos), además de equimosis y otras manifestaciones hemorrágicas que se presentan con mayor frecuencia que las trombóticas. Diagnóstico Los resultados de la biometría hemática al inicio de la enfermedad son hemoglobina y hematócrito normales, recuento de leucocitos normal y recuento plaquetario alto, que en la mayoría de los pacientes es mayor de 1 000 000/μl. Trombocitosis esencial Trastorno mieloproliferativo de origen clonal de las células pluripotenciales, que tiene expresión fenotípica, sobre todo en la línea de megacariocitos y plaquetas, pero que afecta a todas las células sanguíneas. UNIDAD III. Neoplasias hematológicas 3 . Síndromes mieloproliferativos crónicos En el frotis de sangre periférica se observan agregados de plaquetas con morfología anormal. En el aspirado de médula ósea hay hiperplasia megacariocítica, por lo general sin grandes cambios displásicos. El cariotipo generalmente es normal y son raras las alteraciones citogenéticas. Como la trombocitosis esencial no tiene un marcador biológico característico, su diagnóstico se basa en criterios de exclusión . Recientemente se ha encontrado la mutación V617F del gen JAK-2 hasta en el 50% de los casos, lo cual, a pesar de no ser un marcador específico de la enfermedad, ayuda en el diagnóstico y más aún en su evolución, ya que su presencia es un indicador de mal pronóstico. Diagnóstico diferencial Incluye entidades que causan trombocitosis secundaria, como síndromes mieloproliferativos crónicos (LGC, mielofibrosis idiopática y policitemia vera), síndromes mielodisplásicos (anemia sideroblástica y síndrome 5q-) y trombocitosis reactiva por esplenectomía, defi ciencia de hierro, hemorragia aguda, hemólisis, infecciones, neoplasias epiteliales, alguna intervención quirúrgica, vincristina, linfoma y enfermedades inflamatorias crónicas Tratamiento El inicio del tratamiento depende más del cuadro clínico que del recuento plaquetario, ya que no se debe olvidar el potencial leucemógeno del uso crónico de algunos agentes utilizados para el tratamiento a largo plazo. Aunque el criterio es controvertido, hay consenso para iniciar el tratamiento cuando el recuento de plaquetas se encuentra entre 1.0 y 1.5 millones/ μl, debido al riesgo alto de hemorragias. UNIDAD II. Neoplasias hematológicas 3 . Síndromes mieloproliferativos crónicos La mejor opción para tratar presentaciones agudas es la plaquetoféresis; sin embargo, su efecto es limitado y transitorio, por lo cual debe acompañarse de la administración de algún agente terapéutico. Las opciones son administrar hidroxiurea, melfalán, clorambucilo, busulfán o pipobromán. Las alternativas son interferón y anagrelida (antiagregante para el control de la trombocitosis resistente a hidroxiurea e interferón). Los criterios para definir a los pacientes como de alto o bajo riesgo y con base en ello definir su tratamiento son: Alto riesgo: mayores de 60 años, antecedente de trombosis previa. Bajo riesgo: menores de 60 años, sin antecedentes previos de trombosis. Mielofibrosis Esta enfermedad, también conocida como metaplasia mieloide agnógena, es un trastorno clonal de las células precursoras hematopoyéticas caracterizado por la presencia en la sangre periférica de granulocitos inmaduros, precursores eritroides y dacriocitos, con grados variables de fibrosis en la médula ósea y esplenomegalia. El nombre se refiere al depósito excesivo de colágeno en la médula ósea. Etiopatogenia La causa de la mielofi brosis se desconoce, no así su origen, el cual tiene lugar en una célula madre pluripotencial común a las tres series hematopoyéticas. Aunque hay proliferación de fibroblastos en la médula ósea, éstos no forman parte de la proliferación neoplásica, ya que son normales y policlonales. La proliferación fi broblástica intramedular ocurre como consecuencia de la liberación del factor de crecimiento relacionado con las plaquetas, producido por los megacariocitos y almacenada en los gránulos α de las plaquetas. Esta proliferación da lugar a la formación de depósitos de colágeno tipos I y II, fibronectina, proteoglicanos, a neoformación ósea, fibrosis reticulínica y dilatación de sinusoides en la médula ósea con hematopoyesis intrasinusoidal. Los pacientes considerados de alto riesgo deben recibir alguna de las opciones terapéuticas descritas y en los de bajo riesgo asintomáticos se recomienda sólo ácido acetilsalicílico (80 a 100 mg/día). La supervivencia de los pacientes con trombocitosis esencial no difiere mucho de la de la población sana y la evolución de la enfermedad hacia leucemia aguda es rara; cuando esto UNIDAD II. Neoplasias hematológicas ocurre, suele ser hacia LMA M4 o M7. 3 . Síndromes mieloproliferativos crónicos Cuadro clínico La sintomatología es de inicio insidioso y refleja tres aspectos fundamentales: anemia, que se manifiesta con astenia, palidez, palpitaciones, disnea de esfuerzo y edema; estado hipermetabólico, con febrícula, diaforesis y pérdida de peso, y esplenomegalia, con dolor en hipocondrio izquierdo, distensión abdominal y plenitud posprandial. También puede haber manifestaciones hemorrágicas o trombóticas, y prurito en algunos casos. En la exploración física, el hallazgo más constante es la presencia de esplenomegalia (en 90% de los pacientes); en la mayoría de los casos es moderada, pero puede llegar a ser masiva. Datos de laboratorio La anemia tipo normocítica normocrómica es la manifestación más frecuente (en 80% de los pacientes); la valoración morfológica de la sangre periférica muestra anisocitosis, poiquilocitosis y dacriocitos (eritrocitos en lágrima), y es característico un cuadro leucoeritroblástico, es decir, la presencia de eritroblastos y células mieloides inmaduras, como mielocitos y metamielocitos. Puede encontrarse trombocitopenia o trombocitosis. En 85% de los casos, el hallazgo más constante en el perfi l bioquímico es el aumento de la deshidrogenasa láctica (DHL). El aspirado de médula ósea es difícil y en muchas ocasiones no se puede obtener muestra (punción seca). En la biopsia de la médula ósea en la fase inicial de la enfermedad, se encuentra hiperplasia eritroide, granulocítica y megacariocítica, además de fi brosis reticulínica y fi brosis colagena, que se pone de manifiesto mediante tinción argéntica. Diagnóstico Se basa en la existencia de fibrosis de la médula ósea sin una causa que la justifique, además de esplenomegalia, síndrome leucoeritroblástico y presencia de dacriocitos. Debido a que se trata de una enfermedad heterogénea, el diagnóstico diferencial es muy amplio e incluye otros síndromes mieloproliferativos crónicos (como LGC, policitemia vera y trombocitosis esencial), así como síndromes mielodisplásicos, reacciones leucemoides, leucemia de células pilosas, linfomas e infecciones crónicas (como tuberculosis). La determinación de la mutación V617F del gen JAK2está presente en el 50% de los casos. UNIDAD II. Neoplasias hematológicas 4 . Síndromes inmunoproliferativos 3 . Síndromes mieloproliferativos crónicos Tratamiento El único tratamiento eficaz para curar la mielofibrosis es el trasplante de células progenitoras hematopoyéticas, el cual, de no realizarse en las etapas tempranas de la enfermedad, puede incluso resultar difícil por la fibrosis medular extensa. Además de esta opción, hay medidas terapéuticas para tratar las citopenias como el uso de transfusiones, eritropoyetina, andrógenos e incluso talidomida que se ha usado en algunos estudios piloto en donde se demostró mejoría significativa en las citopenias. El tratamiento de los síntomas relacionados con la mieloproliferación incluye el tratamiento de la esplenomegalia, para lo cual se puede recurrir a la radiación, que es útil cuando hay esplenomegalia masiva, o si el dolor es intenso, la cirugía, que por lo general implica riesgos importantes, y el uso de quimioterápicos como la hidroxiurea, 2-clorodesoxiadenosina y alquilantes como el melfalán o lenalidomida. La mediana de supervivencia después del diagnóstico es de cinco años; menos de 20% sobrevive más de 10 años. Los signos de mal pronóstico son: presencia de anemia, trombocitopenia, hepatomegalia, fi ebre inexplicada y hemólisis importante. Las principales causas de muerte son infecciones, hemorragias, complicaciones postesplenectomía y evolución a leucemia aguda. Enfermedades de las cadenas pesadas Las enfermedades de las cadenas pesadas son trastornos de las células plasmáticas que por lo general son malignos. En la mayoría de los trastornos de las células plasmáticas, las proteínas M (proteína inmunoglobulina monoclonal) son estructuralmente similares a las moléculas de anticuerpo normales. Por el contrario, en las enfermedades de la cadena pesada, se producen inmunoglobulinas monoclonales incompletas (paraproteínas verdaderas). Solo están formadas por las cadenas pesadas (alfa [α], gamma [γ], mu [μ] o delta [δ]), sin cadenas livianas (no se ha descrito una enfermedad por cadenas pesadas épsilon [ε]). La mayoría de las proteínas de las cadenas pesadas son fragmentos de las proteínas normales correspondientes, con deleciones internas de longitud variable; estas deleciones parecen deberse a mutaciones estructurales. El cuadro clínico es más similar al linfoma que al mieloma múltiple. Se consideran enfermedades de las cadenas pesadas en pacientes con manifestaciones clínicas que sugieren trastornos linfoproliferativos. UNIDAD II. Neoplasias hematológicas 4 . Síndromes inmunoproliferativos Enfermedad de las cadenas pesadas de IgA (enfermedad de la cadena alfa) Por lo general, la enfermedad de las cadenas pesadas de IgA aparece entre los 10 y 30 años, y se concentra geográficamente en Oriente Medio. La causa puede ser una respuesta inmunitaria aberrante a un parásito o a un microorganismo. Por lo general, se observa atrofia vellositaria e infiltración de la mucosa yeyunal, y a veces, infiltración de los ganglios linfáticos mesentéricos. Habitualmente, no afecta ganglios linfáticos periféricos, médula ósea, hígado ni bazo. Rara vez se ha comunicado una forma de la enfermedad que compromete las vías respiratorias. Las manifestaciones comunes incluyen fiebre, anemia leve, dificultad para tragar (disfagia), infecciones recurrentes de las vías aéreas superiores y agrandamiento del hígado y el bazo. No hay lesiones osteolíticas. Casi todos los pacientes presentan linfoma abdominal difuso y malabsorción. El hemograma completo puede mostrar anemia, leucopenia, trombocitopenia, eosinofilia y células plasmáticas o linfocitos atípicos circulantes. En la mitad de los casos, la electroforesis de proteínas séricas es normal; a menudo, hay aumento de las fracciones alfa-2 y beta o disminución de la fracción gamma. El diagnóstico requiere la detección de una cadena alfa monoclonal en la electroforesis con inmunofijación. En ocasiones se encuentra esta cadena en orina concentrada. Si no puede hallarse en suero o en orina, se requiere una biopsia intestinal. En ocasiones, puede hallarse la proteína anormal en las secreciones intestinales. El infiltrado celular intestinal puede ser pleomorfo y no francamente maligno. No hay proteinuria de Bence Jones. La evolución es sumamente variable: algunos pacientes mueren en 1 o 2 años, mientras que otros tienen remisiones que pueden persistir muchos años, en particular después del tratamiento con corticoides, fármacos citotóxicos y antibióticos de amplio espectro. Enfermedad de las cadenas pesadas de IgG (enfermedad de la cadena gamma) La enfermedad de las cadenas pesadas de IgG afecta fundamentalmente a hombres ancianos, pero puede diagnosticarse en niños. Los trastornos crónicos asociados son artritis reumatoide, síndrome de Sjögren, lupus eritematoso sistémico, tuberculosis, miastenia grave, síndrome hipereosinofílico, anemia hemolítica autoinmunitaria y tiroiditis. Se observan reducciones de las concentraciones de inmunoglobulinas normales. Las lesiones osteolíticas son infrecuentes. A veces aparece una amiloidosis. UNIDAD II. Neoplasias hematológicas 4 . Síndromes inmunoproliferativos Las manifestaciones comunes son linfadenopatías y hepatoesplenemegalia, fiebre e infecciones recurrentes. Se observa edema palatino en alrededor del 25% de los pacientes. El hemograma completo puede mostrar anemia, leucopenia, trombocitopenia, eosinofilia y células plasmáticas o linfocitos atípicos circulantes. El diagnóstico requiere la demostración por inmunofijación de fragmentos de cadenas pesadas monoclonales libres de IgG en suero y orina. De los pacientes afectados, la mitad tiene componentes monoclonales séricos > 1 g/dL (10 g/L) (a menudo, amplios y heterogéneos), y la mitad presenta proteinuria > 1 g/24 horas. Si bien las proteínas de las cadenas pesadas pueden corresponder a cualquier subclase de IgG, la subclase G3 es especialmente frecuente. La biopsia de médula ósea o ganglio linfático, que se realiza si otras pruebas no son diagnósticas, revela histopatología variable. La mediana de la supervivencia en caso de enfermedad agresiva es de alrededor de 1 año. Por lo general, la muerte se debe a una infección bacteriana o una enfermedad maligna progresiva. Los agentes alquilantes, la vincristina o los corticoides y la radioterapia pueden inducir remisiones transitorias. Enfermedad de las cadenas pesadas de IgM (enfermedad de la cadena mu) La enfermedad de las cadenas pesadas de IgM afecta, la mayoría de las veces, a adultos > 50 años. El compromiso de órganos viscerales (bazo, hígado, ganglios linfáticos abdominales) es frecuente, pero no así las abundantes linfadenopatías periféricas. Puede haber fracturas patológicas y amiloidosis. Por lo general, la electroforesis de proteínas séricas es normal o muestra una hipogammaglobulinemia. Se observa proteinuria de Bence Jones (tipo κ) en el 10-15% de los pacientes. El hemograma completo puede mostrar anemia, leucopenia, trombocitopenia, eosinofilia y células plasmáticas o linfocitos atípicos circulantes. Por lo general, el diagnóstico requiere examen de médula ósea; se observan células plasmáticas vacuoladas en dos tercios de los pacientes y, cuando están presentes, son prácticamente patognomónicas. La muerte puede sobrevenir en algunos meses o tras muchos años. La causa habitual de muerte es la proliferación incontrolable de células de leucemia linfocítica crónica. El tratamiento depende del estado del paciente, pero puede consistir en agentes alquilantes más corticoides o ser similar al del trastorno linfoproliferativo al que más se asemeje. UNIDAD II. Neoplasias hematológicas 4 . Síndromes inmunoproliferativos Cuadro clínico Macroglobulinemia de Waldenström (MW) Es una neoplasia de la célula B caracterizada por un infiltrado La mayoría de los pacientes son asintomáticos, pero pueden linfoplasmocítico de la médula ósea, asociado a la presencia de debutar con anemia o manifestaciones de síndrome de una paraproteína monoclonal de clase IgM, misma que se hiperviscosidad: cansancio, debilidad, depósitos cutáneos. encuentra en el plasma en una alta concentración. En su hemorragia cutánea y mucosa, alteraciones visuales, síntomas de descripción original de 1944, Waldenström informó de dos neuropatía periférica y otras manifestaciones neurológicas pacientes con hemorragia oronasal, linfadenopatía, tasa de cambiantes. Un aumento del volumen plasmático puede sedimentación alta, hiperviscosidad, frotis de sangre periférica precipitar insuficiencia cardíaca. Puede haber sensibilidad al normal, citopenias y un infiltrado de predominio linfoide en la frío, síndrome de Raynaud o infecciones bacterianas recurrentes. El examen físico puede mostrar linfadenopatías, médula ósea. El origen de la macroglobulinemia de Waldenström se desconoce; hepatoesplenomegalia y púrpura (que pocas veces puede ser la sin embargo, hay algunos informes que la han asociado con la primera manifestación). La marcada ingurgitación y la estenosis exposición a radiaciones ionizantes y a la infección por el virus de localizada de las venas retinianas, que se asemejan a ristras de la hepatitis C; también se han descrito anormalidades salchichas, sugieren un síndrome de hiperviscosidad. Las citogenéticas, aunque no hay alguna que sea característica de esta hemorragias retinianas, los exudados, los microaneurismas y el enfermedad. En muchos de los pacientes, la presencia de la IgM edema de papila aparecen en estadios tardíos. anormal induce trastornos serios de la coagulación, con alteración Diagnóstico en la agregabilidad plaquetaria, por lo cual la hemorragia es una de •Hemograma completo con plaquetas, índices hematimétricos y las principales manifestaciones. Con respecto a la célula de origen, frotis de sangre periférica no se ha identificado de manera precisa, aunque los datos señalan •Electroforesis de proteínas séricas, seguida de inmunofijación al linfocito T de memoria. Las células en la MW expresan niveles sérica y urinaria y niveles cuantitativos de inmunoglobulinas altos de interleucina 6 (IL-6), lo que concuerda con las cifras altas •Análisis de viscosidad plasmática en suero de la proteína C reactiva que se suele encontrar en estos casos. UNIDAD II. Neoplasias hematológicas 4 . Síndromes inmunoproliferativos La electroforesis con inmunofijación de orina concentrada suele •Examen de médula ósea, incluidas pruebas de mutaciones revelar una cadena liviana monoclonal (con mayor frecuencia específicas como MYD88 y CXCR4 kappa [κ]), pero la proteinuria de Bence Jones macroscópica es •En ocasiones, biopsia de ganglios linfáticos inusual.Los estudios de médula ósea muestran un aumento Se sospecha una macroglobulinemia en pacientes con síntomas de variable de células plasmáticas, linfocitos, linfocitos hiperviscosidad u otros síntomas típicos, en particular si hay plasmocitoides y mastocitos. Las células linfoides pueden tener anemia. Sin embargo, a menudo se la diagnostica incidentalmente material ácido peryódico-Schiff–positivo. La biopsia de ganglio cuando la electroforesis de proteínas muestra una proteína M que linfático, que se realiza si el examen de médula ósea es normal, se se identifica como IgM por inmunofijación. La evaluación de interpreta con frecuencia como un linfoma difuso bien laboratorio incluye pruebas para evaluar trastornos de las células diferenciado o uno linfocítico plasmocítico. Se determina la plasmáticas , así como la determinación de crioglobulinas, factor viscosidad plasmática para confirmar la presunta hiperviscosidad reumatoideo y crioaglutininas; estudios de coagulación; y prueba que, cuando está presente, suele ser > 4 (normal, 1,4-1,8 mPa.s de Coombs (antiglobulina) directa. Puede ocurrir anemia [miliPascal segundo]). normocítica normocrómica moderada, marcada formación en Tratamiento "pilas de monedas" de eritrocitos y una eritrosedimentación •Plasmaféresis (solo cuando existe hiperviscosidad o antes acelerada. En ocasiones se identifica leucopenia, linfocitosis del rituximab para pacientes con niveles altos de IgM) relativa y trombocitopenia. Pueden encontrarse crioglobulinas, •Corticosteroides, agentes alquilantes, análogos de nucleósidos, factor reumatoideo o crioaglutininas. Si hay crioaglutininas, la anticuerpos monoclonales (en especial rituximab) prueba de Coombs directa suele ser positiva. Pueden observarse •Otros fármacos, como los inhibidores del proteasoma diversas alteraciones de coagulación y plaquetarias funcionales. (bortezomib o carfilzomib), agentes inmunomoduladores Los resultados de los estudios sistemáticos en sangre pueden ser (talidomida, pomalidomida, o lenalidomida), ibrutinib, idelalisib o falsos en caso de crioglobulinemia o marcada hiperviscosidad. En combinaciones (en especial rituximab con ibrutinib) la mitad de los pacientes, hay disminución de las inmunoglobulinas normales. UNIDAD II. Neoplasias hematológicas 5 . Trasplante de medula ósea Cuando la médula ósea es incapaz de producir de manera adecuada las células sanguíneas y los tratamientos comunes son incapaces de corregir el problema, se requiere entonces un trasplante que restituya las células hematopoyéticas, el trasplante de progenitores hematopoyéticos (TPH). Las células hematopoyéticas (CH) pluripotenciales (células madre, progenitoras o troncales) son capaces de restablecer de manera duradera la inmunohematopoyesis después de terapia mieloablativa. Al trasplante de estas células se le conocía hasta hace pocos años como trasplante de médula ósea, debido a que para obtener células hematopoyéticas era indispensable puncionar el hueso de las crestas iliacas anterior y posterior, llegar a la médula ósea y obtener el tejido hematopoyético. Aun que esta técnica se sigue utilizando por excepción, gradualmente ha sido sustituida por la obtención de células hematopoyéticas de la sangre periférica, o bien de la sangre contenida en el cordón umbilical y la placenta. Por lo anterior, al trasplante de médula ósea en la actualidad se prefiere llamarlo trasplante de células hematopoyéticas (TCH) y agregar sólo el origen de éstas, es decir, médula ósea, sangre periférica o cordón umbilical. El cordón umbilical es rico en células hematopoyéticas, por lo que se han creado bancos de sangre de cordón para contar con una fuente de células que trasplantar en enfermos que no cuentan con un donador en su familia. Estas células se obtienen mediante la punción del cordón umbilical en el momento del parto o la cesárea y por lo regular se obtienen 50 a 100 ml de sangre que se procesan, estudian y congelan en nitrógeno líquido para su uso subsiguiente. Entonces, en la actualidad el trasplante más utilizado es el de células hematopoyéticas obtenidas de la sangre periférica. En este tipo de trasplante, estas células se obtienen de la sangre previa estimulación mediante administración subcutánea de factor estimulante de colonias de granulocitos (G-CSF, siglas en inglés de granulocyte colony stimulating factor). Este medicamento, obtenido por recombinación en bacterias, principalmente E. coli, aumenta en hasta 10 veces la producción y circulación hacia la sangre periférica de las células hematopoyéticas, con lo que se optimiza la extracción de éstas mediante hemoféresis automatizada con un procesador celular o máquina de aféresis, que de manera selectiva retira la capa de células mononucleares enriquecida con las CH. UNIDAD II. Neoplasias hematológicas 5 . Trasplante de medula ósea Tipos de trasplante Los trasplantes se clasifican según el origen de las células hematopoyéticas en: • Autólogo • cuando las células provienen del mismo enfermo Alogénico • cuando las células provienen de otro individuo. Singénico • cuando las células provienen de un gemelo idéntico. Los trasplantes autólogos se utilizan cuando el trata miento requiere quimioterapia a dosis muy altas para destruir o disminuir al máximo la enfermedad de la médula ósea. Estos trasplantes también se conocen como autotrasplantes, ya que el enfermo es estimulado para que produzca una gran cantidad de progenitores hematopoyéticos que se retiran mediante leucoféresis UNIDAD II. Neoplasias hematológicas 5 . Trasplante de medula ósea Las células hematopoyéticas obtenidas de esta manera son Por ello, este tipo de trasplante da mejores resultados si el congeladas o refrigeradas y se procede a administrar dosis altas de enfermo sufre de una enfermedad resistente a la quimioterapia, fármacos mieloablativos. Este trata miento destruye las células como suele ser el caso de leucemias en recaída o de alto riesgo . malignas al máximo, pero también destruye las células normales de la médula ósea, aunque el enfermo no muere debido a la aplasia medular resultante, ya que es “rescatado” por sus propias células hematopoyéticas, que le son transfundidas al término de la quimioterapia. Este tipo de trasplantes es útil en algunas neoplasias sólidas, mieloma múltiple, linfomas, y en menor grado, en las leucemias. Hay pruebas firmes de que esta modalidad de trasplante es útil en el tratamiento de enfermedades autoinmunes, como el lupus eritematoso diseminado o la esclerosis múltiple, entre otras. Los trasplantes alogénicos requieren de la obtención de células a partir de un donador sano, o bien de sangre del cordón umbilical. En este caso interesa no sólo el efecto de la quimioterapia a dosis altas, sino también el hecho de que los linfocitos del donador pueden atacar a la médula ósea residual del enfermo y a la misma enfermedad, lo que se conoce como efecto del injerto contra el tumor. Por ejemplo, si el enfermo sufre de leucemia, la quimioterapia destruye casi todas las células malignas, pero rara vez esto se hace de manera completa, por lo que los linfocitos del donador llevan a cabo la destrucción final del tejido enfermo del receptor completando así la curación por medio del efecto injerto contra leucemia. UNIDAD II. Neoplasias hematológicas 5 . Trasplante de medula ósea Los trasplantes alogénicos tienen la desventaja de que se requiere una compatibilidad especial para realizarlos. Curiosamente, aunque deseable, no es indispensable la compatibilidad de los grupos sanguíneos entre el donador y el receptor, la compatibilidad que interesa es la del grupo de antígenos codificados por el grupo de genes denominado complejo mayor de histocompatibilidad (CMH), que regula la síntesis de los antígenos leucocitarios humanos o HLA (siglas en inglés de human leucocyte antigens). Los genes de este complejo se localizan en el brazo corto del cromosoma 6 y codifican dos clases de antígenos: los de la clase I, denominados HLA-A, HLA-B y HLA-C, y los de la clase II, llamados HLA-DP, HLA-DQ y HLA-DR, por lo que es de interés estudiar seis antígenos en total (tres de la clase I y tres de la clase II). A causa de que el número de combinaciones posibles entre los alelos de estos antígenos es prácticamente infinito, salvo los hermanos del paciente, resulta difícil contar con donadores. Cada hermano tiene una probabilidad del 25% de compartir los mismos antígenos HLA, que se heredan como rasgos mendelianos simples. Las indicaciones terapéuticas del trasplante de células hematopoyéticas se pueden resumir en las siguientes tres generalizaciones : Restituir la hematopoyesis, después de la quimiotera pia o radiación corporal total, utilizada en el trata miento de enfermedades malignas hematológicas. Establecer una reacción de injerto contra leucemia. Reemplazar el tejido hematológico o inmune enfermo. UNIDAD II. Neoplasias hematológicas 5 . Trasplante de medula ósea Métodos para el trasplante autólogo Una vez que se cuenta con las células hematopoyéticas, se En el caso de los trasplantes autólogos, el mismo enfermo es el procede a la quimioterapia a dosis altas y subletales con el fi n de proveedor de las células hematopoyéticas. Por lo general, el destruir la médula ósea y en consecuencia la enfermedad del paciente se encuentra en remisión completa o parcial de su paciente, quien después será rescatado de esta aplasia yatrógena enfermedad, aunque esto no es un requisito absoluto. El individuo con sus propios progenitores hematopoyéticos. Realizado por un es estimulado durante cinco días con G-CSF, lo cual produce equipo médico experimentado y en condiciones óptimas, la leucocitosis granulocítica, además de aumento en el número de las recuperación postrasplante de la hematopoyesis tarda alrededor células hematopoyéticas circulantes. de 15 días, tiempo en que los neutrófilos llegan a un número En el quinto y sexto días, mediante leucoféresis, se extraen las superior a los 500/μl y las plaquetas alcanzan una cifra mayor a las células progenitoras en una cantidad mínima ideal de 2.5 20 000/μl. Reunidas estas dos condiciones se puede hablar de un millones/kg de peso corporal del paciente; se cuantifican por injerto exitoso. Por razones obvias, en el autotrasplante no existe citometría de flujo y se determina el porcentaje de células la enfermedad de injerto contra hospedador. mononucleares recolectadas que expresan sobre su superficie el antígeno de diferenciación (CD) CD34 y que se denominan células Métodos para el trasplante alogénico CD34+. En el proceso se trata de obtener la mayor cantidad En este tipo de trasplante se cuenta con un donador sano, posible de éstas, ya que un número grande permite una generalmente un hermano, cuyos antígenos del sistema HLA son recuperación rápida y predecible de la hematopoyesis. En idénticos a los del paciente, si bien es posible utilizar con los condiciones óptimas, la dosis de células CD34+ debe ser ≥ 2.5 × mismos buenos resultados otros donadores sin parentesco, 106/kg del receptor, aunque dosis de 0.75 × 106/kg se han siempre y cuando haya identidad hística entre los antígenos del trasplantado con buenos resultados. Las células CD34+ pueden sistema HLA. En ninguno de los casos es indispensable que haya refrigerarse a una temperatura entre 1 y 4°C, si se van a utilizar en compatibilidad entre los antígenos de grupo sanguíneo del los tres a cuatro días siguientes a su extracción. Cuando el sistema ABO, aunque dicha compatibilidad es altamente trasplante se va a realizar posteriormente, las células se conservan deseable. UNIDAD II. Neoplasias hematológicas por criopreservación en nitrógeno líquido 5 . Trasplante de medula ósea En el trasplante alogénico, también se estimula o moviliza al donador con G-CSF para aumentar la cantidad de las células hematopoyéticas circulantes, lo cual se logra después de cinco días. Durante los días 5 y 6 se lleva a cabo la recolección de las células hematopoyéticas del donador mediante leucoféresis de larga duración (4 a 5 h) y alto volumen (18 L de sangre procesada), con lo cual generalmente se obtienen tres a seis millones de células CD34+/kg de peso del paciente, suficientes para lograr el éxito en gran parte de los trasplantes. Al enfermo se le prepara para recibir las células hematopoyéticas con quimioterapia o radioterapia a dosis altas con el propósito de inhibir su capacidad de rechazo inmune, así como destruir la médula ósea enferma o la neoplasia que la afecta. Una vez que el enfermo recibe este “régimen de acondicionamiento”, durante los días −7 a −1, se le infunden las células del donador de modo similar a una transfusión sanguínea o por un catéter venoso central, en el llamado día cero. Para evitar las infecciones se usan de manera profiláctica antibióticos, antimicóticos y antivirales. De igual manera, con el propósito de evitar que las células del donador sean rechazadas, se utiliza la inmunoprofilaxis con ciclosporina y metotrexato (régimen inmunosupresor), medicamentos que a su vez protegen al enfermo de los linfocitos T del donador que pueden “desconocer” al hospedador y atacar hígado, piel e intestino, en un fenómeno que se conoce como enfermedad de injerto contra huésped (GVHD, siglas en inglés de graftversus host disease) aguda o crónica, que puede ser leve y controlable o grave y mortal. Hasta hace poco, este tipo de trasplante se consideraba como de alto riesgo y alto costo, por lo que su uso, con algunas excepciones, resultaba prohibitivo. Sin embargo, en Estados Unidos e Israel se diseñó una nueva técnica de trasplante que utiliza quimioterapia menos intensiva pero capaz de inmunosuprimir al receptor para que no rechace las células trasplantadas. A esta modalidad de trasplante se le denomina de intensidad reducida, o “no mieloablativo”, mismo que ha sido extensamente estudiado y difundido por investigadores mexicanos. Así, mediante la combinación de la quimioterapia y el efecto del injerto contra tumor o enfermedad, se erradica el padecimiento de manera gradual. Este tipo de trasplante es factible y en ocasiones superior al trasplante ordinario, tóxico y complicado. UNIDAD II. Neoplasias hematológicas 5 . Trasplante de medula ósea Complicaciones del trasplante de células hematopoyéticas Efectos tóxicos no hematológicos del régimen de acondicionamiento • Tempranos: alopecia, náuseas y vómito, mucositis bucofaríngea, diarrea, enfermedad venooclusiva hepática, convulsiones, pericarditis, miocardiopatía, neumonitis intersticial, cistitis hemorrágica, exantema cutáneo o hiperpigmentación, o ambos. • Tardíos: hipotiroidismo, esterilidad, trastornos del crecimiento, mucosas bucal y ocular secas, cataratas, osteopenia, osteoporosis y segundas neoplasias. Fracaso del injerto o rechazo • Primario o secundario en < 5% de trasplante de células hematopoyéticas (TCH). Enfermedad de injerto contra huésped (GVHD) aguda,factores de riesgo • Disparidad HLA entre donador y receptor. • • Donador sin parentesco. • • Edad avanzada del receptor, del donador, o de ambos. • • Alosensibilización del donador a antígenos HLA por embarazo o transfusión. • • Géneros diferentes en el par donador/receptor. • • Injerto no purgado de linfocitos T. • Dificultad para cumplir con el régimen de inmunosupresión. Enfermedad de injerto contra huésped (GVHD) crónica: manifestaciones • Esclerodermia, liquen plano, reacción liquenoide bucal o genital, síndrome seco con queratoconjuntivitis seca y pe riodontitis, trastornos de la motilidad del tubo digestivo principalmente del esófago, malabsorción, pérdida de peso, infecciones recurrentes, miositis. • La GVHD crónica ocurre en el 50% de TCH alogénicos • sin parentesco. UNIDAD II. Neoplasias hematológicas Complicaciones del régimen inmunosupresor • Osteoporosis; necrosis avascular de la cadera o el hombro; hipertensión arterial; diabetes mellitus; hiperlipidemia; ateroesclerosis prematura; insuficiencia renal; infecciones bacterianas, virales y por oportunistas; miopatías; segundas en fermedades malignas, como linfomas; depresión; obesidad; hirsutismo; acné; estrías cutáneas. 5 . Trasplante de medula ósea Mortalidad a 12 meses Es del 20 al 30% en el trasplante de células hematopoyéticas (TCH) Las recomendaciones de seguimiento pueden resumirse en: cuando el donador es un hermano HLA-idéntico y hasta de 50% en vigilancia semanal los primeros tres meses, seguimiento el TCH de donador sin parentesco o no relacionado. La recaída trimestral de la GVHD crónica; examen dental cada seis meses; sucede por lo general en los primeros dos años después del examen físico completo anual, que incluya examen de la próstata trasplante; en la leucemia aguda ocurre en el 25% de los casos. En y antígeno prostático específico, examen de mama y mamografía, el TCH autólogo existe el riesgo de que aparezcan síndrome Papanicolaou, búsqueda de melanoma y de cáncer colorrectal, mielodisplásico y leucemia mieloblástica aguda(SMD/LMA), examen oftalmológico, BH, parámetros sanguíneos, pruebas de trastornos linfoproliferativos y tumores sólidos. función tiroidea y hepática, colesterol sérico, radiografía de tórax Seguimiento de pacientes sometidos a trasplante de células y densitometría ósea. hematopoyéticas Los pacientes que reciben un trasplante de células hematopoyéticas (TCH) y sobreviven 10 o más años tienen una inmunidad casi normal, pero durante los primeros cinco años sufren de hipogammaglobulinemia persistente, trastornos en la inmunidad celular e hipoesplenismo. En los primeros dos años, son frecuentes las infecciones sinobronquiales, así como la reactivación del virus varicela zoster y el citomegalovirus hasta en el 50% de los sobrevivientes. Aunque los pacientes son inmunocompetentes a los dos años del TCH, los títulos de anticuerpo generados por vacunación previa declinan durante los primeros cuatro años. No se deben administrar vacunas durante el periodo de inmunoprofilaxis, ni a los pacientes con enfermedad de injerto contra hospedador activa. Las vacunas con virus vivos están UNIDAD II. Neoplasias hematológicas contraindicadas.