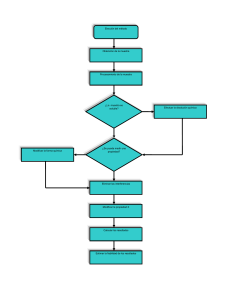
Seminario Química: Magnitud, Precisión y Datos
Anuncio

Lab. Química I (Grado Química) Curso 2012-2013 Seminario 1 - 1 SEMINARIO 1. Presentación de resultados: Magnitud, precisión, exactitud y organización de datos. Elaboración: Begoña García Magnitud física: Es la propiedad o cualidad de un fenómeno, un cuerpo o sustancia que puede ser distinguida cualitativamente y determinada cuantitativamente. En términos generales, es toda propiedad de los cuerpos que puede ser medida directa o indirectamente. Una magnitud particular sujeta a medición se conoce también como Mensurando. Ejemplo: volumen de una pipeta; masa de un vaso; temperatura del agua; concentración de HCl en una disolución, etc. Pueden clasificarse en: Magnitudes físicas fundamentales o básicas. Magnitudes físicas derivadas se expresan como combinación de las primeras. p.ej. volumen, densidad, superficie, frecuencia, periodo, fuerza, presión, trabajo, etc. Sistema de unidades: conjunto de unidades de medida a partir del cual derivan el resto. El más usado es el Sistema Internacional de Unidades o SI. Tabla 1. Unidades de magnitudes básicas en el Sistema Internacional (SI). Magnitud Unidad (Símbolo) Longitud Metro (m) Masa Kilogramo (kg) Cantidad de sustancia Mol (mol) Tiempo Segundo (s) Temperatura Kelvin (K) Corriente eléctrica Amperio (A) Intensidad luminosa Candela (cd) Ángulo plano Radián (rad) Ángulo plano Esteroradián (sr) Las demás unidades derivan de ellas, como el litro (L), volumen equivalente a un decímetro cúbico (10-3 m3). Es aceptado en el SI aunque no pertenece estrictamente a él. Además del SI, hay otros sistemas de unidades que también se utilizan en muchos casos como el gramo (g) para masa (sistema cegesimal) y el grado centígrado (ºC) para la temperatura. Lab. Química I (Grado Química) Curso 2012-2013 Seminario 1 - 2 Tabla 2. Unidades derivadas del sistema internacional. Magnitud Unidad Símbolo Frecuencia Fuerza Hertzio Newton Hz N Presión Energía, trabajo, cantidad de calor Potencia Pascal Pa Julio J Watio W Culombio C Potencial eléctrico Voltio V Resistencia eléctrica Ohmio Ω Capacidad de calor faradio F Cantidad de electricidad Frecuentemente se utilizan sus múltiplos y submúltiplos. Tabla 3. Factores de conversión Factores deSímbolo conversiónEquivalente en SI Cantidad Unidad -3 Tabla 4. Prefijos Prefijo Símbolo Factor L mL 3 10 m 10-6 m3 Peta Tera P T 1015 1012 Volumen litro mililitro Longitud angstrom Å 10-10 m Giga G 109 pulgada In. 0.0254 m Mega M 106 Fuerza dina dyn 10-5 N Kilo k 103 Presión bar bar 105 Pa Hecto h 102 atmósferas atm 101325 Pa Deca da 101 torr (=1 mmHg) Torr 133,322 Pa deci d 10-1 ergio erg 10-7 J centi c 10-2 Electronvoltio eV 1.602176462·10-19 J mili m 10-3 caloría cal 4.184 J micro µ 10-6 kilocaloría Cal 1000 cal nano n 10-9 Temperatura centígrado ºC K -273, 15 pico p 10-12 Fahrenheit ºF 1.8(K-273.15)+32 femto f 10-15 Energía Ejemplo: la concentración molar [A] (moles/litro) de un sólido o líquido A disuelto en un volumen V será mA: masa pesada del sólido o líquido A (g) [A] = (mA x PA)/MMA V PA: pureza de A (gA / gsólido ó líquido), normalmente expresada en %. MMA: masa molecular de A (g/mol) V es el volumen final de la disolución de A (L). Si A es líquido y se toma un volumen VA de densidad dA, la concentración [A] será [A] = (VA x dA x PA)/MMA V Pregunta: ¿En qué unidades debe expresarse [A]? Lab. Química I (Grado Química) Curso 2012-2013 Seminario 1 - 3 Medición y error experimental Medición: Conjunto de operaciones que tienen por objeto determinar el valor de una magnitud. Método de medición: Secuencia de operaciones descritas de manera genérica usada en la ejecución de una medición. Ejemplo: ver Uso de la Balanza y el Granatario. Todas las medidas experimentales vienen afectadas de una imprecisión inevitable o error experimental que además incluye: -imperfecciones del instrumento de medida -limitaciones impuestas por nuestros sentidos al registrar la información. Los errores experimentales pueden clasificarse en dos grandes grupos: Errores sistemáticos Errores aleatorios o indeterminados Tienen valor definido. Son impredecibles en magnitud y signo, Originados por causas identificables. Causas probables pueden ser: Variaciones entre observaciones sucesivas realizadas por un mismo operador. Para un número grande de mediciones se obtienen tantas desviaciones positivas como negativas. Empleando métodos estadísticos se puede llegar a algunas conclusiones sobre el valor más probable. Errores instrumentales. P. Ej. de calibrado. Error personal. P. Ej.: un problema visual. Error de la elección del método. P.ej. vaso de precipitados frente a pipeta. Constantes a lo largo del proceso de medida. Son incontroladas y tienen lugar siempre. Se pueden corregir y eliminar No se pueden anular pero si disminuir. Producen una desviación positiva o negativa Originan dispersión en los resultados y afectan con respecto al valor verdadero y afectan a la a la precisión de las medidas. exactitud de la medida. Por tanto, dado que el valor de las magnitudes físicas se obtiene experimentalmente por medida, bien directa o indirectamente (aplicación de fórmulas), debe admitirse como postulado físico el hecho de que Es imposible llegar a conocer el valor exacto de ninguna magnitud Sin embargo si que se pueden establecer los límites dentro de los cuales se encuentra dicho valor y esto es el principal objetivo de la denominada teoría de errores: acotar el valor de los errores experimentales. Normalmente, para tener más confianza en el resultado de la medición de una magnitud repetimos la medición varias veces (réplicas). Lab. Química I (Grado Química) Curso 2012-2013 Seminario 1 - 4 DEFINICIONES Y CONCEPTOS Error absoluto (E): está asociado a una medida y se define como la diferencia entre el valor medido experimentalmente (xi) y el valor “verdadero” de la magnitud (). Como no es posible conocer el valor real de , en su lugar se suele utilizar la media aritmética x (o promedio) de una serie de medidas independientes. Esta media estima el valor central de la distribución de medidas. Las diferencias entre cada valor y la media son errores aleatorios. P. ej. Para una serie de tres medidas: De modo práctico también se usa como valor de referencia el valor nominal de un equipo, por ej. El volumen nominal de una pipeta o de un matraz aforado. Error relativo (Er): cociente entre el error absoluto y la media x. Se suele expresar como %. Se usa cuando interesa resaltar la importancia relativa de la desviación ya que aporta más información (no es lo mismo error de 1 sobre 500 que sobre 5000, dan distinto %) Incertidumbre absoluta (U): expresa el margen de incertidumbre asociado a una medida y se define como el valor de la semiamplitud de un intervalo alrededor del valor resultante de la medida. Indica una zona de valores entre los cuales es "casi seguro" que se encuentre el valor verdadero, usualmente con un nivel de confianza de 95% (lo que equivale a asumir un riesgo de equivocarnos en 5 de cada 100 veces). Puede tener signo + o – y por tanto el resultado de la medida se expresa como xi ± U Ejemplo: una medida de 12,5 ± 0,2 indica que el valor verdadero está entre 12,3 y 12,7 cm, y puede ser igualmente 12,45 o 12,63 cm, por ejemplo. 0,2 es la incertidumbre. Incertidumbre relativa: es el cociente entre el valor de la incertidumbre absoluta y el valor de la medición, o el valor medio en su caso. Puede expresarse como %. Incluir la incertidumbre al expresar los resultados permite comparar resultados obtenidos por varios laboratorios o con diferentes metodologías. Por ejemplo si dos laboratorios A y B al expresar la cantidad de mercurio en una muestra dan valores de 14 μg/kg y 15 μg/kg, respectivamente, no podremos decir si son comparables. En cambio si se dan como: Laboratorio A: 14 ± 1 μg/kg, indica que el valor está comprendido entre 13-15 μg/kg. Laboratorio B: 15,0 ± 0,5 μg/kg indica que el valor está comprendido entre 14,5–15,5 μg/kg. Podemos contrastar si ambos resultados son comparables. El error tiene un valor concreto ------------- La incertidumbre es un intervalo Sin embargo en muchos textos utilizan indistintamente ambos términos Lab. Química I (Grado Química) Curso 2012-2013 Seminario 1 - 5 Dispersión (D): diferencia entre los valores extremos de las medidas. Si la dispersión es grande se dice que hay poca precisión y viceversa. Se puede expresar como tanto por ciento de dispersión, T. Para tres medidas: Desviación estándar (s, ): para n medidas que se distribuyen en torno a la media. n es el número de medidas realizadas. xi cada uno de los valores medidos x es la media aritmética xi – x es el error aleatorio Desviación estándar relativa o coeficiente de variación: es el cociente entre el valor de la desviación estándar y el valor medio expresado como %. Distribución de las mediciones: Mediante funciones matemáticas se pueden modelar las distribuciones de frecuencia de las distintas mediciones, lo que nos dará la probabilidad de que el valor real este entre unos valores dados. Esto es el principal objetivo de la denominada Teoría de errores: acotar el valor de los errores experimentales. Por ejemplo, la Distribución normal dada por la ecuación de Gauss corresponde a una curva del tipo indicado en la Figura 1, Ecuación de Gauss Figura 1 y: probabilidad con que se puede encontrar el valor x (variable) en la población. x: media aritmética : Desviación estándar El valor máximo de y corresponde a x = x, es decir la media es el valor más probable. La anchura de la curva depende de (desviación standard). El área bajo la curva entre dos valores dados de abcisas da la probabilidad de encontrar un valor de x entre ambos límites. Lab. Química I (Grado Química) Curso 2012-2013 Seminario 1 - 6 EXACTITUD Y PRECISIÓN Exactitud Un instrumento es exacto si la medida (x) o la media de las medidas (x) realizadas son muy próximas al valor “real" Precisión Concordancia entre una medida y otras de la misma magnitud realizadas en condiciones sensiblemente iguales Un instrumento será preciso cuando la diferencia entre medidas de una misma magnitud sea muy pequeña Se estima mediante el Error absoluto. Se estima con la Desviación estándar (s) Grado de concordancia entre el valor real y el experimental. La exactitud implica normalmente precisión, pero la precisión no implica exactitud (los instrumentos pueden ser inexactos debido a errores sistemáticos tales como error de cero, etc.) Errores Sistemáticos Aleatorios X (relac. con la exactitud) Elevados (relac. con la precisión) Elevados Inexacto Impreciso X Elevados Bajos Inexacto Preciso Bajos Elevados Exacto Impreciso Bajos Bajos X X La exactitud esta relacionada con los errores sistemáticos La precisión está relacionada con los errores aleatorios. Exacto Preciso Valor verdadero En este contexto se utilizan los términos. Repetibilidad: Se refiere a la variación de los resultados de la misma persona con el mismo equipo o instrumento, en el mismo lugar y con diferencias de tiempo pequeñas. Reproducibilidad: Se refiere a la variabilidad en las condiciones de la medida de una misma magnitud, como p.ej. si la efectúan distintos operadores, en distintos días, en distintos lugares y con distintos equipos de medida. VER Cuestión 1. Lab. Química I (Grado Química) Curso 2012-2013 Seminario 1 - 7 Incertidumbre en magnitudes directas El valor de una magnitud se expresa mediante la indicación del equipo o instrumento medidor seguido de la incertidumbre U con el signo mas/menos. Ese valor puede ser un valor individual o la media de varias medidas: a) Si se realiza una única medida. Se considera que la incertidumbre absoluta U es el valor de la Sensibilidad (S) del instrumento, es decir el valor mínimo que es capaz de medir. xi ±S (xi ±U) Normalmente, se admite que la sensibilidad de un aparato viene indicada por el valor de la división más pequeña de la escala de medida o por la última cifra significativa (escalas digitales). b) Se realizan varias medidas que pueden presentarse poco o muy dispersas, se sigue el siguiente procedimiento: 1. Se realizan siempre tres medidas y se calcula el valor medio (x), la dispersión D, y el tanto por ciento de dispersión, T. Si D ≤S Valor verdadero = Valor medio (x) Incertidumbre absoluta = Sensibilidad (S) . Si D >S se aumenta el número de medidas según el valor del porcentaje de dispersión T T en las tres primeras medidas nº total de medidas necesarias T ≤ 2% Bastan las 3 medidas realizadas 2% < T ≤ 8% Hay que hacer 3 medidas más, hasta un total de 6 8% < T ≤ 15% Hay que hacer un total de 15 medidas 15% < T Hay que hacer 50 medidas como mínimo 2. Una vez realizadas las medidas necesarias se toma: Valor verdadero = valor medio (x); Imprecisión = desviación estándar (s o ). Incertidumbre en magnitudes indirectas Normalmente es necesario hacer operaciones aritméticas con números que tienen cada uno de ellos un error. Para calcularlo seguiremos las siguientes pautas: Función Sumas y Restas Multiplicación y División Para otras funciones Incertidumbre (U) ó Desviación estándar (s, ) La incertidumbre (U) o la desviación estándar (s o ) se obtiene a partir de las incertidumbres (Ui) o desviaciones estándar (si o i) de cada término individual según la fórmula. Por ejemplo para tres valores: Se expresa U ó s () como porcentajes y después se aplica la fórmula: y= xa y = log x y = ln x y = 10 x y =e x % Uy = a % Ux Uy = 0.43429 (Ux / x) Uy = (Ux / x) (Uy / y) = 2.3026 Ux (Uy / y) = Ux Lab. Química I (Grado Química) Curso 2012-2013 Seminario 1 - 8 CIFRAS SIGNIFICATIVAS Al presentar resultados cuantitativos se ha de tener presente tanto el valor del resultado como la incertidumbre del mismo. El valor de una magnitud debe tener las cifras necesarias para que su última cifra significativa sea del mismo orden decimal que la última de la incertidumbre. Cifras significativas son todos los dígitos relevantes que sean fiables más el primero afectado por la incertidumbre (la cifra situada más a la derecha). NOTA: Si un valor de medida es leído de una tabla u otro lugar, sin indicación de su error, se toma como error la última cifra significativa. Ejemplo: 74,48 tiene 4 cifras significativas. Las tres primeras de la izquierda son seguras, la cuarta (8) es insegura. Si escribimos 74,480 estamos considerando que el 8 es seguro y el cero no. Hay unas reglas para saber cuando los ceros son o no significativos: ►Los ceros entre dígitos distintos de cero son siempre significativos. ►Los ceros situados a la izquierda del primer dígito distinto de cero en números decimales no son significativos ya que solo indican la posición de la coma. Ejemplo: 100006 (6) 12,03 (4) 0,000455 (3) 0,099 (2) ►Los ceros a la derecha del último número distinto de cero son significativos si el último de ellos corresponde al último dígito inseguro. Cuando el orden de magnitud es extremo (grande o pequeño) se debe utilizar la notación exponencial. Ejemplo: 7500 ¿Cuantas cifras significativas tiene este número? ¿2, 3 o 4? 7,5 x103 (2 cifras) 7,50 x 103 (3 cifras) 7,500 x 103 (4 cifras) VER Cuestión 2. 1. Cifras significativas de la magnitud medida: viene dada por la sensibilidad del instrumento de medida. Ejemplo: Pesamos un vaso en dos balanzas distintas: a) En una balanza de resolución 0,1 g leemos una masa de 10,3 g. ¿Cuantas cifras significativas tiene? 3. ¿Podemos escribirlo como 10,30 g? No. El 3 es la última cifra insegura ya que la balanza solo aprecia decigramos. b) En una balanza analítica con resolución de 1 mg la lectura es 10,310 g. ¿Cuántas cifras significativas tiene esta medida? 5 ¿Podemos expresar la medida como 10,31 g ya que el cero no influirá en los cálculos? No, la incertidumbre esta en los miligramos. En buretas y pipetas cuando el menisco del líquido se encuentre entre dos de las marcas de la bureta podemos tratar de estimar la segunda cifra decimal por interpolación visual. Por ejemplo, supongamos una bureta graduada en décimas de mL (0,1 mL) y en una lectura vemos que la parte inferior del menisco del líquido está entre 13,5 y 13,6 mL .Podemos tratar de estimar la segunda cifra decimal por interpolación visual y asignarle el valor 13,56 mL. El número de cifras significativas es 4, siendo el 6 el valor no seguro, ya que podría ser 13,58 o 13,54. 2. Cifras significativas después de operaciones matemáticas sencillas A) Sumas y restas: el contribuyente con menos cifras decimales determina el número de cifras significativas del resultado. Lab. Química I (Grado Química) Curso 2012-2013 Seminario 1 - 9 ► Valores con igual número de dígitos: el resultado tendrá el mismo número de decimales. Ej.: 0,0374 + 0,0235 + 0,0892 = 0,1501 ► Valores con distinto número de dígitos: el número de dígitos viene limitado por el número con menos cifras decimales. Para expresar el resultado es necesario aplicar Reglas de redondeo. a) Último dígito > 5: redondeo hacia arriba: se añade una unidad al dígito anterior: b) Último dígito < 5: redondeo hacia abajo: se deja el dígito anterior. c) Último dígito = 5: redondeo al dígito par más próximo. 0,0585; Resp.: 0,058 El redondeo se aplica secuencialmente de derecha a izquierda hasta llegar a la última cifra que se debe conservar por ser significativa. Ejemplos: a) Redondee una vez los números: 0.0785 0.078 ; 0.0795 0.080; 8552,005 8552,00; 25,658 25,66; 25,643 25,64 b) Redondee a dos cifras decimales el siguiente número 25,64489 25,6449 25,645 25,64 Si ningún número tiene cifras decimales es necesario conocer la incertidumbre de cada término o usar la notación científica. B) Multiplicación y División: El número de dígitos del resultado será generalmente el mismo número que el valor con menos cifras significativas Es importante no redondear en los cálculos intermedios, manteniendo un número de cifras significativas superior a lo que cabría esperar y efectuar el redondeo al final. Ejemplo: Para las siguientes operaciones matemáticas: a) ¿Cuántas cifras significativas tiene cada número? b) ¿Cuántas cifras significativas debe tener el resultado? c) Indique el resultado con las cifras significativas adecuadas: 88,935 x 0,121 x 290 = 3120,72915 ≈ ¿3120,7 o 3121 o 3120 o 3,12 x 103? Hay dos factores con 3 cifras significativas (0,121, 290). El resultado debe tener 3 cifras significativas; como el cero de 3120 no es significativo es mejor expresar el resultado como 3,12 x 10 3. Sin embargo, la regla tiene excepciones: si al aplicar esta regla el resultado tiene un error relativo mucho mayor que el número con mayor error relativo se le dará otra cifra significativa. Ejemplo: 45 x 23,45 x 108,0098/ (0,00192 x 56,89) = 1,092288 ¿1,1 o 1,09? Suponiendo que la incertidumbre de la última cifra es una unidad en todos los números y calculando la incertidumbre relativa para cada uno: 1/45 > 0,01/23,45 > 0,0001/108,0098 Luego el valor con mayor incertidumbre es 45 con una incertidumbre relativa de 1/45 0,02 (2%) y el resultado tendrá una incertidumbre relativa de ese orden y una incertidumbre absoluta (multiplicando por el resultado) de 1,09 x 1/45 = 0,024 0,02. La incertidumbre absoluta afecta a la segunda cifra decimal y se deben dar 3 cifras significativas. El redondeo a 1,1 es excesivo y produce mayor distorsión en el resultado ya que la incertidumbre relativa en tal caso sería de 0,1/1,1 0,1 (10%> 2%). VER Cuestión 3. Lab. Química I (Grado Química) Curso 2012-2013 Seminario 1 - 10 3. Cifras significativas del error o Propagación de la incertidumbre: Normalmente tiene una sola cifra significativa que se obtiene redondeando por arriba en una unidad si la segunda cifra es ≥ 5. Puede tener dos cifras significativas si: a) la primera cifra es 1 b) la primera cifra es 2 y la segunda no llega 5. Valores Incorrectos Valores Correctos 3.418 ± 0.123 3.42 ± 0.12 6.3 ± 0.09 6.30 ± 0.09 46288 ± 1551 46300 ± 1600 428.351 ± 0.27 428.4 ± 0.3 0.01683 ± 0.0058 0.017 ± 0.006 Incertidumbre de material o instrumentos de laboratorio Para medir volúmenes: Material volumétrico (pipetas, buretas, aforados) El material volumétrico puede ser: ● de contenido (matraces aforados y probetas) ● de vertido (pipetas, buretas, dosificadores). Existen dos calidades: A y B. La A es mejor que la B (menor incertidumbre/tolerancia) a igual volumen). Volumen (mL) 5 10 25 50 100 250 500 1000 Volumen (mL) 0,5 1 2 3 4 5 10 15 20 25 50 100 Matraces aforados Clase A Incertidumbre (mL) Volumen (mL) ±0.02 5 ±0.02 10 ±0.03 25 ±0.05 50 ±0.08 100 ±0.12 250 ±0.20 500 ±0.30 1000 Pipetas aforadas Clase A Incertidumbre (mL) Volumen (mL) ±0.006 0,5 ±0.006 1 ±0.006 2 ±0.01 3 ±0.01 4 ±0.01 5 ±0.02 10 ±0.03 15 ±0.03 20 ±0.03 25 ±0.05 50 ±0.08 100 Tolerancia (mL) (ASTM E 288) ±0.02 ±0.02 ±0.03 ±0.05 ±0.08 ±0.12 ±0.20 ±0.30 Tolerancia (mL) (ASTM E 969) ±0.006 ±0.006 ±0.006 ±0.01 ±0.01 ±0.01 ±0.02 ±0.03 ±0.03 ±0.03 ±0.05 ±0.08 Lab. Química I (Grado Química) Curso 2012-2013 Seminario 1 - 11 Buretas Clase A Volumen bureta (mL) Graduación mínima (mL) Incertidumbre (mL) 5 10 25 50 100 0.01 0.05 o 0.02 0.1 0.1 0.2 ±0.01 ±0.02 ±0.03 ±0.03 ±0.10 Volumen Graduación Tolerancia (mL) (mL) mínima (mL) (UNE-EN-ISO 385) 5 10 25 50 100 0.01 0.05 o 0.02 0.1 0.1 0.2 ±0.01 ±0.02 ±0.03 ±0.03 ±0.10 Los fabricantes imprimen sobre el vidrio información sobre el material: Volumen: Para matraces y probetas graduadas la cantidad de líquido que contendrá se indica con el prefijo ‘In’ y el volumen p. ej. 50 mL. Para pipetas y buretas la cantidad de líquido vertida se indica con ‘In’ y el volumen p. ej. 5 mL. Error máximo admisible (tolerancia), que es una estimación de la incertidumbre máxima hasta que se calibre el material. En muchos casos también aparece la temperatura de calibrado en fábrica, que suele ser 20 ºC (a tener en cuenta para trabajos rigurosos ya que el volumen contenido o vertido depende de la temperatura) y el tiempo de espera en el vaciado para pipetas el (por ej. 5 s, 5 segundos). Por ejemplo la marca BLAUGRAND indica en su catálogo para las pipetas aforadas de un enrase las siguientes indicaciones: Para medir masa: Balanzas (básculas, granatarios, balanzas analíticas, de precisión, microbalanzas, etc.). Difieren fundamentalmente en cuanto a la masa máxima que admiten y en cuanto a la resolución (la cifra más baja que podemos leer). Su incertidumbre depende del tipo de balanza y de su clase. P. ej. las balanzas analíticas aprecian hasta la décima de miligramo pero la incertidumbre depende de la masa que se pesa. La incertidumbre absoluta aumenta con la masa, pero la incertidumbre relativa es menor a medida que aumenta la masa. Bibliografía. El texto esta basado en gran medida en el documento sobre “Presentación de resultados: Magnitud, precisión, exactitud y organización de datos” elaborado por Rufino Mateo. Curso 2011-2012. Cuestiones 1. Un volumen de 10 mL se ha medido con tres tipos de material. Complete la tabla siguiente y clasifique el material según su precisión y exactitud: Lab. Química I (Grado Química) Curso 2012-2013 Seminario 1 - 12 Material 1 10.2 9.7 9.8 10.3 Material 2 10.4 10.3 10.2 10.3 Material 3 9.9 10.1 9.9 10.1 Medida 1 Medida 2 Medida 3 Medida 4 Media (x) Dispersión (D) Clasificación (exacto, preciso) 2. Determinar el número de cifras significativas de los siguientes números y redondee (en su caso) hasta 2 cifras significativas : Nº de cifras Redondeo 0,00753 77,028 0,04004500 0,0004004500 3,11 3,1134 23,742 0,0230 0,0000205 Nº de cifras Redondeo 2003 2003,00 0,00200085 120,00 1,2 x103 7,3400 x 10-6 7,340 x 10-6 7,34 x 10-6 10,0 3. Para las siguientes operaciones matemáticas indique: 0,0374 + 1,0097 + 27,3 = 28,3471 270 + 0,00257 + 20,405 = 290,40757 3,4 + 0,020 + 7,31 = 10,73 a) ¿Qué número limita el número de cifras significativas del resultado? b) Indique el resultado con las cifras significativas adecuadas: 4. Para la siguiente operación matemática: 45,89 x 0,0037 / 1008 = 1,6844 a) ¿Qué incertidumbre corresponde a cada número de la operación matemática? b) ¿Cuál es la incertidumbre relativa? ¿Y la absoluta? c) ¿Cuántas cifras significativas deben darse? 6. a) Indique la información que se deduce de lo impreso por el fabricante en las pipetas x), y) y z). b) Un estudiante ha medido el volumen que se puede ver en la imagen I-1 con una pipeta graduada de 5 mL en la cual el fabricante ha impreso la información que se puede apreciar en la imagen I-2. ¿Qué indica el fabricante? ¿Qué volumen ha medido el estudiante? x) z) y) I-1 I-2 Lab. Química I (Grado Química) Curso 2012-2013 Práctica 6 -1 PRÁCTICA 6: Preparación de disoluciones y medida de pH. Elaboración: Begoña García. OBJETIVOS: • Revisión de los conceptos de acidez, basicidad, equilibrio y pH. • Cálculo del pH de disoluciones de ácidos, bases y sales a partir de sus molaridades y según sus características químicas. • Preparación de disoluciones de distintas concentraciones: 1. A partir de una disolución acuosa concentrada de cloruro de hidrógeno gas (HCl conc.). 2. A partir de un ácido líquido (HOAc). • Preparación de disoluciones a partir de sales sólidas. • Determinación de las cifras significativas para la Molaridad a partir de la precisión de las medidas implicadas. • Uso del pH-metro y medidas de pH. INTRODUCCIÓN Las ideas sobre los ácidos y bases están presentes de forma habitual en la vida cotidiana, y durante mucho tiempo los químicos han clasificado las sustancias en ácidos y bases, y desarrollado diferentes teorías sobre las mismas. Una de las más útiles es la de Brönsted y Lowry que definieron: Esto implica que: Ácido (HA): dador de protones Base (B): aceptor de protones ►Siempre nos vamos a encontrar con pares ácido-base, ya que para que un compuesto pueda dar se necesita otro que acepte. HA + B A– + HB+ ►Las especies que se generan en el intercambio de protones son a partir del ácido HA base A– , que se denomina base conjugada de HA. a partir de la base B ácido HB+, que se denomina ácido conjugado de B. ►La acidez o basicidad de una sustancia depende del compuesto con el que interacciona. Puesto que normalmente los compuestos se disuelven, las características del disolvente pueden influir en las propiedades ácido-base del compuesto. Así, cuando el fluoruro de hidrógeno (ácido) o el amoniaco (base) se disuelven en agua HF (aq) + H2O (l) H3O+ (aq) + F- (aq) NH3 (aq) + H2O (l) NH4+ (aq) + OH- (aq) Si analizamos estos equilibrios con detalle vemos que el agua puede actuar como ácido o base Lab. Química I (Grado Química) Curso 2012-2013 Práctica 6 -2 El agua también puede actuar como ácido o base frente a sí misma, en un equilibrio que se conoce como autoionización del agua, y a su constante de equilibrio, como Kw (constante del producto iónico del agua). En términos generales la ecuación de Kw se considera válida para cualquier disolución acuosa diluida, y su valor puede utilizarse para calcular la [H3O+] si se conoce [OH-] y viceversa. Por otra parte, como en disolución acuosa la [H3O+] y la [OH─] suelen ser muy pequeñas, se utilizan preferentemente los términos pH y pOH, que se definen como: pH = log [H3O+] pOH = log [OH─] y pH + pOH = 14 En una disolución neutra [H3O+] = [OH–] = 10-7 M y pH = 7,00 (dos cifras decimales) y será: Disolución ácida [H3O+] > 10-7 M pH < 7,00 Disolución básica [H3O+] < 10-7 M y su pH > 7,00 La medida del pH de disoluciones acuosas es una práctica corriente en el laboratorio. Para medidas rápidas aproximadas se utilizan indicadores, compuestos que cambian de color en un intervalo estrecho de pH, o papel de pH, papel impregnado con mezcla de distintos indicadores. Para medidas más exactas se utiliza el pHmetro (ver Anexo I). El pH de ácidos y bases fuertes Son electrolitos fuertes que en disolución acuosa están completamente disociados en sus iones. HCl (g) H2O H+ (aq H3O+) + Cl- (aq) NaOH H2O Na+ (aq) + OH- (aq) Los ácidos fuertes más comunes son: HCl, HBr, HI, HNO3, HClO3, HClO4, H2SO4. En sus disoluciones acuosas son la única fuente importante de protones por lo que para los ácidos monopróticos: [H3O+] = [ácido] pH = log [ácido] Las bases fuertes solubles más comunes son NaOH y KOH, y en disolución acuosa: [OH–] = [base] pOH = log [base] Estos cálculos son válidos para concentraciones de ácido o base mayores de 10-6 M ya que si la concentración es muy baja hay que tener en cuenta el aporte de la autoionización del agua. El pH de ácidos y bases débiles La mayoría de los ácidos y bases sólo se encuentran parcialmente ionizados en disolución acuosa y su grado de ionización viene indicado por su constante de equilibrio. Para cualquier ácido débil HA la ecuación será La [H2O] se supone constante ya que es el disolvente y la constante de ionización ácida Ka de HA se define como: + - Ka = [H3O ] [A ] [HA] La magnitud de Ka indica la tendencia del ácido a ionizarse en agua (su valor será más alto Lab. Química I (Grado Química) Curso 2012-2013 Práctica 6 -3 cuanto más fuerte sea el ácido) y conociendo su valor y la concentración inicial de la disolución de ácido se puede calcular la concentración [H3O+] y el pH. Consideremos por ejemplo una disolución 0,30 M de ácido acético (CH3COOH, HOAc) con una constante Ka = 1.8 x 10-5. Un procedimiento sencillo para calcular su [H3O+] y pH es el siguiente: a) Definir la evolución de la concentración de las distintas especies al disolverse: Concentración inicial Variación de la concentración al ionizarse Concentración en el equilibrio b) Sustituir la constante de acidez Ka Ka = [HA] [H3O+] [A–] 0,30 M 0 0 xM (0,30 x) M 0+xM xM 0+xM xM [H3O+] [A-] [HA] = (x) (x) 0,30 - x = 1.8 x 10-5 A partir de esta ecuación se puede calcular el valor de x resolviendo la ecuación cuadrática correspondiente, pero se puede simplificar si, c) Suponemos que x <<< 0,30 y por tanto 0,30 – x ≈ 0,30 -5 ya que el valor bajo de Ka (1,8 x 10 ) nos indica que el equilibrio está muy desplazado hacia la izquierda y x es muy pequeño. Así , Ka = x2 0,30 = 1.8 x 10-5 x = 2,3 x 10-3 = [H3O+] pH = -log (2.3 x 10-3) = 2.64 d) Comprobar que el supuesto x << 0,30 y 0,30 – x ≈ 0,30 es correcto x = 2,3 x 10-3 << 0,30, [Nota: es mejor utilizar la fórmula cuadrática si el valor de x es mayor que aprox. 5% del valor de la concentración inicial.] De forma similar para una base débil B la ecuación será: [HB+] [OH-] Y su constante de ionización básica Kb será: Kb= [B] El valor de Kb será más alto cuanto más fuerte sea la base. A partir de Kb y la concentración inicial de B se puede calcular [OH–] y el pOH siguiendo el procedimiento descrito para el ácido débil. El pH de las sales En el caso de las sales nos podemos encontrar con distintas situaciones según las características del anión y catión implicados. Al analizar los equilibrios al principio de esta introducción hemos visto que los iones pueden actuar también como ácidos y bases, por lo que: ● Si el anión de la sal es una base más fuerte que el agua puede interaccionar con ésta en una reacción ácido-base p.ej. el anión OAc– en los acetatos, OAc- + H2O HOAc + OH- Y la disolución será básica (pH > 7). Es el caso de aniones bases conjugadas de ácidos débiles [P.ej. Ka (HOAc)= 1.8 x 10-5] ● Si el catión es un ácido más fuerte que el agua p.ej. NH4+ en las sales de amonio NH4+ + H2O NH3 + H3O+ Y la disolución será ácida (pH < 7) Es el caso de cationes ácidos conjugados de bases débiles [P.ej. Kb (NH3)= 1.8 x 10-5] En ambos casos se dice que el anión y el catión se hidrolizan. De forma general NO SE HIDROLIZAN las sales de ácidos y bases fuertes. El pH de sus disoluciones acuosas es pH = 7. Lab. Química I (Grado Química) Curso 2012-2013 Práctica 6 -4 Las restantes sales SE HIDROLIZAN y el pH de la disolución será: Ácido en sales de ácido fuerte y base débil, p. ej. NH4Cl (de HCl y NH3). Básico en sales de ácido débil y base fuerte, p. ej. NaOAc (de NaOH y HOAc). A determinar en las sales de ácido y base débiles ya que depende de los valores relativos de Ka y Kb de los iones implicados. Se pueden calcular los valores de pH o pOH de las sales que se hidrolizan por un método similar al de los ácidos y bases débiles. Sin embargo, no suelen aparecer en los listados usuales los valores de Ka del catión o Kb del anión, por lo que deben determinarse previamente. Como ejemplo, veamos las líneas generales del procedimiento para calcular el pH de una disolución acuosa de NH4Cl. Esta sal se disocia completamente, y el ión NH4+ se hidroliza con una constante de ionización ácida Ka que no conocemos. Sin embargo, sí conocemos la constante de basicidad Kb (NH3)= 1,8 x 10-5. Si escribimos las ecuaciones de Ka y Kb y las comparamos se observan similitudes entre ellas, especialmente si comparamos Ka con la inversa de Kb NH4+ + H2O NH3 + H3O+ + NH3 + H2O NH4 + OH - Ka = Kb = [NH3] [H3O+] [NH4+] [NH4+] [OH-] 1 [NH3] Kb = [NH3] [NH4+] [OH-] Si multiplicamos numerador y denominador de Ka por [OH–] y sustituimos [H3O+] [OH--] por el producto iónico del agua Kw, el resto es 1/Kb, lo que nos permite calcular Ka = 5.6 x 10-10, Ka = Ka = [NH3] [H3O+] [NH4+] Kw Kb Y de forma general = = [NH3] [H3O+] [OH-] 1.0 x 10-14 1.8 x 10-5 [NH4+] [OH-] = [NH3] [NH4+] [OH-] Kw = Kw Kb = 5.6 x 10-10 Ka (ácido) x Kb (su base conjugada) = Kw Kb (base) x Ka (su ácido conjugado) = Kw EXPERIMENTAL Material Vaso de precipitados de 50 mL (3) Vaso de precipitados de 100 mL Matraz aforado de 100 mL (2) Matraz aforado de 50 mL Matraz aforado de 25 mL Pipetas de 1 mL, 5 mL y 10 mL Propipeta Vial para medidas de pH Varilla Cuentagotas Gradilla + tubos de ensayo (2) Barquilla pesasustancias Espátula Embudo Reactivos Cloruro amónico Ácido acético Cloruro sódico Acetato sódico (trihidrato) HCl (concentrado) Aparatos pHmetro Balanza ● Antes de llevar a cabo el trabajo experimental revisando en los Anexos la información sobre Utilización del material volumétrico (pipetas, propipetas, aforados). Preparación de disoluciones con material aforado. Calibrado y uso del pHmetro. ● Asegúrese de que dispone en el diario de laboratorio una tabla para anotar los datos del tipo: Lab. Química I (Grado Química) Curso 2012-2013 Disolución Volumen a preparar (mL) Práctica 6 -5 A partir de Cantidad calculada Cantidad utilizada pH medido Se utilizará agua desionizada1 para preparar las disoluciones y las cantidades de compuestos calculadas en las Cuestiones previas. I. Preparación de disoluciones de ácido clorhídrico y medida de su pH: a) Tome nota de las características y precisión del material que va a utilizar. b) Prepare 100 mL de una disolución de HCl 0,1M a partir del volumen calculado de HCl (conc.) (Cuestión previa 1a,b: VHCl = ………mL). b) Prepare 50 mL de disolución HCl 0,01M a partir del volumen calculado de HCl 0,1 M (Cuestión previa 1c: (VHCl = ………mL). c) Mida el pH de las dos disoluciones de HCl. II. Preparación de disoluciones de ácido acético y medida de su pH: a) Tome nota de las características y precisión del material que va a utilizar. b) Prepare 100 mL de disolución de ácido acético 0,1M a partir de la cantidad calculada de HOAc (Cuestión previa 2a: VHOAc = ………mL). c) Prepare 100 mL de disolución de ácido acético 0,01M a partir del volumen calculado de HOAc 0,1 M (Cuestión previa 2b: (VHCl = ………mL). d) Mida el pH de las dos disoluciones de ácido acético. III. Preparación de disoluciones de sales y medida de su pH: a) Para cada una de las disoluciones tome nota de las características y precisión del material que va a utilizar. b) Prepare las siguientes disoluciones a partir de las cantidades calculadas en las Cuestiones previas: • 25 mL de acetato sódico 0,5 M a partir de mNaOAc = …………g (Cuestión previa 3). • 50 mL de cloruro sódico 0,1 M a partir de mNaCl = …………g (Cuestión previa 4). • 100 mL de cloruro amónico 0,1 M. a partir de mNH4Cl = …………g (Cuestión previa 5). c) Mida el pH de las dos disoluciones anteriores. Residuos: Deposite todos los productos y residuos obtenidos en los recipientes destinados a tal fin en el laboratorio. Limpieza del material ● Borre el etiquetado con el etanol reservado a tal fin. ● Enjuague el material con agua del grifo. ● Enjuague el material con una pequeña cantidad de agua desionizada del frasco lavador. ● Deje secar el material boca arriba en su puesto de trabajo. AL FINALIZAR PASE LOS DATOS DEL DIARIO A LA HOJA DE RESULTADOS, COMPLÉTELO CON LOS CÁLCULOS NECESARIOS Y AÑADA SUS OBSERVACIONES Y COMENTARIOS 1 Agua destilada vs. Agua desionizada El agua destilada se obtiene destilando agua corriente, normalmente con una columna de fraccionamiento para purificarla al máximo (ver Práctica 9). Al condensar puede retener CO2 del aire, por lo que no debe usarse sin hervir para preparar disoluciones básicas. El agua desionizada se obtiene haciendo pasar agua del grifo por columnas de intercambio iónico (catiónico y aniónico), + de modo que los cationes del agua se cambian por protones (H ) y los aniones por hidroxilos (OH¯). Esta agua puede contener trazas de materia orgánica, pero en cambio su contenido en CO 2 es mínimo o nulo. En el Laboratorio se utiliza agua desionizada, y el grifo específico está claramente indicado. Lab. Química I (Grado Química) Curso 2012-2013 Práctica 6 -6 BIBLIOGRAFÍA Y VÍDEOS Química. La ciencia central T.L. Brown, H.E. LeMay Jr., B.E. Bursten, C.J. Murphy, P. Woodward . Ed. Pearson, 11ª Edicion, 2009. Química, R. Chang, Ed. Mc Graw Hill, 10ª Edición, 2010, Química General R.H. Petrucci, W.S. Harwood, F.G. Herring, Ed. Pearson Educación (Prentice Hall), 8ª Edicion, 2003. Royal Chemical Society: Practical Chemistry for Schools and Colleges: http://www.rsc.org/Education/Teachers/Resources/practical/index3.htm Ver: Making up solutions; Using a burette; Using a pipette, Doing a titration, Univ. de Valencia. Material volumétrico: http://mmedia.uv.es/buildhtml?user=tcliment&path=/LABORATORIO/&name=material_basico_labo.mp4 Calibración del pHmetro: http://mmedia.uv.es/buildhtml?lang=es_ES&user=tcliment&name=calibrado_phmetro.mp4&path=/LABORATORIO/&id=6580 Puede encontrar los enlaces a los vídeos en la web del laboratorio: http://www.uv.es/fqlabo/ CUESTIONES Cuestiones previas Los cálculos que se proponen en este apartado serán los utilizados en el procedimiento experimental. 1. a) ¿Qué cantidad de HCl concentrado (37% en peso; d=1.19 g/mL) se necesita para preparar 100 mL de una disolución de HCl 0,1M? b) ¿Y si se utiliza HCl 35% (d=1.19 g/mL)?. c) ¿Qué cantidad de las anteriores disoluciones se necesita para preparar 50 mL de HCl 0,01M? d) Calcule el valor de pH para las disoluciones de HCl 0,1M y 0,01M. 2. a) ¿Qué cantidad de ácido acético puro (99-100 % en peso; d=1.050 g/mL) se necesita para preparar 100 mL de disolución de ácido acético 0,1M? b) ¿Qué cantidad de la anterior disolución se necesita para preparar 100 mL de disolución de ácido acético 0,01M? c) Calcule el valor de pH para las disoluciones de ácido acético 0,1M y 0,01M. 3. a) Calcule la cantidad que acetato sódico trihidrato que se debe pesar para preparar 25 mL de acetato sódico 0,5 M. b) Escriba los equilibrios iónicos que tienen lugar en la disolución anterior. c) Calcule el valor de pH para esta disolución. 4. a) Calcule la cantidad que cloruro sódico que se debe pesar para preparar 50 mL de cloruro sódico 0,1 M. b) Escriba los equilibrios iónicos que tienen lugar en la disolución anterior. c) Calcule el valor de pH para esta disolución. 5. a) Calcule la cantidad que cloruro amónico que se debe pesar para preparar 100 mL de cloruro amónico 0,1 M. b) Escriba los equilibrios iónicos que tienen lugar en la disolución anterior. c) Calcule el valor de pH para esta disolución. Cuestiones posteriores al trabajo experimental 1. Consulte las etiquetas de los frascos de los distintos compuestos y tome nota de los datos sobre pH que indica el proveedor. Coméntelos brevemente. 2. Los valores de pH medidos para las disoluciones ¿son los que había calculado en las cuestiones previas? Si no es así, busque una explicación. Lab. Química I (Grado Química) Curso 2012-2013 LABORATORIO QUÍMICA I Práctica 6 -7 RESULTADOS PRÁCTICA 6 APELLIDOS Y NOMBRE GRUPO Y PUESTO: Una vez completado recuerde entregar una copia al profesor/profesora. I. PREPARACIÓN DE DISOLUCIONES DE HCl Y MEDIDA DE SU pH. Indique las características del material volumétrico que ha utilizado. Disolución Volumen a preparar (mL) Se prepara a partir de Calculado Cantidad pH que debe necesaria (mL) dar Experimental Volumen pH medido (mL) medido HCl 0,1M HCl 0,01M II. PREPARACIÓN DE DISOLUCIONES DE HOAc Y MEDIDA DE SU pH. Indique las características del material volumétrico que ha utilizado. Disolución Volumen a preparar (mL) Se prepara a partir de Calculado Cantidad pH que necesaria (mL) debe dar Experimental Volumen pH medido (mL) medido HOAc 0,1M HOAc 0,01M III. PREPARACIÓN DE LAS DISOLUCIONES DE SALES Y MEDIDA DE SU pH. Indique las características del material volumétrico que ha utilizado. Disolución Volumen a preparar (mL) Se prepara a partir de Calculado Cantidad pH que necesaria (g) debe dar Experimental Cantidad pesada (g) NaOAc 0,5 M NaCl 0,1 M (NH4)Cl 0,1 M OBSERVACIONES Y COMENTARIOS (Añada las hojas que considere necesarias) pH medido Lab. Química I (Grado Química) Curso 2012-2013 Práctica 6 -8 Lab. Química I (Grado Química) Curso 2012-2013 Práctica 7 -1 PRÁCTICA 7: Valoración ácido-base y uso de patrones primarios. Elaboración: José Vicente Gimeno OBJETIVOS • Revisión de los principios de estequiometría y neutralización que rigen las reacciones ácidobase. • Preparación de disoluciones de NaOH 0,1M y de HCl 0,1M. • Utilización de un indicador en valoraciones* ácido-base. • Utilización de patrones primarios: estandarización de una disolución de NaOH con ftalato ácido de potasio, u otro patrón adecuado. • Utilización de patrones secundarios: estandarización de una disolución de HCl con la disolución estandarizada de NaOH. • Determinación de las cifras significativas para la Molaridad a partir de la precisión de las medidas implicadas. • Cálculo de valores medios y desviación estándar. INTRODUCCIÓN Las disoluciones que habitualmente se utilizan en el laboratorio se preparan al disolver una cantidad medida de soluto en el disolvente adecuado, frecuentemente agua, y así, a partir de: • la cantidad de soluto obtenida por: Pesada en el caso de sólidos (aunque también pueden pesarse líquidos). Medida de volumen en el caso de sustancias líquidas o disoluciones (p.ej. cloruro de hidrógeno en agua). • el volumen final de la disolución, se puede determinar la concentración de la disolución, expresada como MOLARIDAD (M, moles/litro), lo que es fundamental si se quiere efectuar posteriormente cálculos estequiométricos. El problema que se plantea es que, para la mayoría de los solutos, los valores calculados para la concentración son sólo aproximados, incluso utilizando instrumentos de precisión para pesar o medir volúmenes. Esto es debido a que en la mayoría de los casos el soluto presenta un grado desconocido de impurezas, y además, en algunos casos puede ser: Higroscópico, y tomar agua del ambiente. Delicuescente, y tomar agua del ambiente, disolviéndose en ella. Inestable térmicamente, y descomponerse al variar la temperatura. Reactivo con los componentes del aire, etc. Así pues, la incertidumbre que acompaña a la medida de la cantidad de soluto se transmite al resultado de la concentración, que sólo podrá considerarse como aproximada y estas disoluciones no serán apropiadas para llevar a cabo medidas químicas de precisión, como es el caso del análisis químico. No obstante, hay también sustancias químicas con características fisicoquímicas favorables que permiten determinar con precisión la cantidad de sustancia directamente a partir de la pesada utilizando la balanza analítica (precisión 0,001, 0,0001, e incluso 0,00001 g) o de medidas de volumen utilizando material volumétrico de precisión. A estas sustancias se las conoce como patrón primario, o sustancia patrón tipo primario, y constituyen la base para preparar disoluciones con una * Valorar: Determinar la concentración exacta de un compuesto en una disolución. Se utilizarán los términos valorar, estandarizar y titular de forma indistinta. Lab. Química I (Grado Química) Curso 2012-2013 Práctica 7 -2 concentración molar perfectamente conocida. Los patrones primarios son compuestos que cumplen las siguientes características: 1. Son sólidos 2. Tienen composición conocida y recomendablemente serán anhidros 3. Deben tener elevada pureza 4. Son estables a temperatura ambiente 5. Deben permitir su secado en estufa 6. No deben absorber gases 7. Deben reaccionar rápida y estequiométricamente con la especie valorante 8. Deben poseer una elevada masa molecular, por lo que se puede entender que no hay muchas sustancias que cumplan todos estos requisitos. Una cantidad conocida del patrón primario actúa como reactivo frente a una disolución D que se desea valorar. Según el volumen consumido de D y la reacción química que tenga lugar (ácido-base, redox, …) se puede calcular la concentración de D (incluso con la precisión de la cuarta cifra decimal). La disolución así valorada se denomina disolución estándar y puede utilizarse, a su vez, como patrón secundario para la valoración de otras disoluciones. Disolución D Patrón primario Disolución del patrón Valoración Disolución D (patrón secundario) Disolución X Valoración Determinación de la concentración de X Determinación de la concentración de D D disolución estándar Patrones primarios Las valoraciones ácido-base constituyen la metodología para preparar disoluciones estándar ácidas o básicas. La valoración consiste en el seguimiento de una reacción ácido-base entre el patrón primario (ácido o básico) y la disolución (básica o ácida) que es objeto de la estandarización. En el caso de valoraciones ácido-base, los patrones primarios más frecuentemente utilizados se describen a continuación: A) Sustancias de carácter ácido (para estandarizar bases): Nombre Ftalato ácido de potásico Ácido benzoico Ácido sulfanílico Ácido sulfámico Yodato ácido de potasio Ácido oxálico Fórmula KH(C6H4)C2O4 C6H5CO2H NH2C6H5SO3H NH2SO3H KH(IO3)2 C2O4H2 MM (g/mol) 204,221 122,12 173,19 97,08 389,918 90,03 pKa 5,408 4,202 3,227 0,988 0,77 1,252; 4,266 Lab. Química I (Grado Química) Curso 2012-2013 Práctica 7 -3 B) Sustancias de carácter básico (para estandarizar ácidos): Nombre Tris(hidroximetil)aminometano (TRIS) Carbonato de sodio Bórax Fórmula H2NC(CH2OH)3 Na2CO3 Na2B4O7·10H2O MM (g/mol) 121,135 105,99 381,37 pKb 5,975 3,671; 7,648 4,764 Valoración ácido-base Para realizar una valoración ácido-base, se añade un ácido (o base) a una base (o ácido) de forma que reaccionen en proporción estequiométrica. A este punto se le conoce como punto de equivalencia (valor teórico). Por otra parte, se conoce como punto final (valor experimental) de una valoración a aquél en el que tiene lugar un cambio físico observable, o medible, y que nos permite relacionarlo con la condición de equivalencia química. Se debe hacer todo lo posible para asegurar que cualquier diferencia entre el punto de equivalencia y el punto final sea pequeña. Sin embargo, tales diferencias existen como consecuencia de las limitaciones en los cambios físicos y en nuestra capacidad para observarlos. La diferencia entre el punto de equivalencia y el punto final es el error de valoración. En una valoración ácido-base se pueden utilizar indicadores ácido-base, un pH-metro, o la combinación de ambos. Los indicadores son compuestos donde la especie ácida (HIn) presenta color 1 y la especie básica (In─) color 2. Éstos se añaden a la disolución a valorar y varían claramente de color con el cambio de pH. En la región del punto de equivalencia hay grandes cambios de la concentración relativa del ácido y la base que hacen que el indicador cambie su color al modificar la relación [HIn]/[In─]. En una valoración realizada con un indicador sólo observaremos un viraje en el color de la disolución, correspondiente al punto final de dicha valoración, y que puede distar del punto de equivalencia dependiendo del indicador elegido o de la propia manipulación llevada a cabo por el experimentador. Figura 1. Valoración de un ácido fuerte frente a una base fuerte Figura 2. Curvas de valoración para ácidos fuerte (curva I) y débil (curva II) frente a base fuerte Una forma de minimizar el error de valoración consiste en monitorizar el cambio de pH con un pHmetro y representarlo frente al volumen de valorante añadido (Figura1). Para llevar a cabo una valoración ácido-base se sitúa un reactivo en un matraz erlenmeyer y con el otro reactivo se rellena una bureta desde la cual se controlará la adición. Por ejemplo, para valorar un ácido se coloca éste en el erlenmeyer y en la bureta una disolución de la base. Lab. Química I (Grado Química) Curso 2012-2013 Práctica 7 -4 Cuando se inicia la valoración, el pH de la disolución del matraz será ácido. Al añadir la base tiene lugar la reacción y el pH aumenta ya que el ácido se consume. Este cambio es muy lento al principio, y rápido cuando se aproxima el punto de equivalencia. En este punto la adición debe ser muy lenta para poder observar con seguridad el cambio de color del indicador. En la Figura 1 se puede ver que el punto de equivalencia coincide con el de neutralización (pH=7) y se incluyen los indicadores más habituales y el intervalo de pH en el que cambian de color. Este cambio indica el punto final de la valoración cuando ésta se lleva a cabo con un indicador, y por tanto, éste es el momento en que se debe tomar la medida del volumen consumido. Si la valoración se realizase con un pH-metro se continuaría añadiendo valorante, y se observaría que el pH sigue aumentando hasta que se estabiliza, tal como se muestra en la Figura 1 (a pH>11). Si se valora un ácido débil con una base fuerte se obtiene una curva de valoración similar a la de la figura 1, donde la mayor diferencia se encuentra en su primer tramo de la curva de valoración, el cual se traslada hacia valores de pH tanto más elevados cuanto mayor es el pKa del ácido valorado (Figura 2), hecho que da lugar a una reducción del salto de pH de la curva de valoración, lo que conduce a una mayor imprecisión del punto de equivalencia. Por tanto, en estos casos, la elección del indicador empleado para realizar la valoración será crucial para obtener un menor error en la valoración. EXPERIMENTAL Material Bureta de 10 mL Probeta de 10 mL Vaso ppdos de plástico de 100 mL (2) Matraz Erlenmeyer 100 mL (3) Vaso precipitados de vidrio 50 mL Pipeta aforada de 10 mL+ propipeta Barquilla + espátula Común para el grupo Jarra de plástico de 1L Botella de plástico de 1L Vaso de vidrio de 1L Varillas (2) + tubo ensayo + gradilla Reactivos NaOH HCl concentrado Ftalato ácido de potasio Yodato ácido de potasio Fenolftaleína (disol.) Aparatos Balanza I. Preparación previa ● Antes de llevar a cabo el trabajo experimental asegúrese, revisando los Anexos, de que conoce adecuadamente la información sobre Utilización del material volumétrico. La bureta. Protocolo técnico de manejo de la bureta. Valoración con indicador. Pipetas y propipetas. ● Las cantidades de compuestos a utilizar son las calculadas en las Cuestiones previas. ● Asegúrese de que dispone en el diario de laboratorio de una tabla adecuada para anotar los datos del tipo: Valoración Patrón primario (g) V (mL) B1 B2 B3 NaOH ● Revise cuidadosamente el material asegurándose de que está limpio y enjuagado con agua desionizada para evitar contaminaciones. ● Tome nota de las características y precisión de la bureta que va a utilizar. ● Llene la bureta con agua desionizada y practique su manejo para enrasar y controlar la descarga de líquido, comprobando que no gotea y que elimina las burbujas de aire al vaciarla. Para evitar que vuelva a entrar aire cierre la llave antes de vaciarla totalmente, dejando un pequeño volumen de agua por encima de la llave. ● Etiquete los matraces erlenmeyer como B1, B2, B3. Lab. Química I (Grado Química) Curso 2012-2013 Práctica 7 -5 II. Preparación de disoluciones Las disoluciones de este apartado se prepararán y utilizarán por todo el Grupo de Prácticas. Preparación de 0,5 L de hidróxido de sodio 0,14 M aprox. (por una pareja del grupo) En una balanza pese, utilizando una barquilla de plástico, la cantidad de NaOH calculada previamente (Cuestión previa 2a, mNaOH = …………g), o el valor más próximo posible. Anote el valor pesado. Introduzca el sólido en un vaso de precipitados de plástico o jarra de plástico de 1000 mL, añada unos 100 mL de agua desionizada y disuelva el sólido con ayuda de una varilla. La disolución se diluye con agua desionizada hasta un volumen de 500 mL aprox., se homogeneíza agitando con la varilla, se deja enfriar y se guarda, hasta su reparto, en botella de plástico cerrada y etiquetada como NaOH 0,14 M, Grupo y fecha. Cada pareja tomará, en un vaso de plástico de 100 mL, la cantidad necesaria para rellenar su bureta. Preparación de 0,5 L de HCl 0,1 M aprox. (por una pareja del grupo) En la vitrina de gases tome, con ayuda del dosificador, el volumen de ácido clorhídrico concentrado calculado en la Cuestión previa 2b (VHCl = ………mL), sobre un vaso de precipitados de vidrio de 600 mL donde previamente se habrán introducido unos 100 mL de agua desionizada. Ya fuera de la vitrina diluya la mezcla con agua hasta 500 mL aprox., homogeneíce la disolución agitando con la varilla y etiquete como HCl 0,1 M, Grupo y fecha. Cada pareja tomará, en un vaso de vidrio de 50 mL, la cantidad necesaria para realizar su estandarización. III. Estandarización de una disolución de NaOH frente a un patrón primario Llenado de la bureta Lave la bureta 2-3 veces con pequeñas porciones de la disolución NaOH. En cada paso, ciérrela dejando un pequeño volumen de disolución por encima de la llave para impedir que entre aire y se formen burbujas. La bureta tiene una boca muy estrecha por lo que es más fácil introducir el líquido con una ligera inclinación de la bureta. Las valoraciones se efectuarán sucesivamente, rellenando y enrasando la bureta para cada valoración. Se debe evitar que la disolución de NaOH permanezca en la bureta mientras ésta no sea utilizada (más de 5 min.). Preparación de las muestras Pese con balanza analítica, y en una barquilla de plástico, la cantidad calculada de ftalato ácido de potasio u otro patrón primario idóneo (Cuestión previa 3b, mpatrón = ..........g). Nota: la cantidad pesada no tiene que coincidir con el valor calculado ni entre las distintas pesadas, pero si debe ser obtenida y anotada con precisión. Introduzca cuantitativamente el patrón primario en el erlenmeyer B1 y añada unos 10 mL de agua desionizada (tomados con la probeta) y disuelva el sólido mediante una ligera rotación manual del matraz. Nota: Puede utilizar parte del agua para lavar la barquilla y asegurarse de que toda la muestra pesada se ha introducido en el matraz, pero asegúrese de que está seca antes de volver a utilizarla. Repita el proceso para las muestras correspondientes a los erlenmeyer B2 y B3. Añada a cada erlenmeyer 1-2 gotas del indicador (fenolftaleína) y diluya hasta unos 20 mL. Valoración Coloque el erlenmeyer debajo de la bureta. Añada disolución de NaOH manteniendo el erlenmeyer en constante agitación. Al principio de la valoración la adición puede ser relativamente rápida hasta haber añadido aprox. 1-2 mL por debajo del volumen esperado (ver Cuestión previa 3b). A partir de este punto la adición debe ser lenta y con Lab. Química I (Grado Química) Curso 2012-2013 Práctica 7 -6 agitación constante hasta que el cambio de color del indicador que indica el final de la valoración se mantenga al menos 10 seg. Tome la lectura del volumen de NaOH consumido (VNaOH) con precisión y anótelo en la tabla. ATENCIÓN: Deben obtenerse al menos tres resultados concordantes. Descarte los valores con una dispersión mayor del 5% y repita la valoración hasta disponer de tres medidas coherentes. Cálculo: la diferencia del valor alejado respecto de su más próximo dividido por este último deberá ser menor de +/- 0,05 IV. Estandarización de disolución de HCl 0,1 M frente a un patrón secundario Preparación previa ● Asegúrese de que dispone en el diario de laboratorio de una tabla adecuada para anotar los datos del tipo: Valoración V (mL) V (mL) C1 C2 C3 HCl NaOH ● En estas valoraciones se utilizará la disolución estándar de NaOH obtenida en la experiencia anterior como patrón secundario. ● Lave el material y enjuáguelo con agua desionizada para evitar contaminaciones. ● Etiquete los matraces erlenmeyer como C1, C2, C3. ● Tome nota de las características y precisión de la pipeta de 10 mL que va a utilizar. Llénela con agua desionizada y practique el manejo de pipeta+propipeta para enrasar y controlar la descarga de líquido. Lávela cuidadosamente 2-3 veces con una pequeña cantidad de la disolución de HCl que se va a valorar. Tire el HCl del lavado en el recipiente de residuos. Preparación de las muestras Tome sucesivamente tres alicuotas de 10 mL de la disolución de HCl descargando cada alícuota en el correspondiente matraz erlenmeyer. Añada a cada erlenmeyer 1-2 gotas del indicador y diluya con agua destilada hasta aprox. 30 mL. Valoración Las valoraciones se llevarán a cabo siguiendo el procedimiento descrito en el apartado III. Residuos: Deposite todos los productos y residuos obtenidos en los recipientes destinados a tal fin en el laboratorio. Limpieza del material: ● Borre el etiquetado con el etanol reservado a tal fin. ● Enjuague el material con agua del grifo. ● Enjuague el material con una pequeña cantidad de agua desionizada del frasco lavador. ● Deje secar el material boca arriba en su puesto de trabajo. AL FINALIZAR PASE LOS DATOS DEL DIARIO DE LABORATORIO A LA HOJA DE RESULTADOS, COMPLÉTELO CON LOS CÁLCULOS NECESARIOS Y AÑADA SUS OBSERVACIONES Y COMENTARIOS Lab. Química I (Grado Química) Curso 2012-2013 Práctica 7 -7 BIBLIOGRAFÍA Y VÍDEOS Análisis Químico Cuantitativo, D.C. Harris, Ed. Reverté, 2001, pp. 260-268 y 283-284 Química General, R.H. Petrucci et al., Pearson, 2011, pp.160-165; 724-727 Química, R. Chang, Ed. McGraw Hill, 2010, pp. 660-697 Principios de Química: Los caminos del descubrimiento P. Atkins y L. Jones. Ed. Médica Panamericana. 2006. 3ª Edición), Capítulo 10. Royal Chemical Society: Practical Chemistry for Schools and Colleges http://www.rsc.org/Education/Teachers/Resources/practical/index3.htm Universidad de Valencia: Material volumétrico: http://mmedia.uv.es/buildhtml?user=tcliment&path=/LABORATORIO/&name=material_basico_labo.mp4 Valoración: http://mmedia.uv.es/buildhtml?user=martiny&path=/&name=valoracion.flv Puede encontrar los enlaces a los vídeos en la web del laboratorio: http://www.uv.es/fqlabo/ CUESTIONES Cuestiones previas. Los cálculos que se proponen en este apartado serán los utilizados en el procedimiento experimental. Cópielos para completarlo. 1. Defina los conceptos: a) Valoración ácido-base. b) Punto de equivalencia. c) Indicador químico. d) Punto final de la valoración. 2. Con los datos obtenidos a partir de las Fichas simplificadas: a) Calcule la cantidad de NaOH(s) que es necesario pesar para preparar 0,5 L de una disolución 0,14 M de dicho reactivo. b) Calcule el volumen requerido de HCl concentrado 35% (d= 1,18 g/cm3) para preparar 0,5 L de una disolución HCl 0,1 M. ¿Cuál sería el volumen si dispone de HCl 37% (d= 1,19 g/cm3)? 3. Si se utiliza ftalato ácido de potasio (o cualquier otro compuesto) como patrón primario: a) Escriba la reacción entre el ácido y NaOH (fórmulas semidesarrolladas). b) ¿Qué cantidad de patrón propuesto será necesaria para consumir unos 7-8 mL de NaOH 0,14 M? c) Deduzca la fórmula matemática para calcular la concentración molar de la disolución de NaOH, a partir de la cantidad de patrón (mpatrón) y el volumen de NaOH consumido (VNaOH). d) Idem para la disolución de HCl, a partir del VHCl utilizado y del VNaOH consumido. 4. Indicadores. a) Complete la tabla siguiente anotando para cada uno de los indicadores: Cambio de color Intervalo de pH Indicador (de menor a mayor) De ácido básico De básico ácido Fenolftaleína Tornasol Rojo de metilo b) Se va a utilizar fenolftaleína como indicador tanto en la valoración de NaOH como de HCl. ¿Es el más adecuado? ¿Sería mejor utilizar alguno de los otros? Justifique la respuesta. 5. Justifique: a) El uso de la balanza granatario para pesar el patrón primario. Lab. Química I (Grado Química) Curso 2012-2013 Práctica 7 -8 b) La utilización de material de plástico en la preparación de la disolución de NaOH. c) El uso de dosificador en vez de pipeta en la preparación de disolución de HCl. d) La no utilización de matraces aforados para preparar las disoluciones. Cuestiones posteriores al trabajo experimental 1. Explique: a) Los requisitos que deben cumplir los patrones primarios. b) El significado de cantidad perfectamente conocida. c) Por qué se disuelve el patrón en una cantidad aproximada de agua. d) Por qué el cálculo de la cantidad de patrón primario se suele referir a unos 7-8 mL de la disolución en la valoración. 2. Justifique: a) Por qué se utilizan volúmenes de 10 mL de HCl para su valoración. b) Por qué se mide dicho volumen con pipeta aforada en vez de pipeta graduada. c) Por qué se diluye la disolución del erlenmeyer hasta 20 mL aproximadamente. d) Por qué se efectúa la valoración por triplicado. 3. Complete una tabla del tipo siguiente dibujando la fórmula semidesarrollada de los patrones primarios e identifique los grupos ácidos y básicos con un círculo (diferente para ácido y base). Nombre Ácido benzoico Ácido oxálico Ácido sulfanílico Ácido sulfámico Ftalato ácido de potasio Bórax Carbonato de sódio Tris(hidroximetil)aminometano Yodato ácido de potasio Fórmula semidesarrollada y grupos ácido o básico 4. Supongamos que podemos disponer de cualquier compuesto que queramos. a) Proponga justificadamente el patrón primario más adecuado para estandarizar la disolución en la valoración (VNaOH). b) Seleccione el indicador más adecuado para detectar el punto final de cada valoración para la estandarización de la base y del ácido. 5. En general, como habrá visto en el Anexo, en una valoración se introduce en la bureta el valorante, y éste se vierte sobre una disolución de la muestra a valorar. Sin embargo, en una de las partes de esta práctica no se ha procedido de este modo. Indique la razón/razones que lo justifican. Lab. Química I (Grado Química) Curso 2012-2013 Práctica 7 -9 LABORATORIO QUÍMICA I APELLIDOS Y NOMBRE RESULTADOS PRÁCTICA 7 GRUPO Y PUESTO: Una vez completado recuerde entregar una copia al profesor/profesora. A) PREPARACIÓN DE DISOLUCIONES Indique las características del material volumétrico que ha utilizado Para la disolución de NaOH 0,14 M Disolución Volumen a preparar (mL) Para la disolución de HCl 0,1 M A partir de Peso (g) Calculado Pesado Molaridad (M) Esperada Real NaOH 0,14 M Disolución 0,14 Volumen a preparar (mL) A partir de Volumen (mL) Calculado Medido Molaridad (M) Esperada Real HCl 0,1 M 0,1 NOTA: Calcule las cifras significativas para la Molaridad a partir de la precisión de las medidas implicadas. B) ESTANDARIZACIÓN DE LA DISOLUCIÓN DE NaOH Indique las características del material volumétrico que ha utilizado Valoración Ftalato ácido de potasio Peso (g) mmol VNaOH (mL) Molaridad (MNaOH) MNaOH (valor medio) Desviación estándar 1 2 3 NOTA: Calcule las cifras significativas para la Molaridad a partir de la precisión de las medidas implicadas. Lab. Química I (Grado Química) Curso 2012-2013 Práctica 7 -10 C) ESTANDARIZACIÓN DE LA DISOLUCIÓN DE HCl Indique las características del material volumétrico que ha utilizado Valoración HCl VHCl (mL) mmol VNaOH (mL) Molaridad (MHCl) MHCl (valor medio) Desviación estandar 1 2 3 NOTA: Calcule las cifras significativas para la Molaridad a partir de la precisión de las medidas implicadas. OBSERVACIONES Y COMENTARIOS Anote las Observaciones y Comentarios de las dos estandarizaciones por separado. Incluya al final un análisis comparativo de las dos. Continúe en el reverso de la hoja y añada más si es necesario. Lab. Química I (Grado Química) Curso 2012-2013 Práctica 8 - 1 PRÁCTICA 8: Preparación de disoluciones y tratamiento de datos. Efecto de la dilución. OBJETIVOS Preparación de disoluciones acuosas de CuSO4 por dilución a partir de una disolución madre. Preparación y utilidad de una disolución blanco. Utilización del espectrómetro visible y registro del espectro del CuSO4. Selección de la longitud de onda para el análisis espectroscópico. Estudio del efecto de la dilución en la absorbancia de disoluciones de sulfato de cobre. Tratamiento de datos: Realización de la recta de calibrado y determinación de la concentración de una disolución por interpolación de la lectura de su absorbancia en la recta. INTRODUCCIÓN El espectro electromagnético es el conjunto de ondas electromagnéticas que se propagan de manera ondulatoria y con la velocidad constante de la luz (aprox. 300.000 km/s). Las ondas electromagnéticas se caracterizan por su longitud de onda (distancia entre dos ondas sucesivas) y el espectro electromagnético se divide en distintas zonas dependiendo de las longitudes de onda: desde menor (rayos gamma y rayos X), a mayor (radiofrecuencia y microondas) pasando por la luz visible, infrarroja y ultravioleta. Cada onda lleva asociada una energía y todas las sustancias absorben radiación en alguna región del espectro electromagnético pasando de un estado fundamental a otro de mayor energía o excitado. La radiación visible (luz blanca) es una mezcla de todas las longitudes de onda () desde 400 nm (violeta) a los 700 nm (rojo) y los compuestos que absorben radiación en esta región son coloreados. El color de un compuesto sólido o en disolución indica la existencia de un estado excitado de energía relativamente baja, sobre el estado fundamental (aprox. 35-70 Kcal, 150-300 KJ/mol). Cuando un haz de luz blanca incide sobre un compuesto coloreado, parte de la energía de la radiación es absorbida por el mismo, por ejemplo, si se absorbe la radiación roja, el color que percibimos (radiación no absorbida) es verde, y se dice que el verde y el rojo son complementarios. Espectro de absorción y Ley de Lambert-Beer Lambert y Beer demostraron que, a una longitud de onda determinada, la fracción de intensidad de radiación absorbida o Absorbancia (A) de la disolución de una sustancia, es directamente proporcional a su concentración (c), a la distancia recorrida por la de luz en la disolución (l) y a una constante denominada coeficiente de extinción (), característico para cada sustancia, según la fórmula: A= -log I/Io = c l En la que: Io = Intensidad de la radiación incidente I = Intensidad de la radiación transmitida. l = distancia recorrida por la radiación dentro de la disolución (cm). c= concentración de la substancia en mol/L. = coeficiente de absorción molar (L/mol•cm) Siempre que nos encontremos en absorbancias comprendidas entre 0 y 1, se cumple la ley de Lambert-Beer y la relación entre la concentración y la absorbancia es de tipo lineal. En este contexto se define como Transmitancia (T) la fracción de intensidad de radiación transmitida, es decir no absorbida. La relación de T con A viene dada por A = -log T, y puede expresarse también en %. Lab. Química I (Grado Química) Curso 2012-2013 Práctica 8 - 2 Análisis espectrofotométrico La absorbancia es una medida de la proporción de luz monocromática (un solo color) que es absorbida cuando la luz pasa a través de una disolución. Un espectro de absorción es un gráfico de la absorbancia en función de la longitud de onda. Las absorbancias altas corresponden a proporciones grandes de luz absorbida cuando pasa a través de una disolución. Absorbancias bajas significan que la luz se transmite en grandes proporciones. En la figura 1 se representa el espectro de absorción del CuSO4. Para medir la absorbancia, se utiliza el espectrofotómetro, un instrumento que permite medir la intensidad de la radiación absorbida a distintas longitudes de onda. Los equipos disponen de portacubetas en los que se coloca una cubeta con la disolución en el paso del haz de luz. Una vez seleccionada la longitud de onda, el espectrofotómetro da la lectura directa de la absorbancia (A) o bien, el porcentaje de transmitancia (%T). Figura 1: Espectro de disolución CuSO4 0,04 M y determinación de su longitud de onda analítica. (nm) Abs 450 0,007 500 550 600 650 0,007 0,013 0,040 700 750 760 770 0,289 0,438 0,462 0,481 780 790 795 800 0,494 0,504 0,510 0,512 805 810 815 820 0,512 0,509 0,509 0,508 850 900 950 1000 0,484 0,426 0,360 0,300 0,130 * Espectro elaborado en el Laboratorio de Química General- Fac. Química UV. Espectrofotómetro Jenway-6300. Nota: Aunque el ojo humano no detecta longitudes de onda por debajo de 400nm y por encima de 700nm, los espectrofotómetros VISIBLES tienen un intervalo de detección de 320 a 1000nm. El método de análisis espectrofotométrico permite, también, conocer la concentración de un compuesto, B, comparando la absorbancia de una disolución de concentración desconocida de B (CX) con la absorbancia de una disolución patrón o estándar de concentración conocida de B (CP), realizadas ambas en el mismo disolvente. Se elige la longitud de onda de máxima absorción del espectro, denominada longitud de onda analítica. Además de la sustancia a cuantificar, tanto el disolvente como otros compuestos de la disolución pueden absorber en el mismo intervalo de longitudes de onda que la sustancia. Para descartar estas interferencias, se prepara una disolución con todos los componentes de dicha disolución excepto el compuesto a cuantificar, disolución llamada blanco que se utiliza para calibrar el espectrofotómetro ajustando los valores de A = 0 ó T = 100%. Lab. Química I (Grado Química) Curso 2012-2013 Práctica 8 - 3 Curva y recta de calibrado Midiendo la absorbancia de distintas disoluciones estándar de B, a la misma y con el mismo instrumento, se pueden obtener los valores de su absorbancia AB en función de la concentración (CP) que se representan como una gráfica que se denomina curva de calibrado. Para absorbancias comprendidas entre 0 y 1, la relación entre la concentración y A es de tipo lineal y hablamos de recta de calibrado. A partir de la curva de calibrado se puede determinar la concentración CX de una muestra desconocida de B midiendo su absorbancia AX e interpolando en la recta de calibrado. La fiabilidad de la determinación dependerá de la precisión de las medidas. Existen distintos programas, implementados en ordenadores y calculadoras, que nos ayudan a obtener la gráfica y su ecuación matemática sin demasiadas complicaciones. Uno de los más conocidos es el que se incluye en el paquete de Microsoft-office: Excel. EXPERIMENTAL Material 3 matraces aforados de 25 mL 1 matraz aforado de 50 mL 1 matraz aforado de 100 mL 1 pipeta graduada de 1 mL 1 pipeta graduada de 5 mL 1 pipeta de 10mL Barquilla + espátula Vaso vidrio 50 mL + varilla Vaso + cuentagotas plástico Cubetas Vaso de plástico de 250 mL para limpiar cubetas Reactivos CuSO4·5H2O NH3 (conc.) para lavar Aparatos Balanza Espectrofotómetro Preparación previa ● Antes de llevar a cabo el trabajo experimental asegúrese revisando los Anexos de que conoce adecuadamente la información sobre Utilización del material volumétrico. Pipetas y propipetas. y de que dispone en el diario de laboratorio de las tablas necesarias para los distintos apartados. Recuerde que deberá anotar los datos con todas las cifras significativas. ● Las cantidades de compuestos a utilizar son las calculadas en las Cuestiones previas. ● Marque los matraces aforados con las concentraciones que va a preparar. ● No marque las cubetas. Busque la parte translúcida, por donde cogerlas, y la parte transparente, por donde ha de pasar el haz de luz. I. Preparación de las disoluciones. A. Preparación de la disolución madre de CuSO4 0,5 M. Se prepara en primer lugar 25 mL de una disolución 0,5 M de CuSO4 en agua desionizada según la cantidad calculada. (Cuestión previa 2: CuSO4·5H2O:………..g). Anote el valor experimental real y si no coincide exactamente con la masa calculada recalcule la concentración de la disolución madre. B. Preparación de las disoluciones diluidas de CuSO4. A partir de la disolución madre prepare las disoluciones de CuSO4 cada vez más diluidas (0,07; 0,05; 0,03; 0,02 y 0,01M) utilizando los volúmenes calculados en la Cuestión previa 3. Si la concentración de la disolución madre difiere de 0,50 M o la cantidad medida no coincide con la necesaria, recalcule las concentraciones de las diluciones de la tabla. [CuSO4] Volumen a preparar Cantidad necesaria (mL) Cantidad medida (mL) Molaridad real 0,07 M 25 mL 0,05 M 25 mL 0,03 M 25 mL 0,02 M 50 mL 0,01 M 100 mL Lab. Química I (Grado Química) Curso 2012-2013 Práctica 8 - 4 II. Obtención de los datos para el espectro de absorción del sulfato de cobre. Antes de llevar a cabo el trabajo experimental lea las instrucciones y asegúrese de que conoce el funcionamiento del espectrofotómetro asignado. Las instrucciones para el manejo del espectrofotómetro las encontrará junto al aparato (recuerde anotar el espectrofotómetro utilizado). Para obtener el espectro de absorción se debe medir la absorbancia de la disolución de CuSO4 0,03 M a diferentes longitudes de onda entre 450 nm y 950 nm. Para realizar estas medidas, se necesitan dos cubetas, una con agua desionizada y otra con disolución 0,03 M de CuSO4. En términos generales, una vez seleccionada la longitud de onda para cada medida se coloca la cubeta con agua desionizada (blanco) en el haz de luz y se ajusta a cero la absorbancia. Se retira esta cubeta y se coloca la que contiene la disolución, anotando el valor de la A que se muestra en la pantalla para esa longitud de onda. Se repite el proceso completo para cada longitud de onda. En un primer barrido se mide a intervalos de 50 nm para localizar la zona de máxima absorción. max (nm) 550 600 650 700 750 800 850 900 Absorbancia Para precisar con mayor exactitud el máximo, o longitud de onda analítica, se completa con medidas a intervalos de 5 o 10 nm por arriba y por abajo de la máxima absorción observada. max (nm) Absorbancia III. Medida de la Absorbancia a max de distintas disoluciones para la recta de calibrado. En este apartado se trabaja exclusivamente a la longitud de onda analítica (max del apartado anterior) utilizando una única cubeta. Una vez seleccionada la longitud de onda se ajusta el cero de absorbancia con el blanco (agua desionizada) y con la misma cubeta se mide la absorbancia de cada una de las disoluciones preparadas anteriormente a max. Para ello, se vacía la cubeta y se lava al menos dos veces con cada una de las disoluciones antes de efectuar la medida definitiva. Anote los resultados en una tabla tipo: [CuSO4] 0,07 M 0,05 M 0,03 M 0,02 M 0,01 M Mezcla desconocida Absorbancia Finalmente, se prepara una disolución de concentración no conocida mezclando las disoluciones 0,05 y 0,03M de CuSO4, en una proporción volumen/volumen conocida e indicada por el profesor. Mida su absorbancia a max y anote el valor. IV. Representación de la recta de calibrado Absorbancia = f (concentración) y determinación de una concentración desconocida por interpolación. Las parejas de datos obtenidos absorbancia/concentración se utilizan para representar una gráfica Absorbancia = f (concentración) y se ajusta la ecuación de una recta que pasa por el origen. Durante la sesión de laboratorio se utilizará papel milimetrado para un primer registro de la recta y comprobar si los valores obtenidos son aceptables. Si algún valor se desvía de forma inaceptable se debe descartar y repetir la medida. Con la recta de calibrado obtenida se determinara aproximadamente la concentración de la mezcla de CuSO4 que se ha preparado por interpolación. Posteriormente (Cuestiones post) se repetirá la gráfica, el ajuste de la recta y el cálculo de la concentración desconocida con el programa Excel. Lab. Química I (Grado Química) Curso 2012-2013 Práctica 8 - 5 Residuos: Las disoluciones de CuSO4 se recogerán en dos recipientes de residuos marcados como “CuSO4 0,5 M” y “CuSO4 diluidas”. Limpieza del material ● Borre el etiquetado con el etanol reservado a tal fin. ● Enjuague el material con agua del grifo. Si no queda totalmente traslúcido lávelo con unas gotas de amoniaco concentrado y enjuague de nuevo con agua del grifo. ● Enjuague el material con una pequeña cantidad de agua desionizada del frasco lavador. ● Deje secar el material boca arriba en su puesto de trabajo. AL FINALIZAR PASE LOS DATOS DEL DIARIO A LA HOJA DE RESULTADOS, COMPLÉTELO CON LOS CÁLCULOS NECESARIOS Y AÑADA SUS OBSERVACIONES Y COMENTARIOS BIBLIOGRAFIA Y VIDEOS Química General, R.H. Petrucci et al., Pearson, 2011. Química, R. Chang, Ed. McGraw Hill, 2010, (7ª Edición) Principios de Química: Los Caminos del descubrimiento P. Atkins y L. Jones. Ed. Médica Panamericana. 2006. 3ª Edición), Capítulo 10. Royal Chemical Society: Practical Chemistry for Schools and Colleges http://www.rsc.org/Education/Teachers/Resources/practical/index3.htm Universidad de Valencia: Material volumétrico: http://mmedia.uv.es/buildhtml?user=tcliment&path=/LABORATORIO/&name=material_basico_labo.mp4 Valoración: http://mmedia.uv.es/buildhtml?user=martiny&path=/&name=valoracion.flv CUESTIONES (no necesita copiar los enunciados, indique el nº para la respuesta) Cuestiones previas. Los cálculos que se proponen en este apartado serán los utilizados en el procedimiento experimental. 1. La Absorbancia viene definida como A = c l y es adimensional. Justifíquelo teniendo en cuenta las unidades que se deben utilizar para c, y l. 2. ¿Qué cantidad de CuSO4·5H2O se necesita para preparar 25 mL de una disolución 0,5M? 3. Complete la tabla siguiente indicando: a) Los volúmenes de disolución de CuSO4 0,5M necesarios para obtener cada disolución diluida. Sume todos los volúmenes que va a gastar de dicha disolución 0,5 M y compruebe que tendrá bastante con 25 mL. b) Teniendo en cuenta el Material disponible ¿Qué material debe utilizar para medir el volumen necesario de CuSO4 0,5 M para preparar cada disolución? c) ¿Con qué incertidumbre se podrá medir en cada caso el volumen de CuSO4 0,5M? [CuSO4]/ mol/L V a preparar V(CuSO4 0.5 M) (mL) Material a utilizar Incertidumbre 0,07 M 25 mL 0,05 M 25 mL 0,03 M 25 mL 0,02 M 50 mL 0,01 M 100 mL Lab. Química I (Grado Química) Curso 2012-2013 Práctica 8 - 6 4. Si no dispusiéramos de la disolución madre de CuSO4 0,5M deberíamos pesar la cantidad de sólido necesaria para cada disolución. Complete la tabla siguiente indicando: a) La cantidad de CuSO4·5H2O que necesitaría pesar para preparar cada una de las disoluciones. [CuSO4]/ mol/L V a preparar CuSO4·5H2O (g) 0,07 M 25 mL 0,05 M 25 mL 0,03 M 25 mL 0,02 M 50 mL 0,01 M 100 mL b) ¿Con qué incertidumbre puede pesar dichas cantidades teniendo en cuenta las balanzas disponibles? Cuestiones posteriores al trabajo experimental 1. a) Represente el espectro de absorción del CuSO4 con los valores obtenidos de A frente a la longitud de onda (ver Figura 1). b) Indique sobre el espectro max, la longitud de onda donde la A (absorbancia) es máxima. c) ¿Por qué trabajamos en el apartado III a esta longitud de onda? 2. Relacione las gráficas que se obtienen al representar la absorbancia frente a la concentración de CuSO4 con la ecuación de Lambert-Beer. ¿Qué indica la pendiente de la recta? 3. a) Utilizando el programa Excel dibuje y ajuste la recta de calibrado Absorbancia = f (concentración) con los valores obtenidos. Atención: se deben usar los valores reales (experimentales) de las concentraciones con sus cifras significativas. b) Con la recta de calibrado obtenida determine por interpolación la concentración de la mezcla de concentración desconocida de CuSO4 que ha preparado. c) Teniendo en cuenta los volúmenes y molaridades de las dos disoluciones utilizadas para preparar la mezcla desconocida calcule matemáticamente cuál debe ser la concentración resultante y compárela con los valores obtenidos con el programa Excel a partir de la recta de calibrado. Lab. Química I (Grado Química) Curso 2012-2013 Práctica 8 - 7 LABORATORIO QUÍMICA I APELLIDOS Y NOMBRE RESULTADOS PRÁCTICA 8 GRUPO Y PUESTO: Una vez completado recuerde entregar una copia al profesor/profesora. I. PREPARACIÓN DE DISOLUCIONES. [CuSO4] 0,5 M 0,07 M 0,05 M 0,03 M 0,02 M 0,01 M Volumen a preparar 25 mL 25 mL 25 mL 25 mL 50 mL 100 mL Preparada a partir de Cantidad necesaria (g o mL) Cantidad pesada o medida Molaridad real NOTA: Calcule las cifras significativas para la Molaridad a partir de la precisión de las medidas implicadas. II. VALORES DE ABSORBANCIA DEL CuSO4 0,03 M. max (nm) 550 600 650 700 750 800 850 900 950 Absorbancia Determinación de la longitud de onda analítica. max (nm) Absorbancia III. VALORES DE ABSORBANCIA max. [CuSO4] Absorbancia a max: 0,07 M 0,05 M 0,03 M 0,02 M 0,01 M nm Disoluciones mezcladas Mezcla Disolución 1 Disolución 2 Vmezcla (mL) [CuSO4] [CuSO4] [CuSO4]mezcla calculada V (mL) V (mL) Absorbancia (mezcla) IV. RECTA DE CALIBRADO Absorbancia = f (concentración) Y DETERMINACIÓN DE LA CONCENTRACIÓN DE LA MEZCLA. Dibuje la recta de calibrado Absorbancia = f (concentración) en el reverso de la hoja. A partir de la Absorbancia de la mezcla indique sobre la gráfica el camino para determinar su concentración. Compare el valor obtenido con el calculado. Lab. Química I (Grado Química) Curso 2012-2013 OBSERVACIONES Y COMENTARIOS (puede añadir las hojas que necesite): Práctica 8 - 8 Lab. Química I (Grado Química) Curso 2012-2013 Práctica 9 -1 PRÁCTICA 9: Destilación de mezclas de líquidos miscibles: destilación acetona-ácido acético. Estimación de la proporción de acético en el destilado por valoración ácido-base. Redacción: Begoña García. OBJETIVOS: Destilación simple de una mezcla de acetona-ácido acético. Destilación con columna de fraccionamiento de una mezcla acetona-ácido acético. Determinación de la cantidad de ácido en el destilado por valoración ácido-base. Comparación de la eficacia de las dos destilaciones. Determinación de la densidad de mezclas acetona-ácido acético por pesada. INTRODUCCIÓN Hemos visto en la Práctica 4 que un líquido hierve cuando su presión de vapor iguala a la presión ejercida sobre él, y que la destilación consiste en vaporizar un líquido y condensar los vapores. En todas las destilaciones de mezclas de líquidos se cumple la ley de Dalton que establece que la presión de vapor de una mezcla es la suma de las presiones parciales de vapor de sus componentes individuales. Así, cuando una mezcla de líquidos A y B (con presiones de vapor PA y PB, respectivamente) hierve, su presión de vapor P es la suma de las presiones parciales que ejercen cada uno de los componentes (PpA, PpB respectivamente): P = PpA + PpB (Fíjese en que PpA y PpB no corresponden necesariamente a PA y PB) Se observan marcadas diferencias según sean: Líquidos inmiscibles: En este caso cada líquido ejerce una presión parcial igual que si estuviera puro y: P = P A + PB El punto de ebullición de la mezcla es por tanto inferior al de cualquiera de sus componentes y la composición de los vapores, aunque distinta de la de la mezcla líquida, es constante durante la destilación hasta que uno de los dos componentes se agota. Éste no va a ser nuestro caso, aunque tiene gran utilidad, p.ej. para separar aceites esenciales de las plantas para su utilización en farmacología, perfumería y cosmética por codestilación con agua (arrastre o corriente de vapor). Líquidos miscibles: Suponiendo que cada líquido se comporta como si fuera puro (no hay interacciones intermoleculares entre ellos) estas mezclas siguen la ley de Raoult que establece que, a una temperatura y presión determinada, la presión parcial de vapor de un compuesto A (PpA) en una mezcla es directamente proporcional a la presión de vapor del compuesto puro en esas condiciones (PA) multiplicada por su fracción molar en la mezcla líquida (XA): PpA = PA XA Por tanto, la presión total de vapor de una mezcla de líquidos volátiles A y B depende de las presiones de vapor de cada componente puro y de sus fracciones molares en la mezcla líquida, es decir: P = P A X A + P B XB Lab. Química I (Grado Química) Curso 2012-2013 Práctica 9 -2 Diagramas de fases: Se utilizan para analizar la destilación de mezclas de líquidos (Figuras 1 y 2). Representan la composición molar en el equilibrio del vapor (línea superior) y del líquido (línea inferior) en función de la temperatura y nos permiten predecir la composición de la fase de vapor, y por tanto del destilado. Supongamos dos líquidos miscibles A y B con p.eb A < p.eb. B y dos posibles situaciones: que haya una gran diferencia entre los p.eb. (Figura 1), o una diferencia menor (Figura 2). En las Figuras 1a y 2a se ve que a una temperatura T la composición de las fases vapor V y líquida L son distintas. La fracción molar en la fase vapor (YA, YB, respectivamente) depende de la relación presión parcial-presión total del sistema, es decir YA = PpA/P = PA XA/P Yb = Ppb/P= PB XB/P a) Si p.eb. A << p.eb. B la Figura 1a muestra que: ►A una temperatura T el componente A esta casi todo en fase vapor V y B esta fundamentalmente en fase líquida L. ►A mayor temperatura (T‘), aumenta B en fase vapor (V‘) y disminuye A en fase líquida (L’). Si calentamos una mezcla 1:1 A+B (50% molar en A y B), (Figura 1b) la composición del vapor se puede determinar partiendo de la del líquido inicial (1), dibujando una línea horizontal líquidovapor (2) y leyendo en abcisas el valor correspondiente (3). ►Al comenzar la destilación el vapor es fundamentalmente A. ►Al destilar disminuye A en el líquido (nos desplazamos a la derecha), pero aún así, el vapor, y por tanto el destilado esta constituido fundamentalmente por el A ((4) (5) (6)). ►Cuando la mayoría de A haya destilado la temperatura aumentará hasta el p.eb. de B que destilará también prácticamente puro. A y B se pueden separar fácilmente, o lo que es lo mismo líquidos con diferencias en sus p.eb. de 80-100 ºC pueden separarse de forma casi completa por destilación simple. (a) Figura 1 (b) b) Si p.eb. A ≈ p.eb. B la Figura 2 muestra que: ►A las dos temperaturas T y T’, A y B están en proporción apreciable en ambas fases (V, L; V’, L’) ►Pequeñas variaciones de temperatura resultan en cambios destacados en las composiciones de V, V’y L, L’. Práctica 9 -3 vapor T´ T p.eb. B líquido Temperatura Temperatura Lab. Química I (Grado Química) Curso 2012-2013 p.eb. A A 100 B 0 vapor (5) (2) líquido (3) V L´0 50 L 50 Composición molar (%) V´ 100 (a) A 100 B 0 (6) (1) (4) 50 50 Composición (%) Figura 2 (b) En cuanto a la destilación (Figura 2b), el vapor inicial [(1) (2) (3)] es A:B aprox. 85:15 y no se puede obtener directamente A puro aunque el residuo del matraz también se enriquece en B según avanza la destilación (4) (5) (6). Como consecuencia, las mezclas de líquidos con puntos de ebullición próximos (<80-100 ºC) no se pueden separar por destilación simple. Sin embargo, si se volviese a destilar la fracción con 85% de A, se podría obtener A más puro, que a su vez se podría volver a destilar, etc. Esto no es útil ya que consumiría mucho tiempo, por lo que se emplea en su lugar una técnica denominada destilación fraccionada o rectificación. Esta técnica consiste básicamente en intercalar entre el matraz y la cabeza de destilación de un montaje de destilación simple un dispositivo llamado columna de fraccionamiento o de rectificación. Estas columnas tienen una superficie irregular o un relleno sobre el que el vapor se condensa y evapora sucesivamente, de forma que a la salida hacia el refrigerante llega el vapor rico en el componente más volátil. Normalmente se recoge el destilado en distintas fracciones. Finalmente veamos un caso en el que las interacciones moleculares hacen que la destilación de una mezcla no siga la ley de Raoult. La mayoría de estas mezclas presentan diagramas similares a la Figura 3 con un mínimo a una temperatura (menor que los p.eb. de sus componentes) y composición determinada. Estas mezclas se denominan azeótropos o mezclas azeotrópicas. Cuando se calienta un azeótropo de este tipo, la mezcla se enriquece en el componente más volátil hasta alcanzar la composición del mínimo, destilando al p.eb. del azeótropo hasta que uno de los componentes se agota. El componente restante destila después como líquido puro. Por tanto, las mezclas de líquidos que forman azeótropos no pueden separarse en sus componentes puros ni por destilación fraccionada. Un ejemplo muy conocido es el alcohol etílico común, mezcla etanol:agua 96:4, que por ello se comercializa como alcohol etílico 96% o 96º. p.eb. B Temp. vapor p.eb. A vapor líquido líquido p.eb. azeotropo A 100 B 0 Composición del azeotropo 50 50 Composición molar (%) 0 100 Figura 3. Diagrama de fases de un mezcla azeotrópica de p.eb. mínimo 0 100 Lab. Química I (Grado Química) Curso 2012-2013 Práctica 9 -4 EXPERIMENTAL Material Manta calefactora + regulador Taco de madera Matraz fondo redondo (m.f.r.) 100 mL + tapón Aro de corcho Pinza+nuez (2) Soporte Cabeza de destilación + rosca + aro silicona Termómetro Refrigerante + gomas Colector acodado Clips (2) Columna de fraccionamiento Germen de ebullición Vial + tapón (3) Tubos Eppendorf (6) Erlenmeyer 50 mL (3) Probeta 10 mL Pipeta de 0,1mL Pipeta de 1 mL Pipeta de 5 mL Propipeta Micropipeta Bureta de 10 mL Reactivos Ácido acético Acetona NaOH 0,4 M Fenolftaleína (disol.) Aparatos Balanza Secador (1 por mesa) Antes de llevar a cabo el trabajo experimental asegúrese revisando la Práctica 4 y los Anexos de que conoce adecuadamente la información sobre: Procedimiento general para una destilación. Utilización del material volumétrico. La bureta. Protocolo técnico de manejo de la bureta. Valoración con indicador. Pipetas y propipetas. I. Preparación. ● Asegúrese de que dispone en el diario de una tabla del tipo: Fracción Volumen NaOH (mL) [HOAc] (M) ● Revise el material: El material para la destilación, los viales y los tubos Eppendorf deben estar limpios y secos. Pipetas, buretas y erlenmeyers deben estar limpios y enjuagados con agua desionizada. ● Etiquete correctamente: Viales (Mezclas): M-1 (de partida), M-2 (tras destilación simple), destilación+rectificación) Tubos Eppendorf (Fracciones): A-1, A-2, A-3 (Destilación simple) B-1, B-2, B-3 (Destilación + rectificación) Erlenmeyers (para las valoraciones): 1, 2, 3. M-3 (tras ¡ATENCIÓN!: Cuando tome las muestras debe taparlas rápida y firmemente para evitar evaporación de los componentes. ● Tome nota de las características y precisión de la bureta que va a utilizar. Llénela con agua desionizada y practique su manejo para enrasar y controlar la descarga de líquido. II. Preparación de la mezcla y valoración del ácido en la mezcla de partida: Utilizando los dosificadores correspondientes introduzca en el matraz f.r. 20 mL de ácido acético y 20 mL de acetona. Tape el matraz, remueva para homogeneizar y tome 2 mL de la mezcla reservándola en un vial tapado (M-1). III. Destilación simple de la mezcla acético-acetona y valoración del ácido en las fracciones. a) Destilación: Haga el montaje siguiendo las instrucciones de la Práctica 4. Recuerde poner el germen de ebullición. Comience con el regulador al 30% y regúlelo para que destile 1-2 gotas/seg. aproximadamente. Lab. Química I (Grado Química) Curso 2012-2013 Práctica 9 -5 Recoja fracciones de 1,5-2 mL de destilado en 3 tubos Eppendorf (fracciones A-1, A-2, A-3), anotando el intervalo de temperaturas de destilación de cada una y cerrando bien cada tubo. Sustituya el tercer tubo por un erlenmeyer, desenchufe la manta y, con mucho cuidado, retire taco y manta. Aleje la manta de la zona de trabajo y deje enfriar el sistema antes de desmontar. Recuerde desmontar en orden inverso al montaje y tape de nuevo el matraz f.r. Tome 2 mL de la mezcla que queda en el matraz (M-2) y resérvela en un vial tapado. b) Valoración: Prepare la bureta con disolución de NaOH 0,4 M para valorar (ver Práctica 7). Tome con una pipeta 0,1 mL de mezcla M-1 y transfiérala a un matraz erlenmeyer. Añada 10 mL de agua y 2 gotas de fenolftaleína y valore el ácido. Anote en la tabla el volumen de NaOH consumido. Repita con la fracción M-2. Reserve el resto de la mezcla de ambos viales para el apartado V. Tome con una pipeta 1 mL de la fracción A-1, añada 10 mL de agua y 2 gotas de indicador y valore con NaOH 0,4 M. Anote en la tabla el volumen de NaOH consumido. Repita con las fracciones A-2 y A-3. IV. Destilación con columna de rectificación y valoración del ácido en las fracciones. a) Destilación: Antes de hacer el montaje lave con un poco de acetona el refrigerante, la cabeza de destilación y el colector acodado y elimine los restos de acetona con el secador. Para el montaje basta intercalar la columna de rectificación (o fraccionamiento) entre el matraz y la cabeza de destilación (ver Foto). Recuerde poner de nuevo germen de ebullición. La destilación se lleva a cabo de forma similar al apartado III, regulando la calefacción, recogiendo tres fracciones (B-1, B-2 y B-3) y anotando el intervalo de temperaturas de destilación de cada una. Al final se enfría y desmonta del mismo modo y se reservan 2 mL de la mezcla que queda en el matraz (M-3) en un vial tapado. b) Valoración: Valore 0,1 mL de M-3, y 1 mL de B-1, B-2 y B-3 con NaOH como en el apartado III. V. Determinación de la densidad de las mezclas acetona-ácido acético M-1, M-2 y M-3. Para cada mezcla: Tare un vial limpio y seco en una balanza de tres cifras decimales. Utilizando la pipeta de 1 mL añada al vial un volumen exactamente medido de la mezcla. Pese el vial lleno y calcule la densidad de la mezcla en g/mL. Residuos: Deposite todos los productos y residuos obtenidos en los recipientes destinados a tal fin en el laboratorio. Limpieza del material (ver Anexo II) ● Borre el etiquetado con el etanol reservado a tal fin. ● NO FRIEGUE EL MATERIAL UTILIZADO EN LA DESTILACIÓN. Enjuáguelo con una pequeña cantidad de acetona y déjelo secar al aire. ● Enjuague el material de las valoraciones con agua del grifo y después con una pequeña cantidad de agua desionizada y déjelo secar al aire boca arriba. No lave este material con acetona. AL FINALIZAR PASE LOS DATOS DEL DIARIO DE LABORATORIO A LA HOJA DE RESULTADOS, COMPLÉTELO CON LOS CÁLCULOS NECESARIOS Y AÑADA SUS OBSERVACIONES Y COMENTARIOS Lab. Química I (Grado Química) Curso 2012-2013 Práctica 9 -6 BIBLIOGRAFÍA Y VIDEOS Química General, R.H. Petrucci et al., Pearson, 2011. Química, R. Chang, Ed. McGraw Hill, 2010. Química. La ciencia central T.L. Brown et al. Ed. Pearson, 11ª Edición, 2009 Principios de Química: Los caminos del descubrimiento P. Atkins y L. Jones. Ed. Médica Panamericana. 2006. 3ª Edición), Capítulo 10. Técnicas experimentales en Síntesis Orgánica, M.A Martínez Grau, A. G. Csákÿ. Ed. Síntesis. Royal Chemical Society: Practical Chemistry for Schools and Colleges http://www.rsc.org/Education/Teachers/Resources/practical/index3.htm Universidad de Valencia: http://mmedia.uv.es/buildhtml?lang=es_ES&user=tcliment&name=destilacion.mp4&path=/LABORATORIO/&id=6578 CUESTIONES (no necesita copiar los enunciados, indique el nº para la respuesta) Cuestiones previas 120 110 100 Temperatura (ºC) 1. La figura siguiente muestra el diagrama de fases de la mezcla acetona-ácido acético: a) Señale sobre el diagrama los puntos de ebullición de acetona y ácido acético puros. b) Calcule para la mezcla a destilar la fracción molar de acetona y de ácido acético. c) Teniendo en cuenta la composición de la mezcla de partida indique sobre el diagrama el camino para determinar la composición del destilado al inicio de la destilación simple. d) ¿Qué composición tendrá el destilado en fracción molar para los dos componentes? 90 80 70 60 50 Acetona 1 ............................................... 0,5 .......................................... 0 HOAc 0 ............................................... 0,5 .......................................... 1 Fracción molar 2. Una mezcla preparada mezclando volúmenes iguales de acetona y ácido acético se destila. Un mililitro del destilado disuelto en agua desionizada consume 2,5 mL de disolución de NaOH 0,4 M valorando con fenolftaleína como indicador. Calcule con estos datos cuál es la composición de líquido destilado. 3. La densidad de los líquidos puros es: acetona = 0,79 g·cm-3; ácido acético = 1,05 g·cm-3. Calcule la densidad de la mezcla preparada a partir de volúmenes iguales, suponiendo que no hay cambio de volumen al mezclar ambos líquidos. ¿Puede ser la medida de densidad un método para determinar la composición de la mezcla? Justifique la respuesta. 4. ¿Qué es un azeótropo? Cuestiones posteriores al trabajo experimental 1. A la vista de los resultados obtenidos, compare la eficiencia de la destilación en ambas experiencias. 2. Exprese el resultado calculando en cada caso la cantidad de ácido acético en la acetona en: a) g/L. b) fracción molar. Lab. Química I (Grado Química) Curso 2012-2013 Práctica 9 -7 LABORATORIO QUÍMICA I RESULTADOS PRÁCTICA 9 APELLIDOS Y NOMBRE GRUPO Y PUESTO: Una vez completado recuerde entregar una copia al profesor/profesora CONTENIDO DE ÁCIDO ACÉTICO EN LAS MEZCLAS V a valorar Fracción (mL) Mezcla de partida Mezcla tras destilación simple Mezcla tras destilación fraccionada Volumen NaOH (mL) Concentración [HOAc] (M) M-1 M-2 M-3 DESTILACIÓN SIMPLE Fracción V a valorar (mL) Volumen NaOH (mL) Concentración [HOAc] (M) Volumen NaOH (mL) Concentración [HOAc] (M) A-1 A-2 A-3 DESTILACIÓN FRACCIONADA Fracción V a valorar (mL) B-1 B-2 B-3 Fracción Densidad (g/mL) Cantidad de HOAc (g/mL) Concentración [HOAc] (M) M-1 M-2 M-3 OBSERVACIONES Y COMENTARIOS (Añada las hojas que considere necesario) Lab. Química I (Grado Química) Curso 2012-2013 Práctica 9 -8 PRÁCTICA 10: Cálculos estequiométricos: reacción entre el carbonato de calcio y el ácido clorhídrico. Elaboración: Rosa Valero OBJETIVOS: La práctica tiene dos objetivos fundamentales: Determinación de la masa molecular del carbonato cálcico (CaCO3). Estimación de la riqueza en peso de carbonato cálcico en una muestra problema. En ambos casos se utilizaron dos métodos para la determinación Método volumétrico. Método gravimétrico INTRODUCCIÓN En esta práctica se dirigirá la atención hacia el concepto de reacción química. Toda reacción es una transformación de la materia en la que una o varias sustancias, denominadas reactivos, se transforman total o parcialmente en otras, denominadas productos, con propiedades diferentes tanto químicas como físicas. Sin embargo, el hecho de formular una ecuación, por ejemplo: no implica que la reacción ocurra en la práctica, ya que son las propiedades químicas de los reactivos (A y B) las que determinarán si en la práctica la reacción es o no viable. La tendencia a combinarse de una sustancia, o reactividad, viene modulada por la naturaleza de otras sustancias que haya en su entorno y por las condiciones en las que se encuentre, especialmente por su presión y temperatura. Dos son las ramas fundamentales de la Química que se centran en el análisis de la reactividad de los compuestos químicos: La Termodinámica, que estudia los cambios o transformaciones de la energía que acompañan a los cambios físico-químicos y nos indica si es posible el paso de los reactivos a productos, aunque no el tiempo que tarda en producirse el cambio. La Cinética, que estudia cuantitativamente la rapidez de descomposición de los reactivos y formación de los productos, es decir, la velocidad de reacción, y de cómo cambia bajo condiciones variables. Hay que tener presente que, en el caso en que una reacción ocurra, los reactivos pueden no transformarse completamente en productos, alcanzándose la situación de equilibrio químico. La constante de equilibrio K indica la relación entre las concentraciones de productos y de los reactivos que quedan en el sistema. Si la concentración final de alguno de los reactivos es tan pequeña que a efectos de cálculo podamos admitir que los reactivos se han transformado completamente en productos, decimos que la reacción se ha producido de forma cuantitativa (por ejemplo, si [B] ≈ 0, K = ∞). Este caso se produce en aquellas situaciones en las que: a) La constante de equilibrio adopta valores muy altos. b) Uno de los productos escapa del sistema, por ejemplo, cuando uno o más productos son gaseosos y salen del sistema facilitando de este modo el avance de la reacción química. Por otra parte, cuando una reacción tiene lugar con participación de varios reactivos es frecuente que no todos ellos se encuentren en proporciones estequiométricas, es decir en las proporciones que nos indican los coeficientes de la ecuación química. En estos casos, se considera como reactivo limitante aquel que, por la cantidad inicial en la que está presente, limita el avance de la reacción. El reactivo o reactivos en exceso seguirá presente en cantidades significativas en el medio de reacción incluso en el supuesto de que la reacción se produjera de forma cuantitativa. Por ejemplo, en la reacción entre el CaCO3 y el ácido clorhídrico que se puede representar mediante la ecuación: La ecuación química informa de que cada mol de CaCO3 se combina con un volumen de disolución tal que contenga 2 moles de HCl. Si se añadiera un volumen de disolución conteniendo 3 moles de ácido, el carbonato será el reactivo limitante y el HCl el reactivo en exceso. El reactivo limitante se consume por completo en una reacción cuantitativa. El CO2 formado es un gas que escapará del medio contribuyendo de esta forma a que la reacción se complete, es decir será cuantitativa, y en este caso podríamos formularla como: Si partimos de una cantidad conocida de CaCO3 y la tratamos con un volumen suficiente de HCl para que reaccione cuantitativamente, la cuantificación de la cantidad de CO2 desprendido y la estequiometría de la reacción que relaciona los moles de CO2 con los de carbonato: a) Conociendo la cantidad de CaCO3 utilizada se puede calcular fácilmente la masa molecular del CaCO3. b) Se puede determinar la cantidad de CaCO3 en una muestra impura. Para determinar la cantidad de CO2 vamos a utilizar dos métodos: A. Método gravimétrico: aprovecha la pérdida de peso que se produce en el sistema al liberarse el CO2 (g) al medio. La reacción se lleva a cabo con un volumen suficiente de HCl para que el carbonato reaccione cuantitativamente, por lo que pesando el sistema al inicio y al final de la reacción la diferencia entre las dos pesadas nos dará los gramos de CO2 liberados y el número de moles conociendo la masa molecular del CO2. B. Método volumétrico: se basa en la medida del volumen de CO2 desprendido. El dispositivo necesario para recoger el gas y medir su volumen es el eudiómetro (o bureta invertida) (Figura 1). Inicialmente el eudiómetro debe estar lleno de agua y la reacción se lleva a cabo en un sistema que permite introducir el gas desprendido dentro de él. El gas desplaza en parte al agua, por lo que al finalizar la reacción podemos leer sobre la escala del eudiómetro el volumen de agua desplazado. El número de moles de CO2 producidos se puede calcular a través de la ecuación de los gases ideales: PCO2 • V = nCO2 • R • T [R = 0.082 atm·L/mol·K, VCO2 (L), PCO2 (atm) y T (K)] El volumen que ocupa el CO2 se puede leer directamente sobre el eudiómetro, pero para conocer PCO2 debemos relacionarla con la presión atmosférica, ya que ésta puede medirse con un barómetro. Figura 1. Montaje de recogida de gases con eudiómetro Según el Principio Fundamental de la Hidrostática los puntos situados a una misma altura en un líquido tienen idéntica presión, por lo tanto: P1 (dentro del eudiómetro) = P2 (en la superficie del H2O de la cubeta, Patmosférica) Pero P1 no es debida únicamente al CO2 ya que: 1. El CO2 se recoge en contacto con agua por lo que debe considerarse la contribución del vapor de agua. Según la ley de Dalton de las presiones parciales la presión total del gas será: P1 = Pgas = PCO2 + Pvapor H2O = Patmosférica PCO2 = Patmosférica - Pvapor H2O En la que Pgas = presión total del gas PvaporH2O = presión de vapor del agua a la temperatura del agua (ver Tabla 1). Tabla 1. Datos sobre la variación de la presión de vapor del agua con la temperatura. Temperatura (ºC) Presión de vapor del H2O (mmHg) 20,0 17,5 21,0 18,7 22,0 19,8 23,0 21,1 24,0 22,4 25,0 23,8 26,0 25,2 27,0 26,7 28,0 28,3 29,0 30,0 30,0 31,8 2. Normalmente el nivel del agua en la cubeta y dentro del eudiómetro no es el mismo, por lo que debe tenerse en cuenta la contribución de la columna de agua. Por tanto, la presión dentro del eudiómetro será: P1 = Pgas = PCO2 + Pvapor H2O + PhH2O = Patmosférica PCO2 = Patmosférica - Pvapor H2O - PhH2O PhH2O se puede calcular a través de la ecuación: PhH2O = dH2O • g • hH2O La columna de agua es equivalente a una columna de mercurio de distinta altura y relacionadas ambas por la ecuación: PhH2O = dH2O • g • hH2O = dHg • g • hHg hHg = dH2O hH2O / dHg Donde: dH20 = densidad del agua en el SI: 103 Kg/m3 hH2O =altura de la columna de agua (medida con la regla y transformada en metros) dHg = densidad del mercurio en el SI: 13,6 x 103 Kg/m3 hHg =altura de la columna de mercurio (m) Transformando el dato de hHg a mm, se obtiene la presión de la columna de agua en “mmHg”. Pasando el valor de la PCO2 a atm (1 atm =760 mmHg) y substituyendo dicho valor en la ecuación de los gases ideales calcularemos el número de moles de CO2 producidos. EXPERIMENTAL Material y aparatos Soporte + Pinza doble de bureta Eudiómetro de 50 mL Cubeta con agua Erlenmeyer 100 mL con boca esmerilada Esmerilado con oliva + goma + tubo colector Pesasustancias tipo tapón de vial (3) Pinza de disección larga Espátula de cuchara pequeña Vaso de precipitados 50 mL Pipeta 10 mL + Propipeta Papel de aluminio para tapar matraz Regla milimetrada Reactivos CaCO3 HCl 1M HCl 0,2 M Muestra problema Aparatos Barómetro Balanza Cronómetro Antes de llevar a cabo el trabajo experimental asegúrese de que conoce adecuadamente: Utilización del material volumétrico. Pipetas y propipetas. Funcionamiento del cronómetro. Utilización del eudiómetro. A. DETERMINACIÓN DE LA MASA MOLECULAR DEL CaCO3 A.1. Método gravimétrico I. Preparación: ● Asegúrese de que dispone en el diario de laboratorio de una tabla del tipo: Peso del montaje (g) Ensayo CaCO3 (g) HCl 1M (mL) Tiempo (min) 0 min final CO2 (g) ● Revise el material: Los pesasustancias (tapones de vial) deben estar limpios y secos. El erlenmeyer debe estar limpio y enjuagado con agua desionizada. II. Reacción: a) Pese unos 0,450 g de CaCO3 utilizando un tapón de vial como pesasustancias. Recuerde anotar el valor de la pesada con todos los decimales. b) Introduzca en el Erlenmeyer esmerilado de 100 mL un volumen de disolución de HCl 1M calculada previamente para que haya un exceso de 1% sobre la cantidad estequiométrica (Cuestión previa 3, VHCl = …………mL). c) Con ayuda de las pinzas, introduzca el pesasustancias con el carbonato cálcico en el fondo del matraz Erlenmeyer, con cuidado de no volcarlo (puede entrar en contacto con el HCl e iniciarse la reacción antes de lo deseado), y tape bien el matraz con papel de aluminio. d) Traslade el montaje (matraz + pesasustancia + reactivos + papel de aluminio) hasta la balanza, con mucho cuidado para que los reactivos NO entren en contacto, y pese el montaje. e) Prepare el cronómetro (o en su defecto un reloj). Deberá ponerlo en marcha al inicio del paso siguiente. f) Agite el montaje con un ligero movimiento de muñeca para poner en contacto los reactivos, cronometrando el tiempo de reacción que debe ser de 7-8 min. El CO2 que se desprende es más denso que el aire por lo que para facilitar que salga del matraz se debe destapar el sistema cada minuto de reacción. g) Anote el tiempo de reacción y pese de nuevo el sistema. h) Repita la experiencia completa 2 veces para obtener resultados reproducibles. Recuerde lavar con agua del grifo el material utilizado y enjuagar el Erlenmeyer con agua desionizada para evitar contaminaciones. III. Cálculos: Aplicando la fórmula obtenida en la Cuestión previa 4 calcule la masa molecular del CaCO3. Si algún valor se desvía de forma inaceptable se debe descartar y repetir la medida. A.2. Método volumétrico I. Preparación: ● Asegúrese de que dispone en el diario de laboratorio de una tabla del tipo: Ensayo CaCO3 (g) HCl 0,2M (mL) Tiempo (min) hcolumna H2O (cm) PH2O (mmHg) ● Revise el material: Los pesasustancias (tapones de vial) deben estar limpios y secos. El erlenmeyer debe estar limpio y enjuagado con agua desionizada. ● Tome nota de la Pat (mmHg) y de la temperatura (ºC) del laboratorio. ● Prepare el sistema de recogida de gas (ver Figura 1): Llene la cubeta de plástico con agua del grifo. Llene el eudiómetro completamente con agua del grifo y tápelo con la mano. Introduzca el eudiómetro invertido en la cubeta con agua de forma que no entre nada de aire en la columna y sujételo firmemente con la pinza. La parte inferior debe quedar sumergida aunque sin tocar el fondo. Introduzca el extremo del colector de gas en el eudiómetro dentro del baño de agua. III. Reacción: a) Pese aproximadamente 0,1 g de CaCO3 utilizando un tapón de vial como pesasustancias. Recuerde anotar el valor de la pesada con todos los decimales. b) Introduzca en el Erlenmeyer esmerilado de 100 mL un volumen de disolución de HCl 0,2M calculada previamente para que haya un exceso de 1% sobre la cantidad estequiométrica (Cuestión previa 6, VHCl = …………mL). c) Con ayuda de las pinzas, introduzca el pesasustancias con el CaCO3 en el fondo del matraz Erlenmeyer, con cuidado de no volcarlo (puede entrar en contacto con el HCl e iniciarse la reacción antes de lo deseado), y tape bien el matraz con el esmerilado con oliva. d) Agite el montaje con un ligero movimiento de muñeca para poner en contacto los reactivos. e) Una vez finalizado el desprendimiento de gas tome nota: - del volumen de gas por lectura directa en la escala del eudiómetro. - de la altura de la columna de agua con ayuda de una regla milimetrada. f) Repita la experiencia completa 2 veces para obtener resultados reproducibles. Recuerde limpiar cuidadosamente el material utilizado y enjuague el Erlenmeyer con agua desionizada para evitar contaminaciones III. Cálculos: Aplicando las fórmulas descritas en la Introducción calcule la masa molecular del CaCO3. Si algún valor se desvía de forma inaceptable se debe descartar y repetir la medida. B. DETERMINACIÓN DE LA RIQUEZA EN CaCO3 DE UNA MUESTRA IMPURA B.1. Método gravimétrico: Opere de la misma forma que en el Apartado A.1 utilizando la muestra impura de CaCO3. Realice tres medidas, calcule la pureza en CaCO3 expresada como % y promedie los resultados. B.2. Método volumétrico: Opere de la misma forma que en el Apartado A.2 utilizando la muestra impura de CaCO3. Realice tres medidas y a partir del volumen de CO2 calcule la pureza en CaCO3 expresada como % y promedie los resultados. EXPERIENCIA OPTATIVA: Coloque en la vitrina un vaso de precipitados de 100 mL lleno de HCl 2M o CH 3COOH (mitad glacial), introduzca con cuidado 1 huevo crudo previamente lavado y espere a que se acabe la reacción que tiene lugar. Explique que reacción ha tenido lugar y observe el estado en que queda la cáscara del huevo. Residuos: Deposite todos los productos y residuos obtenidos en los recipientes destinados a tal fin en el laboratorio. Limpieza del material: ● Borre el etiquetado con el etanol reservado a tal fin. ● Enjuague el material con agua del grifo y posteriormente con una pequeña cantidad de agua desionizada del frasco lavador y déjelo secar al aire boca arriba en su puesto de trabajo. AL FINALIZAR PASE LOS DATOS DEL DIARIO DE LABORATORIO A LA HOJA DE RESULTADOS, COMPLÉTELO CON LOS CÁLCULOS NECESARIOS Y AÑADA SUS OBSERVACIONES Y COMENTARIOS BIBLIOGRAFÍA Y VÍDEOS Química General, R.H. Petrucci et al., Pearson, 2011. Química, R. Chang, Ed. McGraw Hill, 2010. Química. La ciencia central T.L. Brown et al. Ed. Pearson, 11ª Edición, 2009 Principios de Química: Los caminos del descubrimiento P. Atkins y L. Jones. Ed. Médica Panamericana. 2006. 3ª Edición), Capítulo 10. Texto que ha servido de base para redactar la presente práctica: Universidad de Cádiz. http://www2.uca.es/grup-invest/corrosion/integrado/P16.pdf CUESTIONES (no necesita copiar los enunciados, indique el nº para la respuesta) Cuestiones previas 1. Enuncia la Ley de conservación de la masa en una reacción química. 2. a) ¿Qué entiende como reactivo limitante? b) En el experimento que va a llevar a cabo entre el CaCO3 y el HCl indique cual es el reactivo limitante. Justifique la respuesta. 3. a) Calcule el volumen de HCl 1M que debe añadir a 0,450 g de CaCO3 (MM = 100,09 g/mol) para que reaccione estequiométricamente todo el CaCO3. b) ¿Qué cantidad se deberá utilizar de HCl 1M si queremos disponer de un exceso del 1%? 4. Componga o diseñe una fórmula sencilla que permita calcular con rapidez la MM del carbonato cálcico a partir de los datos del método gravimétrico. 5. Describa las características y finalidad del eudiómetro. 6. a) Calcule el volumen de HCl 0,2M que debe añadir a 0,1 g de CaCO3 (MM = 100,09 g/mol) para que reaccione estequiométricamente todo el CaCO3. b) ¿Qué cantidad se deberá utilizar si queremos disponer de un exceso del 1%? 7. ¿Qué volumen de CO2 cabe esperar que recogerá al hacer reaccionar 0,1 g de CaCO3 con HCl en exceso? Cuestiones posteriores al trabajo experimental 1. Como sabe, el CaCO3 es el componente de las estalactitas y estalagmitas existente en diversas cuevas de interés turístico en España. Actualmente se está pensando en limitar la entrada al público debido al deterioro que experimentan estas precipitaciones calcáreas por la respiración humana. ¿Mediante que reacción química explicaría este hecho? 2. ¿Qué reacción química emplean los fontaneros para limpiar las cañerías de un calentador de agua obstruido por depósitos de cal? LABORATORIO QUÍMICA I RESULTADOS PRÁCTICA 10 APELLIDOS Y NOMBRE GRUPO Y PUESTO: Una vez completado recuerde entregar una copia al profesor/profesora A. DETERMINACIÓN DE LA MASA MOLECULAR DEL CaCO3 A.1. Método gravimétrico. Ensayo CaCO3 (g) HCl 1M (mL) Tiempo (min) Peso del montaje (g) 0 min final CO2 (g) desprendido 1 2 3 Ensayo 1 MASA MOLECULAR DEL CaCO3 CO2 mmol CaCO3 (g) MM CaCO3 (g/mol) CO2 (g) Media (g/mol) 2 3 OPCIONAL: De forma similar, los datos obtenidos en el apartado A.1 se pueden utilizar para determinar la masa molecular del CO2 si se conoce la masa molecular del CaCO3 Ensayo CaCO3 (g) Determinación de la masa molecular del CO2 CaCO3 (mmol) CO2 (g) MM CO2 (g/mol) Media (g/mol) 1 2 3 A.2. Método volumétrico. Pat (mmHg) DATOS NECESARIOS Ensayo CaCO3 (g) HCl 0,2M (mL) Tiempo (min) T (ºC) Pvapor (H2O) (mmHg) hcolumna H2O (cm) PH2O (mmHg) 1 2 3 MASA MOLECULAR DEL CaCO3 Ensayo 1 2 3 PCO2 (mmHg) CO2 (mmol) CaCO3 (g) MM CaCO3 (g/mol) Media (g/mol) B. DETERMINACIÓN DE LA RIQUEZA EN CaCO3 DE UNA MUESTRA IMPURA. B.1. Método gravimétrico. Ensayo Muestra (g) HCl 1M (mL) Tiempo (min) Peso del montaje (g) 0 min final CO2 desprendido g mmol 1 2 3 Ensayo RIQUEZA DE LA MUESTRA EN CaCO3 (% de CaCO3) CaCO3 (mmol) CaCO3 (g) Muestra (g) % de CaCO3 Media (%) 1 2 3 B.2. Método volumétrico. Ensayo Muestra (g) HCl 0,2M (mL) Tiempo (min) hcolumna H2O (cm) PH2O (mmHg) PCO2 (mmHg) 1 2 3 RIQUEZA DE LA MUESTRA EN CaCO3 (% de CaCO3) Ensayo CO2 (mmol) CaCO3 (mmol) CaCO3 (g) Muestra (g) % de CaCO3 Media (%) 1 2 3 OBSERVACIONES Y COMENTARIOS (Añada las hojas que considere necesarias) Agradecimientos A María Villalonga Sánchez, Enrique Gómez Cutillas y Sergio Plaza Martín, personal técnico del Laboratorio de Química General por su contribución a la revisión de los textos y su gran apoyo y dedicación en la puesta a punto del trabajo experimental. A los profesores Francisco Rafael Tortonda García, Ofelia Vila Bueso, José Vicente Gimeno, Miguel Clemente y Mercedes Rubio Mas por sus aportaciones a la elaboración y concreción de los contenidos del Seminario 1.