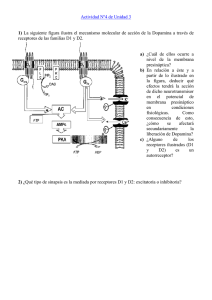
efecto farmacológico Cambios producidos por el fármaco en los procesos fisiológicos y la conducta del animal, lugares de acción Localizaciones donde las moléculas del compuesto interaccionan con moléculas situadas en la superficie o el interior de las células del organismo, afectando así a ciertos procesos bioquímicos de esas células. farmacocinética Proceso por el cual los fármacos son absorbidos, distribuidos por el organismo, metabolizados y excretados. inyección intravenosa (i.v.) Inyección de una sustancia directamente en una vena. inyección intraperitoneal (i.p.) Inyección de una sustancia en la cavidad peritoneal, espacio que rodea el estómago, intestino, hígado y otros órganos abdominales. inyección intramuscular (i.m.) Inyección de una sustancia en un músculo. inyección subcutánea (s.c.) Inyección de una sustancia en el espacio situado bajo la piel. administración oral: Administración de una sustancia en la boca para que sea deglutida. administración sublingual: Administración de una sustancia colocándola debajo de la lengua. administración intrarrectal: Administración de una sustancia en el recto. inhalación Administración de una sustancia vaporizada en los pulmones. administración tópica: Administración de una sustancia directamente sobre la piel o membrana mucosa administración intracerebral : Administración de una sustancia directamente en el encéfalo. administración intracerebroventricular (i.c.v.): Administración de una sustancia en uno de ios ventrículos encefálicos. curva dosis-respuesta: Gráfico de la magnitud del efecto de un fármaco como función de la dosis administrada. índice terapéutico: Cociente entre la dosis que produce el efecto deseado en el 50 % de los animales y la dosis que produce efectos tóxicos en el 50 % de los animales. Afinidad: Facilidad con la que se unen dos moléculas. Tolerancia: Reducción de la eficacia de una sustancia que es administrada repetidamente. Sensibilización: Aumento en la eficacia de un compuesto que es administrado repetidamente. Síntomas de abstinencia: Síntomas contrarios a los producidos por una sustancia cuanto esta se ha administrado en repetidas ocasiones y se interrumpe bruscamente. Placebo: Sustancia inerte administrada a un organismo en lugar de un compuesto fisiológicamente activo; se usa experimentalmente para controlar La psicofarmacología es el estudio de los efectos de los compuestos (fármacos, drogas, y venenos)sobre el sistema nervioso y la conducta. Los compuestos se definen en este contexto como sustancias químicas exógenas innecesarias para el funcionamiento celular normal que alteran significativamente las funciones de determinadas células del organismo cuando se administran en dosis relativamente bajas. Los compuestos tienen efectos, fisiológicos y conductuales, y lugares de acción, moléculas situadas en alguna región del organismo con las que interaccionan para producir esos efectos. La farmacocinética es el destino de un compuesto cuando se absorbe en el organismo, circula por todo él y alcanza sus lugares de acción. Los compuestos pueden administrarse por inyección intravenosa, intraperitoneal, intramuscular y subcutánea; también por vía oral, sublingual, intrarrectal, inhalación y administración tópica (sobre la piel o membranas mucosas); y, en ocasiones, por inyección ¡ntracerebral o intracerebroventricular. Los compuestos liposolubles atraviesan fácilmente la barrera hematoencefálica, mientras que otros la atraviesan lentamente o son incapaces de hacerlo. El curso temporal de las distintas vías de administración es diferente. En último término, los compuestos desaparecen del organismo. Algunos son desactivados por enzimas, especialmente en el hígado, y otros son simplementeexcretados. La curva dosis-respuesta representa la eficacia de un fármaco; relaciona la cantidad administrada (habitualmente en miligramos por kilogramo de peso corporal del individuo) con el efecto resultante. La mayoría de los fármacos tiene más de un lugar de acción y, por tanto, más de un efecto. La seguridad de un fármaco se mide por la diferencia entre la dosis que produce los efectos deseables y aquella que provoca efectos secundarios tóxicos. Los fármacos tienen una eficacia variable por lascaracterísticas de sus lugares de acción y la afinidad de las moléculas del fármaco por esos lugares. La administración repetida de una sustancia causa en ocasiones tolerancia, que a menudo resulta en síntomas de abstinencia, o sensibilización. La tolerancia puede deberse a una menor afinidad del compuesto por sus receptores, reducción del número de receptores o disminución del acoplamiento de los receptores con los pasos bioquímicos que controlan. Algunos de los efectos de una sustancia presentan tolerancia, y otros no, o incluso muestran sensibilización. CUESTIONES PARA REFLEXIONAR 1. Elija un compuesto con cuyos efectos esté familiarizado e indique dónde podrían estar los lugares de acción de ese compuesto en el organismo. 2. Algunas sustancias pueden dañar al hígado si se toman en dosis grandes durante un periodo de tiempo prolongado. ¿Qué aspecto de la farmacocinética de esos compuestos causaría el daño hepático? La mayoría de los fármacos que afectan a la conducta ejercen este efecto influyendo sobre la transmisión sináptica. Los fármacos que afectan a la transmisión sináptica se clasifican en dos grandes categorías. Los que bloquean o inhiben los efectos postsinápticos se denominan antagonistas. Aquellos que los facilitan se llaman agonistas. Antagonista: Compuesto que se opone o inhibe los efectos de un neurotransmisor determinado sobre la célula postsináptica. Agonista: Compuesto que facilita los efectos de un neurotransmisor determinado sobre la célula postsináptica. Agonista directo: Compuesto que se une a un receptor y lo activa. Bloqueante de receptores: Sustancia que se une a un receptor, pero no lo activa; impide que el ligando natural se una al receptor. Antagonista directo: Sinónimo de bloqueante de receptores. Unión no competitiva: Unión de un compuesto a un lugar del receptor; no interfiere con ei lugar de unión del ligando principal. Antagonista indirecto: Compuesto que se une a un lugar de unión del receptor e interfiere con la acción de dicho receptor; no Interfiere con el lugar de unión del ligando principal. Agonista indirecto: Compuesto que se une a un lugar de unión del receptor y facilita la acción del mismo; no interfiere con el lugar de unión del ligando principal. Heterorreceptor presináptico: Receptor situado en la membrana de una terminal nerviosa que recibe la aferencia de otra terminal nerviosa mediante una sinapsis axoaxónica; se une al neurotransmisor liberado por la terminal nerviosa presináptica. acetil-CoA: Cofactor que aporta acetato para la síntesis de acetilcolina. colina-acetiltransferasa (ChAT): Enzima que transfiere el ión acetato déla acetil coenzima A a la colina, produciendo el neurotransmisor acetilcolina. Toxina botulínica: Antagonista de la acetilcolina; impide su liberación por parte de las terminales nerviosas Veneno de la araña viuda negra: Veneno producido por esta araña que desencadena la liberación de acetilcolina. Neostigmina: Fármaco que inhibe la actividad de la acetilcolinesterasa. Receptor nicotínico: Receptor colinérgico ionotrópico estimulado por la nicotina y bloqueado por el curare. Receptor muscarínico: Receptor colinérgico metabotrópico estimulado por la muscarina y bloqueado por la atropina. Atropina: Fármaco que bloquea los receptores muscarínicos de acetilcolina. Curare: Sustancia que bloquea los receptores nicotínicos de acetilcolina. Monoaminas: Clase de aminas que comprende ¡ndolaminas, como la serotonina, y catecolaminas, como la dopamina, la noradrenalina y la adrenalina. Catecolaminas: Clase de aminas que Incluye los neurotransmisores dopamina, noradrenalina y adrenalina. Dopamina: Neurotransmisor; una de las catecolaminas. l-DOPA: Isómero levógiro de la DOPA; precursor de las catecolaminas; usado a menudo para tratar la enfermedad Sistema nigroestriado: Sistema de neuronas que se origina en la sustancia negra y termina en el neoestriado (núcleo caudado y putamen). Sistema mesolímbico: Sistema de neuronas dopaminérgicas que se origina en el área tegmental ventral y termina en el núcleo accumbens, la amígdala y el hipocampo. Sistema mesocortical: Sistema de neuronas dopaminérgicas que se origina en el área tegmental ventral y termina en la corteza prefrontal. Enfermedad de Parkinson: Trastorno neurológico caracterizado por temblor, rigidez de las extremidades, problemas de equilibrio y dificultades para iniciar movimientos; causado por la degeneración del sistema nigroestriado. Metilfenidato: Fármaco que inhibe la recaptación de dopamina. : monoamino-oxidasa (MAO) Clase de enzimas que degradan las monoaminas: dopamina, noradrenalina y serotonina. selegilina: Fármaco que bloquea la actividad de la MAO-B; actúa como agonista dopaminérgico. Clorpromacina: Fármaco que alivia los síntomas de la esquizofrenia mediante el bloqueo de los receptores dopaminérgicos D2. Noradrenalina (NAd): Una de las catecolaminas; neurotransmisor presente en el encéfalo y la división simpática del sistema nervioso autónomo. Adrenalina: Una de las catecolaminas; hormona secretada por la médula suprarrenal; también actúa como neurotransmisor en el encéfalo. Idazoxán: Compuesto que bloquea los receptores noradrenérgicos a2 presinápticos, actuando así como agonista, al facilitar la síntesis y liberación de NAd. serotonina (5-HT): Neurotransmisor indolamínico, también llamado 5-hidroxitriptamina. PCPA: Compuesto que inhibe la actividad de la triptófano- hidroxilasa, interfiriendo así con la síntesis de 5-HT. Fluoxetina: Fármaco que inhibe la recaptación de 5-HT. Fenfluramina: Fármaco que estimula la liberación de 5-HT. LSD: Droga que estimula los receptores 5-HT2aMDMA: Droga que funciona como agonista serotoninérgico y noradrenérgico, también conocida como «éxtasis»; tiene efectos excitadores y alucinógenos. PCP Fenciclidina; droga que se une al lugar de unión de PCP en el receptor de NMDA y funciona como antagonista indirecto. GABA Un aminoácido; neurotransmisor inhibidor más importante del encéfalo. alilglicina Compuesto que inhibe la actividad de la GAD, bloqueando así la síntesis de GABA. muscinol Agonista directo del lugar de unión del GABA en el receptor GABAa. Bicuculina: Antagonista directo del lugar de unión del GABA en el receptor GABAa. Benzodiacepinas: Grupo de fármacos ansiolíticos; agonistas indirectos del receptor GABAa. Ansiolítico: Efecto reductor de la ansiedad Glicina: Un aminoácido; neurotransmisor Inhibidor Importante en la parte inferior del tronco del encéfalo y la médula espinal Estricnina: Antagonista directo del receptor de glicina. la secuencia de la actividad sináptica: • los neurotransmisores son sintetizados y se almacenan en vesículas sinápticas. • Las vesículas se desplazan a la membrana presináptica, donde atracan. • Cuando se activa un axón, se abren canales de calcio dependientes del voltaje en la membrana presináptica, permitiendo la entrada de iones calcio. • Los iones calcio interaccionan con las proteínas de atraque y ponen en marcha la liberación de neurotransmisores a la hendidura sináptica. • Las moléculas del neurotransmisor se unen a receptores postsinápticos, provocando la apertura de determinados canales iónicos, lo que genera potenciales postsinápticos excitadores o inhibidores. • Los efectos del neurotransmisor son relativamente breves por su recaptación mediante moléculas transportadoras a la membrana presináptica o por su destrucción enzimática. • Además, la estimulación de autorreceptores presinápticos en las terminales nerviosas regula la síntesis y liberación del neurotransmisor. • La descripción de los efectos farmacológicos en esta sección seguirá esta misma secuencia básica. Efectos sobre la producción de neurotransmisores Paso1: es la síntesis del neurotransmisor a partir de sus precursores. En ocasiones, la tasa de síntesis y liberación de un neurotransmisor aumenta cuando se administra un precursor; en esos casos, el propio precursor funciona como agonista (véase el paso 1 en la Figura 4.4). Paso 2: Los pasos de la síntesis de neurotransmisores son controlados por enzimas. Por tanto, si un fármaco inactiva una de esas enzimas, impedirá que se produzca el neurotransmisor. Esos fármacos son antagonistas (véase el paso 2 en la Figura 4.4). Efectos sobre el almacenamiento y la liberación de neurotransmisores Paso 3: Los neurotransmisores se almacenan en vesículas sinápticas, que son transportadas a la membrana presináptica, donde se liberan las sustancias. El almacenamiento de neurotransmisores en vesículas lo realiza el mismo tipo de moléculas transportadoras responsables de la recaptación de un neurotransmisor a la terminal nerviosa. Las moléculas transportadoras están situadas en la membrana de las vesículas sinápticas, y su acción consiste en bombear moléculas del neurotransmisor a través de la membrana, llenando las vesículas. Algunas de las moléculas transportadoras que llenan las vesículas sinápticas son susceptibles de ser bloqueadas por fármacos. Las moléculas del fármaco se unen a un lugar concreto del transportador y lo inactivan. Como las vesículas sinápticas se quedan vacías, no se libera nada cuando las vesículas se rompen finalmente en la membrana presináptica. El fármaco funciona como antagonista (véase el paso 3 en la Figura 4.4). Pasos 4 y 5: Algunos compuestos actúan como antagonistas, impidiendo la liberación de neurotransmisores de la terminal nerviosa. Lo consiguen desactivando las proteínas que hacen que las vesículas sinápticas atracadas se fusionen con la membrana presináptica y expulsen su contenido a la hendidura sináptica. Otros fármacos ejercen el efecto contrario: son agonistas, al unirse a estas proteínas y desencadenar directamente la liberación del neurotransmisor (véanse los pasos 4 y 5 en la Figura 4.4). Efectos sobre los receptores El lugar de acción más importante (y más complejo) de los compuestos en el sistema nervioso son los receptores, pre-y postsinápticos. Estudiemos primero los receptores presinápticos (aquí es donde debe comenzar la lectura atenta). Una vez liberado un neurotransmisor, tiene que estimular los receptores postsinápticos. Algunos fármacos se unen a estos receptores, igual que el neurotransmisor. Cuando un fármaco se ha unido con el receptor, puede funcionar como agonista o como antagonista. Un fármaco que remeda los efectos de un neurotransmisor actúa como agonista directo. Las moléculas de fármaco se acoplan al lugar de unión al que se liga normalmente el neurotransmisor. Este acoplamiento provoca que se abran canales iónicos controlados por el receptor, del mismo modo que sucede cuando está presente el neurotransmisor. A continuación, los iones atraviesan estos canales y producen potenciales postsinápticos (véase el paso 6 en la Figura 4.4). Los fármacos que se unen a receptores postsinápticos también pueden funcionar como antagonistas. Sus moléculas se unen a los receptores, pero no abren el canal iónico. Como ocupan el lugar de unión del receptor, impiden que el neurotransmisor abra el canal iónico. Estos fármacos se denominan bloqueantes de receptores o antagonistas directos (véase el paso 7 en la Figura 4.4). Algunos receptores tienen múltiples lugares de unión, a los que pueden acoplarse distintos ligandos. Las moléculas del neurotransmisor se unen a un lugar, y otras sustancias (como neuromoduladores y varios fármacos) se unen a los otros. La unión de una molécula en uno de estos lugares alternativos se conoce como unión no competitiva, porque la molécula no compite con las moléculas moléculas del neurotransmisor por el mismo lugar de unión. Si un fármaco se une a uno de estos lugares alternativos e impide la apertura del canal iónico, el fármaco se califica de antagonista indirecto. El efecto final de un antagonista indirecto es similar al del antagonista directo, pero su sitio de acción es distinto. Si un fármaco se une a uno de los lugares alternativos y facilita la apertura del canal iónico, se le denomina agonista indirecto (véase la Figura 4.5). Como señalamos en el Capítulo 2, las membranas presinápticas de algunas neuronas contienen autorreceptores que regulan la cantidad de neurotransmisor liberada. Puesto que la estimulación de estos receptores provoca que se libere menos neurotransmisor, aquellos fármacos que activan selectivamente los receptores presinápticos actúan como antagonistas. Los fármacos que bloquean los autorreceptores presinápticos tienen el efecto contrario: aumentan la liberación del neurotransmisor, funcionando como agonistas (véanse los pasos 8 y 9 de la Figura 4.4). También vimos en el Capítulo 2 que algunas terminales nerviosas forman sinapsis axoaxónicas, sinapsis de una terminal nerviosa con otra. La activación de la primera terminal causa inhibición o facilitación presináp- tica de la segunda. La segunda terminal nerviosa contiene heterorreceptores presinápticos, sensibles al neurotransmisor liberado por la primera terminal (auto significa «mismo»; hetera quiere decir «otro»). Los heterorreceptores presinápticos que producen inhibición presináptica lo hacen inhibiendo la liberación del neu- rotransmisor. Y a la inversa, los heterorreceptores presinápticos responsables de la facilitación presináptica facilitan la liberación del neurotransmisor. De modo que los fármacos son capaces de bloquear o fomentar la inhibición o facilitación presináptica, dependiendo de si bloquean o activan los heterorreceptores presinápticos (véase la Figura 4.6). Figura 4.6: La facilitación presináptica está causada por la activación de receptores que facilitan la apertura de canales de calcio cercanos a la zona activa de la terminal nerviosa postsináptica, lo que promueve la liberación del neurotransmisor. La inhibición presináptica se debe a la activación de receptores que inhiben la apertura de esos canales de calcio. Figura 4.7 Autorreceptores dendríticos. Las dendritas de algunas neuronas liberan cierta cantidad de neurotransmisor cuando la célula está activa. La activación de los autorreceptores dendríticos por el neurotransmisor (o por un compuesto que se una a esos receptores) hiperpolariza la membrana, reduciendo la tasa de activación de la neurona. El bloqueo de los autorreceptores dendríticos por un compuesto impide este efecto. Efectos sobre la recaptación o destrucción de neurotransmisores El paso siguiente a la estimulación del receptor postsináptico es la finalización del potencial postsináptico. Dos procesos son los encargados de esta misión: las moléculas del neurotransmisor vuelven a la terminal nerviosa mediante el proceso de recaptación, o bien son destruidas por una enzima. Los fármacos pueden interferir con ambos procesos. En el primer caso, las moléculas del fármaco se unen a las moléculas transportadoras responsables de la recaptación y las inactivan, bloqueando así la recaptación. En el segundo, las moléculas del fármaco se unen a la enzima que habitualmente destruye el neurotransmisor e impide su funcionamiento. El ejemplo más importante de esas enzimas es la acetilcolinesterasa, que degrada la acetilcolina. Como ambos tipos de fármacos prolongan la presencia de las moléculas del neurotransmisor en la hendidura sinóptica (y, por tanto, en un lugar donde esas moléculas pueden estimular receptores postsinápticos), funcionan como agonistas (véanse los pasos 10 y 11 de la Figura 4.4). RESUMEN INTERMEDIO Lugares de acción de los compuestos El proceso de la transmisión sináptica supone la síntesis del neurotransmisor, su almacenaje en vesículas sinápticas, liberación a la hendidura sináptica, interacción con receptores postsinápticos, y la consiguiente apertura de canales iónicos en la membrana postsináptica. A continuación, se pone fin a los efectos del neurotransmisor mediante su recaptación a la terminal nerviosa o por inactivación enzimàtica. Con cada uno de los pasos necesarios para la transmisión sináptica pueden interferir compuestos que actúan como antagonistas, y unos pocos pasos son estimulados por sustancias que sirven de agonistas. Así pues, los compuestos pueden aumentar la cantidad de precursor disponible, bloquear una enzima de la biosíntesis, impedir el almacenamiento del neurotransmisor en vesículas sinápticas, estimular o bloquear la liberación del neurotransmisor, estimular o bloquear receptores pre- o postsinápticos, retrasar la recaptación o inactivar enzimas que degradan el neurotransmisor. Un compuesto que active receptores postsinápticos actúa como agonista, mientras que otro que active autorreceptores presinápticos o dendríticos funciona como antagonista. Una sustancia que bloquee receptores postsinápticos sirve de antagonista, pero otra que bloquee autorreceptores actúa como agonista. Un compuesto que active o bloquee heterorreceptores presinápticos funciona como agonista o antagonista, dependiendo de si los heterorreceptores causan facilitación o inhibición presináptica. ■ CUESTIONES PARA REFLEXIONAR Explique cómo un compuesto que bloquea receptores puede actuar como agonista. Neurotransmisores y neuromoduladores Como los neurotransmisores tienen dos efectos globales sobre las membranas presinápticas (despolarización [PPSE] o inhibición [PPSI]), cabe esperar que existan dos tipos de neurotransmisores, excitadores e inhibidores. Más bien, hay muchos tipos diferentes, al menos varias docenas. En el encéfalo, la mayor parte de la comunicación sináptica se efectúa mediante dos neurotransmisores, uno con efectos excitadores (glutamato), y el otro con efectos inhibidores (GABA). (Otro neurotransmisor inhibidor, la glicina, se encuentra en la médula espinal y porción inferior del tronco del encéfalo.) La mayor parte de la actividad de los circuitos neuronales locales supone un equilibrio entre los efectos excitadores e inhibidores de estas sustancias, responsables de casi toda la información transmitida de un lugar a otro del encéfalo. De hecho, probablemente no haya ninguna neurona encefálica que carezca de aferencias excitadoras recibidas de terminales nerviosas secretoras de glutamato y de aferencias inhibidoras de neuronas que liberan GABA o glicina. Y con la excepción de las neuronas que detectan estímulos dolorosos, todos los órganos sensitivos transmiten información al encéfalo a través de axones cuyas terminales liberan glutamato (las neuronas detectoras del dolor secretan un péptido). ¿Qué hacen todos los demás neurotransmisores? Por lo general, ejercen efectos moduladores en vez de transmisores de información. Es decir, la liberación de neurotransmisores distintos de GABA o glutamato tiende a activar o inhibir circuitos completos de neuronas que participan en determinadas funciones encefálicas. Por ejemplo, la secreción de acetilcolina activa la corteza cerebral y facilita el aprendizaje, pero la información aprendida y recordada se transmite mediante neuronas que secretan glutamato y GABA. La secreción de noradrena- lina aumenta la vigilancia y propicia la disposición a la acción cuando se detecta una señal. La secreción de his- tamina fomenta la vigilia. La secreción de serotonina suprime algunos tipos de conductas típicas de la especie y reduce la probabilidad de que el animal actúe impulsivamente. La secreción de dopamina en ciertas regiones encefálicas activa habitualmente movimientos voluntarios, sin especificar cuáles se producirán. En otras regiones, la secreción de dopamina refuerza conductas presentes y aumenta la probabilidad de que aparezcan posteriormente. Como ciertos compuestos afectan selectivamente a las neuronas que secretan determinados neurotransmisores, pueden ejercer efectos específicos sobre la conducta. Acetilcolina La acetilcolina es el neurotransmisor principal secretado por los axones eferentes del sistema nervioso periférico. Todo el movimiento muscular lo realiza la liberación de acetilcolina, y este neurotransmisor también está presente en los ganglios del sistema nervioso autónomo y los órganos diana de la rama parasimpática del SNA. Como la ACh se encuentra fuera del sistema nervioso central en lugares sencillos de estudiar, este neurotransmisor fue el primero descubierto y ha recibido mucha atención por parte de los neurocientíficos. Vayamos con la terminología: estas sinapsis se califican de colinérgicas. En griego, ergon significa «trabajo». Así pues, las sinapsis dopaminérgicas liberan dopamina, las serotoninérgicas liberan serotonina, etc. Los axones y las terminales nerviosas de las neuronas colinérgicas están ampliamente distribuidos por todo el encéfalo. Los neurocientíficos han prestado la máxima atención a tres sistemas: aquellos que se originan en la protuberancia dorsolateral, el prosencéfalo basal y el septo medial. Los efectos de la liberación de ACh en el colinérgicas situadas en la protuberancia dorsolateral participan en el sueño REM (la fase en la que se sueña). Las localizadas en el prosencéfalo basal activan la corteza cerebral y facilitan el aprendizaje, especialmente el aprendizaje perceptivo. Y aquellas presentes en el septo medial controlan los ritmos eléctricos del hipocampo y modulan sus funciones, que incluyen la formación de tipos específicos de memoria. La Figura 4.8 muestra una representación esquemática de un corte sagital medio del encéfalo de rata. En ella están indicados los lugares más importantes de los cuerpos celulares colinérgicos y las regiones donde se proyectan las ramas de sus axones. La figura presenta un encéfalo de rata, porque la mayor parte de los estudios topográficos neuroanatómicos se ha realizado con ratas. Presumiblemente, la localización y las proyecciones de las neuronas colinérgicas en el encéfalo humano se parecen a las presentes en el encéfalo de rata, pero aún no podemos asegurarlo. El Capítulo 5 describe los métodos usados para establecer la topografía de sistemas neuronales específicos en el encéfalo y la dificultad de llevar a cabo esos estudios en el encéfalo humano (véase la Figura 4.8). La acetilcolina tiene dos componentes: colina, sustancia derivada de la degradación de los lípidos, y acetato, el anión presente en el vinagre, también llamado ácido acético. El acetato no puede unirse directamente a la colina, sino que se transfiere a partir de una molécula de acetil- CoA. La CoA (coenzima A) es una molécula compleja, consistente en una porción del ácido pantoténico (una vitamina del grupo B). La CoA se produce en las mitocon- drias y participa en muchas reacciones del organismo. Acetil-CoA consiste simplemente en CoA con un ión acetato unido. La ACh se genera en la siguiente reacción: en presencia de la enzima colina-acetiltransferasa (ChAT), el ión acetato se transfiere de la molécula de acetil-CoA a la molécula de colina, dando lugar a una molécula de ACh y otra de CoA normal (véase la Figura 4.9). Una analogía sencilla ilustra la función de las coenzimas en las reacciones químicas. Consideremos el acetato un perrito caliente, y la colina, el panecillo. La tarea de la persona (enzima) encargada del puesto de perritos es poner la salchicha dentro del panecillo (producir acetilcolina). Para hacerlo, el vendedor necesita un tenedor (coenzima) con el que sacar la salchicha del agua caliente. El vendedor pincha la salchicha con el tenedor (une el acetato a la CoA) y transfiere el perrito del tenedor al panecillo. Dos sustancias, la toxina botulínica y el veneno de la araña viuda negra, afectan a la liberación de acetilco- lina. La toxina botulínica la produce Clostridium botulinum, una bacteria que crece en comida envasada incorrectamente. Esta sustancia impide la liberación de ACh (paso 5 de la Figura 4.4). Como vimos en el caso inicial del capítulo, la toxina botulínica es un veneno extremadamente potente, porque la parálisis que induce da lugar a asfixia. Por el contrario, el veneno de la araña viuda negra tiene el efecto contrario: estimula la liberación de ACh (paso 4 de la Figura 4.4). Aunque este veneno también puede ser mortal, resulta mucho menos tóxico que la toxina botulínica. De hecho, la mayoría de los adultos sanos tendría que ser mordido varias veces, pero lactantes y ancianos frágiles son más vulnerables. Es posible que se esté preguntando por qué la visión doble fue el primer síntoma de botulismo en el caso inicial. La respuesta es que el exquisito equilibrio entre los músculos que mueven los ojos se ve alterado por cualquier interferencia con la transmisión colinèrgica. Recordará del Capítulo 2 que, una vez liberada por la terminal nerviosa, la ACh es inactivada por la enzima acetilcolinesterasa (AChE), presente en la membrana postsináptica (véase la Figura 4.10). Las sustancias que inactivan la AChE (paso 11 de la Figura 4.4) se emplean con varios fines. Algunas sirven de insecticidas. Estos productos matan fácilmente a los insectos, pero no a las personas y otros mamíferos, porque nuestra sangre contiene enzimas que los destruyen (los insectos carecen de esa enzima). Otros inhibidores de la AChE tienen un uso médico. Por ejemplo, un trastorno hereditario llamado miastenia grave está causado por un ataque del sistema inmunitario a los receptores de acetilcolina situados en músculos esqueléticos (Kathryn D., cuyos síntomas describí en el caso inicial del Capítulo 2, sufría esta enfermedad). La persona se siente cada vez más débil, a medida que los músculos responden menos al neurotransmisor. Si se le administra un inhibidor de la AChE, neostigmina, por ejemplo, la persona recuperará parte de su fuerza, porque la acetilcolina liberada tiene un efecto más prolongado sobre los receptores presentes (la neostigmina no atraviesa la barrera hematoencefálica, de modo que no afecta a la AChE del sistema nervioso central). Hay dos tipos de receptores de ACh, uno ionotrópico y otro metabotrópico. Estos receptores se identificaron al descubrir los investigadores que les afectaban sustancias distintas (paso 6 de la Figura 4.4). El receptor ionotrópico de ACh es estimulado por la nicotina, droga presente en las hojas de tabaco (el nombre en latín de la planta es Nicotiniana tabacum). El receptor metabotrópico de ACh es estimulado por la muscarina, compues- tode la seta venenosa Amonita muscaria. En consecuencia, estos dos receptores de ACh se denominan receptores nicotínicos y receptores muscarínicos, respectivamente. Como las fibras musculares tienen que ser capaces de contraerse rápidamente, contienen los receptores nico- tínicosionotrópicos, muy veloces. Puesto que los receptores muscarínicos son metabo- trópicos y, por tanto, controlan canales iónicos a través de la producción de segundos mensajeros, sus acciones son más lentas y prolongadas que las propias de los receptores nicotínicos. El sistema nervioso central contiene ambos tipos de receptores de ACh, pero predominan los muscarínicos. Hay algunos receptores nicotínicos en las sinapsis axoaxónicas del encéfalo, en las que producen facilitación presináptica. La activación de estos nicotina presente en el humo del tabaco. Al igual que dos compuestos distintos estimulan los dos tipos de receptores de acetilcolina, dos fármacos diferentes los bloquea (paso 7 de la Figura 4.4). Ambos fármacos fueron descubiertos hace mucho tiempo en la naturaleza, y los dos siguen utilizándose en la medicina moderna. El primero, la atropina, bloquea los receptores muscarínicos. Su nombre deriva de Atropos, la parca de la mitología griega que cortaba el hilo de la vida (algo que consigue, sin duda, una dosis suficiente de atropina). La atropina es uno de los alcaloides de la belladona extraídos de la planta denominada Atropa belladona, y su nombre contiene una historia. Hace muchos años, las mujeres que querían mostrarse más atractivas a los ojos de los hombres se echaban gotas que contenían alcaloides de belladona en los ojos. De hecho, belladona significa en italiano «mujer guapa». ¿Por qué se usaba el fármaco con este fin? Lina de las respuestas inconscientes que se produce cuando algo nos interesa es la dilatación de las pupilas. Al bloquear los efectos de la acetilcolina sobre la pupila, los alcaloides de la belladona, como la atropina, causan que las pupilas se dilaten. Este cambio provoca que la mujer parezca más interesada en un hombre cuando le mira y, por supuesto, este signo de presunto interés hace que él la considere más atractiva. Otra sustancia, el curare, bloquea los receptores nicotínicos. Como estos receptores están en los músculos, el curare, igual que la toxina botulínica, produce parálisis. Sin embargo, los efectos del curare son mucho más rápidos. La sustancia se extrae de varias especies distintas de plantas presentes en Sudamérica, donde se descubrió hace mucho tiempo, y se usó para impregnar las puntas de flechas y dardos. A los pocos minutos de ser alcanzado por una de esas puntas, el animal cae, deja de respirar y muere. Hoy en día, el curare (y otros fármacos con el mismo lugar de acción) se emplea para paralizar a los pacientes que van a someterse a operaciones quirúrgicas, de modo que sus músculos se relajen por completo y no se contraigan cuando sean seccionados por el bisturí. También hay que usar un anestésico, porque la persona que solo recibe curare permanecerá totalmente consciente y sensible al dolor, aunque esté paralizada. Y, por supuesto, se empleará la ventilación mecánica para hacer llegar el aire a los pulmones. Monoaminas Dopamina, noradrenalina, adrenalina, histamina y serotonina son cinco sustancias químicas que pertenecen a una familia de compuestos denominados monoaminas. Como las estructuras moleculares de estas sustancias son similares, algunos fármacos afectan a la actividad de todas ellas en cierto grado. Las tres primeras (dopamina, noradrenalina y adrenalina) constituyen el subgrupo de las monoaminas llamado catecolaminas. Merece la pena aprender los términos de la Tabla 4.1, porque aparecerán en el resto del libro (véase la Tabla 4.1). Las monoaminas son producidas por varios sistemas neuronales del encéfalo. La mayoría de estos sistemas consiste en un número relativamente pequeño de cuerpos celulares situados en el tronco del encéfalo, cuyos axones se ramifican extensamente y dan lugar a un número enorme de terminales nerviosas distribuidas por muchas regiones encefálicas. Las neuronas monoaminérgicas, por tanto, modulan la función de múltiples regiones del encéfalo, aumentando o disminuyendo la actividad de funciones encefálicas específicas. DOPAMINA La primera catecolamina de la Tabla 4.1, dopamina (DA), produce potenciales postsinápticos excitadores o inhibidores, según el receptor postsináptico. La dopamina es uno de los neurotransmisores más interesantes, porque se ha visto implicada en varias funciones importantes, como movimiento, atención, aprendizaje y efectos reforzadores de drogas de las que los humanos tendemos a abusar. Aparece en los Capítulos 8, 9, 13, 16 y 18. La síntesis de las catecolaminas es algo más complicada que la de ACh, pero cada paso es sencillo. La molécula precursora se modifica ligeramente, paso a paso, hasta llegar a su forma final. Una enzima diferente controla cada paso, añadiendo o quitando una pequeña parte. El precursor de los dos neurotransmisores catecolamínicos principales (dopamina y noradrenalina) es la tirosina, aminoácido esencial que debemos obtener de la dieta. La tirosina recibe un grupo hidroxilo (OH, un átomo de oxígeno y otro de hidrógeno) y se convierte en l-DOPA (L-3,4-dihidroxifenilalanina). La enzima que añade el grupo hidroxilo se llama tirosina-hidroxilasa. A continuación, la l-DOPA pierde un grupo carboxilo (COOH, un átomo de carbono, dos de oxígeno y uno de hidrógeno) gracias a la actividad de la enzima DOPA-descarboxilasa, y se convierte en dopamina. Por último, la enzima dopamina-fi- hidroxilasa une un grupo hidroxilo a la dopamina, que pasa a ser noradrenalina. La Figura 4.11 muestra estas reacciones. El encéfalo contiene varios sistemas de neuronas dopaminérgicas. Los tres más importantes tienen su origen en el mesencèfalo: en la sustancia negra y el área tegmental ventral. (La sustancia negra aparecía en la Figura 3.21; el área tegmental ventral está situada inmediatamente por debajo de esta región.) Los cuerpos celulares de las neuronas del sistema nigroestriado están en la sustancia negra y proyectan sus axones al neoestriado (núcleo caudado y putamen). El neoestriado es una parte importante de los núcleos básales que participa en el control del movimiento. Los cuerpos celulares de las neuronas del sistema mesolímbico están situados en el área tegmental ventral, y proyectan sus axones a varias partes del sistema límbico, incluidos el núcleo accumbens, la amígdala y el hipocampo. núcleo accumbens es muy importante en los efectos de refuerzo (recompensa) de ciertos tipos de estímulos, incluidos los de las drogas de abuso. Los cuerpos celulares de las neuronas del sistema mesocortical también están en el área tegmental ventral. Sus axones se proyectan a la corteza prefrontal. Estas neuronas tienen un efecto excitador sobre la corteza frontal y afectan a funciones como la generación de memoria a corto plazo, la planificación y la preparación de estrategias para la resolución de problemas. La Figura 4.12 muestra estos tres sistemas de neuronas dopaminérgicas. La degeneración de las neuronas dopaminérgicas que conectan la sustancia negra con el núcleo caudado causa la enfermedad de Parkinson, un trastorno del movimiento caracterizado por temblor, rigidez de las extremidades, problemas de equilibrio y dificultades para iniciar movimientos. Los cuerpos celulares de estas neuronas se encuentran en una región encefálica denominada sustancia negra. Esta región se tiñe normalmente de negro con melanina, la sustancia que da color a la piel. Este compuesto resulta de la degradación de dopamina. (La lesión cerebral causante de la enfermedad de Parkinson fue descubierta por anatomopatólogos, al observar que la sustancia negra de una persona fallecida que había padecido esta enfermedad estaba pálida en vez de negra.) A las personas con enfermedad de Parkinson se les administra l-DOPA, el precursor de la dopamina. Aunque la dopamina no atraviesa la barrera hematoencefálica, la l-DOPA sí lo hace. Una vez que llega al encéfalo, es captada por las neuronas dopaminérgicas y convertida en dopamina (paso 1 de la Figura 4.4). El aumento de la síntesis provoca que las neuronas dopaminérgicas supervivientes liberen más dopamina en los pacientes con enfermedad de Parkinson. La consecuencia es que los síntomas mejoran. Otra sustancia, la AMPT (o a-metil-jb-tirosina), inactiva la tirosina-hidroxilasa, enzima que convierte la tiro- sina en l-DOPA (paso 2 de la Figura 4.4). Como este compuesto interfiere con la síntesis de dopamina (y también de noradrenalina), es un antagonista de las catecolaminas. Por lo general no se emplea en medicina, pero sí se ha usado como herramienta de investigación en animales de laboratorio. El fármaco reserpina impide el almacenamiento de las monoaminas en vesículas sinápticas, bloqueando los transportadores de membrana de las vesículas en neuronas monoaminérgicas (paso 3 de la Figura 4.4). Como las vesículas sinápticas se quedan vacías, no se libera neurotransmisor cuando el potencial de acción llega a la terminal nerviosa. Por tanto, la reserpina es un antagonista de las monoaminas. Este fármaco, que proviene de la raíz de un arbusto, fue descubierto hace más de 3.000 años en la India, y se encontró que resultaba útil para tratar las mordeduras de serpiente y que parecía tener un efecto sedante. En los mercados de las áreas rurales indias aún se vende la raíz en porciones. En la medicina occidental, la reserpina se usaba para tratar la hipertensión arterial, pero ha sido sustituida por otros fármacos con menos efectos secundarios. Se han identificado varios tipos distintos de receptores de dopamina, todos ellos metabotrópicos. Los más frecuentes son los receptores D1 y los receptores D2. Aparentemente, los receptores D1 son solo postsinápticos, mientras que los D2, se encuentran en localizaciones pre- y postsinápticas del encéfalo. La estimulación de los receptores D1 aumenta la producción de AMP cíclico, un segundo mensajero, pero la estimulación de los D2 la reduce, al igual que la estimulación de los receptores D3 y D4. Varios fármacos estimulan o bloquean tipos concretos de receptores dopaminérgicos. En las dendritas, el soma y las terminales nerviosas de las neuronas dopaminérgicas hay autorreceptores. La activación de los autorreceptores de la membrana dendrítica y somática reduce la activación neuronal, al producir hiperpolarizaciones. Los autorreceptores presinápticos situados en las terminales nerviosas suprimen la actividad de la enzima tirosina-hidroxilasa, disminuyendo así la producción de dopamina, y en último término su liberación. Los autorreceptores dopaminérgicos se parecen a los receptores D2, pero aparentemente existen varias diferencias. Por ejemplo, el fármaco apomorfina es un agonista D2, pero parece tener más afinidad por los receptores D2 presinápticos que por los D2 postsinápticos. Una dosis baja de apomorfina funciona como antagonista, porque estimula los receptores presinápticos e inhibe la producción y liberación de dopamina. Dosis más altas empiezan a estimular los receptores D2, postsinápticos, y el fármaco comienza a funcionar como agonista directo (véase la Figura 4.13). Varios compuestos inhiben la recaptación de dopamina, actuando así como agonistas dopaminérgicos potentes (paso 10 de la Figura 4.4). Los mejor conocidos son la anfetamina, la cocaína y el metilfenidato. La anfetamina tiene un efecto interesante: causa liberación de dopamina (DA) y noradrenalina (NAd), haciendo que los transportadores de estos neurotransmisores operen al revés, impulsando DA y NAd a la hendidura sináptica. Por supuesto, esta acción también bloquea la recaptación de los neurotransmisores. La cocaína y el metilfenidato solo bloquean la recaptación de dopamina. Dado que la cocaína también bloquea los canales de sodio dependientes del voltaje, en ocasiones se usa como anestésico tópico, especialmente en forma de gotas oftálmicas para cirugía ocular. El metilfenidato se emplea en niños para tratar el trastorno por déficit de atención. La producción de catecolaminas está regulada por una enzima llamada monoamino-oxidasa (MAO). Esta enzima se encuentra en las terminales nerviosas monoaminérgicas, donde degrada cantidades excesivas de los neurotransmisores. Un fármaco, la selegilina, destruye la forma específica de monoamino-oxidasa (MAO-B) presente en las terminales nerviosas dopaminérgicas. Como la selegilina impide la destrucción de la dopamina, se libera más dopamina cuando un potencial de acción alcanza la terminal nerviosa. Así pues, la selegilina actúa como agonista dopaminérgico (véase la Figura 4.14). También hay MAO en la sangre, donde desactiva aminas procedentes de alimentos como el chocolate y el queso. Sin esa desactivación, estas aminas podrían provocar aumentos peligrosos de la presión arterial. Se ha planteado que la dopamina podría ser un neurotransmisor implicado en la esquizofrenia, trastorno mental grave cuyos síntomas incluyen alucinaciones, delirios y alteración de los procesos de pensamiento lógico normales. Fármacos como la clorpromacina, que bloquea los receptores D2, mejoran estos síntomas (paso 7 de la Figura 4.4). Por este motivo, los investigadores han especulado si la esquizofrenia está producida por la hiperactividad de neuronas dopaminérgicas. Otros fármacos descubiertos hace menos tiempo (los denominados antipsicóticos atípicos) tienen acciones más complejas, descritas en el Capítulo 16. NORADRENALINA Como la noradrenalina (NAd), al igual que la ACh, se encuentra en neuronas del sistema nervioso autónomo, ha recibido mucha atención experimental. Los términos adrenalina y epinefrina son sinónimos, al igual que nora- drenalinay norepinefrina. Veamos por qué. La adrenalina (epinefrina) es una hormona producida por la médula suprarrenal, la zona central de las glándulas suprarrenales, situadas inmediatamente por encima de los riñones. La adrenalina también actúa como neurotransmisor en el encéfalo, pero es menos importante que la noradrenalina. En latín, ad renal significa «hacia el riñón». En griego, diríamos epi nephron («encima del riñón»), de donde procede el término epinefrina. Esta última palabra es la adoptada por los farmacólogos de habla inglesa, probablemente porque una compañía farmacéutica se apropió del término Adrenalin como nombre comercial; en castellano se utilizan con más frecuencia los términos adrenalina y noradrenalina. Los adjetivos correspondientes son adrenérgicoy noradrenérgico (también en inglés —noradrenergic—, porque cuesta mucho pronunciar norepinefrinérgico). Hemos visto anteriormente la vía de biosíntesis de la noradrenalina en la Figura 4.11. El compuesto AMPT, que impide la conversión de tirosina en l-DOPA, bloquea la producción de noradrenalina, además de la síntesis de dopamina (paso 2 de la Figura 4.4). La mayoría de los neurotransmisores se sintetiza en el citoplasma de la terminal nerviosa y después se almacena en vesículas sinápticas de nueva creación. Sin embargo, el último paso de la síntesis de noradrenalina tiene lugar en las propias vesículas. Al principio, las vesículas están llenas de dopamina. Después, la dopamina se convierte en noradrenalina por la acción de la enzima dopamina-βhidroxilasa presente en el interior de las vesículas. El fármaco ácido fusárico inhibe la actividad de la enzima dopamina-β-hidroxilasa, y por este motivo bloquea la producción de noradrenalina sin afectar a la síntesis de dopamina. La noradrenalina en exceso presente en la terminal nerviosa es degradada por la monoamino-oxidasa tipo A. Un fármaco, la moclobemida, bloquea específicamente la MAO-A, y es, por tanto, un agonista noradrenérgico. Prácticamente todas las regiones encefálicas reciben aferencias de neuronas noradrenérgicas. Los cuerpos celulares de la mayoría de estas neuronas están situados en siete regiones de la protuberancia y el bulbo, y una región del tálamo. Los cuerpos celulares del sistema noradrenérgico más importante están en el locus cerúleo, un núcleo situado en la protuberancia dorsal. Los axones de estas neuronas proyectan a las regiones mostradas en la Figura 4.15. Como veremos posteriormente, el efecto fundamental de la activación de estas neuronas es un incremento de la vigilancia/alerta, la atención a los acontecimientos del entorno (véase la Figura 4.15). La mayoría de las neuronas que liberan noradrenalina no lo hacen a través de terminales nerviosas en los extremos de los axones, sino que habitualmente la liberan en las varicosidades axónicas, protuberancias en forma de cuenta de las ramas axónicas. Estas varicosidades otorgan un aspecto de collar de cuentas a los axones de las neuronas noradrenérgicas. Hay varios tipos de receptores noradrenérgicos, identificados por su distinta sensibilidad a algunos fármacos. De hecho, estos receptores suelen denominarse adrenérgicos en vez de noradrenérgicos, porque son sensibles a la adrenalina además de a la noradrenalina. Las neuronas del sistema nervioso central contienen receptores adrenérgicos α1 y β2, y receptores adrenérgicos β1 y β2. Estos cuatro tipos de receptores también se encuentran en varios órganos además del encéfalo, y son los responsables de los efectos de la adrenalina y noradrenalina cuando estas actúan como hormonas fuera del sistema nervioso central. En el encéfalo, todos los autorreceptores parecen ser del tipo α2. (El fármaco idazoxán bloquea los autorreceptores α2, funcionando así como agonista.) Todos los receptores adrenérgicos son metabotrópicos; están acoplados a proteínas G que controlan la producción de segundos mensajeros. Los receptores adrenérgicos producen efectos excitadores e inhibidores. Por lo general, los efectos conductuales de la liberación de NAd son excitadores. En el encéfalo, los receptores α1 producen un efecto despolarizador (excitador) lento sobre la membrana postsináptica, mientras que los receptores α2 causan una hiperpolarización lenta. Los dos tipos de receptores β aumentan la capacidad de respuesta de la neurona post- sináptica a sus aferencias excitadoras, lo que presumiblemente está relacionado con la función de este neurotransmisor en la vigilancia/alerta. Las neuronas noradrenérgicas y, concretamente, los receptores α2, también participan en la conducta sexual y el control del hambre. SEROTONINA El tercer neurotransmisor monoaminérgico, la serotonina (también denominada 5-HT, o 5-hidroxitriptamina), también ha recibido mucha atención en la investigación. Sus efectos conductuales son complejos. La serotonina participa en la regulación del estado de ánimo, el control de la ingesta, el sueño y arousal, y en la regulación del dolor. Las neuronas serotoninérgicas están implicadas de alguna manera en el control de los sueños. El precursor de la serotonina es el aminoácido triptófano. La enzima triptófano-hidroxilasa añade un grupo hidroxilo, produciendo 5-HTP (5-hidroxitriptófano). La enzima 5-HTP-descarboxilasa elimina un grupo carboxilo del 5-HTP, y el resultado es 5-HT (serotonina) (véase la Figura 4.16). El fármaco PCPA (pclorofenilalanina) bloquea la actividad de la triptófano-hidroxilasa, y es, pollo tanto, un antagonista serotoninérgico. Los cuerpos celulares de las neuronas serotoninérgicas se encuentran en nueve grupos, la mayoría localizados en los núcleos del rafe del mesencèfalo, la protuberancia y el bulbo. Al igual que la noradrenalina, la 5-HT es liberada de varicosidades en vez de terminales nerviosas. Los dos grupos más importantes de cuerpos celulares serotoninérgicos están en los núcleos del rafe mediano y dorsal, y a estos limitaré mi descripción. La palabra rafe significa «costura» o «pliegue», y hace referencia al hecho de que la mayoría de los núcleos del rafe están situados en la línea media (o sus proximidades) del tronco del encéfalo. Los núcleos del rafe dorsal y mediano proyectan axones a la corteza cerebral. Además, las neuronas del rafe dorsal inervan los núcleos básales, y las del rafe mediano envían axones a la circunvolución dentada, componente de la formación hipocámpica. La Figura 4.17 ilustra estas conexiones y algunas otras. Los investigadores han identificado al menos nueve tipos distintos de receptores de serotonina: 5HT1A_1B, 5-HT1D_1F, 5-HT2A_2C y 5-HT3. De estos, los receptores 5-HT1B y 5-HT1D funcionan como autorreceptores pre- sinápticos. En el núcleo del rafe dorsal y el mediano, los receptores 5-HT1A sirven de autorreceptores en la membrana de dendritas y soma. Todos los receptores serotoninérgicos son metabotrópicos, excepto el receptor 5-HT3, ionotrópico. El receptor 5-HT3 controla un canal de cloruro, lo que significa que produce potenciales postsinápticos inhibidores. Estos receptores parece que están implicados en las náuseas y el vómito, porque los antagonistas 5-HT3 han demostrado ser útiles para tratar los efectos secundarios de la quimioterapia y radioterapia usadas en el tratamiento del cáncer. Los farmacólogos han descubierto compuestos que funcionan como agonistas o antagonistas de algunos de los subtipos de receptores serotoninérgicos, pero no de todos ellos. Los fármacos que inhiben la recaptación de serotonina han alcanzado un lugar muy importante en el tratamiento de los trastornos mentales. El más conocido de ellos, la fluoxetina, se usa para tratar la depresión, algunos tipos de trastornos de ansiedad y el trastorno obsesivo-compulsivo. Los Capítulos 16 y 17 se ocupan de estos trastornos y de su tratamiento. Otro fármaco, la fenfluramina, que provoca liberación de serotonina además de inhibir su recaptación, se usó anteriormente como supresor del hambre en el tratamiento de la obesidad. El Capítulo 12 está dedicado al tema de la obesidad y su control farmacológico. Varias sustancias alucinógenas producen sus efectos interaccionando con la transmisión serotoninérgica. La sustancia LSD (dietilamida del ácido lisérgico) provoca distorsiones de la percepción visual que algunas personas encuentran maravillosas y fascinantes, pero que solo consiguen aterrorizar a otros. Este compuesto, eficaz en dosis extremadamente pequeñas, es un agonista directo de los receptores 5HT2A postsinápticos del prosencéfalo. Otra droga, la MDMA (metilendioximetanfetamina), es un agonista noradrenérgico y serotoninérgico, con efectos excitadores y alucinógenos. Al igual que la anfetamina, fármaco con el que está relacionado, la MDMA (popularmente conocido como «éxtasis») provoca que los transportadores noradrenérgicos operen en dirección contraria, causando la liberación de NAd e inhibiendo su recaptación. Este lugar de acción parece ser el responsable del efecto excitador de la droga. La MDMA también hace que los transportadores serotoninérgicos funcionen al revés, y este lugar de acción es presuntamente el responsable de sus efectos alucinógenos. Por desgracia, la investigación indica que la MDMA puede dañar las neuronas serotoninérgicas y causar déficits cognitivos. Aminoácidos Todos los neurotransmisores descritos hasta ahora se sintetizan dentro de las neuronas: acetilcolina a partir de colina, catecolaminas, a partir del aminoácido tirosina, y serotonina, a partir del aminoácido triptófano. Algunas neuronas secretan aminoácidos simples como neurotransmisores. Como todas las células del encéfalo usan aminoácidos para sintetizar proteínas, es difícil demostrar que un aminoácido concreto es un neuro- transmisor. No obstante, los investigadores sospechan que al menos ocho aminoácidos podrían actuar como neurotransmisores en el sistema nervioso central de los mamíferos. Como vimos en la introducción de esta sección, tres de ellos resultan especialmente importantes, porque son los neurotransmisores más abundantes del SNC: glutamato, ácido gamma-aminobutírico (GABA) y glicina. GLUTAMATO Puesto que el glutamato (también llamado ácido glutámico) y el GABA se encuentran en organismos muy sencillos, muchos investigadores piensan que estos neurotransmisores fueron los primeros en evolucionar. Además de producir potenciales postsinápticos mediante la activación de receptores postsinápticos, también poseen efectos excitadores (ácido glutámico) e inhibidores (GABA) directos sobre los axones; aumentan o disminuyen el umbral de excitación, afectando así a la tasa de aparición de potenciales de acción. Estos efectos directos indican que estas sustancias ejercían una función moduladora global antes incluso del desarrollo evolutivo de moléculas receptoras específicas. El glutamato es el neurotransmisor excitador principal del encéfalo y la médula espinal. Se produce abundantemente en los procesos metabólicos celulares. No hay una forma eficaz de impedir su síntesis sin alterar otras actividades de la célula. Los investigadores han descubierto cuatro tipos principales de receptores del glutamato. Tres de ellos son ionotrópicos y su nombre proviene de los ligandos artificiales que los estimulan: receptor de NMDA, receptor de AMPA y receptor de cainato. El otro receptor del glutamato, receptor metabotrópico de glutamato, es metabo- trópico, obviamente. En realidad, parece ser que existen al menos ocho subtipos de receptores metabotrópicos de glutamato, pero sus funciones apenas se conocen, excepto que algunos de ellos funcionan como autorreceptores presinápticos. El receptor de AMPA es el más frecuente. Controla un canal de sodio, de modo que cuando el glutamato se acopla al lugar de unión, produce PPSE. El receptor de cainato tiene efectos similares. El receptor de NMDA posee ciertas características especiales y muy importantes. Contiene al menos seis lugares de unión distintos, cuatro situados en el exterior del receptor y dos en el interior del canal iónico. Cuando está abierto, el canal iónico controlado por el receptor de NMDA permite que entren iones sodio y calcio a la célula. La entrada de ambos iones causa una despolarización, por supuesto, pero la entrada de calcio (Ca2+) resulta especialmente importante. El calcio actúa como segundo mensajero, uniéndose a varias enzimas celulares y activándolas. Estas enzimas ejercen efectos significativos sobre las propiedades bioquímicas y estructurales de la célula. Como veremos, un resultado importante es la alteración en las características de las sinapsis que constituyen uno de los cimientos de un recuerdo recién formado. Estos efectos de los receptores de NMDA se describen con mucho más detalle en el Capítulo 13. El compuesto AP5 (2-amino-5-fosfonopentanoato) bloquea el lugar de unión del glutamato en el receptor de NMDA y afecta negativamente a la plasticidad sináptica y a ciertos tipos de aprendizaje. La Figura 4.18 presenta el dibujo equemático de un receptor de NMDA y sus lugares de unión. Obviamente, el glutamato se une a uno de esos lugares, o no lo llamaríamos receptor del glutamato. Sin embargo, el glutamato por sí mismo no puede abrir el canal de calcio. Para que esto suceda, una molécula de glicina debe estar acoplada al lugar de unión de la glicina, situado en el exterior del receptor. (Aún no sabemos por qué la glicina, que también funciona como neurotransmisor inhibidor en algunas partes del sistema nervioso central, es necesaria para la apertura de este canal) (véase la Figura 4.18). Un requisito adicional para la apertura del canal del calcio es que no haya un ion magnesio en el lugar de unión del magnesio, situado en la profundidad del canal. En condiciones normales, cuando la membrana postsináptica está en el potencial de reposo, un ión magnesio (Mg2+) es atraído al lugar de unión del magnesio y bloquea el canal de calcio. Si una molécula de glutamato se acopla en su lugar de unión, el canal se ensancha, pero el ión magnesio sigue bloqueándolo, de modo que no puede entrar calcio en la neurona postsináptica. Sin embargo, si la membrana postsináptica está parcialmente despolarizada, el ión magnesio resulta repelido de su lugar de unión. Así pues, el receptor de NMDA solo se abre si hay glutamato y la membrana postsináptica está despolarizada. Por tanto, el receptor de NMDA es un canal iónico dependiente del voltaje y del neurotransmisor (véase la Figura 4.18). ¿Qué sucede con los otros tres lugares de unión? Si un ión zinc (Zn2+) se une al lugar de unión del zinc, la actividad del receptor de NMDA disminuye. Por otra parte, el lugar de poliamina tiene un efecto facilitador. (Las poliaminas son sustancias químicas que, según se ha demostrado, son importantes en el crecimiento y desarrollo de los tejidos. Aún no conocemos la relevancia del lugar de unión de poliamina.) El lugar de PCP, situado en la profundidad del canal iónico cerca del lugar de unión del magnesio, acoge una droga alucinó- gena, la PCP (fenciclidina, también denominada «polvo de ángel»). La PCP funciona como antagonista indirecto; cuando se acopla a su lugar de unión, los iones calcio no pueden atravesar el canal iónico. La PCP es una droga sintética, no la produce el encéfalo. Por este motivo, no es el ligando natural del lugar de unión de PCP. Todavía no conocemos cuál es ese ligando y qué función útil desempeña. Varios compuestos afectan a las sinapsis glutamatérgicas. Como ya sabe, NMDA, AMPA y cainato (más exactamente, ácido caínico) son agonistas directos en los receptores nombrados por ellos. Además, una de las drogas más comunes, el alcohol, actúa como antagonista de los receptores de NMDA. Como veremos en el Capítulo 18, este efecto es el responsable de las crisis epilépticas que pueden aparecer por la interrupción brusca del consumo de alcohol crónico en grandes cantidades. GABA El GABA (ácido gamma-aminobutírico) se sintetiza a partir del ácido glutámico mediante la acción de una enzima (ácido glutámico-descarboxilasa, o GAD) que elimina un grupo carboxilo. El compuesto alilglicina inactiva la GAD, impidiendo así la síntesis de GABA (paso 2 de la Figura 4.4). El GABA es un neurotransmisor inhibidor, y parece estar ampliamente distribuido por todo el encéfalo y la médula espinal. Se han identificado dos receptores: GABAa y GABAb. El receptor GABAa es ionotrópico y controla un canal de cloruro; el receptor GABAb es metabotrópico y controla un canal de potasio. Como sabe, las neuronas del encéfalo están muy interconectadas. Sin la actividad de las sinapsis inhibidoras, estas interconexiones harían que el encéfalo fuera inestable. Es decir, mediante sinapsis excitadoras, las neuronas excitarían a sus vecinas, que, a su vez, excitarían a sus vecinas, que entonces excitarían a las neuronas originalmente activas, y así sucesivamente, hasta que la mayoría de las neuronas encefálicas estuviera activándose incontroladamente. En realidad, esto sucede en ocasiones, y lo llamamos crisis epiléptica. (La epilepsia es un trastorno neurològico caracterizado por la presencia de crisis epilépticas.) Normalmente, las neuronas secretoras de GABA, presentes en gran número en el encéfalo, suponen una influencia inhibidora. Algunos investigadores creen que una de las causas de la epilepsia es una anomalía de la bioquímica de las neuronas secretoras de GABA o de los receptores del GABA. Al igual que los receptores de NMDA, los receptores del GABAa son muy complejos; contienen al menos cinco lugares de unión distintos. El lugar de unión principal es para el GABA, por supuesto. El fármaco muscimol (derivado de la muscarina, agonista colinérgico) actúa como agonista directo en este lugar (paso 6 de la Figura 4.4). Otro compuesto, la bicuculina, bloquea este lugar de unión del GABA, sirviendo de antagonista directo (paso 7 de la Figura 4.4). Un segundo lugar del receptor GABAa se une a un grupo de fármacos tranquilizantes llamados benzodiacepinas. Este grupo contiene el diacepam y el clordiacepóxido, usados para aliviar la ansiedad, inducir el sueño, reducir las crisis epilépticas y producir relajación muscular. Los barbitúricos se unen al tercer lugar. El cuarto está reservado para varios esteroides, incluidos algunos usados para conseguir anestesia general. El quinto lugar se une a la picrotoxina, veneno presente en un arbusto de la India oriental. Además, el alcohol se acopla a un lugar aún desconocido del receptor GABAa (véase la Figura 4.19). Los barbitúricos, los fármacos que se unen al lugar esteroideo y las benzodiacepinas promueven la actividad del receptor GABAa; por tanto, todos esos fármacos funcionan como agonistas indirectos. Las benzodiacepinas son ansiolíticos muy eficaces, o fármacos «que disuelven la ansiedad». Se usan a menudo en el tratamiento de las personas con trastornos de ansiedad. Además, algunas benzodiacepinas son somníferos útiles, y otras se emplean para tratar ciertos tipos de trastornos epilépticos. La picrotoxina tiene efectos contrarios a los de benzodiacepinas y barbitúricos: inhibe la actividad del receptor GABAa, actuando así como antagonista indirecto. En dosis suficientemente altas, causa convulsiones. El organismo produce normalmente varias hormonas esteroideas, y algunas hormonas relacionadas con la progesterona (la hormona principal de la gestación) actúan sobre el lugar de unión de esteroides del receptor GABAa, produciendo un efecto sedante, ansiolítico y relajante. Sin embargo, el encéfalo no produce diacepam, barbitúricos ni picrotoxina. Aún no se han descubierto los ligandos naturales de estos lugares de unión. ¿Qué sucede con el receptor GABAb? Este receptor metabotrópico, acoplado a una proteína G, funciona como receptor postsináptico y autorreceptor presináp-tico. Un agonista del GABAb, el baclofeno, actúa como relajante muscular. Otro compuesto, CGP 335348, es un antagonista. La activación de los receptores GABAb abre canales de potasio, produciendo potenciales postsinápticos inhibidores hiperpolarizadores. GLICINA El aminoácido glicina parece ser el neurotransmisor inhibidor en la médula espinal y las partes inferiores del encéfalo. Apenas se conoce su vía de síntesis; hay varias posibles, pero no sabemos lo suficiente como para determinar con cuál de ellas producen glicina las neuronas. La bacteria causante del tétanos libera una sustancia química que impide la liberación de glicina (y también de GABA); la eliminación del efecto inhibidor de estas sinapsis provoca que los músculos se contraigan continuamente. El receptor de glicina es ionotrópico y controla un canal de cloruro. Así pues, cuando está activo, produce potenciales postsinápticos inhibidores. La estricnina, un alcaloide presente en las semillas del Strychnos nux vómica, árbol de la India, funciona como antagonista de la glicina. La estricnina es muy tóxica, y en dosis relativamente pequeñas provoca convulsiones y la muerte. No se han encontrado aún compuestos que actúen como agonistas específicos de la glicina. Los investigadores han descubierto que algunas terminales nerviosas encefálicas liberan a la vez glicina y GABA. La ventaja aparente de la liberación conjunta de estos dos neurotransmisores inhibidores es la producción de potenciales postsinápticos rápidos y duraderos: la glicina estimula receptores ionotrópicos rápidos, y el GABA activa receptores metabotrópicos de duración prolongada. Obviamente, la membrana postsináptica de estas sinapsis contiene receptores de glicina y de GABA. Acetilcolina (ACh) Agonista - Estimula Veneno de la Araña viuda negra Nicotina Muscarina Neostigmina – inhibe DOPAMINA (DA) Agonista - Estimula L-Dopa – Promueve Cocaína – metilfenidato Anfetamina Selegilina NORADRENALINA (NAD) Agonista - Estimula Efecto del Compuesto Liberación de ACh Receptores Nicotínicos Receptores muscarínicos Acetilcolinesterasa Recaptación de colina Antagonista - Bloquea Toxina Botulínica Curare Atropina Efecto del Compuesto Síntesis de DA Almacenamiento de DA en vesículas Receptores D2 Antagonista - Bloquea AMPT – Inhibe Reserpina - inhibe Clorpromacina - Bloquea Clozapina – Bloquea Hemicolinio - inhibe Bloquea Recaptación de DA Estimula liberación de DA Bloquea la MAO-B Efecto del Compuesto Inhibe la síntesis de NAd Almacenamiento de NAd en vesículas Bloquea los receptores α2 Inhibe la recaptación de NAd Inhibe la MAO-A Estimula liberación de NAd Antagonista - Bloquea Ácido fusárico Reserpina Antagonista - Bloquea PCPA Reserpina FENFLURAMINA FLUOXETINA LSD MDMA Efecto del Compuesto Inhibe la síntesis de 5-HT Almacenamiento de 5-ht en vesículas Estimula liberación de 5-HT Inhibe la recaptación de 5-HT Estimula los receptores 5-HT2a Estimula la liberación de 5-HT GLUTAMATO Agonista – Estimula AMPA ACIDO CAÍNICO NMDA Efecto del Compuesto RECEPTOR AMPA RECEPTOR CAINATO RECEPTOR NMDA Antagonista - Bloquea Efecto del Compuesto SINTESIS DE GABA RECEPTORES DEL GABA ACTÚAN COMO AGONISTAS INDIRECTOS Antagonista - Bloquea ALILGLICINA – INHIBE BICUCULINA Efecto del Compuesto Bloquea receptores de glicina Antagonista - Bloquea Estricnina IDAZOXÁN DESIPRAMINA MOCLOBEMIDA MDMA, ANFETAMINA SERATONINA (5-HT) Agonista – Estimula GABA Agonista – Estimula MUSCINAL BENZODIACEPINAS GLICINA Agonista – Estimula AP5