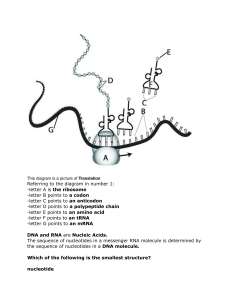
Como la ADN polimerasa cataliza la repricacion y repara con gran fidelidad
Anuncio

REVIEWS How DNA polymerases catalyse replication and repair with contrasting fidelity Wen-Jin Wu1, Wei Yang2 and Ming-Daw Tsai1,3 Abstract | DNA polymerases were named for their function of catalysing DNA replication, a process that is necessary for growth and propagation of life. DNA involving Watson–Crick base-pairing can be synthesized with high fidelity, the structural and mechanistic origins of which have been investigated for many decades. Despite this, new chemical insights continue to be uncovered, including recent findings that may explain newly discovered functions for many DNA polymerases in DNA repair and mutation. Some of these reactions involve non-Watson–Crick base-pairing. In addition, certain DNA polymerases have been engineered for a wide variety of applications in biotechnology and biomedicine. This Review describes the molecular basis for the diverse and contrasting functions of different DNA polymerases, providing an up‑to‑date understanding of how these tasks are accomplished and the means by which we can benefit from them. Institute of Biological Chemistry, Academia Sinica, 128 Academia Road Sec. 2, Nankang, Taipei 115, Taiwan. 2 Laboratory of Molecular Biology, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland 20892, USA. 3 Institute of Biochemical Sciences, National Taiwan University, Taipei 106, Taiwan. 1 Correspondence to M.-D.T. [email protected] doi:10.1038/s41570-017-0068 Published online 6 Sep 2017 The first known DNA polymerase, DNA polymerase I (Pol I), was isolated from Escherichia coli 1,2 and was shown to faithfully copy template DNA sequences3. This discovery, made by Arthur Kornberg and co‑workers2 in 1958, stimulated decades of intensive research in iden‑ tifying new polymerases and studying their chemical mechanisms and biological functions. Scientists have comprehensively catalogued DNA polymerases from all three kingdoms of life, with the enzymes being classified into six major families (A, B, C, D, X and Y) according to their sequence homology4. DNA polymerases are known to most chemists for their role in catalysing DNA replication with high accu‑ racy. Less well known is the function that these enzymes have in DNA repair, with some polymerases, perhaps counter-intuitively, even effecting DNA mutations, some of which are important for life processes5,6. As a consequence, the fidelity (BOX 1) of deoxyribonucleoside triphosphate (dNTP) incorporation is highly depend‑ ent on the polymerase catalyst and varies from being very high (107–108 for replicative polymerases)7 to very low (close to 2 for mutagenic polymerases)8 (FIG. 1a). For example, incorporation of a correct Watson–Crick (W–C) base pair (hereafter referred to as a ‘match’) mediated by Pol β is governed by Kd,app and kpol (refer to BOX 1 for definitions), the values for which fall in the ranges of 10–100 μM and 10–25 s−1, respectively. For incorporation of an incorrect base pair (hereafter referred to as ‘mismatch’), Kd,app values increase by a factor of ~20 (weaker binding) and kpol values decrease by a factor of ~103–104 (slower reaction). Taken together, these thermodynamic and kinetic parameters for the match and mismatch situations result in Pol β fidelity on the order of 103–105 (REF. 9). It has been known for some time that there is a degree of structural variation between polymerases10 (BOX 2). In this Review, we delineate how these small differences in polymerase structure enable the enzymes to perform diverse biological functions, each of which may require a very different fidelity. This variation is perhaps surpris‑ ing given that polymerases operate through very similar chemical mechanisms. Early studies focused on high-fidelity replicative poly‑ merases from bacteria or yeast; however, the mammalian Pol β, a main player in DNA repair, is the most wellcharacterized polymerase. Although Pol β is not a rep‑ licative polymerase, its fidelity is comparable to that of replicative polymerases without exonuclease proof‑ reading activity (FIG. 1a), and its kinetic and structural properties are very similar to replicative polymerases. A useful understanding of DNA chemistry can be gained by studying the catalytic cycle (FIG. 1b) and conformational changes (FIG. 1c) of Pol β11, a system that also serves as a point of comparison for other enzymes. When in its apo form (that is, free of substrate), Pol β adopts an extended conformation (I), which undergoes significant structural change on binding to DNA, after which it assumes an open conformation (II). A ternary complex (III) is formed once the enzyme binds to metal-bound dNTP (MdNTP), with conformational closure of the N subdomain of Pol β NATURE REVIEWS | CHEMISTRY VOLUME 1 | ARTICLE NUMBER 0068 | 1 . d e v r e s e r s t h g i r l l A . e r u t a N r e g n i r p S f o t r a p , d e t i m i L s r e h s i l b u P n a l l i m c a M 7 1 0 2 © REVIEWS Box 1 | Fidelity and error frequency The fidelity (accuracy of DNA synthesis) of a DNA polymerase (pol) has been defined as [(kpol/Kd,app)c + (kpol/Kd,app)i]/(kpol/Kd,app)i (REF. 230) or simply (kpol/Kd,app)c/(kpol/Kd,app)i, with the subscripts c and i referring to the incorporation of correct and incorrect bases, respectively231,115. Here, kpol is the pseudo-first-order catalytic rate constant, and Kd,app is the apparent dissociation constant for the dissociation of dNTP (deoxyribonucleoside triphosphate) from the polymerase–DNA–MdNTP ternary complex, as determined from pre-steady-state kinetic measurements. Fidelity can also be reported as (kcat/Km)c/(kcat/Km)i (REF. 232), for which steady-state kinetic measurements allow determination of kcat and Km, the first-order rate constant (the turnover number) and Michaelis constant, respectively. Both methods produce quantitatively similar values233. The inverse of fidelity has been referred to as ‘error frequency’ (REF. 113). MdNTP, metal-bound dNTP. resulting in the third distinct arrangement, the closed conformation (IV). It is here that the enzyme effects dNTP incorporation with concomitant generation of metal-bound pyrophosphate (MPPi). This product state (V) undergoes conformational change (V → VI) to release MPPi (VI →VII), after which the DNA substrate undergoes translocation and further elongation. Structural data reported for Pol β in its I12,13, II14,15, III 16, IV 14,17–21, V 19,20 and VII 14,19,22 states have been summarized11, and a structure of VI has just been dis‑ closed23. Multiple structures exist for certain states, with data collection having been performed under slightly different conditions. Although these results are highly informative regarding the molecular mechanism by which DNA polymerases operate, we are continually deriving important new insights by characterizing the possible transition state (TS) and intermediate struc‑ tures for each reaction, and by considering how and why the behaviour of some polymerases deviates from the established mechanistic paradigms. This Review first summarizes the chemistry that is involved in polymerase-catalysed match incorporation, the mechanism of which is likely to be common to most polymerases. This discussion is followed by a description of the mismatch incorporation mechanism, a process that may proceed in a different manner for each poly‑ merase. With these two aspects covered, we then address how the conserved structural and mechanistic features of polymerases facilitate different biological functions, including repair and bypass of damaged DNA. These specific functions lend themselves to various applica‑ tions, and we conclude by describing how engineering polymerases allows these to be realized. Incorporation of Watson–Crick pairs Direct monitoring of phosphodiester bond formation. The formation and cleavage of chemical bonds can be very rapid and dynamic processes. As a consequence, they are difficult to observe directly, although a recent report on femtosecond X‑ray scattering may pave the way for pro‑ gress in this area24. For enzymatic reactions, including the nucleotidyl transfer reaction catalysed by polymerases, the active site geometry in the TS can only be inferred from biochemical and computational analyses25–29. A recent breakthrough in monitoring phosphodiester bond for‑ mation was achieved using a revised time-resolved (also referred to as time-lapse) X‑ray crystallography method30. In this case, it is possible to initiate nucleotidyl transfer by immersing a non-reactive ground-state crystal of the Pol η–DNA–CadATP ternary complex in 1 mM aqueous Mg2+. The incorporation of the 3ʹ‑dA can thus be followed at atomic resolution because the reaction proceeds much more slowly in crystallo than it would in solution. At each time point, the reaction was quenched by freezing the crystals at 77 K, after which X‑ray diffraction data were collected. Structural snapshots along the time course for the phosphodiester bond formation can thus be obtained (FIG. 2a–c); this approach has also been applied to match and mismatch incorporation by Pol β19. Three metal ions are essential for nucleotidyl transfer. Analysis of the crystal structures of numerous polymer‑ ases led many to believe that each of these enzymes oper‑ ates by a catalytic mechanism that involves two metal ions10,31 (FIG. 2d). In this mechanism, one site (metal B) binds to all three phosphate groups of dNTP, two of the three active site carboxylate groups, as well as a H2O molecule or a backbone carbonyl (Met14 in the case of Pol η)32. Thus, metal B has a dual role: to correctly position the triphosphate and to stabilize the negative charge that accrues on the oxygen atoms of the PPi leav‑ ing group. Another divalent cation (metal A) binds to all three carboxylate groups, the α‑phosphate and another H2O ligand, activating the 3ʹ‑OH moiety of the upstream primer. Deprotonation of this now acidic 3ʹ‑OH group (probably by water 27,30 or by the Asp256 carboxylate in Pol β33) affords a nucleophilic alkoxide that attacks Pα when the two atoms are collinear with the oxygen atom that bridges the Pα and Pβ atoms (in accordance with an SN2‑type in‑line mechanism)10,11,14,18,19,31–34. Despite the general acceptance of the above mecha‑ nism, recent work has shown that a third metal ion (metal C) must enter the active site to induce phosphodiester bond formation (as highlighted in FIG. 2a). The binding site for metal C can be accurately located by replacing Mg 2+ with Mn2+, a metal ion that also catalyses DNA syn‑ thesis and is readily detected by X‑ray diffraction even at low occupancy (FIG. 2b). Phosphoryl transfer takes place only when the enzyme–substrate complex captures the third divalent cation through thermal activation35. Considering the structures of the reaction intermediates (FIG. 2c), the conversion of conformation 2 to 3 involves an Arg61 side chain rotating upwards and away from the third metal binding site, such that metal C can enter the active site. This metal cation binds to the PPi group and one of the oxygen atoms of the departing α‑phosphate, thereby stabilizing the reaction intermediate. These find‑ ings form the basis of a newly proposed three-metal-ion mechanism for Pol η, in which the third metal ion initi‑ ates the reaction by breaking the existing phosphodiester bond in dNTP and driving the nucleotidyl transfer 35,36 (FIG. 2e). The native 3ʹ‑OH must be well aligned with the substrate and the three metal ions for deprotonation to occur. The catalytic role of a third metal ion is also supported by quantum mechanics/molecular mechan‑ ics (QM/MM) calculations, which confirm that metal C can stabilize the negative charge of the PPi product during DNA replication catalysed by Pol η37. 2 | ARTICLE NUMBER 0068 | VOLUME 1 www.nature.com/natrevchem . d e v r e s e r s t h g i r l l A . e r u t a N r e g n i r p S f o t r a p , d e t i m i L s r e h s i l b u P n a l l i m c a M 7 1 0 2 © REVIEWS A third metal ion has also been identified in timelapse studies of match incorporation by Pol β, although no snapshot yet exists of the three metal sites being occu‑ pied at the same time as the phosphodiester bond forms. Instead, the third metal was observed only in the prod‑ uct state (hence, it is referred to as the product metal) in which the Mg 2+ ion at site A had already been replaced by Na+ (REF. 19). As is discussed later, metal C has been observed concomitant with phosphodiester bond forma‑ tion in translesion syntheses mediated by Pol β38,39. Thus, it is likely that the third divalent metal ion is also present in the Pol β mechanism for match incorporation. These A−, B−, C− pols a A+, B+, C+ pols Fidelity 108 107 Y pols X pols 106 105 104 103 Pol β b Conf. Δ MdNTP DNAn DNAn DNAn MdNTP 10 Pol λ Conf. Δ Chemistry DNAn MdNTP 102 DNAn + 1 MPPi 1 ASFV Pol X − MPPi − DNAn + 1 DNAn + 1 MPPi DNAn + 1 VI VII Translocation I II III IV V Intermediate c Apo Pol β (extended, form I) Pol β–DNA Binary complex (open, form II) Pol β–DNA–MgddCTP Ternary complex (closed, form IV) Lyase domain D (fingers) C (palm) N (thumb) αN Figure 1 | Polymerase fidelity. The fidelities of DNA polymerases vary greatly, despite | Chemistry their mechanisms and structures being largely similar. a | The Nature range ofReviews fidelity in DNA polymerases is broad. The + and − signs for polymerases in the A-, B-, and C-families denote the presence and absence, respectively, of the proofreading exonuclease activity7. The polymerases with very high fidelity feature this domain, whereas the lower-fidelity polymerases do not. The dashed line extending past 107 denotes that polymerase fidelity may exceed this value, although confirmation of this is difficult owing to the limited accuracy associated with biochemical quantification of very low error rates. b | A simplified catalytic cycle for DNA synthesis mediated by a polymerase11. The intermediates are denoted using Roman numerals, with M designating a metal ion, usually in a divalent state. Apo polymerases typically bind to DNA first, followed by the incoming dNTP (deoxyribonucleoside triphosphate), usually as the metal complex MdNTP. After the enzyme closes around the substrates, nucleotidyl transfer — referred to as the ‘chemical’ step — results in elongation of DNAn to give DNAn + 1. A more detailed scheme is provided in FIG. 4c. c | X-Ray crystal structures of forms I (Protein Databank identifier: 1BPD), II (PDB ID: 1BPX) and IV (PDB ID: 1BPY) of DNA polymerase β (Pol β), the conformations that are involved in catalysis, with subdomains labelled in both the original notation (with one’s right hand facing the page) and Wilson’s function-based nomenclature (the DCN system as outlined in BOX 2). The αN helix of the N subdomain is shown in dark green to illustrate closure of the N subdomain upon formation of the nascent base pair (grey region). DNA is shown in yellow. ASFV, African swine fever virus; Conf. Δ, conformational change; ddCTP, 2ʹ,3ʹ‑dideoxy-CTP; MPPi, metal-bound pyrophosphate. experimental findings have been further probed and extended through QM/MM calculations that focused on the specific functions of the third metal40,41. Of the four structures proposed to be involved in the key nucleotidyl transfer reaction (FIG. 2c), structure 3 may represent the most important TS or near‑TS form. Structures 1 and 2 correspond to substrate complex IV in FIG. 1b, whereas structure 4 corresponds to the product complex V. The capture of the intermediate with metal B alone (structure 1) lends support to the early structural and kinetic studies with Cr(iii)dNTP in the absence of Mg 2+; these works showed that binding of Cr(iii)dNTP to Pol β is sufficient to induce the closure of the N sub­ domain of the polymerase20,42. In addition, the Kd value of the Mg2+ ion (0.5–1 mM) was substantially higher than that of MgdNTP (30–50 μM)43–45, implying the existence of a low-affinity metal site. On the basis of the results from titrations of Pol η with metal ions in crystallo, Gao and Yang have unequivocally shown that site C is the one with low affinity for metal ions35,36. The most common metal ion involved in cataly‑ sis mediated by polymerases is Mg 2+, and it is likely that Mg2+ is the preferred metal at each binding site. However, Mn2+ has also been suggested as a natural metal ion that may be important for some polymerase functions. The Mn2+ ion has been observed for Pol λ46,47 and Pol μ48–51 in the X‑family, and has been implicated as an unnatural ‘mutator’ for other polymerases, which is discussed in the section below on non-W–C incor‑ poration. By contrast, polymerase-bound Ca2+ does not catalyse the reaction but does result in the formation of polymerase–DNA–CadNTP ternary complexes that are inert, such that they are amenable to crystallographic characterization19,30,35,39,52. The effects that different metal ions have on the kinetics and fidelity of polymerases have been reviewed53, but the chemistry governing why certain polymerases use particular metal ions at specific sites remains to be elucidated. A common intramolecular hydrogen bond in the incoming dNTP. The structures and conformations of nucleo­bases can also play important roles in the catalysis of DNA polymerases (FIG. 3). For example, the intraand intermolecular hydrogen-bonding motifs involv‑ ing nucleo­b ases can provide clues regarding the fidelities of the polymerase enzymes that house these substrates. Analysis of crystal structures of DNA poly­ merase–DNA–dNTP and RNA polymerase–RNA– rNTP ternary complexes (where rNTP denotes a ribonucleoside triphosphate) revealed the existence of a common intramolecular hydrogen bond between the 3ʹ‑OH and the β‑phosphate of the incoming dNTP or rNTP37 (FIG. 3a). This interaction has been sug‑ gested to promote deprotonation of the primer 3ʹ‑OH by facilitating PP i departure. 2ʹ,3ʹ‑Dideoxy-NTP (ddNTP) can also be incorporated into DNA by poly‑ merases and is commonly used as a chain terminator in DNA sequencing. That ddNTP is unable to form this intramolecular hydrogen bond may contribute to the catalytic efficiency of the enzyme being reduced by a factor of >100 when processing this substrate54. NATURE REVIEWS | CHEMISTRY VOLUME 1 | ARTICLE NUMBER 0068 | 3 . d e v r e s e r s t h g i r l l A . e r u t a N r e g n i r p S f o t r a p , d e t i m i L s r e h s i l b u P n a l l i m c a M 7 1 0 2 © REVIEWS This result further underscores the importance of the hydrogen bond with 3ʹ‑OH, which has been found to form stereospecifically with the pro‑S oxygen atom of the β‑phosphate in dNTP or rNTP. In addition, this hydrogen bond is also present in the structure of the Pol β–DNA–Cr(iii)dTMPPCP (where dTMPPCP is 2ʹ-deoxy­thymidine 5ʹ-(β,γ-methylene)triphosphate) ter‑ nary complex bearing metal B alone20 (FIG. 3b). By contrast, the intramolecular hydrogen bond is not observed in the complexes that feature a dNTP with l‑stereochemistry, such as the unnatural Pol λ–DNA–l‑dCTP complex 55. Watson–Crick base-pairing alone is insufficient to ensure match incorporation. With the exception of Pol ν56–58 and Pol θ59–61, most enzymes in the A- and B-families are replicative polymerases that exhibit fidelities as high as 106–108 (REFS 7,62) (FIG. 1a). 3ʹ‑5ʹ Exonuclease proofreading increases fidelity by up to two orders of magnitude7, such that polymerases without proofreading (as is the case for most of the polymerases described in this Review) have fidelities no greater than ~105–106. One longstanding topic of debate (and one that has been summarized recently 63) is whether fidelity can be entirely attributed to the difference in Gibbs free energy changes between base-pairing of a match and a mismatch. Even when just considering matches, extensive early work sought to address whether hydrogen bonding or shape complementarity is more important 64–68. Recently, the difference in the reaction free energy between the incorporation of a match and a mismatch (ΔΔGinc) has been computed by measur‑ ing the kinetics of the forward and reverse reactions in both cases. These additional data make it clear that polymerase fidelity does not depend solely on intrinsic properties of DNA69 and may well be affected by specific enzyme–substrate interactions. The prevailing evidence suggests that the dNTP-induced conformational closure is fast and that the rate-limiting step of nucleotidyl trans‑ fer is probably the chemical step (IV→V in FIG. 1b) for Pol β11,20,45,70–77, HIV reverse transcriptase 78, human Box 2 | Domain and subdomain nomenclatures for polymerases DNA polymerases comprise multiple independently functioning domains. Of most mechanistic interest is the catalytic domain, which itself consists of three interdependent subdomains. The right-hand-based nomenclature — consisting of fingers, palm and thumb subdomains from the amino (N) to the carboxyl (C) terminus — is widely used234, and the structures of DNA polymerase β (Pol β) in FIG. 1c are labelled in this manner. It has also been suggested, based on functional alignment, that the fingers and thumb subdomains in Pol β should be swapped such that the nomenclature is left-handed235. The confusion that can result from adopting two forms of nomenclature for Pol β led to the proposal of function-based (DCN) nomenclature for the three subdomains of Pol β. From the N to the C terminus, these are D (duplex DNA binding), C (catalytic) and N (nascent base-pair binding). In this Review, we use the right-hand based structural alignment for all polymerases and include the DCN nomenclature for Pol β. In addition to the catalytic domain, polymerases often comprise additional domains or subdomains that are involved in enzymatic function. For example, Pol β has an 8 kDa lyase domain at its N terminus (FIG. 1c), whereas many replicative polymerases feature exonuclease domains for editing at either their N or C termini, and Y‑family polymerases contain an additional little finger subdomain after the thumb subdomain at their C termini. Furthermore, some polymerases contain multiple regulatory domains that are important for their in vivo functions. Pol η35,36, P2 DNA polymerase IV from Sulfolobus solfa‑ taricus79, KlenTaq1 (REF. 80) and bacteriophage T7 poly‑ merase81; thus, it seems that fidelity arises from extensive enzyme–substrate interactions and W–C base-pairing in the TS of the chemical step. This becomes more evident when comparing these results to the contrasting situation in which non-W–C incorporation takes place. Incorporation of non-Watson–Crick pairs Spontaneous errors in Watson–Crick pairing. Watson and Crick82,83 observed that deviation from W–C pairing can arise when tautomeric forms of the bases are present. For example, a dA–dCTP mismatch, in which A under‑ goes keto–enol-like tautomerization to a structure with an exocyclic imine, mimics the shape of a W–C base pair (FIG. 3c). Indeed, this mismatch can be incorporated in DNA by the high-fidelity Bacillus stearothermophilus Pol I large fragment84. The incorporation of an ionized dG–dTTP mismatch has been observed in the reac‑ tion catalysed by avian myeloblastosis virus reverse transcriptase; the efficiency of dTTP misincorporation increased as the pH was increased from 6.5 to 9.5 (REF. 85). The crystal structure of a human Pol λ variant bound to a DNA substrate with dGTP opposite to a template T (dT–dGTP) has been solved, confirming that the bases, despite being mismatched, are nevertheless arranged in a W–C‑like geometry 86 (FIG. 3d). The pH dependence of the misincorporation is consistent with the presence of an ionized base pair. Relaxation dispersion NMR spec‑ troscopy enabled observation, in free duplex DNA, of transient Hoogsteen pairings87–89 — another form of nonW–C base-pairing that can form with DNA polymerases as shown in later examples. Departures from W–C pair‑ ing also occur with RNA. Such mismatches — referred to as wobble pairs — have been observed in dynamic equilibrium with trace amounts of short-lived W–C‑like enol tautomers or ionized bases90. It is important to note that the above deviations from W–C‑like pairing can arise owing to additional factors: use of the mutator metal ion Mn2+ for dA–dCTP incor‑ poration and engineering of a polymerase active site for dT–dGTP. As we describe below, both approaches have been used in conjunction with other polymerases to facilitate mismatch formation. Mn 2+ facilitates mismatched ternary complex formation of Pol β. The presence of Mn2+ at a polymerase active site has been observed to enhance the efficiency of DNA synthesis, although this is often at the expense of fidelity. Thus, Mn2+ is a mutator metal ion for some poly­ merases19,91–99, probably because it has, relative to Mg2+, relaxed coordination requirements and greater tolerance of substrate misalignment94. An example is Pol β, which lacks the common exonuclease 3ʹ‑5ʹ proofreading domain and is the smallest eukaryotic polymerase (39 kDa, with 335 residues) as well as the main polymerase responsi‑ ble for base excision repair (BER)11. Owing to the rela‑ tively high fidelity of Pol β, it was difficult to obtain the structures of mismatched ternary complexes unless Mn2+ was used21. Thus, time-resolved X‑ray crystallography studies were conducted on ternary complexes that 4 | ARTICLE NUMBER 0068 | VOLUME 1 www.nature.com/natrevchem . d e v r e s e r s t h g i r l l A . e r u t a N r e g n i r p S f o t r a p , d e t i m i L s r e h s i l b u P n a l l i m c a M 7 1 0 2 © REVIEWS a 40 s T (3′) 80 s Mg MgC2+ 2+ C MgB2+ MgA2+ D115 E116 D13 M14 b 30 s 60 s 90 s New bond WatN MnA2+ MnC2+ MnC2+ M14 D13 c MgA2+ MgC2+ 1 3 2.1 Å MgB2+ DNAn + 1 MgB2+ O O e 4 R61 O O − O 2+ MB O Pγ O − O O O O O MgB2+ − O O Reaction coordinate O + Pα O− H2O MgA2+ MA2+ O O MC2+ − H O PPi Ea, uncat Figure 2 | Mechanism of phosphodiester bond formation. The mechanism of phosphodiester bond formation catalysed by DNA polymerase η (Pol η) involves three metal ions. a | The DNA synthesis reaction can be followed in crystallo by immersing crystals of the Pol η– DNA–CadATP ternary complex in a 1 mM solution of MgCl2. Omit maps (4.0σ; shown in green mesh) — corresponding to the difference between the observed structure factors (at time intervals between 40 and 230 s) and the calculated structure factor for the system at 40 s (Fo(40 – 230 s) − Fc(40 s)) — are superimposed on to structures of the reaction intermediates. Arrows indicate regions where there is a local increase in electron density, and the third Mg2+ (MgC2+) cation is circled. The apparent delayed binding of MgC2+ after product formation is due to the low electron density of Mg2+ and low occupancy of MgC2+ at 80 s. b | In crystallo catalysis with Pol η also proceeds in 10 mM MnCl2. The Fo − Fc omit map for the new phosphodiester bond (blue mesh), the third Mn2+ cation (MnC2+; blue mesh) and the water molecule WatN (pink O− β nucleobase nucleobase MgC − − P 2+ Ea, cat MgB2+ MA2+ O O O O H2O − O Pα − O WatN 3.2 Å Å 4.2 O .3 Å 2.3 Å 2 MgA H O MgC2+ 2.1 Å R61 MgA2+ O B R61 2+ R61 nucleobase 4 MgC2+ 1 nucleobase O 3 2 dATP d MgA2+ 2 Thermal motion Uncatalysed reaction Catalysed reaction DNAn New bond New bond MnB2+ D115 E116 600 s MnC2+ dATP Energy 230 s dATP 3′O S113 140 s O O P O− 2+ − − O O MB β O − O Pγ O O− O mesh; WatN is hydrogen bonded to the nucleophilic 3ʹ‑OH) are Nature Reviews | Chemistry contoured at 3σ and superimposed on to the structures at each time point. c | The reaction coordinate of the DNA synthesis catalysed by Pol η highlights the roles of the three metal ions, the third of which promotes phosphoryl transfer to the nucleophilic 3ʹ‑OH by overcoming the energy barrier to the product state. d | The previously accepted two-metal-ion mechanism involves deprotonation of 3ʹ‑OH (where :B is a general base) and nucleophilic attack at Pβ. e | The newly proposed three-metal-ion mechanism involves metal-induced polarization of the phosphate and attack of 3ʹ‑OH on Pβ, the latter being a strong electrophile on account of its binding to MC2+. In parts a–c, hydrogen atoms have been omitted for clarity. Ea, activation energy. Part a is adapted with permission from REF. 30, Macmillan Publishers Limited. Parts b and c are adapted with permission from REF. 35, AAAS. Parts d and e are adapted with permission from REF. 36, under the Creative Commons License CC BY 4.0. NATURE REVIEWS | CHEMISTRY VOLUME 1 | ARTICLE NUMBER 0068 | 5 . d e v r e s e r s t h g i r l l A . e r u t a N r e g n i r p S f o t r a p , d e t i m i L s r e h s i l b u P n a l l i m c a M 7 1 0 2 © REVIEWS a β α γ b γ β MgB2+ α CrB3+ MgA2+ H H c Anti Anti H N N N A* N R N N H N Anti O O Anti T* N – H N N G N R N N R H O N H W–C-like dT–dGTP e O H H N N G Anti Anti R O W–C-like dA–dCTP d C N H O N N N N H N G N R R N H N H Distorted dG–dGTP H H N Anti Anti C O H N N R R N H O N H dC–dGTP f N G N N H Anti Syn N G N R N H N O Hoogsteen dG–dGTP N H G O N N H N H N N R H Figure 3 | Nucleobases in various ionization states and/or Nature conformations. Reviews | DNA Chemistry polymerase (Pol)–DNA–MdNTP ternary complexes (where MdNTP is a metal-bound dNTP) can feature diverse substrates in varied conformations. a | When MgdUMPNPP is in the Pol β–DNA–MgdUMPNPP ternary complex (Protein Databank identifier: 2FMS), it forms an intramolecular hydrogen bond (green dashed line) between 3ʹ‑OH and the pro‑S oxygen of Pβ. The use of this N‑containing and non-hydrolysable analogue of dUTP enables the intermediate to be characterized. b | The structure of Cr(iii)dTMPPCP in the Pol β–DNA–Cr(iii)dTMPPCP ternary complex (PDB ID: 1HUO). dTMPPCP is a non-hydrolysable dTTP analogue in which the O atom that bridges Pβ and Pγ is replaced with a CH2 group. The B‑site Cr3+ ion is shown in grey. c | The structure of a Watson–Crick (W–C)‑like dA–dCTP mismatch accommodated by the Bacillus stearothermophilus DNA polymerase I large fragment (PDB ID: 3PX6) features an adenine tautomer with an exocyclic imine (A*). d | A W–C‑like dT–dGTP mismatch bound to a Pol λ variant (PDB ID: 3PML), featuring a deprotonated thymine ring (T*). e | The top panel shows a dG–dGTP mismatch (PDB ID: 4FK4) and the bottom panel shows a dC–dGTP match (PDB ID: 4FJH), accommodated by an RB69 L415A/L561A/S565G/Y567A quadruple mutant. f | A dG–dGTP mismatch with an anti–syn Hoogsteen base pair in ASFV Pol X (PDB ID: 2M2W). The incoming syn dGTP uses its Hoogsteen face (see FIG. 6a) to bind to the template G. Hydrogen atoms have been omitted for clarity. dNTP, deoxyribonucleoside triphosphate; dTMPPCP, 2ʹ-deoxythymidine-5ʹ-(β,γ-methylene)triphosphate; dUMPNPP, 2ʹ‑deoxyuridine‑5ʹ-(α,β-imido)triphosphate. featured either Mg 2+ or Mn2+ ions19, which enabled com‑ parison between near‑TS intermediate structures (with mixtures of bound reactants and products) for match (with Mg 2+) and mismatch (with Mn2+) incorporation catalysed by Pol β. As shown in the case of dG–dCTP (FIG. 4a), the intermediate structure observed in the matched ternary complex of Pol β is similar to that of Pol η (FIG. 2b), except that metal C was undetectable at this time point. The complex for the dG–dATP mismatch (FIG. 4b) also lacks metal C, but its features provide insight into the structural basis of fidelity: the mismatched base-pairing is significantly distorted from planarity, and the distance between the substrate Pα and the product 3ʹ‑OP is longer in the mismatched relative to the matched structure. This distortion leads to a large increase in the activation energy that is required for the chemical step of the mismatch (IV→V, red trace, FIG. 4c), which should be responsible for the substantially reduced rate of mismatch relative to match incorporation. Enlarging the dNTP binding pocket converts the high-fidelity RB69 polymerase to a low-fidelity polymerase. One may expect a decrease in fidelity if the constraints on a polymerase active site are made less stringent. In this regard, the nascent dNTP binding pocket of a high-fidelity replicative polymerase from bacteriophage RB69 was modified by replacing four bulky amino acid residues with smaller ones to afford the quadruple mutant L415A/L561A/S565G/Y567A100. Pre-steady-state kinetic analyses of the mutantcatalysed reaction indicated that its fidelity is lowered by a factor of 103–106 (REF. 101). Consequently, this mutant can form stable ground-state ternary complexes with all 12 mismatches in the presence of the (catalytically inactive) Ca2+ ion, with the resulting structures featuring distorted base-pairing at the active site (the structures of mismatched dG–dGTP and matched dC–dGTP can be compared in FIG. 3e). Some low-fidelity polymerases use an enzyme side chain to select a specific dNTP. The African swine fever virus (ASFV) Pol X, at 174 residues, is a very small polymerase that does not feature the lyase domain and duplex DNA binding subdomain that are present in its mammalian homologue Pol β, which is twice as large102. In terms of mismatch incorporation, ASFV Pol X is perhaps the most extreme case as it catalyses the formation of a dG–dGTP (G–G) mismatch in addition to the four W–C matches8. Structures of the free protein have been determined using solution NMR spectroscopy 103,104, as have those of the ASFV Pol X–MgdGTP binary complex and ASFV Pol X–MgdGTP–DNA ternary complex, which indicate that ASFV Pol X can use either gapped DNA or MgdNTP as the first substrate105,106. ASFV Pol X can bind to MgdGTP in a syn configuration in the absence of DNA and form a dG–dGTP mismatch with an anti–syn Hoogsteen basepair conformation (FIG. 3f). The His115 residue is key to the catalytic incorporation of dG–dGTP mismatches (FIG. 5a) , which has recently been confirmed inde‑ pendently by crystallography 107. A double hairpin DNA with two GAA stem loops was used in the NMR studies 6 | ARTICLE NUMBER 0068 | VOLUME 1 www.nature.com/natrevchem . d e v r e s e r s t h g i r l l A . e r u t a N r e g n i r p S f o t r a p , d e t i m i L s r e h s i l b u P n a l l i m c a M 7 1 0 2 © REVIEWS to acquire high quality NMR spectra106, whereas a natural one-nucleotide gap DNA was used in the crystallographic study. The latter analysis located a unique binding pocket for the 5ʹ‑phosphate group of the downstream primer 107, which affects dG–dGTP mismatch formation107,108. In the case of Pol β11 and Pol λ109, this binding is strengthened by the presence, in the lyase domain, of three cationic residues that are missing in Pol X. The role of an enzyme side chain in selecting a spe‑ cific dNTP has also been confirmed in the case of Rev1, a Y-family polymerase that repairs human DNA. Indeed, the Rev1–DNA–MgdCTP ternary complex has been characterized, and the enzyme, initially identified as a deoxycytidyl transferase110, mediates incorporation of dCTP opposite to template G with very high specificity. To a lesser extent, Rev1 can also install dCTP opposite to an abasic site or an O6‑methylguanine during transle‑ sion DNA synthesis110,111. The origin of the very high specificity that Rev1 shows for dCTP became evident after the crystal structure of yeast Rev1 bound with template G and dCTP112 was determined. The dCTP substrate does not form a base pair with the template G, but instead hydrogen bonds to the Arg324 side chain of Rev1. Furthermore, Rev1 sets aside the template base and uses Arg324 as a template to form an Arg–dCTP pair that mimics a DNA base pair (FIG. 5b). Thus, Rev1 uses its protein side chain to dictate the identity of not only the incoming dNTP, but also the template base112. Understanding fidelity attenuation with the mediumfidelity Pοl λ. In the X‑family pols, Pol λ exhibits a fidelity (calculated from the reported error frequencies to be 30–9,100)113 between those of Pol β (1,700–93,000)114 and ASFV Pol X (1.9–7,700)8. Thorough structure– function studies have been conducted on Pol λ109, and a recent report indicates that, similar to ASFV Pol X, Pol λ possesses high MgdNTP affinity in the absence of DNA52. Analogous to the stabilizing interaction provided by b Helix N a Helix N Open Open Closed PPi dG dCTP Reactant Product Closed MgB2+ D192 MgA2+ PPi dATP MnB2+ D192 dG D190 MnA2+ D256 D256 c D190 TS Gibbs free energy WT mismatch I260Q mismatch WT match EDn II N EDnN III E′DnN IV MA2+ E′DnNMA IV E′Dn + 1PPiMA V − MA2+ E′Dn + 1PPi V EDn + 1PPi VI − PPi EDn + 1 VII Intermediate Reaction coordinate Figure 4 | Structural and energetic differences near the transition state of the nucleotidyl transfer mediated by Pol β. Nature Reviews | Chemistry a | A near‑TS intermediate structure of a dG–dCTP match (yellow; Protein Databank Identifier: 4KLF) has been determined from time-resolved X‑ray crystallography by immersing a crystal of the ternary complex Pol β–DNA–CadCTP in MgCl2 (200 mM, 20 s). The αN helix is closed; the open αN helix of the DNA binary complex (cyan; PDB ID: 3ISB) is shown for comparison. b | Similarly, the near‑TS intermediate structure of a dG–dATP mismatch (magenta; PDB ID: 4KLS) has been determined from a crystal immersed in MnCl2 (200 mM, 10 min). The αN helix is also closed, although not as fully as that in part a. The structures depicted in parts a and b feature 1:1 mixtures of bound reactants and products. c | Free energy diagrams, based on pre-steady-state kinetic analyses, have been proposed for wild-type (WT) and mutant Pol β. The corresponding intermediates from FIG. 1b (where the roles of metal A and metal B are not dissected) are shown below the reaction scheme here. E, enzyme in open conformation; Eʹ, enzyme in closed conformation; Dn, DNA; M, metal; N, MdNTP with MB only; Pol β, DNA polymerase β; PPi, metal-bound pyrophosphate; TS, transition state. Part c is adapted with permission from REF. 119, American Chemical Society. NATURE REVIEWS | CHEMISTRY VOLUME 1 | ARTICLE NUMBER 0068 | 7 . d e v r e s e r s t h g i r l l A . e r u t a N r e g n i r p S f o t r a p , d e t i m i L s r e h s i l b u P n a l l i m c a M 7 1 0 2 © REVIEWS a dG (anti) dGTP (syn) dG (anti) H115 dGTP (syn) H115 b c dG ddC dG Y505 L431 Mg2+ dCTP R324 Mg2+ F506 A431 I492 d Pathway A Pol (ASFV Pol X, Pol λ) Pathway B Watson–Crick base pairing Pol–DNAn Pol–dNTP Pol–DNAn–dNTP Pol–DNAn + 1–PPi Pol–DNAn–dNTP Pol–DNAn + 1–PPi Mismatch base pairing Figure 5 | Polymerases overcome Watson–Crick pairing byNature engaging in multiple Reviews | Chemistry interactions with substrates. a | Stereo views of the ASFV DNA polymerase X (Pol X) active site, highlighting the interactions between MgdGTP and His115 in the Pol X– MgdGTP binary complex (thin lines; Protein Databank identifier: 2M2U), and the Pol X– DNA–MgdGTP ternary complex (sticks; PDB ID: 2M2W). In each case, His115 helps to keep the incoming dGTP in a syn conformation. b | Rev1 uses its Arg324 side chain as a DNA template base to pair up with incoming dCTP, with template G having been set aside from the DNA helix (PDB ID: 2AQ4). c | The structures of the tyrosine–phenylalanine (YF) motifs and surrounding regions for the MnMgdCTP complex of Pol λ (dark blue; PDB ID: 5DDY), as well as the MgdCTP complex of the Pol λ L431A mutant (cyan; PDB ID: 5CR0). The L431A mutation provides space for Ile492 to move away from the Phe506 ring, allowing Phe506 to orient itself parallel to Tyr505, a conformation that facilitates dNTP binding. d | The canonical reaction mechanism for polymerase enzymes involves initial binding of DNA (pathway A; see also FIG. 1b). ASFV Pol X and human Pol λ, and possibly other lower-fidelity polymerases, may also bind dNTP first (pathway B). ASFV, African swine fever virus; dNTP, deoxyribonucleoside triphosphate. His115 in ASFV Pol X, Pol λ makes use of its Tyr505 side chain to bind to the dNTP substrate through π–π inter‑ actions; mutation of Tyr505 into a smaller Ala reduced the affinity significantly. Structural analysis suggested that Pol λ maintains its medium fidelity by binding the sub‑ strate in a well-defined hydrophobic pocket that features Leu431, Ile492, Tyr505 and Phe506 residues. In support, it was predicted and subsequently demonstrated that the L431A mutation enhances MgdNTP pre-binding (FIG. 5c) and lowers fidelity. The mechanism of fidelity. Having established the struc‑ tures and mechanisms that are involved in match and mismatch incorporations, we now address the main factors that are responsible for the high fidelity of cer‑ tain DNA polymerases. The most extensively studied effect is the MdNTP-induced conformational change (closure of the N subdomain or thumb subdomain for Pol β but the fingers subdomain for other high-fidelity poly­merases10). The closed conformation has been well studied in many intermediate structures of polymerase– DNA–MdNTP complexes. Work on the R61A mutant of Pol η also indicated that the closed conformation is a prerequisite for aligning the primer and dNTP such that a third metal ion can bind30,35,36. A major point of interest is the role of conformational closure in differentiating correct and incorrect dNTP. This conformational change was once believed to be the rate-limiting step and thus the main fidelity-controlling step115. However, as men‑ tioned above, it is the chemical step that is now recog‑ nized as being rate limiting. Nevertheless, recent studies have revealed that mismatched MgdNTP can induce only partial conformational closure or none at all116–118. Further studies suggested that the conformational clo‑ sure differentiates dNTP by a thermodynamic rather than a kinetic effect. As becomes evident on considering the free energy profile for the Pol β catalytic cycle (FIG. 4c), mis‑ match incorporation induces conformational closure at a rate comparable to that induced by match incorporation (III → IV, FIG. 4c). However, the correct dNTP can better stabilize the closed form IV119; this has been supported by recent single-molecule studies120,121 and is logical given that the N subdomain (represented by the characteristic αN helix) is closed in the near‑TS intermediate structures in reactions that lead to both match (FIG. 4a) and mismatch incorporation (FIG. 4b). Taken together, the results indicate that both match and mismatch incorporations (if the latter does occur) proceed through analogous conformational trajectories that involve closure of the N subdomain119. Consistent with this conclusion, some lower-fidelity poly­merases, including Pol μ51, Pol λ52 and terminal deoxynucleotidyl transferase (TdT)122,123, exist in a closed conformation even in their substrate-free forms. The DinB homologue (Dbh) polymerase from S. solfataricus, an error-prone enzyme from the Y-family, is also closed in its apo form, and human Pol η is closed when in the Pol η–DNA binary complex124. Furthermore, as described above, several lower-fidelity polymerases are able to bind with MdNTP before binding DNA, which led to the pro‑ posal of a partially random sequential mechanism (FIG. 5d) for some polymerases52,106. Different polymerases may operate through a combination of pathways A (in which DNA is bound first) and B (in which MdNTP is bound first) to achieve their specific functions. As shown in FIG. 4c, the main influence on fidelity, based mainly on the studies of Pol β, is the nature of the TS of the nucleotidyl transfer reaction70. This is supported by kinetic analyses119, as well as by consideration of the near‑TS structures19. In catalysis mediated by Pol η, bind‑ ing of metal C is the rate-limiting sub-step of the chemi‑ cal step35,36. The near‑TS structures of Pol β complexes of matched and mismatched substrates differ in many ways, including the interactions involving active site residues, metal ions, W–C pairing and DNA binding (FIG. 4a,b). These differences are reflected in the higher relative energies of the TS and the intermediates that are close to the TS in the case of mismatch incorporation (FIG. 4c). Indeed, small structural perturbations can affect fidelity, and mismatches may occur more often if the enzyme relaxes its stringency for correct dNTP incorporation (for example, by enlarging the active site) or is inherently 8 | ARTICLE NUMBER 0068 | VOLUME 1 www.nature.com/natrevchem . d e v r e s e r s t h g i r l l A . e r u t a N r e g n i r p S f o t r a p , d e t i m i L s r e h s i l b u P n a l l i m c a M 7 1 0 2 © REVIEWS selective for binding a specific dNTP (as described above for Rev1 and dCTP). In addition, binding of the A‑site M2+ is a key step in the discrimination against an incor‑ rect incoming dNTP, and Mn2+ is less selective than is Mg 2+ in this regard34. Similar reasoning can explain why the mutagenic I260Q variant of Pol β has a lower fidelity than the native Pol β125, as kinetic119, small angle X‑ray scattering 117 and recent crystallographic analyses13 all sug‑ gest that the main cause for the lower fidelity of I260Q is its ability to form a relatively stable mismatched ternary complex (FIG. 4c). Further elaboration of the mechanisms is provided in BOX 3. DNA repair, damage bypass and mutation Roles of DNA polymerases in DNA damage responses. DNA can be damaged by exogenous agents such as reac‑ tive oxygen species (ROS), alkylating agents or UV light. More than 50,000–70,000 DNA sites can be damaged per cell per day 126–128. If left unrepaired, DNA lesions can lead to mutations and cancer formation. To defend against these possibilities, living systems make use of various DNA repair mechanisms that involve many other pro‑ teins in addition to polymerases. DNA damage and repair are broad fields of active research5,11,109,128–132, with the 2015 Nobel Prize in Chemistry having been awarded to Thomas Lindahl, Paul Modrich and Aziz Sancar for their work on the molecular mechanisms of DNA repair. Our Box 3 | Kinetic origins of fidelity The question as to which step is rate limiting for match incorporation, mainly for high-fidelity DNA polymerases, has been the subject of debate over the past two decades. This is despite it being known, for four decades now, that in the evolutionary process, enzymes have evolved to stabilize the transition state of the chemical step (FIG. 1b, IV → V) in their catalytic cycle, to a point where this energy approaches that of the transition states associated with the physical steps, such as substrate binding, product release and conformational changes. At this point, any further stabilization of the transition state of the chemical step would not have a significant effect on the overall rate236. Thus, for match incorporation catalysed by DNA polymerases, the chemical step should be either rate limiting (for less well-evolved enzymes) or very close to the physical steps (for more evolved enzymes). An important point is that fidelity is determined by the difference between parameters for match and mismatch incorporation. As high-fidelity DNA polymerases have not evolved to incorporate mismatches, the energy of the transition state of the chemical step should be much higher in the case of mismatches (FIG. 4c, red trace) relative to matches (green trace). This is evidenced by values for kpol, the pseudo-first-order catalytic rate constant for incorporation, being a few orders of magnitude lower in the case of mismatches. Thus, the chemical step (including its sub-steps) is the key step for the origin of fidelity, although other steps could also contribute partially. Low-fidelity DNA polymerases may have evolved to facilitate mismatch incorporation depending on their specific functions, for example, by exploiting greater control of nucleotide binding. This could also be achieved by artificially engineering the high-fidelity enzyme to facilitate nucleotide binding or by replacing Mg2+ with Mn2+. Nonetheless, there are different viewpoints and exceptions, as well as many computational studies that are largely beyond the scope of this Review. For future studies, we remind the reader that the ‘thio effect’ (that is, the ratio of the rate of a phosphate substrate versus a phosphorothioate substrate) — often used in deciphering whether the conformational closure or the nucleotidyl transfer is rate limiting — is not a definitive approach because the full thio effect when the chemical step is fully rate limiting is often unknown and is highly variable70, and the thio effect measured for enzymatic phosphoryl transfer reactions can vary from 1 to 100,000 (REF. 237). In addition, the effects of site-specific mutation on kinetics should be examined critically and interpreted cautiously in both experimental238 and computational studies239. discussion here largely focuses on human polymerases, as we express more repair polymerases than do lower spe‑ cies, and our enzymes have been the subjects of recent discoveries concerning new mechanisms and functions. When DNA damage occurs, cells usually initiate specific repair mechanisms before the synthesis phase (S phase) of the cell cycle, in which DNA is replicated. These mechanisms, often in conjunction with activation of checkpoint proteins, are generally referred to as DNA damage response. Common repair mechanisms include, but are not limited to, BER, nucleotide excision repair (NER), ribonucleotide excision repair (RER) and mis‑ match repair 128. The roles of polymerases in these repair pathways are usually to catalyse ‘re‑synthesis’ after the damaged nucleobases have been excised through mul‑ tiple steps. BER usually involves the use of Pol β to fill a single-nucleotide gap, whereas NER involves Pol δ and possibly Pol κ133, and RER involves mainly Pol δ to fill longer gaps131. Another role of polymerases in response to DNA damage is translesion synthesis, which usually occurs either before the lesion can be repaired by one of the mechanisms mentioned above or after the lesion escapes the repair mechanisms. The aim is to try to insert a correct base to avoid mutation, and, if this is not possible, a mismatched dNTP is incorporated and then fixed in the post-replication repair. New functions of human DNA polymerases. The 17 human DNA polymerases that have been identified to date belong to the A-, B-, X- and Y-families, with none being in the C- or D-families. Of the A‑family poly­merases (mainly replicative), Pol γ functions in high-fidelity DNA synthesis in mitochondria134,135, the nuclear Pol θ (which features polymerase and helicase domains) also partici‑ pates in double-strand break repair (DSBR)61,136–138 and the nuclear Pol ν can perform translesion synthesis58,139. Of the B‑family polymerases (also replicative), Pol α can use dNTP to extend an RNA primer before it can be used by Pol δ or Pol ε4. Pol δ also participates in DNA repair and binds weakly to the sliding clamp proliferating cell nuclear antigen during replication. Pol δ dissociates on reach‑ ing a stalled site, leaving the antigen on the DNA140 and allowing a polymerase capable of translesion synthesis, such as Pol η, to synthesize past a stalled site such as a T–T dimer 141. Very recently, Pol δ has also been shown to participate in alternative telomere maintenance142. It is generally accepted that the main role of Pol ε is in leading-strand DNA replication, although it is still under debate whether Pol ε or Pol δ is the main polymerase in this function143–145. The X‑family polymerases126,146, includ‑ ing Pol β11,12,17, Pol λ147,148, Pol μ48,51 and TdT122, specialize in BER, translesion synthesis, somatic hypermutation4 and DSBR or V(D)J recombination (the latter involving random rearrangement of variable, diverse and joining DNA regions). The Y‑family polymerases, including Pol κ, Pol ι, Pol η and Rev1, are mainly responsible for translesion synthesis of bulky adducts129,149,150. Telomerase, which belongs to the reverse transcriptase family, is responsible for replication of the chromosome end151,152. An exciting newly discovered polymerase is PrimPol 153,154, which belongs to the archaeal and NATURE REVIEWS | CHEMISTRY VOLUME 1 | ARTICLE NUMBER 0068 | 9 . d e v r e s e r s t h g i r l l A . e r u t a N r e g n i r p S f o t r a p , d e t i m i L s r e h s i l b u P n a l l i m c a M 7 1 0 2 © REVIEWS eukaryotic primase superfamily, and is so named because it can function as both a primase and a polymerase. Uniquely, PrimPol can use both dNTP and rNTP sub‑ strates to initiate DNA and RNA synthesis, respectively, which is in contrast to primase, for which only rNTP can be used. PrimPol can also function in translesion synthesis153,154 but is highly error-prone155. a H R N N H 2N 9 8 7 3 4 5 2 6 1 N H N O Hoogsteen face Approaches used by DNA polymerases to deal with the 8‑oxo‑dG lesion and mutation. 8‑Oxo‑ 7,8‑dihydroxy‑2ʹ‑deoxyguanosine (8‑oxo‑dG) is an abundant mutagenic oxidative DNA lesion, occurring nearly 2,800 times per human cell per day 128. In addi‑ tion, dGTP can also be oxidized to 8‑oxo-dGTP and misincorporated into DNA by many polymerases, such as Pol α156, mitochondrial Pol γ157, Pol β38,156,158,159, Pol λ160, N Oxidation N H 2N dG 3 4 5 2 6 1 N H N 2 3 H 9 8 7 N O H N H dATP (anti) 8-oxo-dG dAMP (anti) (syn) MgC2+ MgB2+ NaA+ N N 4 1 6 5 c PPi 8-oxo-dGMP (syn) R N 9 8 7 8-oxo-dG (syn) b dA (anti) O R D190 D192 MnC2+ MnB2+ MnA2+ D256 d 8-oxo-dG (syn) dCTP (anti) 8-oxo-dG (syn) Mg2+ dATP (syn) Mg2+ e Fingers Palm DNA Little finger Thumb Figure 6 | Certain polymerases, by virtue of the size and nature their active sites, Natureof Reviews | Chemistry are active in translesion syntheses. a | 8‑Oxo‑7,8‑dihydroxy‑2ʹ‑deoxyguanosine (8‑oxo‑dG) uses its Hoogsteen face to pair with an incorrect dATP in syn–anti conformations. b,c | Structures of dA–8‑oxo-dGTP (anti–syn; Protein Databank identifier: 4UAY) and 8‑oxo‑dG–dATP (syn–anti; PDB ID: 4RQ8) Hoogsteen base pairs, respectively, accommodated by DNA polymerase β (Pol β). d | Pol ι uses its exceptionally narrow active site to force the purine bases of 8‑oxo‑G or dATP to adopt a syn conformation. The 8‑oxo‑dG–dCTP pair shown on the left (syn–anti; PDB ID: 3Q8P) is more stable than 8‑oxo‑dG–dATP shown on the right (syn–syn; PDB ID: 3Q8Q), as the former has one more hydrogen bond (dashed lines). e | Space-filling representations of the narrow active site of Pol ι (PDB ID: 3Q8P), shown on the left, and the wide active site of Pol η, shown on the right. The narrow active site of Pol ι forces a template 8‑oxo‑dG to adopt a more compact syn conformation (yellow spheres) to provide space for incoming dCTP (wheat-coloured spheres). The wide active site of Pol η accommodates DNA featuring a bulky cis-syn thymine dimer (yellow spheres), as well as an incoming dAMPNPP (2ʹ‑deoxyadenosine‑5ʹ-(α,β-imido)triphosphate, wheat-coloured spheres). HIV‑1 reverse transcriptase156, Bacillus stearothermophilus Pol I large fragment161 and even telomerase162. Once incor‑ porated, most of the lesions can be repaired by the BER mechanism, but the sheer abundance of the lesions means that some will persist into the S phase. As 8‑oxo‑dG can use its Hoogsteen face to pair with an incorrect dATP (syn–anti, FIG. 6a) in addition to the W–C‑like match with dCTP (anti–anti)163, it can generate dG to dT transver‑ sions161 and lead to cancers. Recent studies have elucidated the reasons for the variation in the response of different polymerases to the 8‑oxo‑dG lesion. Pol β has been shown to insert 8‑oxo-dGTP opposite to a template dA in preference to dC158. The structures of the intermediates in the Pol β‑catalysed reaction for 8‑oxo-dGTP insertion opposite to a template dA or dC have been determined using time-lapse crystallography 38. The dA–8‑oxo-dGMP mismatch forms a good anti–syn Hoogsteen base pair (FIG. 6b), which induces structural changes in the active site such that the mismatch, although compromising the subsequent DNA ligation process159, cannot be identified as a damage site by Pol β. Timelapse crystallography has also been used to show that the 8‑oxo‑dG–dAMP mismatch also exists in a syn–anti (Hoogsteen) conformation39 (FIG. 6c). The corresponding dC–8‑oxo-dGMP and 8‑oxo‑dG–dCMP complexes from the two studies both exist in an anti–anti conformation, as is usually adopted by matched structures. Importantly, the N subdomain was closed39,159 and the third metal ion was also observed in the complexes involving 8‑oxo‑dG or 8‑oxo-dGTP described above, although it was missing in the mismatch complexes of undamaged dNTP19. In contrast to Pol β, the Y‑family enzyme Pol ι pref‑ erentially incorporates a correct dCTP opposite to an 8‑oxo‑dG lesion, a reaction that defines the unique biological role of Pol ι in protection against oxidative stress164. In comparing the four structures of Pol ι with the 8‑oxo‑dG–dCTP match and the dATP, dGTP, or dTTP mismatch163, it was found that the exception‑ ally narrow active site of Pol ι forces the purine bases of template 8‑oxo‑dG and incoming dGTP or dATP to each adopt a syn conformation. This stereochem‑ istry and an extra hydrogen bond favour the smaller 8‑oxo‑dG–dCTP (syn–anti Hoogsteen) base pair over the 8‑oxo‑dG–dATP (syn–syn) base pair (FIG. 6d) and normal W–C (anti–anti) base pairs, leading to correct dCTP incorporation opposite to the template 8‑oxo‑dG. The origin of Pol λ fidelity in promoting error‑free bypass of 8‑oxo‑dG has recently been reported165. Seven novel crystal structures and kinetic data point to Pol λ having a flexible active site that can tolerate 8‑oxo‑dG in either the anti‑ or syn‑conformation, with discrimination against the pro‑mutagenic syn‑conformation occurring at the extension step. Lesion bypass across O6‑methylguanine. The O6‑methylguanine (O6Me‑dG) moiety is a methylated DNA lesion that is produced by various alkylating agents. When left unrepaired, O6Me‑dG causes G to A muta‑ tions, owing to Pol β pairing the methylated base with an incorrect dTTP much more frequently (~30‑fold) than with a correct dCTP166. By using the mutator Mn2+ to 10 | ARTICLE NUMBER 0068 | VOLUME 1 www.nature.com/natrevchem . d e v r e s e r s t h g i r l l A . e r u t a N r e g n i r p S f o t r a p , d e t i m i L s r e h s i l b u P n a l l i m c a M 7 1 0 2 © REVIEWS promote formation of an O6Me‑dG–dTTP mismatched ternary Pol β complex, it has been shown that Pol β adopts a catalytically competent closed conformation, with the O6Me‑dG–dTTP pair recognized as a pseudo W–C base pair 97. By contrast, the enzyme adopts an open conformation in the O6Me‑dG–dCTP ternary complex 97,167. These results provide the structural basis for the carcinogenic O6Me‑dG lesion. Lesion bypass across an abasic site. Abasic DNA sites are estimated to occur approximately 10,000 times per day in each human cell168. These are referred to as being apurinic or apyrimidinic (AP) owing to the absence of purine or pyrimidine bases, respectively. Abasic sites may arise spontaneously or can be induced by chemo­ therapeutics. The chemistry behind the deleterious effects of AP sites has been reviewed169. Referred to as the ‘A rule’, polymerases from families A and B are most likely to install a dATP substrate opposite to an abasic site170,171, which leads to the transversion mutations that are found in cancer cells172. It has been shown that the A‑family polymerase KlenTaq (a Klenow-fragment ana‑ logue of Taq polymerase) follows the A rule by using the side chain of Tyr671 to bind incoming dATP173. By con‑ trast, Pol β from the X‑family uses its lyase subdomain to remove the abasic site from DNA following incision of its 5ʹ‑phosphate, and an irreversible inhibitor of a 2‑phosphato‑1,4‑dioxobutane derivative that mimics an AP DNA lesion has been shown to shut down the lyase activity of Pol β (with a half-maximal inhibitory concentration, IC50, of ~21 μM)174. The enlarged active site of Pοl η enables bypass of the bulky T–T dimer. DNA synthesis often stalls when a rep‑ licative polymerase encounters a bulky adduct such as a cyclobutane–pyrimidine dimer (CPD)175, which forms by UV‑induced [2+2] cycloadditions between pyrimidine bases. In many species, including bacteria, fungi, plants and some mammals (marsupials and the species below), a T–T dimer can be repaired by photoactivation involving photolyase176–179. The apo structure of the catalytic core of yeast (Saccharomyces cerevisiae) Y‑family Pol η features an active site that is more open than that of replicative poly‑ merases, such that Pol η could potentially accommodate a bulky DNA lesion, for example, a T–T dimer 180. Crystal structures have been determined at four time points in the Pol-η-catalysed DNA synthesis on a substrate featuring CPDs32. The structures reveal a uniquely enlarged active site in Pol η, enabling the enzyme to accommodate the bulky T–T dimer while maintaining excellent stereochem‑ ical control during catalysis. In addition, Pol η acts as a ‘molecular splint’ to stabilize the damaged DNA. A com‑ parison between the narrow active site of Pol ι bound with an 8‑oxo‑dG lesion and the wide active site of Pol η bound with a CPD lesion is shown in FIG. 6e. A deficiency in Pol η can cause UV sensitivity and a variant of the human syndrome xeroderma pigmentosum181,182. Misincorporation of rNTP. It has long been recog‑ nized that DNA polymerases can make the mistake of processing rNTPs instead of dNTPs183,184, which is NATURE REVIEWS | CHEMISTRY unsurprising given that the former are present, on aver‑ age, at 30–200‑fold higher cellular concentrations185,186. Most polymerases use a steric gate183,184,187 that would clash with the 2ʹ‑OH group of an rNTP188. However, it has recently been demonstrated that even high-fidelity replicative polymerases, such as Pol α, Pol δ and Pol ε, incorporate more than 10,000 rNTPs into the yeast (S. cerevisiae) nuclear genome in each round of repli‑ cation186, with the estimated value being greater in the human genome131. Furthermore, the X‑family enzymes Pol β and, to a lesser extent, Pol λ can also incorpo‑ rate rNTPs opposite to normal bases or 8‑oxo-dG189. Incorporation of rNTP into DNA, if not repaired, can cause genomic instability and serious diseases. Like other forms of damage, living systems have multiple pathways to repair DNA, with RER being the primary one in this case131. DNA polymerases involved in mutagenic functions. As described above, replicative polymerases minimize mutations and achieve very high fidelity, whereas repair polymerases are expressed to fix and bypass life-threat‑ ening DNA damage and mutation. There are also some polymerases that are involved in mutagenesis, a normal life process. For example, the low-fidelity ASFV Pol X has been implicated in a mutagenic BER pathway8,71,104,190, which could be a survival mechanism for the virus under stress. Mutagenesis is an important part of our immune response. In order to produce specific antibodies to fight against diverse pathogens or other foreign sub‑ jects, B cells must first generate a diverse repertoire of B cell receptors, which involves a key randomization step called V(D)J recombination. Furthermore, activation of B cell receptors by a foreign antigen triggers somatic hypermutation, a process by which the immune system adapts to combat pathogens. Recent studies indicate that three of the X‑family polymerases, Pol λ, Pol μ and TdT, participate in V(D)J recombination, whereas two Y‑family pols, Pol η and Rev1 (and possibly additional polymerases), are involved in somatic hypermutation. Although the detailed biochemical mechanisms of the processes that involve these polymerases are still sub‑ jects of intensive study 6,191, it is intuitively clear that that these polymerases have characteristically low fidelities, as described in the preceding section, because their likely role is to synthesize mismatched DNA. Applications of DNA polymerases Polymerases have diverse and essential biological roles, but further, and more than any other class of enzymes, they have found varied applications in biotechnology and health-related industries. The most obvious appli‑ cations are in DNA amplification and manipulation, including the polymerase chain reaction (PCR) and its error-prone process. Polymerases are also useful in DNA and RNA sequencing, as well as in other emerg‑ ing applications that are described below. For brevity, we introduce only the most sophisticated applications, plac‑ ing emphasis on recent developments that may either enhance current technologies or spawn new ones. VOLUME 1 | ARTICLE NUMBER 0068 | 11 . d e v r e s e r s t h g i r l l A . e r u t a N r e g n i r p S f o t r a p , d e t i m i L s r e h s i l b u P n a l l i m c a M 7 1 0 2 © REVIEWS DNA amplification and manipulation. Many poly‑ merases have been widely used in applications such as the amplification of DNA by PCR192,193, site-directed mutagenesis, error-prone PCR, DNA sequencing, diag‑ nosis by detection of DNA or RNA with methy­lation (for example, 5ʹ‑methylated cytidine) and aptamer selection by systematic enrichment of ligands by expo‑ nential amplification194. In particular, the bacteriophage T7 polymerase has found use in the cloning of genes. Some of these many applications involve polymerases incorporating modified dNTPs into growing DNA. For example, the A‑family KlenTaq as well as B‑family KOD (Thermococcus kodakarensis polymerase) and 9°N (Thermococcus sp. 9°N-7 polymerase) can process dNTP analogues with very bulky groups; the struc‑ tural basis for their surprisingly wide substrate scope is now well known194. The high temperatures required to denature double-stranded DNA demand that PCR use only very thermally stable polymerases195. In this regard, both high-fidelity and error-prone archaeal DNA polymerases with high thermostability have been developed. The use of archaeal polymerases in biotechnology, particularly in different types of PCR, has recently been reviewed195. Recent development in the use of polymerases in DNA sequencing. Certain polymerases can faithfully copy a DNA strand; thus, it makes sense to use these enzymes to sequence genes of interest. The approach most widely adopted is the chain terminator method, which was introduced by Sanger and co-workers196,197 but has now been revised and improved in many ways. One strategy for improving signal detection and throughput makes use of reversible terminator dNTP analogues that fea‑ ture fluorophore labels198, and this technology is now being put to commercial use, for example, by Illumina Cambridge Ltd. However, dNTP bearing a bulky label may be difficult for a polymerase to process; this prob‑ lem has been addressed by the development of mutants such as the 9°N penta-mutant of D141A/E143A/L408S/ Y409A/P410V, an enzyme engineered by New England Biolabs and termed Therminator III. In this 3ʹ‑5ʹ‑exo− polymerase variant, the bulky amino acids are replaced by smaller ones, such that bulky dNTP analogues can be incorporated efficiently. The reversible terminator approach is now widely used in second-generation sequencing, but although these methods constitute tre‑ mendous improvements over the Sanger approach, they are limited in the lengths of DNA that can be read in each step. Some of these problems can be overcome using the single-molecule real-time sequencing method developed by PacBio199,200. This method, unlike second-generation sequencing, does not require a pause between read steps to deprotect the 3ʹ‑OR group or to remove the fluoro‑ phore from the base. This new method is now classified as a third-generation sequencing tool. A very different strategy is key to fourth-genera‑ tion DNA sequencing technologies such as the Oxford Nanopore Technologies Nanopore sequencers. These devices feature a nanopore, formed by the bacteriophage φ29 DNA packaging motor or other materials201, through which a single strand of DNA passes. Changes in electri‑ cal current are measured and can be related back to the sequence of bases that were drawn through the nanopore. Recently, the nanopore technology has been combined with sequencing-by‑synthesis, with the resulting nano­ pore-sequencing-by-synthesis methodology report‑ edly having a false positive background detection rate below 1.2%202. Over the past decade, the next-generation DNA sequencing technology 200 has reduced the cost of sequencing genomes by five orders of magnitude203,204; a person’s whole genome can now be sequenced for as little as US$999 (REF. 205). Engineering polymerases to synthesize DNA with unnatural base pairs. Attempts to expand the genetic alphabet involve the development of unnatural base pairs, which include nucleobase shape mimics65, hydro‑ phobic pairs66,67,206–209, metal-containing base pairs210 and base pairs with altered hydrogen-bonding motifs68,211,212. An unnatural base pair between 2‑amino‑6-(2‑thienyl) purine and pyridin‑2‑one can lead to the incorporation of unnatural amino acids into proteins213. A nucleo‑ tide analogue featuring the unnatural hydrophobic base 7-(2‑thienyl)imidazo[4,5‑b]pyridine increases the chemical and structural diversity of DNA in generating DNA aptamers. Aptamers with this base target proteins with an affinity that is two orders of magnitude greater than do aptamers that contain only natural bases214,215. Remarkably, many DNAs that contain unnatural hydro‑ phobic bases can be replicated by various polymer‑ ases216–218, such that one can even create semi-synthetic organisms216,219,220. A directed evolution approach can be used to prepare polymerase variants with desirable and novel functions, some of which we now mention221,222. Applications in mRNA diagnosis, such as pathogen detection or gene expression analysis, motivated the conversion of a DNAtemplate polymerase into an RNA-reading polymerase, which was achieved by screening thermostable KlenTaq variants in which amino acids in immediate proximity to the 2ʹ‑O of the RNA template base paired with the incoming dNTP are mutated223–225. The KlenTaq mutant transcribes RNA into DNA, and the latter can be ampli‑ fied using reverse transcription-PCR224. Similarly, a KOD1 enzyme variant has been prepared that discrim‑ inates between 2ʹ‑O‑methylated and unmethylated RNA226. Directed evolution221 also enabled engineering of a thermostable Stoffel fragment in Taq polymerase such that the resulting enzyme is an efficient RNA polymerase227. Taq polymerase variants that can rec‑ ognize and amplify C2ʹ‑modified DNA228 also exist. Evolution of the high-fidelity polymerase KOD results in it being able to use RNA templates efficiently and gives it proofreading activity for both DNA and RNA templates222. Lastly, we note that one bacteriophage T4 polymerase variant, an exonuclease-deficient L412M mutant, can replicate difficult DNA sequences. The evolved enzyme can process sequences with high GC content or tracks of mononucleotide repeats with an enhanced ability to incorporate fluorophore-labelled nucleotides229. 12 | ARTICLE NUMBER 0068 | VOLUME 1 www.nature.com/natrevchem . d e v r e s e r s t h g i r l l A . e r u t a N r e g n i r p S f o t r a p , d e t i m i L s r e h s i l b u P n a l l i m c a M 7 1 0 2 © REVIEWS Conclusion The study of DNA polymerases — the enzymes most essential for the existence and understanding of life — brings chemists and biologists together. This Review describes the molecular basis for the function of these enzymes, emphasizing that high-fidelity polymerases have well-aligned TSs for match incorporation but distorted TSs for mismatches. Fidelity is lowered when 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. 14. 15. 16. 17. 18. 19. Lehman, I. R., Bessman, M. J., Simms, E. S. & Kornberg, A. Enzymatic synthesis of deoxyribonucleic acid. I. Preparation of substrates and partial purification of an enzyme from Escherichia coli. J. Biol. Chem. 233, 163–170 (1958). Bessman, M. J., Lehman, I. R., Simms, E. S. & Kornberg, A. Enzymatic synthesis of deoxyribonucleic acid. II. General properties of the reaction. J. Biol. Chem. 233, 171–177 (1958). Lehman, I. R. et al. Enzymatic synthesis of deoxyribonucleic acid. V. Chemical composition of enzymatically synthesized deoxyribonucleic acid. Proc. Natl Acad. Sci. USA 44, 1191–1196 (1958). Hübscher, U., Spadari, S., Villani, G. & Maga, G. DNA Polymerases: Discovery, Characterization, and Functions in Cellular DNA Transactions 1st edn (World Scientific, 2010). A comprehensive reference in which detailed biochemical and functional information can be found. Jaszczur, M. et al. Mutations for worse or better: lowfidelity DNA synthesis by SOS DNA polymerase V is a tightly regulated double-edged sword. Biochemistry 55, 2309–2318 (2016). Zhao, Y. et al. Mechanism of somatic hypermutation at the WA motif by human DNA polymerase η. Proc. Natl Acad. Sci. USA 110, 8146–8151 (2013). Kunkel, T. A. DNA replication fidelity. J. Biol. Chem. 279, 16895–16898 (2004). Showalter, A. K. & Tsai, M.‑D. A. DNA polymerase with specificity for five base pairs. J. Am. Chem. Soc. 123, 1776–1777 (2001). Ahn, J., Werneburg, B. G. & Tsai, M.‑D. DNA polymerase β: structure−fidelity relationship from presteady-state kinetic analyses of all possible correct and incorrect base pairs for wild type and R283A mutant. Biochemistry 36, 1100–1107 (1997). Steitz, T. A. DNA polymerases: structural diversity and common mechanisms. J. Biol. Chem. 274, 17395–17398 (1999). Beard, W. A. & Wilson, S. H. Structure and mechanism of DNA polymerase β. Biochemistry 53, 2768–2780 (2014). Reviews the research on the structure and mechanism of Pol β up until 2014. Sawaya, M. R., Pelletier, H., Kumar, A., Wilson, S. H. & Kraut, J. Crystal structure of rat DNA polymerase β: evidence for a common polymerase mechanism. Science 264, 1930–1935 (1994). Gridley, C. L. et al. Structural changes in the hydrophobic hinge region adversely affect the activity and fidelity of the I260Q mutator DNA polymerase β. Biochemistry 52, 4422–4432 (2013). Sawaya, M. R., Prasad, R., Wilson, S. H., Kraut, J. & Pelletier, H. Crystal structures of human DNA polymerase β complexed with gapped and nicked DNA: evidence for an induced fit mechanism. Biochemistry 36, 11205–11215 (1997). Beard, W. A., Shock, D. D., Batra, V. K., Pedersen, L. C. & Wilson, S. H. DNA polymerase β substrate specificity: side chain modulation of the “A‑rule”. J. Biol. Chem. 284, 31680–31689 (2009). Freudenthal, B. D., Beard, W. A. & Wilson, S. H. Structures of dNTP intermediate states during DNA polymerase active site assembly. Structure 20, 1829–1837 (2012). Pelletier, H., Sawaya, M. R., Kumar, A., Wilson, S. H. & Kraut, J. Structures of ternary complexes of rat DNA polymerase β, a DNA template-primer and ddCTP. Science 264, 1891–1903 (1994). Batra, V. K. et al. Magnesium-induced assembly of a complete DNA polymerase catalytic complex. Structure 14, 757–766 (2006). Freudenthal, B. D., Beard, W. A., Shock, D. D. & Wilson, S. H. Observing a DNA polymerase choose right from wrong. Cell 154, 157–168 (2013). NATURE REVIEWS | CHEMISTRY 20. 21. 22. 23. 24. 25. 26. 27. 28. 29. 30. 31. 32. 33. 34. mismatched substrates are able to form well-aligned inter‑ mediates, as can occur in rationally designed or naturally evolved enzymes. In this way, most low-fidelity or muta‑ genic polymerases achieve specific biological functions, including translesion DNA synthesis and mutagenesis. These diverse properties of DNA polymerases see them amenable to various applications, which further motivates the study of these remarkable enzymes. Time-resolved crystallography enabled the first direct comparison of structural intermediates involved in the incorporation of matched and mismatched dNTP. Arndt, J. W. et al. Insight into the catalytic mechanism of DNA polymerase β: structures of intermediate complexes. Biochemistry 40, 5368–5375 (2001). The authors of this study show that it is metal B (in this case Cr(iii)dNTP) alone that, in addition to inducing rapid conformational change, also induces closure of the N subdomain. This work provides the first direct evidence against conformational closure being rate limiting. Batra, V. K., Beard, W. A., Shock, D. D., Pedersen, L. C. & Wilson, S. H. Structures of DNA polymerase β with active-site mismatches suggest a transient abasic site intermediate during misincorporation. Mol. Cell 30, 315–324 (2008). Krahn, J. M., Beard, W. A. & Wilson, S. H. Structural insights into DNA polymerase β deterrents for misincorporation support an induced-fit mechanism for fidelity. Structure 12, 1823–1832 (2004). Reed, A. J., Vyas, R., Raper, A. T. & Suo, Z. Structural insights into the post-chemistry steps of nucleotide incorporation catalyzed by a DNA polymerase. J. Am. Chem. Soc. 139, 465–471 (2017). Kim, K. H. et al. Direct observation of bond formation in solution with femtosecond X‑ray scattering. Nature 518, 385–389 (2015). Florián, J., Goodman, M. F. & Warshel, A. Computer simulations of protein functions: searching for the molecular origin of the replication fidelity of DNA polymerases. Proc. Natl Acad. Sci. USA 102, 6819–6824 (2005). Lin, P. et al. Energy analysis of chemistry for correct insertion by DNA polymerase β. Proc. Natl Acad. Sci. USA 103, 13294–13299 (2006). Wang, L., Broyde, S. & Zhang, Y. Polymerase-tailored variations in the water-mediated and substrateassisted mechanism for nucleotidyl transfer: insights from a study of T7 DNA polymerase. J. Mol. Biol. 389, 787–796 (2009). Lior-Hoffmann, L. et al. Preferred WMSA catalytic mechanism of the nucleotidyl transfer reaction in human DNA polymerase κ elucidates error-free bypass of a bulky DNA lesion. Nucleic Acids Res. 40, 9193–9205 (2012). Li, Y., Freudenthal, B. D., Beard, W. A., Wilson, S. H. & Schlick, T. Optimal and variant metal-ion routes in DNA polymerase β’s conformational pathways. J. Am. Chem. Soc. 136, 3630–3639 (2014). Nakamura, T., Zhao, Y., Yamagata, Y., Hua, Y. J. & Yang, W. Watching DNA polymerase η make a phosphodiester bond. Nature 487, 196–201 (2012). A breakthrough in studying phosphodiester bond formation was achieved with the use of a revised method of time-resolved X‑ray crystallography. Monitoring this reaction revealed the role of a third metal ion in DNA synthesis. Brautigam, C. A. & Steitz, T. A. Structural and functional insights provided by crystal structures of DNA polymerases and their substrate complexes. Curr. Opin. Struct. Biol. 8, 54–63 (1998). Biertümpfel, C. et al. Structure and mechanism of human DNA polymerase η. Nature 465, 1044–1048 (2010). Batra, V. K. et al. Amino acid substitution in the active site of DNA polymerase β explains the energy barrier of the nucleotidyl transfer reaction. J. Am. Chem. Soc. 135, 8078–8088 (2013). Yang, W., Lee, J. Y. & Nowotny, M. Making and breaking nucleic acids: two-Mg2+-ion catalysis and substrate specificity. Mol. Cell 22, 5–13 (2006). 35. Gao, Y. & Yang, W. Capture of a third Mg2+ is essential for catalyzing DNA synthesis. Science 352, 1334–1337 (2016). This work used time-resolved crystallography to observe an intermediate, possibly near-TS structure involving a third metal ion during DNA synthesis. 36. Yang, W., Weng, P. J. & Gao, Y. A new paradigm of DNA synthesis: three-metal-ion catalysis. Cell Biosci. 6, 51 (2016). 37. Genna, V., Vidossich, P., Ippoliti, E., Carloni, P. & De Vivo, M. A self-activated mechanism for nucleic acid polymerization catalyzed by DNA/RNA polymerases. J. Am. Chem. Soc. 138, 14592–14598 (2016). Analysis of crystal structures enabled identification of a common intramolecular hydrogen bond between the 3ʹ‑OH and β‑phosphate of incoming dNTP or rNTP. This interaction is proposed to assist activation of the nucleophilic 3ʹ‑OH. 38. Freudenthal, B. D. et al. Uncovering the polymeraseinduced cytotoxicity of an oxidized nucleotide. Nature 517, 635–639 (2015). Describes a time-resolved crystallographic study showing that 8‑oxo-dGTP is preferably incorporated opposite to a template dA. This reaction, which leads to mutation and cancer, is facilitated by anti–syn Hoogsteen base-pairing and the presence of a third metal ion. 39. Vyas, R., Reed, A. J., Tokarsky, E. J. & Suo, Z. Viewing human DNA polymerase β faithfully and unfaithfully bypass an oxidative lesion by time-dependent crystallography. J. Am. Chem. Soc. 137, 5225–5230 (2015). This study mirrors the study undertaken in reference 38 by showing that the preferred incorporation of dATP opposite to a 8‑oxo‑dG lesion is facilitated by 8‑oxo‑dG–dATP syn–anti Hoogsteen base-pairing as well as the third metal ion. 40. Perera, L. et al. Requirement for transient metal ions revealed through computational analysis for DNA polymerase going in reverse. Proc. Natl Acad. Sci. USA 112, E5228–E5236 (2015). 41. Perera, L., Freudenthal, B. D., Beard, W. A., Pedersen, L. G. & Wilson, S. H. Revealing the role of the product metal in DNA polymerase β catalysis. Nucleic Acids Res. 45, 2736–2745 (2017). 42. Zhong, X., Patel, S. S. & Tsai, M.‑D. DNA polymerase β. 5. Dissecting the functional roles of the two metal ions with Cr(iii)dTTP1. J. Am. Chem. Soc. 120, 235–236 (1998). 43. Zhong, X., Patel, S. S., Werneburg, B. G. & Tsai, M.‑D. DNA polymerase β: multiple conformational changes in the mechanism of catalysis. Biochemistry 36, 11891–11900 (1997). 44. Dunlap, C. A. & Tsai, M.‑D. Use of 2‑aminopurine and tryptophan fluorescence as probes in kinetic analyses of DNA polymerase β. Biochemistry 41, 11226–11235 (2002). The activation constant of Mg2+ is found to be substantially higher than that of MgdNTP; this work thus provides functional support for the additional low-affinity metal-binding site now known to be occupied by metal C. 45. Bakhtina, M. et al. Use of viscogens, dNTPαS, and rhodium(iii) as probes in stopped-flow experiments to obtain new evidence for the mechanism of catalysis by DNA polymerase β. Biochemistry 44, 5177–5187 (2005). 46. Garcia-Diaz, M., Bebenek, K., Kunkel, T. A. & Blanco, L. Identification of an intrinsic 5ʹ‑deoxyribose‑5‑phosphate lyase activity in human DNA polymerase λ: a possible role in base excision repair. J. Biol. Chem. 276, 34659–34663 (2001). VOLUME 1 | ARTICLE NUMBER 0068 | 13 . d e v r e s e r s t h g i r l l A . e r u t a N r e g n i r p S f o t r a p , d e t i m i L s r e h s i l b u P n a l l i m c a M 7 1 0 2 © REVIEWS 47. Garcia-Diaz, M., Bebenek, K., Krahn, J. M., Pedersen, L. C. & Kunkel, T. A. Role of the catalytic metal during polymerization by DNA polymerase lambda. DNA Repair 6, 1333–1340 (2007). 48. Dominguez, O. et al. DNA polymerase μ (Pol μ), homologous to TdT, could act as a DNA mutator in eukaryotic cells. EMBO J. 19, 1731–1742 (2000). 49. Moon, A. F. et al. Structural insight into the substrate specificity of DNA Polymerase μ. Nat. Struct. Mol. Biol. 14, 45–53 (2007). 50. Andrade, P., Martín, M. J., Juárez, R., López de Saro, F. & Blanco, L. Limited terminal transferase in human DNA polymerase μ defines the required balance between accuracy and efficiency in NHEJ. Proc. Natl Acad. Sci. USA 106, 16203–16208 (2009). 51. Moon, A. F. et al. Sustained active site rigidity during synthesis by human DNA polymerase μ. Nat. Struct. Mol. Biol. 21, 253–260 (2014). 52. Liu, M.‑S. et al. Structural mechanism for the fidelity modulation of DNA polymerase λ. J. Am. Chem. Soc. 138, 2389–2398 (2016). 53. Vashishtha, A. K., Wang, J. & Konigsberg, W. H. Different divalent cations alter the kinetics and fidelity of DNA polymerases. J. Biol. Chem. 291, 20869–20875 (2016). 54. Gardner, A. F., Joyce, C. M. & Jack, W. E. Comparative kinetics of nucleotide analog incorporation by vent DNA polymerase. J. Biol. Chem. 279, 11834–11842 (2004). 55. Vyas, R., Zahurancik, W. J. & Suo, Z. Structural basis for the binding and incorporation of nucleotide analogs with l‑stereochemistry by human DNA polymerase λ. Proc. Natl Acad. Sci. USA 111, E3033–E3042 (2014). 56. Arana, M. E., Potapova, O., Kunkel, T. A. & Joyce, C. M. Kinetic analysis of the unique error signature of human DNA polymerase ν. Biochemistry 50, 10126–10135 (2011). 57. Gowda, A. S. P., Moldovan, G.‑L. & Spratt, T. E. Human DNA polymerase ν catalyzes correct and incorrect DNA synthesis with high catalytic efficiency. J. Biol. Chem. 290, 16292–16303 (2015). 58. Lee, Y.‑S., Gao, Y. & Yang, W. How a homolog of highfidelity replicases conducts mutagenic DNA synthesis. Nat. Struct. Mol. Biol. 22, 298–303 (2015). 59. Arana, M. E., Seki, M., Wood, R. D., Rogozin, I. B. & Kunkel, T. A. Low-fidelity DNA synthesis by human DNA polymerase theta. Nucleic Acids Res. 36, 3847–3856 (2008). 60. Takata, K.‑i., Shimizu, T., Iwai, S. & Wood, R. D. Human DNA polymerase N (POLN) is a low fidelity enzyme capable of error-free bypass of 5S‑thymine glycol. J. Biol. Chem. 281, 23445–23455 (2006). 61. Wood, R. D. & Doublié, S. DNA polymerase θ (POLQ), double-strand break repair, and cancer. DNA Repair 44, 22–32 (2016). 62. Loeb, L. A. & Monnat, R. J. DNA polymerases and human disease. Nat. Rev. Genet. 9, 594–604 (2008). 63. Tsai, M.‑D. How DNA polymerases catalyze DNA replication, repair, and mutation. Biochemistry 53, 2749–2751 (2014). 64. Lee, H. R., Helquist, S. A., Kool, E. T. & Johnson, K. A. Importance of hydrogen bonding for efficiency and specificity of the human mitochondrial DNA polymerase. J. Biol. Chem. 283, 14402–14410 (2008). 65. Winnacker, M. & Kool, E. T. Artificial genetic sets composed of size-expanded base pairs. Angew. Chem. Int. Ed. 52, 12498–12508 (2013). 66. Chen, T., Hongdilokkul, N., Liu, Z., Thirunavukarasu, D. & Romesberg, F. E. The expanding world of DNA and RNA. Curr. Opin. Chem. Biol. 34, 80–87 (2016). 67. Hirao, I. & Kimoto, M. Unnatural base pair systems toward the expansion of the genetic alphabet in the central dogma. Proc. Jpn Acad. Ser. B 88, 345–367 (2012). 68. Benner, S. A. et al. Alternative Watson–Crick synthetic genetic systems. Cold Spring Harb. Perspect. Biol. http://dx.doi.org/10.1101/cshperspect.a023770 (2016). 69. Oertell, K. et al. Kinetic selection versus free energy of DNA base pairing in control of polymerase fidelity. Proc. Natl Acad. Sci. USA 113, E2277–E2285 (2016). 70. Showalter, A. K. & Tsai, M.‑D. A reexamination of the nucleotide incorporation fidelity of DNA polymerases. Biochemistry 41, 10571–10576 (2002). The authors of this article posit, for the first time, that the main step that controls the fidelity of DNA polymerase catalysis should be the chemical step. 71. Showalter, A. K. et al. Mechanistic comparison of highfidelity and error-prone DNA polymerases and ligases involved in DNA repair. Chem. Rev. 106, 340–360 (2006). 72. Bakhtina, M., Roettger, M. P., Kumar, S. & Tsai, M.‑D. A unified kinetic mechanism applicable to multiple DNA polymerases. Biochemistry 46, 5463–5472 (2007). 73. Sucato, C. A. et al. Modifying the β, γ leaving-group bridging oxygen alters nucleotide incorporation efficiency, fidelity, and the catalytic mechanism of DNA polymerase β. Biochemistry 46, 461–471 (2007). 74. Sucato, C. A. et al. DNA polymerase β fidelity: halomethylene-modified leaving groups in pre-steadystate kinetic analysis reveal differences at the chemical transition state. Biochemistry 47, 870–879 (2008). 75. Bakhtina, M., Roettger, M. P. & Tsai, M.‑D. Contribution of the reverse rate of the conformational step to polymerase β fidelity. Biochemistry 48, 3197–3208 (2009). 76. Balbo, P. B., Wang, E. C.‑W. & Tsai, M.‑D. Kinetic mechanism of active site assembly and chemical catalysis of DNA polymerase β. Biochemistry 50, 9865–9875 (2011). 77. Oertell, K. et al. Transition state in DNA polymerase β catalysis: rate-limiting chemistry altered by base-pair configuration. Biochemistry 53, 1842–1848 (2014). 78. Kellinger, M. W. & Johnson, K. A. Nucleotidedependent conformational change governs specificity and analog discrimination by HIV reverse transcriptase. Proc. Natl Acad. Sci. USA 107, 7734–7739 (2010). 79. Fiala, K. A. & Suo, Z. Mechanism of DNA polymerization catalyzed by Sulfolobus solfataricus P2 DNA polymerase IV. Biochemistry 43, 2116–2125 (2004). 80. Rothwell, P. J., Mitaksov, V. & Waksman, G. Motions of the fingers subdomain of Klentaq1 are fast and not rate limiting: implications for the molecular basis of fidelity in DNA polymerases. Mol. Cell 19, 345–355 (2005). 81. Johnson, K. A. The kinetic and chemical mechanism of high-fidelity DNA polymerases. Biochim. Biophys. Acta 1804, 1041–1048 (2010). 82. Watson, J. D. & Crick, F. H. C. Genetical implications of the structure of deoxyribonucleic acid. Nature 171, 964–967 (1953). 83. Watson, J. D. & Crick, F. H. C. Molecular structure of nucleic acids: a structure for deoxyribose nucleic acid. Nature 171, 737–738 (1953). 84. Wang, W., Hellinga, H. W. & Beese, L. S. Structural evidence for the rare tautomer hypothesis of spontaneous mutagenesis. Proc. Natl Acad. Sci. USA 108, 17644–17648 (2011). 85. Yu, H., Eritja, R., Bloom, L. B. & Goodman, M. F. Ionization of bromouracil and fluorouracil stimulates base mispairing frequencies with guanine. J. Biol. Chem. 268, 15935–15943 (1993). 86. Bebenek, K., Pedersen, L. C. & Kunkel, T. A. Replication infidelity via a mismatch with Watson–Crick geometry. Proc. Natl Acad. Sci. USA 108, 1862–1867 (2011). 87. Nikolova, E. N. et al. Transient Hoogsteen base pairs in canonical duplex DNA. Nature 470, 498–502 (2011). 88. Alvey, H. S., Gottardo, F. L., Nikolova, E. N. & Al‑Hashimi, H. M. Widespread transient Hoogsteen base pairs in canonical duplex DNA with variable energetics. Nat. Commum. 5, 4786 (2014). 89. Zhou, H. et al. New insights into Hoogsteen base pairs in DNA duplexes from a structure-based survey. Nucleic Acids Res. 43, 3420–3433 (2015). 90. Kimsey, I. J., Petzold, K., Sathyamoorthy, B., Stein, Z. W. & Al‑Hashimi, H. M. Visualizing transient Watson–Crick-like mispairs in DNA and RNA duplexes. Nature 519, 315–320 (2015). 91. Sirover, M. & Loeb, L. Infidelity of DNA synthesis in vitro: screening for potential metal mutagens or carcinogens. Science 194, 1434–1436 (1976). 92. Weymouth, L. A. & Loeb, L. A. Mutagenesis during in vitro DNA synthesis. Proc. Natl Acad. Sci. USA 75, 1924–1928 (1978). 93. Werneburg, B. G. et al. DNA polymerase β: pre-steadystate kinetic analysis and roles of arginine‑283 in catalysis and fidelity. Biochemistry 35, 7041–7050 (1996). 94. Vaisman, A., Ling, H., Woodgate, R. & Yang, W. Fidelity of Dpo4: effect of metal ions, nucleotide selection and pyrophosphorolysis. EMBO J. 24, 2957–2967 (2005). 95. Frank, E. G. & Woodgate, R. Increased catalytic activity and altered fidelity of human DNA polymerase ι in the presence of manganese. J. Biol. Chem. 282, 24689–24696 (2007). 96. Bebenek, K. et al. Substrate-induced DNA strand misalignment during catalytic cycling by DNA polymerase λ. EMBO Rep. 9, 459–464 (2008). 97. Koag, M.‑C. & Lee, S. Metal-dependent conformational activation explains highly promutagenic replication across O6‑methylguanine by human DNA polymerase β. J. Am. Chem. Soc. 136, 5709–5721 (2014). 98. Choi, J.‑Y. et al. Kinetic and structural impact of metal ions and genetic variations on human DNA polymerase ι. J. Biol. Chem. 291, 21063–21073 (2016). 99. Vashishtha, A. K. & Konigsberg, W. H. Effect of different divalent cations on the kinetics and fidelity of RB69 DNA polymerase. Biochemistry 55, 2661–2670 (2016). 100. Xia, S. & Konigsberg, W. H. RB69 DNA polymerase structure, kinetics, and fidelity. Biochemistry 53, 2752–2767 (2014). 101. Xia, S., Wang, J. & Konigsberg, W. H. DNA mismatch synthesis complexes provide insights into base selectivity of a B family DNA polymerase. J. Am. Chem. Soc. 135, 193–202 (2013). 102. Oliveros, M. et al. Characterization of an African swine fever virus 20‑kDa DNA polymerase involved in DNA repair. J. Biol. Chem. 272, 30899–30910 (1997). 103. Maciejewski, M. W. et al. Solution structure of a viral DNA repair polymerase. Nat. Struct. Mol. Biol. 8, 936–941 (2001). 104. Showalter, A. K., Byeon, I.‑J. L., Su, M.‑I. & Tsai, M.‑D. Solution structure of a viral DNA polymerase X and evidence for a mutagenic function. Nat. Struct. Biol. 8, 942–946 (2001). 105. Kumar, S., Bakhtina, M. & Tsai, M.‑D. Altered order of substrate binding by DNA polymerase X from African swine fever virus. Biochemistry 47, 7875–7887 (2008). 106. Wu, W.‑J. et al. How a low-fidelity DNA polymerase chooses non-Watson–Crick from Watson–Crick incorporation. J. Am. Chem. Soc. 136, 4927–4937 (2014). This study uses NMR structural determination to demonstrate how a mutagenic DNA polymerase achieves its low fidelity by overcoming the forces that govern Watson–Crick base-pairing. 107. Chen, Y. et al. Unique 5ʹ‑P recognition and basis for dG:dGTP misincorporation of ASFV DNA polymerase X. PLoS Biol. 15, http://dx.doi.org/10.1371/journal. pbio.1002599 (2017). 108. García-Escudero, R., García-Díaz, M., Salas, M. L., Blanco, L. & Salas, J. DNA polymerase X of African swine fever virus: insertion fidelity on gapped DNA substrates and AP lyase activity support a role in base excision repair of viral DNA. J. Mol. Biol. 326, 1403–1412 (2003). 109. Bebenek, K., Pedersen, L. C. & Kunkel, T. A. Structure– function studies of DNA polymerase λ. Biochemistry 53, 2781–2792 (2014). 110. Nelson, J. R., Lawrence, C. W. & Hinkle, D. C. Deoxycytidyl transferase activity of yeast Rev1 protein. Nature 382, 729–731 (1996). 111. Haracska, L., Prakash, S. & Prakash, L. Yeast Rev1 protein is a G template-specific DNA polymerase. J. Biol. Chem. 277, 15546–15551 (2002). 112. Nair, D. T., Johnson, R. E., Prakash, L., Prakash, S. & Aggarwal, A. K. Rev1 employs a novel mechanism of DNA synthesis using a protein template. Science 309, 2219–2222 (2005). 113. Fiala, K. A., Abdel-Gawad, W. & Suo, Z. Pre-steadystate kinetic studies of the fidelity and mechanism of polymerization catalyzed by truncated human DNA polymerase λ. Biochemistry 43, 6751–6762 (2004). 114. Ahn, J., Kraynov, V. S., Zhong, X., Werneburg, B. G. & Tsai, M.‑D. DNA polymerase β: effects of gapped DNA substrates on dNTP specificity, fidelity, processivity and conformational changes. Biochem. J. 331, 79–87 (1998). 115. Johnson, K. A. Conformational coupling in DNA polymerase fidelity. Annu. Rev. Biochem. 62, 685–713 (1993). 116. Johnson, S. J. & Beese, L. S. Structures of mismatch replication errors observed in a DNA polymerase. Cell 116, 803–816 (2004). 117. Tang, K.‑H. et al. Mismatched dNTP incorporation by DNA polymerase β does not proceed via globally different conformational pathways. Nucleic Acids Res. 36, 2948–2957 (2008). 118. Moscato, B., Swain, M. & Loria, J. P. Induced fit in the selection of correct versus incorrect nucleotides by DNA polymerase β. Biochemistry 55, 382–395 (2016). 14 | ARTICLE NUMBER 0068 | VOLUME 1 www.nature.com/natrevchem . d e v r e s e r s t h g i r l l A . e r u t a N r e g n i r p S f o t r a p , d e t i m i L s r e h s i l b u P n a l l i m c a M 7 1 0 2 © REVIEWS 119. Roettger, M. P., Bakhtina, M. & Tsai, M.‑D. Mismatched and matched dNTP incorporation by DNA polymerase β proceed via analogous kinetic pathways. Biochemistry 47, 9718–9727 (2008). 120. Santoso, Y. et al. Conformational transitions in DNA polymerase I revealed by single-molecule FRET. Proc. Natl Acad. Sci. USA 107, 715–720 (2010). 121. Rothwell, P. J. et al. dNTP-dependent conformational transitions in the fingers subdomain of Klentaq1 DNA polymerase: insights into the role of the “nucleotidebinding” state. J. Biol. Chem. 288, 13575–13591 (2013). 122. Delarue, M. et al. Crystal structures of a templateindependent DNA polymerase: murine terminal deoxynucleotidyltransferase. EMBO J. 21, 427–439 (2002). 123. Gouge, J., Rosario, S., Romain, F., Beguin, P. & Delarue, M. Structures of intermediates along the catalytic cycle of terminal deoxynucleotidyltransferase: dynamical aspects of the two-metal ion mechanism. J. Mol. Biol. 425, 4334–4352 (2013). 124. Ummat, A. et al. Human DNA polymerase η is prealigned for dNTP binding and catalysis. J. Mol. Biol. 415, 627–634 (2012). 125. Starcevic, D., Dalal, S. & Sweasy, J. Hinge residue Ile260 of DNA polymerase β is important for enzyme activity and fidelity. Biochemistry 44, 3775–3784 (2005). 126. Yamtich, J. & Sweasy, J. B. DNA polymerase family X: function, structure, and cellular roles. Biochim. Biophys. Acta 1804, 1136–1150 (2010). 127. Lindahl, T. & Barnes, D. E. Repair of endogenous DNA damage. Cold Spring Harbor Symp. Quant. Biol. 65, 127–134 (2000). 128. Tubbs, A. & Nussenzweig, A. Endogenous DNA damage as a source of genomic instability in cancer. Cell 168, 644–656 (2017). 129. Yang, W. An overview of Y‑family DNA polymerases and a case study of human DNA polymerase η. Biochemistry 53, 2793–2803 (2014). 130. Maxwell, B. A. & Suo, Z. Recent insight into the kinetic mechanisms and conformational dynamics of Y‑family DNA polymerases. Biochemistry 53, 2804–2814 (2014). 131. Williams, J. S., Lujan, S. A. & Kunkel, T. A. Processing ribonucleotides incorporated during eukaryotic DNA replication. Nat. Rev. Mol. Cell. Biol. 17, 350–363 (2016). 132. Fang, E. F. et al. Nuclear DNA damage signalling to mitochondria in ageing. Nat. Rev. Mol. Cell. Biol. 17, 308–321 (2016). 133. Marteijn, J. A., Lans, H., Vermeulen, W. & Hoeijmakers, J. H. J. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell. Biol. 15, 465–481 (2014). 134. Kunkel, T. A. & Soni, A. Exonucleolytic proofreading enhances the fidelity of DNA synthesis by chick embryo DNA polymerase‑γ. J. Biol. Chem. 263, 4450–4459 (1988). 135. Longley, M. J., Nguyen, D., Kunkel, T. A. & Copeland, W. C. The fidelity of human DNA polymerase γ with and without exonucleolytic proofreading and the p55 accessory subunit. J. Biol. Chem. 276, 38555–38562 (2001). 136. Yousefzadeh, M. J. et al. Mechanism of suppression of chromosomal instability by DNA polymerase POLQ. PLoS Genet. 10, e1004654 (2014). 137. Zahn, K. E., Averill, A. M., Aller, P., Wood, R. D. & Doublié, S. Human DNA polymerase θ grasps the primer terminus to mediate DNA repair. Nat. Struct. Mol. Biol. 22, 304–311 (2015). 138. Mateos-Gomez, P. A. et al. Mammalian polymerase θ promotes alternative NHEJ and suppresses recombination. Nature 518, 254–257 (2015). 139. Yang, W. & Lee, Y.‑S. A. DNA-hairpin model for repeataddition processivity in telomere synthesis. Nat. Struct. Mol. Biol. 22, 844–847 (2015). 140. Hedglin, M., Pandey, B. & Benkovic, S. J. Stability of the human polymerase δ holoenzyme and its implications in lagging strand DNA synthesis. Proc. Natl Acad. Sci. USA 113, E1777–E1786 (2016). 141. Hedglin, M., Pandey, B. & Benkovic, S. J. Characterization of human translesion DNA synthesis across a UV‑induced DNA lesion. eLife 5, e19788 (2016). 142. Dilley, R. L. et al. Break-induced telomere synthesis underlies alternative telomere maintenance. Nature 539, 54–58 (2016). 143. Johnson, R. E., Klassen, R., Prakash, L. & Prakash, S. A major role of DNA polymerase δ in replication of NATURE REVIEWS | CHEMISTRY both the leading and lagging DNA strands. Mol. Cell 59, 163–175 (2015). 144. Burgers, P. M. J., Gordenin, D. & Kunkel, T. A. Who is leading the replication fork, Pol ε or Pol δ? Mol. Cell 61, 492–493 (2016). 145. Johnson, R. E., Klassen, R., Prakash, L. & Prakash, S. Response to Burgers et. al. Mol. Cell 61, 494–495 (2016). 146. Moon, A. F. et al. The X family portrait: structural insights into biological functions of X family polymerases. DNA Repair 6, 1709–1725 (2007). 147. García-Díaz, M. et al. DNA polymerase λ, a novel DNA repair enzyme in human cells. J. Biol. Chem. 277, 13184–13191 (2002). 148. García-Díaz, M., Bebenek, K., Krahn, J. M., Kunkel, T. A. & Pedersen, L. C. A closed conformation for the Pol λ catalytic cycle. Nat. Struct. Mol. Biol. 12, 97–98 (2005). 149. Wei, Y. Portraits of a Y‑family DNA polymerase. FEBS Lett. 579, 868–872 (2005). 150. Prakash, S., Johnson, R. E. & Prakash, L. Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu. Rev. Biochem. 74, 317–353 (2005). 151. Nugent, C. I. & Lundblad, V. The telomerase reverse transcriptase: components and regulation. Genes Dev. 12, 1073–1085 (1998). 152. Cong, Y.‑S., Wright, W. E. & Shay, J. W. Human telomerase and its regulation. Microbiol. Mol. Biol. Rev. 66, 407–425 (2002). 153. García-Gómez, S. et al. PrimPol, an archaic primase/ polymerase operating in human cells. Mol. Cell 52, 541–553 (2013). 154. Bianchi, J. et al. PrimPol bypasses UV photoproducts during eukaryotic chromosomal DNA replication. Mol. Cell 52, 566–573 (2013). 155. Guilliam, T. A. et al. Human PrimPol is a highly errorprone polymerase regulated by single-stranded DNA binding proteins. Nucleic Acids Res. 43, 1056–1068 (2015). 156. Kamath-Loeb, A. S., Hizi, A., Kasai, H. & Loeb, L. A. Incorporation of the guanosine triphosphate analogs 8‑oxo-dGTP and 8‑NH2-dGTP by reverse transcriptases and mammalian DNA polymerases. J. Biol. Chem. 272, 5892–5898 (1997). 157. Pursell, Z. F., McDonald, J. T., Mathews, C. K. & Kunkel, T. A. Trace amounts of 8‑oxo-dGTP in mitochondrial dNTP pools reduce DNA polymerase γ replication fidelity. Nucleic Acids Res. 36, 2174–2181 (2008). 158. Batra, V. K. et al. Mutagenic conformation of 8‑oxo‑7,8‑dihydro‑2ʹ‑dGTP in the confines of a DNA polymerase active site. Nat. Struct. Mol. Biol. 17, 889–890 (2010). 159. Çağlayan, M., Horton, J. K., Dai, D.‑P., Stefanick, D. F. & Wilson, S. H. Oxidized nucleotide insertion by pol β confounds ligation during base excision repair. Nat. Commun. 8, 14045 (2017). 160. Burak, M. J., Guja, K. E. & Garcia-Diaz, M. Nucleotide binding interactions modulate dNTP selectivity and facilitate 8‑oxo-dGTP incorporation by DNA polymerase lambda. Nucleic Acids Res. 43, 8089–8099 (2015). 161. Hsu, G. W., Ober, M., Carell, T. & Beese, L. S. Errorprone replication of oxidatively damaged DNA by a high-fidelity DNA polymerase. Nature 431, 217–221 (2004). 162. Fouquerel, E. et al. Oxidative guanine base damage regulates human telomerase activity. Nat. Struct. Mol. Biol. 23, 1092–1100 (2016). 163. Kirouac, K. N. & Ling, H. Unique active site promotes error-free replication opposite an 8‑oxo-guanine lesion by human DNA polymerase iota. Proc. Natl Acad. Sci. USA 108, 3210–3215 (2011). 164. Petta, T. B. et al. Human DNA polymerase iota protects cells against oxidative stress. EMBO J. 27, 2883–2895 (2008). 165. Burak, M. J., Guja, K. E., Hambardjieva, E., Derkunt, B. & Garcia-Diaz, M. A fidelity mechanism in DNA polymerase lambda promotes error-free bypass of 8‑oxo-dG. EMBO J. 35, 2045–2059 (2016). 166. Reha-Krantz, L. J., Nonay, R. L., Day, R. S. III & Wilson, S. H. Replication of O6-methylguaninecontaining DNA by repair and replicative DNA polymerases. J. Biol. Chem. 271, 20088–20095 (1996). 167. Koag, M.‑C., Nam, K. & Lee, S. The spontaneous replication error and the mismatch discrimination mechanisms of human DNA polymerase β. Nucleic Acids Res. 42, 11233–11245 (2014). 168. Lindahl, T. Instability and decay of the primary structure of DNA. Nature 362, 709–715 (1993). 169. Greenberg, M. M. Looking beneath the surface to determine what makes DNA damage deleterious. Curr. Opin. Chem. Biol. 21, 48–55 (2014). 170. Schaaper, R. M., Kunkel, T. A. & Loeb, L. A. Infidelity of DNA synthesis associated with bypass of apurinic sites. Proc. Natl Acad. Sci. USA 80, 487–491 (1983). 171. Sagher, D. & Strauss, B. Insertion of nucleotides opposite apurinic apyrimidinic sites in deoxyribonucleic acid during in vitro synthesis: uniqueness of adenine nucleotides. Biochemistry 22, 4518–4526 (1983). 172. Hoeijmakers, J. H. Genome maintenance mechanisms for preventing cancer. Nature 411, 366–374 (2001). 173. Obeid, S. et al. Replication through an abasic DNA lesion: structural basis for adenine selectivity. EMBO J. 29, 1738–1747 (2010). 174. Arian, D. et al. Irreversible inhibition of DNA polymerase β by small-molecule mimics of a DNA lesion. J. Am. Chem. Soc. 136, 3176–3183 (2014). 175. Setlow, R. B. Cyclobutane-type pyrimidine dimers in polynucleotides. Science 153, 379–386 (1966). 176. Todo, T. et al. A new photoreactivating enzyme that specifically repairs ultraviolet light-induced (6–4) photoproducts. Nature 361, 371–374 (1993). 177. Essen, L. O. & Klar, T. Light-driven DNA repair by photolyases. Cell. Mol. Life Sci. 63, 1266–1277 (2006). 178. Tan, C. et al. The molecular origin of high DNA-repair efficiency by photolyase. Nat. Comm. 6, 7302 (2015). 179. Faraji, S. & Dreuw, A. Insights into light-driven DNA repair by photolyases: challenges and opportunities for electronic structure theory. Photochem. Photobiol. 93, 37–50 (2017). 180. Trincao, J. et al. Structure of the catalytic core of S. cerevisiae DNA polymerase η: implications for translesion DNA synthesis. Mol. Cell 8, 417–426 (2001). 181. Masutani, C. et al. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase η. Nature 399, 700–704 (1999). 182. Johnson, R. E., Kondratick, C. M., Prakash, S. & Prakash, L. hRAD30 Mutations in the variant form of xeroderma pigmentosum. Science 285, 263–265 (1999). 183. Joyce, C. M. Choosing the right sugar: how polymerases select a nucleotide substrate. Proc. Natl Acad. Sci. USA 94, 1619–1622 (1997). 184. Astatke, M., Ng, K., Grindley, N. D. F. & Joyce, C. M. A single side chain prevents Escherichia coli DNA polymerase I (Klenow fragment) from incorporating ribonucleotides. Proc. Natl Acad. Sci. USA 95, 3402–3407 (1998). 185. Traut, T. W. Physiological concentrations of purines and pyrimidines. Mol. Cell. Biochem. 140, 1–22 (1994). 186. Nick McElhinny, S. A. et al. Abundant ribonucleotide incorporation into DNA by yeast replicative polymerases. Proc. Natl Acad. Sci. USA 107, 4949–4954 (2010). 187. Donigan, K. A., McLenigan, M. P., Yang, W., Goodman, M. F. & Woodgate, R. The steric gate of DNA polymerase ι regulates ribonucleotide incorporation and deoxyribonucleotide fidelity. J. Biol. Chem. 289, 9136–9145 (2014). 188. Brown, J. A. & Suo, Z. Unlocking the sugar “steric gate” of DNA polymerases. Biochemistry 50, 1135–1142 (2011). 189. Crespan, E. et al. Impact of ribonucleotide incorporation by DNA polymerases β and λ on oxidative base excision repair. Nat. Commun. 7, 10805 (2016). 190. Lamarche, B. J., Showalter, A. K. & Tsai, M.‑D. An error-prone viral DNA ligase. Biochemistry 44, 8408–8417 (2005). 191. Zanotti, K. J. & Gearhart, P. J. Antibody diversification caused by disrupted mismatch repair and promiscuous DNA polymerases. DNA Repair 38, 110–116 (2016). 192. Bartlett, J. M. S. & Stirling, D. in PCR Protocols (eds Bartlett, J. M. S. & Stirling, D.) 3–6 (Humana, 2003). 193. Mullis, K. B. et al. Process for amplifying, detecting, and/or‑cloning nucleic acid sequences. US Patent 4683195 A (1986). 194. Hottin, A. & Marx, A. Structural insights into the processing of nucleobase-modified nucleotides by DNA polymerases. Acc. Chem. Res. 49, 418–427 (2016). 195. Zhang, L., Kang, M., Xu, J. & Huang, Y. Archaeal DNA polymerases in biotechnology. Appl. Microbiol. Biotechnol. 99, 6585–6597 (2015). VOLUME 1 | ARTICLE NUMBER 0068 | 15 . d e v r e s e r s t h g i r l l A . e r u t a N r e g n i r p S f o t r a p , d e t i m i L s r e h s i l b u P n a l l i m c a M 7 1 0 2 © REVIEWS 196. Sanger, F. & Coulson, A. R. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J. Mol. Biol. 94, 441–448 (1975). 197. Sanger, F., Nicklen, S. & Coulson, A. R. DNA sequencing with chain-terminating inhibitors. Proc. Natl Acad. Sci. USA 74, 5463–5467 (1977). 198. Bentley, D. R. et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456, 53–59 (2008). 199. Rhoads, A. & Au, K. F. PacBio sequencing and its applications. Genomics Proteomics Bioinformatics 13, 278–289 (2015). 200. Goodwin, S., McPherson, J. D. & McCombie, W. R. Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet. 17, 333–351 (2016). A useful Review on next-generation sequencing technology. 201. Deamer, D., Akeson, M. & Branton, D. Three decades of nanopore sequencing. Nat. Biotechnol. 34, 518–524 (2016). 202. Stranges, P. B. et al. Design and characterization of a nanopore-coupled polymerase for single-molecule DNA sequencing by synthesis on an electrode array. Proc. Natl Acad. Sci. USA 113, E6749–E6756 (2016). 203. Lander, E. S. Initial impact of the sequencing of the human genome. Nature 470, 187–197 (2011). 204. Soon, W. W., Hariharan, M. & Snyder, M. P. Highthroughput sequencing for biology and medicine. Mol. Syst. Biol. 9, 640 (2013). 205. Regalado, A. For $999, Veritas Genetics will put your genome on a smartphone app. MIT Technology Review https://www.technologyreview.com/s/600950/for-999veritas-genetics-will-put-your-genome-on-asmartphone-app/ (2016). 206. Malyshev, D. A. & Romesberg, F. E. The expanded genetic alphabet. Angew. Chem. Int. Ed. 54, 11930–11944 (2015). 207. McMinn, D. L. et al. Efforts toward expansion of the genetic alphabet: DNA polymerase recognition of a highly stable, self-pairing hydrophobic base. J. Am. Chem. Soc. 121, 11585–11586 (1999). 208. Matsuda, S., Henry, A. A. & Romesberg, F. E. Optimization of unnatural base pair packing for polymerase recognition. J. Am. Chem. Soc. 128, 6369–6375 (2006). 209. Hirao, I. Unnatural base pair systems for DNA/RNAbased biotechnology. Curr. Opin. Chem. Biol. 10, 622–627 (2006). 210. Clever, G. H., Kaul, C. & Carell, T. DNA–metal base pairs. Angew. Chem. Int. Ed. 46, 6226–6236 (2007). 211. Zhang, L. et al. Evolution of functional six-nucleotide DNA. J. Am. Chem. Soc. 137, 6734–6737 (2015). 212. Georgiadis, M. M. et al. Structural basis for a six nucleotide genetic alphabet. J. Am. Chem. Soc. 137, 6947–6955 (2015). 213. Hirao, I. et al. An unnatural base pair for incorporating amino acid analogs into proteins. Nat. Biotechnol. 20, 177–182 (2002). 214. Matsunaga, K.‑i., Kimoto, M. & Hirao, I. High-affinity DNA aptamer generation targeting von Willebrand factor A1‑domain by genetic alphabet expansion for systematic evolution of ligands by exponential enrichment using two types of libraries composed of five different bases. J. Am. Chem. Soc. 139, 324–334 (2017). 215. Kimoto, M., Yamashige, R., Matsunaga, K.‑i., Yokoyama, S. & Hirao, I. Generation of high-affinity DNA aptamers using an expanded genetic alphabet. Nat. Biotechnol. 31, 453–457 (2013). 216. Malyshev, D. A. et al. Efficient and sequenceindependent replication of DNA containing a third base pair establishes a functional six-letter genetic alphabet. Proc. Natl Acad. Sci. USA 109, 12005–12010 (2012). 217. Li, L. et al. Natural-like replication of an unnatural base pair for the expansion of the genetic alphabet and biotechnology applications. J. Am. Chem. Soc. 136, 826–829 (2014). 218. Delaney, J. C. et al. Efficient replication bypass of size-expanded DNA base pairs in bacterial cells. Angew. Chem. Int. Ed. 48, 4524–4527 (2009). 219. Malyshev, D. A. et al. A semi-synthetic organism with an expanded genetic alphabet. Nature 509, 385–388 (2014). A semi-synthetic organism can be constructed by engineering DNA polymerases and other proteins, and exposing these to unnatural dNTP substrates. 220. Zhang, Y. et al. A semisynthetic organism engineered for the stable expansion of the genetic alphabet. Proc. Natl Acad. Sci. USA 114, 1317–1322 (2017). 221. Packer, M. S. & Liu, D. R. Methods for the directed evolution of proteins. Nat. Rev. Genet. 16, 379–394 (2015). 222. Ellefson, J. W. et al. Synthetic evolutionary origin of a proofreading reverse transcriptase. Science 352, 1590–1593 (2016). 223. Sauter, K. B. M. & Marx, A. Evolving thermostable reverse transcriptase activity in a DNA polymerase scaffold. Angew. Chem. Int. Ed. 45, 7633–7635 (2006). 224. Blatter, N. et al. Structure and function of an RNAreading thermostable DNA polymerase. Angew. Chem. Int. Ed. 52, 11935–11939 (2013). 225. Aschenbrenner, J. & Marx, A. Direct and sitespecific quantification of RNA 2ʹ‑O-methylation by PCR with an engineered DNA polymerase. Nucleic Acids Res. 44, 3495–3502 (2016). 226. Huber, C., von Watzdorf, J. & Marx, A. 5‑Methylcytosine-sensitive variants of Thermococcus kodakaraensis DNA polymerase. Nucleic Acids Res. 44, 9881–9890 (2016). 227. Xia, G. et al. Directed evolution of novel polymerase activities: mutation of a DNA polymerase into an efficient RNA polymerase. Proc. Natl Acad. Sci. USA 99, 6597–6602 (2002). 228. Chen, T. et al. Evolution of thermophilic DNA polymerases for the recognition and amplification of C2ʹ‑modified DNA. Nat. Chem. 8, 556–562 (2016). The authors demonstrate that a polymerase evolution system can produce thermostable enzymes that efficiently interconvert C2ʹ‑OMe-modified oligonucleotides and their DNA counterparts through transcription and reverse transcription. This seems to be a powerful tool for tailoring polymerases to have other types of novel functions. 229. Reha-Krantz, L. J., Woodgate, S. & Goodman, M. F. Engineering processive DNA polymerases with maximum benefit at minimum cost. Front. Microbiol. 5, 380 (2014). 230. Wong, I., Patel, S. S. & Johnson, K. A. An induced-fit kinetic mechanism for DNA replication fidelity: direct measurement by single-turnover kinetics. Biochemistry 30, 526–537 (1991). 231. Johnson, K. A. 1 Transient-state kinetic analysis of enzyme reaction pathways. Enzymes 20, 1–61 (1992). 232. Fersht, A. Enzyme Structure and Mechanism 2nd edn, 350 (W. H. Freeman, 1985). 233. Bertram, J. G., Oertell, K., Petruska, J. & Goodman, M. F. DNA polymerase fidelity: comparing direct competition of right and wrong dNTP substrates with steady state and pre-steady state kinetics. Biochemistry 49, 20–28 (2010). 234. Kohlstaedt, L. A., Wang, J., Friedman, J. M., Rice, P. A. & Steitz, T. A. Crystal structure at 3.5 Å resolution of HIV‑1 reverse transcriptase complexed with an inhibitor. Science 256, 1783–1790 (1992). 235. Steitz, T. A., Smerdon, S. J., Jäger, J. & Joyce, C. M. A unified polymerase mechanism for nonhomologous DNA and RNA polymerase. Science 266, 2022–2025 (1994). 236. Knowles, J. R. & Albery, W. J. Perfection in enzyme catalysis: the energetics of triosephosphate isomerase. Acc. Chem. Res. 10, 105–111 (1977). 237. Kravchuk, A. V., Zhao, L., Kubiak, R. J., Bruzik, K. S. & Tsai, M.‑D. Mechanism of phosphatidylinositol-specific phospholipase C: origin of unusually high nonbridging thio effects. Biochemistry 40, 5433–5439 (2001). 238. Tsai, M.‑D. & Yan, H. Mechanism of adenylate kinase: site-directed mutagenesis versus X‑ray and NMR. Biochemistry 30, 6806–6818 (1991). 239. Matute, R. A., Yoon, H. & Warshel, A. Exploring the mechanism of DNA polymerases by analyzing the effect of mutations of active site acidic groups in Polymerase β. Proteins 84, 1644–1657 (2016). Acknowledgements The authors acknowledge financial support from the Ministry of Science and Technology (Grant Nos MOST103‑2113‑M0 01 ‑ 01 6 ‑ M Y 3 , M O S T 10 5 ‑ 0 210 ‑ 01 ‑ 1 2 ‑ 01 a n d MOST106‑0210‑01‑15‑04) to M.-D.T. and a US National Institutes of Health intramural grant (DK036146‑08) to W.Y. Competing interests statement The authors declare no competing interests. Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. How to cite this article Wu, W.-J., Yang, W. & Tsai, M.-D. How DNA polymerases catalyse replication and repair with contrasting fidelity. Nat. Rev. Chem. 1, 0068 (2017). DATABASES RCSB Protein Data Bank: http://www.rcsb.org/pdb/home/ home.do ALL LINKS ARE ACTIVE IN THE ONLINE PDF 16 | ARTICLE NUMBER 0068 | VOLUME 1 www.nature.com/natrevchem . d e v r e s e r s t h g i r l l A . e r u t a N r e g n i r p S f o t r a p , d e t i m i L s r e h s i l b u P n a l l i m c a M 7 1 0 2 ©