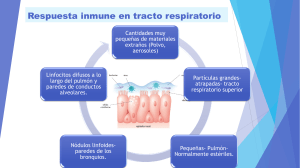
SÍNDROMES POR INMUNODEFICIENCIA Manuel Guillermo de la Garza Rodríguez 172581 Sara Maythe Perales Rivas 172522 Maria Fernanda Siller 175632 Georgina Prieto Carrasco 171379 Josue Sanchez Vazquez 117641 ● Primarias (congénitas) → Causa genética ● Secundarias (adquiridas) Complicaciones de cánceres Infecciones Irradiación Se manifiestan en la clínica por un aumento de las infecciones nuevas o reactivaciones de infecciones latentes INMUNODEFICIENCIAS PRIMARIAS ● La mayoría tienen origen genético ● Afectan a los mecanismos de la inmunidad innata o de la inmunidad adaptativa ● Se detectan mayormente en la lactancia DEFECTOS DE LA INMUNIDAD INNATA Afectación de funciones de los leucocitos o del sistema de complemento Aumento de vulnerabilidad de infecciones DEFECTOS DE LA FUNCIÓN DEL LEUCOCITO ● Defectos hereditarios de la adhesión del leucocito. Tipo1: Defecto en la biosíntesis de la cadena β2 compartida por las integrinas LFA-1 y Mac-1. Tipo 2: Falta de sialil-Lewis X, ligando con fucosa para selectinas E y P, como resultado de un defecto de en la fucosiltransferasa Principal problema clínico → Infecciones bacterianas recurrentes debido a una función inadecuada del granulocito ● Defectos hereditarios en la función del fagolisosoma. Un ejemplo es el síndrome de Chédiak-Higashi. Este se caracteriza por una función defectuosa de los fagocitos y a una tendencia a infecciones. Hay anomalías en los melanocitos, células del sistema nervioso y plaquetas. ● Defectos hereditarios en la actividad microbicida. Enfermedad granulomatosa crónica. Pacientes con disposición a infecciones bacterianas recurrentes. Defectos hereditarios en los genes que codifican componentes de la oxidasa del fagocito (generación de superóxido. ● Defectos de las señales producidas por el TLR. Defectos en TLR3 dan lugar a una encefalitis de repetición por el herpes simple. Defectos en MyD88 se asocian a neumonías bacterianas destructivas. DEFECTOS DE LA INMUNIDAD ADAPTATIVA ● Se subclasifican en función del componente afectado ● Se deben a anomalías en maduración o activación del linfocito DEFECTOS DE LA ACTIVACION Y MADURACION DE LINFOCITOS • Sindrome del linfocito T desnudo. Se debe habitualmente a mutaciones en los factores de transcripcion necesarios para la expresion del CPH de la clase II. • DEFECTOS EN LAS RESPUESTAS DE TH1 Y TH17 INMUNODEFICIENCIA COMBINADA GRAVE (IDCG) • Es una constelación de síndromes con características genéticas diferentes que tienen defectos comunes en las respuestas inmunitarias humorales y celulares. • Las personas con IDCG son sumamente proclives a las infecciones graves y recurrentes de una amplia variedad de microorganismos patógenos como son Cándida albicans, pneumocystitis jiroveci, pseudomonas, cytomegalovirus, varicela y una gran variedad de bacterias. • Los defectos subyacentes son muy variados en las diferentes formas de IDCG y en muchos de los casos se desconoce la alteración genética, el defecto mas común en esta enfermedad reside en el compartimiento de los linfocitos T, con un deterioro secundario de la inmunidad humoral. • Los lactantes afectados se presentan con un muguet (candidiasis oral) prominente, un exantema en la zona del pañal extenso y retraso del crecimiento. • Algunos pacientes sufren un exantema morbiliforme poco después del nacimiento, por que los linfocitos T maternos atraviesan la placenta y atacan al feto lo que provoca las lesiones. Trasplante de células madre hematopoyéticas IDCG LIGADA AL CROMOSOMA X • Es la forma mas frecuente responsable del 50 al 60% de los casos, es mas frecuente en los varones. • El defecto genético de esta IDCG es una mutación en la cadena gamma común de los receptores de citocinas. IDCG AUTOSOMICA RECESIVA • La forma mas común de IDCG autosómica recesiva es la deficiencia de la enzima adenosina desaminasa (ADA.) • Se ha propuesto que la deficiencia de ADA lleva a acumulación de desoxiadenosina (desoxi-ATP) que son tóxicos para los linfocitos inmaduros que se dividen con rapidez especialmente los linfocitos T por lo que hay una mayor disminución de linfocitos T que de los linfocitos B. CAUSAS MENOS FRECUENTES DE IDCG • Mutaciones en los genes activadores de la recombinasa (RAG) Bloquea desarrollo de linfocitos T y B • Mutaciones en la cinasa intracelular jak3 • Mutaciones en las moléculas transmisoras de señales Principalmente los necesarios para la entrada de calcio La severidad del defecto depende de que tanto tejido del timo desarrolle el niño. Tratamiento: específico para cada sintomatología. AGAMMAGLOBULINEMIA LIGADA AL CROMOSOMA X ● ● Se caracteriza por la falta de desarrollo de los precursores del linfocito B (pro linfocitos B y pre linfocitos B) en los linfocitos B maduros Se debe a la mutación de la tirosina cinasa de Bruton (btk) ¿QUÉ ES LA BTK? ● Tirosina cinasa que se asocia al complejo receptor de Ig de los linfocitos pre-B y B maduros ● Se necesita para traducir señales desde el receptor EFECTOS DE LA MUTACIÓN BTK El receptor del linfocito pre-B no puede producir señales y la maduración se detiene en esta fase ● No podrán ensamblarse al receptor completo para la molécula de antígeno, ni estar en la membrana ● Los pacientes son más proclives a infecciones por microorganismos ● CARACTERÍSTICAS ● Linfocitos B faltan o están muy reducidos y las concentraciones séricas de todas las clases de Ig están reducidas ● Poco desarrollo de los centros germinales, placas de Peyer, el apéndice y las amígdalas ● No hay células plasmáticas ● Reacciones por linfocitos T son normales ● Se presentan después de los 6 meses al agotarse las Ig maternas TRATAMIENTO ● Tratamiento restitutivo con Ig ● Tratamiento con Ig intravenosas profilácticas HIPOPLASIA TÍMICA: SÍNDROME DE DIGEORGE -Deficiencia de linfocitos T debido a un fallo en el desarrollo de la tercera y cuarta bolsas faríngeas. -Pérdida variable de inmunidad celular -La zonas de Linfocitos T de los órganos linfoides están vacías, por lo que las concentraciones de Ig pueden ser normales o bajas -Se debe a una deleción situada en el cromosoma 22q11 DIAGNÓSTICO -Basado en señas o síntomas que presentan al nacimiento, como: ● Rasgos faciales característicos ● Niveles bajos de calcio en sangre (hipoparatiroidismo) ● Inquietud o convulsiones ● Defectos cardiacos (soplos,IC, Cianosis) Análisis FISH (de Hibridación Fluorescente in Situ). SÍNDROME DE HIPERINMUNOGLOBULINEMIA M Los pacientes afectados producen Ac IgM, pero no pueden producir anticuerpos IgG, IgA e IgE. Esta enfermedad afecta la capacidad de los T CD4 colaboradores de enviar señales activadoras de linfocitos B y macrofagos. 70% de los sujetos con este síndrome tienen una forma ligada al cromosoma X, causada por mutaciones en el q=gen que codifica CD40L localizada en Xq26autosómica recesiva Desde el punto de vista clínico los pacientes presentan: ● ● ● ● ● ● Infecciones Piogenas Neumonía por Pneumocystis jiroveci Anemia hemolítica Trombocitopenia Infiltración de las mucosas del tubo digestivo Neutropenia Diagnóstico: identificación de una mutación que afecta al gen ligando CD40 Tratamiento: s infusiones regulares de IVIG cada 3 o 4 semanas son efectivas en disminuir la cantidad de infecciones Profilaxis: trimetoprim-sulfametoxazol INMUNODEFICIENCIA VARIABLE COMUN Grupo heterogéneo de trastornos por hipogammaglobulinemia Alteración de las respuestas de anticuerpos a la infección (o vacunación) y aumento de la susceptibilidad a las infecciones MANIFESTACIONES CLÍNICAS Recuerdan superficialmente a las de la ALX, pero en la inmunodeficiencia variable común, los dos sexos se ven afectados por igual los síntomas comienzan mucho más tarde en la segunda o tercera década de vida DIAGNÓSTICO ● ● Suele ser de exclusión (después de haber descartado otras causas de inmunodeficiencia) Se calcula una prevalencia de 1 caso por 50,000 habitantes Aunque la mayoría de los pacientes tienen tienen cifras normales de linfocitos B maduros ------------> Las células plasmáticas están ausentes --------> lo que sugiere un bloqueo en la diferenciación de los linfocitos B estimulado por ANTÍGENOS LOS PACIENTES SON PROPENSOS A: ● ● Padecer anemia hemolítica o perniciosa tumores linfoides Algunos Afectados tienen mutaciones en los receptores de linfocitos B Para ciertos factores de crecimiento o en moléculas implicadas en las interacciones de los linfocitos T y B Sin embargo en la mayoría de los casos no se conoce la base genética DÉFICIT AISLADO DE IGA ● ● La más frecuente de todas las enfermedades de inmunodeficiencia primaria Afecta a 1 de cada 700 individuos blancos La IgA principal en las secreciones de las mucosas y está implicada en la defensa de las vías respiratorias y del tubo digestivo ● ● ● ● La mayoría de los individuos son asintomáticos La debilidad de las defensas de las mucosas predispone a presentar infecciones Sinopulmonares diarrea PATOGENIA DE LA DEFICIENCIA DE IGA Esta implicada en un bloqueo en la diferenciación terminal de los linfocitos B Secretores de la inmunoglobulina IgA a células plasmáticas Las Subclases IgM e IgG de anticuerpos se hallan presentes en concentraciones normales o elevadas Se desconoce la base molecular de este defecto SÍNDROME LINFOPROLIFERATIVO LIGADO AL CROMOSOMA X Incapacidad de eliminar el virus de Epstein-Barr Conduce a una mononucleosis infecciosa fulminante y desarrollo de tumores de linfocito B Pacientes capaces de producir anticuerpos de afinidad alta INMUNODEFICIENCIAS ASOCIADAS A ENFERMEDADES SISTÉMICAS ● Síndrome de Wiskott-Aldrich Se caracteriza por trombocitopenia, eccema y vulnerabilidad infecciones Sufren linfomas de linfocitos B Concentraciones de inmunoglobulinas: ➢ IgM - Baja ➢ IgG - Normal ➢ IgA e IgE - Elevada ATAXIA-TELANGIECTASIA • Es un trastorno autosómico recesivo caracterizado por ataxia, malformaciones vasculares, deficiencias neurológicas, un aumento de la incidencia de tumores e inmunodeficiencia. INMUNODEFICIENCIAS SECUNDARIAS