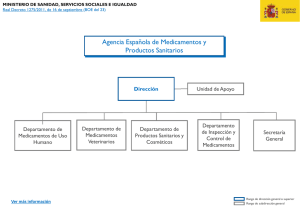
Códigos electrónicos Código de Derecho Farmacéutico Selección y ordenación: GARRIGUES Edición actualizada a 17 de abril de 2019 BOLETÍN OFICIAL DEL ESTADO La última versión de este Código en PDF y ePUB está disponible para su descarga gratuita en: www.boe.es/legislacion/codigos/ Alertas de actualización en BOE a la Carta: www.boe.es/a_la_carta/ Para adquirir el Código en formato papel: tienda.boe.es © Agencia Estatal Boletín Oficial del Estado NIPO (PDF): 007-14-138-8 NIPO (Papel): 007-15-030-0 NIPO (ePUB): 007-14-137-2 ISBN: 978-84-340-2170-9 Depósito Legal: M-8673-2015 Catálogo de Publicaciones de la Administración General del Estado publicacionesoficiales.boe.es Agencia Estatal Boletín Oficial del Estado Avenida de Manoteras, 54 28050 MADRID www.boe.es CÓDIGO DE DERECHO FARMACÉUTICO SUMARIO NORMATIVA GENERAL § 1. Real Decreto Legislativo 1/2015, de 24 de julio, por el que se aprueba el texto refundido de la Ley de garantías y uso racional de los medicamentos y productos sanitarios . . . . . . . . . . . . . . . . . . . . 1 § 2. Ley 29/2006, de 26 de julio, de garantías y uso racional de los medicamentos y productos sanitarios. [Inclusión parcial] . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 89 § 3. Ley 14/1986, de 25 de abril, General de Sanidad. [Inclusión parcial] . . . . . . . . . . . . . . . . . . . . . . . . 91 § 4. Ley 16/2003, de 28 de mayo, de cohesión y calidad del Sistema Nacional de Salud. [Inclusión parcial] . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 97 § 5. Ley 24/2015, de 24 de julio, de Patentes. [Inclusión parcial] . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 99 § 6. Real Decreto 316/2017, de 31 de marzo, por el que se aprueba el Reglamento para la ejecución de la Ley 24/2015, de 24 de julio, de Patentes. [Inclusión parcial] . . . . . . . . . . . . . . . . . . . . . . . . . . . . 140 MEDICAMENTOS DE USO HUMANO § 7. Real Decreto 1345/2007, de 11 de octubre, por el que se regula el procedimiento de autorización, registro y condiciones de dispensación de los medicamentos de uso humano fabricados industrialmente . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 165 § 8. Real Decreto 824/2010, de 25 de junio, por el que se regulan los laboratorios farmacéuticos, los fabricantes de principios activos de uso farmacéutico y el comercio exterior de medicamentos y medicamentos en investigación . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 251 § 9. Real Decreto 177/2014, de 21 de marzo, por el que se regula el sistema de precios de referencia y de agrupaciones homogéneas de medicamentos en el Sistema Nacional de Salud, y determinados sistemas de información en materia de financiación y precios de los medicamentos y productos sanitarios . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 287 § 10. Real Decreto 1416/1994, de 25 de junio, por el que se regula la publicidad de los medicamentos de uso humano . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 303 § 11. Real Decreto 1907/1996, de 2 de agosto, sobre publicidad y promoción comercial de productos, actividades o servicios con pretendida finalidad sanitaria . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 316 § 12. Real Decreto 782/2013, de 11 de octubre, sobre distribución de medicamentos de uso humano . . . 321 § 13. Real Decreto 954/2015, de 23 de octubre, por el que se regula la indicación, uso y autorización de dispensación de medicamentos y productos sanitarios de uso humano por parte de los enfermeros 341 § 14. Real Decreto 1785/2000, de 27 de octubre, sobre la circulación intracomunitaria de medicamentos de uso humano . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 353 – III – CÓDIGO DE DERECHO FARMACÉUTICO SUMARIO § 15. Real Decreto 577/2013, de 26 de julio, por el que se regula la farmacovigilancia de medicamentos de uso humano . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 358 § 16. Real Decreto 870/2013, de 8 de noviembre, por el que se regula la venta a distancia al público, a través de sitios web, de medicamentos de uso humano no sujetos a prescripción médica . . . . . . . . 384 § 17. Real Decreto 1015/2009, de 19 de junio, por el que se regula la disponibilidad de medicamentos en situaciones especiales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 392 MEDICAMENTOS DE USO VETERINARIO § 18. Real Decreto 109/1995, de 27 de enero, sobre medicamentos veterinarios . . . . . . . . . . . . . . . . . . . 402 § 19. Real Decreto 1246/2008, de 18 de julio, por el que se regula el procedimiento de autorización, registro y farmacovigilancia de los medicamentos veterinarios fabricados industrialmente . . . . . . . . 441 PRODUCTOS SANITARIOS § 20. Real Decreto 1591/2009, de 16 de octubre, por el que se regulan los productos sanitarios . . . . . . . 523 § 21. Real Decreto 1616/2009, de 26 de octubre, por el que se regulan los productos sanitarios implantables activos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 589 § 22. Real Decreto 1662/2000, de 29 de septiembre, sobre productos sanitarios para diagnóstico "in vitro" . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 630 § 23. Real Decreto 437/2002, de 10 de mayo, por el que se establecen los criterios para la concesión de licencias de funcionamiento a los fabricantes de productos sanitarios a medida . . . . . . . . . . . . . . . 675 § 24. Real Decreto 1085/2009, de 3 de julio, por el que se aprueba el Reglamento sobre instalación y utilización de aparatos de rayos X con fines de diagnóstico médico . . . . . . . . . . . . . . . . . . . . . . . . 679 ENSAYOS CLÍNICOS Y ESTUDIOS POSAUTORIZACIÓN § 25. Real Decreto 1090/2015, de 4 de diciembre, por el que se regulan los ensayos clínicos con medicamentos, los Comités de Ética de la Investigación con medicamentos y el Registro Español de Estudios Clínicos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 702 § 26. Orden SAS/3470/2009, de 16 de diciembre, por la que se publican las directrices sobre estudios posautorización de tipo observacional para medicamentos de uso humano . . . . . . . . . . . . . . . . . . 742 OFICINAS DE FARMACIA § 27. Ley 16/1997, de 25 de abril, de Regulación de Servicios de las Oficinas de Farmacia . . . . . . . . . . . 756 RECETA MÉDICA § 28. Real Decreto 1718/2010, de 17 de diciembre, sobre receta médica y órdenes de dispensación . . . . 761 AGENCIA ESPAÑOLA DE MEDICAMENTOS Y PRODUCTOS SANITARIOS § 29. Real Decreto 1275/2011, de 16 de septiembre, por el que se crea la Agencia estatal "Agencia Española de Medicamentos y Productos Sanitarios" y se aprueba su Estatuto . . . . . . . . . . . . . . . . – IV – 785 CÓDIGO DE DERECHO FARMACÉUTICO ÍNDICE SISTEMÁTICO NORMATIVA GENERAL § 1. Real Decreto Legislativo 1/2015, de 24 de julio, por el que se aprueba el texto refundido de la Ley de garantías y uso racional de los medicamentos y productos sanitarios . . . . . . . . . . . . . Preámbulo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Artículos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones adicionales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones derogatorias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones finales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . TÍTULO PRELIMINAR. Disposiciones generales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . TÍTULO I. Garantías y obligaciones generales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . TÍTULO II. De los medicamentos. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO I. De los medicamentos reconocidos por la ley y sus clases . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO II. De las garantías exigibles a los medicamentos de uso humano elaborados industrialmente y de las condiciones de prescripción y dispensación de los mismos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO III. De las garantías exigibles a los medicamentos veterinarios elaborados industrialmente y de las condiciones de prescripción y dispensación de los mismos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO IV. De las garantías sanitarias de las fórmulas magistrales y preparados oficinales. . . . . . . . . . . . CAPÍTULO V. De las garantías sanitarias de los medicamentos especiales . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO VI. De las garantías de seguimiento de la relación beneficio/riesgo en los medicamentos . . . . . . . TÍTULO III. De la investigación de los medicamentos de uso humano y sus garantías . . . . . . . . . . . . . . . . . . . TÍTULO IV. De las garantías exigibles en la fabricación y distribución de medicamentos. . . . . . . . . . . . . . . . . . CAPÍTULO I. De la fabricación de medicamentos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO II. De la distribución de medicamentos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . TÍTULO V. De las garantías sanitarias del comercio exterior de medicamentos . . . . . . . . . . . . . . . . . . . . . . . TÍTULO VI. Del registro de laboratorios farmacéuticos y del registro de fabricantes, importadores o distribuidores de principios activos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . TÍTULO VII. Del uso racional de los medicamentos de uso humano. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO I. De las garantías de formación e información independiente y de calidad para la utilización adecuada de los medicamentos y productos sanitarios . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO II. Del uso racional de medicamentos en la atención primaria a la salud. . . . . . . . . . . . . . . . . . . CAPÍTULO III. Del uso racional de los medicamentos en la atención hospitalaria y especializada . . . . . . . . . . CAPÍTULO IV. Del uso racional de medicamentos en las oficinas de farmacia . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO V. De la trazabilidad de los medicamentos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . TÍTULO VIII. De la financiación pública de los medicamentos y productos sanitarios . . . . . . . . . . . . . . . . . . . . TÍTULO IX. Régimen sancionador . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO I. Inspección y medidas cautelares . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO II. Infracciones y sanciones . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . TÍTULO X. De la acción de cesación . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . TÍTULO XI. Tasas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones adicionales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones transitorias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones finales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . § 2. Ley 29/2006, de 26 de julio, de garantías y uso racional de los medicamentos y productos sanitarios. [Inclusión parcial] . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . [...] –V– 1 1 7 7 7 7 7 9 12 12 13 22 30 31 34 36 39 39 41 43 44 44 44 48 49 50 52 52 63 63 64 74 76 83 87 88 89 CÓDIGO DE DERECHO FARMACÉUTICO ÍNDICE SISTEMÁTICO § 3. Ley 14/1986, de 25 de abril, General de Sanidad. [Inclusión parcial] . . . . . . . . . . . . . . . . . . . . . 91 [...] TÍTULO V. De los productos farmacéuticos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO ÚNICO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 91 91 [...] [...] [...] § 4. Ley 16/2003, de 28 de mayo, de cohesión y calidad del Sistema Nacional de Salud. [Inclusión parcial] . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 97 [...] CAPÍTULO II. De la farmacia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 1.ª Organización y ejercicio de las competencias del estado en materia de farmacia . . . . . . . . . . . Sección 2.ª Colaboración de las oficinas de farmacia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 97 97 98 [...] § 5. Ley 24/2015, de 24 de julio, de Patentes. [Inclusión parcial] . . . . . . . . . . . . . . . . . . . . . . . . . . . 99 [...] TÍTULO II. Patentabilidad . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 99 [...] TÍTULO IV. Invenciones realizadas en el marco de una relación de empleo o de servicios . . . . . . . . . . . TÍTULO V. Solicitud y procedimiento de concesión . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO I. Presentación y requisitos de la solicitud de patente . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO II. Procedimiento de Concesión . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO III. Oposiciones y recursos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO IV. Certificados complementarios de protección de medicamentos y productos fitosanitarios. CAPÍTULO V. Disposiciones comunes a todos los procedimientos y a la información de los terceros . . . TÍTULO VI. Efectos de la patente y de la solicitud de la patente . . . . . . . . . . . . . . . . . . . . . . . . . . . . TÍTULO VII. Acciones por violación del derecho de patente . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . TÍTULO VIII. La solicitud de patente y la patente como objetos del derecho de propiedad. . . . . . . . . . . . CAPÍTULO I. Inscripción registral, cotitularidad y expropiación . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO II. Transferencias, Licencias y Gravámenes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO III. Licencias de pleno derecho . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . TÍTULO IX. Obligación de explotar y licencias obligatorias. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO I. Obligación de explotar la invención y requisitos para la concesión de licencias obligatorias CAPÍTULO II. Procedimiento de concesión de las licencias obligatorias . . . . . . . . . . . . . . . . . . . . . CAPÍTULO III. Régimen de las licencias obligatorias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . TÍTULO X. Nulidad, revocación y caducidad de la patente. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO I. Nulidad . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO II. Revocación o limitación a instancia del titular de la patente . . . . . . . . . . . . . . . . . . . . CAPÍTULO III. Caducidad . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 101 104 104 107 110 111 111 115 118 120 120 122 123 124 124 127 128 129 129 130 131 . . . . . . . . . . . . . . . . . . . . . . . . . 133 133 135 136 138 [...] TÍTULO XII. Jurisdicción y normas procesales . . . . . . . CAPÍTULO I. Disposiciones generales . . . . . . . . . . CAPÍTULO II. Diligencias de comprobación de hechos CAPÍTULO III. Medidas cautelares . . . . . . . . . . . . . CAPÍTULO IV. Solución extrajudicial de controversias. . . . . . . . . . . [...] – VI – . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CÓDIGO DE DERECHO FARMACÉUTICO ÍNDICE SISTEMÁTICO § 6. Real Decreto 316/2017, de 31 de marzo, por el que se aprueba el Reglamento para la ejecución de la Ley 24/2015, de 24 de julio, de Patentes. [Inclusión parcial] . . . . . . . . . . . . . . . REGLAMENTO DE EJECUCIÓN DE LA LEY 24/2015, DE 24 DE JULIO, DE PATENTES . . . . . . . TÍTULO I. Patentes de invención. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO I. Solicitud de patente . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO II. Procedimiento de concesión . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 1.ª Admisión a trámite y examen de oficio . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 2.ª Informe sobre el estado de la técnica y opinión escrita . . . . . . . . . . . . . . . . . . . Sección 3.ª Examen sustantivo y resolución . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO III. Procedimiento de oposición . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO IV. Procedimiento de revocación o limitación . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO V. Otros procedimientos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 1.ª Solicitudes divisionales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 2.ª Cambio de modalidad. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 3.ª Tramitación secreta de patentes que interesan a la defensa nacional . . . . . . . . . TÍTULO II. Certificados complementarios de protección de medicamentos o de su prórroga fitosanitarios . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . y . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . productos . . . . . . . 140 140 140 140 146 146 150 153 155 158 159 159 160 161 163 [...] MEDICAMENTOS DE USO HUMANO § 7. Real Decreto 1345/2007, de 11 de octubre, por el que se regula el procedimiento de autorización, registro y condiciones de dispensación de los medicamentos de uso humano fabricados industrialmente . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Preámbulo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO I. Disposiciones generales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO II. Autorización de medicamentos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 1.ª Solicitudes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 2.ª Procedimiento de autorización . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO III. Etiquetado y prospecto . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 1.ª Disposiciones generales del etiquetado y prospecto . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Seccion 2.ª Garantías de identificación del medicamento: Etiquetado . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 3.ª Garantías de información del medicamento: Prospecto . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Seccion 4.ª Disposiciones particulares para determinados formatos de medicamentos . . . . . . . . . . . . . . . . . CAPÍTULO IV. Disposiciones particulares para determinadas clases de medicamentos . . . . . . . . . . . . . . . . Sección 1.ª Medicamentos hemoderivados. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 2.ª Vacunas y alérgenos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 3.ª Medicamentos radiofármacos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 4.ª Medicamentos tradicionales a base de plantas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 5.ª Medicamentos homeopáticos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 6.ª Gases medicinales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO V. Obligaciones del titular del medicamento. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO VI. Modificaciones de las condiciones de autorización de medicamentos . . . . . . . . . . . . . . . . . . CAPÍTULO VII. Procedimientos para la suspensión y revocación de la autorización . . . . . . . . . . . . . . . . . . . CAPÍTULO VIII. Procedimientos comunitarios . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones adicionales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones transitorias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones derogatorias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones finales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ANEXO I. Normas y protocolos analíticos, farmacotoxicológicos y clínicos relativos a la realización de pruebas de medicamentos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ANEXO II. Contenido de la ficha técnica o resumen de características del producto . . . . . . . . . . . . . . . . . . . . ANEXO III. Contenido del etiquetado de los medicamentos que se fabrican industrialmente . . . . . . . . . . . . . . . ANEXO IV. Símbolos, siglas y leyendas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ANEXO V. Contenido mínimo del prospecto de los medicamentos de fabricación industrial . . . . . . . . . . . . . . . . – VII – 165 165 167 171 171 175 182 182 183 184 185 185 185 186 187 188 190 192 193 193 197 198 201 201 203 204 204 244 245 247 249 CÓDIGO DE DERECHO FARMACÉUTICO ÍNDICE SISTEMÁTICO § 8. Real Decreto 824/2010, de 25 de junio, por el que se regulan los laboratorios farmacéuticos, los fabricantes de principios activos de uso farmacéutico y el comercio exterior de medicamentos y medicamentos en investigación . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Preámbulo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO I. Disposiciones generales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO II. Fabricación e importación de medicamentos y medicamentos en investigación . . . . . . . . . . . . Sección 1.ª Procedimiento de autorización . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 2.ª Modificaciones de la autorización . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 3.ª Procedimiento para la suspensión y revocación de la autorización, y medidas cautelares . . . . . . Sección 4.ª Obligaciones del titular de la autorización de laboratorio farmacéutico fabricante o importador . . . Sección 5.ª Director Técnico . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 6.ª Fabricación por terceros . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO III. Fabricación de principios activos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO IV. Normas de correcta fabricación . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO V. Laboratorios titulares de la autorización de comercialización . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO VI. Registro de laboratorios farmacéuticos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO VII. Inspección, toma de muestras y problemas de calidad . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO VIII. Comercio exterior de medicamentos y envío de medicamentos autorizados en España a otros Estados miembros . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO IX. Controles en la importación de principios activos utilizados en la fabricación de medicamentos de uso humano. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones adicionales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones transitorias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones derogatorias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones finales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . § 9. Real Decreto 177/2014, de 21 de marzo, por el que se regula el sistema de precios de referencia y de agrupaciones homogéneas de medicamentos en el Sistema Nacional de Salud, y determinados sistemas de información en materia de financiación y precios de los medicamentos y productos sanitarios . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Preámbulo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO I. Disposiciones de carácter general . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO II. Sistema de precios de referencia de medicamentos. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO III. Sistema de agrupaciones homogéneas de medicamentos . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO IV. Sistemas de información en materia de financiación y precios de los medicamentos y productos sanitarios . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 1.ª El nomenclátor oficial de la prestación farmacéutica del Sistema Nacional de Salud . . . . . . . . . Sección 2.ª Sistema de información de apoyo a la gestión de fijación de precios y decisión de financiación de los medicamentos y productos sanitarios . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 3.ª Sistema de información sobre consumo de medicamentos en la red pública hospitalaria del sistema nacional de salud . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones adicionales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones transitorias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones finales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . § 10. Real Decreto 1416/1994, de 25 de junio, por el que se regula la publicidad de los medicamentos de uso humano . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Preámbulo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO I. Disposiciones generales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPITULO II. Publicidad destinada al público . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPITULO III. Publicidad dirigida a las personas facultadas para prescribir o dispensar medicamentos . Sección 1.ª Principios generales. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 2.ª Información técnica del medicamento. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 3.ª Publicidad documental . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 4.ª Muestras gratuitas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 5.ª Otros medios de publicidad . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO IV. Obligaciones del titular de la autorización sanitaria del medicamento en relación publicidad . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO V. Control de la publicidad. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 1.ª Control de la publicidad dirigida al publico . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . – VIII – . . . . . . . . . . . . . . . . . . . . . . . . . . . con . . . . . . . . . . . . . . . . . . . . . . . . . . . la . . . . . . 251 251 254 257 257 259 260 261 262 264 266 267 276 277 278 282 284 285 285 286 286 287 287 289 290 293 296 296 298 298 298 301 301 303 303 304 305 307 307 307 308 309 310 310 311 311 CÓDIGO DE DERECHO FARMACÉUTICO ÍNDICE SISTEMÁTICO Sección 2.ª Control de la publicidad dirigida medicamentos . . . . . . . . . . . . . . . . . . . . Sección 3.ª Inspección y sanciones. . . . . . . . . Disposiciones adicionales . . . . . . . . . . . . . . . . . . Disposiciones transitorias . . . . . . . . . . . . . . . . . . Disposiciones derogatorias . . . . . . . . . . . . . . . . . Disposiciones finales . . . . . . . . . . . . . . . . . . . . . dispensar . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 312 313 313 314 314 315 § 11. Real Decreto 1907/1996, de 2 de agosto, sobre publicidad y promoción comercial de productos, actividades o servicios con pretendida finalidad sanitaria . . . . . . . . . . . . . . . . . . . 316 Preámbulo . . . . . . . . . . . Artículos . . . . . . . . . . . . Disposiciones adicionales . Disposiciones derogatorias Disposiciones finales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . a . . . . . . . . . . . . . . . . . . . . . . las . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . personas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . facultadas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . para . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . prescribir . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . o . . . . . . . . . . . . . . . . . . . . . . . . . . . 316 317 320 320 320 § 12. Real Decreto 782/2013, de 11 de octubre, sobre distribución de medicamentos de uso humano . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 321 Preámbulo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO I. Disposiciones generales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO II. Requisitos materiales y de personal de las entidades de distribución de medicamentos CAPÍTULO III. Obligaciones de los titulares de una autorización de distribución . . . . . . . . . . . . . . CAPÍTULO IV. Intermediación de medicamentos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO V. Autorización de entidades de distribución de medicamentos . . . . . . . . . . . . . . . . . CAPÍTULO VI. Buenas prácticas de distribución . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO VII. Inspección . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones adicionales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones transitorias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones derogatorias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones finales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 321 323 325 327 328 330 332 333 334 334 334 335 § 13. Real Decreto 954/2015, de 23 de octubre, por el que se regula la indicación, uso y autorización de dispensación de medicamentos y productos sanitarios de uso humano por parte de los enfermeros . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 341 Preámbulo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO I. Disposiciones de carácter general . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO II. Indicación, uso y autorización de dispensación de medicamentos y productos sanitarios de uso humano relacionados con su ejercicio profesional por parte de los enfermeros . . . . . . . . . . . . . . . . . . . . CAPÍTULO III. Elaboración y validación de protocolos y guías de práctica clínica y asistencial para la indicación, uso y autorización de dispensación por parte de los enfermeros de medicamentos sujetos a prescripción médica . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO IV. Acreditación de los enfermeros para la indicación, uso y autorización de dispensación de medicamentos y productos sanitarios de uso humano . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones adicionales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones transitorias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones finales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ANEXO I. Competencias necesarias para indicar, usar y autorizar la dispensación de medicamentos y productos sanitarios de uso humano y características de los programas formativos . . . . . . . . . . . . . . . . . . . . . . . . . . § 14. Real Decreto 1785/2000, de 27 de octubre, sobre la circulación intracomunitaria de medicamentos de uso humano . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Preámbulo . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO I. Disposiciones generales . . . . . CAPÍTULO II. Requisitos . . . . . . . . . . . . . . CAPÍTULO III. Procedimiento de autorización . Disposiciones adicionales . . . . . . . . . . . . . . . Disposiciones transitorias . . . . . . . . . . . . . . . Disposiciones derogatorias . . . . . . . . . . . . . . Disposiciones finales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . – IX – . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 341 343 343 345 346 347 348 348 351 353 353 354 354 355 357 357 357 357 CÓDIGO DE DERECHO FARMACÉUTICO ÍNDICE SISTEMÁTICO § 15. Real Decreto 577/2013, de 26 de julio, por el que se regula la farmacovigilancia de medicamentos de uso humano . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Preámbulo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO I. Disposiciones generales . . . . . . . . . . . . . . . . . . . . CAPÍTULO II. De las administraciones sanitarias . . . . . . . . . . . . . . CAPÍTULO III. De los profesionales sanitarios y los ciudadanos . . . . CAPÍTULO IV. De los titulares de la autorización de comercialización . CAPÍTULO V. De la intervención administrativa . . . . . . . . . . . . . . . CAPÍTULO VI. De los estudios posautorización . . . . . . . . . . . . . . . Disposiciones adicionales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones transitorias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones derogatorias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones finales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 358 361 364 367 368 373 378 379 380 381 381 § 16. Real Decreto 870/2013, de 8 de noviembre, por el que se regula la venta a distancia al público, a través de sitios web, de medicamentos de uso humano no sujetos a prescripción médica . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 384 Preámbulo . . . . . . . . . . Artículos . . . . . . . . . . . Disposiciones adicionales Disposiciones transitorias Disposiciones finales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 392 . . . . . . . . . . . . . . . . . . . . . . . . § 17. Real Decreto 1015/2009, de 19 de junio, por el que se regula la disponibilidad de medicamentos en situaciones especiales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 384 385 390 391 391 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Preámbulo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO I. Disposiciones generales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO II. Uso compasivo de medicamentos en investigación . . . . . . . . . . . . CAPÍTULO III. Acceso a medicamentos en condiciones diferentes a las autorizadas CAPÍTULO IV. Medicamentos no autorizados en España. . . . . . . . . . . . . . . . . . Disposiciones adicionales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones transitorias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones finales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 358 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 392 394 395 397 399 400 401 401 § 18. Real Decreto 109/1995, de 27 de enero, sobre medicamentos veterinarios . . . . . . . . . . . . . . . . 402 MEDICAMENTOS DE USO VETERINARIO Preámbulo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . TÍTULO I. Disposiciones generales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . TÍTULO II. Medicamentos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO I. Medicamentos veterinarios legalmente reconocidos y sus clases. . . . . . . . . . . . CAPÍTULO II. Comisión Nacional de Evaluación de Medicamentos Veterinarios . . . . . . . . . . . CAPÍTULO III. Evaluación, autorización y registro de medicamentos veterinarios . . . . . . . . . . CAPÍTULO IV. Reconocimientos mutuos y procedimientos coordinado y centralizado. . . . . . . . CAPÍTULO V. Requisitos sanitarios de los demás medicamentos veterinarios . . . . . . . . . . . . CAPÍTULO VI. Medicamentos especiales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 1.ª Medicamentos estupefacientes y Psicotropos . . . . . . . . . . . . . . . . . . . . . . . Sección 2.ª Medicamentos de plantas medicinales con destino a los animales . . . . . . . . . . Sección 3.ª Radiofármacos de aplicación veterinaria . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 4.ª Productos homeopáticos destinados a los animales . . . . . . . . . . . . . . . . . . . CAPÍTULO VII. Farmacopea y control de calidad . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO VIII. Farmacovigilancia veterinaria . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . TÍTULO III. De los ensayos clínicos veterinarios . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO ÚNICO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . TÍTULO IV. De los fabricantes de medicamentos veterinarios. . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO ÚNICO. De los laboratorios preparadores de medicamentos veterinarios . . . . . . . . TÍTULO V. De las garantías sanitarias del comercio intracomunitario y con terceros países de veterinarios. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . –X– . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . medicamentos . . . . . . . . . . 402 403 404 404 406 406 406 406 409 409 409 409 410 410 411 411 411 414 414 414 CÓDIGO DE DERECHO FARMACÉUTICO ÍNDICE SISTEMÁTICO CAPÍTULO I. Importación de terceros países y envío desde otros Estados miembros. . . . . . . . CAPÍTULO II. Exportación a terceros países y envío a otros Estados miembros . . . . . . . . . . . TÍTULO VI. Comercialización, prescripción y utilización de los medicamentos veterinarios . . . . . . CAPÍTULO I. Presentación comercial e información . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO II. Distribución . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO III. Prescripción . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO IV. Dispensación de medicamentos veterinarios . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO V. Aplicación y uso de medicamentos veterinarios . . . . . . . . . . . . . . . . . . . . . . TÍTULO VII. Vigilancia y régimen sancionador . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO I. Inspección y medidas cautelares . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO II. Infracciones y sanciones . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO III. Suspensión o extinción de las autorizaciones y retirada del mercado de veterinarios . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . TÍTULO VIII. Tasa . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO ÚNICO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones adicionales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones transitorias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones derogatorias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones finales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ANEXO I . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ANEXO II. Relación de materias colorantes autorizados para la coloración de los medicamentos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . medicamentos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . § 19. Real Decreto 1246/2008, de 18 de julio, por el que se regula el procedimiento de autorización, registro y farmacovigilancia de los medicamentos veterinarios fabricados industrialmente. . . Preámbulo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO I. Disposiciones generales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO II. Autorización de medicamentos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 1.ª Solicitudes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 2.ª Procedimiento de autorización . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO III. Etiquetado y prospecto . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 1.ª Disposiciones generales del etiquetado y del prospecto . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 2.ª Garantías de identificación del medicamento: etiquetado . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 3.ª Garantías de información del medicamento: prospecto. . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPITULO IV. Disposiciones particulares para determinadas clases de medicamentos . . . . . . . . . . . . . . . . Sección 1.ª Medicamentos homeopáticos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 2.ª Gases medicinales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO V. Obligaciones del titular del medicamento. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO VI. Modificaciones de las condiciones de autorización de medicamentos . . . . . . . . . . . . . . . . . . CAPÍTULO VII. Procedimientos para la suspensión y revocación de la autorización . . . . . . . . . . . . . . . . . . . CAPÍTULO VIII. Procedimientos comunitarios . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO IX. Farmacovigilancia veterinaria . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones adicionales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones transitorias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones derogatorias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones finales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ANEXO I. Normas químicas, farmacéuticas y analíticas, estudios de seguridad y de residuos, estudios preclínicos y clínicos relacionados con los análisis de medicamentos veterinarios . . . . . . . . . . . . . . . . . . . . . . . . . . . TITULO I. Requisitos relativos a los medicamentos veterinarios no inmunológicos . . . . . . . . . . . . . . . . . . . . . TÍTULO II. Requisitos relativos a los medicamentos veterinarios inmunológicos . . . . . . . . . . . . . . . . . . . . . . . TÍTULO III. Requisitos relativos a solicitudes específicas de autorización de comercialización . . . . . . . . . . . . . . TÍTULO IV. Requisitos relativos a solicitudes de autorización de comercialización. . . . . . . . . . . . . . . . . . . . . . ANEXO II. Ficha técnica o resumen de las características del producto . . . . . . . . . . . . . . . . . . . . . . . . . . . . ANEXO III. Etiquetado y prospecto . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 414 414 414 414 415 417 423 427 431 431 435 436 436 436 437 437 439 439 439 439 441 441 443 447 447 451 458 458 458 459 460 460 462 463 463 467 468 471 476 477 478 478 479 480 501 514 517 518 519 PRODUCTOS SANITARIOS § 20. Real Decreto 1591/2009, de 16 de octubre, por el que se regulan los productos sanitarios. . . . Preámbulo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO I. Disposiciones generales . . . . . . . . . . . CAPÍTULO II. Instalaciones . . . . . . . . . . . . . . . . . . CAPÍTULO III. Clasificación y marcado de conformidad. CAPÍTULO IV. Productos con una finalidad especial. . . – XI – . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 523 523 526 532 534 537 CÓDIGO DE DERECHO FARMACÉUTICO ÍNDICE SISTEMÁTICO CAPÍTULO V. Organismos notificados. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO VI. Comercialización y puesta en servicio . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO VII. Comercio intracomunitario y exterior . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO VIII. Investigaciones clínicas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO IX. Sistema de vigilancia. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO X. Inspección y medidas de protección de la salud . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO XI. Publicidad y exhibiciones . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO XII. Infracciones y sanciones . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones adicionales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones transitorias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones derogatorias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones finales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ANEXO I. Requisitos esenciales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ANEXO II. Declaración CE de conformidad . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ANEXO III. Examen CE de tipo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ANEXO IV. Verificación CE . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ANEXO V. Declaración CE de conformidad . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ANEXO VI. Declaración CE de conformidad. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ANEXO VII. Declaración CE de conformidad . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ANEXO VIII. Declaración relativa a los productos que tengan una finalidad especial . . . . . . . . . . . . . . . . . . . . ANEXO IX. Criterios de clasificación . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ANEXO X. Evaluación clínica . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ANEXO XI. Criterios mínimos que deben observarse para la designación de los organismos notificados . . . . . . . ANEXO XII. Marcado CE de conformidad . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ANEXO XIII. Análisis del riesgo y estrategia de gestión del riesgo de los productos sanitarios en cuya elaboración se utilizan tejidos de origen animal . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . § 21. Real Decreto 1616/2009, de 26 de octubre, por el que se regulan los productos sanitarios implantables activos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Preámbulo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO I. Disposiciones generales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO II. Instalaciones . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO III. Marcado de conformidad . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO IV. Productos con una finalidad especial. . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO V. Organismo notificados . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO VI. Comercialización y puesta en servicio . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO VII. Comercio intracomunitario y exterior . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO VIII. Investigaciones clínicas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO IX. Sistema de vigilancia. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO X. Inspección y medidas de protección de la salud . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO XI. Publicidad y exhibiciones . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO XII. Infracciones y sanciones . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones adicionales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones transitorias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones derogatorias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones finales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ANEXO 1. Requisitos esenciales. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ANEXO 2. Declaración CE de conformidad . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ANEXO 3. Examen CE de tipo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ANEXO 4. Verificación CE . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ANEXO 5. Declaración CE de conformidad con el tipo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ANEXO 6. Declaración relativa a los productos con una finalidad especial . . . . . . . . . . . . . . . . ANEXO 7. Evaluación clínica . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ANEXO 8. Requisitos mínimos que deberán reunir los organismos notificados para su designación ANEXO 9. Marcado de conformidad CE . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . § 22. Real Decreto 1662/2000, de 29 de septiembre, sobre productos sanitarios para diagnóstico "in vitro". . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 630 . . . . . . . . . . – XII – . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 589 589 592 596 598 599 600 602 604 605 606 608 609 611 611 611 611 611 612 616 619 621 622 624 626 628 629 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 585 . . . . . . . . . . . . . . . . . . . . . . . . . . Preámbulo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO I. Disposiciones generales . . . . . . . . . CAPÍTULO II. Garantías sanitarias de los productos . CAPÍTULO III. Comercialización y puesta en servicio CAPÍTULO IV. Distribución y venta . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 538 540 544 544 546 547 549 551 551 551 552 552 554 561 565 567 569 572 575 576 578 582 584 585 . . . . . . . . . . 630 631 633 638 639 CÓDIGO DE DERECHO FARMACÉUTICO ÍNDICE SISTEMÁTICO CAPÍTULO V. Transacciones comunitarias y comercio exterior . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO VI. Actuaciones de las Administraciones públicas . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO VII. Publicidad y exhibiciones. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO VIII. Infracciones y sanciones . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones adicionales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones transitorias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones derogatorias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones finales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ANEXO I. Requisitos esenciales relativos al diseño y a la fabricación. . . . . . . . . . . . . . . . . . . . . . . . . ANEXO II. Lista de los productos a que se refieren los apartados 1.1 y 1.2 del artículo 7 . . . . . . . . . . . . ANEXO III. Declaración CE de conformidad . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ANEXO IV. Declaración CE de conformidad (sistema de garantía de calidad total) . . . . . . . . . . . . . . . . ANEXO V. Examen CE de tipo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ANEXO VI. Verificación CE . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ANEXO VII. Declaración CE de conformidad (sistema de garantía de calidad de la producción) . . . . . . . . ANEXO VIII. Declaración y procedimientos relativos a los productos para la evaluación del funcionamiento ANEXO IX. Criterios para la designación de organismos notificados . . . . . . . . . . . . . . . . . . . . . . . . . ANEXO X. Marcado CE de conformidad . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 641 641 645 647 647 656 657 657 657 663 663 665 668 669 671 672 673 674 § 23. Real Decreto 437/2002, de 10 de mayo, por el que se establecen los criterios para la concesión de licencias de funcionamiento a los fabricantes de productos sanitarios a medida . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 675 Preámbulo . . . . . . . . . . Artículos . . . . . . . . . . . Disposiciones adicionales Disposiciones transitorias Disposiciones finales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 675 675 678 678 678 § 24. Real Decreto 1085/2009, de 3 de julio, por el que se aprueba el Reglamento sobre instalación y utilización de aparatos de rayos X con fines de diagnóstico médico . . . . . . . . . . . . . . . . . . . 679 Preámbulo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Artículos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones adicionales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones transitorias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones derogatorias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones finales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . REGLAMENTO SOBRE INSTALACIÓN Y UTILIZACIÓN DE APARATOS DE RAYOS X CON FINES DE DIAGNÓSTICO MÉDICO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO I. Disposiciones generales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO II. De las empresas de venta y asistencia técnica . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO III. Procedimiento de declaración y registro de los equipos e instalaciones de rayos X de diagnóstico médico . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO IV. Operación de las instalaciones de rayos X de diagnóstico médico . . . . . . . . . . . . . . . . . . . . CAPÍTULO V. De los Servicios y Unidades Técnicas de Protección Radiológica que prestan sus servicios en instalaciones de rayos X de diagnóstico médico . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO VI. Régimen sancionador . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ANEXO I.a. DECLARACIÓN DEL TITULAR PARA EL REGISTRO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ANEXO I.b. DESCRIPCIÓN DEL PROYECTO Y PLANOS DE LAS SALAS O VEHÍCULOS . . . . . . . . . . . . . . . ANEXO II. CERTIFICADO DE CONFORMIDAD DE LOS EQUIPOS PARA SU REGISTRO (EVAT) . . . . . . . . . . ANEXO III. CERTIFICADO DE CONFORMIDAD DE LA INSTALACIÓN PARA SU REGISTRO (SPR/UTPR) . . . . 679 681 681 682 682 682 683 683 684 687 688 693 695 696 698 699 701 ENSAYOS CLÍNICOS Y ESTUDIOS POSAUTORIZACIÓN § 25. Real Decreto 1090/2015, de 4 de diciembre, por el que se regulan los ensayos clínicos con medicamentos, los Comités de Ética de la Investigación con medicamentos y el Registro Español de Estudios Clínicos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Preámbulo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO I. Disposiciones generales . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO II. Protección de los sujetos del ensayo y consentimiento informado . . CAPÍTULO III. Indemnización por daños y perjuicios y régimen de responsabilidad – XIII – . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 702 702 705 710 713 CÓDIGO DE DERECHO FARMACÉUTICO ÍNDICE SISTEMÁTICO CAPÍTULO IV. Comités de Ética de la Investigación con medicamentos . . . . . . . . . . CAPÍTULO V. Presentación, validación y procedimiento de evaluación y autorización con medicamentos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 1.ª Disposiciones comunes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 2.ª Procedimiento de autorización de un ensayo clínico . . . . . . . . . . . . . CAPÍTULO VI. De la continuación del tratamiento tras el ensayo . . . . . . . . . . . . . . . CAPÍTULO VII. Aspectos económicos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO VIII. Medicamentos utilizados en un ensayo clínico . . . . . . . . . . . . . . . . CAPÍTULO IX. Normas de buena práctica clínica. . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO X. Verificación del cumplimiento de las normas de buena práctica clínica . . CAPÍTULO XI. Comunicaciones . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO XII. Vigilancia de la seguridad de los medicamentos en investigación . . . . CAPÍTULO XIII. Infracciones. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones adicionales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones transitorias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones derogatorias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones finales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ANEXO. Datos que incluirá el REec . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . de los ensayos clínicos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . § 26. Orden SAS/3470/2009, de 16 de diciembre, por la que se publican las directrices sobre estudios posautorización de tipo observacional para medicamentos de uso humano . . . . . . . Preámbulo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Artículos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones finales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ANEXO. Directrices de estudios posautorización de tipo observacional para medicamentos de uso humano . 1. Antecedentes y base legal . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2. Ámbito de aplicación de las directrices . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3. Definiciones . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4. Objetivos de los estudios posautorización . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5. Consideraciones éticas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6. Identificación de los responsables y elementos que debe contener el protocolo del estudio . . . . . . . . . . 7. Procedimientos administrativos para los estudios posautorización de tipo observacional . . . . . . . . . . . . 8. Seguimiento de los estudios posautorización . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 714 718 718 719 722 722 723 725 729 730 732 735 735 736 738 738 740 742 . . . . . . . . . . . . 742 742 743 743 743 744 744 745 745 746 748 752 § 27. Ley 16/1997, de 25 de abril, de Regulación de Servicios de las Oficinas de Farmacia . . . . . . . . 756 OFICINAS DE FARMACIA Preámbulo . . . . . . . . . . . Artículos . . . . . . . . . . . . Disposiciones transitorias . Disposiciones derogatorias Disposiciones finales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 756 757 759 759 759 § 28. Real Decreto 1718/2010, de 17 de diciembre, sobre receta médica y órdenes de dispensación 761 RECETA MÉDICA Preámbulo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO I. Definiciones y ámbito de aplicación . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO II. Requisitos comunes de las recetas médicas públicas y privadas. . . . . . . . . . . CAPÍTULO III. Las recetas médicas oficiales del Sistema Nacional de Salud en soporte papel . CAPÍTULO IV. La receta médica electrónica oficial del Sistema Nacional de Salud . . . . . . . . CAPÍTULO V. La receta médica privada . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO VI. Dispensación de recetas médicas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO VII. Orden de dispensación hospitalaria pública y privada. . . . . . . . . . . . . . . . . CAPÍTULO VIII. De la custodia y protección de datos . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO IX. Régimen sancionador . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones adicionales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones transitorias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones derogatorias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . – XIV – . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 761 762 763 765 767 771 771 774 775 776 776 778 778 CÓDIGO DE DERECHO FARMACÉUTICO ÍNDICE SISTEMÁTICO Disposiciones finales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ANEXO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 778 779 AGENCIA ESPAÑOLA DE MEDICAMENTOS Y PRODUCTOS SANITARIOS § 29. Real Decreto 1275/2011, de 16 de septiembre, por el que se crea la Agencia estatal "Agencia Española de Medicamentos y Productos Sanitarios" y se aprueba su Estatuto . . . . . . . . . . . . Preámbulo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Artículos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones adicionales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones transitorias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones derogatorias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Disposiciones finales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ESTATUTO DE LA AGENCIA ESPAÑOLA DE MEDICAMENTOS Y PRODUCTOS SANITARIOS. CAPÍTULO I. Disposiciones generales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO II. Objeto y competencias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO III. Órganos y estructura de la Agencia. . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 1.ª Órganos de Gobierno . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 2.ª Órgano Ejecutivo. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 3.ª Comisión de Control . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 4.ª Órganos complementarios. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 5.ª Estructura administrativa . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Sección 6.ª Red de Expertos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO IV. Régimen del personal de la Agencia . . . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO V. El Contrato de gestión y el Plan de Acción Anual . . . . . . . . . . . . . . . . . . . . CAPÍTULO VI. Régimen patrimonial y de contratación . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO VII. De la Asistencia Jurídica a la Agencia . . . . . . . . . . . . . . . . . . . . . . . . . . CAPÍTULO VIII. Régimen económico-financiero, presupuestario, de contabilidad y de control . – XV – . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 785 785 788 789 789 790 791 791 791 792 796 796 798 799 800 812 817 817 819 820 821 821 CÓDIGO DE DERECHO FARMACÉUTICO §1 Real Decreto Legislativo 1/2015, de 24 de julio, por el que se aprueba el texto refundido de la Ley de garantías y uso racional de los medicamentos y productos sanitarios Ministerio de Sanidad, Servicios Sociales e Igualdad «BOE» núm. 177, de 25 de julio de 2015 Última modificación: 6 de agosto de 2018 Referencia: BOE-A-2015-8343 I La disposición final cuarta de la Ley 10/2013, de 24 de julio, por la que se incorporan al ordenamiento jurídico español las Directivas 2010/84/UE del Parlamento Europeo y del Consejo, de 15 de diciembre de 2010, sobre farmacovigilancia, y 2011/62/UE del Parlamento Europeo y del Consejo, de 8 de junio de 2011, sobre prevención de la entrada de medicamentos falsificados en la cadena de suministro legal, y se modifica la Ley 29/2006, de 26 de julio, de garantías y uso racional de los medicamentos y productos sanitarios, autoriza al Gobierno para elaborar un texto refundido de la Ley de garantías y uso racional de los medicamentos y productos sanitarios. Esta autorización, por un plazo de dos años a partir de la entrada en vigor del texto legal habilitante, tiene por objeto consolidar, en un texto único, las sucesivas modificaciones que se han ido incorporando, desde su entrada en vigor, en la citada ley e incluye la facultad de regularizar, aclarar y armonizar los textos legales que deben ser refundidos. La autorización que da cobertura al presente texto refundido tiene su razón de ser en la necesidad de dotar de una mayor seguridad jurídica a una regulación que se ha caracterizado por una continua sucesión de normas que han completado o modificado, de forma muy dispar, el texto original de la Ley 29/2006, de 26 de julio, lo que aconseja la aprobación de un texto único en el que se incluyan, debidamente armonizadas, todas las disposiciones aplicables en el ámbito de esta ley. El texto resultante debería tener, así, una vocación de estabilidad, una vez que se han culminado con éxito los necesarios procesos de consolidación y adaptación imprescindibles para asegurar la continuidad de la prestación pública sanitaria y mejorado los mecanismos de farmacovigilancia y de protección de la cadena de suministro. La Ley 29/2006, de 26 de julio, pretendió, al igual que la Ley 25/1990, de 20 de diciembre, del Medicamento, derogada por ella, dotar a la sociedad española de un instrumento institucional que permitiera que los problemas relativos a los medicamentos fueran abordados por cuantos agentes sociales se vieran involucrados en su manejo, en la perspectiva del perfeccionamiento de la atención a la salud. El tiempo transcurrido desde la aprobación de ambos textos legales permite afirmar que se ha alcanzado en gran parte el –1– CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios objetivo pretendido consagrándose la prestación farmacéutica como una prestación universal. La prestación farmacéutica comprende los medicamentos y los productos sanitarios, así como el conjunto de actuaciones encaminadas a que los pacientes los reciban y los utilicen de forma adecuada a sus necesidades clínicas y en las dosis precisas según sus requerimientos individuales, durante el período de tiempo adecuado, con la información necesaria para su correcto uso y al menor coste posible. Es necesario hacer una valoración positiva de lo que son y de lo que representan los medicamentos y los productos sanitarios para el Sistema Nacional de Salud por lo que la política farmacéutica desarrollada en las últimas décadas se ha orientado en la dirección de asegurar su disponibilidad para cubrir las necesidades de los pacientes. En este aspecto, el papel de los profesionales del sector ha sido fundamental para alcanzar estos logros. El médico es una figura central en las estrategias de impulso de la calidad en la prestación farmacéutica dado el papel que se le atribuye en el cuidado de la salud del paciente y, por tanto, en la prevención y en el diagnóstico de la enfermedad, así como en la prescripción, en su caso, del tratamiento con medicamentos. El trabajo que los farmacéuticos y otros profesionales sanitarios realizan en los procedimientos de atención farmacéutica también tiene una importancia esencial ya que asegura la accesibilidad al medicamento ofreciendo, en coordinación con el médico, consejo sanitario, seguimiento farmacoterapéutico y apoyo profesional a los pacientes. El desafío sigue siendo, mucho más en la situación económica actual, asegurar la calidad de la prestación en todo el Sistema Nacional de Salud en un marco descentralizado capaz de impulsar el uso racional de los medicamentos y en el que el objetivo central sea que todos los ciudadanos sigan teniendo acceso al medicamento que necesiten, cuando y donde lo necesiten, en condiciones de efectividad y seguridad. II La transferencia de competencias a las comunidades autónomas en materia de sanidad es hoy una realidad al haberse completado la descentralización sanitaria prevista en la Ley 14/1986, de 25 de abril, General de Sanidad. Así, desde comienzos del año 2002, todas las comunidades autónomas han asumido las funciones que venía desempeñando y los servicios que venía prestando el Instituto Nacional de la Salud (INSALUD), lo que supone una descentralización completa de la asistencia sanitaria del Sistema Nacional de Salud, incluida la de la prestación farmacéutica. La gestión de las comunidades autónomas en materia de sanidad comprende un amplio espectro de políticas en cuanto a prioridades en el tratamiento de los problemas de salud, introducción de nuevas tecnologías y nuevos tratamientos, promoción de las alternativas más eficientes en los procesos diagnósticos y terapéuticos desarrollados por los profesionales de las respectivas comunidades autónomas, así como en políticas de rentas que afectan a los sistemas retributivos y de incentivos económicos a profesionales y centros sanitarios, todo ello dentro del amplio margen que corresponde al ejercicio de las competencias asumidas en el marco de los criterios establecidos por la Ley 16/2003, de 28 de mayo, de cohesión y calidad del Sistema Nacional de Salud, y demás normativa estatal sobre la materia. Las políticas farmacéuticas han establecido estrategias orientadas a intensificar el uso racional de los medicamentos, pudiendo destacar las orientadas a ofrecer una información de calidad, periódica e independiente, a los profesionales sanitarios para garantizar una formación adecuada sobre el uso racional de los medicamentos, reforzando la exigencia de la receta médica como documento imprescindible para la seguridad del paciente, o las referidas a la modificación de los prospectos de los medicamentos para hacerlos inteligibles a los ciudadanos, ayudando a la consecución de la necesaria adherencia al tratamiento para que pueda alcanzarse el éxito terapéutico previsto por el médico con la imprescindible cooperación del farmacéutico. Es necesario que nuestro Sistema Nacional de Salud garantice a los profesionales sanitarios que la información, la formación y la promoción comercial de los medicamentos tengan como elementos centrales de su desarrollo el rigor científico, la transparencia y la ética en la práctica de estas actividades. –2– CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios Aunque los medicamentos han contribuido decisivamente a la mejora de la esperanza y al aumento de la calidad de vida de los ciudadanos, en ocasiones plantean problemas de efectividad y de seguridad que han de ser conocidos por los profesionales sanitarios. Por este motivo, cobra especial relevancia el protagonismo que esta ley otorga al sistema español de farmacovigilancia del Sistema Nacional de Salud, con un enfoque más innovador, que incorpora el concepto de farmacoepidemiología y gestión de los riesgos, así como la garantía de seguimiento continuado del balance beneficio/riesgo de los medicamentos autorizados. Los próximos años dibujan un panorama caracterizado por un sensible aumento de la población, pero con un marcado envejecimiento de la misma y, por tanto, con unas mayores necesidades sanitarias derivadas de este fenómeno, así como de la cronificación de numerosas patologías. Estas necesidades tienen que garantizarse en un marco riguroso en cuanto a las exigencias de seguridad y eficacia de los medicamentos en beneficio de la calidad asistencial para los ciudadanos. El crecimiento sostenido de las necesidades en materia de prestación farmacéutica tendrá, por tanto, que enmarcarse necesariamente en estrategias de uso racional de los medicamentos y de control del gasto farmacéutico que permitan seguir asegurando una prestación universal de calidad contribuyendo a la sostenibilidad del Sistema Nacional de Salud. En este sentido, la ley considera necesario que la financiación selectiva y no indiscriminada de medicamentos se realice en función de la utilidad terapéutica de los mismos y de su necesidad para mejorar la salud de los ciudadanos. Cabe recordar que la aparición en estos años de los medicamentos genéricos, de eficacia clínica demostrada y más económicos, al haber expirado el periodo de exclusividad de datos del medicamento original, asegura idénticas condiciones de calidad, seguridad y eficacia a menor precio. III Este texto refundido aborda todos estos aspectos al incorporar las modificaciones producidas en la materia desde la entrada en vigor de la Ley 29/2006, de 26 de julio. La primera modificación de esta ley se introdujo mediante la Ley 51/2007, de 26 de diciembre, de Presupuestos Generales del Estado para el año 2008, con el exclusivo objeto de fijar las diversas tarifas de la tasa por prestación de servicios y realización de actividades de la Administración General del Estado en materia de medicamentos. Dos años más tarde, la Ley 25/2009, de 22 de diciembre, de modificación de diversas leyes para su adaptación a la Ley sobre el libre acceso a las actividades de servicios y su ejercicio, modificó, entre otras muchas normas, la Ley 29/2006, de 26 de julio, con el objeto de adaptar la misma a lo dispuesto en la Ley 17/2009, de 23 de noviembre, sobre el libre acceso a las actividades de servicios y su ejercicio, y de suprimir requisitos o trabas no justificados o desproporcionados con el claro objetivo de impulsar la actividad económica. Prácticamente sin solución de continuidad se aprobó la Ley 28/2009, de 30 de diciembre, de modificación de la Ley 29/2006, de 26 de julio, de garantías y uso racional de los medicamentos y productos sanitarios, con el objeto de contemplar la participación en la prescripción de medicamentos y productos sanitarios de otros profesionales sanitarios distintos de los médicos y odontólogos, como era el caso de los enfermeros y podólogos. Como consecuencia de la crisis económica iniciada en el año 2008, la Ley 29/2006, de 26 de julio, ha seguido experimentando diversas modificaciones. Algunas de ellas han sido de tipo técnico, como las relativas a las garantías de eficacia, seguridad y calidad de los medicamentos y productos sanitarios, pero las más significativas se han producido sobre los aspectos económicos, siendo el más notorio el atinente a las iniciativas de control del gasto farmacéutico. La necesidad de este control no obedecía sólo a la obligada eficiencia en la gestión del gasto público, máxime en una situación de grave crisis, sino que dicho control era también necesario en cuanto a la mejora tecnológica de los propios medicamentos, así como por la aparición de nuevos medicamentos, algunos de los cuales introdujeron sustanciales avances en el tratamiento terapéutico y, por consiguiente, en el estado de salud. La positiva evolución tecnológica se produjo cuando la mayor parte de los países del mundo habían incorporado a sus ordenamientos jurídicos los Acuerdos sobre los aspectos de los Derechos de Propiedad Intelectual relacionados con el Comercio (ADPIC), lo que en el terreno –3– CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios farmacéutico significa la plena protección de los descubrimientos patentados. Este fenómeno propició un incremento en los precios exigidos para las innovaciones farmacológicas lo que, junto a otros factores, determinó, a la larga, un incremento de los presupuestos de gasto farmacéutico que crecieron por encima de los parámetros que caracterizan la riqueza de las naciones –Producto Interior Bruto (PIB) per cápita– o del nivel de desarrollo del Estado de bienestar –porcentaje sobre PIB dedicado a asistencia sanitaria–. España, donde la cobertura del gasto farmacéutico por parte del Sistema Nacional de Salud es muy elevada, sufrió más que otros países los embates de la crisis económica, lo que obligó a incorporar políticas de eficiencia en el gasto sanitario. De este modo, la contención del gasto farmacéutico requirió de reformas urgentes y, a este efecto, se promulgaron varias normas, en concreto, el Real Decreto-ley 4/2010, de 26 de marzo, de racionalización del gasto farmacéutico con cargo al Sistema Nacional de Salud, el Real Decreto-ley 8/2010, de 20 de mayo, por el que se adoptan medidas extraordinarias para la reducción del déficit público, y un año más tarde, el Real Decreto-ley 9/2011, de 19 de agosto, de medidas para la mejora de la calidad y cohesión del Sistema Nacional de Salud, de contribución a la consolidación fiscal y de elevación del importe máximo de los avales del Estado para 2011, que introdujeron descuentos y limitaciones de orden general, afectando a la oferta de medicamentos. El Real Decreto-ley 4/2010, de 26 de marzo, constituyó la primera reacción ante la crisis económica iniciada años antes y perseguía el objetivo inaplazable de modificar la financiación pública de los medicamentos y productos sanitarios prevista en la Ley 29/2006, de 26 de julio, para facilitar la aplicación del sistema de precios de referencia e introducir descuentos y limitaciones que redujeran el gasto farmacéutico con cargo al Sistema Nacional de Salud. El posterior Real Decreto-ley 8/2010, de 20 de mayo, abordó el establecimiento de medidas complementarias a las ya adoptadas en el marco de la prestación farmacéutica para establecer nuevas deducciones y reducciones de precios. El sistema de precios de referencia contemplado en la Ley 29/2006, de 26 de julio, experimentó una nueva variación por medio de la Ley 34/2010, de 5 de agosto, de modificación de las Leyes 30/2007, de 30 de octubre, de Contratos del Sector Público, 31/2007, de 30 de octubre, sobre procedimientos de contratación en los sectores del agua, la energía, los transportes y los servicios postales, y 29/1998, de 13 de julio, reguladora de la Jurisdicción Contencioso-Administrativa para adaptación a la normativa comunitaria de las dos primeras, mientras que el régimen de incompatibilidades de los profesionales sanitarios también se modificó por la Ley 14/2011, de 1 de junio, de la Ciencia, la Tecnología y la Innovación. Por último, el Real Decreto-ley 9/2011, de 19 de agosto, tuvo por objeto generalizar la prescripción de medicamentos por principio activo, modificar el sistema de precios de referencia, haciéndolo más ágil y fácil de gestionar, y mejorar los criterios para la financiación selectiva de medicamentos, incorporando a la prestación aquellos que ofrecen mejoras sustanciales en los tratamientos. Por su especial trascendencia en el ámbito de la consolidación de la prestación pública sanitaria, y no únicamente en relación con la modificación de la Ley 29/2006, de 26 de julio, a la que dedicó su capítulo IV, destaca el Real Decreto-ley 16/2012, de 20 de abril, de medidas urgentes para garantizar la sostenibilidad del Sistema Nacional de Salud y mejorar la calidad y seguridad de sus prestaciones, norma que abordó una reforma sustancial de carácter global introduciendo reformas sobre la demanda. La reforma introducida racionaliza la financiación farmacéutica mediante la exclusión de la financiación pública de aquellos medicamentos destinados al tratamiento de síntomas menores, introduce una modificación estructural al sistema de fijación de precios de los medicamentos, con un esquema innovador de precios seleccionados, marcando un cambio hacia la financiación selectiva con criterios como el coste-efectividad y la valoración del impacto presupuestario, al lado de otros elementos cualitativos que han permitido modular la cartera de medicamentos financiados. Y, sin duda, cabe destacar, como medida de mayor trascendencia social, la consistente en establecer la aportación de los beneficiarios al gasto farmacéutico en función de su capacidad económica, buscando un uso más responsable de la prestación así como un reparto más equitativo y sostenible del esfuerzo de financiación, mejorando con ello –4– CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios el sistema mantenido inicialmente por la Ley 29/2006, de 26 de julio, que era el vigente desde los años ochenta del siglo pasado. Una nueva modificación de la Ley 29/2006, de 26 de julio, tuvo lugar mediante el Real Decreto-ley 28/2012, de 30 de noviembre, de medidas de consolidación y garantía del sistema de la Seguridad Social, que redefinió la prestación farmacéutica ambulatoria considerando como tal la que se dispensa al paciente mediante receta médica u orden de dispensación hospitalaria a través de oficinas o servicios de farmacia. La última reforma de calado de la Ley 29/2006, de 26 de julio, fue la operada por la Ley 10/2013, de 24 de julio, que precisamente incorporó la autorización de las Cortes Generales al Gobierno para la aprobación de este texto refundido. La finalidad principal de dicha ley fue la de incorporar al ordenamiento jurídico español las Directivas 2010/84/UE del Parlamento Europeo y del Consejo, de 15 de diciembre de 2010, sobre farmacovigilancia, y 2011/62/UE del Parlamento Europeo y del Consejo, de 8 de junio de 2011, sobre prevención de la entrada de medicamentos falsificados en la cadena de suministro legal. Pero, más allá de dar satisfacción a dicha finalidad, se modificaron otros aspectos sustanciales de la ley ajenos a las mencionadas normas europeas, entre los que cabe destacar los de la adecuación técnica del procedimiento sancionador y el régimen de los ingresos públicos por actuaciones de la Agencia Española de Medicamentos y Productos Sanitarios o la introducción de una serie de mejoras consistentes en extender el régimen hasta ahora aplicable a los medicamentos de uso humano también a los medicamentos veterinarios, a los productos sanitarios, a los cosméticos y a los productos de cuidado personal, a fin de ofrecer una regulación general completa en el marco de la Ley 29/2006, de 26 de julio, sin perjuicio de los correspondientes desarrollos reglamentarios. Asimismo, al objeto de agilizar el sector farmacéutico se incorporaron algunas previsiones que, hasta entonces referidas a autorizaciones, establecieron la posibilidad de realizar notificaciones, en la medida en que así resultase posible, pues la autorización no es, ciertamente, el único mecanismo de control de las actividades a que se refiere la Ley 29/2006, de 26 de julio. Por último, se introdujo un cambio relevante en el informe de posicionamiento terapéutico como herramienta clave en la utilización correcta y eficiente de los nuevos medicamentos. Finalmente, la última modificación de la Ley 29/2006, de 26 de julio, previa a la aprobación de este texto refundido, ha sido la operada mediante la Ley 36/2014, de 26 de diciembre, de Presupuestos Generales del Estado para el año 2015, para modificar dos tasas por prestación de servicios y realización de actividades de la Administración General del Estado en materia de medicamentos, productos sanitarios, productos cosméticos y productos de cuidado personal. IV La apretada síntesis expuesta da cuenta de la conveniencia de enmarcar el crecimiento sostenido de las necesidades en materia de prestación farmacéutica dentro de estrategias de uso racional de los medicamentos y de control del gasto farmacéutico que permitan seguir asegurando una prestación universal de calidad, contribuyendo a la sostenibilidad del Sistema Nacional de Salud en unos momentos específicos de dificultad económica y cambio tecnológico. La integración normativa de todos los cambios operados en esta materia requiere de una imprescindible unificación sistemática por lo que, de acuerdo con la autorización contenida en la disposición final cuarta de la Ley 10/2013, de 24 de julio, se aprueba este texto refundido siguiendo los criterios que a continuación se exponen. En primer lugar, se ha procedido a integrar en un texto único todas las modificaciones vigentes introducidas en la Ley 29/2006, de 26 de julio, a través de diversas leyes que han dado una nueva redacción a determinados preceptos o que han introducido nuevas disposiciones a aquella. Como consecuencia de lo anterior, y al amparo de la facultad concedida para regularizar, aclarar y armonizar los textos legales que deben ser refundidos, se ha ajustado la ubicación y numeración de los artículos, así como las remisiones y concordancias entre ellos, y se han homogeneizado algunos términos empleados en la misma. Debe aclararse que, en cumplimiento de esta delegación legislativa, solo se regularizan, aclaran y armonizan aquellas disposiciones de los textos legales que han modificado –5– CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios expresamente la Ley 29/2006, de 26 de julio, dando nueva redacción o incluyendo nuevos preceptos a dicha ley, pues el legislador ha establecido claramente como marco normativo de la refundición la consolidación de las distintas normas que han modificado la Ley 29/2006, de 26 de julio, lo que no es extensible, en consecuencia, a normas legales que no han dado nueva redacción o que no han incorporado nuevos preceptos a la misma. Por otro lado, y al amparo también de dicha facultad concedida al Gobierno, se han tenido en cuenta otras normas cuya aplicación exigía la adaptación de determinados aspectos contenidos en la Ley 29/2006, de 26 de julio. Se trata de aspectos tales como la eliminación de la previsión de actualizar determinadas cantidades o importes con arreglo al Índice de Precios al Consumo, en aplicación de la Ley 2/2015, de 30 de marzo, de desindexación de la economía española, o la actualización del importe de las tasas conforme a lo dispuesto por la Ley 36/2014, de 26 de diciembre. No ha ocurrido lo mismo, sin embargo, con la adaptación a la Ley 20/2013, de 9 de diciembre, de garantía de la unidad de mercado, pues una revisión integral de la Ley 29/2006, de 26 de julio, para adaptarla a aquella hubiera excedido de la habilitación concedida para regularizar, aclarar y armonizar los textos legales que debían ser refundidos. Ahora bien, debe quedar claro que la aprobación de este texto refundido no supone un obstáculo, desde luego, para que se continúe trabajando en una revisión generalizada del mismo con objeto de adaptarlo a los criterios señalados por la Ley 20/2013, de 9 de diciembre, revisión que, en todo caso, deberá someterse a los cauces y trámites ordinarios previstos para la modificación de disposiciones legales. Finalmente, y al amparo de la autorización recibida, se han revisado y modificado las disposiciones adicionales, transitorias y finales de la Ley 29/2006, de 26 de julio, por la necesidad de adaptar su contenido al tiempo transcurrido desde su aprobación y de sus sucesivas modificaciones. Así, el contenido de la disposición adicional duodécima, relativa a la participación de los enfermeros en el ámbito de los medicamentos sujetos a prescripción médica, ha sido incorporado, por razones sistemáticas, en el artículo 79 de este texto refundido por su íntima conexión con el mismo. En cuanto se refiere a las disposiciones transitorias de la Ley 29/2006, de 26 de julio, algunas de ellas, en concreto, las numeradas como cuarta, quinta, sexta, séptima y novena, no se han incluido en el texto refundido al haber agotado su vigencia por el transcurso del espacio temporal previsto en ellas. Se trata, respectivamente, de las relativas a la adaptación de autorizaciones para ejercer actividades de distribución, al plazo establecido para incluir las indicaciones en alfabeto braille en los envases y prospectos de los medicamentos, de la relativa a las innovaciones galénicas, de la relativa a la indicación del período en el que debía comenzar a aplicarse el régimen de aportaciones al Sistema Nacional de Salud al que se refiere la disposición adicional sexta y, por último, de la relativa al período de adaptación de determinadas garantías de información. Por otra parte, el contenido de otras disposiciones que figuraban como transitorias en la citada ley, en concreto, las numeradas como tercera y octava, aparecen incorporadas en el texto refundido como disposiciones adicionales decimoquinta y decimosexta, al ser su contenido más propio de una disposición de estas características. Se trata, respectivamente, de las relativas a la aplicación del régimen de los informes periódicos de seguridad a las autorizaciones de medicamentos renovadas tras la entrada en vigor de la Ley 29/2006, de 26 de julio, y a la aplicación del sistema de fijación de precios en los productos sanitarios autorizados con anterioridad a la entrada en vigor de dicha ley. Por último, se ha efectuado la correspondiente adecuación de la disposición final primera para adaptar la referencia de los títulos competenciales en base a los cuales se regula cada una de las materias objeto de esta norma a la nueva estructura de la misma. Este real decreto legislativo, sobre el que se ha concedido audiencia a los sectores afectados, ha sido sometido a consulta tanto de las Comunidades Autónomas y Ciudades de Ceuta y Melilla, como de la Federación Española de Municipios y Provincias. Además, ha sido informado por el Consejo Económico y Social, por la Comisión Nacional de los Mercados y la Competencia, por la Agencia Española de Protección de Datos, por el Consejo de Consumidores y Usuarios y por el Comité Consultivo y el Consejo Interterritorial del Sistema Nacional de Salud. –6– CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios En su virtud, a propuesta del Ministro de Sanidad, Servicios Sociales e Igualdad, de acuerdo con el Consejo de Estado y previa deliberación del Consejo de Ministros, en su reunión del día 24 de julio de 2015, DISPONGO: Artículo único. Aprobación del texto refundido de la Ley de garantías y uso racional de los medicamentos y productos sanitarios. Se aprueba el texto refundido de la Ley de garantías y uso racional de los medicamentos y productos sanitarios, cuyo texto se inserta a continuación. Disposición adicional única. Remisiones normativas. Las referencias normativas efectuadas en otras disposiciones a la Ley 29/2006, de 26 de julio, de garantías y uso racional de los medicamentos y productos sanitarios, se entenderán efectuadas a los preceptos correspondientes del texto refundido que se aprueba. Disposición derogatoria única. Derogación normativa. Quedan derogadas cuantas disposiciones, de igual o inferior rango, se opongan a lo establecido en esta ley y, en particular, la Ley 29/2006, de 26 de julio, de garantías y uso racional de los medicamentos y productos sanitarios, a excepción de sus disposiciones finales segunda, tercera y cuarta. Disposición final única. Entrada en vigor. El presente real decreto legislativo y el texto refundido que aprueba entrarán en vigor el mismo día de su publicación en el «Boletín Oficial del Estado». TÍTULO PRELIMINAR Disposiciones generales Artículo 1. Objeto y ámbito de aplicación de la ley. Esta ley regula, en el ámbito de las competencias que corresponden al Estado: a) Los medicamentos de uso humano y productos sanitarios, su investigación clínica, su evaluación, autorización, registro, fabricación, elaboración, control de calidad, almacenamiento, distribución, circulación, trazabilidad, comercialización, información y publicidad, importación y exportación, prescripción y dispensación, seguimiento de la relación beneficio-riesgo, así como la ordenación de su uso racional y el procedimiento para, en su caso, la financiación con fondos públicos. La regulación también se extiende a las sustancias, excipientes y materiales utilizados para su fabricación, preparación o envasado. b) La actuación de las personas físicas o jurídicas en cuanto intervienen en la circulación industrial o comercial y en la prescripción o dispensación de los medicamentos y productos sanitarios. c) Los criterios y exigencias generales aplicables a los medicamentos veterinarios y, en particular, a los especiales, como las fórmulas magistrales, y los relativos a los elaborados industrialmente, incluidas las premezclas para piensos medicamentosos. d) Los cosméticos y productos de cuidado personal y, en particular, las medidas cautelares y el régimen de infracciones y sanciones aplicables a estos. Artículo 2. Definiciones. A los efectos de esta ley se entenderá por: a) «Medicamento de uso humano»: Toda sustancia o combinación de sustancias que se presente como poseedora de propiedades para el tratamiento o prevención de –7– CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios enfermedades en seres humanos o que pueda usarse en seres humanos o administrarse a seres humanos con el fin de restaurar, corregir o modificar las funciones fisiológicas ejerciendo una acción farmacológica, inmunológica o metabólica, o de establecer un diagnóstico médico. b) «Medicamento veterinario»: Toda sustancia o combinación de sustancias que se presente como poseedora de propiedades curativas o preventivas con respecto a las enfermedades animales o que pueda administrarse al animal con el fin de restablecer, corregir o modificar sus funciones fisiológicas ejerciendo una acción farmacológica, inmunológica o metabólica, o de establecer un diagnóstico veterinario. También se considerarán «medicamentos veterinarios» las «premezclas para piensos medicamentosos» elaboradas para ser incorporadas a un pienso. c) «Principio activo» o «sustancia activa»: Toda sustancia o mezcla de sustancias destinadas a la fabricación de un medicamento y que, al ser utilizadas en su producción, se convierten en un componente activo de dicho medicamento destinado a ejercer una acción farmacológica, inmunológica o metabólica con el fin de restaurar, corregir o modificar las funciones fisiológicas, o de establecer un diagnóstico. d) «Excipiente»: Todo componente de un medicamento distinto del principio activo y del material de acondicionamiento. e) «Materia prima»: Toda sustancia –activa o inactiva– empleada en la fabricación de un medicamento, ya permanezca inalterada, se modifique o desaparezca en el transcurso del proceso. f) «Forma galénica» o «forma farmacéutica»: La disposición a que se adaptan los principios activos y excipientes para constituir un medicamento. Se define por la combinación de la forma en la que el producto farmacéutico es presentado por el fabricante y la forma en la que es administrado. g) «Medicamento genérico»: Todo medicamento que tenga la misma composición cualitativa y cuantitativa en principios activos y la misma forma farmacéutica, y cuya bioequivalencia con el medicamento de referencia haya sido demostrada por estudios adecuados de biodisponibilidad. Las diferentes sales, ésteres, éteres, isómeros, mezclas de isómeros, complejos o derivados de un principio activo se considerarán un mismo principio activo, a menos que tengan propiedades considerablemente diferentes en cuanto a seguridad y/o eficacia. Las diferentes formas farmacéuticas orales de liberación inmediata se considerarán una misma forma farmacéutica. El solicitante podrá estar exento de presentar los estudios de biodisponibilidad si puede demostrar que el medicamento genérico satisface los criterios pertinentes definidos en las correspondientes directrices detalladas. h) «Producto intermedio»: El destinado a una posterior transformación industrial por un fabricante autorizado. i) «Fórmula magistral»: El medicamento destinado a un paciente individualizado, preparado por un farmacéutico, o bajo su dirección, para cumplimentar expresamente una prescripción facultativa detallada de los principios activos que incluye, según las normas de correcta elaboración y control de calidad establecidas al efecto, dispensado en oficina de farmacia o servicio farmacéutico y con la debida información al usuario en los términos previstos en el artículo 42.5. j) «Preparado oficinal»: Aquel medicamento elaborado según las normas de correcta elaboración y control de calidad establecidas al efecto y garantizado por un farmacéutico o bajo su dirección, dispensado en oficina de farmacia o servicio farmacéutico, enumerado y descrito por el Formulario Nacional, destinado a su entrega directa a los enfermos a los que abastece dicha farmacia o servicio farmacéutico. k) «Medicamento en investigación»: Forma farmacéutica de un principio activo o placebo que se investiga o se utiliza como referencia en un ensayo clínico, incluidos los productos con autorización cuando se utilicen o combinen, en la formulación o en el envase, de forma diferente a la autorizada, o cuando se utilicen para tratar una indicación no autorizada o para obtener más información sobre un uso autorizado. l) «Producto sanitario»: Cualquier instrumento, dispositivo, equipo, programa informático, material u otro artículo, utilizado solo o en combinación, incluidos los programas informáticos destinados por su fabricante a finalidades específicas de diagnóstico y/o terapia y que –8– CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios intervengan en su buen funcionamiento, destinado por el fabricante a ser utilizado en seres humanos con fines de: 1.º Diagnóstico, prevención, control, tratamiento o alivio de una enfermedad; 2.º diagnóstico, control, tratamiento, alivio o compensación de una lesión o de una deficiencia; 3.º investigación, sustitución o modificación de la anatomía o de un proceso fisiológico; 4.º regulación de la concepción, y que no ejerza la acción principal que se desee obtener en el interior o en la superficie del cuerpo humano por medios farmacológicos, inmunológicos ni metabólicos, pero a cuya función puedan contribuir tales medios. m) «Producto de cuidado personal»: Sustancias o mezclas que, sin tener la consideración legal de medicamentos, productos sanitarios, cosméticos o biocidas, están destinados a ser aplicados sobre la piel, dientes o mucosas del cuerpo humano con finalidad de higiene o de estética, o para neutralizar o eliminar ectoparásitos. n) «Producto cosmético»: Toda sustancia o mezcla destinada a ser puesta en contacto con las partes superficiales del cuerpo humano (epidermis, sistema piloso y capilar, uñas, labios y órganos genitales externos) o con los dientes y las mucosas bucales, con el fin exclusivo o principal de limpiarlos, perfumarlos, modificar su aspecto, protegerlos, mantenerlos en buen estado o corregir los olores corporales. o) «Medicamento falsificado»: Cualquier medicamento cuya presentación sea falsa con respecto a: 1.º Su identidad, incluidos el envase y etiquetado, el nombre o composición en lo que respecta a cualquiera de sus componentes, incluidos los excipientes, y la dosificación de dichos componentes; 2.º su origen, incluidos el fabricante, el país de fabricación, el país de origen y el titular de la autorización de comercialización; o, 3.º su historial, incluidos los registros y documentos relativos a los canales de distribución empleados. La presente definición no comprende los defectos de calidad involuntarios y se entiende sin perjuicio de las violaciones de los derechos de propiedad intelectual. p) «Distribución mayorista de medicamentos»: Toda actividad que consista en obtener, almacenar, conservar, suministrar o exportar medicamentos, excluida la dispensación al público de los mismos. q) «Almacén por contrato»: Entidad que actúa como tercero, con la cual un laboratorio o un almacén mayorista suscribe un contrato para realizar determinadas actividades de distribución de medicamentos. r) «Intermediación de medicamentos»: Todas las actividades relativas a la venta o compra de medicamentos, a excepción de aquellas incluidas en la definición de distribución mayorista, tal y como se define en este artículo, que no incluyen contacto físico con los mismos y que consisten en la negociación de manera independiente y en nombre de otra persona jurídica o física. TÍTULO I Garantías y obligaciones generales Artículo 3. Garantías de abastecimiento y dispensación. 1. Los laboratorios farmacéuticos, entidades de distribución, importadores, oficinas de farmacia, servicios de farmacia de hospitales, centros de salud y demás estructuras de atención a la salud están obligados a suministrar o a dispensar los medicamentos y productos sanitarios que se les soliciten en las condiciones legal y reglamentariamente establecidas. –9– CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios 2. Los responsables de la producción, distribución, venta y dispensación de medicamentos y productos sanitarios deberán respetar el principio de continuidad en la prestación del servicio a la comunidad. 3. El Gobierno, para asegurar el abastecimiento de medicamentos, podrá adoptar medidas especiales en relación con su fabricación, importación, distribución y dispensación. En el caso de los «medicamentos huérfanos», según lo dispuesto en el Reglamento (CE) n.º 141/2000, y de los «medicamentos sin interés comercial» el Gobierno podrá adoptar, además de las medidas señaladas, las relativas al régimen económico y fiscal de dichos medicamentos. A estos efectos, así como a los previstos en el artículo 121.1, se entiende por «medicamentos sin interés comercial» aquéllos de los que existe ausencia o insuficiencia de suministro en el mercado nacional, siendo necesarios para el tratamiento de determinadas enfermedades o patologías. 4. La prescripción y dispensación de medicamentos y productos sanitarios deberá realizarse de acuerdo con los criterios básicos de uso racional que se establecen en esta ley. 5. Se prohíbe la venta, por correspondencia y por procedimientos telemáticos, de medicamentos y productos sanitarios sujetos a prescripción. La normativa de desarrollo establecerá los requisitos aplicables y regulará dichas modalidades de venta con respecto a los medicamentos no sujetos a prescripción garantizando, en todo caso, que los medicamentos de uso humano se dispensen por una oficina de farmacia autorizada, con la intervención de un farmacéutico, previo asesoramiento personalizado conforme previenen los artículos 19.4 y 86.1, y con cumplimiento de la normativa aplicable en función de los medicamentos objeto de venta o de la modalidad de venta y cumplimiento de los requisitos en materia de información recogidos en la Ley 34/2002, de 11 de julio, de servicios de la sociedad de la información y de comercio electrónico y, en el caso de los medicamentos veterinarios, se dispensen por uno de los establecimientos descritos en los párrafos a) y b) del artículo 38.2, con la intervención de un farmacéutico, debiendo asimismo cumplir con los requisitos establecidos en la Ley 34/2002, de 11 de julio. Se prohíbe, asimismo, la venta a domicilio y cualquier tipo de venta indirecta al público de medicamentos. Las Administraciones sanitarias, por razones de salud pública o seguridad de las personas, podrán limitar, condicionar o prohibir la venta a domicilio y cualquier tipo de venta indirecta al público de productos sanitarios. Lo establecido en este apartado se entiende sin perjuicio del reparto, distribución o suministro a las entidades legalmente autorizadas para la dispensación al público. La normativa de desarrollo establecerá los requisitos para que puedan venderse directamente a profesionales de la medicina, odontología, veterinaria y podología, exclusivamente, los medicamentos necesarios para el ejercicio de su actividad profesional. 6. La custodia, conservación y dispensación de medicamentos de uso humano corresponderá exclusivamente: a) A las oficinas de farmacia abiertas al público, legalmente autorizadas. b) A los servicios de farmacia de los hospitales, de los centros de salud y de las estructuras de atención primaria del Sistema Nacional de Salud para su aplicación dentro de dichas instituciones o para los medicamentos que exijan una particular vigilancia, supervisión y control del equipo multidisciplinar de atención a la salud, de conformidad con la calificación otorgada por la Agencia Española de Medicamentos y Productos Sanitarios para tales medicamentos. c) En el ámbito del Sistema Nacional de Salud, además de los medicamentos especificados en el párrafo b), corresponderá a los servicios de farmacia de los hospitales la custodia, conservación y dispensación de los medicamentos de uso humano en los que el Ministerio de Sanidad, Servicios Sociales e Igualdad acuerde establecer reservas singulares, limitando su dispensación sin necesidad de visado a los pacientes no hospitalizados. No obstante, en el caso de ensayos clínicos que se realicen en centros de investigación que no posean servicios de farmacia será posible el envío de los medicamentos en investigación por el promotor al centro de investigación, asumiendo el investigador de dicho centro las responsabilidades relativas a la correcta administración, custodia y entrega de dichos medicamentos de acuerdo con lo especificado en el protocolo del estudio. – 10 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios 7. Se prohíbe la dispensación, venta o comercialización de cualquier medicamento que sea devuelto o entregado por los pacientes, o el público en general, a las oficinas de farmacia. Artículo 4. Garantías de independencia. 1. Sin perjuicio de las incompatibilidades establecidas para el ejercicio de actividades públicas, el ejercicio clínico de la medicina, de la odontología, de la veterinaria, así como de otras profesiones sanitarias con facultad para prescribir o indicar la dispensación de los medicamentos, será incompatible con cualquier clase de intereses económicos directos derivados de la fabricación, elaboración, distribución, intermediación y comercialización de los medicamentos y productos sanitarios. Se exceptúa de lo anterior lo establecido en la Ley 14/2011, de 1 de junio, de la Ciencia, la Tecnología y la Innovación, respecto a la participación del personal de los centros de investigación dependientes de las Administraciones Públicas en las entidades creadas o participadas por aquellos, con el objeto previsto en la misma. 2. Asimismo, el ejercicio profesional del farmacéutico en oficina de farmacia, en establecimiento comercial detallista, en entidades o agrupaciones ganaderas o en un servicio de farmacia hospitalaria y demás estructuras asistenciales será incompatible con cualquier clase de intereses económicos directos de los laboratorios farmacéuticos, entidades de intermediación y/o entidades de distribución. 3. El ejercicio clínico de la medicina, odontología, veterinaria y otras profesiones sanitarias con facultad para prescribir o indicar la dispensación de los medicamentos serán incompatibles con el desempeño de actividad profesional o con la titularidad de oficina de farmacia. 4. La pertenencia a los comités de la Agencia Española de Medicamentos y Productos Sanitarios, a los Comités Éticos de Investigación Clínica o a los comités u órganos asesores o consultivos establecidos por las Administraciones sanitarias de las comunidades autónomas será incompatible con cualquier clase de intereses derivados de la fabricación y venta de los medicamentos y productos sanitarios. 5. El ejercicio de los profesionales sanitarios implicados en el ciclo de prescripción, dispensación y administración de medicamentos será incompatible con las funciones de delegados de visita médica, representantes, comisionistas o agentes informadores de los laboratorios farmacéuticos. 6. A efectos de garantizar la independencia de las decisiones relacionadas con la prescripción, dispensación, y administración de medicamentos respecto de intereses comerciales se prohíbe el ofrecimiento directo o indirecto de cualquier tipo de incentivo, bonificaciones, descuentos, primas u obsequios, por parte de quien tenga intereses directos o indirectos en la producción, fabricación y comercialización de medicamentos a los profesionales sanitarios implicados en el ciclo de prescripción, dispensación y administración de medicamentos o a sus parientes y personas de convivencia. Esta prohibición será asimismo de aplicación cuando el ofrecimiento se realice a profesionales sanitarios que prescriban productos sanitarios. Se exceptúan de la anterior prohibición los descuentos por pronto pago o por volumen de compras, que realicen los distribuidores a las oficinas de farmacia, siempre que no se incentive la compra de un producto frente al de sus competidores y queden reflejados en la correspondiente factura. Estos descuentos podrán efectuarse para los medicamentos financiados con cargo al Sistema Nacional de Salud, siempre que se lleve un registro mensual de tales descuentos en las empresas titulares de los mismos y en las entidades de distribución, interconectado telemáticamente con el Ministerio de Sanidad, Servicios Sociales e Igualdad. Artículo 5. Garantías de defensa de la salud pública. 1. Se prohíbe la elaboración, fabricación, importación, exportación, distribución, comercialización, prescripción y dispensación de productos, preparados, sustancias o combinaciones de las mismas que se presenten como medicamentos sin estar legalmente reconocidos como tales. 2. Queda expresamente prohibida la promoción, publicidad o información destinada al público de los productos incluidos en el apartado 1. – 11 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios 3. Lo establecido en los apartados anteriores será de aplicación a los productos que se presenten como productos sanitarios o como productos cosméticos sin que tengan tal consideración, así como a los productos sanitarios y a los productos cosméticos que se comercialicen sin haber seguido los procedimientos establecidos en sus normativas específicas. 4. El incumplimiento de las prohibiciones anteriores dará lugar a las responsabilidades y sanciones previstas en el capítulo II del título IX, con independencia de las medidas cautelares que procedan y de las responsabilidades civiles o penales a que haya lugar. Artículo 6. Obligaciones de las Administraciones Públicas y participación de los profesionales. 1. A efectos de salvaguardar las exigencias de salud y seguridad pública, las Administraciones públicas están obligadas a comunicarse cuantos datos, actuaciones o informaciones se deriven del ejercicio de sus competencias y resulten necesarias para la correcta aplicación de esta ley. 2. Todos los profesionales que presten sus servicios en el Sistema Nacional de Salud o en el sistema público de investigación científica y desarrollo tecnológico español tienen el derecho a participar y el deber de colaborar con las Administraciones sanitarias en la evaluación y control de medicamentos y productos sanitarios. 3. Las comisiones y comités previstos en esta ley se ajustarán a lo dispuesto sobre órganos colegiados en las disposiciones vigentes. Artículo 7. Transparencia en la adopción de decisiones por las administraciones sanitarias. Las administraciones sanitarias garantizarán la máxima transparencia en los procesos de adopción de sus decisiones en materia de medicamentos y productos sanitarios, sin perjuicio del derecho de la propiedad industrial. La participación en dichos procesos de toma de decisión será incompatible con cualquier clase de intereses personales derivados de la fabricación, comercialización, representación, distribución y venta relacionados con los medicamentos y productos sanitarios. TÍTULO II De los medicamentos CAPÍTULO I De los medicamentos reconocidos por la ley y sus clases Artículo 8. Medicamentos legalmente reconocidos. 1. Sólo serán medicamentos los que se enumeran a continuación: a) Los medicamentos de uso humano y veterinarios elaborados industrialmente o en cuya fabricación intervenga un proceso industrial. b) Las fórmulas magistrales. c) Los preparados oficinales. d) Los medicamentos especiales previstos en esta ley. 2. Tendrán el tratamiento legal de medicamentos, a efectos de la aplicación de esta ley y de su control general, las sustancias o combinaciones de sustancias autorizadas para su empleo en ensayos clínicos o para investigación en animales. 3. Corresponde a la Agencia Española de Medicamentos y Productos Sanitarios resolver sobre la atribución de la condición de medicamento. 4. Los remedios secretos están prohibidos. Serán considerados secretos aquellos productos respecto de los que se desconozca su composición y características. 5. Es obligatorio declarar a la autoridad sanitaria todas las características y propiedades conocidas de los medicamentos. – 12 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios 6. En caso de duda, cuando un producto pueda responder a la definición de medicamento se le aplicará esta ley, incluso si a dicho producto se le pudiera aplicar la definición contemplada en otra norma. CAPÍTULO II De las garantías exigibles a los medicamentos de uso humano elaborados industrialmente y de las condiciones de prescripción y dispensación de los mismos Artículo 9. Autorización y registro. 1. Ningún medicamento elaborado industrialmente podrá ser puesto en el mercado sin la previa autorización de la Agencia Española de Medicamentos y Productos Sanitarios e inscripción en el Registro de Medicamentos o sin haber obtenido la autorización de conformidad con lo dispuesto en las normas europeas que establecen los procedimientos comunitarios para la autorización y control de los medicamentos de uso humano y veterinario y que regula la Agencia Europea de Medicamentos. 2. Cuando un medicamento haya obtenido una autorización de acuerdo con el apartado anterior, toda dosificación, forma farmacéutica, vía de administración y presentaciones adicionales, así como cualesquiera otras modificaciones y ampliaciones al expediente de autorización que se introduzcan, deberán ser objeto de autorización o notificación conforme se disponga en la normativa que regula el procedimiento de autorización, registro y condiciones de dispensación de los medicamentos de uso humano fabricados industrialmente. Todas estas modificaciones se considerarán pertenecientes a la misma autorización global de comercialización, en particular a los efectos de la aplicación de los periodos de exclusividad de datos. 3. Toda modificación, transmisión y extinción de las autorizaciones de los medicamentos deberá constar en el Registro de Medicamentos que a estos efectos tendrá, del mismo modo que la inscripción, carácter constitutivo. 4. La Agencia Española de Medicamentos y Productos Sanitarios procederá de oficio a la incorporación al Registro de Medicamentos de las autorizaciones otorgadas por la Comisión Europea conforme al Reglamento (CE) n.º 726/2004/CE. Artículo 10. Garantías exigibles para la autorización de medicamentos. 1. La Agencia Española de Medicamentos y Productos Sanitarios otorgará la autorización a un medicamento si satisface las siguientes condiciones: a) Alcanzar los requisitos de calidad que se establezcan. b) Ser seguro, no produciendo en condiciones normales de utilización efectos tóxicos o indeseables desproporcionados al beneficio que procura. c) Ser eficaz en las indicaciones terapéuticas para las que se ofrece. d) Estar correctamente identificado. e) Suministrar la información precisa, en formato accesible y de forma comprensible por el paciente, para su correcta utilización. 2. La evaluación de los efectos terapéuticos positivos del medicamento se apreciarán en relación con cualquier riesgo relacionado con la calidad, la seguridad y la eficacia del medicamento para la salud del paciente o la salud pública, entendido como relación beneficio/riesgo. 3. Lo establecido en este artículo será asimismo de aplicación a las modificaciones que se produzcan en la autorización y habrá de observarse durante toda la vida del medicamento. 4. El titular de la autorización o, en su caso, el fabricante deben contar con los medios materiales y personales, la organización y la capacidad operativa suficientes para su correcta fabricación. – 13 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios Artículo 11. Garantías de calidad. 1. Todo medicamento deberá tener perfectamente establecida su composición cualitativa y cuantitativa. Alternativamente, en el caso de sustancias como las biológicas en las que esto no sea posible, sus procedimientos de preparación deben ser reproducibles. 2. La persona titular del Ministerio de Sanidad, Servicios Sociales e Igualdad establecerá el tipo de controles exigibles al laboratorio titular de la autorización de comercialización y al fabricante para garantizar la calidad de las materias primas, de los productos intermedios, del proceso de fabricación y del producto final, incluyendo envasado y conservación, a efectos de la autorización y registro, manteniéndose dichos controles mientras dure la producción y/o comercialización del medicamento. Los procedimientos de control de calidad habrán de modificarse conforme al avance de la técnica. 3. La Real Farmacopea Española es el código que establece la calidad que deben cumplir los principios activos y excipientes que entran en la composición de los medicamentos de uso humano y veterinarios. Se actualizará y publicará periódicamente. El Ministerio de Sanidad, Servicios Sociales e Igualdad, a través de la Agencia Española de Medicamentos y Productos Sanitarios, fijará y publicará en el «Boletín Oficial del Estado» la fecha de la puesta en vigor de los sucesivos volúmenes de la Real Farmacopea Española. 4. La Real Farmacopea Española está constituida por las monografías contenidas en la Farmacopea Europea del Consejo de Europa y, en casos justificados, por las monografías peculiares españolas. Para las sustancias fabricadas en países pertenecientes a la Unión Europea rige, en defecto de la Farmacopea Europea, la monografía de la farmacopea del país fabricante y, en su defecto, la de un tercer país. La Farmacopea incluirá monografías convenientemente ordenadas y codificadas con las especificaciones de identidad, pureza y riqueza de, como mínimo, los principios activos y excipientes, así como los métodos analíticos oficiales y textos generales necesarios para la correcta aplicación de las monografías. Las especificaciones definidas en las monografías constituyen exigencias mínimas de obligado cumplimiento. Toda materia prima presentada bajo una denominación científica o común de la Farmacopea en vigor debe responder a las especificaciones de la misma. El Ministerio de Sanidad, Servicios Sociales e Igualdad podrá reconocer la vigencia en España a monografías concretas de Farmacopeas extranjeras. 5. Las oficinas de farmacia, servicios farmacéuticos, entidades de distribución y laboratorios farmacéuticos deben garantizar que disponen de acceso a la Real Farmacopea Española. 6. La Agencia Española de Medicamentos y Productos Sanitarios y las comunidades autónomas establecerán programas de control de calidad de los medicamentos para comprobar la observancia de las condiciones de la autorización y de las demás que sean de aplicación. A efectos de coordinación de dichos programas, en el marco del Consejo Interterritorial del Sistema Nacional de Salud se establecerán criterios unitarios relativos a la extensión, intensidad y frecuencia de los controles a realizar. 7. Las autoridades y profesionales sanitarios y los laboratorios y distribuidores están obligados a colaborar diligentemente en los referidos programas de control de calidad y comunicar las anomalías de las que tuvieran conocimiento. Artículo 12. Garantías de seguridad. 1. Los medicamentos, principios activos y materias primas que compongan aquéllos serán objeto de los estudios toxicológicos y clínicos que permitan garantizar su seguridad en condiciones normales de uso y que estarán en relación con la duración prevista del tratamiento. 2. Los estudios toxicológicos comprenderán ensayos de toxicidad aguda y crónica, ensayos de teratogenia, embriotoxicidad, fertilidad, ensayos de mutagénesis y, en su caso, de carcinogénesis y, en general, aquellos otros que se consideren necesarios para una correcta evaluación de la seguridad y tolerancia de un medicamento en condiciones normales de uso y en función de la duración del tratamiento. En todo caso, se cumplirá la normativa en materia de protección de animales utilizados para fines científicos. Estos estudios deberán realizarse de acuerdo con las buenas prácticas de laboratorio establecidas. – 14 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios 3. Complementariamente a los estudios toxicológicos y a los ensayos clínicos, los solicitantes de la autorización deberán acreditar la capacidad de realizar una adecuada vigilancia post-comercialización de la seguridad del medicamento. Asimismo, deberán presentar los planes específicos de farmacovigilancia y de gestión de riesgos que, de acuerdo con las directrices nacionales y europeas, se consideren necesarios, así como el compromiso fehaciente de desarrollo y ejecución de los mismos. 4. Las garantías de seguridad del medicamento se extenderán a los riesgos relativos a su utilización y, en particular, a cualquier riesgo de efectos no deseados para el medio ambiente. 5. Sin perjuicio de su propia responsabilidad, todas las autoridades y profesionales sanitarios así como los laboratorios farmacéuticos y entidades de distribución están obligados a colaborar diligentemente en el conocimiento de la seguridad del producto. Asimismo, los profesionales sanitarios, los laboratorios farmacéuticos y las entidades de distribución están obligados a comunicar a las autoridades sanitarias las anomalías de las que tuvieran noticia, conforme a lo establecido en el capítulo VI de este título. Artículo 13. Garantías de eficacia. 1. La eficacia de los medicamentos para cada una de sus indicaciones deberá establecerse con base en la realización previa de estudios preclínicos y ensayos clínicos que se ajustarán a las exigencias normativas y a las que se deriven de los avances en el conocimiento científico de la materia. 2. Los estudios en animales deberán diseñarse y realizarse de forma que permitan conocer el perfil farmacológico global de la sustancia. En todo caso, se cumplirá la normativa en materia de protección de animales utilizados para fines científicos. 3. Los ensayos clínicos estarán planificados y se realizarán de tal modo que permitan obtener la información necesaria para conocer el comportamiento de la sustancia en el organismo y evaluar la eficacia del medicamento. El efecto terapéutico debe cuantificarse para las distintas dosis y en todas las indicaciones solicitadas. En todos los ensayos se respetarán los requisitos éticos establecidos para la investigación con seres humanos. Artículo 14. Garantías de identificación. 1. A cada principio activo le será atribuida una denominación oficial española (DOE) por la Agencia Española de Medicamentos y Productos Sanitarios. La denominación oficial española será de uso obligatorio, sin perjuicio de que pueda expresarse, además, en las correspondientes lenguas oficiales de las comunidades autónomas. La denominación oficial española deberá ser igual, o lo más aproximada posible, salvadas las necesidades lingüísticas, a la denominación común internacional (DCI) fijada por la Organización Mundial de la Salud. Las denominaciones oficiales españolas de los principios activos serán de dominio público. La Agencia Española de Medicamentos y Productos Sanitarios publicará una lista con las denominaciones oficiales españolas de los principios activos autorizados en España, que se actualizará periódicamente. Los organismos públicos promoverán la utilización de las denominaciones oficiales españolas, si existen, o, en su defecto, de las denominaciones comunes internacionales o, a falta de éstas, de las denominaciones usuales o científicas. 2. La denominación del medicamento podrá consistir en un nombre de fantasía que no pueda confundirse con la denominación común, o una denominación común o científica acompañada de una marca o del nombre del titular de la autorización de comercialización. La denominación del medicamento no podrá confundirse con una denominación oficial española o una denominación común internacional ni inducir a error sobre las propiedades terapéuticas o la naturaleza del medicamento. Los medicamentos genéricos deberán designarse con una denominación oficial española de principio activo y, en su defecto, con la denominación común internacional o bien, si esta no existiese, con la denominación común usual o científica de dicha sustancia, acompañada, en su caso, del nombre o marca del titular o fabricante; asimismo, podrán denominarse con una marca siempre que no pueda confundirse con una denominación oficial española o una denominación común internacional ni inducir a error sobre las propiedades terapéuticas o la – 15 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios naturaleza del medicamento. Se identificarán, además, con las siglas EFG (Equivalente Farmacéutico Genérico). 3. El Ministerio de Sanidad, Servicios Sociales e Igualdad establecerá un Código Nacional de Medicamentos de general aplicación que facilite su pronta identificación y podrá exigir que sus números o claves figuren en el etiquetado de los medicamentos. Artículo 15. Garantías de información. 1. El Ministerio de Sanidad, Servicios Sociales e Igualdad regulará los aspectos relativos a las garantías de información y, en concreto, las características, extensión, pormenores y lugares donde deba figurar. En todo caso, para la elaboración de esta información sobre el medicamento, su titular proporcionará información escrita suficiente sobre su identificación, indicaciones y precauciones a observar en su empleo. Esta información se presentará, al menos, en la lengua española oficial del Estado y con ella se elaborará la ficha técnica, el prospecto y el etiquetado. Los textos y demás características de la ficha técnica, el prospecto y el etiquetado forman parte de la autorización de los medicamentos y han de ser previamente autorizados por la Agencia Española de Medicamentos y Productos Sanitarios. Sus modificaciones requerirán asimismo autorización previa o notificación, según proceda. 2. La ficha técnica o resumen de las características del producto reflejará las condiciones de uso autorizadas para el medicamento y sintetizará la información científica esencial para los profesionales sanitarios. La Agencia Española de Medicamentos y Productos Sanitarios aprobará la ficha técnica en la que constarán datos suficientes sobre la identificación del medicamento y su titular, así como las indicaciones terapéuticas para las que el medicamento ha sido autorizado, de acuerdo con los estudios que avalan su autorización. A la ficha técnica se acompañará, preceptivamente, información actualizada del precio del medicamento y, cuando sea posible, la estimación del coste del tratamiento. La Agencia Española de Medicamentos y Productos Sanitarios pondrá la ficha técnica a disposición de los servicios de salud de las comunidades autónomas, de los colegios u organizaciones profesionales, de los médicos, odontólogos, podólogos y farmacéuticos en ejercicio y, en su caso, de los veterinarios en ejercicio. El titular de la autorización estará obligado a poner la ficha técnica actualizada a disposición de las Administraciones sanitarias y de los profesionales en todas sus actividades de promoción e información en los términos establecidos reglamentariamente. 3. El prospecto, que se elaborará de acuerdo con el contenido de la ficha técnica, proporcionará a los pacientes información suficiente sobre la denominación del principio activo, identificación del medicamento y su titular e instrucciones para su administración, empleo y conservación, así como sobre los efectos adversos, interacciones, contraindicaciones, en especial los efectos sobre la conducción de vehículos a motor, y otros datos que se determinen reglamentariamente con el fin de promover su más correcto uso y la observancia del tratamiento prescrito, así como las medidas a adoptar en caso de intoxicación. El prospecto deberá ser legible, claro, asegurando su comprensión por el paciente y reduciendo al mínimo los términos de naturaleza técnica. 4. En el etiquetado figurarán los datos del medicamento, como la denominación del principio activo, del titular de la autorización, vía de administración, cantidad contenida, número de lote de fabricación, fecha de caducidad, precauciones de conservación, condiciones de dispensación y demás datos que reglamentariamente se determinen. En cada embalaje figurarán codificados los datos del Código Nacional del Medicamento, el lote y unidad que permitan su identificación de forma individualizada por medios mecánicos, electrónicos e informáticos, en la forma que se determine reglamentariamente. En el embalaje deberá incluirse un espacio en blanco a rellenar por el farmacéutico donde éste podrá describir la posología, duración del tratamiento y frecuencia de tomas. Al dispensar un medicamento, las oficinas de farmacia deberán emitir un recibo en el que se haga constar el nombre del medicamento, su precio de venta al público y la aportación del paciente. En el caso de los medicamentos que sean objeto de la publicidad prevista en el artículo 80, el recibo hará constar, además, el descuento que, en su caso, se hubiese efectuado. 5. A fin de garantizar el acceso a la información de las personas invidentes o con discapacidad visual, en todos los envases de los medicamentos deberán figurar impresos en – 16 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios alfabeto braille los datos necesarios para su correcta identificación. El titular de la autorización garantizará que, previa solicitud de las asociaciones de pacientes afectados, el prospecto esté disponible en formatos apropiados para las personas invidentes o con visión parcial. 6. Los medicamentos se elaborarán y presentarán de forma que se garantice la prevención razonable de accidentes, especialmente en relación con la infancia y personas con discapacidad. Los envases llevarán, en su caso, algún dispositivo de precinto que garantice al usuario que el medicamento mantiene la composición, calidad y cantidad del producto envasado por el laboratorio. Asimismo, los embalajes incluirán el símbolo autorizado por la Agencia Española de Medicamentos y Productos Sanitarios a efectos de facilitar la aplicación y desarrollo del sistema de recogida de residuos de medicamentos y favorecer la protección del medio ambiente. Artículo 16. Procedimiento de autorización y sus modificaciones. Requisitos y garantías de transparencia. 1. La Agencia Española de Medicamentos y Productos Sanitarios podrá requerir al solicitante para que aporte documentación, estudios, datos o informaciones complementarias, siendo de aplicación la normativa comunitaria, la específica de desarrollo de esta ley y, en su defecto, la normativa reguladora del procedimiento administrativo común. 2. En el procedimiento de evaluación de los medicamentos, la Agencia Española de Medicamentos y Productos Sanitarios contará, a efectos de la emisión de los informes que correspondan, con comités u órganos de asesoramiento que incorporen a expertos cualificados del mundo científico y profesional. 3. En el procedimiento de autorización se podrá someter el medicamento, sus materias primas, productos intermedios y otros componentes a examen de los laboratorios oficiales de control de la Agencia Española de Medicamentos y Productos Sanitarios, que podrá solicitar la colaboración de otro laboratorio nacional acreditado a tal efecto por la propia Agencia, de un laboratorio oficial de control comunitario o de un tercer país. 4. Para garantizar la transparencia de sus actuaciones, la Agencia Española de Medicamentos y Productos Sanitarios asegurará el acceso público de sus decisiones sobre las autorizaciones de medicamentos, sus modificaciones, suspensiones y revocaciones, cuando todas ellas sean firmes, así como el resumen de las características del producto. Será, asimismo, de acceso público el informe de evaluación motivado, previa supresión de cualquier información comercial de carácter confidencial. La confidencialidad no impedirá la publicación de los actos de decisión de los órganos colegiados de asesoramiento técnico y científico del Ministerio de Sanidad, Servicios Sociales e Igualdad relacionados con la autorización de medicamentos, sus modificaciones, suspensiones y revocaciones. Artículo 17. Expediente de autorización. 1. El expediente para la autorización de un medicamento constará de toda la documentación relativa a información administrativa, resúmenes de expertos, información química, farmacéutica y biológica para medicamentos que contengan principios activos químicos y/o biológicos, el resultado de las pruebas farmacéuticas, preclínicas y clínicas, y cualquier otra que se determine reglamentariamente. El solicitante o titular de una autorización será responsable de la exactitud de los documentos y datos presentados. 2. En la solicitud de autorización de los medicamentos figurará, entre los datos de identificación, la completa y exacta composición cualitativa y cuantitativa, incluyendo no sólo principios activos, sino también todos los excipientes y los disolventes, aunque estos últimos desaparezcan en el proceso de fabricación. Asimismo, en la solicitud figurarán las indicaciones sobre las medidas de precaución y de seguridad que han de adoptarse al almacenar el medicamento, al administrarlo a los pacientes y al eliminar los productos residuales, junto con la indicación de cualquier riesgo potencial que el medicamento podría presentar para el medio ambiente. En la solicitud se acreditará que el solicitante dispone de una persona cualificada responsable de la farmacovigilancia, así como de la infraestructura necesaria para informar – 17 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios sobre toda reacción adversa que se sospeche que ya se haya producido o se pueda producir. 3. El solicitante no tendrá obligación de facilitar los resultados de los ensayos preclínicos y clínicos establecidos si puede demostrar que el medicamento es genérico de un medicamento de referencia que está o ha sido autorizado desde hace ocho años como mínimo en cualquier Estado miembro de la Unión Europea, o por la Unión Europea, incluso cuando el medicamento de referencia no estuviera autorizado en España, sin perjuicio del derecho relativo a la protección de la propiedad industrial y comercial. 4. Cuando un medicamento biológico que sea similar a un producto biológico de referencia no cumpla las condiciones de la definición de medicamento genérico, debido en particular a diferencias relacionadas con las materias primas o diferencias en el proceso de fabricación del medicamento biológico y del medicamento biológico de referencia, deberán aportarse los resultados de los ensayos preclínicos o clínicos adecuados relativos a dichas condiciones y demás requisitos establecidos reglamentariamente. 5. El solicitante podrá sustituir los resultados de los ensayos clínicos y de los estudios preclínicos por una documentación bibliográfica-científica adecuada, si puede demostrar que los principios activos del medicamento han tenido un uso médico bien establecido al menos durante diez años dentro de la Unión Europea y presentan una eficacia reconocida, así como un nivel aceptable de seguridad. 6. Cuando se trate de un medicamento que tenga la misma composición cualitativa y cuantitativa en principios activos y la misma forma farmacéutica que otro ya autorizado e inscrito, el solicitante podrá usar la documentación farmacéutica, preclínica y clínica que obre en el expediente del medicamento autorizado siempre que cuente con el consentimiento del titular. 7. Los medicamentos que contengan principios activos que entren en la composición de medicamentos autorizados, pero que no hayan sido combinados con fines terapéuticos, deberán aportar los resultados de los nuevos ensayos clínicos y de los estudios preclínicos relativos a la combinación sin necesidad de aportar la documentación relativa a cada principio activo individual. 8. Otorgada la autorización de un medicamento, cualquier modificación que se solicite en relación con la misma deberá cumplir los requisitos documentales que reglamentariamente se establezcan. 9. El titular de la autorización de un medicamento deberá mantener actualizado el expediente aportado para obtener aquélla, incorporando al mismo cuantos datos, informes o modificaciones tecnológicas impongan los avances de la ciencia y las normas de correcta fabricación y control. Asimismo, deberá presentar los informes periódicos de seguridad establecidos en la legislación vigente con el fin de mantener actualizado el expediente en materia de seguridad. 10. Las Administraciones públicas competentes podrán exigir, en cualquier momento, del laboratorio farmacéutico que justifique la realización de los controles de calidad y cuantos otros se encuentren establecidos en la normativa vigente. Artículo 18. Exclusividad de datos. 1. Sin perjuicio del derecho relativo a la protección de la propiedad industrial y comercial, los medicamentos genéricos autorizados con arreglo a lo dispuesto en el artículo 17.3 no podrán ser comercializados hasta transcurridos diez años desde la fecha de la autorización inicial del medicamento de referencia. Este período de diez años de exclusividad de datos se ampliará hasta un máximo de once años si, durante los primeros ocho años del período de diez, el titular de la autorización del medicamento de referencia obtiene una autorización para una o varias nuevas indicaciones terapéuticas y, durante la evaluación científica previa a su autorización, se establece que dichas indicaciones aportarán un beneficio clínico significativo en comparación con las terapias existentes. 2. En el supuesto de que para una sustancia de uso médico bien establecido se autorice una nueva indicación, con base en ensayos clínicos o estudios preclínicos significativos, se concederá un periodo no acumulativo de exclusividad de datos de un año. – 18 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios 3. Cuando, con base en ensayos clínicos o estudios preclínicos significativos, la autorización de medicamento sujeto a prescripción médica se haya modificado por la de medicamento no sujeto a prescripción médica o viceversa, se concederá un período de un año de exclusividad de datos para los mismos. Artículo 19. Condiciones de prescripción y dispensación de medicamentos. 1. En la autorización del medicamento, la Agencia Española de Medicamentos y Productos Sanitarios determinará sus condiciones de prescripción clasificándolo, según corresponda, en las siguientes categorías: a) Medicamento sujeto a prescripción médica. b) Medicamento no sujeto a prescripción médica. 2. Estarán en todo caso sujetos a prescripción médica los medicamentos que se encuentren en alguno de los siguientes supuestos: a) Puedan presentar un peligro, directa o indirectamente, incluso en condiciones normales de uso, si se utilizan sin control médico. b) Se utilicen frecuentemente, y de forma muy considerable, en condiciones anormales de utilización, y ello pueda suponer, directa o indirectamente, un peligro para la salud. c) Contengan sustancias o preparados a base de dichas sustancias, cuya actividad y/o reacciones adversas sea necesario estudiar más detalladamente. d) Se administren por vía parenteral, salvo casos excepcionales, por prescripción médica. 3. La Agencia Española de Medicamentos y Productos Sanitarios podrá establecer, en los medicamentos que sólo pueden dispensarse bajo prescripción médica, las siguientes subcategorías: a) Medicamentos de dispensación bajo prescripción médica renovable o no renovable. b) Medicamentos sujetos a prescripción médica especial. c) Medicamentos de dispensación bajo prescripción médica restringida, de utilización reservada a determinados medios especializados. Reglamentariamente se establecerán los criterios para su aplicación. 4. La Agencia Española de Medicamentos y Productos Sanitarios podrá calificar como medicamentos no sujetos a prescripción médica aquéllos que vayan destinados a procesos o condiciones que no necesiten un diagnóstico preciso y cuyos datos de evaluación toxicológica, clínica o de su utilización y vía de administración no exijan prescripción médica, de modo que dichos medicamentos puedan ser utilizados para autocuidado de la salud, mediante su dispensación en la oficina de farmacia por un farmacéutico, que informará, aconsejará e instruirá sobre su correcta utilización. 5. Los prospectos y el etiquetado de los medicamentos que no requieran prescripción médica, además de lo previsto en el artículo 15.1, contendrán aquellas advertencias que convengan a su naturaleza y, en especial, las orientadas a prevenir su uso indebido. 6. El Ministerio de Sanidad, Servicios Sociales e Igualdad establecerá los requisitos mínimos, características y plazo de validez de las recetas médicas y prescripciones hospitalarias, así como los requisitos especiales para la prescripción y dispensación de los medicamentos de sustancias psicoactivas y otros que por su naturaleza lo requieran o para tratamientos peculiares. 7. La dispensación de medicamentos se ajustará a las condiciones de prescripción establecidas. 8. La Agencia Española de Medicamentos y Productos Sanitarios, con el fin de ajustar las unidades dispensadas por oficinas de farmacia a la duración del tratamiento, podrá autorizar la dispensación de unidades concretas en un plazo de seis meses a contar desde la determinación de los correspondientes grupos de medicamentos y/o patologías. Estas unidades podrán dispensarse a partir del fraccionamiento de un envase de un medicamento autorizado e inscrito, respetando la integridad del acondicionamiento primario, excepto cuando, en el marco de proyectos o programas autorizados por la mencionada Agencia, sea – 19 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios procedente su reacondicionamiento protocolizado y garantizando las condiciones de conservación del medicamento, así como la información al paciente. Para los casos previstos en este apartado, el Ministerio de Sanidad, Servicios Sociales e Igualdad establecerá el sistema de cálculo de precio de venta al público y los márgenes de comercialización correspondientes. 9. En orden a asegurar el uso racional de los medicamentos, la Agencia Española de Medicamentos y Productos Sanitarios adoptará cuantas medidas sean necesarias para adecuar el contenido de todos los envases de los nuevos medicamentos autorizados a la duración de los tratamientos en la práctica clínica. Igualmente, realizará revisiones anuales, a los mismos efectos, de los medicamentos ya autorizados, dando cuenta semestralmente al Consejo Interterritorial del Sistema Nacional de Salud. Artículo 20. Denegación de la autorización. La autorización de un medicamento podrá ser denegada por las siguientes razones: a) Cuando la relación beneficio-riesgo no sea favorable. b) Cuando no se justifique suficientemente la eficacia terapéutica. c) Cuando el medicamento no tenga la composición cualitativa y cuantitativa declarada o carezca de la calidad adecuada. d) Cuando los datos e informaciones contenidos en la documentación de la solicitud de autorización sean erróneos o incumplan la normativa de aplicación en la materia. Artículo 21. Validez de la autorización. 1. La autorización de medicamentos tendrá una duración de cinco años. 2. La autorización podrá renovarse transcurridos cinco años, previa reevaluación de la relación beneficio/riesgo. La renovación de la autorización tendrá carácter indefinido, salvo que razones de farmacovigilancia justifiquen su sometimiento a un nuevo procedimiento de renovación. 3. El titular de una autorización comunicará de forma expresa a la Agencia Española de Medicamentos y Productos Sanitarios la puesta en el mercado por vez primera de un medicamento autorizado e inscrito por dicha Agencia y efectuará anualmente una declaración de comercialización en los términos que reglamentariamente se establezcan. 4. La autorización de un medicamento se entenderá caducada si, en un plazo de tres años, el titular no procede a la comercialización efectiva del mismo o una vez autorizado, inscrito y comercializado deja de encontrarse de forma efectiva en el mercado durante tres años consecutivos. Lo anterior no será de aplicación cuando concurran razones de salud o de interés sanitario, en cuyo caso la Agencia Española de Medicamentos y Productos Sanitarios mantendrá la validez de la autorización y podrá exigir la comercialización efectiva del producto. Artículo 22. Suspensión y revocación de la autorización. 1. La autorización será temporalmente suspendida o definitivamente revocada por la Agencia Española de Medicamentos y Productos Sanitarios en los siguientes casos: a) Cuando el medicamento no tenga la composición cuantitativa o cualitativa autorizada o cuando se incumplan las garantías de calidad o cuando no se ejecuten los controles de calidad exigidos en esta ley. b) Cuando, basándose en datos de seguridad y/o eficacia el medicamento tenga una relación beneficio/riesgo desfavorable. c) Cuando el medicamento resulte no ser terapéuticamente eficaz. d) Cuando los datos e informaciones contenidos en la documentación de la solicitud de autorización sean erróneos o incumplan la normativa de aplicación en la materia. e) Cuando, por cualquier otra causa, suponga un riesgo previsible para la salud o seguridad de las personas o animales. f) En cualquier otro caso en el que la Agencia Europea de Medicamentos así lo hubiere acordado. – 20 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios g) Cuando se incumpla con los requisitos establecidos en las condiciones de autorización de comercialización en materia de farmacovigilancia. 2. Asimismo, la Agencia Española de Medicamentos y Productos Sanitarios, a solicitud del titular de la autorización, podrá suspender temporalmente o revocar la autorización de un medicamento, previa justificación en motivos tecnológicos, científicos o cualesquiera otros que resulten proporcionados y siempre que la decisión no origine laguna terapéutica en la prestación farmacéutica del Sistema Nacional de Salud y no colisione con los criterios establecidos en esta ley para la inclusión de medicamentos en la citada prestación. Artículo 23. Modificaciones de la autorización por razones de interés general. La Agencia Española de Medicamentos y Productos Sanitarios podrá modificar, de forma justificada y notificándolo al titular de la autorización de comercialización, la autorización de los medicamentos que lo requieran por razones de interés público o defensa de la salud o seguridad de las personas. Ello, sin perjuicio de la obligación del titular de la autorización de comercialización de asegurar que la información de sus productos esté actualizada en función de los últimos conocimientos científicos, incluidas las conclusiones de las evaluaciones y las recomendaciones publicadas en el portal Web europeo sobre medicamentos y en el portal Web de la Agencia Española de Medicamentos y Productos Sanitarios. Artículo 24. Garantías de disponibilidad de medicamentos en situaciones específicas y autorizaciones especiales. 1. En circunstancias excepcionales, la Agencia Española de Medicamentos y Productos Sanitarios podrá conceder una autorización supeditada a la obligación por parte del solicitante de cumplir determinadas condiciones revisables anualmente. Dichas condiciones quedarán, en especial, referidas a la seguridad del medicamento, a la información a las autoridades competentes de todo incidente relacionado con su utilización y a las medidas que deben adoptarse. Reglamentariamente se establecerán los criterios para la concesión de estas autorizaciones. 2. La Agencia Española de Medicamentos y Productos Sanitarios podrá, de oficio o a solicitud de las comunidades autónomas interesadas, por razones sanitarias objetivas y debidamente motivadas, sujetar a reservas singulares la autorización de medicamentos que así lo requieran por su naturaleza o características, así como sus condiciones generales de prescripción y dispensación. 3. La prescripción y la aplicación de medicamentos no autorizados a pacientes no incluidos en un ensayo clínico con el fin de atender como uso compasivo necesidades especiales de tratamiento de situaciones clínicas de pacientes concretos se regulará reglamentariamente, con pleno respeto a lo establecido en la legislación vigente en materia de autonomía del paciente y de los derechos y obligaciones en materia de información y documentación clínica. La persona titular del Ministerio de Sanidad, Servicios Sociales e Igualdad establecerá las condiciones para la prescripción de medicamentos autorizados cuando se utilicen en condiciones distintas a las autorizadas, que en todo caso tendrá carácter excepcional. 4. La Agencia Española de Medicamentos y Productos Sanitarios podrá autorizar la importación de medicamentos no autorizados en España siempre que estén legalmente comercializados en otros Estados, cuando esta importación resulte imprescindible para la prevención, el diagnóstico o el tratamiento de patologías concretas por no existir en España alternativa adecuada autorizada para esa indicación concreta o por situaciones de desabastecimiento que lo justifiquen. 5. La Agencia Española de Medicamentos y Productos Sanitarios podrá autorizar temporalmente la distribución de medicamentos no autorizados, en respuesta a la propagación supuesta o confirmada de un agente patógeno o químico, toxina o radiación nuclear capaz de causar daños. En estas circunstancias, si se hubiere recomendado o impuesto por la autoridad competente el uso de medicamentos en indicaciones no autorizadas o de medicamentos no autorizados, los titulares de la autorización y demás profesionales que intervengan en el proceso estarían exentos de responsabilidad civil o – 21 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios administrativa por todas las consecuencias derivadas de la utilización del medicamento, salvo por los daños causados por productos defectuosos. 6. La Agencia Española de Medicamentos y Productos Sanitarios autorizará con carácter excepcional la elaboración y distribución de muestras gratuitas en las condiciones que reglamentariamente se establezcan. En todo caso, no se autorizarán muestras gratuitas de medicamentos de sustancias psicoactivas que causen dependencia y de aquellas otras que determine la Agencia Española de Medicamentos y Productos Sanitarios. 7. La Agencia Española de Medicamentos y Productos Sanitarios podrá establecer modalidades de autorización especiales para medicamentos que, de acuerdo con una resolución expresa de la misma, se consideren necesarios para atender requerimientos especiales, siempre y cuando sean destinados para uso de un paciente individual bajo prescripción de un facultativo acreditado y bajo su responsabilidad directa. CAPÍTULO III De las garantías exigibles a los medicamentos veterinarios elaborados industrialmente y de las condiciones de prescripción y dispensación de los mismos Artículo 25. Autorización y registro. 1. Ningún medicamento veterinario elaborado industrialmente podrá ser puesto en el mercado sin la previa autorización de la Agencia Española de Medicamentos y Productos Sanitarios e inscripción en el Registro de Medicamentos o sin haber obtenido la autorización de conformidad con lo dispuesto en las normas europeas que establecen los procedimientos comunitarios para la autorización y control de los medicamentos de uso humano y veterinarios y que regulan la Agencia Europea de Medicamentos. A efectos de lo establecido en este artículo y, en general, en este capítulo, la Agencia Española de Medicamentos y Productos Sanitarios actuará de acuerdo con los criterios emanados del Ministerio de Agricultura, Alimentación y Medio Ambiente y conforme a la normativa de sanidad animal. 2. Cuando un medicamento veterinario haya obtenido una autorización de acuerdo con el apartado anterior, toda dosificación, forma farmacéutica, vía de administración y presentaciones adicionales, así como cualesquiera otras modificaciones y ampliaciones al expediente de autorización que se introduzcan, deberán ser objeto de autorización o notificación, conforme se disponga en la normativa que regula el procedimiento de autorización, registro y farmacovigilancia de los medicamentos veterinarios fabricados industrialmente. Todas estas modificaciones se considerarán pertenecientes a la misma autorización global de comercialización, en particular a los efectos de la aplicación de los periodos de exclusividad de datos. 3. Toda modificación, transmisión y extinción de las autorizaciones de los medicamentos veterinarios deberá constar en el Registro de Medicamentos que, a estos efectos, tendrá, del mismo modo que la inscripción, carácter constitutivo. 4. La Agencia Española de Medicamentos y Productos Sanitarios procederá de oficio a la incorporación al Registro de Medicamentos de las autorizaciones otorgadas por la Comisión Europea conforme al Reglamento (CE) n.º 726/2004/CE. Artículo 26. Garantías exigibles para la autorización de medicamentos. 1. La Agencia Española de Medicamentos y Productos Sanitarios autorizará un medicamento veterinario si satisface las siguientes condiciones: a) Alcanzar los requisitos mínimos de calidad que se establezcan. b) Ser seguro. c) Ser eficaz en las indicaciones terapéuticas para las que se ofrece. d) Estar correctamente identificado. e) Suministrar la información precisa. 2. La evaluación de los efectos terapéuticos positivos del medicamento se apreciarán en relación con cualquier riesgo relacionado con la calidad, la seguridad y la eficacia del medicamento para la salud del animal o la salud pública, entendido como relación beneficio/ – 22 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios riesgo. Reglamentariamente se adecuará esta evaluación a las necesidades específicas en el caso de medicamentos destinados a los animales de terrario, pájaros domiciliarios, peces de acuario, pequeños roedores y otros que no requieran prescripción veterinaria. 3. Lo establecido en este artículo será, asimismo, de aplicación a las modificaciones que se produzcan en la autorización del medicamento. Artículo 27. Garantías de calidad. 1. Todo medicamento deberá tener perfectamente establecida su composición cualitativa y cuantitativa. Alternativamente, en el caso de sustancias como las biológicas en las que esto no sea posible, sus procedimientos de preparación deben ser reproducibles. 2. El Ministerio de Sanidad, Servicios Sociales e Igualdad, en coordinación con el Ministerio de Agricultura, Alimentación y Medio Ambiente, establecerá el tipo de controles exigibles al laboratorio titular de la autorización de comercialización y al fabricante para garantizar la calidad de las materias primas, de los productos intermedios, del proceso de fabricación y del producto final, a efectos de la autorización y registro, manteniéndose dichos controles mientras dure la producción y/o comercialización del medicamento. Los procedimientos de control de calidad habrán de modificarse conforme al avance de la técnica. 3. Las Administraciones sanitarias competentes realizarán controles periódicos de calidad de los medicamentos existentes en el mercado, de las materias primas y de los productos intermedios, así como del material de envasado y de las condiciones de conservación, transporte y venta. Artículo 28. Garantías de seguridad. 1. Los medicamentos, sustancias medicinales y los excipientes que compongan aquéllos serán objeto de los estudios toxicológicos y clínicos que permitan garantizar su seguridad en condiciones normales de uso y que estarán en relación con la duración prevista del tratamiento. 2. Los estudios comprenderán ensayos de toxicidad aguda y crónica, ensayos de teratogenia, embriotoxicidad, fertilidad, ensayos de mutagénesis y, en su caso, de carcinogénesis y, en general, aquellos otros que se consideren necesarios para una correcta evaluación de la seguridad y tolerancia de un medicamento en condiciones normales de uso y en función de la duración del tratamiento. En todo caso, se cumplirá la normativa en materia de protección de animales utilizados para fines científicos. 3. Los medicamentos veterinarios serán objeto de estudios y ensayos complementarios que permitan garantizar su seguridad, en los que se tendrá en cuenta: a) Que cuando se administran a animales productores de alimentos destinados al consumo humano debe conocerse el tiempo de espera adecuado para eliminar los riesgos para las personas que se deriven de los residuos o metabolitos de aquéllos. b) Las repercusiones sobre las personas que los manejan, principalmente para los productos destinados a la mezcla con los piensos. c) Las influencias sobre el medio ambiente, cuando puedan dar lugar a una acción residual a través de los productos de desecho. d) Tratándose de productos biológicos y de las vacunas en particular, las repercusiones epizoóticas. 4. Sin perjuicio de su propia responsabilidad, todas las autoridades y profesionales sanitarios, así como los laboratorios farmacéuticos y entidades de distribución, están obligados a colaborar diligentemente en el conocimiento de la seguridad del producto. Asimismo, los profesionales sanitarios, los laboratorios farmacéuticos y las entidades de distribución están obligados a comunicar a las autoridades sanitarias las anomalías de las que tuvieran noticia, conforme a lo establecido en el capítulo VI de este título. Artículo 29. Garantías de eficacia. 1. La eficacia de los medicamentos veterinarios deberá establecerse de un modo adecuado para cada una de las especies e indicaciones para las que estén destinados – 23 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios mediante la previa realización de ensayos clínicos y estudios preclínicos controlados por personas suficientemente cualificadas. 2. Dichos estudios deberán reproducir los efectos de las distintas dosis solicitadas para la sustancia de la forma que reglamentariamente se establezca e incluir, asimismo, uno o más grupos de control tratados o no con un producto de referencia. Artículo 30. Garantías de identificación. 1. Cada principio activo de uso veterinario utilizará la correspondiente denominación oficial española (DOE) conforme a lo establecido en el artículo 14. 2. Podrá designarse a un medicamento veterinario con una marca o con una denominación oficial española de principio activo y, en su defecto, con la denominación común internacional o bien, si ésta no existiese, con la denominación común usual o científica de dicha sustancia. Reglamentariamente podrán regularse los supuestos en los que podrá designarse a un medicamento genérico con una denominación comercial o con una marca. La denominación del medicamento, cuando sea una marca o una denominación comercial, no podrá confundirse con una denominación oficial española de principio activo o una denominación común internacional ni inducir a error sobre las propiedades terapéuticas o la naturaleza del medicamento. 3. La Agencia Española de Medicamentos y Productos Sanitarios establecerá un Código Nacional de Medicamentos veterinarios de general aplicación que facilite su pronta identificación y, asimismo, podrá exigir que sus números o claves figuren en el envase, etiquetado y embalaje de los medicamentos veterinarios. Artículo 31. Garantías de información. 1. Los Ministerios de Agricultura, Alimentación y Medio Ambiente, y de Sanidad, Servicios Sociales e Igualdad regularán los aspectos relativos a las garantías de información y, en concreto, las características, extensión, pormenores y lugares donde deba figurar. En todo caso, para la elaboración de esta información sobre el medicamento veterinario su titular proporcionará información escrita suficiente sobre su identificación, indicaciones y precauciones a observar en su empleo. Esta información se presentará, al menos, en la lengua española oficial del Estado y con ella se elaborará la ficha técnica, el prospecto y el etiquetado. Los textos y demás características de la ficha técnica, el prospecto y el etiquetado forman parte de la autorización de los medicamentos veterinarios y han de ser previamente autorizados por la Agencia Española de Medicamentos y Productos Sanitarios. Sus modificaciones requerirán, asimismo, autorización previa o notificación, según proceda. 2. La ficha técnica resumirá la información científica esencial sobre el medicamento veterinario a que se refiere. La Agencia Española de Medicamentos y Productos Sanitarios aprobará la ficha técnica en la que constarán datos suficientes sobre la identificación del medicamento veterinario y su titular, la información que se requiera para una actuación terapéutica y una atención farmacéutica correctas, de acuerdo con los estudios que avalan su autorización. La Agencia Española de Medicamentos y Productos Sanitarios pondrá la ficha técnica a disposición de las comunidades autónomas, de los colegios u organizaciones profesionales y de los veterinarios y farmacéuticos en ejercicio. El titular de la autorización estará obligado a poner la ficha técnica actualizada a disposición de los profesionales en todas sus actividades de promoción e información en los términos establecidos reglamentariamente, así como cuando la misma le sea solicitada. 3. El prospecto proporcionará información suficiente sobre la identificación del medicamento veterinario y su titular e instrucciones para su administración, empleo y conservación, así como sobre los efectos adversos, interacciones, contraindicaciones, tiempo de espera, si procede, y otros datos que se determinen reglamentariamente con el fin de promover su más correcto uso y la observancia del tratamiento prescrito, así como las medidas a adoptar en caso de intoxicación. Asimismo, el prospecto no deberá contener términos de naturaleza técnica a fin de asegurar su fácil lectura y comprensión. 4. En el envase y en el embalaje figurarán los datos del medicamento veterinario, del titular de la autorización, vía de administración, cantidad contenida, número de lote de – 24 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios fabricación, fecha de caducidad, precauciones de conservación, condiciones de dispensación, tiempo de espera, si procede, y demás datos que reglamentariamente se determinen. Al dispensar un medicamento, las oficinas de farmacia, los establecimientos comerciales detallistas autorizados y los servicios de farmacia de las entidades o agrupaciones ganaderas deberán emitir un recibo en el que se haga constar el nombre del medicamento y su precio de venta al público. 5. A fin de garantizar el acceso a la información de las personas invidentes o con discapacidad visual, reglamentariamente se desarrollarán las disposiciones necesarias para que en los envases de los medicamentos destinados a animales de compañía figuren impresos en alfabeto braille los datos necesarios para su correcta identificación, así como que el titular de la autorización garantice que, previa solicitud de las asociaciones de afectados, el prospecto esté disponible en formatos apropiados para las personas invidentes o con visión parcial. 6. Reglamentariamente se establecerán los requisitos necesarios para facilitar la aplicación y desarrollo de un sistema de recogida de los medicamentos veterinarios no utilizados o que hayan caducado. Artículo 32. Procedimiento de autorización y sus modificaciones. Requisitos y garantías de transparencia. 1. De acuerdo con lo dispuesto en esta ley, el Gobierno reglamentará el procedimiento para la obtención de la autorización e inscripción en el Registro de Medicamentos, de conformidad con los trámites y plazos que la Unión Europea establezca en virtud de la armonización comunitaria. Asimismo, el Gobierno reglamentará, conforme a la normativa comunitaria, el procedimiento para la notificación y autorización de cuantas modificaciones se produzcan en la autorización inicial. 2. La Agencia Española de Medicamentos y Productos Sanitarios podrá requerir al solicitante para que aporte documentación, estudios, datos o informaciones complementarias, siendo de aplicación la normativa específica de desarrollo de esta ley y, en su defecto, la normativa reguladora del procedimiento administrativo común. 3. En el procedimiento de evaluación de los medicamentos veterinarios, la Agencia Española de Medicamentos y Productos Sanitarios, en coordinación con el Ministerio de Agricultura, Alimentación y Medio Ambiente, contará, a efectos de la emisión de los informes que correspondan, con comités u órganos de asesoramiento que incorporen a expertos cualificados de las comunidades autónomas y del mundo científico y profesional. 4. En el procedimiento de autorización se podrá someter el medicamento, sus materias primas, productos intermedios y otros componentes a examen de los laboratorios oficiales de control de la Agencia Española de Medicamentos y Productos Sanitarios o, en su caso, de otro laboratorio nacional acreditado a tal efecto por la propia Agencia o de un laboratorio oficial de control comunitario o de un tercer país. 5. Las autorizaciones de medicamentos veterinarios, sus modificaciones, suspensiones y revocaciones, cuando todas ellas sean firmes, así como el resumen de las características del producto, serán de acceso público. Será, asimismo, de acceso público el informe de evaluación motivado, previa supresión de cualquier información comercial de carácter confidencial. Sin perjuicio de lo anterior, el contenido de los expedientes de autorización de los medicamentos veterinarios tendrá carácter confidencial. No obstante, los inspectores para el desarrollo de sus funciones podrán tener acceso a toda la información que precisen. Artículo 33. Expediente de autorización. 1. El expediente para la autorización de un medicamento veterinario constará de toda la documentación relativa a información administrativa, resúmenes de expertos, información química, farmacéutica y biológica para medicamentos veterinarios que contengan principios activos químicos y/o biológicos, pruebas de inocuidad y de estudios de residuos y ensayos preclínicos y clínicos, y cualquier otra que se determine reglamentariamente. El solicitante o titular de una autorización será responsable de la exactitud de los documentos y datos presentados. – 25 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios 2. En la solicitud de autorización de los medicamentos veterinarios figurará, entre los datos de identificación, la completa y exacta composición cualitativa y cuantitativa, incluyendo no sólo las sustancias medicinales, sino también todos los excipientes y los disolventes, aunque estos últimos desaparezcan en el proceso de fabricación. 3. Sin perjuicio del derecho relativo a la protección de la propiedad industrial y comercial, el solicitante no tendrá obligación de facilitar los resultados de las pruebas de inocuidad y de estudios de residuos ni los ensayos preclínicos y clínicos establecidos si puede demostrar que el medicamento es genérico de un medicamento veterinario de referencia que está o ha sido autorizado e inscrito, desde hace ocho años como mínimo, en cualquier Estado miembro de la Unión Europea, o por la Unión Europea, incluso cuando el medicamento de referencia no estuviera autorizado en España. 4. Cuando un medicamento veterinario biológico que sea similar a un producto biológico de referencia no cumpla las condiciones de la definición de medicamento genérico, debido en particular a diferencias relacionadas con las materias primas o diferencias en el proceso de fabricación del medicamento biológico y del medicamento biológico de referencia, deberán aportarse los resultados de los ensayos preclínicos o clínicos adecuados relativos a dichas condiciones y demás requisitos establecidos reglamentariamente. 5. El solicitante podrá sustituir los resultados de las pruebas de inocuidad y de los estudios de residuos y de los ensayos preclínicos o clínicos por una documentación bibliográfica-científica adecuada si puede demostrar que los principios activos del medicamento veterinario han tenido un uso veterinario bien establecido, al menos durante diez años, dentro de la Unión Europea y presentan una eficacia reconocida, así como un nivel aceptable de seguridad. 6. Cuando se trate de un medicamento veterinario que tenga la misma composición cualitativa y cuantitativa en principios activos y la misma forma farmacéutica que otro ya autorizado, el solicitante podrá usar la documentación farmacéutica, de inocuidad, de estudio de residuos, preclínica y clínica que obre en el expediente del medicamento veterinario autorizado siempre que cuente con el consentimiento del titular. 7. Los medicamentos veterinarios que contengan principios activos que entren en la composición de medicamentos veterinarios autorizados pero que no hayan sido combinados con fines terapéuticos deberán aportar los resultados de las pruebas de inocuidad y estudios de residuos, en caso necesario, y los resultados de nuevos ensayos clínicos y preclínicos relativos a la combinación sin necesidad de aportar la documentación relativa a cada principio activo individual. 8. Otorgada la autorización de un medicamento veterinario cualquier modificación que se solicite en relación con la misma deberá estar debidamente documentada conforme se establezca reglamentariamente. 9. El titular de la autorización de un medicamento veterinario deberá mantener actualizado el expediente aportado para obtener aquélla, incorporando al mismo cuantos datos, informes o modificaciones tecnológicas impongan los avances de la ciencia y las normas de correcta fabricación y control. Asimismo, deberá presentar los informes periódicos de seguridad establecidos reglamentariamente con el fin de mantener actualizado el expediente en materia de seguridad. 10. La Agencia Española de Medicamentos y Productos Sanitarios o las comunidades autónomas podrán exigir, en cualquier momento, del laboratorio farmacéutico que justifique la realización de los controles de calidad y cuantos otros se encuentren establecidos en la autorización y registro de aquél. Artículo 34. Exclusividad de datos. 1. Sin perjuicio del derecho relativo a la protección de la propiedad industrial y comercial, los medicamentos genéricos veterinarios autorizados con arreglo a lo dispuesto en el artículo 33.3 no podrán ser comercializados hasta transcurridos diez años desde la fecha de la autorización inicial del medicamento de referencia. No obstante, los expedientes de los medicamentos veterinarios destinados a peces, abejas y otras especies que se determine en el ámbito de la Unión Europea, contarán con un periodo de exclusividad de datos de trece años. – 26 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios Para los medicamentos veterinarios destinados a especies productoras de alimentos el período de diez años al que se refiere el párrafo anterior se podrá ampliar hasta un máximo de trece años conforme a las condiciones que reglamentariamente se establezcan. 2. En el supuesto de que a una sustancia de uso veterinario bien establecido se le otorgue, con base en nuevos estudios de residuos y nuevos ensayos clínicos, una autorización para otra especie productora de alimentos, se concederá a esa especie un periodo de exclusividad de datos de tres años. Artículo 35. Causas de denegación, suspensión o revocación de la autorización. 1. Serán causas de denegación, suspensión o revocación de la autorización de un medicamento veterinario: a) Que la relación beneficio-riesgo del medicamento veterinario no sea favorable en las condiciones de empleo autorizadas. Cuando se refiera a medicamentos veterinarios de uso zootécnico se tendrán especialmente en cuenta los beneficios en materia de salud y bienestar de los animales, así como de inocuidad para el consumidor. b) Que el medicamento veterinario no tenga efecto terapéutico o que esté insuficientemente justificado respecto de la especie animal que deba someterse a tratamiento. c) Que el medicamento veterinario no tenga la composición cualitativa o cuantitativa declarada. d) Que el tiempo de espera indicado sea insuficiente para que los productos alimenticios procedentes del animal tratado no contengan residuos que puedan presentar peligros para la salud del consumidor, o esté insuficientemente justificado. e) Que el medicamento veterinario se presente a la venta para una utilización no autorizada. f) El incumplimiento de las obligaciones establecidas reglamentariamente. g) Cualquiera otro supuesto en el que la Agencia Europea de Medicamentos así lo hubiere acordado. 2. La suspensión y revocación a que se refiere el apartado anterior se producirá, según lo establecido en el mismo, previas las correspondientes actuaciones de inspección y control realizadas por la Administración General del Estado, en su caso, o por las comunidades autónomas. 3. Las resoluciones de denegación, suspensión o revocación de la autorización de un medicamento veterinario serán motivadas y se adoptarán previo informe del comité competente de la Agencia Española de Medicamentos y Productos Sanitarios en materia de evaluación de medicamentos veterinarios, en el que deberá estar representado el Ministerio de Agricultura, Alimentación y Medio Ambiente. Artículo 36. Validez de la autorización. 1. La autorización de medicamentos veterinarios tendrá una duración de cinco años. 2. La autorización podrá renovarse transcurridos cinco años, previa reevaluación de la relación beneficio/riesgo. La renovación de la autorización tendrá carácter indefinido, salvo que razones de farmacovigilancia justifiquen su sometimiento a un nuevo procedimiento de renovación. 3. El titular de una autorización comunicará, de forma expresa, a la Agencia Española de Medicamentos y Productos Sanitarios la puesta en el mercado por vez primera de un medicamento autorizado y efectuará anualmente una declaración de comercialización en los términos que reglamentariamente se establezcan. 4. La autorización de un medicamento veterinario se entenderá caducada si, en un plazo de tres años, el titular no procede a la comercialización efectiva del mismo o una vez autorizado, inscrito y comercializado deja de encontrarse de forma efectiva en el mercado durante tres años consecutivos. Lo anterior no será de aplicación cuando concurran razones de salud o de interés sanitario o circunstancias excepcionales; en tal caso, la Agencia Española de Medicamentos y Productos Sanitarios mantendrá la validez de la autorización y podrá exigir la comercialización efectiva del producto. – 27 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios Artículo 37. Prescripción de medicamentos veterinarios. 1. Al objeto de proteger la salud humana y la sanidad animal, se exigirá prescripción veterinaria para dispensar al público los siguientes medicamentos veterinarios: a) Los medicamentos respecto de los que los veterinarios deban adoptar precauciones especiales con objeto de evitar riesgos innecesarios a las especies a que se destinan, a la persona que administre dichos medicamentos a los animales y al medio ambiente. b) Los destinados a tratamientos o procesos patológicos que requieran un diagnóstico preciso previo o de cuyo uso puedan derivarse consecuencias que dificulten o interfieran las acciones diagnósticas o terapéuticas posteriores. c) Los medicamentos de sustancias psicoactivas cuyo suministro o utilización estén sujetos a restricciones derivadas de la aplicación de los pertinentes convenios de la Organización de las Naciones Unidas contra el tráfico ilícito de sustancias estupefacientes y psicotrópicas o las derivadas de la legislación comunitaria. d) Los medicamentos veterinarios destinados a animales productores de alimentos. No obstante, el Ministerio de Sanidad, Servicios Sociales e Igualdad, tras consulta al Ministerio de Agricultura, Alimentación y Medio Ambiente, podrá establecer excepciones a este requisito de acuerdo con las decisiones adoptadas por la Comisión Europea en esta materia. e) Los medicamentos utilizados en los supuestos de prescripción excepcional por vacío terapéutico, incluidos los preparados oficinales, fórmulas magistrales y autovacunas. f) Los inmunológicos. 2. Asimismo, se exigirá prescripción para todos aquellos medicamentos veterinarios nuevos que contengan un principio activo cuya utilización en los medicamentos veterinarios lleve menos de cinco años autorizada. 3. Reglamentariamente se establecerá el régimen de prescripciones excepcionales. 4. Sin perjuicio de los supuestos previstos al efecto en la normativa vigente, será precisa la administración, directamente por veterinario o bajo su responsabilidad, de todos aquellos medicamentos veterinarios en que así se prevea en la autorización de comercialización y en los contemplados en los párrafos a), c) y f) del apartado 1. 5. La receta veterinaria será válida en todo el territorio nacional y se editará en la lengua española oficial del Estado y en las respectivas lenguas cooficiales en las comunidades autónomas que dispongan de ella. Reglamentariamente se establecerán los datos que deban constar en la receta veterinaria. Artículo 38. Distribución y dispensación de medicamentos veterinarios. 1. El Gobierno desarrollará la normativa de carácter básico relativa a la distribución y dispensación de medicamentos veterinarios. 2. La dispensación al público de los medicamentos se realizará exclusivamente por: a) Las oficinas de farmacia legalmente establecidas, que además serán las únicas autorizadas para la elaboración y dispensación de fórmulas magistrales y preparados oficinales. b) Los establecimientos comerciales detallistas autorizados, siempre que cuenten con un servicio farmacéutico responsable de la custodia, conservación y dispensación de estos medicamentos. c) Las entidades o agrupaciones ganaderas autorizadas que cuenten con servicio farmacéutico responsable de la custodia, conservación y dispensación de estos medicamentos para el uso exclusivo de sus miembros. Reglamentariamente se regulará la actuación profesional del farmacéutico en cada uno de los establecimientos anteriormente descritos en los párrafos b) y c) como condición y requisito para garantizar el control efectivo en la dispensación al público de los medicamentos veterinarios. No obstante lo anterior, los medicamentos destinados a perros, gatos, animales de terrario, pájaros domiciliarios, peces de acuario y pequeños roedores que no requieran prescripción veterinaria podrán distribuirse y venderse en otros establecimientos, en los términos previstos reglamentariamente. – 28 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios 3. Por razones de urgencia y lejanía de las oficinas de farmacia podrán utilizarse botiquines de medicamentos veterinarios en las condiciones que reglamentariamente se determinen. 4. Reglamentariamente se establecerá el régimen de adquisición, distribución y dispensación de medicamentos veterinarios por parte de las entidades o agrupaciones ganaderas autorizadas que cuenten con servicios farmacéuticos y veterinarios, para el uso exclusivo de sus miembros. Asimismo, reglamentariamente se establecerá el régimen por el que las industrias de alimentación animal y explotaciones ganaderas podrán adquirir directamente las premezclas medicamentosas autorizadas, destinadas a la elaboración de piensos medicamentosos. 5. Las Administraciones públicas, en el ejercicio de sus competencias, podrán adquirir los medicamentos veterinarios, en especial las vacunas, que sean precisos, directamente de los laboratorios farmacéuticos o de cualquier centro de distribución autorizado. Artículo 39. Garantías de disponibilidad de medicamentos veterinarios en situaciones específicas y autorizaciones especiales. 1. En circunstancias excepcionales, la Agencia Española de Medicamentos y Productos Sanitarios podrá conceder una autorización supeditada a la obligación por parte del solicitante de cumplir determinadas condiciones revisables anualmente. Dichas condiciones quedarán, en especial, referidas a la seguridad del medicamento, a la información a las autoridades competentes de todo incidente relacionado con su utilización y a las medidas que deben adoptarse. Reglamentariamente se establecerán los criterios para la obtención de estas autorizaciones. 2. Sin perjuicio del régimen de prescripciones excepcionales, el Ministerio de Sanidad, Servicios Sociales e Igualdad, de acuerdo con el Ministerio de Agricultura, Alimentación y Medio Ambiente, regulará, con carácter excepcional, la utilización de medicamentos por los veterinarios en condiciones distintas a las autorizadas, con el fin de asegurar el bienestar animal y evitar sufrimientos innecesarios a los animales o por motivos de sanidad animal. Esta regulación deberá establecerse, en todo caso, de conformidad con lo dispuesto en la legislación sobre sanidad animal. 3. En caso de epizootias graves, la Agencia Española de Medicamentos y Productos Sanitarios, previa solicitud e informe preceptivo del Ministerio de Agricultura, Alimentación y Medio Ambiente, podrá permitir provisionalmente la utilización de medicamentos inmunológicos veterinarios sin autorización, si no existe el medicamento adecuado, informando previamente sobre sus condiciones de utilización a la Comisión Europea. Artículo 40. Ensayos clínicos con medicamentos veterinarios. 1. A los efectos de esta ley se entiende por ensayo clínico en animales con un medicamento en investigación, a toda investigación efectuada a través de su administración o aplicación a la especie de destino, o a una categoría particular de la misma, a la que se pretende destinar el futuro tratamiento, orientado a confirmar cuando se estime oportuno los efectos farmacodinámicos y/o farmacocinéticos y/o establecer la eficacia para una indicación terapéutica y/o conocer el perfil de sus reacciones adversas y establecer la seguridad y/o tolerancia en las condiciones normales de uso. 2. Los ensayos clínicos en animales con medicamentos en investigación estarán sometidos a régimen de autorización de la Agencia Española de Medicamentos y Productos Sanitarios, conforme al procedimiento reglamentariamente establecido. Deberá cumplirse, además, la normativa aplicable en materia de protección de los animales utilizados para experimentación y otros fines científicos. Artículo 41. Sistema Español de Farmacovigilancia de medicamentos veterinarios. 1. El Sistema Español de Farmacovigilancia de medicamentos veterinarios tiene por objeto la identificación, cuantificación, evaluación y prevención de los riesgos del uso de los medicamentos veterinarios y seguimiento de los posibles efectos adversos de éstos en los animales, las personas o el medio ambiente, así como la presunta falta de eficacia y la detección de tiempos de espera inadecuados. – 29 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios 2. Reglamentariamente se desarrollará el Sistema Español de Farmacovigilancia de medicamentos veterinarios que, coordinado por la Agencia Española de Medicamentos y Productos Sanitarios y conforme establece el artículo 57, integrará las actividades que las Administraciones sanitarias realicen para recoger y elaborar la información sobre reacciones adversas a los medicamentos veterinarios. 3. Los laboratorios farmacéuticos, los veterinarios, los farmacéuticos y demás profesionales sanitarios tienen el deber de comunicar a la Agencia Española de Medicamentos y Productos Sanitarios, de la manera que se determine reglamentariamente, las sospechas de reacciones adversas de las que tengan conocimiento y que puedan haber sido causadas por medicamentos veterinarios. CAPÍTULO IV De las garantías sanitarias de las fórmulas magistrales y preparados oficinales Artículo 42. Requisitos de las fórmulas magistrales. 1. Las fórmulas magistrales serán preparadas con sustancias de acción e indicación reconocidas legalmente en España, de acuerdo con el artículo 44.1 y según las directrices del Formulario Nacional. 2. Las fórmulas magistrales se elaborarán en las oficinas de farmacia y servicios farmacéuticos legalmente establecidos que dispongan de los medios necesarios para su preparación de acuerdo con las exigencias establecidas en el Formulario Nacional. No obstante, las oficinas de farmacia y servicios farmacéuticos que no dispongan de los medios necesarios, excepcionalmente y sin perjuicio de lo establecido en el artículo 66.2, podrán encomendar a una entidad de las previstas en esta ley, autorizada por la Administración sanitaria competente, la realización de una o varias fases de la elaboración y/o control de fórmulas magistrales. 3. En la preparación de fórmulas magistrales se observarán las normas de correcta elaboración y control de calidad de fórmulas magistrales y preparados oficinales. 4. Las fórmulas magistrales destinadas a los animales estarán prescritas por un veterinario y se destinarán a un animal individualizado o a un reducido número de animales de una explotación concreta que se encuentren bajo el cuidado directo de dicho facultativo. Se prepararán por un farmacéutico, o bajo su dirección, en su oficina de farmacia. 5. Las fórmulas magistrales irán acompañadas del nombre del farmacéutico que las prepare y de la información suficiente que garantice su correcta identificación y conservación, así como su segura utilización. 6. La formulación magistral de sustancias o medicamentos no autorizados en España se ajustará al régimen previsto en el artículo 24. Artículo 43. Requisitos de los preparados oficinales. 1. Los preparados oficinales deberán cumplir las siguientes condiciones: a) Estar enumerados y descritos en el Formulario Nacional. b) Cumplir las normas de la Real Farmacopea Española. c) Ser elaborados y garantizados por un farmacéutico de la oficina de farmacia o del servicio farmacéutico que los dispense. d) Presentarse y dispensarse necesariamente bajo principio activo o, en su defecto, bajo una denominación común o científica o la expresada en el formulario nacional y, en ningún caso, bajo marca comercial. e) Ir acompañados del nombre del farmacéutico que los prepare y de la información suficiente que garantice su correcta identificación y conservación, así como su segura utilización. 2. Excepcionalmente, y sin perjuicio de lo establecido en el artículo 66.2, las oficinas de farmacia y servicios farmacéuticos que no dispongan de los medios necesarios podrán encomendar a una entidad legalmente autorizada para tal fin por la Administración sanitaria competente la realización de una o varias fases de la elaboración y/o control de, – 30 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios exclusivamente, aquellos preparados oficinales que respondan a una prescripción facultativa. 3. Los preparados oficinales destinados a los animales serán elaborados en oficinas de farmacia de acuerdo con las indicaciones de un formulario y serán entregados directamente al usuario final. Artículo 44. Formulario Nacional. 1. El Formulario Nacional contendrá las fórmulas magistrales tipificadas y los preparados oficinales reconocidos como medicamentos, sus categorías, indicaciones y materias primas que intervienen en su composición o preparación, así como las normas de correcta preparación y control de aquéllos. 2. Las oficinas de farmacia y servicios farmacéuticos deben garantizar que disponen de acceso a la documentación correspondiente al Formulario Nacional. 3. Queda expresamente prohibida la publicidad de fórmulas magistrales y preparados oficinales. CAPÍTULO V De las garantías sanitarias de los medicamentos especiales Artículo 45. Vacunas y demás medicamentos biológicos. 1. Las vacunas y los productos biológicos utilizables como medicamentos estarán sujetos al régimen de éstos con las particularidades previstas en esta ley o que se establezcan reglamentariamente según su naturaleza y características de aplicación propia. 2. Queda exceptuada de lo dispuesto en el apartado anterior la preparación individualizada de vacunas y alérgenos para un solo paciente, la cual solo podrá efectuarse en las condiciones y establecimientos que reúnan las particularidades que reglamentariamente se establezcan. 3. En el caso de los productos biológicos, cuando sea necesario por interés de la salud pública, la Agencia Española de Medicamentos y Productos Sanitarios podrá someter a autorización previa cada lote de fabricación de producto terminado y condicionar la comercialización a su conformidad. También podrá someter a autorización previa los materiales de origen, productos intermedios y graneles y condicionar a su conformidad su empleo en la fabricación. Los referidos controles se considerarán realizados cuando se acredite documentalmente que han sido efectuados en el país de origen, con idénticas exigencias a las previstas en esta ley y siempre que se hayan mantenido las condiciones originales del producto. Artículo 46. Medicamentos de origen humano. 1. Los derivados de la sangre, del plasma y el resto de sustancias de origen humano (fluidos, glándulas, excreciones, secreciones, tejidos y cualesquiera otras sustancias), así como sus correspondientes derivados, cuando se utilicen con finalidad terapéutica, se considerarán medicamentos y estarán sujetos al régimen previsto en esta ley, con las particularidades que se establezcan reglamentariamente según su naturaleza y características. 2. La sangre, plasma y sus derivados, así como el resto de sustancias de origen humano mencionadas en el apartado 1 y sus correspondientes derivados, deberán ser obtenidos en centros autorizados y bajo control y vigilancia de estos centros, procederán en todo caso de donantes identificados a través del correspondiente registro de donantes. Estos centros autorizados deberán adoptar las medidas precisas de control, vigilancia y trazabilidad que impidan la transmisión de enfermedades infecciosas. 3. La importación y la autorización como medicamentos de los derivados de la sangre y del plasma podrá ser denegada o revocada cuando aquélla no proceda de donaciones altruistas realizadas en bancos de sangre o centros de plasmaféresis, ubicados en los Estados miembros de la Unión Europea que reúnan las debidas garantías. – 31 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios 4. La importación y la autorización como medicamentos del resto de sustancias de origen humano mencionadas en el apartado 1, así como de sus correspondientes derivados, será denegada o revocada cuando no proceda de donantes identificados mediante el correspondiente registro o cuando no se haya obtenido en centros autorizados que reúnan las medidas precisas de control, vigilancia y trazabilidad exigidas en el apartado 2. 5. La autorización como medicamentos de los derivados de la sangre y del plasma podrá condicionarse a la presentación por el solicitante de documentación que acredite que el precio del medicamento no incluye beneficio ilegítimo sobre la sangre donada de forma altruista. Las Administraciones sanitarias promoverán las donaciones de sangre altruistas así como el desarrollo de la producción y utilización de los hemoderivados provenientes de estas donaciones. Artículo 47. Medicamentos de terapia avanzada. 1. Se considera «medicamento de terapia génica», el producto obtenido mediante un conjunto de procesos de fabricación destinados a transferir, in vivo o ex vivo, un gen profiláctico, de diagnóstico o terapéutico, tal como un fragmento de ácido nucleico, a células humanas/animales y su posterior expresión in vivo. La transferencia genética supone un sistema de expresión contenido en un sistema de distribución conocido como vector, que puede ser de origen viral o no viral. El vector puede incluirse asimismo en una célula humana o animal. 2. Se considera «medicamento de terapia celular somática», la utilización en seres humanos de células somáticas vivas, tanto autólogas, procedentes del propio paciente, como alogénicas, procedentes de otro ser humano, o xenogénicas, procedentes de animales, cuyas características biológicas han sido alteradas sustancialmente como resultado de su manipulación para obtener un efecto terapéutico, diagnóstico o preventivo por medios metabólicos, farmacológicos e inmunológicos. Dicha manipulación incluye la expansión o activación de poblaciones celulares autólogas ex vivo, tal como la inmunoterapia adoptiva, y la utilización de células alogénicas y xenogénicas asociadas con productos sanitarios empleados ex vivo o in vivo, tales como microcápsulas, matrices y andamiajes intrínsecos, biodegradables o no biodegradables. 3. Los criterios y exigencias generales de esta ley, así como la normativa europea relativa a las garantías exigibles y condiciones de autorización, serán de aplicación a los medicamentos de terapia avanzada a que se refiere este artículo, siempre que se fabriquen industrialmente. El Gobierno determinará reglamentariamente la aplicación de esta ley a los medicamentos de terapia avanzada cuando, aun concurriendo en ellos las características y condiciones establecidas en las definiciones de «medicamento de terapia génica» o de «medicamento de terapia celular somática», no hayan sido fabricados industrialmente. Artículo 48. Radiofármacos. 1. A los efectos de esta ley se entenderá por: a) «Radiofármaco»: Cualquier producto que, cuando esté preparado para su uso con finalidad terapéutica o diagnóstica, contenga uno o más radionucleidos (isótopos radiactivos). b) «Generador»: Cualquier sistema que incorpore un radionucleido (radionucleido padre) que en su desintegración origine otro radionucleido (radionucleido hijo) que se utilizará como parte integrante de un radiofármaco. c) «Equipo reactivo»: Cualquier preparado industrial que deba combinarse con el radionucleido para obtener el radiofármaco final. d) «Precursor»: Todo radionucleido producido industrialmente para el marcado radiactivo de otras sustancias antes de su administración. 2. Sin perjuicio de las demás obligaciones que vengan impuestas por disposición legal o reglamentaria, la fabricación industrial y la autorización y registro de los generadores, equipos reactivos, precursores y radiofármacos requerirá la autorización previa de la Agencia – 32 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios Española de Medicamentos y Productos Sanitarios, otorgada de acuerdo con los principios generales de esta ley y según las exigencias y procedimientos que reglamentariamente se establezcan. 3. La autorización prevista en el apartado anterior no será exigida para la preparación extemporánea de un radiofármaco, en una unidad de radiofarmacia autorizada, bajo la supervisión y control de un facultativo especialista en radiofarmacia, para su aplicación en un centro o institución legalmente facultados para ello, si se realiza exclusivamente a partir de generadores, equipos reactivos y precursores autorizados y con arreglo a las instrucciones del fabricante. 4. La autorización prevista en el apartado 2 no será exigida para la preparación de muestras autólogas dónde participen radionucleidos, así como la extracción de dosis individuales de radiofármacos listos para su uso en una unidad de radiofarmacia autorizada, bajo la supervisión y control de un facultativo especialista en radiofarmacia, para su aplicación en un centro o institución legalmente facultados para ello. 5. La autorización prevista en el apartado 2 podrá no ser exigida para la preparación de radiofármacos PET (Tomografía de emisión de positrones) en una unidad de radiofarmacia autorizada, bajo la supervisión y control de un facultativo especialista en radiofarmacia, siempre que se realice en las condiciones y con los requisitos determinados reglamentariamente. 6. Los preceptos de esta ley se entenderán sin perjuicio de lo dispuesto por la legislación sobre protección contra las radiaciones de las personas sometidas a exámenes o tratamientos médicos o sobre protección de la salud pública y de los trabajadores. Artículo 49. Medicamentos con sustancias psicoactivas con potencial adictivo. 1. Las sustancias psicoactivas incluidas en las listas anexas a la Convención Única de 1961 sobre Estupefacientes y al Convenio de 1971 sobre Sustancias Psicotrópicas, así como los medicamentos que las contengan, se regirán por esta ley y por su normativa específica. 2. Se someterán dichas sustancias a restricciones derivadas de las obligaciones adquiridas ante la Organización de Naciones Unidas en la lucha contra el tráfico ilícito de sustancias estupefacientes y psicotrópicas. Artículo 50. Medicamentos homeopáticos. 1. Se considera medicamento homeopático, de uso humano o veterinario, el obtenido a partir de sustancias denominadas cepas homeopáticas con arreglo a un procedimiento de fabricación homeopático descrito en la Farmacopea Europea o en la Real Farmacopea Española o, en su defecto, en una farmacopea utilizada de forma oficial en un Estado miembro de la Unión Europea. Un medicamento homeopático podrá contener varios principios activos. 2. Reglamentariamente se establecerán los requisitos de autorización de medicamentos homeopáticos atendiendo a sus condiciones especiales. En particular se establecerá un procedimiento simplificado para aquellos productos cuyas garantías de calidad y seguridad lo permitan. 3. La Agencia Española de Medicamentos y Productos Sanitarios podrá autorizar la comercialización y distribución de las preparaciones homeopáticas que contengan algún componente de los denominados «nosodes», siempre que el titular del producto acredite, de manera suficiente, que la relación beneficio-riesgo de tales preparaciones resulta favorable. A tal efecto, se entenderán por «nosodes» aquellos productos patológicos de origen humano o animal así como los agentes patógenos o sus productos metabólicos y los productos de descomposición de órganos de origen humano o animal. 4. El Ministerio de Sanidad, Servicios Sociales e Igualdad establecerá un código nacional que facilite su pronta identificación y, asimismo, exigirá que sus números o claves figuren en el envase, etiquetado y embalaje de los medicamentos homeopáticos con el mismo criterio que en los demás medicamentos. – 33 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios Artículo 51. Medicamentos de plantas medicinales. 1. Las plantas y sus mezclas, así como los preparados obtenidos de plantas en forma de extractos, liofilizados, destilados, tinturas, cocimientos o cualquier otra preparación galénica que se presente con utilidad terapéutica, diagnóstica o preventiva seguirán el régimen de las fórmulas magistrales, preparados oficinales o medicamentos industriales, según proceda y con las especificidades que reglamentariamente se establezcan. 2. El Ministerio de Sanidad, Servicios Sociales e Igualdad establecerá una lista de plantas cuya venta al público estará restringida o prohibida por razón de su toxicidad. 3. Podrán venderse libremente al público las plantas, tradicionalmente consideradas como medicinales y que se ofrezcan sin referencia a propiedades terapéuticas, diagnósticas o preventivas, quedando prohibida su venta ambulante. Artículo 52. Gases medicinales. 1. Los gases medicinales se consideran medicamentos y están sujetos al régimen previsto en esta ley, con las particularidades que reglamentariamente se establezcan. 2. Sin perjuicio de lo dispuesto en el artículo 3.6, las empresas titulares, fabricantes, importadoras y comercializadoras de gases medicinales licuados podrán suministrarlos, conforme determinen las autoridades sanitarias competentes, a los centros de asistencia sanitaria, de atención social a los pacientes con terapia respiratoria a domicilio, así como a los establecimientos clínicos veterinarios legalmente autorizados. A tales efectos, se entenderá por gases medicinales licuados el oxígeno líquido, nitrógeno líquido y protóxido de nitrógeno líquido así como cualesquiera otros que, con similares características y utilización, puedan fabricarse en el futuro. CAPÍTULO VI De las garantías de seguimiento de la relación beneficio/riesgo en los medicamentos Artículo 53. Farmacovigilancia y obligación de declarar. 1. La Farmacovigilancia es la actividad de salud pública que tiene por objetivo la identificación, cuantificación, evaluación y prevención de los riesgos del uso de los medicamentos una vez comercializados, permitiendo así el seguimiento de los posibles efectos adversos de los medicamentos. 2. Los profesionales sanitarios tienen la obligación de comunicar con celeridad a los órganos competentes en materia de farmacovigilancia de cada Comunidad Autónoma las sospechas de reacciones adversas de las que tengan conocimiento y que pudieran haber sido causadas por medicamentos. 3. Las comunidades autónomas trasladarán la información recibida a la Agencia Española de Medicamentos y Productos Sanitarios. 4. Los titulares de la autorización también están obligados a poner en conocimiento de las autoridades sanitarias competentes en materia de farmacovigilancia las sospechas de reacciones adversas de las que tengan conocimiento y que pudieran haber sido causadas por los medicamentos que fabrican o comercializan, de conformidad con las directrices europeas sobre Buenas Prácticas de Farmacovigilancia. Asimismo, estarán obligados a la actualización permanente de la información de seguridad del producto, a la ejecución de los planes de farmacovigilancia y programas de gestión de riesgos y a la realización de una evaluación continuada de la relación beneficio/riesgo del medicamento, conforme a las directrices nacionales y europeas en la materia. Cuando las autoridades sanitarias consideren que dicha información sobre seguridad interesa de forma relevante a la salud pública, garantizarán el acceso público a la misma. Artículo 54. Sistema Español de Farmacovigilancia. 1. El Sistema Español de Farmacovigilancia, que coordina la Agencia Española de Medicamentos y Productos Sanitarios, integra las actividades que las Administraciones – 34 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios sanitarias realizan de manera permanente y continuada para recoger, elaborar y, en su caso, procesar toda la información útil para la supervisión de medicamentos y, en particular, la información sobre reacciones adversas a los medicamentos, así como para la realización de cuantos estudios se consideren necesarios para evaluar la seguridad de los medicamentos. 2. La Agencia Española de Medicamentos y Productos Sanitarios evaluará la información recibida del Sistema Español de Farmacovigilancia así como de otras fuentes de información. Los datos de reacciones adversas detectadas en España se integrarán en las redes europeas e internacionales de farmacovigilancia, de las que España forme parte, con la garantía de protección de los datos de carácter personal exigida por la normativa vigente. 3. En el Sistema Español de Farmacovigilancia están obligados a colaborar todos los profesionales sanitarios. 4. Las autoridades sanitarias podrán suspender aquellos programas de farmacovigilancia en los que se aprecien defectos graves en los procedimientos de obtención de datos y tratamiento de la información obtenida. Dicha suspensión requerirá el previo informe favorable del comité competente en materia de seguridad de medicamentos de la Agencia Española de Medicamentos y Productos Sanitarios. Artículo 55. Farmacoepidemiología y gestión de los riesgos. La Agencia Española de Medicamentos y Productos Sanitarios promoverá la realización de los estudios de farmacoepidemiología necesarios para evaluar la seguridad de los medicamentos autorizados e inscritos en condiciones reales de uso. Asimismo, establecerá las medidas oportunas tendentes a la gestión de los riesgos identificados, incluyendo la formación e información necesarias. Las autoridades sanitarias de las comunidades autónomas y los profesionales sanitarios participarán en la realización de estos estudios y colaborarán en la difusión de conocimiento sobre la seguridad de los medicamentos en el ámbito asistencial. Artículo 56. Objetividad en la evaluación de la seguridad. La Agencia Española de Medicamentos y Productos Sanitarios contará, para el desarrollo de las tareas relacionadas con la farmacovigilancia en el Sistema Nacional de Salud, con un comité de expertos independientes que asesorará y participará en la evaluación de nuevas evidencias sobre seguridad de medicamentos autorizados e inscritos. El comité propondrá las medidas necesarias para minimizar los riesgos asociados al uso de los medicamentos y para mantener el adecuado equilibrio en la relación beneficio/riesgo de los mismos, especialmente en lo que se refiere a nuevos medicamentos. Los informes de evaluación de las nuevas evidencias sobre seguridad de medicamentos autorizados y las recomendaciones del comité serán de carácter público. Artículo 57. Farmacovigilancia veterinaria. 1. La Agencia Española de Medicamentos y Productos Sanitarios velará por el mantenimiento de las garantías de seguridad de los medicamentos veterinarios, tanto para los animales como para las personas o el medio ambiente. 2. La Agencia Española de Medicamentos y Productos Sanitarios evaluará la información de sospechas de reacciones adversas atribuibles a medicamentos veterinarios recibida de los profesionales implicados en su prescripción, distribución y utilización, así como de los laboratorios titulares de medicamentos veterinarios. Asimismo, promoverá la realización de programas de farmacovigilancia veterinaria e integrará en las correspondientes redes europeas e internacionales la información sobre reacciones adversas detectadas. Asimismo, promoverá la realización de los estudios de farmacoepizootiología necesarios para evaluar la seguridad de los medicamentos veterinarios autorizados en condiciones reales de uso. 3. A efectos de evaluar la información relativa a la seguridad de los medicamentos, la Agencia Española de Medicamentos y Productos Sanitarios contará con un comité de expertos independientes que asesorará y participará en la valoración de nuevas evidencias sobre seguridad de medicamentos veterinarios. El comité propondrá las medidas necesarias para minimizar los riesgos asociados al uso de los medicamentos de forma que sea posible – 35 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios mantener el adecuado equilibrio en la relación beneficio/riesgo de los mismos, tanto para los animales como para la salud pública. TÍTULO III De la investigación de los medicamentos de uso humano y sus garantías Artículo 58. Ensayos clínicos y estudios observacionales. 1. A los efectos de esta ley, se entiende por «ensayo clínico» toda investigación efectuada en seres humanos con el fin de determinar o confirmar los efectos clínicos, farmacológicos y/o demás efectos farmacodinámicos, y/o de detectar las reacciones adversas, y/o de estudiar la absorción, distribución, metabolismo y eliminación de uno o varios medicamentos en investigación con el fin de determinar su seguridad y/o su eficacia. Todos los ensayos clínicos, incluidos los estudios de biodisponibilidad y bioequivalencia, serán diseñados, realizados y comunicados de acuerdo con las normas de «buena práctica clínica» y con respeto a los derechos, la seguridad y el bienestar de los sujetos del ensayo, que prevalecerán sobre los intereses de la ciencia y la sociedad. Las autoridades sanitarias deberán facilitar la realización de los ensayos clínicos en el Sistema Nacional de Salud, tanto en el ámbito de la atención primaria como de la hospitalaria. Las condiciones de desarrollo de los ensayos clínicos en los servicios sanitarios del Sistema Nacional de Salud se establecerán en virtud de los acuerdos que se establezcan entre el promotor y los servicios de salud de las comunidades autónomas con criterios de transparencia y según lo establecido en esta ley. Dichos acuerdos incluirán todos los aspectos necesarios para la correcta realización del ensayo, incluidos los profesionales participantes, los recursos implicados y las compensaciones que se establezcan. 2. A los efectos de esta ley, se entiende por «estudio observacional» el estudio en el que los medicamentos se prescriben de la manera habitual, de acuerdo con las condiciones establecidas en la autorización. La asignación de un paciente a una estrategia terapéutica concreta no estará decidida de antemano por el protocolo de un ensayo, sino que estará determinada por la práctica habitual de la medicina. La decisión de prescribir un medicamento determinado estará claramente disociada de la decisión de incluir al paciente en el estudio. No se aplicará a los pacientes ninguna intervención, ya sea diagnóstica o de seguimiento, que no sea la habitual de la práctica clínica. Se utilizarán métodos epidemiológicos para el análisis de los datos recogidos. En todo caso, los estudios observacionales no estarán sometidos a lo establecido en este título. Artículo 59. Garantías de idoneidad. 1. Los ensayos clínicos con medicamentos en investigación estarán sometidos a régimen de autorización por la Agencia Española de Medicamentos y Productos Sanitarios, conforme al procedimiento reglamentariamente establecido. 2. La Agencia Española de Medicamentos y Productos Sanitarios podrá interrumpir en cualquier momento la realización de un ensayo clínico o exigir la introducción de modificaciones en su protocolo, en los casos siguientes: a) Si se viola la ley. b) Si se alteran las condiciones de su autorización. c) Si no se cumplen los postulados éticos recogidos en el artículo 60. d) Para proteger la salud de los sujetos del ensayo. e) En defensa de la salud pública. 3. Las Administraciones sanitarias tendrán facultades inspectoras en materia de ensayos clínicos, pudiendo investigar incluso las historias clínicas individuales de los sujetos del ensayo, guardando siempre su carácter confidencial. Asimismo, podrán realizar la interrupción cautelar del ensayo por cualquiera de las causas señaladas en el apartado anterior, comunicándolo de inmediato a la Agencia Española de Medicamentos y Productos Sanitarios. – 36 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios 4. Las Administraciones sanitarias velarán por el cumplimiento de las normas de «buena práctica clínica», realizando las inspecciones oportunas, con personas de la debida cualificación y formación universitaria en medicina, farmacia, farmacología, toxicología u otras materias pertinentes. 5. A los efectos previstos en el apartado 2, el investigador de un ensayo deberá notificar inmediatamente al promotor todos los acontecimientos adversos graves, salvo cuando se trate de los señalados en el protocolo como acontecimientos que no requieren comunicación inmediata. El promotor, a su vez, notificará en el menor plazo posible a la Agencia Española de Medicamentos y Productos Sanitarios las reacciones adversas graves e inesperadas que surjan a lo largo del ensayo y adicionalmente enviará informes periódicos de seguridad. Asimismo, el promotor deberá llevar un registro detallado de todos los acontecimientos adversos que le sean notificados, cuya comunicación a las Administraciones sanitarias y al Comité Ético de Investigación Clínica deberá realizarse en los términos y plazos que reglamentariamente se establezcan. 6. El método de los ensayos clínicos deberá ser tal que la evaluación de los resultados que se obtengan con la aplicación de la sustancia o medicamento objeto del ensayo quede controlada por comparación con el mejor patrón de referencia, en orden a asegurar su objetividad, salvo las excepciones impuestas por la naturaleza de su propia investigación. 7. La realización del ensayo deberá ajustarse, en todo caso, al contenido del protocolo de investigación de cada ensayo de acuerdo con el cual se hubiera otorgado la autorización, así como a sus modificaciones posteriores. 8. Los resultados favorables o desfavorables de cada ensayo clínico, tanto si éste llega a su fin como si se abandona la investigación, deberán ser comunicados a la Agencia Española de Medicamentos y Productos Sanitarios, sin perjuicio de su comunicación a las comunidades autónomas en las que se hayan realizado dichos ensayos clínicos. Artículo 60. Garantías de respeto a los postulados éticos. 1. Los ensayos clínicos deberán realizarse en condiciones de respeto a los derechos fundamentales de la persona y a los postulados éticos que afectan a la investigación biomédica en la que resultan afectados seres humanos, siguiéndose, a estos efectos, lo contenido en la Declaración de Helsinki y cualesquiera otros instrumentos internacionales suscritos por España en esta materia. 2. No podrá iniciarse ningún ensayo clínico en tanto no se disponga de suficientes datos científicos y, en particular, ensayos farmacológicos y toxicológicos en animales, que garanticen que los riesgos que implica en la persona en que se realiza son admisibles. 3. Con el fin de evitar investigaciones obsoletas o repetitivas, sólo se podrán iniciar ensayos clínicos para demostrar la eficacia y seguridad de las modificaciones terapéuticas propuestas, siempre que sobre las mismas existan dudas razonables. 4. El sujeto del ensayo prestará su consentimiento libremente, expresado por escrito, tras haber sido informado sobre la naturaleza, importancia, implicaciones y riesgos del ensayo clínico. Si el sujeto del ensayo no está en condiciones de escribir, podrá dar, en casos excepcionales, su consentimiento verbal en presencia de, al menos, un testigo mayor de edad y con capacidad de obrar. El sujeto participante en un ensayo clínico o su representante podrá revocar, en todo momento, su consentimiento sin expresión de causa. En el caso de personas que no puedan emitir libremente su consentimiento, éste deberá ser otorgado por su representante legal previa instrucción y exposición ante el mismo del alcance y riesgos del ensayo. Será necesario, además, la conformidad del representado si sus condiciones le permiten comprender la naturaleza, importancia, alcance y riesgos del ensayo. 5. Lo establecido en el apartado anterior se entenderá sin perjuicio de lo previsto en el artículo 9.2 de la Ley 41/2002, de 14 de noviembre, reguladora de la autonomía del paciente y de derechos y obligaciones en materia de información y documentación clínica, en los términos que reglamentariamente se determinen. 6. Ningún ensayo clínico podrá ser realizado sin informe previo favorable de un Comité Ético de Investigación Clínica, que será independiente de los promotores e investigadores y de las autoridades sanitarias. El Comité deberá ser acreditado por el órgano competente de la Comunidad Autónoma que corresponda, el cuál asegurará la independencia de aquél. La – 37 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios acreditación será comunicada a la Agencia Española de Medicamentos y Productos Sanitarios por el órgano competente de la respectiva Comunidad Autónoma. 7. Los Comités Éticos de Investigación Clínica estarán formados, como mínimo, por un equipo interdisciplinar integrado por médicos, farmacéuticos de atención primaria y hospitalaria, farmacólogos clínicos, personal de enfermería y personas ajenas a las profesiones sanitarias de las que al menos uno será licenciado en Derecho especialista en la materia. 8. El Comité Ético de Investigación Clínica ponderará los aspectos metodológicos, éticos y legales del protocolo propuesto, así como el balance de riesgos y beneficios anticipados dimanantes del ensayo. 9. Los Comités Éticos de Investigación Clínica podrán requerir información completa sobre las fuentes y cuantía de la financiación del ensayo y la distribución de los gastos en, entre otros, los siguientes apartados: reembolso de gastos a los pacientes, pagos por análisis especiales o asistencia técnica, compra de aparatos, equipos y materiales, pagos debidos a los hospitales o a los centros en que se desarrolla la investigación por el empleo de sus recursos y compensación a los investigadores. 10. Reglamentariamente se establecerá el procedimiento para la designación del Comité Ético de referencia y para la obtención del dictamen único con validez en todo el territorio, con el objetivo de impulsar la investigación clínica en el Sistema Nacional de Salud. El Ministerio de Sanidad, Servicios Sociales e Igualdad desarrollará acciones que permitan que los Comités Éticos de Investigación Clínica acreditados puedan compartir estándares de calidad y criterios de evaluación adecuados y homogéneos. Artículo 61. Garantías de asunción de responsabilidades. 1. La realización de un ensayo clínico exigirá que, mediante la contratación de un seguro o la constitución de otra garantía financiera, se garantice previamente la cobertura de los daños y perjuicios que, para la persona en la que se lleva a efecto, pudieran derivarse de aquél. 2. Cuando, por cualquier circunstancia, el seguro no cubra enteramente los daños causados, el promotor del ensayo, el investigador responsable del mismo y el hospital o centro en que se hubiere realizado responderán solidariamente de aquéllos, aunque no medie culpa, incumbiéndoles la carga de la prueba. Ni la autorización administrativa ni el informe del Comité Ético de Investigación Clínica les eximirán de responsabilidad. 3. Se presume, salvo prueba en contrario, que los daños que afecten a la salud de la persona sujeta al ensayo, durante la realización del mismo y durante el plazo de un año contado desde su finalización, se han producido como consecuencia del ensayo. Sin embargo, una vez concluido el año, el sujeto del mismo está obligado a probar el daño y nexo entre el ensayo y el daño producido. 4. Es promotor del ensayo clínico la persona física o jurídica que tiene interés en su realización, firma la solicitud de autorización dirigida al Comité Ético de Investigación Clínica y a la Agencia Española de Medicamentos y Productos Sanitarios y se responsabiliza de él. 5. Es investigador principal quien dirige la realización del ensayo y firma en unión del promotor la solicitud, corresponsabilizándose con él. La condición de promotor y la de investigador principal pueden concurrir en la misma persona física. Artículo 62. Garantías de transparencia. 1. Los ensayos clínicos autorizados por la Agencia Española de Medicamentos y Productos Sanitarios formarán parte de un registro nacional de ensayos clínicos público y libre que será accesible en las condiciones que reglamentariamente se determine. 2. El promotor está obligado a publicar los resultados del ensayo clínico, sean positivos o no. La publicación se realizará, previa supresión de cualquier información comercial de carácter confidencial, preferentemente en revistas científicas y, de no ser ello posible, a través de los medios y en los plazos máximos que se establezcan reglamentariamente. En la publicación se mencionará el Comité Ético de Investigación Clínica que los informó. 3. Cuando se hagan públicos estudios y trabajos de investigación sobre medicamentos dirigidos a la comunidad científica, se harán constar los fondos obtenidos por el autor por o para su realización y la fuente de financiación. – 38 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios 4. En caso de no publicarse los resultados de los ensayos clínicos y cuando los mismos permitan concluir que el medicamento presenta modificaciones de su perfil de eficacia o seguridad, la Agencia Española de Medicamentos y Productos Sanitarios hará públicos los resultados. 5. Toda la información sobre el ensayo clínico deberá registrarse, tratarse y conservarse de forma que pueda ser comunicada, interpretada y comprobada de manera precisa, protegiendo al mismo tiempo el carácter confidencial de los registros de los sujetos del ensayo. 6. El Gobierno, previo informe del Consejo Interterritorial del Sistema Nacional de Salud, y con carácter básico, regulará los requisitos comunes para la realización y financiación de los ensayos clínicos, asegurando la buena práctica clínica y las condiciones de su realización. Los centros, servicios, establecimientos y profesionales sanitarios participarán en la realización de ensayos clínicos de acuerdo con estos requisitos comunes y condiciones de financiación y los que en su desarrollo puedan establecer las Administraciones sanitarias competentes. TÍTULO IV De las garantías exigibles en la fabricación y distribución de medicamentos CAPÍTULO I De la fabricación de medicamentos Artículo 63. Autorización del laboratorio farmacéutico. 1. A efectos de esta ley, las personas físicas o jurídicas que se dediquen a la fabricación de medicamentos o a cualquiera de los procesos que ésta pueda comprender, incluso los de fraccionamiento, acondicionamiento y presentación para la venta, deberán estar autorizadas previamente por la Agencia Española de Medicamentos y Productos Sanitarios. Esta autorización será asimismo necesaria para la importación y comercialización de medicamentos e incluso para el supuesto de que el medicamento se fabrique exclusivamente para su exportación. La Agencia Española de Medicamentos y Productos Sanitarios hará pública la autorización así como sus modificaciones y la extinción de la misma. 2. Para obtener la autorización de laboratorio farmacéutico, el solicitante deberá cumplir los siguientes requisitos: a) Detallar los medicamentos y las formas farmacéuticas que pretenda fabricar, así como el lugar, establecimiento o laboratorio de fabricación y control. b) Disponer de locales, equipo técnico y de control, adecuados y suficientes para una correcta fabricación, control y conservación que responda a las exigencias legales. c) Disponer de un director técnico responsable, de un responsable de fabricación y de un responsable de control de calidad. Todos ellos deberán cumplir las condiciones profesionales y funcionales que reglamentariamente se establezcan. Los laboratorios que fabriquen pequeñas cantidades o productos simples podrán atribuir la función de control al director técnico, pero la dirección de fabricación deberá corresponder a persona distinta. Artículo 64. Garantías para la correcta fabricación de medicamentos y de materias primas. 1. Sin perjuicio de las demás obligaciones que vengan impuestas por disposición legal o reglamentaria, el laboratorio farmacéutico deberá cumplir las siguientes obligaciones: a) Disponer de personal suficiente y con la cualificación técnica necesaria para garantizar la calidad de los medicamentos y la ejecución de los controles procedentes con arreglo a lo dispuesto en la ley. b) Suministrar los medicamentos de acuerdo con la legislación vigente. c) Tener abastecido el mercado con los productos registrados, de modo adecuado y continuado para posibilitar el cumplimiento de las exigencias de funcionamiento que se – 39 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios señalan en el artículo 69.1, pudiendo suspenderse tal abastecimiento sólo en casos excepcionales debidamente justificados tras disponer de la correspondiente autorización de la Agencia Española de Medicamentos y Productos Sanitarios. d) Permitir, en todo momento, el acceso a sus locales y archivos a las autoridades competentes para realizar inspecciones. e) Facilitar el cumplimiento de sus funciones al director técnico y cuidar de que disponga de los medios necesarios para ello. f) Responder de las obligaciones que les sean exigibles durante el tiempo de su actividad, incluso en caso de suspensión de la misma, y durante los cinco años posteriores a su clausura. g) Garantizar que el transporte de los medicamentos hasta destino, sea a entidades de distribución o servicios u oficinas de farmacia, se realiza cumpliendo tanto las obligaciones impuestas en la autorización de los mismos como las normas de correcta distribución de los medicamentos. h) Ajustar a lo establecido por la normativa de las comunidades autónomas las actividades de promoción, publicidad y patrocinio realizadas por los laboratorios. i) Comunicar al Ministerio de Sanidad, Servicios Sociales e Igualdad las unidades de medicamentos vendidas para ser dispensadas en el territorio nacional, incluyendo los números de lote independientemente del destino final. j) Comunicar la suspensión o cese de sus actividades. 2. Los laboratorios farmacéuticos deberán cumplir las normas de correcta fabricación publicadas por el Ministerio de Sanidad, Servicios Sociales e Igualdad, conforme a las directrices detalladas sobre prácticas de correcta fabricación de medicamentos establecidas en el marco comunitario. Asimismo, los fabricantes y los distribuidores de principios activos utilizados como materias primas deberán cumplir las normas de correcta fabricación de principios activos y las buenas prácticas de distribución de principios activos, publicadas por el Ministerio de Sanidad, Servicios Sociales e Igualdad. A tales efectos, se entiende por «fabricación de principios activos utilizados como materias primas» la fabricación completa o parcial o la importación de un principio activo utilizado como materia prima, tal y como se define en el artículo 2, así como los diversos procesos de división, acondicionamiento y presentación previos a su incorporación en un medicamento, incluidos el reacondicionamiento y reetiquetado. El laboratorio farmacéutico únicamente podrá utilizar como materias primas principios activos fabricados de conformidad con las normas de correcta fabricación de principios activos y distribuidos de conformidad con las buenas prácticas de distribución de principios activos. Para este fin, el laboratorio farmacéutico verificará el cumplimiento por parte del fabricante y de los distribuidores de principios activos de las normas correctas de fabricación y de las buenas prácticas de distribución, mediante la realización de auditorías en las instalaciones de fabricación y distribución de los fabricantes y distribuidores de los mismos. El laboratorio farmacéutico verificará tal cumplimiento por sí mismo o, sin perjuicio de su responsabilidad de conformidad con lo dispuesto en esta ley, a través de una entidad que actúe por cuenta de él en virtud de un contrato. 3. El laboratorio farmacéutico garantizará que los excipientes son aptos para su utilización en un medicamento mediante la determinación de las prácticas de fabricación apropiadas, con una evaluación formal de riesgos realizada con arreglo a las directrices aplicables contempladas en las normas de correcta fabricación de medicamentos así como las establecidas en el marco comunitario. 4. El laboratorio farmacéutico realizará los controles de calidad que procedan sobre las materias primas, los productos intermedios de fabricación y el producto terminado de acuerdo con los métodos y técnicas generalmente aceptados. 5. A efectos de cumplir lo establecido en el apartado anterior, cada laboratorio farmacéutico contará con una unidad de control y garantía de calidad de los productos, procesos y procedimientos con la autoridad y responsabilidad de aceptar o rechazar materias primas, intermedios y productos finales. Los procesos y procedimientos de fabricación deberán estar validados. – 40 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios 6. Los fabricantes de medicamentos y productos sanitarios deberán contar con un seguro, aval o garantía financiera equivalente para responder de los daños sobre la salud derivados de problemas de seguridad de los medicamentos, en los términos que reglamentariamente se disponga. Artículo 65. Modificación, suspensión y revocación de la autorización. 1. Cualquier modificación de los requisitos a que se refieren los párrafos a) y b) del artículo 63.2 o del objeto de la autorización deberá ser previamente aprobada por la Agencia Española de Medicamentos y Productos Sanitarios. 2. La sustitución del director técnico se comunicará a la Agencia Española de Medicamentos y Productos Sanitarios y al órgano competente de la Comunidad Autónoma. 3. La Agencia Española de Medicamentos y Productos Sanitarios podrá suspender o revocar la autorización del laboratorio, para una categoría determinada de productos o para todos ellos, cuando no se cumplan los requisitos y/o las obligaciones establecidas en este capítulo. Asimismo, podrá suspenderla o revocarla cuando el laboratorio no cumpla las buenas prácticas de farmacovigilancia o no realice en tiempo y forma los estudios que, a tales efectos, se exigen en esta ley. Artículo 66. Fabricación por terceros. 1. Los laboratorios farmacéuticos podrán encomendar a terceros la realización de actividades de fabricación o controles previstos en esta ley para los medicamentos, si se cumplen los requisitos siguientes: a) El tercero contratado deberá disponer de la autorización a que se refiere el artículo 63. b) La Agencia Española de Medicamentos y Productos Sanitarios deberá autorizar específicamente la fabricación por terceros. 2. Excepcionalmente, y cuando así lo requiera la atención a sus pacientes, los servicios de farmacia hospitalaria y oficinas de farmacia podrán encomendar a una entidad legalmente autorizada por la Agencia Española de Medicamentos y Productos Sanitarios la realización de alguna fase de la producción de una preparación concreta o de su control analítico. CAPÍTULO II De la distribución de medicamentos Artículo 67. Garantías de accesibilidad y disponibilidad de los medicamentos. 1. La distribución de los medicamentos autorizados se realizará a través de entidades de distribución o directamente por el laboratorio titular de la autorización de comercialización de los mismos. 2. La actividad de distribución deberá garantizar un servicio de calidad, siendo su función prioritaria y esencial el abastecimiento a las oficinas de farmacia y a los servicios de farmacia legalmente autorizados en el territorio nacional. 3. La utilización de terceros por parte de un laboratorio o una entidad de distribución para la distribución de medicamentos deberá incluirse en la correspondiente autorización como laboratorio o entidad de distribución. Artículo 68. Control administrativo de la distribución mayorista. 1. Los almacenes mayoristas, así como los almacenes por contrato, estarán sometidos a la autorización previa de la comunidad autónoma donde esté domiciliado el almacén. Ello no obstante, el almacén deberá comunicar la realización de sus actividades a las autoridades sanitarias de las comunidades autónomas donde, no estando domiciliado, tales actividades se realicen. La autorización de entidad de distribución podrá incluir la actividad de almacén por contrato. 2. Sin perjuicio de las competencias de las comunidades autónomas, la entidad de distribución y, en su caso, el laboratorio titular de la autorización de comercialización – 41 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios deberán comunicar directamente a la Agencia Española de Medicamentos y Productos Sanitarios el inicio de sus actividades. 3. Sin perjuicio de lo dispuesto en el apartado 1, los almacenes de medicamentos bajo control o vigilancia aduanera estarán sometidos a la autorización previa como entidad de distribución de medicamentos que será otorgada por la Agencia Española de Medicamentos y Productos Sanitarios. Artículo 69. Exigencias de funcionamiento. 1. Las entidades de distribución y, en su caso, los laboratorios farmacéuticos que distribuyan directamente sus productos estarán obligados: a) A disponer de locales y equipos dotados de medios personales, materiales y técnicos para garantizar la correcta conservación y distribución de los medicamentos, con plena garantía para la salud pública. b) A garantizar la observancia de las condiciones generales o particulares de conservación de los medicamentos y especialmente el mantenimiento de la cadena de frío en toda la red de distribución mediante procedimientos normalizados. c) A mantener unas existencias mínimas de medicamentos que garanticen la adecuada continuidad del abastecimiento. d) A asegurar plazos de entrega, frecuencia mínima de repartos, asesoramiento técnico farmacéutico permanente y medios de apoyo a oficinas y servicios de farmacia. e) A cumplir servicios de guardia y prevención de catástrofes. f) A disponer de un plan de emergencia que garantice la aplicación efectiva de cualquier retirada del mercado ordenada por las autoridades sanitarias competentes. g) A tener implantado un sistema de alertas que cubra todas las farmacias del territorio de su ámbito de actuación. h) A cumplir con las normas de buenas prácticas de distribución que hayan sido promovidas o autorizadas por las Administraciones sanitarias competentes y a colaborar con éstas para asegurar una prestación farmacéutica de calidad. i) Al cumplimiento de las demás obligaciones que vengan impuestas por disposición legal o reglamentaria. 2. El Gobierno, con carácter básico, podrá establecer los requisitos y condiciones mínimos de estos establecimientos a fin de asegurar las previsiones contenidas en el apartado anterior. El Gobierno velará por preservar el derecho de la entidad de distribución a ser suministrada por los laboratorios. Artículo 70. Director técnico. Todas las entidades de distribución autorizadas de acuerdo con el artículo 68 dispondrán de un director técnico farmacéutico, cuyo cargo será incompatible con otras actividades de carácter sanitario que supongan intereses directos con la fabricación o dispensación de medicamentos o que vayan en detrimento del adecuado cumplimiento de sus funciones. El Gobierno establecerá las funciones del director técnico. Artículo 71. Intermediación en la distribución de medicamentos de uso humano. 1. Las personas que se dediquen a tareas de intermediación en la distribución de medicamentos de uso humano establecidas en España deberán inscribirse, de forma previa al inicio de su actividad, en un registro que la Agencia Española de Medicamentos y Productos Sanitarios mantendrá a tal efecto, que incluirá todos los datos que se fijen de forma reglamentaria. Las autoridades sanitarias de las comunidades autónomas tendrán acceso a los datos completos de este registro a efectos de inspección. Este registro será de acceso público. 2. Las personas que se dediquen a la intermediación en el comercio de medicamentos deberán cumplir las obligaciones que vengan impuestas en la normativa vigente así como las disposiciones específicas incluidas en las buenas prácticas de distribución de medicamentos publicadas por el Ministerio de Sanidad, Servicios Sociales e Igualdad. – 42 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios TÍTULO V De las garantías sanitarias del comercio exterior de medicamentos Artículo 72. Importaciones. 1. Sin perjuicio de otras exigencias legal o reglamentariamente establecidas, sólo podrán importarse medicamentos autorizados e inscritos en el Registro de Medicamentos de acuerdo con las exigencias previstas en esta ley. 2. La distribución de los medicamentos se ajustará a las exigencias previstas en el título IV. A tal efecto el importador podrá utilizar los canales farmacéuticos legalmente habilitados para ello o constituirse en entidad de distribución previa la correspondiente autorización otorgada de acuerdo con el capítulo II del título IV. 3. El director técnico de la entidad importadora garantiza la conformidad de los lotes importados y responde de que cada lote de fabricación importado ha sido objeto en España de un análisis cualitativo completo, de un análisis cuantitativo referido, por lo menos, a todos los principios activos y de los demás controles que resulten necesarios para garantizar su calidad según los términos de la autorización y registro del medicamento. A tal efecto se deberá facilitar la documentación y muestras que reglamentariamente se determinen para su control por el Ministerio de Sanidad, Servicios Sociales e Igualdad. 4. Los controles mencionados en el apartado anterior se considerarán realizados cuando a juicio del Ministerio de Sanidad, Servicios Sociales e Igualdad se acredite documentalmente haberse efectuado, en el país de origen, con idénticas exigencias a las previstas en esta ley, sin perjuicio de las obligaciones derivadas de la pertenencia a la Unión Europea y demás tratados internacionales suscritos por España. 5. La importación de «medicamentos en investigación» requerirá autorización previa de la Agencia Española de Medicamentos y Productos Sanitarios. 6. El titular de un medicamento en España no podrá impedir su importación y comercialización por terceros siempre que lo introduzcan en el mercado español con las garantías establecidas por esta ley con las adaptaciones que reglamentariamente se determinen. 7. Las personas físicas o jurídicas que se dediquen a la importación de medicamentos, materias primas o productos sanitarios deberán contar, en los mismos términos que los fabricantes, con un seguro, aval o garantía financiera equivalente para responder de los daños para la salud derivados de problemas de seguridad de los medicamentos, de acuerdo a lo que reglamentariamente se disponga. Artículo 73. Exportaciones. 1. Podrán exportar medicamentos los laboratorios y entidades de distribución que cumplan los requisitos legalmente establecidos. 2. La exportación de medicamentos autorizados e inscritos se notificará por el exportador a la Agencia Española de Medicamentos y Productos Sanitarios en los casos y términos que reglamentariamente se determinen. 3. No se exigirán al producto a exportar los requisitos establecidos por esta ley para su autorización como medicamento en España, en lo que se refiere a formato o presentación, textos, etiquetado y características de los envases, siempre que se respeten los principios que esta ley establece sobre garantías de información a los profesionales y los usuarios. Artículo 74. Medicamentos destinados al tratamiento de los viajeros. 1. Los medicamentos que acompañen a los viajeros destinados a su propia administración o tratamiento quedan excluidos de las exigencias establecidas en los artículos anteriores, sin perjuicio de las medidas de control cuando dichos medicamentos pudieran representar una desviación por su cuantía o destino especialmente en prevención de su utilización ilícita. – 43 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios 2. Las Administraciones públicas adoptarán las medidas oportunas para impedir que los productos objeto de esta ley, en régimen de tránsito hacia un tercer país, puedan ser desviados para su uso en España sin cumplimiento de las exigencias previstas en esta ley. 3. De acuerdo con lo previsto en la legislación sobre protección de la salud y lucha contra el dopaje en el deporte, los deportistas, equipos o grupos deportivos y los directivos extranjeros que los representen están obligados, cuando entren en España para participar en una actividad deportiva, a remitir debidamente cumplimentados a la Agencia Española de Protección de la Salud en el Deporte los formularios que la misma establezca, en los que se identifiquen los productos que transportan para su uso, las unidades de los mismos y el médico responsable de su prescripción o, en el caso de animales que participen en eventos deportivos, el veterinario. TÍTULO VI Del registro de laboratorios farmacéuticos y del registro de fabricantes, importadores o distribuidores de principios activos Artículo 75. Registro de laboratorios farmacéuticos. 1. La Agencia Española de Medicamentos y Productos Sanitarios mantendrá un registro de laboratorios farmacéuticos que incluirá todos los datos que estén obligados a suministrar para el cumplimiento de las previsiones de esta ley. Este registro será de acceso público. 2. Es obligatoria la inscripción en este registro de la autorización inicial, así como de cualquier transmisión, modificación o extinción. Artículo 76. Registro de fabricantes, importadores o distribuidores de principios activos. 1. La Agencia Española de Medicamentos y Productos Sanitarios mantendrá un registro de fabricantes, importadores o distribuidores de principios activos que incluirá todos los datos que se fijen de forma reglamentaria. Las autoridades sanitarias de las comunidades autónomas tendrán acceso a los datos completos de este registro, a efectos de inspección. Los datos de este registro serán de acceso público. 2. Es obligatoria la inscripción en este registro de forma previa al inicio de la actividad de fabricación, importación o distribución así como la remisión inmediata de cualquier cambio en los datos proporcionados que pueda repercutir en la calidad, seguridad o eficacia de los principios activos. Asimismo, de forma anual se actualizarán los datos remitidos. TÍTULO VII Del uso racional de los medicamentos de uso humano CAPÍTULO I De las garantías de formación e información independiente y de calidad para la utilización adecuada de los medicamentos y productos sanitarios Artículo 77. Garantías de las Administraciones públicas. 1. Las Administraciones públicas competentes en los órdenes sanitario y educativo dirigirán sus actuaciones a promover la formación universitaria y post-universitaria continuada y permanente sobre medicamentos, terapéutica y productos sanitarios de los profesionales sanitarios. 2. Las Administraciones públicas sanitarias dirigirán sus actuaciones a instrumentar un sistema ágil, eficaz e independiente que asegure a los profesionales sanitarios información científica, actualizada y objetiva de los medicamentos y productos sanitarios. 3. Las Administraciones públicas dirigirán sus actuaciones a impulsar la constitución de centros propios de información de medicamentos y productos sanitarios, mediante la promoción y coordinación en la utilización de recursos y tecnologías de la información que – 44 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios permitan a las instituciones sanitarias profesionales y otras entidades acceder a la información sobre dichos productos. 4. Las Administraciones públicas sanitarias promoverán la publicación de guías farmacológicas y/o farmacoterapéuticas para uso de los profesionales sanitarios. 5. Las Administraciones públicas sanitarias realizarán programas de educación sanitaria sobre medicamentos dirigidos al público en general impulsando actuaciones que favorezcan un mejor conocimiento de los medicamentos para mejorar el cumplimiento terapéutico, evitar los riesgos derivados de un uso incorrecto y concienciar a los ciudadanos del valor económico del medicamento. Artículo 78. Objetividad y calidad de la información y promoción dirigida a los profesionales sanitarios. 1. La información y promoción dirigida a los profesionales sanitarios, bajo control de las Administraciones sanitarias en los términos previstos en el artículo 102.1 de la Ley 14/1986, de 25 de abril, General de Sanidad, deberá estar de acuerdo con la información técnica y científica autorizada por la Agencia Española de Medicamentos y Productos Sanitarios y deberá ser rigurosa, bien fundada y objetiva y no inducir a error, de acuerdo con la legislación vigente, y ajustarse a la ficha técnica. 2. La información y promoción podrá realizarse a través de soportes escritos, audiovisuales o de otra naturaleza, dirigidos con exclusividad a profesionales sanitarios y tendrá carácter científico. En el caso de informes o artículos financiados por un laboratorio farmacéutico o entidad relacionada con el mismo, deberá especificarse esta circunstancia en la publicación. 3. Cuando se trate de información o promoción distribuida por medios informáticos, las Administraciones sanitarias podrán acceder a ella a los efectos de inspección. 4. Las ofertas de premios, becas, contribuciones y subvenciones a reuniones, congresos, viajes de estudio y actos similares por cualquier persona, física o jurídica, relacionada con la fabricación, elaboración, distribución, prescripción y dispensación de medicamentos y productos sanitarios, se harán públicas en la forma que se determine reglamentariamente y se aplicarán exclusivamente a actividades de índole científica cuando sus destinatarios sean profesionales sanitarios o las entidades en que se asocian. En los programas, publicaciones de trabajos y ponencias de reuniones, congresos y actos similares se harán constar la fuente de financiación de los mismos y los fondos obtenidos de cada fuente. La misma obligación alcanzará al medio de comunicación por cuya vía se hagan públicos y que obtenga fondos por o para su publicación. Artículo 79. La receta médica y la prescripción hospitalaria. 1. La receta médica, pública o privada, y la orden de dispensación hospitalaria son los documentos que aseguran la instauración de un tratamiento con medicamentos por instrucción de un médico, un odontólogo o un podólogo, en el ámbito de sus competencias respectivas, únicos profesionales con facultad para recetar medicamentos sujetos a prescripción médica. Sin perjuicio de lo anterior, los enfermeros de forma autónoma, podrán indicar, usar y autorizar la dispensación de todos aquellos medicamentos no sujetos a prescripción médica y los productos sanitarios relacionados con su ejercicio profesional, mediante la correspondiente orden de dispensación. Los fisioterapeutas también podrán indicar, usar y autorizar, de forma autónoma, la dispensación de medicamentos no sujetos a prescripción médica y de productos sanitarios relacionados con el ejercicio de su profesión, mediante orden de dispensación. El Gobierno regulará la indicación, uso y autorización de dispensación de determinados medicamentos sujetos a prescripción médica por los enfermeros, en el marco de los principios de la atención integral de salud y para la continuidad asistencial, mediante la aplicación de protocolos y guías de práctica clínica y asistencial, de elaboración conjunta, acordados con las organizaciones colegiales de médicos y enfermeros y validados por la Dirección General de Salud Pública, Calidad e Innovación del Ministerio de Sanidad, Servicios Sociales e Igualdad. – 45 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios Igualmente el Gobierno regulará la indicación, uso y autorización de dispensación de determinados medicamentos sujetos a prescripción médica por los enfermeros, en el ámbito de los cuidados tanto generales como especializados, y fijará, con la participación de las organizaciones colegiales de enfermeros y de médicos, los criterios generales, requisitos específicos y procedimientos para la acreditación de dichos profesionales, con efectos en todo el territorio del Estado, en las actuaciones previstas en este apartado. El Ministerio de Sanidad, Servicios Sociales e Igualdad, con la participación de las organizaciones colegiales correspondientes, acreditará con efectos en todo el Estado a los enfermeros y a los fisioterapeutas para las actuaciones previstas en este artículo. Téngase en cuenta que se declara la inconstitucionalidad y nulidad de la referencia al «Ministerio de Sanidad, Servicios Sociales e Igualdad», del párrafo 5 del apartado 1, en los términos del fj 9, por Sentencia 76/2018, de 5 de julio. Ref. BOE-A-2018-11274 2. El farmacéutico dispensará con receta aquellos medicamentos que la requieran. Dicho requisito deberá especificarse expresamente en el embalaje del medicamento. 3. La receta médica será válida en todo el territorio nacional y se editará en la lengua española oficial del Estado y en las respectivas lenguas cooficiales en las comunidades autónomas que dispongan de ella. 4. Las recetas médicas y órdenes hospitalarias de dispensación deberán contener los datos básicos de identificación de prescriptor, paciente y medicamentos. 5. En las recetas y órdenes hospitalarias de dispensación, el facultativo incluirá las pertinentes advertencias para el farmacéutico y para el paciente, así como las instrucciones para un mejor seguimiento del tratamiento a través de los procedimientos de la atención farmacéutica, con el fin de garantizar la consecución de los objetivos sanitarios de aquéllas. 6. El Gobierno podrá regular con carácter básico lo dispuesto en los apartados anteriores y establecer la exigencia de otros requisitos que por afectar a la salud pública o al sistema sanitario hayan de ser de general aplicación en las recetas médicas u órdenes hospitalarias. 7. Los trámites a que sean sometidas las recetas y órdenes médicas y especialmente en su tratamiento informático, respetarán lo dispuesto en el artículo 10 de la Ley 14/1986, de 25 de abril. 8. El Gobierno determinará con carácter básico los requisitos mínimos que han de cumplir las recetas médicas extendidas y/o editadas en soporte informático con el fin de asegurar la accesibilidad de todos los ciudadanos, en condiciones de igualdad efectiva en el conjunto del territorio español, a la prestación farmacéutica del Sistema Nacional de Salud. No será necesario el consentimiento del interesado para el tratamiento y la cesión de datos que sean consecuencia de la implantación de sistemas de información basados en receta médica en soporte papel o electrónico, de conformidad con lo dispuesto en los artículos 7, apartados 3 y 6; 8; y 11, apartado 2.a), de la Ley Orgánica 15/1999, de 13 de diciembre, de Protección de Datos de Carácter Personal. Las citadas actuaciones deberán tener por finalidad facilitar la asistencia médica y farmacéutica al paciente y permitir el control de la prestación farmacéutica del Sistema Nacional de Salud. 9. Las Administraciones públicas sanitarias realizarán programas de educación sanitaria destinados a la población general, orientados a destacar la importancia de la receta médica como garantía de calidad y seguridad de los pacientes. 10. Lo dispuesto en este artículo será asimismo de aplicación a la receta veterinaria, en cuyo caso las referencias al médico y odontólogo se entenderán hechas al veterinario. Artículo 80. Garantías en la publicidad de medicamentos y productos sanitarios destinada al público en general. 1. Podrán ser objeto de publicidad destinada al público los medicamentos que cumplan todos los requisitos que se relacionan a continuación: a) Que no se financien con fondos públicos. – 46 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios b) Que, por su composición y objetivo, estén destinados y concebidos para su utilización sin la intervención de un médico que realice el diagnóstico, la prescripción o el seguimiento del tratamiento, aunque requieran la intervención de un farmacéutico. Este requisito podrá exceptuarse cuando se realicen campañas de vacunación aprobadas por las autoridades sanitarias competentes. c) Que no constituyan sustancias psicotrópicas o estupefacientes con arreglo a lo definido en los convenios internacionales. 2. La publicidad de un medicamento que sea objeto de publicidad al público, cumplirá con los requisitos establecidos en el apartado 1 de este artículo; por su parte, los mensajes publicitarios deberán reunir los siguientes requisitos: a) Que resulte evidente el carácter publicitario del mensaje y quede claramente especificado que el producto es un medicamento. b) Que se incluya la denominación del medicamento en cuestión, así como la denominación común cuando el medicamento contenga una única sustancia activa. c) Que se incluyan todas las informaciones indispensables para la utilización correcta del medicamento así como una invitación expresa y claramente visible a leer detenidamente las instrucciones que figuren en el prospecto o en el embalaje externo, según el caso, y la recomendación de consultar al farmacéutico sobre su correcta utilización. d) No incluir expresiones que proporcionen seguridad de curación, ni testimonios sobre las virtudes del producto ni de profesionales o personas cuya notoriedad pueda inducir al consumo. e) No utilizar como argumento publicitario el hecho de haber obtenido autorización sanitaria en cualquier país o cualquier otra autorización, número de registro sanitario o certificación que corresponda expedir, ni los controles o análisis que compete ejecutar a las autoridades sanitarias con arreglo a lo dispuesto en esta ley. f) Los mensajes publicitarios de los medicamentos que se emitan en soporte audiovisual deberán cumplir las condiciones de accesibilidad para personas con discapacidad establecidas en el ordenamiento jurídico para la publicidad institucional. 3. La publicidad de medicamentos no sujetos a prescripción médica no requerirá de autorización administrativa previa, si bien las Administraciones sanitarias competentes efectuarán los controles necesarios para garantizar que los contenidos publicitarios cumplan con las normas legales y reglamentarias, que les sean de aplicación y que se ajusten fielmente a las condiciones científicas y técnicas recogidas en la autorización de comercialización. 4. Las Administraciones sanitarias, por razones de salud pública o seguridad de las personas, podrán limitar, condicionar o prohibir la publicidad de los medicamentos y de los productos sanitarios. 5. Se prohíben las primas, obsequios, premios, concursos, bonificaciones o similares como métodos vinculados a la promoción o venta al público de estos medicamentos. 6. En el caso de los productos sanitarios queda excluida la posibilidad de realizar publicidad directa o indirecta, dirigida al público en el caso de que un producto esté financiado por el Sistema Nacional de Salud. Esta prohibición de publicidad afecta a las empresas fabricantes, distribuidoras o comercializadoras, así como a todas aquellas entidades que puedan mantener un contacto directo con el paciente. Asimismo, se prohíben las primas, obsequios, descuentos, premios, concursos, bonificaciones o similares como métodos vinculados a la promoción o venta al público de dichos productos. 7. No podrán ser objeto de publicidad destinada al público los productos sanitarios que estén destinados a ser utilizados o aplicados exclusivamente por profesionales sanitarios. 8. La publicidad de las técnicas o procedimientos médicos o quirúrgicos ligados a la utilización de productos sanitarios específicos respetará los criterios contemplados en la publicidad de productos sanitarios. Artículo 81. Garantías en la publicidad de productos con supuestas propiedades sobre la salud. La publicidad y promoción comercial de los productos, materiales, sustancias o métodos a los que se atribuyan efectos beneficiosos sobre la salud se regulará reglamentariamente. – 47 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios Artículo 82. Utilización racional de los medicamentos en el deporte. La importación, exportación, distribución, comercialización, prescripción y dispensación de medicamentos, legalmente reconocidos, no tendrán por finalidad aumentar las capacidades físicas de los deportistas o modificar los resultados de las competiciones en las que participan, debiendo ajustarse en su desarrollo y objetivos a la normativa de aplicación en la materia. CAPÍTULO II Del uso racional de medicamentos en la atención primaria a la salud Artículo 83. Estructuras de soporte para el uso racional de medicamentos y productos sanitarios en atención primaria. 1. Sin perjuicio de la responsabilidad que todos los profesionales sanitarios tienen en el uso racional de los medicamentos, las estructuras de gestión de atención primaria deberán disponer de servicios o unidades de farmacia de atención primaria. 2. Para contribuir al uso racional de los medicamentos las unidades o servicios de farmacia de atención primaria realizarán las siguientes funciones: a) Garantizar y asumir la responsabilidad técnica de la adquisición, calidad, correcta conservación, cobertura de las necesidades, custodia, preparación de fórmulas magistrales o preparados oficinales y dispensación de los medicamentos para ser aplicados dentro de los centros de atención primaria y de aquéllos para los que se exija una particular vigilancia, supervisión y control, según se establece en el artículo 103 de la Ley 14/1986, de 25 de abril, General de Sanidad y en las disposiciones reglamentarias que lo desarrollan. b) Establecer un sistema eficaz y seguro de distribución de medicamentos y productos sanitarios en los centros y estructuras a su cargo. c) Establecer sistemas de información sobre gestión de la farmacoterapia que incluya aspectos clínicos, de efectividad, seguridad y eficiencia de la utilización de los medicamentos y proporcionar una correcta información y formación sobre medicamentos y productos sanitarios a los profesionales sanitarios. d) Desarrollar protocolos y guías farmacoterapéuticas que garanticen la correcta asistencia farmacoterapéutica a los pacientes, en especial lo referente a la selección de medicamentos y la continuidad de los tratamientos y sistemas de apoyo a la toma de decisiones clínicas en farmacoterapia. e) Impulsar la coordinación en farmacoterapia entre diferentes estructuras sanitarias y niveles asistenciales y promover una investigación clínica en farmacoterapia de calidad y adecuada a las necesidades de los pacientes, garantizando la correcta custodia y dispensación de los productos en fase de investigación clínica. f) Establecer un sistema para el seguimiento de los tratamientos a los pacientes que contribuya a garantizar el cumplimiento terapéutico así como programas que potencien un uso seguro de los medicamentos. g) Impulsar y participar en programas de educación de la población sobre medicamentos, su empleo racional y la prevención de su abuso y formar parte de las comisiones relacionadas con el uso racional de medicamentos y productos sanitarios. h) Impulsar la coordinación y trabajo en equipo y colaboración con los hospitales y servicios de atención especializada, con la finalidad de asegurar la calidad de la prestación farmacéutica mediante el seguimiento de los tratamientos prescritos por el médico. i) Realizar cuantas funciones puedan redundar en un mejor uso y control de los medicamentos, mediante estrategias de colaboración entre los profesionales sanitarios de los equipos de atención primaria. 3. Todo lo anterior será asimismo de aplicación para los productos sanitarios excepto en aquellos supuestos donde resulte imposible su aplicación por la propia naturaleza del producto. – 48 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios CAPÍTULO III Del uso racional de los medicamentos en la atención hospitalaria y especializada Artículo 84. hospitales. Estructuras de soporte para el uso racional de los medicamentos en los 1. Sin perjuicio de la responsabilidad que todos los profesionales sanitarios tienen en el uso racional de los medicamentos, los hospitales deberán disponer de servicios o unidades de farmacia hospitalaria con arreglo a las condiciones mínimas establecidas por esta ley. Los hospitales del más alto nivel y aquellos otros que se determinen deberán disponer de servicios o unidades de Farmacología Clínica. 2. Para contribuir al uso racional de los medicamentos las unidades o servicios de farmacia hospitalaria realizarán las siguientes funciones: a) Garantizar y asumir la responsabilidad técnica de la adquisición, calidad, correcta conservación, cobertura de las necesidades, custodia, preparación de fórmulas magistrales o preparados oficinales y dispensación de los medicamentos precisos para las actividades intrahospitalarias y de aquellos otros para tratamientos extrahospitalarios, conforme a lo establecido en el artículo 3.6. b) Establecer un sistema eficaz y seguro de distribución de medicamentos, tomar las medidas para garantizar su correcta administración, custodiar y dispensar los productos en fase de investigación clínica y velar por el cumplimiento de la legislación sobre medicamentos de sustancias psicoactivas o de cualquier otro medicamento que requiera un control especial. c) Formar parte de las comisiones hospitalarias en que puedan ser útiles sus conocimientos para la selección y evaluación científica de los medicamentos y de su empleo. d) Establecer un servicio de información de medicamentos para todo el personal del hospital, un sistema de farmacovigilancia intrahospitalario, estudios sistemáticos de utilización de medicamentos y actividades de farmacocinética clínica. e) Llevar a cabo actividades educativas sobre cuestiones de su competencia dirigidas al personal sanitario del hospital y a los pacientes. f) Efectuar trabajos de investigación propios o en colaboración con otras unidades o servicios y participar en los ensayos clínicos con medicamentos. g) Colaborar con las estructuras de atención primaria y especializada de la zona en el desarrollo de las funciones señaladas en el artículo 83. h) Realizar cuantas funciones puedan redundar en un mejor uso y control de los medicamentos. i) Participar y coordinar la gestión de las compras de medicamentos y productos sanitarios del hospital a efectos de asegurar la eficiencia de la misma. 3. Las funciones definidas en los párrafos c) a h) del apartado anterior serán desarrolladas en colaboración con farmacología clínica y demás unidades o servicios clínicos del hospital. Artículo 85. Farmacia hospitalaria. 1. Los servicios de farmacia hospitalaria estarán bajo la titularidad y responsabilidad de un farmacéutico especialista en farmacia hospitalaria. 2. Las Administraciones sanitarias con competencias en ordenación farmacéutica realizarán tal función en la farmacia hospitalaria manteniendo los siguientes criterios: a) Fijación de requerimientos para su buen funcionamiento, acorde con las funciones establecidas. b) Que las actuaciones se presten con la presencia y actuación profesional del o de los farmacéuticos necesarios para una correcta asistencia. c) Los farmacéuticos de las farmacias hospitalarias deberán haber cursado los estudios de la especialidad correspondiente. – 49 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios 3. Los hospitales que no cuenten con servicios farmacéuticos deberán solicitar de las comunidades autónomas autorización para, en su caso, mantener un depósito de medicamentos bajo la supervisión y control de un farmacéutico. Las condiciones, requisitos y normas de funcionamiento de tales depósitos serán determinadas por la autoridad sanitaria competente. CAPÍTULO IV Del uso racional de medicamentos en las oficinas de farmacia Artículo 86. Oficinas de farmacia. 1. En las oficinas de farmacia, los farmacéuticos, como responsables de la dispensación de medicamentos a los ciudadanos, velarán por el cumplimiento de las pautas establecidas por el médico responsable del paciente en la prescripción y cooperarán con él en el seguimiento del tratamiento a través de los procedimientos de atención farmacéutica, contribuyendo a asegurar su eficacia y seguridad. Asimismo, participarán en la realización del conjunto de actividades destinadas a la utilización racional de los medicamentos, en particular a través de la dispensación informada al paciente. Una vez dispensado el medicamento podrán facilitar sistemas personalizados de dosificación a los pacientes que lo soliciten, en orden a mejorar el cumplimiento terapéutico, en los tratamientos y con las condiciones y requisitos que establezcan las administraciones sanitarias competentes. 2. Las Administraciones sanitarias realizarán la ordenación de las oficinas de farmacia, debiendo tener en cuenta los siguientes criterios: a) Planificación general de las oficinas de farmacia en orden a garantizar la adecuada asistencia farmacéutica. b) La presencia y actuación profesional del farmacéutico como condición y requisito inexcusable para la dispensación al público de medicamentos, teniendo en cuenta el número de farmacéuticos necesarios en función de la actividad de la oficina. c) Las exigencias mínimas materiales, técnicas y de medios, incluida la accesibilidad para personas con discapacidad, que establezca el Gobierno con carácter básico para asegurar la prestación de una correcta asistencia sanitaria, sin perjuicio de las competencias que tengan atribuidas las comunidades autónomas en esta materia. 3. Las oficinas de farmacia vienen obligadas a dispensar los medicamentos que se les demanden tanto por los particulares como por el Sistema Nacional de Salud en las condiciones reglamentarias establecidas. 4. Por razones de emergencia y lejanía de la oficina de farmacia u otras circunstancias especiales que concurran, en ciertos establecimientos podrá autorizarse, excepcionalmente, la creación de botiquines en las condiciones que reglamentariamente se determinen con carácter básico, sin perjuicio de las competencias que tengan atribuidas las comunidades autónomas en esta materia. 5. Las Administraciones públicas velarán por la formación continuada de los farmacéuticos y la adecuada titulación y formación de los auxiliares y ayudantes técnicos de farmacia. 6. Las oficinas de farmacia tienen la consideración de establecimientos sanitarios privados, de interés público. Artículo 87. Prescripción de medicamentos y productos sanitarios. 1. La prescripción de medicamentos y productos sanitarios en el Sistema Nacional de Salud se efectuará en la forma más apropiada para el beneficio de los pacientes, a la vez que se protege la sostenibilidad del sistema. 2. En el Sistema Nacional de Salud, las prescripciones de medicamentos incluidos en el sistema de precios de referencia o de agrupaciones homogéneas no incluidas en el mismo, se efectuarán de acuerdo con el siguiente esquema: a) Para procesos agudos, la prescripción se hará, de forma general, por principio activo. – 50 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios b) Para los procesos crónicos, la primera prescripción, correspondiente a la instauración del primer tratamiento, se hará, de forma general, por principio activo. c) Para los procesos crónicos cuya prescripción se corresponda con la continuidad de tratamiento, podrá realizarse por denominación comercial, siempre y cuando ésta se encuentre incluida en el sistema de precios de referencia o sea la de menor precio dentro de su agrupación homogénea. 3. No obstante, la prescripción por denominación comercial de medicamentos será posible siempre y cuando se respete el principio de mayor eficiencia para el sistema y en el caso de los medicamentos considerados como no sustituibles. 4. Cuando la prescripción se realice por principio activo, el farmacéutico dispensará el medicamento de precio más bajo de su agrupación homogénea. 5. En todo caso, la prescripción de un medicamento para su utilización en condiciones diferentes a las establecidas en su ficha técnica deberá ser autorizada, previamente, por la comisión responsable de los protocolos terapéuticos u órgano colegiado equivalente en cada comunidad autónoma. Artículo 88. Sistemas de información para apoyo a la prescripción. 1. Los órganos competentes de las comunidades autónomas dotarán a sus prescriptores de un sistema de prescripción electrónica común e interoperable y que permitirá el registro de la información sobre el número de dosis ajustada a las necesidades del tratamiento, en el que se incorporarán subsistemas de apoyo a la prescripción, tales como: nomenclátor de medicamentos en línea; correspondencia entre principios activos, medicamentos disponibles y patologías en las que están indicados; protocolos de tratamiento por patología recomendados desde las instituciones sanitarias y las sociedades médicas, con indicación de los estándares de elección y los beneficios esperados; coste del tratamiento prescrito y alternativas de elección terapéutica, según criterios de eficiencia; base de datos de interacciones; base de datos de ensayos clínicos en su provincia o comunidad autónoma; información periódica en línea (autorización y retirada de medicamentos y productos sanitarios, alertas y comunicaciones de interés para la protección de la salud pública); difusión de noticias sobre medicamentos que, sin ser alertas en sentido estricto, contribuyan a mejorar el nivel de salud de la población. 2. Los sistemas de apoyo a la prescripción recogerán la información correspondiente a los precios seleccionados vía aportación reducida, de modo que el médico pueda tomar en consideración el impacto económico durante la prescripción de medicamentos y productos sanitarios. 3. Los sistemas de apoyo a la prescripción serán gestionados desde los órganos competentes a nivel de comunidad autónoma. El Consejo Interterritorial del Sistema Nacional de Salud velará por que los mismos se articulen de modo eficiente y contribuyan a mantener la equidad del sistema sanitario. 4. El Ministerio de Sanidad, Servicios Sociales e Igualdad, en coordinación con las comunidades autónomas, establecerá protocolos asistenciales de carácter básico de modo que se oriente la prescripción y utilización de aquellos medicamentos que, por sus características singulares, requieran especial atención y cautela en su prescripción y dispensación. Artículo 89. Sustitución por el farmacéutico. 1. El farmacéutico dispensará el medicamento prescrito por el médico. 2. Con carácter excepcional, cuando por causa de desabastecimiento no se disponga en la oficina de farmacia del medicamento prescrito o concurran razones de urgente necesidad en su dispensación, el farmacéutico podrá sustituirlo por el de menor precio. En todo caso, deberá tener igual composición, forma farmacéutica, vía de administración y dosificación. El farmacéutico informará en todo caso al paciente sobre la sustitución y se asegurará de que conozca el tratamiento prescrito por el médico. 3. En estos casos, el farmacéutico anotará, en el lugar correspondiente de la receta, el medicamento de la misma composición, forma farmacéutica, vía de administración y dosificación que dispense, la fecha, su firma y su rúbrica. – 51 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios 4. Quedarán exceptuados de esta posibilidad de sustitución aquellos medicamentos que, por razón de sus características de biodisponibilidad y estrecho rango terapéutico, determine el Ministerio de Sanidad, Servicios Sociales e Igualdad. 5. Cuando la prescripción se realice por denominación comercial, si el medicamento prescrito tiene un precio superior al precio menor de su agrupación homogénea, el farmacéutico sustituirá el medicamento prescrito por el de precio más bajo de su agrupación homogénea. En el caso de los medicamentos biosimilares, se respetarán las normas vigentes según regulación específica en materia de sustitución e intercambiabilidad. CAPÍTULO V De la trazabilidad de los medicamentos Artículo 90. Garantías de trazabilidad. 1. Con el fin de lograr un adecuado abastecimiento del mercado y establecer garantías de seguridad para los ciudadanos, los laboratorios, las entidades de distribución, las oficinas de farmacia, los establecimientos comerciales detallistas y las entidades o agrupaciones ganaderas autorizadas para la dispensación de medicamentos veterinarios, están sujetos a las obligaciones de información a que se refiere este artículo. 2. Los laboratorios farmacéuticos deberán comunicar, en los términos que se fijen, reglamentariamente, al Ministerio de Sanidad, Servicios Sociales e Igualdad, las unidades de presentaciones identificadas por lotes de medicamentos y destinatario, vendidas en territorio nacional, así como las que sean objeto de devolución. Asimismo, garantizarán, en los términos que se fijen reglamentariamente, la identificación de cada unidad a lo largo de su recorrido, de acuerdo con lo dispuesto en el artículo 15.4. 3. Las entidades de distribución comunicarán, en los términos que se fijen reglamentariamente, a la Comunidad Autónoma en la que tengan su domicilio social y al Ministerio de Sanidad, Servicios Sociales e Igualdad las unidades suministradas y las devueltas, con indicación del lote al que pertenezcan así como el destinatario, tanto se trate de oficinas o servicios de farmacia como de otras entidades de distribución, con independencia de la Comunidad Autónoma en la que radiquen. 4. Sin perjuicio de los conciertos que se pudieran suscribir, los titulares de las oficinas de farmacia comunicarán al órgano competente de la Comunidad Autónoma en la que tengan su ámbito de actuación las unidades de medicamentos dispensadas. Los órganos competentes de las comunidades autónomas remitirán dicha información al Ministerio de Sanidad, Servicios Sociales e Igualdad, en los términos que se fijen reglamentariamente. 5. Las oficinas de farmacia, los establecimientos comerciales detallistas y las entidades o agrupaciones ganaderas autorizadas para dispensar medicamentos veterinarios, comunicarán, en los términos que se fijen reglamentariamente, a la Agencia Española de Medicamentos y Productos Sanitarios las unidades de medicamentos veterinarios dispensados. 6. La recogida y tratamiento de datos a que se refiere este artículo deberá adecuarse a la normativa vigente en materia de seguridad y protección de datos de carácter personal, en cumplimiento de la Ley Orgánica 15/1999, de 13 de diciembre, de Protección de Datos de Carácter Personal, teniendo la consideración de responsables de sus respectivos ficheros de titularidad pública la Administración General del Estado, las Administraciones sanitarias competentes de las comunidades autónomas y, en su caso, las Administraciones corporativas correspondientes. TÍTULO VIII De la financiación pública de los medicamentos y productos sanitarios Artículo 91. Principio de igualdad territorial y coordinación. 1. Se reconoce el derecho de todos los ciudadanos a obtener medicamentos en condiciones de igualdad en todo el Sistema Nacional de Salud, sin perjuicio de las medidas – 52 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios tendentes a racionalizar la prescripción y la utilización de medicamentos y productos sanitarios que puedan adoptar las comunidades autónomas en ejercicio de sus competencias. 2. Las disposiciones normativas del Gobierno o del Ministerio de Sanidad, Servicios Sociales e Igualdad y las resoluciones emitidas por el centro directivo competente de dicho Ministerio, en materia de financiación de medicamentos y productos sanitarios del Sistema Nacional de Salud, surtirán efecto en todo el territorio español desde la fecha en que resulten aplicables. 3. El precio industrial de financiación pública, establecido por el órgano competente del Ministerio de Sanidad, Servicios Sociales e Igualdad, para los medicamentos dispensados en oficinas de farmacia mediante receta médica oficial del Sistema Nacional de Salud, no podrá ser objeto de modificación o bonificación, salvo en el caso de que la misma consista en un descuento porcentual o lineal aplicable en todo el territorio nacional, sin perjuicio de lo regulado en el artículo 4.6. Las previsiones contenidas en el párrafo anterior también serán de aplicación a los productos sanitarios, una vez se desarrolle reglamentariamente el sistema de financiación de precios y márgenes de dichos productos, incluidos en la prestación farmacéutica del Sistema Nacional de Salud. 4. Toda modificación del precio de un medicamento o producto sanitario financiado por el Sistema Nacional de Salud surtirá efecto en la misma fecha en todo el territorio español. 5. Las medidas tendentes a racionalizar la prescripción y utilización de medicamentos y productos sanitarios que puedan adoptar las comunidades autónomas no producirán diferencias en las condiciones de acceso a los medicamentos y productos sanitarios financiados por el Sistema Nacional de Salud, catálogo y precios. Dichas medidas de racionalización serán homogéneas para la totalidad del territorio español y no producirán distorsiones en el mercado único de medicamentos y productos sanitarios. 6. El Consejo Interterritorial del Sistema Nacional de Salud podrá acordar las condiciones generales de planificación, coordinación, contratación, adquisición y suministro de medicamentos y productos sanitarios de las estructuras y servicios de titularidad pública integrados en el Sistema Nacional de Salud. Artículo 92. Procedimiento para la financiación pública. 1. Para la financiación pública de los medicamentos y productos sanitarios será necesaria su inclusión en la prestación farmacéutica mediante la correspondiente resolución expresa de la unidad responsable del Ministerio de Sanidad, Servicios Sociales e Igualdad, estableciendo las condiciones de financiación y precio en el ámbito del Sistema Nacional de Salud. Del mismo modo se procederá cuando se produzca una modificación de la autorización que afecte al contenido de la prestación farmacéutica, con carácter previo a la puesta en el mercado del producto modificado, bien por afectar la modificación a las indicaciones del medicamento, bien porque, sin afectarlas, la Agencia Española de Medicamentos y Productos Sanitarios así lo acuerde por razones de interés público o defensa de la salud o seguridad de las personas. La inclusión de medicamentos en la financiación del Sistema Nacional de Salud se posibilita mediante la financiación selectiva y no indiscriminada teniendo en cuenta criterios generales, objetivos y publicados y, concretamente, los siguientes: a) Gravedad, duración y secuelas de las distintas patologías para las que resulten indicados. b) Necesidades específicas de ciertos colectivos. c) Valor terapéutico y social del medicamento y beneficio clínico incremental del mismo teniendo en cuenta su relación coste-efectividad. d) Racionalización del gasto público destinado a prestación farmacéutica e impacto presupuestario en el Sistema Nacional de Salud. e) Existencia de medicamentos u otras alternativas terapéuticas para las mismas afecciones a menor precio o inferior coste de tratamiento. f) Grado de innovación del medicamento. – 53 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios Sin perjuicio de lo dispuesto en el artículo 24, y con objeto de garantizar el uso racional de los medicamentos y productos sanitarios, el Ministerio de Sanidad, Servicios Sociales e Igualdad podrá someter a reservas singulares las condiciones específicas de prescripción, dispensación y financiación de los mismos en el Sistema Nacional de Salud, de oficio o a propuesta de las comunidades autónomas en la Comisión Permanente de Farmacia del Consejo Interterritorial del Sistema Nacional de Salud. Con el fin de garantizar el derecho de todas las personas que gocen de la condición de asegurado y beneficiario en el Sistema de un acceso a la prestación farmacéutica en condiciones de igualdad en todo el Sistema Nacional de Salud, las comunidades autónomas no podrán establecer, de forma unilateral, reservas singulares específicas de prescripción, dispensación y financiación de fármacos o productos sanitarios. No obstante lo anterior, en el seno de la Comisión Permanente de Farmacia del Consejo Interterritorial del Sistema Nacional de Salud podrá decidirse la excepción motivada por una o varias comunidades autónomas en razón de sus propias particularidades. 2. El Ministerio de Sanidad, Servicios Sociales e Igualdad revisará los grupos, subgrupos, categorías y/o clases de medicamentos cuya financiación no se estime necesaria para cubrir las necesidades sanitarias básicas de la población española. En todo caso, no se incluirán en la prestación farmacéutica medicamentos no sujetos a prescripción médica, medicamentos que no se utilicen para el tratamiento de una patología claramente determinada, ni los productos de utilización cosmética, dietéticos, aguas minerales, elixires, dentífricos y otros productos similares. Tampoco se financiarán los medicamentos indicados en el tratamiento de síndromes y/o síntomas de gravedad menor, ni aquellos que, aun habiendo sido autorizados de acuerdo a la normativa vigente en su momento, no respondan a las necesidades terapéuticas actuales, entendiendo por tal un balance beneficio/riesgo desfavorable en las enfermedades para las que estén indicados. 3. La decisión de excluir, total o parcialmente, o someter a condiciones especiales de financiación, los medicamentos ya incluidos en la prestación farmacéutica del Sistema Nacional de Salud, se hará con los criterios establecidos en los apartados anteriores y teniendo en cuenta el precio o el coste del tratamiento de los medicamentos comparables existentes en el mercado y las orientaciones del Consejo Interterritorial del Sistema Nacional de Salud. 4. De forma equivalente se procederá en el caso de los productos sanitarios que vayan a ser incluidos en la prestación farmacéutica del Sistema Nacional de Salud y que se dispensen, a través de receta oficial, en territorio nacional. 5. El Gobierno revisará periódicamente y actualizará la relación de los medicamentos y productos sanitarios incluidos en la prestación farmacéutica del Sistema Nacional de Salud, de acuerdo con la evolución de los criterios de uso racional, los conocimientos científicos, la aparición de nuevos medicamentos de mayor utilidad terapéutica o la aparición de efectos adversos que hagan variar la relación beneficio/riesgo y los criterios incluidos en los apartados anteriores. 6. Los productos sanitarios que vayan a ser incluidos en la prestación farmacéutica del Sistema Nacional de Salud y que se dispensen, a través de receta oficial, en territorio nacional, seguirán los criterios indicados para los medicamentos. En todo caso, deberán cumplir con las especificaciones y prestaciones técnicas contrastadas que hubiera previamente determinado el Ministerio de Sanidad, Servicios Sociales e Igualdad, teniendo en cuenta criterios generales, objetivos y publicados y en concreto los siguientes: a) Gravedad, duración y secuelas de las distintas patologías para las que resulten indicadas. b) Necesidades específicas de ciertos colectivos. c) Valor diagnóstico, de control, de tratamiento, prevención, alivio o compensación de una discapacidad. d) Valor social del producto sanitario y beneficio clínico incremental del mismo teniendo en cuenta su relación coste-efectividad. e) Existencia de productos sanitarios u otras alternativas terapéuticas para las mismas afecciones a menor precio o inferior coste de tratamiento. – 54 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios 7. Reglamentariamente se desarrollará el procedimiento de la incorporación a la prestación farmacéutica del Sistema Nacional de Salud de los medicamentos que, según lo dispuesto en el artículo 14, lleven las siglas EFG en razón de su intercambiabilidad. 8. Para la decisión de financiación de nuevos medicamentos, además del correspondiente análisis coste-efectividad y de impacto presupuestario, se tendrá en cuenta el componente de innovación, para avances terapéuticos indiscutibles por modificar el curso de la enfermedad o mejorar el curso de la misma, el pronóstico y el resultado terapéutico de la intervención y su contribución a la sostenibilidad del Sistema Nacional de Salud si, para un mismo resultado en salud, contribuye positivamente al Producto Interior Bruto. Artículo 93. sanitarios. Exclusión de la prestación farmacéutica de medicamentos y productos 1. El órgano responsable de la prestación farmacéutica del Ministerio de Sanidad, Servicios Sociales e Igualdad actualizará, mediante resolución motivada, la lista de medicamentos que quedan excluidos de la prestación farmacéutica en el Sistema Nacional de Salud. 2. La motivación de la exclusión responderá a alguno de los siguientes criterios: a) El establecimiento de precios seleccionados. b) La convivencia con un medicamento no sujeto a prescripción médica con la que comparte principio activo y dosis. c) La consideración del medicamento como susceptible de publicitarse, directamente al público, en la Unión Europea. d) Que el principio activo cuente con un perfil de seguridad y eficacia favorable y suficientemente documentado a través de años de experiencia y un uso extenso. e) Por estar indicados en el tratamiento de síntomas menores. f) Por cumplir cualquiera de los criterios de no inclusión en financiación pública recogido en el artículo 92.2. 3. Los responsables de los productos excluidos de la financiación comunicarán al órgano competente los precios a los que van a comercializar dichos medicamentos. La misma obligación se extiende a las variaciones en los precios. 4. En el mes siguiente a la entrada en el registro del órgano competente de las comunicaciones a las que se refiere el apartado anterior, éste resolverá sobre su conformidad o no a los precios propuestos. En caso de disconformidad, dicho órgano elevará la discrepancia a la Comisión Interministerial de Precios de los Medicamentos, la cual resolverá sobre dicha cuestión. Dicha decisión será notificada mediante resolución del órgano competente al interesado. La decisión administrativa recogida en el párrafo anterior se basará en razones de protección de la salud pública, de igualdad de acceso a los medicamentos por parte de los pacientes o de lesión real o potencial de los intereses de colectivos desfavorecidos. 5. En tanto en cuanto se mantenga la disconformidad mencionada en el apartado anterior, se mantendrá la vigencia del precio industrial máximo. Artículo 94. Fijación de precios. 1. Corresponde al Gobierno establecer los criterios y procedimiento para la fijación de precios de medicamentos y productos sanitarios financiables por el Sistema Nacional de Salud, tanto para los medicamentos de dispensación por oficina de farmacia a través de receta oficial, como para los medicamentos de ámbito hospitalario, incluidos los envases clínicos, o dispensados por servicios de farmacia a pacientes no ingresados. Se tendrán en consideración, los mecanismos de retorno (descuentos lineales, revisión de precio) para los medicamentos innovadores. La Comisión Interministerial de Precios de los Medicamentos tendrá en consideración los análisis coste-efectividad y de impacto presupuestario. 2. Para la comercialización de un medicamento en territorio español será imprescindible haber tramitado la oferta del mismo al Sistema Nacional de Salud. Se procederá, de igual modo, si se producen variaciones sustanciales en las condiciones de autorización del medicamento. – 55 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios 3. El Gobierno podrá regular el mecanismo de fijación de los precios de los medicamentos y productos sanitarios no sujetos a prescripción médica que se dispensen en territorio español, siguiendo un régimen general objetivo y transparente. 4. En todo caso, los titulares de autorizaciones de comercialización de los mismos podrán comercializar los medicamentos que se dispensen en territorio español en régimen de precios notificados, entendiendo por tal la comunicación del precio al Ministerio de Sanidad, Servicios Sociales e Igualdad, de modo que el departamento pueda objetar el mismo por razones de interés público. 5. Corresponde a la Comisión Interministerial de Precios de los Medicamentos, adscrita al Ministerio de Sanidad, Servicios Sociales e Igualdad, fijar, de modo motivado y conforme a criterios objetivos, los precios de financiación del Sistema Nacional de Salud de medicamentos y productos sanitarios para los que sea necesario prescripción médica, que se dispensen en territorio español. Cuando estos mismos productos no resulten financiados, si son dispensados en territorio nacional operará lo establecido en el apartado 4. 6. En todo caso, los medicamentos y productos sanitarios que se decida puedan ser financiados por el Sistema Nacional de Salud podrán también comercializarse para su prescripción fuera del mismo. 7. Como regla general, el precio de financiación por el Sistema Nacional de Salud será inferior o igual al precio industrial del medicamento aplicado cuando sea dispensado fuera del Sistema Nacional de Salud. Los laboratorios farmacéuticos, las entidades de distribución y las oficinas de farmacia a través de la Organización Farmacéutica Colegial, deben aportar la información necesaria para hacer efectivo el reembolso debido por las oficinas de farmacia a laboratorios farmacéuticos y entidades de distribución en aquellos medicamentos que hayan sido dispensados fuera del Sistema Nacional de Salud. Dicha información se obtendrá a través del Sistema que se determine para dar cumplimiento en España a lo dispuesto por la Comisión Europea en virtud del artículo 54 bis de la Directiva 2001/83/CE. 8. Para la toma de decisiones, la Comisión Interministerial de Precios de los Medicamentos tendrá en consideración los informes que elabore el Comité Asesor para la Financiación de la Prestación Farmacéutica del Sistema Nacional de Salud. 9. Las cuantías económicas correspondientes a los conceptos de la distribución y dispensación de los medicamentos y de los productos sanitarios y, en su caso, de las deducciones aplicables a la facturación de los mismos al Sistema Nacional de Salud serán fijados por el Gobierno, previo acuerdo de la Comisión Delegada del Gobierno para Asuntos Económicos, de forma general o por grupos o sectores, tomando en consideración criterios de carácter técnico-económico y sanitario. 10. El Ministerio de Sanidad, Servicios Sociales e Igualdad establecerá el precio de venta al público de los medicamentos y productos sanitarios financiados mediante la agregación del precio industrial autorizado, que tiene carácter de máximo, y de los márgenes correspondientes a las actividades de distribución mayorista y dispensación al público. Artículo 95. Del Comité Asesor para la Financiación de la Prestación Farmacéutica del Sistema Nacional de Salud. 1. El Comité Asesor para la Financiación de la Prestación Farmacéutica del Sistema Nacional de Salud es el órgano colegiado, de carácter científico-técnico, adscrito al órgano ministerial competente en materia de prestación farmacéutica del Ministerio de Sanidad, Servicios Sociales e Igualdad, encargado de proporcionar asesoramiento, evaluación y consulta sobre la pertinencia, mejora y seguimiento de la evaluación económica necesaria para sustentar las decisiones de la Comisión Interministerial de Precios de los Medicamentos. 2. El Comité Asesor para la Financiación de la Prestación Farmacéutica del Sistema Nacional de Salud estará compuesto por un número máximo de siete miembros designados por la persona titular del Ministerio de Sanidad, Servicios Sociales e Igualdad, de entre profesionales de reconocido prestigio, con experiencia y trayectoria acreditadas en evaluación farmacoeconómica. 3. Asimismo, en función de los asuntos que se debatan, podrán asistir a las sesiones del Comité los evaluadores del órgano competente en materia de medicamentos y productos – 56 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios sanitarios que hayan elaborado las evaluaciones de los medicamentos y productos sanitarios objeto de debate. 4. En todo caso, la creación y el funcionamiento del Comité Asesor para la Financiación de la Prestación Farmacéutica del Sistema Nacional de Salud será atendido con los medios personales, técnicos y presupuestarios asignados al órgano al que se encuentre adscrito. Artículo 96. Revisión del precio. 1. El precio fijado conforme a lo establecido en el artículo 94 será revisable de oficio o a instancia de parte de acuerdo con lo previsto en los artículos 102 y siguientes de la Ley 30/1992, de 26 de noviembre, de Régimen Jurídico de las Administraciones Públicas y del Procedimiento Administrativo Común. 2. Fuera de los supuestos previstos en el apartado anterior, el precio de un medicamento podrá ser modificado cuando lo exijan cambios en las circunstancias económicas, técnicas, sanitarias o en la valoración de su utilidad terapéutica. 3. El Consejo de Ministros, previo acuerdo de la Comisión Delegada del Gobierno para Asuntos Económicos, podrá revisar globalmente o fijar las condiciones de revisión periódica de los precios industriales o, en su caso, de los precios de venta al público, para todos o una parte de los medicamentos y productos sanitarios incluidos en la prestación farmacéutica del Sistema Nacional de Salud. 4. Corresponde igualmente al Consejo de Ministros, previo acuerdo de la Comisión Delegada del Gobierno para Asuntos Económicos, la revisión de las cuantías económicas correspondientes a la distribución y dispensación de los medicamentos y productos sanitarios. 5. Los medicamentos excluidos de la financiación con cargo a fondos públicos y que tienen indicaciones no excluidas de la misma, se considerarán financiados por dichos fondos, a efectos de la fijación y de la revisión de su precio intervenido. 6. A los efectos de las revisiones de precios menores a petición de parte previstas en el apartado 1, sólo se tendrán en cuenta las que supongan, como mínimo, una reducción del 10 % sobre el precio industrial máximo en vigor autorizado para la financiación con fondos públicos. Artículo 97. Información económica. 1. A los efectos de la fijación de precios, los laboratorios farmacéuticos deberán facilitar al Ministerio de Sanidad, Servicios Sociales e Igualdad toda la información sobre los aspectos técnicos, económicos y financieros. El Ministerio podrá efectuar comprobaciones sobre la información facilitada. 2. En el caso de que la empresa esté integrada en un grupo que realice otras actividades, además de las relacionadas con medicamentos, o las desarrolle fuera de España, el Ministerio de Sanidad, Servicios Sociales e Igualdad podrá requerir la información que permita conocer la imputación para determinar los gastos afectados a la actividad farmacéutica en España. 3. La información que en virtud de este artículo obtenga la Administración General del Estado será confidencial. 4. El Ministerio de Sanidad, Servicios Sociales e Igualdad elevará anualmente a la Comisión Delegada del Gobierno para Asuntos Económicos un informe sobre sus actuaciones en materia de precios. Artículo 98. Sistema de precios de referencia. 1. La financiación pública de medicamentos estará sometida al sistema de precios de referencia. El precio de referencia será la cuantía máxima con la que se financiarán las presentaciones de medicamentos incluidas en cada uno de los conjuntos que se determinen, siempre que se prescriban y dispensen con cargo a fondos públicos. 2. Los conjuntos incluirán todas las presentaciones de medicamentos financiadas que tengan el mismo principio activo e idéntica vía de administración, entre las que existirá incluida en la prestación farmacéutica del Sistema Nacional de Salud, al menos, una presentación de medicamento genérico o biosimilar, salvo que el medicamento o su – 57 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios ingrediente activo principal hayan sido autorizados con una antelación mínima de diez años en un Estado miembro de la Unión Europea, en cuyo caso no será indispensable la existencia de un medicamento genérico o biosimilar para establecer un conjunto. Las presentaciones indicadas para tratamientos en pediatría, así como las correspondientes a medicamentos de ámbito hospitalario, incluidos los envases clínicos, constituirán conjuntos independientes. 3. El precio de referencia de cada conjunto se calculará en base al coste/tratamiento/día menor de las presentaciones de medicamentos en él agrupadas y, en todo caso, deberá garantizarse el abastecimiento a las oficinas de farmacia para los medicamentos de precio menor. Los medicamentos no podrán superar el precio de referencia del conjunto al que pertenezcan. 4. Se establecerán los nuevos conjuntos y se revisarán los precios de los conjuntos ya existentes con carácter anual. No obstante, los precios menores de las nuevas agrupaciones homogéneas serán fijados, automáticamente, en el Nomenclátor que corresponda, y los precios menores de las ya existentes serán revisados con carácter trimestral. 5. El Ministerio de Sanidad, Servicios Sociales e Igualdad establecerá un sistema similar de precios para los productos sanitarios. Artículo 99. Sistema de precios seleccionados. 1. El Ministerio de Sanidad, Servicios Sociales e Igualdad podrá proponer a la Comisión Interministerial de Precios de los Medicamentos la aplicación del sistema de precios seleccionados a los medicamentos y productos sanitarios financiables. 2. A tales efectos, el Ministerio de Sanidad, Servicios Sociales e Igualdad elaborará una propuesta motivada, de acuerdo a los criterios recogidos en este artículo, que contendrá el precio máximo seleccionado aplicable en cada caso. 3. Una vez autorizado por la Comisión Interministerial de Precios de los Medicamentos, el Ministerio de Sanidad, Servicios Sociales e Igualdad publicará la decisión por resolución de la unidad responsable de la prestación farmacéutica. 4. En el caso de los medicamentos financiados, el sistema de precios seleccionados se aplicará a medicamentos sujetos a precios de referencia, teniendo en cuenta: a) El consumo del conjunto. b) El impacto presupuestario. c) La existencia de, al menos, tres medicamentos en el conjunto. d) Que no se produzca riesgo de desabastecimiento. 5. Para el caso de productos sanitarios se aplicarán criterios análogos. 6. Valorados los criterios anteriores, el Ministerio de Sanidad, Servicios Sociales e Igualdad, a través del órgano competente en materia de prestación farmacéutica, procederá a comunicar a los proveedores el inicio de un procedimiento de precio seleccionado, con comunicación del precio máximo de financiación que se propone para que manifiesten sus intenciones. 7. En base a las comunicaciones recibidas, el Ministerio de Sanidad, Servicios Sociales e Igualdad elaborará la propuesta a que hace referencia el apartado 2. 8. Aquellos medicamentos y/o productos sanitarios que superen el precio máximo financiable quedarán excluidos de la financiación por el Sistema Nacional de Salud. 9. El precio seleccionado tendrá una vigencia de dos años durante los cuales no podrá ser modificado. 10. El sistema de precios seleccionados se actualizará, para los casos en los que no haya sido aplicado con anterioridad, con periodicidad anual, de forma simultánea a la actualización del sistema de precios de referencia. 11. La aplicación de este sistema supondrá la exclusión de la financiación pública de aquellas presentaciones que no resulten seleccionadas, por el tiempo de vigencia del precio seleccionado. 12. En cualquier caso, las presentaciones de los medicamentos que resulten afectadas por lo regulado en este artículo quedarán exentas, a partir de dicho momento, de la aplicación de las deducciones reguladas en los artículos 8, 9 y 10 del Real Decreto-ley – 58 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios 8/2010, de 20 de mayo, por el que se adoptan medidas extraordinarias para la reducción del déficit público. 13. Los laboratorios titulares de la autorización de comercialización de las presentaciones de los medicamentos y las empresas ofertantes de las presentaciones de los productos sanitarios que resulten finalmente seleccionadas deberán asumir el compromiso de garantizar su adecuado abastecimiento mediante declaración expresa al efecto. 14. El sistema de precios seleccionados podrá aplicarse a medicamentos y productos sanitarios que, no estando financiados, se consideren de interés para la salud pública en los términos expresados en la Ley 33/2011, de 4 de octubre, General de Salud Pública. 15. A este respecto, el Ministerio de Sanidad, Servicios Sociales e Igualdad para la determinación del precio seleccionado tendrá en cuenta las especiales características de distribución y aplicación de estos productos. 16. Asimismo, se podrá extender el sistema de precios seleccionados a través de la fijación de una aportación reducida por agrupaciones homogéneas. Artículo 100. Fomento de la competencia y la competitividad. 1. Para la consecución de los fines de eficiencia y sostenibilidad de la prestación farmacéutica del Sistema Nacional de Salud se implementarán las medidas administrativas y regulatorias que en cada ejercicio presupuestario se consideren apropiadas para estimular la competencia entre proveedores de insumos farmacéuticos, redundando en descensos de precios unitarios. 2. Toda actuación limitativa de la competencia se considerará contraria a los principios de eficiencia y sostenibilidad y será perseguida de oficio por los órganos competentes. Artículo 101. Obligaciones de los pacientes. 1. El Gobierno revisará periódicamente la participación en el pago a satisfacer por los ciudadanos por la prestación farmacéutica incluida en la cartera común suplementaria del Sistema Nacional de Salud, y los supuestos de financiación íntegra con cargo a fondos públicos. La revisión se publicará en el «Boletín Oficial del Estado» mediante orden de la persona titular del Ministerio de Sanidad, Servicios Sociales e Igualdad. 2. La participación en el pago podrá modularse por el Gobierno con criterios que tengan en cuenta: a) La capacidad de pago. b) La utilidad terapéutica y social de los medicamentos o de los productos sanitarios. c) Las necesidades específicas de ciertos colectivos. d) La gravedad, duración y secuelas de las distintas patologías para los que resulten indicados. e) Racionalización del gasto público destinado a prestación farmacéutica. f) Existencia de medicamentos o productos sanitarios ya disponibles y otras alternativas mejores o iguales para las mismas afecciones. 3. Los usuarios estarán obligados a justificar su derecho a la correspondiente modalidad de pago cuando así les sea requerido por el personal facultativo del Sistema Nacional de Salud o en las oficinas de farmacia dispensadoras. Artículo 102. Aportación de los usuarios y sus beneficiarios en la prestación farmacéutica ambulatoria. 1. Se entiende por prestación farmacéutica ambulatoria la que se dispensa al paciente mediante receta médica u orden de dispensación hospitalaria, a través de oficinas o servicios de farmacia. 2. Solo la prestación farmacéutica ambulatoria que se dispense por medio de receta médica oficial u orden de dispensación a través de oficinas de farmacia estará sujeta a aportación del usuario. 3. La aportación del usuario se efectuará en el momento de la dispensación del medicamento o producto sanitario. – 59 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios 4. La aportación del usuario será proporcional al nivel de renta que se actualizará, como máximo, anualmente. 5. Con carácter general, el porcentaje de aportación del usuario seguirá el siguiente esquema: a) Un 60 % del PVP para los usuarios y sus beneficiarios cuya renta sea igual o superior a 100.000 euros consignada en la casilla de base liquidable general y del ahorro de la declaración del Impuesto sobre la Renta de las Personas Físicas. b) Un 50 % del PVP para las personas que ostenten la condición de asegurado activo y sus beneficiarios cuya renta sea igual o superior a 18.000 euros e inferior a 100.000 euros consignada en la casilla de base liquidable general y del ahorro de la declaración del Impuesto sobre la Renta de las Personas Físicas. c) Un 40 % del PVP para las personas que ostenten la condición de asegurado activo y sus beneficiarios y no se encuentren incluidos en los apartados a) o b) anteriores. d) Un 10 % del PVP para las personas que ostenten la condición de asegurado como pensionistas de la Seguridad Social y sus beneficiarios, con excepción de las personas incluidas en el apartado a). e) Un 40 % del PVP para las personas extranjeras no registradas ni autorizadas como residentes en España a los que se refiere el artículo 3 ter de la Ley 16/2003, de 28 de mayo. 6. Con el fin de garantizar la continuidad de los tratamientos de carácter crónico y asegurar un alto nivel de equidad a los pacientes pensionistas con tratamientos de larga duración, los porcentajes generales estarán sujetos a topes máximos de aportación en los siguientes supuestos: a) Un 10 % del PVP en los medicamentos pertenecientes a los grupos ATC de aportación reducida, con una aportación máxima de 4,24 euros. b) Para las personas que ostenten la condición de asegurado como pensionistas de la Seguridad Social y sus beneficiarios cuya renta sea inferior a 18.000 euros consignada en la casilla de base liquidable general y del ahorro de la declaración del Impuesto sobre la Renta de las Personas Físicas o que no estén incluidos en los siguientes apartados c) o d), hasta un límite máximo de aportación mensual de 8,23 euros. c) Para las personas que ostenten la condición de asegurado como pensionistas de la Seguridad Social y sus beneficiarios cuya renta sea igual o superior a 18.000 euros e inferior a 100.000 euros consignada en la casilla de base liquidable general y del ahorro de la declaración del Impuesto sobre la Renta de las Personas Físicas, hasta un límite máximo de aportación mensual de 18,52 euros. d) Para las personas que ostenten la condición de asegurado como pensionista de la Seguridad Social y sus beneficiarios cuya renta sea superior a 100.000 euros consignada en la casilla de base liquidable general y del ahorro de la declaración del Impuesto sobre la Renta de las Personas Físicas, hasta un límite máximo de aportación mensual de 61,75 euros. 7. El importe de las aportaciones que excedan de las cuantías mencionadas en el apartado anterior será objeto de reintegro por la comunidad autónoma correspondiente, con una periodicidad máxima semestral. 8. Estarán exentos de aportación los usuarios y sus beneficiarios que pertenezcan a una de las siguientes categorías: a) Afectados de síndrome tóxico y personas con discapacidad en los supuestos contemplados en su normativa específica. b) Personas perceptoras de rentas de integración social. c) Personas perceptoras de pensiones no contributivas. d) Parados que han perdido el derecho a percibir el subsidio de desempleo en tanto subsista su situación. e) Personas con tratamientos derivados de accidente de trabajo y enfermedad profesional. 9. El nivel de aportación de las personas encuadradas en la Mutualidad General de Funcionarios Civiles del Estado, el Instituto Social de las Fuerzas Armadas y la Mutualidad – 60 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios General Judicial será del 30 % con carácter general, resultándoles de aplicación lo dispuesto en el párrafo a) del apartado 6 y en el párrafo e) del apartado 8. Artículo 103. Protección de datos personales. 1. El Instituto Nacional de la Seguridad Social o, en su caso, el Instituto Social de la Marina podrá tratar los datos obrantes en los ficheros de las entidades gestoras y servicios comunes de la Seguridad Social y de las entidades que colaboran con las mismas que resulten imprescindibles para determinar la cuantía de la aportación de los beneficiarios en la prestación farmacéutica. Dicho tratamiento, que no requerirá el consentimiento del interesado, se someterá plenamente a lo dispuesto en la Ley Orgánica 15/1999, de 13 de diciembre, y sus disposiciones de desarrollo. 2. Del mismo modo, y con la finalidad a la que se refiere el apartado anterior, la administración competente en materia tributaria podrá comunicar al Instituto Nacional de la Seguridad Social o, en su caso, al Instituto Social de la Marina, sin contar con el consentimiento del interesado, los datos que resulten necesarios para determinar el nivel de renta requerido. Igualmente, los órganos de las administraciones públicas que resulten competentes para determinar la concurrencia de los requisitos establecidos para la exención de la aportación previstos en el artículo 102.8, podrán comunicar esta circunstancia al Instituto Nacional de la Seguridad Social o, en su caso, al Instituto Social de la Marina sin contar con el consentimiento del interesado. 3. El Instituto Nacional de la Seguridad Social o, en su caso, el Instituto Social de la Marina comunicará al Ministerio de Sanidad, Servicios Sociales e Igualdad y éste, a su vez, a las demás administraciones sanitarias competentes el dato relativo al nivel de aportación que corresponda a cada usuario de conformidad con lo establecido en la normativa reguladora de las recetas médicas y órdenes de dispensación. En ningún caso, dicha información incluirá el dato de la cuantía concreta de las rentas. Los datos comunicados de conformidad con lo dispuesto en el párrafo anterior serán objeto de tratamiento por la administración sanitaria correspondiente a los solos efectos de su incorporación al sistema de información de la tarjeta sanitaria individual. Artículo 104. Valoración de la prescripción. 1. En el ámbito del Sistema Nacional de Salud corresponde a las Administraciones Públicas sanitarias la evaluación de las prescripciones por áreas, zonas, terapias, grupos poblacionales y otras circunstancias. 2. El Ministerio de Sanidad, Servicios Sociales e Igualdad establecerá los mecanismos de coordinación que permitan conocer la utilización de medicamentos y productos sanitarios, optimizar la investigación de su evolución y adoptar las medidas de información y promoción del uso racional de los medicamentos y productos sanitarios y, en su caso, las medidas cautelares y de control correspondientes con exigencia de las responsabilidades administrativas y penales a que hubiere lugar. Artículo 105. Colaboración entre oficinas de farmacia y el Sistema Nacional de Salud. 1. Las oficinas de farmacia, como establecimientos sanitarios que son, colaborarán a los fines de esta ley para garantizar el uso racional de los medicamentos en la atención primaria a la salud. 2. Con independencia de las obligaciones establecidas en esta ley y las que se determinen en la normativa de desarrollo, las oficinas de farmacia podrán ser objeto de concertación en el Sistema Nacional de Salud, de acuerdo con el sistema general de contratación administrativa y conforme a los criterios generales a que se refiere el artículo 91.6. Artículo 106. Gestión de información sobre prestación farmacéutica del Sistema Nacional de Salud. 1. Al objeto de ejecutar las acciones necesarias para la valoración de la prescripción y de la política farmacéutica general, las Administraciones Públicas competentes facilitarán la – 61 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios información agregada o desagregada relativa al consumo de medicamentos tanto por receta como a nivel de centros hospitalarios y cualesquiera otros ámbitos incluidos dentro de la prestación farmacéutica del Sistema Nacional de Salud. Como mínimo, dicha información se presentará con periodicidad mensual; se facilitará desde las Consejerías responsables de las comunidades autónomas al Ministerio de Sanidad, Servicios Sociales e Igualdad, que efectuará la agregación y depuración correspondiente antes de hacerla pública. 2. La información agregada resultante del procesamiento de las recetas del Sistema Nacional de Salud, incluyendo las de la Mutualidad de Funcionarios de la Administración Civil del Estado, las de la Mutualidad General Judicial y las de Instituto Social de las Fuerzas Armadas, es de dominio público, salvando siempre la confidencialidad de la asistencia sanitaria y de los datos comerciales de empresas individualizadas. Su gestión corresponde a los Servicios de Salud de las comunidades autónomas en su ámbito territorial y a la Administración General del Estado en la información del conjunto del Sistema Nacional de Salud. Lo establecido en el párrafo anterior será, asimismo, de aplicación a la información relativa a las compras de medicamentos y de productos sanitarios realizadas a través de los correspondientes servicios de farmacia por los hospitales del Sistema Nacional de Salud. 3. La información agregada resultante del procesamiento de las recetas a que se refiere el apartado anterior será tratada y remitida en formato electrónico por los organismos responsables del mismo. El Gobierno, mediante real decreto, establecerá el procedimiento de remisión de la información a las administraciones responsables de la gestión de la prestación farmacéutica de forma que permita aplicar a la factura mensual de cada oficina de farmacia, por recetas de medicamentos de uso humano fabricados industrialmente con cargo a fondos públicos de las comunidades autónomas y del Instituto Nacional de Gestión Sanitaria, MUFACE, ISFAS y MUGEJU, una escala conjunta de deducción sobre los márgenes aplicables. 4. La información a que se hace referencia en este artículo será facilitada con periodicidad mensual y estará referida a un período no superior a los tres meses inmediatamente anteriores a la fecha en que sea facilitada. Artículo 107. Fundamentos de los sistemas de información para el control de la prestación farmacéutica. 1. La intervención del Estado en materia de medicamentos y productos sanitarios financiados por el Sistema Nacional de Salud exige la plena disposición de información sólida sobre el consumo de los insumos sanitarios objeto de dicha información. A tal efecto, tanto el Ministerio de Sanidad, Servicios Sociales e Igualdad como las consejerías competentes de las comunidades autónomas y, en su caso, las empresas proveedoras y sus órganos de representación profesional, aportarán la siguiente información relativa al tráfico y consumo de los mismos: a) Datos de facturación de recetas oficiales del Sistema Nacional de Salud con periodicidad mensual, dispensadas por oficinas de farmacia y agregadas por provincia y comunidad autónoma. b) Datos de adquisiciones por servicios farmacéuticos de centros y servicios sanitarios o sociosanitarios del Sistema Nacional de Salud y, en su caso, abonos de medicamentos y productos sanitarios, al menos con periodicidad mensual y con nivel de agregación por provincia y comunidad autónoma. 2. La Mutualidad General de Funcionarios Civiles del Estado, la Mutualidad General Judicial y el Instituto Social de las Fuerzas Armadas aportarán el mismo tipo de información, con las salvedades propias de las modalidades asistenciales que les son propias. 3. Los medicamentos dispensados por servicios farmacéuticos de centros y servicios sanitarios o sociosanitarios del Sistema Nacional de Salud a pacientes ambulatorios serán recogidos en una aplicación informática específica. 4. El tratamiento informático al que se refiere el apartado anterior podrá ser extendido a otros medicamentos y productos sanitarios de uso exclusivo hospitalario a los que la Comisión Interministerial de Precios de los Medicamentos considere oportuno aplicar un régimen de cautelas singulares. – 62 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios TÍTULO IX Régimen sancionador CAPÍTULO I Inspección y medidas cautelares Artículo 108. Inspección. 1. Corresponde a las Administraciones sanitarias en el ámbito de sus competencias la realización de las inspecciones necesarias para asegurar el cumplimiento de lo previsto en esta ley. 2. Corresponde a la Administración General del Estado la realización de la función inspectora en los siguientes casos: a) Cuando se trate de las actuaciones necesarias para las oportunas autorizaciones o registros que, de acuerdo con esta ley, corresponden a la Administración General del Estado. b) En todo caso, cuando se trate de inspecciones a realizar en el territorio de las comunidades autónomas que no ostenten competencias de ejecución de la legislación de productos farmacéuticos o no hubieren recibido los correspondientes traspasos. c) Cuando se trate de medicamentos, productos o artículos destinados al comercio exterior o cuya utilización o consumo pudiera afectar a la seguridad pública. 3. El personal al servicio de las Administraciones públicas que desarrolle las funciones de inspección, cuando ejerza tales funciones y acredite su identidad, estará autorizado para: a) Entrar libremente y sin previa notificación, en cualquier momento, en todo centro o establecimiento sujeto a esta ley. b) Proceder a las pruebas, investigaciones o exámenes necesarios para comprobar el cumplimiento de esta ley y de las normas que se dicten para su desarrollo. c) Tomar o sacar muestras, en orden a la comprobación del cumplimiento de lo previsto en esta ley y en las disposiciones para su desarrollo. d) Realizar cuantas actuaciones sean precisas en orden al cumplimiento de las funciones de inspección que desarrollen. Artículo 109. Medidas cautelares. 1. En el caso de que exista o se sospeche razonablemente la existencia de un riesgo inminente y grave para la salud, las autoridades sanitarias podrán adoptar las siguientes medidas cautelares en el ámbito de esta ley: a) La puesta en cuarentena, la retirada del mercado y la prohibición de utilización de medicamentos, fórmulas magistrales y preparados oficinales, así como la suspensión de actividades, publicidad y la clausura provisional de establecimientos, centros o servicios. La puesta en cuarentena supondrá el bloqueo inmediato en el establecimiento farmacéutico en que se encuentren o al que se destinen, en caso de transporte no concluido, por el tiempo que se determine o hasta nueva orden, a cargo de su responsable. b) La suspensión de la elaboración, prescripción, dispensación y suministro de medicamentos y productos sanitarios en investigación. c) La limitación, prohibición, suspensión o sujeción a condiciones especiales de la fabricación, importación, comercialización, exportación, publicidad, puesta en servicio o utilización de los productos sanitarios, cosméticos o productos de cuidado personal, así como la puesta en cuarentena, la retirada del mercado y la recuperación de dichos productos. – 63 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios 2. La duración de las medidas a que se refiere el apartado anterior, que se fijarán para cada caso, sin perjuicio de las prórrogas sucesivas acordadas por resoluciones motivadas, no excederá de lo que exija la situación de riesgo inminente y grave que la justificó. 3. La Agencia Española de Medicamentos y Productos Sanitarios deberá ser informada de modo inmediato por la autoridad sanitaria que adoptó la medida cautelar. 4. De las medidas cautelares la Agencia Española de Medicamentos y Productos Sanitarios dará conocimiento por los medios idóneos y con la rapidez adecuada a cada caso, a los servicios sanitarios, entidades responsables o público en general, según proceda. 5. El coste de las medidas cautelares será sufragado por la persona física o jurídica que hubiese dado lugar a su adopción. CAPÍTULO II Infracciones y sanciones Artículo 110. Disposiciones generales. 1. Las infracciones en materia de medicamentos, productos sanitarios, cosméticos y productos de cuidado personal serán objeto de las sanciones administrativas correspondientes, previa instrucción del oportuno expediente, sin perjuicio de las responsabilidades civiles, penales o de otro orden que puedan concurrir. 2. La instrucción de causa penal ante los Tribunales de Justicia suspenderá la tramitación del expediente administrativo sancionador que hubiera sido incoado por los mismos hechos y, en su caso, la eficacia de los actos administrativos de imposición de sanción. Las medidas administrativas que hubieran sido adoptadas para salvaguardar la salud y seguridad de las personas se mantendrán en tanto la autoridad judicial se pronuncie sobre las mismas. 3. En ningún caso se impondrá una doble sanción por los mismos hechos y en función de los mismos intereses públicos protegidos, si bien deberán exigirse las demás responsabilidades que se deduzcan de otros hechos o infracciones concurrentes. 4. Con respecto al régimen sancionador y en lo no previsto por esta ley será de aplicación lo establecido por el título IX de la Ley 30/1992, de 26 de noviembre. Artículo 111. Infracciones en materia de medicamentos. 1. Las infracciones se califican como leves, graves y muy graves atendiendo a los criterios de riesgos para la salud, cuantía del eventual beneficio obtenido, gravedad de la alteración sanitaria y social producida, generalización de la infracción y reincidencia. 2. Constituirán faltas administrativas y serán sancionadas en los términos previstos en el artículo 114, las infracciones que a continuación se tipifican: a) Infracciones leves: 1.ª No aportar, las entidades o personas responsables, los datos, declaraciones así como cualesquiera información que estén obligados a suministrar por razones sanitarias, técnicas, económicas, administrativas y financieras. 2.ª Incumplir el deber de colaborar con la administración sanitaria en la evaluación y control de medicamentos. 3.ª No disponer, los establecimientos obligados a ello, de acceso a la Real Farmacopea Española y al Formulario Nacional. 4.ª No proporcionar, los laboratorios farmacéuticos, a los facultativos sanitarios en ejercicio que lo soliciten la ficha técnica de medicamentos antes de su comercialización. 5.ª Realizar publicidad de fórmulas magistrales o de preparados oficinales. 6.ª Incumplir los requisitos que para la realización de la visita médica, establezca la normativa de las Administraciones sanitarias competentes en la gestión de la prestación farmacéutica. 7.ª No cumplimentar correctamente los datos y advertencias que deben contener las recetas normalizadas. 8.ª Dispensar medicamentos transcurrido el plazo de validez de la receta. – 64 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios 9.ª Realizar la sustitución de un medicamento, en los casos que ésta sea posible, incumpliendo los requisitos establecidos en esta ley. 10.ª Incumplir los requisitos, obligaciones o prohibiciones establecidas en esta ley y disposiciones que la desarrollan de manera que, en razón de los criterios contemplados en este artículo, tales incumplimientos merezcan la calificación de leves o no proceda su calificación como faltas graves o muy graves. 11.ª No incluir en los envases de los medicamentos la información en alfabeto braille para su correcta identificación por las personas invidentes y con discapacidad visual, conforme a lo dispuesto en el artículo 15.5. b) Infracciones graves: 1.ª No realizar en la elaboración, fabricación, importación, exportación y distribución de medicamentos o de principios activos, los controles de calidad exigidos en la legislación sanitaria o incumplir las directrices detalladas sobre normas de correcta fabricación o buenas prácticas de distribución establecidas en el marco comunitario o efectuar los procesos de fabricación o control mediante procedimientos no validados. 2.ª Elaborar, fabricar, importar, exportar, dispensar o distribuir medicamentos por personas físicas o jurídicas que no cuenten con la preceptiva autorización. 3.ª Dificultar la labor inspectora mediante cualquier acción u omisión que perturbe o retrase la misma. 4.ª Preparar de forma individualizada vacunas y alérgenos en establecimientos distintos de los autorizados. 5.ª Prescribir y preparar fórmulas magistrales y preparados oficinales incumpliendo los requisitos legales establecidos. 6.ª Modificar por parte del titular, sin autorización previa o notificación, según proceda, cualquiera de las condiciones de autorización del medicamento. 7.ª No disponer, un laboratorio farmacéutico o entidad de distribución, de director técnico o del resto del personal exigido en cada caso. 8.ª Incumplir, el director técnico y demás personal, las obligaciones que competen a sus cargos. 9.ª Incumplir, el promotor o investigador de un ensayo clínico, las obligaciones establecidas para cada uno de ellos en la legislación vigente, cuando el hecho en razón de los criterios contemplados en este artículo no merezca la calificación de muy grave. 10.ª Incumplir, el promotor de ensayos clínicos, los plazos de comunicación a las autoridades sanitarias de las reacciones adversas graves e inesperadas ocurridas en un ensayo clínico. 11.ª Facilitar, al Comité Ético de Investigación Clínica o a las autoridades sanitarias, información y/o documentación, relacionada con un ensayo clínico, no veraz o que dé lugar a conclusiones inexactas. 12.ª Incumplir, el promotor, la obligación de publicación de los resultados de un ensayo clínico según lo establecido en el artículo 62. 13.ª Actuar, los integrantes del Comité Ético de Investigación Clínica, sin ajustarse a los requisitos de funcionamiento establecidos legalmente o sin estar debidamente acreditados. 14.ª Incumplir, los laboratorios farmacéuticos, entidades de distribución o personal sanitario, el deber de farmacovigilancia. 15.ª Negarse a dispensar medicamentos sin causa justificada. 16.ª Dispensar medicamentos sin receta, cuando ésta resulte obligada. 17.ª Suministrar, adquirir o vender medicamentos a entidades no autorizadas para la realización de tales actividades. 18.ª Actuar, los profesionales sanitarios implicados en el ciclo de prescripción, dispensación y administración y siempre que estén en ejercicio, en funciones de delegados de visita médica, representantes, comisionistas o agentes informadores de los laboratorios de medicamentos. 19.ª Incumplir, el personal sanitario, el deber de garantizar la confidencialidad y la intimidad de los pacientes en la tramitación de las recetas y órdenes médicas. 20.ª Funcionar, los servicios farmacéuticos y oficinas de farmacia, sin la presencia y actuación profesional del farmacéutico responsable. – 65 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios 21.ª Incumplir, las oficinas de farmacia, las exigencias que conlleva la facturación al Sistema Nacional de Salud de los productos contemplados en esta ley. 22.ª Defraudar, las oficinas de farmacia, al Sistema Nacional de Salud o al beneficiario del mismo con motivo de la facturación y cobro de recetas oficiales. 23.ª Dispensar o suministrar medicamentos en establecimientos distintos a los autorizados. 24.ª No ajustar los precios de los medicamentos a lo determinado por la Administración. 25.ª Sustituir medicamentos en la dispensación, contraviniendo lo dispuesto en el artículo 89. 26.ª Coartar la libertad del usuario en la elección de la oficina de farmacia mediante cualquier acto u omisión. 27.ª Ofrecer directa o indirectamente cualquier tipo de incentivo, bonificaciones, descuentos prohibidos, primas u obsequios, efectuados por quien tenga intereses directos o indirectos en la producción, fabricación y comercialización de medicamentos, a los profesionales sanitarios, con motivo de la prescripción, dispensación y administración de los mismos, o a sus parientes y personas de su convivencia. 28.ª Aceptar, los profesionales sanitarios, con motivo de la prescripción, dispensación y administración de medicamentos con cargo al Sistema Nacional de Salud, o sus parientes y personas de su convivencia, cualquier tipo de incentivo, bonificaciones, descuentos prohibidos, primas u obsequios efectuados por quien tenga intereses directos o indirectos en la producción, fabricación y comercialización de medicamentos. 29.ª No comunicar los laboratorios farmacéuticos al Ministerio de Sanidad, Servicios Sociales e Igualdad las unidades de medicamentos vendidas para ser dispensadas en territorio nacional. 30.ª No informar las entidades de distribución a las autoridades sanitarias de las comunidades autónomas en las que tengan su domicilio social y al Ministerio de Sanidad, Servicios Sociales e Igualdad, de las unidades suministradas a oficinas de farmacia o servicios de farmacia que radiquen en territorio nacional, así como, en su caso, a otras entidades de distribución, con independencia de la Comunidad Autónoma en que estas últimas radiquen. 31.ª No comunicar las oficinas de farmacia la información sobre medicamentos dispensados a que se refiere esta ley. 32.ª Aportar u ocultar las entidades o personas responsables, datos, declaraciones o cualquier información que estén obligados a suministrar a las administraciones sanitarias competentes de forma que no resulten veraces o den lugar a conclusiones inexactas, con la finalidad de obtener con ello algún beneficio, ya sea económico o de cualquier otra índole. 33.ª Incumplir, las personas que se dediquen a la intermediación de medicamentos, los requisitos establecidos en la normativa vigente y en las buenas prácticas de distribución de medicamentos. 34.ª Incumplir, el fabricante de los medicamentos, las obligaciones en materia de excipientes que se utilicen en la fabricación de medicamentos. 35.ª Realizar, por parte del titular de la autorización de laboratorio o del titular de una autorización de distribución, actividades que no se ajusten a la misma. 36.ª Incumplir, las oficinas de farmacia y servicios de farmacia, lo establecido legalmente en materia de aportación del usuario en la prestación farmacéutica del Sistema Nacional de Salud. 37.ª Realizar la oficina de farmacia cualquier acto que induzca al usuario a adquirir una mayor cantidad de medicamentos dentro de la prestación farmacéutica del Sistema Nacional de Salud que el verdaderamente necesario o demandado por este último. c) Infracciones muy graves: 1.ª Poner en el mercado medicamentos de cualquier naturaleza sin haber obtenido la preceptiva autorización sanitaria para ello. 2.ª Fabricar, importar, exportar, intermediar, distribuir, dispensar y vender medicamentos falsificados. Esta infracción también se aplicará en el caso de que esta venta se efectúe a distancia. – 66 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios 3.ª Incumplir, el titular de la autorización, la obligación de presentar los informes periódicos de seguridad 4.ª Preparar remedios secretos. 5.ª Importar y exportar sangre, fluidos, glándulas y tejidos humanos y sus componentes y derivados sin la previa autorización. 6.ª Realizar ensayos clínicos sin la previa autorización administrativa. 7.ª Realizar ensayos clínicos sin contar con el consentimiento del sujeto del ensayo o, en su caso, de su representante legal, o el incumplimiento, por parte del investigador, del deber de información sobre el ensayo clínico a quien participa como sujeto del mismo. 8.ª No comunicar, el promotor de un ensayo clínico, a las autoridades sanitarias las reacciones adversas ocurridas en el desarrollo del mismo o los informes periódicos de seguridad. 9.ª Incumplir, el promotor o investigador de un ensayo clínico, las obligaciones establecidas para cada uno de ellos en la legislación vigente cuando suponga perjuicio en los derechos, seguridad y bienestar de los sujetos. 10.ª Distribuir o conservar los medicamentos sin observar las condiciones exigidas, así como poner a la venta medicamentos alterados, en malas condiciones o, cuando se haya señalado, pasado el plazo de validez. 11.ª Vender medicamentos a domicilio o a través de internet o de otros medios telemáticos o indirectos, en contra de lo previsto en esta ley o incumpliendo las disposiciones que regulen dicha modalidad de venta. 12.ª Incumplir las entidades de distribución y las oficinas de farmacia sus obligaciones legales y, en particular, no disponer de las existencias de medicamentos adecuadas para la normal prestación de sus actividades o servicios. 13.ª Incumplir las entidades de distribución y las oficinas de farmacia sus obligaciones legales y, en particular, no disponer de existencias mínimas de medicamentos para supuestos de emergencia o catástrofes, en los casos que resulte obligado. 14.ª Elaborar, fabricar, importar, exportar, distribuir, comercializar, prescribir y dispensar productos, preparados, sustancias o combinaciones de las mismas, que se presenten como medicamentos sin estar legalmente reconocidos como tales. 15.ª Incumplir con la obligación de suscribir un seguro, aval o garantía financiera equivalente en los supuestos exigidos por esta ley. 16.ª Realizar promoción, información o publicidad de medicamentos no autorizados o sin que tales actividades se ajusten a lo dispuesto en esta ley o en la legislación general sobre publicidad. 17.ª Efectuar promoción, publicidad o información destinada al público de productos o preparados, con fines medicinales, aun cuando el propio producto no haga referencia explícita a dichos fines, incluidas las sustancias medicinales y sus combinaciones, que no se encuentren autorizados como medicamentos. 18.ª Ofrecer primas, obsequios, premios, concursos, bonificaciones, descuentos o similares como métodos vinculados a la promoción o venta al público de los productos regulados en esta ley. 19.ª Incumplir las medidas cautelares y definitivas sobre medicamentos que las autoridades sanitarias competentes acuerden por causa grave de salud pública. 20.ª No cumplir los requisitos y condiciones reglamentariamente exigidos en materia de publicidad y promoción comercial de los productos, materiales, sustancias, energías o métodos a los que se atribuyan efectos beneficiosos sobre la salud. 21.ª Cesar el suministro de un medicamento por parte del titular de autorización de comercialización, en el caso en el que concurran razones de salud o de interés sanitario, como en el supuesto de originarse laguna terapéutica, ya sea en el mercado en general o en la prestación farmacéutica del Sistema Nacional de Salud. 22.ª Distribuir fuera del territorio nacional medicamentos para los que existan problemas de desabastecimiento con repercusión asistencial. 23.ª Realizar, por parte de las oficinas de farmacia, actividades de distribución de medicamentos a otras oficinas de farmacia, entidades de distribución autorizadas, u otras entidades, centros o personas físicas sin autorización para la actividad de distribución o bien la realización de envíos de medicamentos fuera del territorio nacional. – 67 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios 24.ª Dispensar, vender o comercializar los medicamentos devueltos o entregados por los pacientes o el público en general a las oficinas de farmacia. 25.ª Incumplir, el titular de la autorización de comercialización, su obligación de tener suficientemente abastecido el mercado, de modo adecuado y continuado para posibilitar el cumplimiento de las exigencias legalmente establecidas en materia de prestación farmacéutica del Sistema Nacional de Salud y garantizar el abastecimiento a las oficinas de farmacia y servicios de farmacia de los medicamentos incluidos en agrupaciones homogéneas, de precio más bajo y precio menor. 26.ª Impedir la actuación de los inspectores debidamente acreditados, en los centros en los que se elaboren, fabriquen, distribuyan y dispensen medicamentos. 3. La comisión de una infracción, precedida de otras dos de grado inmediatamente inferior o igual sancionadas en firme en el plazo de un año previo a dicha comisión, incrementará de leve a grave, o de grave a muy grave, dicha infracción. Artículo 112. Infracciones en materia de productos sanitarios. 1. Las infracciones se califican como leves, graves y muy graves atendiendo a los criterios de riesgo para la salud, cuantía del eventual beneficio obtenido, gravedad de la alteración sanitaria y social producida, generalización de la infracción y reincidencia. 2. Constituirán infracciones administrativas y serán sancionadas en los términos previstos en el artículo 114 las conductas que a continuación se tipifican: a) Infracciones leves: 1.ª No aportar, las entidades o personas responsables, los datos, declaraciones, así como cualquier información que estén obligados a suministrar por razones sanitarias, técnicas, económicas, administrativas y financieras. 2.ª Incumplir el deber de colaborar con las autoridades sanitarias en la evaluación, vigilancia y control de los productos sanitarios. 3.ª Dificultar la labor inspectora mediante cualquier acción u omisión que perturbe o retrase la misma. 4.ª Presentar en ferias, exposiciones y demostraciones productos no aptos para la puesta en el mercado o en servicio sin la correspondiente indicación de su no conformidad o imposibilidad de puesta en servicio. 5.ª No mantener a disposición del paciente la declaración prevista para los productos a medida, no informarle al respecto o no entregársela a su requerimiento. 6.ª No identificar como tales los productos destinados exclusivamente a exportación. 7.ª Incumplir los requisitos, obligaciones o prohibiciones establecidos en la reglamentación aplicable que, en razón de los criterios contemplados en este artículo, merezcan la calificación de leves o cuando no proceda su calificación como faltas graves o muy graves. b) Infracciones graves: 1.ª No facilitar al paciente y/o no incluir en su historia clínica la información preceptiva sobre el producto que ha recibido o la tarjeta de implantación cuando así se haya establecido, así como no remitir dicha tarjeta a la empresa suministradora o al registro nacional que se haya dispuesto. 2.ª Fabricar, agrupar y esterilizar los productos en territorio nacional sin la licencia sanitaria previa de funcionamiento de la instalación, así como importar productos sanitarios sin la licencia previa de establecimiento. 3.ª Fabricar, agrupar, esterilizar o importar productos sin respetar los requisitos exigidos o sin ajustarse a las condiciones en que se otorgó la licencia de funcionamiento. 4.ª Incumplir el responsable técnico las obligaciones que competen a su cargo. 5.ª Comercializar productos sanitarios sin «marcado CE» cuando éste sea preceptivo, usar cualquier otro marcado que pueda inducir a confusión con el «marcado CE», así como colocar el «marcado CE» en los productos en condiciones distintas de las establecidas, salvo lo dispuesto en la infracción 5.ª de la letra c) de este apartado. – 68 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios 6.ª No mantener a disposición de las autoridades competentes y por el tiempo señalado la documentación preceptiva, así como negarse a facilitar dicha documentación a las autoridades sanitarias. 7.ª Incumplir el deber de comunicación de comercialización en los productos en los que dicha comunicación sea requerida, así como no comunicar las modificaciones producidas o el cese de la comercialización. 8.ª Incumplir el deber de comunicación de los responsables establecidos en España cuando dicha comunicación sea requerida así como no comunicar las modificaciones producidas. 9.ª Incumplir el fabricante, representante autorizado, importador o distribuidor las obligaciones relativas a la identificación de los agentes que les preceden o les siguen en la cadena de comercialización. 10.ª Incumplir el importador o el distribuidor las obligaciones que les incumben para asegurarse de que los productos han seguido los procedimientos de evaluación de conformidad correspondientes y se ha elaborado la documentación preceptiva. 11.ª Distribuir o vender productos de forma ambulante o en establecimientos que no han sido debidamente comunicados o autorizados, o que no dispongan del técnico o del profesional cualificado que corresponda. 12.ª Distribuir, instalar, mantener y utilizar productos sin observar las condiciones exigidas, así como poner a la venta productos sanitarios alterados, en malas condiciones o cuando se haya sobrepasado el plazo de validez. 13.ª Distribuir productos implantables sin proporcionar la correspondiente tarjeta de implantación cuando ésta sea preceptiva, así como no dar el tratamiento debido a dichas tarjetas. 14.ª Vender al público productos sanitarios en los casos no permitidos, así como sin exigir la correspondiente prescripción cuando ésta resulte obligada, salvo lo contemplado en la infracción 7.ª, de la letra c) de este apartado. 15.ª Realizar investigaciones clínicas sin atenerse a los procedimientos y condiciones previstos salvo lo contemplado en las infracciones 8.ª y 9.ª de la letra c) de este apartado. 16.ª Ejecutar incorrectamente el organismo notificado las actuaciones que se le encomiendan sin que tenga repercusiones para la seguridad de los productos certificados. 17.ª Negarse a facilitar el organismo notificado la documentación solicitada por el Ministerio de Sanidad, Servicios Sociales e Igualdad a fin de verificar el cumplimiento de sus requisitos y obligaciones. 18.ª Incumplir el fabricante, representante autorizado, importador o distribuidor el deber de notificación de los incidentes adversos y acciones de seguridad al Sistema de Vigilancia de Productos Sanitarios, así como negarse a modificar o suspender las acciones en las condiciones requeridas por la autoridad sanitaria. 19.ª Incumplir el deber de notificación en el transcurso de las investigaciones clínicas de las circunstancias requeridas. 20.ª Impedir la actuación de los inspectores, debidamente acreditados, en los centros en que se fabriquen, almacenen, distribuyan, vendan o utilicen productos sanitarios. 21.ª Violar el principio de confidencialidad en relación con las informaciones de pacientes y productos por quienes están obligados a mantenerla. 22.ª Poner en servicio en España productos que no incluyan en el etiquetado e instrucciones de uso los datos e informaciones requeridos, al menos en español. 23.ª Incumplir los requisitos y condiciones relativos a la publicidad y promoción de los productos sanitarios. 24.ª Efectuar publicidad dirigida al público de los productos en los que no está permitida, excepto lo contemplado en la infracción 12.ª de la letra c) de este apartado. 25.ª Ofrecer, otorgar o prometer primas, ventajas pecuniarias o ventajas en especie a los profesionales sanitarios o a cualquier otro cualificado, relacionados con la utilización o prescripción de los productos sanitarios, así como a sus parientes y personas de su convivencia. También el solicitarlas o aceptarlas. 26.ª Utilizar un profesional productos en condiciones y para usos distintos de los indicados por el fabricante, o por personal no cualificado o debidamente adiestrado. – 69 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios 27.ª Utilizar en pacientes productos que no hayan satisfecho los procedimientos de evaluación de la conformidad que les sean de aplicación. 28.ª Negarse a dispensar productos sanitarios sin causa justificada. 29.ª Actuar, los profesionales sanitarios implicados en el ciclo de prescripción, dispensación y administración y siempre que estén en ejercicio, en funciones de delegados de visita médica, representantes, comisionistas o agentes informadores de los laboratorios de productos sanitarios. 30.ª Fabricar productos sanitarios a medida sin contar con la correspondiente prescripción escrita por un facultativo. 31.ª Incumplir el profesional sanitario el deber de notificación de los incidentes adversos al Sistema de Vigilancia de Productos Sanitarios. 32.ª Incumplir, las oficinas de farmacia y servicios de farmacia, lo establecido legalmente en materia de aportación del usuario en la prestación farmacéutica del Sistema Nacional de Salud. c) Infracciones muy graves: 1.ª Poner en el mercado y/o en servicio productos que no cumplan con los requisitos esenciales que les sean de aplicación. 2.ª Poner en el mercado y/o en servicio productos que no hayan satisfecho los procedimientos de evaluación de la conformidad o que no hayan efectuado las declaraciones que, en su caso, les resulten de aplicación. 3.ª Comercializar y/o poner en servicio productos que comprometan la salud o la seguridad de los pacientes, usuarios o, en su caso, de terceros. 4.ª Instalar y/o mantener inadecuadamente productos, de forma que comprometan la salud o la seguridad de los pacientes, usuarios o, en su caso, de terceros. 5.ª Usar indebidamente el «marcado CE» en productos no conformes o que no hayan satisfecho los procedimientos de evaluación de la conformidad correspondientes, así como en los productos que no tienen la condición de productos sanitarios. 6.ª Incumplir el deber de ejecución de las medidas y acciones necesarias para reducir o eliminar riesgos para la salud ocasionados por los productos, así como de las medidas y acciones ordenadas por las autoridades sanitarias. 7.ª Vender al público productos para el diagnóstico genético. 8.ª Realizar investigaciones clínicas incumpliendo las obligaciones establecidas en la legislación vigente, cuando suponga un perjuicio en los derechos, seguridad y bienestar de los pacientes. 9.ª Realizar investigaciones clínicas sin contar con el consentimiento del sujeto de las mismas o, en su caso, de su representante, o incumplir por parte del investigador el deber de información sobre la investigación clínica a quien participa como sujeto de la misma. 10.ª Utilizar por un profesional productos en condiciones y para usos distintos de los indicados por el fabricante, o por personal no cualificado o debidamente adiestrado, con riesgo para la salud y seguridad de las personas. 11.ª Ejecutar incorrectamente, el organismo notificado, las actuaciones que se le encomiendan, cuando quede perjudicada la seguridad de los productos certificados, así como continuar certificando una vez retirada la correspondiente designación. 12.ª Efectuar publicidad dirigida al público de los productos para el diagnóstico genético. 13.ª Falsificar productos sanitarios, así como falsificar los documentos acreditativos de la conformidad. 14.ª Incumplir, la empresa suministradora, su obligación de tener suficientemente abastecido el mercado, de modo adecuado y continuado para posibilitar el cumplimiento de las exigencias legalmente establecidas en materia de prestación con productos sanitarios del Sistema Nacional de Salud y garantizar el abastecimiento. 15.ª Cualquier acto de la oficina de farmacia que induzca al usuario a adquirir una mayor cantidad de productos sanitarios dentro de la prestación farmacéutica del Sistema Nacional de Salud que el verdaderamente necesario o demandado por este último. – 70 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios Artículo 113. Infracciones en materia de productos cosméticos y productos de cuidado personal. 1. Las infracciones se califican como leves, graves y muy graves atendiendo a los criterios de riesgo para la salud, cuantía del eventual beneficio obtenido, gravedad de la alteración sanitaria y social producida, generalización de la infracción y reincidencia. 2. Constituirán infracciones administrativas y serán sancionadas en los términos previstos en el artículo 114 las conductas que a continuación se tipifican: a) Infracciones leves: 1.ª Dificultar la labor inspectora mediante cualquier acción u omisión que perturbe o retrase la misma. 2.ª Incumplir el deber de colaborar con las autoridades sanitarias competentes en la evaluación, vigilancia y control de los cosméticos. 3.ª Incumplir los requisitos, obligaciones o prohibiciones establecidas en la normativa aplicable, cuando en razón a los criterios contemplados en este artículo, tales incumplimientos merezcan la calificación de leves, o no proceda su calificación como falta grave o muy grave. b) Infracciones graves: 1.ª Comercializar como si fueran cosméticos productos que no se ajusten a la definición de tales establecida en la normativa vigente, bien por el lugar de aplicación a que se destina, bien por su finalidad. 2.ª Comercializar como si fueran cosméticos productos destinados a la prevención, diagnóstico y tratamiento de enfermedades, así como los destinados a ser ingeridos, inhalados, inyectados o implantados en el cuerpo humano o a la protección frente a la contaminación o infección por microorganismos, hongos o parásitos. 3.ª Comercializar sustancias o mezclas que se presenten como cosméticos sin cumplir la normativa aplicable. 4.ª No proporcionar a la Administración competente la información que esté obligado a suministrar la persona responsable, así como la falta de comunicación de cualquier modificación de las informaciones iniciales que sea necesario comunicar. 5.ª No facilitar a las autoridades sanitarias competentes la información que les sea requerida sobre las sustancias en las que exista duda en relación con su seguridad, así como cualquier otra información que sea requerida por dichas autoridades con fines de control del mercado. 6.ª La falta de coincidencia entre las menciones requeridas del etiquetado de los productos y la información proporcionada a la Administración competente. 7.ª Comercializar cosméticos que omitan en el etiquetado alguna de las menciones requeridas o no la expresen en la lengua y/o en los términos establecidos. 8.ª Utilizar en el etiquetado, en la comercialización o en la publicidad de los productos cosméticos textos, denominaciones, marcas, imágenes o cualquier otro símbolo figurativo o no, con el fin de atribuir a estos productos características o funciones de las que carecen, así como efectuar reivindicaciones que incumplan los criterios comunes establecidos. 9.ª Comercializar productos cosméticos que induzcan a confusión con alimentos, medicamentos, productos sanitarios, biocidas u otros productos, o bien que hagan referencia al tratamiento de patologías. 10.ª Comercializar cosméticos sin haber realizado la evaluación de la seguridad prevista en la regulación o sin haberla realizado en las condiciones establecidas. 11.ª Realizar las actividades de fabricación de productos cosméticos o alguna de sus fases, como el control, envasado o etiquetado, en territorio nacional, o de importación de cosméticos procedentes de países no comunitarios sin un técnico responsable con cualificación adecuada conforme a la normativa específica. 12.ª Fabricar o importar productos cosméticos, o trasladar, ampliar o modificar sustancialmente las actividades e instalaciones sin haber presentado la declaración responsable de cumplimiento de requisitos para realizar dichas actividades. – 71 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios 13.ª Fabricar o importar productos cosméticos sin atenerse a las condiciones manifestadas en la declaración responsable, así como elaborar los productos cosméticos sin observar los principios de buenas prácticas de fabricación. 14.ª Incumplir el técnico responsable y demás personal las obligaciones que competan a sus cargos. 15.ª Incumplir la persona responsable o el distribuidor las obligaciones que le incumben para poner en conformidad los productos no conformes y negarse a ejecutar las medidas dictadas por las autoridades sanitarias competentes con este fin. 16.ª Incumplir la persona responsable o el distribuidor las obligaciones relativas a la identificación de los agentes que les preceden o les siguen en la cadena de comercialización. 17.ª Incumplir la persona responsable, o el distribuidor las obligaciones que les incumben para asegurarse de que los productos que comercializan cumplen los requisitos establecidos en la normativa. 18.ª Incumplir el deber de notificar la persona responsable o distribuidor a las autoridades sanitarias los efectos graves no deseados, los riesgos que presenten los productos y las medidas correctoras adoptadas. 19.ª Impedir la actuación de los inspectores, debidamente acreditados, en los centros en que se fabriquen, almacenen, distribuyan, vendan o utilicen productos cosméticos. 20.ª Suministrar a los consumidores cosméticos destinados a estudios internos o destinados a ser presentados en ferias, exposiciones o demostraciones y cuya introducción en territorio español se haya autorizado exclusivamente para ese fin. 21.ª Distribuir cosméticos sin observar las condiciones exigidas, así como poner a la venta productos cosméticos alterados, en malas condiciones o cuando se haya sobrepasado la fecha de duración mínima, cuando proceda. 22.ª No mantener a disposición de las autoridades sanitarias competentes alguna/s de las informaciones que se establece/n en el expediente de información del producto, o no expresarlas en español, cuando resulte exigible. 23.ª No facilitar al público por parte de la persona responsable la información que resulta preceptiva de acuerdo con la regulación. 24.ª Introducir en el mercado productos cosméticos fabricados en instalaciones que no hayan sido objeto de declaración responsable. c) Infracciones muy graves: 1.ª Comercializar productos cosméticos o productos que se presenten como cosméticos que perjudiquen la salud humana cuando se apliquen en las condiciones normales o razonablemente previsibles de uso, o en los que no se advierta a los consumidores de los riesgos que previsiblemente pudieran derivarse de su normal utilización por medio de instrucciones, advertencias e indicaciones apropiadas. 2.ª Comercializar productos cosméticos que incluyan: 1.º Sustancias prohibidas para su uso en cosméticos. 2.º Sustancias en concentraciones superiores y/o en condiciones diferentes de las establecidas para su uso en cosméticos. 3.º Colorantes, conservantes o filtros ultravioleta distintos de los autorizados para su uso en cosméticos, o en concentraciones superiores y/o en condiciones diferentes a las establecidas. 4.º Sustancias clasificadas como carcinógenas, mutágenas o tóxicas para la reproducción, fuera de las condiciones establecidas en la normativa de cosméticos. 3.ª Comercializar cosméticos que incumplan los requisitos establecidos relativos a la experimentación animal. 4.ª Falsear la información que debe proporcionarse a la autoridad sanitaria, así como falsear la declaración responsable de cumplimiento de requisitos para la realización de actividades de fabricación e importación. 5.ª Incumplir el deber de ejecución de las medidas y acciones necesarias para eliminar riesgos para la salud ocasionados por los cosméticos, así como de las medidas y acciones ordenadas por las autoridades sanitarias. – 72 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios 6.ª Elaborar los productos cosméticos en condiciones técnico-sanitarias deficientes que afecten a su seguridad. 7.ª Fabricar, introducir en el mercado o comercializar productos falsificados. 3. Lo dispuesto en los apartados anteriores se aplicará a los productos de cuidado personal cuando el objeto de la infracción resulte aplicable a dichos productos. En todo caso se considerarán infracciones muy graves: a) Comercializar los productos de cuidado personal sin la preceptiva autorización sanitaria. b) Elaborar los productos de cuidado personal en condiciones técnico-sanitarias deficientes que afecten a su seguridad. c) Comercializar los productos de cuidado personal que perjudiquen la salud humana cuando se apliquen en las condiciones normales o razonablemente previsibles de uso, o en los que no se advierta a los consumidores de los riesgos que previsiblemente pudieran derivarse de su normal utilización por medio de instrucciones, advertencias e indicaciones apropiadas. 4. La comisión de una infracción, precedida de otras dos de grado inmediatamente inferior o igual cometidas y sancionadas en firme en el plazo de un año previo a dicha comisión, incrementará de leve a grave, o de grave a muy grave, dicha infracción. Artículo 114. Sanciones. 1. Las infracciones en materia de medicamentos serán sancionadas con multa, de conformidad con lo establecido en el artículo 111 aplicando una graduación de mínimo, medio y máximo a cada nivel de infracción, en función de la negligencia e intencionalidad del sujeto infractor, fraude, connivencia, incumplimiento de las advertencias previas, cifra de negocios de la empresa, número de personas afectadas, perjuicio causado, beneficios obtenidos a causa de la infracción, permanencia o transitoriedad de los riesgos y reincidencia por comisión en el término de un año de más de una infracción de la misma naturaleza cuando así haya sido declarado por resolución firme: a) Infracciones leves: Grado mínimo: Hasta 6.000 euros. Grado medio: Desde 6.001 a 18.000 euros. Grado máximo: Desde 18.001 a 30.000 euros. b) Infracciones graves: Grado mínimo: Desde 30.001 a 60.000 euros. Grado medio: Desde 60.001 a 78.000 euros. Grado máximo: Desde 78.001 a 90.000 euros. c) Infracciones muy graves: Grado mínimo: Desde 90.001 a 300.000 euros. Grado medio: Desde 300.001 a 600.000 euros. Grado máximo: Desde 600.001 a 1.000.000 de euros, pudiendo rebasar dicha cantidad hasta alcanzar el quíntuplo del valor de los productos o servicios objeto de la infracción. No obstante, en el caso de infracciones en materia de medicamentos veterinarios, la sanción sólo se impondrá en el grado máximo cuando la actuación infractora haya producido un daño directo o provocado un riesgo grave y directo en la salud pública o en la seguridad alimentaria. 2. Sin perjuicio de la multa que proceda imponer conforme a lo dispuesto en el apartado anterior, las infracciones en materia de medicamentos serán sancionadas con el comiso, en favor del Tesoro Público, del beneficio ilícito obtenido como consecuencia de la perpetración de la infracción. La resolución de la Administración determinará a estos efectos la cuantía del beneficio ilícito obtenido. 3. Sin perjuicio de la multa que proceda imponer conforme a lo dispuesto en el apartado uno de este artículo, la infracción muy grave en materia de medicamentos, recogida en el – 73 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios apartado 23 del párrafo c) del artículo 111.2, podrá conllevar la inhabilitación de la oficina de farmacia implicada para dispensar recetas del Sistema Nacional de Salud por un periodo mínimo de 3 meses y máximo de 1 año. 4. Las sanciones por la comisión de infracciones graves y muy graves serán publicadas en el diario oficial correspondiente una vez que adquieran firmeza. 5. Corresponde el ejercicio de la potestad sancionadora a la Administración General del Estado o a las comunidades autónomas que ostentan la función inspectora, de acuerdo con lo regulado en el artículo 108. 6. Además, en los supuestos de infracciones muy graves podrá acordarse, por el Consejo de Ministros o por los órganos competentes de las comunidades autónomas a las que corresponda la ejecución de la legislación sobre productos farmacéuticos, el cierre temporal del establecimiento, instalación o servicio por un plazo máximo de cinco años. En tal caso, será de aplicación lo previsto en el artículo 53 de la Ley 31/1995, de 8 de noviembre, de Prevención de Riesgos Laborales. 7. Lo previsto en los apartados anteriores resultará de aplicación a las infracciones en materia de productos sanitarios y cosméticos, de conformidad con lo establecido en los artículos 112 y 113. No obstante, en el caso de infracciones en materia de productos sanitarios y cosméticos, la sanción sólo se impondrá en su grado máximo cuando la actuación infractora haya producido un daño directo o provocado un riesgo grave y directo en la salud pública. Artículo 115. Otras medidas. 1. No tendrán carácter de sanción la clausura y cierre de establecimientos, instalaciones o servicios que no cuenten con las previas autorizaciones o registros sanitarios preceptivos, o la suspensión de su funcionamiento hasta tanto se subsanen los defectos o se cumplan los requisitos exigidos por razones de sanidad, higiene o seguridad. 2. La autoridad a que corresponda resolver el expediente podrá acordar el comiso de productos y medicamentos deteriorados, caducados, no autorizados o que puedan entrañar riesgo para la salud. 3. Los gastos de transporte, distribución o destrucción de los productos y medicamentos, así como los derivados de la suspensión, clausura y cierre de establecimientos, instalaciones o servicios señalados en los apartados anteriores, serán por cuenta del infractor. Artículo 116. Prescripción. 1. Las infracciones muy graves prescribirán a los cinco años, las graves a los dos años y las leves al año; en los mismos plazos prescribirán las sanciones. 2. El plazo de prescripción de las infracciones comenzará a contarse desde el día en que la infracción se hubiera cometido. Interrumpirá la prescripción la iniciación, con conocimiento del interesado, del procedimiento sancionador, reanudándose el plazo de prescripción si el expediente sancionador estuviera paralizado durante más de un mes por causa no imputable al presunto responsable. 3. El plazo de prescripción de las sanciones comenzará a contarse desde el día siguiente a aquél en que adquiera firmeza la resolución por la que se impone la sanción. Interrumpirá la prescripción la iniciación, con conocimiento del interesado, del procedimiento de ejecución, volviendo a transcurrir el plazo si aquél está paralizado durante más de un mes por causa no imputable al infractor. TÍTULO X De la acción de cesación Artículo 117. Solicitud previa al ejercicio de la acción de cesación. 1. Cuando una publicidad de medicamentos de uso humano, de productos sanitarios o de productos con supuestas propiedades sobre la salud sea contraria a esta ley, a sus – 74 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios disposiciones de desarrollo o a la Ley 14/1986, de 25 de abril, afectando a los intereses colectivos o difusos de los consumidores y usuarios, podrán solicitar su cesación: a) La Agencia Española de Consumo, Seguridad Alimentaria y Nutrición y los órganos o entidades correspondientes de las comunidades autónomas y de las corporaciones locales competentes en materia de defensa de los consumidores. b) Las asociaciones de consumidores y usuarios que reúnan los requisitos establecidos en el texto refundido de la Ley General para la Defensa de los Consumidores y Usuarios y otras leyes complementarias, aprobado por el Real Decreto Legislativo 1/2007, de 16 de noviembre, o, en su caso, en la legislación autonómica en materia de defensa de los consumidores. c) Las entidades de otros Estados miembros de la Unión Europea a las que alude el artículo 118. d) Los titulares de un derecho o de un interés legítimo. 2. La solicitud se hará en forma que permita tener constancia fehaciente de su fecha, de su recepción y de su contenido. 3. La cesación podrá ser solicitada desde el comienzo hasta el fin de la actividad publicitaria. Asimismo, la acción podrá ejercitarse para prohibir la realización de una conducta cuando ésta haya finalizado al tiempo de ejercitar la acción, si existen indicios suficientes que hagan temer su reiteración de modo inmediato. 4. Dentro de los quince días siguientes a la recepción de la solicitud, el requerido comunicará al requirente en forma fehaciente su voluntad de cesar en la actividad publicitaria y procederá efectivamente a dicha cesación. 5. En los casos de silencio o negativa, o cuando no hubiera tenido lugar la cesación, el requirente, previa justificación de haber efectuado la solicitud de cesación, podrá ejercitar la acción prevista en el artículo siguiente. 6. Tanto la solicitud como la voluntad de cesar, o, en su caso, la negativa a cesar en la actividad publicitaria, deberá ser comunicada a la autoridad sanitaria competente en materia de control de publicidad de medicamentos. Artículo 118. Acción de cesación. 1. Podrá ejercitarse la acción de cesación frente a las siguientes conductas, siempre que sean contrarias a esta ley, a sus normas de desarrollo o a la Ley 14/1986, de 25 de abril, y lesionen intereses colectivos o difusos de los consumidores y usuarios: a) Conductas en materia de publicidad de medicamentos de uso humano, en cuyo caso podrá ejercitarse la acción sin necesidad de presentar la solicitud previa contemplada en el artículo 117, que tendrá carácter potestativo. b) Conductas en materia de publicidad de productos sanitarios o productos con supuestas propiedades para la salud, previa la preceptiva presentación de la solicitud contemplada en el artículo 117. 2. La acción de cesación se dirige a obtener una sentencia que condene al demandado a cesar en la conducta contraria a las normas citadas en el apartado anterior y a prohibir su reiteración futura. Asimismo, la acción podrá ejercerse para prohibir la realización de una conducta cuando ésta haya finalizado al tiempo de ejercitar la acción, si existen indicios suficientes que hagan temer su reiteración de modo inmediato. Deberá comunicarse a la autoridad sanitaria competente en materia de control de la publicidad de medicamentos tanto la interposición de la acción, como la sentencia que, en su caso, se dicte. 3. Estarán legitimados para ejercitar la acción de cesación: a) La Agencia Española de Consumo, Seguridad Alimentaria y Nutrición y los órganos o entidades correspondientes de las comunidades autónomas y de las corporaciones locales. b) Las asociaciones de consumidores y usuarios que reúnan los requisitos establecidos en el texto refundido de la Ley General para la Defensa de los Consumidores y Usuarios, o, en su caso, en la legislación autonómica en materia de defensa de los consumidores. c) El Ministerio Fiscal. – 75 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios d) Las entidades de otros Estados miembros de la Unión Europea constituidas para la protección de los intereses colectivos y de los intereses difusos de los consumidores que estén habilitadas mediante su inclusión en la lista publicada a tal fin en el «Diario Oficial de la Unión Europea». Los Jueces y Tribunales aceptarán dicha lista como prueba de la capacidad de la entidad habilitada para ser parte, sin perjuicio de examinar si la finalidad de la misma y los intereses afectados legitiman el ejercicio de la acción. e) Los titulares de un derecho o interés legítimo. Todas las entidades citadas en este artículo podrán personarse en los procesos promovidos por otra cualquiera de ellas, si lo estiman oportuno para la defensa de los intereses que representan. TÍTULO XI Tasas Artículo 119. Creación, normativa y ámbito territorial. 1. Se crea la tasa por prestación de servicios y realización de actividades de la Administración General del Estado en materia de medicamentos, productos sanitarios, productos cosméticos y productos de cuidado personal. 2. El tributo regulado en este título se regirá por lo establecido en esta ley, en su defecto, por la Ley 8/1989, de 13 de abril, de Tasas y Precios Públicos, y disposiciones reglamentarias de desarrollo. 3. Dicha tasa será de aplicación en todo el territorio nacional de acuerdo con lo previsto en el artículo 124, y sin perjuicio de las facultades que correspondan a las comunidades autónomas. Artículo 120. Hecho imponible. Constituye el hecho imponible de la tasa la prestación o realización, por los órganos competentes de la Administración General del Estado, de los servicios o actividades a que se refiere el artículo 123 relativas a medicamentos legalmente reconocidos, productos sanitarios, productos cosméticos y productos de cuidado personal, laboratorios farmacéuticos y entidades de distribución. Artículo 121. Exenciones. 1. Estarán exentas las prestaciones de servicios o realización de actividades relativas a la fabricación de «medicamentos sin interés comercial» a que se refiere el artículo 3.3. 2. Estarán exentos los servicios y actividades por modificaciones en el material de acondicionamiento que tengan como objeto hacer efectiva la impresión en lenguaje braille, de acuerdo con lo previsto en el artículo 15.5. 3. Estarán exentos del pago de la tasa correspondiente los servicios y actividades relativas a medicamentos de terapia avanzada que hayan de ser realizadas por entidades de naturaleza pública integradas en el Sistema Nacional de Salud así como aquellos que no vayan destinados a la comercialización de dichos productos. 4. Estarán exentas parcialmente del pago de la tasa correspondiente las modificaciones o variaciones de autorizaciones concedidas por la Agencia Española de Medicamentos y Productos Sanitarios cuando deriven necesariamente de la aprobación, por norma reglamentaria, de una nueva regulación general. La tasa se reducirá en un 95 % de la cuantía establecida en cada caso. 5. Estarán exentos parcialmente del pago de la tasa correspondiente los titulares de autorizaciones de comercialización en medicamentos autorizados cuando, por razones de interés sanitario, la Agencia Española de Medicamentos y Productos Sanitarios o la Comisión Europea inste su modificación. La tasa se reducirá en un noventa y cinco por ciento de la cuantía establecida en cada caso. – 76 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios 6. Se aplicará a los medicamentos veterinarios destinados exclusivamente a especies menores o usos menores una exención del 70 % de las tasas correspondientes a las autorizaciones de comercialización, a las extensiones de línea a especies menores o usos menores de medicamentos ya autorizados a especies mayores, a las modificaciones de la autorización de comercialización, asesoramientos científicos, productos en fase de investigación clínica veterinaria, ensayos clínicos veterinarios, renovación de la autorización, presentación de la declaración anual simple de intención de comercialización e informes periódicos de seguridad, de medicamentos veterinarios autorizados por procedimiento nacional, de reconocimiento mutuo o descentralizado. No se aplicará a los procedimientos de transmisión de titularidad y/o de representante del titular. 7. Estarán exentas parcialmente del pago de la tasa correspondiente las asesorías científicas sobre medicamentos que incluyan únicamente preguntas relacionadas con el desarrollo pediátrico. La tasa se reducirá en un 95 % de la cuantía establecida en cada caso. Artículo 122. Sujeto pasivo. Serán sujetos pasivos de la tasa las personas físicas o jurídicas que soliciten la prestación de los servicios o la realización de las actividades que constituyen el hecho imponible. Artículo 123. Cuantía. 1. La cuantía de cada tasa en euros será: Grupo I. Medicamentos de uso humano. Epígrafe 1.1 1.2 1.3 1.4 1.5 1.6 1.7 1.8 1.9 1.10 1.11 1.12 1.13 1.14 1.15 1.16 1.17 1.18 1.19 1.20 Descripción Euros Evaluación, autorización e inscripción de nuevos medicamentos Tasa por la evaluación, autorización e inscripción en el registro de un nuevo medicamento de uso humano (expediente de autorización presentado según el artículo 20.734,46 17, excepto el contemplado en el artículo 17.3). Tasa por la evaluación, autorización e inscripción en el registro de un nuevo medicamento de uso humano genérico (expediente de autorización presentado según 8.434,22 el artículo 17.3). Tasa por la evaluación, autorización e inscripción en el registro de un nuevo gas medicinal. 8.434,22 Transmisión de titularidad de un medicamento de uso humano Tasa por el procedimiento de transmisión de la titularidad de la autorización de un medicamento de uso humano, o por modificación del representante del titular. 704,55 Evaluación, autorización e inscripción en el registro de una variación de un medicamento de uso humano Tasa por el procedimiento de modificación de la autorización de un medicamento de uso humano, calificada como de «importancia mayor» tipo II. 7.122,25 Tasa por el procedimiento de modificación de la autorización de un medicamento de uso humano, calificada como tipo IB. 1.249,22 Tasa por el procedimiento de modificación de la autorización de un medicamento de uso humano, calificada de tipo IA (incluida las tipo IA de notificación inmediata). 724,42 Procedimientos de renovación de la autorización de comercialización Tasa por el procedimiento de renovación de la autorización de un medicamento de uso humano. 2.342,71 Tasa anual de mantenimiento de medicamentos autorizados Tasa anual simple de un medicamento de uso humano ya autorizado. 373,70 Tasas por importaciones paralelas Tasa por el procedimiento de autorización para la «importación paralela» de un medicamento de uso humano. 905,45 Tasa por el procedimiento de modificación de la autorización para la « importación paralela» de un medicamento de uso humano. 366,49 Tasa por el procedimiento de renovación de la autorización para la «importación paralela» de un medicamento de uso humano. 366,49 Tasa por notificación de importación. 359,04 Tasas por liberación de lotes de vacunas, hemoderivados y graneles Tasa por la expedición de certificado europeo de liberación de lote para vacunas y hemoderivados de uso humano cuando se requiere el análisis de un 1.212,00 medicamento por lote. Tasa por liberación de lotes de hemoderivados y vacunas de acuerdo con los artículos 41.4 y 43.3 del Real Decreto 1345/2007, de 11 de octubre:. (a) cada solicitud individualizada. 101,00 (b) entre 6 y 10 solicitudes/año (por año). 505,00 (c) entre 11 y 40 solicitudes/año (por año). 1.515,00 (d) entre 41 y 160 solicitudes/año (por año). 3.535,00 (e) por más de 160 solicitudes/año (por año). 5.050,00 Tasa por la expedición certificado europeo de liberación de lote para vacunas y hemoderivados de uso humano cuando se requiere el análisis de un granel (por 339,36 granel). Tasa por la evaluación de innovaciones galénicas Tasa por la evaluación de una solicitud de declaración de innovación galénica de interés terapéutico. 984,04 Tasa aplicable a la exportación Autorizaciones de exportación, a países intracomunitarios y terceros países, de medicamentos estupefacientes y psicótropos. 171,70 Otras Tasa para las actuaciones previstas en el artículo 123.6. 366,49 Tasa por la reserva de una vacante para actuar como Estado miembro de Referencia en un procedimiento descentralizado o de reconocimiento mutuo. 757,50 Grupo II. Medicamentos alérgenos. – 77 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios Epígrafe 2.1 2.2 2.3 2.4 2.5 2.6 2.7 2.8 Descripción Evaluación, autorización e inscripción en el registro de un nuevo medicamento alérgeno de uso humano para uso diagnóstico Tasa por el Procedimiento Nacional. Transmisión de titularidad de un medicamento alérgeno para uso diagnóstico Tasa por el procedimiento de transmisión de la titularidad de la autorización de un medicamento alérgeno para uso diagnóstico. Evaluación, autorización e inscripción en el registro de una variación de un medicamento alérgeno para uso diagnóstico Tasa por el procedimiento de modificación de la autorización de un medicamento alérgeno para uso diagnóstico, calificada como de «importancia mayor» tipo II. Tasa por el procedimiento de modificación de la autorización de un medicamento alérgeno para uso diagnóstico, definida como tipo IB. Tasa por el procedimiento de modificación de la autorización un medicamento alérgeno para uso diagnóstico, calificada de tipo IA (incluida las tipo IA de notificación inmediata). Procedimientos de renovación de la autorización de comercialización Tasa por el procedimiento de renovación de la autorización de un medicamento alérgeno para uso diagnóstico. Tasa anual de mantenimiento de medicamentos alérgenos autorizados Tasa anual simple de un medicamento alérgeno para uso diagnóstico ya autorizado. Tasa por liberación de graneles Tasa por autorización de graneles. Euros 860,93 472,46 502,74 88,19 51,14 307,38 373,70 614,77 Grupo III. Medicamentos de plantas medicinales. Epígrafe 3.1 3.2 3.3 3.4 3.5 3.6 3.7 3.8 3.9 3.10 3.11 Descripción Evaluación, autorización e inscripción en el registro de un nuevo medicamento tradicional a base de plantas (MTP) Tasa por un procedimiento simplificado nacional. Evaluación, autorización e inscripción en el registro de una variación de un medicamento tradicional a base de plantas (MTP) Tasa por el procedimiento de modificación de la autorización un medicamento tradicional a base de plantas (MTP). Evaluación, autorización e inscripción en el registro de un nuevo medicamento de uso humano a base de plantas por uso bien establecido Tasa por el Procedimiento Nacional, excepto epígrafe 3.1. Transmisión de titularidad de medicamentos a base de plantas Tasa por el procedimiento de transmisión de la titularidad de la autorización de un medicamento tradicional a base de plantas (MTP) o un medicamento a base de plantas autorizado por uso bien establecido. Evaluación, autorización e inscripción en el registro de una variación de un medicamento a base de plantas Tasa por el procedimiento de modificación de la autorización de un medicamento de uso humano a base de plantas, calificada como de «importancia mayor» tipo II. Tasa por el procedimiento de modificación de la autorización de un medicamento de uso humano a base de plantas, calificada como tipo IB. Tasa por el procedimiento de modificación de la autorización de un medicamento de uso humano a base de plantas, calificada de tipo IA (incluida las tipo IA de notificación inmediata). Procedimientos de renovación de la autorización de comercialización Tasa por el procedimiento de renovación de la autorización de un medicamento tradicional a base de plantas (MTP). Tasa por el procedimiento de renovación de la autorización de un medicamento a base de plantas autorizado por uso bien establecido. Tasas anuales de mantenimiento de medicamentos a base de plantas autorizados Tasa anual simple de un medicamento tradicional a base de plantas (MTP) ya autorizado. Tasa anual simple de un medicamento a base de plantas autorizado por uso bien establecido. Euros 2.186,94 338,98 8.434,22 704,55 1.249,22 557,67 317,85 307,38 1.522,95 373,70 373,70 Grupo IV. Medicamentos homeopáticos de uso humano y veterinarios. Epígrafe 4.1 4.2 4.3 4.4 4.5 4.6 4.7 4.8 4.9 4.10 4.11 4.12 4.13 Descripción Euros Evaluación, autorización e inscripción en el registro de un nuevo medicamento homeopático sin indicación terapéutica aprobada Tasa por un procedimiento simplificado nacional: Una sola cepa. 596,69 Entre dos y cinco cepas. 745,85 Más de seis cepas. 932,32 Evaluación, autorización e inscripción en el registro de una variación de un medicamento homeopático sin indicación terapéutica aprobada Tasa por el procedimiento de modificación de la autorización de un medicamento homeopático sin indicación terapéutica aprobada. 329,01 Evaluación, autorización e inscripción en el registro de un nuevo medicamento homeopático con indicación terapéutica aprobada Tasa por el Procedimiento Nacional. 8.434,22 Transmisión de titularidad de un medicamento homeopático con o sin indicación terapéutica aprobada Tasa por el procedimiento de transmisión de la titularidad de la autorización de un medicamento homeopático con o sin indicación terapéutica aprobada, o por 704,55 modificación del representante del titular. Evaluación, autorización e inscripción en el registro de una variación de un medicamento homeopático con indicación terapéutica aprobada Tasa por el procedimiento de modificación de la autorización de un medicamento homeopático con indicación terapéutica aprobada, calificada como de «importancia 1.249,22 mayor» tipo II. Tasa por el procedimiento de modificación de la autorización de un medicamento homeopático con indicación terapéutica aprobada, calificada como tipo IB. 557,67 Tasa por el procedimiento de modificación de la autorización de un medicamento homeopático con indicación terapéutica aprobada, calificada de tipo IA (incluida las 317,85 tipo IA de notificación inmediata). Procedimientos de Renovación de la autorización de comercialización Tasa por el procedimiento de renovación de la autorización de un medicamento homeopático sin indicación terapéutica aprobada. 307,38 Tasa por el procedimiento de renovación de la autorización de un medicamento homeopático con indicación terapéutica aprobada. 1.522,95 Tasas anuales de mantenimiento de medicamentos homeopáticos autorizados Tasa anual simple de un medicamento homeopático sin indicación terapéutica aprobada ya autorizado. 90,90 Tasa anual simple de un medicamento homeopático de uso humano con indicación terapéutica aprobada ya autorizado. 373,70 Grupo V. Investigación clínica. – 78 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios Epígrafe 5.1 5.2 5.3 5.4 5.5 Descripción Tasa por un procedimiento de evaluación del primer ensayo clínico con medicamentos no autorizados en un país perteneciente a la Conferencia Internacional de armonización (ICH) con principios activos o combinaciones de principios activos no autorizados en España. Tasa por el procedimiento: a) De autorización de un ensayo clínico con un medicamento autorizado en un país perteneciente a la Conferencia Internacional de armonización (ICH), que no sea España. b) De autorización de ensayos clínicos con medicamentos no autorizados en algún país perteneciente a la Conferencia Internacional de armonización (ICH), posteriores al primer ensayo clínico incluido en el epígrafe 5.1. c) De autorización de un ensayo clínico con las características indicadas en el epígrafe 5.1 en los casos de una reiteración de la solicitud de autorización cuando el resultado de la primera solicitud fue un desistimiento o la no autorización del ensayo. d) De autorización de un ensayo clínico con un medicamento no autorizado en un país perteneciente a la Conferencia Internacional de armonización (ICH) con principios activos autorizados en España. Tasa por el procedimiento: a) De autorización de un ensayo clínico con medicamentos autorizados e inscritos en España, con independencia del etiquetado específico de éstos para el ensayo. b) De autorización de un ensayo clínico cuyo promotor sea un investigador o grupo de investigadores en los que un Servicio de Farmacia sea el encargado de elaborar o enmascarar los medicamentos en investigación. Procedimiento de calificación como producto en fase de investigación clínica de un medicamento veterinario no autorizado en España. Tasa por procedimiento de ensayo clínico veterinario. Euros 4.242,00 404,00 112,30 278,17 112,30 Grupo VI. Laboratorios farmacéuticos, fabricantes, importadores o distribuidores de principios activos y otras entidades que desarrollen actividades con medicamentos o principios activos. Epígrafe Descripción 6.1 Procedimiento de autorización de apertura de un laboratorio farmacéutico. 6.2 Procedimiento de modificación de la autorización de un laboratorio farmacéutico por cambios menores en la misma. Procedimiento de modificación de la autorización de un laboratorio farmacéutico por cambios mayores en la misma cuando las actuaciones inspectoras no incluyan 6.3.a) visita de inspección. Procedimiento de modificación de la autorización de un laboratorio farmacéutico por cambios mayores en la misma cuando las actuaciones inspectoras incluyan 6.3.b) visita de inspección. Actuaciones inspectoras individualizadas, salvo en los supuestos de denuncia o a petición de una asociación de usuarios o consumidores representativa, en el 6.4.a) ámbito nacional. Actuaciones inspectoras individualizadas, salvo en los supuestos de denuncia o a petición de una asociación de usuarios o consumidores representativa, en 6.4.b) terceros países. 6.4.c) Actuaciones inspectoras individualizadas en terceros países, no preceptivas, a solicitud del interesado. 6.5 Procedimiento de autorización de fabricación de medicamentos aprobados en otros países y no registrados en España. 6.6 Procedimiento de autorización de fabricación excepcional por terceros de medicamentos de uso humano y/o veterinario. 6.7 Procedimiento de autorización y/o certificación de almacenes de medicamentos bajo control o vigilancia aduanera. 6.8 Resolución de autorización de cultivos de plantas que puedan destinarse a la fabricación de medicamentos estupefacientes y psicótropos. Inscripción inicial, notificación de modificaciones preceptivas o actualización anual del registro de empresas fabricantes, importadoras o distribuidoras de principios 6.9 activos. 6.10 Inscripción en el registro de personas dedicadas a la intermediación en la distribución de medicamentos de uso humano. Euros 5.916,36 329,01 3.896,36 5.916,36 5.004,97 10.908,00 20.200,00 643,22 329,01 1.313,00 606,00 808,00 252,50 Grupo VII. Certificaciones e informes. Epígrafe Descripción 7.1 Tasa por la expedición de una certificación. Tasa por asesoramientos científicos para medicamentos que incluyan preguntas multidisciplinares sobre (a) calidad, seguridad y desarrollo clínico, o (b) calidad y 7.2 desarrollo clínico, o (c) seguridad y desarrollo clínico, o (d) asesoría pre-remisión de un expediente. Tasa por asesoramientos científicos para medicamentos que incluyan preguntas sobre (a) desarrollo clínico, o (b) calidad y seguridad, o (c) calidad y estudios de 7.3 bioequivalencia en el caso de medicamentos genéricos. Tasa por asesoramientos científicos para medicamentos que incluyan preguntas sobre (a) calidad, o (b) seguridad, o (c) estudios de bioequivalencia en el caso de 7.4 medicamentos genéricos. 7.5 Tasa por asesoramiento de seguimiento de los supuestos incluidos en el epígrafe 7.2. 7.6 Tasa por asesoramiento de seguimiento de los supuestos incluidos en el epígrafe 7.3. 7.7 Tasa por asesoramiento de seguimiento de los supuestos incluidos en el epígrafe 7.4. Tasa por asesoramiento para la clasificación de variaciones no clasificadas según el artículo 5, y para agrupamiento de variaciones, según el artículo 7 del 7.8 Reglamento (CE) n.º 1234/2008 de la Comisión Europea. 7.9 Tasa por asesoramientos científicos sobre medicamentos para uso pediátrico en cualquiera de los supuestos incluidos en los epígrafes anteriores. 7.10 Asesoramiento científico/técnico sobre el diseño de instalaciones y procesos de fabricación de acuerdo con las normas de correcta fabricación. Euros 141,86 4.224,39 2.112,20 633,66 2.112,20 1.056,10 316,83 344,41 205,01 492,01 Grupo VIII. Productos sanitarios, cosméticos y productos de cuidado personal. Epígrafe 8.1 8.2 8.3 8.4 8.5 8.6 8.7 Descripción Procedimiento de declaración especial de cosméticos. Procedimiento de registro y autorización individualizada para productos de cuidado personal y desinfectantes. Procedimiento de registro e inscripción de productos sanitarios. Procedimiento de modificación y convalidación de productos de cuidado personal y desinfectantes. Procedimiento de expedición de una certificación. Procedimiento de comprobación y control de la declaración responsable de la actividad de fabricación de productos cosméticos y de cuidado personal. Procedimiento de comprobación y control de la declaración responsable de la actividad de importación de productos cosméticos y de cuidado personal. – 79 – Euros 487,90 487,90 101,00 170,02 147,82 717,08 369,63 CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios Epígrafe 8.8 8.9 8.10 8.11 8.12 8.13 8.14 8.15 8.16 8.17 8.18 8.19 8.20 8.21 8.22 8.23 8.24 8.25 8.26 8.27 8.28 8.29 8.30 8.31 8.32 8.33 Descripción Procedimiento de comprobación y control de la declaración responsable de modificaciones de la actividad de fabricación de productos cosméticos y de cuidado personal. Procedimiento de comprobación y control de la declaración responsable de modificaciones de la actividad de importación de productos cosméticos y de cuidado personal. Actuaciones inspectoras individualizadas para la comprobación de la declaración responsable. Procedimiento de autorización de confidencialidad de ingredientes cosméticos. Procedimiento de licencia previa de funcionamiento de productos sanitarios y desinfectantes: establecimiento de fabricación, agrupación. Procedimiento de licencia previa de funcionamiento de productos sanitarios y desinfectantes: establecimiento de importación. Procedimiento de modificación de la licencia previa de funcionamiento de establecimientos de productos sanitarios y desinfectantes en lo referente a su emplazamiento: establecimiento de fabricación, agrupación. Procedimiento de modificación de la licencia previa de funcionamiento de establecimientos de productos sanitarios y desinfectantes en lo referente a su emplazamiento: establecimiento de importación. Procedimiento de modificación de la licencia previa de funcionamiento de establecimientos de productos sanitarios y desinfectantes. Procedimiento de revalidación de la licencia de establecimientos de productos sanitarios y desinfectantes: establecimiento de fabricación. Procedimiento de revalidación de la licencia de establecimientos de productos sanitarios y desinfectantes: establecimiento de importación. Autorización de investigaciones clínicas de productos sanitarios. Informe de evaluación de principio activo incorporado en un producto sanitario. Evaluación de expedientes de certificación del «marcado CE» de productos sanitarios pertenecientes a la misma familia por sistema completo de garantía de calidad. Evaluación de expedientes de certificación del «marcado CE» de productos sanitarios por examen «CE» de tipo, combinado con garantía de calidad de la producción, verificación «CE» o garantía de calidad del producto. Evaluación de expediente de certificación del «marcado CE» de productos sanitarios pertenecientes a la misma familia, por declaración «CE» de conformidad combinada con garantía de calidad de la producción, verificación «CE» o garantía de calidad del producto. Verificación de productos y lotes de productos. Evaluación de expediente de certificación del «marcado CE» de productos sanitarios por examen «CE» de diseño. Auditoría inicial conforme a sistema completo de garantía de calidad. Auditoría inicial conforme a garantía de calidad de la producción. Auditoría inicial conforme a garantía de calidad de producto. Auditorias de seguimiento y de prórroga de certificación. Auditorías a local suplementario y de repetición. Modificación de datos administrativos en la certificación del «marcado CE». Prórrogas de las certificaciones del «marcado CE». Procedimiento de modificación de productos sanitarios. Euros 369,63 170,02 717,08 487,90 717,08 369,63 717,08 369,63 170,02 517,47 317,88 808,00 1.478,50 2.460,36 887,10 739,27 230,17 1.626,37 3.232,00 2.686,60 2.154,33 2.154,33 1.077,67 147,82 147,82 60,60 Grupo IX. Medicamentos veterinarios. Epígrafe 9.1 9.2 9.3 9.4 9.5 9.6 9.7 9.8 9.9 9.10 Descripción Tasa por solicitud de autorización de comercialización de un medicamento veterinario, excepto para las solicitudes contempladas en el artículo 17.3. Tasa por solicitud de autorización de comercialización de un medicamento veterinario genérico (expediente presentado según el artículo 17.3). Tasa por el procedimiento de transmisión de la titularidad de la autorización de un medicamento veterinario, o por modificación del representante del titular. Tasa por el procedimiento de modificación de la autorización de un medicamento veterinario, calificada como de «importancia mayor» tipo II. Tasa por el procedimiento de modificación de la autorización de un medicamento veterinario, definida como tipo IB. Tasa por el procedimiento de modificación de la autorización de un medicamento veterinario, calificada de tipo IA (incluidas las tipo IA de notificación inmediata). Tasa por el procedimiento de renovación de la autorización de un medicamento veterinario. Tasa por declaración anual simple de intención de comercializar un medicamento veterinario ya autorizado. Tasa por el procedimiento de autorización para la «importación paralela» de un medicamento veterinario. Tasa por evaluación de informe periódico de seguridad semestral de un medicamento veterinario, esté o no registrado el medicamento en España. 9.11 Tasa por evaluación de informe periódico de seguridad anual de un medicamento veterinario, esté o no registrado el medicamento en España. 9.12 Tasa por evaluación de informe periódico de seguridad trienal o superior a tres años de un medicamento veterinario, esté o no registrado el medicamento en España. Tasa por expedición de certificado europeo de liberación oficial de lote para medicamentos inmunológicos veterinarios según el artículo 81 de la Directiva 2001/82/CE. Tasa por expedición de certificado europeo de liberación oficial de lote para medicamentos inmunológicos veterinarios según el artículo 82 de 9.14 la Directiva 2001/82/CE. Tasa por la reserva de una vacante para actuar España como Estado miembro de Referencia en un procedimiento Descentralizado o de 9.15 Reconocimiento Mutuo. Euros 10.367,22 4.217,10 704,55 3.561,13 1.224,72 724,42 2.342,71 120,20 738,93 382,71 (la cuantía de la tasa 9.10 vigente durante el correspondiente ejercicio presupuestario) x 2 (la cuantía de la tasa 9.10 vigente durante el correspondiente ejercicio presupuestario) x 6 9.13 339,36 1.212,00 404,00 Grupo X. Procedimientos de financiación con cargo a fondos públicos y fijación de precio de productos sanitarios. Epígrafe 10.1 10.2 Descripción Procedimiento de inclusión de un producto sanitario en la prestación farmacéutica del Sistema Nacional de Salud. Procedimiento de exclusión de un producto sanitario en la prestación farmacéutica del Sistema Nacional de Salud. – 80 – Euros 347,90 347,90 CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios 2. A los efectos del apartado anterior, se entenderá por extensión de línea la segunda y sucesivas solicitudes de autorización e inscripción en el registro de otras formas farmacéuticas, vías de administración y concentración de un medicamento ya autorizado e inscrito. La cuantía de la tasa de las extensiones de línea será del setenta por ciento de la primera autorización del medicamento. En el caso de los medicamentos veterinarios, tendrán la consideración de extensión de línea la ampliación de una autorización a nuevas especies de destino siempre que se trate de especies productoras de alimentos. Constituirán una extensión de línea aquellas modificaciones que requieran la presentación de una nueva solicitud de autorización, de acuerdo con la norma europea que regula las modificaciones de autorización de medicamentos otorgadas por la autoridad competente de un Estado miembro. Lo dispuesto en los párrafos anteriores referentes a las extensiones de línea es aplicable también cuando el medicamento no está todavía autorizado y se presentan en paralelo extensiones de línea de una solicitud principal. A los efectos de la tasa descrita en los epígrafes 8.1 y 8.21, tiene la consideración de: a) «Producto cosmético sometido a declaración especial», aquel que, previa la autorización correspondiente de la Agencia Española de Medicamentos y Productos Sanitarios, incluye en su composición colorantes, agentes conservadores o filtros ultravioletas, no incluidos entre las sustancias admitidas como componentes de los productos cosméticos. b) «Familia de productos sanitarios», el conjunto de productos sanitarios que, perteneciendo a la misma categoría, se destinan a aplicaciones sanitarias idénticas o similares. 3. La cuantía de las tasas por los servicios y actividades de la Administración General del Estado en materia de medicamentos, productos sanitarios, productos cosméticos y productos de cuidado personal, de acuerdo con lo previsto en la Ley 8/1989, de 13 de abril, podrá modificarse a través de la Ley de Presupuestos Generales del Estado. 4. Cuando la evaluación y control de un medicamento o producto sanitario requiera actuaciones en el extranjero o costes excepcionales, las correspondientes tasas se liquidarán sobre el coste real del servicio en que consiste el supuesto que determina su exigencia. Igualmente se liquidarán sobre el coste real del servicio los gastos de desplazamiento, estancia y ensayos derivados de las actuaciones previstas en los epígrafes 6.1, 6.4 a), b) y c), 8.22, 8.24, 8.26, 8.27, 8.28, 8.29, 8.30 y 10.1. 5. Cuando en el procedimiento de autorización e inscripción en el registro de un medicamento de uso humano o veterinario, que se corresponde con las tasas previstas en los epígrafes 1.1, 1.2, 1.3, 1.5, 9.1, 9.2 y 9.4, la solicitud presentada sea rechazada en la fase de validación, se procederá a la devolución de un setenta por ciento de la cuantía total de la tasa. A los efectos de lo establecido en la presente ley, se entiende por validación la acción de carácter administrativo, desarrollada con el propósito de verificar que la solicitud reúne todos los requisitos necesarios para realizar la prestación del servicio o realización de la actividad administrativa. 6. Las modificaciones de la autorización de un medicamento, que sean consecuencia de una decisión de la Comisión Europea y que no conlleven actividad de evaluación científica por parte de la Agencia Española de Medicamentos y Productos Sanitarios devengarán la tasa prevista en el epígrafe 1.19. En los casos de agrupación de modificaciones independientes Tipo IA, siendo estas iguales o distintas, que afecten a varios medicamentos pertenecientes al mismo titular y siempre que se presenten al mismo tiempo y en un único formato de solicitud de acuerdo con el artículo 7 del Reglamento (CE) n.º 1234/2008 de la Comisión, de 24 de noviembre de 2008, relativo al examen de las modificaciones de los términos de las autorizaciones de comercialización de medicamentos para uso humano y medicamentos veterinarios, – 81 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios devengarán para cada uno de los tipos de modificaciones iguales una tasa principal y tasas reducidas para el resto de las modificaciones. Cuando una modificación afecte a distintos medicamentos del mismo titular, y que conlleven una única evaluación científica, la segunda y siguientes devengarán la tasa prevista en el epígrafe 1.19. Cuando se produzcan distintas modificaciones en la autorización de comercialización de un medicamento, el importe total de las mismas no podrá ser superior a la tasa prevista para el procedimiento de autorización e inscripción en el Registro del tipo de medicamento de que se trate. En el caso de agrupación de modificaciones independientes que afecten al mismo medicamento y siempre que todas se presenten al mismo tiempo y en un único formato de solicitud, de acuerdo con el artículo 7 del Reglamento (CE) n.º 1234/2008 de la Comisión Europea, se exigirá una tasa por cada una de las modificaciones solicitadas. La tasa correspondiente al epígrafe 7.2, «Tasa por asesoramientos científicos para medicamentos que incluyan preguntas multidisciplinares sobre (a) calidad, seguridad y desarrollo clínico, o (b) calidad y desarrollo clínico, o (c) seguridad y desarrollo clínico, o (d) asesoría pre-remisión de un expediente» se reducirá en un 25 % para los asesoramientos científicos que incluyan preguntas sobre desarrollo sólo de eficacia clínica; o de calidad y seguridad preclínica; o de calidad y bioequivalencias. 7. Las tasas de los procedimientos descentralizados para medicamentos de uso humano o veterinario, que resulten en una autorización nacional en los que España actúe como Estado miembro de referencia (prestaciones de los epígrafes 1.1, 1.2, 1.3, 1.5, 1.6, 1.7, 1.8, 3.1, 3.3, 3.5, 3.6, 3.7, 4.1, 4.2, 4.3, 4.5, 4.7, 4.8, 4.9, 9.1, 9.2, 9.4, 9.5, 9.6 y 9.7) se incrementarán en un 25 % sobre el valor de la tasa correspondiente. En los procedimientos de reconocimiento mutuo para medicamentos de uso humano o veterinario en los que España actúe como Estado miembro de referencia se abonará una tercera parte de la tasa completa de referencia (prestaciones de los epígrafes 1.1, 1.2, 1.3, 3.1, 3.3, 4.1, 4.2, 4.3, 4.5, 9.1 y 9.2). La tasa del epígrafe 1.20, que será de aplicación a cualquier medicamento de uso humano, incluidos los medicamentos especiales, será descontada del importe total que proceda abonar en el caso de que el interesado presente una solicitud relativa a un procedimiento descentralizado o de reconocimiento mutuo, actuando España como Estado miembro de referencia. Asimismo la tasa del epígrafe 9.15, que será de aplicación a cualquier medicamento veterinario, será descontada del importe total que proceda abonar en el caso de que el interesado presente una solicitud relativa a un procedimiento descentralizado o de reconocimiento mutuo, actuando España como Estado miembro de referencia. Artículo 124. Devengo. La tasa se devengará cuando la solicitud, que inicia el expediente, tenga entrada en el registro de la Agencia Española de Medicamentos y Productos Sanitarios o del Ministerio de Sanidad, Servicios Sociales e Igualdad, según su respectiva competencia, momento en el cual se inicia la prestación del servicio o la realización de la actividad administrativa. Artículo 125. Pago. 1. El pago de la tasa deberá efectuarse conforme a lo establecido en la Ley 8/1989, de 13 de abril, la Ley 58/2003, de 17 de diciembre, General Tributaria y demás normas de desarrollo. El pago de las tasas contempladas en esta ley se realizará, preferentemente, por vía o medio electrónico, conforme a lo previsto en la Ley 11/2007, de 22 de junio, de Acceso Electrónico de los Ciudadanos a los Servicios Públicos. 2. No se tramitará solicitud alguna que no vaya acompañada del justificante de pago de la tasa que corresponda. 3. Cuando, abonada la tasa, la Administración no pueda tramitar el procedimiento correspondiente por causa no imputable al sujeto pasivo, la devolución de la misma será de un ochenta por ciento de su cuantía. – 82 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios 4. Abonada la tasa, el sujeto pasivo habrá de presentar la solicitud correspondiente dentro de los diez días siguientes al ingreso. 5. La gestión recaudatoria de las tasas reguladas en esta ley corresponde, en vía voluntaria, al Ministerio de Sanidad, Servicios Sociales e Igualdad y a la Agencia Española de Medicamentos y Productos Sanitarios, según su respectiva competencia. 6. La Agencia Española de Medicamentos y Productos Sanitarios podrá utilizar para obtener la efectividad de sus débitos con naturaleza de derecho público, el procedimiento administrativo de apremio, siempre que dichos débitos se encuentren en periodo ejecutivo. La Agencia Española de Medicamentos y Productos Sanitarios podrá convenir con la Agencia Estatal de Administración Tributaria la gestión recaudatoria de sus ingresos de derecho público en la forma prevista por el Reglamento General de Recaudación, aprobado por Real Decreto 939/2005, de 29 de julio. Artículo 126. Supuestos de devolución de tasas. Procederá la devolución de ingresos por tasas, además de en los supuestos contemplados en el artículo 221 de la Ley 58/2003, de 17 de diciembre, cuando abonada la tasa, el sujeto pasivo no presente la solicitud de la prestación del servicio o realización de actividad correspondiente dentro del plazo de diez días siguientes al ingreso que establece el artículo 125.4, siempre que sea por causa no imputable al sujeto pasivo, acreditada de forma fehaciente. Esta devolución será de un ochenta por ciento de su cuantía. Disposición adicional primera. Garantía de suministro de medicamentos y productos sanitarios y coordinación de disponibilidad de fluidos y otros elementos. 1. Con objeto de desarrollar e impulsar las actividades necesarias en materia de suministros de medicamentos y productos sanitarios y coordinar la adecuada disponibilidad de sangre y demás fluidos, glándulas y tejidos humanos y sus componentes y sus derivados necesarios para la asistencia sanitaria, el Ministerio de Sanidad, Servicios Sociales e Igualdad, además de las misiones que esta ley le encomienda, desarrollará las siguientes funciones: a) Garantizar el depósito de medicamentos de sustancias psicoactivas de acuerdo con lo dispuesto en los tratados internacionales. b) Autorizar la importación de medicación extranjera y urgente no autorizada en España. c) Mantener un depósito estatal estratégico de medicamentos y productos sanitarios para emergencias y catástrofes. d) Realizar la adquisición y distribución de medicamentos y productos sanitarios para programas de cooperación internacional. e) Coordinar el suministro de vacunas, medicamentos y otros productos para campañas sanitarias cuya adquisición y distribución conjunta se decida por las distintas Administraciones sanitarias. f) Promover la fabricación y comercialización de «medicamentos sin interés comercial». 2. También ejercerá la coordinación de los intercambios y del transporte de sangre y demás fluidos, glándulas y tejidos humanos y de sus componentes y derivados. Disposición adicional segunda. Fuerzas Armadas. Aplicación de la ley a los servicios sanitarios de las La aplicación de los criterios y normas establecidos en esta ley a los servicios sanitarios de las fuerzas armadas será determinada reglamentariamente a propuesta conjunta de los Ministerios interesados. Disposición adicional tercera. Aplicación de la ley a los productos sanitarios, cosméticos y productos de cuidado personal. 1. De conformidad con lo dispuesto en esta ley, se determinarán reglamentariamente las condiciones y requisitos que cumplirán los productos sanitarios para su fabricación, importación, investigación clínica, distribución, comercialización, puesta en servicio, – 83 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios dispensación y utilización, así como los procedimientos administrativos respectivos, de acuerdo con lo establecido en la normativa de la Unión Europea. 2. Lo establecido en el apartado anterior se aplicará igualmente, en aquello que proceda, a los productos de cuidado personal y cosméticos. 3. Las actividades de fabricación e importación de cosméticos y productos de cuidado personal se someten al régimen de declaración responsable, regulado en el artículo 71 bis de la Ley 30/1992, de 26 de noviembre. Esta declaración responsable deberá ser presentada ante la Agencia Española de Medicamentos y Productos Sanitarios. La presentación de la declaración responsable permitirá el inicio de las actividades, sin perjuicio de la comprobación posterior por la Agencia Española de Medicamentos y Productos Sanitarios, mediante verificación documental y, en su caso, inspección, de los elementos y circunstancias puestos de manifiesto por el interesado en la declaración responsable. 4. Se devengarán las tasas necesarias para cubrir los costes de comprobación de la declaración responsable y de la inspección que, en su caso, resulte necesaria. Disposición adicional cuarta. Depósito de medicamentos en centros penitenciarios. Los centros penitenciarios podrán solicitar de la Administración competente en cada caso autorización para mantener un depósito de medicamentos para la asistencia a los internos, bajo la supervisión y control de un farmacéutico de los servicios farmacéuticos autorizados del hospital del Sistema Nacional de Salud más cercano. Disposición adicional quinta. Procedimiento para la exclusión total o parcial de medicamentos de la prestación farmacéutica del Sistema Nacional de Salud. El Gobierno por real decreto, previo informe del Consejo Interterritorial del Sistema Nacional de Salud, establecerá la forma, requisitos y condiciones de aplicación de los criterios contenidos en el artículo 92 y determinará las exclusiones totales o parciales de los grupos, subgrupos, categorías o clases de medicamentos de la financiación con cargo a fondos públicos. Disposición adicional sexta. Aportaciones por volumen de ventas al Sistema Nacional de Salud. 1. Las personas físicas, los grupos empresariales y las personas jurídicas no integradas en ellos que se dediquen en España a la fabricación, importación u oferta al Sistema Nacional de Salud de medicamentos y/o productos sanitarios que, financiados con fondos públicos, se dispensen en oficinas de farmacia a través de receta oficial u orden de dispensación del Sistema Nacional de Salud, en territorio nacional, deberán ingresar con carácter cuatrimestral las cantidades que resulten de aplicar sobre su volumen cuatrimestral de ventas a través de dicha receta u orden de dispensación los porcentajes contemplados en la escala siguiente: Ventas cuatrimestrales a PVL Porcentaje de aportación Desde Hasta 0,00 3.000.000,00 1,5 3.000.000,01 En adelante 2,0 En el supuesto de que el volumen total de ventas de medicamentos y productos sanitarios al Sistema Nacional de Salud sea, en términos corrientes anuales, inferior al del año precedente, el Gobierno podrá revisar los anteriores porcentajes de aportación. Las cuantías resultantes de la aplicación de la escala anterior se verán minoradas en función de la valoración de las compañías en el marco del programa Profarma según los porcentajes siguientes: a) No valoradas: 0,00. b) Aceptables: 5 %. c) Buenas: 10 %. d) Muy buenas: 15 %. e) Excelentes: 25 %. – 84 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios Aquellas empresas clasificadas en el programa Profarma como muy buenas o excelentes, que participen en consorcios de I+D o realicen asociaciones temporales con este fin con otras empresas establecidas en España y centros de I+D públicos y privados, para realizar investigación básica y preclínica de relevancia mediante proyectos específicos y determinados, podrán beneficiarse de una minoración adicional de un diez por ciento de la aportación. Las minoraciones que puedan afectar a estos retornos surtirán efecto a partir de la última resolución del programa Profarma. Los grupos empresariales comunicarán al Ministerio de Sanidad, Servicios Sociales e Igualdad, durante el mes de enero de cada año natural, las compañías integradas en ellos. En caso de que se modifique la composición de un grupo empresarial en el transcurso del año, la comunicación se efectuará durante el mes en que dicha modificación haya tenido lugar. A efectos de lo señalado, se considera que pertenecen a un mismo grupo las empresas que constituyan una unidad de decisión, en los términos del artículo 4 de la Ley 24/1988, de 28 de julio, del Mercado de Valores. 2. El Ministerio de Sanidad, Servicios Sociales e Igualdad, en función de lo previsto en el apartado anterior y sobre las ventas del ejercicio corriente, comunicará la cantidad a ingresar a cada fabricante, importador u oferente afectado, así como el plazo de ingreso de dicha cantidad. En el primer plazo del ejercicio siguiente se efectuarán, en su caso, la regularización de las liquidaciones cuatrimestrales, en el supuesto de que hayan de incorporarse al expediente datos no tenidos en cuenta en las citadas liquidaciones parciales. 3. Las cantidades a ingresar se destinarán a la investigación, en el ámbito de la biomedicina, en cantidad suficiente para financiar las necesidades de investigación clínica que se lleva a cabo, a través de la iniciativa sectorial de investigación en biomedicina y ciencias de la salud, ingresándose en la caja del Instituto de Salud Carlos III. El resto de fondos se destinarán al desarrollo de políticas de cohesión sanitaria, de programas de formación para facultativos médicos, odontólogos, farmacéuticos y enfermeros, así como a programas de educación sanitaria de la población para favorecer el uso racional de los medicamentos, según la distribución que determine el Ministerio de Sanidad, Servicios Sociales e Igualdad, previo informe del Consejo Interterritorial del Sistema Nacional de Salud, ingresándose en el Tesoro Público. Disposición adicional séptima. Conservación de órganos para trasplantes. Las soluciones para conservación de órganos para trasplantes, se regirán, en cuanto les sea de aplicación, por lo previsto en esta ley para los medicamentos. Disposición adicional octava. Medicamentos objeto de publicidad. El precio fijado en el envase de los medicamentos que sean objeto de la publicidad prevista en el artículo 80 será considerado como precio máximo de venta al público. Reglamentariamente se establecerá el descuento máximo aplicable por las oficinas de farmacia. Disposición adicional novena. Organismos modificados genéticamente. Las actividades de utilización confinada y liberación voluntaria de organismos modificados genéticamente que se incorporen o puedan incorporarse a medicamentos de uso humano o veterinario estarán sujetas a lo establecido en la Ley 9/2003, de 25 de abril, por la que se establece el régimen jurídico de la utilización confinada, liberación voluntaria y comercialización de organismos modificados genéticamente y en su normativa de desarrollo. Disposición adicional décima. Participación de las comunidades autónomas y de las mutualidades de funcionarios en los procedimientos de decisión en materia de medicamentos y productos sanitarios. Las comunidades autónomas y las Mutualidades de funcionarios participarán en los términos establecidos reglamentariamente, en el Consejo Rector de la Agencia Española de Medicamentos y Productos Sanitarios, como órgano colegiado de dirección del organismo. – 85 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios Asimismo, la Agencia contará con la colaboración de expertos independientes de reconocido prestigio científico propuestos por las comunidades autónomas. El Ministerio de Sanidad, Servicios Sociales e Igualdad facilitará un informe a todas las comunidades autónomas y a las Mutualidades de funcionarios, en cada reunión de la Comisión Permanente de Farmacia del Consejo Interterritorial del Sistema Nacional de Salud, identificando el nombre de los medicamentos y productos sanitarios que hayan sido autorizados por la Agencia Española de Medicamentos y Productos Sanitarios desde la última reunión del Consejo, así como el precio de aquellos medicamentos y productos sanitarios que hayan sido incluidos en la financiación del Sistema Nacional de Salud. Disposición adicional undécima. Garantía de calidad, seguridad y eficacia de los productos farmacéuticos y la protección de los pacientes. Las autoridades, en el ejercicio de sus competencias, velarán por el cumplimiento de lo dispuesto en la presente ley, a los efectos de garantizar la calidad, seguridad y eficacia de los productos farmacéuticos y la protección de los pacientes. En particular, asegurarán, mediante sus funciones de inspección y control, el cumplimiento de los requisitos exigidos en la legislación farmacéutica. Disposición adicional duodécima. Colocación o entrega de productos sanitarios a medida por un facultativo. La colocación o entrega de productos sanitarios a medida por un facultativo, en el ejercicio de sus atribuciones profesionales, no tendrá la consideración de dispensación, comercialización, venta, distribución, suministro o puesta en el mercado de los mismos, a los efectos de los artículos 4.1 y 111. En todo caso, el facultativo deberá separar sus honorarios de los costes de fabricación. Disposición adicional decimotercera. Información de los precios menores de las agrupaciones homogéneas de medicamentos y productos sanitarios. 1. A efectos de aplicar los supuestos de dispensación y sustitución establecidos en los artículos 87 y 89, respectivamente, la Dirección General de Cartera Básica de Servicios del Sistema Nacional de Salud y Farmacia del Ministerio de Sanidad, Servicios Sociales e Igualdad publicará en su página web, junto al Nomenclátor de productos farmacéuticos del Sistema Nacional de Salud, la información relativa a las agrupaciones homogéneas de las presentaciones de los medicamentos y de los productos sanitarios para pacientes no hospitalizados que requieran para su dispensación receta médica oficial u orden de dispensación. La información sobre los precios menores se actualizará el primer día hábil de cada mes y se publicará en la página web del Ministerio de Sanidad, Servicios Sociales e Igualdad. 2. En cada agrupación homogénea de medicamentos se integrarán las presentaciones de los medicamentos financiadas con el/los mismo/s principio/s activo/s en cuanto a dosis, contenido, forma farmacéutica o agrupación de forma farmacéutica, y vía de administración, que puedan ser objeto de intercambio en su dispensación. Se diferenciarán las agrupaciones homogéneas de medicamentos integradas, exclusivamente, por un medicamento y sus licencias con el mismo precio que el medicamento de referencia. 3. Asimismo, en cada agrupación homogénea de productos sanitarios se integrarán, siempre que sea posible, las presentaciones financiadas que, teniendo las mismas características, tipo, tamaño y contenido y estando clasificadas conforme a los grupos relacionados en los anexos I y II del Real Decreto 9/1996, de 15 de enero, por el que se regula la selección de los efectos y accesorios, su financiación con fondos de la Seguridad Social, o fondos estatales afectos a la sanidad y su régimen de suministro y dispensación a pacientes no hospitalizados, puedan ser objeto de intercambio en su dispensación. 4. Si las variaciones experimentadas en los precios de los medicamentos o productos sanitarios así lo aconsejaran y, previo acuerdo de la Comisión Permanente de Farmacia del Consejo Interterritorial del Sistema Nacional de Salud, la mencionada Dirección General podrá actualizar la información de los precios menores, afectando en su caso dicha – 86 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios actualización a todas las agrupaciones, o a las agrupaciones que se estimen pertinentes, así como incorporar nuevas agrupaciones, y establecer plazos de coexistencia de los precios y devolución de existencias, de las presentaciones de medicamentos y productos sanitarios que hubieran reducido voluntariamente su precio en función del precio menor de cada agrupación, pudiéndose autorizar que dicha reducción se realice sin cambio del código nacional. A efectos informativos las actualizaciones de la información de precios menores se incorporarán en el Nomenclátor de productos farmacéuticos del Sistema Nacional de Salud, finalizados los plazos que en su caso se hubieran establecido. Disposición adicional decimocuarta. Excepción del régimen jurídico previsto en el artículo 43.1 de la Ley 30/1992, de 26 de noviembre. 1. Sin perjuicio de los procedimientos relacionados en la disposición adicional vigésima novena de la Ley 14/2000, de 29 de diciembre, de Medidas Fiscales, Administrativas y del Orden Social, correspondientes a la excepción prevista en el artículo 43.1 de la Ley 30/1992, de 26 de noviembre, se entenderán incluidos en la referida excepción, los siguientes: a) Autorización y modificaciones mayores de laboratorios farmacéuticos. b) Autorización de importación, exportación y fabricación de medicamentos no registrados. c) Autorización excepcional de exportación de medicamentos para donaciones humanitarias. d) Declaración de innovación galénica de interés terapéutico. e) Autorización, modificación y renovación de la importación paralela de medicamentos. f) Autorización de medicamentos por procedimiento descentralizado entre Estados de la Unión Europea. 2. El procedimiento para el registro y autorización de productos sanitarios no incluidos en el ámbito de aplicación del Real Decreto 1616/2009, de 26 de octubre, por el que se regulan los productos sanitarios implantables activos, y del Real Decreto 1591/2009, de 16 de octubre, por el que se regulan los productos sanitarios, recogido en la disposición adicional vigésima novena de la Ley 14/2000, de 29 de diciembre, queda excluido de la excepción prevista en el artículo 43.1 de la Ley 30/1992, de 26 de noviembre. Disposición adicional decimoquinta. Renovación de autorizaciones de medicamentos. A los medicamentos cuya autorización haya sido renovada tras la entrada en vigor de la Ley 29/2006, de 26 de julio, les será de aplicación, a partir de dicha renovación, la normativa vigente en relación con los informes periódicos de seguridad. Disposición adicional decimosexta. Aplicación del sistema de fijación de precios en los productos sanitarios autorizados con anterioridad a la entrada en vigor de la Ley 29/2006, de 26 de julio. Los productos sanitarios autorizados previamente a la entrada en vigor de la Ley 29/2006, de 26 de julio, tendrán un precio industrial máximo resultante de la aplicación del sistema de precios regulado en dicha ley, partiendo de su PVP correspondiente y descontando los márgenes de comercialización. Disposición transitoria primera. Períodos de exclusividad de datos aplicables a solicitudes de autorización presentadas antes del 1 de noviembre de 2005. Los períodos de exclusividad de datos de los medicamentos de referencia para los que se hubiese presentado una solicitud de autorización antes del 1 de noviembre de 2005 serán los que regían con anterioridad a la entrada en vigor de la Ley 29/2006, de 26 de julio. Disposición transitoria segunda. Conflicto de intereses. Sin perjuicio del cumplimiento de lo dispuesto en esta ley y, en particular, en su artículo 4, los farmacéuticos en ejercicio profesional con oficina de farmacia, en establecimiento – 87 – CÓDIGO DE DERECHO FARMACÉUTICO § 1 Ley de garantías y uso racional de los medicamentos y productos sanitarios comercial detallista, en entidades o agrupaciones ganaderas o en un servicio de farmacia hospitalaria y demás estructuras asistenciales que, a la entrada en vigor de la Ley 29/2006, de 26 de julio, tuvieran intereses económicos directos en laboratorios farmacéuticos autorizados, podrán mantener esos intereses hasta la extinción de la autorización o transferencia del laboratorio. Asimismo, los farmacéuticos relacionados en el párrafo anterior que formen parte o que puedan entrar a formar parte de cooperativas con un mínimo de 20 cooperativistas o de sociedades mercantiles con un mínimo de 100 accionistas o socios, conformadas en ambos casos exclusivamente por los citados farmacéuticos y ya existentes a la entrada en vigor de la Ley 29/2006, de 26 de julio, podrán participar en éstas hasta su disolución, siempre que la misma no conlleve un posible conflicto de intereses. Disposición transitoria tercera. Régimen transitorio para la identificación automática de cada unidad de medicamento a lo largo de su recorrido. En tanto no se fije reglamentariamente el mecanismo que permita la identificación automática de cada unidad de medicamento a lo largo de su recorrido, de conformidad con lo dispuesto en el artículo 90, los laboratorios farmacéuticos y las entidades de distribución deberán comunicar puntualmente al Ministerio de Sanidad, Servicios Sociales e Igualdad, en los términos que establezca el Ministerio mediante resolución, los datos del lote y del número de unidades vendidas o suministradas, así como las que sean objeto de devolución, en territorio nacional, especificando el destinatario, tanto se trate de oficinas de farmacia o servicios de farmacia o de otras entidades de distribución. Disposición final primera. Título competencial. Esta ley se dicta al amparo de los siguientes títulos competenciales del Estado: 1. El título preliminar; los títulos I; II, excepto el artículo 38; III; IV, excepto los artículos 67 a 70 de su capítulo II; V; VI; los artículos 77.2, 78, 80 a 82, 84 y 87 a 90 del título VII; los artículos 93 a 99 del título VIII; y IX; las disposiciones adicionales primera, segunda, séptima, octava, decimotercera, decimoquinta y decimosexta así como las disposiciones transitorias, tienen la condición de legislación sobre productos farmacéuticos y se dictan al amparo del artículo 149.1.16.ª de la Constitución. 2. Los artículos 38, 67 a 70, 77.1, 3, 4 y 5, 79, 83, 85, 86, 100 y 104 a 107 y las disposiciones adicionales tercera, cuarta, novena a duodécima y decimocuarta, tienen la condición de normativa básica y se dictan al amparo del artículo 149.1.16.ª de la Constitución, que atribuye al Estado competencia exclusiva en materia de bases y coordinación general de la sanidad. 3. Los artículos 91, 92 y 101 a 103, así como las disposiciones adicionales quinta y sexta, se dictan al amparo del artículo 149.1.17.ª de la Constitución, que atribuye al Estado la competencia exclusiva en materia de régimen económico de la Seguridad Social. 4. El título X se dicta al amparo del artículo 149.1.6.ª de la Constitución, que atribuye al Estado competencia exclusiva en materia de legislación procesal. 5. El título XI se dicta al amparo del artículo 149.1.14.ª de la Constitución, que atribuye al Estado competencia exclusiva en materia de Hacienda General. Disposición final segunda. Desarrollo normativo. Se autoriza al Gobierno, en el ámbito de sus competencias, para que apruebe los reglamentos y normas para la aplicación y desarrollo de esta ley. – 88 – CÓDIGO DE DERECHO FARMACÉUTICO §2 Ley 29/2006, de 26 de julio, de garantías y uso racional de los medicamentos y productos sanitarios. [Inclusión parcial] Jefatura del Estado «BOE» núm. 178, de 27 de julio de 2006 Última modificación: 25 de julio de 2015 Referencia: BOE-A-2006-13554 Norma derogada, excepto las disposiciones finales 2, 3 y 4, por la disposición derogatoria única del Real Decreto Legislativo 1/2015, de 24 de julio. Ref. BOE-A-2015-8343. [...] Disposición final segunda. Modificación de la Ley de Patentes. Se modifica el artículo 52.1 de la Ley 11/1986, de 20 de marzo, de Patentes, mediante la siguiente redacción del párrafo b) del apartado 1: «b) A los actos realizados con fines experimentales que se refieran al objeto de la invención patentada, en particular los estudios y ensayos realizados para la autorización de medicamentos genéricos, en España o fuera de España, y los consiguientes requisitos prácticos, incluidos la preparación, obtención y utilización del principio activo para estos fines.» Disposición final tercera. Animal. Modificación de la Ley 8/2003, de 24 de abril, de Sanidad Se modifica el artículo 63 de la Ley 8/2003, de 24 de abril, de Sanidad Animal, que tendrá la siguiente redacción: «En el caso de los productos biológicos, cuando sea necesario por interés de la sanidad animal, el Ministerio de Agricultura, Pesca y Alimentación podrá someter a control oficial los lotes de productos antes de su comercialización, en los términos que reglamentariamente se determine.» Disposición final cuarta. Agencia de Evaluación de Tecnologías Sanitarias. Se da nueva redacción al apartado 2 del artículo 21 de la Ley 16/2003, de 28 de mayo, de cohesión y calidad del Sistema Nacional de Salud, en los siguientes términos: «Las nuevas técnicas, tecnologías o procedimientos serán sometidas a evaluación, con carácter previo a su utilización en el Sistema Nacional de Salud, por – 89 – CÓDIGO DE DERECHO FARMACÉUTICO § 2 Ley de garantías y uso racional de los medicamentos y productos sanitarios [parcial] el Ministerio de Sanidad y Consumo, a través de la Agencia de Evaluación de Tecnologías Sanitarias del Instituto de Salud Carlos III, que se realizará en colaboración con otros órganos evaluadores propuestos por las Comunidades Autónomas, en los términos previstos reglamentariamente.» [...] – 90 – CÓDIGO DE DERECHO FARMACÉUTICO §3 Ley 14/1986, de 25 de abril, General de Sanidad. [Inclusión parcial] Jefatura del Estado «BOE» núm. 102, de 29 de abril de 1986 Última modificación: 6 de diciembre de 2018 Referencia: BOE-A-1986-10499 [...] TÍTULO V De los productos farmacéuticos CAPÍTULO ÚNICO Artículo noventa y cinco. 1. Corresponde a la Administración Sanitaria del Estado valorar la idoneidad sanitaria de los medicamentos y demás productos y artículos sanitarios, tanto para autorizar su circulación y uso como para controlar su calidad. 2. Para la circulación y uso de los medicamentos y productos sanitarios que se les asimilen, se exigirá autorización previa. Para los demás productos y artículos sanitarios se podrá exigir autorización previa individualizada o el cumplimiento de condiciones de homologación. No podrán prescribirse y se reputará clandestina la circulación de medicamentos o productos sanitarios no autorizados u homologados, con las responsabilidades administrativas y penales a que hubiere lugar. 3. Sólo se autorizarán medicamentos seguros y eficaces con la debida calidad y pureza y elaborados por persona física o jurídica con capacidad suficiente. 4. El procedimiento de autorización asegurará que se satisfacen las garantías de eficacia, tolerancia, pureza, estabilidad e información que marquen la legislación sobre medicamentos y demás disposiciones que sean de aplicación. En especial se exigirá la realización de ensayos clínicos controlados. 5. Todas las personas calificadas que presten sus servicios en los Servicios sanitarios y de investigación y de desarrollo tecnológico públicos tienen el derecho de participar y el deber de colaborar en la evaluación y control de medicamentos y productos sanitarios. – 91 – CÓDIGO DE DERECHO FARMACÉUTICO § 3 Ley General de Sanidad [parcial] Artículo noventa y seis. 1. La autorización de los medicamentos y demás productos sanitarios será temporal y, agotada su vigencia, deberá revalidarse. El titular deberá notificar anualmente su intención de mantenerlos en el mercado para que no se extinga la autorización. 2. La autoridad sanitaria podrá suspenderla o revocada por causa grave de salud pública. Artículo noventa y siete. La Administración Sanitaria del Estado, de acuerdo con los tratados internacionales de los que España sea parte, otorgará a los medicamentos una denominación oficial española adaptada a las denominaciones comunes internacionales de la Organización Mundial de la Salud, que será de dominio público y lo identificará apropiadamente en la información a ellos referida y en sus embalajes, envases y etiquetas. Las marcas comerciales no podrán confundirse ni con las denominaciones oficiales españolas ni con las comunes internacionales. Artículo noventa y ocho. 1. El Gobierno codificará las normas de calidad de los medicamentos obligatorias en España. 2. El Formulario Nacional contendrá las directrices según las cuales se prepararán, siempre con sustancias de acción e indicación reconocidas, las fórmulas magistrales por los farmacéuticos en sus oficinas de farmacia. Artículo noventa y nueve. Los importadores, fabricantes y profesionales sanitarios tienen la obligación de comunicar los efectos adversos causados por medicamentos y otros productos sanitarios, cuando de ellos pueda derivarse un peligro para la vida o salud de los pacientes. Artículo ciento. 1. La Administración del Estado exigirá la licencia previa a las personas físicas o jurídicas que se dediquen a la importación, elaboración, fabricación, distribución o exportación de medicamentos y otros productos sanitarios y a sus laboratorios y establecimientos. Esta licencia habrá de revalidarse periódicamente. Lo anterior se entenderá sin perjuicio de las competencias de las Comunidades Autónomas en relación con los establecimientos y las actividades de las personas físicas o jurídicas que se dediquen a la fabricación de productos sanitarios a medida. En todo caso los criterios para el otorgamiento de la licencia previa serán elaborados por el Ministerio de Sanidad y Consumo. 2. La Administración del Estado establecerá normas de elaboración, fabricación, transporte y almacenamiento. 3. Los laboratorios fabricantes y los mayoristas contarán con un Director Técnico, Farmacéutico o Titulado Superior suficientemente cualificado, de acuerdo con las directivas farmacéuticas de la Comunidad Económica Europea. Artículo ciento uno. 1. La licencia de los medicamentos y demás productos sanitarios y de las entidades a que se refiere el artículo 96, a su otorgamiento y anualmente, devengarán las tasas necesarias para cubrir los costes de su evaluación y control. Para evitar solicitudes especulativas de licencias, modificaciones y revalidaciones periódicas, la Administración podrá exigir fianza antes de su admisión a trámite. 2. En la determinación del importe de las tasas y fianzas se tendrán en cuenta reglas objetivas tendentes a estimular la comercialización de medicamentos y productos sanitarios peculiares, para dar acceso al mercado a las Empresas medianas y pequeñas, por razones de política industrial, o para fomentar el empleo. – 92 – CÓDIGO DE DERECHO FARMACÉUTICO § 3 Ley General de Sanidad [parcial] Artículo ciento dos. 1. La información y promoción de los medicamentos y los productos sanitarios dirigida a los profesionales se ajustará a las condiciones técnicas y científicas autorizadas del producto y cumplirá con las exigencias y controles previstos en el artículo 76 de la Ley 29/2006, de 26 de julio, de garantías y uso racional de los medicamentos y productos sanitarios. 2. La publicidad de productos sanitarios dirigida al público requerirá la autorización previa de los mensajes por la autoridad sanitaria. Se procederá a revisar el régimen de control de la publicidad de los productos sanitarios atendiendo a su posible simplificación sin menoscabo de las garantías de protección de la salud pública que ofrece el régimen actual. Artículo ciento tres. 1. La custodia, conservación y dispensación de medicamentos corresponderá: a) A las oficinas de farmacia legalmente autorizadas. b) A los servicios de farmacia de los hospitales, de los Centros de Salud y de las estructuras de Atención Primaria del Sistema Nacional de Salud para su aplicación dentro de dichas instituciones o para los que exijan una particular vigilancia, supervisión y control del equipo multidisciplinario de atención a la salud. 2. Las oficinas de farmacia abiertas al público se consideran establecimientos sanitarios a los efectos previstos en el título IV de esta Ley. 3. Las oficinas de farmacia estarán sujetas a la planificación sanitaria en los términos que establezca la legislación especial de medicamentos y farmacias. 4. Sólo los farmacéuticos podrán ser propietarios y titulares de las oficinas de farmacia abiertas al público. [...] Artículo ciento seis. Seguimiento de la sostenibilidad del gasto farmacéutico y sanitario de las Comunidades Autónomas. Las Comunidades Autónomas remitirán periódicamente al Ministerio de Hacienda y Administraciones Públicas, para su seguimiento y publicación a través de la Central de Información Económico-Financiera de las Administraciones Públicas, de acuerdo con lo que se prevea en la Orden HAP/2105/2012, de 1 de octubre, por la que se desarrollan las obligaciones de suministro de información previstas en la Ley Orgánica 2/2012, de 27 de abril, con el principio de transparencia previsto en el artículo 6 de la Ley Orgánica 2/2012, de 27 de abril: a) Los datos relativos a su gasto farmacéutico hospitalario, su gasto en productos farmacéuticos y sanitarios por recetas médicas u orden de dispensación y su gasto en productos sanitarios sin receta médica u orden de dispensación, de acuerdo con lo previsto en esta Ley. b) Datos relativos al gasto en inversiones reales en el ámbito sanitario, especialmente en relación a equipos de alta tecnología sanitaria de uso hospitalario, así como otros datos significativos en relación al gasto sanitario. c) Las medidas adoptadas, así como su grado de avance, para mejorar la eficiencia y sostenibilidad del sistema sanitario. Artículo ciento siete. Delimitación del gasto farmacéutico. A los efectos previstos en este Título, se entiende por gasto farmacéutico la suma del gasto en productos farmacéuticos y sanitarios, derivado de la expedición de la receta oficial u orden de dispensación del Sistema Nacional de Salud en oficinas de farmacia, y del gasto farmacéutico hospitalario por suministro de medicamentos a hospitales del Sistema Nacional de Salud. – 93 – CÓDIGO DE DERECHO FARMACÉUTICO § 3 Ley General de Sanidad [parcial] Artículo ciento ocho. Delimitación del gasto farmacéutico hospitalario. Se entiende por gasto farmacéutico hospitalario el gasto devengado por las unidades clasificadas como Administración Pública en términos de contabilidad nacional derivado de medicamentos financiados con fondos públicos en los hospitales y centros de atención sanitaria y sociosanitaria del Sistema Nacional de Salud. Artículo ciento nueve. Delimitación del gasto en productos farmacéuticos y sanitarios por recetas médicas u orden de dispensación. Se entiende por gasto en productos farmacéuticos y sanitarios por recetas médicas u orden de dispensación el gasto devengado por las unidades clasificadas como Administración Pública en términos de contabilidad nacional derivado de medicamentos y/o productos sanitarios que, financiados con fondos públicos, se dispensen en oficinas de farmacia a través de receta oficial u orden de dispensación del Sistema Nacional de Salud en territorio nacional. Artículo ciento diez. Delimitación del gasto en productos sanitarios sin receta médica u orden de dispensación. Se entiende por gasto en productos sanitarios sin receta médica u orden de dispensación el gasto devengado por las unidades clasificadas como Administración Pública en términos de contabilidad nacional derivado de la adquisición de los productos previstos en el artículo 2, apartado 1, letras a) a e) del Real Decreto 1591/2009, de 16 de octubre, por el que se regulan los productos sanitarios, siempre que no tengan la condición de bienes de capital o de naturaleza inventariable, por quedar los mismos registrados en los gastos o presupuestos de capital de las correspondientes entidades, ni hayan sido dispensados en oficinas de farmacia a través de receta oficial u orden de dispensación del Sistema Nacional de Salud. Artículo ciento once. sanitario. Medidas para mejorar la eficiencia y sostenibilidad del sistema Por Acuerdo de la Comisión Delegada del Gobierno para Asuntos Económicos, que se publicará en el «Boletín Oficial del Estado», se aprobará un conjunto de medidas que contribuyan a mejorar la sostenibilidad y eficiencia del gasto farmacéutico y sanitario para que puedan ser adoptadas por aquellas Comunidades Autónomas que así lo consideren. Artículo ciento doce. Incumplimiento de la obligación de remisión de información. Sin perjuicio de la posible responsabilidad personal que corresponda, el incumplimiento de las obligaciones de remisión de información a las que se refiere este título, en lo referido a los plazos establecidos, al correcto contenido e idoneidad de los datos requeridos o al modo de envío, dará lugar a un requerimiento de cumplimiento. El requerimiento de cumplimiento indicará el plazo, no superior a quince días naturales, para atender la obligación incumplida con apercibimiento de que transcurrido el mencionado plazo se procederá a dar publicidad al incumplimiento y a la adopción de las medidas automáticas de corrección previstas en el artículo 20 de la Ley Orgánica 2/2012, de 27 de abril, de conformidad con lo establecido en el artículo 27.7 de la mencionada Ley. Artículo ciento trece. Creación del instrumento de apoyo a la sostenibilidad del gasto farmacéutico y sanitario. 1. Se crea un instrumento de apoyo a la sostenibilidad del gasto farmacéutico y sanitario de las Comunidades Autónomas, con vigencia durante 2015, salvo que por Acuerdo de la Comisión Delegada del Gobierno para Asuntos Económicos se decida prorrogar este plazo. 2. Para adherirse a este instrumento la Comunidad Autónoma adoptará un Acuerdo de Consejo de Gobierno en el que conste su voluntad de adhesión a este instrumento y su compromiso de cumplir con lo previsto en este título. – 94 – CÓDIGO DE DERECHO FARMACÉUTICO § 3 Ley General de Sanidad [parcial] Artículo ciento catorce. Límites de gasto sanitario. 1. Cuando una Comunidad Autónoma se haya adherido a este instrumento la variación interanual, a ejercicio cerrado, del gasto farmacéutico, tanto hospitalario como en productos farmacéuticos y sanitarios por recetas médicas u orden de dispensación, y del gasto en productos sanitarios sin receta médica u orden de dispensación no podrá ser superior a la tasa de referencia de crecimiento del Producto Interior Bruto de medio plazo de la economía española prevista en el artículo 12.3 de la Ley Orgánica 2/2012, de 27 de abril, de acuerdo con el último informe elaborado por el Ministerio de Economía y Competitividad y publicado en la Central de Información Económico-Financiera de las Administraciones Públicas. 2. Publicada, a cierre del ejercicio, la información referida al gasto farmacéutico hospitalario, al gasto en productos farmacéuticos y sanitarios por recetas médicas u orden de dispensación, y al gasto en productos sanitarios sin receta médica u orden de dispensación a la que se refieren los artículos 107 a 110, la Comisión Delegada del Gobierno para Asuntos Económicos evaluará el grado de cumplimiento de lo previsto en el apartado 1. Anualmente en el Consejo interterritorial del Sistema Nacional de Salud se informará sobre el grado de cumplimiento previsto en el párrafo anterior. Artículo ciento quince. Consecuencias de la superación del límite de gasto farmacéutico o del gasto en productos sanitarios. Cuando el gasto farmacéutico o el gasto en productos sanitarios sin receta médica u orden de dispensación de una Comunidad Autónoma adherida a este instrumento supere el límite previsto en el artículo 114: a) La Comunidad Autónoma no podrá aprobar la cartera de servicios complementaria de acuerdo con lo previsto en el artículo 8 quinquies.tres de la Ley 16/2003, de 28 de mayo, de cohesión y calidad del Sistema Nacional de Salud y no podrán prestar servicios distintos de la cartera común de servicios del sistema nacional de salud. b) El acceso de la Comunidad Autónoma al reparto de recursos económicos que en materia sanitaria se realice por parte de la Administración General del Estado, estará sujeto al informe previsto en el artículo 20.3 de la Ley Orgánica 2/2012, de 27 de abril. c) La Comunidad Autónoma deberá aplicar las medidas de mejora de la eficiencia y sostenibilidad del sistema sanitario que sean acordadas por la Comisión Delegada del Gobierno para Asuntos Económicos. Artículo ciento dieciséis. Transparencia y sostenibilidad del gasto sanitario estatal. 1. El Instituto Nacional de Gestión Sanitaria, la Mutualidad General de Funcionarios Civiles del Estado, las Instituciones penitenciarias y la Mutualidad General Judicial deberán calcular y hacer público a través de la Central de Información Económico-Financiera de las Administraciones Públicas, dependiente del Ministerio de Hacienda y Administraciones Públicas, su gasto farmacéutico hospitalario, su gasto en productos farmacéuticos y sanitarios por recetas médicas u orden de dispensación y el gasto en productos sanitarios sin receta médica u orden de dispensación, de acuerdo con la delimitación definida en los artículos 108 a 110 de esta Ley. 2. La variación interanual del gasto farmacéutico, tanto hospitalario como en productos farmacéuticos y sanitarios por recetas médicas u orden de dispensación extrahospitalario, y del gasto en productos sanitarios sin receta médica u orden de dispensación no farmacéuticos del Instituto Nacional de Gestión Sanitaria, la Mutualidad General de Funcionarios Civiles del Estado y la Mutualidad General Judicial, no podrá ser superior a la tasa de referencia de crecimiento del Producto Interior Bruto de medio plazo de la economía española prevista en el artículo 12.3 de la Ley Orgánica 2/2012, de 27 de abril. 3. Cuando alguno de los sujetos citados en el apartado 2 supere el límite del gasto farmacéutico o el gasto en productos sanitarios sin receta médica u orden de dispensación previsto en el citado apartado 2, aplicará las medidas de mejora de la eficiencia y sostenibilidad del sistema sanitario que sean acordadas por la Comisión Delegada del Gobierno para Asuntos Económicos. – 95 – CÓDIGO DE DERECHO FARMACÉUTICO § 3 Ley General de Sanidad [parcial] [...] Sexta. Remisión de información y publicación del gasto farmacéutico y del gasto en productos sanitarios no farmacéuticos de las Comunidades Autónomas. 1. Mientras no se produzca la modificación de la Orden HAP/2105/2012, de 1 de octubre, las Comunidades Autónomas remitirán al Ministerio de Hacienda y Administraciones Públicas, para su publicación y seguimiento, antes del día 15 de cada mes, la información referida al mes anterior de su gasto farmacéutico hospitalario, de su gasto en productos farmacéuticos y sanitarios por recetas médicas u orden de dispensación y su gasto en productos sanitarios sin receta médica u orden de dispensación a la que se refiere los artículos 107 a 110, junto a la información relativa al mismo mes del ejercicio anterior. La información relativa al mes de diciembre de cada ejercicio se remitirá hasta el 31 de enero siguiente, siendo dicha información la base del cómputo del cumplimiento del límite establecido en el artículo 114. 2. La primera remisión de información mensual relativa al ejercicio 2015 se producirá el 30 de junio de 2015, comprensiva de los cinco primeros meses del ejercicio 2015, junto con los mismos meses de 2014. La publicación en la Central de Información EconómicoFinanciera de las Administraciones Públicas se producirá en el mes siguiente a la finalización del plazo para la remisión de los datos mensuales, salvo los datos relativos al cierre del ejercicio que se publicarán antes del 1 de abril. 3. Esta información será remitida por la intervención general o unidad equivalente que tenga competencias en materia de contabilidad por medios electrónicos a través de los modelos normalizados y sistema que el Ministerio de Hacienda y Administraciones Públicas habilite al efecto, y mediante firma electrónica avanzada basada en un certificado reconocido, de acuerdo con la Ley 59/2003, de 19 de diciembre, de firma electrónica, salvo en aquellos casos en los que el Ministerio de Hacienda y Administraciones Públicas considere que no es necesaria su utilización. [...] Decimosexta. Habilitación normativa. Por Orden Conjunta del Ministro de Hacienda y Administraciones Públicas y del Ministro de Sanidad, Servicios Sociales e Igualdad, por acuerdo de la Comisión Delegada del Gobierno para Asuntos Económicos, previa consulta a las Comunidades Autónomas, se podrá modificar lo previsto en los artículos 107 a 110 sobre la delimitación del gasto farmacéutico hospitalario, gasto en productos farmacéuticos y sanitarios por recetas médicas u orden de dispensación y gasto en productos sanitarios sin receta médica u orden de dispensación. [...] – 96 – CÓDIGO DE DERECHO FARMACÉUTICO §4 Ley 16/2003, de 28 de mayo, de cohesión y calidad del Sistema Nacional de Salud. [Inclusión parcial] Jefatura del Estado «BOE» núm. 128, de 29 de mayo de 2003 Última modificación: 30 de julio de 2018 Referencia: BOE-A-2003-10715 [...] CAPÍTULO II De la farmacia Sección 1.ª Organización y ejercicio de las competencias del estado en materia de farmacia Artículo 30. Competencias de la Administración General del Estado en materia de farmacia. Corresponde al Ministerio de Sanidad y Consumo el ejercicio de las competencias del Estado en materia de evaluación, registro, autorización, vigilancia y control de los medicamentos de uso humano y veterinario y de los productos sanitarios, así como la decisión sobre su financiación pública y la fijación del precio correspondiente, en los términos previstos en la Ley 25/1990, de 20 de diciembre, del Medicamento, sin perjuicio de las competencias ejecutivas de las comunidades autónomas. Artículo 31. Ejercicio de las competencias del Estado en materia de farmacia. 1. El ejercicio de las competencias del Estado en materia de farmacia corresponde al Ministerio de Sanidad y Consumo, a través de la Dirección General de Farmacia y Productos Sanitarios y del organismo autónomo Agencia Española de Medicamentos y Productos Sanitarios. 2. Corresponde a la Dirección General de Farmacia y Productos Sanitarios la dirección, desarrollo y ejecución de la política farmacéutica del departamento, el ejercicio de las funciones que competen al Estado en materia de financiación pública y fijación del precio de medicamentos y productos sanitarios, así como las condiciones especiales de prescripción y dispensación de medicamentos en el Sistema Nacional de Salud. 3. La Agencia Española de Medicamentos y Productos Sanitarios asume, como organismo técnico especializado, las actividades de evaluación, registro, autorización, inspección, vigilancia y control de medicamentos de uso humano y veterinario y productos sanitarios, cosméticos y de higiene personal, y la realización de los análisis económicos – 97 – CÓDIGO DE DERECHO FARMACÉUTICO § 4 Cohesión y calidad del Sistema Nacional de Salud [parcial] necesarios para la evaluación de estos productos, sin perjuicio de las competencias ejecutivas de las comunidades autónomas. 4. El Ministerio de Sanidad y Consumo, junto con las comunidades autónomas, acometerá acciones encaminadas al uso racional del medicamento que comprenderán entre otras: a) Programas de educación sanitaria dirigidos a la población general para la prevención de la automedicación, el buen uso de los medicamentos y la concienciación social e individual sobre su coste. b) Programas de formación continua de los profesionales, que les permita una constante incorporación de conocimientos sobre nuevos medicamentos y la actualización sobre la eficacia y efectividad de éstos. Artículo 32. Órganos de dirección, control y de asesoramiento técnico-científico de la Agencia Española de Medicamentos y Productos Sanitarios. 1. Los órganos de dirección de la Agencia Española de Medicamentos y Productos Sanitarios son el Consejo Rector y el Director de la Agencia. El Consejo Rector estará presidido por el Subsecretario de Sanidad y Consumo. Sus funciones, composición y régimen de funcionamiento se establecerán reglamentariamente. En todo caso, formarán parte del Consejo Rector representantes de los Ministerios de Sanidad y Consumo, de Agricultura, Pesca y Alimentación y de Ciencia y Tecnología, así como de las comunidades autónomas. La dirección y la representación legal de la Agencia Española de Medicamentos y Productos Sanitarios corresponden a su Director. Reglamentariamente se determinarán sus funciones. 2. La Agencia Española de Medicamentos y Productos Sanitarios contará con un Consejo Asesor integrado por expertos. Su composición, funciones y régimen de funcionamiento se determinarán reglamentariamente. 3. La Agencia Española de Medicamentos y Productos Sanitarios contará con los órganos de asesoramiento técnico-científico en materia de evaluación de medicamentos y productos sanitarios que se regulen en su estatuto. Sección 2.ª Colaboración de las oficinas de farmacia Artículo 33. Colaboración de las oficinas de farmacia. 1. Las oficinas de farmacia colaborarán con el Sistema Nacional de Salud en el desempeño de la prestación farmacéutica a fin de garantizar el uso racional del medicamento. Para ello los farmacéuticos actuarán coordinadamente con los médicos y otros profesionales sanitarios. 2. En el marco de la Ley 25/1990, de 20 de diciembre, del Medicamento, el Ministerio de Sanidad y Consumo, previo acuerdo del Consejo Interterritorial del Sistema Nacional de Salud, establecerá los criterios generales y comunes para el desarrollo de la colaboración de las oficinas de farmacia, por medio de conciertos que garanticen a los ciudadanos la dispensación en condiciones de igualdad efectiva en todo el territorio nacional, independientemente de su comunidad autónoma de residencia. Se tenderá a la dispensación individualizada de medicamentos y a la implantación de la receta electrónica, en cuyo desarrollo participarán las organizaciones colegiales médica y farmacéutica. 3. Entre los criterios del apartado anterior se definirán los datos básicos de farmacia, para la gestión por medios informáticos de la información necesaria para el desempeño de las actividades anteriormente mencionadas y para la colaboración con las estructuras asistenciales del Sistema Nacional de Salud. Se ajustarán a lo dispuesto en la Ley Orgánica 15/1999, de 13 de diciembre, de Protección de Datos de Carácter Personal, y a las especificaciones establecidas por los servicios de salud de las comunidades autónomas. [...] – 98 – CÓDIGO DE DERECHO FARMACÉUTICO §5 Ley 24/2015, de 24 de julio, de Patentes. [Inclusión parcial] Jefatura del Estado «BOE» núm. 177, de 25 de julio de 2015 Última modificación: 4 de julio de 2018 Referencia: BOE-A-2015-8328 [...] TÍTULO II Patentabilidad Artículo 4. Invenciones patentables. 1. Son patentables, en todos los campos de la tecnología, las invenciones que sean nuevas, impliquen actividad inventiva y sean susceptibles de aplicación industrial. Las invenciones a que se refiere el párrafo anterior podrán tener por objeto un producto compuesto de materia biológica o que contenga materia biológica, o un procedimiento mediante el cual se produzca, transforme o utilice materia biológica. 2. La materia biológica aislada de su entorno natural o producida por medio de un procedimiento técnico podrá ser objeto de una invención, aun cuando ya exista anteriormente en estado natural. 3. A los efectos de la presente Ley, se entenderá por «materia biológica» la materia que contenga información genética autorreproducible o reproducible en un sistema biológico y por «procedimiento microbiológico» cualquier procedimiento que utilice una materia microbiológica, que incluya una intervención sobre la misma o que produzca una materia microbiológica. 4. No se considerarán invenciones en el sentido de los apartados anteriores, en particular: a) Los descubrimientos, las teorías científicas y los métodos matemáticos. b) Las obras literarias, artísticas o cualquier otra creación estética, así como las obras científicas. c) Los planes, reglas y métodos para el ejercicio de actividades intelectuales, para juegos o para actividades económico-comerciales, así como los programas de ordenadores. d) Las formas de presentar informaciones. 5. Lo dispuesto en el apartado anterior excluye la patentabilidad de las materias o actividades mencionadas en el mismo solamente en la medida en que la solicitud de patente o la patente se refiera exclusivamente a una de ellas considerada como tal. – 99 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] Artículo 5. Excepciones a la patentabilidad. No podrán ser objeto de patente: 1. Las invenciones cuya explotación comercial sea contraria al orden público o a las buenas costumbres, sin que pueda considerarse como tal la explotación de una invención por el mero hecho de que esté prohibida por una disposición legal o reglamentaria. En particular, no se considerarán patentables en virtud de lo dispuesto en el párrafo anterior: a) Los procedimientos de clonación de seres humanos. b) Los procedimientos de modificación de la identidad genética germinal del ser humano. c) Las utilizaciones de embriones humanos con fines industriales o comerciales. d) Los procedimientos de modificación de la identidad genética de los animales que supongan para estos sufrimientos sin utilidad médica o veterinaria sustancial para el hombre o el animal, y los animales resultantes de tales procedimientos. 2. Las variedades vegetales y las razas animales. Serán, sin embargo, patentables las invenciones que tengan por objeto vegetales o animales si la viabilidad técnica de la invención no se limita a una variedad vegetal o a una raza animal determinada. 3. Los procedimientos esencialmente biológicos de obtención de vegetales o de animales. A estos efectos se considerarán esencialmente biológicos aquellos procedimientos que consistan íntegramente en fenómenos naturales como el cruce o la selección. Lo dispuesto en el párrafo anterior no afectará a la patentabilidad de las invenciones cuyo objeto sea un procedimiento microbiológico o cualquier otro procedimiento técnico o un producto obtenido por dichos procedimientos. 4. Los métodos de tratamiento quirúrgico o terapéutico del cuerpo humano o animal, y los métodos de diagnóstico aplicados al cuerpo humano o animal. Esta disposición no será aplicable a los productos, en particular a las sustancias o composiciones, ni a las invenciones de aparatos o instrumentos para la puesta en práctica de tales métodos. 5. El cuerpo humano en los diferentes estadios de su constitución y desarrollo, así como el simple descubrimiento de uno de sus elementos, incluida la secuencia total o parcial de un gen. Sin embargo un elemento aislado del cuerpo humano u obtenido de otro modo mediante un procedimiento técnico, incluida la secuencia o la secuencia parcial de un gen, podrá considerarse como una invención patentable, aun en el caso de que la estructura de dicho elemento sea idéntica a la de un elemento natural. La aplicación industrial de una secuencia total o parcial de un gen deberá figurar explícitamente en la solicitud de patente. 6. Una mera secuencia de ácido desoxirribonucleico (ADN) sin indicación de función biológica alguna. Artículo 6. Novedad. 1. Se considera que una invención es nueva cuando no está comprendida en el estado de la técnica. 2. El estado de la técnica está constituido por todo lo que antes de la fecha de presentación de la solicitud de patente se ha hecho accesible al público en España o en el extranjero por una descripción escrita u oral, por una utilización o por cualquier otro medio. 3. Se entiende igualmente comprendido en el estado de la técnica el contenido de las solicitudes españolas de patentes o de modelos de utilidad, de solicitudes de patentes europeas que designen a España y de solicitudes de patente internacionales PCT que hayan entrado en fase nacional en España, tal como hubieren sido originariamente presentadas, cuya fecha de presentación sea anterior a la que se menciona en el apartado precedente y que hubieren sido publicadas en español en aquella fecha o lo sean en otra posterior. 4. Lo dispuesto en los apartados 2 y 3 no excluirá la patentabilidad de cualquier sustancia o composición comprendida en el estado de la técnica para ser usada en alguno de los métodos mencionados en el artículo 5.4 siempre que su utilización para cualquiera de esos métodos no esté comprendida en el estado de la técnica. – 100 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] 5. Lo dispuesto en los apartados 2 y 3 no excluirá la patentabilidad de una sustancia o composición de las señaladas en el apartado 4 para una utilización determinada en alguno de los métodos mencionados en el artículo 5.4 siempre que dicha utilización no esté comprendida en el estado de la técnica. Artículo 7. Divulgaciones inocuas. No se tomará en consideración para determinar el estado de la técnica una divulgación de la invención que, acaecida dentro de los seis meses anteriores a la fecha de presentación de la solicitud, haya sido consecuencia directa o indirecta: a) De un abuso evidente frente al solicitante o su causante. b) Del hecho de que el solicitante o su causante hubieren exhibido la invención en exposiciones oficiales u oficialmente reconocidas en el sentido del Convenio Relativo a Exposiciones Internacionales, firmado en París el 22 de noviembre de 1928 y revisado por última vez el 30 de noviembre de 1972. En este caso será preciso que el solicitante, al presentar la solicitud, declare que la invención ha sido realmente exhibida y que, en apoyo de su declaración, aporte el correspondiente certificado dentro del plazo y en las condiciones que se determinen reglamentariamente. Artículo 8. Actividad inventiva. 1. Se considera que una invención implica una actividad inventiva si aquélla no resulta del estado de la técnica de una manera evidente para un experto en la materia. 2. Si el estado de la técnica comprende documentos de los mencionados en el artículo 6.3 no serán tomados en consideración para decidir sobre la existencia de la actividad inventiva. Artículo 9. Aplicación industrial. Se considera que una invención es susceptible de aplicación industrial cuando su objeto puede ser fabricado o utilizado en cualquier clase de industria, incluida la agrícola. [...] TÍTULO IV Invenciones realizadas en el marco de una relación de empleo o de servicios Artículo 15. Invenciones pertenecientes al empresario. 1. Las invenciones realizadas por el empleado o prestador de servicios durante la vigencia de su contrato o relación de empleo o de servicios con el empresario que sean fruto de una actividad de investigación explícita o implícitamente constitutiva del objeto de su contrato pertenecen al empresario. 2. El autor de la invención no tendrá derecho a una remuneración suplementaria por su realización, excepto si su aportación personal a la invención y la importancia de la misma para el empresario exceden de manera evidente del contenido explícito o implícito de su contrato o relación de empleo. Artículo 16. Invenciones pertenecientes al empleado o prestador de servicios. Las invenciones en cuya realización no concurran las circunstancias previstas en el artículo 15.1 pertenecen al autor de las mismas. Artículo 17. Invenciones asumibles por el empresario. 1. No obstante lo dispuesto en el artículo 16, cuando el empleado realizase una invención relacionada con su actividad profesional en la empresa y en su obtención hubiesen – 101 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] influido predominantemente conocimientos adquiridos dentro de la empresa o la utilización de medios proporcionados por ésta, el empresario tendrá derecho a asumir la titularidad de la invención o a reservarse un derecho de utilización de la misma. 2. Cuando el empresario asuma la titularidad de una invención o se reserve un derecho de utilización de la misma, el empleado tendrá derecho a una compensación económica justa fijada en atención a la importancia industrial y comercial del invento y teniendo en cuenta el valor de los medios o conocimientos facilitados por la empresa y las aportaciones propias del empleado. Dicha compensación económica podrá consistir en una participación en los beneficios que obtenga la empresa de la explotación o de la cesión de sus derechos sobre dicha invención. Artículo 18. empleado. Deber de información y ejercicio de los derechos por el empresario y el 1. El empleado que realice alguna de las invenciones a que se refieren los artículos 15 y 17 deberá informar de ello al empresario mediante comunicación escrita, con los datos e informes necesarios para que éste pueda ejercitar los derechos que le correspondan. Esta comunicación deberá realizarse en el plazo de un mes a contar desde la fecha en que se haya concluido la invención. El incumplimiento de esta obligación llevará consigo la pérdida de los derechos que se reconocen al empleado en este Título. 2. Cuando se trate una invención asumible por el empresario de acuerdo con lo dispuesto en el artículo 17, el empresario, en el plazo de tres meses contados a partir del día siguiente al de la recepción de la comunicación a que se refiere el apartado anterior, deberá evaluar la invención de que se trate y comunicar por escrito al empleado su voluntad de asumir la titularidad de la invención o de reservarse un derecho de utilización sobre la misma. Si el empresario, no comunica al empleado su voluntad de asumir la titularidad de la invención en los plazos previstos caducará su derecho, pudiendo el empleado presentar la solicitud de patente. Si el empresario, habiendo comunicado al empleado su voluntad de asumir la titularidad de la invención, no presentase la solicitud de propiedad industrial dentro de un plazo adicional razonable fijado con el empleado, podrá este presentar la solicitud de patente en nombre y por cuenta del empresario. 3. Las mejoras técnicas no patentables obtenidas por el empleado en el desarrollo de las actividades previstas en los artículos 15 y 17 que mediante su explotación como secreto industrial ofrezcan al empleador una posición ventajosa similar a la obtenida a partir de un derecho de propiedad industrial, darán derecho a reclamar del empleador una compensación razonable fijada de acuerdo con los criterios establecidos en los artículos citados tan pronto como este último explote la propuesta. 4. Tanto el empresario como el empleado deberán prestar su colaboración en la medida necesaria para la efectividad de los derechos reconocidos en este Título, absteniéndose de cualquier actuación que pueda redundar en detrimento de tales derechos. Artículo 19. Carga de la prueba y renuncia de derechos. 1. Salvo prueba en contrario, las invenciones para las que se presente una solicitud de patente o de otro título de protección exclusiva dentro del año siguiente a la extinción de la relación de empleo o de servicios, se presumen realizadas durante la vigencia de ésta. 2. Será nula toda renuncia anticipada del empleado a los derechos que la Ley le otorga en este Título. Artículo 20. Ámbito de aplicación. Las normas del presente Título serán asimismo aplicables a los funcionarios, empleados y trabajadores del Estado, Comunidades Autónomas, Provincias, Municipios y demás Entes Públicos, sin perjuicio de lo previsto en el artículo siguiente. – 102 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] Artículo 21. Invenciones realizadas por el personal investigador de las Universidades Públicas y de los Entes Públicos de Investigación. 1. Las invenciones realizadas por el personal investigador de los Centros y Organismos Públicos de Investigación de la Administración General del Estado, de los Centros y Organismos de investigación de otras Administraciones Públicas, de las Universidades Públicas, de las Fundaciones del Sector Público Estatal y de las Sociedades Mercantiles Estatales pertenecerán a las entidades cuyos investigadores las hayan obtenido en el ejercicio de las funciones que les son propias, cualquiera que sea la naturaleza de la relación jurídica por la que estén vinculados a ellas. A estos efectos se considera en todo caso personal investigador el definido como tal en el artículo 13 de la Ley 14/2011, de 1 de junio, de la Ciencia, la Tecnología y la Innovación, el personal técnico considerado en dicha Ley como personal de investigación y el personal técnico de apoyo que, conforme a la normativa interna de las universidades y de los centros de investigación, también tenga la consideración de personal de investigación. 2. Las invenciones contempladas en el apartado 1 deberán ser comunicadas por escrito a la entidad pública a cuyo servicio se halle el investigador autor de la misma en el plazo de tres meses desde la conclusión de la invención. La falta de comunicación por parte del personal investigador llevará consigo la pérdida de los derechos que se le reconocen en los apartados siguientes. 3. El organismo o la entidad pública, en el plazo de tres meses contados desde la recepción de la notificación a que se refiere el apartado precedente, deberán comunicar por escrito al autor o autores de la invención su voluntad de mantener sus derechos sobre la invención, solicitando la correspondiente patente, o de considerarla como secreto industrial reservándose el derecho de utilización sobre la misma en exclusiva. No podrá publicarse el resultado de una investigación susceptible de ser patentada antes de que transcurra dicho plazo o hasta que la entidad o el autor hayan presentado la solicitud de patente. Si el organismo o entidad pública no comunica en el plazo indicado su voluntad de mantener sus derechos sobre la invención, el autor o autores de la misma podrán presentar la solicitud de patente de acuerdo con lo previsto en el artículo 18.2. 4. El investigador tendrá en todo caso derecho a participar en los beneficios que obtengan las entidades en las que presta sus servicios de la explotación o de la cesión de sus derechos sobre dichas invenciones, cuando la patente se solicite a nombre de la entidad o se decida el secreto industrial. Estas entidades podrán también ceder la titularidad de dichas invenciones al autor de las mismas, reservándose una licencia no exclusiva, intransferible y gratuita de explotación o una participación de los beneficios que se obtengan de la explotación de esas invenciones determinada de conformidad con lo dispuesto en los apartados 6 y 7. 5. En los contratos o convenios que las entidades a que se refiere el apartado 1 celebren con entes públicos o privados, se deberá estipular a quién corresponderá la titularidad de las invenciones que el personal investigador pueda realizar en el marco de dichos contratos o convenios, así como todo lo relativo a los derechos de uso y explotación comercial y al reparto de los beneficios obtenidos. 6. El Consejo de Gobierno de la Universidad determinará las modalidades y la cuantía de la participación del personal investigador de la universidad en los beneficios que se obtengan con la explotación de las invenciones contempladas en este artículo, y en su caso, de la participación de la Universidad en los beneficios obtenidos por el investigador con la explotación de las mismas, sin perjuicio de lo establecido en el artículo 83 de la Ley Orgánica 6/2001, de 21 de diciembre, de Universidades y en el artículo 64 de la Ley 2/2011, de 4 de marzo, de Economía Sostenible. 7. Las modalidades y cuantía de la participación del personal investigador de los Entes Públicos de Investigación en los beneficios que se obtengan de la explotación o cesión de las invenciones contempladas en este artículo se establecerán por el Gobierno atendiendo a las características concretas de cada Ente Público de Investigación. Esta participación no tendrá en ningún caso naturaleza retributiva o salarial. Las Comunidades Autónomas podrán desarrollar por vía reglamentaria regímenes específicos de participación en beneficios para el personal investigador de Entes Públicos de Investigación de su competencia. – 103 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] TÍTULO V Solicitud y procedimiento de concesión CAPÍTULO I Presentación y requisitos de la solicitud de patente Artículo 22. Presentación de la solicitud. 1. La solicitud de patente se presentará en la Oficina Española de Patentes y Marcas o en el órgano competente de cualquier Comunidad Autónoma. 2. La solicitud de patente también podrá presentarse en los lugares previstos en el artículo 38.4 de la Ley 30/1992, de 26 de noviembre, de Régimen Jurídico de las Administraciones Públicas y del Procedimiento Administrativo Común, dirigida a cualquiera de los órganos que, de conformidad con el apartado precedente, son competentes para recibir la solicitud. 3. A la presentación electrónica de solicitudes será de aplicación la Ley 11/2007, de 22 de junio, de acceso electrónico de los ciudadanos a los Servicios Públicos. Artículo 23. Requisitos de la solicitud. 1. La solicitud de patente deberá contener: a) Una instancia de solicitud, según el modelo oficial, dirigida al Director de la Oficina Española de Patentes y Marcas. b) Una descripción de la invención para la que se solicita la patente. c) Una o varias reivindicaciones. d) Los dibujos a los que se refieran la descripción o las reivindicaciones y, en su caso, las secuencias biológicas presentadas en el formato que se establezca reglamentariamente. e) Un resumen de la invención. 2. Cuando la invención se refiera a materia biológica de origen vegetal o animal la solicitud deberá incluir la mención de su origen geográfico o la fuente de procedencia de dicha materia si estos datos fueran conocidos. Esta información no prejuzgará la validez de la patente. En los supuestos previstos en el Reglamento (UE) n.º 511/2014 del Parlamento Europeo y del Consejo, de 16 de abril de 2014, relativo a las medidas de cumplimiento de los usuarios del Protocolo de Nagoya sobre el acceso a los recursos genéticos y participación justa y equitativa en los beneficios que se deriven de su utilización en la Unión, la solicitud de patente deberá asimismo contener, en la medida en que reglamentariamente se determine, la información que los usuarios de tales recursos vienen obligados a conservar con arreglo a lo previsto en la norma citada. La referida información tampoco prejuzgará la validez de la patente. 3. Tanto la solicitud como los demás documentos que hayan de presentarse en la Oficina Española de Patentes y Marcas deberán estar redactados en castellano y cumplir los requisitos que se establezcan reglamentariamente. Sin perjuicio de lo dispuesto en el artículo 24.1.c), en las Comunidades Autónomas donde exista otra lengua oficial, tales documentos podrán redactarse en dicha lengua debiendo ir acompañados de la correspondiente traducción al castellano, que se considerará auténtica en caso de duda entre ambas. 4. La presentación de la solicitud dará lugar al pago de la tasa correspondiente, así como de la tasa por realización del informe sobre el estado de la técnica. – 104 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] Artículo 24. Fecha de presentación. 1. La fecha de presentación de la solicitud será la del momento en que el solicitante entregue a las oficinas autorizadas para la recepción de solicitudes de patente la documentación que contenga al menos los siguientes elementos: a) La indicación de que se solicita una patente. b) Las informaciones que permitan identificar al solicitante y contactar con él. c) Una descripción de la invención para la que se solicita la patente, aunque no cumpla con los requisitos formales establecidos en la Ley, o la remisión a una solicitud presentada con anterioridad. A los efectos de obtención de una fecha de presentación, la descripción podrá redactarse en cualquier idioma debiendo presentarse la correspondiente traducción al castellano en el plazo reglamentariamente establecido. 2. La remisión a una solicitud anterior debe indicar el número de ésta, su fecha de presentación y la oficina en la que haya sido presentada. En la referencia habrá de hacerse constar que la misma sustituye a la descripción y, en su caso, a los dibujos. 3. Si la solicitud se remite a una anterior según lo previsto en el apartado precedente deberá presentarse una copia certificada de la solicitud anterior, acompañada, en su caso, de la correspondiente traducción al castellano, en el plazo fijado en el reglamento de ejecución. 4. La fecha de presentación de las solicitudes depositadas en una oficina de correos será la del momento en que dicha oficina reciba la documentación que contenga los elementos mencionados en los apartados anteriores, siempre que sean presentados por correo certificado en la forma prevista por el artículo 31 del Reglamento por el que se regula la prestación de los servicios postales, aprobado por Real Decreto 1829/1999, de 3 de diciembre. En todo caso la documentación deberá estar dirigida al órgano competente para recibir la solicitud. Artículo 25. Designación del inventor. La solicitud deberá designar al inventor. En el caso de que el solicitante no sea el inventor o no sea el único inventor, la designación deberá ir acompañada de una declaración en la que se exprese cómo ha adquirido el solicitante el derecho a la patente. Artículo 26. Unidad de invención. 1. La solicitud de patente no podrá comprender más que una sola invención o un grupo de invenciones relacionadas entre sí de tal manera que integren un único concepto inventivo general. 2. Las solicitudes que no cumplan lo dispuesto en el apartado anterior habrán de ser divididas de acuerdo con lo que se disponga reglamentariamente. 3. Las solicitudes divisionales tendrán la misma fecha de presentación que la solicitud inicial de la que procedan, en la medida en que su objeto estuviere ya contenido en aquella solicitud. Artículo 27. Descripción de la Invención. 1. La invención debe ser descrita en la solicitud de patente de manera suficientemente clara y completa para que un experto sobre la materia pueda ejecutarla. 2. Cuando la invención se refiera a una materia biológica no accesible al público, o a su utilización, y cuando la materia biológica no pueda ser descrita en la solicitud de patente de manera tal que un experto pueda reproducir la invención, sólo se considerará que la descripción cumple con lo dispuesto en el apartado anterior si, concurren los siguientes requisitos, tal como hayan sido desarrollados reglamentariamente: a) Que la materia biológica haya sido depositada no más tarde de la fecha de presentación de la solicitud de patente en una institución reconocida legalmente para ello, en condiciones iguales a las establecidas por el Tratado de Budapest, sobre el reconocimiento – 105 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] internacional del depósito de microorganismos a los fines del procedimiento en materia de patentes, hecho en Budapest el 28 de abril de 1977 (en lo sucesivo Tratado de Budapest). En todo caso, se considerarán reconocidas las autoridades internacionales de depósito que hayan adquirido dicho rango de conformidad con el artículo 7 de dicho Tratado. b) Que la solicitud, tal como ha sido presentada, contenga la información relevante de que disponga el solicitante sobre las características de la materia biológica depositada. c) Que de conformidad con lo previsto en el Reglamento, se indique el nombre de la institución de depósito y el número del mismo. 3. Si la materia biológica depositada de acuerdo con lo previsto en el apartado anterior, dejase de estar disponible en la institución de depósito reconocida, se autorizará un nuevo depósito de esa materia, en condiciones análogas a las previstas en el Tratado de Budapest. 4. Todo nuevo depósito deberá ir acompañado de una declaración firmada por el depositante que certifique que la materia biológica objeto del nuevo depósito es la misma que se depositó inicialmente. Artículo 28. Reivindicaciones. Las reivindicaciones definen el objeto para el que se solicita la protección. Deben ser claras y concisas y han de fundarse en la descripción. Artículo 29. Resumen. El resumen de la invención servirá exclusivamente para una finalidad de información técnica. No podrá ser tomado en consideración para ningún otro fin, y en particular no podrá ser utilizado ni para la determinación del ámbito de la protección solicitada, ni para delimitar el estado de la técnica a los efectos de lo dispuesto en el artículo 6.3. Artículo 30. Prioridad. 1. Quienes hayan presentado regularmente una primera solicitud de patente de invención, de modelo de utilidad, o de certificado de utilidad en o para alguno de los Estados parte en el Convenio de París para la Protección de la Propiedad Industrial, hecho en París el 20 de marzo de 1883 o del Acuerdo por el que se establece la Organización Mundial del Comercio, o sus causahabientes, gozarán, para la presentación de una solicitud de patente en España sobre la misma invención, de un derecho de prioridad de doce meses a partir de la fecha de presentación de dicha primera solicitud, nacional o extranjera, en las condiciones establecidas en el artículo 4 del Convenio de París. 2. Tendrán el mismo derecho de prioridad mencionado en el apartado anterior quienes hubieren presentado una primera solicitud de protección en o para un Estado no mencionado en el apartado 1 que reconozca a las solicitudes presentadas en España un derecho de prioridad en condiciones y con efectos equivalentes a los previstos en el Convenio de París. 3. En virtud del ejercicio del derecho de prioridad se considerará como fecha de presentación de la solicitud, a efectos de lo dispuesto en los artículos 6, 10.3 y 139, la fecha de presentación de la solicitud anterior cuya prioridad hubiere sido válidamente reivindicada. Artículo 31. Reivindicación de la prioridad. 1. El solicitante que desee reivindicar la prioridad de una solicitud anterior deberá presentar, en la forma y plazos que reglamentariamente se establezcan, una declaración de prioridad y una copia certificada por la oficina de origen de la solicitud anterior acompañada de su traducción al español cuando esté redactada en otro idioma. 2. No será sin embargo necesaria la presentación de la copia de la solicitud anterior ni de la traducción cuando la reivindicación de prioridad no se considere relevante para determinar la patentabilidad de la invención, o la solicitud anterior o su traducción obren ya en poder de la Oficina Española de Patentes y Marcas o estén disponibles en una biblioteca digital. 3. Para una misma solicitud y, en su caso, para una misma reivindicación podrán reivindicarse prioridades múltiples, aunque tengan su origen en Estados diferentes. Si se reivindican prioridades múltiples, los plazos que hayan de computarse a partir de la fecha de prioridad se contarán desde la fecha de prioridad más antigua. – 106 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] 4. Cuando se reivindiquen una o varias prioridades, el derecho de prioridad sólo amparará a los elementos de la solicitud que estuvieren contenidos en la solicitud o solicitudes cuya prioridad hubiere sido reivindicada. 5. Aun cuando determinados elementos de la invención para los que se reivindique la prioridad no figuren entre las reivindicaciones formuladas en la solicitud anterior, podrá otorgarse la prioridad para los mismos si el conjunto de los documentos de aquella solicitud anterior revela de manera suficientemente clara y precisa tales elementos. 6. La reivindicación de prioridad implicará el pago de la tasa correspondiente. CAPÍTULO II Procedimiento de Concesión Artículo 32. Recepción de la solicitud y remisión a la OEPM. 1. El órgano competente de acuerdo con lo previsto en el artículo 22 hará constar en el momento de recibir la solicitud su número de registro, el día, la hora y minuto de su presentación, y expedirá un recibo acreditativo o copia sellada de la documentación presentada en la forma que reglamentariamente se determine. Si no se hubiese hecho constar la hora, se le asignará la última del día. Si no se hubiese hecho constar el minuto, se le asignará el último de la hora. 2. Cuando el órgano competente para recibir la solicitud lo sea de una Comunidad Autónoma remitirá a la Oficina Española de Patentes y Marcas la documentación presentada dentro de los tres días siguientes al de su recepción. El incumplimiento de este plazo en ningún caso perjudicará al solicitante. Artículo 33. Establecimiento de fecha de presentación y admisión a trámite. 1. Dentro de los 10 días siguientes a su recepción, la Oficina Española de Patentes y Marcas comprobará si la solicitud de patente cumple los requisitos para que se le otorgue una fecha de presentación y, si es así, la admitirá a trámite y procederá de acuerdo con lo previsto en el artículo 34. 2. Si la faltara alguno de los requisitos necesarios para obtener fecha de presentación, se notificarán los defectos al interesado para que los subsane en el plazo establecido. La fecha de presentación será en ese caso la del momento en que la Oficina Española de Patentes y Marcas reciba la documentación con los defectos debidamente corregidos. Si los defectos no se subsanan en plazo, la solicitud no será admitida a trámite y así se comunicará, con indicación de los motivos, al solicitante. 3. Si las tasas de solicitud y la tasa por realización del informe sobre el estado de la técnica no hubieran sido abonadas con la solicitud, o no lo hubieren sido en su totalidad, se notificará esta circunstancia al solicitante para que realice o complete el pago en el plazo establecido. Transcurrido dicho plazo sin efectuar o completar el pago, se le tendrá por desistido de la solicitud. 4. Los plazos mencionados en los apartados anteriores son los fijados en el reglamento de ejecución de la presente Ley. Artículo 34. Patentes de interés para la defensa nacional. La Oficina Española de Patentes y Marcas pondrá a disposición del Ministerio de Defensa, a los efectos previstos en el Título XI de esta Ley, todas las solicitudes de patentes que puedan ser de interés para la defensa nacional, estableciendo para ello la necesaria coordinación con dicho Ministerio. Artículo 35. Examen de oficio. 1. Admitida a trámite la solicitud, la Oficina Española de Patentes y Marcas verificará: a) Si el objeto de la misma no está manifiestamente y en su totalidad excluido de la patentabilidad por aplicación de los artículos 4.4 y 5 de esta Ley. – 107 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] b) Si se cumplen los requisitos relativos a la representación y a la reivindicación de prioridad en su caso, así como cualquier otro referido a la regularidad formal de la solicitud cuya comprobación haya de realizarse, de acuerdo con lo establecido en el Reglamento, antes de la publicación de la solicitud. 2. La presencia de defectos formales en la documentación no suspenderá la realización del informe sobre el estado de la técnica a que se refiere el artículo siguiente, siempre que aquellos no sean de tal naturaleza que impidan su realización. 3. Si de la comprobación resulta que el objeto de la solicitud está excluido de patentabilidad conforme al apartado 1.a), o no se cumplen los requisitos mencionados en el apartado 1 b), se comunicarán éstas circunstancias al interesado para que formule sus alegaciones o subsane los defectos en el plazo establecido. Si los obstáculos persisten o los defectos no fueran corregidos en plazo, se denegará la solicitud mediante resolución motivada. Cuando los defectos se refieran al derecho de prioridad el solicitante perderá este derecho. Artículo 36. Emisión del informe sobre el estado de la técnica y de la opinión escrita. 1. La Oficina Española de Patentes y Marcas emitirá, tal como se establezca reglamentariamente, un informe sobre el estado de la técnica y una opinión escrita, preliminar y no vinculante, relativos a la solicitud de patente, realizados sobre la base de las reivindicaciones, teniendo debidamente en cuenta la descripción y, en su caso los dibujos o secuencias biológicas. Tanto del informe sobre el estado de la técnica como de la opinión escrita se dará traslado al solicitante. 2. El informe sobre el estado de la técnica se fundará en una búsqueda que, de acuerdo con lo previsto en el artículo 6.2 de la presente Ley, se extenderá a todo lo que se haya hecho accesible al público en España o en el extranjero por una descripción escrita u oral, por una utilización o por cualquier otro medio. 3. Cuando la falta de claridad o coherencia de la descripción o de las reivindicaciones impida proceder en todo o en parte a la elaboración del informe, la Oficina Española de Patentes y Marcas efectuará la oportuna notificación al solicitante para que formule sus alegaciones o subsane los defectos en el plazo reglamentariamente establecido. Para subsanar los defectos el solicitante podrá modificar las reivindicaciones. Si el solicitante no responde en el plazo establecido, o no precisa el objeto de la búsqueda en la medida suficiente para subsanar los defectos señalados, dicha Oficina realizará un informe sobre el estado de la técnica basado en una búsqueda parcial. Si ello no fuere posible, se denegará la solicitud mediante resolución motivada, y así se le notificará. 4. Si falta unidad de invención y el solicitante, a requerimiento de la Oficina Española de Patentes y Marcas, no divide su solicitud o no paga tasas adicionales, el procedimiento continuará para la invención o grupo de invenciones reivindicadas en primer lugar que cumplan las condiciones del artículo 26, teniéndole por desistido respecto de las restantes. 5. No serán objeto del informe sobre el estado de la técnica ni de la opinión escrita las solicitudes cuyo informe de búsqueda internacional haya sido realizado por la Oficina Española de Patentes y Marcas en su calidad de Administración encargada de la búsqueda internacional. Artículo 37. Publicación de la solicitud y del informe. 1. Transcurridos dieciocho meses desde la fecha de presentación de la solicitud o desde la fecha de prioridad que se hubiera reivindicado y superado el examen de oficio, la Oficina Española de Patentes y Marcas publicará lo antes posible la solicitud de patente, haciendo el correspondiente anuncio en el «Boletín Oficial de la Propiedad Industrial» y poniendo a disposición del público los documentos obrantes en el expediente de la solicitud de patente publicada. Asimismo, se editará un folleto que contendrá el texto de la descripción con las reivindicaciones y, en su caso, los dibujos o las secuencias biológicas, en la forma y con los elementos que se establezcan en el Reglamento de ejecución. 2. A petición del solicitante podrá publicarse la solicitud de patente, aun cuando no hubiera transcurrido el plazo mencionado en el apartado 1. – 108 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] 3. No tendrá lugar la publicación cuando la solicitud haya sido denegada, o se considere retirada o desistida, o haya sido retirada por el solicitante antes de que concluyan los preparativos técnicos para su publicación. 4. El informe sobre el estado de la técnica se publicará, haciendo el correspondiente anuncio en el «Boletín Oficial de la Propiedad Industrial», y emitiendo un folleto con dicho informe junto con la solicitud de patente o posteriormente, si ésta hubiera sido ya publicada. Artículo 38. Observaciones de terceros. Una vez publicada la solicitud cualquier persona podrá formular observaciones debidamente razonadas y documentadas sobre la patentabilidad de la invención objeto de la misma en la forma y plazo que reglamentariamente se establezca, sin que se interrumpa la tramitación. Estos terceros no se considerarán parte en el procedimiento. Artículo 39. Examen sustantivo. 1. La Oficina Española de Patentes y Marcas examinará previa petición del solicitante y de acuerdo con lo establecido en el Reglamento, si la solicitud de patente y la invención que constituye su objeto cumplen los requisitos formales, técnicos y de patentabilidad establecidos en la Ley. 2. La petición, que podrá formular el solicitante desde el momento del depósito de la solicitud, habrá de presentarse, en todo caso, antes de que transcurra el plazo de tres meses contados desde la publicación del informe sobre el estado de la técnica, no se considerará realizada hasta que se haya efectuado el pago de la tasa de examen. La revocación de la petición de examen equivaldrá a la retirada de la solicitud de patente. 3. Junto con la petición de examen sustantivo el solicitante podrá presentar observaciones al informe sobre el estado de la técnica, a la opinión escrita y a las observaciones de terceros y modificar, si lo estima oportuno, las reivindicaciones y los restantes documentos de la solicitud con sujeción a lo dispuesto en el artículo 48. 4. Transcurrido el plazo mencionado en el apartado 2 sin que el solicitante haya presentado su petición de examen, se entenderá que su solicitud ha sido retirada. Artículo 40. Tramitación y resolución. 1. Cuando el examen no revele la falta de ningún requisito que lo impida, la Oficina Española de Patentes y Marcas concederá la patente solicitada. 2. Si como resultado del examen se apreciasen motivos que impiden en todo o en parte la concesión de la patente, se comunicarán éstos al solicitante para que en el plazo reglamentariamente establecido conteste a las objeciones señaladas por la Oficina Española de Patentes y Marcas o modifique, si lo estima oportuno, las reivindicaciones. 3. En caso de que el solicitante no realice ningún acto para obviar las objeciones formuladas por la Oficina Española de Patentes y Marcas, la patente deberá ser denegada. En los demás casos la Oficina Española de Patentes y Marcas resolverá una vez recibida la contestación del solicitante. 4. Si una vez recibida la contestación del solicitante y pese a las alegaciones o modificaciones aportadas, la Oficina Española de Patentes y Marcas considera que persisten motivos que impiden en todo o en parte la concesión de la patente se comunicarán estos al solicitante dándole nuevas oportunidades de corregir su solicitud o formular alegaciones, en las condiciones y plazos establecidos en el Reglamento, antes de resolver definitivamente sobre la concesión o denegación de la patente. Artículo 41. Anuncio de la concesión y publicación de la patente. La concesión de la patente se anunciará en el «Boletín Oficial de la Propiedad Industrial», poniendo a disposición del público los documentos obrantes en el expediente de la patente concedida. – 109 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] Artículo 42. Edición del folleto de la patente. Para cada patente concedida se editará un folleto que contendrá el texto de la descripción, con las reivindicaciones y los dibujos y en su caso, las secuencias biológicas, tal como se hubieren finalmente concedido. El folleto, cuyo contenido se establecerá reglamentariamente, mencionará también el «Boletín Oficial de la Propiedad Industrial» en que se hubiere anunciado la concesión. En el folleto se hará constar que la patente se concede sin perjuicio de tercero y sin garantía del Estado en cuanto a la validez de la misma y a la utilidad del objeto sobre el que recae. CAPÍTULO III Oposiciones y recursos Artículo 43. Oposiciones. 1. Dentro de los seis meses siguientes a la publicación de la concesión en el «Boletín Oficial de la Propiedad Industrial», cualquier persona podrá oponerse a la concesión por alguno de los siguientes motivos: a) La invención reivindicada no reúne alguno de los requisitos de patentabilidad establecidos en el Título II de esta Ley. b) Su descripción no es lo suficientemente clara y completa para que un experto en la materia pueda ejecutarla. c) El objeto de la patente concedida excede del contenido de la solicitud tal como fue presentada. 2. La oposición deberá dirigirse a la Oficina Española de Patentes y Marcas en escrito motivado, acompañado de los correspondientes documentos probatorios y previo pago de la tasa correspondiente. 3. Admitido a trámite el escrito de oposición se comunicará al titular de la patente registrada para que éste presente sus alegaciones y modifique, si lo estima oportuno, las reivindicaciones. La Oficina Española de Patentes y Marcas dará traslado a cada una de las partes de las alegaciones y propuestas de modificación presentadas por la otra, concediéndoles un trámite de réplica en cada caso, todo ello en los plazos y condiciones establecidos en el reglamento. 4. Transcurridos los plazos mencionados en el apartado precedente la Oficina Española de Patentes y Marcas resolverá estimando en todo o en parte las oposiciones presentadas, cuando concurra alguno de los motivos de oposición señalados en el apartado 1 o desestimándolas en caso contrario. No obstante, cuando pese a las modificaciones o alegaciones aportadas persistan motivos que impidan en todo o en parte el mantenimiento de la patente, se otorgará al titular al menos una oportunidad de subsanar el defecto, o presentar nuevas alegaciones, antes de resolver con carácter definitivo sobre la oposición planteada. 5. La resolución de la Oficina Española de Patentes y Marcas se publicará en el «Boletín Oficial de la Propiedad Industrial», recogiendo, en su caso, las modificaciones que se hubieran introducido en la patente. La protección conferida por esta Ley se extenderá retroactivamente a la patente así modificada. 6. Al efecto retroactivo de la revocación se le aplicará, en su caso, lo previsto en el artículo 104 respecto de la nulidad. Artículo 44. Recursos. 1. El recurso administrativo contra la concesión de una patente solo podrá interponerse por quienes hayan sido parte en un procedimiento de oposición y se dirigirá contra el acto resolutorio de la oposición planteada. A estos efectos podrá entenderse desestimada la oposición si transcurrido el plazo para resolverla y notificarla no hubiese recaído resolución expresa. – 110 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] 2. El solicitante de una patente podrá presentar recurso administrativo contra la resolución denegatoria de la solicitud de patente por parte de la Oficina Española de Patentes y Marcas. 3. En el procedimiento de recurso el titular de la patente podrá modificar la solicitud con sujeción a lo dispuesto en el artículo 48. CAPÍTULO IV Certificados complementarios de protección de medicamentos y productos fitosanitarios Artículo 45. Solicitudes. 1. Las solicitudes de certificados complementarios de protección de medicamentos, las de prórroga de los mismos, y las de certificados complementarios de protección de productos fitosanitarios, se dirigirán a la Oficina Española de Patentes y Marcas en el modelo normalizado puesto a disposición de los usuarios por la Oficina en el que se harán constar las declaraciones y datos previstos en la normativa comunitaria tal como hayan sido desarrollados reglamentariamente. 2. Las solicitudes de certificados complementarios de protección y de su prórroga estarán sujetas al pago de la tasa correspondiente. Artículo 46. Tramitación. 1. La Oficina Española de Patentes y Marcas comprobará si la solicitud de certificado y el producto a que se refiere, o la de prórroga en su caso, cumplen los requisitos establecidos en la normativa comunitaria. La Oficina no investigará de oficio si la autorización de comercialización es la primera como medicamento o producto fitosanitario en la Unión Europea. 2. Si la solicitud y el producto objeto del certificado o la de su prórroga cumplen las condiciones establecidas en la normativa comunitaria, la Oficina los concederá. En caso contrario se comunicarán los defectos al solicitante para que los subsane o formule sus alegaciones en el plazo que reglamentariamente se establezca. Cuando los defectos no se corrijan en plazo y la Oficina considere que persisten las objeciones señaladas en la notificación, se denegará la solicitud. Tanto la solicitud como la resolución final se publicarán en el «Boletín Oficial de la Propiedad Industrial», de acuerdo con lo que se disponga reglamentariamente. Contra las resoluciones de la Oficina podrá interponerse recurso de alzada de acuerdo con lo previsto en el régimen jurídico de las Administraciones Públicas, y del Procedimiento Administrativo Común y la disposición adicional primera de esta Ley. Artículo 47. Mantenimiento. La tasa de mantenimiento del certificado complementario de protección se efectuará en un solo pago, cuya cuantía se fijará en función de la duración del certificado. CAPÍTULO V Disposiciones comunes a todos los procedimientos y a la información de los terceros Artículo 48. Modificaciones. 1. Salvo en los casos en que se trate de subsanar errores manifiestos, el interesado solo podrá modificar las reivindicaciones en aquellos trámites del procedimiento de concesión en que así lo permita la presente Ley, y con sujeción a lo que se establezca reglamentariamente. La posibilidad de modificar las reivindicaciones incluye la de modificar la descripción y, en su caso, los dibujos o las secuencias biológicas. – 111 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] 2. El solicitante podrá modificar las reivindicaciones, conforme a lo dispuesto en el apartado anterior, sin necesidad de contar con el consentimiento de quienes tengan derechos inscritos sobre su solicitud en el Registro de Patentes. 3. Toda modificación deberá ir acompañada de un escrito en el que el solicitante especifique las diferencias entre el texto nuevo y el texto sustituido indicando la razón de las modificaciones y el alcance de las mismas. 4. Cuando la patente haya sido modificada como consecuencia de un procedimiento de oposición, de limitación o de recurso o de una resolución judicial se editará un nuevo folleto, que contendrá el texto íntegro del documento de patente tal como haya quedado modificado siendo aplicable lo previsto en el artículo 43.5. A falta de pago de la tasa la patente no producirá efectos. Si el procedimiento es de limitación, ésta se tendrá por no realizada. 5. La solicitud de la patente o la patente no podrán modificarse de manera que su objeto exceda del contenido de la solicitud tal como se haya presentado inicialmente. 6. En el procedimiento de oposición, o en su caso en el de limitación, la patente no podrá modificarse de modo que se amplíe la protección que confiere. Artículo 49. Rectificación de errores. A instancia del solicitante se admitirán las modificaciones de los defectos de expresión o transcripción o de los errores contenidos en cualquier documento de la solicitud con sujeción a las limitaciones establecidas reglamentariamente. No obstante, si la petición de rectificación afecta a la descripción, las reivindicaciones, los dibujos o las secuencias biológicas, la rectificación deberá resultar evidente en el sentido de que se deduzca inmediatamente que ningún otro texto, dibujo o secuencia que el que resulte de la modificación puede haber sido propuesto por el solicitante. Artículo 50. Suspensión de los procedimientos. La presencia de defectos en la documentación interrumpirá el procedimiento desde que se notifique al solicitante la existencia de los mismos, mediante el correspondiente suspenso en la tramitación, hasta que dichos defectos se subsanen o expire el plazo para ello. Artículo 51. Cambio de modalidad. 1. En cualquier momento anterior a la finalización del examen sustantivo previsto en el artículo 39, el interesado podrá pedir la transformación de su solicitud de modo que el objeto de la misma quede protegido bajo un título distinto de Propiedad Industrial. 2. La Oficina Española de Patentes y Marcas, como consecuencia del examen que debe realizar en virtud de lo dispuesto en los artículos 35 y 40, podrá proponer al interesado el cambio de modalidad de la solicitud. El solicitante podrá aceptar o rechazar la propuesta, entendiéndose que la rechaza si en el plazo previsto en el Reglamento no pide expresamente el cambio de modalidad. Si la propuesta es rechazada continuará la tramitación del expediente en la modalidad solicitada. 3. En el caso de que el solicitante pida el cambio de modalidad, se acordará el cambio, notificándosele los documentos que haya de presentar dentro del plazo reglamentariamente establecido para la nueva tramitación a que haya de someterse la solicitud. A falta de presentación oportuna de la nueva documentación se le tendrá por desistido y así se le comunicará. 4. Cuando la resolución acordando el cambio de modalidad se produzca después de la publicación de la solicitud de la patente, deberá publicarse en el «Boletín Oficial de la Propiedad Industrial». Artículo 52. Retirada de la solicitud. 1. La solicitud de patente podrá ser retirada por el solicitante en cualquier momento antes de que la patente sea concedida. – 112 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] 2. Cuando figuren inscritos en el Registro de Patentes derechos de terceros sobre la solicitud, ésta sólo podrá ser retirada con el consentimiento de los titulares de tales derechos. Artículo 53. Restablecimiento de derechos. 1. El solicitante o el titular de una patente o cualquier otra parte en un procedimiento que, aun habiendo demostrado toda la diligencia requerida por las circunstancias, no hubiera podido cumplir un plazo en alguno de los procedimientos previstos en esta Ley será, previa petición, restablecido en sus derechos si la omisión hubiera tenido como consecuencia directa, en virtud de las disposiciones de esta Ley o de su Reglamento, la pérdida de un derecho. En el caso de que el plazo corresponda a la interposición de un recurso, tendrá como consecuencia su admisión a trámite, salvo lo previsto en el apartado 5. 2. La petición deberá presentarse por escrito en el plazo que primero expire de los siguientes: i) dos meses contados a partir del cese del impedimento; ii) doce meses contados a partir de la fecha de expiración del trámite omitido o, cuando una petición guarde relación con la falta de pago de una tasa de mantenimiento, doce meses contados a partir de la fecha de expiración del período de seis meses de pago con recargos al que se refiere el artículo 185. El trámite incumplido deberá realizarse dentro de ese plazo. No obstante, en el caso de que el restablecimiento de derechos se solicite para el plazo previsto en el artículo 30 la petición deberá presentarse dentro de los dos meses siguientes a la expiración del mismo o antes de que concluyan los preparativos técnicos de la publicación de la solicitud posterior, aplicándose el plazo que expire antes. 3. La petición deberá motivarse, indicándose los hechos y las justificaciones que se aleguen en su apoyo. Sólo se tendrá por presentada cuando se haya pagado la tasa de restablecimiento de derechos. 4. Será competente para resolver la petición el órgano que lo sea para pronunciarse sobre el acto que no se hubiere cumplido. 5. Las disposiciones del presente artículo no serán aplicables a los plazos mencionados en el apartado 2 de este artículo y en los artículos 43.1 y 144. Tampoco serán de aplicación al plazo de interposición del recurso administrativo contra un acto declarativo de derechos. 6. El titular de la solicitud o del registro restablecido en sus derechos no podrá invocar éstos frente a un tercero que, de buena fe, en el período comprendido entre la pérdida del derecho y la publicación de la mención de restablecimiento de ese derecho, hubiere comenzado a explotar la invención objeto de la solicitud o de la patente, o hubiese hecho preparativos serios y efectivos con esa finalidad, siempre que el tercero se limite a iniciar o continuar esa explotación en su empresa o para las necesidades de su empresa. 7. Contra la resolución que restablezca en sus derechos al solicitante podrán interponer recurso de alzada, tanto el tercero que pueda beneficiarse del derecho a continuar o a iniciar la explotación de la invención prevista en el apartado 6, como el tercero frente a quien puedan invocarse los derechos anteriores derivados de la solicitud objeto del restablecimiento de derechos. 8. La resolución de restablecimiento de derechos se inscribirá en el Registro de Patentes y se publicará en el «Boletín Oficial de la Propiedad Industrial». Artículo 54. Revisión de los actos en vía administrativa y contencioso-administrativa. 1. Los actos y resoluciones dictados por los órganos de la Oficina Española de Patentes y Marcas serán recurribles de conformidad con lo dispuesto en esta Ley y en la Ley 30/1992, de 26 de noviembre, de Régimen Jurídico de las Administraciones Públicas y del Procedimiento Administrativo Común. 2. Las resoluciones de los recursos administrativos dictados por los órganos competentes de la Oficina Española de Patentes y Marcas que pongan fin a la vía administrativa serán recurribles ante la Jurisdicción Contencioso-administrativa. 3. Frente a la resolución de concesión de una patente la Oficina Española de Patentes y Marcas no podrá ejercer de oficio o a instancia de parte la potestad revisora prevista en el – 113 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] artículo 102 de la Ley 30/1992, de 26 de noviembre, si la nulidad de la patente se funda en alguna de las causas previstas en el artículo 102 de la presente Ley. Dichas causas de nulidad sólo se podrán hacer valer ante los tribunales. Artículo 55. Consulta de expedientes. 1. Los expedientes relativos a solicitudes de patente, modelos de utilidad o de certificados complementarios de protección todavía no publicados sólo podrán ser consultados con el consentimiento del solicitante. Después de su publicación podrán ser consultados con sujeción a las condiciones que se establezcan reglamentariamente. 2. Cualquiera que pruebe que el solicitante de una patente, de un modelo de utilidad o de un certificado complementario de protección ha pretendido hacer valer frente a él los derechos derivados de su solicitud podrá consultar el expediente antes de su publicación sin el consentimiento del solicitante. 3. Cuando se publique una solicitud divisional, una nueva solicitud de patente presentada en virtud de lo dispuesto en el artículo 11.1 o la solicitud derivada de un cambio de modalidad de la protección según lo establecido en el artículo 51 cualquier persona podrá consultar el expediente de la solicitud inicial antes de su publicación y sin el consentimiento del solicitante. 4. Los expedientes correspondientes a solicitudes que hayan sido denegadas, retiradas o se tengan por desistidas antes de su publicación no serán accesibles al público. 5. En el caso de que se vuelva a presentar una de las solicitudes mencionadas en el apartado anterior, se considerará como una solicitud nueva sin perjuicio del posible derecho de prioridad que pudiera derivarse de la solicitud anterior. Artículo 56. Accesibilidad de la materia biológica. 1. La materia biológica depositada a que se refiere el artículo 27 será accesible: a) Antes de la primera publicación de la solicitud de patente, sólo a quien tenga derecho a consultar el expediente de acuerdo con lo establecido en el artículo anterior. b) Entre la primera publicación de la solicitud y la concesión de la patente, a toda persona que lo solicite o únicamente a un experto independiente si así lo pide el solicitante de la patente. c) Tras la concesión de la patente, y aunque la patente caduque o se anule, a toda persona que lo solicite. 2. El acceso se realizará mediante la entrega de una muestra de la materia biológica depositada, siempre y cuando la persona que lo solicite se comprometa mientras duren los efectos de la patente: a) A no suministrar a terceros ninguna muestra de la materia biológica depositada o de una materia derivada de la misma, y b) A no utilizar muestra alguna de la materia biológica depositada, o derivada de la misma, excepto con fines experimentales, salvo renuncia expresa del solicitante o del titular de la patente a dicho compromiso. 3. En caso de denegación o de retirada de la solicitud, el acceso a la materia depositada quedará limitado, a petición del solicitante y durante veinte años contados a partir de la fecha de presentación de la solicitud de la patente, a un experto independiente. En este caso será de aplicación lo dispuesto en el apartado 2. 4. Las peticiones del solicitante a que se refieren el párrafo b) del apartado 1 y el apartado 3 sólo podrán presentarse hasta la fecha en que se consideren concluidos los preparativos técnicos para la publicación de la solicitud de patente. Artículo 57. Obligación de facilitar información a terceros. 1. Cualquiera que pretenda hacer valer frente a un tercero derechos derivados de una solicitud de patente o de una patente ya concedida deberá darle a conocer el número de la misma. – 114 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] 2. Quien incluya en un producto, en sus etiquetas o embalajes, o en cualquier clase de anuncio o impreso, cualesquiera menciones tendentes a producir la impresión de que existe la protección de una solicitud de patente o de una patente ya concedida deberá hacer constar el número de las mismas. TÍTULO VI Efectos de la patente y de la solicitud de la patente Artículo 58. Duración y cómputo de los efectos. La patente tiene una duración de veinte años improrrogables, contados a partir de la fecha de presentación de la solicitud y produce sus efectos desde el día en que se publica la mención de que ha sido concedida. Artículo 59. Prohibición de explotación directa de la invención. 1. La patente confiere a su titular el derecho a impedir a cualquier tercero que no cuente con su consentimiento: a) La fabricación, el ofrecimiento para la venta, la introducción en el comercio o la utilización de un producto objeto de la patente o la importación o posesión del mismo para alguno de los fines mencionados. b) La utilización de un procedimiento objeto de la patente o el ofrecimiento de dicha utilización, cuando el tercero sabe, o las circunstancias hacen evidente que la utilización del procedimiento está prohibida sin el consentimiento del titular de la patente. c) El ofrecimiento para la venta, la introducción en el comercio o la utilización del producto directamente obtenido por el procedimiento objeto de la patente o la importación o posesión de dicho producto para alguno de los fines mencionados. 2. Cuando la patente tenga por objeto una materia biológica que, por el hecho de la invención, posea propiedades determinadas, los derechos conferidos por la patente se extenderán a cualquier materia biológica obtenida a partir de la materia biológica patentada por reproducción o multiplicación, en forma idéntica o diferenciada y que posea esas mismas propiedades. 3. Cuando la patente tenga por objeto un procedimiento que permita producir una materia biológica que, por el hecho de la invención, posea propiedades determinadas, los derechos conferidos por la patente se extenderán a la materia biológica directamente obtenida por el procedimiento patentado y a cualquier otra materia biológica obtenida a partir de ella por reproducción o multiplicación, en forma idéntica o diferenciada, y que posea esas mismas propiedades. 4. Cuando la patente tenga por objeto un producto que contenga información genética o que consista en información genética, los derechos conferidos por la patente se extenderán, sin perjuicio de lo dispuesto en el apartado 5 del artículo 5, a toda materia a la que se incorpore el producto y en la que se contenga y ejerza su función la información genética. Artículo 60. Prohibición de explotación indirecta de la invención. 1. La patente confiere igualmente a su titular el derecho a impedir que sin su consentimiento cualquier tercero entregue u ofrezca entregar medios para la puesta en práctica de la invención patentada relativos a un elemento esencial de la misma a personas no habilitadas para explotarla, cuando el tercero sabe o las circunstancias hacen evidente que tales medios son aptos para la puesta en práctica de la invención y están destinados a ella. 2. Lo dispuesto en el apartado anterior no es aplicable cuando los medios a que el mismo se refiere sean productos que se encuentren corrientemente en el comercio, a no ser que el tercero incite a la persona a la que realiza la entrega a cometer actos prohibidos en el artículo anterior. – 115 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] 3. No tienen la consideración de personas habilitadas para explotar la invención patentada, en el sentido del apartado 1, quienes realicen los actos previstos en los párrafos a) a d) del artículo siguiente. Artículo 61. Límites generales y agotamiento del derecho de patente. 1. Los derechos conferidos por la patente no se extienden: a) A los actos realizados en un ámbito privado y con fines no comerciales. b) A los actos realizados con fines experimentales que se refieran al objeto de la invención patentada. c) A la realización de los estudios y ensayos necesarios para obtener la autorización de comercialización de medicamentos en España o fuera de España, y los consiguientes requisitos prácticos, incluida la preparación, obtención y utilización del principio activo para estos fines. d) A la preparación de medicamentos realizada en las farmacias extemporáneamente y por unidad en ejecución de una receta médica ni a los actos relativos a los medicamentos así preparados. e) Al empleo del objeto de la invención patentada a bordo de buques de países de la Unión de París para la protección de la Propiedad Industrial, en el cuerpo del buque, en las máquinas, en los aparejos, en los aparatos y en los restantes accesorios, cuando esos buques penetren temporal o accidentalmente en las aguas españolas, siempre que el objeto de la invención sea utilizado exclusivamente para las necesidades del buque. f) Al empleo del objeto de la invención patentada en la construcción o el funcionamiento de medios de locomoción, aérea o terrestre, que pertenezcan a países miembros de la Unión de París para la protección de la Propiedad Industrial o de los accesorios de los mismos, cuando esos medios de locomoción penetren temporal o accidentalmente en el territorio español. g) A los actos previstos por el artículo 27 del Convenio sobre Aviación Civil Internacional, hecho en Chicago el 7 de diciembre de 1944, cuando tales actos se refieran a aeronaves de un Estado al cual sean aplicables las disposiciones del mencionado artículo. 2. Los derechos conferidos por la patente no se extienden a los actos relativos a un producto protegido por ella después de que ese producto haya sido puesto en el comercio en el Espacio Económico Europeo por el titular de la patente o con su consentimiento a menos que existan motivos legítimos que justifiquen que el titular de la patente se oponga a la comercialización ulterior del producto. 3. Los derechos conferidos por la patente no se extenderán a los actos relativos a la materia biológica obtenida por reproducción o multiplicación de una materia biológica protegida objeto de la patente, después de que ésta haya sido puesta en el mercado en el Espacio Económico Europeo por el titular de la patente o con su consentimiento, cuando la reproducción o multiplicación sea el resultado necesario de la utilización para la que haya sido comercializada dicha materia biológica, y a condición de que la materia obtenida no se utilice posteriormente para nuevas reproducciones o multiplicaciones. Esta limitación no se aplicará cuando existan motivos legítimos que justifiquen que el titular de la patente se oponga a la comercialización ulterior de la materia biológica. Artículo 62. Excepciones del ganadero y del agricultor. 1. No obstante lo dispuesto en el artículo 59, la venta, o cualquier otra forma de comercialización de material de reproducción vegetal realizada por el titular de la patente o con su consentimiento a un agricultor para su explotación agrícola, implicará el derecho de éste último a utilizar el producto de su cosecha para ulterior reproducción o multiplicación realizada por él mismo en su propia explotación. El alcance y las modalidades de esta excepción corresponderán a las previstas en el artículo 14 del Reglamento (CE) n.º 2100/94, del Consejo, de 27 de julio, relativo a la protección comunitaria de las obtenciones vegetales y a la Ley 3/2000, de 7 de enero, de régimen jurídico de la protección de obtenciones vegetales. 2. No obstante lo dispuesto en el artículo 59, la venta o cualquier otra forma de comercialización de animales de cría o de material de reproducción animal realizada por el – 116 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] titular de la patente o con su consentimiento a un agricultor o ganadero, implicará la autorización a estos últimos para utilizar el ganado protegido con fines agrícolas o ganaderos. Ello incluirá la puesta a disposición del ganado o de otro material de reproducción animal para que el agricultor o ganadero pueda proseguir su actividad agrícola o ganadera, pero no la venta en el marco de una actividad de reproducción comercial o con esa finalidad. El alcance y las modalidades de esta excepción corresponderán con las que se fijen reglamentariamente. Artículo 63. Derechos derivados de la utilización anterior. 1. El titular de una patente no tiene derecho a impedir que quienes de buena fe y con anterioridad a la fecha de prioridad de la patente hubiesen venido explotando en España lo que resulte constituir el objeto de la misma, o hubiesen hecho preparativos serios y efectivos para explotar dicho objeto, prosigan o inicien su explotación en la misma forma en que la venían realizando hasta entonces o para la que habían hecho los preparativos y en la medida adecuada para atender a las necesidades razonables de su empresa. Los derechos de explotación solo son transmisibles juntamente con las empresas que los vengan ejerciendo. 2. Los derechos conferidos por la patente no se extienden a los actos relativos a un producto amparado por ella después de que ese producto haya sido puesto en el comercio por la persona que disfruta del derecho de explotación establecido en el apartado anterior. Artículo 64. Falta de cobertura frente a patentes anteriores. El titular de una patente no podrá invocarla para defenderse frente a las acciones dirigidas contra él por infracción de otras patentes que tengan una fecha de prioridad anterior a la de la suya. Artículo 65. Patentes dependientes. El hecho de que el invento objeto de una patente no pueda ser explotado sin utilizar la invención protegida por una patente anterior perteneciente a distinto titular no será obstáculo para la validez de aquélla. En este caso ni el titular de la patente anterior podrá explotar la patente posterior durante la vigencia de ésta sin consentimiento de su titular, ni el titular de la patente posterior podrá explotar ninguna de las dos patentes durante la vigencia de la patente anterior, a no ser que cuente con el consentimiento del titular de la misma o haya tenido una licencia obligatoria. Artículo 66. Limitaciones legales. La explotación del objeto de una patente no podrá llevarse a cabo en forma abusiva o contraria a la Ley, la moral, el orden público o la salud pública, y estará supeditada, en todo caso, a las prohibiciones o limitaciones, temporales o indefinidas, establecidas o que se establezcan por las disposiciones legales. Artículo 67. Protección provisional. 1. A partir de la fecha de su publicación, la solicitud de patente confiere a su titular una protección provisional consistente en el derecho a exigir una indemnización, razonable y adecuada a las circunstancias, de cualquier tercero que, entre aquella fecha y la fecha de publicación de la mención de que la patente ha sido concedida hubiera llevado a cabo una utilización de la invención que después de ese período estaría prohibida en virtud de la patente. 2. Esa misma protección provisional será aplicable aun antes de la publicación de la solicitud frente a la persona a quien se hubiera notificado la presentación y el contenido de ésta. 3. Cuando el objeto de la solicitud de patente esté constituido por un procedimiento relativo a un microorganismo, la protección provisional comenzará solamente desde que el microorganismo haya sido hecho accesible al público. – 117 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] 4. Se entiende que la solicitud de patente no ha tenido nunca los efectos previstos en los apartados anteriores cuando hubiera sido o se considere retirada, o cuando hubiere sido denegada o revocada en virtud de una resolución firme. Artículo 68. Alcance de la protección. 1. El alcance de la protección conferida por la patente o por la solicitud de patente se determina por las reivindicaciones. La descripción y los dibujos servirán para interpretar las reivindicaciones. 2. Para el período anterior a la concesión de la patente, el alcance de la protección se determinará por las reivindicaciones de la solicitud, tal como haya sido publicada. Esto no obstante, la patente, tal como hubiera sido concedida, o modificada en el curso de un procedimiento de oposición, de recurso, de limitación o de nulidad, determinará con carácter retroactivo la protección mencionada, siempre que ésta no haya resultado ampliada. 3. Para determinar el alcance de la protección conforme a los apartados 1 y 2 anteriores deberá tenerse debidamente en cuenta todo elemento equivalente a un elemento indicado en las reivindicaciones. Artículo 69. Alcance de la protección en las patentes de procedimiento. 1. Cuando se introduzca en España un producto con relación al cual exista una patente de procedimiento para la fabricación de dicho producto, el titular de la patente tendrá con respecto al producto introducido los mismos derechos que la presente Ley le concede en relación con los productos fabricados en España. 2. Si una patente tiene por objeto un procedimiento para la fabricación de productos o sustancias nuevos, se presume, salvo prueba en contrario, que todo producto o sustancia de las mismas características ha sido obtenido por el procedimiento patentado. 3. En la práctica de las diligencias para la prueba en contrario prevista en el apartado anterior se tomarán en consideración los legítimos intereses del demandado para la protección de sus secretos de fabricación y negocios. TÍTULO VII Acciones por violación del derecho de patente Artículo 70. Defensa del derecho. El titular de una patente podrá ejercitar ante los órganos judiciales las acciones que correspondan, cualquiera que sea su clase y naturaleza, contra quienes lesionen su derecho y exigir las medidas necesarias para su salvaguardia. Artículo 71. Acciones civiles. 1. El titular cuyo derecho de patente sea lesionado podrá, en especial, solicitar: a) La cesación de los actos que violen su derecho, o su prohibición si éstos todavía no se han producido. b) La indemnización de los daños y perjuicios sufridos. c) El embargo de los objetos producidos o importados con violación de su derecho y de los medios exclusivamente destinados a tal producción o a la realización del procedimiento patentado. d) La atribución en propiedad de los objetos o medios embargados en virtud de lo dispuesto en el apartado anterior cuando sea posible, en cuyo caso se imputará el valor de los bienes afectados al importe de la indemnización de daños y perjuicios. Si el valor mencionado excediera del importe de la indemnización concedida, el titular de la patente deberá compensar a la otra parte por el exceso. e) La adopción de las medidas necesarias para evitar que prosiga la infracción de la patente y, en particular, la transformación de los objetos o medios embargados en virtud de – 118 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] lo dispuesto en el párrafo c), o su destrucción cuando ello fuera indispensable para impedir la infracción de la patente. f) Excepcionalmente el órgano judicial podrá también, a petición del titular de la patente, ordenar la publicación de la sentencia condenatoria del infractor de la patente, a costa del condenado, mediante anuncios y notificaciones a las personas interesadas. Las medidas comprendidas en los párrafos c) y e) serán ejecutadas a cargo del infractor, salvo que se aleguen razones fundadas para que no sea así. 2. Las medidas contempladas en los párrafos a) y e) del apartado precedente podrán también solicitarse, cuando sean apropiadas, contra los intermediarios a cuyos servicios recurra un tercero para infringir derechos de patente, aunque los actos de dichos intermediarios no constituyan en sí mismos una infracción, sin perjuicio de lo dispuesto en la Ley 34/2002, de 11 de julio, de servicios de la sociedad de la información y de comercio electrónico. Dichas medidas habrán de ser objetivas, proporcionadas y no discriminatorias. Artículo 72. Presupuestos de la indemnización de daños y perjuicios. 1. Quien, sin consentimiento del titular de la patente, fabrique, importe objetos protegidos por ella o utilice el procedimiento patentado, estará obligado en todo caso a responder de los daños y perjuicios causados. 2. Todos aquellos que realicen cualquier otro acto de explotación del objeto protegido por la patente sólo estarán obligados a indemnizar los daños y perjuicios causados si hubieren actuado a sabiendas o mediando culpa o negligencia. En todo caso, se entenderá que el infractor ha actuado a sabiendas si hubiera sido advertido por el titular de la patente acerca de la existencia de ésta, convenientemente identificada y de su infracción, con el requerimiento de que cesen en la misma. Artículo 73. Exhibición de documentos para el cálculo de la indemnización. 1. A fin de fijar la cuantía de los daños y perjuicios sufridos por la explotación no autorizada del invento, el titular de la patente podrá exigir la exhibición de los documentos del responsable que puedan servir para aquella finalidad. 2. En la ejecución de esta medida se tomarán en consideración los legítimos intereses del demandado para la protección de sus secretos empresariales de fabricación y negocios, sin perjuicio del derecho del titular de la patente a disponer de la información necesaria para poder concretar el alcance de la indemnización a su favor cuando la investigación a estos efectos se realice en fase de ejecución de la resolución sobre el fondo que haya apreciado la existencia de infracción. Artículo 74. Cálculo de los daños y perjuicios e indemnizaciones coercitivas. 1. La indemnización de daños y perjuicios debida al titular de la patente comprenderá no sólo el valor de la pérdida que haya sufrido, sino también el de la ganancia que haya dejado de obtener el titular a causa de la violación de su derecho. La cuantía indemnizatoria podrá incluir, en su caso, los gastos de investigación en los que se haya incurrido para obtener pruebas razonables de la comisión de la infracción objeto del procedimiento judicial. 2. Para fijar la indemnización por daños y perjuicios se tendrán en cuenta, a elección del perjudicado: a) Las consecuencias económicas negativas, entre ellas los beneficios que el titular habría obtenido previsiblemente de la explotación de la invención patentada si no hubiera existido la competencia del infractor o alternativamente, los beneficios que este último haya obtenido de la explotación del invento patentado. En el caso de daño moral procederá su indemnización, aun no probada la existencia de perjuicio económico. b) Una cantidad a tanto alzado que al menos comprenda la cantidad que el infractor hubiera debido pagar al titular de la patente por la concesión de una licencia que le hubiera permitido llevar a cabo su explotación conforme a derecho. Para su fijación se tendrá en cuenta especialmente, entre otros factores, la importancia económica del invento patentado, el tiempo de vigencia que le reste a la patente en el momento en que comenzó la infracción y el número y clase de licencias concedidas en ese momento. – 119 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] 3. Cuando el órgano jurisdiccional estime que el titular no cumple con la obligación de explotar la patente establecida en el artículo 90 la ganancia dejada de obtener se fijará de acuerdo con lo establecido en el párrafo b) del apartado anterior. 4. Cuando se condene a la cesación de los actos que infrinjan una patente el Tribunal fijará una indemnización coercitiva a favor del demandante adecuada a las circunstancias por día transcurrido hasta que se produzca la cesación efectiva de la infracción. El importe definitivo de esta indemnización, que se acumulará a la que le corresponda percibir con carácter general en aplicación del apartado 2, así como el día a partir del cual surgirá la obligación de indemnizar, se fijarán en ejecución de sentencia, de acuerdo con lo previsto en la Ley 1/2000, de 7 de enero, de Enjuiciamiento Civil. 5. Las diligencias relativas al cálculo o cuantificación y liquidación de daños de acuerdo con los criterios establecidos en este artículo se llevarán a cabo a partir de las bases fijadas en la sentencia conforme al procedimiento previsto en el Capítulo IV del Título V del Libro III de la Ley de Enjuiciamiento Civil. Artículo 75. Incidencia de los beneficios comerciales. 1. Para fijar la ganancia dejada de obtener según los criterios establecidos en el artículo 74.2 podrán incluirse en el cálculo de los beneficios, en la proporción que el órgano jurisdiccional estime razonable, los producidos por la explotación de aquellas cosas de las que el objeto inventado constituya parte esencial desde el punto de vista comercial. 2. Se entiende que el objeto inventado constituye parte esencial de un bien desde el punto de vista comercial cuando la consideración del invento incorporado suponga un factor determinante para la demanda de dicho bien. Artículo 76. Indemnización por desprestigio. El titular de la patente podrá exigir también la indemnización del perjuicio que suponga el desprestigio de la invención patentada causado por el infractor por cualquier causa y, en especial, como consecuencia de una realización defectuosa o una presentación inadecuada de aquélla en el mercado. Artículo 77. Deducción de las indemnizaciones ya percibidas. De la indemnización debida por quien hubiera producido o importado sin consentimiento del titular de la patente los objetos protegidos por la misma se deducirán las indemnizaciones que éste haya percibido por el mismo concepto de quienes explotaron de cualquier otra manera el mismo objeto. Artículo 78. Prescripción y límite al ejercicio de las acciones. 1. Las acciones civiles derivadas de la infracción del derecho de patente prescriben a los cinco años, contados desde el momento en que pudieron ejercitarse. 2. El titular de la patente no podrá ejercitar las acciones establecidas en este Título frente a quienes exploten los objetos que hayan sido introducidos en el comercio por personas que le hayan indemnizado en forma adecuada los daños y perjuicios causados. TÍTULO VIII La solicitud de patente y la patente como objetos del derecho de propiedad CAPÍTULO I Inscripción registral, cotitularidad y expropiación Artículo 79. Inscripción en el Registro de Patentes. 1. En el Registro de Patentes se inscribirán, en la forma que se disponga reglamentariamente, tanto las solicitudes de patente como las patentes ya concedidas. – 120 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] 2. Salvo en el caso previsto en el artículo 13.1, la transmisión, las licencias y cualesquiera otros actos o negocios jurídicos, tanto voluntarios como necesarios, que afecten a las solicitudes de patentes o a las patentes ya concedidas, sólo surtirán efectos frente a terceros de buena fe desde que hubieren sido inscritos en el Registro de Patentes. Reglamentariamente se establecerá la forma y documentación necesaria para dichas inscripciones. 3. No podrán invocarse frente a terceros derechos sobre solicitudes de patente o sobre patentes que no estén debidamente inscritos en el Registro de Patentes. Tampoco podrá mencionar en sus productos una solicitud de patente o una patente quien no tenga inscrito un derecho suficiente para hacer esa mención. Los actos realizados con infracción de lo dispuesto en este apartado serán sancionados como actos de competencia desleal. 4. La Oficina Española de Patentes y Marcas calificará la legalidad, validez y eficacia de los actos que hayan de inscribirse en el Registro de Patentes. El Registro de Patentes será público. 5. Inscrito en el Registro de Patentes alguno de los derechos o gravámenes contemplados en el artículo 82.1, no podrá inscribirse ningún otro de igual o anterior fecha que resulte opuesto o incompatible con aquél. Si solo se hubiere anotado la solicitud de inscripción, tampoco podrá inscribirse ningún otro derecho o gravamen incompatible hasta que se resuelva aquella. Artículo 80. Cotitularidad. 1. Cuando la solicitud de patente o la patente ya concedida pertenezcan pro indiviso a varias personas, la comunidad resultante se regirá por lo acordado entre las partes, en su defecto por lo dispuesto en este artículo y en último término por las normas del derecho común sobre la comunidad de bienes. 2. Sin embargo, cada uno de los partícipes por sí solo podrá: a) Disponer de la parte que le corresponda notificándolo a los demás comuneros que podrán ejercitar los derechos de tanteo y retracto. El plazo para el ejercicio del derecho de tanteo será de dos meses, contados a partir desde el envío de la notificación, y el del retracto, de un mes a partir de la inscripción de la cesión en el Registro de Patentes. b) Explotar la invención previa notificación a los demás cotitulares. c) Realizar los actos necesarios para la conservación de la solicitud o de la patente. d) Ejercitar acciones civiles o criminales contra los terceros que atenten de cualquier modo a los derechos derivados de la solicitud o de la patente común. El partícipe que ejercite tales acciones queda obligado a notificar a los demás comuneros la acción emprendida, a fin de que éstos puedan sumarse a la acción. 3. La concesión de licencia a un tercero para explotar la invención deberá ser otorgada conjuntamente por todos los partícipes, a no ser que el órgano jurisdiccional por razones de equidad, dadas las circunstancias del caso, faculte a alguno de ellos para otorgar la concesión mencionada Artículo 81. Expropiación. 1. Cualquier solicitud de patente o patente ya concedida podrá ser expropiada por causa de utilidad pública o de interés social, mediante la justa indemnización. 2. La expropiación podrá hacerse con el fin de que la invención caiga en el dominio público y pueda ser libremente explotada por cualquiera, sin necesidad de solicitar licencias, o con el fin de que sea explotada en exclusiva por el Estado, el cual adquirirá, en este caso, la titularidad de la patente. 3. La utilidad pública o el interés social será declarado por la Ley que ordene la expropiación, la cual dispondrá si la invención ha de caer en el dominio público o si ha de adquirir el Estado la titularidad de la patente o de la solicitud. El expediente que haya de instruirse se ajustará en todo, incluida la fijación del justiprecio, al procedimiento general establecido en la Ley de 16 de diciembre de 1954 sobre Expropiación Forzosa. – 121 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] CAPÍTULO II Transferencias, Licencias y Gravámenes Artículo 82. Principios generales. 1. Tanto la solicitud de patente como la patente son transmisibles y podrán darse en garantía o ser objeto de otros derechos reales, licencias, opciones de compra, embargos, otros negocios jurídicos o medidas que resulten del procedimiento de ejecución. En el supuesto de que se constituya una hipoteca mobiliaria, ésta se regirá por sus disposiciones específicas y se inscribirá en la sección cuarta del Registro de Bienes Muebles con notificación de dicha inscripción al Registro de Patentes para su inscripción en el mismo. A estos efectos ambos registros estarán coordinados para comunicarse telemáticamente los gravámenes inscritos o anotados en ellos. 2. Los actos a que se refiere el apartado anterior, cuando se realicen entre vivos, deberán constar por escrito para que sean válidos. 3. A los efectos de su cesión o gravamen la solicitud de patente o la patente ya concedida son indivisibles, aunque pueden pertenecer en común a varias personas. 4. Las disposiciones de este Capítulo se entienden sin perjuicio de las normas referidas al contenido y límites de los contratos de cesión y licencia sobre bienes inmateriales impuestos en otras Leyes nacionales que resulten aplicables, o de la aplicación, por los órganos nacionales o comunitarios correspondientes, de las disposiciones establecidas en los reglamentos comunitarios relativos a la aplicación del apartado 3 del artículo 101 del Tratado de Funcionamiento de la Unión Europea a determinadas categorías de acuerdos de transferencia de tecnología. Artículo 83. Licencias contractuales. 1. Tanto la solicitud de patente como la patente pueden ser objeto de licencias en su totalidad o en alguna de las facultades que integran el derecho de exclusiva, para todo el territorio nacional o para una parte del mismo. Las licencias pueden ser exclusivas o no exclusivas. 2. Podrán ser ejercitados los derechos conferidos por la patente o por la solicitud frente a un licenciatario que viole alguno de los límites de su licencia establecidos en virtud de lo dispuesto en el apartado anterior. 3. Los titulares de licencias contractuales no podrán cederlas a terceros, ni conceder sublicencias, a no ser que se hubiere convenido lo contrario. 4. Salvo pacto en contrario, el titular de una licencia contractual tendrá derecho a realizar todos los actos que integran la explotación de la invención patentada, en todas sus aplicaciones, en todo el territorio nacional y durante toda la duración de la patente. 5. Se presumirá que la licencia no es exclusiva y que el licenciante podrá conceder otras licencias y explotar por sí mismo la invención. 6. La licencia exclusiva impide el otorgamiento de otras licencias y el licenciante sólo podrá explotar la invención si en el contrato se hubiera reservado expresamente ese derecho. Artículo 84. Conocimientos técnicos. 1. Salvo pacto en contrario, quien transmita una solicitud de patente o una patente o conceda una licencia sobre las mismas, está obligado a poner a disposición del adquirente o del licenciatario los conocimientos técnicos que posea y que resulten necesarios para poder proceder a una adecuada explotación de la invención. 2. El adquirente o licenciatario a quien se comuniquen conocimientos secretos estará obligado a adoptar las medidas necesarias para evitar su divulgación. Artículo 85. Responsabilidad del transmitente y del licenciante. 1. Quien transmita a título oneroso una solicitud de patente o una patente ya concedida u otorgue una licencia sobre las mismas responderá, salvo pacto en contrario, si – 122 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] posteriormente se declarara que carecía de la titularidad o de las facultades necesarias para la realización del negocio de que se trate. Cuando se retire o se deniegue la solicitud, se revoque la patente o se declare su nulidad se aplicará en todo caso lo dispuesto en el artículo 104.3, a no ser que se hubiera pactado una responsabilidad mayor para el transmitente o el licenciante. 2. El transmitente o licenciante responderá siempre, cuando hubiere actuado de mala fe. La mala fe se presume, salvo prueba en contrario, cuando no hubiere dado a conocer al otro contratante, haciéndolo constar en el contrato con mención individualizada de tales documentos, los informes o resoluciones, españoles o extranjeros, de que disponga o le conste su existencia, referente a la patentabilidad de la invención objeto de la solicitud o de la patente. 3. Las acciones a que se refieren los apartados anteriores prescribirán a los seis meses, contados desde la fecha de la resolución definitiva o de la sentencia firme que les sirva de fundamento. Serán de aplicación a las mismas las normas del Código Civil sobre saneamiento por evicción. Artículo 86. Responsabilidad frente a terceros. 1. Quien transmita una solicitud de patente o una patente ya concedida u otorgue una licencia sobre las mismas, responderá solidariamente con el adquirente o con el licenciatario de las indemnizaciones a que hubiere lugar como consecuencia de los daños y perjuicios ocasionados a terceras personas por defectos inherentes a la invención objeto de la solicitud o de la patente. 2. La parte que efectúe el pago de la indemnización a que se refiere el apartado anterior podrá repetir del declarado responsable las cantidades abonadas, a no ser que se hubiere pactado lo contrario, que hubiere procedido de mala fe o que, dadas las circunstancias del caso y por razones de equidad, deba ser él quien soporte en todo o en parte la indemnización establecida a favor de los terceros. CAPÍTULO III Licencias de pleno derecho Artículo 87. Licencias de pleno derecho. Son licencias de pleno derecho las que resultan de un ofrecimiento público de licencias contractuales no exclusivas, realizado por el titular de la patente, de acuerdo con lo previsto en este Capítulo. Artículo 88. Ofrecimiento de licencias de pleno derecho. 1. Si el titular de la patente hace un ofrecimiento de licencias de pleno derecho, declarando por escrito a la Oficina Española de Patentes y Marcas que está dispuesto a autorizar la utilización de la invención a cualquier interesado, en calidad de licenciatario, se reducirá a la mitad el importe de las tasas anuales que devengue la patente después de recibida la declaración. Cuando se produzca un cambio total de la titularidad de la patente como consecuencia del ejercicio de la acción judicial prevista en el artículo 12, el ofrecimiento se considerará que ha sido retirado al inscribirse al nuevo titular en el Registro de Patentes. La Oficina Española de Patentes y Marcas inscribirá en el Registro de Patentes y dará la adecuada publicidad a los ofrecimientos de licencias de pleno derecho. 2. El ofrecimiento podrá ser retirado en cualquier momento por medio de una notificación escrita dirigida a la Oficina Española de Patentes y Marcas siempre que nadie haya comunicado todavía al titular de la patente su intención de utilizar la invención. La retirada del ofrecimiento será efectiva a partir del momento de su notificación. 3. El importe de la reducción de tasas que hubiere tenido lugar desde que se comunicó el ofrecimiento hasta la retirada del mismo deberá abonarse dentro del mes siguiente a la retirada del ofrecimiento. Será aplicable a este caso lo dispuesto en el artículo 184.3, computándose el plazo de seis meses que en él se prevé a partir de la terminación del plazo anteriormente mencionado. – 123 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] 4. No podrá hacerse el ofrecimiento de licencias de pleno derecho cuando figure inscrita en el Registro de Patentes una licencia exclusiva o cuando hubiere sido presentada una solicitud de inscripción de una licencia de esa clase. Una vez presentado el ofrecimiento de licencias de pleno derecho, no podrá admitirse ninguna solicitud de inscripción de una licencia exclusiva en el Registro de Patentes, a menos que se retire o se considere retirado el ofrecimiento. 5. La aceptación de un ofrecimiento público de licencias de pleno derecho legitima a cualquier persona para utilizar la invención en calidad de licenciatario no exclusivo. Artículo 89. Obtención de licencias de pleno derecho. 1. Cualquiera que desee utilizar la invención sobre la base del ofrecimiento de licencias de pleno derecho deberá notificárselo a la Oficina Española de Patentes y Marcas indicando la utilización que vaya a hacerse de la invención. La Oficina Española de Patentes y Marcas remitirá la notificación tanto al titular de la patente como al solicitante. 2. El solicitante de la licencia estará legitimado para utilizar la invención en la forma indicada por él en el plazo de un mes contado desde la recepción de la notificación que le haya sido remitida por la Oficina Española de Patentes y Marcas. 3. A falta de pacto entre las partes en el plazo indicado, la Oficina Española de Patentes y Marcas, a petición escrita de cualquiera de ellas y previa audiencia de ambas, fijará el importe adecuado de la compensación que haya de pagar el licenciatario o la modificará si hubieren acaecido o se hubieren conocido hechos que hagan aparecer como manifiestamente inadecuado el importe establecido. Sólo podrá pedirse que sea modificada la compensación establecida de este modo después de transcurrido un año desde que aquélla hubiere sido fijada por última vez. Para que la petición de fijar o modificar la compensación se considere presentada será preciso que haya sido abonada la tasa correspondiente. 4. Al término de cada trimestre del año natural, el licenciatario deberá informar al titular de la patente sobre la utilización que hubiere hecho de la invención y deberá abonarle la correspondiente compensación. Si no cumpliere las obligaciones mencionadas, el titular de la patente podrá otorgarle un plazo suplementario que sea razonable para que las cumpla. Transcurrido el plazo infructuosamente, se cancelará la licencia, previa petición justificada por el titular de la patente. TÍTULO IX Obligación de explotar y licencias obligatorias CAPÍTULO I Obligación de explotar la invención y requisitos para la concesión de licencias obligatorias Artículo 90. Obligación de explotar. 1. El titular de la patente está obligado a explotar la invención patentada bien por sí o por persona autorizada por él mediante su ejecución en España o en el territorio de un Estado miembro de la Organización Mundial del Comercio, de forma que dicha explotación resulte suficiente para abastecer la demanda en el mercado español. 2. La explotación deberá realizarse dentro del plazo de cuatro años desde la fecha de presentación de la solicitud de patente, o de tres años desde la fecha en que se publique su concesión en el «Boletín Oficial de la Propiedad Industrial», aplicándose automáticamente el plazo que expire más tarde. 3. La prueba de que la invención está siendo explotada de conformidad con lo dispuesto en el apartado 1 incumbe al titular de la patente. – 124 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] Artículo 91. Supuestos de concesión de licencias obligatorias. Procederá la concesión de licencias obligatorias sobre una determinada patente cuando concurra alguno de los supuestos siguientes: a) Falta o insuficiencia de explotación de la invención patentada. b) Dependencia entre las patentes, o entre patentes y derechos de obtención vegetal. c) Necesidad de poner término a prácticas que una decisión administrativa o jurisdiccional firme haya declarado contrarias a la legislación nacional o comunitaria de defensa de la competencia. d) Existencia de motivos de interés público para la concesión. e) Fabricación de productos farmacéuticos destinados a la exportación en aplicación del Reglamento (CE) n.º 816/2006 del Parlamento Europeo y del Consejo, de 17 de mayo de 2006, sobre la concesión de licencias obligatorias sobre patentes relativas a la fabricación de productos farmacéuticos destinados a la exportación a países con problemas de salud pública. Artículo 92. Licencias obligatorias por falta o insuficiencia de explotación. 1. Una vez finalizado el plazo previsto en el artículo 90 para iniciar la explotación de la invención patentada, cualquier persona podrá solicitar la concesión de una licencia obligatoria si en el momento de la solicitud, y salvo excusas legítimas, no se ha iniciado la explotación de la patente o cuando tal explotación, una vez transcurrido dicho plazo, haya sido interrumpida durante más de un año. 2. Se considerarán como excusas legítimas las dificultades objetivas de carácter técnico legal, ajenas a la voluntad y a las circunstancias del titular de la patente, que hagan imposible la explotación del invento o que impidan que esa explotación sea mayor de lo que es. Artículo 93. Licencias obligatorias por dependencia. 1. Cuando no sea posible explotar el invento protegido por una patente sin menoscabo de los derechos conferidos por una patente o por un derecho de obtención vegetal anterior, el titular de la patente posterior podrá solicitar una licencia obligatoria, para la explotación del objeto de la patente o de la variedad objeto del derecho de obtención vegetal anterior, mediante el pago de un canon adecuado. 2. Cuando no sea posible explotar un derecho de obtención vegetal sin menoscabo de los derechos conferidos por una patente anterior, el obtentor podrá solicitar una licencia obligatoria, para la explotación del invento protegido por la patente, mediante el pago de un canon adecuado. 3. Si una patente tuviera por objeto un procedimiento para la obtención de una sustancia química o farmacéutica protegida por una patente en vigor, tanto el titular de la patente de procedimiento como el de la patente de producto, tendrán derecho a la obtención de una licencia obligatoria sobre la patente del otro titular. 4. Los solicitantes de las licencias a que se refieren los apartados anteriores deberán demostrar: a) Que la invención o la variedad representa un progreso técnico significativo de considerable importancia económica con relación a la invención reivindicada en la patente anterior o a la variedad protegida por el derecho de obtención vegetal anterior. b) Que han intentado, sin conseguirlo en un plazo prudencial, obtener del titular de la patente o del derecho de obtención vegetal anterior, una licencia contractual en los términos previstos en el artículo 97.1. 5. Cuando proceda la concesión de una licencia obligatoria por dependencia, también el titular de la patente o del derecho de obtención vegetal anterior podrá solicitar el otorgamiento, en condiciones razonables, de una licencia para utilizar la invención o la variedad protegida por la patente o por el derecho de obtención vegetal posterior. 6. Las licencias obligatorias por dependencia se otorgarán solamente con el contenido necesario para permitir la explotación de la invención protegida por la patente, o de la – 125 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] variedad protegida por el derecho de obtención vegetal de que se trate, y quedarán sin efecto al declararse la nulidad o la caducidad de alguno de los títulos entre los cuales se dé la dependencia. 7. La tramitación y la resolución de las solicitudes de licencias obligatorias por dependencia para el uso no exclusivo de una invención patentada, se regirá por lo dispuesto en la presente Ley. La tramitación y la resolución de las solicitudes de licencias obligatorias por dependencia para el uso de la variedad protegida por un derecho de obtentor se regirán por su legislación específica. Artículo 94. Licencias obligatorias para poner remedio a prácticas anticompetitivas. 1. La resolución administrativa o jurisdiccional firme que haya declarado la violación del derecho de la competencia por parte del titular de la patente se comunicará a la Oficina Española de Patentes y Marcas por la Comisión Nacional de los Mercados y la Competencia o por el Juez o Tribunal que la haya emitido. 2. Cuando la resolución decrete directamente la sujeción de la patente al régimen de licencias obligatorias, la Oficina Española de Patentes y Marcas la publicará en el «Boletín Oficial de la Propiedad Industrial» y procederá de acuerdo con lo previsto en los artículos 98 y 99 de esta Ley. 3. No será precisa en este caso la justificación de la negociación previa entre el titular de la patente y el potencial usuario, solicitante de la licencia obligatoria. La necesidad de corregir las prácticas anticompetitivas se podrá tener en cuenta al determinar el canon de la licencia. 4. Sin perjuicio de lo previsto en los apartados precedentes, cuando el Gobierno considere que existen razones de interés público para poner término a prácticas anticompetitivas, la sujeción de la patente al régimen de licencias obligatorias podrá acordarse por real decreto de acuerdo con lo previsto en el artículo siguiente. Artículo 95. Licencias obligatorias por motivos de interés público. 1. Por motivo de interés público, el Gobierno podrá someter, en cualquier momento, una solicitud de patente o una patente ya otorgada, al régimen de licencias obligatorias, disponiéndolo así por real decreto. 2. Se considerará en todo caso que existen motivos de interés público cuando: a) La iniciación, el incremento o la generalización de la explotación del invento, o la mejora de las condiciones en que tal explotación se realiza, sean de primordial importancia para la salud pública o para la defensa nacional. b) La falta de explotación o la insuficiencia en calidad o en cantidad de la explotación realizada implique grave perjuicio para el desarrollo económico o tecnológico del país. c) Las necesidades de abastecimiento nacional así lo exijan. 3. El real decreto al que se hace referencia en el apartado 1 deberá ser acordado a propuesta del Ministerio de Industria, Energía y Turismo. En los casos en que la importancia de la explotación del invento se relacione con la salud pública o con la defensa nacional, la propuesta deberá formularse conjuntamente con el Ministro competente en materia de sanidad o de defensa, respectivamente. 4. El real decreto que disponga la sujeción de la patente al régimen de licencias obligatorias podrá establecer directamente, en todo o en parte, el alcance, condiciones y canon de licencia en los supuestos previstos en el artículo 97.2, o remitir la fijación de tales condiciones al oportuno procedimiento ante la Oficina Española de Patentes y Marcas previsto en el capítulo siguiente para su concreción en la resolución que conceda la licencia. 5. Cuando la sujeción al régimen de licencias obligatorias por motivos de interés público se deba a su importancia para la defensa nacional, podrá reservarse la posibilidad de solicitar tales licencias a una o varias empresas determinadas. – 126 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] Artículo 96. Licencias obligatorias para la fabricación de medicamentos destinados a países con problemas de salud pública. 1. Las solicitudes de licencias obligatorias presentadas en aplicación del Reglamento (CE) n.º 816/2006 del Parlamento Europeo y del Consejo, de 17 de mayo de 2006, sobre la concesión de licencias obligatorias sobre patentes relativas a la fabricación de productos farmacéuticos destinados a la exportación a países con problemas de salud pública, se dirigirán a la Oficina Española de Patentes y Marcas, en los modelos normalizados que se establezcan al efecto. Las licencias se tramitarán conforme a lo dispuesto en el citado Reglamento (CE) n.º 816/2006 y se regirán por lo dispuesto en el mismo. 2. La licencia surtirá efecto a partir de la fecha en la que la resolución que la conceda se notifique al solicitante y al titular del derecho, aplicándose la que sea posterior. La resolución que acuerde la licencia establecerá el canon de la misma. La licencia podrá ser revocada por la Oficina Española de Patentes y Marcas si el licenciatario no cumple las condiciones bajo las que fue otorgada de acuerdo con lo previsto en el artículo 16 del citado Reglamento (CE) n.º 816/2006. 3. Sin perjuicio de cualquier otra consecuencia legalmente prevista toda infracción de la prohibición prevista en el artículo 13 del Reglamento (CE) n.º 816/2006 y en el artículo 2 del Reglamento (CE) n.º 953/2003 del Consejo, de 26 de mayo de 2003, destinado a evitar el desvío comercial hacia la Unión Europea de determinados medicamentos esenciales, se considerará una infracción de la patente sobre la que recae la licencia. CAPÍTULO II Procedimiento de concesión de las licencias obligatorias Artículo 97. Justificación previa del solicitante de la licencia. 1. Previamente a la solicitud de una licencia obligatoria el interesado deberá probar que ha intentado, sin conseguirlo en un plazo prudencial, obtener del titular de la patente una licencia contractual en términos y condiciones comerciales razonables. Para las licencias previstas en el artículo 96, y salvo que se den las circunstancias previstas en el artículo 9.2 del Reglamento (CE) n.º 816/2006 al que se refiere el apartado 1 del artículo precedente, este plazo será en todo caso de treinta días, anteriores a la presentación de la solicitud. 2. Lo dispuesto en el apartado anterior no será aplicable: a) En los casos de emergencia nacional o en otras circunstancias de extrema urgencia. b) En los casos de uso público no comercial. c) En el supuesto previsto en la letra c) del artículo 91. Artículo 98. Solicitud de la licencia. 1. La solicitud de licencia obligatoria, dirigida a la Oficina Española de Patentes y Marcas en el modelo normalizado que se establezca al efecto, deberá ir acompañada de la prueba que acredite el intento previo de licencia contractual, salvo en los casos previstos en el apartado 2 del artículo anterior. La solicitud estará sujeta al pago de la tasa correspondiente. 2. El solicitante, además de concretar su petición, deberá exponer las circunstancias que la justifiquen, aportar las pruebas de que disponga en apoyo de sus afirmaciones, y acreditar que cuenta con los medios y garantías suficientes para llevar a cabo una explotación real y efectiva de la invención patentada acorde con la finalidad de la licencia. Artículo 99. Tramitación y resolución. 1. La Oficina Española de Patentes y Marcas dará traslado de una copia de la solicitud con los documentos que la acompañen al titular de la patente, a fin de que conteste en el plazo máximo de un mes. La contestación deberá ir acompañada de las pruebas que justifiquen las alegaciones realizadas. Si el titular de la patente no contestara dentro del plazo, dicha Oficina procederá a la concesión de la licencia. – 127 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] 2. Cuando, valoradas las alegaciones y pruebas presentadas, la Oficina Española de Patentes y Marcas considere que se dan las circunstancias que justifican la concesión de la licencia, invitará a las partes para que en el plazo de dos meses designen un mediador común o, en su defecto, nombre cada una un experto que, junto a un tercer experto nombrado por la mencionada Oficina, acuerden las condiciones de aquélla. 3. A falta de acuerdo sobre la designación de mediador o experto, o sobre las condiciones de la licencia en el plazo de dos meses adicionales, la Oficina Española de Patentes y Marcas decidirá sobre la concesión de la licencia y resolverá en consecuencia. 4. La resolución que otorgue la licencia deberá determinar el contenido de ésta. En particular habrá de fijar el ámbito de la licencia, el canon, la duración, las garantías que deba prestar el licenciatario, y cualesquiera otras cláusulas que aseguren el cumplimiento por su parte de las condiciones que justifican la concesión de la licencia. 5. Durante la tramitación del expediente, la Oficina Española de Patentes y Marcas podrá realizar de oficio las actuaciones que sean pertinentes y puedan ser de utilidad para resolver sobre la concesión de la licencia. Dicha Oficina podrá suspender por una sola vez la tramitación a petición justificada de ambas partes, en las circunstancias previstas en el Reglamento de ejecución de esta Ley. 6. La resolución determinará los gastos que hayan de ser sufragados por cada parte, que serán los causados a instancia suya. Los gastos comunes serán pagados por mitad. Podrá imponerse el pago de todos los gastos a una de las partes cuando se declare que ha actuado con temeridad o mala fe. 7. La interposición de un recurso administrativo o jurisdiccional contra la resolución que ponga término al expediente no suspenderá la ejecución del acto impugnado, pero la Oficina Española de Patentes y Marcas podrá autorizar al licenciatario previa petición fundada de éste, a demorar el comienzo de la explotación hasta que sea firme la concesión de la licencia. CAPÍTULO III Régimen de las licencias obligatorias Artículo 100. Características de las licencias obligatorias. 1. Las licencias obligatorias no serán exclusivas. 2. La licencia llevará aparejada una remuneración adecuada según las circunstancias propias de cada caso, habida cuenta de la importancia económica de la invención. 3. Si la patente recae sobre tecnología de semiconductores las licencias obligatorias solo podrán tener por objeto un uso público no comercial o utilizarse para rectificar una práctica declarada anticompetitiva tras un procedimiento judicial o administrativo. 4. Las relaciones que mantengan el titular de la patente y el licenciatario con motivo de la concesión de una licencia obligatoria deberán atenerse a la buena fe. Para el titular de la patente, la aplicación de este principio incluirá la obligación de poner a disposición del licenciatario los conocimientos técnicos que posea y resulten necesarios para poder proceder a una adecuada explotación comercial del invento. En caso de violación de este principio, declarada por sentencia judicial, por parte del titular de la patente, el licenciatario podrá pedir a la Oficina Española de Patentes y Marcas que reduzca el canon fijado para la licencia, en proporción a la importancia que tenga para la explotación del invento la obligación incumplida. Si en las mismas condiciones se declarase la actuación del licenciatario contraria a la buena fe contractual, el licenciante podrá instar de la mencionada Oficina la extinción de la licencia obligatoria. 5. La licencia obligatoria comprenderá los certificados complementarios de protección que al concederse la licencia o posteriormente, recaigan sobre el objeto de la patente de base incluido en el ámbito de la licencia obligatoria. 6. En cuanto no se opongan a lo dispuesto en este Título o en la normativa comunitaria, serán de aplicación a las licencias obligatorias las normas establecidas para las licencias contractuales previstas en el Título VIII, Capítulo II, de esta Ley. – 128 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] Artículo 101. Cesión, modificación y cancelación de las licencias obligatorias. 1. Para que la cesión de una licencia obligatoria sea válida, será preciso que la licencia se transmita junto con la empresa o parte de la empresa que la explote y que la cesión sea expresamente anotada por la Oficina Española de Patentes y Marcas. Tratándose de licencias por dependencia de patentes será preciso, además, que la licencia se transmita junto con la patente dependiente. 2. Será nula, en todo caso, la concesión de sublicencias por parte del titular de una licencia obligatoria. 3. Tanto el licenciatario como el titular de la patente podrán solicitar de la Oficina Española de Patentes y Marcas la modificación del canon u otras condiciones de la licencia obligatoria cuando existan nuevos hechos que justifiquen el cambio y, en especial, cuando el titular de la patente otorgue, con posterioridad a la licencia obligatoria, licencias contractuales en condiciones injustificadamente más favorables a las de aquella. 4. Si el licenciatario incumpliera grave o reiteradamente algunas de las obligaciones que le corresponden en virtud de la licencia obligatoria, la Oficina Española de Patentes y Marcas, previa audiencia de la parte afectada, de oficio o a instancia de parte interesada, podrá cancelar la licencia. TÍTULO X Nulidad, revocación y caducidad de la patente CAPÍTULO I Nulidad Artículo 102. Causas de nulidad. 1. Se declarará la nulidad de la patente: a) Cuando se justifique que no concurre, respecto del objeto de la patente, alguno de los requisitos de patentabilidad contenidos en el Título II de esta Ley. b) Cuando no describa la invención de forma suficientemente clara y completa para que pueda ejecutarla un experto en la materia. c) Cuando su objeto exceda del contenido de la solicitud de patente tal como fue presentada, o en el caso de que la patente hubiere sido concedida como consecuencia de una solicitud divisional o como consecuencia de una solicitud presentada con base en lo dispuesto en el artículo 11, cuando el objeto de la patente exceda del contenido de la solicitud inicial tal como ésta fue presentada. d) Cuando se haya ampliado la protección conferida por la patente tras la concesión. e) Cuando el titular de la patente no tuviera derecho a obtenerla conforme a lo dispuesto en el artículo 10. 2. Si las causas de nulidad sólo afectan a una parte de la patente ésta quedará limitada mediante la modificación de la o las reivindicaciones afectadas y se declarará parcialmente nula. A estos efectos, en el escrito de contestación a las alegaciones de nulidad el titular de la patente, sin perjuicio de poder defender con carácter principal la validez de las reivindicaciones concedidas, podrá defender con carácter subsidiario el juego o los juegos de reivindicaciones que proponga en la contestación. Artículo 103. Ejercicio de la acción de nulidad. 1. Será pública la acción para impugnar la validez de la patente. Esto no obstante, en el caso previsto en el apartado 1, párrafo e), del artículo precedente sólo podrá solicitar la declaración de nulidad la persona legitimada para obtener la patente. 2. La acción de nulidad podrá ejercitarse durante toda la vida legal de la patente y durante los cinco años siguientes a la caducidad de ésta. – 129 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] 3. La acción se dirigirá siempre contra quien sea titular registral de la patente en el momento de la interposición de la demanda, y ésta deberá ser notificada a todas las personas titulares de derechos sobre la patente debidamente inscritos en el Registro de Patentes con el fin de que puedan personarse e intervenir en el proceso. 4. En el procedimiento de nulidad el titular de la patente podrá limitar el alcance de la misma modificando las reivindicaciones. La patente, así limitada, servirá de base al procedimiento. 5. No podrá demandarse ante la Jurisdicción civil la nulidad de una patente, invocando la misma causa de nulidad que hubiera sido ya objeto de pronunciamiento, en cuanto al fondo de la cuestión, en sentencia dictada en la vía contencioso-administrativa, sobre los mismos hechos invocados como causa de nulidad. Artículo 104. Efectos de la declaración de nulidad. 1. La declaración de nulidad implica que la patente no fue nunca válida, considerándose que ni la patente ni la solicitud que la originó han tenido nunca los efectos previstos en el Título VI de esta Ley, en la medida en que hubiere sido declarada la nulidad. 2. La nulidad de la patente determinará la de sus certificados complementarios en la medida en que afecte al derecho sobre el producto protegido por la patente de base que fundamentó la concesión de aquéllos. 3. Sin perjuicio de la indemnización de daños y perjuicios a que hubiere lugar cuando el titular de la patente, hubiera actuado de mala fe, el efecto retroactivo de la nulidad no afectará: a) A las resoluciones sobre infracción de la patente que hubieran adquirido fuerza de cosa juzgada y hubieran sido ejecutadas con anterioridad a la declaración de nulidad. b) A los contratos concluidos antes de la declaración de nulidad, en la medida en que hubieran sido ejecutados con anterioridad a la misma. Esto no obstante, por razones de equidad y en la medida que lo justifiquen las circunstancias, será posible reclamar la restitución de sumas pagadas en virtud del contrato. 4. Una vez firme, la declaración de nulidad de la patente tendrá fuerza de cosa juzgada frente a todos. 5. La sentencia que declare la nulidad, total o parcial, de la patente será, en todo caso, comunicada a la Oficina Española de Patentes y Marcas para que se proceda a la cancelación de su inscripción o a la modificación del título inscrito. CAPÍTULO II Revocación o limitación a instancia del titular de la patente Artículo 105. Petición de revocación o de limitación. 1. A petición de su titular, la patente cuya concesión sea firme podrá ser revocada o limitada modificando las reivindicaciones en cualquier momento de su vida legal, incluido el periodo de vigencia de los certificados complementarios en su caso. 2. La solicitud de revocación o de limitación dirigida a la Oficina Española de Patentes y Marcas, se formulará en el impreso oficial establecido al efecto y solo se considerará válidamente formulada tras el pago de la tasa correspondiente. 3. No se admitirá la revocación o la limitación de una patente sobre la que existan derechos reales, opciones de compra, embargos o licencias inscritos en el Registro de Patentes sin que conste el consentimiento de los titulares de esos derechos. Tampoco se admitirá la solicitud de revocación o limitación si figurase inscrita en el Registro de Patentes la presentación de una demanda judicial reivindicando la titularidad de la patente o el reconocimiento de otros derechos patrimoniales sobre la misma en tanto no conste el consentimiento del demandante. 4. Cuando esté pendiente un procedimiento judicial sobre la validez de la patente y sin perjuicio de lo previsto en el artículo 120, la petición de limitación, dirigida a la Oficina Española de Patentes y Marcas, habrá de ser autorizada por el Juez o Tribunal que conozca del procedimiento. – 130 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] Artículo 106. Procedimiento. 1. La Oficina Española de Patentes y Marcas comprobará la regularidad de los documentos presentados y examinará si, en su caso, las reivindicaciones modificadas se ajustan a lo dispuesto en los artículos 28 y 48. 2. Si la documentación presenta defectos o si el nuevo juego de reivindicaciones no limita el objeto de la patente, se comunicarán las objeciones al interesado, indicando los motivos, para que éste corrija los defectos o presente sus alegaciones en el plazo establecido reglamentariamente. La solicitud será denegada si los defectos no son subsanados en plazo. No existiendo objeciones, o superadas éstas, se resolverá acordando la revocación o la limitación solicitada. Artículo 107. Efectos de la revocación o de la limitación. 1. Los efectos de la revocación o de la limitación son los mismos que los de la nulidad total o parcial. Las reivindicaciones modificadas determinarán retroactivamente el alcance de la protección conferida por la patente. 2. Los efectos de la revocación o de la limitación sobre resoluciones anteriores y los contratos concluidos con anterioridad a la resolución que la declare serán los previstos en el artículo 104. CAPÍTULO III Caducidad Artículo 108. Causas de caducidad. 1. Las patentes caducan: a) Por la expiración del plazo para el que hubieren sido concedidas. b) Por renuncia del titular. c) Por falta de pago en tiempo oportuno de una anualidad y, en su caso, de la sobretasa correspondiente. d) Si la invención no es explotada en los dos años siguientes a la concesión de la primera licencia obligatoria. e) Por incumplimiento de la obligación de explotar prevista en el artículo 90, cuando el titular de la patente no pueda beneficiarse de las disposiciones del referido Convenio de París para la Protección de la Propiedad Industrial, de 20 de marzo de 1883 o del Acuerdo sobre los Aspectos de los Derechos de Propiedad Intelectual Relacionados con el Comercio, hecho en Marrakech el 15 de abril de 1994 y resida habitualmente o tenga su establecimiento industrial o comercial en un país cuya legislación admita la adopción de una medida similar. 2. Sin perjuicio de su declaración por la Oficina Española de Patentes y Marcas y su publicación en el «Boletín Oficial de la Propiedad Industrial», la caducidad de una patente incorpora el objeto patentado al dominio público desde el momento en que se produjeron los hechos u omisiones que dieron lugar a ella, salvo en la parte en que ese mismo objeto estuviere amparado por otra patente anterior y vigente. Será aplicable a la caducidad de la patente de base por alguna de las causas previstas en los apartados 1.b) a 1.e), y desde el momento en que esta se produzca, lo dispuesto en el artículo 104.2 respecto a la nulidad. 3. En los supuestos de falta de pago de una anualidad, se entiende que la omisión que da lugar a la caducidad se produce al comienzo del año de la vida de la patente para el cual no hubiere sido abonada la anualidad. No obstante lo previsto en el apartado precedente, la caducidad no se producirá en este caso antes de que transcurran los seis meses de demora sin que se haya pagado la anualidad y la sobretasa correspondiente, o en su caso, la correspondiente tasa de regularización. 4. En el supuesto del apartado 1, párrafo d), la caducidad será declarada previa instrucción por la Oficina Española de Patentes y Marcas del correspondiente expediente administrativo. – 131 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] Artículo 109. Caducidad por falta de pago en tiempo oportuno de una anualidad. 1. Cuando existan embargos inscritos sobre una patente o una acción reivindicatoria en curso y su titular no hubiere pagado en tiempo oportuno una anualidad, no caducará dicha patente hasta el levantamiento del embargo o la desestimación definitiva de la acción reivindicatoria. El titular de la patente embargada podrá no obstante evitar la caducidad abonando las anualidades devengadas en el plazo de dos meses contados desde la fecha en la que se le comunique la cancelación del embargo. 2. Si como consecuencia de los procedimientos a que se refiere el apartado anterior se produjera un cambio en la titularidad de la patente el nuevo titular podrá abonar las anualidades devengadas en el plazo de dos meses a contar desde la fecha en la que la sentencia sobre la acción reivindicatoria hubiera ganado firmeza o desde que la autoridad o tribunal competente hubieran notificado a la Oficina Española de Patentes y Marcas la adjudicación definitiva de la patente embargada. 3. Transcurridos los plazos previstos en los apartados 1 y 2, la patente caducará si no se hubiere efectuado el correspondiente pago. 4. Tampoco caducará una patente por falta de pago en tiempo oportuno de una anualidad cuando se encuentre inscrita en el Registro de Patentes una hipoteca mobiliaria sobre la misma. El titular hipotecario podrá efectuar el pago en nombre de su propietario en el plazo de un mes a contar desde la finalización del plazo de recargos previsto en el artículo 185. Podrán también efectuar el pago en las mismas condiciones los titulares de otros derechos inscritos sobre la patente que pudieran verse afectados por su caducidad, sin perjuicio de su derecho a repetir frente al titular de la patente las cantidades abonadas. Cuando la hipoteca se haya constituido a favor de la Hacienda Pública el pago quedará suspendido hasta la cancelación de la misma, sin que se produzca la caducidad de la patente por falta de pago de las anualidades pendientes, que deberán ser abonadas, bien por el titular de la patente que hipotecó la misma, bien por quien resulte nuevo propietario tras la ejecución de la garantía hipotecaria por el procedimiento administrativo de apremio. Artículo 110. Renuncia. 1. El titular podrá renunciar a toda la patente o a una o varias reivindicaciones de la misma. 2. La renuncia, dirigida a la Oficina Española de Patentes y Marcas, deberá presentarse por escrito y solo tendrá efectos frente a terceros una vez inscrita en el Registro de Patentes. 3. Cuando la renuncia sea parcial, la patente seguirá en vigor con referencia a las reivindicaciones no comprendidas en la renuncia, siempre que la renuncia no suponga la ampliación del objeto de la patente. 4. No se admitirá la renuncia de una patente sobre la que existan derechos reales, opciones de compra, embargos o licencias inscritos en el Registro de Patentes sin que conste el consentimiento de los titulares de esos derechos. Tampoco se admitirá la renuncia si existiera en curso una acción reivindicatoria o de nulidad sobre la patente y no constara el consentimiento del demandante. 5. La renuncia a la patente se publicará en el «Boletín Oficial de la Propiedad Industrial». Cuando la renuncia sea parcial se editará, previo pago de la tasa correspondiente, un nuevo folleto de la patente de acuerdo con lo que se disponga reglamentariamente. [...] – 132 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] TÍTULO XII Jurisdicción y normas procesales CAPÍTULO I Disposiciones generales Artículo 116. Jurisdicción. El conocimiento de todos los litigios que se susciten como consecuencia del ejercicio de acciones de cualquier clase y naturaleza que sean, derivadas de la aplicación de los preceptos de esta Ley, corresponderá en función de su delimitación competencial, al orden jurisdiccional civil, penal o contencioso-administrativo. Artículo 117. Legitimación para el ejercicio de las acciones. 1. Estarán legitimados para el ejercicio de las acciones a que se refiere el artículo 2.3 de esta Ley, además de los titulares de los derechos inscritos en el Registro de Patentes, quienes acrediten haber solicitado debidamente la inscripción en dicho registro del acto o negocio del que traiga causa el derecho que se pretenda hacer valer, siempre que dicha inscripción llegue a ser concedida. 2. Salvo pacto en contrario, el titular de una licencia exclusiva podrá ejercitar en su propio nombre todas las acciones que en la presente Ley se reconocen al titular de la patente frente a los terceros que infrinjan su derecho, pero no podrá ejercitarlas el concesionario de una licencia no exclusiva. 3. El licenciatario, que conforme a lo dispuesto en el apartado anterior, no esté legitimado para ejercitar las acciones por infracción de la patente, podrá requerir fehacientemente al titular de la misma para que entable la acción judicial correspondiente. Si el titular se negara o no ejercitara la oportuna acción dentro de un plazo de tres meses, podrá el licenciatario entablarla en su propio nombre, acompañando el requerimiento efectuado. Con anterioridad al transcurso del plazo mencionado, el licenciatario podrá pedir al Juez la adopción de medidas cautelares urgentes cuando justifique la necesidad de las mismas para evitar un daño importante, con presentación del referido requerimiento. 4. El licenciatario que ejercite una acción en virtud de lo dispuesto en alguno de los apartados anteriores deberá notificárselo fehacientemente al titular de la patente, el cual podrá personarse e intervenir en el procedimiento, ya sea como parte en el mismo o como coadyuvante. Artículo 118. Competencia. 1. Los litigios civiles que puedan surgir al amparo de la presente Ley se resolverán en el juicio que corresponda conforme a la Ley 1/2000, de 7 de enero, de Enjuiciamiento Civil. 2. Será objetivamente competente el Juez de lo Mercantil de la ciudad sede del Tribunal Superior de Justicia de aquellas Comunidades Autónomas en las que el Consejo General del Poder Judicial haya acordado atribuir en exclusiva el conocimiento de los asuntos de patentes. 3. En particular será territorialmente competente el Juez de lo Mercantil especializado a que se refiere el apartado anterior correspondiente al domicilio del demandado o, en su defecto, del lugar de residencia del representante autorizado en España para actuar en nombre del titular, si en la Comunidad Autónoma de su domicilio existieran Juzgados de lo Mercantil especializados en asuntos de patentes conforme al apartado 2. De no existir, a elección del actor, será competente cualquier Juez de lo Mercantil a quien corresponda el conocimiento de asuntos de patentes de conformidad con el apartado 2. 4. En caso de acciones por violación del derecho de patente también será competente, a elección del demandante, el mismo juzgado a que se refiere el apartado anterior de la – 133 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] Comunidad Autónoma donde se hubiera realizado la infracción o se hubieran producido sus efectos, siempre que en dicha Comunidad Autónoma existieran Juzgados de lo Mercantil especializados en asuntos de patentes conforme al apartado 2. De no existir, a elección del actor será competente cualquier Juez de lo Mercantil a quien correspondiera el conocimiento de asuntos de patentes de conformidad con el apartado 2. Artículo 119. Plazos en litigios en materia de patentes. 1. El demandado por cualquier acción civil regulada en la presente Ley dispondrá de un plazo de dos meses para contestar a la demanda y, en su caso, formular reconvención. El mismo plazo regirá para contestar la reconvención, así como para contestar a la limitación de la patente solicitada por el titular de la misma a resultas de la impugnación de la validez del título realizada por vía de reconvención o de excepción por el demandado. 2. La previsión contenida en el artículo 337 de la Ley de Enjuiciamiento Civil no operará, salvo que la demandada justifique cumplidamente la imposibilidad de aportar el o los informes de que pretenda valerse al contestar a la demanda o, en su caso, a la reconvención. Artículo 120. Nulidad de la patente del actor. 1. La persona frente a la que se ejercite una acción por violación de los derechos derivados de una patente podrá alegar, en toda clase de procedimientos, por vía de reconvención o por vía de excepción, la nulidad total o parcial de la patente del actor, de conformidad con las normas del Derecho procesal común. A tales efectos, se tendrá en cuenta lo dispuesto en el artículo 103. 2. Si la nulidad se planteara mediante excepción, el titular de la patente dispondrá de 8 días, desde la recepción de la contestación a la demanda, para solicitar del Juez o Tribunal que la excepción sea tratada como reconvención. 3. En el caso de que el titular de la patente, con carácter principal o subsidiario, optara por limitar la patente modificando sus reivindicaciones, deberá aportar el nuevo juego o juegos de reivindicaciones y su justificación en el trámite de contestación a la demanda de nulidad, de contestación a la reconvención o de contestación a la excepción de nulidad. El titular de la patente que haya ejercitado acción por infracción de la misma deberá, en el mismo trámite de contestación a la impugnación de su patente, razonar, y en su caso probar, en qué modo afectan las limitaciones propuestas a la acción de infracción ejercitada frente al demandado. 4. Sin perjuicio de lo establecido en el apartado anterior, cuando por circunstancias sobrevenidas la patente resulte modificada fuera del proceso, el titular de la misma podrá solicitar que la patente así modificada sirva de base al proceso. En estos casos el Juez o Tribunal deberá conceder trámite de alegaciones a las demás partes del proceso. 5. El Juez o Tribunal dará traslado de la solicitud de limitación al instante de la nulidad en turno de alegaciones, para que mantenga o modifique sus pretensiones, a la vista de la limitación propuesta. El cómputo del plazo de dos meses previsto en el artículo 119.1 se iniciará desde el momento de la recepción de la solicitud de limitación de la patente presentada por el actor. 6. Presentada la solicitud de limitación con carácter principal o subsidiario, el Juez o Tribunal librará oficio a la Oficina Española de Patentes y Marcas para su inscripción como anotación preventiva. La resolución firme que resuelva sobre la limitación de la patente se notificará de oficio a dicha Oficina para su anotación registral y, en su caso, modificación de la patente. 7. En los procesos en que se ejerciten acciones por las que se cuestione la validez de una patente, el Juez o Tribunal podrá acordar, de acuerdo con lo previsto en la Ley de Enjuiciamiento Civil y previo pago de la tasa correspondiente cuando se solicite a instancia de parte, la emisión de un informe pericial de la Oficina Española de Patentes y Marcas para que dictamine por escrito sobre aquellos extremos concretos en los que los informes periciales aportados por las partes resulten contradictorios. El autor del informe podrá ser llamado a declarar sobre el contenido del informe cuando sea requerido para ello por el Juez o Tribunal que conozca del asunto. En ningún caso se entenderá que esta disposición limita – 134 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] la discrecionalidad del Juez o Tribunal para recabar este dictamen del centro o institución que considere más conveniente dadas las circunstancias del caso. Artículo 121. Acción negatoria. 1. Cualquier interesado podrá ejercitar una acción contra el titular de una patente, para que el Juez competente declare que una actuación determinada no constituya una infracción de esa patente. 2. El interesado, con carácter previo a la presentación de la demanda, requerirá fehacientemente al titular de la patente para que se pronuncie sobre la oponibilidad entre la misma y la explotación industrial que el requirente lleve a cabo sobre territorio español o frente a los preparativos serios y efectivos que desarrolle a tales efectos. Transcurrido un mes desde la fecha del requerimiento sin que el titular de la patente se hubiera pronunciado o cuando el requirente no esté conforme con la respuesta, podrá ejercitar la acción prevista en el apartado anterior. 3. No podrá ejercitar la acción mencionada en el apartado 1 quien hubiere sido demandado por la infracción de la patente de que se trate. 4. Si el demandante prueba que la actuación a que se refiere su demanda no constituye una infracción de la patente, el Juez hará la declaración requerida. 5. La demanda deberá ser notificada a todas las personas titulares de derechos sobre la patente debidamente inscritos en el Registro de Patentes, con el fin de que puedan personarse e intervenir en el proceso. Esto no obstante, no podrán personarse en autos los licenciatarios contractuales cuando así lo disponga su contrato de licencia. 6. La acción a que se refiere el presente artículo podrá ser ejercitada junto con la acción para que se declare la nulidad de la patente. Artículo 122. Tratamiento de la información confidencial. Cuando para el esclarecimiento de los hechos objeto de los procedimientos judiciales a que se refiere este capítulo, ya sea a través de la práctica de diligencias preliminares o de medidas para el aseguramiento de la prueba, fuese necesario recabar información que a juicio del Juez o Tribunal sea de carácter confidencial, el órgano jurisdiccional adoptará la decisión de obtener o requerir la misma y dispondrá, a petición de las partes, las medidas y actuaciones necesarias para garantizar la confidencialidad de la información requerida y el derecho a la tutela judicial efectiva de la parte procesal que demande la información. CAPÍTULO II Diligencias de comprobación de hechos Artículo 123. Petición de las diligencias. 1. La persona legitimada para ejercitar las acciones derivadas de la patente podrá pedir al Juez que con carácter urgente acuerde la práctica de diligencias para la comprobación de hechos que pueden constituir violación del derecho exclusivo otorgado por la patente, sin perjuicio de las que puedan solicitarse al amparo del artículo 256.1 de la Ley 1/2000, de 7 de enero, de Enjuiciamiento Civil. 2. Antes de resolver sobre la petición formulada, el Juez podrá requerir los informes y ordenar las investigaciones que estime oportunas. 3. Solamente podrá acordarse la práctica de las diligencias cuando, dadas las circunstancias del caso, sea presumible la infracción de la patente y no sea posible comprobar la realidad de la misma sin recurrir a las diligencias solicitadas. 4. Al acordar, en su caso, la práctica de las diligencias solicitadas, el Juez, de acuerdo con lo previsto en la Ley de Enjuiciamiento Civil, fijará la caución que deberá prestar el peticionario para responder de los daños y perjuicios que eventualmente puedan ocasionarse. 5. Si el Juez no considerara suficientemente fundada la pretensión, la denegará por medio de auto que será apelable en ambos efectos. – 135 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] Artículo 124. Práctica de las diligencias. 1. Sin que medie en ningún caso notificación previa de su práctica a quien deba soportarla, en la diligencia de comprobación el Juez, con intervención del perito o peritos que a tal efecto haya designado, y oídas las manifestaciones de la persona con quien se entienda la diligencia, determinará si a la vista del examen practicado se puede estar llevando a cabo la infracción alegada de la patente. Cuando la invención no se fabrique o se ejecute en España, el examen y comprobación se referirán a los productos importados y/o comercializados objeto de la diligencia. 2. Cuando el Juez considere que a la vista del examen practicado no es presumible que se esté llevando a cabo la infracción de la patente, dará por terminada la diligencia, ordenará que se forme una pieza separada en la que se incluirán las actuaciones, que se mantendrá secreta, y dispondrá que el Secretario Judicial notifique al peticionario que no procede darle a conocer el resultado de las diligencias realizadas. 3. En los demás casos, el Juez, con intervención del perito o peritos designados al efecto, efectuará una detallada descripción de las máquinas, dispositivos, productos, procedimientos, instalaciones o actuaciones mediante la utilización de los cuales se lleve presumiblemente a cabo la infracción alegada. 4. En todo caso cuidará el Juez de que la diligencia de comprobación no sirva como medio para violar secretos industriales o para realizar actos que constituyan competencia desleal. 5. Contra la decisión del Juez sobre el resultado de la diligencia practicada no se dará recurso alguno. Artículo 125. Certificaciones y copias de las diligencias. 1. De las diligencias de comprobación realizadas no podrán expedirse otras certificaciones ni copias que la destinada a la parte afectada y la precisa para que el solicitante de las mismas inicie la correspondiente acción judicial. El solicitante sólo podrá utilizar esta documentación para plantear dicha acción, con prohibición de divulgarla o comunicarla a terceros. 2. Si en el plazo de 30 días hábiles a partir de la fecha de la entrega al solicitante de la certificación de las diligencias no se hubiere presentado la correspondiente demanda ejercitando la acción judicial, quedarán aquéllas sin efecto y no podrán ser utilizados en ninguna otra acción judicial. Artículo 126. Compensación de la parte afectada. La parte afectada por las diligencias de comprobación podrá reclamar en todo caso, de quien la hubiere solicitado, el afianzamiento de los gastos y daños que se le hubieren ocasionado, incluido el lucro cesante. El pago solo deberá realizarse si no se ejercita la acción principal o esta es rechazada, todo ello sin perjuicio de la responsabilidad general por daños y perjuicios en que pudiera haber incurrido el solicitante de las medidas en los casos que a ello hubiere lugar. CAPÍTULO III Medidas cautelares Artículo 127. Petición de medidas cautelares. Quien ejercite o vaya a ejercitar una acción de las previstas en la presente Ley podrá solicitar del órgano judicial que haya de entender de la misma la adopción de medidas cautelares tendentes a asegurar la eficacia de dichas acciones, de conformidad con lo previsto en la misma y en la Ley 1/2000, de 7 de enero, de Enjuiciamiento Civil. – 136 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] Artículo 128. Posibles medidas cautelares. 1. Se podrán adoptar como medidas cautelares contra el presunto infractor las que aseguren debidamente la completa efectividad del eventual fallo que en su día recaiga, y en especial las siguientes: a) La cesación de los actos que pudieran infringir el derecho del peticionario o su prohibición, cuando existan indicios racionales para suponer la inminencia de dichos actos. b) La retención y depósito de las mercancías presuntamente infractoras del derecho del titular de la patente y de los medios exclusivamente destinados a tal producción o a la realización del procedimiento patentado. c) El afianzamiento de la eventual indemnización de daños y perjuicios. d) Las anotaciones registrales que procedan. 2. Las medidas cautelares previstas en el apartado 1 podrán también solicitarse, cuando sean apropiadas, contra los intermediarios a cuyos servicios recurra un tercero para infringir derechos de patente, aunque los actos de dichos intermediarios no constituyan en sí mismos una infracción, sin perjuicio de lo dispuesto en la Ley 34/2002, de 11 de julio, de servicios de la sociedad de la información y de comercio electrónico. Dichas medidas habrán de ser objetivas, proporcionadas y no discriminatorias. 3. No procederá la adopción de medidas cautelares cuando resultare que el demandado está amparado por un derecho fundado en una utilización anterior según el artículo 63. Artículo 129. Fianzas. 1. Al acordar, en su caso, las medidas cautelares solicitadas, el Juez fijará la caución que deberá prestar el peticionario para responder de los daños y perjuicios que eventualmente puedan ocasionarse. Si éste no prestara la caución en el plazo al efecto señalado por el Juez, que en ningún caso será inferior a 5 días hábiles, se entenderá que renuncia a las medidas. 2. En caso de que las medidas solicitadas impliquen restricciones para la actividad industrial o comercial del demandado, el Juez podrá señalar al tiempo de acordarlas, el importe de la fianza mediante la prestación de la cual dicho demandado podrá sustituir en cualquier momento la efectividad de dichas medidas restrictivas acordadas. 3. En todo caso, las fianzas que con carácter principal o sustitutorio se decreten para el demandado, se fijarán siempre en un tanto por período de tiempo que transcurra, cuando las mismas deriven de unos actos de explotación industrial o comercial que puedan tener continuidad indefinida. 4. La fianza podrá consistir en un aval bancario. No se admitirán las fianzas personales. 5. Para la fijación del importe de las fianzas el Juez deberá oír a ambas partes durante la tramitación de las medidas, sin perjuicio de la aplicación del artículo 733.2 de la Ley de Enjuiciamiento Civil. Artículo 130. Medidas cautelares en caso de apelación. 1. Si la sentencia de primera instancia dictada en el procedimiento civil de fondo estableciera pronunciamientos condenatorios para alguna de las partes y fuera objeto de apelación, se dará cuenta del recurso a la parte apelada para que ésta pueda, dentro del plazo de 3 días, exigir del Juez la adopción de las correspondientes medidas cautelares o la prestación de la oportuna fianza sustitutoria, tendentes al aseguramiento de la efectividad del fallo recaído, siempre que estas medidas no se hubieren adoptado previamente o fueren insuficientes. 2. El Juez de instancia mantendrá la competencia para tramitar y resolver lo pertinente sobre este incidente de aseguramiento, con independencia de la admisión de la apelación y la elevación de los autos principales al Tribunal al que corresponda conocer de los recursos de apelación. – 137 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] Artículo 131. Levantamiento de las medidas cautelares. 1. En el caso de formularse la petición de medidas cautelares antes de ejercitarse la acción principal, quedarán sin efecto en su totalidad si la demanda no se presentara en el plazo previsto por el artículo 730.2 de la Ley 1/2002, de 7 de enero, de Enjuiciamiento Civil. 2. Las medidas cautelares que se hubieran acordado en su caso, quedarán siempre sin efecto, si la sentencia dictada en primera instancia no fuere favorable a los pedimentos para el aseguramiento de cuya efectividad hubieren sido aquellas medidas solicitadas, o se revocara la sentencia de primera instancia, en el supuesto de que ésta hubiera sido favorable a los referidos pedimentos. Será aplicable en todo caso lo previsto en el artículo 744 de la citada Ley de Enjuiciamiento Civil. 3. El Secretario Judicial procederá a devolver las garantías al solicitante de las medidas cautelares transcurridos dos meses desde su levantamiento, sin perjuicio del plazo al que tenga derecho el perjudicado por las medidas cautelares para solicitar la indemnización por daños y perjuicios que aquellas le hubieran ocasionado. 4. La indemnización deberá determinarse de conformidad con lo establecido en el artículo 712 y concordantes de la Ley de Enjuiciamiento Civil, y si la fianza no fuera suficiente para hacer frente a todos los perjuicios ocasionados, se seguirá la vía de apremio contra el responsable. Artículo 132. Escritos preventivos. 1. La persona que prevea la interposición de una solicitud de medidas cautelares sin audiencia previa en su contra, podrá comparecer en legal forma ante el órgano o los órganos judiciales que considere competentes para conocer de dichas posibles medidas y justificar su posición mediante un escrito preventivo. El Juez o Tribunal acordará la formación de un procedimiento de medidas cautelares que notificará al titular de la patente y, si en el plazo de tres meses las medidas cautelares fueran presentadas, aquél podrá dar al procedimiento el curso previsto en los artículos 733.1 y 734.3 de la Ley de Enjuiciamiento Civil, sin que ello sea obstáculo a la posibilidad de acordarlas sin más trámite mediante auto en los términos y plazos del artículo 733.2 de dicha Ley. 2. El titular que considere que el Juez o Tribunal ante el que se presentó el escrito preventivo no es el competente, podrá presentar su solicitud de medidas cautelares ante aquél que entiende realmente competente, debiendo hacer constar en su solicitud la existencia del escrito preventivo y el órgano judicial ante el que éste se hubiere presentado. CAPÍTULO IV Solución extrajudicial de controversias Artículo 133. Conciliación en materia de invenciones de empleados. Antes de iniciar acciones judiciales basadas en la aplicación de las normas del Título IV de esta Ley relativo a las invenciones realizadas en el marco de una relación de empleo o de servicios la cuestión litigiosa podrá ser sometida, si las partes así lo acuerdan, a un acto de conciliación ante la Oficina Española de Patentes y Marcas. Artículo 134. Comisión de conciliación. 1. Para la conciliación se constituirá, de acuerdo con lo que se disponga reglamentariamente, una comisión presidida por un experto de la Oficina Española de Patentes y Marcas designado por su Director y otros dos elegidos respectivamente por cada una de las partes en conflicto, o cuando el inventor sea una persona al servicio de cualquiera de las Administraciones Públicas, y por lo que se refiere al representante de éstas, en la forma que reglamentariamente se establezca dentro del marco de la legislación laboral o estatutaria aplicable a la relación de empleo. 2. Cuando se trate de invenciones realizadas por el personal al que se refiere el apartado 1 del artículo 21, el miembro de la comisión que represente a la Universidad o al – 138 – CÓDIGO DE DERECHO FARMACÉUTICO § 5 Ley 24/2015, de Patentes [parcial] organismo o centro de investigación se designará en la forma que dispongan los estatutos u otra normativa interna de la Universidad o la normativa reguladora del organismo o centro de investigación. En su defecto su designación corresponderá, en el supuesto de las Universidades al Consejo de Gobierno y, en el caso de los organismos o centros de investigación, a su máximo órgano de gobierno. Artículo 135. Propuesta de acuerdo y conformidad. 1. Una propuesta de acuerdo deberá dictarse por la comisión en un plazo máximo de dos meses desde que se solicitó la conciliación, y las partes deberán manifestar en el plazo máximo de 15 días si están o no conformes con dicha propuesta. En caso de que no sea posible constituir la comisión de conciliación por incomparecencia de alguna de las partes, o alguna de ellas no acepte la propuesta de acuerdo dentro de los plazos respectivos, se dará por concluido el procedimiento. La aceptación deberá ser expresa. En caso de silencio se entenderá que no existe conformidad. 2. Si hubiere conformidad, el Director de la Oficina Española de Patentes y Marcas emitirá una certificación del acuerdo según la propuesta aceptada por las partes. A los efectos previstos en el artículo 517, apartado 2.9.º, de la Ley de Enjuiciamiento Civil, la certificación del Director de la Oficina Española de Patentes y Marcas del acuerdo, según la propuesta aceptada por las partes, llevará aparejada ejecución. 3. La ejecución del acuerdo se llevará a cabo conforme a lo establecido en la Ley de Enjuiciamiento Civil para la ejecución de sentencias y convenios judicialmente aprobados. Artículo 136. Arbitraje y mediación. 1. Los interesados podrán recurrir a la mediación o someter a arbitraje las cuestiones litigiosas surgidas entre ellos con ocasión del ejercicio de los derechos reconocidos en esta Ley, en aquellas materias no excluidas de la libre disposición de las partes conforme a derecho. 2. No son de libre disposición, y quedan excluidas de la mediación o el arbitraje, las cuestiones relativas a los procedimientos de concesión, oposición o recursos referentes a los títulos regulados en esta Ley, cuando el objeto de la controversia sea el cumplimiento de los requisitos exigidos para su concesión, su mantenimiento o su validez. 3. El laudo arbitral firme producirá efectos de cosa juzgada de acuerdo con lo establecido en el artículo 43 de la Ley 60/2003, de 23 de diciembre, de Arbitraje, que será de aplicación en todo lo no previsto por el presente artículo, y la Oficina Española de Patentes y Marcas procederá a realizar las actuaciones necesarias para su ejecución. 4. El acuerdo de mediación suscrito por el mediador y las partes, una vez que elevado a escritura pública u homologado por el Juez se constituya como título ejecutivo de acuerdo con lo dispuesto en la Ley 5/2012, de 6 de julio, de mediación en asuntos civiles y mercantiles, se comunicará a la Oficina Española de Patentes y Marcas para proceder a la ejecución del mismo. 5. Deberán comunicarse a la Oficina Española de Patentes y Marcas la presentación de los recursos que se interpongan frente al laudo arbitral o el ejercicio de una acción de nulidad contra lo convenido en el acuerdo de mediación. Una vez firmes las correspondientes resoluciones se comunicarán fehacientemente a la Oficina Española de Patentes y Marcas a los efectos previstos en el apartado anterior. [...] – 139 – CÓDIGO DE DERECHO FARMACÉUTICO §6 Real Decreto 316/2017, de 31 de marzo, por el que se aprueba el Reglamento para la ejecución de la Ley 24/2015, de 24 de julio, de Patentes. [Inclusión parcial] Ministerio de la Presidencia y para las Administraciones Territoriales «BOE» núm. 78, de 1 de abril de 2017 Última modificación: sin modificaciones Referencia: BOE-A-2017-3550 [...] REGLAMENTO DE EJECUCIÓN DE LA LEY 24/2015, DE 24 DE JULIO, DE PATENTES TÍTULO I Patentes de invención CAPÍTULO I Solicitud de patente Artículo 1. Solicitud de patente. Para la obtención de una patente de invención deberá formularse la solicitud a que se refiere el artículo 23 y siguientes de la Ley 24/2015, de 24 de julio, de Patentes (en adelante, la Ley), con sujeción a lo que se determina en los artículos siguientes. Artículo 2. Requisitos de la instancia de la solicitud de patente. 1. La instancia por la que se solicita la patente, que se formalizará en un modelo oficial, deberá dirigirse al Director de la Oficina Española de Patentes y Marcas y contener los siguientes datos: a) Indicación de que se solicita una patente de invención. b) La identidad del solicitante. Si hubiera varios solicitantes, se hará constar la identidad de cada uno de ellos. Cuando el solicitante sea una persona física se identificará con su nombre y apellido o apellidos, documento de identidad, domicilio y nacionalidad; y cuando sea una persona jurídica se identificará por su denominación social completa o de acuerdo con las disposiciones legales por las que se rija, su NIF, domicilio y nacionalidad. – 140 – CÓDIGO DE DERECHO FARMACÉUTICO § 6 Reglamento para la ejecución de la Ley 24/2015, de Patentes [parcial] En el supuesto de que hubiera varios solicitantes, se especificará la dirección o medio de comunicación de uno de ellos a efectos de notificaciones; de no hacerse así, las notificaciones se dirigirán al solicitante mencionado en primer lugar en la solicitud. c) Sin perjuicio de lo dispuesto en el artículo 175.2 de la Ley, en el supuesto de que el solicitante actúe por sí mismo y no tenga domicilio ni sede o establecimiento comercial serio y efectivo en territorio del Estado español deberá designar, a efectos de notificaciones, una dirección postal en España o indicar que las notificaciones le sean dirigidas por cualquier otro medio técnico de comunicación admitido por la Oficina Española de Patentes y Marcas. d) Título de la invención, en el que, sin denominaciones de fantasía y de la manera más clara y concisa posible, aparezca la designación técnica de la invención que deberá ser congruente con las reivindicaciones. e) La designación del inventor o inventores, con indicación de su nombre y apellido o apellidos; en el supuesto de que el solicitante no fuera el inventor o el único inventor, se indicará cómo ha adquirido el derecho a la patente en relación con cada uno de los inventores. f) Relación de documentos que acompañan a la solicitud. g) La firma del solicitante o de su representante. 2. En su caso, la instancia deberá ser completada con los siguientes datos: a) Cuando el solicitante actúe por medio de un representante, se indicará su identidad, de conformidad con el párrafo b) anterior. En el supuesto de que el representante fuera un Agente de la Propiedad Industrial al que se refiere el artículo 176 de la Ley, solo se indicará el nombre y apellidos del Agente, persona física, o la denominación social de la persona jurídica a través de la cual realice su actividad el Agente de la Propiedad Industrial, mencionando el código de Agente otorgado por la Oficina Española de Patentes y Marcas. b) En el caso en que se solicite una patente divisional, un cambio de modalidad, una transformación de una solicitud de patente europea o una entrada en fase nacional de una solicitud internacional PCT, se indicará el número y la fecha de la solicitud de origen. Igualmente se indicará que el solicitante tiene derecho a presentar dicha solicitud. c) Cuando la solicitud se remita a una solicitud presentada con anterioridad según lo dispuesto en los apartados 1.c) y 2 del artículo 24 de la Ley, deberá indicarse el número de la citada solicitud anterior, la fecha de presentación y la oficina en la que o para la que se haya presentado. Igualmente se indicará que el solicitante tiene derecho a presentar dicha solicitud. d) En el caso de que el inventor o inventores renuncien a su derecho a ser mencionados como tales, se indicará en la instancia o, si el inventor o inventores no coinciden con el solicitante, se aportará una declaración de renuncia firmada por ellos. e) En el supuesto de que se reivindique una o varias prioridades extranjeras o nacionales, la instancia deberá contener el número de cada una de las solicitudes anteriores sobre las que se basa la prioridad, así como el Estado y la fecha de prioridad reivindicada. Igualmente, se hará constar que el solicitante tiene derecho a reivindicar la prioridad indicada. f) Si la invención hubiera sido exhibida en exposiciones oficiales u oficialmente reconocidas en el sentido del artículo 7.b) de la Ley, la instancia deberá contener la indicación del nombre de la exposición, así como el lugar y fecha de exhibición. g) Cuando la invención se refiera a una materia biológica no accesible al público, o a su utilización, y no pueda ser descrita en la solicitud de patente, y esta haya sido depositada en una institución reconocida legalmente para ello, deberá indicarse la institución de depósito, el país, la fecha de depósito y el número de depósito otorgado por la institución de depósito. h) Cuando la invención se refiera a materia biológica, se indicará su origen geográfico o la fuente de procedencia de dicha materia, si estos datos fueran conocidos. Cuando la invención se refiera a un recurso genético o a un conocimiento tradicional asociado a dicho recurso cubierto por el Reglamento (UE) n.º 511/2014 del Parlamento Europeo y del Consejo, de 16 de abril de 2014, relativo a las medidas de cumplimiento de los usuarios del Protocolo de Nagoya sobre el acceso a los recursos genéticos y participación justa y equitativa en los beneficios que se deriven de su utilización en la Unión, se indicará si se ha utilizado un recurso genético o un conocimiento tradicional asociado a dicho recurso. – 141 – CÓDIGO DE DERECHO FARMACÉUTICO § 6 Reglamento para la ejecución de la Ley 24/2015, de Patentes [parcial] En caso afirmativo, cuando proceda, se hará constar el número de registro que justifique la presentación de la declaración de diligencia debida de conformidad con el artículo 14. 3 el Real Decreto 124/2017, 24 de febrero, relativo al acceso a los recursos genéticos procedentes de taxones silvestres y al control de la utilización. En cualquier caso, estas informaciones no prejuzgarán la validez de la patente, tal como dispone el artículo 23.2 de la Ley. i) Cuando la solicitud contenga listas de secuencias de aminoácidos y ácidos nucleicos, se indicará esta circunstancia. j) Si se solicitara la reducción de tasas prevista en el artículo 186 de la Ley, deberá mencionarse este extremo. k) Si el solicitante es universidad pública, deberá mencionarse este extremo. Artículo 3. Contenido de la descripción. 1. La descripción estará redactada en la forma más concisa y clara posible, sin repeticiones inútiles, y en congruencia con las reivindicaciones. 2. En la misma se indicarán los siguientes datos: a) La indicación del sector de la técnica al que se refiera la invención. b) La indicación del estado de la técnica anterior a la fecha de prioridad, conocido por el solicitante y que pueda ser útil para la comprensión de la invención y para la elaboración del informe sobre el estado de la técnica y para el examen, citando, en la medida de lo posible, los documentos que sirvan para reflejar el estado de la técnica anterior. c) Una explicación de la invención, tal como es caracterizada en las reivindicaciones, que permita la comprensión del problema técnico planteado, aunque no se designe expresamente de este modo, así como la solución al mismo, indicándose, en su caso, las ventajas de la invención en relación con el estado de la técnica anterior. d) Una breve descripción del contenido de los dibujos, si los hubiera. e) Una exposición detallada de, al menos, un modo de realización de la invención, que podrá ilustrarse con ejemplos y referencias, en su caso, a los dibujos, si los hubiera. f) La indicación de la manera en que la invención es susceptible de aplicación industrial, a no ser que ello resulte de una manera evidente de la descripción o de la naturaleza de la invención. En el supuesto de que la invención consista en una secuencia total o parcial de un gen o de una secuencia de ácido nucleico, tal como disponen el párrafo tercero del artículo 5.5 y el artículo 5.6 de la Ley, respectivamente, la aplicación industrial deberá figurar explícitamente. 3. La descripción deberá ser presentada de la manera y en el orden indicados en el apartado 2 de este artículo, a menos que, por causa debida a la naturaleza de la invención, una manera o un orden diferente permitan una mejor comprensión y una presentación más concisa. Artículo 4. Requisitos de la descripción en las invenciones referidas a materia biológica. 1. Cuando la invención se refiera a una materia biológica, el solicitante deberá indicar, en la descripción, cuál es el nombre de la institución autorizada donde haya depositado una muestra de la materia biológica y consignar el número o clave de identificación de dicha materia biológica por la Institución autorizada. 2. Si la materia biológica depositada deja de estar disponible en la autoridad de depósito reconocida, se considerará que no se ha interrumpido la accesibilidad a condición de que se haya realizado un nuevo depósito en las mismas condiciones que las previstas en el Tratado de Budapest sobre reconocimiento internacional de depósito de microorganismos a los fines del procedimiento en materia de patentes (hecho en Budapest, el 28 de abril de 1977) y de que, en un plazo de cuatro meses a partir de la fecha del nuevo depósito, se haya comunicado a la Oficina Española de Patentes y Marcas una copia del recibo de ese nuevo depósito expedido por la autoridad de depósito, acompañada de la indicación del número de solicitud de patente o de la patente. 3. La comunicación de esta información implica el consentimiento irrevocable del solicitante de que la materia biológica sea accesible al público de acuerdo con el artículo 56 de la Ley. – 142 – CÓDIGO DE DERECHO FARMACÉUTICO § 6 Reglamento para la ejecución de la Ley 24/2015, de Patentes [parcial] Artículo 5. Condiciones para el acceso público a la materia biológica. 1. El acceso a la materia biológica depositada se realizará, en los plazos previstos en el artículo 56 de la Ley, mediante el envío de una muestra de la materia biológica solicitada, siempre y cuando la persona que solicite el acceso a la materia biológica se comprometa frente al solicitante o titular de la patente: a) A no comunicar ni entregar a terceros la materia biológica objeto de la patente o un cultivo derivado de ella, antes que la solicitud de patente haya sido denegada o retirada, o sea considerada retirada o que la patente haya caducado. b) A no utilizar la materia biológica objeto de la patente o un cultivo derivado de ella, más que con fines experimentales hasta la fecha en que la solicitud de patente sea rechazada o retirada, o considerada retirada, o hasta la fecha de la publicación de la mención de la concesión de la patente. 2. Cuando, por cualquier razón, la institución autorizada no pueda remitir muestras de la materia biológica depositada, será de aplicación lo dispuesto en el Tratado de Budapest sobre reconocimiento internacional de depósito de microorganismos a los fines del procedimiento en materia de patentes y su Reglamento de ejecución (hechos en Budapest el 28 de abril de 1977). 3. Lo dispuesto en los apartados anteriores se entenderá sin perjuicio de la aplicación de los artículos 6 y 7 del Real Decreto 124/2017, de 24 de febrero, relativo al acceso a los recursos genéticos procedentes de taxones silvestres y al control de la utilización, cuando se trate de material regulado por los artículos 71, 72, 74, 80 y 81 de la Ley 42/2007, de 13 de diciembre, del Patrimonio Natural y de la Biodiversidad. Artículo 6. Figura del experto independiente. 1. El solicitante podrá realizar una petición a la Oficina Española de Patentes y Marcas hasta la finalización de los preparativos técnicos para la publicación de la solicitud de patente, para que el acceso a la materia biológica depositada al que se refiere el artículo 56 de la Ley se lleve a cabo únicamente a través del suministro de la muestra a un experto independiente. Este acceso se lleve a cabo en los siguientes plazos: a) Hasta la publicación de la mención de la concesión de la patente o, cuando sea aplicable, b) Durante veinte años desde la fecha de presentación, si la solicitud se deniega o se retira, o se considera retirada. 2. Podrá ser nombrado como experto independiente, a los fines de lo previsto en el artículo 56 de la Ley: a) Cualquier persona física, siempre que el peticionario demuestre en el momento de hacer la petición que dicho nombramiento tiene la aprobación del solicitante de la patente. b) Cualquier persona física que tenga el reconocimiento de experto independiente por el Director de la Oficina Española de Patentes y Marcas. El nombramiento deberá ir acompañado de una declaración del experto independiente por la cual se compromete frente al solicitante a respetar lo establecido en el artículo 56 de la Ley, bien hasta que caduque la patente o bien hasta la fecha indicada en el párrafo b) del apartado 1 del presente artículo si la solicitud se deniega o se retira, o se considera retirada. En este sentido, el peticionario de la muestra deberá ser considerado como un tercero y se aplicará lo dispuesto en el artículo 56.2 de la Ley. 3. La petición a la que se refiere el apartado 1 se presentará ante la Oficina Española de Patentes y Marcas. Se comprobará que se ha presentado una solicitud de patente que se refiere a una materia biológica depositada y que el peticionario o el experto independiente nombrado por el solicitante están autorizados a recibir una muestra de dicho material. – 143 – CÓDIGO DE DERECHO FARMACÉUTICO § 6 Reglamento para la ejecución de la Ley 24/2015, de Patentes [parcial] Artículo 7. Contenido y forma de las reivindicaciones. 1. Las reivindicaciones definirán el objeto para el que se solicita la protección en términos de características técnicas de la invención. Cuando sea apropiado, las reivindicaciones deberán contener: a) Un preámbulo mencionando el objeto de la invención y las características técnicas necesarias para la definición de los elementos reivindicados pero que, combinadas entre ellas, forman parte del estado de la técnica. b) Una parte caracterizadora que, comenzando con una expresión del tipo «caracterizado por», exponga las características técnicas que, en combinación con las mencionadas en el párrafo a), se desean proteger. 2. Sin perjuicio de lo dispuesto en el artículo 26 de la Ley, una misma solicitud puede comprender más de una reivindicación independiente de una misma categoría (producto, procedimiento, dispositivo o utilización), siempre que el objeto de la solicitud consista en: a) Una pluralidad de productos interrelacionados. b) Diferentes utilizaciones de un producto o dispositivo. c) Soluciones alternativas a un problema particular, cuando no sea adecuado incluir estas alternativas en una misma reivindicación. 3. Cualquier reivindicación independiente deberá incluir las características esenciales de la invención y podrá ir seguida de una o varias reivindicaciones dependientes relativas a realizaciones particulares de dicha invención. 4. Cualquier reivindicación dependiente, esto es, que incluya todas las características contenidas en cualquier otra reivindicación, deberá contener, preferiblemente al comienzo, una referencia a la reivindicación de la que depende y, a continuación, las características adicionales que se desean proteger. Será también admisible una reivindicación dependiente referida a una o más reivindicaciones dependientes. Todas las reivindicaciones dependientes referidas a una reivindicación anterior única, y todas las reivindicaciones dependientes que se refieran a varias reivindicaciones anteriores, deberán agruparse en la medida y forma más apropiada posible. 5. El número de reivindicaciones deberá ser adecuado y razonable atendiendo a la naturaleza de la invención cuya protección se solicita. 6. Las reivindicaciones no realizarán remisiones a la descripción o a los dibujos para definir las características técnicas de la invención, salvo que fuera absolutamente necesario. En particular, no deberán contener expresiones del tipo «tal como se describe en la parte… de la descripción» o «tal como se ilustra en la figura… de los dibujos». 7. Cuando la solicitud de patente contenga dibujos que incluyan signos de referencia, las características técnicas reivindicadas deberán, preferiblemente, venir seguidas de tales signos de referencia que las identifiquen siempre que con ello se contribuya a la comprensión de la reivindicación. Tales signos se representarán entre paréntesis y no se considerará que limitan las reivindicaciones. Artículo 8. Presentación de dibujos. 1. Los dibujos deberán realizarse con los requisitos que se especifican en el anexo de este Reglamento. 2. Los esquemas de etapas de un proceso y los diagramas se consideran como dibujos. Artículo 9. Resumen de la invención. 1. El resumen al que se refiere el artículo 29 de la Ley tendrá una extensión máxima de ciento cincuenta palabras, deberá indicar el título de la invención y contener una exposición concisa del contenido de la descripción, reivindicaciones y, en su caso, dibujo más característico que deberá situarse separadamente del texto del resumen. Asimismo, se podrá indicar la fórmula química que, entre las que figuran en la solicitud de patente, caracterice mejor la invención. El resumen deberá permitir una fácil comprensión del problema técnico planteado, la solución aportada y el uso o usos principales de la invención. – 144 – CÓDIGO DE DERECHO FARMACÉUTICO § 6 Reglamento para la ejecución de la Ley 24/2015, de Patentes [parcial] 2. Tanto el título como el resumen de la invención podrán ser modificados por la Oficina Española de Patentes y Marcas cuando lo estime necesario para la mejor información de los terceros. De dicha modificación se dará traslado al solicitante en la opinión escrita. Artículo 10. Normas generales relativas a la presentación de los documentos de la solicitud. Los requisitos formales para la presentación de la solicitud y documentos que la acompañan se recogen en el anexo al presente Reglamento. Artículo 11. Elementos prohibidos. La solicitud de patente no podrá contener: a) Elementos contrarios al orden público y a las buenas costumbres. b) Declaraciones denigratorias relativas a productos o procedimientos de terceros o al mérito o validez de las solicitudes de patentes o patentes de terceros. Las simples comparaciones con el estado de la técnica no serán consideradas en sí mismas como denigrantes. c) Elementos manifiestamente extraños a la solicitud o superfluos. Artículo 12. Designación del inventor. 1. La designación del inventor o inventores contenida en la solicitud de patente se incluirá en las publicaciones de la solicitud de patente y de la concesión, así como en los folletos a los que se refieren respectivamente los artículos 31 y 35.3 del presente Reglamento. 2. Si el inventor o inventores renuncian a ser mencionados como tales, la declaración firmada a que se refiere el artículo 2.2.d) del presente Reglamento deberá ser aportada antes de que concluyan los preparativos técnicos para la publicación de la solicitud de patente. Artículo 13. Prioridad de la solicitud de patente. 1. La declaración por la que se reivindique una prioridad nacional o extranjera prevista en el artículo 31 de la Ley, indicará, tal como se establece en el artículo 2.2.e) del presente Reglamento, la fecha de la solicitud anterior, el Estado en el cual o para el cual ha sido efectuada, así como el número que le ha sido atribuido. La reivindicación de prioridad implicará el pago de la tasa correspondiente. 2. Cuando la reivindicación de prioridad se considere relevante para determinar la patentabilidad de la invención, la Oficina Española de Patentes y Marcas podrá exigir al solicitante que aporte, en el plazo de dos meses desde la comunicación o dieciséis meses desde la fecha de prioridad más antigua reivindicada, aplicándose el plazo que expire más tarde, una copia certificada de la solicitud anterior emitida por la oficina de origen, salvo que dicho documento obre en los archivos de la Oficina Española de Patentes y Marcas o cuando esté disponible desde una biblioteca digital aceptada por la Oficina Española de Patentes y Marcas. Si la solicitud anterior no estuviera redactada en español el solicitante aportará igualmente una traducción al español de dicho documento dentro del mismo plazo. Si no se aporta la copia certificada y, en su caso, la traducción al español en el plazo prescrito, el derecho de prioridad no se considerará válidamente reivindicado. Artículo 14. Corrección o adición de una reivindicación de prioridad. 1. Un solicitante podrá pedir la corrección o adición de la reivindicación de prioridad respecto de una solicitud de patente presentada ante la Oficina Española de Patentes y Marcas. La petición de corrección o adición deberá ir firmada y presentarse dentro del plazo que expire más tarde de entre los siguientes: a) En el plazo de dieciséis meses a partir de la fecha de prioridad más antigua o, cuando la corrección o adición suponga un cambio en la fecha de prioridad más antigua, en el plazo de dieciséis meses a partir de la fecha de prioridad más antigua modificada, aplicándose el plazo de dieciséis meses que expire antes; – 145 – CÓDIGO DE DERECHO FARMACÉUTICO § 6 Reglamento para la ejecución de la Ley 24/2015, de Patentes [parcial] b) en el plazo de cuatro meses desde la fecha de presentación de la solicitud de patente. 2. Si una petición de corrección o adición se recibe en la Oficina Española de Patentes y Marcas después de que el solicitante haya pedido una publicación anticipada en virtud del artículo 37.2 de la Ley, se considerará como no presentada la petición de corrección o adición, salvo que la petición de publicación anticipada se retirara antes de que hayan finalizado los preparativos técnicos para la publicación de la solicitud de patente. 3. En el caso en que se solicite una patente divisional, un cambio de modalidad, una transformación de una solicitud de patente europea o una entrada en fase nacional de una solicitud internacional PCT, el plazo para pedir la corrección o adición de la reivindicación de prioridad será de cuatro meses desde la fecha de depósito de la solicitud de que se trate o de dieciséis meses de la fecha de prioridad, aplicándose el plazo que expire más tarde. 4. Los plazos previstos en los párrafos precedentes no serán susceptibles de prórroga ni de petición de restablecimiento de derechos. 5. Antes de que la Oficina Española de Patentes y Marcas proceda a la denegación de una adición o una corrección de una reivindicación de prioridad, concederá al peticionario un plazo de diez días a contar desde la publicación de la intención de denegar en el «Boletín Oficial de la Propiedad Industrial» para que formule observaciones. Artículo 15. Exhibición en exposiciones oficiales u oficialmente reconocidas. 1. En el caso previsto en el párrafo segundo del artículo 7.b) de la Ley y en el artículo 2.2.f) del presente Reglamento, el solicitante deberá aportar una certificación expedida por la persona que se designe como autoridad encargada de asegurar la protección de la propiedad industrial en dicha exposición, que acredite que la invención ha sido realmente exhibida en la misma durante el período de su celebración. Esta certificación deberá también mencionar la fecha de apertura de la exposición y, en su caso, la de la primera divulgación de la invención si estas dos fechas no fueran coincidentes. A la certificación deberán acompañarse los documentos que permitan identificar la invención, debidamente autenticada por la autoridad mencionada. 2. El plazo para presentar esta certificación, así como la documentación adjunta, será de cuatro meses a contar desde la fecha de presentación de la solicitud o hasta la finalización del plazo previsto en el artículo 24 del presente Reglamento, aplicándose el plazo que expire más tarde. CAPÍTULO II Procedimiento de concesión Sección 1.ª Admisión a trámite y examen de oficio Artículo 16. Marcas. Recepción de la solicitud y remisión a la Oficina Española de Patentes y 1. El órgano competente para recibir la solicitud conforme a lo dispuesto en el artículo 22 de la Ley hará constar el número de registro, así como el día, hora y minuto de depósito tanto en el lugar destinado para ello en la instancia de la solicitud, como en la documentación que la acompañe, en su caso. 2. En el momento del depósito, el órgano competente expedirá al depositante un recibo acreditativo de la presentación de la solicitud, en el que se hará constar el número de registro y el lugar, día, hora y minuto de depósito. Si la solicitud fuese acompañada de una copia, el recibo consistirá en la entrega de dicha copia, en la que se hará constar el número de registro y el lugar, día, hora y minuto del depósito. 3. Cuando la solicitud de patente se haya presentado ante el órgano competente de una comunidad autónoma, este remitirá la solicitud, junto con toda la documentación aportada, a la Oficina Española de Patentes y Marcas en el plazo prescrito en el artículo 32.2 de la Ley. 4. Una vez recibida la solicitud por la Oficina Española de Patentes y Marcas, se le asignará número de solicitud de patente que se le notificará al solicitante. – 146 – CÓDIGO DE DERECHO FARMACÉUTICO § 6 Reglamento para la ejecución de la Ley 24/2015, de Patentes [parcial] Artículo 17. Requisitos para obtener fecha de presentación. 1. A los efectos de lo dispuesto en el artículo 24 de la Ley y el artículo 18 del presente Reglamento, será indispensable presentar, para obtener una fecha de presentación de la solicitud de patente, los siguientes documentos: a) Una indicación expresa o implícita de que se solicita una patente, b) indicaciones que permitan identificar o contactar con el solicitante y c) una parte que, a primera vista, parezca constituir una descripción, aunque no cumpla con los requisitos formales establecidos en la Ley o el Reglamento, o una incorporación por referencia, es decir, una remisión a una solicitud presentada con anterioridad. 2. A los efectos de obtener fecha de presentación, las indicaciones de los párrafos a) y b) del apartado anterior deberán presentarse en español. Sin embargo, la descripción podrá redactarse en cualquier idioma, debiendo presentarse una traducción al español en el plazo de dos meses desde la fecha de depósito de la solicitud de patente o hasta la finalización del plazo previsto en el artículo 24 del presente Reglamento, aplicándose el plazo que expire más tarde. 3. A los efectos de obtener fecha de presentación, una remisión a una solicitud presentada con anterioridad reemplazará a la descripción y, en su caso, a cualesquiera dibujos. Para realizar esta remisión, el solicitante, en el momento de presentar la solicitud de patente, deberá efectuar una petición de incorporación por referencia a una solicitud anterior, en la que deberá indicar en español: a) Que la remisión a la solicitud anterior reemplaza a la descripción y, en su caso, a los dibujos. b) El número de la solicitud anterior, su fecha de presentación y la oficina en la que o para la que haya sido presentada. c) Que la solicitud anterior ha sido presentada por el propio solicitante, su antecesor en derecho o su derechohabiente. 4. Si la solicitud de patente se remite a una solicitud anterior según lo previsto en el apartado precedente, el solicitante deberá aportar, en el plazo de dos meses desde la fecha del depósito de la solicitud, una copia certificada de la solicitud anterior y, en su caso, una traducción al español. No será necesario aportar la copia certificada de la solicitud anterior o la traducción al español si dicha copia o traducción obran en los archivos de la Oficina Española de Patentes y Marcas o están disponibles en una biblioteca digital aceptada por la Oficina Española de Patentes y Marcas. Artículo 18. Otorgamiento de la fecha de presentación y admisión a trámite. 1. Dentro de los diez días siguientes a la recepción de la solicitud de patente en la Oficina Española de Patentes y Marcas, esta examinará si la misma reúne los requisitos necesarios para obtener fecha de presentación según lo dispuesto en el artículo 24 de la Ley y el artículo 17 del presente Reglamento. 2. Si al examinar los requisitos necesarios para obtener una fecha de presentación se advirtieran defectos, se comunicarán al solicitante para que los subsane y formule alegaciones en el plazo de dos meses a contar desde su notificación, con indicación de que, si así no lo hiciera, no se admitirá a trámite y se resolverá teniendo por desistida la solicitud de patente. 3. Si los defectos son corregidos en plazo, se otorgará como fecha de presentación la que corresponda al día en que se hayan cumplido todos los requisitos y así se comunicará al solicitante. Si los defectos no se subsanaran en la debida forma y en el plazo prescrito, la solicitud no será admitirá como solicitud de patente y se tendrá por desistida. La resolución de desistimiento se notificará al solicitante con indicación de los motivos y se publicará en el «Boletín Oficial de la Propiedad Industrial». 4. Una vez otorgada fecha de presentación, se examinará si las tasas de solicitud y por realización del informe sobre el estado de la técnica han sido abonadas. Si se comprobara la falta de pago de las tasas o el pago insuficiente, se comunicará al solicitante para que realice o complete el pago en el plazo de un mes a contar desde la publicación del defecto en el – 147 – CÓDIGO DE DERECHO FARMACÉUTICO § 6 Reglamento para la ejecución de la Ley 24/2015, de Patentes [parcial] «Boletín Oficial de la Propiedad Industrial», con indicación de que si así no lo hiciera se tendrá por desistida la solicitud. La resolución de desistimiento se notificará al solicitante y se publicará en el «Boletín Oficial de la Propiedad Industrial». Artículo 19. Incorporación por referencia a una solicitud anterior. 1. En el supuesto de que el solicitante hubiera pedido la incorporación por referencia en virtud de lo dispuesto en los artículos 24.1.c) de la Ley y 17.3 del presente Reglamento, pero no hubiera aportado la copia certificada de la solicitud anterior según lo dispuesto en el apartado 4 del citado artículo 17, y este documento no esté a disposición de la Oficina Española de Patentes y Marcas, esta circunstancia se notificará al solicitante para que aporte dicha documentación en el plazo de dos meses a contar desde su notificación, con indicación de que si así no lo hiciera no será admitida a trámite y se resolverá teniendo por desistida la solicitud de patente. 2. Si los defectos son corregidos en plazo, se mantendrá como fecha de presentación aquella en la que se hubiesen cumplido todos los requisitos del artículo 17.1 del presente Reglamento y así se comunicará al solicitante. Si los defectos no se subsanaran en la debida forma y en el plazo prescrito, la solicitud no se admitirá como solicitud de patente y se tendrá por desistida. La resolución de desistimiento será notificada al solicitante y publicada en el «Boletín Oficial de la Propiedad Industrial». Artículo 20. Partes omitidas de la descripción o dibujos omitidos. 1. En el caso de que, al examinar si la solicitud de patente reúne los requisitos necesarios para obtener una fecha de presentación, la Oficina Española de Patentes y Marcas detectara que parece faltar una parte de la descripción o parecen faltar dibujos a los que hace referencia la descripción, este defecto se comunicará al solicitante para que, en el plazo de dos meses desde su notificación, complete la solicitud o indique si se remite a una solicitud anterior cuya prioridad se reivindica. 2. Si el solicitante completa la solicitud en el plazo de dos meses desde la fecha de depósito de la solicitud de patente o desde la notificación mencionada en el apartado anterior, se otorgará como fecha de presentación la fecha en la que se reciba la parte omitida de la descripción o los dibujos omitidos, o la fecha en la que se cumplan todos los requisitos prescritos en el artículo 24 de la Ley y el artículo 17 del presente Reglamento, aplicándose la fecha que sea posterior. Esta fecha se comunicará al solicitante. Se mantendrá como fecha de presentación aquella en la que se cumplan todos los requisitos prescritos en el artículo 24 de la Ley y el artículo 17 del presente Reglamento, en el supuesto de que el solicitante retire, en el plazo de un mes desde su aportación, la parte omitida de la descripción o el dibujo omitido. 3. Si la solicitud de patente reivindica la prioridad de una solicitud anterior, el solicitante podrá indicar que se remite a esa solicitud anterior para incorporar la parte omitida de la descripción o los dibujos omitidos. En este caso, la parte omitida de la descripción o los dibujos omitidos deberán estar contenidos íntegramente en la solicitud anterior. Se mantendrá como fecha de presentación la fecha en la que se hubieran cumplido todos los requisitos prescritos en el artículo 24 de la Ley y el artículo 17 del presente Reglamento, si el solicitante presenta en el plazo prescrito en el apartado 2: a) Una petición indicando que el contenido de la solicitud anterior se incorpore por referencia en la solicitud. b) Una copia certificada de la solicitud anterior y, en su caso, una traducción al español, salvo que dichos documentos estén a disposición de la Oficina. c) Una indicación del lugar en el que la parte omitida de la descripción o el dibujo omitido figuran en la solicitud anterior o en la traducción, en su caso. 4. En caso de que el solicitante no conteste a la comunicación del apartado 1, la fecha de presentación será la fecha en la que se hayan cumplido todos los requisitos prescritos en el artículo 24 de la Ley y el artículo 17 del presente Reglamento. Sin embargo, no se tendrán en cuenta la parte omitida de la descripción ni los dibujos omitidos. – 148 – CÓDIGO DE DERECHO FARMACÉUTICO § 6 Reglamento para la ejecución de la Ley 24/2015, de Patentes [parcial] Artículo 21. Notificación conjunta de defectos y plazo de subsanación. Los defectos previstos en los artículos 18, 19 y 20 del presente Reglamento podrán ser comunicados al solicitante conjuntamente mediante una única notificación, otorgando un plazo común de dos meses a contar desde su notificación para su subsanación, con indicación de que si no lo hiciera se tendrá por desistida la solicitud. Artículo 22. Patentes de interés para la defensa nacional. 1. En la admisión a trámite de la solicitud, la Oficina Española de Patentes y Marcas examinará si el objeto de la invención pudiera ser de interés para la defensa nacional. En caso afirmativo, en aplicación de los artículos 33 y 34 de la Ley, la Oficina Española de Patentes y Marcas pondrá a disposición del Ministerio de Defensa la solicitud de patente una vez admitida a trámite. 2. Si el Ministerio de Defensa emitiera un informe motivado considerando que la invención interesa a la defensa nacional, será de aplicación la tramitación secreta de la solicitud de patente conforme a lo dispuesto en los artículos 47 y siguientes del presente Reglamento. Artículo 23. Examen de oficio. 1. Otorgada fecha de presentación y abonadas las tasas correspondientes, la Oficina Española de Patentes y Marcas examinará, a los efectos de la publicación de la solicitud: a) Si la instancia se ajusta a lo establecido en el artículo 2 del presente Reglamento. b) En el supuesto de que la descripción se presentara en un idioma distinto al español, si el solicitante ha aportado la correspondiente traducción a la que se refiere el artículo 17.2 del presente Reglamento. c) Si la solicitud contiene una o varias reivindicaciones según lo dispuesto en el artículo 23.1.c) de la Ley o una referencia a una solicitud presentada con anterioridad, en virtud del artículo 17.3 del presente Reglamento, indicando que también reemplaza las reivindicaciones. d) En el supuesto de que el solicitante hubiera pedido una incorporación por referencia según lo dispuesto en los artículos 24.1.c) de la Ley y 17.3 del presente Reglamento, si el solicitante ha aportado la traducción a la que se refiere el artículo 17.4 del presente Reglamento. e) Si la descripción, las reivindicaciones, los dibujos y el resumen guardan las formalidades previstas en el anexo del presente Reglamento, únicamente en la medida en que su cumplimiento sea necesario a los fines de una publicación uniforme. f) Si la solicitud reivindica la prioridad de una solicitud anterior, nacional o extranjera, o la divulgación inocua derivada de la exhibición en una exposición oficial u oficialmente reconocida se examinará si cumplen los requisitos exigidos en los artículos 13 a 15 del presente Reglamento. g) En el supuesto de solicitudes de patentes divisionales, de cambio de modalidad, de transformación de una solicitud de patente europea o de entrada en fase nacional de una solicitud internacional PCT, si las menciones al número y a la fecha de solicitud de la patente de origen, han sido realizadas. h) Si cumple los requisitos relativos a la representación según lo dispuesto en el artículo 175 de la Ley y en los artículos 107 y 108 del presente Reglamento. i) Si el objeto de la solicitud no está manifiestamente y en su totalidad excluido de patentabilidad por aplicación de los artículos 4.4 y 5 de la Ley. 2. La presencia de defectos formales en la documentación no suspenderá la realización del informe sobre el estado de la técnica siempre que aquellos no sean de tal naturaleza que impidan su realización o desvirtúen de tal modo el objeto de búsqueda que su resultado resulte inservible. – 149 – CÓDIGO DE DERECHO FARMACÉUTICO § 6 Reglamento para la ejecución de la Ley 24/2015, de Patentes [parcial] Artículo 24. Notificación de defectos. 1. Si la solicitud de patente presentase alguno de los defectos mencionados en el artículo anterior, la Oficina Española de Patentes y Marcas notificará al solicitante todas las objeciones para que este, en el plazo de dos meses a contar desde la publicación de defectos en el «Boletín Oficial de la Propiedad Industrial», subsane los defectos o efectúe las alegaciones que estime oportunas en defensa de la solicitud de patente. En la medida en la que sea necesario para corregir los defectos notificados, el solicitante podrá modificar la descripción, reivindicaciones y dibujos o secuencias biológicas, en los términos previstos en el artículo 48 de la Ley. 2. La contestación a la notificación de defectos implicará el pago de la tasa correspondiente. Artículo 25. Denegación de la solicitud. 1. Transcurrido el plazo para la subsanación de defectos o para la presentación de alegaciones previsto en el artículo 24 del presente Reglamento, la Oficina Española de Patentes y Marcas examinará si los defectos han sido debidamente subsanados y si se ha pagado la tasa correspondiente. En caso contrario, se denegará la solicitud. Dicha resolución, que deberá estar motivada, se notificará al solicitante, publicándose asimismo en el «Boletín Oficial de la Propiedad Industrial» una mención relativa a la denegación con los datos necesarios para la identificación de la solicitud de patente. 2. En el caso de que los defectos se refieran al derecho de prioridad previsto en el artículo 13 o a la divulgación inocua derivada de la exhibición en una exposición oficial u oficialmente reconocida prevista en el artículo 15, ambos del presente Reglamento, se notificará al solicitante la pérdida de este derecho. Sección 2.ª Informe sobre el estado de la técnica y opinión escrita Artículo 26. Contenido del informe sobre el estado de la técnica y de la opinión escrita. 1. El informe sobre el estado de la técnica mencionará los elementos del estado de la técnica de que disponga la Oficina Española de Patentes y Marcas en el momento de establecer el informe, que puedan ser tomados en consideración para apreciar la novedad y la actividad inventiva de la invención objeto de la solicitud, sobre la base de las reivindicaciones, teniendo debidamente en cuenta la descripción y, en su caso, los dibujos o las secuencias biológicas. 2. Cada mención será realizada en relación con las reivindicaciones correspondientes. En la medida de lo posible, se identificará la parte concreta del documento citado. 3. El informe sobre el estado de la técnica deberá distinguir en los documentos mencionados entre los que han sido publicados antes de la fecha de prioridad, entre la fecha de prioridad y la fecha de presentación y en la fecha de presentación o posteriormente. El informe sobre el estado de la técnica mencionará la clasificación de la solicitud de patente, según la clasificación internacional de patentes. 4. Todo documento que se refiera a una divulgación oral, a un uso o a cualquier otra divulgación que haya sido anterior a la fecha de presentación de la solicitud de patente, será mencionado en el informe sobre el estado de la técnica, precisando, si existe, la fecha de la publicación del documento y de la divulgación no escrita. 5. El informe sobre el estado de la técnica se acompañará de una opinión escrita, preliminar y no vinculante, acerca de si la invención objeto de la solicitud de patente parece ser nueva, implicar actividad inventiva y ser susceptible de aplicación industrial, así como si la solicitud reúne las condiciones de la Ley y del presente Reglamento. 6. Sin perjuicio de lo dispuesto en el artículo 36.5 de la Ley, se realizará el informe sobre el estado de la técnica, acompañado de la opinión escrita, en el supuesto de que no se hubiera realizado el informe de búsqueda internacional para la totalidad de la solicitud internacional o que la solicitud nacional incluyera elementos sobre los cuales no se hubiera realizado búsqueda en la fase internacional. – 150 – CÓDIGO DE DERECHO FARMACÉUTICO § 6 Reglamento para la ejecución de la Ley 24/2015, de Patentes [parcial] Artículo 27. Falta de claridad o de coherencia. 1. Si la Oficina Española de Patentes y Marcas apreciara falta de claridad o de coherencia en la descripción o en las reivindicaciones, o defectos que impidieran total o parcialmente realizar una búsqueda significativa, se notificará al solicitante, para que en el plazo de dos meses a contar desde la publicación de los defectos en el «Boletín Oficial de la Propiedad Industrial» formule las alegaciones que estime oportunas, subsane los defectos, modificando, en su caso, la descripción o las reivindicaciones y, en su caso, los dibujos, en los términos previstos en el artículo 48 de la Ley, o precise el objeto de la búsqueda. 2. Transcurrido ese plazo, si el solicitante no contesta, o los defectos no hubieran sido subsanados y persistiera una falta de claridad o de coherencia o de precisión en el objeto de la búsqueda, la Oficina Española de Patentes y Marcas realizará, en la medida de lo posible, una búsqueda parcial y así se reflejará en el informe sobre el estado de la técnica y la opinión escrita. 3. Si persistiera una falta de claridad o de coherencia de la descripción o de las reivindicaciones que impidiera totalmente realizar una búsqueda significativa, la Oficina Española de Patentes y Marcas no procederá a realizar el informe sobre el estado de la técnica ni la opinión escrita y denegará la solicitud de patente, con indicación de los motivos, notificándoselo al interesado. La mención de la resolución de denegación se publicará en el «Boletín Oficial de la Propiedad Industrial». Artículo 28. Solicitudes que comprendan una pluralidad de reivindicaciones independientes. 1. Si la Oficina Española de Patentes y Marcas considera que las reivindicaciones, tal como han sido presentadas, no se ajustan a lo dispuesto en el artículo 7.2 del presente Reglamento, se notificará al solicitante para que, en el plazo de dos meses a contar desde la publicación de los defectos en el «Boletín Oficial de la Propiedad Industrial», presente las alegaciones que estime convenientes o aporte un nuevo juego de reivindicaciones que servirá de base para la realización de la búsqueda. 2. Si, en el plazo prescrito, el solicitante no procediera a la aportación del nuevo juego de reivindicaciones o este siguiera sin ajustarse a lo dispuesto en el artículo 7.2 del presente Reglamento, la búsqueda se realizará en relación con la primera reivindicación de cada categoría. Artículo 29. Falta de unidad de invención. 1. Si, al iniciar la búsqueda, la Oficina Española de Patentes y Marcas apreciara que la solicitud de patente no satisface la exigencia de la unidad de invención prevista en el artículo 26 de la Ley, emitirá un informe sobre el estado de la técnica parcial respecto de las partes de la solicitud que se refieren a la invención o al grupo de invenciones mencionadas en primer lugar en las reivindicaciones. Dicho informe parcial, acompañado de la opinión escrita, se trasladará al solicitante para que, en el plazo de dos meses a contar desde la publicación en el «Boletín Oficial de la Propiedad Industrial» del defecto de la falta de unidad de invención, efectúe las correspondientes alegaciones frente a dicha objeción de falta de unidad de invención, divida la solicitud o pague una tasa adicional por solicitud de informe sobre el estado de la técnica por cada invención adicional reivindicada. En el momento de pagar las tasas adicionales el solicitante podrá hacer igualmente alegaciones sobre la objeción de falta de unidad de invención. 2. No obstante lo dispuesto en el apartado anterior, por razones de economía procedimental, al mismo tiempo que se realiza la búsqueda en relación con la invención principal, se podrá efectuar la búsqueda de una o más invenciones adicionales, si dicha búsqueda implicara poco o ningún esfuerzo complementario. 3. Si, a la vista de las alegaciones del solicitante presentadas en el plazo prescrito en el apartado 1, la Oficina Española de Patentes y Marcas finalmente considerara que hay unidad de invención, se realizará la búsqueda para las reivindicaciones de la solicitud de patente inicialmente no buscadas y se emitirá un informe sobre el estado de la técnica y una – 151 – CÓDIGO DE DERECHO FARMACÉUTICO § 6 Reglamento para la ejecución de la Ley 24/2015, de Patentes [parcial] opinión escrita definitivos para la totalidad de la solicitud. En el caso de haberse pagado tasas adicionales, estas serán devueltas al solicitante. 4. Si, en el plazo prescrito, el solicitante presentara una o más solicitudes divisionales, se considerará el informe parcial y la opinión escrita como definitivos para la invención o grupo de invenciones respecto de los que se hubieran realizado para la invención de origen. 5. Si el solicitante hubiera pagado las tasas adicionales en el plazo prescrito, la Oficina Española de Patentes y Marcas realizará una búsqueda sobre las partes de la solicitud que guarden relación con las invenciones o grupo de invenciones para las cuales se hubieran pagado tasas y emitirá el informe sobre el estado de la técnica y opinión escrita definitivos. 6. Si en el plazo prescrito el solicitante no subsanara los defectos o no pagara las tasas adicionales o no dividiera la solicitud, la Oficina Española de Patentes y Marcas considerará el informe parcial acompañado de la opinión escrita como definitivos para la invención o grupo de invenciones en relación con los cuales se hayan realizado. La tramitación solo continuará para las reivindicaciones respecto de las cuales se haya realizado el informe y así se indicará en la opinión escrita. Artículo 30. Traslado del informe sobre el estado de la técnica y de la opinión escrita. Una vez elaborados el informe sobre el estado de la técnica y la opinión escrita, la Oficina Española de Patentes y Marcas dará traslado de los mismos al solicitante de la patente. Al mismo tiempo se le dará acceso a los documentos citados. Artículo 31. Publicación de la solicitud y del informe sobre el estado de la técnica. 1. Sin perjuicio de lo dispuesto en el artículo 37.2 de la Ley, transcurridos dieciocho meses desde la fecha de presentación de la solicitud o desde la fecha de prioridad que se hubiera reivindicado, una vez superado el examen de oficio, la Oficina Española de Patentes y Marcas publicará, lo antes posible, la mención en el «Boletín Oficial de la Propiedad Industrial» de que la solicitud de patente se pone a disposición del público. 2. A petición del solicitante, la solicitud podrá publicarse antes del plazo de dieciocho meses mencionado en el artículo 37.1 de la Ley, siempre que la solicitud hubiera superado el examen de oficio. 3. La mención en el «Boletín Oficial de la Propiedad Industrial» a la que se refiere el apartado 1 incluirá las siguientes indicaciones: a) El número de la solicitud y de publicación. b) La fecha de presentación de la solicitud. c) Los datos completos de la prioridad o prioridades reivindicadas. d) La clasificación internacional de patentes. e) El título de la invención. f) La identificación del solicitante y de su representante, en su caso. g) La identificación del inventor o inventores, salvo que hubieran renunciado a ser mencionados como tales. h) El resumen. i) El dibujo más representativo, en su caso. 4. Al mismo tiempo se editará un folleto de la solicitud de patente que, además de las indicaciones incluidas en el apartado 3, contendrá la descripción, las reivindicaciones y, en su caso, los dibujos. Mencionará también el «Boletín Oficial de la Propiedad Industrial» en el cual se publica la solicitud de patente. Las secuencias biológicas se harán accesibles al público y así se mencionará en el folleto. 5. Si en el momento de la publicación de la solicitud de patente estuviera disponible el informe sobre el estado de la técnica, la Oficina Española de Patentes y Marcas publicará simultáneamente en el «Boletín Oficial de la Propiedad Industrial» la mención de la publicación de la solicitud y de la puesta a disposición del público tanto del informe como de la opinión escrita. Asimismo, el folleto de la solicitud de patente al que se refiere el apartado precedente incluirá el informe sobre el estado de la técnica. 6. Si, en aplicación del artículo 36.5 de la Ley, no se realizara el informe sobre el estado de la técnica, se publicará en el «Boletín Oficial de la Propiedad Industrial» una mención a la publicación del informe de búsqueda internacional. A partir de dicha publicación en el – 152 – CÓDIGO DE DERECHO FARMACÉUTICO § 6 Reglamento para la ejecución de la Ley 24/2015, de Patentes [parcial] «Boletín Oficial de la Propiedad Industrial» se abrirá el cómputo del plazo previsto en el artículo 39.2 de la Ley. Cuando proceda, se emitirá una opinión escrita respecto del objeto de la solicitud, de la cual se dará traslado al solicitante y se pondrá a disposición del público. Artículo 32. Observaciones de terceros a la solicitud. 1. Una vez efectuado el anuncio de publicación de la solicitud de patente en el «Boletín Oficial de la Propiedad Industrial», cualquier persona podrá formular observaciones debidamente razonadas y documentadas sobre la patentabilidad de la invención objeto de la solicitud hasta el momento anterior a la finalización del examen sustantivo. 2. Las observaciones de terceros se presentarán ante la Oficina Española de Patentes y Marcas, no interrumpirán la tramitación de la solicitud y serán trasladadas al solicitante quien podrá realizar alegaciones si lo estima conveniente. Sección 3.ª Examen sustantivo y resolución Artículo 33. Petición de examen sustantivo. 1. El solicitante podrá formular la petición de examen sustantivo desde el momento del depósito de la solicitud y hasta el transcurso del plazo de tres meses contado a partir de la fecha de publicación en el «Boletín Oficial de la Propiedad Industrial» de la mención de la puesta a disposición del público del informe sobre el estado de la técnica. La petición del examen sustantivo implicará el pago de la tasa correspondiente. 2. El solicitante podrá presentar observaciones al informe sobre el estado de la técnica y a la opinión escrita y, en su caso, a las observaciones de terceros, así como modificar la solicitud de patente en los términos previstos en el artículo 48 de la Ley, hasta finalizar el plazo mencionado en el apartado anterior. 3. Transcurrido el plazo prescrito sin que se hubiera formulado petición de examen sustantivo o sin que se hubiera pagado la tasa correspondiente, la solicitud de patente se considerará retirada. La resolución por la que se declare retirada la solicitud se comunicará al solicitante y se publicará en el «Boletín Oficial de la Propiedad Industrial». 4. De acuerdo con el artículo 39.2 de la Ley, la petición de examen se podrá revocar en cualquier momento del procedimiento. En este caso, la Oficina Española de Patentes y Marcas considerará retirada la solicitud de patente y así se publicará en el «Boletín Oficial de la Propiedad Industrial». Esta revocación estará sujeta a las mismas limitaciones de la retirada de la solicitud de patente, conforme al artículo 67 de este Reglamento. Si se hubiera iniciado el examen, no procederá la devolución de la tasa de examen sustantivo. Artículo 34. Examen sustantivo y resolución. 1. Publicado el informe sobre el estado de la técnica y presentada en plazo la petición de examen y pagada la tasa correspondiente, así como, en su caso, presentadas las observaciones y modificaciones pertinentes, se dará inicio al examen. 2. La Oficina Española de Patentes y Marcas considerará el informe sobre el estado de la técnica y la opinión escrita como primera comunicación al solicitante sobre si la invención reúne los requisitos formales, técnicos y de patentabilidad previstos en la Ley. Ello, no obstante, se podrá llevar a cabo una búsqueda complementaria con el fin de descubrir la existencia de los documentos que hayan sido publicados o se hayan puesto a disposición del público con posterioridad a la fecha en que se haya realizado el informe sobre el estado de la técnica. 3. Cuando del examen no resulte la falta de ningún requisito que lo impida, se concederá la patente solicitada conforme a lo dispuesto en el artículo 35 del presente Reglamento. Si el solicitante hubiera modificado su solicitud de patente, se comprobará que tales modificaciones cumplen con los requisitos de los artículos 48 de la Ley y 64 del presente Reglamento. 4. La Oficina Española de Patentes y Marcas denegará la patente en el caso de que el solicitante no hubiera realizado ningún acto para obviar las objeciones contenidas en la primera comunicación. La resolución de denegación deberá notificarse, indicando los – 153 – CÓDIGO DE DERECHO FARMACÉUTICO § 6 Reglamento para la ejecución de la Ley 24/2015, de Patentes [parcial] motivos, y se publicará en el «Boletín Oficial de la Propiedad Industrial» una mención relativa a la denegación. 5. En los demás casos si, a la vista de la contestación recibida y pese a las alegaciones o modificaciones aportadas, la Oficina Española de Patentes y Marcas considera que persisten motivos que impiden en todo o en parte la concesión de la patente, se comunicarán estos al solicitante dándole la oportunidad de realizar observaciones o corregir su solicitud en el plazo de dos meses a partir de la publicación de la mención de objeciones en el «Boletín Oficial del de la Propiedad Industrial». Al corregir la solicitud, el solicitante podrá modificar, en los términos previstos en el artículo 48 de la Ley, la descripción, las reivindicaciones y los dibujos o secuencias biológicas, en su caso, redactando la patente tal como pretenda que sea concedida. 6. La Oficina Española de Patentes y Marcas podrá repetir la comunicación de objeciones enviando nuevas comunicaciones de defectos, dando nuevas oportunidades al solicitante para subsanar en el plazo de dos meses en cada una de ellas, a contar desde la publicación en el «Boletín Oficial del de la Propiedad Industrial», si a pesar de sus contestaciones, no hubiera aún logrado corregir completamente todos los defectos que impidieran la concesión de la patente, siempre y cuando se estime que los defectos restantes son subsanables y que el solicitante ha tratado manifiestamente de corregirlos. 7. Las nuevas oportunidades a las que se refiere el apartado anterior podrán consistir en uno o varios trámites escritos o concentrarse en una única vista oral cuando se estime conveniente o sea solicitado por el solicitante. En el caso de ausencia del solicitante de la patente, el trámite se tendrá por concluido y se continuará la tramitación. De los asuntos tratados en la vista oral se elevará una breve acta, a la que se adjuntarán los textos acordados. El solicitante deberá aportar la descripción y las reivindicaciones, tal como hayan sido acordadas cumpliendo los requisitos formales exigidos en el presente Reglamento en el plazo de diez días hábiles a contar desde el día siguiente a la publicación del anuncio de la elevación del acta en el «Boletín Oficial de la Propiedad Industrial». 8. Finalizadas las actuaciones de los tres apartados anteriores, la Oficina Española de Patentes y Marcas resolverá definitivamente sobre la concesión o denegación de la patente, teniendo en cuenta el texto aportado por el solicitante. 9. Contra la resolución denegatoria de la solicitud de patente podrá formularse recurso de alzada por el solicitante de la patente. El plazo para la interposición del recurso de alzada será de un mes a contar desde la fecha de publicación de la denegación en el «Boletín Oficial de la Propiedad Industrial». No obstante, este plazo es susceptible de restablecimiento de derechos en las condiciones y supuestos previstos en el artículo 53 de la Ley. 10. En el procedimiento de recurso el titular de la patente podrá modificar la solicitud con sujeción a lo dispuesto en el artículo 48 de la Ley. 11. La resolución del recurso de alzada pondrá fin a la vía administrativa. Artículo 35. Concesión de la patente. 1. La concesión de la patente y la mención de que el expediente está a disposición del público se publicará en el «Boletín Oficial del de la Propiedad Industrial». 2. La mención en el «Boletín Oficial de la Propiedad Industrial» a que se refiere el apartado 1 incluirá las siguientes indicaciones: a) El número de la solicitud y de publicación. b) La fecha de presentación de la solicitud. c) Los datos completos de la prioridad o prioridades reivindicadas. d) La fecha de publicación de la solicitud y, en el caso de no ser coincidentes, la fecha de publicación del informe sobre el estado de la técnica o de la mención a la publicación del informe de búsqueda internacional. e) Referencia a las modificaciones introducidas en las reivindicaciones. f) La clasificación internacional de patentes. g) El título de la invención. h) La identificación del titular y de su representante, en su caso. – 154 – CÓDIGO DE DERECHO FARMACÉUTICO § 6 Reglamento para la ejecución de la Ley 24/2015, de Patentes [parcial] i) La identificación del inventor o inventores, salvo que hubieran renunciado a ser mencionados como tales. j) La fecha de concesión. k) El resumen. 3. Al mismo tiempo se editará un folleto de la patente que, además de las menciones incluidas en el apartado anterior, se indicará el «Boletín Oficial del de la Propiedad Industrial» en el que se anunció la concesión, contendrá la descripción, las reivindicaciones y, en su caso, los dibujos tal como hubieran sido concedidos. Las secuencias biológicas se harán accesibles al público y así se mencionará en el folleto. En el folleto se hará constar que la patente se concede sin perjuicio de tercero y sin garantía del Estado en cuanto a la validez de la misma y a la utilidad del objeto sobre el que recae. CAPÍTULO III Procedimiento de oposición Artículo 36. Oposición a la concesión. 1. Conforme a lo previsto en el apartado 1 del artículo 43 de la Ley cualquier persona podrá oponerse a la concesión de una patente mediante la presentación de un escrito de oposición dentro de los seis meses siguientes a la publicación de la concesión en el «Boletín Oficial de la Propiedad Industrial». 2. El escrito de oposición, al que se refiere el artículo 43 de la Ley, suficientemente motivado, deberá presentarse a la Oficina Española de Patentes y Marcas. Dicho escrito deberá acompañarse de las correspondientes alegaciones, hechos y pruebas invocadas en apoyo de las mismas. Si las pruebas aportadas no estuvieran redactadas en español, deberá aportarse una traducción al español. La presentación del escrito de oposición implicará el pago de la tasa correspondiente. 3. El escrito de oposición deberá contener los siguientes datos: a) La identidad del oponente, de acuerdo con lo dispuesto en los párrafos b) y c) de artículo 2.1 del presente Reglamento. b) Cuando el oponente actúe por medio de un representante, se indicará la identidad de este, de acuerdo con lo dispuesto en el párrafo a) del artículo 2.2 del presente Reglamento. c) El número de la solicitud de patente contra la que se formula oposición, así como la identificación del titular. d) Los motivos en los que se funda dicha oposición de acuerdo con lo dispuesto en el artículo 43.1 de la Ley, así como una declaración que especifique en qué medida la oposición planteada afecta a la patente, detallando las reivindicaciones afectadas por la oposición. e) La firma del oponente o de su representante. 4. La oposición no se admitirá cuando no haya sido presentada dentro del plazo de seis meses previsto en el artículo 43.1 de la Ley. 5. En los supuestos en que el escrito de oposición no se ajustara a las disposiciones de los apartados 1 y 2 o cuando no se hubiera abonado la tasa de oposición o el pago fuese insuficiente, se notificarán las irregularidades observadas al oponente para que las subsane en el plazo de un mes a contar desde la publicación de defectos en el «Boletín Oficial de la Propiedad Industrial», con indicación de que si así no lo hiciera se le tendrá por desistido de la oposición. 6. La resolución por la que se admita, inadmita o se tenga por desistida la oposición se notificará al oponente, indicando los motivos, y se publicará en el «Boletín Oficial de la Propiedad Industrial» una mención relativa a la resolución. Asimismo, la resolución por la que se inadmita o se tenga por desistida la oposición se notificará al titular de la patente. Artículo 37. Presentación y tramitación de las oposiciones. 1. Podrá presentarse oposición contra una patente, aunque su titular haya renunciado a la patente o ésta haya caducado. – 155 – CÓDIGO DE DERECHO FARMACÉUTICO § 6 Reglamento para la ejecución de la Ley 24/2015, de Patentes [parcial] 2. Si el titular renuncia a la patente o esta caduca durante la tramitación de una oposición se pondrá en conocimiento del oponente, que podrá solicitar por escrito la continuación del procedimiento de oposición en el plazo de un mes a contar desde la publicación de la puesta en conocimiento al oponente en el «Boletín Oficial de la Propiedad Industrial». 3. En cualquier caso, una vez admitida a trámite la oposición, esta podrá ser tramitada por la Oficina Española de Patentes y Marcas, aunque el oponente falleciese o retirase el escrito de oposición. Artículo 38. Traslado de las oposiciones al titular de la patente. 1. Transcurrido el plazo de presentación de oposiciones, se dará traslado al titular de la patente de aquellas oposiciones admitidas a trámite, poniendo a su disposición la documentación anexa para que, en el plazo de tres meses desde la publicación del traslado de las oposiciones en el «Boletín Oficial de la Propiedad Industrial», presente alegaciones y, en la medida que sea necesario para subsanar los defectos notificados, modifique las reivindicaciones, descripción y dibujos o secuencias biológicas, en los términos previstos en los artículos 48 de la Ley y 64 del presente Reglamento. 2. En el caso de que el titular de la patente conteste a las oposiciones, la Oficina Española de Patentes y Marcas dará traslado simultáneamente a todos los oponentes, de existir varios, de las alegaciones y propuestas de modificación presentadas por el titular de la patente, otorgándoles un trámite de réplica en cada caso por un plazo común de dos meses desde la publicación del traslado de la contestación a las oposiciones en el «Boletín Oficial de la Propiedad Industrial». Artículo 39. Examen de las oposiciones y resolución. 1. Para examinar los escritos de oposición, así como, en su caso, la contestación y réplicas, se creará una Comisión compuesta por tres expertos de la Oficina Española de Patentes y Marcas técnicamente cualificados, siendo uno de ellos Presidente. La Comisión se completará con un jurista de la misma Oficina si se considera que la naturaleza de la decisión así lo exige. En caso de empate de votos, el Presidente tendrá voto de calidad. Los miembros de la Comisión serán designados por el Director del Departamento de Patentes e Información Tecnológica atendiendo a criterios de experiencia y especialización. 2. La Oficina Española de Patentes y Marcas examinará los motivos de oposición, así como las alegaciones de todas las partes, incluidas, en su caso, las modificaciones presentadas por el titular de la patente. En el supuesto de que considere que ningún motivo de oposición impide el mantenimiento de la patente tal como fue concedida, desestimará la oposición o las oposiciones, en su caso. La resolución de desestimación se notificará al titular y a los oponentes, con indicación de los motivos y rechazando la propuesta de modificación. Dicha resolución se publicará en el «Boletín Oficial de la Propiedad Industrial» y se incluirá una mención relativa al mantenimiento de la patente tal como fue concedida. 3. Cuando, a la vista de las alegaciones recibidas, incluidas las modificaciones presentadas por el titular de la patente, la Oficina Española de Patentes y Marcas considere que la patente puede mantenerse concedida en la forma modificada propuesta por el titular, resolverá estimando total o parcialmente las oposiciones, manteniendo la concesión de la patente de forma modificada. 4. Cuando, a pesar de las alegaciones o modificaciones efectuadas por el titular, persistan los motivos que impidan el mantenimiento de la patente, se otorgará al titular un nuevo plazo de un mes a contar desde la publicación de su mención en el «Boletín Oficial de la Propiedad Industrial», para presentar nuevas alegaciones o modificaciones de las reivindicaciones, de la descripción y, en su caso, de los dibujos o secuencias biológicas, en los términos previstos en los artículos 48 de la Ley y 64 del presente Reglamento. Se otorgarán nuevas oportunidades, siempre y cuando se estime que las objeciones son subsanables y que el titular ha tratado manifiestamente de corregirlas. Antes de resolver definitivamente, se otorgará un plazo de 10 días a los oponentes para que presenten las alegaciones finales que estimen pertinentes. 5. El trámite de nuevas oportunidades al titular y de alegaciones finales a los oponentes al que se refiere el apartado anterior podrá consistir en uno o varios trámites escritos o concentrarse en una única vista oral, cuando se estime conveniente, o a petición del titular – 156 – CÓDIGO DE DERECHO FARMACÉUTICO § 6 Reglamento para la ejecución de la Ley 24/2015, de Patentes [parcial] de la patente o de alguno de los oponentes. La ausencia de los oponentes no impedirá la celebración de la vista. En el caso de ausencia del titular de la patente, el trámite se tendrá por concluido y se continuará la tramitación. De los asuntos tratados en la vista oral se elevará una breve acta, a la que se adjuntarán los textos propuestos por el titular. El titular deberá aportar la descripción, las reivindicaciones y, en su caso, los dibujos o secuencias biológicas, tal como hayan sido propuestos cumpliendo los requisitos formales exigidos en el presente Reglamento en el plazo de diez días hábiles a contar desde el día siguiente a la publicación del anuncio de la elevación del acta en el «Boletín Oficial de la Propiedad Industrial». 6. Si, finalmente, la Oficina Española de Patentes y Marcas resolviera estimando total o parcialmente las oposiciones, se revocará la concesión de la patente o se mantendrá la concesión de la patente de forma modificada. 7. La resolución motivada por la que se revoque la patente se notificará al titular y a los oponentes, indicando los motivos, y se publicará en el «Boletín Oficial de la Propiedad Industrial» una mención relativa a la revocación. 8. La mención en el «Boletín Oficial de la Propiedad Industrial» a que se refiere el apartado anterior incluirá las siguientes menciones: a) El número de solicitud y de publicación. b) La fecha de presentación de la solicitud. c) Los datos completos de la prioridad o prioridades reivindicadas. d) La clasificación internacional de patentes. e) El título de la invención. f) La identificación del titular y de su representante, en su caso. g) Identificación de que la patente se ha revocado y la fecha de la resolución de revocación. 9. La resolución motivada por la que se acuerde mantener la patente de forma modificada, se notificará al titular y a los oponentes. Asimismo, se publicará en el «Boletín Oficial de la Propiedad Industrial» la mención de que la patente se mantiene de forma modificada y de que está a disposición del público. 10. La mención en el «Boletín Oficial del de la Propiedad Industrial» a que se refiere el apartado anterior incluirá las siguientes indicaciones: a) El número de solicitud y de publicación. b) La fecha de presentación de la solicitud. c) Los datos completos de la prioridad o prioridades reivindicadas. d) La fecha de publicación de la solicitud y, en caso de no ser coincidentes, la fecha de publicación del informe sobre el estado de la técnica o de la mención a la publicación del informe de búsqueda internacional. e) Referencia a las modificaciones introducidas en las reivindicaciones. f) La clasificación internacional de patentes. g) El título de la invención. h) La identificación del titular y de su representante, en su caso. i) La identificación del inventor o inventores, salvo que hubieran renunciado a ser mencionados como tales. j) La fecha de resolución por la que se acuerda mantener la patente de forma modificada. k) La fecha de modificación de las reivindicaciones. l) El resumen. 11. Al mismo tiempo se editará un folleto de la patente que, además de las indicaciones incluidas en el apartado anterior, contendrá la descripción, las reivindicaciones y, en su caso, los dibujos tal como hubieran sido modificados. Las secuencias biológicas se harán accesibles al público y así se mencionará en el folleto. 12. Contra la concesión de una patente solo podrá interponerse recurso de alzada por quienes hayan sido parte en un procedimiento de oposición y se dirigirá contra el acto resolutorio de la oposición planteada. A estos efectos podrá entenderse desestimada la oposición si transcurrido el plazo para resolverla y notificarla no hubiese recaído resolución expresa. El plazo para la interposición del recurso de alzada será de un mes a contar desde – 157 – CÓDIGO DE DERECHO FARMACÉUTICO § 6 Reglamento para la ejecución de la Ley 24/2015, de Patentes [parcial] la fecha de publicación de la resolución en el «Boletín Oficial de la Propiedad Industrial». No obstante, este plazo es susceptible de restablecimiento de derechos en las condiciones y supuestos previstos en el artículo 53 de la Ley. 13. En el procedimiento de recurso el titular de la patente podrá modificar la solicitud con sujeción a lo dispuesto en el artículo 48 de la Ley de Patentes. 14. La resolución del recurso de alzada pondrá fin a la vía administrativa. Artículo 40. Concurrencia de procedimiento judicial y de procedimiento de oposición. 1. En el caso de que conste anotado en el Registro de Patentes la tramitación de un procedimiento judicial sobre la validez de la patente o de infracción contra la que se ha presentado la oposición, la Oficina Española de Patentes y Marcas pondrá en conocimiento del Juez o Tribunal, a los efectos oportunos, la tramitación de un procedimiento de oposición. 2. Transcurrido el plazo para presentar oposiciones cualquier tercero podrá solicitar intervenir en el procedimiento de oposición siempre y cuando presente escrito de oposición en la forma prevista en el artículo 36 de este Reglamento y acredite que el titular ha entablado una acción de violación contra él o que después de haber sido requerido por el titular de la patente para cesar en la presunta violación de dicha patente ha ejercitado una acción negatoria, conforme a lo establecido en el artículo 121 de la Ley. La declaración de intervención se presentará en un plazo de tres meses desde la fecha en que se entable la acción judicial correspondiente. Admitida la solicitud de intervención de un tercero se tramitará como una oposición. CAPÍTULO IV Procedimiento de revocación o limitación Artículo 41. Solicitud de revocación o limitación. 1. El titular podrá solicitar la revocación total o limitación de su patente tal como fue concedida o limitada en un procedimiento de oposición o de limitación anterior. 2. La solicitud de revocación o limitación, que implicará el pago de la tasa correspondiente, deberá presentarse por escrito ante la Oficina Española de Patentes y Marcas mediante modelo oficial que deberá contener los siguientes datos: a) La identidad del titular de la patente, conforme a lo dispuesto en el párrafo b) del artículo 2.1 del presente Reglamento. c) Si el titular hubiera designado un representante, la identidad de este, conforme a lo dispuesto en el párrafo a) del artículo 2.2 del presente Reglamento. c) El número de la solicitud, el número de publicación, la fecha de publicación y el código de tipo de documento de la patente cuya revocación o limitación se solicita. d) Si el titular solicitara la limitación de la patente, indicación de que aporta un juego de reivindicaciones modificadas y, en su caso, una modificación de la descripción, de los dibujos o de las secuencias biológicas, en los términos previstos en los artículos 48 de la Ley y 64 del presente Reglamento. e) Si existen inscritos derechos reales, opciones de compra, embargos, licencias o una demanda judicial, indicación de que se aporta el consentimiento de los titulares de estos derechos o del demandante. f) Firma del solicitante o de su representante. 3. El titular no podrá presentar una solicitud de limitación durante el plazo para presentar oposiciones previsto en el artículo 43.1 de la Ley, ni mientras se halle en tramitación una oposición contra la concesión de la patente o se esté tramitando una limitación anteriormente solicitada. 4. La solicitud deberá ir acompañada de los documentos que se citan en los párrafos d) y e) del apartado 2 de este artículo. – 158 – CÓDIGO DE DERECHO FARMACÉUTICO § 6 Reglamento para la ejecución de la Ley 24/2015, de Patentes [parcial] Artículo 42. Procedimiento de revocación o limitación. 1. La Oficina Española de Patentes y Marcas examinará si la documentación presentada cumple los requisitos establecidos en los artículos 105 de la Ley y 41 del presente Reglamento. Si del examen efectuado resultara alguna irregularidad o defecto, se suspenderá la tramitación, notificando al titular las objeciones observadas para que, en el plazo de dos meses, a contar desde la publicación del suspenso en el «Boletín Oficial de la Propiedad Industrial», las subsane o presente sus alegaciones. 2. Transcurrido el plazo de dos meses, si los defectos han sido debidamente corregidos la Oficina Española de Patentes y Marcas resolverá sobre lo solicitado. En caso contrario, se denegará la solicitud de revocación o limitación. En ambos casos la resolución correspondiente deberá notificarse, indicando los motivos, y se publicará en el «Boletín Oficial de la Propiedad Industrial» una mención relativa, en su caso, a la revocación, limitación o denegación de la misma. La mención de la limitación contendrá las indicaciones a las que se refiere el artículo 39.8 del presente Reglamento. Al mismo tiempo se editará un folleto de la patente con las indicaciones citadas en el artículo 39.11 del presente Reglamento, sustituyendo las referencias a la concesión de forma modificada por la limitación. Artículo 43. Petición de limitación estando pendiente un procedimiento judicial. 1. Cuando esté en trámite un procedimiento judicial sobre la validez de la patente que conste inscrito en el Registro de Patentes, la Oficina Española de Patentes y Marcas comunicará al Juez o Tribunal la petición de limitación, a los efectos oportunos. La denegación de la autorización facultará al titular de la patente para solicitar a la Oficina Española de Patentes y Marcas la devolución de la tasa de limitación. 2. Si iniciado un procedimiento de limitación se notifica e inscribe en el Registro de Patentes un procedimiento judicial sobre la validez de la patente, la Oficina Española de Patentes y Marcas comunicará al Juez o Tribunal la existencia de un procedimiento de limitación en trámite, a los efectos oportunos. 3. Una vez tramitado el procedimiento de limitación, la Oficina Española de Patentes y Marcas pondrá en conocimiento del Juez o Tribunal la resolución, aportando la patente tal como hubiera quedado modificada. CAPÍTULO V Otros procedimientos Sección 1.ª Solicitudes divisionales Artículo 44. Petición de división. 1. Las solicitudes que no cumplan el requisito de unidad de invención establecido en el artículo 26 de la Ley, podrán ser divididas por el solicitante, previo requerimiento por parte de la Oficina Española de Patentes y Marcas, según se regula en los artículos 29 y 59 de este Reglamento. 2. El solicitante de una patente podrá solicitar, a iniciativa propia, la división de su solicitud en cualquier momento anterior a la finalización del examen sustantivo. El solicitante de un modelo de utilidad podrá solicitar, a iniciativa propia, la división de su solicitud en cualquier momento anterior a la resolución a la que se refieren los apartados 3 y 4 del artículo 62 del presente Reglamento. 3. Al presentar la solicitud divisional, el solicitante deberá justificar en qué medida el objeto de protección de la solicitud divisional es una parte y no es esencialmente el mismo que el de la solicitud originaria. – 159 – CÓDIGO DE DERECHO FARMACÉUTICO § 6 Reglamento para la ejecución de la Ley 24/2015, de Patentes [parcial] Artículo 45. Formalización y tramitación de la solicitud divisional. 1. Al momento de pedir la división de la solicitud de patente o de modelo de utilidad, el solicitante deberá formalizar su solicitud divisional, que deberá cumplir los requisitos prescritos en el Capítulo I del Título I del presente Reglamento. 2. A los efectos de mantener como fecha de presentación la asignada a la solicitud inicial, la Oficina Española de Patentes y Marcas verificará si el objeto de la solicitud divisional se encuentra comprendido en la solicitud inicial. 3. En el caso de que se solicite la división de una solicitud de patente, la tasa de solicitud y la tasa de informe sobre el estado de la técnica deberán abonarse dentro del plazo de un mes desde el depósito de la solicitud divisional. Si, en relación con la solicitud inicial, se hubieran pagado tasas adicionales referidas en el artículo 29 del presente Reglamento, el solicitante no tendrá que pagar, respecto de la solicitud divisional, la tasa de solicitud del informe sobre el estado de la técnica, en la medida en que el objeto de la solicitud divisional ya hubiera sido objeto de búsqueda. En este caso, se emitirá una opinión escrita respecto del objeto de la solicitud divisional, de la cual se dará traslado al solicitante y se pondrá a disposición del público. Asimismo, se publicará en el «Boletín Oficial de la Propiedad Industrial» una mención a la publicación del informe sobre el estado de la técnica realizado respecto de la solicitud inicial. A partir de dicha publicación en el «Boletín Oficial de la Propiedad Industrial» se abrirá el cómputo del plazo previsto en el artículo 39.2 de la Ley para solicitar el examen sustantivo. 4. En el caso de que no se hubiera abonado la tasa de informe sobre el estado de la técnica o no se hubiera abonado en su totalidad o si, a pesar de haberse pagado tasas adicionales referidas en el artículo 29 del presente Reglamento resultara que el objeto o parte del objeto de la solicitud divisional no hubiera sido objeto de búsqueda, se comunicará al solicitante la necesidad de abonar la tasa o completar el pago en el plazo de un mes a contar desde la publicación del defecto en el «Boletín Oficial de la Propiedad Industrial», con indicación de que si así no se hiciera se resolverá teniendo por desistida la solicitud divisional. La resolución de desistimiento se notificará al solicitante y se publicará en el «Boletín Oficial de la Propiedad Industrial». 5. La descripción y los dibujos, tanto en la solicitud inicial de patente o de modelo de utilidad, como de cualquier solicitud divisional, sólo deben referirse, en principio, a los elementos que se pretenden proteger en dicha solicitud. Sin embargo, cuando sea necesario describir en una solicitud los elementos para los que se ha pedido protección en otra solicitud, deberá hacerse referencia a esta otra solicitud. Sección 2.ª Cambio de modalidad Artículo 46. Cambio de modalidad. 1. El solicitante de una patente podrá pedir, en cualquier momento anterior a la finalización del examen sustantivo, que se transforme su solicitud de patente en una solicitud para la protección del objeto de su invención bajo otra modalidad de Propiedad Industrial. El solicitante de un modelo de utilidad podrá pedir, en cualquier momento anterior a la resolución a la que se refieren los apartados 3 y 4 del artículo 62 del presente Reglamento, que se transforme su solicitud de modelo de utilidad en una solicitud para la protección del objeto de su invención bajo otra modalidad de Propiedad Industrial. La petición de cambio de modalidad implicará el pago de la tasa correspondiente. 2. La Oficina Española de Patentes y Marcas, como consecuencia del examen de oficio del artículo 35 de la Ley o del examen sustantivo del artículo 40 de la Ley o del examen de oficio del artículo 142 de la Ley, podrá proponer al solicitante el cambio de modalidad de la solicitud, notificándoselo para que, en los plazos previstos en los artículos 24, 34.5 y 59.3, respectivamente del presente Reglamento, acepte o rechace la propuesta, entendiéndose que la rechaza si en el plazo mencionado no pide expresamente el cambio de modalidad. Si la propuesta es rechazada, continuará la tramitación del expediente en la modalidad originariamente solicitada. – 160 – CÓDIGO DE DERECHO FARMACÉUTICO § 6 Reglamento para la ejecución de la Ley 24/2015, de Patentes [parcial] 3. Cuando el solicitante pida el cambio de modalidad, la Oficina Española de Patentes y Marcas acordará el cambio y comunicará el acuerdo al interesado con indicación de la documentación que debe presentar, indicándole que posee para ello un plazo de dos meses a contar desde la publicación del anuncio de acuerdo de cambio de modalidad en el «Boletín Oficial de la Propiedad Industrial». Si se detectara la falta del pago de la tasa o el pago insuficiente, se comunicará, igualmente al solicitante que realice o complete el pago en dicho plazo. La falta de presentación de la nueva documentación o del pago de la tasa en el plazo indicado hará que se tenga por desistida la solicitud de cambio de modalidad, se anulará la solicitud correspondiente a la nueva modalidad y se continuará la tramitación de la solicitud originaria. 4. Si el solicitante aporta en el plazo prescrito la documentación indicada o subsana el pago de la tasa, la Oficina Española de Patentes y Marcas le dará el trámite oportuno, manteniendo, en su caso, la fecha de presentación de la solicitud originaria. Sección 3.ª Tramitación secreta de patentes que interesan a la defensa nacional Artículo 47. Patentes de interés para la defensa nacional. 1. El contenido de todas las solicitudes de patentes se mantendrá secreto hasta que transcurra un mes desde la fecha de su presentación. Antes de que finalice, la Oficina Española de Patentes y Marcas prorrogará este plazo, según lo dispuesto en el artículo 111.1 de la Ley, hasta cuatro meses si considera que la invención objeto de la misma puede ser de interés para la defensa nacional. 2. La Oficina Española de Patentes y Marcas notificará la prórroga al solicitante y enviará al Ministerio de Defensa copia de la solicitud de la patente para que se pronuncie sobre si el objeto de la solicitud de patente es de interés para la defensa nacional. 3. En caso de que el Ministerio de Defensa considere que la invención interesa a la defensa nacional, requerirá a la Oficina Española de Patentes y Marcas para que, antes de que finalice el plazo de cuatro meses, decrete la tramitación secreta de la misma. El acuerdo por el que se decrete la tramitación secreta de la solicitud de patente se notificará al solicitante, dando traslado del mismo al Ministerio de Defensa. Artículo 48. Solicitudes que reivindiquen la prioridad de una solicitud extranjera declarada secreta. 1. Si una solicitud de patente presentada ante la Oficina Española de Patentes y Marcas reivindica la prioridad de una solicitud de patente declarada secreta por un país perteneciente a la Organización del Tratado del Atlántico Norte (OTAN) o con el que España haya suscrito un acuerdo internacional en materia de defensa, se le otorgará al menos el mismo nivel de secreto que aquel otorgado por el país de origen. 2. De conformidad con el Artículo III del Acuerdo de la OTAN para la salvaguardia mutua del secreto de las invenciones relacionadas con la defensa respecto de las cuales se hayan presentado solicitudes de patentes (hecho en París el 21 de septiembre de 1960), se salvaguardará el secreto de la invención si el solicitante renuncia a toda reclamación de indemnización por daños o pérdidas debidos exclusivamente a la imposición del secreto sobre la invención por parte del país de origen. En caso de que no presente la renuncia a la indemnización, la Oficina Española de Patentes y Marcas rechazará la solicitud de patente y devolverá al solicitante los documentos remitidos. Artículo 49. Solicitudes de patente en el extranjero que reivindiquen la prioridad de una solicitud nacional declarada secreta. 1. El solicitante no podrá presentar solicitudes de protección en el extranjero reivindicando la prioridad de una solicitud de patente presentada ante la Oficina Española de Patentes y Marcas, antes del transcurso del plazo de un mes desde la fecha de presentación, salvo autorización expresa de la Oficina Española de Patentes y Marcas. 2. La Oficina Española de Patentes y Marcas no podrá conceder esta autorización para aquellas solicitudes de patentes que hayan sido puestas a disposición del Ministerio de – 161 – CÓDIGO DE DERECHO FARMACÉUTICO § 6 Reglamento para la ejecución de la Ley 24/2015, de Patentes [parcial] Defensa en virtud de lo dispuesto en el artículo 111.1 de la Ley o que estén sujetas a régimen de secreto, salvo que el Ministerio de Defensa lo autorice expresamente. Artículo 50. Primera solicitud de patente en el extranjero de invenciones en España. 1. Cuando se trate de invenciones realizadas en España, el interesado no podrá presentar una solicitud de patente como primera solicitud en el extranjero, salvo autorización expresa de la Oficina Española de Patentes y Marcas. La petición de autorización deberá presentarse por el interesado ante la Oficina Española de Patentes y Marcas. 2. Para evaluar si la invención interesa a la defensa nacional, el interesado deberá aportar, en condiciones de secreto, una copia de la solicitud de patente tal como la pretenda presentar en el extranjero, junto con la descripción, reivindicaciones y dibujos y, en su caso, una traducción al español de dicha documentación. 3. La Oficina Española de Patentes y Marcas autorizará, en el plazo máximo de un mes, la presentación de una primera solicitud en el extranjero cuando considere que la invención no es de interés para la defensa nacional y su presentación en el extranjero no contraviniera lo previsto en los Acuerdos Internacionales en materia de Defensa suscritos por España. 4. Sin embargo, si la Oficina Española de Patentes y Marcas considera que la invención pudiera interesar a la defensa nacional, denegará, en el mismo plazo de un mes del apartado anterior, la autorización a presentar una primera solicitud en el extranjero y así se lo notificará al interesado. En este caso, solo se concederá la autorización si el interesado aporta autorización expresa del Ministerio de Defensa. Artículo 51. Tramitación de las solicitudes de patentes sujetas a régimen de secreto. 1. Los Capítulos I, II y III del presente Título I serán de aplicación a las solicitudes de patentes declaradas en régimen de secreto, salvo en lo referente a la publicación y la divulgación. Las notificaciones se realizarán directamente al solicitante o a su representante. 2. Los trámites relativos a solicitudes de patentes tramitadas en régimen de secreto se anotarán en el Registro de Patentes Secretas, que solo será accesible al personal habilitado de acuerdo con la normativa vigente en materia de protección de información clasificada del Ministerio de Energía, Turismo y Agenda Digital. 3. Una vez levantado el secreto, la Oficina Española de Patentes y Marcas continuará con- los trámites correspondientes previstos en el Título I del presente Reglamento. Las anotaciones practicadas en el Registro de Patentes Secretas se trasladarán al Registro de Patentes al que hace referencia el artículo 79 de la Ley. En el caso de que el levantamiento del secreto hubiera tenido lugar una vez concedida la patente, se publicará en el «Boletín Oficial de la Propiedad Industrial» la concesión y se emitirá el correspondiente folleto, tal como prevé el artículo 35 del presente Reglamento. A partir de esta publicación se abrirá el plazo de presentación de oposiciones recogido en el artículo 43 de la Ley. Artículo 52. Levantamiento de secreto. 1. Las solicitudes de patente o las patentes sujetas a régimen de secreto declarado por un país perteneciente a la Organización del Tratado del Atlántico Norte o con el que España haya suscrito un acuerdo internacional en materia de defensa, mantendrán dicho régimen hasta que la Oficina Española de Patentes y Marcas reciba la comunicación de levantamiento del secreto. 2. Tanto las solicitudes de patente que se encuentren en tramitación en régimen de secreto como aquellas solicitudes de patente que hubieran sido denegadas bajo esta forma de tramitación, mantendrán este régimen secreto hasta que el Ministerio de Defensa acuerde el levantamiento del secreto. 3. Las patentes secretas cuya concesión se hubiera producido durante su tramitación bajo el régimen de secreto se mantendrán en ese mismo régimen a partir de la fecha de concesión, por años renovables automáticamente, hasta que el Ministerio de Defensa comunique el levantamiento del secreto. A continuación, la Oficina Española de Patentes y Marcas se lo notificará al titular de la patente. – 162 – CÓDIGO DE DERECHO FARMACÉUTICO § 6 Reglamento para la ejecución de la Ley 24/2015, de Patentes [parcial] Artículo 53. Régimen de las solicitudes de patentes o de las patentes sujetas a secreto. 1. Las solicitudes de patente o las patentes sujetas a régimen de secreto no podrán ser retiradas, renunciadas, revocadas o limitadas sin la autorización expresa de la autoridad que declaró el secreto. 2. Las patentes secretas no estarán sujetas al pago de anualidades. Una vez levantado el secreto, según lo previsto en el artículo 52 del presente Reglamento, el titular de las patentes deberá abonar las anualidades que devenguen a partir de la publicación de concesión en el «Boletín Oficial de la Propiedad Industrial». TÍTULO II Certificados complementarios de protección de medicamentos o de su prórroga y productos fitosanitarios Artículo 54. Presentación de la solicitud. 1. La solicitud de certificado complementario de protección o de su prórroga, se presentará en el modelo oficial, y deberá contener: a) Cuando la solicitud se refiera a medicamentos: i. Lo establecido en el artículo 8 del Reglamento (CE) n.º 469/2009 del Parlamento Europeo y del Consejo, de 6 de mayo de 2009, relativo al certificado complementario de protección para los medicamentos (en adelante, Reglamento (CE) n.º 469/2009). ii. Información de que el producto está protegido por la patente de base designada por su titular a los fines del procedimiento de obtención del certificado. b) Cuando la solicitud se refiera a productos fitosanitarios: i. Lo establecido en el artículo 8 del Reglamento (CE) n.º 1610/96 del Parlamento Europeo y del Consejo, de 23 de julio de 1996, por el que se crea un certificado complementario de protección para los productos fitosanitarios (en adelante, Reglamento (CE) n.º 1610/96). ii. Información de que el producto está protegido por la patente de base designada por su titular a los fines del procedimiento de obtención del certificado. c) Cuando la solicitud se refiera a una prórroga de un certificado complementario de protección de medicamento: i. Lo establecido en el artículo 8, apartado d), del Reglamento (CE) n.º 469/2009. ii. Declaración responsable sobre el contenido de los documentos aportados con indicación de los Estados miembros de la Unión Europea a los que correspondan. d) La firma del solicitante o de su representante. 2. La presentación de la solicitud de certificado complementario de protección o de su prórroga implicará el pago de la tasa correspondiente. Artículo 55. Examen de formalidades y publicación de la solicitud. 1. La Oficina Española de Patentes y Marcas comprobará si se ha abonado la tasa por la solicitud de certificado complementario de protección o su prórroga y si se reúnen los datos necesarios para la publicación previstos en el artículo 9 del Reglamento (CE) n.º 1610/96 o del artículo 9 del Reglamento (CE) n.º 469/2009. En caso de que se detecte algún defecto, se notificarán al solicitante, otorgándole un plazo de diez días desde la publicación del defecto en el «Boletín Oficial de la Propiedad Industrial» para que mejore la solicitud, con indicación de que si así no lo hiciera se denegará la solicitud. 2. Una vez superado el examen, dentro del plazo de tres meses la Oficina Española de Patentes y Marcas publicará en el «Boletín Oficial de la Propiedad Industrial» la solicitud de certificado complementario de protección o de prórroga de acuerdo con lo establecido en el – 163 – CÓDIGO DE DERECHO FARMACÉUTICO § 6 Reglamento para la ejecución de la Ley 24/2015, de Patentes [parcial] artículo 9 del Reglamento (CE) n.º 469/2009 y en el artículo 9 del Reglamento (CE) n.º 1610/96. Artículo 56. Examen de la solicitud. 1. Una vez publicada la solicitud, la Oficina Española de Patentes y Marcas comprobará si la solicitud de certificado complementario de protección o su prórroga y el producto a que se refiere cumplen los requisitos establecidos en el Reglamento (CE) n.º 469/2009 y el Reglamento (CE) n.º 1610/96. No se comprobará de oficio si la autorización de comercialización es la primera autorización como medicamento o producto fitosanitario en la Unión Europea. 2. Si se detectaran irregularidades en la documentación o si la solicitud o el objeto de la misma no reúnen las condiciones respectivamente establecidas en el Reglamento (CE) n.º 469/2009 o el Reglamento (CE) n.º 1610/96, se comunicarán los defectos al solicitante para que los subsane o formule sus alegaciones en el plazo de dos meses a contar desde su publicación en el «Boletín Oficial de la Propiedad Industrial», con indicación de que si no los subsana se denegará la solicitud. 3. Cuando los defectos no se subsanen o se considere que persisten las objeciones señaladas en la notificación, se denegará la solicitud de certificado complementario de protección o la solicitud de prórroga, indicando los motivos, y se publicará la resolución en el «Boletín Oficial de la Propiedad Industrial». Artículo 57. Concesión del certificado complementario de protección o de la prórroga. Si la solicitud del certificado complementario de protección o de la prórroga y el producto al que se refiere cumplen las condiciones establecidas en la normativa comunitaria, la Oficina Española de Patentes y Marcas concederá el certificado o la prórroga y publicará en el «Boletín Oficial de la Propiedad Industrial» la resolución de concesión según lo establecido en el artículo 11 del Reglamento (CE) n.º 469/2009 y en el artículo 11 del Reglamento (CE) n.º 1610/96. [...] – 164 – CÓDIGO DE DERECHO FARMACÉUTICO §7 Real Decreto 1345/2007, de 11 de octubre, por el que se regula el procedimiento de autorización, registro y condiciones de dispensación de los medicamentos de uso humano fabricados industrialmente Ministerio de Sanidad y Consumo «BOE» núm. 267, de 7 de noviembre de 2007 Última modificación: 17 de septiembre de 2013 Referencia: BOE-A-2007-19249 La normativa actual en materia de medicamentos, ha contribuido a que en el mercado se encuentren medicamentos con probadas garantías de calidad, seguridad y eficacia. No obstante, a la luz de la experiencia adquirida, la Unión Europea ha considerado necesario adoptar nuevas medidas para favorecer el funcionamiento del mercado interior, sin olvidar en ningún momento la consecución de un elevado nivel de protección de la salud humana, para lo cual se avanza en la incorporación de criterios y procedimientos armonizados para la evaluación y autorización de medicamentos y se profundiza en medidas orientadas a la evaluación continuada de la seguridad de los mismos. La Ley 29/2006, de 26 de julio, de garantías y uso racional de medicamentos y productos sanitarios recoge estos criterios y procedimientos, siendo el instrumento por el que se transponen en gran parte las últimas disposiciones comunitarias sobre los medicamentos. Este real decreto completa la transposición de la Directiva 2004/27/CE del Parlamento Europeo y del Consejo, de 31 de marzo, por la que se modifica la Directiva 2001/83/CE, del Parlamento Europeo y del Consejo, de 6 de noviembre de 2001, por la que se establece un código comunitario sobre medicamentos para uso humano, que armoniza y recopila en un solo texto la normativa comunitaria sobre medicamentos de uso humano, y la Directiva 2004/24/CE, del Parlamento Europeo y del Consejo, de 31 de marzo, por la que se modifica, en lo que se refiere a los medicamentos tradicionales a base de plantas, la Directiva 2001/83/CE. La transposición de la Directiva 2004/27/CE implica la modificación, entre otras normas, del Real Decreto 767/1993, de 21 de mayo, por el que se regula la evaluación, autorización, registro y condiciones de dispensación de especialidades farmacéuticas de uso humano y otros medicamentos fabricados industrialmente, y otras disposiciones en materia de medicamentos especiales. Por ello, y dado que la modificación afecta a una gran cantidad de preceptos, se hace necesaria la elaboración de una nueva disposición que integre las normas originarias y sus posteriores modificaciones. Los aspectos fundamentales de la Directiva 2004/27/CE objeto de transposición en este real decreto, se refieren a la necesidad de mejorar el funcionamiento de los procedimientos de autorización de medicamentos, por lo que se ha revisado el procedimiento de autorización nacional y especialmente el de reconocimiento mutuo, con el fin de reforzar la posibilidad de cooperación entre Estados miembros y, asimismo, con el mismo fin, se – 165 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano incorpora un nuevo procedimiento de autorización comunitario denominado descentralizado y se establecen garantías de confidencialidad en la evaluación y transparencia y publicidad de las decisiones. La necesidad de garantizar un adecuado seguimiento de los efectos terapéuticos y del perfil de seguridad de cada nuevo medicamento, hace que la autorización de comercialización deba renovarse cinco años después de concedida. Una vez ratificada esa autorización, el periodo de validez debe ser, normalmente, ilimitado sin perjuicio de la evaluación continuada de los riesgos a través de sistemas adecuados de farmacovigilancia y de estudios de utilización de medicamentos en condiciones reales de uso. Por otra parte, el marco legislativo europeo prevé la posibilidad de que un medicamento autorizado no sea comercializado, estableciendo que toda autorización que no haga efectiva la comercialización del medicamento durante tres años consecutivos pierda la validez. No obstante, deben establecerse excepciones a esta norma cuando estén justificadas por razones de salud pública o interés general. Con el fin evitar una duplicidad de normas, se decide aplicar a las modificaciones de las autorizaciones de comercialización nacionales los mismos criterios de tipificación de los procedimientos comunitarios y se adopta el Reglamento (CE) 1084/2003 de la Comisión,, de 3 de junio de 2003, relativo al examen de las modificaciones de los términos de las autorizaciones de comercialización de medicamentos para uso humano y medicamentos veterinarios concedidas por la autoridad competente de un Estado miembro, así como sus sucesivas actualizaciones. Otro aspecto fundamental del medicamento es su identificación e información que ha de constar en el etiquetado y en el prospecto del mismo, como garantía de su correcto empleo, promoviendo la seguridad y la eficacia en su utilización. La Directiva 2004/27/CE, en cuanto a etiquetado y prospecto persigue definir normas comunes en la materia, dejando un amplio margen a las legislaciones nacionales, sobre todo en lo relacionado con las garantías de autenticidad y correcta identificación, para garantizar un elevado nivel de protección de los consumidores y permitir el uso correcto de los medicamentos a partir de una información completa y comprensible. La transposición de esta directiva implica la modificación del Real Decreto 2236/1993, de 17 de diciembre, por el que se regula el etiquetado y prospecto de los medicamentos de uso humano, afectando a una gran cantidad de preceptos, por lo que se ha considerado oportuno y siguiendo el criterio comunitario de unificar la legislación en un texto, recoger esta materia con el procedimiento de autorización, registro y condiciones de dispensación de los medicamentos de uso humano. Con el mismo criterio de unificación, se recogen en este real decreto los medicamentos especiales que contaban con su propia legislación y que quedan ahora integrados en esta misma norma. Así, se derogan, los Reales Decretos 479/1993, de 2 abril, por el que se regulan los medicamentos radiofármacos de uso humano, 478/1993, de 2 de abril, por el que se regulan los medicamentos derivados de la sangre y plasma humano, 288/1991, de 8 de marzo, por el que se regulan los medicamentos inmunológicos de uso humano, 2208/1994, de 16 de noviembre, que regula los medicamentos homeopáticos de uso humano de fabricación industrial y el 1800/2003, de 26 de diciembre, que regula los gases medicinales, este último fue sometido al procedimiento de información en materia de normas y reglamentaciones técnicas, previsto en el Directiva 98/34/CE y cuyo contenido se ha incluido en esta disposición. La Directiva 2004/24/CE del Parlamento Europeo y del Consejo de 31 de marzo de 2004, modifica, en lo que se refiere a los medicamentos tradicionales a base de plantas, la Directiva 2001/83/CE, armonizando las legislaciones de los Estados miembros en lo relativo a los medicamentos tradicionales a base de plantas y asegura las necesarias garantías de calidad, seguridad y eficacia de estos medicamentos, evitando las diferencias existentes que podrían repercutir sobre la protección de la salud pública. La principal novedad consiste en establecer un procedimiento de registro simplificado para los medicamentos tradicionales a base de plantas. La peculiaridad de este procedimiento se halla en que para obtener un registro como medicamento tradicional a base de plantas, se tendrá en cuenta el amplio uso tradicional, por lo que las pruebas clínicas y preclínicas que se suelen exigir con carácter general para la inscripción de un – 166 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano medicamento de uso humano, no serán necesarias aunque las autoridades competentes podrían solicitar información adicional para evaluar la seguridad, si se considerara necesario. Para la efectiva aplicación de este procedimiento se establece un periodo transitorio hasta el 30 de abril de 2011, en línea con las exigencias comunitarias. Esta adecuación a la nueva regulación supone una derogación de la legislación vigente hasta el momento, sin que por ello quede afectado negativamente el sector, puesto que las plantas tradicionalmente consideradas como medicinales cualquiera que sea su forma de presentación siempre que no tengan la consideración de medicamentos y se ofrezcan sin referencia a propiedades terapéuticas, diagnósticas o preventivas, podrán venderse libremente, quedando sometidas, en caso de reunir los criterios exigidos, a la legislación alimentaria. Por otra parte, la Orden SCO/3461/2003, de 26 de noviembre, mediante la cual se modificó el anexo II del Real Decreto 767/1993, incorporando al ordenamiento jurídico español la Directiva 2003/63/CE de la Comisión, de 25 de junio de 2003 se deroga y su contenido se incorpora como anexo I de este real decreto. La Directiva 2003/63/CE tiene una gran importancia por establecer requisitos normalizados para el expediente de autorización de comercialización de los medicamentos en todos los Estados miembros. Esto se consiguió con la implementación del documento técnico común (DTC). Actualmente este documento armonizado constituye la base fundamental para la consiguiente implantación de instrumentos telemáticos para la tramitación automatizada de solicitudes. La utilización de herramientas informáticas de gestión es a su vez un elemento esencial para el objetivo de dar acceso público a las decisiones de la Agencia Española de Medicamentos y Productos Sanitarios, así como a la información de medicamentos rigurosa y objetiva que acompaña a cada autorización. Con el presente real decreto queda derogada la legislación nacional sobre el procedimiento de autorización de comercialización de los medicamentos, que transponía diversas directivas comunitarias que fueron derogadas y codificadas por la Directiva 2001/83/CE. Como anexo II se recogen los datos que deben de figurar en la ficha técnica del medicamento. En el anexo III se incluye la información que debe incluirse en el etiquetado de los medicamentos. En el anexo IV se establecen los símbolos, siglas y leyendas que deben aparecer en el etiquetado de los medicamentos. El anexo V recoge los datos que debe contener como mínimo el prospecto y que se elabora de acuerdo con la ficha técnica del medicamento. El presente real decreto se adopta en desarrollo de la Ley 29/2006, de 26 de julio, de garantías y uso racional de los medicamentos y productos sanitarios, y tiene carácter de legislación de productos farmacéuticos a los efectos previstos en el artículo 149.1.16.ª de la Constitución; garantizando, en lo que concierne al tratamiento de los datos personales el respecto a la Ley Orgánica 15/1999, de 13 de diciembre, de Protección de Datos de Carácter Personal, y su normativa de desarrollo. Finalmente, en el proceso de elaboración de esta norma se ha consultado, entre otros, a las Comunidades Autónomas y a los sectores afectados. En su virtud, a propuesta del Ministro de Sanidad y Consumo, con la aprobación previa de la Ministra de Administraciones Públicas, de acuerdo con el Consejo de Estado y previa deliberación del Consejo de Ministros en su reunión del día 11 de octubre de 2007, DISPONGO: CAPÍTULO I Disposiciones generales Artículo 1. Objeto. Este real decreto tiene por objeto regular los medicamentos de uso humano fabricados industrialmente y en particular: – 167 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano a) Los requisitos de la solicitud para la autorización de comercialización. b) Los procedimientos de autorización, suspensión y revocación de la autorización, así como de las modificaciones de las condiciones de autorización. c) La ficha técnica, el etiquetado y prospecto. d) Las condiciones particulares para determinadas clases de medicamentos. e) Las obligaciones del titular. f) Los procedimientos comunitarios. g) La inscripción en el registro de medicamentos, incluidos los medicamentos especiales regulados en el capítulo IV. Artículo 2. Definiciones. A los efectos de la presente disposición se entenderá por: 1. Medicamento: toda sustancia o combinación de sustancias que se presente como poseedora de propiedades para el tratamiento o prevención de enfermedades en seres humanos, o que pueda usarse, o administrarse a seres humanos con el fin de restaurar, corregir o modificar las funciones fisiológicas ejerciendo una acción farmacológica, inmunológica o metabólica, o de establecer un diagnóstico médico; 2. Principio activo o sustancia activa: Toda sustancia o mezcla de sustancias destinadas a la fabricación de un medicamento y que, al ser utilizadas en su producción, se convierten en un componente activo de dicho medicamento destinado a ejercer una acción farmacológica, inmunológica o metabólica con el fin de restaurar, corregir o modificar las funciones fisiológicas, o de establecer un diagnóstico. 3. Excipiente: Todo componente de un medicamento distinto del principio activo y del material de acondicionamiento. 4. Materia prima: toda sustancia -activa o inactiva- empleada en la fabricación de un medicamento, ya permanezca inalterada, se modifique o desaparezca en el transcurso del proceso. 5. Forma galénica o forma farmacéutica: la disposición a que se adaptan los principios activos y excipientes para constituir un medicamento. Se define por la combinación de la forma en la que el producto farmacéutico es presentado por el fabricante y la forma en la que es administrada. 6. Presentación: cada una de las combinaciones en las que el medicamento está dispuesto para su utilización incluyendo composición, forma farmacéutica, dosis, y formato. 7. Formato: número de unidades contenidas en el envase y/o el contenido del mismo. 8. Nombre del medicamento: identifica al medicamento y consta de la denominación del medicamento, dosis y forma farmacéutica y cuando proceda, la mención de los destinatarios: lactantes, niños o adultos. 9. Denominación común: la Denominación Oficial Española (D.O.E) atribuida a cada principio activo por la Agencia Española de Medicamentos y Productos Sanitarios, en su defecto, la Denominación Común Internacional (D.C.I.) recomendada por la Organización Mundial de la Salud o, en su defecto, la denominación común usual. 10. Dosis del medicamento: el contenido de principio activo, expresado en cantidad por unidad de toma, por unidad de volumen o de peso en función de la presentación. 11. Ficha Técnica o resumen de las características del producto: documento autorizado por la Agencia, donde se reflejan las condiciones de uso autorizadas para el medicamento y recoge la información científica esencial para los profesionales sanitarios. 12. Acondicionamiento primario: el envase o cualquier otra forma de acondicionamiento que se encuentre en contacto directo con el medicamento. 13. Embalaje exterior: el embalaje en que se encuentre el acondicionamiento primario. 14. Etiquetado: las informaciones que constan en el embalaje exterior y en el acondicionamiento primario. 15. Prospecto: la información escrita dirigida al paciente o usuario, que acompaña al medicamento. 16. Medicamentos especiales: son aquellos medicamentos que requieren un tratamiento especial a efectos de demostrar su calidad, seguridad y eficacia. – 168 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano 17. Extensión de línea: la segunda y sucesivas solicitudes de autorización e inscripción en el registro de otras formas farmacéuticas, vías de administración y/o dosis de un medicamento ya autorizado, así como aquellas modificaciones que requieran la presentación de una nueva solicitud de autorización, de acuerdo con la norma europea que regula las modificaciones de autorización de medicamentos otorgadas por la autoridad competente de un Estado miembro. 18. Medicamento hemoderivado: medicamentos a base de constituyentes sanguíneos preparados industrialmente por establecimientos públicos o privados; dichos medicamentos comprenden, en particular, albúmina, factores de coagulación e inmunoglobulinas de origen humano 19. Medicamento inmunológico: Es todo medicamento consistente en vacunas, toxinas, sueros y alérgenos: a) Las vacunas, toxinas o sueros, que comprenden en particular: 1.º Los agentes utilizados para provocar una inmunidad activa como la vacuna anticolérica, el BCG, la vacuna antipoliomelítica, la vacuna antivariólica. 2.º Los agentes utilizados para diagnosticar el estado de inmunidad, en particular la tuberculina y la tuberculina PPD, las toxinas utilizadas en los test de Schick y de Dick, la brucelina. 3.º Los agentes utilizados para provocar una inmunidad pasiva, como la antitoxina diftérica, la globulina antivariólica, la globulina antilinfocítica. b) Los productos alergénicos comprendiendo cualquier medicamento destinado a detectar o provocar una alteración adquirida y específica en la respuesta inmunológica a un agente alergizante. 20. Vacunas individualizadas: son las preparadas con agentes inmunizantes, a concentración y dilución especifica en base a la correspondiente prescripción facultativa para un paciente determinado. 21. Alérgeno: todo producto destinado a identificar o provocar una modificación específica y adquirida de la respuesta inmunológica a un agente alergizante. 22. Radiofármaco: cualquier producto que, cuando esté preparado para su uso con finalidad terapéutica o diagnóstica, contenga uno o más radionucleidos (isótopos radiactivos). 23. Generador: cualquier sistema que incorpore un radionucleido (radionucleido padre) que en su desintegración origine otro radionucleido (radionucleido hijo) que se utilizará como parte integrante de un radiofármaco. 24. Equipo reactivo: cualquier preparado industrial que deba combinarse con el radionucleido para obtener el radiofármaco final. 25. Precursor: todo radionucleido producido industrialmente para el marcado radioactivo de otras sustancias antes de su administración. 26. Preparación extemporánea de un radiofármaco: es la preparación en el momento de su uso de un radiofármaco listo para su uso a partir del marcaje radioisotópico de un equipo o de muestras autólogas del propio paciente (células, proteínas), con un radionucleido precursor o un radionucleido producido por un generador de radionucleido. Esta preparación sólo podrá realizarse bajo petición mediante prescripción médica y si se cumplen las normas de correcta preparación extemporánea de radiofármacos que se publicarán posteriormente. 27. Medicamentos a base de plantas: el medicamento que contenga exclusivamente como principios activos, sustancias vegetales, preparados vegetales o combinaciones de estos. 28. Medicamento tradicional a base de plantas: el medicamento a base de plantas que cumpla las condiciones establecidas en el artículo 51. 29. Sustancias vegetales: las plantas, principalmente enteras, fragmentadas o cortadas, las partes de plantas, algas, hongos y líquenes no tratados, normalmente en forma seca pero también frescos. Determinados exudados que no han sido sometidos a un tratamiento específico se consideran también sustancias vegetales. Las sustancias vegetales se definen precisamente por la parte de la planta utilizada y la denominación botánica de acuerdo con el sistema binomial que incluye género, especie, variedad y autor. – 169 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano 30. Preparados vegetales: los que se obtienen sometiendo las sustancias vegetales a tratamientos como extracción, destilación, prensado, fraccionamiento, purificación, concentración o fermentación. Se incluyen las sustancias vegetales trituradas o pulverizadas, las tinturas, los extractos, los aceites esenciales, los zumos exprimidos y los exudados tratados. 31. Medicamento homeopático: El obtenido a partir de sustancias denominadas cepas homeopáticas, con arreglo a un procedimiento de fabricación homeopático descrito en la Farmacopea Europea, o en la Real Farmacopea Española o, en su defecto, en una farmacopea utilizada de forma oficial en un país de la Unión Europea. Un medicamento homeopático podrá contener varios principios activos. 32. Gases medicinales: Es el gas o mezcla de gases destinado a entrar en contacto directo con el organismo humano y que, actuando principalmente por medios farmacológicos, inmunológicos o metabólicos, se presente dotado de propiedades para prevenir, diagnosticar, tratar, aliviar o curar enfermedades o dolencias. Se consideran gases medicinales los utilizados en terapia de inhalación, anestesia, diagnóstico «in vivo» o para conservar y transportar órganos, tejidos y células destinados al transplante, siempre que estén en contacto con ellos. Se entenderá por gases medicinales licuados, el oxígeno líquido, nitrógeno líquido y protóxido de nitrógeno líquido, así como cualquier otro que con similares características y utilización, puedan fabricarse en el futuro. 33. Titular de la autorización de comercialización: es la persona física o jurídica responsable de la comercialización del medicamento para el que ha obtenido la preceptiva autorización de comercialización. A tal efecto se creará un registro de titulares de autorizaciones de comercialización. 34. Representante del titular de la autorización de comercialización: la persona física o jurídica, normalmente conocida como representante local, designada por el titular de la autorización de comercialización para representarle en España. 35. Medicamento genérico: el medicamento que tenga la misma composición cualitativa y cuantitativa en principios activos y la misma forma farmacéutica, y cuya bioequivalencia con el medicamento de referencia haya sido demostrada por estudios adecuados de biodisponibilidad. Artículo 3. Ámbito de aplicación y exclusiones. 1. Este real decreto es de aplicación a los medicamentos de uso humano y a los medicamentos especiales de uso humano, fabricados industrialmente o en cuya fabricación intervenga un proceso industrial en lo referente a los procedimientos y requisitos para la presentación de solicitudes de autorización, la evaluación de las mismas, requisitos para la autorización de comercialización, ficha técnica, etiquetado y prospecto incluyendo condiciones de prescripción y dispensación, así como la inclusión de los medicamentos autorizados en el correspondiente registro oficial. 2. Quedan excluidos del ámbito de aplicación del presente real decreto: a) Los medicamentos de terapia avanzada recogidos en el articulo 47 de la Ley 29/2006, de 26 de julio, tal como se definen en el Reglamento (CE) n.º 1394/2007 del Parlamento Europeo y del Consejo, de 13 de noviembre de 2007, sobre medicamentos de terapia avanzada y por el que se modifican la Directiva 2001/83/CE y el Reglamento (CE) n.º 726/2004, preparados ocasionalmente, de acuerdo con normas de calidad específicas, y empleados en España, en una institución hospitalaria y bajo la responsabilidad profesional exclusiva de un médico colegiado, con el fin de cumplir una prescripción facultativa individual de un producto hecho a medida destinado a un solo paciente, y sean medicamentos en fase de investigación clínica o sean medicamentos que la Agencia Española de Medicamentos y Productos Sanitarios considere que satisfacen las garantías de calidad, seguridad, eficacia, identificación e información. b) La sangre completa, el plasma y las células sanguíneas de origen humano. 3. La presente disposición no es de aplicación en lo referido a las solicitudes, evaluación y autorización, para los medicamentos contemplados en el anexo I del Reglamento (CE) 726/2004 del Parlamento Europeo y del Consejo, de 31 de marzo de 2004, por el que se – 170 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano establecen procedimientos comunitarios para la autorización y supervisión de medicamentos de uso humano y veterinario y por el que se crea la Agencia Europea de Medicamentos. No obstante, sí les será de aplicación lo previsto en el artículo 21.3 y los anexos III y IV. Artículo 4. Carácter de la autorización de comercialización. 1. Ningún medicamento fabricado industrialmente podrá ser puesto en el mercado sin la previa autorización de comercialización otorgada por la Agencia Española de Medicamentos y Productos Sanitarios o por la Comisión Europea, e inscripción en el registro de medicamentos, de acuerdo con los procedimientos establecidos para cada caso. Toda modificación, transmisión, suspensión y revocación de la autorización de comercialización de un medicamento deberá ser notificada, o solicitada y autorizada por la Agencia Española de Medicamentos y Productos Sanitarios, según proceda, debiendo constar en todos los casos en el registro de medicamentos autorizados que, a estos efectos, tendrá, del mismo modo que la inscripción, carácter constitutivo, salvo en el caso de los medicamentos autorizados por la Comisión Europea. 2. Cuando un medicamento haya obtenido una autorización de comercialización inicial, toda dosificación, forma farmacéutica, vía de administración y presentación adicionales, así como cualesquiera modificaciones y ampliaciones que se introduzcan habrán también de ser notificadas, o solicitadas y autorizadas. Todas estas autorizaciones de comercialización se considerarán pertenecientes a la misma autorización global de comercialización, en particular, a los efectos de la aplicación de los períodos de exclusividad de datos, así como para las modificaciones posteriores de la autorización que afecten a todo un conjunto de medicamentos de un mismo titular conteniendo el mismo principio activo. CAPÍTULO II Autorización de medicamentos Sección 1.ª Solicitudes Artículo 5. Requisitos del solicitante de una autorización de comercialización. El solicitante de la autorización de comercialización de un medicamento ha de estar establecido en la Unión Europea. El solicitante podrá designar un representante legal con el que se entenderán las actuaciones derivadas de la tramitación de la solicitud de autorización del medicamento o de sus posteriores modificaciones. Asimismo, el solicitante podrá indicar en la solicitud el titular de la autorización de comercialización propuesto para España. Artículo 6. Solicitud de autorización de comercialización de medicamentos fabricados industrialmente. 1. Las solicitudes de autorización de comercialización de medicamentos se presentarán en cualquiera de los lugares previstos en el artículo 38.4 de la Ley 30/1992, de 26 de noviembre, de Régimen Jurídico de las Administraciones Públicas y del Procedimiento Administrativo Común, dirigidas a la Agencia Española de Medicamentos y Productos Sanitarios. Asimismo y conforme a lo previsto en el artículo 38.9 de la mencionada Ley, las solicitudes podrán presentarse por medios telemáticos. 2. El modelo de solicitud habrá de ajustarse al que en cada momento establezca la Agencia Española de Medicamentos y Productos Sanitarios, de acuerdo a los modelos normalizados aprobados por la Comisión Europea. 3. La documentación se presentará, al menos, en castellano. No obstante lo anterior, la Agencia Española de Medicamentos y Productos Sanitarios podrá establecer que una o varias partes de la documentación científico-técnica puedan presentarse en otro idioma. 4. Las solicitudes de autorización de medicamentos habrán de ir acompañadas del documento acreditativo del pago de la tasa. – 171 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano 5. La solicitud, cuyo modelo podrá ser obtenido a través de la página web de la Agencia Española de Medicamentos y Productos Sanitarios, deberá comprender al menos los datos y documentos que a continuación se relacionan. El expediente se presentará en formato normalizado de acuerdo con lo establecido en el Anexo I de este Real Decreto: a) nombre o razón social y domicilio o sede social del solicitante y, en su caso, del fabricante, DNI/NIE o CIF; b) nombre del medicamento; c) composición cualitativa y cuantitativa de todos los componentes del medicamento, incluyendo la de su denominación común internacional (DCI) recomendada por la Organización Mundial de la Salud, y su equivalencia con la denominación oficial española (DOE), cuando la tenga, o la mención de la denominación química pertinente en ausencia de las anteriores. En el caso de sustancias y preparados vegetales se declararán de acuerdo con lo establecido para los mismos; d) evaluación del riesgo que el medicamento podría representar para el medio ambiente. Este impacto se deberá estudiar y se deberán prever, caso por caso, las disposiciones particulares destinadas a limitarlo; e) descripción del modo de fabricación; f) indicaciones terapéuticas, contraindicaciones y reacciones adversas; g) posología, forma farmacéutica, forma y vía de administración y período o plazo de validez previsto; h) indicaciones sobre las medidas de precaución y de seguridad que han de adoptarse al almacenar el medicamento, al administrarlo a los pacientes y al eliminar los productos residuales, junto con la indicación de cualquier riesgo potencial que el medicamento pudiera presentar para el medio ambiente; i) descripción de los métodos de control utilizados por el fabricante; j) resultado de las pruebas: 1.º Farmacéuticas (fisicoquímicas, biológicas o microbiológicas). 2.º Preclínicas (toxicológicas y farmacológicas). 3.º Clínicas. Los documentos e información relativos a los resultados de las pruebas farmacéuticas, preclínicas y clínicas deberán ir acompañados de resúmenes detallados e informes de expertos, que formarán parte de la correspondiente solicitud y quedarán integrados en el expediente de autorización. Estos informes han de ser elaborados y firmados por personas que posean las cualificaciones técnicas y profesionales necesarias, avaladas en un currículum que se acompañará al informe; k) un resumen del sistema de farmacovigilancia del solicitante, que incluya: 1.º Prueba de que el solicitante dispone de los servicios de una persona cualificada responsable de la farmacovigilancia, 2.º Los Estados miembros en los que reside y desempeña sus funciones dicha persona cualificada, 3.º Datos de contacto de la persona cualificada, 4.º Declaración firmada por el solicitante que certifique que dispone de los medios necesarios para desempeñar las funciones y asumir las responsabilidades definidas en la normativa sobre farmacovigilancia, 5.º Referencia a la ubicación del archivo maestro del sistema de farmacovigilancia del medicamento; l) El plan de gestión de riesgos con la descripción del sistema de gestión de riesgos que el solicitante vaya a elaborar para el medicamento, junto con un resumen. El sistema de gestión de riesgos deberá ser proporcionado a los riesgos identificados o potenciales del medicamento, y a la necesidad de datos sobre seguridad en la fase posautorización, debiendo actualizarse la información cuando proceda; m) una declaración del solicitante según la cual los ensayos clínicos llevados a cabo fuera de la Unión Europea cumplen los principios éticos y normas de buena práctica clínica previstos en el Real Decreto 223/2004, de 6 de febrero, por el que se regulan los ensayos clínicos con medicamentos; – 172 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano n) ficha técnica o resumen de las características del producto de acuerdo con el anexo II, una maqueta del diseño y contenido del embalaje exterior y del acondicionamiento primario, así como el prospecto de acuerdo con la normativa al respecto, a efectos de garantizar la adecuada comprensión por los ciudadanos; ñ) documento acreditativo de que el fabricante está autorizado en su país para fabricar medicamentos; o) Copias de la documentación siguiente: 1.º En su caso, copia de la autorización de comercialización obtenida en otro Estado miembro o en un tercer país, junto con la ficha técnica y un resumen de los datos de seguridad, incluidos los datos contenidos en los informes periódicos actualizados en materia de seguridad, cuando se disponga de ellos, y las notificaciones de sospechas de reacciones adversas, junto con la lista de los Estados miembros en los que se esté estudiando una solicitud de autorización, 2.º La ficha técnica propuesta por el solicitante en virtud del anexo II o aprobada por las autoridades competentes del Estado miembro, y el prospecto propuesto con arreglo a los anexos IV y V o aprobado por la autoridad competente del Estado miembro, 3.º Detalles de cualquier decisión de denegación de autorización, tanto en la Unión Europea como en un tercer país, y los motivos de tal decisión. p) Documento acreditativo de que el fabricante del medicamento ha verificado mediante auditorías el cumplimiento, por parte del fabricante del principio activo, de los principios y directrices de las normas de correcta fabricación. Este documento contendrá una referencia a la fecha de la auditoría, así como a que su resultado confirma que la fabricación se atiene a los principios y directrices de las referidas normas de correcta fabricación. Artículo 7. Requisitos específicos de la autorización de medicamentos genéricos. 1. Respecto a los medicamentos genéricos definidos en el artículo 2.35, las diferentes sales, ésteres, éteres, isómeros, mezclas de isómeros, complejos o derivados de un principio activo se considerarán un mismo principio activo, a menos que tengan propiedades considerablemente diferentes en cuanto a seguridad y/o eficacia, en cuyo caso el solicitante deberá facilitar datos suplementarios para demostrar la seguridad y/o eficacia de la diversidad de sales, ésteres o derivados de un principio activo autorizado. Las diferentes formas farmacéuticas orales de liberación inmediata se considerarán una misma forma farmacéutica. El solicitante podrá estar exento de presentar los estudios de biodisponibilidad si puede demostrar que el medicamento genérico satisface los criterios pertinentes definidos en las correspondientes directrices detalladas. 2. Sin perjuicio del derecho relativo a la protección de la propiedad industrial y comercial, el solicitante no tendrá obligación de facilitar los resultados de los ensayos preclínicos y clínicos si puede demostrar que el medicamento es genérico de un medicamento de referencia que está o ha sido autorizado con arreglo a la presente disposición, desde hace ocho años como mínimo por un Estado miembro o en la Unión Europea por procedimiento centralizado. A estos efectos, se entiende por medicamento de referencia aquel autorizado en base a un expediente completo. 3. Los medicamentos genéricos de un medicamento de referencia, autorizado con arreglo a la presente disposición, no se comercializarán hasta transcurridos diez años desde la fecha de la autorización inicial del medicamento de referencia. Este período de diez años se ampliará hasta un máximo de once años si, durante los primeros ocho años del período de diez años, el titular de la autorización de comercialización del medicamento de referencia obtiene una autorización para una o varias indicaciones terapéuticas nuevas y, durante la evaluación científica previa a su autorización, se establece que dichas indicaciones aportarán un beneficio clínico significativo en comparación con las terapias existentes. 4. Cuando el medicamento de referencia no esté autorizado en España el solicitante deberá indicar en la solicitud el nombre del Estado miembro en que esté o haya sido autorizado y la fecha de autorización. La Agencia Española de Medicamentos y Productos Sanitarios, solicitará a la autoridad competente del otro Estado miembro una confirmación, en el plazo de un mes, de que el medicamento de referencia está o ha sido autorizado, junto – 173 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano con la composición completa del medicamento de referencia y, en caso necesario, cualquier otra documentación que considere pertinente. 5. Los medicamentos genéricos deberán designarse con la denominación oficial española del principio activo y, en su defecto, con la denominación común usual o científica de dicha sustancia, acompañada, en su caso, del nombre o marca del titular o fabricante; asimismo, podrán denominarse con una marca siempre que no pueda confundirse con una denominación oficial española o una denominación común internacional ni inducir a error sobre las propiedades terapéuticas o la naturaleza del medicamento. Los medicamentos genéricos se identificarán por llevar a continuación de su nombre las siglas EFG. Artículo 8. Requisitos específicos de la autorización de medicamentos con solicitud combinada con datos suplementarios. Cuando el medicamento no se ajuste a los requisitos de medicamento genérico del apartado 1 del artículo anterior, cuando la bioequivalencia no pueda ser demostrada por medio de estudios de biodisponibilidad o cuando haya diferencias en los principios activos, las indicaciones terapéuticas, la dosificación, la forma farmacéutica o la vía de administración con respecto a las del medicamento de referencia, deberán facilitarse los resultados de los ensayos preclínicos y/o clínicos adecuados suplementarios. Artículo 9. Requisitos específicos de la autorización de medicamentos biológicos similares a otro de referencia. 1. Las solicitudes de autorización deberán incluir los resultados de los ensayos preclínicos y clínicos adecuados cuando un medicamento biológico que sea similar a un producto biológico de referencia no cumpla las condiciones de la definición de medicamentos genéricos, debido a diferencias relacionadas con las materias primas o diferencias en el proceso de fabricación del medicamento biológico y del medicamento biológico de referencia. 2. La documentación deberá ajustarse a los criterios establecidos en el documento técnico común (DTC) acordado en la Unión Europea y recogido en el Anexo I así como a lo dispuesto en las directrices detalladas especificas para cada materia. Artículo 10. Requisitos específicos de la autorización de medicamentos basados en principios activos suficientemente comprobados. 1. El solicitante no tendrá obligación de facilitar los resultados de ensayos preclínicos y clínicos propios si puede demostrar que el principio activo del medicamento ha tenido un uso médico bien establecido al menos durante diez años dentro de la Unión Europea y presenta una eficacia reconocida, así como un nivel aceptable de seguridad en virtud de las condiciones previstas en el anexo I. 2. En este caso, los resultados de los ensayos se sustituirán por una documentación bibliográfico-científica que aporte evidencia científica adecuada. 3. Lo establecido en este artículo no será de aplicación a productos que deban cumplir las condiciones señaladas en los artículos 7, 8 y 9. Artículo 11. Solicitudes de nuevas asociaciones de principios activos autorizados. Las solicitudes de medicamentos que contengan asociación de principios activos presentes en la composición de medicamentos autorizados, pero que no hayan sido combinadas todavía con fines terapéuticos, deberán aportar los resultados de los ensayos preclínicos y/o clínicos relativos a la nueva asociación, sin necesidad de facilitar la documentación relativa a cada principio activo individual. Artículo 12. Solicitudes de autorización de medicamentos con consentimiento expreso del titular de una autorización previa o de un expediente en trámite de registro. 1. Tras la concesión de una autorización de comercialización, el titular de la misma podrá consentir que otro solicitante haga uso de la documentación farmacéutica, preclínica y clínica – 174 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano que obre en el expediente de su medicamento, para el estudio de una solicitud posterior de un medicamento que tenga la misma composición cualitativa y cuantitativa en sustancias activas y la misma forma farmacéutica. Esta situación deberá certificarse por ambas partes en la documentación que acompañe a la solicitud, significando la exactitud de ambos expedientes en todos los aspectos farmacéuticos, preclínicos y clínicos, excepto en los aspectos de identificación y diseño del etiquetado del medicamento. 2. El solicitante de un expediente en trámite podrá así mismo, consentir a otro solicitante la presentación de otra solicitud basada en idéntica documentación farmacéutica, preclínica y clínica, acompañando en el segundo expediente certificación por ambas partes de esta autorización y de la exactitud de ambas documentaciones en todos los aspectos farmacéuticos, preclínicos y clínicos, excepto en los aspectos de identificación y diseño del etiquetado del medicamento. Artículo 13. Periodo de exclusividad para nuevas indicaciones de principios activos suficientemente conocidos. Cuando se autorice una nueva indicación para un principio activo suficientemente conocido, se concederá un período de un año de exclusividad de datos, no acumulativo a otros periodos de protección de datos, siempre y cuando se hayan llevado a cabo estudios clínicos y/o preclínicos significativos en relación con la nueva indicación. Sección 2.ª Procedimiento de autorización Artículo 14. Objetivos del procedimiento de autorización. 1. El procedimiento de autorización tiene por objeto comprobar que el medicamento: a) Alcanza los requisitos de calidad establecidos. b) Es seguro, no produciendo en condiciones normales de utilización efectos tóxicos o indeseables desproporcionados al beneficio que procura. c) Es eficaz en las indicaciones terapéuticas aprobadas. d) Está correctamente identificado y va acompañado de la información precisa para su utilización. 2. La evaluación de los efectos terapéuticos positivos del medicamento se apreciarán en relación con cualquier riesgo relacionado con la calidad, la seguridad y la eficacia del medicamento para la salud del paciente o la salud pública, entendido como relación beneficio-riesgo 3. Lo establecido en este artículo será asimismo de aplicación a las modificaciones que se produzcan en la autorización y seguirá siendo aplicable, en tanto el producto esté en el mercado, de acuerdo a las nuevas evidencias que con respecto a su seguridad y efectividad se vayan obteniendo. 4. En cualquier momento la Agencia Española de Medicamentos y Productos Sanitarios, podrá comprobar que se cumplen los requisitos del apartado 1 anterior. Artículo 15. Garantías de confidencialidad. La documentación de la solicitud de autorización y los informes de experto tendrán carácter confidencial. Artículo 16. Admisión a trámite y validación de la solicitud. 1. La Agencia Española de Medicamentos y Productos Sanitarios, en el plazo de diez días naturales desde la presentación de la solicitud, verificará que ésta reúne los requisitos previstos, y notificará al solicitante su admisión a trámite con indicación del procedimiento aplicable y la identificación del expediente, así como del plazo para la notificación de la resolución. 2. En el caso de que la solicitud no reúna los requisitos establecidos, se requerirá al solicitante que subsane las deficiencias en el plazo máximo de diez días naturales, con – 175 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano indicación de que si así no lo hiciera se archivará su solicitud, previa resolución que se dictará en los términos establecidos en el artículo 42 de la Ley 30/1992, de 26 de noviembre. 3. El plazo máximo para la notificación de la resolución del procedimiento de autorización de medicamentos será de 210 días naturales, que comenzarán a computarse a partir del día siguiente a la fecha de presentación de una solicitud válida. 4. En caso de que el medicamento objeto de la solicitud haya sido previamente autorizado en otro Estado miembro, el mismo titular no podrá presentar solicitud ante la Agencia Española de Medicamentos y Productos Sanitarios, ni será admitida a trámite, salvo si la solicitud se presenta de acuerdo con el procedimiento de reconocimiento mutuo establecido en el artículo 72, o si se trata de una extensión de línea de un medicamento autorizado en España a través del procedimiento nacional. 5. En caso de que el medicamento objeto de la solicitud esté siendo evaluado en otro Estado miembro deberá ser comunicado por el solicitante a la Agencia Española de Medicamentos y Productos Sanitarios. La Agencia informará al interesado que deberá tramitar su solicitud por procedimiento de reconocimiento mutuo o bien por procedimiento descentralizado, de acuerdo con lo establecido en los artículos 72 y 73, archivando su solicitud si procede. Artículo 17. Evaluación de la documentación farmacéutica, preclínica y clínica y emisión del correspondiente informe. 1. Admitida a trámite la solicitud, la Agencia Española de Medicamentos y Productos Sanitarios realizará la evaluación del expediente y emitirá informe de evaluación. A tal efecto, podrá requerir documentación complementaria o aclaraciones al solicitante sobre cualquier extremo objeto de la solicitud, estableciendo un plazo de tres meses, que excepcionalmente podrá ampliarse a seis, para la presentación de dicha documentación. Cuando la Agencia Española de Medicamentos y Productos Sanitarios haga uso de esta facultad, el plazo previsto en el apartado tercero del artículo anterior quedará suspendido hasta que se proporcionen los datos complementarios requeridos. 2. El informe de evaluación será motivado y contemplará los aspectos farmacéuticos, preclínicos y clínicos del medicamento. 3. En el proceso de evaluación, la Agencia Española de Medicamentos y Productos Sanitarios podrá someter el medicamento, sus materias primas, productos intermedios y otros componentes a examen de sus Laboratorios Oficiales de Control; asimismo, podrá solicitar la colaboración de otro laboratorio nacional acreditado a tal efecto por la propia Agencia, a un laboratorio oficial de control comunitario o de un tercer país. 4. Como complemento de lo anterior, podrá concederse una autorización de comercialización de un medicamento sujeta adicionalmente al cumplimiento de una o varias de las condiciones siguientes: a) Se adopten determinadas medidas para garantizar el uso seguro del medicamento que se incluyan en el sistema de gestión de riesgos; b) Se realicen estudios de seguridad posautorización; c) Se cumplan las obligaciones sobre el registro o la notificación de sospechas de reacciones adversas que sean más estrictas que las contempladas en la normativa vigente sobre farmacovigilancia de medicamentos de uso humano. d) Cualquier otra condición o restricción relacionada con el uso seguro y eficaz del medicamento; e) El sistema de farmacovigilancia sea adecuado; f) Se realicen estudios de eficacia posautorización, cuando se planteen cuestiones sobre la eficacia del medicamento que solo puedan resolverse después de la comercialización de este. La obligación de realizar tales estudios se basará en los actos delegados adoptados de conformidad con la normativa europea. Artículo 18. Dictamen del Comité de Evaluación de Medicamentos de Uso Humano. La Agencia Española de Medicamentos y Productos Sanitarios podrá solicitar al Comité de Evaluación de Medicamentos de Uso Humano su dictamen sobre las solicitudes de nuevas autorizaciones de medicamentos, y solicitudes de modificaciones mayores de – 176 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano autorizaciones de comercialización de acuerdo con lo establecido en artículo 63, para lo cual remitirá a dicho Comité el informe de evaluación y en su caso, propuesta de la ficha técnica y el prospecto. En todo caso, los dictámenes del Comité de Evaluación de Medicamentos de Uso Humano no tendrán carácter vinculante. Articulo 19. Causas y procedimiento de denegación. 1. La solicitud de autorización de un medicamento podrá ser denegada por las siguientes razones, cuando: a) la relación beneficio-riesgo no sea favorable; b) no se justifique suficientemente la eficacia terapéutica; c) el medicamento no tenga la composición cualitativa y cuantitativa declarada o carezca de la calidad adecuada; d) los datos e informaciones contenidos en la documentación de la solicitud de autorización sean erróneos o incumplan la normativa de aplicación en la materia. 2. En caso de que el resultado de la evaluación sea desfavorable por alguna de las causas previstas en el apartado anterior, o existan diferencias sustanciales de la información del medicamento con respecto a la propuesta realizada por el solicitante, la Subdirección General de Medicamentos de Uso Humano de la Agencia Española de Medicamentos y Productos Sanitarios lo pondrá de manifiesto al interesado a fin de que, en un plazo de quince días, pueda efectuar las alegaciones y presentar la documentación que considere oportuna. 3. Efectuadas alegaciones por parte del solicitante, la Agencia modificará el informe de evaluación y si procede, de acuerdo con el artículo anterior, será remitido al Comité de Evaluación de Medicamentos de Uso Humano, a fin de la emisión del oportuno dictamen. Artículo 20. Resolución. 1. Finalizada la instrucción del procedimiento se dictará resolución motivada que se notificará al interesado con expresión de los recursos que procedan de acuerdo con lo previsto en la normativa vigente. 2. Cuando los resultados de la evaluación sean favorables, la Agencia Española de Medicamentos y Productos Sanitarios emitirá resolución autorizando la comercialización, sin perjuicio de las obligaciones derivadas de las normas sobre financiación pública. 3. La resolución de autorización de un medicamento contendrá las condiciones de autorización y formarán parte de la misma los datos administrativos, la ficha técnica, el etiquetado y el prospecto. 4. En el documento de autorización deberán figurar, al menos, los siguientes datos: a) Nombre del medicamento. b) Número de registro. c) Grupo terapéutico. d) Forma farmacéutica. e) Vía de administración. f) Presentaciones autorizadas con sus respectivos Códigos Nacionales. g) Condiciones de conservación y caducidad. h) Condiciones de prescripción y dispensación. i) Nombre y dirección del titular de la autorización. j) Nombre y dirección del representante del titular de la autorización de comercialización, en su caso. k) Nombre y dirección del fabricante, tanto del principio activo como del medicamento en caso de que difieran. l) Composición cualitativa y cuantitativa completa. m) Estudios posautorización cuando proceda y los plazos para su cumplimiento. n) Frecuencia para la presentación de los informes periódicos actualizados en materia de seguridad. – 177 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano Artículo 20 bis. Requisitos posautorización. 1. Con posterioridad a la concesión de una autorización de comercialización, la Agencia Española de Medicamentos y Productos Sanitarios podrá obligar al titular de la autorización de comercialización: a) A que realice un estudio de seguridad posautorización en caso de existir preocupación por los riesgos del medicamento autorizado. Cuando la misma preocupación afecte a más de un medicamento, la Agencia Española de Medicamentos y Productos Sanitarios previa consulta al Comité de Seguridad de Medicamentos de Uso Humano, cuando proceda, instará al titular o titulares de la autorización de comercialización de que se trate a realizar conjuntamente un estudio de seguridad posautorización; b) A que realice un estudio de eficacia posautorización cuando el conocimiento de la enfermedad o la metodología clínica indiquen que las evaluaciones de eficacia anteriores podrían tener que revisarse de forma significativa. La Agencia Española de Medicamentos y Productos Sanitarios notificará la imposición de la obligación, la cual estará debidamente justificada, y en la que se especificarán los objetivos y el calendario de presentación y realización del estudio. 2. El titular de la autorización de comercialización en los 30 días naturales siguientes a la recepción de la notificación de la obligación podrá presentar sus objeciones. 3. Revisadas las objeciones presentadas por el titular de la autorización de comercialización, la Agencia Española de Medicamentos y Productos Sanitarios retirará o confirmará la obligación. En caso de confirmación de la obligación el titular solicitará la modificación de la autorización de comercialización para incluir la obligación como condición de la autorización y en consecuencia se actualizará el sistema de gestión de riesgos. Artículo 21. Inscripción en el registro de medicamentos. 1. La autorización del medicamento se inscribirá de oficio en el Registro de Medicamentos de la Agencia Española de Medicamentos y Productos Sanitarios. 2. Cada número de registro se referirá a una composición, una forma farmacéutica, una dosis por unidad de administración incluyendo todas las presentaciones para la venta. Cada una de las presentaciones será identificada por su correspondiente Código Nacional. En el caso de un medicamento que deba administrarse con un dispositivo aplicador exclusivo que permita ser utilizado repetidas veces, podrá admitirse en el mismo registro una presentación con un dispositivo aplicador y otra sin él, asignando un Código Nacional a cada una de las presentaciones. También podrán admitirse bajo el mismo número de registro otros supuestos cuando así lo determine la Agencia Española de Medicamentos y Productos Sanitarios. 3. En el caso de los medicamentos relacionados en el Anexo del Reglamento (CE) 726/2004, la puesta en el mercado deberá ser comunicada a la Agencia Española de Medicamentos y Productos Sanitarios a efectos de su inclusión en el registro de medicamentos autorizados. Artículo 21 bis. Comunicación a la Agencia Europea. La Agencia Española de Medicamentos y Productos Sanitarios comunicará a la Agencia Europea de Medicamentos las autorizaciones de comercialización que haya concedido con las condiciones contempladas en los artículos 17.4, 20 bis y 26. Artículo 22. Transparencia y publicidad. 1. Para cada medicamento que haya autorizado, la Agencia Española de Medicamentos y Productos Sanitarios pondrá a disposición del público sin dilación la autorización de comercialización, el prospecto, la ficha técnica y todas las condiciones establecidas con arreglo a los artículos 17.4, 20 bis y 26 junto con todos los plazos para el cumplimiento de dichas condiciones, de conformidad con lo dispuesto en la Ley Orgánica 15/1999, de 13 de diciembre, de Protección de Datos de Carácter Personal, y previa disociación de datos de carácter personal. – 178 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano La Agencia Española de Medicamentos y Productos Sanitarios elaborará un informe de evaluación y realizará comentarios sobre el expediente en lo referente a los resultados de las pruebas farmacéuticas y preclínicas, los ensayos clínicos, el sistema de gestión de riesgos y el sistema de farmacovigilancia del medicamento de que se trate. El informe de evaluación se actualizará cuando se disponga de nuevos datos que sean importantes para la evaluación de la calidad, la seguridad o la eficacia del medicamento. La Agencia Española de Medicamentos y Productos Sanitarios pondrá a disposición del público sin dilación el informe de evaluación y los motivos del dictamen, previa supresión de cualquier información comercial de carácter confidencial, y disociación de datos de carácter personal. Así mismo, se facilitará una justificación por separado para cada una de las indicaciones solicitadas. El informe público de evaluación contendrá un resumen redactado de forma comprensible para el público. El resumen incluirá, en particular, una sección relativa a las condiciones de utilización del medicamento. 2. La Agencia Española de Medicamentos y Productos Sanitarios creará un portal web con información de los medicamentos autorizados en España. Así mismo, dicho portal enlazará con el portal web creado con arreglo al artículo 26 del Reglamento (CE) n.º 726/2004 del Parlamento Europeo y del Consejo, de 31 de marzo de 2004, por el que se establecen procedimientos comunitarios para la autorización y el control de los medicamentos de uso humano y veterinario y por el que se crea la Agencia Europea de Medicamentos. En el portal web de la Agencia Española de Medicamentos y Productos Sanitarios estará disponible la siguiente información: a) Informes públicos de evaluación, junto con un resumen. b) Fichas técnicas y prospectos de los medicamentos autorizados. c) Resúmenes de los planes de gestión de riesgos de los medicamentos autorizados. d) Lista de los medicamentos contemplados en el artículo 23 del Reglamento (CE) n.º 726/2004 del Parlamento Europeo y del Consejo, de 31 de marzo de 2004. e) Información sobre los distintos medios para que los profesionales sanitarios y los pacientes puedan notificar al Sistema Español de Farmacovigilancia las sospechas de reacciones adversas a los medicamentos, incluidos los formularios web estructurados contemplados en el artículo 25 del Reglamento (CE) n.º 726/2004 del Parlamento Europeo y del Consejo, de 31 de marzo de 2004. Artículo 23. Responsabilidad del titular y del fabricante. 1. El titular de la autorización de comercialización de cada medicamento será el responsable del cumplimiento de las obligaciones derivadas de la autorización y deberá contar con los medios materiales y personales necesarios para cumplir las obligaciones derivadas de la misma. 2. La autorización de un medicamento se concederá sin perjuicio de la responsabilidad civil o penal del fabricante o fabricantes y así mismo, del fabricante o fabricantes implicados en el proceso de fabricación del producto o de su materia prima, y en su caso del titular de la autorización de comercialización. Artículo 24. Condiciones de prescripción y dispensación de los medicamentos. 1. La Agencia Española de Medicamentos y Productos Sanitarios clasificará el medicamento como: a) Medicamento sujeto a prescripción médica. b) Medicamento no sujeto a prescripción médica. Dentro de los medicamentos, cuya dispensación requiera prescripción médica, existirán las siguientes subcategorías: 1.º Medicamentos sujetos a prescripción médica de dispensación renovable o no renovable. 2.º Medicamentos sujetos a prescripción médica especial. – 179 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano 3.º Medicamentos de prescripción médica restringida, de utilización reservada a determinados medios especializados. 2. Los medicamentos se someterán a prescripción médica especial cuando: a) Contengan, en dosis no exentas, una sustancia clasificada como estupefaciente o psicótropo de acuerdo a los convenios internacionales sobre la materia. b) Puedan ser objeto, en caso de utilización anormal, de riesgo considerable de abuso medicamentoso, puedan provocar toxicodependencia o ser desviados para usos ilegales. c) Contengan alguna sustancia que, por su novedad o propiedades, se considere necesaria su inclusión en este grupo como medida de precaución. 3. Los medicamentos se someterán a prescripción médica restringida cuando: a) Se trate de medicamentos que exigen particular vigilancia, supervisión y control del equipo multidisciplinar de atención a la salud, los cuales a causa de sus características farmacológicas o por su novedad, o por motivos de salud pública, se reserven para tratamientos que solo pueden utilizarse o seguirse en medio hospitalario o centros asistenciales autorizados (Medicamentos de Uso Hospitalario). b) Se utilicen en el tratamiento de enfermedades que deban ser diagnosticadas en medio hospitalario, o en establecimientos que dispongan de medios de diagnóstico adecuados o por determinados médicos especialistas, aunque la administración y seguimiento pueda realizarse fuera del hospital (Medicamentos de Diagnóstico Hospitalario de prescripción por determinados médicos especialistas). c) Estén destinados a pacientes ambulatorios, pero cuya utilización pueda producir reacciones adversas muy graves, lo que requerirá, en su caso, prescripción por determinados médicos especialistas y una vigilancia especial durante el tratamiento (Medicamentos de Especial Control Médico). 4. No obstante lo dispuesto en los apartados anteriores, la Agencia Española de Medicamentos y Productos Sanitarios, podrá establecer excepciones a los mismos teniendo en cuenta lo siguiente: a) la dosis máxima única o la dosis máxima diaria, la dosificación, la forma farmacéutica, determinados envases y/o b) otras condiciones de utilización que garantice el uso adecuado del medicamento. 5. La Agencia Española de Medicamentos y Productos Sanitarios podrá modificar de oficio la clasificación otorgada a un medicamento, de acuerdo con los criterios expuestos en este artículo, cuando de la reevaluación del expediente se desprendan nuevos datos que lo justifiquen. 6. La Agencia Española de Medicamentos y Productos Sanitarios podrá calificar como medicamentos no sujetos a prescripción médica a aquellos que vayan destinados a procesos o condiciones que no necesiten un diagnóstico preciso y cuyos datos de evaluación toxicológica, clínica o de su utilización y vía de administración no exijan prescripción médica. 7. Cuando, con base en ensayos clínicos o estudios preclínicos significativos, la autorización de medicamento sujeto a prescripción médica se haya modificado por la de medicamento no sujeto a prescripción médica o viceversa, se concederá un periodo de un año de exclusividad de datos para los mismos desde la autorización de la modificación. 8. El Ministerio de Sanidad y Consumo establecerá las condiciones y requisitos específicos para la aplicación de cada una de estas categorías de prescripción y dispensación. Artículo 25. Medicamentos objeto de publicidad destinada al público. Podrán ser objeto de publicidad destinada al público los medicamentos que cumplan todos los requisitos que se relacionan a continuación: a) Que no se financien con fondos públicos. b) Que por su composición y objetivo estén concebidos y destinados para ser utilizados sin la intervención de un médico que realice el diagnóstico, la prescripción o el seguimiento del tratamiento. – 180 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano c) Que no contengan en su composición sustancias psicotrópicas ni estupefacientes. Artículo 26. Autorizaciones sometidas a condiciones especiales. 1. En circunstancias excepcionales, la Agencia Española de Medicamentos y Productos Sanitarios podrá autorizar un medicamento basado en una solicitud cuyos datos preclínicos o clínicos estén incompletos, cuando el solicitante pueda justificar por razones objetivas y verificables que no puede suministrar datos completos sobre la eficacia y seguridad en las condiciones normales de uso del producto, por alguna de las razones siguientes: a) los casos para los que está indicado el medicamento se presentan tan raramente que el solicitante no puede razonablemente estar obligado a proporcionar las evidencias detalladas; b) el estado actual de desarrollo de la ciencia no permite proporcionar información completa; c) los principios de deontología médica comúnmente admitidos prohíben recoger esta información. 2. En estas circunstancias, la autorización concedida por la Agencia Española de Medicamentos y Productos Sanitarios será revisable anualmente y supeditada a la obligación por parte del solicitante de cumplir las siguientes condiciones según proceda: a) Realizar, dentro del plazo establecido por la Agencia Española de Medicamentos y Productos Sanitarios, un programa de estudios determinado cuyos resultados constituirán la base de una nueva evaluación de la relación beneficio/riesgo. b) Calificar el medicamento como sujeto a prescripción médica y, en caso necesario, autorizar únicamente su administración si se efectúa bajo estricto control médico, a ser posible en un centro hospitalario. c) Incluir la información disponible en la ficha técnica explicando las limitaciones de los datos, así como en el prospecto y en cualquier otra información médica, destacando que, en relación con determinados aspectos, no existen aún datos concluyentes sobre el medicamento en cuestión. Artículo 27. Plazo de validez y renovación de la autorización. 1. La autorización de un medicamento tendrá una validez de cinco años, pudiendo ser objeto de renovación. A tal efecto, el titular de la autorización de comercialización facilitará a la Agencia Española de Medicamentos y Productos Sanitarios una versión consolidada del expediente en relación con la calidad, la seguridad y la eficacia, incluyendo la evaluación de los datos consignados en las notificaciones de sospechas de reacciones adversas y los informes periódicos actualizados en materia de seguridad presentados de acuerdo con la normativa específica de farmacovigilancia, así como información sobre todas las modificaciones introducidas desde la concesión de la autorización de comercialización, al menos nueve meses antes de que la autorización de comercialización deje de tener validez. 2. Una vez renovada, la autorización de comercialización tendrá una validez ilimitada, salvo que la Agencia Española de Medicamentos y Productos Sanitarios decida su renovación adicional por cinco años, con arreglo al apartado 1, por motivos justificados de farmacovigilancia, incluida la exposición de un número insuficiente de pacientes al medicamento de que se trate. 3. La solicitud de renovación se presentará en cualquiera de los lugares previstos en el artículo 38.4 de la Ley 30/1992, de 26 de noviembre, y de acuerdo con el formato de solicitud publicado por la Agencia Española de Medicamentos y Productos Sanitarios. 4. La Agencia Española de Medicamentos y Productos Sanitarios notificará la resolución antes de la expiración de la validez de la autorización. En caso de que la resolución sea negativa se seguirá lo dispuesto en el artículo 68. 5. La autorización quedará extinguida si no se presenta solicitud de renovación de la autorización, en el plazo establecido, salvo que la Agencia Española de Medicamentos y Productos Sanitarios mediante resolución acuerde mantener la vigencia de la autorización. – 181 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano Artículo 28. Comercialización efectiva. 1. El titular de la autorización comunicará de forma expresa a la Agencia Española de Medicamentos y Productos Sanitarios la fecha de comercialización efectiva de cada medicamento. Dicha comunicación se realizará por cada una de las autorizaciones de comercialización, como mínimo quince días antes de efectuarse esa comercialización. 2. El titular de la autorización efectuará anualmente una declaración de intención de comercialización del medicamento de forma expresa. Ésta comunicación se efectuará ante la Agencia Española de Medicamentos y Productos Sanitarios durante el mes de octubre del año anterior, acompañando justificación del pago de la correspondiente tasa. En el caso de no presentar esta declaración, se entenderá que se solicita la suspensión de la autorización de comercialización de acuerdo con el artículo 69.1, iniciándose el correspondiente procedimiento. 3. Cada autorización de comercialización de un medicamento perderá su validez si, en un plazo de tres años, el titular no procede a la comercialización efectiva del mismo. El periodo de los tres años empezará a contarse a partir del día siguiente de la fecha de la notificación de la resolución de autorización emitida por la Agencia Española de Medicamentos y Productos Sanitarios. 4. La autorización de comercialización de un medicamento perderá también su validez, si una vez autorizado y comercializado deja de encontrarse de forma efectiva en el mercado durante tres años consecutivos. 5. Cuando un titular de una autorización de comercialización manifieste a la Agencia su intención de no continuar la comercialización de un medicamento, la Agencia podrá hacer pública esta situación, instando a otros laboratorios que puedan estar interesados a solicitar una autorización de comercialización de ese medicamento, con base en los artículos 7, 8, 10 y 12, según proceda. 6. No obstante, cuando concurran razones de salud o de interés sanitario, como en el supuesto de originarse laguna terapéutica, ya sea en el mercado en general o en la prestación farmacéutica del Sistema Nacional de Salud, la Agencia Española de Medicamentos y Productos Sanitarios mantendrá la validez de la autorización y exigirá la comercialización efectiva del medicamento. 7. La Agencia Española de Medicamentos y Productos Sanitarios procederá a anotar las anteriores circunstancias en el Registro de Medicamentos. CAPÍTULO III Etiquetado y prospecto Sección 1.ª Disposiciones generales del etiquetado y prospecto Artículo 29. Objetivos del etiquetado y prospecto: Garantías de identificación e información para el uso racional del medicamento. 1. El etiquetado y el prospecto del medicamento habrán de ser conformes a la información de su ficha técnica. 2. El etiquetado y el prospecto garantizarán la inequívoca identificación del medicamento, proporcionando la información necesaria para su correcta administración y uso por los pacientes o usuarios y, en su caso, por los profesionales sanitarios. 3. El etiquetado y el prospecto, en su diseño y contenido, facilitarán la adecuada comprensión y conocimiento del medicamento por el ciudadano. El prospecto deberá ser legible, claro, asegurando su comprensión por el paciente y reduciendo al mínimo los términos de naturaleza técnica. – 182 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano Artículo 30. Autorización de la información contenida en el etiquetado y prospecto. 1. Los textos y demás características del etiquetado y del prospecto forman parte de la solicitud de autorización del medicamento y cuando proceda su modificación, esta se realizará de acuerdo con el procedimiento establecido para cada tipo de modificación. 2. Los textos se presentarán, al menos, en castellano. Además, también se podrán redactar en otros idiomas, siempre que en todos ellos figure la misma información. En estos casos, con la solicitud se acompañará la documentación acreditativa de la fidelidad de la traducción. 3. En el caso de los medicamentos huérfanos, las informaciones previstas en el etiquetado podrán redactarse, previa solicitud debidamente motivada, en una lengua oficial de la Unión Europea, en aquellos casos que determine la Agencia Española de Medicamentos y Productos Sanitarios. 4. Así mismo, cuando el destino del medicamento no sea la dispensación directa al paciente, o cuando existan problemas graves respecto de su disponibilidad, la Agencia Española de Medicamentos y Productos Sanitarios podrá dispensar de la obligación de hacer figurar determinadas informaciones en el etiquetado y el prospecto, sin perjuicio de adoptar las medidas que considere necesarias para salvaguardar la salud pública. También podrán establecer una exención total o parcial de la obligación de que el etiquetado y el prospecto estén redactados en castellano. 5. Sin perjuicio de lo anterior, en los casos de los apartados 3 y 4, el titular de la autorización de comercialización pondrá a disposición de la Agencia Española de Medicamentos y Productos Sanitarios la información del etiquetado y/o del prospecto en castellano, de manera que pueda hacerse disponible a los ciudadanos y profesionales interesados. Seccion 2.ª Garantías de identificación del medicamento: Etiquetado Artículo 31. Requisitos generales. 1. El etiquetado del medicamento deberá incluir la información detallada en el anexo III. 2. Los datos que han de mencionarse obligatoriamente en el etiquetado de los medicamentos estarán expresados en caracteres fácilmente legibles, claramente comprensibles e indelebles. Estos datos no inducirán a error sobre la naturaleza del producto ni sobre las propiedades terapéuticas del mismo. Artículo 32. Garantías de autenticidad y trazabilidad del etiquetado. El embalaje exterior o en su defecto el acondicionamiento primario incorporará los elementos que permitan la autentificación del producto, así como la información necesaria para determinar la trazabilidad del medicamento desde su fabricación hasta su dispensación al ciudadano, incluyendo para ello la identificación que se establezca reglamentariamente. Artículo 33. Incorporación de símbolos y motivos gráficos. 1. Será obligatorio incluir en el etiquetado los símbolos recogidos en el anexo IV. 2. Se podrá autorizar la inclusión de otros motivos gráficos que, siendo conformes a la ficha técnica, y no teniendo carácter publicitario, sean adecuados para facilitar la interpretación por los pacientes y usuarios de determinadas menciones del anexo III. Artículo 34. Obligación de declarar determinados excipientes. En el etiquetado, en la declaración de la composición del medicamento, se incluirán los excipientes de declaración obligatoria cuyo conocimiento resulte necesario para una correcta administración y uso del medicamento. Los excipientes de declaración obligatoria se irán actualizando conforme a los avances científicos y técnicos y de acuerdo con lo que se establezca en la Unión Europea. – 183 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano Artículo 35. Garantía de correcta identificación: nombre del medicamento. 1. El nombre con el que se comercialice el medicamento definido en el artículo 2.8, habrá de reunir los requisitos establecidos legalmente y en ningún caso podrá inducir a error sobre las propiedades terapéuticas o la naturaleza del medicamento. La denominación podrá ser un nombre de fantasía que no pueda confundirse con la denominación común, definida en el artículo 2.9 o bien la denominación común o científica del principio activo, acompañada de una marca comercial o del nombre del titular o fabricante de la autorización de comercialización. 2. Se evitarán aquellas denominaciones que puedan inducir a error en la prescripción o dispensación a causa de denominaciones ya existentes en el mercado farmacéutico, del empleo de otras denominaciones anteriores o de los hábitos de prescripción. 3. En general, y de acuerdo con lo establecido en los dos apartados anteriores, no serán admisibles las denominaciones de medicamentos cuando: a) Su prescripción o dispensación pueda dar lugar a confusión fonética u ortográfica con el de otro medicamento o con productos sanitarios, cosméticos o alimentarios. b) Haya sido utilizada en un medicamento cuya autorización haya sido revocada y no hubieran transcurrido cinco años desde su revocación, excepto que tengan la misma composición en principios activos. c) Tenga parecido ortográfico con una Denominación Oficial Española, con una Denominación Común Internacional recomendada o propuesta por la Organización Mundial de la Salud, o con una denominación común usual o científica. d) Se trate de medicamentos objeto de publicidad dirigida al público, cuya denominación no podrá ser igual o inducir a confusión con la de otro medicamento sujeto a prescripción médica o financiado con fondos públicos. Sección 3.ª Garantías de información del medicamento: Prospecto Artículo 36. Requisitos generales del prospecto. 1. El prospecto es la información escrita que acompaña al medicamento, dirigida al paciente o al usuario. En él se identifica al titular de la autorización y en su caso, el nombre del representante del titular de la autorización de comercialización y al responsable de la fabricación del medicamento, se declara su composición y se dan instrucciones para su administración, empleo y conservación, así como sus efectos adversos, interacciones, contraindicaciones y demás datos que se determinan en el anexo V, con el fin de proponer su más correcto uso y la observancia del tratamiento prescrito, así como las medidas a adoptar en caso de intoxicación. 2. El prospecto deberá estar redactado y concebido en términos claros y comprensibles para permitir que los pacientes y usuarios actúen de forma adecuada, cuando sea necesario con ayuda de los profesionales sanitarios. 3. El prospecto deberá reflejar los resultados de las consultas con los grupos de pacientes o de usuarios para garantizar su legibilidad, claridad y facilidad de comprensión para favorecer el uso correcto del medicamento. 4. Como norma general, el prospecto sólo contendrá la información concerniente al medicamento al que se refiera. No obstante, la Agencia Española de Medicamentos y Productos Sanitarios podrá autorizar, en determinadas circunstancias, que se incluya información relativa a distintas dosis y formas farmacéuticas disponibles de un mismo medicamento. 5. Es obligatoria la inserción del prospecto en todos los medicamentos, salvo si toda la información exigida se incluye en el embalaje exterior o, en su defecto, en el acondicionamiento primario. 6. El titular de la autorización de comercialización garantizará que, previa solicitud de las organizaciones de pacientes, el prospecto esté disponible en formatos apropiados para las personas invidentes o con visión parcial. – 184 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano Artículo 37. Omisión de indicaciones terapéuticas. La Agencia Española de Medicamentos y Productos Sanitarios podrá decidir que ciertas indicaciones terapéuticas no figuren en el prospecto o en la ficha técnica, en particular cuando el solicitante de un medicamento genérico comunique que éstas indicaciones estuvieran cubiertas por el derecho de patentes o de protección de datos en el momento en que el medicamento genérico se autorice. Artículo 38. Motivos gráficos. Se podrá autorizar la inclusión en el prospecto de dibujos, y otros motivos gráficos, que complementen la información escrita del prospecto así como otras informaciones, siempre que, siendo conformes con la ficha técnica, se justifiquen por razones de educación sanitaria o favorezcan una mayor comprensión para el consumidor o usuario al que se dirijan, y no respondan a criterios de promoción o publicidad del medicamento. Seccion 4.ª Disposiciones particulares para determinados formatos de medicamentos Artículo 39. Material de acondicionamiento de los envases clínicos. 1. En el embalaje exterior habrán de figurar los datos establecidos en la parte primera del anexo III, con las siguientes excepciones: a) Supresión del cupón precinto del Sistema Nacional de Salud. b) Supresión del recuadro o espacio en blanco que permita indicar la posología recetada, duración del tratamiento y frecuencia de tomas». c) Inclusión de forma destacada de la leyenda: «Envase clínico, prohibida su venta al detalle». 2. En el acondicionamiento primario constarán los datos reflejados en la parte segunda del anexo III. 3. El número de prospectos que se incluyan en el embalaje serán los suficientes, dependiendo del número de unidades del envase clínico, para garantizar la información de los posibles pacientes o usuarios, y contendrán la información que se establece en el anexo V. Artículo 40. Material de acondicionamiento de las muestras gratuitas. El material de acondicionamiento de las muestras gratuitas, cualquiera que sea éste, habrá de reunir las mismas características y condiciones que las autorizadas para los envases de venta al público, con las siguientes excepciones: a) Se suprimirá o anulará el cupón precinto del Sistema Nacional de Salud. b) En el embalaje exterior se indicará de manera indeleble y bien visible la leyenda: «Muestra gratuita, prohibida su venta». CAPÍTULO IV Disposiciones particulares para determinadas clases de medicamentos Sección 1.ª Medicamentos hemoderivados Artículo 41. Autorización previa de lotes de fabricación de medicamentos hemoderivados. 1. Por motivos de salud pública, la Agencia Española de Medicamentos y Productos Sanitarios, de acuerdo con el artículo 45.3 de la Ley 29/2006, de 26 de julio, someterá a autorización previa cada lote de fabricación del producto terminado y condicionará la comercialización a su conformidad. – 185 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano 2. Se exceptúan de lo anterior los derivados del plasma que intervengan como excipiente o como reactivo en la producción de otro medicamento o producto sanitario, los productos en fase de ensayos clínicos y los medicamentos señalados en el apartado 2 del artículo 24 de la Ley 29/20006, de 26 de julio. 3. La autorización previa del lote de fabricación, implicará la revisión de los protocolos de producción y control que acompañaran a la solicitud y, en su caso, la realización de los ensayos analíticos que se consideren oportunos. 4. Cuando se acredite documentalmente ante la Agencia Española de Medicamentos y Productos Sanitarios que el lote ha sido certificado por la autoridad competente de otro Estado miembro, se otorgará la mencionada autorización sin realizar nuevos análisis. 5. Cuando no se precise realizar análisis, se entenderá autorizado el lote de fabricación si en el plazo de cinco días hábiles desde la recepción de la solicitud de autorización en la Agencia Española de Medicamentos y Producto Sanitarios ésta no requiere al solicitante que subsane o mejore la solicitud. La Agencia Española de Medicamentos y Productos Sanitarios determinará el medio telemático para efectuar dicha solicitud. 6. En caso que la solicitud implique la realización de análisis del lote por no contar con la certificación señalada anteriormente, dicha solicitud se resolverá en el plazo máximo de 60 días desde su presentación. Artículo 42. Comercio exterior de los medicamentos hemoderivados. 1. La entrada y salida de medicamentos hemoderivados, de sus materias primas y de sus intermedios, del territorio español está sometido a autorización previa por la Agencia Española de Medicamentos y Productos Sanitarios. 2. En el supuesto de salida de medicamentos hemoderivados, se requerirá informe previo favorable de la Dirección General de Salud Publica, del Ministerio de Sanidad y Consumo. Sección 2.ª Vacunas y alérgenos Artículo 43. Autorización previa de lotes de fabricación de vacunas y alérgenos. 1. Por motivos de salud pública, la Agencia Española de Medicamentos y Productos Sanitarios, de acuerdo con el artículo 45.3 de la Ley 29/2006, de 26 de julio, someterá a autorización previa cada lote de fabricación de producto terminado y condicionará la comercialización a su conformidad, de las siguientes vacunas: a) Las vacunas víricas. b) Las vacunas frente al tétanos, difteria y tosferina tanto monovalentes como polivalentes. c) La vacuna antitífica atenuada. 2. La referida autorización previa implicará la revisión de los protocolos de producción y control y, en su caso, la realización de los ensayos analíticos que se consideren oportunos. 3. Cuando se acredite documentalmente ante la Agencia Española de Medicamentos y Productos Sanitarios que el lote ha sido certificado por la autoridad competente de otro Estado miembro de la Unión europea, se otorgará la mencionada autorización sin realizar nuevos análisis. Transcurrido el plazo del apartado 4 del presente artículo sin pronunciamiento negativo, y teniendo en cuenta la especial necesidad de estos productos estratégicos, se entenderá conforme el lote para su comercialización. 4. En el plazo máximo de 60 días naturales desde la recepción de la solicitud se resolverá el expediente. 5. En el caso de la vacuna de la gripe los plazos contemplados en los apartados 3 y 4 serán de 30 días naturales. Artículo 44. Vacunas individualizadas. Para las vacunas de uso individual se podrán establecer limitaciones del alcance de lo indicado en el Anexo I de acuerdo con las características de estos productos. – 186 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano Artículo 45. Recomendaciones de uso. En aquellas vacunas que se autoricen por procedimientos centralizado, descentralizado y reconocimiento mutuo, y conste en su ficha técnica que se utilizarán de acuerdo con las recomendaciones oficiales, dichas recomendaciones oficiales de uso deberán acompañarse a la ficha técnica durante las acciones de promoción de la vacuna. Sección 3.ª Medicamentos radiofármacos Artículo 46. Autorización de medicamentos radiofármacos. Los generadores de radionucleidos, equipos, radionucleidos precursores y radiofármacos fabricados industrialmente tienen la consideración de medicamentos y están sometidos a autorización y registro por parte la Agencia Española de Medicamentos y Productos Sanitarios Artículo 47. Exenciones. 1. No será exigida la autorización para los casos siguientes: a) La preparación extemporánea de un medicamento radiofármaco, entendida como preparación de un radiofármaco en el momento de su uso, en una unidad de radiofarmacia autorizada, bajo la supervisión y control de un facultativo especialista en radiofarmacia, para su aplicación en un centro o institución legalmente facultados para ello, si se realiza exclusivamente a partir de generadores de radionucleidos, radionucleidos precursores y equipos debidamente autorizados y con arreglo a las instrucciones del fabricante. b) La preparación en el momento de su uso de muestras autólogas donde participen radionucleidos, así como la extracción de dosis individuales de radiofármacos listos para su uso, en una unidad de radiofarmacia autorizada, bajo la supervisión y control de un facultativo especialista en radiofarmacia, para su aplicación en un centro o institución legalmente facultados para ello, si se realiza exclusivamente a partir de generadores de radionucleidos, radionucleidos precursores, radiofármacos fabricados industrialmente y equipos debidamente autorizados y con arreglo a las instrucciones del fabricante. c) Los radiofármacos utilizados para tomografía por emisión de positrones (radiofármacos PET) preparados en una unidad de radiofarmacia autorizada bajo la supervisión y control de un facultativo especialista en radiofarmacia, siempre que cumplan los siguientes requisitos: 1.º Elaborados íntegramente y utilizados, sin ánimo de lucro, en centros vinculados al Sistema Nacional de Salud, 2.º Sean sustancias en fase de investigación clínica o sean medicamentos que la Agencia Española de Medicamentos y Productos Sanitarios considera que satisfacen las garantías de calidad, seguridad, eficacia, identificación e información, y que se elaboren en instalaciones adecuadas. Artículo 48. Requisitos específicos de la autorización de medicamentos radiofármacos. 1. Las solicitudes de autorización de medicamentos radiofármacos, además de cumplir el artículo 6, deberán incluir una explicación detallada completa de la dosimetría interna de la radiación. En caso de los generadores de radionucleidos además deberá incluirse una descripción general del sistema, junto con una descripción detallada de los componentes del mismo que puedan afectar a la composición o calidad del radionucleido hijo, así como las características cualitativas y cuantitativas del eluído o del sublimado. En caso de radiofármacos que precisen una preparación extemporánea, deben incluirse instrucciones detalladas suplementarias para la preparación extemporánea y el control de calidad de esta preparación y en su caso, tiempo máximo de almacenamiento durante el cual cualquier preparado intermedio, como un eluído, o el radiofármaco listo para su empleo cumplen las especificaciones previstas. – 187 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano 2. La documentación deberá ajustarse a los criterios establecidos en el documento técnico común (DTC) acordado en la Unión Europea y recogido en el anexo I. así como a lo dispuesto en las directrices detalladas específicas para cada materia. Artículo 49. Cumplimiento de la legislación sobre protección sanitaria. Los preceptos de este real decreto se entenderán sin perjuicio de lo dispuesto por la legislación sobre protección sanitaria de la población y de los trabajadores expuestos, así como de las personas con ocasión de exposiciones médicas, contra los riesgos de las radiaciones ionizantes. Sección 4.ª Medicamentos tradicionales a base de plantas Artículo 50. Registro de medicamentos tradicionales a base de plantas. Sin perjuicio de lo establecido en el artículo 51.3 de la Ley 29/2006, de 26 de julio, los medicamentos tradicionales a base de plantas no podrán comercializarse sin la previa inscripción en el registro de medicamentos tradicionales a base de plantas creado por la Agencia Española de Medicamentos y Productos Sanitarios. Artículo 51. Criterios que deben de cumplir los medicamentos tradicionales a base de plantas para registrarse por el procedimiento simplificado. 1. Para obtener el registro simplificado de un medicamento tradicional a base de plantas se tendrán que cumplir las siguientes condiciones: a) Que los medicamentos tengan indicaciones apropiadas exclusivamente para medicamentos tradicionales a base de plantas, que por su composición y finalidad, estén destinados y concebidos para su utilización sin el control de un médico a efectos de diagnóstico, prescripción o seguimiento de un tratamiento. b) Que se administren siempre de acuerdo con una dosis o posología determinada. c) Que se trate de preparados para uso por vía oral, externo o por inhalación. d) Que haya transcurrido el periodo de uso tradicional, consistente en un periodo mínimo de treinta años, de los cuales al menos quince, se haya utilizado en la Unión Europea. e) Que la información sobre uso tradicional sea suficiente y en particular que el producto demuestre no ser nocivo en las condiciones de uso establecidas y la acción farmacológica o la eficacia del medicamento a base de plantas, se pueda deducir de la experiencia en la utilización tradicional. 2. Sin perjuicio de lo dispuesto en el artículo 2.28, un medicamento tradicional a base de plantas podrá contener vitaminas o minerales cuya seguridad esté bien documentada podrá ser registrado de acuerdo con el artículo 50. En estos casos, la acción de las vitaminas y minerales ha de ser secundaria con respecto a las sustancias activas vegetales en lo referente a las indicaciones específicas autorizadas. Artículo 52. Procedimiento de registro simplificado para medicamentos tradicionales a base de plantas. 1. La solicitud irá acompañada de los datos y documentos siguientes: a) Los recogidos en las letras a) a la i), inclusive y la n) del artículo 6.5. b) Los resultados de las pruebas farmacéuticas (fisicoquímicas, biológicas o microbiológicas). c) La ficha técnica del producto, sin la información sobre las propiedades farmacológicas en su caso, una maqueta del envase y del etiquetado, así como el prospecto de acuerdo con la normativa al respecto, a efectos de garantizar la adecuada comprensión por los usuarios. d) Documentación acreditativa de las autorizaciones o denegaciones obtenidas por el solicitante en otro Estado miembro o en un tercer país para el medicamento especificando, en su caso, los motivos correspondientes a la decisión. – 188 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano e) En caso de asociación de sustancias vegetales, preparados vegetales o combinaciones de ambos, se incluirá información sobre el uso tradicional de la combinación, en particular que el producto demuestre no ser nocivo en las condiciones de uso y que la acción farmacológica o eficacia se deduzcan del uso y experiencia de larga tradición y si las sustancias activas individuales no fueran suficientemente conocidas, la información se referirá asimismo a éstas. f) Referencias bibliográficas o los informes de experto en los que se demuestre que el medicamento en cuestión o un producto equivalente, tal como se cita en este apartado ha tenido un uso farmacológico durante un periodo mínimo de treinta años con anterioridad a la fecha de solicitud, de los que, al menos durante quince, se haya utilizado en la Unión Europea o que el medicamento haya obtenido un dictamen favorable del Comité de Medicamentos de Plantas de la Agencia Europea de Medicamentos que lo considere como medicamento tradicional a base de plantas. El requisito de presentar pruebas del uso farmacológico durante un periodo de treinta años, se cumplirá incluso cuando la comercialización del producto no se haya basado en una autorización específica. Asimismo, se cumplirá si el número o la cantidad de ingredientes del medicamento se hubieran reducido durante ese periodo. g) Documentación bibliográfica, acompañada de un informe de experto, sobre la seguridad del medicamento. La Agencia Española de Medicamentos y Productos Sanitarios podrá solicitar información adicional para evaluar la seguridad del medicamento tradicional a base de plantas. 2. No será necesario presentar la documentación relacionada en los apartados d), f) y g) de este artículo, cuando las sustancias o preparados vegetales o sus combinaciones estén incluidos en la lista de sustancias y preparados vegetales y de combinaciones de éstos, para su uso en medicamentos tradicionales a base de plantas elaborada por el Comité de Medicamentos de Plantas de la Agencia Europea de Medicamentos. Las monografías comunitarias elaboradas por el Comité de Medicamentos de Plantas de la Agencia Europea del Medicamento, serán de referencia en la preparación de la documentación acreditativa del uso tradicional. 3. Cuando la solicitud de registro de medicamentos tradicionales a base de plantas sea de un medicamento que haya estado en uso en la Unión Europea durante menos de 15 años, pero que pueda acogerse por otros motivos al registro simplificado, la Agencia Española de Medicamentos y Productos Sanitarios solicitará un dictamen sobre el uso tradicional del medicamento presentado en la solicitud al Comité de Medicamentos de Plantas de la Agencia Europea de Medicamentos. Cuando exista una monografía comunitaria sobre las plantas que forman parte del medicamento propuesto, esta será tenida en cuenta en la resolución de la solicitud de registro. 4. La Agencia Española de Medicamentos y Productos Sanitarios, cuando evalúe la solicitud de registro de un medicamento a base de plantas tendrá en consideración los registros de medicamentos tradicionales a base de plantas concedidos en otros Estados miembros en base a la presente normativa. 5. En caso de que se solicite el registro de un medicamento tradicional a base de plantas, que haya sido registrado como tal en otro Estado miembro se aplicará el procedimiento de reconocimiento mutuo, siempre que se haya publicado una monografía comunitaria de la planta medicinal de uso tradicional, o el medicamento a base de plantas esté compuesto por sustancias, preparados o combinaciones de estos que figuren en la lista elaborada por el Comité de Medicamentos de Plantas de la Agencia Europea de Medicamentos y publicada por la Comisión Europea. 6. El plazo máximo para la notificación de la resolución del procedimiento será de seis meses desde el día siguiente a la fecha de la presentación de una solicitud válida. En caso contrario, se entenderá desestimada la solicitud, pudiéndose interponer los recursos administrativos y contencioso-administrativos que resulten procedentes. – 189 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano Artículo 53. Causas de denegación. 1. Sin perjuicio de las causas de denegación establecidas en el artículo 19, se denegará la solicitud de registro simplificado de medicamentos tradicionales a base de plantas cuando no se cumpla lo establecido en los artículos 51 y 52, y cuando la información sobre el uso tradicional sea insuficiente, especialmente si los aspectos farmacológicos o la eficacia no se deducen de su utilización y experiencia de larga tradición o el producto sea nocivo en las condiciones normales de uso. 2. Si una solicitud de registro hace referencia a una sustancia o preparado vegetal o a una combinación de estos que figure en la lista publicada por la Comisión Europea, no podrá denegarse la solicitud de registro por las dos últimas causas recogidas en el apartado anterior. 3. La Agencia Española de Medicamentos y Productos Sanitarios notificará al solicitante, a la Comisión y a toda autoridad competente que lo requiera cualquier decisión que adopte relativa a la denegación de un registro para uso tradicional y las razones de esta última. Artículo 54. etirada del mercado. Además de las causas establecidas en el articulo 68.1, cuando una sustancia, preparado vegetal o combinación de éstos deje de figurar en la lista elaborada por el Comité de medicamentos a base de plantas de la Agencia Europea de Medicamentos, se dejarán sin efecto las respectivas inscripciones anotando estas circunstancias en el registro de medicamentos tradicionales a base de plantas, a menos que en el plazo de tres meses presenten la documentación acreditativa referida en el artículo 52.1 y se procederá a la retirada del mercado de los medicamentos tradicionales de plantas que contengan alguno de estos componentes. Sección 5.ª Medicamentos homeopáticos Artículo 55. Clases de medicamentos homeopáticos. Los medicamentos homeopáticos podrán ser: a) Con indicación terapéutica aprobada, cuyo procedimiento de autorización y registro, seguirá el establecido en el capítulo II, teniendo en cuenta su naturaleza homeopática. b) Sin indicaciones terapéuticas aprobadas, cuyo procedimiento de autorización y registro, será el simplificado especial de medicamentos homeopáticos, creado a tal efecto por la Agencia Española de Medicamentos y Productos Sanitarios, siempre y cuando cumplan con los requisitos establecidos para ese procedimiento. En caso contrario, deberán seguir el procedimiento establecido en el capítulo II, teniendo en cuenta su naturaleza homeopática. Artículo 56. Criterios que han de cumplir los medicamentos homeopáticos para registrarse por el procedimiento simplificado especial. Para obtener el registro simplificado de un medicamento homeopático se tendrán que cumplir las siguientes condiciones: a) Que su vía de administración sea oral o externa. b) Ausencia de indicación terapéutica particular en la etiqueta o en cualquier información relativa al medicamento. c) Que su grado de dilución garantice la inocuidad del medicamento, en particular, el preparado no deberá contener más de una parte por 10.000 de tintura madre ni más de una centésima parte de la dosis mas baja que eventualmente se emplee en medicina alopática de aquellos principios activos cuya presencia en un medicamento alopático implique la obligatoriedad de presentar receta médica. – 190 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano Artículo 57. Procedimiento de registro simplificado especial para los medicamentos homeopáticos. 1. La solicitud de registro, que podrá abarcar toda la serie de medicamentos obtenidos a partir de la misma cepa o cepas homeopáticas, irá acompañada de los datos y documentos siguientes: a) Denominación científica de la cepa o cepas homeopáticas u otra denominación reconocida en la Real Farmacopea Española, en la Farmacopea Europea, o en su defecto, en una farmacopea utilizada de forma oficial en un país de la Unión Europea. b) Vías de administración, formas farmacéuticas y grados de dilución que se pretenden registrar. c) Memoria descriptiva de la obtención y control de la cepa o cepas homeopáticas. d) Justificación de su uso homeopático, en base en una bibliografía adecuada. e) Descripción del procedimiento de fabricación y control para cada forma farmacéutica, así como de los métodos de dilución y de dinamización utilizados. f) Información sobre la estabilidad del medicamento. g) Pruebas preclínicas o justificación de su ausencia. h) Documentación bibliográfica, acompañada de un informe de experto, que demuestre la seguridad del medicamento. 2. En caso de que se solicite el registro simplificado de un medicamento homeopático que haya sido registrado como tal en otros Estados miembros se aplicará el procedimiento de reconocimiento mutuo, cuya solicitud deberá observar lo establecido en el artículo 16.4. 3. El plazo máximo para la notificación de la resolución del procedimiento será de 6 meses desde el día siguiente a la fecha de la presentación de una solicitud válida. En caso contrario, se entenderá desestimada la solicitud, pudiéndose interponer los recursos administrativos y contencioso-administrativos que resulten procedentes. Artículo 58. Etiquetado de los medicamentos homeopáticos. 1. El etiquetado y, en su caso, el prospecto de los medicamentos homeopáticos con indicación terapéutica, se ajustarán a las disposiciones generales relativas al etiquetado y prospecto contemplados en el presente real decreto y a las disposiciones particulares que se adopten reglamentariamente. 2. El etiquetado y, en su caso, el prospecto de los medicamentos homeopáticos sin indicación terapéutica aprobada, deben incluir, obligatoriamente, los siguientes datos: a) Denominación científica de la cepa o cepas seguida del grado y tipo de dilución empleando los símbolos de la farmacopea utilizada; si el medicamento homeopático se compone de varias cepas, la denominación científica de las mismas, con su grado de dilución en el etiquetado, podrá completarse por un nombre de fantasía. b) Nombre y dirección del titular de la autorización sanitaria y, en su caso, del fabricante. c) Forma y vía de administración. d) Fecha de caducidad expresada claramente (mes y año). Además, los medicamentos con una estabilidad reducida después de su reconstitución, dilución o su apertura, indicarán el tiempo de validez de la preparación reconstituida, diluida o tras su apertura e incluirán un recuadro para su consignación por los usuarios. e) Forma farmacéutica. f) Contenido del envase de venta, en peso, volumen o unidades de administración. g) Precauciones particulares de conservación, en su caso. h) Advertencias especiales, cuando el medicamento las requiera. i) Lote de fabricación. j) Código Nacional del Medicamento. k) La leyenda «Medicamento homeopático sin indicaciones terapéuticas aprobadas». l) Una advertencia que aconseje al usuario que consulte a un médico si los síntomas persisten. – 191 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano Sección 6.ª Gases medicinales Artículo 59. Autorización de comercialización de los gases medicinales. Los gases medicinales se autorizan conforme a lo dispuesto en el presente real decreto, teniendo en cuenta las siguientes especificidades: 1. Deberán cumplir con las características técnicas de calidad exigidas en la Real Farmacopea Española, en la Farmacopea Europea o, en su defecto, en otras farmacopeas oficiales de los Estados miembros de la Unión Europea o de otro país, al que el Ministerio de Sanidad y Consumo reconozca unas exigencias de calidad equivalentes a las referidas farmacopeas. 2. Los gases medicinales que contengan el mismo componente con calidades ajustadas a farmacopeas diferentes, serán considerados productos distintos a efectos de su autorización de comercialización. 3. Cualquier otro gas medicinal que se pretenda utilizar con finalidad terapéutica antes de estar reconocido por alguna farmacopea de las previstas en el apartado 1 de este artículo será sometido, a efectos de la autorización de comercialización, a la evaluación de su calidad, seguridad y eficacia. Artículo 60. Condiciones particulares. Con carácter excepcional y para la atención de sus pacientes, los centros sanitarios podrán solicitar a un laboratorio farmacéutico debidamente autorizado, la fabricación de una composición distinta de las autorizadas cuando se cumplan las siguientes condiciones: a) Que obedezca a la prescripción escrita y motivada de un médico para un paciente concreto. b) Que se empleen en su elaboración gases medicinales cuyas especificaciones estén descritas en las farmacopeas previstas en el apartado 1 del artículo anterior y en concentraciones distintas de las autorizadas. c) Que la elaboración se efectúe con las mismas garantías de calidad que los productos autorizados. d) Que en el etiquetado del envase se consigne, como mínimo, la composición porcentual, la identificación del prescriptor y del centro asistencial en el que se utilizará, el código de identificación del paciente, la razón social del laboratorio fabricante, el director técnico del laboratorio fabricante, la fecha de caducidad y las condiciones de conservación, si proceden, y el número de protocolo de fabricación y control. El laboratorio deberá notificar dicha circunstancia a la Agencia Española de Medicamentos y Productos Sanitarios en el plazo máximo de quince días a partir de la recepción de la solicitud, y archivará la petición escrita del prescriptor junto con el protocolo de fabricación y el certificado de liberación del producto. Artículo 61. Suministro, entrega o dispensación. 1. Durante el transporte de los gases medicinales licuados a los depósitos de almacenamiento de centros hospitalarios, otros centros asistenciales, o centros de investigación o experimentación, se acompañará un certificado firmado y fechado donde consten los datos del etiquetado, que estará a disposición de las autoridades sanitarias. El destinatario archivará un ejemplar de la certificación por envío. 2. El suministro de los gases medicinales licuados para uso humano, de conformidad con lo dispuesto en el apartado 2 del artículo 52 de la Ley 29/2006, de 26 de julio, podrá realizarse conforme determinen las autoridades sanitarias competentes, observándose las necesarias medidas de seguridad y calidad en la aplicación de los gases medicinales por los centros sanitarios o asistenciales correspondientes. 3. La entrega directa a los pacientes en los casos de terapia a domicilio exigirá la presentación de la correspondiente orden médica debidamente cumplimentada por el – 192 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano facultativo prescriptor. Las condiciones específicas de dispensación se desarrollarán reglamentariamente. CAPÍTULO V Obligaciones del titular del medicamento Artículo 62. Obligaciones del titular de la autorización. El titular de la autorización de un medicamento está obligado a respetar las normas sobre farmacovigilancia y, durante la vigencia de la autorización de comercialización, a: 1. Observar las condiciones en las que se concedió la autorización de comercialización, además de las obligaciones generales que señala la legislación vigente, así como las de cualquier modificación de las condiciones de la autorización establecidas en el capítulo siguiente, incluidas las de los procedimientos de fabricación y de control. 2. Respetar la continuidad en el servicio. El titular de la autorización de comercialización tiene obligación de tener abastecido el mercado de los medicamentos autorizados. 3. Mantener permanente actualización el expediente. El titular de la autorización deberá presentar los informes periódicos de seguridad establecidos reglamentariamente, con el fin de mantener actualizado el expediente en materia de seguridad y en particular la información dirigida a los profesionales incluida en la ficha técnica del medicamento y la información del prospecto garantizando la adecuada comprensión del mismo. 4. Contribuir al adecuado conocimiento del medicamento y promover su uso racional. El titular de la autorización está obligado a poner a disposición pública, en particular de los profesionales sanitarios la información actualizada de la ficha técnica del medicamento con la información legalmente establecida, así como a hacer públicos los resultados de los ensayos clínicos, independientemente del resultado favorable o no de sus conclusiones. 5. Colaborar en los programas de control, garantizar la adecuación de los productos en el mercado e informar de cualquier posible retirada de lotes del mercado. El titular de la autorización de un medicamento deberá comunicar a la Agencia Española de Medicamentos y Productos Sanitarios, a las comunidades autónomas y a las autoridades de todos los países donde se haya distribuido, con la rapidez adecuada a cada caso y exponiendo los motivos, toda acción emprendida para retirar un lote del mercado. 6. Participar en sistemas que garanticen la recogida de los residuos de medicamentos que se generen en los domicilios. 7. Cualquier otra obligación legal o reglamentariamente establecida. CAPÍTULO VI Modificaciones de las condiciones de autorización de medicamentos Artículo 63. Modificaciones de las condiciones de autorización del medicamento. 1. Las modificaciones de las condiciones de autorización serán en su caso notificadas, o solicitadas a la Agencia Española de Medicamentos y Productos Sanitarios para su posterior resolución de acuerdo con el procedimiento establecido para cada una de ellas. 2. Son modificaciones de las condiciones de una autorización de comercialización los cambios en el contenido de los detalles y documentos contenidos en el artículo 6 de este real decreto. Se considerarán: a) Modificaciones de importancia menor de tipo IA, aquellas que tengan solamente un impacto mínimo, o ninguno, en la calidad, seguridad o eficacia del medicamento en cuestión, y estén contempladas dentro del anexo II, apartado 1 del Reglamento (CE) n.º 1234/2008 de la Comisión, de 24 de noviembre de 2008, relativo al examen de las modificaciones de las autorizaciones de comercialización de medicamentos para uso humano y de medicamentos veterinarios. b) Modificaciones de importancia mayor tipo II, aquellas que, sin ser una extensión de línea, puedan tener repercusiones en la calidad, seguridad o eficacia del medicamento en cuestión y estén contempladas dentro del anexo II, apartado 2 del Reglamento (CE) n.º – 193 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano 1234/2008 de la Comisión, de 24 de noviembre de 2008, así como las transmisiones de titularidad de las autorizaciones de comercialización. c) Extensión de línea, extensión de una autorización de comercialización, o extensión, aquella modificación indicada en el anexo I, apartados 1 y 2 del Reglamento (CE) n.º 1234/2008 de la Comisión, de 24 de noviembre de 2008. d) Modificaciones de importancia menor tipo IB, aquellas que no sean una modificación de importancia menor tipo IA, ni una modificación de importancia mayor tipo II, ni una extensión de línea. 3. No se admitirán modificaciones durante la tramitación de las solicitudes de autorización de comercialización, excepto las impuestas de oficio por la Agencia Española de Medicamentos y Productos Sanitarios y/o por la Comisión Europea. 4. La Agencia Española de Medicamentos y Productos Sanitarios, notificará a la Agencia Europea de Medicamentos las resoluciones de modificaciones de las autorizaciones que se consideren relevantes. Artículo 63 bis. Agrupación de modificaciones. 1. Cuando se notifiquen o soliciten varias modificaciones, para cada una de ellas se presentará una notificación o solicitud independiente. 2. No obstante a lo anterior, se podrán agrupar modificaciones en un solo formato de notificación o solicitud, siempre y cuando todas se presenten al mismo tiempo y por el mismo titular, de acuerdo con los siguientes criterios generales: a) Modificaciones de importancia menor tipo IA de una o varias autorizaciones de comercialización. b) Modificaciones de la misma autorización de comercialización, siempre y cuando la agrupación de las mismas se encuentre dentro de los casos recogidos en el anexo III del Reglamento (CE) n.º 1234/2008 de la Comisión, de 24 de noviembre de 2008. c) Siempre que la Agencia Española de Medicamentos y Productos Sanitarios previamente haya aceptado la propuesta de presentar una agrupación de modificaciones que no esté contemplada en el anexo III del Reglamento (CE) n.º 1234/2008 de la Comisión, de 24 de noviembre de 2008. Artículo 63 ter. Obligaciones de comunicación de las modificaciones de la autorización de comercialización. 1. El titular de la autorización de comercialización deberá tener en cuenta en lo referente a los métodos de fabricación y de control del artículo 6, apartado 5, letras e) e i), los avances científicos y técnicos e introducir las modificaciones necesarias para que el medicamento sea fabricado y controlado por métodos científicos generalmente aceptados. Dichas modificaciones se tramitarán según proceda de acuerdo con lo previsto en este real decreto. 2. El titular de la autorización de comercialización comunicará de forma inmediata a la Agencia Española de Medicamentos y Productos Sanitarios cualquier información nueva que pueda implicar una modificación de los datos o documentos a que se refieren los artículos 6, 7, 8, 9, 10, 11 y 12, así como los anexos de este real decreto. En particular, el titular de la autorización de comercialización informará de manera inmediata a la Agencia Española de Medicamentos y Productos Sanitarios de cualquier prohibición o restricción impuesta por las autoridades competentes de cualquier país en el que se comercialice el medicamento y les transmitirá cualquier otra información nueva que pueda influir en la evaluación del beneficio-riesgo del medicamento en cuestión. La información incluirá los resultados positivos y negativos de ensayos clínicos u otros estudios en todas las indicaciones y poblaciones, estén o no incluidos en la autorización de comercialización, así como los datos sobre el uso del medicamento cuando tal uso no se ajuste a los términos de la autorización de comercialización. 3. El titular de una autorización de comercialización se asegurará de que la información del medicamento esté actualizada en función de los últimos conocimientos científicos, incluidas las conclusiones de las evaluaciones y las recomendaciones publicadas en el portal web europeo sobre medicamentos creado de conformidad con el artículo 26 del Reglamento – 194 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano (CE) n.º 726/2004 del Parlamento Europeo y del Consejo, de 31 de marzo de 2004, sin perjuicio de la información que la Agencia Española de Medicamentos y Productos Sanitarios publicará en su portal web. Artículo 64. Procedimiento de notificación para las modificaciones de importancia menor tipo IA y tipo IB. 1. El procedimiento para las modificaciones de importancia menor se regirá, por lo establecido en este real decreto y por las instrucciones sobre la materia que apruebe la Comisión Europea y/o la Agencia Española de Medicamentos y Productos Sanitarios. 2. Para las modificaciones de importancia menor tipo IA el titular presentará, en cualquier momento dentro de los 12 meses siguientes a la aplicación de la modificación, una notificación a la Agencia Española de Medicamentos y Productos Sanitarios con la información enumerada en el anexo IV del Reglamento (CE) n.º 1234/2008 de la Comisión, de 24 de noviembre de 2008, y en las instrucciones que la Comisión Europea y/o la Agencia Española de Medicamentos y Productos Sanitarios elaboren al respecto. No obstante lo anterior, la notificación se presentará inmediatamente tras la aplicación de la modificación, en el caso de modificaciones de importancia menor que requieran notificación inmediata (tipo IAin) para la supervisión continua del medicamento en cuestión. 3. En un plazo de 30 días naturales tras la recepción de una notificación válida de una modificación tipo IA, la Agencia Española de Medicamentos y Productos Sanitarios comunicará al titular la aceptación o no de la modificación, realizando al mismo tiempo, de oficio, la actualización de las autorizaciones de comercialización, si procede. De no producirse dicha comunicación, podrá considerarse aceptada la modificación. Las modificaciones tipo IA no necesitan la aprobación previa de la Agencia Española de Medicamentos y Productos Sanitarios para su aplicación. Si una modificación tipo IA es rechazada, el titular deberá cesar inmediatamente en la aplicación de la misma, una vez recibida la comunicación motivada de la Agencia Española de Medicamentos y Productos Sanitarios. 4. Para las modificaciones de importancia menor tipo IB, el titular presentará una notificación a la Agencia Española de Medicamentos y Productos Sanitarios con la información enumerada en el anexo IV del Reglamento (CE) n.º 1234/2008 de la Comisión, de 24 de noviembre de 2008, y en las instrucciones que la Comisión Europea y/o la Agencia Española de Medicamentos y Productos Sanitarios elaboren al respecto. En un plazo de 30 días naturales tras la recepción de una notificación válida de una modificación tipo IB, la Agencia Española de Medicamentos y Productos Sanitarios comunicará al titular la aceptación o no de la modificación propuesta. De no producirse dicha comunicación, podrá considerarse que la modificación propuesta ha sido aceptada. 5. En caso de no ser aceptada la modificación tipo IB propuesta, la Agencia Española de Medicamentos y Productos Sanitarios comunicará al solicitante los motivos de la no aceptación y le otorgará un plazo de 30 días naturales para la subsanación de las deficiencias. Recibidas las aclaraciones presentadas por el titular, la Agencia Española de Medicamentos y Productos Sanitarios dispondrá de un nuevo plazo de 30 días naturales para finalizar el procedimiento. La no presentación de las aclaraciones o la presentación de las mismas fuera del plazo establecido para ello, dará lugar a la no aceptación de la modificación propuesta. 6. Cuando la modificación tipo IB no haya sido aceptada, se comunicarán al titular los motivos del rechazo. 7. Cuando la aceptación de la modificación tipo IB conlleve la modificación de la autorización de comercialización, ésta se realizará de oficio en un plazo no superior a 6 meses. Artículo 65. Procedimiento para las modificaciones de importancia mayor tipo II. 1. El procedimiento para las modificaciones de importancia mayor tipo II se regirá, por lo establecido en este real decreto y por las instrucciones sobre la materia que apruebe la Comisión Europea y/o la Agencia Española de Medicamentos y Productos Sanitarios. – 195 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano 2. El titular presentará a la Agencia Española de Medicamentos y Productos Sanitarios una solicitud de modificación con la información enumerada en el anexo IV del Reglamento (CE) n.º 1234/2008 de la Comisión, de 24 de noviembre de 2008, y en las instrucciones que la Comisión Europea y/o la Agencia Española de Medicamentos y Productos Sanitarios elaboren al respecto. 3. Tras la recepción de una solicitud válida, la Agencia Española de Medicamentos y Productos Sanitarios en un plazo de 60 días naturales elaborará un informe de evaluación y comunicará al titular el resultado de la evaluación. 4. En caso de que el resultado de la evaluación fuera desfavorable, y en todo caso, antes de la finalización del plazo establecido en el párrafo anterior, se podrá requerir al titular la presentación de aclaraciones, lo que dará lugar a la suspensión del procedimiento por un plazo máximo de 60 días naturales. La no presentación de las aclaraciones o la presentación de las mismas fuera del plazo establecido para ello, dará lugar a la no aceptación de la modificación propuesta. Recibidas las aclaraciones presentadas por el titular, la Agencia Española de Medicamentos y Productos Sanitarios dispondrá de un plazo de 60 días naturales para emitir una propuesta de resolución positiva o negativa que comunicará al titular. 5. En el caso de propuesta de resolución positiva que conlleve la modificación de la autorización de comercialización, se abrirá un periodo de 30 días naturales para que el titular remita los textos definitivos objeto de la modificación y la Agencia Española de Medicamentos y Productos Sanitarios acepte dichos textos. La Agencia Española de Medicamentos y Productos Sanitarios dispondrá de un plazo no superior a dos meses para emitir el documento de resolución. La no presentación de los textos definitivos o su presentación fuera del plazo establecido, o, en su caso, la no aceptación de los textos presentados por el titular, dará lugar a la emisión de una resolución negativa motivada por la que se denegará la modificación de la autorización de comercialización solicitada. 6. La Agencia Española de Medicamentos y Productos Sanitarios notificará al titular la resolución definitiva, con indicación de los recursos que contra el mismo procedan. Transcurrido el plazo máximo para la resolución del procedimiento sin haberse notificado la resolución, el solicitante podrá entender desestimada su solicitud. 7. El periodo de 60 días naturales para la elaboración del informe de evaluación se podrá ampliar hasta 90 días naturales para el caso de las modificaciones contempladas en el anexo V, parte I del Reglamento (CE) n.º 1234/2008 de la Comisión, de 24 de noviembre de 2008. Asimismo, se podrá reducir a 30 días naturales en las modificaciones de carácter urgente o que resulten de las decisiones del Comité de Seguridad de Medicamentos de Uso Humano. Artículo 66. Modificaciones especiales. Tendrán la consideración de modificaciones especiales las siguientes: 1. Modificaciones urgentes por razones de seguridad. Cuando se tenga conocimiento de una nueva información que indique un riesgo importante para la salud pública asociada al uso del medicamento o tenga un impacto relevante en la seguridad del mismo, se podrá realizar un cambio provisional de la información del medicamento que afectará especialmente a algunos de los siguientes datos de la ficha técnica: indicaciones, posología, contraindicaciones o advertencias, precauciones especiales de empleo y reacciones adversas. Para ello, se seguirán los procedimientos específicos establecidos en la normativa de farmacovigilancia de medicamentos de uso humano. 2. Modificación anual para las vacunas de la gripe humana. a) Las modificaciones generales que puedan afectar a las vacunas de la gripe humana, se regirán por lo previsto en el artículo 65. b) En el caso particular de la solicitud de adecuación de las cepas a las recomendaciones anuales establecidas por la Organización Mundial de la Salud, se seguirá el procedimiento establecido para las modificaciones de importancia mayor, con las – 196 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano especificidades documentales para la vacuna de la gripe, emitiéndose resolución en el plazo máximo de 45 días naturales. 3. Modificación de la autorización por razones de interés general. a) La Agencia Española de Medicamentos y Productos Sanitarios por razones de interés público, defensa de la salud o seguridad de las personas, podrá modificar las condiciones de dispensación y/o prescripción de un medicamento. b) Este procedimiento se tramitará con audiencia al interesado y el plazo máximo para la notificación de la resolución será de 90 días naturales. Asimismo será preceptivo el dictamen del Comité de Evaluación de Medicamentos de Uso Humano. c) En caso de que el procedimiento se base en motivos de seguridad del medicamento, el procedimiento se tramitará de acuerdo con lo establecido en la normativa específica sobre farmacovigilancia. Artículo 66 bis. Extensión de la autorización de comercialización. La extensión de línea de la autorización de comercialización se evaluará siguiendo el procedimiento seguido por la autorización de comercialización inicial a la que hace referencia. A la extensión de línea se le otorgará una nueva autorización de comercialización. Artículo 67. Cambio de titular del medicamento. 1. El cambio de titular de la autorización de comercialización del medicamento está sometido a autorización por parte de la Agencia Española de Medicamentos y Productos Sanitarios. 2. Las modificaciones de la autorización del medicamento que sean consecuencia del cambio de titular, se regirán por el procedimiento establecido para las modificaciones. CAPÍTULO VII Procedimientos para la suspensión y revocación de la autorización Artículo 68. Causas de suspensión y revocación. La Agencia Española de Medicamentos y Productos Sanitarios, podrá acordar la suspensión, revocación o modificación de la autorización de un medicamento cuando: 1. Se considere que el medicamento es nocivo. 2. El medicamento resulte no ser terapéuticamente eficaz. La carencia de eficacia terapéutica se valorará cuando se llegue a la conclusión de que no se pueden obtener resultados terapéuticos del medicamento. 3. Con base a los datos de seguridad, el medicamento tenga una relación beneficioriesgo desfavorable. 4. El medicamento no tenga la composición cuantitativa o cualitativa autorizada, o se incumplan las garantías de calidad, o no se ejecuten los controles de calidad exigidos. 5. Los datos e informaciones contenidos en la documentación sean erróneos o incumplan la normativa de aplicación en la materia. 6. El modo de fabricación del medicamento o los métodos de control utilizados por el fabricante no se ajusten a los descritos en la autorización. 7. Por cualquier otra causa, que suponga un riesgo previsible para la salud o seguridad de las personas o animales. 8. En cualquier otro caso en el que la Comisión Europea, así lo hubiera acordado. Artículo 69. Procedimiento de suspensión y revocación de oficio. 1. La Agencia Española del Medicamento y Productos Sanitarios podrá suspender o revocar la autorización de un medicamento, por las causas previstas en el artículo anterior. En el supuesto de la suspensión de la autorización de comercialización, esta no interrumpirá el plazo previsto en los apartados 3 y 4 del artículo 28. – 197 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano 2. El procedimiento se incoará mediante acuerdo de iniciación y audiencia al interesado, trámite tras el que se dictará resolución que se notificará al interesado en el plazo máximo de seis meses, indicando los recursos procedentes. En el caso de que los motivos de la suspensión o revocación sean los señalados en los apartados b), d) o e) y conciernan a la seguridad del medicamento, se seguirán los procedimientos establecidos en la normativa de farmacovigilancia de medicamentos de uso humano. 3. La Agencia Española de Medicamentos y Productos Sanitarios notificará a la Agencia Europea de Medicamentos las resoluciones de suspensión y revocación de las condiciones de la autorización que se consideren relevantes. Artículo 70. Procedimiento de suspensión y revocación a instancia de parte. 1. Cuando el titular de una autorización de un medicamento pretenda suspender o cesar la comercialización del mismo, deberá notificar a la Agencia Española de Medicamentos y Productos Sanitarios, al menos dos meses antes de la fecha en la que tenga previsto cesar en la comercialización del medicamento, motivando esa solicitud. En el supuesto de la suspensión de la autorización de comercialización, esta no interrumpirá el plazo previsto en los apartados 3 y 4 del artículo 28. 2. No obstante lo anterior, cuando concurran razones de salud o de interés sanitario, como en el supuesto de originarse laguna terapéutica, ya sea en el mercado en general o en la prestación farmacéutica del Sistema Nacional de Salud, la Agencia Española de Medicamentos y Productos Sanitarios mantendrá la validez de la autorización y exigirá la comercialización efectiva del medicamento. CAPÍTULO VIII Procedimientos comunitarios Artículo 71. Definiciones y requisitos generales de los procedimientos comunitarios. 1. Se entiende por reconocimiento mutuo, el procedimiento comunitario establecido para la concesión de una autorización de comercialización de un medicamento en más de un Estado miembro cuando el medicamento ya ha sido evaluado y autorizado en alguno de los Estados miembros. 2. Se entiende por descentralizado, el procedimiento comunitario establecido para la concesión de una autorización de comercialización de un medicamento en más de un Estado miembro cuando el medicamento no disponga de una autorización en ningún Estado miembro de la Unión Europea en el momento de la solicitud. 3. Ambos procedimientos exigen al solicitante presentar una solicitud basada en un expediente idéntico en todos los Estados miembros implicados en el procedimiento. El expediente incluirá la información y documentos referidos en los artículos 6, 7, 8, 9, 10, 11, 12 y anexo II. Los documentos presentados incluirán una lista de los Estados miembros afectados por la solicitud. Artículo 71 bis. Grupo de coordinación. 1. La Agencia Española de Medicamentos y Productos Sanitarios nombrará un representante en el Grupo de coordinación, por un periodo de tres años. A su vez, la Agencia Española de Medicamentos y Productos Sanitarios podrá nombrar un suplente para dicho grupo. Este representante podrá asistir a las reuniones acompañado de expertos, en virtud de los asuntos a tratar en el Grupo de coordinación. 2. Las funciones principales del representante de la Agencia Española de medicamentos y Productos Sanitarios en el Grupo de coordinación son: a) Participar en las discusiones sobre cuestiones relacionadas con autorizaciones de comercialización de medicamentos en dos o más Estados miembros. – 198 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano b) Participar en las decisiones sobre las fechas de presentación de los informes periódicos de seguridad, y sobre el mantenimiento, la suspensión o la revocación de las autorizaciones de comercialización como resultado de los datos de farmacovigilancia. c) Tratar de consensuar una posición sobre las medidas que deban tomarse a nivel europeo. 3. Asimismo, el representante de la Agencia Española de Medicamentos y Productos Sanitarios y el suplente en el Grupo de coordinación garantizarán la coordinación apropiada entre las labores del Grupo de coordinación y las Unidades de la Agencia Española de Medicamentos y Productos Sanitarios responsables de la validación, evaluación y autorización de los medicamentos y sus órganos consultivos. 4. Además, el representante de la Agencia Española de Medicamentos y Productos Sanitarios y el suplente en el Grupo de coordinación están obligados a cumplir con el artículo 63 del Reglamento (CE) n.º 726/2004 del Parlamento Europeo y del Consejo, de 31 de marzo de 2004, en lo relacionado con la transparencia, estando obligados, incluso después de haber cesado en sus funciones a no divulgar ninguna información que esté cubierta por el secreto profesional. 5. La Agencia Española de Medicamentos y Productos Sanitarios pondrá a disposición del miembro del Grupo de coordinación, del suplente y de los expertos los recursos científicos y reguladores de que dispone, supervisando en todo momento la competencia de las evaluaciones llevadas a cabo y también facilitará las actividades de sus representantes en el Grupo de coordinación y de sus expertos. Artículo 72. Procedimiento de reconocimiento mutuo. 1. Cuando el titular de un medicamento ya autorizado en España pretenda solicitar la autorización del mismo en otro u otros Estados miembros, España podrá actuar como Estado miembro de referencia en el procedimiento. 2. En el caso de que España actúe como Estado miembro de referencia, el titular de la autorización de comercialización solicitará a la Agencia Española de Medicamentos y Productos Sanitarios que elabore un informe de evaluación del medicamento o que actualice el informe de evaluación existente para ese medicamento. 3. La Agencia Española de Medicamentos y Productos Sanitarios elaborará o actualizará dicho informe en el plazo de 90 días a partir de la recepción de una solicitud válida. 4. El informe de evaluación, así como la ficha técnica del medicamento autorizado, el etiquetado y el prospecto, se enviarán a los Estados miembros afectados y al solicitante. 5. En un plazo de 90 días a partir de la recepción de los documentos a que se refiere el apartado anterior, los Estados miembros concernidos aprobarán los documentos remitidos por la Agencia Española de Medicamentos y Productos Sanitarios, a la que informarán de su aceptación. La Agencia garantizará el acuerdo general y finalizará el procedimiento e informará de ello al solicitante. En el plazo de 30 días, todos los Estados miembros implicados deberán resolver de conformidad con el acuerdo general. 6. Cuando se presente ante la Agencia Española de Medicamentos y Productos Sanitarios una solicitud de autorización de comercialización de un medicamento autorizado en otro Estado miembro, se le aplicará el procedimiento de reconocimiento mutuo, entendiéndose que España es Estado miembro concernido en el procedimiento. En este caso se aplicará el procedimiento descrito anteriormente, a partir de la documentación remitida por el Estado miembro que actúe como Estado miembro de referencia, de acuerdo con lo dispuesto en el apartado anterior. Artículo 73. Procedimiento descentralizado. 1. Cuando se pretenda conseguir una autorización de comercialización de un medicamento en más de un Estado miembro, el solicitante deberá pedir a uno de ellos que actúe como Estado miembro de referencia. 2. Cuando España actúe como Estado miembro de referencia, la Agencia Española de Medicamentos y Productos Sanitarios preparará un proyecto de informe de evaluación, un proyecto de ficha técnica y un proyecto de etiquetado y prospecto, en un plazo de 120 días a – 199 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano partir de la recepción de una solicitud válida, y los enviará al resto de los Estados miembros afectados y al solicitante. 3. En un plazo de 90 días, a partir de la recepción de los documentos a que se refiere el apartado anterior, los Estados miembros concernidos aprobarán los documentos remitidos por la Agencia Española de Medicamentos y Productos Sanitarios, informándola de su aceptación. La Agencia garantizará el acuerdo general y finalizará el procedimiento e informará de ello al solicitante. En el plazo de 30 días todos los Estados miembros implicados deberán resolver de conformidad con el acuerdo general. 4. Cuando en un procedimiento descentralizado sea otro Estado el que actúe como Estado miembro de referencia, estando España implicada como Estado concernido, la Agencia Española de Medicamentos y Productos Sanitarios aplicará el procedimiento descrito anteriormente, a partir de la documentación remitida por el Estado miembro de referencia, de acuerdo con lo dispuesto en el apartado anterior. Artículo 74. Discrepancia en las decisiones y procedimiento de arbitraje. 1. Cuando en un procedimiento comunitario España, como estado miembro concernido, no pueda aprobar en el plazo de 90 días previsto en los artículos 72.5 y 73.3 el informe de evaluación, el resumen de las características del producto, el etiquetado y el prospecto por considerar que existe un riesgo potencial grave para la salud pública, motivará su decisión de forma detallada y comunicará sus razones al Estado miembro de referencia así como a los demás Estados Miembros concernidos y al solicitante. 2. En el caso de que sea España estado miembro de referencia y reciba una comunicación de desacuerdo de otro Estado Miembro, la Agencia Española de Medicamentos y Productos Sanitarios comunicará el desacuerdo a los demás Estados miembros concernidos y al solicitante. Asimismo, comunicará inmediatamente los motivos de desacuerdo al Grupo de Coordinación, para examinar las cuestiones relacionadas con la autorización de comercialización de medicamentos en dos o más Estados miembros. 3. Todos los Estados miembros implicados en el procedimiento procurarán ponerse de acuerdo en el marco del Grupo de coordinación, sobre las medidas que deban adoptarse. Ofrecerán al solicitante la posibilidad de emitir consideraciones oralmente o por escrito. Si en el plazo de 60 días a partir de la comunicación al grupo de coordinación, los Estados miembros llegan a un acuerdo, el Estado miembro de referencia, en su caso la Agencia Española de Medicamentos y Productos Sanitarios, garantizará el acuerdo general y finalizará el procedimiento e informará de ello al solicitante. 4. Si en el plazo establecido en el apartado anterior los Estados miembros no llegan a un acuerdo, el Estado miembro de referencia, en su caso la Agencia Española de Medicamentos y Productos Sanitarios, informará a la Agencia Europea de Medicamentos con el fin de iniciar el procedimiento de arbitraje, remitiendo una descripción pormenorizada de las cuestiones sobre las que los Estados miembros no han podido alcanzar un acuerdo y los motivos de su desacuerdo. Se enviará al solicitante una copia de esta información, el cual deberá remitir sin demora una copia del expediente a la Agencia Europea de Medicamentos. 5. No obstante lo anterior, en el caso de que la Agencia Española de Medicamentos y Productos Sanitarios haya aprobado el informe de evaluación, la ficha técnica, el etiquetado y el prospecto, podrá, a petición del solicitante, autorizar la comercialización del medicamento previa a la decisión de la Comisión Europea. En este caso la autorización se concederá a reserva del resultado del referido procedimiento de arbitraje. Artículo 75. Armonización de las autorizaciones comunitarias. 1. Cuando un mismo medicamento haya sido objeto de diferentes solicitudes de autorización y los Estados miembros hayan adoptado decisiones discrepantes en relación con la autorización, suspensión o revocación, España, cualquier Estado miembro, la Comisión, el solicitante o titular de la autorización podrá dirigirse al Comité de Medicamentos de uso humano de la Agencia Europea de Medicamentos a fin de que se aplique el procedimiento de arbitraje. – 200 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano 2. A fin de fomentar la armonización de los medicamentos autorizados en la Unión Europea, la Agencia Española de Medicamentos y Productos Sanitarios remitirá anualmente al Grupo de coordinación una lista de los medicamentos para los cuales considere que deben elaborarse fichas técnicas armonizadas. Este Grupo de coordinación tendrá en cuenta las diferentes propuestas presentadas por todos los Estados miembros y remitirá una lista a la Comisión Europea para su armonización. Artículo 76. Decisiones de la Unión. 1. En casos específicos en los que estén en juego los intereses de la Unión Europea, cualquier Estado miembro, en su caso, la Agencia Española de Medicamentos y Productos Sanitarios, la Comisión, el solicitante o el titular de la autorización recurrirán al Comité de Medicamentos de Uso Humano de la Agencia Europea de Medicamentos para que aplique el procedimiento de arbitraje antes de que se adopte una decisión sobre una solicitud de autorización, sobre una suspensión o revocación de una autorización o de cualquier modificación de una autorización de comercialización que sea necesaria. A dicha petición deberá acompañarse toda la información disponible. De lo anterior se informará al solicitante o al titular de la autorización cuando la Comisión o cualquier Estado miembro recurra al Comité de Medicamentos de Uso Humano de la Agencia Europea de Medicamentos. 2. Cuando el recurso derive de la evaluación realizada por la Agencia Española de Medicamentos y Productos Sanitarios de datos de farmacovigilancia de un medicamento autorizado, ésta remitirá el asunto, previo asesoramiento del Comité de Seguridad de Medicamentos de Uso Humano, al Comité Europeo para la Evaluación de Riesgos en Farmacovigilancia de la Agencia Europea de Medicamentos. El Comité Europeo para la Evaluación de Riesgos en Farmacovigilancia emitirá una recomendación que se tramitará al Comité de Evaluación de Medicamentos de la Agencia Europea de Medicamentos o al Grupo de coordinación. Todas las recomendaciones adoptadas en el marco del Grupo de coordinación serán de aplicación obligatoria en los términos recogidos en las mismas. Disposición adicional industrialmente. primera. Aplicación a otros medicamentos fabricados El presente real decreto se aplicará, en lo que no se establezca en su norma específica, a los medicamentos con sustancias psicoactivas con potencial adictivo. Disposición adicional segunda. Aplicación del Reglamento (CE) n.º 1901/2006 del Parlamento Europeo y del Consejo, de 12 de diciembre de 2006, sobre medicamentos para uso pediátrico. Lo regulado en el presente real decreto será de aplicación a los medicamentos de uso pediátrico, sin perjuicio de lo establecido en el Reglamento (CE) n.º 1901/2006 del Parlamento Europeo y del Consejo, de 12 de diciembre de 2006, sobre medicamentos para uso pediátrico. Disposición adicional tercera. Obligaciones para el sistema de gestión de riesgos. La obligación de disponer de un sistema de gestión de riesgos para los titulares de autorizaciones de comercialización se regirá por lo dispuesto en el artículo 11 del Real Decreto 577/2013, de 26 de julio, por el que se regula la farmacovigilancia de medicamentos de uso humano. Disposición transitoria primera. Aplicación de periodos de protección de datos. De conformidad con la disposición transitoria primera de la Ley 29/2006, el periodo de exclusividad de datos establecido en el artículo 7.2 y 7.3 de este real decreto se aplicará solamente a los medicamentos de referencia que hayan presentado una solicitud de autorización después del 1 de noviembre de 2005. Los períodos de exclusividad de datos de los medicamentos de referencia para los que se ha presentado una solicitud de autorización – 201 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano antes del 1 de noviembre de 2005 serán los que regían con anterioridad a la entrada en vigor de la ley 29/2006, otorgándose las siglas EFG siempre que hayan transcurrido 10 años desde la autorización en España del medicamento de referencia o esté autorizado como medicamento genérico en otro país de la Unión Europea. Disposición transitoria segunda. Renovación de autorizaciones de medicamentos. Los medicamentos autorizados a la entrada en vigor de la Ley 29/2006, de 26 de julio, deberán proceder a su renovación en la fecha que les corresponda. A partir de dicha renovación les será de aplicación lo dispuesto en la normativa vigente en relación con los informes periódicos de seguridad. Disposición transitoria tercera. Plazo de adecuación del etiquetado y prospecto. 1. Los titulares de una autorización de comercialización deberán solicitar las modificaciones correspondientes al etiquetado y prospecto, según las previsiones contenidas en el capítulo III, a la Agencia Española del Medicamento y Productos Sanitarios. 2. Con carácter general la adecuación del etiquetado y prospecto de los medicamentos se deberá realizar con la solicitud de cualquier modificación de tipo II, excepto aquellas modificaciones que afecten exclusivamente a la calidad del medicamento y, en todo caso, con cualquier modificación que afecte al etiquetado y prospecto, así como con las modificaciones que se encuentren pendientes de aprobación. 3. En caso de no haberse realizado ninguna de las modificaciones contempladas en los apartados anteriores, se procederá a su solicitud junto con la solicitud de renovación. En todo caso, se deberá solicitar la modificación correspondiente para adecuar su etiquetado y prospecto siempre antes de haber transcurrido cinco años desde la entrada en vigor de la Ley 29/2006, de 26 de julio. Disposición transitoria cuarta. Aplicación del Capítulo III sobre etiquetado y prospecto a las solicitudes en trámite. El Capítulo III del mismo será aplicable a las solicitudes de autorización de comercialización que estén en trámite, así como a las modificaciones de los medicamentos de uso humano elaborados industrialmente y a los medicamentos que se encuentren en situación de suspensión temporal. No obstante, lo dispuesto en el artículo 36.3 de este reglamento, podrá ser cumplimentado como documentación adicional antes de la resolución de la solicitud o, como máximo, seis meses después de concedida la autorización, mediante la modificación correspondiente. Disposición transitoria quinta. Conservación de órganos para transplantes. Las soluciones para la conservación de órganos recogidos en la disposición adicional séptima de la Ley 29/2006, de 26 de julio, que estuvieran comercializadas en España a la entrada en vigor de la misma, podrán continuar comercializándose hasta la resolución del expediente y a resultas del mismo, si dentro del año siguiente a la entrada en vigor del presente real decreto se presenta en la Agencia Española de Medicamentos y Productos Sanitarios la solicitud de autorización correspondiente. Disposición transitoria sexta. Medicamentos homeopáticos. 1. Los medicamentos homeopáticos acogidos a la disposición transitoria segunda del Real Decreto 2208/1994, de 16 de noviembre, por el que se regulan los medicamentos homeopáticos de uso humano de fabricación industrial, deberán adecuarse a las previsiones de este real decreto, conforme a lo previsto en los apartados siguientes. 2. Los titulares de medicamentos afectados por la disposición transitoria segunda del Real Decreto 2208/1994, de 16 de noviembre, deberán comunicar a la Agencia Española de Medicamentos y Productos Sanitarios su intención de adecuarse a este real decreto. La comunicación se realizará en el soporte informático que la Agencia Española de Medicamentos y Productos Sanitarios pondrá a disposición de los titulares a través de su página web y deberá producirse en el plazo de tres meses desde la entrada en vigor de la – 202 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano orden por la que el Ministerio de Sanidad, Servicios Sociales e Igualdad determinará los requisitos mínimos y el procedimiento para la comunicación. Junto con la comunicación se abonará la tasa prevista en los apartados 4.12 o 4.13 del artículo 111 de la Ley 29/2006, de 26 de julio, según corresponda. Transcurrido dicho plazo, los medicamentos acogidos a la disposición transitoria segunda del Real Decreto 2208/1994, de 16 de noviembre, para los que no se hubiera comunicado su intención de adecuarse, conforme a lo establecido en este apartado, no podrán ser comercializados, debiendo ser retirados del mercado. 3. La Agencia Española de Medicamentos y Productos Sanitarios fijará un calendario para que los titulares de los medicamentos homeopáticos que hubieran realizado la comunicación prevista en el apartado anterior presenten las solicitudes de nuevo registro y documentación necesaria para adecuar su situación provisional y evaluar la relación beneficio/riesgo del producto. Esta solicitud habrá de acompañarse del comprobante del pago de tasas correspondiente a los epígrafes 4.10 y 4.11, del artículo 111 de la Ley 29/2006, de 26 de julio, según corresponda. 4. En todo caso, respecto de los medicamentos homeopáticos que la Agencia Española de Medicamentos y Productos Sanitarios considere de revisión prioritaria para garantizar la adecuada relación beneficio/riesgo, el procedimiento de adecuación previsto en esta disposición transitoria deberá finalizar en el plazo de un año a contar desde la entrada en vigor de la orden mencionada en el apartado 2. Disposición transitoria séptima. Medicamentos a base de plantas medicinales. 1. Los medicamentos tradicionales a base de plantas que actualmente se comercializan al amparo de la Orden Ministerial de 3 de octubre de 1973, por la que se establece el registro especial para los preparados de especies vegetales medicinales, podrán adecuarse a las previsiones de este real decreto, antes del 30 de abril de 2011. Finalizado el periodo de adecuación, todas las autorizaciones concedidas o registros practicados conforme a la Orden de 1973 quedarán sin efecto, quedando prohibida la comercialización como medicamentos, sin perjuicio de que las plantas tradicionalmente consideradas como medicinales cualquiera que sea su forma de presentación siempre que no tengan la consideración de medicamento y se ofrezcan sin referencia a propiedades terapéuticas, diagnósticas o preventivas, puedan venderse libremente, en los términos del artículo 51.2 y 3 de la Ley 29/2006, de 26 de julio, de garantías y uso racional de los medicamentos y productos sanitarios. 2. Las solicitudes de autorización o registro de medicamentos tradicionales a base de plantas a las que se refiere esta disposición, deberán ser presentadas en el plazo máximo de tres años a contar desde la entrada en vigor de este real decreto. Disposición derogatoria única. Derogación normativa. Quedan derogadas cuantas disposiciones de igual o inferior rango se opongan a lo establecido en este real decreto y en particular: a) Real Decreto 767/1993, de 21 de mayo, sobre evaluación, autorización, registro y condiciones de dispensación de especialidades farmacéuticas de uso humano fabricadas industrialmente. b) Real Decreto 2236/1993, de 17 de diciembre, por el que se regula el etiquetado y el prospecto de los medicamentos de uso humano. c) Real Decreto 288/1991, de 8 de marzo, por el que se regulan los medicamentos inmunológicos de uso humano. d) Real decreto 478/1993, de 2 de abril, por el que se regulan los medicamentos derivados de la sangre y plasma humano. e) Real Decreto 479/1993, de 2 de abril, por el que se regulan los medicamentos radiofármacos de uso humano. f) Real Decreto 2730/1981, de 19 de octubre, sobre características y registro de las especialidades farmacéuticas publicitarias. g) Real Decreto 2208/1994, de 16 de noviembre, que regula los medicamentos homeopáticos de uso humano de fabricación industrial. – 203 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano h) Real Decreto 1800/2003, de 26 de diciembre, que regula los gases medicinales, con excepción de lo relativo a los medicamentos de uso veterinario. i) Orden ministerial de 3 de octubre de 1973, por la que se establece el registro especial para los preparados de especies vegetales medicinales. j) Artículos 28 y 29 del Real Decreto 223/2004, de 6 de febrero, por el que se regulan los ensayos clínicos con medicamentos. Disposición final primera. Legislación sobre productos farmacéuticos. Este real decreto se dicta al amparo del artículo 149.1.16 de la Constitución Española, que atribuye al Estado competencia exclusiva en materia de legislación sobre productos farmacéuticos. Disposición final segunda. Incorporación de derecho de la Unión Europea. Mediante este real decreto se incorpora al derecho español la Directiva 2004/27/CE del Parlamento Europeo y del Consejo, de 31 de marzo de 2004, por la que se modifica la Directiva 2001/83/CE por la que se establece un código comunitario sobre medicamentos de uso humano, y la Directiva 2004/24/CE del Parlamento Europeo y del Consejo, de 31 de marzo de 2004, por la que se modifica, en lo que se refiere a los medicamentos tradicionales a base de plantas, la Directiva 2001/83/CE. Disposición final tercera. Desarrollo normativo. Se autoriza al Ministro de Sanidad y Consumo para dictar cuantas disposiciones sean necesarias para la aplicación y desarrollo de este real decreto, así como para actualizar sus anexos conforme al avance de los conocimientos científicos y técnicos de acuerdo con las orientaciones y directrices de la Unión Europea. Disposición final cuarta. Entrada en vigor. El presente real decreto entrará en vigor el día siguiente al de su publicación en el «Boletín Oficial del Estado». ANEXO I Normas y protocolos analíticos, farmacotoxicológicos y clínicos relativos a la realización de pruebas de medicamentos INTRODUCCIÓN Y PRINCIPIOS GENERALES 1. Los datos y la documentación que han de acompañar a toda solicitud de autorización de comercialización con arreglo a la Sección 1.ª del Capitulo II del presente real decreto deberán presentarse según los requisitos que se exponen en el presente anexo, siguiendo las orientaciones publicadas por la Comisión en las Normas sobre medicamentos de la Unión Europea, volumen 2 B, Nota explicativa para los solicitantes, Medicamentos de uso humano, Presentación y contenido del expediente, Documento Técnico Común (DTC). 2. Los datos y documentos deben presentarse en cinco módulos: el módulo 1 recoge los datos administrativos específicos para la Comunidad Europea; en el módulo 2 se incluyen los resúmenes de la calidad, clínicos y no clínicos; el módulo 3 ofrece información química, farmacéutica y biológica; el módulo 4 recoge los informes no clínicos; y el módulo 5 contiene los informes de estudio clínico. En dicha presentación se aplica un formato común para todas las regiones de la Conferencia Internacional sobre Armonización (International Conference on Harmonization, ICH): la Unión Europea, Estados Unidos y Japón. Los cinco módulos mencionados han de presentarse estrictamente con arreglo al formato, contenido y sistema de numeración que se definen pormenorizadamente en el volumen 2 B de la mencionada Nota explicativa para los solicitantes. 3. La presentación del DTC de la Unión Europea es aplicable a todos los tipos de solicitud de autorización de comercialización para cualquier procedimiento que se aplique – 204 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano (centralizado, reconocimiento mutuo o nacional) y tanto si se basa en una solicitud completa o abreviada. También es aplicable a todos los tipos de productos, incluidas las Nuevas Entidades Químicas (NEQ), radiofármacos, derivados del plasma, vacunas, medicamentos a base de plantas, etcétera. 4. Al constituir el expediente de solicitud de autorización de comercialización, los solicitantes deberán tener asimismo en cuenta las directrices científicas sobre calidad, seguridad y eficacia de los medicamentos de uso humano adoptadas por el Comité de medicamentos de uso humano farmacéuticas y publicadas por la Agencia Europea de Medicamentos (EMEA), así como las demás directrices farmacéuticas comunitarias publicadas por la Comisión en los distintos volúmenes de las Normas sobre medicamentos de la Unión Europea. 5. Por lo que respecta a la parte de calidad (química, farmacéutica y biológica) del expediente, son aplicables la totalidad de las monografías, incluidos los capítulos y monografías generales de la Farmacopea europea y de la Real Farmacopea Española. 6. El proceso de fabricación deberá cumplir los requisitos del Real Decreto 1564/1992, de 18 de diciembre, por el que se desarrolla y regula el régimen de autorización de los laboratorios farmacéuticos e importadores de medicamentos y la garantía de calidad de su fabricación industrial y con los principios y directrices relativos a las prácticas correctas de fabricación, publicados por la Comisión en las Normas sobre medicamentos de la Unión Europea, volumen 4. 7. Deberá incluirse en la solicitud toda la información pertinente para la evaluación del medicamento correspondiente, tanto si resulta favorable como desfavorable al producto. En concreto, deberán ofrecerse todos los datos pertinentes acerca de todas las pruebas o ensayos farmacotoxicológicos o clínicos incompletos o abandonados relativos al medicamento y/o ensayos completos relacionados con indicaciones terapéuticas no cubiertas por la solicitud. 8. Todos los ensayos clínicos que se realicen en la Unión Europea deberán ajustarse a los requisitos que figuran en la Directiva 2001/20/CE del Parlamento Europeo y del Consejo, relativa a la aproximación de las disposiciones legales, reglamentarias y administrativas de los Estados miembros sobre la aplicación de las buenas prácticas clínicas en la realización de ensayos clínicos de medicamentos de uso humano y en España al Real Decreto 223/2004, de 6 de febrero, por el que se regulan los ensayos clínicos con medicamentos. Para poder ser tenidos en cuenta durante la evaluación de una solicitud, los ensayos clínicos realizados fuera de la Unión Europea relacionados con medicamentos destinados a ser utilizados en la misma deberán concebirse, realizarse y notificarse, por lo que respecta a las prácticas clínicas y principios éticos, con arreglo a principios equivalentes a los expuestos en Real Decreto 223/2004, de 6 de febrero. Deberán llevarse a cabo con arreglo a los principios éticos que se recogen, por ejemplo, en la Declaración de Helsinki. 9. Los estudios no clínicos (farmacotoxicológicos) deberán realizarse de acuerdo con las disposiciones sobre prácticas correctas de laboratorio establecidas en el Real Decreto 822/1993, de 28 de mayo por el que se establecen los principios buenas prácticas de laboratorio y su aplicación en la realización de estudios no clínicos sobre sustancias y productos químicos, sobre inspección y verificación de buenas prácticas de laboratorio. 10. Las pruebas realizadas con animales han de llevarse a cabo de acuerdo con el Real Decreto 1201/2005, de 10 de octubre, sobre Protección de los Animales Utilizados para Experimentación y otros Fines Científicos. 11. Con el fin de hacer un seguimiento de la evaluación de beneficios/riesgos, deberá presentarse a la autoridad competente toda nueva información que no figure en la solicitud original y todos los datos sobre farmacovigilancia. Una vez concedida la autorización de comercialización, todas las modificaciones de los datos del expediente deberán someterse a las autoridades competentes con arreglo a los requisitos que figuran en los Reglamentos (CE) n.º 1084/2003 y (CE) n.º 1085/2003 de la Comisión, así como los requisitos expuestos en el volumen 9 de la publicación de la Comisión Normas sobre medicamentos de la Unión Europea. El presente anexo se divide en cuatro partes: – 205 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano En la parte I se expone el formato de la solicitud, el resumen de características del producto, el etiquetado, el prospecto y los requisitos de presentación de las solicitudes normalizadas (módulos 1 a 5). En la parte II se exponen las excepciones que se aplicarán a las «solicitudes específicas», a saber: uso médico suficientemente comprobado, medicamentos esencialmente similares, medicamentos de combinación fija, medicamentos biológicos similares, circunstancias excepcionales y solicitudes mixtas (parte bibliográfica y parte de estudios propios). En la parte III se abordan los «Requisitos particulares de las solicitudes de autorización de comercialización» de medicamentos biológicos (archivo principal sobre plasma; archivo principal sobre antígenos de vacuna), radiofármacos, medicamentos homeopáticos, medicamentos a base de plantas y medicamentos huérfanos. La parte IV, que trata de los «medicamentos de terapia avanzada», aborda los requisitos específicos de los medicamentos de terapia génica (mediante un sistema autólogo o alogénico humano, o mediante sistema xenogénico) y medicamentos de terapia celular, tanto de origen humano como animal, y medicamentos para transplantes xenogénicos. PARTE I. REQUISITOS DE LOS EXPEDIENTES NORMALIZADOS AUTORIZACIÓN DE COMERCIALIZACIÓN DE 1. Módulo 1: Información administrativa 1.1 Índice.–Deberá presentarse un índice exhaustivo de los módulos 1 a 5 del expediente presentado para solicitar la autorización de comercialización. 1.2 Formulario de solicitud.–El medicamento para el que se presenta la solicitud deberá identificarse mediante su nombre y el nombre de la(s) sustancia(s) activa(s), junto con su forma farmacéutica, vía de administración, dosificación y presentación final, incluido el envase. Deberá hacerse constar el nombre y dirección del solicitante, así como el nombre y la dirección de los fabricantes y los lugares donde se realizan las distintas fases de fabricación (incluido el fabricante del producto acabado y el fabricante o fabricantes de las sustancias activas) y, cuando proceda, el nombre y dirección del importador. El solicitante deberá identificar el tipo de solicitud e indicar, en su caso, las muestras que facilita. Deberán adjuntarse con los datos administrativos copias de la autorización de fabricación que se define en el artículo 18 del Real Decreto 1564/1992, junto con una lista de países en los que se ha concedido la autorización, copias de los resúmenes de características del producto aprobadas por los Estados miembros y la lista de países en los que se ha presentado la solicitud. Tal como se señala en el formulario de solicitud, los solicitantes deberán facilitar, entre otros elementos, datos detallados sobre el medicamento objeto de la misma, el fundamento jurídico de la solicitud, el titular propuesto de la autorización de comercialización y el fabricante o fabricantes, información sobre la situación jurídica de los medicamentos huérfanos, dictámenes científicos y un programa de desarrollo pediátrico. 1.3 Ficha técnica, etiquetado y prospecto. 1.3.1 Ficha técnica.–El solicitante deberá proponer una ficha técnica o resumen de las características del producto, con arreglo al artículo 6. 1.3.2 Etiquetado y prospecto.–Deberá facilitarse el texto de etiquetado propuesto para el acondicionamiento primario y el embalaje exterior, así como para el prospecto. Todos ellos deberán ajustarse al Capitulo III y anexos III,IV y V. 1.3.3 Maquetas y muestras.–El solicitante deberá facilitar muestras y/o maquetas del acondicionamiento primario y del embalaje exterior, las etiquetas y los prospectos del medicamento correspondiente. 1.3.4 Fichas técnica ya aprobadas.–Con los datos administrativos del formulario de solicitud se adjuntarán copias de todas fichas técnicas del producto con arreglo al artículo 6 y una lista de países en los que se ha presentado solicitud. – 206 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano 1.4 Información acerca de los expertos.–Con arreglo al artículo 6.5.j), los expertos deberán facilitar informes detallados de sus comprobaciones sobre los documentos y los datos que constituyen el expediente de autorización de comercialización, en concreto los módulos 3, 4 y 5 (documentación química, farmacéutica y biológica, documentación no clínica y documentación clínica, respectivamente). Los expertos deberán abordar los puntos decisivos relacionados con la calidad del medicamento y de los estudios realizados en animales y seres humanos y notificar todos los datos pertinentes para la evaluación. Estos requisitos deberán cumplirse facilitando un resumen global de la calidad, una visión general de la parte no clínica (datos extraídos de estudios realizados en animales) y una visión general de la parte clínica que se incluirá en el módulo 2 del expediente de solicitud de autorización de comercialización. En el módulo 1 se presentará una declaración firmada por los expertos, junto con una síntesis de sus datos académicos, su formación y su experiencia laboral. Los expertos deberán poseer la adecuada cualificación técnica o profesional. Deberá declararse la relación profesional entre el experto y el solicitante. 1.5 Requisitos especiales para los distintos tipos de solicitudes.–En la parte II del presente anexo se exponen los requisitos específicos para los distintos tipos de solicitudes. 1.6 Evaluación del riesgo para el medio ambiente.–Si procede, en las solicitudes de autorización de comercialización se incluirá una evaluación general de los posibles riesgos para el medio ambiente debido a la utilización y/o eliminación del medicamento y se formularán las propuestas de disposiciones relativas al etiquetado que procedan. Deberán abordarse los riesgos para el medio ambiente relacionados con la liberación de medicamentos que contengan o consistan en organismos modificados genéticamente (OMG) con arreglo a la Ley 9/2003, de 25 de abril, por la que se establece el régimen jurídico de la utilización confinada, liberación voluntaria y comercialización de organismos modificados genéticamente y al Real Decreto 178/2004, de 30 de enero por el que se aprueba el Reglamento para el desarrollo y ejecución de la Ley 9/2003. La información relacionada con el riesgo para el medio ambiente deberá figurar como anexo del módulo 1. La información se presentará con arreglo a lo dispuesto en las disposiciones anteriores, teniendo en cuenta todos los documentos de orientación publicados por la Comisión acerca de la aplicación de la mencionada Directiva. La información constará de los elementos siguientes: una introducción; una copia de todos los posibles consentimientos por escrito a la liberación intencional en el medio ambiente de OMG con fines de investigación y desarrollo con arreglo al título II de la Ley 9/2003, de 25 de abril; la información que se exige en los anexos II a IV de la Directiva 2001/18/CE, incluidos los métodos de detección e identificación y el identificador único de los OMG, más toda información suplementaria sobre los OMG o el producto que resulte pertinente para la evaluación del riesgo para el medio ambiente; un informe de evaluación del riesgo para el medio ambiente elaborado a partir de la información que se especifica en los anexos III y IV de la Directiva 2001/18/CE y con arreglo al anexo II de la Directiva 2001/18/CE; una conclusión en la que se tenga en cuenta la información anterior y la evaluación del riesgo para el medio ambiente y se proponga una estrategia adecuada de gestión de riesgos que incluya, en lo que concierne a los OMG y el producto correspondiente, un plan de seguimiento de la fase de postcomercialización y la determinación de toda indicación especial que deba figurar en el resumen de características del producto, el etiquetado o el prospecto; medidas adecuadas para informar a los ciudadanos. Deberá incluirse la firma con fecha del autor, los datos académicos, de formación y experiencia laboral del autor y una declaración de la relación entre el autor y el solicitante. 2. Módulo 2: Resúmenes El objeto del presente módulo es resumir los datos químicos, farmacéuticos y biológicos y los datos no clínicos y clínicos presentados en los módulos 3, 4 y 5 del expediente de – 207 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano autorización de comercialización, y proporcionar los informes y síntesis señalados descritos en el artículo 6.5.j). Deberán tratarse y analizarse los puntos decisivos. Se ofrecerán resúmenes objetivos en los que se incluirán tablas. En los informes se remitirá a las tablas o a la información que contenga la documentación principal presentada en el módulo 3 (documentación química, farmacéutica y biológica), el módulo 4 (documentación no clínica) y el módulo 5 (documentación clínica). La información que contenga el módulo 2 deberá presentarse con arreglo al formato, contenido y sistema de numeración que se definen en el volumen 2 de la Nota explicativa para los solicitantes. Las síntesis y resúmenes deberán ajustarse a los principios y requisitos básicos que se establecen a continuación: 2.1 Índice general.–En el módulo 2 deberá figurar un índice de la documentación científica presentada en los módulos 2 a 5. 2.2 Introducción.–Deberá indicarse la clase farmacológica, el modo de acción y la utilización clínica propuesta del medicamento para el que se solicita la autorización de comercialización. 2.3 Resumen global de la calidad.–Se presentará un resumen global de la calidad, en el que se examinará la información relacionada con los datos químicos, farmacéuticos y biológicos. Deberá hacerse hincapié en los parámetros críticos y cuestiones fundamentales en relación con aspectos de calidad, así como en la justificación de los casos en los que no se sigan las directrices pertinentes. En este documento se expondrán las líneas generales de los datos detallados correspondientes que se presentan en el módulo 3. 2.4 Visión general de la parte no clínica.–Deberá presentarse una valoración integrada y crítica de la evaluación no clínica del medicamento en animales/in vitro. Deberá incluirse la discusión y justificación de la estrategia de ensayo y de la desviación respecto a las directrices pertinentes. Excepto para los medicamentos biológicos, deberá incluirse una evaluación de las impurezas y productos de degradación, así como de sus potenciales efectos farmacológicos y toxicológicos. Deberán discutirse las repercusiones de cualquier posible diferencia en la quiralidad, la forma química y el perfil de impurezas entre el compuesto utilizado en los estudios no clínicos y el producto que se desea comercializar. Para los medicamentos biológicos, se evaluará la comparabilidad del material utilizado en los estudios no clínicos, los estudios clínicos y el medicamento que se desea comercializar. Deberá realizarse una evaluación específica de la seguridad de todo nuevo excipiente. Se definirán las características del medicamento demostradas en los estudios no clínicos y se discutirá las repercusiones de las conclusiones en relación con la seguridad del medicamento para la utilización clínica prevista en el ser humano. 2.5 Visión general de la parte clínica.–La visión general de la parte clínica tiene por objeto ofrecer un análisis crítico de los datos clínicos incluidos en el resumen clínico y el módulo 5. Se expondrán el enfoque del desarrollo clínico del medicamento, incluyendo el diseño del estudio crítico, las decisiones relacionadas con los estudios y la realización de los mismos. Se ofrecerá una breve visión general de las conclusiones clínicas, en la que se tratarán las limitaciones importantes y se evaluarán los riesgos y beneficios a partir de las conclusiones de los estudios clínicos. Deberá interpretarse de qué modo las conclusiones relativas a la eficacia y a la seguridad justifican la dosis propuesta y las indicaciones y una evaluación de cómo la ficha técnica del producto y otros optimizarán los beneficios y afrontarán los riesgos. Se expondrán las cuestiones relativas a la eficacia o la seguridad que se planteen en el desarrollo, así como los problemas pendientes de resolución. 2.6 Resumen no clínico.–Los resultados de los estudios de farmacología, farmacocinética y toxicología realizados en animales/in vitro se presentarán como resúmenes objetivos escritos y tabulados, que se presentarán en el orden siguiente: Introducción. Resumen escrito de farmacología. – 208 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano Resumen tabulado de farmacología. Resumen escrito de farmacocinética. Resumen tabulado de farmacocinética. Resumen escrito de toxicología. Resumen tabulado de toxicología. 2.7 Resumen clínico.–Se ofrecerá un resumen objetivo detallado de la información clínica relativa al medicamento que se incluye en el módulo 5. Comprenderá los resultados de todos los estudios biofarmacéuticos, de los estudios clínicos de farmacología y de los estudios clínicos sobre eficacia y seguridad. Deberá presentarse una sinopsis de cada estudio. La información clínica resumida se presentará en el orden siguiente: Resumen de los estudios biofarmacéuticos y los métodos analíticos relacionados. Resumen de los estudios clínicos de farmacología. Resumen sobre eficacia clínica. Resumen sobre seguridad clínica. Sinopsis de cada estudio. 3. Módulo 3: Información química, farmacéutica y biológica para medicamentos que contengan sustancias activas químicas y/o biológicas 3.1 Formato y presentación.–El esquema general del módulo 3 es el siguiente: Índice. Conjunto de datos: Principio activo: Información general: Nomenclatura. Estructura. Propiedades generales. Fabricación: Fabricante(s). Descripción del proceso de fabricación y de los controles en proceso. Control de materiales. Control de las etapas críticas y los productos intermedios. Validación y/o evaluación del proceso. Desarrollo del proceso de fabricación. Caracterización: Elucidación de la estructura y otras características. Impurezas. Control del principio activo: Especificaciones. Procedimientos analíticos. Validación de los procedimientos analíticos. Análisis de lotes. Justificación de las especificaciones. Estándares o materiales de referencia. Sistema de cierre del envase. Estabilidad: Resumen y conclusiones sobre estabilidad. Protocolo de estabilidad después de la autorización y compromiso de estabilidad. Datos de estabilidad. Producto terminado: Descripción y composición del medicamento. Desarrollo farmacéutico: Componentes del medicamento: – 209 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano Principio activo. Excipientes. Medicamento: Desarrollo de la formulación. Sobredosificación. Propiedades físico-químicas y biológicas. Desarrollo del proceso de fabricación. Sistema de cierre del envase. Atributos microbiológicos. Compatibilidad. Fabricación: Fabricante(s). Fórmula del lote. Descripción del proceso de fabricación y de los sistemas de control del proceso. Control de etapas críticas y de los productos intermedios. Validación y/o evaluación del proceso. Control de los excipientes: Especificaciones. Procedimientos analíticos. Validación de los procedimientos analíticos. Justificación de las especificaciones. Excipientes de origen humano o animal. Nuevos excipientes. Control del producto terminado: Especificación (-ones). Procedimientos analíticos. Validación de los procedimientos analíticos. Análisis de lotes. Caracterización de las impurezas. Justificación de la especificación (-ones). Estándares o materiales de referencia. Sistema de cierre del envase. Estabilidad: Resumen y conclusiones sobre estabilidad. Protocolo de estabilidad después de la autorización y compromiso de estabilidad. Datos de estabilidad. Anexos: Instalaciones y equipo (únicamente medicamentos biológicos). Evaluación de la seguridad respecto a los agentes extraños/externos. Excipientes. Información suplementaria para la Unión Europea: Esquema de la validación del proceso para el producto terminado. Producto sanitario. Certificado(s) de idoneidad. Medicamentos que contengan o utilicen en el proceso de fabricación materiales de origen animal y/o humano (procedimiento relativo a las encefalopatías espongiformes transmisibles, EET). Referencias bibliográficas. 3.2 Contenido.–Principios básicos y requerimientos. 1. En los datos químicos, farmacéuticos y biológicos que se faciliten deberán incluir, en relación con el (los) principio(s) activo(s) y el producto terminado, toda la información pertinente acerca del desarrollo, el proceso de fabricación, la caracterización y propiedades, – 210 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano operaciones y requisitos de control de calidad estabilidad, así como una descripción de la composición y presentación del producto terminado. 2. Se presentarán dos conjuntos principales de datos, respectivamente relacionados con el (los) principio(s) activo(s) y con el producto terminado. 3. Este módulo deberá, además, proporcionar información detallada sobre los materiales de partida y materias primas utilizados durante las operaciones de fabricación del principio(s) activo(s), y los excipientes incorporados en la formulación del producto terminado. 4. Todos los procedimientos y métodos utilizados para la fabricación y control del principio activo y el producto terminado deberán describirse de manera suficientemente pormenorizada para que puedan reproducirse en los ensayos realizados a petición de la autoridad competente. Todos los ensayos estarán en consonancia con el estado actual del progreso científico y deberán estar validados. Se proporcionarán los resultados de los estudios de validación. En el caso de los procedimientos de ensayo incluidos en la Farmacopea Europea, esta descripción deberá sustituirse por la referencia correspondiente a la(s) monografía(s) y capítulo(s) general(es). 5. Las monografías de la Real Farmacopea Española y de la Farmacopea Europea deberán ser aplicables a todas las sustancias, preparados y formas farmacéuticas que figuren en ellas. No obstante, cuando un material de la Farmacopea Europea o de la farmacopea de un Estado miembro haya sido preparado mediante un método susceptible de dejar impurezas no controladas en la monografía de la farmacopea, se deberán declarar dichas impurezas y sus límites máximos de tolerancia y deberá describirse un procedimiento de ensayo adecuado. En aquellos casos en que una especificación que figure en una monografía de la Farmacopea Europea o en la farmacopea de un Estado miembro pueda resultar insuficiente para garantizar la calidad de la sustancia, las autoridades competentes podrán solicitar al titular de la autorización de comercialización especificaciones más adecuadas. Las autoridades competentes deberán informar a las autoridades responsables de la farmacopea de que se trate. El titular de la autorización de comercialización proporcionará a las autoridades responsables de dicha farmacopea los detalles de la presunta insuficiencia y las especificaciones adicionales aplicadas. En el caso de los procedimientos analíticos incluidos en la Farmacopea Europea, podrá sustituirse tal descripción en cada apartado pertinente por la referencia pormenorizada que proceda a la(s) monografía(s) y capítulo(s) general(es). 6. En caso de que los materiales de partida, materias primas, principio(s) activo(s) o excipiente(s) no estén descritos en la Farmacopea Europea ni en la farmacopea de un Estado miembro, podrá aceptarse el cumplimiento con la monografía de la farmacopea de un tercer país. En estos casos, el solicitante presentará una copia de la monografía, acompañada por la validación de los procedimientos analíticos contenidos en la monografía y por una traducción, cuando proceda. 7. En caso de que el principio activo y/o material de partida, materia prima o los excipientes sean objeto de una monografía de la Farmacopea Europea, el solicitante podrá hacer referencia a un certificado de idoneidad, que, cuando haya sido expedido por la Dirección Europea para la Calidad del Medicamento (EDQM), se presentará en el apartado que corresponda del presente Módulo. Se considerará que dichos certificados de idoneidad de la monografía de la Farmacopea Europea sustituyen los datos pertinentes de los apartados correspondientes descritos en este módulo. El fabricante garantizará por escrito al solicitante que el proceso de fabricación no se ha modificado desde la concesión del certificado de idoneidad por parte de la Dirección Europea para la Calidad del Medicamento. 8. Para un principio activo bien definido, su fabricante o el solicitante podrán disponer que: a) la descripción del proceso de fabricación, b) el control de calidad durante la fabricación, y c) la validación del proceso se faciliten en un documento separado (parte cerrada) dirigido directamente a las autoridades competentes por el fabricante del principio activo, en calidad de archivo maestro del principio activo. En este caso, sin embargo, el fabricante deberá proporcionar al solicitante todos los datos que puedan resultar necesarios para que éste asuma la responsabilidad del – 211 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano medicamento. El fabricante deberá comprometerse por escrito frente al solicitante a garantizar la homogeneidad de los lotes y a no modificar el proceso de fabricación o las especificaciones sin haberle previamente informado. Se deberán presentar a las autoridades competentes los documentos en apoyo de esta solicitud de modificación; dichos documentos también se proporcionarán al solicitante cuando se refieran a la parte abierta del archivo maestro. 9. Medidas específicas concernientes a la prevención de la transmisión de encefalopatías espongiformes animales (materiales procedentes de rumiantes): en cada fase del proceso de fabricación, el solicitante deberá demostrar el cumplimiento de los materiales utilizados con la Nota Explicativa sobre Minimización del Riesgo de Transmisión de los Agentes de la Encefalopatía Espongiforme Animal a través de Medicamentos y sus actualizaciones, publicada por la Comisión en el «Diario Oficial de la Unión Europea». La demostración del cumplimiento con lo dispuesto en la mencionada Nota Explicativa, podrá realizarse presentando, preferiblemente, un certificado de idoneidad en relación con la monografía pertinente de la Farmacopea Europea expedido por la Dirección Europea para la Calidad del Medicamento, o bien los datos científicos que corroboren dicho cumplimiento. 10. En relación con los agentes extraños/externos, deberá facilitarse información que evalúe el riesgo con respecto a la contaminación potencial por dicho tipo de agentes, bien sean virales o no virales, tal como se establece en las directrices correspondientes y en la monografía y el capítulo generales pertinentes de la Farmacopea Europea. 11. Se describirán con los detalles necesarios todos los aparatos y equipos especiales que puedan utilizarse en alguna fase del proceso de fabricación y las operaciones de control del producto terminado. 12. En los casos en que proceda y sea necesario, se presentará la marca CE requerida por la legislación comunitaria sobre productos sanitarios. Deberá prestarse particular atención a los elementos seleccionados siguientes. 3.2.1 Principio o principios activos. 3.2.1.1 Información general e información sobre los materiales de partida y materias primas. a) Se proporcionará información sobre la nomenclatura del principio activo incluyendo la denominación común internacional recomendada (DCI), la denominación de la Farmacopea Europea si procede y la(s) denominación (-ones) química(s). Se proporcionará la fórmula estructural, incluyendo la estereoquímica relativa y absoluta, la fórmula molecular y la masa molecular relativa. En el caso de los medicamentos biotecnológicos, si procede, deberá comunicarse la secuencia de aminoácidos esquemática y la masa molecular relativa. Se presentará una lista de propiedades físico-químicas y otras propiedades relevantes de la sustancia activa, incluyendo la actividad biológica en el caso de los medicamentos biológicos. b) A efectos del presente anexo, se entenderá por materiales de partida todos los materiales a partir de los cuales se fabrica o de los que se extrae el principio activo. En el caso de los medicamentos biológicos, se entenderá por materiales de partida toda sustancia de origen biológico, tales como los microorganismos, órgano s y tejidos de origen vegetal o animal, las células o fluidos (incluyendo sangre y plasma) de origen humano o animal y los diseños celulares biotecnológicos (sustratos celulares, sean o no recombinantes, incluidas las células primarias). Un medicamento biológico es un producto cuyo principio activo es biológico. Una sustancia biológica es aquella que se produce o se extrae a partir de una fuente biológica y que necesita, para su caracterización y determinación de su calidad, una combinación de ensayos físico-químico y biológico junto con el proceso de producción y su control. Se considerarán medicamentos biológicos: los medicamentos inmunológicos y los medicamentos derivados de la sangre o el plasma humanos; los medicamentos que pertenezcan al ámbito de aplicación del apartado 1 del anexo del Reglamento (CE) n.º 726/2004; los medicamentos de terapia avanzada, definidos en la parte IV del presente anexo. – 212 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano Cualquier otra sustancia utilizada para la fabricación o extracción del (los) principio(s) activo(s), pero de las que no deriva directamente dicho principio activo, como los reactivos, los medios de cultivo, suero de ternera fetal, aditivos y soluciones tampón utilizadas para la cromatografía, etc., se consideran materias primas. 3.2.1.2 Proceso de fabricación del principio o principio activos. a) La descripción del proceso de fabricación del principio activo representa el compromiso del solicitante respecto a la fabricación del principio activo. Para describir de manera adecuada el proceso de fabricación y los controles en proceso, se facilitará la información adecuada que se establece en las directrices publicadas por la Agencia Europea de Medicamentos. b) Se presentará una relación de todos los materiales necesarios para fabricar el(los) principio(s) activo(s), identificando en qué parte del proceso se utiliza cada material. Se facilitará información sobre la calidad y el control de dichos materiales. También se presentará información que demuestre que los materiales cumplen los estándares apropiados para su utilización prevista. Se presentará una relación de las materias primas y se documentarán también su calidad y sus procedimientos de control. Se proporcionarán el nombre, la dirección y la responsabilidad de cada fabricante, incluyendo sus contratistas, y cada una de las sedes de producción o instalaciones propuestas dedicadas a la fabricación y control. c) Para los medicamentos biológicos se aplicarán los siguientes requisitos adicionales. Se describirá y documentará el origen y la historia de los materiales de partida. Respecto a las medidas específicas de prevención de la transmisión de las encefalopatías espongiformes animales, el solicitante deberá demostrar que el principio activo cumple con lo dispuesto en la Nota Explicativa sobre Minimización del Riesgo de Transmisión de los Agentes de la Encefalopatía Espongiforme Animal a través de Medicamentos y sus actualizaciones, publicada por la Comisión en el «Diario Oficial de la Unión Europea». Cuando se usen bancos celulares, deberá demostrarse que las características de las células se han mantenido inalteradas en los pasos empleados para la producción y posteriormente. Los materiales de siembra, los bancos de células, las mezclas de suero o plasma sin elaborar y demás materias de origen biológico, así como, siempre que sea posible, los materiales de los que se hayan obtenido, deberán someterse a ensayos para comprobar que están libres de agentes extraños/externos. Cuando la presencia de agentes extraños/externos potencialmente patogénicos es inevitable, el material correspondiente deberá utilizarse si un tratamiento posterior garantiza su eliminación y/o inactivación, y esto deberá ser validado. Siempre que sea posible, la producción de vacunas deberá basarse en un sistema de lotes de siembra y de bancos celulares establecidos. En el caso de vacunas bacterianas y virales, las características del agente infeccioso deberán demostrarse en los materiales de siembra. Además, para las vacunas vivas, la estabilidad de las características de atenuación deberán ser demostradas en el material de siembra, si esta prueba no es suficiente, las características de atenuación deberán también demostrarse en la etapa de producción. Cuando se trate de medicamentos derivados de la sangre o del plasma humano, deberán describirse y documentarse, con arreglo a lo dispuesto en la parte III del presente anexo, el origen y los criterios de recogida, transporte y conservación de los materiales de partida. Se describirán las instalaciones y el equipo de fabricación. d) Deberán facilitarse, si procede, los ensayos y los criterios de aceptación realizados en cada una de las etapas críticas, información sobre la calidad y el control de los productos intermedios, así como sobre la validación del proceso y/o los estudios de evaluación. e) Si la presencia de agentes extraños/externos potencialmente patógenos es inevitable, el material correspondiente deberá utilizarse únicamente si un tratamiento posterior garantiza su eliminación y/o inactivación, y esto deberá ser validado en el apartado en que se aborde la evaluación de la seguridad viral. – 213 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano f) Se facilitará una descripción y discusión de los cambios significativos introducidos en el proceso de fabricación durante el desarrollo y/o el lugar de fabricación del principio activo. 3.2.1.3 Caracterización del principio o principios activos.–Deberán presentarse datos que pongan de manifiesto la estructura y otras características del principio(s) activo(s).Se facilitará la confirmación de la estructura del principio(s) activo(s) a partir de algún método físico-químico y/o inmuno-químico y/o biológico, así como información sobre las impurezas. 3.2.1.4 Control del principio(s) activo(s).–Se presentará información detallada sobre las especificaciones utilizadas para los controles de rutina del(los) principio(s) activo(s), la justificación de la elección de dichas especificaciones, métodos de análisis y su validación. Se presentarán los resultados del control efectuado en lotes fabricados durante el desarrollo. 3.2.1.5 Estándares o materiales de referencia.–Se identificarán y describirán detalladamente los estándares y preparaciones de referencia. Cuando sea relevante, se utilizará material de referencia químico y biológico de la Farmacopea Europea. 3.2.1.6 Envase y sistema de cierre del principio activo.–Se presentará la descripción del envase y el sistema o sistemas de cierre y sus especificaciones. 3.2.1.7 Estabilidad del principio(s) activo(s). a) Deberán resumirse los tipos de estudios realizados, los protocolos empleados y los resultados de los estudios. b) Se presentarán con el formato adecuado los resultados detallados de los estudios de estabilidad, incluyendo información relativa a los procedimientos analíticos para obtener dichos datos, así como la validación de estos procedimientos. c) Se facilitarán el protocolo de estabilidad tras la autorización y el compromiso de estabilidad. 3.2.2 Producto terminado. 3.2.2.1 Descripción y composición del producto terminado.–Deberá describirse el producto terminado y su composición. La información deberá incluir la descripción de la forma farmacéutica y su composición con todos los componentes del producto terminado, la cantidad de los mismos por unidad y la función: Del principio(s) activo(s). Componente(s) los excipientes, cualquiera que sea su naturaleza o la cantidad utilizada, incluyendo los colorantes, conservantes, adyuvantes, estabilizadores, espesantes, emulsionantes, correctores del sabor, aromatizantes, etcétera. Los componentes de la cubierta externa de los medicamentos (cápsulas duras, cápsulas blandas, supositorios, comprimidos recubiertos, comprimidos recubiertos con cubierta pelicular, etc.) que vayan a ser ingeridos o administrados al paciente de otra forma. Estos aspectos deberán completarse con cualquier otro dato relevante relacionado con el tipo de envase y, si procede, su sistema de cierre, junto con detalle de los dispositivos que serán utilizados para la administración del medicamento y que se suministrarán con él. La «terminología usual», a utilizar en la descripción de los componentes del medicamento, deberá ser: Cuando se trate de productos que figuren en la Farmacopea Europea o, en su defecto, en la farmacopea nacional de un Estado Miembro, la denominación principal recogida en el encabezamiento de la correspondiente monografía con referencia a la farmacopea de que se trate; para los restantes productos, la denominación común internacional recomendada por la Organización Mundial de la Salud o, en su defecto, la denominación científica exacta; las sustancias que carezcan de denominación común internacional o de denominación científica exacta se describirán declarando a su origen y modo de obtención, completándose estos datos con cualquier otro detalle relevante, en caso necesario; con respecto a los colorantes, la designación por el código «E» que se les atribuya en el Real Decreto 2001/1995, de 7 de diciembre, por el que se aprueba la lista positiva de aditivos colorantes autorizados para su uso en la elaboración de productos alimenticios, así como sus condiciones de utilización. Para proporcionar la «composición cuantitativa» de los principios activos del medicamento, será preciso, según la forma farmacéutica, especificar la masa o el número de – 214 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano unidades de actividad biológica, bien sea por dosis o por unidad de masa o de volumen, de cada principio activo. Los principios activos presentes en forma de compuestos o derivados se designarán cuantitativamente mediante su masa total y, en caso necesario o relevante, mediante la masa de las fracciones activas de la molécula. En el caso de los medicamentos que contengan un principio activo cuya autorización se haya solicitado en cualquier Estado miembro por primera vez, la declaración cuantitativa de un principio activo que sea una sal o hidrato se expresará sistemáticamente en términos de masa de los fragmentos activos de la molécula. Todas las autorizaciones posteriores de medicamentos en los Estados miembros dispondrán de su composición cualitativa expresada de la misma manera para el mismo principio activo. Las unidades de actividad biológica se emplearán en las sustancias que no pueden definirse en términos moleculares. Cuando la Organización Mundial de la Salud haya definido una unidad de actividad biológica, es ésta la que deberá usarse. En los casos en los que no se haya definido una unidad internacional, las unidades de actividad biológica se expresarán de forma que proporcionen información inequívoca sobre la actividad de la sustancia, utilizando, cuando proceda, las unidades de la Farmacopea Europea. 3.2.2.2 Desarrollo farmacéutico.–El presente capítulo se dedicará a la información sobre los estudios de desarrollo realizados para establecer que la forma farmacéutica, la formulación, el proceso de fabricación, el sistema de cierre del envase, los atributos microbiológicos y las instrucciones de uso son los adecuados para el uso previsto especificado en el expediente de solicitud de autorización de comercialización. Los estudios descritos en el presente capítulo son distintos de las pruebas de controles de rutina que se realizan según las especificaciones. Se determinarán y describirán los parámetros críticos de la formulación y los atributos del proceso que puedan influir en la reproducibilidad del lote, la eficacia del medicamento y su calidad. Los datos de apoyo adicionales deberán remitir, cuando proceda, a los capítulos relevantes del Módulo 4 (Informes de estudios no clínicos) y del Módulo 5 (Informes de estudios clínicos) del expediente de solicitud de autorización de comercialización. a) Deberá documentarse la compatibilidad del principio activo con los excipientes, así como las características físico-químicas clave del principio activo que puedan influir en la eficacia del producto terminado o en la compatibilidad de los distintos principios activos entre sí en el caso de los productos en los que se combinen. b) Se documentará la elección de los excipientes, especialmente en relación con sus funciones respectivas y su concentración. c) Se describirá el desarrollo del producto terminado, teniendo en consideración la vía de administración y la utilización propuestas. d) Deberá justificarse cualquier sobredosificación en la formulación(es). e) En lo que respecta a las propiedades físico-químicas y biológicas, deberán tratarse y documentarse todos los parámetros que conciernan al comportamiento del producto terminado. f) Se presentará la selección y optimización del proceso de fabricación, así como las diferencias entre el (los) proceso(s) de fabricación utilizados para producir lotes clínicos pivotales y el proceso empleado para la fabricación del producto terminado propuesto. g) Se documentará la idoneidad del envase y el sistema de cierre empleado para el almacenamiento, el transporte y la utilización del producto terminado. Puede ser necesario considerar la posible interacción entre el medicamento y el envase. h) Los atributos microbiológicos de la forma farmacéutica en relación con productos no estériles y estériles deberán ajustarse a lo prescrito en la Farmacopea Europea y documentarse con arreglo a ello. i) Con el fin de ofrecer información útil y adecuada para el etiquetado, se documentará la compatibilidad del producto terminado con los diluyentes de reconstitución y los dispositivos de administración. 3.2.2.3 Proceso de fabricación del producto terminado. a) La descripción del método de fabricación que deberá acompañar a la solicitud de autorización se redactará de forma que ofrezca una idea clara del carácter de las operaciones efectuadas. – 215 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano Con este fin, dicha descripción deberá incluir, como mínimo: Referencia a las diferentes fases del proceso de fabricación, incluidos los sistemas de control del proceso y los criterios de aceptación correspondientes, de modo que se pueda evaluar si los procesos utilizados en la producción de la forma farmacéutica puedan producir un cambio adverso en los componentes; en caso de fabricación en serie, información completa sobre las precauciones tomadas para asegurar la homogeneidad del producto terminado; estudios experimentales de validación del procedimiento de fabricación cuando se emplee un método de fabricación no estándar o cuando sea crítico para el producto; en el caso de medicamentos estériles, detalles de los procesos de esterilización y/o asépticos utilizados; la fórmula detallada del lote. Se presentará el nombre, la dirección y la responsabilidad de cada fabricante, incluidos sus contratistas, y cada una de las sedes de producción o instalaciones propuestas dedicadas a la fabricación y ensayo. b) Se incluirán los datos relativos a los ensayos de control del producto que puedan realizarse en una fase intermedia del proceso de fabricación, con el fin de asegurar la consistencia de la producción. Estos ensayos son esenciales para comprobar la conformidad del medicamento con la fórmula cuando, excepcionalmente, el solicitante proponga un método analítico para analizar el producto terminado que no incluya la determinación de todos los principios activos (o de todos los componentes del excipiente sujetos a los mismos requerimientos que las sustancias activas). Lo anterior será igualmente aplicable cuando el control de calidad del producto terminado dependa de los controles en proceso, especialmente en el caso de que el medicamento se defina principalmente por su proceso de preparación. c) Se presentará descripción, documentación y resultados de los estudios de validación para las etapas o ensayos críticos utilizados en el proceso de fabricación. 3.2.2.4 Control de los excipientes. a) Se presentará una relación de todos los materiales necesarios para fabricar el (los) excipiente(s), identificando cuándo se emplea cada material en el proceso. Se facilitará información sobre la calidad y el control de dichos materiales. También se presentará información que demuestre que los materiales cumplen los estándares apropiados para su utilización prevista. En todos los casos, los colorantes deberán reunir los requisitos que se establecen en el Real Decreto 2001/1995, de 7 de diciembre. Además, los colorantes deberán cumplir los criterios de pureza establecidos en el Real Decreto 2107/1996, de 20 de septiembre, por el que se establecen las normas de identidad y pureza de los colorantes utilizados en los productos alimenticios. b) Deberán detallarse las especificaciones de cada excipiente, así como su justificación. Se describirán y validarán debidamente los procedimientos analíticos. c) Se prestará atención específica a los excipientes de origen humano o animal. Respecto a las medidas específicas relativas a la prevención de la transmisión de encefalopatías espongiformes animales, el solicitante deberá demostrar asimismo que el medicamento está fabricado con arreglo a la Nota Explicativa sobre Minimización del Riesgo de Transmisión de los Agentes de la Encefalopatía Espongiforme Animal a través de Medicamentos y sus actualizaciones, publicada por la Comisión en el «Diario Oficial de la Unión Europea». Para demostrar el cumplimiento de lo dispuesto en la mencionada Nota Explicativa, se podrá presentar, preferiblemente, un certificado de idoneidad con la monografía relativa a Encefalopatías Espongiformes Transmisibles de la Farmacopea Europea, o bien los datos científicos que corroboran dicho cumplimiento. d) Nuevos excipientes. Para los excipientes utilizados por primera vez en un medicamento o para una nueva vía de administración, se presentarán con arreglo al formato del principio activo previamente – 216 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano descrito todos los datos de fabricación, caracterización y controles, haciendo referencia cruzada a los datos de apoyo relativos a seguridad, tanto clínicos como no clínicos. Se presentará un documento en el que figurará la información pormenorizada de carácter químico, farmacéutico y biológico. Dicha información deberá presentarse en el mismo orden que el capítulo dedicado al (los) principio(s) activo(s) del módulo 3. La información relativa a nuevos excipientes podrá presentarse como documento independiente según el formato descrito en los párrafos anteriores. En caso de que el solicitante no sea el fabricante del nuevo excipiente, el mencionado documento independiente deberá ponerse a disposición del solicitante para su presentación a la autoridad competente. En el módulo 4 del expediente se ofrecerá información suplementaria sobre los estudios de toxicidad con el excipiente novedoso. En el módulo 5 se presentarán los estudios clínicos. 3.2.2.5 Control del producto terminado.–A efectos de control del producto terminado, se entenderá por lote de un medicamento una entidad que comprenda todas las unidades de una forma farmacéutica que provengan de una misma cantidad inicial de material y hayan sido sometidas a la misma serie de operaciones de fabricación y esterilización o, en el caso de un proceso de producción continuo, todas las unidades fabricadas en un lapso de tiempo determinado. Salvo debida justificación, la desviación máxima tolerable del contenido del principio activo en el producto acabado no podrá ser superior a ± 5% en el momento de fabricación. Se presentará información detallada sobre las especificaciones, la justificación (liberación y período de validez) de su elección, los métodos de análisis y su validación. 3.2.2.6 Estándares o materiales de referencia.–Se determinarán y describirán detalladamente los estándares y materiales de referencia utilizados para poner a prueba el producto terminado, en caso de que no se hayan presentado anteriormente en el apartado relativo al principio activo. 3.2.2.7 Envase y cierre del producto terminado.–Se entregará la descripción del envase y el sistema o sistemas de cierre, incluyendo la identidad de cada material de acondicionamiento primario y sus especificaciones. En las especificaciones se incluirán la descripción e identificación. Se incluirán, cuando proceda, los métodos no recogidos en la farmacopea (con validación). Para los materiales de acondicionamiento exterior no funcionales únicamente se ofrecerá una breve descripción. Para los materiales de embalaje exterior funcionales se ofrecerá información suplementaria. 3.2.2.8 Estabilidad del producto terminado. a) Deberán resumirse los tipos de estudios realizados, los protocolos empleados y los resultados de los estudios. b) Se presentarán con el formato adecuado los resultados pormenorizados de los estudios sobre estabilidad, incluida la información relativa a los procedimientos de análisis seguidos para obtener los datos, así como la validación de dichos procedimientos; para las vacunas, se proporcionará la información sobre la estabilidad acumulativa en aquellos casos en que sea pertinente. c) Se facilitarán el protocolo de estabilidad tras la aprobación y el compromiso de estabilidad. 4. Módulo 4: Informes no clínicos 4.1 Formato y presentación.–El esquema general del módulo 4 es el siguiente: Índice. Informes de estudios. Farmacología: Farmacodinámica primaria. Farmacodinámica secundaria. Farmacología de seguridad. Interacciones farmacodinámicas. Farmacocinética: – 217 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano Métodos analíticos e informes de validación. Absorción. Distribución. Metabolismo. Excreción. Interacciones farmacocinéticas (no clínicas). Otros estudios de farmacocinética. Toxicología: Toxicidad por dosis única. Toxicidad por administración continuada. Genotoxicidad. In vitro. In vivo (incluidas las evaluaciones toxicocinéticas de apoyo). Carcinogénesis: Estudios a largo plazo. Estudios a corto o medio plazo. Otros estudios. Toxicidad en la reproducción y el desarrollo: Fertilidad y desarrollo embrionario inicial. Desarrollo embrionario y fetal. Desarrollo prenatal y postnatal. Estudios en los que se administran dosis a las crías (animales jóvenes) y/o se evalúan posteriormente. Tolerancia local. Otros estudios sobre toxicidad: Antigenicidad. Inmunotoxicidad. Estudios mecanicistas. Dependencia. Metabolitos. Impurezas. Otros. Referencias bibliográficas. 4.2 Contenido:Principios y requisitos básicos.–Deberá prestarse particular atención a los elementos seleccionados siguientes: 1. Las pruebas toxicológicas y farmacológicas deberán poner de manifiesto lo siguiente: a) la toxicidad potencial del producto y los efectos peligrosos o no deseables que pudieran producirse en seres humanos en las condiciones de uso propuestas, valorándose estos efectos en función del proceso patológico de que se trate; b) sus propiedades farmacológicas, en relación a la posología y la actividad farmacológica con el uso indicado en seres humanos. Todos los resultados deberán ser fiables y de aplicación general. En la medida en que sea conveniente, se utilizarán procedimientos matemáticos y estadísticos para la elaboración de los métodos experimentales y la valoración de los resultados. Además, será necesario informar a los clínicos sobre el potencial terapéutico y toxicológico del producto. 2. En el caso de medicamentos biológicos tales como medicamentos inmunológicos y medicamentos derivados de la sangre o el plasma humanos, puede ser necesario adaptar los requisitos del presente módulo para algunos productos determinados; por esta razón, el solicitante deberá justificar el programa de las pruebas. Al fijar el programa de las pruebas, deberán tenerse en cuenta los siguientes puntos: Todas las pruebas que requieran la administración reiterada del producto se diseñarán de modo que tengan en consideración la posible inducción de anticuerpos e interferencia por parte de éstos; deberá preverse un estudio de la función reproductora, de la toxicidad embrionaria, fetal y perinatal, del potencial mutágeno así como del potencial carcinógeno. – 218 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano Cuando los efectos sean atribuibles a componentes distintos del principio o principios activos, el estudio podrá sustituirse por la validación de la eliminación de aquéllos. 3. Se deberá investigar la toxicología y la farmacocinética de un excipiente que se utilice por primera vez en el ámbito farmacéutico. 4. Cuando se dé la posibilidad de una degradación significativa durante el almacenamiento del medicamento, deberá tomarse en consideración la toxicología de los productos de la degradación. 4.2.1 Farmacología.–El estudio de farmacología deberá efectuarse siguiendo dos planteamientos distintos: En primer lugar, las acciones relacionadas con el uso terapéutico propuesto deberán estudiarse y describirse de manera adecuada. Siempre que sea posible se realizarán ensayos reconocidos y validados, tanto in vivo como in vitro. Deberán describirse técnicas experimentales novedosas de manera suficientemente pormenorizada para que puedan reproducirse. Los resultados se expresarán en términos cuantitativos, utilizando, por ejemplo, curvas dosis-efecto y tiempo-efecto, etc. En la medida de lo posible, se establecerán comparaciones con los datos correspondientes a una sustancia o sustancias con una acción terapéutica análoga. En segundo lugar, el solicitante deberá investigar las posibles repercusiones farmacodinámicas no deseadas de la sustancia en las funciones fisiológicas. Tales investigaciones se realizarán en exposiciones correspondientes a la gama terapéutica prevista y por encima de la misma. Las técnicas experimentales, a no ser que sean las que se utilicen habitualmente, se describirán de forma tal que permitan su reproducción, debiendo el investigador demostrar su validez. Deberá estudiarse todo indicio de modificación de las respuestas derivadas de la administración reiterada de la sustancia. Respecto a la interacción farmacodinámica de los medicamentos, las pruebas de combinaciones de principios activos podrán justificarse bien por necesidades farmacológicas, bien por indicaciones clínicas. En el primer caso, el estudio farmacodinámico deberá poner de manifiesto aquellas interacciones que hagan recomendable la combinación para el uso clínico. En el segundo caso, cuando la experimentación clínica tenga por fin justificar científicamente la combinación de sustancias, la investigación deberá determinar si los efectos esperados de la combinación pueden demostrarse en animales y, como mínimo, la importancia de las reacciones adversas. 4.2.2 Farmacocinética.–Se entiende por farmacocinética el estudio del conjunto de procesos que sufre el principio activo y sus metabolitos en el organismo. Comprende el estudio de la absorción, la distribución, el metabolismo (biotransformación) y la excreción de las sustancias. El estudio de estas distintas fases se puede llevar a cabo principalmente con métodos físicos, químicos o en su caso biológicos, y mediante la observación de la actividad farmacodinámica real de la propia sustancia. Los datos referentes a la distribución y eliminación serán necesarios en todos los casos en que dichos datos resulten indispensables para determinar las dosis administrables a seres humanos, así como en las sustancias quimioterapéuticas (antibióticos, etc.) y en las sustancias cuyo uso se base en efectos no farmacodinámicos (por ejemplo, numerosos agentes de diagnóstico, etc.). También pueden realizarse estudios in vitro, con la ventaja de la utilización de material humano para su comparación con material animal (es decir, fijación con proteínas, metabolismo, interacción entre medicamentos). Es necesario el estudio farmacocinético de todas las sustancias farmacológicamente activas. Cuando se trate de nuevas combinaciones de sustancias conocidas que hayan sido estudiadas con arreglo a las disposiciones del presente real decreto, no será necesario exigir las investigaciones farmacocinéticas si las pruebas de toxicidad y la experimentación clínica justifican su omisión. El programa farmacocinético se elaborará de modo que sean posibles la comparación y la extrapolación entre animales y seres humanos. 4.2.3 Toxicología. – 219 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano a) Toxicidad por dosis única.–Una prueba de toxicidad por dosis única es un estudio cualitativo y cuantitativo de las reacciones tóxicas que pueden derivarse de una administración única del principio o principios activos contenidos en el medicamento, en las proporciones y en el estado físico-químico en que están presentes en el producto. La prueba de toxicidad por dosis única puede realizarse con arreglo a las orientaciones pertinentes publicadas por la Agencia Europea de Medicamentos. b) Toxicidad por administración continuada.–Las pruebas de toxicidad por administración continuada tendrán por objeto revelar las alteraciones funcionales y/o anatomo-patológicas subsiguientes a la administración repetida del principio activo o de la combinación de principios activos en cuestión y establecer de qué modo se relacionan dichas alteraciones con la posología. Generalmente es aconsejable realizar dos pruebas: una a corto plazo, durante dos a cuatro semanas, y la otra a largo plazo. La duración de la segunda prueba dependerá de las condiciones de la utilización clínica. Su objeto es describir los posibles efectos nocivos, a los que deberá prestarse atención en los estudios clínicos. La duración se define en las directrices correspondientes publicadas por la Agencia Europea de Medicamentos. c) Genotoxicidad.–El objeto del estudio del potencial mutagénico y clastogénico es revelar las alteraciones que puede causar una sustancia en el material genético de las personas y las células. Las sustancias mutagénicas pueden representar un riesgo para la salud, ya que la exposición a un mutágeno supone el riesgo de inducir una mutación germinal, con la posibilidad de trastornos hereditarios, y el riesgo de mutaciones somáticas, que incluso pueden ser causa de cáncer. Dicho estudio será obligatorio para cualquier sustancia nueva. d) Carcinogénesis.–Se exigirá habitualmente efectuar pruebas dirigidas a revelar efectos carcinógenos: 1. Estos estudios se realizarán con todos los medicamentos cuya utilización clínica se prevea para un período prolongado de la vida del paciente, bien de manera continuada, bien de manera reiterada e intermitente. 2. Los estudios relativos a determinados medicamentos se recomiendan si se piensa que representan un potencial carcinogenético, por ejemplo tomando como referencia un producto de la misma clase o estructura, o a raíz de pruebas obtenidas en estudios de toxicidad por administración continuada. 3. No son necesarios los estudios con componentes inequívocamente genotóxicos, ya que se supone que son carcinógenos que afectan a distintas especies y suponen un riesgo para el ser humano. Si se pretende administrar un medicamento de este tipo de manera crónica a seres humanos, puede resultar necesario un estudio crónico para detectar efectos tumorigénicos precoces. e) Toxicidad en la reproducción y el desarrollo.–La investigación acerca de posibles alteraciones de la función reproductora masculina o femenina, así como los efectos nocivos para los descendientes, deberá realizarse mediante las pruebas pertinentes. En ellas se incluyen los estudios sobre la repercusión en la función reproductora masculina y femenina, sobre los efectos tóxicos y teratógenos en todas las fases de desarrollo desde la concepción a la madurez sexual, así como los efectos latentes, cuando el medicamento investigado ha sido administrado a la mujer durante el embarazo. Deberá justificarse de manera adecuada la omisión de tales pruebas. En función de la utilización indicada del medicamento, podrá justificarse la realización de estudios suplementarios acerca del desarrollo de la descendencia cuando se administra el medicamento. Los estudios de toxicidad embrionaria y fetal se realizarán normalmente con dos especies de mamíferos, una de las cuales deberá no ser un roedor. Los estudios perinatales y postnatales se llevarán a cabo con al menos una especie. Si se sabe que el metabolismo de un medicamento en determinada especie es similar al del hombre, es deseable incluir esa especie. También es deseable que una de las especies sea la misma que la de los estudios de toxicidad por administración continuada. La concepción del estudio se determinará teniendo en cuenta el estado de los conocimientos científicos en el momento de presentarse la solicitud. – 220 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano f) Tolerancia local.–El objetivo de los estudios de tolerancia local es determinar si los medicamentos (tanto los principios activos como los excipientes) se toleran en los lugares del cuerpo que pueden entrar en contacto con el medicamento como consecuencia de su administración durante el uso clínico. El procedimiento de prueba debe ser tal que todo efecto mecánico de la administración, o las acciones puramente fisicoquímicas del producto, puedan distinguirse de los efectos toxicológicos o farmacodinámicos. Deberán realizarse las pruebas sobre tolerancia local con el preparado que se está desarrollando para su uso humano, utilizando el vehículo y/o los excipientes en el tratamiento del grupo o grupos de control. Si es preciso, se incluirán controles y sustancias de referencia positivos. La concepción de las pruebas de tolerancia local (elección de la especie, duración, frecuencia y vía de administración, dosificación) dependerá del problema que deba investigarse y las condiciones propuestas de administración en la utilización clínica. Deberá realizarse la reversibilidad de las lesiones locales cuando resulte pertinente. Los estudios en animales podrán sustituirse por pruebas validadas in vitro, siempre que los resultados de las pruebas sean de calidad y utilidad análogas para los fines de la evaluación de la seguridad. En el caso de las sustancias químicas aplicadas a la piel (por ejemplo, dérmicas, rectales, vaginales) se evaluará el potencial de sensibilización como mínimo en uno de los sistemas de prueba actualmente disponibles (ensayo con cobayas o ensayo de ganglio linfático local). 5. Módulo 5: Informes de estudios clínicos 5.1 Formato y presentación.–El esquema general del módulo 5 es el siguiente: Índice de informes de estudios clínicos. Listado en forma de tabla de todos los estudios clínicos. Informes de los estudios clínicos. Informes de estudios biofarmacéuticos. Informes de estudios de biodisponibilidad. Informes de estudios comparativos de biodisponibilidad y bioequivalencia. Informes de estudios de correlación in vitro-in vivo. Informes de métodos bioanalíticos y analíticos. Informes de estudios sobre farmacocinética mediante biomateriales humanos. Informes de estudios de fijación con proteínas del plasma. Informes de estudios sobre metabolismo hepático e interacción. Informes de estudios mediante otros biomateriales humanos. Informes de estudios de farmacocinética humana. Informes de estudios de farmacocinética y tolerancia inicial en sujetos sanos. Informes de estudios de farmacocinética y tolerancia inicial en pacientes. Informes de estudios de farmacocinética de factores intrínsecos. Informes de estudios de farmacocinética de factores extrínsecos. Informes de estudios de farmacocinética en la población. Informes de estudios de farmacodinámica humana. Informes de estudios de farmacodinámica y farmacocinética/farmacodinámica en sujetos sanos. Informes de estudios de farmacodinámica y farmacocinética/farmacodinámica en pacientes. Informes de estudios sobre eficacia y seguridad. Informes de estudios clínicos controlados relativos a la indicación declarada. Informes de estudios clínicos no controlados. Informes de análisis de datos procedentes de diversos estudios, incluido cualquier metaanálisis, análisis comparativo (bridging analyses) y análisis integrado formal. Otros informes de estudio. Informes de experiencia posterior a la comercialización. Referencias bibliográficas. – 221 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano 5.2 Contenido: Principios y requisitos básicos.–Deberá prestarse particular atención a los elementos seleccionados siguientes. a) Los datos clínicos que se suministren en cumplimiento de lo dispuesto en el artículo 6.5.j), deberán permitir formarse una opinión suficientemente fundada y científicamente válida acerca de si la especialidad responde a los criterios previstos para la concesión de la autorización de comercialización. Por este motivo, es preceptivo que se comuniquen los resultados de todos los ensayos clínicos que se hayan realizado, tanto favorables como desfavorables. b) Los ensayos clínicos deberán ir siempre precedidos de las necesarias pruebas farmacológicas y toxicológicas en animales, efectuadas con arreglo a lo dispuesto en el módulo 4 del presente anexo. El investigador deberá conocer las conclusiones de los exámenes farmacológico y toxicológico y, por tanto, el solicitante deberá proporcionarle, como mínimo, el manual del investigador, que consistirá en toda la información pertinente conocida antes del inicio de un ensayo clínico, e incluirá datos químicos, farmacéuticos y biológicos, datos toxicológicos, farmacocinéticos y farmacodinámicos en animales, así como los resultados de ensayos clínicos anteriores, con datos útiles que justifiquen la naturaleza, la escala y la duración del ensayo propuesto; a petición del investigador se deberán suministrar los informes farmacológicos y toxicológicos completos. Cuando se trate de materias de origen humano o animal, se emplearán todos los medios disponibles antes del inicio del ensayo para garantizar que no se transmiten agentes infecciosos. c) Los titulares de la autorización de comercialización deberán tomar las medidas necesarias para que los documentos de los ensayos clínicos esenciales (incluidos los impresos de recogida de datos) distintos del expediente médico del sujeto sean custodiados por los propietarios de los datos: Durante un mínimo de 15 años tras la finalización o interrupción del ensayo, o durante un mínimo de dos años tras la concesión de la última autorización de comercialización en la Unión Europea y en aquellos casos en que no haya solicitudes de comercialización pendientes o previstas en la Unión Europea, o durante un mínimo de dos años tras la interrupción oficial del desarrollo clínico del producto objeto de investigación. El expediente médico del sujeto deberá ser custodiado con arreglo a la normativa aplicable y conforme al período máximo permitido por el hospital, institución o consulta privada. No obstante, podrán retenerse los documentos durante un período más largo, si así lo exigen las disposiciones normativas aplicables o el acuerdo con el promotor. Corresponderá al promotor informar al hospital, institución o consulta privada acerca del momento en que no será preciso continuar conservando dichos documentos. El promotor o el propietario de los datos conservará toda la restante documentación relativa al ensayo durante el período de validez del medicamento. Entre dicha documentación deberán figurar: el protocolo, incluidos la justificación, los objetivos, el diseño estadístico y la metodología del ensayo, con las condiciones en las que se efectúe y gestione, así como los pormenores del medicamento de investigación, el medicamento de referencia y/o el placebo que se empleen; los procedimientos normalizados de trabajo; todos los informes escritos sobre el protocolo y los procedimientos; el manual del investigador; el cuaderno de recogida de datos de cada sujeto; el informe final; el(los) certificado(s) de auditoría, cuando se disponga de él (ellos). El promotor o el propietario subsiguiente conservará el informe final hasta pasados cinco años tras haberse agotado el plazo de validez del medicamento. Además de los ensayos que se realicen dentro de la Unión Europea, el titular de la autorización de comercialización tomará todas las medidas suplementarias para el archivo de la documentación con arreglo a lo dispuesto en la Directiva 2001/20/CE y en España el Real Decreto 223/2004, de 6 de febrero y en las directrices detalladas de aplicación. Deberá documentarse todo cambio que se produzca en la propiedad de los datos. Todos los datos y documentos deberán ponerse a disposición de las autoridades competentes, si éstas así lo solicitan. d) Los datos sobre cada ensayo clínico deberán estar suficientemente detallados para permitir un juicio objetivo, y contendrán, en particular: – 222 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano El protocolo, incluyendo la justificación, los objetivos, el diseño estadístico y la metodología del ensayo, con las condiciones en las que se efectúa y gestiona, así como los pormenores del medicamento objeto de estudio que se emplee; el (los) certificado(s) de auditoría, cuando se disponga de él (ellos); la lista de investigadores; cada investigador deberá indicar su nombre, domicilio, cargo, titulación y obligaciones clínicas, hacer constar dónde se llevó a cabo el ensayo y reunir la información relativa a cada uno de los pacientes, incluyendo los impresos de recogida de datos de cada sujeto; el informe final, firmado por el investigador y para ensayos multicéntricos por todos los investigadores o por el investigador responsable de la coordinación. e) Los anteriores datos sobre los ensayos clínicos se remitirán a las autoridades competentes. No obstante, el solicitante podrá omitir parte de esta información con el consentimiento de dichas autoridades. A petición de éstas, deberá enviar sin demora la documentación completa. El investigador deberá pronunciarse, en sus conclusiones de la experimentación, sobre la seguridad del producto en las condiciones normales de utilización, su tolerancia y su eficacia, aportando todas las precisiones que resulten útiles sobre las indicaciones y contraindicaciones, la posología y la duración media del tratamiento, así como, en caso necesario, las precauciones particulares de uso y los signos clínicos de sobre dosificación. Cuando informe sobre los resultados de un estudio multicéntrico, el investigador principal deberá expresar, en sus conclusiones, su opinión sobre la seguridad y eficacia del medicamento que es objeto del estudio en nombre de todos los centros. f) Se resumirán las observaciones clínicas de cada ensayo, indicando: 1) El número de los sujetos tratados, distribuidos por sexo; 2) la selección y la distribución por edad de los grupos de pacientes que son objeto de investigación y las pruebas comparativas; 3) el número de pacientes que hayan sido retirados prematuramente de los ensayos, así como los motivos para ello; 4) en caso de que se hayan llevado a cabo ensayos controlados según lo dispuesto anteriormente, si el grupo control: No ha sido sometido a tratamiento, ha recibido un placebo, ha recibido otro medicamento de efecto conocido, ha recibido un tratamiento no medicamentoso; 5) la frecuencia de las reacciones adversas observadas; 6) todas las precisiones sobre los pacientes que presenten una especial sensibilidad (ancianos, niños, mujeres embarazadas o en período de menstruación) o cuyo estado fisiológico o patológico exija una especial consideración; 7) parámetros o criterios para evaluar la eficacia, así como los resultados referentes a estos parámetros; 8) una evaluación estadística de los resultados, en la medida en que se requiera por el diseño de los ensayos y las variables implicadas. g) Además, el investigador deberá en todo caso señalar sus observaciones sobre: 1) Todo indicio de habituación, adicción o dificultad en pacientes que dejan de tomar el medicamento; 2) las interacciones observadas con otros medicamentos que se administren simultáneamente; 3) los criterios con arreglo a los cuales se excluyó a determinados pacientes de los ensayos; 4) toda muerte que se haya producido durante el ensayo o durante el período de seguimiento. h) Los datos relativos a una nueva combinación de sustancias medicamentosas deberán ser idénticos a los que se exigen en el caso de medicamentos nuevos, y deberán justificar la seguridad y la eficacia de la combinación. i) Será obligatorio justificar la ausencia parcial o total de datos. Si se producen resultados imprevistos a lo largo de los ensayos, deberán realizarse y documentarse ensayos preclínicos, toxicológicos y farmacológicos adicionales. – 223 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano Habrá que suministrar datos sobre toda modificación de la acción farmacológica tras una administración reiterada, así como sobre la determinación de una dosificación a largo plazo. 5.2.1 Informes de estudios biofarmacéuticos.–Deberán presentarse informes de estudios de biodisponibilidad, biodisponibilidad comparativa, bioequivalencia, correlación in vitro-in vivo y métodos bioanalíticos y analíticos. Además, deberá evaluarse la biodisponibilidad cuando sea necesario para demostrar la bioequivalencia de los medicamentos a los que se refiere los artículos 7, 8 y 9. 5.2.2 Informes de estudios sobre farmacocinética mediante biomateriales humanos.–A efectos del presente anexo, se entenderá por biomateriales humanos todas las proteínas, células, tejidos y materiales conexos de origen humano que se utilizan in vivo o ex vivo para evaluar las propiedades farmacocinéticas de las sustancias medicamentosas. A este respecto, se entregarán informes de estudios de fijación con proteínas del plasma, estudios de metabolismo hepático e interacción de sustancias activas y estudios que utilicen otros biomateriales humanos. 5.2.3 Informes de estudios de farmacocinética humana. a) Se describirán las siguientes características farmacocinéticas: Absorción (velocidad y magnitud), distribución, metabolismo, excreción. Deberán describirse los aspectos significativos desde el punto de vista clínico, incluyendo la implicación de los datos cinéticos para el régimen de dosificación, especialmente para los pacientes de riesgo, y las diferencias entre el hombre y las especies animales utilizadas en los estudios preclínicos. Además de los estudios normales de farmacocinética de muestras múltiples, los análisis farmacocinéticos de la población basados en un muestreo disperso durante los estudios clínicos también pueden abordar las cuestiones relativas a la contribución de los factores intrínsecos y extrínsecos a la variabilidad de la relación dosis-respuesta farmacocinética. Se entregarán informes de estudios de farmacocinética y tolerancia inicial en sujetos sanos y en pacientes, informes de estudios farmacocinéticos destinados a evaluar la repercusión de los factores intrínsecos y extrínsecos e informes de estudios farmacocinéticos de la población. b) Cuando el medicamento vaya a administrarse, de forma habitual, simultáneamente con otros medicamentos, deberán proporcionarse datos sobre las pruebas de administración conjunta realizadas para demostrar posibles modificaciones de la acción farmacológica. Se investigarán las interacciones farmacocinéticas entre los principios activos y otros medicamentos o sustancias. 5.2.4 Informes de estudios de farmacodinámica humana. a) Deberá demostrarse la acción farmacodinámica correlacionada con la eficacia, incluyendo: La relación dosis-respuesta y su curso temporal, la justificación de la posología y las condiciones de administración, cuando sea posible, el modo de acción. Se describirá la acción farmacodinámica no relacionada con la eficacia. La demostración de efectos farmacodinámicos en seres humanos no bastará por sí misma para establecer conclusiones en cuanto a un posible efecto terapéutico. b) Cuando el medicamento vaya a administrarse, de forma habitual, simultáneamente con otros medicamentos, deberán proporcionarse datos sobre las pruebas de administración conjunta realizadas para demostrar posibles modificaciones de la acción farmacológica. Se investigarán las interacciones farmacodinámicas entre los principios activos y otros medicamentos o sustancias. 5.2.5 Informes de estudios sobre eficacia y seguridad. 5.2.5.1 Informes de estudios clínicos controlados relativos a la indicación declarada.–En general, los ensayos clínicos se efectuarán en forma de ensayos clínicos controlados siempre que sea posible, aleatorizados y, según convenga, en comparación con un placebo y un medicamento conocido, cuyo valor terapéutico esté bien establecido; cualquier otro – 224 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano diseño deberá justificarse. El tratamiento asignado al grupo control variará según los casos y dependerá también de consideraciones éticas y del ámbito terapéutico. En este sentido, en ocasiones puede resultar más conveniente comparar la eficacia de un nuevo medicamento con el efecto de un medicamento conocido, cuyo valor terapéutico esté bien establecido, y no con el efecto de un placebo. 1. En la medida de lo posible, y muy especialmente en ensayos en los que el efecto del producto no pueda medirse objetivamente, se tomarán medidas para evitar un sesgo, incluyendo métodos de aleatorización y métodos ciegos (de doble ciego). 2. El protocolo del ensayo deberá incluir una descripción pormenorizada de los métodos estadísticos a los que se recurra, del número de pacientes y las razones por las que se incluyen (con el cálculo del valor estadístico del ensayo), el nivel de significación que se use y una descripción de la unidad estadística. Deben documentarse las medidas que se adopten para evitar el sesgo, en particular los métodos de aleatorización. La inclusión de un gran número de pacientes a lo largo de un ensayo no deberá considerarse en ningún caso el sustituto válido de un ensayo controlado bien ejecutado. Los datos sobre seguridad deberán examinarse teniendo en cuenta las directrices publicadas por la Comisión, prestando especial atención a hechos que den como resultado la alteración de la dosis o la necesidad de medicación concomitante, hechos nocivos graves, hechos que provoquen la retirada y fallecimientos. Deberán determinarse todos los pacientes o grupos que corren mayor riesgo y se prestará especial atención a los pacientes potencialmente vulnerables que puedan resultar poco numerosos, por ejemplo, niños, embarazadas, personas de edad avanzada delicadas, personas con fuertes anomalías de metabolismo o excreción, etc. Se describirá la repercusión de la evaluación de la seguridad para los posibles empleos del medicamento. 5.2.5.2 Informes de estudios clínicos no controlados, informes de análisis de datos obtenidos en diversos estudios y otros informes de estudios clínicos.–Deberán facilitarse todos estos informes. 5.2.6 Informes de experiencia posterior a la comercialización.–Si el medicamento ya está autorizado en terceros países, deberá proporcionarse información sobre reacciones adversas al medicamento en cuestión y a medicamentos que contengan los mismos principios activos, a ser posible en relación con la tasa de utilización. 5.2.7 Cuadernos de recogida de datos y listados de pacientes.–Al presentar los cuadernos de recogida de datos y las listas de pacientes con arreglo a las directrices pertinentes publicadas por la Agencia Europea de Medicamentos, deberán facilitarse y presentarse en el mismo orden que los informes de estudios clínicos e indexarse por estudio. PARTE II. EXPEDIENTES DE AUTORIZACIÓN DE COMERCIALIZACIÓN Y REQUISITOS ESPECÍFICOS Algunos medicamentos presentan características específicas que hacen necesaria la adaptación de todos los requisitos del expediente de solicitud de autorización de comercialización que se establecen en la parte I del presente anexo. Con el fin de tener en cuenta estas situaciones especiales, los solicitantes utilizarán una presentación adaptada y adecuada del expediente. 1. Solicitudes bibliográficas.–Se aplicarán las normas específicas que se exponen a continuación a los medicamentos cuyo(s) principio(s) activo(s) tengan, tal como se menciona en el artículo 10 un «uso medicinal claramente establecido» (o suficientemente comprobado), con una eficacia reconocida y un nivel aceptable de seguridad. El solicitante deberá presentar los módulos 1, 2 y 3 tal como se describen en la parte I del presente anexo. Para los módulos 4 y 5, deberán abordarse las características clínicas y no clínicas mediante una bibliografía científica detallada. Las siguientes normas específicas serán de aplicación para demostrar la existencia de un uso médico suficientemente comprobado: a) Los factores que han de tenerse en cuenta para determinar un uso médico suficientemente comprobado de los componentes del medicamento son los siguientes: El período durante el que se ha utilizado una sustancia, – 225 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano los aspectos cuantitativos del empleo de la misma, el grado de interés científico de su utilización (que se refleja en la bibliografía científica publicada), la coherencia de las evaluaciones científicas. Por tanto, pueden ser necesarios períodos de tiempo diferentes a fin de establecer el uso médico suficientemente comprobado de las diferentes sustancias. En todo caso, el período de tiempo necesario para establecer que un componente de un medicamento tiene un uso medicinal suficientemente comprobado no podrá ser inferior a diez años, contados a partir de la primera utilización sistemática y documentada de esa sustancia como medicamento dentro de la Unión Europea. b) La documentación presentada por el solicitante deberá cubrir todos los aspectos de la evaluación de la seguridad y/o de la eficacia e incluir o hacer referencia a un estudio bibliográfico pertinente, que tenga en cuenta los estudios previos y posteriores a la comercialización y la literatura científica publicada relativa a la experimentación en forma de estudios epidemiológicos y, en particular, de estudios epidemiológicos comparativos. Deberán comunicarse todos los documentos existentes, tanto favorables como desfavorables. Respecto a las disposiciones sobre «uso médico suficientemente comprobado», es particularmente necesario aclarar que la «referencia bibliográfica» a otras pruebas (estudios posteriores a la comercialización, estudios epidemiológicos, etc.), y no sólo los datos relacionados con ensayos, puede servir como prueba válida de la seguridad y eficacia de un medicamento si una solicitud explica y justifica satisfactoriamente la utilización de estas fuentes de información. c) Se prestará atención particular a cualquier información omitida y se justificará porqué puede afirmarse la existencia de un nivel aceptable de seguridad y/o eficacia, pese a la ausencia de determinados estudios. d) En la visión general de las partes no clínicas y/o clínicas deberá explicarse la relevancia de todos los datos presentados relativos a un producto diferente de aquel que será comercializado. Se deberá valorar si el producto examinado puede considerarse similar al producto cuya autorización de comercialización se ha solicitado a pesar de las diferencias existentes. e) La experiencia posterior a la comercialización de otros productos que contengan los mismos componentes revestirá particular importancia y los solicitantes deberán insistir especialmente en este aspecto. 2. Medicamentos esencialmente similares. a) Las solicitudes basadas en el del artículo 7 deberán contener los datos descritos en los módulos 1, 2 y 3 de la parte I del presente anexo, siempre que el solicitante haya obtenido el consentimiento del titular de la autorización de comercialización original para hacer referencia cruzada al contenido de sus módulos 4 y 5. b) Las solicitudes basadas en el artículo 7 incluirán los datos descritos en los módulos 1, 2 y 3 de la parte I del presente anexo, junto con los datos que demuestren la biodisponibilidad y la bioequivalencia con el medicamento original, siempre que éste no sea un medicamento biológico (véase el punto 4 de la parte II, medicamentos biológicos similares). Los resúmenes visiones generales no clínicas/clínicas de dichos productos se centrarán especialmente en los siguientes elementos: Los motivos por los que se reclama la similaridad esencial; un resumen de las impurezas presentes en lotes del (los) principio(s) activo(s) así como las del producto terminado (y, cuando proceda, los productos de descomposición que se forman durante el almacenamiento) tal como se propone para ser comercializador, acompañado de una evaluación de dichas impurezas; una evaluación de los estudios de bioequivalencia o una justificación por no haber realizado los estudios siguiendo las directrices sobre la «Investigación de labiodisponibilidad y bioequivalencia»; una actualización de la bibliografía publicada sobre la sustancia y la presente solicitud; se aceptarán las anotaciones con este fin de artículos de publicaciones especializadas; – 226 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano cada afirmación que figure en el resumen de las características del producto no conocida o deducida a partir de las propiedades del medicamento y/o su grupo terapéutico deberá discutirse en los resúmenes/visiones generales de las partes no clínicas/clínicas y justificarse mediante la bibliografía publicada o estudios suplementarios; si procede, el solicitante deberá aportar datos adicionales para probar la equivalencia de las propiedades de seguridad y eficacia de las diferentes sales, ésteres o derivados de un principio activo autorizado, en caso de que reclame la similaridad esencial. 3. Datos suplementarios exigidos en situaciones específicas.–Cuando el principio activo de un medicamento esencialmente similar contenga la misma fracción terapéutica que el medicamento autorizado original, asociada a un complejo/derivado de sales/ésteres diferentes, habrá de probarse que no se produce alteración alguna de la farmacocinética de la fracción, la farmacodinamia y/o la toxicidad que pueda modificar su perfil de seguridad/ eficacia. De lo contrario, se considerará que tal asociación constituye un nuevo principio activo. En los casos en que el medicamento esté destinado a un uso terapéutico diferente o se presente en una forma farmacéutica distinta o deba administrarse por vías diferentes o con dosificación diferente, deberán suministrarse los resultados de las pruebas toxicológicas y farmacológicas apropiadas y/o de los ensayos clínicos. 4. Medicamentos biológicos similares.–En el caso de los medicamentos biológicos lo dispuesto en el artículo 9 puede no ser suficiente. Si la información exigida en el caso de medicamentos genéricos no permite la demostración de la naturaleza análoga de dos medicamentos biológicos, se deberán facilitar datos suplementarios, en particular el perfil toxicológico y clínico. Cuando un solicitante independiente solicite, una vez concluido el período de protección de datos, una autorización de comercialización de un medicamento biológico definido en el punto 3.2 de la parte I del presente anexo que se relacione con un medicamento original que haya obtenido la autorización de comercialización en la Unión Europea, se aplicará el enfoque que se expone a continuación. La información que habrá de facilitarse no se limitará a los módulos 1, 2 y 3 (datos farmacéuticos, químicos y biológicos), complementada con los datos sobre bioequivalencia y biodisponibilidad. El tipo y la cantidad de datos suplementarios (esto es, datos toxicológicos, datos no clínicos y datos clínicos pertinentes) se determinará en cada caso, conforme a todas las directrices científicas pertinentes. Debido a la diversidad de medicamentos biológicos, la autoridad competente determinará si es necesario exigir los estudios identificados previstos en los módulos 4 y 5, teniendo en cuenta las características especiales de cada medicamento. Los principios generales que han de aplicarse se recogen en las directrices publicadas por la Agencia Europea de Medicamentos, en las que se tienen en cuenta las características de los medicamentos biológicos en cuestión. En caso de que el medicamento autorizado originalmente tenga más de una indicación, deberán justificarse la eficacia y la seguridad del medicamento que se afirma es similar o, si es necesario, deberán demostrarse por separado respecto a cada una de las indicaciones declaradas. 5. Asociaciones de principios activos autorizados.–Las solicitudes basadas en el artículo 11, se referirán a medicamentos nuevos compuestos por dos principios activos como mínimo que no han sido autorizadas anteriormente como asociaciones a dosis fijas. En el caso de esas solicitudes se presentará un expediente completo (módulos 1 a 5) para la asociación a dosis fija. Si procede, se facilitará información relativa a los lugares de fabricación y la evaluación de la seguridad de los agentes extraños/externos. 6. Documentación para las solicitudes de autorización en condiciones especiales.– Cuando, como se establece en el artículo 26, el solicitante pueda demostrar que no puede suministrar datos completos sobre la eficacia y seguridad en las condiciones normales de uso del producto, por alguna de las razones siguientes: Los casos para los que están indicados los productos en cuestión se presentan tan raramente que el solicitante no puede razonablemente estar obligado a proporcionar las evidencias detalladas; – 227 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano el estado actual de desarrollo de la ciencia no permite proporcionar información completa; principios de deontología médica comúnmente admitidos prohíben recoger esta información, podrá concederse la autorización de comercialización en función de determinadas obligaciones específicas. Entre dichas obligaciones podrán figurar las siguientes: El solicitante concluirá, dentro de un plazo especificado por la autoridad competente, un programa de estudios determinado cuyos resultados constituirán la base de una nueva evaluación de la relación riesgo/beneficio; la especialidad de que se trate sólo se expedirá con receta médica y, en caso necesario, sólo se autorizará su administración si se efectúa bajo estricto control médico, a ser posible en un centro hospitalario y, cuando se trate de un radiofármaco, por parte de una persona autorizada; el prospecto y cualquier otra información médica destacará que, en relación con determinados aspectos, no existen aún datos fiables sobre el medicamento en cuestión. 7. Solicitudes mixtas de autorización de comercialización.–Por solicitudes mixtas de autorización de comercialización se entenderán los expedientes de solicitud en los que los módulos 4 y/o 5 constan de una combinación de informes de estudios limitados no clínicos y/o clínicos realizados por el solicitante y de referencias bibliográficas. Todos los demás módulos se ajustarán a la estructura descrita en la parte I del presente anexo. La autoridad competente deberá aceptar el formato propuesto que presente el solicitante considerando individualmente cada caso. PARTE III. MEDICAMENTOS ESPECIALES En esta parte se establecen los requisitos relacionados con la naturaleza de determinados medicamentos. 1. Medicamentos biológicos. 1.1 Medicamentos hemoderivados.–Respecto a los medicamentos derivados de sangre humana o plasma y no obstante lo dispuesto en el módulo 3, los requisitos de los expedientes mencionados en «información sobre los materiales de partida y materias primas» referentes a los materiales de partida derivados de sangre o plasma humanos podrán ser sustituidos por un Archivo Principal sobre Plasma Certificado con arreglo a lo expuesto en la presente parte. a) Principios: A efectos del presente anexo: Se entenderá por «Archivo Principal sobre Plasma (PMF)» aquella documentación independiente e independiente del expediente de autorización de comercialización que contenga toda la información pormenorizada pertinente sobre las características de todo el plasma humano empleado como material de partida y/o materia prima para la fabricación de subfracciones o fracciones, que forman parte de los medicamentos o productos sanitarios mencionados en el Real Decreto 710/2002, de 19 de julio, por el que se modifica el Real Decreto 414/1996, de 1 de marzo, por el que se regulan los productos sanitarios, en lo referente a los que incorporen derivados estables de la sangre o plasma humanos, como excipiente y principio(s) activo(s). Cada centro o establecimiento de fraccionamiento/procesamiento de plasma humano deberá preparar y mantener al día el conjunto de información pormenorizada pertinente a la que se hace referencia en el archivo principal sobre plasma. El solicitante de una autorización de comercialización o el titular de la autorización de comercialización presentará el archivo principal sobre plasma a la Agencia Europea de Medicamentos o a la autoridad competente. En caso de que el solicitante de una autorización de comercialización o el titular de la misma no sean el titular del archivo principal sobre plasma, este archivo deberá ponerse a disposición del solicitante o del titular de la autorización de comercialización para su presentación a la autoridad competente. En cualquier caso, el solicitante o titular de la autorización de comercialización asumirá la responsabilidad del medicamento. – 228 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano La autoridad competente que evalúa la autorización de comercialización esperará a que la Agencia Europea de Medicamentos expida el certificado antes de tomar una decisión sobre la solicitud. Todos los expedientes de autorización de comercialización que contengan algún componente derivado de plasma humano deberán remitir al archivo principal sobre plasma correspondiente al plasma utilizado como material de partida o materia prima. b) Contenido: Con arreglo a lo dispuesto en el Real Decreto 1088/2005, de 16 de septiembre, por el que se establecen los requisitos técnicos y condiciones mínimas de la hemodonación y de los centros y servicios de transfusión, referente a los requisitos que deben reunir los donantes y a la verificación de las donaciones, el archivo principal sobre plasma incluirá información sobre el plasma utilizado como material de partida o materia prima, en concreto: 1. Origen del plasma. a) Información acerca de los centros o establecimientos en los que se recoja la sangre o plasma, incluidas la inspección y aprobación, tal como se recoge en el artículo 45 del Real decreto 1088/2005, y datos epidemiológicos sobre infecciones transmisibles por la sangre. b) Centros o establecimientos de información en los que se realizan análisis de las donaciones, incluida la categoría de la inspección y aprobación. c) Criterios de selección/exclusión de los donantes de sangre y plasma. d) Sistema implantado que permite la trazabilidad de cada donación desde el establecimiento de recogida de sangre y plasma hasta los productos terminados y viceversa. 2. Calidad y seguridad del plasma. a) Cumplimiento de las monografías de la Farmacopea europea. b) Realización de análisis de las donaciones en los centros de Transfusión para detectar agentes infecciosos, incluida la información sobre los métodos de análisis y, en el caso de los bancos de plasma, datos de validación acerca de los métodos de análisis empleados. c) Características técnicas de las bolsas de recogida de sangre y plasma, incluidos los datos sobre las soluciones anticoagulantes empleadas. d) Condiciones de almacenamiento y transporte de plasma. e) Procedimientos para el mantenimiento de inventarios y/o períodos de cuarentena. f) Caracterización de la mezcla de plasma original. 3. Sistema en funcionamiento entre el fabricante de medicamentos derivados de plasma y/o la entidad que se ocupa del fraccionamiento o tratamiento del plasma, por un parte, y los centros de transfusión sanguínea, por otra, que define las condiciones de su interacción y las especificaciones acordadas entre ellos. Además, en el archivo principal sobre plasma se ofrecerá una lista de los medicamentos para los que es válido el archivo, tanto los medicamentos que han obtenido una autorización de comercialización como los que están en vías de obtenerla, incluidos los medicamentos mencionados en el artículo 2 del Real Decreto 223/2004, de 6 de febrero. c) Evaluación y certificación: En el caso de los medicamentos aún no autorizados, el solicitante de la autorización de comercialización presentará un expediente completo a la autoridad competente, que deberá ir acompañado por un archivo principal sobre plasma aparte en caso de que éste no exista ya. El archivo principal sobre plasma estará sujeto a una evaluación científica y técnica que realiza la Agencia Europea de Medicamentos. La evaluación positiva supondrá la expedición de un certificado de cumplimiento de la legislación comunitaria relativo al archivo principal sobre plasma, que irá acompañado del informe de evaluación. El certificado que se expedirá será válido en toda la Unión Europea. El archivo principal sobre plasma se actualizará y certificará de nuevo anualmente. Las modificaciones introducidas posteriormente en la formulación del archivo principal sobre plasma deberán seguir el procedimiento de evaluación establecido en el Reglamento (CE) n.º 542/95, relativo al examen de las modificaciones de los términos de las autorizaciones de comercialización pertenecientes al ámbito de aplicación del Reglamento (CEE) n.º 2309/93 del Consejo, de 22 de julio de 1993, por el que se establecen procedimientos comunitarios para la autorización y supervisión de medicamentos de uso – 229 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano humano y veterinario y por el que se crea la Agencia Europea de Medicamentos. Las condiciones para la evaluación de dichas modificaciones se establecen en el Reglamento (CE) 1085/2003. En un segundo paso, tras lo dispuesto en los guiones primero, segundo, tercero y cuarto, la autoridad competente que conceda o que haya concedido la autorización de comercialización del medicamento tendrá en cuenta la certificación, la recertificación o las modificaciones del archivo principal sobre plasma sobre los medicamentos de que se trate. No obstante lo dispuesto en el segundo guión del presente punto (evaluación y certificación), en caso de que un archivo principal sobre plasma corresponda únicamente a medicamentos derivados de sangre o plasma cuya autorización de comercialización se limite a un solo Estado miembro, la evaluación científica y técnica de dicho archivo principal sobre plasma deberá realizarla la autoridad nacional competente de ese Estado miembro. 1.2 Vacunas.–Respecto a las vacunas de uso humano y no obstante lo dispuesto en el módulo 3 sobre «principio(s) activo(s)», serán de aplicación los siguientes requisitos, cuando se basen en la utilización de un sistema de archivo principal sobre antígenos de la vacuna. El expediente de solicitud de autorización de comercialización de toda vacuna distinta de la de la gripe humana deberá incluir un archivo principal sobre cada antígeno de la vacuna que constituya un principio activo de la misma. a) Principios: A efectos del presente anexo: Se entenderá por «archivo principal sobre un antígeno de vacuna (VaMF)» una parte independiente del expediente de solicitud de autorización de comercialización de una vacuna que contendrá toda la información pertinente de naturaleza biológica, farmacéutica y química relativa a cada uno de los principios activos que forman parte del medicamento. La parte independiente podrá ser común a una o varias vacunas monovalentes y/o combinadas que presente el mismo solicitante o titular de autorización de comercialización. Cada vacuna puede contener uno o varios antígenos distintos. Cada vacuna contiene tantos principios activos como antígenos. Una vacuna combinada contiene como mínimo dos antígenos distintos destinados a la prevención de una o varias enfermedades infecciosas. Una vacuna monovalente contiene un solo antígeno destinado a la prevención de una sola enfermedad infecciosa. b) Contenido: El archivo principal sobre antígeno de vacuna contendrá la información siguiente extraída de la parte correspondiente (sustancia activa) del módulo 3 sobre «calidad de los datos», tal como se define en la parte I del presente anexo: Principio activo. 1. Información general, incluido el seguimiento de las monografías pertinentes de la Farmacopea Europea. 2. Información sobre la fabricación del principio activo: ha de abarcar el proceso de fabricación, la información sobre los materiales de partida y las materias primas, las medidas específicas sobre evaluación de la seguridad respecto a las EET y los agentes extraños/ externos e instalaciones y equipo. 3. Caracterización del principio activo. 4. Control de calidad del principio activo. 5. Estándares y materiales de referencia. 6. Envase y sistema de cierre del principio activo. 7. Estabilidad del principio activo. c) Evaluación y certificación: En el caso de las nuevas vacunas que contengan un nuevo antígeno, el solicitante presentará a una autoridad competente un expediente completo de solicitud de autorización de comercialización que incluya todos los archivos principales sobre antígeno de vacuna correspondientes a cada uno de los antígenos que forman parte de la nueva vacuna, en el caso de que no exista ya ningún archivo principal de cada antígeno. La Agencia Europea de Medicamentos realizará la evaluación científica y técnica del mencionado archivo principal sobre antígeno de vacuna. La evaluación positiva de un medicamento supondrá la expedición de un certificado de cumplimiento de la legislación comunitaria relativo a cada archivo principal sobre antígeno de vacuna, que irá acompañado – 230 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano del informe de evaluación. El certificado que se expedirá tendrá validez en toda la Unión Europea. Lo dispuesto en el primer guión será aplicable a cada vacuna consistente en una nueva combinación de antígenos de vacuna, independientemente de que alguno de dichos antígenos pueda formar parte de vacunas ya autorizadas en la Unión Europea. Las modificaciones del contenido de un archivo principal sobre antígeno de vacuna correspondiente a una vacuna autorizada en la Unión Europea estarán sujetas a una evaluación científica y técnica que realizará la Agencia Europea de Medicamentos con arreglo al procedimiento establecido en el Reglamento (CE) núm. 1085/2003. En caso de evaluación positiva, la Agencia Europea de Medicamentos expedirá un certificado de cumplimiento de la legislación comunitaria del archivo principal sobre el antígeno de vacuna. El certificado que se expedirá tendrá validez en toda la Unión Europea. No obstante lo dispuesto en el primero, segundo y tercer guiones del presente apartado (evaluación y certificación), en caso de que un archivo principal sobre antígeno de vacuna corresponda únicamente a una vacuna que ha sido objeto de una autorización de comercialización que no se ha concedido (o que no se concederá) según un procedimiento comunitario y siempre que la vacuna autorizada incluya antígenos de vacuna no evaluados mediante un procedimiento comunitario, la autoridad nacional competente que concedió la autorización de comercialización realizará la evaluación científica y técnica del mencionado archivo principal sobre antígeno de vacuna y sus posteriores modificaciones. En un segundo paso, tras lo dispuesto en los guiones primero, segundo, tercero y cuarto, la autoridad competente que conceda o que haya concedido la autorización de comercialización tendrá en cuenta la certificación, la recertificación o las modificaciones del archivo principal sobre antígeno de vacuna relativa a los medicamentos de que se trate. 2. Radiofármacos y precursores. 2.1. Radiofármacos.–A efectos del presente apartado, deberá presentarse un expediente completo en el que se incluirán los siguientes datos específicos: Módulo 3. a) Cuando se trate de equipos reactivos radiofarmacéuticos que deban ser marcados radiactivamente tras el suministro por el fabricante, se considerará que el principio activo es aquella parte de la formulación cuyo propósito es transportar o unirse al radionucleido. La descripción del método de fabricación de equipos reactivos radiofarmacéuticos incluirá datos sobre la propia fabricación del equipo y datos sobre el tratamiento final recomendado para producir el radiofármaco. Las especificaciones necesarias del radionucleido se describirán, si es pertinente, con arreglo a la monografía general o las monografías específicas de la Farmacopea Europea. Además, se describirá cualquier compuesto esencial para el marcaje radiactivo. También se describirá la estructura del compuesto marcado radiactivamente. En cuanto a los radionucleidos, se discutirán las reacciones nucleares que producen. En un generador, tanto los radionucleidos padre como hijo se considerarán principios activos. b) Se ofrecerán datos sobre la naturaleza del radionucleido, la identidad del isótopo, las impurezas probables, el portador, el uso y la actividad específica. c) Las materias diana para la irradiación se incluyen entre los materiales de partida. d) Se incluirán consideraciones acerca de la pureza química/radioquímica y su relación con la biodistribución. e) Se describirá la pureza radionucleídica, la pureza radioquímica y la actividad específica. f) Para los generadores se requiere información detallada sobre las pruebas de los radionucleidos padre e hijo. En el caso de los eluidos de un generador deben realizarse pruebas del radionucleido padre y de los demás componentes del generador. g) El requisito de expresar el contenido de principio activo en función de la masa de las fracciones activas sólo se aplicará a los equipos reactivos radiofarmacéuticos. Cuando se trate de radionucleidos la radiactividad se expresará en bequerelios, fijando una fecha y, si fuera necesario, una hora determinada, haciendo referencia al huso horario. Deberá indicarse el tipo de radiación emitida. – 231 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano h) En el caso de equipos reactivos, las especificaciones del producto terminado incluirán pruebas de la eficacia de los produc tos tras el marcaje radiactivo. Deberán incluirse controles apropiados de pureza radioquímica y radionucleídica del producto marcado radiactivamente. Se identificarán y controlarán todos los materiales esenciales para el marcaje radiactivo. i) Se ofrecerá información sobre la estabilidad en el caso de los generadores de radionucleidos, los equipos reactivos combinados con radionucleidos y los productos marcados radiactivamente. La estabilidad en uso de los radiofármacos en viales multidosis deberá documentarse. Módulo 4.–Se estima que la toxicidad puede ir asociada a la dosis de radiación. Cuando se utilizan los radiofármacos con fines diagnósticos, ésta es consecuencia del uso de los mismos; cuando se utilizan con fines terapéuticos, es la propiedad deseada. Por tanto, la evaluación de la seguridad y eficacia de los radiofármacos tendrá en cuenta los requisitos exigidos para los medicamentos en general y la dosimetría de la radiación. Deberá documentarse la exposición a las radiaciones de órganos y tejidos. Las estimaciones de las dosis de radiación absorbida se calcularán con arreglo a un sistema especificado reconocido internacionalmente para una determinada vía de administración. Módulo 5.–Cuando proceda, se facilitarán los resultados de los ensayos clínicos; si no se hace, deberá justificarse en el informe de experto de la documentación clínica. 2.2 Precursores radiofarmacéuticos para marcaje radiactivo.–En el caso específico de un precursor radiofarmacéutico que tenga únicamente por objeto el marcaje radiactivo, el objetivo primario será presentar información acerca de las posibles consecuencias de una escasa eficiencia del marcaje radiactivo o de la disociación «in vivo» del conjugado marcado radiactivamente, es decir, los aspectos relacionados con los efectos del radionucleido libre sobre el paciente. Por otra parte, también es necesario presentar toda la información pertinente en relación con los riesgos profesionales, como la exposición a la radiación de los trabajadores profesionalmente expuestos y del entorno. En concreto, se facilitará cuando proceda la información que se especifica a continuación: Módulo 3.–Lo dispuesto en el módulo 3, definido en las letras a) a i), se aplicará, cuando proceda, al registro de los precursores radiofarmacéuticos. Módulo 4.–En lo que respecta a la toxicidad por dosis única y por administración continuada, deberán facilitarse los resultados de los estudios realizados de conformidad con los principios de las buenas prácticas de laboratorio que se establecen en el Real Decreto 1369/2000, de 19 de julio, por el que se modifica el Real Decreto 822/1993, de 28 de mayo, que establece los principios de buenas prácticas de laboratorios y su aplicación en la realización de estudios no clínicos sobre sustancias y productos químicos, y en el Real Decreto 2043/1994, de 14 de octubre, sobre inspección y verificación de buenas prácticas de laboratorio, salvo justificación de lo contrario. Los estudios de mutagenicidad del radionucleido no se consideran útiles en este caso concreto. Deberá presentarse información relacionada con la toxicidad química y la distribución del nucleido no radiactivo correspondiente. Módulo 5.–La información clínica obtenida a partir de estudios clínicos utilizando el precursor no se considera adecuada en el caso específico de un precursor radiofarmacéutico que tenga únicamente por objeto el marcado radiactivo. No obstante, se aportará información que demuestre la utilidad clínica del precursor radiofarmacéutico cuando esté ligado a moléculas portadoras apropiadas. 3. Medicamentos homeopáticos.–En este apartado se exponen las disposiciones específicas sobre la aplicación de los módulos 3 y 4 a los medicamentos homeopáticos definidos en el artículo 2.31. Módulo 3.–Lo dispuesto en el módulo 3 se aplicará a los documentos presentados para las solicitudes de autorización de comercialización de medicamentos homeopáticos definidos en el artículo 55, con las modificaciones que se exponen a continuación: – 232 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano a) Terminología.–La denominación latina de la cepa homeopática descrita en el expediente de solicitud de autorización de comercialización deberá ser acorde con la denominación latina de la Farmacopea Europea o, en su ausencia, con la de una farmacopea oficial de un Estado Miembro. Se incluirá, cuando resulte pertinente, el nombre o nombres tradicionales usados en cada Estado Miembro. b) Control de los materiales de partida.–Los detalles y documentos sobre los materiales de partida, es decir, todos los materiales utilizados incluidas las materias primas e intermedias hasta la dilución final que haya de incorporarse al producto terminado, que se adjunten a la solicitud, se complementarán con datos adicionales sobre la cepa homeopática. Los requisitos generales de calidad se aplicarán a todos los materiales de partida y materias primas, así como a los pasos intermedios del proceso de fabricación hasta la dilución final que será incorporada al producto terminado. Si es posible, se requerirá una determinación cuantitativa si hay presencia de componentes tóxicos y si, debido al elevado grado de dilución, la calidad no puede ser controlada en la dilución final que será incorporada. Se describirá minuciosamente cada paso del proceso de fabricación desde los materiales de partida hasta la dilución final que será incorporada al producto terminado. Las diluciones deberán realizarse de acuerdo con los métodos homeopáticos de fabricación establecidos en la monografía pertinente de la Farmacopea Europea o, en su defecto, en una farmacopea oficial de un Estado Miembro. c) Métodos de control del producto terminado.–Los requisitos generales de calidad serán aplicables a los medicamentos homeopáticos acabados; el solicitante deberá justificar debidamente cualquier excepción. Se efectuará la identificación y determinación cuantitativa de todos los componentes pertinentes desde un punto de vista toxicológico. En caso de que pueda justificarse que no es posible la identificación y/o la cuantificación de todos los componentes pertinentes desde un punto de vista toxicológico (por ejemplo, debido a su dilución en el producto terminado), la calidad deberá demostrarse mediante la validación completa de los procesos de fabricación y dilución. d) Pruebas de estabilidad.–Deberá demostrarse la estabilidad del producto terminado. Los datos de estabilidad de las cepas homeopáticas generalmente son transferibles a las diluciones/trituraciones obtenidas de las mismas. Si no es posible la identificación o determinación cuantitativa del principio activo debido al grado de dilución, podrán considerarse los datos de estabilidad de la forma farmacéutica. Módulo 4.–Las disposiciones del presente módulo serán aplicables al registro simplificado de los medicamentos homeopáticos mencionados en el artículo 55 b), con las especificaciones siguientes. Se justificará la ausencia de cualquier dato; por ejemplo, se justificará por qué puede afirmarse la existencia de un nivel aceptable de seguridad, pese a la ausencia de determinados estudios. 4. Medicamentos a base de plantas.–Las solicitudes relativas a medicamentos a base de plantas se presentarán con un expediente completo en el que figurarán los detalles específicos siguientes. Módulo 3.–Lo dispuesto en el módulo 3, incluido el seguimiento de las monografías pertinentes de la Farmacopea Europea, se aplicará a la autorización de medicamentos a base de plantas. Al presentar la solicitud se tendrá en cuenta el estado de los conocimientos científicos. Habrán de considerarse los siguientes aspectos específicos de los medicamentos a base de plantas. 1. Sustancias y preparados vegetales.–A efectos del presente anexo, el término «sustancias vegetales y preparados vegetales» se considerará equivalente al término «herbal drugs and herbal drug preparations», tal y como aparece definido en la Farmacopea Europea. Respecto a la nomenclatura de las sustancias vegetales, se incluirá la denominación científica binomial de la planta (género, especie, variedad y autor), así como su quimiotipo – 233 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano (cuando proceda), las partes de las plantas utilizadas, la definición de la sustancia vegetal, los otros nombres (sinónimos mencionados en las otras farmacopeas) y el código de laboratorio. Respecto a la nomenclatura del preparado vegetal, se incluirá la denominación científica binomial de la planta (género, especie, variedad y autor), así como su quimiotipo (cuando proceda), las partes de las plantas utilizadas, la definición del preparado vegetal, la proporción entre la sustancia vegetal y el preparado vegetal, el (los) disolvente(s) para extracción, otros nombres (sinónimos mencionados en otras farmacopeas) y el código de laboratorio. Para documentar el apartado de la estructura de la(s) sustancia(s) vegetal(es) y el (los) preparado(s) vegetal(es) cuando proceda, se incluirán la forma física, la descripción de los componentes con actividad terapéutica conocida o los marcadores (fórmula molecular, masa molecular relativa, fórmula estructural, incluidas la estereoquímica relativa y absoluta, la fórmula molecular y la masa molecular relativa), así como las de otros constituyentes. Con el fin de documentar el apartado sobre el fabricante de la sustancia vegetal, se incluirán, cuando proceda, el nombre, la dirección y la responsabilidad de cada proveedor, incluidos contratistas, y cada lugar o instalación propuestos para la producción/recogida y control de la sustancia vegetal. Con el fin de documentar el apartado sobre el fabricante del preparado vegetal, se incluirán, cuando proceda, el nombre, la dirección y la responsabilidad de cada fabricante, incluidos contratistas, y cada lugar de fabricación o instalación propuestos para la fabricación y ensayo del preparado vegetal. En relación con la descripción del proceso de fabricación y de los controles del proceso de la sustancia vegetal, se ofrecerá información para describir adecuadamente la producción y recogida de plantas, incluidas la procedencia geográfica de la planta medicinal y sus condiciones de cultivo, cosecha, secado y almacenamiento. En relación con la descripción del proceso de fabricación y de los controles del proceso del preparado vegetal, se ofrecerá información para describir adecuadamente el proceso de fabricación del preparado vegetal, incluida la descripción del tratamiento, los disolventes y reactivos, las fases de purificación y la estandarización. Por lo que se refiere al desarrollo del proceso de fabricación, se presentará cuando proceda un breve resumen en el que se describa el desarrollo de la(s) sustancia(s) vegetal(es) y el(los) preparado(s) vegetal(es), teniendo en cuenta la vía de administración y utilización propuestas. Deberán discutirse, cuando proceda, los resultados en que se compare la composición fitoquímica de las sustancias vegetales y preparados vegetal(es), según el caso, reseñado(s) en los datos bibliográficos de apoyo y las sustancias vegetales y preparados vegetales, según el caso, que contiene como sustancias activas el medicamento a base de plantas para el que se solicita la autorización. Respecto a la dilucidación de la estructura y otras características de la(s) sustancia(s) vegetal(es), se facilitará información sobre la caracterización botánica, macroscópica, microscópica y fitoquímica, así como sobre su actividad biológica si fuera necesario. Respecto a la dilucidación de la estructura y otras características de los preparados vegetales, se facilitará información sobre la caracterización fitoquímica y físicoquímica, así como sobre su actividad biológica si fuera necesario. Se presentarán cuando proceda las especificaciones de la(s) sustancia(s) vegetal(es) y de los preparado(s) vegetales. También se informará si procede acerca de los procedimientos analíticos empleados para controlar la(s) sustancia(s) vegetal(es) y los preparado(s) vegetal(es). Por lo que se refiere a la validación de los procedimientos analíticos, cuando proceda, se ofrecerá información sobre validación analítica, incluyendo los datos experimentales de los procedimientos analíticos empleados para controlar la(s) sustancia(s) vegetal(es) y los preparado(s) vegetal(es). En relación con el análisis de los lotes, se describirán si procede los lotes y los resultados de los análisis de los mismos en relación con la(s) sustancia(s) vegetal(es) y los preparado(s) vegetal(es), incluyendo los de las sustancias de farmacopea. Habrán de justificarse, cuando sea pertinente, las especificaciones de la(s) sustancia(s) vegetal(es) y de los preparado(s) vegetal(es). – 234 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano Asimismo se informará, en su caso, sobre las normas y materiales de referencia empleados para probar la(s) sustancia(s) vegetal(es) y los preparado(s) vegetal(es). Cuando la sustancia vegetal o el preparado vegetal sea objeto de una monografía, el solicitante podrá solicitar un certificado de idoneidad expedido por la Dirección Europea de la Calidad del Medicamento. 2. Medicamentos a base de plantas.–Respecto al desarrollo de la formulación, se presentará un resumen sucinto en el que se describirá el desarrollo del medicamento a base de plantas, teniendo en cuenta la vía de administración y la utilización propuestas. Deberán discutirse, cuando proceda, los resultados en los que se compare la composición fitoquímica de los productos reseñados en los datos bibliográficos de apoyo y el medicamento a base de plantas para el que se solicita autorización. 5. Medicamentos huérfanos.–En el caso de un medicamento huérfano que se regirán por el Reglamento (CE) n.º 726//2004 de 31 de marzo, así como el Reglamento (CE) n.º 141/2000, se pueden aplicar las disposiciones generales que figuran en el punto 6 de la parte II (condiciones especiales). El solicitante deberá justificar en los resúmenes no clínicos y clínicos las razones que impiden facilitar la información completa, así como el balance riesgo/beneficios del medicamento huérfano de que se trate. PARTE IV. MEDICAMENTOS DE TERAPIA AVANZADA 1. Introducción Las solicitudes de autorización de comercialización de medicamentos de terapia avanzada, tal como se definen en el artículo 2, apartado 1, letra a), del Reglamento (CE) n.º 1394/2007 del Parlamento Europeo y del Consejo, de 13 de noviembre de 2007, sobre medicamentos de terapia avanzada y por el que se modifican la Directiva 2001/83/CE del Parlamento Europeo y del Consejo, de 6 de noviembre de 2001, por la que se establece un código comunitario sobre medicamentos para uso humano y el Reglamento (CE) n.º 726/2004 del Parlamento Europeo y del Consejo, de 31 de marzo de 2004, por el que se establecen procedimientos comunitarios para la autorización y el control de los medicamentos de uso humano y veterinario, deberán cumplir los requisitos de formato (módulos 1, 2, 3, 4 y 5) descritos en la parte I del presente anexo. En el caso de medicamentos biológicos deberán aplicarse los requisitos técnicos de los módulos 3, 4 y 5, según lo descrito en la parte I del presente anexo. Los requisitos específicos para los medicamentos de terapia avanzada descritos en las secciones 3, 4 y 5 de esta parte explican cómo deben aplicarse los requisitos de la parte I a los medicamentos de terapia avanzada. Además, teniendo en cuenta las especificidades de los medicamentos de terapia avanzada, se han establecido, en su caso, requisitos adicionales. Debido a la naturaleza específica de los medicamentos de terapia avanzada, puede llevarse a cabo un plan basado en un análisis del riesgo para determinar la extensión de los datos de calidad, no-clínicos y clínicos que deben incluirse en la solicitud de autorización de comercialización, con arreglo a las directrices científicas sobre calidad, seguridad y eficacia de los medicamentos indicadas en el punto 4 de la sección “Introducción y principios generales”. El análisis del riesgo puede cubrir todo el proceso. Los factores de riesgo que pueden considerarse incluyen los siguientes: el origen de las células (autólogo, alogénico, xenogénico), la capacidad de proliferación o diferenciación y de iniciar una respuesta inmunitaria, el nivel de manipulación celular, la combinación de células con moléculas bioactivas o materiales estructurales, la naturaleza de los medicamentos de terapia génica, el grado de capacidad replicativa de los virus o microorganismos utilizados in vivo, el nivel de integración de los genes o las secuencias de ácidos nucleicos en el genoma, la funcionalidad a largo plazo, el riesgo de carcinogenicidad y el modo de administración o uso. También pueden tenerse en cuenta para el análisis del riesgo la experiencia o los datos no-clínicos y clínicos disponibles y pertinentes relativos a otros medicamentos de terapia avanzada relacionados. – 235 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano Cualquier desviación de los requisitos de este anexo deberá justificarse científicamente en el módulo 2 del expediente de solicitud. Cuando se aplique el análisis del riesgo antes descrito, también se incluirá y se describirá en el módulo 2. En tal caso se discutirá la metodología seguida, la naturaleza de los riesgos identificados y las implicaciones del planteamiento basado en el análisis del riesgo para el programa de desarrollo y evaluación, y se describirá cualquier desviación de los requisitos del presente anexo que resulte del análisis del riesgo. 2. Definiciones A efectos del presente anexo, además de las definiciones establecidas en el Reglamento (CE) n.º 1394/2007, se aplicarán las definiciones establecidas en las secciones 2.1 y 2.2. 2.1 Medicamento de terapia génica.–Un medicamento de terapia génica es un medicamento biológico con las características siguientes: a) incluye un principio activo que contiene un ácido nucleico recombinante, o está constituido por él, utilizado en seres humanos, o administrado a los mismos, con objeto de regular, reparar, sustituir, añadir o eliminar una secuencia génica; b) su efecto terapéutico, profiláctico o diagnóstico depende directamente de la secuencia del ácido nucleico recombinante que contenga, o del producto de la expresión genética de dicha secuencia. Los medicamentos de terapia génica no incluyen las vacunas contra enfermedades infecciosas. 2.2 Medicamento de terapia celular somática.–Un medicamento de terapia celular somática es un medicamento biológico con las características siguientes: a) contiene células o tejidos, o está constituido por ellos, que han sido objeto de manipulación sustancial de modo que se hayan alterado sus características biológicas, funciones fisiológicas o propiedades estructurales pertinentes para el uso clínico previsto, o por células o tejidos que no se pretende destinar a la misma función esencial en el receptor y en el donante; b) se presenta con propiedades para ser usado por seres humanos, o administrado a los mismos, con objeto de tratar, prevenir o diagnosticar una enfermedad mediante la acción farmacológica, inmunológica o metabólica de sus células o tejidos. A efectos de la letra a), no se considerarán manipulaciones sustanciales las enumeradas en concreto en el anexo I del Reglamento (CE) n.º 1394/2007. 3. Requisitos específicos relativos al módulo 3 3.1 Requisitos específicos para todos los medicamentos de terapia avanzada.–Debe proporcionarse una descripción del sistema de trazabilidad que el titular de una autorización de comercialización se propone establecer y mantener para garantizar que pueda seguirse el rastro de cada medicamento y sus materiales de partida y materias primas, incluidas todas las sustancias que entren en contacto con las células o los tejidos que contenga, durante el abastecimiento, la fabricación, el empaquetado, el almacenamiento, el transporte y el suministro al hospital, institución o consulta privada en que vaya a utilizarse. El sistema de trazabilidad será complementario y compatible con los requisitos prescritos en el Real Decreto 1301/2006, de 10 de noviembre, por el que se establecen las normas de calidad y seguridad para la donación, la obtención, la evaluación, el procesamiento, la preservación, el almacenamiento y la distribución de células y tejidos humanos y se aprueban las normas de coordinación y funcionamiento para su uso en humanos, por lo que se refiere a células y tejidos humanos, con excepción de las células sanguíneas, y en el Real Decreto 1088/2005, de 16 de septiembre, por el que se establecen los requisitos técnicos y condiciones mínimas de la hemodonación y de los centros y servicios de transfusión, por lo que se refiere a células sanguíneas humanas. 3.2 Requisitos específicos para medicamentos de terapia génica. 3.2.1 Introducción: producto terminado, principio activo y materiales de partida. – 236 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano 3.2.1.1 Medicamento de terapia génica que contenga una secuencia de ácido nucleico recombinante o un microorganismo o virus modificado genéticamente.–El producto terminado será la secuencia de ácido nucleico o el microorganismo o virus modificado genéticamente, formulado en su envase inmediato final para su uso médico previsto. El producto terminado podrá combinarse con un producto sanitario o con un producto sanitario implantable activo. El principio activo será la secuencia del ácido nucleico o el microorganismo o virus modificado genéticamente. 3.2.1.2 Medicamento de terapia génica que contenga células modificadas genéticamente.–El producto terminado contendrá las células modificadas genéticamente, formuladas en el envase inmediato final para su uso médico previsto. El producto terminado podrá combinarse con un producto sanitario o con un producto sanitario implantable activo. El principio activo serán células modificadas genéticamente por algún producto de los descritos en el apartado 3.2.1.1. 3.2.1.3 En el caso de medicamentos que consistan en virus o vectores virales, los materiales de partida serán los componentes a partir de los cuales se obtiene el vector, es decir, la semilla maestra del vector viral o los plásmidos utilizados para transfectar las células empaquetadoras y el banco celular maestro de la línea celular empaquetadora. 3.2.1.4 En el caso de medicamentos que consistan en plásmidos, vectores no virales o microorganismos modificados genéticamente, excepto los virus o vectores virales, los materiales de partida serán los componentes utilizados para generar la célula encargada de la producción, es decir, el plásmido, la bacteria huésped y el banco celular maestro de las células microbianas recombinantes. 3.2.1.5 En el caso de células modificadas genéticamente, los materiales de partida serán los componentes utilizados para obtener las células modificadas genéticamente, es decir, las materias primas para producir el vector, el propio vector y las células humanas o animales. Las normas de correcta fabricación deberán aplicarse a partir del sistema de banco utilizado para producir el vector. 3.2.2 Requisitos específicos: Además de los requisitos establecidos en los apartados 3.2.1 y 3.2.2 de la parte I del presente anexo, deberán aplicarse los requisitos siguientes: a) se facilitará información acerca de todos los materiales de partida utilizados para fabricar el principio activo, incluidos los productos necesarios para la modificación genética de las células humanas o animales y, en su caso, del cultivo y la preservación subsiguientes de las células modificadas genéticamente, tomando en consideración la posible falta de etapas de purificación; b) en el caso de medicamentos que utilicen un microorganismo o un virus, se facilitarán los datos sobre la modificación genética, el análisis de la secuencia, la atenuación de la virulencia, el tropismo por tipos específicos de tejidos y células, la dependencia del ciclo celular del microorganismo o virus, la patogenicidad y las características genéticas de la cepa parental; c) las impurezas relacionadas con el proceso y con el medicamento se describirán en las secciones pertinentes del expediente, y en especial los virus contaminantes capaces de replicarse si el vector ha sido diseñado para no ser replicativo; d) en el caso de plásmidos, la cuantificación de las diversas formas de plásmido se llevará a cabo a lo largo del periodo de validez del medicamento; e) en el caso de células modificadas genéticamente, deberán comprobarse sus características antes y después de la modificación genética, así como antes y después de cualquier procedimiento subsiguiente de congelación o almacenamiento. En el caso de células modificadas genéticamente, además de los requisitos específicos para medicamentos de terapia génica, se aplicarán los requisitos de calidad para medicamentos de terapia celular somática y de productos de ingeniería tisular (véase la sección 3.3). 3.3 Requisitos específicos para medicamentos de terapia celular somática y productos de ingeniería tisular. – 237 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano 3.3.1 Introducción: producto terminado, principio activo y materiales de partida.–El producto terminado estará constituido por el principio activo formulado en su envase inmediato para el uso médico previsto, y en su combinación final para medicamentos combinados de terapia avanzada. El principio activo estará compuesto por las células o los tejidos manipulados por ingeniería. Las sustancias adicionales (como soportes, matrices, productos sanitarios, biomateriales, biomoléculas u otros componentes) que se combinan con las células manipuladas formando una parte integrante de ellas se considerarán materiales de partida, aunque no sean de origen biológico. Los materiales utilizados para fabricar el principio activo (como los medios de cultivo y los factores de crecimiento) que en principio no van a formar parte del mismo se considerarán materias primas. 3.3.2 Requisitos específicos: Además de los requisitos establecidos en los puntos 3.2.1 y 3.2.2 de la parte I del presente anexo, deberán aplicarse los requisitos siguientes: 3.3.2.1 Materiales de partida. a) se facilitará una información resumida sobre la donación y adquisición de tejidos y células humanos utilizados como materiales de partida con arreglo al Real Decreto 1301/2006, de 10 de noviembre, así como de los ensayos realizados sobre ellos; si se utilizan células o tejidos no sanos (como tejidos cancerosos) como materiales de partida, deberá justificarse su uso; b) si se mezclan poblaciones de células alogénicas, deberán describirse las estrategias seguidas para obtener la mezcla y las medidas para garantizar la trazabilidad; c) la variabilidad potencial introducida mediante tejidos y células humanos o animales se abordará como parte de la validación del proceso de fabricación, de la caracterización del principio activo y del producto terminado, del desarrollo de ensayos, del establecimiento de especificaciones y de la estabilidad; d) en el caso de medicamentos a base de en células xenogénicas, se facilitarán los datos de la fuente animal (como el origen geográfico, la explotación ganadera y la edad), los criterios específicos de aceptación, las medidas para prevenir y controlar infecciones en los animales donantes, ensayos para agentes infecciosos en los animales, incluyendo microorganismos transmitidos verticalmente y virus, y las pruebas de la adecuación de las instalaciones para animales; e) en el caso de medicamentos a base de células procedentes de animales modificados genéticamente, deberán describirse las características específicas de las células respecto a la modificación genética; deberá aportarse una detallada descripción del método de creación y de caracterización del animal transgénico; f) en caso de modificación genética de las células, deberán aplicarse los requisitos técnicos especificados en la sección 3.2; g) deberá describirse y justificarse la estrategia de control de cualquier sustancia adicional (soportes, matrices, productos sanitarios, biomateriales, biomoléculas u otros componentes) que se combine con las células manipuladas genéticamente de las cuales forme parte integrante; h) en el caso de soportes, matrices y productos definidos como productos sanitarios o como productos sanitarios implantables activos, se facilitará la información requerida con arreglo a la sección 3.4 para evaluar el medicamento combinado de terapia avanzada. 3.3.2.2 Proceso de fabricación. a) se validará el proceso de fabricación para garantizar la consistencia de lotes y del proceso, la integridad funcional de las células durante la fabricación y el transporte hasta el momento de la aplicación o administración, y el estado apropiado de diferenciación; b) si las células han crecido directamente en el interior o sobre una matriz, un soporte o un producto sanitario, se facilitará la información sobre la validación del proceso de cultivo celular en lo que respecta al crecimiento de las células, la función y la integridad de la combinación. 3.3.2.3 Estrategia de caracterización y control. – 238 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano a) se facilitará información pertinente sobre la caracterización de la población de células o mezclas de células en cuanto a su identidad, pureza (por ejemplo, agentes microbianos adventicios y contaminantes celulares), viabilidad, potencia, estudio del cariotipo, tumorigenicidad y adecuación para el uso terapéutico previsto; deberá demostrarse la estabilidad genética de dichas células; b) se facilitará información cualitativa y, si es posible, cuantitativa de las impurezas relacionadas con el medicamento y con el proceso, así como de cualquier material capaz de introducir productos de degradación durante la producción; deberá justificarse el nivel de cualificación de las impurezas; c) deberá justificarse, en cada caso, si no pueden llevarse a cabo determinados ensayos de liberación en el principio activo o el producto terminado, sino solo en productos intermedios clave o como ensayos durante el proceso; d) en el caso de que moléculas biológicamente activas (como factores de crecimiento o citocinas) estén presentes como componentes del producto celular, deberá caracterizarse su impacto e interacción con otros componentes del principio activo; e) en el caso de que una estructura tridimensional forme parte de la función prevista, formarán parte de la caracterización de estos productos celulares el estado de diferenciación, la organización estructural y funcional de las células y, en su caso, la matriz extracelular generada; en caso necesario, la caracterización fisicoquímica deberá complementarse mediante estudios no-clínicos. 3.3.2.4 Excipientes.–A los excipientes utilizados en el medicamento a base de células o tejidos (como los componentes del medio de transporte) se les aplicarán los requisitos para excipientes nuevos, según dispone la parte I del presente anexo, a menos que existan datos acerca de las interacciones entre las células o los tejidos y los excipientes. 3.3.2.5 Estudios de desarrollo farmacológico.–La descripción del programa de desarrollo abordará la elección de los materiales y procesos. En concreto, se discutirá la integridad de la población celular en la formulación final. 3.3.2.6 Materiales de referencia.–Deberá documentarse y caracterizarse un estándar de referencia pertinente y específico para el principio activo o el producto terminado. 3.4 Requisitos específicos para los medicamentos de terapia avanzada que contengan productos sanitarios. 3.4.1 Medicamentos de terapia avanzada que contengan productos sanitarios contemplados en el artículo 7 del Reglamento (CE) n.º 1394/2007: Se describirán las características físicas y la eficacia del medicamento, así como los métodos de diseño del mismo. Se describirá la interacción y compatibilidad entre los genes, células o tejidos y los componentes estructurales. 3.4.2 Medicamentos combinados de terapia avanzada, tal como se definen en el artículo 2, apartado 1, letra d), del Reglamento (CE) n.º 1394/2007: En el caso de la parte celular o de tejido del medicamento combinado de terapia avanzada, se aplicarán los requisitos específicos para medicamentos de terapia celular somática y productos de ingeniería tisular contemplados en la sección 3.3; en el caso de las células modificadas genéticamente, se aplicarán los requisitos específicos a los medicamentos de terapia génica contemplados en la sección 3.2. El producto sanitario o el producto sanitario implantable activo podrán ser parte integrante del principio activo. En el caso de que el producto sanitario o el producto sanitario implantable activo se combinen con las células en el momento de la fabricación, la aplicación o la administración del producto terminado, se considerarán parte integrante de este. Se facilitará la información relacionada con el producto sanitario o el producto sanitario implantable activo (que sea parte integrante del principio activo o del producto terminado) pertinente para la evaluación del medicamento combinado de terapia avanzada. Dicha información incluirá los elementos siguientes: a) información sobre la selección y función previstas del producto sanitario o el producto sanitario implantable activo y demostración de la compatibilidad de dicho producto con otros componentes del medicamento; – 239 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano b) pruebas de la conformidad de la parte del producto sanitario con los requisitos esenciales establecidos en el anexo I del Real Decreto 1591/2009, de 16 de octubre, por el que se regulan los productos sanitarios, o de la conformidad de la parte del producto sanitario implantable activo con los requisitos esenciales establecidos en el anexo 1 del Real Decreto 1616/2009, de 26 de octubre, por el que se regulan los productos sanitarios implantables activos; c) en su caso, pruebas de que el producto sanitario o el producto sanitario implantable activo cumple los requisitos relativos a la EEB y las EET establecidos en el Real Decreto 1591/2009, de 16 de octubre, en el caso de los productos sanitarios, o del Real Decreto 1616/2009, de 26 de octubre, para los productos sanitarios implantables activos; d) en su caso, los resultados de cualquier evaluación de la parte del producto sanitario o de la parte del producto sanitario implantable activo por un organismo notificado con arreglo al Real Decreto 1591/2009, de 16 de octubre, en el caso de los productos sanitarios, o del Real Decreto 1616/2009, de 26 de octubre, para los productos sanitarios implantables activos. A petición de la autoridad competente que evalúe la solicitud, el organismo notificado que lleve a cabo la evaluación contemplada en la letra d) del presente punto deberá poner a la disposición de dicha autoridad toda la información relativa a los resultados de la evaluación, con arreglo al Real Decreto 1591/2009, de 16 de octubre, o del Real Decreto 1616/2009, de 26 de octubre. Podrá incluir información y documentación presente en la solicitud de evaluación de la conformidad en cuestión si es necesaria para evaluar el medicamento combinado de terapia avanzada en su conjunto. 4. Requisitos específicos relativos al módulo 4 4.1 Requisitos específicos para todos los medicamentos de terapia avanzada.–Los requisitos del módulo 4 de la parte I del presente anexo relativos a los ensayos farmacológicos y toxicológicos de los medicamentos pueden no ser siempre apropiados dadas las propiedades estructurales y biológicas únicas y diversas de los medicamentos de terapia avanzada. Los requisitos técnicos de las secciones 4.1, 4.2 y 4.3 explican cómo deben aplicarse los requisitos de la parte I del presente anexo a los medicamentos de terapia avanzada. Teniendo en cuenta las especificidades de los medicamentos de terapia avanzada, se han establecido, en su caso, requisitos adicionales. En el informe general de la parte no-clínica deberá debatirse y justificarse el fundamento para el desarrollo no-clínico y los criterios utilizados para elegir las especies relevantes y los modelos (in vitro e in vivo). En los modelos animales elegidos pueden incluirse animales inmunodeprimidos, con genes desactivados, humanizados o transgénicos. Se tendrá en cuenta el uso de modelos homólogos (como las células de ratón analizadas en ratones) o de imitación de enfermedades, especialmente en los estudios de inmunogenicidad e inmunotoxicidad. Además de los requisitos de la parte I, deberá establecerse la seguridad, idoneidad y biocompatibilidad de todos los componentes estructurales (como matrices, soportes y productos sanitarios) y de cualquier sustancia adicional (como productos celulares, biomoléculas, biomateriales, y sustancias químicas) presentes en el producto terminado. Se tendrán en cuenta sus propiedades físicas, mecánicas, químicas y biológicas. 4.2 Requisitos específicos para medicamentos de terapia génica.–Se tendrá en cuenta el diseño y el tipo de medicamento de terapia génica al establecer la extensión y el tipo de estudios no-clínicos necesarios para determinar el nivel apropiado de datos no-clínicos de seguridad. 4.2.1 Farmacología. a) los estudios in vitro e in vivo de las acciones relacionadas con el uso terapéutico propuesto (es decir, los estudios farmacodinámicos de «prueba de concepto») se establecerán con modelos y especies animales relevantes diseñados para mostrar que la secuencia de ácido nucleico alcanza su objetivo (órgano o células diana) y cumple la función prevista (nivel de expresión y actividad funcional); se establecerá la duración de la función de la secuencia de ácido nucleico y la pauta posológica propuesta en los estudios clínicos; – 240 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano b) selectividad por la diana: si el medicamento de terapia génica se ha diseñado para tener una funcionalidad de destino selectiva o restringida, se facilitarán los estudios para confirmar la especificidad y la duración de la funcionalidad y actividad en las células y tejidos de destino. 4.2.2 Farmacocinética. a) los estudios de biodistribución deberán incluir investigaciones sobre la persistencia, eliminación y la movilización; Los estudios de biodistribución deberán además abordar riesgo de transmisión a la línea germinal; b) la evaluación del riesgo ambiental deberá acompañarse con investigaciones sobre diseminación y el riesgo de transmisión a terceros, salvo si se justifica debidamente en solicitud en función del tipo de medicamento. la el la la 4.2.3 Toxicología. a) deberá evaluarse la toxicidad del medicamento de terapia génica terminado; además, en función del tipo de medicamento, se tendrán en cuenta ensayos individuales del principio activo y los excipientes, y deberá evaluarse el efecto in vivo de los productos expresados relacionados con la secuencia de ácido nucleico no previstos para la función fisiológica; b) los estudios de toxicidad por administración única podrán combinarse con los de farmacología de seguridad y los de farmacocinética (por ejemplo, para investigar la persistencia); c) cuando se prevea la administración múltiple a seres humanos deberán proporcionarse los estudios de toxicidad de administración repetida; el modo y la pauta de administración deberán reflejar fielmente la posología clínica prevista; los estudios de toxicidad múltiple se considerarán en los casos en los que la administración única pueda dar lugar a una funcionalidad prolongada de la secuencia de ácido nucleico en seres humanos; la duración de los estudios podrá ser mayor que la de los de toxicidad estándar en función de la persistencia del medicamento de terapia génica y de los riesgos potenciales previstos; en tal caso, dicha duración deberá justificarse; d) deberá estudiarse la genotoxicidad; no obstante, solo se realizarán estudios estándar de genotoxicidad cuando sean necesarios para estudiar una impureza específica o un componente del sistema de liberación; e) deberá estudiarse la carcinogenicidad; no se requerirán estudios estándar de carcinogenicidad en roedores; sin embargo, en función del tipo de producto, deberá evaluarse el potencial tumorigénico en modelos relevantes in vivo/in vitro; f) toxicidad para la reproducción y el desarrollo: deberán proporcionarse estudios sobre los efectos en la fertilidad y en la función reproductiva general. Se proporcionarán estudios de toxicidad embriofetal y perinatal y de transmisión a la línea germinal, salvo si se justifica debidamente en la solicitud en función del tipo de medicamento; g) estudios adicionales de toxicidad: Estudios de integración: deberán proporcionarse estudios de integración para todo medicamento de terapia génica, a no ser que la ausencia de estos estudios esté científicamente justificada (por ejemplo, porque las secuencias de ácido nucleico no penetren en el núcleo de la célula); en el caso de medicamentos de terapia génica que no se supongan capaces de integración, sólo se llevarán a cabo estudios de integración si los datos de biodistribución indican un riesgo de transmisión a la línea germinal. Inmunogenicidad e inmunotoxicidad: deberán estudiarse los efectos potencialmente inmunogénicos e inmunotóxicos. 4.3 Requisitos específicos para medicamentos de terapia celular somática y productos de ingeniería tisular. 4.3.1 Farmacología. a) los estudios farmacológicos primarios serán pertinentes para demostrar la prueba de concepto; deberá estudiarse la interacción de los productos a base de células con el tejido adyacente; b) se establecerá la cantidad de medicamento necesaria para lograr el efecto deseado/la dosis efectiva y, en función del tipo de medicamento, la frecuencia de la administración; – 241 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano c) deberán tenerse en cuenta los estudios de farmacología secundaria para evaluar los efectos fisiológicos potenciales no relacionados con el efecto terapéutico deseado del medicamento de terapia celular somática, del producto de ingeniería tisular o de sustancias adicionales, pues podrían secretarse moléculas biológicamente activas además de la proteína o proteínas de interés, o esta proteína o proteínas de interés podrían tener dianas no deseadas. 4.3.2 Farmacocinética. a) no se requerirán estudios de farmacocinética convencionales para investigar la absorción, la distribución, el metabolismo y la excreción; no obstante, se investigarán parámetros como la viabilidad, la longevidad, la distribución, el crecimiento, la diferenciación y la migración salvo si se justifica debidamente en la solicitud en función del tipo de medicamento; b) en el caso de medicamentos de terapia celular somática y productos de ingeniería tisular que produzcan biomoléculas activas sistémicas, deberá estudiarse la distribución, la duración y la magnitud de la expresión de dichas moléculas. 4.3.3 Toxicología. a) deberá evaluarse la toxicidad del producto terminado; se tendrán en cuenta los ensayos individuales sobre cada principio activo, excipiente, sustancia adicional y cualquier impureza relacionada con el proceso; b) la duración de las observaciones podrá ser mayor que la de los estudios de toxicidad estándar y se tendrá en cuenta la vida útil prevista del medicamento, así como su perfil farmacodinámico y farmacocinético; en tal caso, la duración deberá justificarse; c) no se requerirán estudios convencionales de carcinogenicidad y genotoxicidad, salvo los del potencial tumoral del medicamento; d) deberán estudiarse los posibles efectos inmunogénicos e inmunotóxicos; e) en el caso de medicamentos a base de células que contengan células animales, deberán abordarse las cuestiones específicas de seguridad asociadas, como la transmisión a las personas de organismos patógenos xenogénicos. 5. Requisitos específicos relativos al módulo 5 5.1 Requisitos específicos para todos los medicamentos de terapia avanzada. 5.1.1 Los requisitos específicos de esta sección de la parte IV se añadirán a los que figuran en el módulo 5 de la parte I del presente anexo. 5.1.2 En los casos en que la aplicación clínica de los medicamentos de terapia avanzada requiera un tratamiento concomitante específico e implique procedimientos quirúrgicos, deberá investigarse y describirse el procedimiento terapéutico completo. Durante el desarrollo clínico se facilitará la información sobre la normalización y optimización de dichos procedimientos. Deberá informarse sobre los productos sanitarios utilizados en los procedimientos quirúrgicos para aplicar, implantar o administrar el medicamento de terapia avanzada si esos productos pueden tener un impacto en la eficacia o seguridad de dicho medicamento. Se definirá la experiencia específica que se exija para poder aplicar, implantar, administrar o realizar las actividades de seguimiento. En caso necesario, se presentará el plan de formación de los profesionales sanitarios sobre los procedimientos de uso, aplicación, implantación o administración de dichos medicamentos. 5.1.3 Dado que, por la naturaleza de los medicamentos de terapia avanzada, es posible que su proceso de fabricación cambie durante el desarrollo clínico, podrán requerirse estudios adicionales para demostrar la comparabilidad. 5.1.4 Durante el desarrollo clínico deberán tratarse los riesgos que surjan de agentes infecciosos potenciales o del uso de material derivado de fuentes animales, y deberán tomarse medidas para reducir dicho riesgo. 5.1.5 Se realizarán estudios de búsqueda de dosis a fin de definir la posología y la pauta de administración. – 242 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano 5.1.6 La eficacia de las indicaciones propuestas estará sustentada por los resultados pertinentes de estudios clínicos que utilicen variables principales de evaluación clínicamente significativas para el uso previsto. En determinadas condiciones clínicas podrán requerirse pruebas de la eficacia a largo plazo. Deberá establecerse la estrategia para evaluar dicha eficacia. 5.1.7 Deberá incluirse en el plan de gestión de riesgos una estrategia para el seguimiento a largo plazo de la seguridad y la eficacia. 5.1.8 En el caso de medicamentos combinados de terapia avanzada, los estudios de seguridad y eficacia se diseñarán y realizarán para el medicamento combinado en su conjunto. 5.2 Requisitos específicos para medicamentos de terapia génica. 5.2.1 Estudios de farmacocinética humana: Los estudios de farmacocinética humana deberán incluir los aspectos siguientes: a) estudios de diseminación para determinar la excreción de los medicamentos de terapia génica; b) estudios de biodistribución; c) estudios farmacocinéticos del medicamento y de las moléculas de expresión del gen (como las proteínas expresadas o características genómicas). 5.2.2 Estudios de farmacodinámica humana: Los estudios de farmacodinámica humana abordarán la expresión y función de la secuencia de ácido nucleico tras la administración del medicamento de terapia génica. 5.2.3 Estudios de seguridad: Los estudios de seguridad deberán abordar los aspectos siguientes: a) la aparición de vectores capaces de replicarse; b) la aparición de cepas nuevas; c) el reagrupamiento de las secuencias genómicas existentes; d) la proliferación neoplásica por mutagénesis insercional. 5.3 Requisitos específicos para medicamentos de terapia celular somática. 5.3.1 Medicamentos de terapia celular somática cuyo modo de acción se basa en la producción de biomoléculas activas definidas: En el caso de medicamentos de terapia celular somática cuyo modo de acción se base en la producción de biomoléculas activas definidas, se abordará, si es posible, el perfil farmacocinético de dichas moléculas (sobre todo su distribución, su duración y la magnitud de su expresión). 5.3.2 Biodistribución, persistencia e injerto a largo plazo de los componentes de medicamento de terapia celular somática: La biodistribución, la persistencia y el injerto a largo plazo de los componentes del medicamento de terapia celular somática se abordarán durante el desarrollo clínico. 5.3.3 Estudios de seguridad: Los estudios de seguridad deberán abordar los aspectos siguientes: a) la distribución y el injerto tras la administración; b) el injerto ectópico; c) la transformación oncogénica y la fidelidad a la estirpe de la célula o el tejido. 5.4 Requisitos específicos para los productos de ingeniería tisular. 5.4.1 Estudios farmacocinéticas: En los casos en que los estudios farmacocinéticos convencionales no sean pertinentes para los productos de ingeniería tisular, deberá abordarse durante el desarrollo clínico la biodistribución, la persistencia y la degradación de los componentes de dichos productos. 5.4.2 Estudios farmacodinámicos: Los estudios farmacodinámicos se diseñarán según las especificidades de los productos de ingeniería tisular y se adaptarán a ellas. Se aportarán los resultados de la prueba de concepto y la cinética del producto para obtener la regeneración, reparación o sustitución prevista. Se tendrán en cuenta los marcadores farmacodinámicos apropiados, relacionados con la función y la estructura previstas. 5.4.3 Estudios de seguridad: Se aplicará el punto 5.3.3. – 243 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano ANEXO II Contenido de la ficha técnica o resumen de características del producto La ficha técnica o el resumen de características del producto, contendrá, por este orden los datos siguientes: 1. Nombre del medicamento. 2. Composición cualitativa y cuantitativa. 3. Forma farmacéutica. 4. Datos clínicos. 4.1 Indicaciones terapéuticas. 4.2 Posología y forma de administración. 4.3 Contraindicaciones. 4.4 Advertencias y precauciones especiales de empleo. 4.5 Interacciones con otros medicamentos y otras formas de interacción. 4.6 Embarazo y lactancia. 4.7 Efectos sobre la capacidad para conducir y utilizar máquinas. 4.8 Reacciones adversas. 4.9 Sobredosis. 5. Propiedades farmacológicas: 5.1 Propiedades farmacodinámicas. 5.2 Propiedades farmacocinéticas. 5.3 Datos preclínicos sobre seguridad. 6. Datos farmacéuticos: 6.1 Lista de excipientes. 6.2 Incompatibilidades. 6.3 Periodo de validez. 6.4 Precauciones especiales de conservación. 6.5 Naturaleza y contenido del envase. 6.6 Precauciones especiales de eliminación, y «otras manipulaciones», en su caso. 7. Titular de la autorización de comercialización. 8. Número de la autorización de comercialización. 9. Fecha de la primera autorización /renovación de la autorización. 10. Fecha de la revisión del texto.–Además, en caso de medicamentos radiofármacos: 11. Dosimetría para los radiofármacos, con una explicación detallada completa de la dosimetría interna de la radiación. 12. Instrucciones para la preparación de radiofármacos, instrucciones detalladas suplementarias para la preparación extemporánea y el control de calidad de esta preparación y, en su caso, tiempo máximo de almacenamiento durante el cual cualquier preparado intermedio, como un eluido, o el radiofármaco listo para su empleo cumplan las especificaciones previstas. 13. Adicionalmente, los medicamentos sometidos a un seguimiento adicional deberán incluir junto al nombre del medicamento la siguiente declaración: «Este medicamento está sujeto a un seguimiento adicional». Esta declaración irá precedida por el símbolo negro a que se refiere el artículo 1 del Reglamento de Ejecución (UE) n.º 198/2013 de la Comisión, de 7 de marzo de 2013, relativo a la selección de un símbolo de identificación de los medicamentos de uso humano sujetos a un seguimiento adicional, y seguida por la frase explicativa normalizada que corresponda. Además, para todos los medicamentos, se incluirá en la sección 4.8 un texto estándar en el que se pida expresamente a los profesionales de la salud que notifiquen toda sospecha de reacción adversa al Sistema Español de Farmacovigilancia (se incluirá enlace al formulario web que la Agencia Española de Medicamentos y Productos Sanitarios establecerá al efecto). – 244 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano ANEXO III Contenido del etiquetado de los medicamentos que se fabrican industrialmente Parte primera. Información que debe incluirse en el embalaje exterior 1. Nombre del medicamento, que estará formado por la denominación del medicamento, seguido de la dosificación y de la forma farmacéutica y, cuando proceda, la mención de los destinatarios lactantes, niños o adultos; cuando el producto contenga hasta tres principios activos, se incluirá la Denominación Oficial Española (DOE), en su defecto, la Denominación Común Internacional (DCI) o, en su defecto, su denominación común. Como norma general las denominaciones de los medicamentos no contendrán abreviaturas ni siglas. No obstante, la Agencia Española de Medicamentos y Productos Sanitarios podrá, por razones de salud pública y a petición del solicitante, autorizar su inclusión. 2. El nombre del medicamento, también deberá indicarse en alfabeto Braille en el embalaje exterior o, en su ausencia, en el acondicionamiento primario, teniendo en cuenta las particularidades de cada medicamento. 3. Composición cualitativa y cuantitativa, en principios activos por unidad de administración o, según la forma de administración para un volumen o peso determinados, utilizando las Denominaciones Oficiales Españolas o las Denominaciones Comunes Internacionales, o, en su defecto, sus denominaciones comunes o científicas. 4. Relación de los excipientes que tengan una acción o efecto conocidos y que sean de declaración obligatoria. Además, deberán indicarse todos los excipientes cuando se trate de un producto inyectable, de una preparación tópica o de un colirio. 5. Forma farmacéutica y contenido en peso, volumen o unidades de administración. 6. Forma de administración y vía de administración. 7. Advertencia: «Mantener fuera del alcance y de la vista de los niños». 8. Advertencias especiales, cuando el medicamento las requiera. 9. En caso de medicamentos que contengan radionucleidos, condiciones de transporte de mercancías peligrosas. 10. En el caso de gases medicinales deberán incluirse las especificaciones técnicas que deben cumplir, las condiciones de suministro y transporte, y en su caso, los símbolos correspondientes. 11. Fecha de caducidad expresada claramente (mes y año). Además, los medicamentos con una estabilidad reducida después de su reconstitución, dilución o su apertura, indicarán el tiempo de validez de la preparación reconstituida, diluida o tras su apertura e incluirán un recuadro para su consignación por los usuarios. En los medicamentos que contengan radionucleidos, se expresará día/mes/año, y en su caso, hora: minutos y país de la referencia horaria. 12. Precauciones particulares de conservación, en su caso. 13. Precauciones especiales de eliminación de los medicamentos no utilizados y de los materiales de desecho derivados de su uso, cuando corresponda, y en su caso los símbolos autorizados por la Agencia Española de Medicamentos y Productos sanitarios, a efectos de facilitar la aplicación y desarrollo de los sistemas de recogida de medicamentos y favorecer la protección del medio ambiente. 14. Nombre y dirección del titular de la autorización de comercialización del medicamento y, en su caso, el nombre del representante local designado por el titular. 15. Código Nacional del Medicamento. 16. Lote de fabricación. 17. Para los medicamentos no sujetos a prescripción médica, la indicación de uso. 18. Condiciones de prescripción y dispensación. 19. Símbolos, siglas y leyendas descritos en el anexo IV. 20. Recuadro o espacio en blanco que permita indicar la posología recetada, duración del tratamiento y frecuencia de uso o tomas, excepto en aquellos casos que la Agencia – 245 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano Española de Medicamentos y Productos Sanitarios determine, teniendo en cuenta las particularidades de cada medicamento. 21. Cupón precinto del Sistema Nacional de Salud, cuando proceda. Parte segunda: Información que debe incluirse en el acondicionamiento primario 1. Los acondicionamientos primarios o en su caso el blindaje de protección de los medicamentos que contengan radionucleidos, que se presenten sin embalaje exterior, habrán de incluir las informaciones recogidas en la parte primera. 2. Los acondicionamientos primarios o en su caso el blindaje de protección de los medicamentos que contengan radionucleidos, distintos de los pequeños envases y blister que se mencionan en los apartados 3 y 4, habrán de incluir las informaciones recogidas en la parte primera, excepto las correspondientes a los apartados 18, 20 y 21, y las leyendas del apartado 19. 3. Cuando el acondicionamiento primario o en su caso el blindaje de protección de los medicamentos que contengan radionucleidos, contenido en un embalaje exterior sea tan pequeño que no permita la inclusión de los datos previstos en la parte primera, deberá llevar como mínimo la información siguiente: a) Nombre del medicamento, tal como se contempla en apartado 1 de la parte primera y, si fuera necesario, la vía de administración, b) fecha de caducidad, c) número de lote de fabricación, d) forma de administración, si fuera necesario, e) contenido en peso, en volumen o en unidades de administración y en bequerelios en caso de medicamentos que contengan radionucleidos, f) cualquier otra información necesaria para la conservación y uso seguro del medicamento. g) símbolo internacional de radiactividad, en el caso de los medicamentos que contengan radionucleidos. h) nombre del fabricante, en el caso de los medicamentos que contengan radionucleidos. 4. Los acondicionamientos primarios de medicamentos presentados en forma de blister y tiras cuando estén contenidos en un embalaje exterior, deberá llevar como mínimo la información siguiente: a) Nombre del medicamento, tal como se contempla en el apartado 1 de la parte primera, b) fecha de caducidad, c) número de lote de fabricación, d) nombre del titular de la autorización de comercialización del medicamento, e) cualquier otra información necesaria para la conservación y uso seguro del medicamento. En el caso de que el acondicionamiento primario esté preparado para cortarse en unidades, la integridad de la identificación del producto, la fecha de caducidad y el número de lote, deberá garantizarse en cada unidad. 5. Información en las ampollas del disolvente: a) Identificación del contenido; b) contenido en volumen; c) nombre del titular de la autorización de comercialización del medicamento; d) número de lote de fabricación; e) fecha de caducidad; f) cualquier otra información necesaria para la conservación y uso seguro del medicamento. – 246 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano ANEXO IV Símbolos, siglas y leyendas Los símbolos, siglas y leyendas que deben aparecer en el etiquetado de los medicamentos serán los siguientes: 1. Símbolos: a) Dispensación sujeta a prescripción médica: b) Dispensación con receta oficial de estupefacientes de la lista I anexa a la Convención Única de 1961: c) Medicamentos que contengan sustancias psicotrópicas incluidas en el anexo I del Real Decreto 2829/1977, de 6 de octubre: d) Medicamentos que contengan sustancias psicotrópicas incluidas en el anexo II del Real Decreto 2829/1977, de 6 de octubre: e) Conservación en frigorífico: f) Medicamentos que pueden reducir la capacidad de conducir o manejar maquinaria peligrosa: Conducción: ver prospecto Sobre fondo blanco, un triángulo equilátero rojo, con el vértice hacia arriba, y con un coche negro en el interior sobre fondo blanco. Su tamaño se adaptará al del envase; en todo caso, el lado del triángulo no será inferior a 10 mm. g) (Suprimida) h) Símbolo Internacional de radiactividad recogido en la norma UNE-73302 de 1991, sobre distintivos para señalización de radiaciones ionizantes: – 247 – CÓDIGO DE DERECHO FARMACÉUTICO § 7 Autorización, registro y condiciones de dispensación de los medicamentos de uso humano Material radiactivo Sobre fondo blanco, un triángulo equilátero negro, con el vértice hacia arriba. En su interior y sobre fondo amarillo, el símbolo establecido por la norma UNE-73302 indicativo de radiactividad en negro. Su tamaño se adaptará al del envase; en todo caso, el lado del triángulo no será inferior a 10 mm. La leyenda se imprimirá sobre el mismo fondo blanco, en negrita y color negro. Se situará debajo o, en caso necesario, al lado del triángulo. i) Símbolo de gas medicinal comburente: Sobre fondo blanco, un rombo negro. En su interior y sobre fondo amarillo anaranjado, una llama de fuego sobre un círculo impreso en negro (símbolo establecido para sustancias comburentes del anexo II del Real Decreto 363/1995, de 10 de marzo por el que se aprueba el Reglamento sobre notificación de sustancias nuevas y clasificación, envasado y etiquetado de sustancias peligrosas). Su tamaño se adaptará al del envase; en todo caso, el lado no será inferior a 10 mm. j) Símbolo de gas medicinal inflamable: Sobre fondo blanco, un rombo negro. En su interior y sobre fondo rojo, una llama de fuego impresa en negro. Su tamaño se adaptará al del envase; en todo caso, el lado no será inferior a 10 mm. 2. Siglas: a) Medicamento de uso hospitalario: H. b) Medicamento de diagnóstico hospitalario o de prescripción por determinados médicos especialistas: DH. c) Medicam