TEMA 1 MÉTODOS DE MINIMIZACIÓN A) INTRODUCCIÓN
Anuncio
 INTRODUCCIÓN")
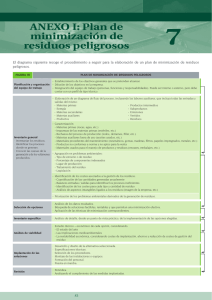
TEMA 1 MÉTODOS DE MINIMIZACIÓN A) INTRODUCCIÓN MATEMÁTICA Generalmente el problema se ceñirá a una minimización de una función real [f(x)] de n variables. Se asume que la función f(x) es una función continua y que además posee la primera y segunda derivada también continuas. Se define gradiente (gi) como la derivada parcial de la función con respecto a la variable x i. r r r ∂ f(x) g i (x) = ∂x i [1.1] Se define la matriz del hessiano como una matriz de n x n cuyas componentes son las segundas derivadas de la función. r ∂ 2 f(x) r H ij (x) = ∂x i ∂x j [1.2] Esta matriz suele ser simétrica (Hij=Hji) ya que en la mayoría de las funciones de interés químico se cumple que las derivadas cruzadas son iguales, es decir que la segunda parcial con respecto a xi y xj es igual a la parcial con respecto a xj y xi . Como ejemplo de gradiente y hessiano, supóngase que se tiene la función: f(x) = x13 + 3 x1 x 2 + 2 x 42 El gradiente de la función es: ∂f(x)/∂x 1 3 x12 + 3 x 2 = g(x) = 3 ∂f(x)/∂x 2 3 x1 + 8 x 2 La matriz del hessiano es igual a: H H(x) = 11 H 21 H 12 6 x 1 = H 22 3 3 24 x 22 El vector gradiente representa la norma (pendiente) de la tangente al hiperplano en el punto x. Existen diferentes tipos de puntos estacionarios en una función. A continuación se detallan los más importantes QFC(Tema 1) 1 14/03/03 Mínimo Máximo Punto de silla Desde el punto de vista químico los más importantes son los mínimos y los puntos de silla. En una superficie de energía potencial, los mínimos corresponderán a moléculas o estructuras estables y los puntos de silla corresponden a estados de transición que unen dos estructuras de mínima energía. Existen diferentes tipos de mínimos, dependiendo del entorno de ese mínimo. f(x) Mínimo local Mínimo local débil Mínimo global x 1) Mínimo global: se cumple que el valor de la función en el mínimo es siempre menor que la función en cualquier otro valor de x. f(xm) < f(x) (xm≠x y xm∈Rn) 2) Mínimo local: cuando sólo se cumple en un intervalo que el valor de la función en ese punto es menor que la función en otro punto f(xm) < f(x) (xm≠x |x – xm| <η y xm∈Rn) 3) Mínimo local débil: como en el caso anterior pero el intervalo es muy pequeño. Para que un punto sea un mínimo, deben cumplirse dos condiciones: • El gradiente ha de ser cero [g(xm)] • Los valores propios de la matriz del hessiano han de ser positivos [H(xm)>0] Los puntos de silla, que tienen gran importancia en el estudio de mecanismos de reacción, son máximos en un sentido (el sentido de la coordenada de reacción) y mínimos en los otros sentidos. Por esta razón, estos puntos se caracterizan por poseer un valor propio de la matriz del hessiano negativo y todos los demás positivos. QFC(Tema 1) 2 14/03/03 B) VALORES Y VECTORES PROPIOS DE UNA MATRIZ Si A es una matriz hermítica de n x n (una matriz cuadrada, con todos los elementos reales y ademá simétrica) existen una serie de constantes reales λ1, λ2, … λn llamados r r r valores propios, asociados a una serie de vectores ( x 1 , x 2 , ... x n ), llamados vectores propios de tal manera que r r A x i = λi x i a 11 a 21 . a n1 a 12 [1.3] . a 1n x 1 x1 a 2n x 2 x =λ 2 . . . a nn x n x2 [1.4] El cálculo de los valores propios de una determinada matriz se puede hacer por diferentes métodos. 1) Método del determinante A partir de la ecuación tres se puede escribir: r r A x -λ x =0 [1.5] r (A - λ I ) x = 0 [1.6] donde I es la matriz identidad. La ecuación 6 representa un sistema homogéneo de ecuaciones que para que tenga una solución diferente de la trivial (x=0) debe de cumplir que |A- λ I| = 0. 1 2 , se obtiene: Si se aplica este procedimiento a la matriz A = 2 − 1 1 2 1 0 1 2 λ 0 1 - λ - λ = - = 2 − 1 0 1 2 − 1 0 λ 2 1- λ 2 2 − 1 − λ 2 = 0 → (1-λ)(-1-λ) – 4 = 0 −1 − λ λ2 - 5 = 0 → λ =± 5 Para calcular los vectores propios, hay que resolver la ecuación: x 1 2 x1 = λ 1 2 − 1 x 2 x2 QFC(Tema 1) 3 14/03/03 Si se considera el valor propio λ = + 5 x 1 2 x1 = 5 1 2 − 1 x 2 x2 La resolución de la ecuación anterior permite obtener el siguiente sistema de ecuaciones: x1 + 2 x 2 = 5 x1 2 x1 - x 2 = 5 x 2 Ya que si un vector es vector propio de una matriz, también lo es ese vector multiplicado por una constante, se puede fijar una de las dos variables. Si se fija x2 = 1 se obtiene: x1 = 1+ 5 2 El vector propio es: 1+ 5 2 1 Si se considera el otro valor propio ( λ = - 5 ) por el mismo procedimiento se puede obtener: 1− 5 2 1 2) Método de Jacobi Si D es una matriz diagonalizada (Dij= 0 si i≠j y Dij ≠ 0 si i=j) Los valores propios coinciden con los elementos de la diagonal Si se tiene la matriz: a 0 0 D = 0 b 0 0 0 c Para obtener los valores propios hay que resolver el determinante: a -λ 0 0 QFC(Tema 1) 0 0 b-λ 0 =0 0 c-λ 4 14/03/03 La resolución del determinante lleva a la ecuación: (a-λ) (b-λ) (c-λ) = 0 Evidentemente las soluciones de la ecuación anterior son: λ1 = a λ2 = b λ3 = c El método de Jacobi es un procedimiento iterativo para diagonalizar una matriz hermítica de n x n. Actualmente ya existen muchos programas de ordenador que permiten diagonalizar matrices. C) MÉTODOS DE MINIMIZACIÓN LOCAL Los métodos de minimización locales son generalmente métodos descendentes, es decir que va descendiendo por la superficie o hipersuperficie de energía potencial hasta alcanzar el mínimo local más próximo. Son procedimientos iterativos mediante los cuales a partir de un valor inicial x0 se va pasando por diferentes valores hasta alcanzar el xm. Generalmente no se suelen permitir pasos que produzcan un aumento de la función. Evidentemente estos valores son muy sensibles al valor inicial así como a la dirección de búsqueda y al algoritmo de minimización empleado. La dirección descendente se establece a partir del gradiente, es decir se desciende por el camino de mayor gradiente. f(x) método local x Existen dos grandes grupos de métodos de minimización local: 1) Métodos no derivativos En estos métodos para la minimización se emplean los valores de la función y en primera instancia no se calculan los valores de la derivada de la misma. Este tipo de métodos se emplea cuando por algún motivo es difícil evaluar la derivada de la función. Generalmente estos métodos son muy fáciles de implementar pero las propiedades de convergencia son bastante pobres. En algunos casos, los gradientes se pueden calcular numéricamente, tomando incrementos finitos QFC(Tema 1) 5 14/03/03 g(x) ≈ 1 [f(x + h) - f(x)] h [1.7] donde hay que elegir convenientemente el valor de h. En este tipo de métodos, los más conocidos son: • Método Powell: este método asegura alcanzar el mínimo exacto de una función cuadrática convexa después de n pasos. • Método Simplex: en este método, a partir de un valor inicial se generan dos nuevos puntos y se hace pivotar el triángulo formado sobre el lado que une los dos puntos más bajos. Así se genera un nuevo punto con el que se forma un nuevo triángulo y se repite el procedimiento hasta alcanzar el mínimo. 2) Métodos del gradiente (o derivativos) Este tipo de métodos extrae información de la primera y a veces de la segunda derivada para acelerar el proceso de minimización. Por término medio son más rápidos que los anteriores. Existen muchos tipos diferentes: • Descenso más inclinado (SD, steepest descent): es uno de los métodos más antiguos y simples, aunque actualmente no tiene mucha importancia. La dirección de búsqueda se toma como menos el gradiente. • Gradiente Conjugado (CG, conjugate gradient): es muy parecido al anterior, pero emplea otro algoritmo para la búsqueda del mínimo. • Métodos de Newton (o Newton-Raphson): en este tipo de métodos se necesita hasta la segunda derivada. Son métodos muy extendidos pero presentan algunos problemas por lo que generalmente se aplican a pequeños problemas o en las proximidades del mínimo. Algunas veces inicialmente se emplea otro método y en las proximidades del mínimo se cambia a éste. Presenta algunas variantes: • TN (Truncated Newton) • QN (quasi-Newton) • Método de Davidov-Fletcher-Powell: es un método muy parecido al anterior pero emplea una fórmula diferente para calcular el siguiente valor de x en la iteración. • Método de Marquart-Levenberg: es un método muy extendido actualmente. Es una mezcla de métodos, lejos del mínimo emplea un método del tipo SD y posteriormente emplea un método basado en el hessiano D) MÉTODOS DE MINIMIZACIÓN GLOBAL Los métodos de optimización global intentan soslayar la existencia de mínimos locales, mediante la exploración de grandes regiones del espacio. Este problema ha sido tratado ampliamente en el análisis conformacional de oligopéptidos, en donde se pueden presentar innumerables mínimos locales dependiendo de los valores de los ángulos diedros. Se puede alcanzar el mínimo global empleando fundamentalmente dos tipos de aproximación, determinística y estocástica. Los métodos determinísticos usualmente requieren una función que cumpla ciertas propiedades de “suavidad”. Se construye una secuencia que permita alcanzar el mínimo global QFC(Tema 1) 6 14/03/03 desde un mínimo local. Estos métodos son relativamente nuevos y su aplicación a problemas físicos y químicos tienen un gran futuro Los métodos estocásticos intentan alcanzar el mínimo global mediante la generación aleatoria de muchos puntos iniciales desde los cuales iniciar la minimización. Es previsible que alguno de los puntos generados al azar se encuentre en la posición adecuada para alcanzar el mínimo global. Estos métodos para alcanzar mínimos globales están basados en el empleo de ordenadores. 1) Optimización de interacciones electrostáticas (SCEF, self consisten electric field) Este método se ha aplicado fundamentalmente al estudio conformacional de pequeños polipéptidos. Es un método determinista. El método consiste en realizar una aproximación inicial despreciando todos los términos energéticos excepto las interacciones electrostáticas. Se calcula así el momento dipolar de cada fragmento y se intenta alinearlo con el momento dipolar de toda la molécula. Esto se consigue modificando las orientaciones de cada residuo hasta que al final se llega a un punto en que cada residuo tiene su momento dipolar perfectamente alineado con el momento dipolar total. Una vez alcanzado este punto se procede a una minimización total de la energía hasta que se alcanza el mínimo energético. El procedimiento se repite iterativamente hasta que se alcanza la autoconsistencia. De hecho este método emplea las interacciones electrostáticas para buscar una geometría inicial a partir de la cual se minimiza la energía. La filosofía inicial del método es que si se realiza una minimización de la energía de la conformación adecuada, se llegará al mínimo global. Un buen ejemplo de la aplicación de este método puede verse en L. Piela and H.A. Scheraga, Biopolymers (1987) 26, 533. 2) Método de Monte Carlo más minimización (MCM) Este método consiste en realizar una alteración al azar de la estructura inicial y posteriormente minimizar la energía de dicha estructura. Posteriormente se realiza otra modificación al azar y subsiguiente minimización. Esto genera una serie de estructuras que posteriormente se minimizan. Es de esperar que alguna o algunas de ellas sean buenas estructuras iniciales para alcanzar el mínimo global. La filosofía de este método es completamente diferente de la del anterior. En este caso no se tiene ningún procedimiento sistemático para generar y modificar las estructuras hasta llegar al mínimo global. Es un método estocástico, ya que se emplean procedimiento al azar para generar muchas estructuras iniciales para alcanzar el mínimo global. En la aplicación del método de Monte Carlo se suelen emplear los criterios del método Metrópolis. El estudio de Li y Scheraga [Z. Li and H.A. Scheraga, THEOCHEM (1988) 179, 33] se puede encontrar una interesante aplicación de este método al estudio conformacional de pequeños polipéptidos. 3) Método de Monte Carlo conducido electrostáticamente (EDMC, “elestrostatically driven Monte Carlo) Es un método que combina los dos métodos anteriores. En este método se realiza un diagnóstico electrostático de la estructura inicial, estudiando la orientación de los momentos dipolares con respecto al momento dipolar total. Posteriormente, combinando estos resultados con un muestreo al azar se generan nuevas conformaciones que posteriormente sufren un proceso de minimización de la energía para alcanzar los mínimos locales. QFC(Tema 1) 7 14/03/03 Estos mínimos locales son comparados entre si para saber si se ha llegado a un mínimo global. Este método es semejante al MCM pero la generación de estructuras está dirigida de tal manera que no se necesitan generar tantas y así el método es más rápido. 4) Método de la ecuación de difusión (DEM, Diffusion equation method) Este método consiste en ir deformando la superficie o hipersuperficie de energía hasta transformarla en una superficie con un solo mínimo (evidentemente es el mínimo global de esa superficie generado) que generalmente está relacionado con el mínimo global de la superficie primitiva. Posteriormente se realiza el proceso contrario para regenerar la superficie primitiva.. La superficie se va deformando con el denominado operador de transformación. Si se supone una función f(x) La función f1(x) se define como: f1(x) f1(x) = f(x) + β f ”(x) (β>0) f(x) En esta transformación los puntos de inflexión no se modifican ya que f ”(x) = 0, mientras que las zonas cóncavas aumentan su valor (f”(x)>0) y las convexas lo disminuyen (f”(x)<0). Esto genera una suavización de la función. Si la transformación se realiza N veces, se obtendrá: d2 f (x) = 1 + β 2 dx N N f(x) [1.8] Esta expresión ya se podría aplicar directamente para provocar la deformación de una superficie, sin embargo el procedimiento es más efectivo cuando β tiendo a cero y N a infinito. Se puede definir una nueva variable t, tal que: β= t N [1.9] Esto permite definir una nueva función: t d2 F(x, t) = lim 1 + 2 N dx N d2 f(x) = exp t 2 f(x) dx 142 4 43 4 [1.10] T(t) El operador T(t) es el denominado operador transformación. QFC(Tema 1) 8 14/03/03 La curva inicial f(x) se transforma en diferentes funciones F(x,t) mediante la ecuación de difusión: ∂ 2 F ∂F = ∂t ∂x 2 [1.11] Las funciones F y f están relacionadas también por las condiciones de entorno, es decir: F(x,0) = f(x) [1.12] El grupo de Scheraga ha realizado interesantes aplicaciones de este método a diferentes problemas de múltiples mínimos [L. Piela, J. Kostrowicki and H.A. Scheraga, J. Phys. Chem. (1989) 93, 3339; J. Kostrowicki and H.A. Scheraga, J. Phys. Chem. (1992) 96,7442] t=0 t = 0.2 t = 0.3 t = 0.5 En la figura anterior se ha representado la superficie de energía potencial del análisis conformacional de un etano sustituido (t=0) y las subsiguientes deformaciones hasta llegar a una superficie con un solo mínimo. QFC(Tema 1) 9 14/03/03