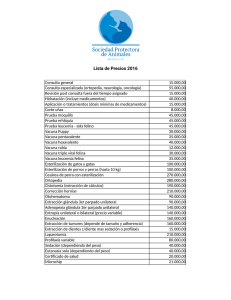
Revista Nº143 | ver
Anuncio

Agosto de 2014 | Nº 143 Volumen 54 Nº 143 Agosto de 2014 ISSN: 0558/1265 Uruguay 469, piso 2º B C1015ABI Buenos Aires, Argentina Tel: (54-11) 4373-0462 / 8900 (54-11) 4372-7389 Fax: (54-11) 4374-3630 www.safybi.org El nuevo film de PE. Su nueva referencia. Revista Revista SAFYBI | Nueva familia de bolsas Flexsafe. Revista SAFYBI Passed ASTM Shipping Test Flexsafe reune los requisitos de robustez excepcional y fácil uso en todos los Entrevista exclusiva SAFYBI Farm. Boni, Directora de la Dirección de Evaluación de Tecnologías Sanitarias de la anmat ONE FILM FOR ALL pasos del proceso de fabricación de sus productos. La resistencia y la flexibilidad del film proporcionan un rendimiento constante y un manejo sencillo incluso en las USP - Envasado farmacéutico: pruebas de permeabilidad a la humedad e integridad aplicaciones más exigentes como el cultivo celular, almacenamiento a largo plazo o el transporte de fármacos. Sartorius Argentina S.A. · Tel. +54.11.4721.0505 · [email protected] Ver vídeos: www.sartorius-stedim.com/flexsafe USP – DSP – F +F Visite nuestra revista online en www.safybi.org Novedades en vacunas Vacuna contra la fiebre hemorrágica argentina, un producto nacional Avances en las técnicas de purificación de vacunas Revista Safybi COMITÉ EDITOR Comisión Directiva Presidente: Dr. Federico E. Montes de Oca Vicepresidente: Dr. Domingo A. García Secretaria: Dra. Mirta B. Fariña Prosecretaria: Dra. Susana B. Muñoz Director: Dra. Magdalena Nannei Consejo Asesor: Dr. Federico E. Montes de Oca Dr. Domingo A. García Dra. Mirta B. Fariña Dr. Germán C. Fernández Otero Dr. Alberto Grimoldi Corresponsal de Asuntos Universitarios: Dra. Susana B. Muñoz Tesorero: Dr. Germán C. Fernández Otero Administración: Paula Mosquera Protesorero: Dr. Guido M. Furer Fotografía: Marcela Marinangeli Vocales Titulares: Dra. Marta Fasanella Dr. Leonardo Fullone Dra. María Celeste González Dr. Bernardo Gutman Dr. Alejandro A. Meneghini Dr. Luis Moyano Diseño y diagramación: Virginia Gallino Comercialización: DKsiclo Group Cel: (011) 15-4474-2426 [email protected] Coordinación General: Lic. Doris Kaplán Vocales Suplentes: Dr. Mariano Arismendi Dr. Martín Dobovsek Dr. Claudio Vilariño Comités de expertos Ver información actualizada en la página 8 Uruguay 469 2º B C1015ABI Bs. As., Argentina Tel.: (54-11) 4373-0462 / 8900 Tel.: (54-11) 4372-7389 Fax: (54-11) 4374-3630 [email protected] / www.safybi.org Cambios de domicilio, teléfono y correo electrónico Se ruega a los socios que notifiquen inmediatamente a la Secretaría todo cambio de domicilio, número telefónico o e-mail con el objeto de que no se interrumpan las comunicaciones de la Institución ni se envíen publicaciones a direcciones no vigentes. ISSN 0558-7265 Inscripta en el Registro Nacional de Propiedad Intelectual Nº 5120647 Propietario y Publicación Trimestral de la Asociación Argentina de Farmacia y Bioquímica Industrial, Uruguay 469 2º B, Buenos Aires, Argentina. Está incorporada al servicio de información bibliográfica internacional Pharmaceutical Abstracts Service. SAFYBI es publicada en Buenos Aires. Circula sin cargo entre profesionales y empresas asociadas a SAFYBI. Las opiniones vertidas en artículos y traducciones son exclusiva responsabilidad de los Señores Autores. Producción integral: DKsiclo Group E-mail: [email protected] Preimpresión e impresión: Artes Gráficas Buschi Ferré 2250/52 - C1437FUP C.A.B.A., Argentina SUMARIO VOL 54 - No 143 - agosto de 2014 SAFYBI Calidad 8 Comités de expertos 10Editorial 12 Palabras del Presidente ANMAT 14 Evaluación de Tecnologías Sanitarias Entrevista a la Farmac. Silvia Boni, Directora de la Dirección de Evaluación de Tecnologías Sanitarias de la ANMAT USP 20 Envasado farmacéutico: pruebas de permeabilidad a la humedad e integridad Desmond G Hunt, Dwain Sparks, Michael N. Eakins y Mary G. Foster Conferencias, cursos, programas y seminarios 30 Programa de capacitación en esterilización para farmacéuticos Módulo II 38 Desarrollo de un Programa de Especialización en Asuntos Regulatorios 1a parte Artículos Biotecnología 42 Vacuna contra la fiebre hemorrágica argentina, un producto nacional 49 Herramientas de la calidad: síntesis y ampliación Dr. Mario Lisnizer Instalaciones y equipos 53 Entrevista al Sr. Daniel A. Ferretti Logística 57 Cold Chain Congress, IPQC, mayo 2014, Panamá Dra. Sandra Rumiano Nuestros anunciantes 59 Avances en las técnicas de purificación de vacunas Sartorius 67 Optimización de las carteras de productos farmacéuticos con sistemas de administración de aerosoles nasales o sprays bucales Aptar 71VROC®- Un dispositivo MEMS capaz de determinar la viscosidad a partir de un pequeño volumen de muestra RHEOSENSE INC. CAS 72 Decontaminación y esterilización de envases Ionics 73 Mediciones de temperatura y humedad relativa en la industria farmacéutica Testo Dra. Laura Riera Foto de tapa: Colocación de cartuchos filtrantes Sartopore® en carcasa de acero inoxidable para la filtración esterilizante de formas líquidas en industria farmacéutica. Sartorius S.A. Safybi Comités de Expertos Aseguramiento de la Calidad Coordinador: Alejandro Meneghini Amneris Gatti Ana Rey Cecilia Sobrero Cristina Wiege Diana Oldani Eduardo Alberto Uran Esteban Pablo Fuentes Guillermo Alberto Duda Gustavo Hernán Aguirre Hernan Martínez Abal Martín Dobovsek Oscar Mauricio Gonzalez Pablo Ponziani Silvina Bessone 8 revista safybi Asuntos Regulatorios Coordinador: Leonardo Fullone Andrea Simanski Carina Rismondo Graciela Luque María Teresa Manzolido Miriam Juarez Mirta Billy Noemí Brunet Ricardo Díaz Roberto Kuktosky Virginia Peluffo Viviana Ureña Biotecnología Coordinador: Federico Montes de Oca Augusto Pich Otero Claudio Vilariño Cristina Wiege Eduardo Spitzer Esteban Fuentes Marina Henrich Patricio R. Santagapita Distribución y Operadores Logísticos Coordinador: Susana Muñoz Adriana Leone Juan Rolandi Liliana Kuharo Luis Moyano Mauricio Rittiner Nancy Suane Norma Amaya Pablo Álvarez Pablo Bedo Materias Primas Farmacéuticas Coordinador: Carlos Chiesa Cecilia Beatriz Sobrero Daniel Roberto Vega Leandro Pintos María Emilia Callisto Melina Bisio Sonia Faudone Viviana Graciela Dabbene Microbiología Coordinador: Marta Fasanella Horacio Frade Juana Rodríguez de Flotta Stella Maris Stagnaro Productos Médicos y Esterilización Coordinador: Nora Canoura Alberto Grimoldi Amalia Barboza Andrea Induni Damián Ramírez Spadaro Laura Grodeky María del Carmen Graziano Marta Centeno Omar Silvetti Química Analítica Coordinador: Bernardo Gutman Eduardo E. Lopez Hernán Invenenato Marcelo Feltrinelli Sergio Kulesnik Tecnología Farmacéutica Coordinador: Germán Fernández Otero Arturo Hoya Daniel Ventura Fabian De Bonis Javier Luca Jorge Budzvicky Jorge Ferrari Laura Maurizio Pablo Musi Rodolfo Díaz Rosana Kelman Sebastian Barber Safybi Editorial En este número de la Revista SAFYBI se presentan artículos que demuestran cómo en nuestro país se han producido desarrollos trascendentales en la industria farmacéutica y biotecnológica, tanto en la producción de medicamentos esenciales como en la provisión de los sofisticados servicios que requiere esta industria para llevar adelante su producción. Además, se lanza en este número la invitación a participar en la EXPOFYBI 2015, EXPOSICIÓN DE FARMACIA Y BIOQUÍMICA INDUSTRIAL, que tendrá lugar los días 4, 5, 6 y 7 de agosto de 2015. Junto a esta exposición se desarrollará el III Congreso Latinoamericano de Farmacia y Bioquímica Industrial, el XIV Congreso Argentino de Farmacia y Bioquímica Industrial y las XII Jornadas JorFyBI de Farmacia y Bioquímica Industrial. Nuevamente tuvimos la oportunidad de contar con la participación de la ANMAT, esta vez por medio de una entrevista realizada a la Farmacéutica Silvia Boni, Directora de la Dirección de Evaluación de Tecnologías Sanitarias. En el paradigma del avance tecnológico continuo, en el cual la población se ve literalmente bombardeada por nuevas tecnologías, no siempre efectivas y beneficiosas, la Dirección que encabeza la Farm. Boni, justamente tiene la misión de evaluar su efectividad y seguridad antes de aprobar su uso en nuestro país y los detalles de esa actividad son ampliamente descriptos. El método original para medir la permeabilidad a la humedad, factor esencial para asegurar la estabilidad de los productos orales sólidos, ha sido cuestionado por su falta de confiabilidad y reproducibilidad cuando se usa en nuevos sistemas de envase-cierre. La USP patrocinó un taller en mayo del año 2013, junto con el Instituto de Investigación sobre Calidad de Productos, en el cual se basa el artículo presentado por la USP y cuyos autores Hunt, Sparks, Eakins y Foster pertenecen a la División de Ciencias y Normas Globales de la USP, al Comité de Expertos en Envasado, Almacenamiento y Distribución y los dos últimos nombrados a Eli Lilly and Company. Los PROGRAMAS DE CAPACITACIÓN especiales que está desarrollando SAFYBI para distintas especialidades han contribuido por medio de un artículo y una entrevista. El artículo, cuyo resumen es presentado por la Dra. María Celeste González, miembro de la Comisión Directiva de nuestra institución, es el resultado del Programa de capacitación en esterilización para farmacéuticos, módulo II, en el cual participaron los Farmacéuticos Letché, Sarubbio, Sigyel, Ramírez, Sayavedra, Pedrini y Enriquez, la Lic. en Biología Figliolo, la ECI Vernazzi, el Ing. Schichi y el Sr. Fer- 10 revista safybi nández. Con respecto al Programa de Especialización en Asuntos Regulatorios, contestó un cuestionario especialmente preparado sobre el mismo, el Dr. Leonardo Fullone, Coordinador del Comité de Expertos en Asuntos Regulatorios de SAFYBI. La Dra. Laura Riera, describe la historia, las estrategias de desarrollo y las características de elaboración, control y garantía de calidad de la vacuna contra la fiebre hemorrágica argentina (FHA), producto elaborado por el Instituto Nacional de Enfermedades Virales Humanas “Dr. Julio. I. Maiztegui” (INEVH), dependiente de la Administración Nacional de Laboratorios e Institutos de Salud (ANLIS) “Dr. Carlos G. Malbrán”, Ministerio de Salud de la Nación. La importancia de esta vacuna, desarrollada por científicos argentinos en colaboración con profesionales de los Estados Unidos, y elaborada en nuestro país bajo estrictas normas de bioseguridad y calidad, cobra especial actualidad en estos tiempos, ya que demuestra como nuestro país supo reaccionar con rapidez y eficacia frente a una amenaza viral específica de nuestra región. En este número de la Revista SAFYBI, el Dr. Mario Lisnizer, describe las Herramientas Básicas de la Calidad, dentro de las que incluye ocho herramientas y las Nuevas Herramientas, en las cuales considera siete y describe las mismas: Diagrama de afinidad, Diagrama de flechas, Matriz de análisis de datos, Diagrama Matriz, Diagrama para el proceso de decisión, Diagrama de relaciones y Diagrama de árbol. Nuevamente en esta edición encaramos una entrevista con los proveedores de nuestra industria, otra vez en el área de Instalaciones y Equipos, pero en esta oportunidad el tema se refiere a los criterios de selección y a la validación de las cabinas de bioseguridad, y para ello se prestó gentilmente el Sr. Daniel Ferretti, socio gerente de Ferretti Validaciones. El Congreso “Cold Chain and Temperature Management Logistics” Central & Latin America, realizado en la ciudad de Panamá en mayo de este año, contó con la participación de los Dres. Montes de Oca, Presidente de SAFYBI y Sandra Rumiano, Chairman de las actividades del Congreso a pedido del International Quality and Productivity Center. La Dra. Rumiano publicó un resumen de las actividades y discusiones de las mesas de trabajo señalando los desafíos estratégicos y operativos que el tema implica. Magdalena Nannei Director de la Revista Nos es grato contactarlos a fin de informarles que en el marco del III Congreso Latinoamericano y XIV Congreso Argentino de Farmacia y Bioquímica Industrial, se desarrollará una renovada edición de la tradicional EXPOFYBI. Dichos eventos, organizados por SAFYBI, la Asociación Argentina de Farmacia y Bioquímica Industrial tendrán lugar los días 4, 5, 6 y 7 de agosto de 2015 en el Pabellón 6 del Centro Costa Salguero. EXPOFYBI, la exposición de proveedores de equipos y servicios de la Industria Farmacéutica, con una trayectoria de más de 30 años en nuestro medio, es el espacio natural de encuentro entre los profesionales y directivos de los laboratorios nacionales e internacionales. En esta oportunidad EXPOFYBI 2015 se realizará en forma integrada con EXPOENVASE, organizada por el Instituto Argentino del Envase, y en cuya última edición han recibido 27.000 visitantes de la Industria a lo largo de los cuatro días de exposición. La oferta conjunta será de más de 250 empresas exponiendo sus pro- 12 revista safybi blicitar sus productos y servicios en un ambiente Científico Tecnológico y de Negocios Empresariales. En busca de una mayor sinergia los invitamos a contratar un paquete de difusión, integrado por: ductos y novedades como así también más de 150 módulos de capacitación, lo que la convierte en uno de los eventos más importantes de Latinoamérica. Se prevé que nos visiten en esta oportunidad un gran número de profesionales con cargos gerenciales y técnicos y con capacidad de decisión, que desarrollan su actividad en las industrias bioquímico-farmacéutica, alimenticia, cosmética, veterinaria, productos médicos y afines provenientes no solo de todo el país, sino también de Brasil, Uruguay, Chile, Bolivia, Paraguay, Perú, Venezuela, Colombia y Centroamérica. En este contexto EXPOFYBI les brindará la posibilidad de contarlos entre sus destacados expositores, ofreciéndoles una oferta de atractivos beneficios para promocionar y pu- 1. Presencia en EXPOFYBI 2015. Con la reserva de un stand, la empresa tiene derecho a dar una charla técnica-empresarial en el marco del programa del Congreso (sin cargo adicional). 2. Publicidad on-line en la PÁGINA WEB OFICIAL DE SAFYBI y en los envíos que realiza la Asociación anunciando sus cursos y novedades en su extensa base de datos. Consultar por precios y fechas de salidas. 3. Publicidad en la renovada y digitalizada REVISTA SAFYBI. Pueden visualizarla on-line en www. safybi.org. Consultar por precios y fechas de salidas. Esperamos contarlos entre nuestros prestigiosos expositores y anunciantes. Lic. Doris Kaplán Directora Ejecutiva DKsiclo Group Comercialización Exclusiva: Revista SAFYBI - EXPOFYBI 2015 [email protected] 15-4474-2426 ANMAT Evaluación de Tecnologías Sanitarias Nota del Editor: El impacto de las nuevas tecnologías afecta profundamente la salud de la población y en consecuencia merece la máxima atención por parte de las autoridades de salud de un país. Con esas perspectivas la Farmac. Silvia Boni, Directora de la Dirección de Evaluación de Tecnologías Sanitarias de la ANMAT, accedió gentilmente ante nuestro requerimiento para tratar el tema de Evaluación de Tecnologías Sanitarias y de las actividades que abarca para la Revista SAFYBI. La Farmacéutica Silvia Boni es Especialista en Sistemas de Salud y Seguridad Social y Magister en Sistemas de Salud y Seguridad Social. Actualmente está a cargo de la Dirección de Evaluación de Tecnologías Sanitarias de la Administración Nacional de Medicamentos, Alimentos y Tecnología Médica. Coordinadora del Grupo de Trabajo de Registro de Medicamentos de la Red Panamericana para la Armonización Regulatoria de Medicamentos, se ha destacado en conferencias y cursos en la temática de los medicamentos en Argentina. Le agradeceríamos nos aclare cuáles son las principales funciones y actividades de la Dirección de Evaluación de Tecnologías Sanitarias. En el marco del nuevo paradigma de Ciencia Reguladora de la actual gestión encabezada por el Dr Carlos Chiale, a través del Decreto 1271/2013 fue puesta en marcha la nueva estructura de la ANMAT. Dentro de este modelo, y bajo el concepto de Vigilancia Sanitaria fue creada la Dirección de Evaluación de Tecnologías Sanitarias (ETS) con la finalidad de hacer una evaluación sistemática de la efectividad de las tecnologías sanitarias a través de la recuperación y del análisis de la información disponible en la literatura científica. Desde esta nueva visión es que asumimos el desafío de usar en cada acto decisorio la mejor evidencia disponible. Se plantean entonces objetivos estratégicos entre los que menciono el de optimizar las herramientas para garantizar a los pacientes/usuarios la seguridad, eficacia y calidad de los productos que son de incumbencia de la ANMAT, además de acompañar y vehiculizar los procesos de renovación tecnológica, entre otros. Funciona también en esta Dirección una unidad denominada Sala de Situación concebida como un espacio de gestión, análisis, interpretación y contextualización sistemática de datos e información orientada a una vigilancia sanitaria integrada de los productos regulados por la Administración. Por otra parte, dentro de la Dirección funciona el Área de Toxicología, de funcionamiento transversal dentro de la Administración, cuya función se orienta a favorecer el trabajo en red intra y extra ANMAT. Allí se aborda la problemática 14 revista safybi de la toxicidad de los productos de nuestra incumbencia. Durante este año, por ejemplo, se ha colaborado con otras áreas de la Administración, como el Observatorio, brindando charlas a la comunidad con el fin de prevenir intoxicaciones evitables y el uso indebido de algunos productos, por ejemplo los alisadores de pelo. Las charlas dadas fueron “Uso seguro de cosméticos y alisantes en el ámbito de la peluquería”, “Cómo prevenir Intoxicaciones en el hogar” y “Minimizando Riesgos en el Jardín de Infantes”. ¿Cuáles son los sectores económicos, sociales y gubernamentales que participan en la Evaluación de Tecnologías Sanitarias? ¿Existe algún trabajo conjunto o interacción? ¿Qué novedades tecnológicas tienen o tendrían en un futuro cercano mayor impacto en la salud de la población, por ejemplo, nuevos modelos de atención, telemedicina, tecnologías informativas, etc.? Ante todo permítame recordar que la Evaluación de Tecnologías Sanitaria (ETS) es una herramienta clave para orientar la toma de decisiones de manera racional, basada en métodos científicos, con el fin de proporcionar respuestas a preguntas que plantean los diferentes actores que operan en el escenario sanitario, siendo de utilidad tanto para los profesionales asistenciales como también para los Poderes Públicos, Aseguradores, Administradores, Financiadores y para los Ciudadanos. Se basa en el proceso de análisis e investigación de las nuevas tecnologías, teniendo además en cuenta su impacto económico y social, pero fundamentalmente adaptándolo al contexto local. Está dirigida a estimar el valor y contribución relativos de cada tecnología sanitaria a la mejora de la salud individual y colectiva. Constituye un puente entre el conocimiento científico y el proceso de toma de decisiones. En la Evaluación de Tecnologías Sanitarias en general participan muchos sectores. Como dijimos, el trabajo que se desarrolla en este sentido en la ANMAT es de desarrollo transversal, donde participan directa o indirectamente todos los sectores de la Administración. www.aptar.com/pharma APtAr PHArmA is yOur glObAl sOlutiOn PrOviDer fOr innOvAtive AnD PrOven AerOsOl, sPrAy AnD DisPensing systems. Our products and services add value to your products in the fields of biotechnology, healthcare and pharmaceuticals. Aptar Pharma has two divisions: Prescription and Consumer Health Care. Delivering solutions, shaping the future. ANMAT Las evaluaciones se realizan no sólo sobre los medicamentos y dispositivos médicos nuevos, también sobre los modelos nuevos de organización, atención a distancia y comunicaciones según el grado de criticidad, con el fin de acompañar los avances. Por ejemplo, es de gran preocupación la introducción de nuevas tecnologías que se agregan a las anteriores sin evaluaciones profundas previas, sobrecargando el sistema si no presentan una clara mejora con respecto al standard del momento. Aunque parezca inocuo el impacto de una tecnología inútil o inofensiva para la salud de la población es negativo aunque no haga daño en forma directa. Porque podría distraer recursos críticos, producir demora en los diagnósticos o tratamientos o generar inequidades. Respecto a novedades tecnológicas que tendrán mayor presencia en los productos de incumbencia de la ANMAT estimo que serán los productos que contengan nanopartículas, los que conllevan una evaluación y análisis muy concreto y especifico. Se trata de una ciencia, la Nanotecnologia que en el área de salud nos proporcionará grandes alcances como las nanocápsulas que permitirán no sólo detectar enfermedades, o anomalía fisico-química sino además poder administrar, de manera selectiva y específica, los medicamentos necesarios para solucionar el problema. ¿Cuáles son las herramientas económicas usuales para la evaluación de tecnologías sanitarias? ¿Cómo impacta en las evaluaciones las desigualdades económicas de la población? ¿En base a qué índices se establecen las prioridades? Para la evaluación de tecnologías sanitarias se usan todas las herramientas adecuadas a las circunstancias y según la disponibilidad de información en el momento de tomar la decisión. En esta disciplina se tiene en cuenta la seguridad, la eficacia, el costo y la relación costo/efectividad. Cuando la tecnología en evaluación es un medicamento se la llama farmacoeconomia como sinónimo de evaluación económica de medicamentos. Los análisis farmacoeconómicos combinan las ciencias de la medicina, la estadística, la economía y la epidemiología para lograr el método científico para la toma de decisiones basadas en evidencias científicas disponibles. Respecto del impacto, justamente la ETS tiende a disminuir las desigualdades en la población, permitiendo que lo científicamente comprobado pueda llegar a la comunidad en forma oportuna. Las prioridades se establecen por patología prevalente o grave o sin tratamiento disponible, por impacto social, por discapacidad o mortalidad. Algunas priorizaciones también pueden obedecer a cuestiones presupuestarias o políticas, finalmente el costo también es un factor de priorización. ¿Cuál es el impacto de los medicamentos para enfermedades poco frecuentes o serias con riesgo de muerte y/o invalidez grave? ¿Qué situación de impacto económico especialmente relevante unida a los nuevos medicamentos desea usted destacar especialmente? La entrada de medicamentos novedosos y de tecnologías sanitarias nuevas en la práctica médica rutinaria exige 16 revista safybi esfuerzos que aportarán beneficios muy relevantes a la sociedad. Por ejemplo, los nuevos medicamentos autorizados con Registros bajo condiciones especiales destinados al diagnóstico o tratamiento de enfermedades poco frecuentes o a enfermedades serias con riesgo de muerte y/o invalidez grave. Algunos de ellos son el carfilzomib indicado para pacientes con mieloma múltiple y tafamidis meglumin indicado para la amiloidosis genética. Las Autoridades Regulatorias son más exigentes en las pruebas que deben acometerse para evidenciar la calidad, la eficacia y la seguridad cuanto mayor sea el grado de innovación de ese producto candidato a registro, y está muy bien que así sea. En este contexto y a la luz de la Disposición 4622/12 se ha conformado para el registro de este tipo de medicamentos especiales un grupo particularmente celoso a la hora de demandar pruebas y garantías, asegurando que su utilización clínica una vez lanzado, reportará beneficios que superen a los riesgos. Puede suceder ocasionalmente que se produzcan dilaciones o demoras en la aprobación de estos nuevos productos. En estos casos, deben ser atribuidas a que la ANMAT siempre priorizará la seguridad para los pacientes. Con el desarrollo de las diversas tecnologías, parecería existir grandes oportunidades para mejorar la atención de la salud ¿Cómo se pueden demostrar los verdaderos beneficios de la mejora tecnológica? El desarrollo de las nuevas tecnologías puede ser considerado por un lado como un potente incentivo al fomento de la innovación y a la mejora de la atención de la salud de los pacientes. Generalmente el mayor número de tratamientos siempre significa más datos clínicos y más rápida acumulación de conocimiento científico, que es tanto como decir mayor velocidad de avance para la ciencia. El gran desafío es no perder la accesibilidad a esos tratamientos, ¿Qué es la UCEETS y como participa la ANMAT de la misma? ¿Qué otros organismos colaboran? La UCEETS es la Unidad Coordinadora de Evaluación y Ejecución de Tecnologías Sanitarias (UCEETS) coordinada por la Dirección de Calidad en Servicios de Salud del Ministerio de Salud de la Nación. Está conformada por nodos representantes de: la DIRECCION NACIONAL DE REGULACION SANITARIA Y CALIDAD EN SERVICIOS DE SALUD, la DIRECCION DE CALIDAD EN SERVICIOS DE SALUD, la ADMINISTRACION NACIONAL DE MEDICAMENTOS, ALIMENTOS Y TECNOLOGIA MEDICA (A.N.M.A.T.), la ADMINISTRACION NACIONAL DE LABORATORIOS E INSTITUTOS DE SALUD (A.N.L.I.S.), la SUPERINTENDENCIA DE SERVICIOS DE SALUD, el INSTITUTO NACIONAL DE SERVICIOS SOCIALES PARA JUBILADOS Y PENSIONADOS (I.N.S.S.J.P.), la ADMINISTRACION DE PROGRAMAS ESPECIALES (APE), el INSTITUTO NACIONAL CENTRAL UNICO COORDINADOR DE ABLACION E IMPLANTE (I.N.C.U.C.A.I.), la COMISION NACIONAL DE SALUD, CIENCIA Y TECNOLOGIA (SACYT) y los Hospitales Nacionales “Dr. Alejandro POSADAS” y Pediatría “Prof. Dr. Juan P. GARRAHAN”. La ANMAT es un miembro activo de esta Red. Por otra parte, la RedARETS es la Red Argentina Pública de Evaluación de Tecnologías Sanitarias. Es una red de centros del ámbito público que desarrollan productos o informes ANMAT de ETS. Tiene como visión el desarrollo sustentable de un sistema de cooperación interjurisdiccional público de ETS en la República Argentina, que permita generar y difundir este tipo de evaluaciones en nuestro país, compartir los recursos públicos y poner a disposición de una manera eficiente los resultados de las ETS a los decisores sanitarios. ¿Existe algún tipo de integración regional? ¿Cuál es el propósito de la Plataforma Regional sobre Acceso e Innovación para Tecnologías Sanitarias, desarrollada por la OPS/OMS? ¿Qué países intervienen? ¿Qué rol desempeña la Argentina en esta Plataforma Regional? La REDETSA, Red de Evaluación de Tecnologías Sanitarias para las Américas, es una red Americana. Convocadas por la OPS, doce países de la Región (Argentina, Bolivia, Brasil, Chile, Colombia, Costa Rica, Cuba, Ecuador, México, Paraguay, Perú, Uruguay) acordaron lanzar la REDETSA con representación de bloques de integración subregionales (MERCOSUR, Región Andina), centros colaboradores de OPS/OMS (CENETEC, Universidad de Ottawa, IEB) y expertos del ámbito de la Evaluación de Tecnologías Sanitarias (ETS) y la salud pública (IECS). La ANMAT se integra a ella a través de la UCEETS. Tiene como misión promover y fortalecer la ETS, a través del intercambio regional, como herramienta que apoya la toma de decisiones sobre incorporación, difusión y uso de tecnologías, contemplando los contextos de los sistemas de salud a nivel país, ampliando el acceso con equidad. En 18 revista safybi este momento se están aprobando reglamentos de funcionamiento y consolidando lazos y ya se ha empezado a compartir documentos de ETS. Finalmente, ¿qué impacto/impactos de trascendencia ha tenido en los últimos años la Evaluación de Tecnologías Sanitarias en nuestro sistema de salud? La implementación y puesta en marcha efectiva de esta mirada es incipiente. Nuestro sistema está creciendo y asentándose de manera de valerse de ETS para tomar decisiones a nivel de macro, meso y micro gestión. n USP Envasado farmacéutico: pruebas de permeabilidad a la humedad e integridad Estímulo al proceso de revisión Desmond G Hunt,a Dwain Sparks,b Michael N. Eakins,c y Mary G. Fosterc Los artículos de estímulo no necesariamente reflejan las políticas de la USPC o del Consejo de Expertos de la USP Artículo de estímulo basado en un taller co-patrocinado por la Convención de la Farmacopea de los EE.UU. (USP, por sus siglas en inglés) y el Instituto de Investigación sobre Calidad de Productos (PQRI, por sus siglas en inglés), que se llevó a cabo del 20 al 21 de mayo de 2013, en las oficinas centrales de la USP, en Rockville, Maryland. Resumen Este artículo de Estímulo provee información actualizada sobre desafíos clave y avances en las pruebas de desempeño de sistemas de envasado para medicamentos en formas farmacéuticas orales sólidas (SODF, por sus siglas en inglés). El análisis de desempeño es esencial para un entendimiento cabal de las propiedades del sistema de envasado, lo cual define más extensamente la identidad, el contenido, la calidad, la pureza y el desempeño del medicamento durante toda su vida útil. Este artículo se enfoca en la medición de la permeabilidad de la humedad al interior de los sistemas de envasado y describe un nuevo método propuesto para cuantificar la velocidad de transmisión de vapor de humedad (MVTR, por sus siglas en inglés). El método propuesto ha demostrado ser más confiable y reproducible que los métodos actuales contenidos en el capítulo 〈671〉 Envases—Pruebas de Desempeño. El Comité de Expertos de la USP en Envasado, Almacenamiento y Distribución (PSD, por sus siglas en inglés) propuso revisiones al capítulo 〈671〉 en el PF 39(2) [marzo– abril 2013], incluyendo un nuevo método de velocidad de transmisión de vapor de humedad para sistemas de envasado de formas farmacéuticas sólidas orales. En mayo de 2013, se realizó un taller para reunir a las partes interesadas y facilitar la discusión de los cambios propuestos y el impacto potencial del nuevo método de velocidad de transmisión de vapor de humedad sobre la industria y las agencias reglamentarias. Las oportunidades presentadas 20 revista safybi y analizadas durante el taller incluyeron la posible reducción de las pruebas de estabilidad y las limitaciones en las clasificaciones de la USP para sistemas de envasado. Se resaltan los resultados del taller así como los temas que requieren una discusión más profunda. El Comité de Expertos de la USP en Envasado, Almacenamiento y Distribución anticipa que el nuevo método, que genera un valor de MVTR/m o por unidad, se oficializará con base en la discusión del taller y los comentarios públicos realizados acerca de la propuesta en el PF 39(2). Los futuros cambios en las definiciones de “impermeable” y “bien cerrado” y en los métodos existentes de la Sección 2, los cuales están dirigidos a los farmacéuticos, se realizarán después de realizar consultas adicionales con las partes interesadas y aparecerán en el PF para comentarios públicos. Introducción Los productos farmacéuticos se fabrican de acuerdo con estrictos estándares de calidad y se envasan en sistemas de envase-cierre que protegen adecuadamente a la forma farmacéutica de factores que pueden ocasionar la degradación de su calidad durante su vida útil. Las causas comunes de la degradación del producto incluyen exposición a la luz, exposición a gases reactivos, pérdida de disolvente y absorción de vapor de agua. Las formas farmacéuticas orales sólidas tales como tabletas y cápsulas por lo regular se almacenan en frascos de plástico o blísteres. El vapor de agua puede pasar al interior del sistema de envase-cierre, ya sea a través de la pared de un envase de plástico o difundiéndose a través del sello en un envase de plástico o blíster. En última instancia, la exposición a la humedad mediante la permeabilidad a la misma puede tener un impacto sobre la calidad del medicamento. Historia del capítulo 〈671〉 de la USP y PQRI sobre permeabilidad a la humedad El método gravimétrico original para medir la permeabilidad a la humedad se desarrolló en la década de 1970 y se oficializó en USP–NF XIX (1975). El método para medir la permeabilidad al vapor de agua en envases de unidades múltiples para cápsulas y tabletas proveía dos criterios de USP aceptación para envases “impermeables” y “bien cerrados”. Aunque la prueba estaba destinada para su uso por parte de farmacéuticos, los fabricantes y reenvasadores de medicamentos la emplean comúnmente. En 1980, se oficializó en la USP–NF XX un método para envases unitarios y envase de dosis única para tabletas y cápsulas, el cual proveía criterios de aceptación para cuatro estándares (Clase A, B, C y D). La Guidance for Industry, Container Closure Systems for Packaging Human Drugs and Biologics (Guía para la Industria, Sistemas de Envase-Cierre para Envasado de Medicamentos y Productos Biológicos de Uso Humano) de 1999 de la FDA, hizo referencia al capítulo 〈671〉. La mención del capítulo general por la FDA promovió el método como una prueba que debería ser realizada por la industria farmacéutica, aun cuando su aplicabilidad estaba inicialmente limitada a farmacéuticos. Los métodos del capítulo 〈671〉 no proveen resultados que sean suficientemente confiables y reproducibles para diseños críticos de sistemas de envase-cierre. En la década pasada el PQRI, un consorcio de organizaciones que trabajan asociadas con el PQRI, diseñó y realizó estudios y recopiló datos sobre la transmisión de vapor de humedad en varios sistemas de envase-cierre. Estos datos se usaron en el desarrollo de un nuevo método propuesto que tratase dicha deficiencia para los fabricantes farmacéuticos. Los resultados del nuevo método se informaron como velocidad de transmisión de vapor de humedad (MVTR) o velocidad de transmisión de vapor de agua (WVTR, por sus siglas en inglés), que se consideran términos intercambiables. Este nuevo método gravimétrico es más confiable y reproducible que los métodos del capítulo 〈671〉. El Comité de Expertos de la USP en Envasado, Almacenamiento y Distribución ha incorporado el nuevo método en una revisión propuesta del capítulo 〈671〉 Envases—Pruebas de Desempeño, que se publicó en el Pharmacopeial Forum 39 (2) [marzo–abril 2013]. A fin de dar una consideración más profunda al nuevo método que fuese más allá de los comentarios públicos realizados a la propuesta del PF, la USP llevó a cabo un taller los días 20–21 de mayo de 2013 en sus oficinas centrales de Rockville, Maryland, EE.UU., el cual se diseñó para tratar el impacto potencial de la implementación del nuevo método para determinar la velocidad de transmisión de vapor de humedad en sistemas de envasado y para analizar diversas formas para mejorar el sistema de clasificación vigente para formas farmacéuticas sólidas orales. Los ponentes y asistentes de la FDA, de la industria farmacéutica, del PQRI y de la USP compartieron sus experiencias con el método gravimétrico actual, además de sus conocimientos relacionados que incluían el uso de agua en lugar de desecante y nuevos métodos para medir la permeabilidad a la humedad en sistemas de envasado de medicamentos. Nuevo método propuesto El nuevo método para medir la velocidad de transmisión de vapor de humedad (MVTR, por sus siglas en inglés) fue desarrollado por el Grupo de Trabajo en Sistemas de Envase Cierre del PQRI para abordar el problema del mé- 22 revista safybi todo gravimétrico original, que no es lo suficientemente robusto y tiende a arrojar resultados poco uniformes. El objetivo del Grupo de Trabajo del PQRI era promover el uso del método de MVTR/m (MVTR/m, donde m = masa de la unidad de dosificación) para cumplir con las expectativas de la FDA en cuanto al sustento de sistemas de envasado en lugar de realizar pruebas de estabilidad. Tanto la técnica anterior como la nueva son métodos gravimétricos, pero el nuevo enfoque es más confiable y reproducible que el método anterior, además de que toma en cuenta materiales de barrera alta y de barrera baja. El método propuesto también se alinea con una nueva norma de ASTM International (D7009), la cual es una adaptación del método del PQRI. Aspectos principales del método El método propuesto provee mejoras al método gravimétrico tradicional, las cuales permiten una determinación más robusta de la velocidad de transmisión de vapor de humedad para su uso en el diseño y la selección de los sistemas apropiados de envase-cierre. La USP reconoce la existencia de métodos alternativos para determinar la velocidad de transmisión de vapor de humedad. Por lo tanto, el método propuesto, con sus mejoras en la determinación gravimétrica tradicional de la velocidad de transmisión de vapor de humedad, se publicará en este capítulo a fin de proveer un enfoque estándar para mejorar la robustez de las determinaciones de la velocidad de transmisión de vapor de humedad usando un método gravimétrico. Este capítulo y los detalles provistos no pueden cubrir todos los aspectos de la preparación de la muestra, manipulación, y demás controles para reducir la variabilidad en los resultados obtenidos con este método. La Tabla 1 siguiente enumera las mejoras con respecto a los métodos vigentes. Los analistas en los laboratorios de prueba y demás usuarios de los datos de velocidad de transmisión de vapor de humedad deberían adquirir un entendimiento cabal de las bases científicas para las determinaciones de la velocidad de transmisión de vapor de humedad y aplicar los controles apropiados para reducir la variabilidad y aumentar la robustez del método. Las mejoras incorporadas en este método gravimétrico son necesarias para el diseño adecuado de los sistemas de envase-cierre y para el mantenimiento continuo de dichos sistemas a medida que se introducen cambios. Uso de desecantes El nuevo método recomienda varios desecantes, incluyendo cloruro de calcio anhidro, gel de sílice y tamices moleculares. En la prueba de velocidad de transmisión de vapor de humedad, la función del desecante es mantener la humedad interna a no más de 10% durante todo el análisis. La humedad y la temperatura externas también deben mantenerse constantes durante la medición de la velocidad de transmisión de vapor de humedad para reducir la variabilidad. Se requiere una cantidad adecuada de desecante secado de manera apropiada con una capacidad de sorción de humedad intrínseca lo suficientemente alta para crear condiciones de exceso (sink) para la prueba de 63 USP Tabla 1. Mejoras en la Determinación Gravimétrica Tradicional de la Velocidad de Transmisión de Vapor de Humedad en el Método Ejemplificativo para su Uso por parte de Fabricantes de Productos Farmacéuticos y Suplementos Dietéticos Atributo del Método Mejora Beneficio Se elucida y elabora la distinción entre tipos de muestras. Selección apropiada de la cantidad de desecante y de la duración de la prueba Eliminación de blancos (o controles) Variabilidad reducida Los cierres se mantienen en su lugar (no se retiran 30 veces antes de la prueba) Permite probar los sellos de inducción Combinación (bundling) de muestras Variabilidad reducida Preparación del desecante Se refinan el tiempo y la temperatura para lograr la condición de humedad más baja posible. Control del contenido de humedad en el desecante para mantener la humedad interna en no más de 10% durante todo el estudio. Número de determinaciones de peso Aumentadas Permite la determinación de la velocidad de transmisión de vapor de humedad usando un análisis de regresión lineal. Condiciones de la cámara Uso recomendado de 40º/ 75% de humedad relativa Variabilidad reducida en sistemas de barrera alta. Cálculo de la velocidad de transmisión de vapor de humedad Eliminación de la determinación de peso inicial (antes de colocar muestras en la cámara) para sistemas de barrera alta y ultra alta. Permite lograr un estado estacionario de permeabilidad a la humedad. Preparación de la muestra 24 revista safybi velocidad de transmisión de vapor de humedad y, por lo tanto, para mantener la humedad interna en no más de 10% durante el análisis. En general, la manipulación, secado y almacenamiento de desecantes juegan un papel importante en la exactitud y la variabilidad de los resultados de las pruebas de velocidad de transmisión de vapor de humedad. Las discusiones en el taller generaron recomendaciones adicionales para mejorar la manipulación de desecantes: 1) Se debe especificar un tiempo mínimo de secado y 2) El desecante debe enfriarse hasta temperatura ambiente en un envase sellado si se va a almacenar durante algún tiempo. Aplicaciones potenciales El nuevo método para medir la permeabilidad a la humedad puede aplicarse para determinar un intervalo o rango aceptable probado (PAR, por sus siglas en inglés) para su uso en presentaciones reglamentarias. Esto puede facilitar la revisión de los cambios en los sistemas de envasado, p.ej. la introducción de nuevos tamaños de empaques, cambios en la cantidad de blísteres por lámina/ tira, entre otros. Este método simplificado se basa en una aplicación cuidadosa y racional de MVTR/m (o MVTR por unidad). La FDA apoya este enfoque e invita a los científicos a discutirlo con dicha1 agencia antes de presentar 01-Carpe-aviso Safybi 2014.pdf 26/02/14 16:22 solicitudes y de realizar estudios de estabilidad. La aplicación más simple es para diferentes tamaños de frascos y contenido de unidades cuando los materiales de construcción son idénticos. La velocidad de transmisión de vapor de humedad para las condiciones más exigentes que proveen un perfil aceptable de estabilidad se puede determinar a través de una serie de estudios de velocidad de transmisión de vapor de humedad. Este valor (MVTR/m o MVTR por unidad) se puede usar para reducir las pruebas de estabilidad para el registro y para demostrar la equivalencia en los cambios de sistemas de envase-cierre. Opinión reglamentaria actual sobre el nuevo método El nuevo método se puede aplicar en varias etapas, tales como las pruebas tempranas de selección de envases, la etapa IND y la etapa NDA o ANDA, incluyendo las presentaciones de Calidad por Diseño (QbD, por sus siglas en inglés) en solicitudes de nuevos medicamentos y cambios en las aplicaciones de medicamentos aprobados. Don Klein, Ph.D., Químico Revisor Principal de la Administración de Alimentos y Medicamentos de los EE.UU. / Centro de Evaluación e Investigación de Medicamentos, expuso ante los asistentes al taller que el valor MVTR/m es un paso adelante positivo en materia reglamentaria Soluciones integrales, incorporando innovación, calidad y servicio desde 1939 RAM E S P E C T R Ó M E T R O R A M A N P O R TÁT I L Proveedor sugerido por la • Farmacopea de Estados Unidos (En versiones: Libro, Online, CD). • Amplio rango de pH (0-12,0) . • Guía de Reactivos Cromatográficos. • Gran variedad de tamaños de partícula, entre 3 y 10 µ • Compendio de Suplementos Dietarios. • Mayor vida últil de las columnas. • Estándares de Referencia. • Alta eficiencia y resolución de las columnas. • Cursos online. • Food Chemical Codex (FCC): Compendio de Normas reconocido internacionalmente para determinar la pureza y la identidad de ingredientes alimenticios Aprobado por la • Identificación de polvos y/o líquidos, a través de su envase traslucido (Bolsas, Blisters y Vidrio). • Identificación según su forma polimorfa. • Rápida verificación (de 3 a 60 seg.) de la materia prima. • Flexibilidad para el desarrollo de métodos. • Tecnología Patentada TE Cooling (aprobada y validada por la USP). • Mejor sensibilidad para LC-MS . • Mayor duración de las columnas. Your Spectroscopy Partner Servicios de entregas regulares e inmediatas. CARPE SCHEIDER & CIA S.A. Godoy Cruz 2769 5 Piso. C1425FQK - CABA Tel.: +54 11 4776-0477 Fax +54 5276 - 9813 Bajos costos, amplia variedad de columnas para desarrollos. [email protected] www.carpescheider.com.ar • Identificación de la materia prima en cualquier lugar del laboratorio. • Gran ahorro de tiempos y recursos del departamento de Control de Calidad. El mejor precio del mercado. AÑOS revista safybi 25 USP debido a que permitirá una reducción en las pruebas de estabilidad. Se puede considerar el siguiente ejemplo. Un fabricante farmacéutico provee datos de estabilidad para cada presentación en frascos de una forma farmacéutica particular, p.ej., materiales idénticos de construcción, tamaños de 10; 30; 90 y 1000 unidades. La Tabla 2 provee dicho ejemplo de datos hipotéticos. Tabla 2. Velocidad de transmisión de vapor de humedad para presentaciones en frasco Tamaño Nominal de Frasco (mL) Contenido MVTR \[mg/(día × empaque)] 40°/75% de humedad relativa MVTR [mg/ (día × unidad)] 75 10 0,43 0,04 125 30 0,74 0,02 180 90 0,94 0,01 950 1000 1,21 0,001 Si se invoca el principio de MVTR/m y se identifica el sistema de condiciones más exigentes, podrían ser suficientes los datos de estabilidad en dicho sistema (la condición más exigente es la de 10 unidades en una presentación en frasco de 75 mL con una MVTR = 0,04 mg/(día × unidad)) después de consultar con la FDA. El Dr. Klein recomendó adicionalmente a la industria planear con antelación y tomar la decisión sobre el sistema de envasado en las etapas tempranas del proceso de revisión para evitar la necesidad de un cambio inesperado en el envasado en una etapa avanzada de la revisión reglamentaria. Asimismo, el Dr. Klein invitó a la industria a tomar ventaja del uso de la WVTR/m para generar Protocolos de Comparabilidad para reducir aún más el volumen de informes y el tiempo de revisión. Sistema de clasificación de la USP En la actualidad, el Sistema de Clasificación de la USP para sistemas de envase-cierre de medicamentos de unidades múltiples se limita a dos categorías: “bien cerrado” e “impermeable.” De éstas dos, “impermeable” se usa comúnmente en la Declaración sobre Envasado y Almacenamiento de las Monografías. Aunque no se usa en la Declaración de Envasado y Almacenamiento de las Monografías, el capítulo USP 〈671〉 también define clases para blísteres (Clases A, B, C y D). No obstante, en el taller, se expuso la preocupación de que dichos términos carecen de rigor para definir los sistemas de envasado farmacéutico, los cuales, por diseño, logran una mayor protección que los viales de las farmacias, a los que estuvieron destinadas originalmente las clases de unidades múltiples. La clase A, que es la clase de blíster más restrictiva, no describe adecuadamente las películas de barrera alta que se usan en la actualidad. Se notó que la mayoría de las monografías de formas farmacéuticas orales sólidas de la USP y su etiquetado correspondiente especifican “conservar en un envase impermeable” (preserve in a tight contai- 26 revista safybi ner), aunque el significado de esta declaración es ambiguo y no describe con exactitud la protección requerida para los medicamentos terminados sensibles a la humedad. Asimismo, se discutieron los desafíos de usar el sistema de clasificación vigente, al igual que las ideas para mejorarlo. Algunos productos farmacéuticos sensibles a la humedad requieren un nivel de protección más estricto que “impermeable” y Clase A. Por lo tanto, se requieren acciones para tratar esta cuestión, en particular cuando las clasificaciones se usan para operaciones de reenvasado y dispensación. Los cambios al sistema de clasificación tendrán un impacto tanto en la USP como en la industria debido al uso generalizado del sistema de clasificación actual en la USP y la industria. Dicho impacto incluye la incorporación de nuevas normas y definiciones y lograr la alineación en todos los segmentos de la industria. Se realizaron dos sugerencias con respecto a cómo se podría tratar el asunto: 1) Eliminar de las monografías la mención sobre la clasificación y permitir que los requisitos de envasado se determinen mediante los datos de estabilidad del fabricante y/o; 2) Volver a definir el sistema de clasificación para farmacéuticos y reenvasadores dado que estas dos partes interesadas son los principales usuarios del sistema. Resultados del taller Durante el taller de dos días, las partes interesadas compartieron información y trataron una gran variedad de temas, preocupaciones e ideas sobre mejoras. Los puntos y cuestiones claves incluyeron lo siguiente: • Un objetivo importante de amplio alcance es determinar un intervalo aceptable probado (PAR) para permeabilidad a la humedad, con valores PAR diferentes para medicamentos individuales. Este método centra la atención en el producto mismo, no en el sistema de envase-cierre, lo que representa un cambio en la perspectiva con respecto a la permeabilidad a la humedad. • Las partes interesadas necesitan acceder a datos exactos sobre MVTR/unidad para medicamentos, los cuales son reenvasados o dispensados, particularmente en el caso del despacho de órdenes por correo. • Resulta importante usar el “envasado óptimo”, evitando el sobreenvasado para reducir costos. • Los dispensadores y reenvasadores necesitan contar con información adicional para comprender los requisitos de protección con respecto a la permeabilidad a la humedad. • La alineación entre las partes interesadas (FDA, USP, compañías farmacéuticas, dispensadores, reenvasadores y fabricantes de componentes de sistemas de envase-cierre) es crítica. • Todas las partes interesadas desean minimizar los riesgos relacionados con las fallas de estabilidad. La Guía de la FDA emitida en 1999 debe ser actualizada para contemplar el uso de MVTR/m a fin de sustentar la reducción en los requisitos de estabilidad y las exigencias reglamentarias para los cambios. • Existe la necesidad de alinear esfuerzos por parte de fa- USP bricantes, reenvasadores (los reenvasadores compran los medicamentos a los fabricantes) y dispensadores. En el pasado, el sistema de clasificación ha representado un medio para describir la selección del sistema de envase-cierre para reenvasado y dispensación, pero se ha determinado que el sistema de clasificación existente no provee una descripción exacta de los requisitos de protección para el producto. • La industria desea mayor flexibilidad que la prescrita en el método propuesto sin tener que presentar sus métodos para medir la velocidad de transmisión de vapor de humedad en sus presentaciones reglamentarias. Por ejemplo, el uso de agua en lugar de desecante ofrece muchas ventajas sobre el desecante y emplea los mismos principios del método gravimétrico. Esto significa que se emplean métodos gravimétricos simples basados en principios idénticos a los métodos con desecante para determinar la velocidad de transmisión de vapor de humedad de frascos y blísteres rellenos de agua. La industria considera que dichas modificaciones deben incluirse dentro del alcance de esta norma USP dado que los datos de la velocidad de transmisión de vapor de humedad se incluyen en la sección de aptitud del CTD (3.2.P.2.4), no en la sección de compromiso (3.2.P.7). Por ende, los datos y métodos no representan especi- 28 revista safybi ficaciones y no es necesario hacer referencia a la USP (en 3.2.P.2.4). La carga de demostrar la aptitud recae sobre el fabricante. Sin embargo, para los propósitos de clasificación de un sistema de envase-cierre (por parte de reenvasadores y dispensadores), resulta apropiado hacer referencia al método de clasificación de la USP. Pasos siguientes para el comité de expertos de la usp en envasado, almacenamiento y distribución Las discusiones en el taller enfatizaron la importancia y el significado de este esfuerzo de revisión. El Comité de Expertos se encuentra revisando los comentarios escritos recibidos sobre el capítulo revisado en el PF 39(2), así como los comentarios y sugerencias recibidas durante el taller. El Comité de Expertos anticipa que la revisión propuesta que aparece en el PF 39(2) se oficializará con cambios mínimos. Los cambios futuros a las definiciones de “impermeable” y “bien cerrado” y a los métodos existentes en la Sección 2 se realizarán después de consultar en mayor detalle con las partes interesadas y aparecerán en el PF para comentarios públicos. a División de Ciencias y Normas Globales de la USP. bComité de Expertos en Envasado, Almacenamiento y Distribución c Eli Lilly and Company n Conferencias, cursos, programas y seminarios Programa de capacitación en esterilización para farmacéuticos Módulo II Resumen preparado por la Dra. María Celeste González La Asociación Argentina de Farmacia y Bioquímica Industrial (SAFYBI) y la Asociación Argentina de Farmacéuticos de Hospital (AAFH) en el marco del acuerdo firmado entre ambas instituciones para desarrollar actividades de capacitación en Esterilización y Productos Médicos presento el segundo módulo del Programa de Capacitación en Esterilización para Farmacéuticos, complementando el iniciado el pasado año con el objeto de nivelar los conocimientos para aquellos farmacéuticos que deseen certificarse como especialistas en Esterilización de acuerdo a lo establecido por la Resolución Ministerial 1186 / 2013. En el primer módulo se abordaron temas relacionados con los métodos de esterilización, su aplicación y validación. En este segundo módulo se abordaron los temas relacionados con la gestión de centrales de esterilización en hospitales e industria, a través de la presentación de nueve bloques temáticos que se desarrollaron poniendo énfasis en un aprendizaje por medio de la aplicación de conocimientos en casos prácticos complementando los contenidos teóricos. El curso estuvo bajo la dirección de la Dra. Nora Graña, y de la Dra. Alicia Avila y Dra. Eugenia Martínez Mónaco como miembros del comité de Docencia. La coordinación estuvo a cargo de la Dra. María Celeste González. Las citas consistieron en una clase semanal de seis horas durante 8 semanas y con evaluación final optativa, pudiendo optarse por asistir a la clase en forma presencial o a través de la web en simultáneo. Los temas presentados se detallan a continuación. 1. Estructura orgánica y funcional de una Central de Esterilización en una institución de salud. Marco normativo. Recursos humanos. Planta física, equipamiento. El contenido estuvo a cargo de la Farmacéutica Virginia Letché, Farmacéutica y Licenciada en Ciencias Farmacéuticas. Perito Farmacéutico. Jefa de Sala Farmacia del Hospital Sor María Ludovica. Integrante del Comité de Control de Infecciones del HIAEP “Sor María Ludovica” de La Plata. Instructora de trabajos prácticos del Curso de auxiliares y técnicos en Esterilización del Ministerio de Salud de la Pcia. de Bs As. Miembro de Comisión Directiva del Colegio de Farmacéuticos de La Plata y coordinadora del Departamento de Educación Profesional en dicha institución. Ex- Miembro de Comisión Directiva de la 30 revista safybi AAFH (Asociación Argentina de Farmacéuticos de Hospital).Disertante y panelista en numerosas jornadas, seminarios, cursos y congresos. Resumen de la presentación La organización y funcionamiento de las Centrales de Esterilización (CE) de establecimientos de salud públicos y privados de nuestro país se enmarca en la Resolución 102/2008 del Ministerio de Salud de la Nación. Esta resolución tiene como propósito unificar criterios para coordinar las actividades de las centrales de esterilización. Las centrales que ya estaban en funcionamiento al momento de la publicación de esta resolución tuvieron y tienen la posibilidad de alinearse a estas directrices de acuerdo a sus posibilidades, mientras que las nuevas Centrales de Esterilización tienen la obligación de hacerlo. La Res. 102 define a la CE como la estructura orgánica y funcional destinada a la recepción, limpieza, acondicionamiento, esterilización y dispensación de elementos estériles utilizados en el tratamiento de los pacientes internados y/o ambulatorios. También establece los aspectos a analizar para garantizar una adecuada calidad de los procesos. .Estos son: la planta física, el marco normativo de funcionamiento, el recurso humano, el equipamiento tecnológico y los indicadores de calidad. De manera destacada la resolución establece que en establecimientos de salud de cualquier nivel de riesgo las centrales de esterilización deben estar a cargo de un profesional farmacéutico. Acompañó esta unidad temática la Farmacéutica Maria Celeste González disertando sobre el tema “Ambientes Controlados” desarrollando conceptos de clasificación de áreas limpias, característica y materiales constructivos, métodos de medición y control microbiológico. 2. Materiales y su respuesta a los métodos de esterilización A cargo de la Farmacéutica Marisol Garcia Sarubbio, Farmacéutica y Licenciada en Ciencias Farmacéuticas. Facultad de Ciencias Exactas. UNLP. Especialista en prótesis, ortesis y dispositivos biomédicos. Facultad de Cs. Exactas. UNLP.Jefe de Residentes de Farmacia. HZGA “San Roque” de Gonnet. Farmacéutico asistente de planta permanente en el HIGA “Gral. San Martín” de La Plata. Asesora de las centrales de Esterilización del Instituto Médico Platense e Instituto de Diagnóstico de la Plata. Conferencias, cursos, programas y seminarios Resumen de la presentación Los biomateriales son sustancias naturales o sintéticas que al ponerse en contacto con los tejidos vivos no le provocan daño o alteración, manteniendo su efectividad física y biológica. A ellos se les exigen dos cualidades fundamentales: Biocompatibilidad y Propiedades Mecánicas acordes al Uso Previsto. Pueden clasificarse según su origen en naturales o sintéticos, según su composición en simples o compuestos, según su estructura en metales, cerámicos o polímeros y según se degraden o no. Los productos médicos (PM) que requieren esterilización terminal en su proceso de fabricación o aquellos reusables, semicriticos o críticos, que la requieren como último paso del proceso de descontaminación, están constituidos por biomateriales. Su exposición a las condiciones propias de cada método de esterilización y a la de los procesos previos como lavado enjuague y secado, si corresponde, puede provocar alteraciones en los mismos dando lugar a cambios en sus propiedades físicas, químicas y/o mecánicas, lo que en algunos casos resulta en la perdida de la funcionalidad del PM e incluso puede hacerle perder la condición de biocompatible. Es fundamental contemplar la compatibilidad de los biomateriales constituyentes de un PM, con el método de esterilización elegido y controlar las condiciones de los procesos a los que se los somete, como por ejemplo: calidad del agua y su sistema de distribución, correcto funcionamiento del sistema de aireación para PM esterilizados por óxido de etileno, entre otros, a fin de lograr el objetivo tanto de la Industria como de la Central de Esterilización Hospitalaria que es obtener PM seguros y eficaces para el uso en pacientes. 3. Limpieza y desinfección de materiales A cargo de la Farmacéutica Especialista Natalia Sigyel, Farmacéutica Especialista en Esterilización. Jefa de Farmacia y Esterilización del Instituto Cardiovascular de Buenos Aires. DT de importadora de productos médicos, American Fiure. Ex Jefa de Farmacia y Esterilización de la Clínica Del Sol y Clínica Bazterrica. Ex jefa de Farmacia y Esterilización del Sanatorio de la Trinidad San Isidro. Resumen de la presentación La condición de limpieza previa a la esterilización y/o desinfección de los materiales es un hecho incuestionable. Esta afirmación se basa en que el éxito del proceso de esterilización se apoya fundamentalmente en esta etapa. Por lo tanto, no se esteriliza lo que está sucio. Lamentablemente en la práctica hospitalaria nos llegan a veces, a la central de esterilización, materiales que no han recibido un correcto proceso de limpieza, contribuyendo de este modo al fracaso de la desinfección y/o esterilización. Para mejorar esto, es necesaria la toma de conciencia, e involucrar a todo el personal que interviene en alguna de las etapas de manipulación de los dispositivos, para que puedan colaborar en el cumplimiento de las buenas prácticas del proceso. Por otro lado, en el ámbito del cuidado de la salud, se utilizan un gran número de desinfectantes. Hay que tener en 32 revista safybi cuenta que cada uno tiene sus características de actividad y así el usuario puede seleccionar el desinfectante apropiado y utilizarlo del modo más seguro y eficiente. Algunos desinfectantes están formulados en combinaciones que pueden alterar su actividad antimicrobiana. Cada formulación de ingredientes activos e inertes es considerada un producto separado y debe someterse al proceso de aprobación y registro de la Agencia de Protección Ambiental, dentro de los EEUU, o de la ANMAT, en la Argentina. Finalmente, las dermatitis de contacto entre el personal de la Salud, se han asociado con el uso de varios desinfectantes. Por lo tanto, deben tomarse medidas de bioseguridad, para proteger al personal que los manipula y para reducir al mínimo la exposición a los mismos. 4. Empaques y relación con la caducidad de producto por perdida de la condición Estéril. Materiales empleados en los empaques. Tipos de empaques. Métodos de empaque. El empaque en función del método de esterilización. A cargo del Farmacéutico Damián J. Ramírez, Farmacéutico egresado de la Universidad de Buenos Aires (UBA). Farmacéutico Especialista en Esterilización, Facultad de Farmacia y Bioquímica (UBA). Magister en Control de Infecciones Hospitalarias. Universidade FAMESP (São Paulo, Brasil). Miembro de la Comisión de Expertos en Productos Médicos y Esterilización, SAFYBI. Disertante invitado en numerosas Jornadas y Congresos, relacionados a Productos Médicos y Esterilización. Director Técnico en la empresa Diavamedic, fabricante de Productos Médicos descartables. Evaluador de ITAES, Instituto Técnico para la Acreditación de Establecimientos de Salud. Amplia trayectoria en Centrales de Esterilización de Instituciones de Salud. Resumen de la presentación Los productos que son esterilizados y posteriormente almacenados hasta el momento de su uso, deben estar envueltos. El material de empaque utilizado para productos sometidos a esterilización terminal, debe cumplir con determinados requisitos. El fabricante del envoltorio y el usuario, deben corroborar mediante ensayos, presentes en nuestra FA VII edición o en normas nacionales e internacionales reconocidas, el cumplimiento de estos requisitos. Se debe corroborar principalmente el cumplimiento de la propiedad de barrera microbiana, su permeabilidad al agente esterilizante, su resistencia a través de ensayos que simulen situaciones de stress cotidiano, ausencia de sustancias tóxicas, repelencia al agua y otros. Sin olvidarnos de ensayos funcionales para el envasado final, tal como la resistencia del sellado. Una batería de normas existentes nos ayudan a incorporar en nuestros procedimientos habituales factores muy importantes para asegurar la calidad tanto del material de envoltorio como de la hermeticidad del paquete: Normas IRAM (3108, 3110, 3112, 3116, 3117), Normas EN ISO 11607:1 y 11607:2, Norma EN 868. La incorporación de nuevas propiedades a los materiales usados como envoltorio para productos con esterilización Conferencias, cursos, programas y seminarios terminal, tal como acontece en la actualidad, debe prestar mucha atención a efectos de evaluar si esas propiedades realmente aportan valor, y en ese caso conocerlas, someterlas a ensayos, divulgarlas profesionalmente, y determinar su posible incorporación a través de los envoltorios de nuestro uso diario. 5. Seguridad, Higiene y bioseguridad. Gestión de residuos sanitarios. Normativa Ambiental. A cargo de la Lic. Carla Figliolo, Bióloga, egresada de la Univ. Nac. de Córdoba, Argentina. Master en Ecoauditorías y Planificación Empresarial del Medio Ambiente (España). Especialista en Residuos sólidos y peligrosos (México). Especialista en Instrumentos y Políticas de Gestión Ambiental (España y Francia). Actual Coordinadora de la Unidad de Investigación y Desarrollo Ambiental de la Sec. de Ambiente y Desarrollo Sustentable de Argentina. Docente universitaria de formulación de proyectos y de evaluación de proyectos de la carrera de Gerenciamiento Ambiental de la Univ. de Ciencias Empresariales y Sociales, Docente titular de salud ambiental de la licenciatura en gestión ambiental de la Univ. Nacional Arturo Jauretche, Docente invitada de la Fac. de Derecho de la UBA y Docente de posgrado de la Univ. ISALUD de Buenos Aires y de la Univ. de la Matanza. Coordinadora del Grupo de Trabajo 4 de la Comisión Nacional de Investigación en Agroquímicos. Ha sido consultora de salud ambiental y desarrollo sustentable en la Representación Argentina de la OPS. Coautora del Proyecto de Ley de Residuos Peligrosos aprobado con modificaciones por la Ciudad de Buenos Aires. Los puntos tratados fueron: Condiciones del ambiente de trabajo en centrales de esterilización. Almacenamiento de sustancias químicas. Peligro de incendio. Riesgo eléctrico. Límites máximos permitidos en ambiente de trabajo para óxido de etileno y formaldehido. Características de las sustancias. Bioseguridad: concepto, diferencia con protección biológica, clasificación de los microorganismos infecciosos según grupos de riesgo, concepto de riesgo biológico, niveles de bioseguridad en laboratorios. Pictogramas de peligro. Ecuación de riesgo. Minimización del riesgo por exposición a sustancias químicas, gestión de residuos de establecimientos de salud: clasificación, principios ambientales, marco legal, contenedores adecuados para la gestión de residuos, señalética, generación de residuos, indicadores de gestión, documentación de gestión de residuos, profilaxis en el manejo de residuos biocontaminados, tratamiento de residuos biocontaminados, elementos de protección personal para las distintas instancias de manejo de residuos, procedimiento en caso de accidente con punzocortantes, manejo de derrames. 6. Control de infecciones hospitalarias. Factores de riesgo. Medidas de aislamiento. Muestras biológicas ambientales. Red de agua potable Dictado por la Farm. Maria Eugenia Sayavedra, Farmacéutica Especialista en Esterilización, Jefa del Servicio de Esterilización del Hospital Central desde febrero 1992. Miembro del Comité de Infecciones Hospital Central Comisión Permanente de Esteriliza- 34 revista safybi ción- Ministerio de Salud, Mendoza, Integrante de la Comisión redactora de la Resolución 2860/07 Ministerio de Salud Provincia de Mendoza, Guía Provincial de Normas de Esterilización. Docente del Posgrado de Especialista en Esterilización - Facultad de Farmacia y Bioquímica, Universidad Juan Agustín Maza. ECI Maria Laura Vernazzi, Especialista en Epidemiología y Enfermería en Control de Infecciones por el Honorable Consejo Deontológico de Enfermería de Mendoza. Certificada por ADECI (Asociación Argentina de Enfermeros en Control de Infecciones) como Especialista en Epidemiología y Control de infecciones. Vocal de la Comisión Directiva de ADECI. Supervisora en Control de Infecciones Hospital Central Mendoza. Integrante del Comité de Prevención y Control de Infecciones y Comité de Investigación del Hospital Central Mendoza. Miembro de Comisión de reglamentación de la Ley 7168 “Residuos patogénicos y farmacéuticos” de la provincia de Mendoza. Miembro de Comisión que redactó el anteproyecto de decreto de reglamentación de la Ley nº 7434 “Tatuajes y/o perforaciones corporales” de la provincia de Mendoza Resumen de la presentación En esta clase las disertantes presentaron una exposición de la problemática del Control de Infecciones en los nosocomios. La Dra. Sayavedra destacó varios puntos. El Farmacéutico Especialista en Esterilización debe participar activamente en el Comité de Prevención y Control de Infecciones. Un aporte esencial es el Sistema de Gestión de Calidad implementado en la Central de Esterilización ofreciendo seguridad y confiabilidad, fundamentales para el cumplimento de los objetivos del Programa de Control de Infecciones. Su intervención en la adquisición de Productos Médicos Reutilizables, búsqueda de procesos microbicidas adecuados para la institución, elaboración de instructivos de trabajos para el Reprocesamiento de endoscopios flexibles, selección de agentes químicos a utilizarse en la higiene de espacios físicos, son algunas de las tantas actividades realizadas por el Farmacéutico Especialista en Esterilización en el marco del Comité de Prevención y Control de infecciones, junto al resto del equipo multidisciplinario. Sin olvidarnos también de su participación en los planes de Capacitación de diversos aspectos vinculados al reprocesamiento de Productos Médicos, como son prelavado de los mismos, Manejo de Productos Médico estériles, entre otros. La ECI Vernazzi desarrolló ampliamente un análisis sobre el origen y consecuencia de las infecciones nosocomiales, y las acciones necesarias para su control, ilustrando con casos y situaciones específicas, y dejando en claro que el abordaje de la problemática deberá ser siempre en una gestión multidisciplinaria. 7. Gestión de la calidad. Registros. Trazabilidad. Indicadores de calidad y de producción. Sistemas de gestión de calidad. Mapa de procesos de una Central de Esterilización. Diagrama de flujo del proceso de esterilización. A cargo de la Farmacéutica Sandra Marcela Pedrini, graduada en la UNT. Profesora adjunta en la Cátedra de Farma- Conferencias, cursos, programas y seminarios cia Hospitalaria y Clínica. Universidad de Belgrano. Coordinadora Docente en la Carrera Tecnicatura Superior en Tecnología de Salud con Especialidad en Farmacia Hospitalaria dependiente del Ministerio de Salud de la Prov. de Buenos Aires. Instructora de Residentes en el Servicio de Farmacia y Jefa de Unidad de Internación de Esterilización del Servicio de Farmacia y Esterilización del H I.G.A. “San Roque” de Gonnet, La Plata. Farmacéutica de Planta Permanente del Servicio de Farmacia del mismo Hospital desde el 11 de julio del 2001 hasta la fecha. Resumen de la presentación El concepto de calidad ha evolucionado significativamente durante los últimos años. De ser concebido como un valor referido a las características físicas de bienes materiales, fue ampliando su contenido al de excelencia de la organización en su capacidad de dar respuestas a las expectativas y necesidades de sus usuarios. La calidad es el conjunto de propiedades y características de un servicio que le confieren la aptitud para satisfacer las necesidades implícitas o explícitas de nuestro clientepaciente. La adopción de un sistema de gestión de calidad es una decisión estratégica de la organización, por lo tanto debe: - Determinar los procesos del SGC (Sistema de Gestión de Calidad), la secuencia e interacción entre ellos, - Determinar los criterios y los métodos para el control de los procesos - Gestionar los recursos de: infraestructura, humano, ambiente de trabajo. - Realizar el seguimiento, control y el análisis de estos procesos El objetivo principal es conseguir que los procesos se lleven a cabo con eficiencia y eficacia, logrando la satisfacción de los pacientes principales destinatarios de los servicios. 8. Estructura, planta física y SGC en una central de Esterilización Industrial. Marco normativo y regulatorio. A cargo del Farmacéutico Gustavo Enriquez, graduado en la UBA. Especializado en Esterilización Industrial de acuerdo a programa AAMI (U.S.A. 2009/2013), Director Técnico de ASISTHOS S.R.L. (Servicios de esterilización para terceros) y miembro de la Subcomisión de Productos Médicos de SAFYBI. Experiencia laboral anterior en Laboratorio de Análisis Químicos, Refinerías de Maíz y Laboratorio de Control de Calidad y Desarrollo de PROQUIMA SAICYF y MOLINOS RIO DE LA PLATA SA (Planta Matarazzo). Disertante invitado en temas de esterilización industrial y relacionados: SAFYBI, Simposio de productos médicos y esterilización, Fudesa, Cladest, Congreso Latinoamericano de Microbiología de Medicamentos, Hospital Zonal General Agudos «Dr. Isidoro Iriarte», Hospital Alemán, Jornadas Regionales de Esterilización y Arquitectura Hospitalaria, Hospital General de Agudos “Dr. Cosme Argerich”, ISPE y otros. 36 revista safybi La exposición del Dr. Enriquez apuntó a presentar generalidades de funcionamiento para una planta de Esterilización Industrial y brindar un panorama sobre los recursos de todo tipo necesarios para su operación efectiva e Indicar requerimientos regulatorios y normativos. 9. Reuso de productos médicos diseñados para un sólo uso. Legislación Nacional e Internacional. Casos de protocolos y procedimientos aplicados en instituciones de salud en Argentina. A cargo de la Farmacéutica Maria Celeste González, Especialista en Esterilización UBA. Especialista en transformación de polímeros y plásticos INTI-UNSAM. Miembro de subcomisión de Productos Médicos de Farmacopea Argentina. Miembro de Comisión Directiva SAFYBI. Miembro de Comité de expertos de Productos Médicos y Esterilización SAFYBI. Miembro del grupo de trabajo en Esterilización de AAFH. Docente Universidad Juan Agustin Maza. carrera de Especialización en Esterilización y Dispositivos Biomédicos, materia “Materiales y Dispositivos Biomédicos II”. Docente en la Facultad de Ciencias Exactas UNLP, Carrera de Especialización en Prótesis, Ortesis y Productos Biomédicos, materias “Estructura y Diseño” y “Dispositivos Biomédicos, Control de Calidad y Legislación.” Directora Técnica de LEXEL SRL, empresa fabricante de Productos Médicos. Resumen de la presentación Se presentó la situación normativa Internacional sobre reuso de Productos Médicos de un solo uso, y el camino regulatorio emprendido por la CE en los últimos seis años, el de Brasil, y la situación normativa de Argentina. Participaron de este encuentro como invitados, miembros del departamento de plásticos del INTI, la Ing. Nora Schichi, y el Sr. Víctor Fernández aportando su experiencia en el estudio de los materiales plásticos y las superficies de estos, ilustrando con fotografías de microscopia electrónica. Participaron también como disertantes las Farmacéuticas Virginia Letche y Marisol García Sarubbio, quienes presentaron al auditorio un trabajo de recopilación de datos y encuestas realizado en la Pcia de Buenos Aires, sobre el tema reuso de productos médicos en los hospitales, además de sus experiencias personales. El auditorio cerró este encuentro proponiendo que desde las instituciones científicas y en conjunto con la autoridad de Salud Pública se trabaje para avanzar en el estudio y la actualización de la regulación del reuso de productos médicos diseñados para un solo uso, en nuestro país En la última clase se efectuó una evaluación final optativa, que fue rendida por la mayoría de los participantes, tanto presenciales como en la modalidad WEBEX. Cabe destacar que la modalidad a través de la Web en simultaneo permitió la inscripción, cursada y evaluación final a participantes de Misiones, Tierra del Fuego, Entre Ríos, así como de diversas localidades de la Pcia. de Buenos Aires. n Conferencias, cursos, programas y seminarios Desarrollo de un Programa de Especialización en Asuntos Regulatorios 1ª parte Nota del Editor: El Dr. Leonardo Fullone, Coordinador del Comité de Expertos en Asuntos Regulatorios, contestó nuestro cuestionario sobre las motivaciones, perspectivas y desafíos de esta actividad fundamental de la industria farmacéutica y de las expectativas que SAFYBI ha tenido en cuenta al desarrollar este importante Programa de Especialización. La Doctora Virginia Peluffo y el Dr. Ricardo Díaz, ambos integrantes del Comité de Asuntos Regulatorios, colaboraron también en la redacción de algunas de las respuestas a este cuestionario. ¿Cuáles fueron los objetivos que se fijó SAFYBI al desarrollar el Programa de Especialización en Asuntos Regulatorios (AARR)? ¿Quiénes diseñaron los contenidos y cómo se organizaron los mismos? Como todos sabemos, SAFYBI es la organización que concentra la mayor oferta de capacitaciones relacionadas con la Industria. Este es uno de nuestros principales objetivos. En relación a la Especialización en AARR, fue una iniciativa de los miembros del comité de expertos de asuntos regulatorios, quienes entendimos la necesidad de generar contenidos en esta actividad tan dinámica y de importancia estratégica para la Industria farmacéutica. Dentro de los objetivos fijados podemos mencionar: • Crear un programa de especialización anual • Abarcar los principales campos de aplicación de asuntos regulatorios • Convocar a especialistas y referentes tanto del sector privado como del público • Generar contenidos que sean de utilidad práctica para los profesionales que realizan actividades en Asuntos Regulatorios • Brindar los lineamientos gerenciales que tiene la actividad regulatoria y las interacciones con los demás departamentos de una organización. Los contenidos fueron diseñados por los integrantes del Comité, dentro de los cuales algunos disertantes fueron integrantes del Comité de Expertos en AARR (ver sección Comités de Expertos) en conjunto con otros disertantes convocados especialmente para este programa. Si bien todos han participado en mayor o menor medida, cabe destacar la importante labor de la Farm. Virginia Peluffo quien es la Coordinadora Académica del Programa de Especialización. ¿Qué actividades abarca Asuntos Regulatorios? Los Asuntos Regulatorios (AARR), también llamados Asuntos Legales, ya son una profesión dentro de las industrias reguladas como las de los productos farmacéuticos, dispositivos médicos, la energía, la banca, las telecomunicaciones, etc. Los Profesionales de Asuntos Regulatorios, en general, tienen las siguientes responsabilidades comunes: • Asegurar que sus empresas cumplan con todas las regulaciones y leyes relacionadas con su negocio (esto tiene un significado muy específico dentro de: las industrias de la salud, los productos farmacéuticos, dispositivos médicos, 38 revista safybi productos biológicos, suplementos nutricionales, etc.). • Trabajar con el estado, con las agencias reguladoras y con su personal sobre cuestiones concretas que afecten su desenvolvimiento, es decir, trabajar con la ANMAT, FDA o la EMA para productos farmacéuticos y dispositivos médicos, con Departamentos de Energía para empresas petroleras o de electricidad, o con la Bolsa de Valores para la banca, etc. • Asesorar a las empresas sobre los aspectos reglamentarios y describirles el «clima regulatorio» en torno a temas generales y/o particulares. ¿Cuál es el rol de la ANMAT e INAME y cómo se compara con las agencias regulatorias de otros países? La ANMAT (Administración Nacional de Medicamentos, Alimentos y Tecnología Médica) es un organismo descentralizado de la Administración Pública Nacional, creado en agosto de 1992. Su jurisdicción abarca todo el territorio nacional. Depende técnica y científicamente de las normas y directivas que le imparte la Secretaria de Políticas, Regulación e Institutos del Ministerio de Salud y posee un régimen de autarquía económica y financiera. Está integrada por Profesionales y Técnicos que realizan los procesos de autorización, registro, normatización, vigilancia y fiscalización de los productos que se utilizan en medicina, alimentación y cosmética humana. Colabora en la protección de la salud humana, asegurando la calidad de: medicamentos, alimentos, productos médicos, reactivos de diagnóstico, cosméticos, suplementos dietarios y productos de uso doméstico. Su principal objetivo es: “garantizar que los medicamentos, alimentos y dispositivos médicos a disposición de la población, posean eficacia (cumplimiento del objetivo terapéutico, nutricional o diagnóstico) seguridad (alto coeficiente beneficio/riesgo) y calidad (respondan a las necesidades y expectativas de la ciudadanía)”. Ejecutivamente, posee tres grandes áreas de administración y control: 1-INAME (Instituto Nacional de Medicamentos): Entiende en todo lo concerniente con medicamentos. 2-INAL (Instituto Nacional de Alimentos): Entiende en todo lo concerniente con alimentos. 3-Dirección Nacional de Tecnología Médica: Entiende en todo lo concerniente con dispositivos médicos y de diagnóstico. A estas tres áreas se agregan también en carácter de Direcciones Generales: Asuntos Jurídicos y Administración. Conferencias, cursos, programas y seminarios Las agencias regulatorias de casi todos los países Latinoamericanos y de América del Norte, e incluso de muchos países de Europa y algunos de Asia, poseen similares estructuras y objetivos de funcionamiento. ¿Cómo describiría la responsabilidad del Director Técnico? La responsabilidad del Director Técnico es clara e indelegable. Es la autoridad de referencia dentro de las empresas y tiene la máxima responsabilidad ante la autoridad sanitaria. De hecho desde cierto punto de vista son los ojos de la autoridad sanitaria en la actividad diaria, velando que se cumpla con todas las normativas vigentes de manera de asegurar que los productos fabricados sean seguros, eficaces y de calidad. A través de los años el rol del Director Técnico fue cambiando, en la medida que las actividades se fueron especializando en forma creciente. En los comienzos el número de profesionales en los diversos sectores era mucho menor y el Director Técnico tenía un grado de involucramiento casi exclusivo en las actividades técnicas. Hoy en día debido al grado de exigencia que demanda la actividad, una compañía requiere un mayor número de profesionales que acompañen al Director Técnico en la gestión. ¿Cómo se pueden atraer profesionales a esta actividad? ¿Cómo se puede demostrar y divulgar mejor todo el contenido científico, tecnológico y gerencial que componen la Actividad Regulatoria? Hay un ejercicio interesante que de vez en cuando deberíamos hacer y es comparar “como uno se percibe” con “como se nos percibe”. En el caso de los que ejercen actividad regulatoria, muchas veces eran percibidos aislados del resto del equipo. Actualmente y dado el creciente nivel de especialización, y actualización continua de las normativas, se requiere que los responsables y colaboradores de los sectores tengan un grado de involucramiento e interacción mucho más activo con el resto de los sectores de una compañía farmacéutica. Una de las maneras de atraer más profesionales a la actividad regulatoria es adoptar en forma definitiva un rol más proactivo en la operación. Justamente uno de los objetivos de este curso es brindar los lineamientos del rol neurálgico que tiene la actividad regulatoria dentro de una organización, aportando los contenidos científicos y técnicos más relevantes. Uno de los problemas más frecuentes hoy en día dada la volatilidad de la situación económica global, es el cambio continuo de las condiciones originales de registro: ¿cómo se encaró este tema en el curso? (De hecho, las palabras “cambio” y “modificación” aparecen en total 16 veces en el temario del Programa) No se encaró especialmente desde las condiciones cambiantes de registro, sino desde la comprensión de que el profesional de AARR es el que debe realizar el nexo entre la realidad social que se vive en el país y en el mundo y la realidad de la empresa para la cual está trabajando. Es el que debe estar al tanto de todos los cambios que se imponen a través de los organismos internacionales, para poder ir adaptando 40 revista safybi de a poco las condiciones de registro de los productos que tiene bajo su responsabilidad con respecto a las nuevas exigencias, y es el que debe transmitir las nuevas modalidades para formar conciencia a la hora de tomar una decisión con respecto a un registro nuevo. No hay otra forma de trabajar en AARR que manteniéndose constantemente actualizado, en cuanto a la normativa en desarrollo o ejecución y en cuanto a la realidad política, económica y social del país, de manera de poder adelantarse a los cambios y sumar productividad y desarrollo, y no simplemente burocracia. AARR no son sólo papeles a cumplir, es una filosofía a llevar a cabo para cumplir el espíritu impartido por la ley sin dejar de lado la necesaria rentabilidad que necesitan las empresas para sustentarse. Y este es el verdadero desafío que debe plantearse cada profesional que toma esta área. ¿Cuáles son las nuevas tendencias de habilitación de laboratorios sin áreas de producción y las actividades en terceros? Las últimas normativas, marcan una tendencia clara desde la autoridad sanitaria para que los laboratorios que no tienen áreas productivas, las incorporen al menos en alguna de las formas farmacéuticas que comercializa ¿Qué avances ha logrado la Red Panamericana para la armonización de la reglamentación farmacéutica? Existen distintos bloques regionales de América que están tendiendo a lograr criterios comunes en temas Regulatorios de Salud, como el Mercado Común del Sur (MERCOSUR) creado en 1991, formado por Argentina, Brasil, Paraguay y Uruguay, y en la actualidad posee como Estados Asociados a Bolivia, Chile, Colombia, Ecuador, Venezuela y Perú que participan en las discusiones técnicas pero no como miembros; el Acuerdo de Libre Comercio de Norteamérica (ALCN o North American Free Trade Agreement: NAFTA) cuyos países integrantes son Canadá, Estados Unidos y México; el Sistema de Integración Centroamericano (SICA) integrado por Guatemala, El Salvador, Honduras, Nicaragua y Costa Rica; la Comunidad del Caribe (CARICOM) cuyos países integrantes son Antigua y Barbuda, Bahamas, Barbados, Belize, Dominicana, Grenada, Guyana, Haití, Jamaica, Montserrat, Saint Lucia, St. Kitts y Nevis, St. Vincent y Grenadines, Surinam y Trinidad y Tobago, y sus miembros asociados Anguilla, Bermuda, Islas Vírgenes Británicas, Islas Caimán e Islas Turcos y Caicos. Todos estos grupos se están reuniendo y tratando de consensuar un acuerdo de registro común a través de la Red PARF (Red Panamericana para la Armonización de la Reglamentación Farmacéutica), la última reunión fue en Washington en junio del 2011 y en ella se efectuaron Recomendaciones para la Evaluación de Productos Bioterapéuticos Similares, pero hasta el momento sólo son propuestas. En resumen, a diferencia de la Comunidad Europea, en América todavía no está implementado un sistema unificado de registro de medicamentos, ni siquiera a nivel de los distintos subbloques regionales (Mercosur, Comunidad Andina, etc.), de manera que los registros de medicamentos se realizan de acuerdo a la normativa de cada país y por eso cada país tiene su organismo regulatorio semejante a nuestra ANMAT.n Biotecnología Vacuna contra la fiebre hemorrágica argentina, un producto nacional Breve reseña histórica y descripción de los procesos implementados para garantizar su calidad Dra. Laura Riera Historia La fiebre hemorrágica argentina (FHA) emergió en la década de los cincuenta como brotes epidémicos caracterizados clínicamente por fiebre, manifestaciones neurológicas, alteraciones renales y hepáticas, leucopenia y plaquetopenia (1). Su agente causal, el virus Junin (VJ), fue aislado en 1958 (2). El ratón de campo Calomys musculinus, reservorio principal del VJ (3), desarrolla infecciones crónicas y elimina virus a través de saliva, orina y materia fecal. La vía de infección más importante es la inhalación de aerosoles de las excretas de los reservorios infectados. En 1965 el índice de mortalidad era cercano al 30%. Se comenzó a organizar y desarrollar un programa cuyo objetivo era controlar la FHA. En la fase preparatoria, se estableció en Pergamino, BA, un centro de estudios sobre FHA. En esta etapa se probaron las técnicas y la organización del sistema en un área limitada y se logró establecer la eficacia del plasma inmune para el tratamiento de la enfermedad (4). Durante la puesta en marcha la primera misión del Instituto Nacional de Estudios sobre Virosis Hemorrágica (INEVH), dirigido por el Dr. Julio I. Maiztegui, fue la organización del Programa Nacional de Lucha contra la FHA con los objetivos de reducir: • la morbilidad mediante la implementación de un sistema de notificación de casos con diagnóstico clínico, un sistema de confirmación laboratorial, actividades continuas de educación para la salud y formación de recursos humanos • la letalidad mediante la promoción de un sistema de diagnóstico clínico precoz y tratamiento temprano de los enfermos, instalación de una red de servicios en el área endémica para la consulta de la población y de bancos de plasma inmune en las cuatro provincias afectadas (Buenos Aires, Santa Fe, Córdoba y La Pampa). El programa ingresó en su fase de consolidación, mantenimiento y actualización en la que se involucraron efectores públicos y privados, la comunidad y organizaciones no gubernamentales. Se organizó la descentralización de la atención médica y la continuidad de actividades dirigidas a reducir la letalidad mediante la capacitación del personal de salud para el diagnóstico precoz y un programa de educación para la salud dirigido a la población. Se decidió comenzar con el proyecto de obtención de una vacuna. 42 revista safybi Proyecto de desarrollo y producción de una vacuna para prevenir la fha El proyecto de obtención de una vacuna contra la FHA fue iniciado en 1978 por un convenio internacional y en 1979 se definieron las siguientes metas: • Enviar a un científico designado por Argentina a Estados Unidos para preparar una Semilla Maestra de virus Junin atenuado dado que el país no se contaba con instalaciones apropiadas. • Construir un laboratorio en Argentina para producir y controlar la vacuna desarrollada. Entre 1979 y 1989 se llevó a cabo la construcción de las primeras instalaciones de un edificio de Nivel 3 de Bioseguridad en el INEVH. En 1983 el Dr. Julio Barrera Oro culminó, en los laboratorio de Estados Unidos el desarrollo de la vacuna Candid #1. Los procedimientos de atenuación incluyeron sucesivos pasajes en huéspedes certificados (animales de laboratorio libres de patógenos específicos (SPF) y cultivos de células diploides certificadas), selección clonal por pseudosingle burst y dilución terminal. Se realizaron ensayos preclínicos donde se demostró seguridad (inocuidad) por ausencia de neurovirulencia (monos Rhesus), ausencia de neurotropismo y de enfermedad, estabilidad genética, ausencia de persistencia viral en monos, inmunogenicidad en cobayos y monos Rhesus y eficacia protectora mediante desafío con cepa aislada de paciente. Se inició la producción en The Salk Institute, Swiftwater, Estados Unidos. Con los lotes obtenidos se realizaron los ensayos clínicos de fase I y II en Estados Unidos y Argentina en los que se demostró la inocuidad de Candid #1 y una inmunogenicidad superior al 90%. Los estudios de fase III se realizaron en Argentina entre 1988 y 1990, “Estudio prospectivo, aleatorio, a doble ciego, utilizando placebo”, en el cual se demostró una eficacia del 95,5 %. A partir de ese momento se comenzó con una vacunación selectiva bajo protocolo de uso compasivo con una efectividad resultante de un 98 % (5). Historia de la producción en Argentina de la vacuna contra la fha Las principales acciones se detallan a continuación (Tabla A) Proceso de elaboración de la vacuna candid # 1 Para la obtención de la vacuna se utilizan cultivos de células FRhL-2 (pulmón de feto de monos Rhesus). La presentación es en frascos de producto liofilizado que contienen 10 dosis Biotecnología Tabla A: Historia de la producción en Argentina de la Vacuna contra la FHA Años Acciones 1995 2002 • • • • 2001 • Habilitación de la planta por el ANMAT (Disp. 3775/01) • Ensayo preclínico en cobayos. 20032004 2004 2005 Remodelación instalaciones INEVH (Figura 1). Transferencia de tecnología desde The Salk Institute al INEVH. Estandarización de los procesos de producción y control. Producción de los primeros lotes experimentales. • Subsidio de la SecTyP gestionado por la ANLIS para producción y liberación interna de los primeros lotes de Vacuna Candid #1 para ensayo clínico (6). • Obtención del registro de agua estéril para inyectables. • Realización del “Estudio Clínico Puente, en fase III, en voluntarios humanos sanos, de entre 15 y 65 años de edad, de ambos sexos, a riesgo de adquirir FHA, aleatorio, a doble ciego, para evaluar la comparabilidad entre la vacuna Candid #1 producida en la Argentina y en los Estados Unidos”, con resultados satisfactorios (7). 2006 2007 • Obtención del Registro de la Vacuna Candid #1(Res. ANMAT 4882/06, Certif. 53205) • Incorporación de la misma en el calendario nacional de inmunización en el área endémica de la FHA. 20082013 • Escalado de producción • Distribución (hasta septiembre de 2013) a los vacunatorios instalados en las provincias afectadas unas 329.330 dosis (8). Tabla B: Liberación del sustrato celular Ensayo de esterilidad para hogos y bacterias (cultivo) Los medios de cultivo son evaluados en su capacidad promotora de crecimiento demostrando que permiten el desarrollo de menos de 100 unidades formadoras de colonias (UFC) de cepas de referencia: Pseudomonas aeruginosa ATCC14502, Micrococus salivarius ATCC 14344, Escherichia coli ATCC 4157, Bacteroides distasonis ATCC 8503, Penicilium notatum ATCC 9478, Aspergillus niger ATCC 34467, Candida albicans ATCC 10231. Ausencia de contaminantes Ensayo de Micoplasmas por cultivo Se utilizan medios sólidos y líquidos y se incuban en condiciones aeróbicas y de microaerofilia. Se verifican las propiedades nutritivas de cada lote con cepas de referencia: Acholeplasma laidlawii ATCC 23206, Mycoplasma arginini ATCC23838, Mycoplasma pneumoniae ATCC 15535. Ensayo de Micoplasmas por método del indicador en cultivo celular o tinción de DNA Permite detectar micoplamas no cultivables. Se ensaya un sustrato, células Vero, inoculándolo con cepas de referencia y verificando la detección de un patrón particulado o filamentoso de fluorescencia sobre la superficie de la célula. Se inocula la muestra sobre el cultivo celular indicador, se realizan subcultivos y se realiza la fijación y coloración con bisbenzamida, se examinan por epifluorescencia. Se compara con los cultivos controles positivos y negativos y se evalúa la fluorescencia extranuclear. Ensayo de micobacterias Se inoculan medios sólidos apropiados: Lowestein – Jensen y Middlebrook 7H10. Se determina la fertilidad de los medios inoculando la cepa BCG de Mycobacterium. Ensayos in vivo Seguridad en animales: en cobayos, inoculados intraperitoneal e intracranealmente, se busca fundamentalmente la presencia de Virus de la Coriomeningitis Linforcitaria y Mycobacterium tuberculosis. En conejo, su busca la presencia de Virus B. En ratón adulto se busca virus de la Coriomeningitis Linforcitaria y virus Coxsackie. En ratones lactantes también se inocula el material intracerebralmente e intraperitonealmente. En huevos embrionados se busca la presencia de agentes adventicios, se inocula una determina cantidad de células viables en la cavidad alantoidea. Seguridad Ensayos in vitro Seguridad en cultivos celulares: se utilizan las siguientes líneas celulares: MRC-5, FRhL-2, RK-13, VERO C76, cultivo primario de embrión de pollo para la detección de potenciales agentes adventicios. Se inoculan las líneas celulares con el material en estudio y se observa la presencia de efecto citopatogénico y al finalizar el test se buscan virus hemadsorbentes mediante el agregado de una suspensión de glóbulos rojos. Ensayo de tumorigenicidad Se inocula una suspensión de la línea celular a probar en ratones nude (Nu/Nu), también se inocula igual cantidad de ratones nude con una línea celular tumorigénica (Hela). Los animales se observan y palpan en el sitio de inoculación a intervalos regulares para detectar aparición progresiva de nódulos, registrándose el tamaño de los mismos. Se realizan necropsias de los animales con toma de muestras del sitio de inoculación y de pulmón para estudios histopatológicos. Identidad Caracterización cromosómica Determinación de número modal. Al cultivo celular se le agrega colchicina para frenar la división celular en metafase, se colorea con Giemsa y se cuenta el número de cromosomas de 100 células en metafase. Características bioquímicas Se determina mediante la utilización de un kit comercial el perfil de isoenzimas. 44 revista safybi de vacuna con una ampolla de diluyente de 5,5 ml de agua estéril para inyectable. En el proceso de producción (Figura 2), se amplifica la línea celular y se inocula Fig. 1- Instalaciones del INEVH en Pergamino con el virus vacunal. Así se obtiene la cosecha única, la cual es sometida a un extenso proceso de control de calidad para su liberación. Las cosechas se mezclan, en función de la cantidad de vacuna a envasar, formando la mezcla de cosechas; luego se formula en un área grado A y se obtiene así la vacuna final a granel, la que será sometida al proceso de envase final en viales multidosis de 20 ml de capacidad para su posterior liofilización. Control y aseguramiento de calidad El proceso de elaboración de la vacuna y las distintas etapas en la liberación de materias primas, productos Figura 2: Esquema de producción de Vacuna Candid # 1 intermedios y producto final se realizan en el INEVH. El control de calidad comienza con las materias primas: controles fisicoquímicos, microbiológicos, ensayos para evaluar la capacidad promotora de crecimiento de los medios de cultivo, toxicidad para la línea celular sus- LABCO - Laboratorio de Control S.A. R Laboratorio de analisis fisico-químico, microbiológico y de efluentes para terceros O! SERVICI O V E U N Dentro de nuestro servicio se incluye: ¡ e Gluten Espectrofotometría de Absorción Atómica Espectrofotometría Infrarroja Análisis por Cromatografía Líquida (IR-DAD-UV-FL-COND) Microbiología Ensayos Fisicoquímicos Desarrollo y Validaciones de técnicas analíticas Análisis de efluentes Cromatografía gaseosa (contamos con varios detectores) Tamaño de Partícula (por Difracción Laser) Análisis d CALIDAD nuestra razón de ser HABILITACIONES - CERTIFICACIONES ISO 9001:2008 RI: 9000-2738 (Disp. Nº0688) Tte. Gral. Guido 1095 (1708) Morón Tel.: (54-11) 4483-4494/97 ó 4627-7774 • [email protected] • www.labco.com.ar revista safybi 45 Biotecnología Tabla C: Liberación de la cosecha única viral Ausencia de contaminantes Ensayo para demostrar esterilidad para bacterias y hongos por método directo. Se utiliza la metodología que se describirá en ensayos de producto terminado. Ensayos para detectar presencia de micoplamas por los métodos de cultivo y tinción y micobacterias descritos para el sustrato celular. Seguridad Ensayos de seguridad in vivo (ratón adulto, ratón lactante y cobayos) e in vitro, ambos descritos para el sustrato celular. En este caso es necesario neutralizar el virus vacunal con un antisuero específico obtenido en conejo para evitar los efectos del mismo en ratones lactantes y en las líneas celulares. Título viral Identidad Para este propósito se utiliza el ensayo de potencia in vitro que se realiza a la vacuna producto terminado. Identidad Se enfrenta una muestra de la cosecha única de fluido vacunal con antisuero específico debiéndose observar reducción del número de Unidades Formadoras de Placa bajo agarosa, se procede de igual forma con un virus de referencia. Tabla D: Liberación de la Vacuna Candid #1 liofilizada Ausencia de contaminantes Esterilidad: para evidenciar la ausencia de contaminación de microorganismos capaces de crecer en las condiciones del ensayo lo que confirma que la vacuna cumple con los requisitos, no siendo suficiente para suponer la esterilidad del lote; el tamaño de la muestra es restrictivo, dando una estimación grosera del estado de esterilidad del mismo. Dada esta limitación, es requisito la realización de la validación de llenado aséptico. A los medios de cultivo se les realiza control de esterilidad y ensayo de promoción de crecimiento desafiándolos con las cepas de referencia detalladas previamente. Seguridad (toxicidad inespecífica) Está indicado para demostrar la presencia de contaminantes tóxicos extraños inoculando ratones y cobayos. La vacuna cumple la especificación si los animales sobreviven el período del test, no exhiben ninguna respuesta y su peso no es menor al final del período de prueba que al momento de la inyección. Potencia Se determina mediante un ensayo in vitro en el que se cuantifica el efecto de lisis provocado por la infección del virus vacunal sobre una monocapas de células susceptibles (Vero 76). La adición de una cubierta de agarosa restringe los cambios del efecto citopatogénico a áreas discretas de la monocapa limitando la diseminación viral de célula a célula. Estas áreas focales de infección pueden visualizarse macroscópicamente como áreas discretas o placas con el agregado de un colorante para contrastar el área afectada (placa) con las células adyacentes sin afectar. El resultado se expresa en Unidades Formadoras de 4 5 Placa por mililitro (UFP/ml) y la vacuna producto terminado debe contener entre 1x10 y 9x10 UFP/ml. Identidad Consiste en enfrentar la vacuna con antisuero específico producido en conejos, adsorber la mezcla virus – suero en células Vero, revelar y calcular el título del antisuero, debiendo observarse que la vacuna ha sido neutralizada. Humedad residual por valoración culombimétrica según Karl Fisher. La vacuna Candid #1 debe contener una humedad residual < 3%. Pureza (se determina mediante la utilización de monitores) pH por potenciometría. El valor de pH requerido para la Vacuna Candid # 1 producto terminado es entre 6,5 y 7,5. Osmolalidad por depresión en el punto de congelamiento. La vacuna debe poseer una osmolalidad entre 260-330 mOsm/kg. Presencia de endotoxinas bacterianas mediante el ensayo de formación de gel del lisado de amebocito de cangrejo (Limulus polyphemus, LAL) el cual forma un coágulo cuando se lo mezcla con endotoxinas de bacilos gram negativos. El límite de endotoxina se calcula tomando en cuenta la dosis, la vía de administración y el peso corporal. El límite de endotoxinas para la vacuna Candid # 1 es de 700 EU/ml. trato e inhibición de la replicación del virus vacunal. Al material de envase, viales de vidrio borosilicato Tipo I incoloro se le realiza controles químicos y a los tapones elastoméricos gris siliconados autoclavables control de reactividad biológica evaluando su toxicidad sobre cultivos celulares por método de difusión en agar. A ambos y a los sellos de aluminio laqueados se les realiza además la verificación de dimensiones provistas por el fabricante En todo el proceso de control se utilizan métodos compendiados en la Farmacopea Argentina, otras farmacopeas reconocidas internacionalmente, métodos estandarizados reconocidos y métodos estandarizados en el INEVH; todos son sometidos a sus correspondientes validaciones. Se describen a continuación las etapas críticas de control. Sustrato celular Se utiliza un sistema de producción en banco el cual garantiza que el costo invertido en la caracterización sea válido a lo largo de la existencia del mismo. Se mantiene un 46 revista safybi registro completo de la genealogía de la línea celular: origen, historia de pasajes y medio usado para su propagación. La Semilla Maestra es establecida a un nivel específico de pasaje y se evalúa la viabilidad celular durante el periodo de almacenamiento. La liberación de los lotes de sustrato celular comprende la demostración de: ausencia de contaminación, seguridad e identidad (Tabla B). Cosecha única de fluido vacunal A cada lote de cosecha única de fluido vacunal se le realizan una serie de controles para su liberación (Tabla C). Vacuna Candid # 1 liofilizada Los estabilizadores que se utilizarán en la formulación, donde se mezclan con las cosechas virales liberadas, deben haber sido sometido a ensayos previos. • Los lotes de vacuna son sometidos, para su liberación a los ensayos descritos en la Tabla D. Controles de procesos Figura 3: Planta de tratamiento de agua para inyectables Esterilización: se utilizan métodos de esterilización por calor seco, calor húmedo y filtración. También se incorporan controles químicos, test de Bowie Dick, integradores e indicadores biológicos (IB). En el INEVH se producen y controlan IB. Los procesos de esterilización por filtración son controlados mediante la prueba de integridad del filtro y test de difusión. Llenado: verificación de la calibración de las bombas y toma de muestras para referencia en distintas etapas del proceso. Liofilización: ensayo de integridad y monitoreo continuo de temperatura de producto y vacío. Visualizaciones del 100 % de los lotes de vacuna y ampollas de agua estéril para inyectables. Equipo de doble ósmosis reversa Programmable Logic Controller y Electrodeionizador Agua estéril para inyectables El sistema de producción de agua purificada y para inyectables del INEVH, las que se utilizan para la preparación de diluyente de vacuna, medios y reactivos y procesos de lavado, está formado por filtro multimedia, columnas de intercambio iónico, equipo de ósmosis reversa, electrodeionizador y un destilador de cuatro efectos (Figura 3). El monitoreo diario de la planta comprende: controles de caudales y caídas de presión de los filtros, de los ablandadores, de la ósmosis inversa y del electrodeionizador; mediciones de conductividad en línea de anillos de distribución de agua purificada y de inyectables; medición de dureza y niveles de cloro. El programa de control de los sitios de uso abarca: controles microbiológicos (recuento en placa estándar y recuento en membrana heterotrópica por método de filtración por membrana; búsqueda de Escherichia coli y de Pseudomonas aeruginosa; determinación de endotoxinas bacterianas por el método de formación de gel de amebocito de limulus (LAL) y controles de pH, conductividad y carbono orgánico total. Animales de experimentación utilizados en los controles de calidad Ratones libres de patógenos específicos -specific pathogen free (SPF)-, cobayos y conejos controlados. Los animales son criados bajo barreras sanitarias. Se realizan controles microbiológicos y químicos de cada lote de alimento balanceado Los ratones SPF se estudian para garantizar la ausencia de una gran variedad de patógenos específicos (bacterias, virus y parásitos) Líneas celulares utilizadas en los controles de calidad Las líneas celulares que se utilizan en los controles de calidad se someten a controles de: esterilidad, búsqueda de mycoplasmas, TB in vitro e identidad. Aseguramiento de la calidad Monitoreo ambiental Durante los procesos se realizan monitoreo ambiental de las áreas grado A, B, C y D: recuento de partículas totales, recuento de partículas viables mediante método volumétrico de impactación sobre placa con medio Agar Tripticasa Destilador de cuatro efectos, tanque de almacenamiento y sistema de distribución Soja (1000 l de aire) y método de placas expuestas. Para el monitoreo del proceso de limpieza se colectan muestras de superficies pre y pos limpieza de puntos representativos de cada ambiente seleccionado utilizando placas RODAC. Se toman muestras de de vestuario estéril con placas RODAC del personal involucrado en tareas de producción de vacuna y cultivos celulares normales, test de esterilidad y manutención de ratones SPF. Los microorganismos son contados e identificados en género y especie. Se cuenta con un banco de contaminantes habituales los que son preservados apropiadamente y utilizados para promoción de crecimiento de medios de cultivo y desafíos de desinfectantes. El monitorio ambiental incluye la comprobación de la calidad de los productos desinfectantes. Se realizan controles químicos y microbiológicos para los que se utiliza una amplia variedad de cepas de referencia adquiridas por el INEVH de ATCC, las que son conservadas en sistema de banco en nitrógeno líquido (cepas de reserva y de trabajo) y controladas periódicamente para verificación de viabilidad y pureza. Calificaciones Se califica el equipamiento, áreas limpias, cámaras de Frío, estufas de esterilización, horno de depirogenación, estufas de incubación, freezers, liofilizador, esterilizadores, instrumentos analíticos. Calibraciones Se realizan calibraciones de conductímetros y pHmetros, revista safybi 47 Biotecnología manómetros, unidad Integritest, micropipetas, balanzas, medidores de temperatura. Certificaciones Se certifican plenos de aire acondicionado, flujos laminares y gabinetes de seguridad biológica (GSB), áreas de producción, campana de flujo laminar, manómetro Magnahelic, puesta a tierra, filtros cartucho, manómetros, balanzas, patrones metrológicos primarios (tercerizado) y secundarios, transductores de presión y temperatura de autoclaves Validaciones Se cuenta con un Plan Maestro de Validación el cual da origen a planes anuales los que contemplan la validación de: utilidades (sistema de producción de agua para inyectables y sistema HVAC), métodos analíticos (microbiológicos, biológicos, fisicoquímicos) y de procesos (limpieza y desinfección, esterilización y despirogenación, liofilización). Validación de llenado aséptico Se realizan los estudios de simulación del proceso de llenado aséptico (media fill) de modo de evaluar la confianza en la esterilidad del proceso. Los estudios de simulación incluyen formulación, filtración, llenado con medio apropiado, manipulación para la liofilización y encasquillado. Sistema de gestión Documentación Existe un sistema de documentación que permite garantizar la trazabilidad. El mismo comprende: Procedimientos Operativos Estándar (POE), Instructivos de trabajo (IT), Protocolos de Validación, Informes de Validación, Especificaciones, Certificados analíticos, Procedimientos de producción y registro de medios y reactivos (MR) y registro de producción de lotes (RPL), rótulos, etiquetas, prospectos. Auditorias Se cuenta con procedimientos de autoinspecciones y auditorías a proveedores. Capacitación La capacitación del personal es una prioridad institucional. El mismo recibe capacitación interna y externa y se lleva un registro del entrenamiento y calificación del personal. Mejora continua El INEVH en su conjunto se encuentra inmerso en una política de mejora continua. A modo de ejemplo, actualmente se adquirió y se está incorporando a la gestión institucional un software que permitirá la gestión segura y trazable de documentos, No Conformidades y Auditorías. A modo de conclusión Candid #1 es considerada una vacuna huérfana ya que está destinada a una población limitada de nuestro país estimada en 5.000.000 de habitantes y su comercialización no recuperaría los costos de producción. Es importante remarcar que en los últimos años ha continuado la extensión del área endémica de la FHA con cambios en los patrones eco epidemiológicos (8). Por otra parte existen estudios que demostraron que la vacuna puede resultar efectiva para la prevención de la fiebre hemorrágica boliviana. La situación de orfandad de la vacuna, la extensión de la zona afectada que comprende a cuatro provincias argentinas, ha otorgado al proyecto de elaboración de Candid #1 una indiscutible incumbencia de la salud pública nacional. Son objetivos del INEVH: sostener vigente la vigilancia de casos y reconocimiento temprano de la enfermedad, diseñar posibles estrategias de vacunación para tener el mejor resultado posible desde el punto de vista de la Salud Pública, realizar estudios de vigilancia de la actividad viral en sus reservorios para anticipar áreas probables de mayor riesgo, continuar con los estudios de fase IV, realizar estudios para vacunar niños y de persistencia inmune para evaluar necesidad de revacunación, y producir la cantidad de dosis de Candid # 1 necesarias para abastecer a la población a riesgo de contraer FHA. La FHA es una enfermedad no erradicable, por ser una zoonosis con reservorio distinto al humano. Con cantidades suficientes de vacuna para toda la población del área endémica, manteniendo una vigilancia epidemiológica activa y continua, la FHA es una enfermedad controlable. El INEVH cuenta, para la incorporación de proyectos estratégicos y nuevos desafíos, con capacidad instalada y personal con un amplio know how en la elaboración y control de biológicos. n Dra. Laura Riera: Bioquímica, doctora en Ciencias Biológicas, ha realizado la Carrera Docente Universitaria y es especialista en Planificación y Gestión en Ciencia y Tecnología en Salud. Se desempeña desde 1990 en el actual Instituto Nacional de Enfermedades Virales Humanas “Dr. Julio. I. Maiztegui” (INEVH) dependiente de la Administración Nacional de Laboratorios e Institutos de Salud ANLIS “Dr. Carlos G. Malbrán” (ANLIS), Ministerio de Salud de la Nación. Desde 1998 es Jefe de Control y Aseguramiento de Calidad del INEVH. Es docente de grado y posgrado de la Universidad Nacional del Noroeste de la Provincia de Buenos Aires (UNNOBA). Es socio activo SAFYBI y miembro de la comisión directiva de la División Alimentos, Medicamentos y Cosméticos de la Asociación Argentina de Microbiología. Referencias 1. Arribalzaga, R.A. Una nueva enfermedad epidémica a germen desconocido: hipertermia nefrotóxica, leucopénica y enantemática. Dia Médico 1955; 27: 1204-1210. 2. Parodi, A.S., Greenway, D.J., Rugiero, H.R., Rivero, E., Frigerio, M.J., De la Barrera, J.M, et al. Sobre la etiología del brote epidémico de Junin. Día Médico 1958; 30: 2300-2. 3. Sabattini, M.S., Gonzalez, L.E., Díaz, G., Vega, V.R. Infección natural y experimental de roedores con virus Junin. Medicina (Buenos Aires) 1977; 37 (Supl. 3): 149- 61. 4. Maiztegui, J.I., Fernández, N., Damilano, A. Efficacy of immune plasma in treatment of Argentine haemorrhagic fever and association between treatment and late neurological syndrome. Lancet 1979; ii: 1216-7. 5. Enria, D.A., Barrera Oro, J.G. Junin Virus Vaccines. Curr Top Microbiol Immunol. 2002; 263: 239-61. 48 revista safybi 6. Ambrosio, A.M., Saavedra, M.C., Riera, L.M., Fassio, R. La producción nacional de vacuna a virus Junin vivo atenuado (Candid #1) anti-fiebre hemorrágica argentina. Acta Bioquímica Clínica Latinoamericana, vol. 40, núm. 1, enero-marzo, 2006, pp. 5-17. 7. Enria, D. A. y GRUPO DE ESTUDIO DE LA VACUNA CONTRA LA FIEBRE HEMORRAGICA ARGENTINA et al. Vacuna contra la fiebre hemorrágica argentina Candid#1 producida en la Argentina: Inmunogenicidad y seguridad. Medicina (B. Aires) [online]. 2010, vol.70, n.3, pp. 215-222. ISSN 0025-7680. 8. Informe para la XXVII reunión anual del programa nacional de control de la fiebre hemorrágica argentina. Córdoba, provincia de Córdoba, 13 de septiembre de 2013. Calidad Herramientas de la calidad: síntesis y ampliación Auxiliares desde el análisis de situación hasta la institucionalización del cambio Dr. Mario Lisnizer Introducción La Calidad, como toda disciplina, necesita y cuenta con herramientas para desarrollar su tarea; lograr su uso frecuente y sistemático requerirá de algunos conocimientos teóricos y por supuesto de la voluntad de aplicarlos. El enfoque de este artículo será fundamentalmente práctico y consistirá en: • Recordar la existencia de un conjunto de herramientas disponibles para facilitar los trabajos en el ámbito de la Calidad. • Ofrecer una guía que ayude a decidir cuál herramienta utilizar en cada caso. • Incorporar al kit algunas herramientas para la toma de decisiones Por otra parte se presentará un nuevo conjunto de herramientas y se describirá una de ellas. Las herramientas de la calidad son metodologías que ayudan a: • Registrar datos • Controlar y mejorar los procesos • Comprender los síntomas de situaciones no deseadas • Identificar las causas de esas situaciones • Tomar decisiones • Verificar la eficacia de las acciones emprendidas En síntesis, su uso facilita y ofrece racionalidad al proceso de mejora continua de la calidad. En dicho proceso existen dos claras etapas, según la visión del maestro Juran de obligada cita en esta columna: El viaje del diagnóstico y El viaje del remedio; para ambas etapas las herramientas de la calidad no pueden faltar en el equipaje. Los datos y la información son el corazón de toda buena investigación y toma de decisiones, siendo las Herramientas de la Calidad los instrumentos recomendables para su obtención y análisis. Las primeras descripciones formales como tales e indicaciones para su uso sistemático pueden atribuirse a Kaoru Ishikawa, sus destinatarios eran principalmente supervisores de fábricas que integraban los círculos de calidad para la mejora continua. Por ello, a algunas de ellas también se las conoce como “Herramientas básicas para la calidad”, pues no requieren conocimientos de estadística y pueden utilizarse para analizar y resolver una amplia variedad de temas relacionados con la calidad. Tal como se mencionó anteriormente, además existe un conjunto de herramientas – complementarias de las anteriores - generalmente denominadas “Las nuevas herramientas de la calidad”- cuyo uso está orientado principalmente a facilitar procesos de gestión y planificación. Las Herramientas Básicas 1, 2 Ishikawa presenta formal o implícitamente las siguientes herramientas: Diagrama de flujo, Tormenta de ideas, Diagrama causa-efecto (particularmente conocido como diagrama de Ishikawa o espina de pescado), Diagrama de barras, Análisis o diagrama de Pareto, Histograma, Diagrama de dispersión, recolección de datos, gráficos y tablas. No es el objetivo de este artículo desarrollar cada una de estas herramientas, que por otra parte fueron analizadas en el “Rincón de la Estadística” de esta misma publicación (Vol. 129 a 137), sino sólo presentar una breve descripción de las mismas, su habitual campo de aplicación y los beneficios de su uso combinado. La propuesta es presentar en esta oportunidad una reseña para recordarlas y tenerlas a mano, de la misma manera que trabaja el mecánico en su taller. Sabe de su existencia y las usa selectivamente. En la tabla A se presenta un conjunto de herramientas básicas, con una síntesis de su descripción y principales usos. Por otra parte, la tabla B ofrece una guía para la utilización de múltiples herramientas para el análisis de causas, la selección y la implementación de acciones en la resolución de problemas o en la búsqueda de la mejora continua. Un uso adecuado y combinado de las herramientas facilita el desarrollo de la tarea, genera sinergia entre ellas y clarifica la situación desde la selección del proyecto hasta la institucionalización del cambio. La tabla B incluye algunas herramientas que no fueron mencionadas anteriormente; dos de ellas podrían sumarse a las clasificadas como básicas y corresponden a la etapa de toma de decisiones, éstas son: Tabla de decisión y Análisis de comparación de pares, mientras que otras como: Diagrama de afinidad y Diagrama de árbol se identifican habitualmente dentro del grupo de las nuevas herramientas. Tabla de decisión Es una tabla en la cual se presentan posibles opciones y se las enfrenta con diferentes criterios de evaluación considerados de importancia. Como criterios de uso general pueden considerarse aspectos como Relación costo-beneficio, Tiempo (duración del proceso de cambio), Resistencia al cambio, Eficacia técnica prevista; sumadas a características propias del proceso y ámbito de trabajo. En una industria regulada como la farmacéutica, varias de las decisiones deben tener en cuenta también otros aspectos, en particular los Legales. revista safybi 49 Calidad Además, es recomendable asociar a esta técnica con un formal análisis de riesgos a través del cual se puedan evaluar potenciales consecuencias no deseadas del cambio. Generalmente esta herramienta utiliza una escala no lineal para asignar los puntajes (1 = bajo, 3 = medio, 9 = alto) y se multiplican los mismos para obtener un resultado final. En algunos casos la relación es inversa (por ejemplo a mayor tiempo y a mayor resistencia al cambio los puntajes serán menores). En la tabla C se presenta un ejemplo, de tres posibles Tabla A: Herramientas básicas para la mejora continua de la calidad Herramienta Descripción Principales usos Histograma Representación gráfica que despliega la frecuencia de distribución de datos cuantitativos • Comunicar información acerca de variaciones de un proceso • Tomar decisiones para conducir las mejoras Diagrama de Pareto Un tipo especial de gráfico de barras, que muestra la importancia relativa de aspectos específicos • Determinar la frecuencia o la importancia relativas de diferentes Diagrama de causa y efecto Representación gráfica de una característica de calidad (efecto y) y las potenciales causas (variables x) • Organizar y desplegar las interrelaciones de varias teorías sobre las Hojas de chequeo Recolección objetiva de datos en una planilla previamente preparada • Clarificar datos y tendencias Estratificación Separación de datos en categorías • Analizar la situación previo a la recolección de datos • Identificar las fuentes de un problema Diagrama de dispersión Representación gráfica de la relación entre dos variables • Confirmar la correlación entre dos variables • Determinar el tipo de correlación Gráfico de control Representación gráfica basada en conceptos estadísticos • Interpretar información sobre el desarrollo de una variable en un problemas o causas • Concentrarse en cuestiones vitales causas de un problema proceso • Determinar con objetividad si un proceso está controlado o fuera de control Diagrama de flujo Representación gráfica de la secuencia de pasos para obtener un resultado • Disponer de una imagen del proceso para su análisis • Encontrar pasos del proceso susceptibles de mejora Tormenta de ideas Técnica de grupo con alta participación • Determinar las posibles causas y/o soluciones a problemas • Generar ideas constructivas y creativas Tabla B: Guía para el uso de múltiples herramientas en el análisis de causas y resolución de problemas Etapas del análisis y selección de las acciones Herramientas usadas frecuentemente Selección del proyecto / definición del problema • Tormenta de ideas • Diagramas de Pareto (sucesivos) Análisis de la situación actual • Gráficos de control • Estratificación Comprensión del proceso • Diagrama de flujo (elaboración del diagrama) Identificación de posibles causas • Diagrama de flujo (análisis del diagrama) • Diagrama de causa-efecto Recolección de datos • Hojas de chequeo Análisis de datos • Histograma • Diagrama de Pareto • Estratificación Identificación y selección de posibles acciones (soluciones) a implementar • Tabla de decisión • Comparación de pares Implementación de la posible solución o mejora Evaluación de los efectos • Hojas de chequeo • Gráficos y tablas varios Institucionalización del cambio / Normalización • Diagrama de flujo (del nuevo proceso) 50 revista safybi • Tormenta de ideas • Diagrama de afinidad • Diagrama de árbol • Gráficos de control • Diagrama de dispersión Tabla C: Tabla de decisión – Ejemplo de uso para facilitar la decisión de una acción a tomar Tabla D: Análisis de comparación de pares – Ejemplo de uso para un caso de 4 opciones evaluadas por 8 personas Criterios a evaluar Acciones posibles Beneficio vs. Costo Tiempo (duración del proceso de cambio) ResisEficacia tencia al técnica cambio prevista Total A/B A/C A/D A 2 4 3 B 6 C Reasignación de responsabilidades 3 9 (corto) 1 (alta) 3 81 Tercerización de los servicios 1 3 (medio) 1 (alta) 3 9 Automatización del proceso 3 3 (medio) 3 (media) 9 243 soluciones para la mejora de un proceso y obtención de un resultado orientativo para facilitar la decisión. Análisis de comparación de pares Es una tabla diagramada para comparar cada opción contra todas las demás, obteniéndose un resultado cuantitativo. Es una herramienta útil para ponderar la importancia relativa de diferentes alternativas, en particular cuando: • No están claras las prioridades • Las opciones son completamente diferentes • Los criterios de evaluación son subjetivos • Se desea clarificar prioridades y lograr consenso Respecto a este último punto, se considera oportuno mencionar que no existe un único método correcto para la toma de decisiones, cada uno presenta sus ventajas y desventajas, y su elección dependerá de un análisis previo. Una simple clasificación puede identificarlos según el 4 8 8 C/D Total 8 19 7 5 11 1 3 9 8 8 48 2 5 8 B/D 9 6 D Total B/C modo en que un individuo o grupo adopta sus decisiones: Autónomos (basado sólo en sus conocimientos y criterios), Consultativos (basado en sus conocimientos y criterios, pero sólo luego de conocer otras opiniones y Consensuados (todos concuerdan que se trata de la mejor decisión). El método aquí presentado facilita el consenso y se basa en asumir que tomar decisiones de a un par por vez es más fácil que seleccionar una opción entre varias. En la tabla D se muestra un ejemplo de cuatro posibles soluciones (A, B, C y D) analizadas por un equipo de ocho personas. Las Nuevas Herramientas 3 Para este grupo es habitual identificar estas siete herramientas: Diagrama de afinidad, Diagrama de flechas, Matriz de análisis de datos, Diagrama Matriz, Diagrama para el proceso de decisión, Diagrama de relaciones y Diagrama de árbol. El origen de esta compilación se debe a un trabajo realizado en el año 1976 por un equipo de profesionales pertenecientes a la Asociación de Científicos e Ingenieros de Japón (JUSE). Su objetivo era contribuir a encontrar formas para facilitar la innovación, la comunicación y la planificación de proyectos. revista safybi 51 Calidad Figura 1: Diagrama de afinidad – Ejemplo de uso en un análisis de posibles causas de errores en un laboratorio de control de calidad. A continuación se describirán las principales características del Diagrama de afinidad. Diagrama de afinidad Es una técnica creativa que requiere de la apertura mental de los participantes. Permite agrupar datos y hallar correlaciones entre los grupos resultantes. Es una herramienta útil para el análisis de un problema, generalmente cuando se dispone de datos cualitativos y se requiera: • Estructurar una cuestión compleja (extensa o complicada) • Desglosar una cuestión compleja en componentes que faciliten su comprensión • Agrupar causas relacionadas en clases, para su posterior análisis • Analizar la relación entre diferentes causas • Obtener consenso respecto a una situación Los pasos habituales para desarrollar un Diagrama de afinidad son los siguientes: 1.Constituir un equipo de trabajo y definir el asunto o problema a analizar. 2.Requerir a cada uno de los participantes que escriba sobre papeles para notas (preferiblemente adhesivos) breves descripciones de probables causas, cada una en un papel diferente. 3.Recoger los papeles completados. 4.Colocar los papeles sobre una mesa o pizarra y ordenarlos en grupos relacionados mediante la participación y consenso de todo el equipo. Es recomendable que los grupos principales (categorías) no sean demasiados, en general entre cinco y diez. 5.Crear títulos para cada grupo y dividir en sub-grupos aquellos conjuntos con mayor número de elementos 6.Analizar el diagrama resultante, formular las conclusiones (Tabla de teorías y sub-teorías) y delinear las próximas etapas del proyecto o mejora a iniciar . 7.Puede complementarse el uso de esta herramienta indicándose con flechas las relaciones identificadas entre grupos y/o sub-grupos. En la figura 1 se presenta el ejemplo de un diagrama 52 revista safybi completado en el cual además se han señalado algunas de las relaciones identificadas, como así mismo dos de las posibles causas principales. Tal como se ha señalado anteriormente, generalmente es recomendable el uso de herramientas en forma combinada, para el caso del Diagrama de afinidad la siguiente secuencia es una de las posibilidades: Tormenta de ideas à Diagrama de afinidad à Tabla de teorías y sub-teorías à Diagrama de causa y efecto à Diagrama de árbol. Conclusiones La evolución del hombre puede asociarse a su inventiva en la creación y uso de herramientas, aunque también podría postularse que fueron éstas las que contribuyeron en forma significativa a su evolución. De la misma forma puede hacerse un parangón entre una gestión profesional de la Calidad, en todos sus aspectos, y un adecuado conocimiento y aplicación sistemática de las herramientas. Quizás una forma risueña, pero no por ello menos cierta, para concluir este artículo sobre las herramientas de la calidad es recordar una máxima de Abraham Maslow “Si la única herramienta que tiene es un martillo, todo comienza a parecerse a un clavo”. n Dr. Mario Lisnizer: Químico y Bioquímico, graduado en la UBA, con estudios de posgrado en el país (Universidad Austral) y en el exterior (Kellogg Graduate School of Management – Northwestern University, USA) y cursos de capacitación en Bélgica, Brasil, Canadá, Estados Unidos e Inglaterra. Profesional con más de 35 años de experiencia en temas relacionados con la Gestión de Calidad. Coordinador y Auditor Líder del Programa de Armonización Industria Farmacéutica del Instituto Profesional Argentino para la Calidad y la Excelencia (IPACE). Auditor de Calidad Certificado por la American Society for Quality (ASQ). Juez del Premio Nacional a la Calidad. Ex Juez Internacional del Premio Iberoamericano de la Calidad (FUNDIBEQ). Consultor Internacional del IPACE con realización de trabajos en Latino América, España y África. Ocupó funciones de Calidad y Desarrollo gerenciales en Janssen Argentina y Johnson & Johnson. Es docente en temas de Calidad y Buenas Prácticas en IPACE e ITBA. Referencias 1. Juran, J.M. Manual de la Calidad – Vol II – pág. AV.1 2. Kaoru Ishikawa. Guía para el Control de la Calidad 3. Shigeru Mizuno. Management for Quality Improvement: The Seven New QC Tools Instalaciones y equipos Daniel A. Ferretti La entrevista al Sr. Daniel A. Ferretti, socio gerente de Ferretti Validaciones, fue realizada por Magdalena Nannei, Directora de la Revista SAFYBI. Daniel Ferretti es socio Gerente de Ferretti Validaciones, empresa dedicada a las actividades de conteo de partículas en áreas limpias y equipos de flujo laminar, determinación de patrones de flujo de aire, ensayo de fuga en filtros HEPA, medición de caudal de aire, medición de iluminación, medición de nivel sonoro, medición de presiones diferenciales, medición de velocidad, temperatura y humedad y verificación de equipos de seguridad biológica. Con el asombroso desarrollo que está teniendo la biotecnología, el tema de la bioseguridad se impone como uno de suma actualidad, por su complejidad e implicancias sociales. En base a su experiencia en la validación de cabinas de bioseguridad, ¿cuál debería ser el elemento más importante a tener en cuenta al decidir la compra de estos equipos? Antes que cualquier otro tema, se debe considerar el Riesgo, entendiendo como tal el Riesgo biológico principalmente para proteger al operador. Ese Riesgo se debe medir de acuerdo a la contaminación que se está manipulando o por el grado de contaminación. Existen distintas clases de gabinetes de bioseguridad, pero los de uso más común en la industria farmacéutica son los de la clase II tipo A2, ya que según la NSF 49, la misma es una cabina desarrollada para la protección del personal, producto y medio ambiente. Estos gabinetes tiene un frente con vidrio de seguridad y una abertura (aprox. 20cm) para ingreso solo de las manos, un flujo descendente de aire filtrado por filtros HEPA para la protección del producto, y una extracción filtrada por HEPA (exaustor) para la protección del medio ambiente, podrán adicionalmente tener un conducto al exterior que ayudará a generar una depresión en el ambiente en el momento de estar funcionando. La cabinas Clase II pueden ser de cuatro tipos distintos: A1, A2, B1 y B2. ¿Cuál es la diferencia entre las cabinas tipos A1, A2, B1 y B2 y qué razones existen para el uso de los distintos tipos? Estas cabinas se diferencian en cuatro tipos y se distinguen entre sí por ejemplo, en la velocidad de entrada del aire por la abertura frontal, en la cantidad de aire recirculado que vuelve a pasar por la superficie de trabajo y que sale de la cámara, en el sistema de extracción de aire, que determina si el aire se evacua hacia la sala o al exterior por su propio sistema de evacuación o por el sistema de evacuación de aire del edificio o forzado, por las presiones en el interior de la cámara (en unas los conductos y los Figura 1: Cabinas de Bioseguridad Clase II Tipo A1 y A2 plenos de distribución cargados de aire biológicamente contaminado están bajo presión , en otras, están rodeados por plenos y cámaras de distribución bajo presión negativa). La diferencia entre las cabinas A1 y A2 se pueden observar en la Fig. 1 En el gabinete Clase II Tipo A1 podría accidentalmente escapar la contaminación de su pleno positivo al ambiente donde está instalada la cabina por ejemplo, por fisuras, fallas en las juntas, etc., cosa que no sucede con la Clase II tipo A2 que como dijera, el pleno positivo está rodeado de otro pleno negativo, esta ultima es la más común en la industria farmacéutica. Los equipos Clase II A2 tienen las siguientes características: • Permite la protección del personal • Permite la protección del producto • Permite trabajar con microorganismos del grupo de riesgo 4 El detalle de los mismos está delineado en la Fig. 2. Las cabinas Clase II Tipo B1 y B2 son adecuadas para trabajar además con sustancia químicas y radionúclidos volátiles y son muy poco comunes en nuestro país por las condiciones especiales de su instalación. En la Fig. 3 se comparan los flujos de aire para ambos tipos. Usted indica en su página web que “La certificación de dichos equipos se realiza según especificaciones del fabricante y/o normas internacionales en los casos y etapas que así lo requieran, tomando como base la NSF 49.” ¿Cuál es la razón de la selección de la NSF 49? La NSF es una organización internacional no gubernamenrevista safybi 53 Instalaciones y equipos Figura 2: Flujo de aire en una cabina de seguridad Clase II Tipo A2 tal, sin fines de lucro, proveedora de soluciones globales de salud y seguridad basadas en la gestión del riesgo. En el Manual de Bioseguridad en el Laboratorio de la Organización Mundial de la Salud, en su tercera edición, se indica entre las referencias el estándar NSF 49, que ha sido designado como estándar ANSI y que se ocupa de las cabinas de bioseguridad en cuanto a su diseño, construcción, performance y certificación; el título del estándar en inglés es Biosafety Cabinetry: Design, Construction, Performance, and Field certification. ¿Según su criterio quiénes deberían formar parte del grupo encargado de gestionar la compra de una cabina de bioseguridad? El usuario debe elegir el equipo de acuerdo al Riesgo. Los departamentos de Calidad, Validaciones, Ingeniería, expertos en Higiene y Seguridad, Medio Ambiente y Compras, son algunas de las áreas que deben intervenir en la decisión de compra. Mi experiencia me indica que generalmente no se efectúan consultas previas y tan solo se solicita una cabina de bioseguridad sin especificar el tipo o lo que es aún peor, una cabina de flujo laminar vertical , que solo protegen al producto y no a las personas. El usuario debe definir la Clase y el Tipo, teniendo en cuenta: • La valoración del Riesgo • La selección del tipo de cabina de bioseguridad y el tipo de extracción • La evaluación del ambiente del laboratorio y el de la ubicación de la cabina en el mismo Say hello to streamlined sterile connections in seconds, anywhere Say goodbye to bulky, expensive tube welders Available in multiple sizes, Kleenpak™ sterile connectors are rigorously validated under worst-case conditions, so you can be sure the connection remains sterile every time... even outside a controlled air environment. w: www.pall.com/allegro e: [email protected] Providing Flexible Solutions © 2010 Pall Corporation. Pall, , and Kleenpak are trademarks of Pall Corporation. The ® indicates a trademark registered in the USA. GN10.3621 Instalaciones y equipos Tabla A: OMS, Manual de Bioseguridad en el Laboratorio, Relación de los grupos de riesgo con los niveles de bioseguridad, las prácticas y el equipo (CBS: cámara de seguridad biológica; TMA: técnicas microbiológicas apropiadas) Grupo de Riesgo Nivel de Bioseguridad Tipo de Laboratorio Prácticas de Laboratorio Equipo de Seguridad 1 Básico Nivel 1 Enseñanza básica, investigación TMA Ninguno; trabajo en Nivel 1 investigación mesa de laboratorio al descubierto 2 Básico Nivel 2 Servicios de atención primaria; diagnóstico, investigación TMA y ropa protectora, señal de riesgo biológico Trabajo en mesa al descubierto y CSB para posibles Aerosoles 3 Contención Nivel 3 Diagnóstico especial, investigación Prácticas de nivel 2 más ropa especial, acceso controlado y flujo direccional del aire CSB además de otros medios de contención primaria para todas las actividades 4 Contención máxima Nivel 4 Unidades de patógenos peligrosos Prácticas de nivel 3 más cámara de entrada con cierre hermético, salida ducha y eliminación especial de residuos CSB de clase III o trajes presurizados junto con CSB de clase II, autoclave de doble puerta (a través de la pared), aire filtrado Figura 3: Flujo de aire en las Cabinas Clase II, B1 y B2 des de flujo del aire (hacia abajo, hacia adentro), determinación de patrones de flujo de aire (mapeo de humo), la performance del ventilador, la clasificación de partículas (conteo) dentro del área de trabajo y muchísimas más. ¡La NSF tiene 14 requerimientos distintos y algunos de ellos son compuestos! ¿Cuántos niveles de bioseguridad existen y a qué están asociados? Los niveles de bioseguridad son cuatro: el nivel 1 es básicamente seguro, el nivel 2 implica algún riesgo, el nivel 3 se refiere a un nivel de riesgo serio y el nivel 4 involucra un riesgo extremo. La Tabla A extraída del Manual de Bioseguridad en el Laboratorio de la Organización Mundial de la Salud anteriormente mencionado detalla estos niveles y los requerimientos para cada uno de ellos. ¿Podría indicarnos cuáles son algunos de los ensayos requeridos por la NSF 49 y la razón de los mismos? La caída de presión en todos los plenos, la falta de pérdidas en los filtros HEPA llamada también integridad de filtros, realizándose en condiciones operativas normales verificándolo con generadores de aerosoles con DOP, PAO o un producto equivalente y detectores fotométricos de partículas, el nivel de ruido, la intensidad luminosa, que la vibración no exceda los límites establecidos, las velocida- 56 revista safybi ¿Cuál es la historia de la empresa a la cual usted dirige? Ferretti Validaciones es una empresa de servicios con absoluta independencia comercial y tecnica de aquellas que venden equipos y filtros, se inició en junio del año 2000 con el objeto de realizar la certificación y validación de áreas limpias, de bioseguridad y equipos asociados en referencia a la calidad de aire ambiental y aire comprimido. Estamos certificados por normas de calidad ISO 9001 y estamos en vías de obtener la acreditación de la norma ISO 17025 que establece los requisitos que deben cumplir los laboratorios de ensayo y calibración. Dicha acreditación del OAA, Organismo Argentino de Acreditación, seria en nuestro caso como laboratorio de ensayo para la verificación de filtros HEPA y conteo de partículas, como primer paso, es decir siempre apuntando a brindar la excelencia en el servicio al cliente. n Logística Cold Chain Congress, IPQC, mayo 2014, Panamá Dra. Sandra Rumiano Nota del Editor: El Dr. Federico Montes de Oca y la Dra. Sandra Olga Rumiano fueron disertantes en el Congreso organizado por el IPQC, International Quality and Productivity Center el pasado mes de mayo, en la ciudad de Panamá. La Dra. Rumiano a solicitud de la representante de IPQC coordinó como Chairman las actividades del Congreso. El mismo fue organizado en dos etapas, work shop pre-Congreso y dos jornadas de plenarios (ver fotos 1 y 2). El Congreso “Cold Chain and Temperature Management Logistics” Central & Latin America, se desarrolló bajo el lema “Best practices and Cost effective Solutions for a GDP compliant temperature controlled life sciences supply chain”, cuya traducción al español es “Mejores Prácticas y Soluciones Costo-beneficio aplicadas a medicamentos temperatura sensibles, en el marco de la cadena de abastecimiento en cumplimiento de las Buenas Prácticas de Distribución.” Los seminarios contaron con disertantes internacionales provenientes de Argentina, Colombia, México, España y Estados Unidos. El evento incluyó un plenario de discusiones, y presentaciones sobre modelos de transporte de productos biológicos, planes de inmunización y rol de la cadena de abastecimiento (Colombia), análisis de casos exitosos, como la aplicación de Six Sigma en GDP para productos sensibles a la temperatura (insulinas), novedades en tecnología de transporte activo vs. pasivo, entre otros temas. Las Configuraciones de Transporte Pasivos son los sistemas que normalmente están aislados con poliestireno, poliuretano o paneles aislantes similares, diseñados para una cierta capacidad de carga durante un período determinado de tiempo: 24, 72 ó 96 horas, al menos. Con estos tipos de configuraciones, quien envía la carga deberá n “Industry Tips Navigate Challenging Regulatory Compliance Requirements throughout Central & Latin America More Effi- Foto 1: Dr. Federico Montes de Oca Foto 2: Dra. Sandra Rumiano acompañarla para crear el “ambiente climatizado” requerido, utilizando paquetes de gel u otros tipos de materiales de cambio de fase con el fin de mantener la temperatura deseada. Por el contrario, las Configuraciones de Transporte Activo, suelen ser alquilados, son contenedores con monitoreo continuo de temperatura que funcionan con electricidad y/o batería; de acuerdo al diseño cuentan con sistema de refrigeración incorporado, o pueden, alternativamente utilizar hielo seco como refrigerante y un sistema de circulación interno de aire para forzar a que el aire frío llegue al área de carga para mantener una temperatura de ajuste específico. Estos tipos de configuraciones están diseñados para distintos volúmenes de carga, siendo de elección sin embargo para cargas de gran volumen. Al inicio y al cierre del Congreso los doctores Montes de Oca y Rumiano tuvieron la oportunidad de coordinar dos paneles de discusión coordinados en mesas de trabajo (ver Figs. 3 y 4), llegando a conclusiones que se compartieron con el resto de los participantes. Los temas tratados en los paneles de discusión fueron: revista safybi 57 Logística Foto 3: Mesas de Trabajo Foto 4: Mesas de Trabajo ciently”: cuyos principales lineamientos de trabajo fueron: 1.Aduanas: calificación de áreas de almacenamiento de acuerdo a los principios del Sistema de Calidad Farmacéutico 2.Componentes de la cadena de abastecimiento “aislados” y poco relacionados entre si: cómo romper las barreras para garantizar el flujo de la información intra e inter empresas 3.Puntos críticos y elaboración de planes de contingencia a aplicar en casos especiales: a.drogas para enfermedades “huérfanas” b.enfermedades raras 4.Aspectos relevantes y críticos en la capacitación de la cadena de abastecimiento de medicamentos y tecnología médica nas es un imperativo: como trabajar en conjunto, propuesta de las Empresas y Cámaras de Laboratorios/Colegios Farmacéuticos (de acuerdo a la estructura organizacional/País) 3.El impacto de la educación y el proceso de entrenamiento dentro de la empresa antes de iniciar un negocio: i. Trabajo en equipo ii. Compartir el conocimiento 4.El pensamiento estratégico es un requerimiento para lograr ahorros reales que van más allá de los costos económicos y considerar los costos de no calidad y de desabastecimiento o abastecimiento fuera de término 5.La exigencia del involucramiento de Marketing y Ventas en la cadena de abastecimiento integral 6.La identificación de los procedimientos que no pueden faltar en el Sistema de Calidad en el momento de controlar la cadena de abastecimiento de productos sensibles 7.¿Cómo capacitar al personal que forma parte de la cadena de abastecimiento y pertenece, por ejemplo, a terceros que brindan servicios logísticos? Roles y responsabilidades del profesional Farmacéutico y/o Bioquímico 8.Necesidad e implementación práctica de Acuerdos de Calidad entre los miembros de la Cadena de Abastecimiento n n “Networking Round Table Discussions”: los planes de trabajo más importantes fueron: 1.Empaque de productos sensibles a la temperatura: análisis de costo-beneficio, impacto en el medio ambiente, rol en la logística inversa, administración de inventarios 2.“Visibilidad” del control de temperatura en productos sensibles: seguimiento práctico y on line de las condiciones de almacenamiento y despacho 3.Inconsistencias detectadas en puntos críticos de despacho: Aduanas, propuestas de trabajo a desarrollar junto con las Autoridades Sanitarias locales y las Aduanas en pos de una harmonización regional 4.Aspectos críticos para garantizar la integridad de la cadena de abastecimientos de medicamentos y tecnología médica, incluyendo aspectos de “seguridad” (prevención contra robos y/o falsificaciones) Se establecieron algunas conclusiones importantes que seguramente originarán acciones en el futuro: 1.La necesidad de un rol pro-activo del farmacéutico/bioquímico en la Industria Farmacéutica en el cuidado de la cadena de frío: decisión en las inversiones, aporte en el diseño del empaque, aporte en el diseño de áreas y de sistemas de control de temperaturas 2.El acercamiento entre Autoridades Sanitarias y las Adua- 58 revista safybi Dra. Sandra Olga Rumiano: Farmacéutica y Bioquímica, UBA. Master en Administración de Empresas, Escuela Internacional de Negocios, Universidad de Belgrano. Senior Manager de Calidad GXP (CGMP, GSP, GDP, GCP, GxP, GLP) sistemas ISO. Auditor Líder ISO: 9001 IRCA / TÜV. Organización Mundial de la Salud como consultor senior QSM desde 2007. Consultor Profesional para la Industria Farmacéutica, Cosmética, Dispositivos Médicos, Alimentos. Especialista en sistemas de calidad y herramientas de calidad/mejora continua, auditorías, capacitación, proceso de transferencia, calificaciones y validaciones, asesora en inversiones en instalaciones y sus actualizaciones. Preparación de documentación y presentación (local/CTD). Anteriormente ocupó diversas posiciones gerenciales y de liderazgo en Laboratorios Andrómaco, Boehringer Ingelheim y Abbott Laboratories Argentina S.A. Nuestros anunciantes Avances en las técnicas de purificación de vacunas Resumen La vacunación ha sido una de las estrategias más eficaces para la prevención de las enfermedades infecciosas. Los programas mundiales de inmunización han permitido el control de las enfermedades graves, como la difteria, el tétanos, la tos ferina, el sarampión, las paperas, la rubéola y la poliomielitis, y han llevado a la erradicación de la viruela. Las vacunas de primera generación usan el agente infeccioso atenuado o inactivado que, aun siendo eficaz, presenta inconvenientes significativos. Las vacunas atenuadas pueden provocar efectos adversos graves, mientras que las inactivadas requieren una dosis de refuerzo y, frecuentemente, adyuvantes. Por otra parte, su caracterización físico-química no es fácil, lo que dificulta su control de calidad. Como respuesta a estas limitaciones, se desarrolló una segunda generación de vacunas compuesta de subunidades del agente infeccioso. Para que presentasen un perfil de seguridad y reactogenicidad adecuados, estos antígenos fueron altamente purificados, introduciéndose métodos avanzados de purificación en los procesos productivos, que aún son necesarios en las vacunas de tercera generación, basados en técnicas recombinantes. Este artículo revisa el progreso de las metodologías de purificación empleadas en la producción de vacunas con el fin de obtener un antígeno dentro de las especificaciones de los organismos reguladores con respecto a los niveles de impurezas, siendo al mismo tiempo robusto y viable técnico-económicamente para su uso a escala industrial. 1. Introducción La lucha contra las enfermedades puede ser de carácter profiláctico (vacuna/inmunobiológicos) o terapéutico (fármacos). En términos de coste-beneficio, la profilaxis de las enfermedades infecciosas mediante la vacunación es la herramienta de salud pública más ventajosa. Con la creación de la primera vacuna por Edward Jenner en 1796, la viruela fue la primera enfermedad humana erradicada con una campaña mundial de inmunización. En Brasil, con el Programa Nacional de Inmunización (PNI), ya se ha conseguido erradicar la viruela (1973) y la poliomielitis (1994). Además de la erradicación de estas dos enfermedades, Brasil ha conseguido un avance significativo en el control del sarampión, el tétanos neonatal, la difteria y la tos ferina. La difteria y la tos ferina disminuyeron un 96,3 % en siete años y un 98 % en 27 años, respectivamente (NHS, 2009). La rubéola es otro ejemplo de una enfermedad prevenible por vacunación que experimentó una disminución significativa (96 %) en el número de casos en tan sólo cuatro años, pasando de 5867 en 2001 a 233 en 2005. Las vacunas se clasifican en: (I) inactivadas; (II) atenuadas; (III) de subunidades, (IV) recombinantes; (V) de ADN. La mayoría de las vacunas virales se basa en la atenuación o inactivación del agente infeccioso, mientras que las células bacterianas en gran parte son de subunidades. Las vacunas atenuadas contienen el agente infeccioso en su forma atenuada pero con capacidad de multiplicarse en el organismo huésped, estimulando así la respuesta inmunológica idéntica a la infección natural debido a que el sistema inmunológico es incapaz de diferenciar virus de la vacuna del virus salvaje. Los ejemplos más comunes de las vacunas de virus atenuados son el sarampión, las paperas, la rubéola, la poliomielitis Sabin, la fiebre amarilla y la varicela. Como ejemplo de vacuna bacteriana atenuada se puede citar la BCG. Este grupo de vacunas presenta como desventaja la posibilidad de reversión de la atenuación, que puede conducir a la enfermedad, especialmente en individuos inmunocomprometidos (Cox, 2012). El virus inactivado, incapaz de multiplicarse, es el antígeno que integra la vacuna de virus inactivados. La respuesta inmunológica es principalmente humoral (producción de anticuerpos) con poca o ninguna respuesta celular. Los ejemplos típicos de este grupo de vacunas son: hepatitis A, rabia, poliomielitis Salk e influenza. Como ejemplo de vacuna bacteriana inactivada se puede citar la tos ferina celular. Como desventaja de las vacunas inactivadas cabe señalar una respuesta inmune más débil que conduce a la necesidad de dosis de refuerzo y/o adyuvantes (Zepp, 2010). Las vacunas de subunidades se componen de fracciones o subunidades del agente infeccioso, seleccionado debido a su capacidad de iniciar una respuesta inmune específica. Presentan mayor seguridad que las vacunas anteriormente mencionadas aunque se reduce su eficacia. Por esta razón, a menudo requieren la adición de adyuvantes o, en el caso de las fracciones de polisacáridos, la conjugación con una proteína transportadora. Los toxoides son antígenos de un grupo importante de vacunas de subunidades bacterianas. Como ejemplo podemos mencionar las producidas contra la difteria y el tétanos, donde la toxina de la bacteria se modifica químicamente por inactivación, manteniendo sus propiedades inmunogénicas (Almond et al., 2011). Otro ejemplo importante de este grupo son las vacunas de polisacáridos. Compuestas por el polisacárido capsular bacteriano, están siendo sustituidas por las vacunas conjugadas en las que estos polisacáridos están unidos químicamente a una proteína transportadora con el fin de provocar una respuesta inmune de larga duración. Las vacunas Haemophilus influenzae tipo b, contra la neumonía para los niños y contra la meningitis meningocócica C, son algunos de los ejemplos más importantes. Como vacunas de polisarevista safybi 59 Nuestros anunciantes cáridos se puede citar la vacuna contra la neumonía para los ancianos (Josefsberg y Buckland, 2012). La tecnología del ADN recombinante (ADNr) desarrollado en la década de 1970 hizo posible la manipulación del ADN y la producción, virtualmente, de cualquier proteína en forma recombinante. A partir de la década de 1980, se produjo una competición entre las industrias biotecnológicas para producir proteínas recombinantes en diversos sistemas de expresión con diferentes fines, incluyendo el diagnóstico, la profilaxis (vacuna) y la terapia (productos biofarmacéuticos), auxiliando en la identificación, prevención o tratamiento enfermedades de alta incidencia en la población (Mellado y Castilho, 2008). Mientras que las primeras vacunas de ADN viral todavía son objeto de ensayos clínicos, las vacunas recombinantes ya se comercializan desde 1980, cuando fue aprobada la vacuna recombinante contra la hepatitis B. Esta vacuna se basa en pseudo-partículas virales (PPVs) que contienen el antígeno de superficie del virus de la hepatitis B (HBsAg), siendo producida en levaduras (McAleer et al., 1984). Las PPVs consisten en agregados de, aproximadamente, un centenar de estos monómeros (HBsAg) y proporcionan una alta inmunidad cuando se aplican en seres humanos (Mello et. Al 2008). El uso de vacunas recombinantes basadas en PPVs para la prevención de enfermedades virales se ha incrementado significativamente en los últimos tiempos, consolidándose más recientemente con la aprobación de las vacunas 60 revista safybi Gardasil™ (Merck & Co) y Cervarix™ (GlaxoSmithKline) para la profilaxis del virus del papiloma, producidas en levaduras y en células de insectos, respectivamente. Sin embargo, existen algunas PPVs en ensayos clínicos, como la de la gripe (cepas H5N1 y H3N2, ambas en fase I y IIa), la del virus Norwalk (Fase I) y múltiples en fase de ensayos preclínicos (rotavirus, VIH, hepatitis C, norovirus, enterovirus, etc.), produciéndose la mayoría en células de insectos infectadas con baculovirus recombinantes (Roldão et al., 2010). En enero de 2013 la FDA (Food and Drug Administration) autorizó la Flublok® (Protein Sciences Corporation), una vacuna contra la gripe estacional que, aunque no es una PPV, por tratarse únicamente de una proteína del virus, también se produce en células de insectos (FDA, 2013). Actualmente, las vacunas virales humanas se pueden producir por tres vías diferentes: (i) la propagación del virus en embriones de pollo (in ovo), (II) la propagación del virus en cultivo celular de células de mamíferos in vitro y (III) la expresión de las proteínas estructurales del virus en las células huésped. El método clásico de producción consiste en realizar el cultivo estático de las células en el soporte adecuado, seguido de la infección viral, la recogida y purificación del virus y la formulación de la vacuna (Mello et al. 2008). La producción de vacunas bacterianas, de forma general, comprende las siguientes macro etapas: (I) propagación de la bacteria en cultivos en biorreactores, (II) separación de las células por microfiltración o centrifugación, seguida de clarificación, (III) purificación, (IV) formulación y (V) tratamiento final. La purificación se puede dividir en varias etapas, definidas por el sistema de expresión utilizado, por la localización del antígeno y por el grado de pureza deseado. Brasil destaca en el escenario de producción de vacunas humanas. Las principales instituciones productoras, la Fundación Oswaldo Cruz (Fiocruz) y Butantan, se crearon en 1900. Desde entonces, ambas han seguido creciendo y fortaleciendo sus estructuras y capacidad productiva. En 2009, estas dos instituciones suministraron el 83% de la demanda del PNI para vacunas humanas (Homma, 2009). Debido a su enorme población de casi 200 millones de habitantes, el gobierno brasileño decidió invertir en la capacidad productiva nacional de vacunas y otros productos biológicos para no depender de la importación de estos insumos esenciales para la salud pública (Homma, 2009). Bio-Manguinhos, unidad de Fiocruz responsable de la producción y desarrollo tecnológico de vacunas, entre otros, es uno de los centros de producción más modernos de América Latina. Desarrolla una actividad relevante en el ámbito internacional, exportando sus excedentes de producción a 71 países a través de la Organización Panamericana de la Salud (OPS) y UNICEF, siendo precalificada por la Organización Mundial de la Salud para el suministro de vacunas contra la fiebre amarilla y la meningitis AC. En este contexto, el presente artículo tiene como objetivo contribuir a la investigación, desarrollo y producción de nuevas vacunas en Brasil, así como a mejorar los procesos ya existentes mediante la revisión de los últimos avances y tecnologías disponibles para la purificación de vacunas humanas. 2. Extracción y clarificación Esta primera etapa del proceso de purificación a menudo se caracteriza por la separación celular, seguida de una filtración para eliminar las partículas. Puede ser necesaria una fase adicional previa de ruptura celular, dependiendo de la localización del antígeno en el sistema de expresión y propagación (Li y Qiu, 2013). La separación de las células comúnmente se realiza por centrifugación continua o intermitente. En la última década, sin embargo, los procesos de separación con membranas se han convertido en una alternativa atractiva para la industria biofarmacéutica: el escalado está garantizado, ya que es fácil aumentar el área de la membrana, aumentando los cartuchos o módulos utilizados en paralelo (Vicente et al., 2011). Dado el creciente interés por las tecnologías desechables que eliminan la necesidad de limpieza y validación, los procesos con membranas representan una clara ventaja sobre la centrifugación, ya que se adaptan fácilmente a los formatos desechables. Los avances en esta etapa eran necesarios debido al aumento de la densidad de células por el desarrollo del cultivo continuo, orientado a una mayor productividad. Los filtros de profundidad son ampliamente utilizados en la eliminación de material en partículas antes de la primera etapa de captura, después de la centrifugación o microfiltración tangencial (Roush y Lu, 2008). En muchos casos estos filtros se utilizan justo después de recoger el biorreactor, por lo general, con disminución de la porosidad, dispuestos en serie. Los filtros de profundidad se componen generalmente de tierra de diatomeas, perlita, fibras de celulosa, aglutinante de resina cargado positivamente y, algunos, también tienen características hidrofóbicas. Los filtros de profundidad a base de celulosa se consideran una técnica de separación eficiente y con buena relación coste/beneficio para la eliminación de las células CHO, desechos celulares y compuestos coloides antes de comenzar la purificación. Estos filtros ofrecen varias ventajas sobre los desafíos técnicos actuales de clarificación (Prashad y Tarrach, 2006): • Capaces de procesar grandes volúmenes de proceso; • Capaces de procesar altas densidades celulares; • Garantizan la transmisión del producto de interés a través del filtro; • Permiten la separación inmediata y rápida de las células; • Se utiliza como tecnología desechable y escalable; • Requieren una inversión inicial baja. Debido a sus características hidrofóbicas y de cargas, los filtros de profundidad han sido ampliamente utilizados en la eliminación de contaminantes, tales como: endotoxinas, ADN, proteínas contaminantes de las células y virus adventicios (Roush y Lu, 2008; Prashad y Tarrach, 2006). En el desarrollo de procesos de clarificación de vacunas también se ha empleado otro tipo de filtro de profundidad a base de polipropileno. Los resultados que se resumen en la Tabla 1 muestran que este tipo de clarificación proporciona rendimientos de hasta un 95 % para diversos productos (intracelular o extracelular) y diferentes tipos de revista safybi 61 Nuestros anunciantes Tabla 1: Aplicaciones de filtros de profundidad de polipropileno para la clarificación de las vacunas y semejantes. Filtro(s) Producto a filtrar Las células eliminadas Aplicación Rendimiento Referencia 3 mm PPV del rotavirus Sf-9 Vacuna viral recombinante humanos 85% Peixoto et al. 2007a 20 mm Ehrlichia ruminantium Células endoteliales de bovino Vacuna bacteriana veterinaria 66% Peixoto et al. 2007b 3 mm seguido de 0,65 mm Baculovírus Sf-9 Terapia genética 95% Vicente et al. 2009 Filtros dispuestos en serie: (i) 8 mm, (ii) 3,0 + 0,8 mm, (iii) 0,45 mm + 0,2 mm Virus de la fiebre amarilla Vero Vacuna de virus inactivados humana n.d. Pato et al. 2012 n.d. no determinado células. En aplicaciones en las que el producto permanece dentro de las células, es necesario añadir detergentes con el fin de romper las células antes de proceder a la filtración per se (Peixoto et al., 2007a). La clarificación va más allá de la filtración o centrifugación para eliminar las células y los restos celulares. Por lo tanto, prevenir la contaminación y mantener la esterilidad del efluente filtrado es de suma importancia. Por esta razón, los filtros esterilizantes también se integran en el proceso de clarificación como una técnica desechable (Tarrach et al. 2008). Los filtros de profundidad con múltiples capas proporcionan un mejor control de los contaminantes debido a la mayor altura del lecho cuando se comparan con los filtros de una sola capa. Por lo tanto, es posible mejorar el rendimiento de la filtración esterilizante posterior y reducir los costos. 3. Etapa de Captura La captura es un paso crítico del proceso de purificación, que tiene como objetivo concentrar, aislar y estabilizar el antígeno de interés, con el fin de preservar su actividad. También es necesario eliminar parcialmente algunos contaminantes, tales como proteínas de la célula huésped o substrato celular, ADN o proteínas virales (Vincent et. Al 2011). Si la ruptura de las células en la etapa de clarificación se realizó con detergente, el uso de la técnica de diafiltración, seguida de concentración, favorece su eliminación junto con los lípidos celulares (Peixoto et al. 2007a). La reducción del volumen de procesos para los pasos posteriores redunda en la reducción de costos de equipos y materiales, y reduce el nivel de bioseguridad requerido por ciertos tipos de vacunas (Vicente et al., 2011). Tradicionalmente, el método de concentración de vacunas virales usado con más frecuencia era la ultracentrifugación. Sin embargo, presenta numerosas limitaciones y, por esta razón, esta técnica ha sido sustituida por métodos más modernos de concentración, tales como la filtración tangencial y la cromatografía por membrana. La Tabla 2 muestra las diferentes ventajas de usar la cromatografía por membrana y la filtración tangencial en la etapa de captura en lugar de la ultracentrifugación. La filtración tangencial (FT), cuyos principios se han descrito anteriormente en detalle (Ghosh, 2006), se puede clasificar como de microfiltración o de ultrafiltración según el tamaño medio de poro de la membrana empleada. Los Tabla 2: Comparación entre la ultrafiltración, cromatografía por membrana, filtración tangencial para concentrar vacunas virales (adaptado Ray 2011). Ultracentrifugación Cromatografía por membrana Filtración tangencial Título viral bajo (106 PV/mL) Título viral alto (1013 PV/mL) Título viral alto (1010 PV/mL) Tiempo de proceso prolongado (~ 36h) Requiere validación contra contaminación procedente de diferentes lotes Requiere la eliminación del cloruro de cesio o sacarosa utilizada en el gradiente Proceso rápido (~ 2 h) Tecnología desechable (no requiere validación) Los módulos en forma de casete se pueden desechar y no requieren validación No requieren la eliminación de ningún residuo contaminante Requiere medidas adicionales para eliminar los contaminantes Elimina contaminantes tales como proteínas y ADN Dependiendo del tamaño del poro tiene capacidad para eliminar algunos contaminantes Requiere un mantenimiento periódico de alto coste Mantenimiento preventivo del cromatógrafo con un coste medio Mantenimiento preventivo del sistema con bajo coste El control de procesos es un reto PV: partícula viral 62 revista safybi Parámetros del proceso fácilmente controlables y optimizados módulos de membrana disponibles comercialmente son de tipo casete, de fibra hueca o espiral. La etapa de captura de la vacuna veterinaria bacteriana de hidropericardio realizada con FT, utilizando membrana de celulosa con 0,2 mm en forma de casete, se tradujo en un rendimiento del 88 % (Peixoto et al. 2007b.). La purificación de polisacáridos bacterianos se describió con éxito en diferentes trabajos. En el primero de ellos, la etapa de captura de la suspensión que contiene el polisacárido de Neisseria meningitis serogrupo C, se produce por filtración tangencial con casete de 100 kDa, con una recuperación de alrededor del 99 % (Pato et al., 2006). En un trabajo más reciente, el mismo polisacárido se concentró en tres etapas seguidas de FT con membranas de 300, 50 y 30 kDa (Robinson et al., 2011). Albani y sus colaboradores obtuvieron elevada pureza del polisacárido de Haemophilus influenzae serotipo b con membrana en espiral y casete de 100 kDa (Albani et al, 2012.). Se han utilizado con éxito casetes con membrana de celulosa de 30 kDa en la ultrafiltración tangencial de las toxinas tetánicas (Meyeroltmams y Schimidt, 2008a) y la difteria (Meyeroltmams y Schimidt, 2008b). En el primer caso, el volumen de proceso se concentró más de 5 veces con una recuperación del 100 % después de la diafiltración. En el segundo caso, la concentración fue de 25 veces con una recuperación del 90 %. Se utilizaron cartuchos de fibras huecas con membranas de polietersulfona (PESU) en la concentración de PPV del rotavirus (Peixoto et al., 2007a), adenovirus (Peixoto et al. 2008) y PPV del VIH (Hammonds et al., 2007). En el primer trabajo, la recuperación alcanzada con el empleo de la membrana de 750 kDa fue del 86 % con eliminación del 97 % del ADN contaminante. En el segundo estudio, sin embargo, los autores compararon las membranas de 750, 500 y 300 kDa y obtuvieron recuperaciones del 42, 71 y 96 %, respectivamente. Hammonds y colaboradores obtuvieron una recuperación del 95 % y un factor de concentración de 20 veces, usando una membrana de 300 kDa. La cromatografía positiva, donde se produce la adsorción del antígeno, constituye una alternativa interesante en esta etapa porque da lugar a un alto factor de concentración si el producto se diluye muy bien y si la matriz cromatográfica proporciona una elevada capacidad dinámica (Vicente et al., 2011). Además de las ventajas con respecto a la ultracentrifugación ya mencionadas en la Tabla 2, la cromatografía con membrana de adsorción tiene otras ventajas atractivas en comparación con la cromatografía convencional con resinas: • Flujo predominantemente convectivo (Figura 1), lo que permite el uso de altas tasas de flujo sin pérdida de capacidad dinámica, redundando en una disminución sustancial del tiempo de proceso; Nuestros anunciantes Figura 1: Comparación de la difusión en resinas cromatográficas y membranas de adsorción. En las resinas, la mayor parte de los aglutinantes se encuentra dentro de los poros. La molécula de interés se adsorbe al aglutinante después de la difusión en los poros. En la membrana de adsorción, los poros son lo suficientemente grandes para que la molécula de interés se adsorba al aglutinante usando convectivo dependiendo muy poco de la difusión. ➧ Flujo convectivo ➡ Resina cromatográfica Membrana de adsorción Difusión en los poros ➡ La difusión en la película • Dispositivo listo para su uso, lo que reduce el tiempo de preparación con el relleno de la columna y los gastos de mano de obra/hora ; • El límite de exclusión por tamaño es de, aproximadamente 107 Da, debido al tamaño de los poros (3-5 mm), mientras que en la cromatografía en columna, este límite está por debajo de 106 Da, lo que limita la adsorción de virus y contaminantes, tales como el ADN y las endotoxinas; • El escalado se logra fácilmente aumentando el área de la membrana; • No requiere validación, ya que se trata de un producto desechable. El uso de membranas de adsorción como captura ya se ha experimentado con varios tipos de vacunas, especialmente las virales. El grupo de investigación dirigido por el profesor Udo Reichl del Instituto Max-Planck, en Alemania, publicó dos trabajos con el virus de la influenza. En el primero, los autores experimentaron con éxito una membrana de adsorción aniónica fuerte con recuperaciones de hasta el 86 % (Kalbfuss et al., 2007). En el segundo estudio, el uso de membrana de celulosa sulfatada en la captura del mismo virus dio lugar a recuperaciones de hasta el 94 % y la reducción del ADN y proteína totales en un 32 % y 43 %, respectivamente (Opitz et al. 2009). El intercambio aniónico fuerte con membrana también se utilizó con éxito en la captura del virus de la fiebre amarilla, en la que los autores describieron recuperaciones de hasta el 95 % y una reducción significativa del ADN contaminante (Pato et al., 2012) y del adenovirus con recuperación del 62 % (Peixoto et al. 2008). La membrana de intercambio aniónico débil también ha sido empleada con éxito en la captura de la PPV del rotavirus (Vicente et al., 2008). A pesar de la recuperación media del 55 % en este último estudio, los autores consiguieron incrementar la pureza al 55 %, un factor de concentración del 29, 90 % de eliminación del ADN contaminante y 1,3 log de reducción de baculovirus contaminante. El intercambio catiónico también se ha empleado con éxito en la captura de herpes virus alfa con recuperación del 86 % (Karger et al. 1998). 4. Purificación intermedia Esta etapa del proceso de purificación se puede omitir si la captura es suficientemente eficaz per se, requiriendo únicamente que se complemente con una etapa de pulido para lograr la pureza deseada. 64 revista safybi Hay dos factores importantes para la eficacia de la etapa de captura, determinando así la necesidad o no de purificación intermedia: (I) especificidad del sistema de expresión, (II) diferencias significativas entre las características físico-químicas del antígeno y las impurezas que se eliminan. La cromatografía líquida es la operación unitaria utilizada con más frecuencia en esta etapa, cuando es necesaria. 5. Purificación final (Pulido) Esta etapa final del proceso de purificación es extremadamente crítica, pues se destina a la eliminación de impurezas para que el producto cumpla las exigencias de las agencias reguladoras cada vez más estrictas. Al llegar a esta etapa, la mayor parte de los contaminantes ya se han eliminado, restando solamente la eliminación de trazas de proteínas, endotoxinas y ácidos nucleicos o agregados/fragmentos del antígeno diana. La técnica seleccionada en este momento debe permitir una alta recuperación, ya que las pérdidas del producto en esta etapa son más importantes económicamente que en las fases anteriores. Otra preocupación es la obtención del producto ya en solución tampón adecuada para la etapa de formulación. La filtración tangencial o cromatografía de exclusión molecular son muy ventajosas porque, además de eliminar fragmentos o agregados y transferir el antígeno a la solución tampón deseada, arrojan excelentes datos de rendimiento. La cromatografía negativa (operada en modo “flowthrough”, en el que los contaminantes se adsorben y el producto de interés no) es otra técnica utilizada en esta etapa (Vincent et al. 2011). Recientemente, las membranas con aglutinante de intercambio aniónico tolerante a la sal – presentes en esta fase, ya que de forma general, la etapa anterior implica la desorción del antígeno en concentraciones elevadas de sal – han proporcionado excelentes resultados en la eliminación de ADN (Leuthold et al 2010.) y proteínas contaminantes (Kang et al. 2012). En los dos trabajos citados se ha utilizado la planificación de experimentos para optimizar la adsorción de los contaminantes. El elevado número de condiciones ensayadas en un único experimento ha sido posible con una placa de 96 pocillos que contenían las membranas de adsorción. El uso del aglutinante resistente a la sal permite la eliminación de una etapa de diafiltración entre dos etapas de cromatografía, lo que contribuye a aumentar el rendimiento global del proceso de purificación (Farid, 2009). La eliminación de contaminantes como los agregados del propio producto de interés, se puede lograr asimismo con membranas hidrofóbicas también en modo negativo (Davies, 2009; Fraud et al, 2009). En la purificación de partículas virales se ha utilizado otro tipo de cromatografía convectiva, a base de columnas (Jungbauer e Hahn, 2008). Recientemente, la purificación final de la PPV de la hepatitis B producida en levadura se ha realizado en columna con aglutinante hidrofóbico (Burden et al., 2012). También existe una resina cuya superficie externa se ha reducido mediante el aumento del diámetro medio de la partícula, combinando la cromatografía de adsorción con exclusión de tamaño (Iyer et al. 2011). La cromatografía negativa con esta resina dio lugar a recuperaciones del 70-80 % del virus de la gripe y el 80 % de eliminación de impurezas. 6. Buenas Prácticas de Fabricación y Aspectos Regulatorios Para garantizar la seguridad y eficacia de las vacunas, los organismos reguladores han elevado las exigencias de forma continua y permanente, provocando que las actividades de producción resulten muy complejas y costosas (Homma et al, 2011). Los laboratorios pequeños han desaparecido del mercado, pues no tienen capacidad para cumplir las normas de Buenas Prácticas de Fabricación, que requieren una elevada inversión para la adecuación de las instalaciones, con áreas clasificadas y certificadas, niveles de bioseguridad apropiados y un flujo adecuado de materiales y personas en el proceso de producción. Además de las instalaciones, también se deben validar las utilidades y los sistemas computerizados (ANVISA, 2010). Los equipos deberán presentar conexiones sanitarias, poseer calificación de instalación y operación, así como permitir el control y registro de los principales parámetros. La calibración y verificación de instrumentos de medición deben ser periódicas. El diseño del proceso se debe planificar de forma a permitir un alto grado de automatización, reduciendo al mínimo los fallos operativos. Debe darse prioridad a los sistemas cerrados, eliminando la posibilidad de contaminación. Cada etapa del proceso de purificación se debe validar identificando los parámetros críticos y las fuentes de variabilidad, indicándose la carga máxima de producto que se puede aplicar por área de membrana o volumen de resina, el porcentaje de recuperación del producto y de eliminación de contaminantes no deseados, entre otros. Por otra parte, se ha exigido la validación de sanitización de membranas, resinas, tanques y otros materiales para su reutilización. Por esta razón, ha aumentado el uso de materiales desechables, lo que también permite una plataforma de producción flexible, que se puede utilizar para la fabricación de más de un producto en diferentes periodos de campaña (Almond et al. 2011). Se han utilizado algunas herramientas con el fin de di- Nuestros anunciantes señar y desarrollar procesos robustos y repetibles que garanticen la obtención de un producto dentro de las especificaciones predefinidas al final del proceso de fabricación. Entre ellas podemos destacar PAT (Process Analytical Technology), QbD (Quality by Design). Por otra parte, el control del proceso ha jugado un papel fundamental para garantizar la calidad del producto, proporcionando datos que también permiten evaluar la consistencia de la producción (Vicente et al., 2011). 7. Conclusión En el último siglo se han conseguido muchos avances en el campo de la vacunología, permitiendo campañas de vacunación exitosas contra las principales enfermedades infecciosas. En la búsqueda de una vacuna con alto grado de seguridad, se han utilizado conocimientos en el área de la inmunología y la biología molecular con el fin de obtener antígenos purificados con alta productividad y rendimiento. Para ello, se están construyendo diversos sistemas de expresión, afectando de manera significativa al desarrollo de las estrategias de purificación, ya que estos determinan los subproductos que se deberán eliminar en las etapas de producción. Se han realizado avances significativos para alcanzar un proceso downstream escalable, robusto, reproducible, con viabilidad técnico-económica y que pueda dar lugar a un producto acorde con las especificaciones de los organis- mos reguladores. Entre estos avances, podemos destacar las nuevas tecnologías para la producción de soportes cromatográficos, que han ampliado la aplicación de la cromatografía líquida a escala industrial, pues han permitido el uso de altas velocidades de flujo, el aumento de la capacidad dinámica y han disminuido el consumo de soluciones tampón. Por otro lado, las membranas de micro e ultrafiltración, que tradicionalmente se utilizaban en etapas que no exigían alta resolución, como clarificación, concentración e intercambio de solución tampón, han mejorado drásticamente en su selectividad, manteniendo la característica de alto rendimiento. En cumplimiento de las Buenas Prácticas de Fabricación de Productos Biológicos, se han desarrollado equipos con conexiones sanitarias, monitorización de parámetros de proceso en línea y alto grado de automoción, para minimizar el impacto de los fallos operacionales. Asimismo, se ha producido un considerable aumento de los materiales desechables con el fin de eliminar la necesidad de validación de los protocolos de desinfección entre lotes. Estos avances, asociados a los nuevos conceptos de PAT (Process Analytical Technology), QbD (Quality by Design), CPP (Critical Process Parameters) y CQA (Critical Quality Attributes), entre otros, han contribuido a que la producción de vacunas sea segura y rentable y con menos trabas para su aprobación por los organismos reguladores, para permitir su rápida distribución a nivel mundial. n Referencias ALBANI, S. M. F.; SILVA, M. R.; TAKAGI, M.; CABRERA-CRESPO, J. Improvement in the Purification Process of the Capsular Polysaccharide from Haemophilus influenzae Type b by Using Tangential Ultrafiltration and Diafiltration. Applied Biochemistry Biotechnology 167, 2068–2075 (2012). ANVISA 2010. ftp://ftp.saude.sp.gov.br/ftpsessp/bibliote/informe_eletronico/2010/iels.abr.10/ Iels73/U_RS-MS-ANVISA-RDC-17_160410.pdf. Consultado en abril de 2013. Burden, C. S.; Jin, J.; Podgornik, A.; Bracewell, D. G. A monolith purification process for virus-like particles from yeast homogenate Journal of Chromatography B 880, 82– 89 (2012). Cox, M. M. J. Recombinant protein vaccines produced in insect cells. Vaccine 30, 1759– 1766 (2012). FDA http://www.fda.gov/downloads/BiologicsBloodVaccines/Vaccines/ApprovedProducts/ UCM338898.pdf. Consultado en mayo de 2013 GHOSH R. Membrane based bioseparation. In: Principles of bioseparations engineering. Singapore. World Scientific Publishing, 199-250 (2006). Hammonds, J.; Chen, X.; Zhang, X.; Lee, F.; Spearman, P. Advances in methods for the production, purification, and characterization of HIV-1 Gag–Env pseudovirion vaccines Vaccine 25, 8036–8048 (2007). Homma, A; Martins, R. M.; Leal, M. L. F.; Freire, M. S.; Couto, A. R. Actualización sobre vacunas, inmunizaciones e innovación tecnológica. Ciência & Saúde Coletiva 16(2), 445-458 (2011). Iyer, G.; Ramaswamy, S.; Asher, D.; Mehta, U.; Leahy, A.; Chung, F.; Cheng, K. S. Reduced surface area chromatography for flow-through purification of viruses and virus like particles. Journal of Chromatography A 1218, 3973–3981 (2011). Josefsberg, J. O.; Buckland, B. Vaccine Process Technology. Biotechnology and Bioengineering 109, 1443–1460 (2012). Jungbauer, A.; Hahn, R. Polymethacrylate monoliths for preparative and industrial separation of biomolecular assemblies. Journal of Chromatography A 1184, 62–79 (2008). Kalbfuss, B.; Wolff, M.; Geisler, L.; Tappe, A.; Wickramasinghe, R.; Thom, V.; Reichl, U. Direct capture of influenza a virus from cell culture supernatant with Sartobind anion-exchange membrane adsorbers. Journal of Membrane Science 299, 251–260 (2007). Karger, A.; Bettin, B.; Granzow, H.; Mettenleiter, T. C. Simple and rapid purification of alphaherpesviruses by chromatography on a cation exchange mambrane. Journal of Virological Methods 70, 219-224 (1998). Leuthold, M.; Faber, R.; Fischer-Fruhholz, S. 96-well Plate Membrane Adsorber Screening for Buffer and Salt Influence. In: BioProduction, Barcelona (2010). Li, M.; Qiu, Y. X. A review on current downstream bio-processing technology of vaccine products. Vaccine 31, 1264– 1267 (2013). MCALEER, W. J.; BUYNAK, E. B.; MAIGETTER, R. Z.; WAMPLER, D. E.; MILLER, W. J.; HILLEMAN, M. R. Human hepatitis B vaccine from recombinant yeast. Nature 307, 178 (1984). MELLADO, M. C. M.; CASTILHO, L. R. Recombinant therapeutic proteins. In: CASTILHO, L. R.; MORAES, A. M.; AUGUSTO, E. F. P.; BUTLER, M. Animal Cell Technology: From Biopharmaceuticals to Gene Therapy. Abingdon, Taylor & Francis, 389 (2008). MELLO, I. M. V. G. C.; CONCEIÇÃO, M. M.; JORJE, S. A. C.; CRUZ, P. E.; ALVES, P. M.; CARRRONDO, J. M. T.; PEREIRA, C. A. Viral Vaccines: concepts, principles and bioprocesses. In: CASTILHO, L. R.; MORAES, A. M.; AUGUSTO, E. F. P.; BUTLER, M. editors. Animal Cell Technology: from Biopharmaceuticals to Gene Therapy. New York: Taylor & Francis. p 435 (2008). 66 revista safybi Meyeroltmanns, F.; Schmidt, P. Concentration and diafiltration of Tetanus vaccine. Publication number SL-1046-e08103 (2008a). Meyeroltmanns, F.; Schmidt, P. Concentration of Diphtheria vaccine in downstream processing. Publication number SL-1047-e08102 (2008b). Mollerup, J. M.; Thomas, B. H.; Kidal, S.; Staby, A. Quality by design - Thermodynamic modelling of chromatographic separation of proteins. Journal of Chromatography A, 1177 (2008) 200–206. Opitz, L.; Lehmann, S.; Reichl, U.; Wolff, M. Sulfated membrane adsorbers for economic pseudoaffinity capture of influenza virus particles. Biotechnology and Bioengineering. PATO, T. P.; BARBOSA, A. P. R.; SILVA JUNIOR, J. G. Purification of capsular polysaccharide from Neisseria meningitidis serogroup C by liquid chromatography. Journal of Chromatography B 832, 2, 177-332 (2006). PATO, T. P.; SOUZA, M. C. O.; YOKOMIZO, A. Y.; SILVA, M. V.; CARIDE, E.; GASPAR, L. P.; CASTILHO, L. R.; FREIRE, M. S. Development of a membrane adsorber based capture step for the purification of yellow fever virus. In: Vaccine Technology IV, Albufeira (2012). Peixoto, C.; Ferreira, T. B.; Sousa, M. F. Q.; Carrondo, M. J. T.; Alves, P. M. Towards Purification of Adenoviral Vectors Based on Membrane Technology. Biotechnology Progress 24, 1290-1296 (2008). Peixoto, C.; Marcelino, I.; AMARAL, A. I.; CARRONDO, M. J. T.; ALVES, P. M. Purification by membrane technology of an intracellular Ehrlichia ruminantium candidate vaccine against heartwater. Process Biochemistry 42, 1084–1089 (2007b). Peixoto, C.; Sousa, M. F. Q.; Silva, A. C.; CARRONDO, M. J. T.; Alves, P. M. Downstream processing of triple layered rotavirus like particles. Journal of Biotechnology 127, 452–61 (2007a). Prashad, M.; TARRACH, K. Depth filtration: cell clarification of bioreactor offloads. Filtration + Separation 43, 28–30 (2006). RAY S. Advances in Vaccine Technologies. BioPharm International supplement, s1-s8 (October 2011). ROBINSON, A.; LEE, S. M.; KRUSE, B.; HU, P. Meningitis vaccine manufacturing: fermentation harvest procedures affect purification. BioPharm International supplement, s21-s26 (April 2011). ROLDÃO, A.; MELLADO, M. C. M.; CASTILHO, L. R.; CARRONDO, M. J. T.; ALVES, P. M. Vírus-like particles in vaccine development. Expert Review of Vaccines 9, 1149-1176 (2010). ROUSH, D. J.; LU, Y. Advances in Primary Recovery: Centrifugation and Membrane Technology. Biotechnology Progress 24, 488-495 (2008). Smith, J.; LIPSITCH, M.; Almond, J. W. New Decade of Vaccines 3: Vaccine production, distribution, access, and uptake. Lancet 378, 428–38 (2011). SUS. 2009. www.datasus.gov.br. Consultado en agosto de 2009. TARRACH, K.; KÖHLER, K.; GRIMM, C. Managing initial recovery processes with multilayer depth filtration, Pharmaceutical Technology (2008). Vicente T.; Peixoto, C.; CARRONDO, M. J. T.; ALVES, P. M. Purification of recombinant baculoviruses for gene therapy using membrane processes. Gene Therapy 16, 766–775 (2009). VICENTE, T.; MOTA, J. P. B.; Peixoto, C.; ALVES, P. M.; CARRONDO, M. J. T. Rational design and optimization of downstream processes of virus particles for biopharmaceutical applications: Current advances, Biotechnology Advances 29, 869–878 (2011). Vicente, T.; Sousa, M. F. Q.; Peixoto, C.; Mota, J. P. B.; Alves, P. M.; Carrondo, M. J. T. Anionexchange membrane chromatography for purification of rotavirus-like particles, Journal of Membrane Science, 311, 270–283 (2008). ZEPP, F. Principles of vaccine design - Lessons from nature. Vaccine 28S, C14–C24 (2010). Nuestros anunciantes Optimización de las carteras de productos farmacéuticos con sistemas de administración de aerosoles nasales o sprays bucales Un mercado dinámico y cómo los aerosoles nasales y los sprays bucales pueden ofrecer una oportunidad Gerallt Williams Director de Asuntos Cientificos en Aptar Pharma, Prescription Division En los últimos años, no ha habido grandes variaciones en cuanto a la cantidad de nuevas entidades moleculares (NEM) que llegan al mercado (1, 2 y 3). Esto se debe mayormente al incremento de los costos para desarrollar los fármacos y a las normas más rigurosas que se aplican a la hora de aprobar los medicamentos. Aún existen muchas oportunidades en este mercado farmacéutico cambiante tanto para las compañías que fabrican fármacos genéricos como para aquellas que se dedican a los innovadores y que estén dispuestas a buscar nuevas maneras de innovar. La clave es encontrar formas novedosas de trabajar con las drogas existentes. Las ventajas científicas y médicas de la administración de fármacos con aerosoles nasales y sprays bucales están bien documentadas (4). Las terapias locales para la boca o la nariz funcionan bien en el lugar de administración: un buen ejemplo es el uso de antihistamínicos y corticoesteroides en sprays nasales para el tratamiento de la rinitis alérgica. Las extensas y muy vascularizadas mucosas nasal y oral (5) también son objetivos atractivos para la administración de drogas sistémicas. Se observa que los fármacos con bajo peso molecular administrados en esos lugares actúan con rapidez y desde allí pueden pasar al torrente sanguíneo sin tener que hacerlo por el tracto gastrointestinal ni por el hígado (donde algunos componentes activos de las drogas se descomponen). La administración de fármacos por vía nasal (IN) o bucal Figura 1. Gráfico concentración vs. tiempo luego de la administración de fármacos, adaptado de Drug Development & Delivery, Vol. 2 No. 1, enero de 2002. puede ofrecer un inicio rápido de la acción debido a su perfil farmacocinético (véase la Figura 1), que se sitúa en algún punto entre la administración intravenosa (IV) y la intramuscular (IM), y resulta mucho más rápida que la vía oral. Los sprays orales y sublinguales son fáciles de usar, ya sea en el caso de la autoadministración o si debe hacerlo un tercero. Esta característica es muy ventajosa para pacientes geriátricos y pediátricos, y puede mejorar el tratamiento ambulatorio y la automedicación, lo que reduciría la carga que pesa sobre los sistemas sanitarios. Encontrar un nuevo propósito para un producto que ya se comercializa puede ofrecer valor agregado a las compañías farmacéuticas y así completar una gama de productos existentes e incrementar la cobertura del mercado. Esto puede hacerse con un costo mucho menor de lo que supondría comercializar una NEM, que ahora se calcula en aproximadamente USD 1800 millones (6), debido a los costos más bajos de desarrollo y a mecanismos reglamentarios menos rigurosos. Ejemplos de fármacos actuales utilizados por vía nasal u oral Actualmente existen diversos medicamentos que se administran a los pacientes por vía nasal y a través de la mucosa bucal. Entre ellos se encuentran los fármacos para controlar el dolor, como la lidocaína, nicotina para dejar de fumar, nitroglicerina en spray nasal para la angina de pecho; drogas para las crisis epilépticas, como las benzodiacepinas (diacepam); drogas para la sobredosis de opioides, es decir, naloxona; tratamientos para la hipoglucemia: glucagón; drogas para la emesis en los postoperatorios o después de la quimioterapia, como ondansetron y metoclopramida; y la hormonas testosterona, progesterona y estradiol. Entre los ejemplos de fármacos que han conseguido con éxito pasar a ser administrados por vía nasal u oral se encuentran las hormonas, como la desmopresina para la enuresis; vitaminas, como la B12, para la hipocobalaminemia; triptanos (por ej., sumatriptán) para las migrañas; opioides, como el fentanilo nasal y bucal; y analgésicos no opioides, ketorolac y THC (tetrahidrocannabinol) para el manejo del dolor asociado al tratamiento de la esclerosis múltiple. Los opioides, como el fentanilo nasal y bucal se vienen revista safybi 67 Nuestros anunciantes comercializando hace varios años y resultan ventajosos dada su probada eficacia en comparación con otras formas farmacéuticas. Si se los compara con las inyecciones, salen muy favorecidos, véase la Tabla A. Tabla A – Fentanilo nasal e intravenoso. Comparación. Fentanilo (7) Biodisponibilidad Intranasal (IN) Intravenoso (IV) >89% >98% Tmax ~12,8mins ~6,0mins AUC equivalente equivalente A título comparativo, el fentanilo bucal (con formato de paleta o “lollipop”) tarda 15 minutos en administrarse y otros 15 minutos para lograr el alivio eficaz del dolor (8). La morfina opioide también ha sido estudiada a fondo (9) respecto de su administración mediante un aerosol nasal para tratar el dolor irruptivo provocado por el cáncer. La morfina, como muchos opioides, tiene poca biodisponibilidad por vía oral a causa del extenso metabolismo de primer paso. Los datos farmacocinéticos humanos para la administración de la morfina por vía nasal, oral e inyecciones se detallan en la Figura 2. Se puede ver que por la vía nasal se logra un inicio de la acción (Tmax ~15mins) similar al de las inyecciones, y que es mucho más rápido que la vía oral (Tmax ~50mins). En el caso de los tratamientos de manejo del dolor como este, resulta vital un inicio rápido de la acción para aliviar el dolor. Figura 2: Tmax PK para Morfina en humanos, IM=intramuscular, IN=intranasal, adaptado de Costantino et ál. (2007). El midazolam se utiliza a diario en hospitales para sedar y relajar a los pacientes bajo tratamiento terapéutico, diagnóstico o endoscópico, y para tratar las crisis epilépticas. Para los pacientes, el método tradicional endovenoso es estresante y doloroso; además, se tarda mucho tiempo y tiene un alto costo. No obstante, por vía nasal no resulta doloroso, no es invasivo, es casi inmediato, su efecto dura la misma cantidad de tiempo que si se administra de manera intravenosa, y la droga puede usarse fuera de los hospitales, por ejemplo, en clínicas odontológicas, consultorios médicos y clínicas privadas. 68 revista safybi Varios estudios clínicos dan cuenta de la farmacocinética y farmacodinamia positivas de la administración nasal del midazolam comparada con otras formas de administración, como la oral y rectal. El midazolam intranasal tiene una biodisponibilidad de 50-80% y tiene un muy rápido inicio de acción, 5 minutos, por lo que puede compararse con la inyección, 2-5 minutos (10). Optimización de las carteras de productos patentados mediante la gestión eficaz del ciclo vida El tratamiento actual de la rinitis alérgica (incluidos los sprays nasales y descongestivos) cubre aproximadamente el 80% de la utilización de fármacos nasales. La gran cantidad de medicamentos genéricos en el mercado de rinitis alérgica y la disminución del número de las NEM que llegan al mercado en los Estados Unidos y la Unión Europea están modificando considerablemente la dinámica del mercado. Esto podría dar como resultado que muchos fármacos indiferenciados inunden el mercado y que se dependa en gran medida de ciertas estrategias, como la competencia agresiva de precios, redes de distribución y calidad de los servicios. El manejo eficaz del ciclo de Figura 3: Ejemplo de un dispositivo vida de los medicamentos exiscon accionador tentes puede marcar una difelateral, Latitude rencia entre las compañías y optimizar la cartera de productos sin involucrar el costo, tiempo ni riesgos relacionados con el desarrollo de nuevas drogas. Un buen ejemplo de esto es el desarrollo de dispositivos con accionador lateral (véase la Figura 3) que se han utilizado para productos, como el Veramyst® (furoato de fluticasona) para tratar la alergia estacional. Estos dispositivos son de gran utilidad para los pacientes que tienen la motricidad de las manos deteriorada o débil. De esta manera, los pacientes geriátricos y pediátricos tienen un mejor acceso al producto. Cambios a las formulaciones existentes y uso de etiquetas Los cambios a las formulaciones existentes aportan a las empresas valor agregado a los productos que ya han percibido una importante inversión financiera y pueden ofrecer un mejor retorno sobre el capital invertido que las nuevas entidades químicas (NEM). La estrategia no es nueva: hasta 2009, entre el 30% y 40% de los medicamentos o productos biológicos aprobados o lanzados por primera vez en los Estados Unidos eran fármacos reposicionados para nuevas indicaciones, reformulaciones o nuevas combinaciones de fármacos existentes (11). Algunas de las mejoras que se han realizado a las formulaciones en sprays nasales incluyen el uso de diferentes ésteres. Un ejemplo de cómo distintos ésteres para el mismo compuesto pueden tener una eficacia diferente en el tratamiento es el caso de los medicamentos nasales utilizados para el tratamiento de la rinitis alérgica y el asma: glucocorticoides, furoato de fluticasona (FF; Veramyst®, GlaxoSmithKline/Avamys®) y propionato de fluticasona (FP; GlaxoSmithKline®/Flonase®, Flixonase®). Teniendo en cuenta que el grupo de ésteres contribuye a las características fisicoquímicas de la molécula, el tipo de éster puede determinar el índice de solubilidad y la afinidad por el tejido. Por ejemplo, el éster en el FF ofrece mayor afinidad para el tejido nasal y pulmonar comparado con el FP (12), y hay datos que sugieren que el FF puede ser mejor que el FP por la mejora de los síntomas (13), con lo cual se puede lograr un tratamiento más eficaz con menos medicación (14). El uso de distintas soluciones para administrar el medicamento, por ejemplo, salina o sacarosa, y el uso de formulaciones sin conservantes pueden ser estrategias eficaces para mejorar la cartera de productos. En particular, la no utilización de conservantes, tales como el cloruro de benzalconio y parabenos, evita daños de largo plazo relacionados con el uso crónico. La combinación de los fármacos existentes puede ofrecer nuevas y mejores soluciones para los tratamientos, por ejemplo, la combinación de un antiinflamatorio y un esteroide en el spray nasal Dymista® para la rinitis alérgica. Los cambios y las mejoras de los métodos de administración de aerosoles nasales y sprays bucales pueden incluir el cambio de formulaciones acuosas con bombas nasales a formulaciones compuestas por propulsores de hidrofluoroalcano (HFA) administradas con inhaladores presurizados de dosis medida (pMDI). Esto ofrece nuevas formas de dosificación para que sean más sencillos y prácticos de administrar; por ejemplo, el dipropionato de beclometasona (BDP, Teva) para tratar la rinitis alérgica y ciclesonida (Sunovion) para la rinitis alérgica y el asma son dos lanzamientos recientes al mercado que demuestran esto. En tales casos, a veces, se prefiere el uso de un pMDI seco para evitar el goteo del producto, lo cual deja un sabor en la boca y permite que se apliquen más fácilmente pequeñas dosis (es decir, 25 µl). La distribución de un folleto con información del producto a distintos grupos de pacientes, como geriátricos o pediátricos, puede ser una forma de ampliar el mercado y optimizar la cartera de productos. No obstante, la mayor prioridad es garantizar que todo cambio que se realice al producto debe ofrecer ventajas a los pacientes o compradores de fármacos, o prestadoras de servicios de salud, poniendo especial atención en lograr una mayor eficacia, menores efectos secundarios, mayor cumplimiento del tratamiento y una mejor capacidad de autoadministración del fármaco. Soluciones de administración de monodosis o multidosis Sprix® (Roxro) es una reformulación, la primera de su clase, del primer analgésico no opioide en la forma de spray nasal, el cual ha sido recientemente introducido en el mer- Figura 4: Ejemplos de dispositivos monodosis cado para aplicarse con un dispositivo multidosis. Instanyl® y Subsys® son otros ejemplos recientes para el tratamiento contra el dolor que utilizan un dispositivo de monodosis (véase la Figura 4). Otros proyectos interesantes de aerosoles nasales que están en desarrollo incluyen un sistema de administración por vía nasal de medicamento en polvo monodosis que contiene Dihidroergotamina para tratar la migraña (Shin Nippon Biomedical Labs), como así también los nuevos dispositivos nasales potenciados, actualmente en etapa de estudios clínicos, para el tratamiento de ciertas enfermedades, como pólipos nasales, migraña y rinosinusitis crónica (Optinose). Plataforma del Contador eDose y Plataforma eLockout Las carteras de productos pueden mejorarse invirtiendo en dispositivos mecánicos para la administración de fármacos por vía nasal u oral con módulos electrónicos o dispositivos ‘e’ que permiten la autodosificación de los tratamientos (véase la figura 5). La autoadministración de los fármacos ayuda a que haya una mayor tendencia a las terapias ambulatorias sin la intervención directa del profesional, protegiendo al paciente con sistemas de trabas y conteo de dosis. Gracias a estos dispositivos, los fabricantes logran un nuevo potencial en el mercado, nuevas aplicaciones del producto y avances tecnológicos, además de responder a las necesidades de los usuarios. Los contadores electrónicos. pueden indicar cuántas dosis quedan en un spray nasal (para renovar la receta y asegurarse de que no se acabará el producto) y brindar información bien visible para pacientes con problemas de visión, por ejemplo, personas mayores. En el caso de los médicos, estas “Plataformas del Contador eDose” les indican cómo y cuándo se ha recibido la dosis, la cual podrá integrarse en un enfoque de telemedicina, donde los proferevista safybi 69 Nuestros anunciantes Figura 5: Ejemplos de dispositivos con Plataformas electrónicas Contador eDose y bloqueo de utilización. sionales de la salud podrán cargar los datos almacenados en los dispositivos electrónicos y hacer los ajustes necesarios para conseguir la máxima eficacia del tratamiento, según cada caso en particular. Dichas Plataformas también pueden facilitar las aprobaciones reglamentarias para las sustancias controladas y las moléculas potentes, y reducir las cuestiones de responsabilidad relacionadas con la desviación. Los dispositivos electrónicos garantizan a los organismos reguladores gubernamentales que no habrá sobredosis ni abuso de fármacos y que se pueden administrar drogas más seguras y eficaces. Innovación, crecimiento y mejor tratamiento para el paciente: el futuro de los aerosoles nasales y sprays bucales • Los métodos nuevos de administración de medicamentos pueden ser un factor importante para el crecimiento y ayudar a las empresas a hacer frente a las presiones que cambian la imagen del sector farmacéutico. Los fármacos existentes y seguros podrían ser más eficaces si se hicieran inversiones para combinarlos con nuevas formas de administración. • Las carteras de productos pueden administrarse durante ciclos más prolongados a través de una estrategia de cartera viable que permita a las empresas consolidar sus productos. Con el uso de aerosoles nasales y sprays bucales, los productos de la cartera de una empresa llegan a una población de pacientes más amplia y variada, con lo cual se puede incrementar el mercado de una empresa, mejorar su imagen y optimizar la relación precio-calidad. • Los ejemplos de este artículo demostraron cómo las drogas existentes pueden utilizarse en los aerosoles nasales y sprays bucales para mejorar el tratamiento de los pacientes, especialmente en cuanto al cumplimiento, conveniencia y seguridad del paciente. La tecnología emergente podría impulsar el rumbo futuro de la medicina, es decir la telemedicina, en donde los tratamientos dejarán de ser hospitalarios para ser ambulatorios. • El cambio de la forma actual de administración de fármacos (oral, inyectable) al uso de sistemas más cómodos para el paciente, como los aerosoles nasales y sprays bucales, puede representar un beneficio real tanto para los pacientes como para todos los involucrados en áreas de la salud. n Referencias 1. FDA (2010) NMEs Approved by CDER. [Online] Available: http://www.fda.gov/ downloads/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/DrugandBiologicApprovalReports/UCM242695.pdf [Accessed 26th March 2013]. 2. Osborne, R. (2013) Figure 1: FDA new molecular entities and biological license application approvals, 1998–2012. Fresh from the biotech pipeline—2012 Nature Biotechnology 31:100-103. 3. FACTBOX-World’s top-selling drugs in 2014 vs 2010. [Online] Available: www.reuters.com/article/2010/04/13/roche-avastin-drugs-idUSLDE63C0 BC20100413. [Accessed 26th March 2013]. 4. Rahisuddin , Sharam, P.K., Garg, G., Salim, M. (2011) Review on nasal drug delivery system with recent advancements. International Journal of Pharmacy and Pharmaceutical Sciences 3 (2): 6-11. 5. Hearnden, V., Sankar, V., Hull, K., Juras, D.V., Greenberg, M., Ross Kerr, A., Lockhart, P.B., Patton, L.L., Porter, S., Thornhill, M.H. (2012) New developments and opportunities in oral mucosal drug delivery for local and systemic disease. Advanced Drug Delivery Review 64: 16-28. 6. Paul, S.M., Mytelka, D.S., Dunwiddie, C.T., Persinger, C.C., Munos, B.H., Lindborg, S.R. and Schacht, A.L. (2010) How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nature Reviews Drug Discovery 9: 203-214. 7. Christrup LL, Foster D, Popper LD, Troen T, Upton R.Clin Ther. 2008 Mar;30(3):469 70 revista safybi 8. Portenoy RK, Payne R, Coluzzi P, Raschko JW, Lyss A, Busch MA, Frigerio V, Ingham J, Loseth DB, Nordbrock E, Rhiner M. Oral transmucosal fentanyl citrate (OTFC) for the treatment of breakthrough pain in cancer patients: a controlled dose titration study. Pain. 1999 Feb;79(2-3):303-12 9. Illum L et al, Intranasal delivery of morphine, J.Pharmacol Exp.Therp., 301, 391-400, 2002 10.Rey E, Delaunay L, Pons G, Murat I, Richard MO, Saint-Maurice C, Olive G. Pharmacokinetics of midazolam in children: comparative study of intranasal and intravenous administration.Eur J Clin Pharmacol. 1991;41(4):355-7. 11.Graul, A.I., Sorbera, L., Pina, P., Tell, M., Cruces, E., Rosa, E., Stringer, M., Castaner, R., Revel, L. (2010) The year’s new drugs & biologics – 2009. Drug News and Perspectives 23 (1): 7. 12.Baumann D, Bachert C, Hogger P. (2009) Dissolution in nasal fluid, retention and anti-inflammatory activity of fluticasone furoate in human nasal tissue ex vivo. Clinical and Experimental Allergy 39: 1540–50. 13.Keith P.K, Scadding G.K (2009). Are intranasal corticosteroids all equally consistent in managing ocular symptoms of seasonal allergic rhinitis? Current Medical Research and Opinion. 25: 2021–41. 14.Garris C, Shah M, D’Souza A, Stanford R. (2009) Comparison of corticosteroid nasal sprays in relation to concomitant use and cost of other prescription medications to treat allergic rhinitis symptoms. Clinical Drug Investigation. 29: 515–26. VROC®- Un dispositivo MEMS capaz de determinar la viscosidad a partir de un pequeño volumen de muestra RHEOSENSE INC. Nota de Aplicación: Estudio de desplegamiento o desnaturalización de proteínas con VROC® Aplicación Esta nota de aplicación aborda la medición de la viscosidad mediante el sistema m-VROC. VROC® es un viscosímetro – reómetro en un chip de precisión, un sensor de viscosidad en escala micrón para pequeños volúmenes de muestra. La viscosidad es un parámetro importante al momento de optimizar drogas inyectables. El anticuerpo monoclonal (mAb) es reconocido como un importante componente biológico terapéutico: ataca la enfermedad con menos efectos secundarios. Para una eficacia terapéutica, su concentración debe mantenerse en un nivel elevado. Sin embargo, la viscosidad aumenta cuando la concentración de mAb aumenta. Al momento de tratar a los pacientes, una viscosidad más elevada resulta ser un problema. Para la administración de la droga, es difícil inyectar una medicación de alta viscosidad sin causar dolor físico al paciente. Esto hace que la formulación de mAb resulte un desafío: se debe lograr una droga estable con valores de viscosidad lo más bajo posibles. La viscosidad es una herramienta efectiva para monitorear la desnaturalización, degradación y/o estabilidad de las drogas. A medida que las moléculas de proteínas se despliegan o “agregan”, la viscosidad aumenta. En esta nota de aplicación las moléculas de proteínas se despliegan agregando urea. Se investiga y estudia el alcance del desplegamiento con datos directamente relacionados con la viscosidad. pliegan y pierden tanto su estructura terciaria natural como su eficacia. La urea entra en contacto con las moléculas de la proteína y éstas últimas se despliegan. A medida que esto sucede, el tamaño de las moléculas aumenta así como el radio hidrodinámico. El incremento del radio hidrodinámico resulta en el incremento de la viscosidad. Para investigar el desplegamiento, se prepararon soluciones de BSA (compradas a Sigma-Aldrich) en PBS. Se prepararon varias series de soluciones de BSA con altas concentraciones de urea. Las concentraciones de BSA se mantenían constantes en 100 mg/ml. El siguiente gráfico muestra claramente que la viscosidad de la solución de BSA (albúmina sérica bovina) se incrementaba cuando aumentaba la concentración de urea. Debido al desplegamiento de moléculas de BSA, el nivel de viscosidad excedía la contribución dada por las elevadas concentraciones de urea. El aumento de viscosidad fue aún mayor cuando la concentración de urea superó los 3 M. Viscosidad de la solución de urea en PBS como función de la concentración La urea es una pequeña molécula que se sintetiza en el cuerpo de varios organismos. Se sabe que interactúa con proteínas y, consecuentemente, despliega las moléculas. En este estudio las viscosidades de la solución de urea en diferentes concentraciones se midieron con el VROC a 25±0,1°C y fueron comparadas con los valores obtenidos con el viscosímetro capilar de vidrio en la literatura11. Como se ilustra a continuación, los resultados muestran un amplio consenso. Desplegamiento de moléculas de proteína y su efecto en la viscosidad La urea es una conocida desnaturalizante de proteínas. Durante el proceso de desnaturalización, las proteínas se des- La concentración de urea se mide en masa Molar A medida que la urea aumentaba su concentración, más moléculas de BSA se desplegaban lo que resultó en una mayor fracción de volumen ocupada por moléculas. Cuando la fracción de volumen de moléculas desplegadas aumenta, la viscosidad aumenta. Este resultado concuerda con el trabajo de micro-reología realizado por Tu y Breedveld2. Otra técnica que se utilizó para monitorear la agregación de las proteínas es la dispersión de luz. Sin embargo, esta técnica está limitada a utilizar una concentración muy diluida. De lo contrario, se debe introducir una interacción de partícula compleja. Una concentración de 100mg/ml probablemente sea demasiado elevada para la técnica de dispersión de la luz. Sin embargo, el rango de detección del VROC® no se limita a bajas concentraciones. n Conclusiones 1.El VROC® cuenta con una alta resolución para distinguir pequeños incrementos de viscosidad, una poderosa herramienta para la investigación cuantitativa. 2.El VROC puede utilizarse para investigar la estabilidad de la droga sin diluir la solución. Referencias 1 K. Kawahara y C. Tanford “J. Biological Chemistry”, Vol.241, N° 13, 3228 (1966) 2 R. Tu y V. Breedveld, “Physical Review”, E72,041914-1, 2005. revista safybi 71 Nuestros anunciantes Decontaminación y esterilización de envases Las prioridades productivas de la industria para lograr bienes de calidad garantizada implican a las materias primas utilizadas y a los procesos de elaboración de los productos. Para los casos en que los bienes requieran ser conservados en condiciones predeterminadas que aseguren su integridad física, química, higiénica y sanitaria, son de de vital importancia los envases empleados. Esta es la realidad de los productos de uso médico, biomédico-descartables, farmacéuticos, cosméticos y alimentarios, para cuyos procesos de elaboración se requiere el cumplimiento de pautas higiénicas apropiadas al uso para el que están destinados. Para ello se establecieron las Buenas Prácticas de Manufactura (BPM), que implican el cumplimiento de todos los requerimientos señalados por los Puntos Críticos del Control, sean éstos referidos a las materias primas como a productos terminados, es decir, los ya dispuestos en su envase final. Con el correr del tiempo los envases o contenedores de vidrio fueron reemplazados por unidades descartables de un solo uso, compuestos por polímeros plásticos que otorgan distintas características físico químicas y propiedades funcionales referidas a su rigidez, elasticidad, tamaño, transparencia, entre otras. No obstante este cambio, los productos a ser envasados siguen requiriendo contenedores estériles o limpios. Los envases, sean de uso biomédico, farmacológico, alimentario o cosmético, guardan entre sí Ventajas comparativas según el tratamiento de esterilización empleado El tratamiento de Ionización Gamma es el indicado para frascos, válvulas, tapas, insertos, goteros, jeringas prellenadas, y todo tipo de envases farmacéuticos. una constante que se corresponde con el requerimiento higiénico del producto que contienen. El grado específico o margen de seguridad sanitaria se define previamente en base a las condiciones finales del uso al que son destinados los contenedores. Para resolver la problemática de la esterilización se seleccionaron inicialmente materiales útiles y capaces de cumplir con las condiciones exigidas, sumado al hecho de que los procesos a los que serían sometidos no indujesen en ellos cambios en sus características químicas y/o bioquímicas. Se promovió así el uso de agentes esterilizantes alternativos al calor. Actualmente, en nuestro país se emplean: gases como el óxido de etileno u otros agentes químicos como por ejemplo el formaldehído, o bien procesos como la ionización, por ejemplo por rayos gamma, emitidos por el Co60 (cobalto sesenta), o los electrones acelerados. Existen ventajas comparativas significativas según el tratamiento de esterilización empleado, y de acuerdo a lo expuesto en el cuadro comparativo presentado en esta nota, las evidentes ventajas que ofrece el tratamiento de esterilización con energía gamma, aseguran que los materiales procesados alcancen realmente el margen de seguridad de esterilización requerido según el uso posterior al que están destinados. A esto se le suma la forma práctica de su aplicación comercial, ya que en el proceso de ionización gamma, los productos (materias primas, semielaborados, terminados) son tratados en sus envases cerrados. Las experiencias prácticas de los últimos años han permitido asegurar que la aplicación de la tecnología gamma no induce daño alguno a los materiales poliméricos tratados. En el orden internacional, la ionización es prácticamente el único método viable para la esterilización de envases farmacéuticos y accesorios (frascos, válvulas, tapas, insertos, goteros, jeringas prellenadas, etc.) n Para mayor información contactar a: IONICS S.A. [email protected] - Tel/fax: 4740-0566/7443 72 revista safybi Mediciones de temperatura y humedad relativa en la industria farmacéutica Las tareas de medir, controlar y registrar las variables ambientales que afectan a la correcta producción, fraccionamiento, estabilidad y almacenamiento de productos químicos y biológicos presentan ciertos desafíos. El objetivo del monitoreo de las condiciones ambientales es asegurar la calidad del producto final y de las distintas etapas del proceso. Instrumentos electrónicos de medición Instrumentos portátiles En el primer caso, los instrumentos están diseñados para ser utilizados de forma manual por el usuario y pueden ser trasladados fácilmente para tomar mediciones de corta duración en distintos puntos de medición. En su forma más básica, están compuestos por el sensor o los sensores correspondientes a las variables medi- Ilustración 1 - Termómetro portátil con das, un visualizador sonda externa de inmersión de las mediciones, la placa electrónica para controlarlos y la fuente de alimentación de energía eléctrica. Para que el instrumento sea portátil, debe emplear baterías. En algunos casos, se dispone de conexión opcional a la red eléctrica. Pueden llegar a almacenar mediciones en una memoria no volátil, o no tener memoria de mediciones. Si poseen memoria de datos, deben disponer además de alguna forma de acceder a los datos almacenados a través de una interfaz (USB, RS232, Bluetooth, infrarrojos, conexión a impresora, tarjeta SD). Pueden proveer herramientas adicionales, como por ejemplo la detección de mínimos, máximos y cálculo de promedios. Los instrumentos portátiles pueden poseer sensores internos o sondas externas, fijas o removibles. Una misma sonda puede contener uno o más elementos sensores de la misma variable o de distintas variables. Aplicaciones: Todo tipo de mediciones de corta duración donde el instrumento pueda ser manipulado por el usuario. Monitores de condiciones ambiente Su objetivo principal es mostrar las condiciones ambientales actuales. Poseen display, y pueden indicar mínimos, máximos, promedios y tendencias. Algunos pueden graficar las últimas mediciones. Aplicaciones: Visualización de las mediciones ambientales en un laboratorio. Registradores de datos Los data loggers o registradores de datos son instrumentos que pueden ejecutar un programa de medición y almacenar las mediciones obtenidas durante un determinado periodo de tiempo en su propia memoria interna, no volátil (EEPROM). Tienen tamaño reducido y funcionan con batería, esto posibilita su uso en aplicaciones de transporte de productos también. La memoria interna suele tener espacio suficiente para almacenar varios días de mediciones y la batería brinda una gran autonomía en comparación a los portátiles. El programa de medición generalmente permite definir un criterio de inicio (por ejemplo: al presionar un botón en el equipo o comenzar a registrar a una determinada hora), el intervalo de registro y el criterio de finalización de la medición (una determinada fecha o hasta completarse la memoria). Los registradores pueden tener display o no. Los sensores del equipo pueden ser internos o pueden ser externos. En el primer caso, los sensores están más resguardados pero el rango de medición está acotado por el rango de temperatura de funcionamiento del instrumento. Por ejemplo: -20 a +70ºC, limitado por la batería y la placa electrónica. Las sondas externas, fijas o removibles, independizan a los sensores del rango de trabajo del instrumento, para poder obtener rangos más amplios de medición. Las conexiones para sondas removibles permiten además aumentar la cantidad de apli- Ilustración 2 - Registrador de datos con caciones en las que conexión para cuatro sondas externas revista safybi 73 Nuestros anunciantes Tabla A: Comparativa entre sensores de temperatura Variable medida Temperatura Tipo de sensor Rango de medición Exactitud Termocupla T -40ºC a 350ºC Clase 1: +/- 0,5ºC ó +/-0,001|t| Sensor NTC -50ºC a 150ºC +/- 0,2ºC a +/- 0,5ºC Sensores RTD (Pt100, Pt1000) -200ºC a 600ºC Clase B: +/-(0,3 + 0,005 |t|) Clase A: +/-(0,15 + 0,002 |t|) pueden ser utilizados, y la disponibilidad de varios canales de conexión para sondas externas permite el monitoreo de varios puntos de medición con un mismo registrador. La adquisición de datos por medio de registradores es semi-desatendida. El usuario debe descargar las mediciones almacenadas en la memoria del registrador a la PC por medio de alguna conexión en el instrumento, en general USB. Para ello, deben programarse rondas de descarga de datos a PC para la generación de informes. Para las aplicaciones de transporte en particular, existen data loggers descartables. Una vez agotada la batería del equipo, el mismo debe ser reemplazado. Aplicaciones: Monitoreo de temperatura y humedad en ensayos de media y larga duración. Monitoreo de temperatura de heladeras, freezers, ultra-freezers, estufas y cámaras de estabilidad. Monitoreo de temperatura en procesos de autoclavado. Monitoreo de las condiciones ambientales en laboratorios. Monitoreo de temperatura de materias primas y productos durante el transporte. Monitoreo de las condiciones ambientales en los depósitos. Instrumentos estacionarios Para situaciones donde se requiere realizar un monitoreo permanente, los instrumentos son instalados de forma fija. Los transmisores no poseen memoria de datos, sino que transmiten las mediciones para ser almacenadas en otro dispositivo que pueda manejar volúmenes grandes de información. Los instrumentos tienen la posibilidad de ser configurados para ejecutar tareas de medición a un ritmo de medición determinado, de forma automatizada. Si la medición se realiza de forma permanente o a largo plazo, es indispensable que el instrumento tenga conexión a la red eléctrica. Como el registro es desatendido, las mediciones son almacenadas para poder ser analizadas. El almacenamiento puede realizarse en un medio externo. Por ejemplo, si el instrumento tiene conexión Ethernet, las mediciones pueden ser enviadas periódicamente a una PC para su registro. Un transmisor con salidas analógicas puede poseer una o varias salidas de tensión o de corriente. Dichas salidas están escaladas para entregar un nivel de tensión o corriente proporcional al parámetro medido, casi siempre en tiempo real. Los transmisores con salidas analógicas deben poseer una salida por cada parámetro que se quiera monitorear simultáneamente. Por ejemplo, un canal para la señal de presión, uno para la temperatura y uno para la humedad relativa. Del otro lado del lazo de transmisión, el dispositivo receptor posee una entrada de similares características y se encargará de volver a convertir el 74 revista safybi nivel de tensión/corriente en el valor del parámetro medido. Para ello, ambos dispositivos deben tener el mismo escalado. El dispositivo que recibe la señal puede ser, por ejemplo, un Controlador Lógico Programable (PLC). Este dispositivo puede almacenar el dato en su memoria, retransmitirlo a una PC o tomar una acción preventiva/correctiva en base al valor recibido. Por ejemplo: activar una unidad de tratamiento de aire si la humedad relativa del aire ambiente excede el 60%. Otras posibilidades de transmisión de las mediciones son a través de Ethernet o de forma inalámbrica a un servidor o una base que centralice las mediciones para su almacenamiento y análisis. Aplicaciones: medición estacionaria de temperatura y humedad en salas limpias y depósitos, monitoreo de la temperatura de punto de rocío en cañerías de aire comprimido para procesos de envasado. Selección del instrumento de medición adecuado Los requerimientos a considerar para seleccionar el instrumento de medición correcto para cualquier aplicación: • Duración y frecuencia de la medición • Rango de medición de los sensores • Precisión y exactitud • Velocidad de respuesta • Tipo de sonda • Histéresis • Características constructivas de los sensores según el medio donde se utilicen • Mantenimiento y recalibración Medición de temperatura por contacto Los sensores más empleados en la industria farmacéutica en instrumentos electrónicos para la medición de temperatura por contacto son los NTC, las termocuplas T y los sensores RTD de platino. Sensores NTC: Los sensores NTC (Negative Temperature Coefficient) brindan un rango de medición amplio combinado con una buena exactitud y tiempo de respuesta. Varían su valor de resistencia eléctrica en función de la temperatura, de forma exponencial. El sensor forma parte de un circuito eléctrico auxiliar que le permite al instrumento medir su valor. Luego el valor medido es convertido al valor de temperatura a través de una tabla en la memoria del instrumento. Termocuplas tipo T: Las termocuplas tipo T presentan una excelente velocidad de respuesta, pero no son tan exactas y precisas como los otros tipos de sensores. La unión de dos metales distintos genera una diferencia de potencial en función de la temperatura (efecto Seebeck). Requieren de compensación de junta fría. El instrumento debe tener aparte otro sensor de temperatura ambiente para realizar la compensación. Sensores RTD: Los sensores RTD o Resistor Temperature Detector son otro tipo de sensores que varían su resistencia eléctrica en función de la temperatura. En este caso la variación es lineal. Son los más exactos y precisos, aunque presentan los tiempos de respuesta más prolongados. El sensor más común es el Pt100 (RTD de platino, valor de resistencia de 100 ohms a 0ºC). |t| = módulo de la temperatura medida en grados Celsius. Ilustración 3 – Algunos modelos de sondas de temperatura Tipos de sonda Una sonda puede contener uno o más sensores, del mismo parámetro o de distintos parámetros. Puede ser un simple soporte para los mismos o puede incluir también la electrónica que realiza la transducción de la señal, de analógica a digital. Algunas sondas también incluyen electrónica de transmisión wireless y display. Las sondas deben tener una estructura acorde a su emplazamiento. Si se necesita monitorear temperatura ambiente o temperatura y humedad ambiente dentro del rango de -20 a 70ºC, pueden utilizarse registradores con sensores internos. Existen sondas de inmersión/penetración donde el sensor está envainado, sondas de superficie para las mediciones por contacto y sondas de aire donde el sensor está descubierto para proporcionar una respuesta más rápida ante las variaciones. Sondas de inmersión y/o penetración: en este tipo de sondas el sensor se encuentra dentro de una vaina metálica sumergible, terminada en punta, que lo protege. Es aconsejable mantener una relación de inmersión vs. diámetro de la sonda de 10 a 15 veces. Sondas de superficie: tienen cabezales diseñados para revista safybi 75 Nuestros anunciantes Electrodo superior - permite el ingreso del vapor de agua a la capa dieléctrica - protege contra el condensado y los contaminantes una óptima transferencia de calor entre la sonda y la superficie. No son aptas para mediciones por inmersión. Sondas de aire ambiente: En este caso, el sensor está expuesto para lograr una velocidad de respuesta más rápida. No son aptas para mediciones por inmersión. Medición de humedad relativa del aire El porcentaje en masa del vapor de agua en el aire está comprendido entre el 0,1% y el 2%. A pesar de esta pequeña cantidad de agua presente en el aire, la calidad de muchos procesos técnicos depende de ella en gran medida. La humedad es indeseable o deseable solamente en concentraciones determinadas, dependiendo de la aplicación. Las sustancias higroscópicas cambian sus propiedades como consecuencia de la humedad ambiente (polvos, granulados, tabletas). La humedad debe ser mantenida a un nivel óptimo y constante, para garantizar la calidad deseada y la procesabilidad de los materiales. Existen varios tipos de sensores de medición de humedad, sin embargo, muchos métodos para determinar la humedad quedan descartados debido a la necesidad de mantenimiento, o por requerimientos de exactitud y velocidad de respuesta o porque son imprácticos para realizar mediciones con una frecuencia alta. Lo más usual para la cuantificación del vapor de agua en el aire es medir la humedad relativa porcentual y la temperatura. A partir de estos valores medidos, los instrumentos pueden mostrar también otros valores calculados como la temperatura de bulbo húmedo, la humedad absoluta, la concentración volumen/volumen, el grado de humedad, etc. La humedad relativa es la relación entre la presión parcial del vapor de agua presente en el aire y la presión de saturación de vapor de agua a esa temperatura. Tabla B: Coeficientes Magnus para el cálculo de presión de vapor de agua Coeficientes Magnus (DIN 50010) C1 [mbar] C2 C3 [ºC] Sobre hielo, -50,9ºC<t<0ºC 6,10714 22,44294 272,44 Sobre agua, -50,9ºC<t<0ºC 6,10780 17,84362 245,425 Sobre agua 0ºC≤t≤100ºC 6,10780 17,08085 234,175 Donde φ es la humedad relativa porcentual, pH2O es la presión parcial del vapor de agua y Psat es la presión de saturación del vapor de agua a la temperatura dada. Sensores capacitivos para medir humedad relativa: Hoy en día, los sensores capacitivos han adquirido una gran popularidad en prácticamente todas las aplicaciones donde se requiera medir humedad. La combinación de buena exactitud, amplio rango de medición, estabilidad a largo plazo, tamaño muy reducido, mantenimiento prácticamente nulo, fácil calibración y posibilidad de ajuste, lo 76 revista safybi Polímero dieléctrico Electrodo inferior Conexiones Sustrato Sustrato cerámico para protección mecánica Ilustración 4 – Sensor capacitivo de humedad relativa convierten en adecuado para una gran variedad de aplicaciones. Los sensores capacitivos ofrecen un rango de medición de 0 a 100% (sin condensación). Están formados por delgadas capas de material poroso, a modo de electrodos, separadas por un material dieléctrico. El conjunto se sitúa sobre un sustrato cerámico para darle protección mecánica. El vapor de agua penetra a través del electrodo en contacto con el aire y afecta a la constante dieléctrica del polímero orgánico que separa a los electrodos. La variación de la constante dieléctrica modifica la capacitancia del sensor, que se encuentra en un circuito oscilador. La frecuencia de oscilación es comparada con una frecuencia de referencia, y el valor de humedad relativa se obtiene en función de dicha frecuencia. Cabe destacar que la respuesta no es lineal y es dependiente de la temperatura, por lo tanto suele encontrarse junto con un sensor de temperatura para realizar la compensación. Las exactitudes de los sensores capacitivos van de +/- 3% a +/- 1% (sondas de alta exactitud). Sondas de humedad y temperatura: son sondas de aire ambiente, los sensores se encuentran expuestos. Ofrecen rangos de trabajo de 0 a 100% de humedad relativa y temperaturas entre -20ºC y 140ºC. Las sondas con exactitud de 1% también son empleadas para la medición de aW (actividad acuosa), siendo insertadas en una cámara de tamaño reducido junto con la muestra y midiendo la humedad de equilibro del aire. Sistemas de monitoreo Un sistema de monitoreo permite la centralización de las mediciones adquiridas de forma automática en diversos puntos de medición y de diversos parámetros, y puede emitir y registrar alarmas cuando se excedan los valores límite. Debe además brindar la posibilidad de acceder a los datos para su análisis. A nivel hardware, un sistema de monitoreo está compuesto por módulos con sensores que envían periódicamente las mediciones a otro dispositivo que las administra y centraliza junto con las alarmas. Muchos sistemas permiten la incorporación de transmisores con salidas analógicas. El dispositivo que centraliza las mediciones puede ser un componente diseñado específicamente para actuar como base central del sistema o de forma directa por una PC. A nivel software, el sistema debe poder enviar alertas al usuario cuando se dispare una alarma, debe brindar opciones de búsqueda y consulta de las mediciones. Es de- seable que el software restrinja el acceso o las capacidades de configuración de los usuarios según niveles de acceso. Es posible realizar el monitoreo a nivel local así como también en múltiples sedes a nivel internacional, con sensores ubicados físicamente en cada sede y vinculados a través de la estructura de red de la empresa. Los sistemas más recientes incorporan la posibilidad de subir a la nube las mediciones recolectadas, aunque emplear el ciberespacio como medio de almacenamiento presenta otros desafíos para la seguridad informática de la empresa. Es preferible tener toda la información en soporte local, en servidores de la empresa destinados a tal fin. n Román Gabriel Lanzillotta Técnico en electrónica Especialista en Aplicaciones, Testo Argentina S.A. Referencias Ilustración 5 - Esquema de un sistema de monitoreo Servidor-Cliente DIN 50010, Part 2 – Climatological terms and definitions. Technical Committee NMP841 Klimate, Begriffe und Messungen. Agosto 1981 revista safybi 77 Asóciese a SAFYBI Requisitos a) Socios Activos: Ser farmacéutico, doctor en farmacia, bioquímico y doctor en bioquímica, con título Nacional o reconocido por la Nación, que estén directamente interesados en la industria bioquímicofarmacéutica, o afín; que sean presentados por dos socios activos y aprobada su solicitud por los 2/3 de votos de la Comisión Directiva. b) Socios Adherentes: Las personas que actúen en la industria bioquímico-farmacéutica o afín y que estén interesados en su progreso, que sean presentados por dos socios activos y ser aceptada su solicitud por las 2/3 partes de votos de la Comisión Directiva. c) Socios Adherentes-Estudiantes: Los estudiantes de farmacia y bioquímica interesados en la industria bioquímico-farmacéutica que sean presentados por dos socios activos y ser aceptada su solicitud por los 2/3 partes de votos de la Comisión Directiva. El derecho a esta categoría será por seis años y mientras mantengan la condición de estudiante. Cuando cumplan con los requisitos para ser aceptados como Socios Activos, deberán comunicarlo a la Comisión directiva para considerar su paso a la categoría de socio activo. Algunos de los beneficios para los Socios son: • Aranceles preferenciales para todas las actividades organizadas por la Asociación: Cursos, Conferencias, Exposiciones y Congresos. • Acceso gratuito a nuestra Biblioteca. • Recibir la Revista SAFYBI en forma gratuita. • Integrar los Comités de Expertos. • Presentación y difusión de trabajos, comunicaciones y novedades científicas en forma gratuita a través de nuestras publicaciones. • Los Socios Adherentes Cooperadores (Laboratorio, Empresa o Institución), con un año de antigüedad y/o pago adelantado de un año de la cuota societaria, tendrán los siguientes beneficios: • Salón Auditorio de SAFYBI: • Uso gratuito de Media Jornada, 1 (una) vez al año. • Uso todo el año con bonificación especial. • Publicación en la Revista SAFYBI de una nota empresarial sin cargo. • Publicación de notas técnicas (previa Autorización de Comisión Directiva) gratuitas. • Inscripción como representantes, de dos personas a los aranceles de socios, en todas las actividades organizadas por SAFYBI. Anunciantes de este número IONICS...................................................... Ret. de tapa CIMA-SINOTEK......................................... 3 EXPOFYBI 2015........................................ 5 COSTER GROUP....................................... 7 Akribis..................................................... 8 GE.............................................................. 9 ROEMMERS.............................................. 11 LABORATORIO AEROFARMA S.A.I.C...... 13 APTAR PHARMA....................................... 15 TSYA ......................................................... 17 KLOCKNER PENTAPLAST de ARGENTINA S.A.................................. 18 FERRETTI VALIDACIONES S.R.L.............. 19 LA POSTAL................................................ 21 Pharma Panel....................................... 23 EUROLAB ................................................. 24 CARPE SCHEIDER..................................... 25 CAS INSTRUMENTAL............................... 27 Securtemp............................................. 28 ANDREANI................................................ 29 78 revista safybi INDUSTRIAS HÖGNER............................. DROMEX................................................... GS1............................................................ KIMS.......................................................... TESTO ARGENTINA S.R.L........................ ASISTHOS................................................. AKRIBIS..................................................... LABCO-Laboratorio de Control S.A....... MATRICOD............................................... Sensaphoneweb.................................. PALL TECHNOLOGIES S.A....................... EDYAFE..................................................... ETICOR...................................................... TETRAFARM.............................................. LAB. ARGENPACK S.A.............................. GUIBA TÉCNICA....................................... GEZZI........................................................ SINAX S.A.................................................. FARMAWALL............................................ SARTORIUS............................................... 31 33 35 37 39 41 43 45 51 54 55 60 61 63 65 75 75 77 Ret. contratapa Contratapa Agosto de 2014 | Nº 143 Volumen 54 Nº 143 Agosto de 2014 ISSN: 0558/1265 Uruguay 469, piso 2º B C1015ABI Buenos Aires, Argentina Tel: (54-11) 4373-0462 / 8900 (54-11) 4372-7389 Fax: (54-11) 4374-3630 www.safybi.org Revista El nuevo film de PE. Su nueva referencia. Revista de la Asociación Argentina de Farmacia y Bioquímica Industrial Revista SAFYBI | Nueva familia de bolsas Flexsafe. Revista SAFYBI Passed ASTM Shipping Test Flexsafe reune los requisitos de robustez excepcional y fácil uso en todos los Entrevista exclusiva SAFYBI Dra. Boni, Directora de la Dirección de xxxx de la anmat ONE FILM FOR ALL pasos del proceso de fabricación de sus productos. La resistencia y la flexibilidad del film proporcionan un rendimiento constante y un manejo sencillo incluso en las USP: Envasado farmacéutico: pruebas de permeabilidad a la humedad e integridad aplicaciones más exigentes como el cultivo celular, almacenamiento a largo plazo o el transporte de fármacos. Sartorius Argentina S.A. · Tel. +54.11.4721.0505 · [email protected] Ver vídeos: www.sartorius-stedim.com/flexsafe USP – DSP – F +F Visite nuestra revista online en www.safybi.org Novedades en vacunas Vacuna contra la fiebre hemorrágica argentina, un producto nacional Avances en las técnicas de purificación de vacunas