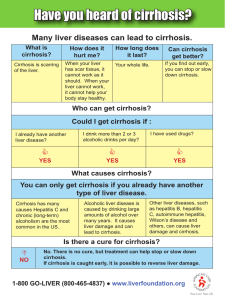
5º Curso para residentes
Anuncio

5º Curso para Residentes Diagnóstico y Tratamiento de las Enfermedades Hepáticas Barcelona, 30 y 31 de octubre de 2015 Director: Jaume Bosch Genover Organiza © 2015 Fundación Española para el Estudio del Hígado © 2015 Asociación Española para el Estudio del Hígado © 2015 Elsevier España, S.L.U. Avda. Josep Tarradellas, 20-30. 28029 Barcelona Zurbano, 76. 28010 Madrid Reservados todos los derechos. El contenido de esta publicación no puede ser reproducido, ni transmitido por ningún procedimiento electrónico o mecánico, incluyendo fotocopia o grabación magnética, ni registrado por ningún medio, sin la previa autorización por escrito del titular de los derechos de explotación. Elsevier y sus asociados no asumen responsabilidad alguna por cualquier lesión y/o daño sufridos por personas o bienes en cuestiones de responsabilidad de productos, negligencia o cualquier otra, ni por uso o aplicación de métodos, productos, instrucciones o ideas contenidos en el presente material. Dados los rápidos avances que se producen en las ciencias médicas, en particular, debe realizarse una verificación independiente de los diagnósticos y las posologías de los fármacos. sumario 5º CURSO PARA RESIDENTES DIAGNÓSTICO Y TRATAMIENTO DE LAS ENFERMEDADES HEPÁTICAS Barcelona, 30 y 31 de octubre de 2015 Director Jaume Bosch Genover SESIÓN 1: HEPATITIS VIRALES Moderadora: María Buti Hepatitis E M. Buti ................ 1 Temas no resueltos en hepatitis B M. Hernández Conde, C. Perelló y J.L. Calleja ................ 4 Tratamiento de la hepatitis C: genotipos 3, 5 y 6 R. Souto Rodríguez y J. Turnes ................ 11 Hepatitis colestásica fibrosante M.C. Londoño ................ 18 ................ 24 TIPS: indicaciones y resultados J.C. García-Pagán ................ 29 Insuficiencia renal en la cirrosis X. Ariza y P. Ginès ................ 32 ................ 39 ................ 45 ................ 50 SESIÓN 2: CIRROSIS E HIPERTENSIÓN PORTAL Moderador: Jaume Bosch Profilaxis y tratamiento de la peritonitis bacteriana espontánea: implicaciones de los cambios en el espectro microbiano G. Soriano Bloqueadores beta: indicaciones, contraindicaciones y riesgos en cirrosis A. Albillos Fracaso hepático agudo sobre crónico R. Bañares Impacto de la encefalopatía hepática mínima en la morbimortalidad del paciente con cirrosis hepática J. Ampuero Herrojo y M. Romero Gómez sumario SESIÓN 3: ENFERMEDADES METABÓLICAS Y SISTÉMICAS Moderador: Raúl J. Andrade Diagnóstico no invasivo de cirrosis e hipertensión portal S. Augustin, M. Pons, B. Santos, M. Ventura y J. Genescà ................ 54 ................ 62 ................ 66 ................ 76 ................ 81 ................ 87 Cirrosis biliar primaria: nuevas opciones de tratamiento A. Parés ................ 91 Hepatocarcinoma: manejo del estadio intermedio M. Varela ................ 98 Colangiocarcinoma intrahepático B. Sangro ................ 104 Hipertensión portal idiopática V. Hernández-Gea ................ 110 Patogenia de la enfermedad hepática por depósito de grasa no alcohólica M.T. Arias Loste y J. Crespo Toxicidad hepática inducida por fármacos R.J. Andrade Riesgo vascular en el paciente con esteatosis hepática: implicaciones en el entorno del trasplante hepático G. Ontanilla Clavijo y M. Romero Gómez Caso clínico: Enfermedad sistémica con manifestaciones hepáticas: fiebre, hepatoesplenomegalia y síndrome poliadenopático J.F. Andrés Cordón y J. Genescà Ferrer SESIÓN 4: OTRAS ENFERMEDADES HEPÁTICAS Moderador: Joan Genescà Inclusión de candidatos en lista de espera de trasplante hepático: evaluación e indicación M. de la Mata SESIÓN 1 HEPATITIS VIRALES Hepatitis E María Buti Servicio de Hepatología-Medicina Interna, Hospital Universitario Vall d’Hebron, Barcelona, España INTRODUCCIÓN El virus de la hepatitis E (VHE) es un virus ARN, de una única hebra de sentido positivo, perteneciente al género Hepevirus y a la familia Hepeviridae. La infección por el VHE causa hepatitis aguda autolimitada, excepto en sujetos no inmunocompetentes, en los que puede evolucionar a hepatitis crónica. La infección por VHE se caracterizó en 1980 a partir de muestras de sangre obtenidas de pacientes que sufrieron una epidemia de hepatitis aguda de transmisión por aguas fecales en Nueva Delhi (India) entre 1955 y 1956. Su agente causal, el “virus de hepatitis entérica no-A”, se identificó en 1983 y se denominó E por sus características de entérico y epidémico. En España, la mayoría de los casos de hepatitis por VHE se trataba de “casos importados”, adquiridos mediante viajes a zonas endémicas. Sin embargo, en los últimos años se ha documentado un número creciente de “casos autóctonos”, sin antecedentes epidemiológicos de viaje a zonas donde la infección por VHE es endémica. La prevalencia de anticuerpos de tipo IgG frente al VHE, que indican exposición a este virus, en España se sitúa en torno al 0,6-7,3% en la población general y alcanza el 19% en individuos con factores de riesgo como la exposición a ganado porcino. infectar a humanos y otros mamíferos, y son los principales responsables de casos esporádicos de hepatitis E en regiones no epidémicas. Las infecciones por genotipos 1 y 2 en países en vías de desarrollo afectan sobre todo a niños mayores y adultos jóvenes, mientras que los genotipos 3 y 4, predominantes en países desarrollados, afectan más a sujetos de edad avanzada o sujetos inmunodeprimidos como aquellos con infección por el virus de inmunodeficiencia humana (VIH), lo que sugiere que los genotipos 3 y 4 causan enfermedad en sujetos inmunológicamente más débiles. El genotipo 5 solo afecta a aves. En voluntarios sanos, tras la inoculación del VHE se ha observado un período de incubación de la infección de 4-5 semanas, que puede oscilar de 2 a 10 semanas en los brotes de hepatitis. Se han reportado diversas vías de transmisión del VHE, que en orden decreciente de importancia son: a) fecal-oral, por contaminación de los suministros de agua potable; b) por alimentos contaminados, crudos o poco cocidos; c) de forma menos frecuente por transfusión de productos sanguíneos infectados; d) transmisión vertical (maternofetal), y e) por contacto directo con sujetos infectados. En algunos casos, sobre todo en los casos esporádicos en regiones no endémicas, es difícil esclarecer el mecanismo de adquisición de la infección. VIRUS DE LA HEPATITIS E: CARACTERÍSTICAS VIROLÓGICAS EPIDEMIOLOGÍA Y CLÍNICA DE LA INFECCIÓN POR EL VIRUS DE LA HEPATITIS E El VHE está formado por una partícula icosaédrica, sin envoltura, de unos 32 nm y resistente a la inactivación por las condiciones ácidas y alcalinas leves del tracto intestinal, que facilitan la vía de transmisión fecal-oral. Existen 4 genotipos del VHE definidos filogenéticamente en mamíferos. Los genotipos 1 y 2, restringidos a humanos, se asocian a grandes epidemias transmitidas por el agua en las regiones donde la infección es endémica. Por el contrario, los genotipos 3 y 4 pueden La infección por el VHE tiene una distribución mundial, pero con prevalencias más elevadas en países en desarrollo, donde la enfermedad clínica puede ser endémica. La infección por VHE tiene 2 patrones claramente diferenciados: un patrón en regiones endémicas (países en vías de desarrollo) y otro muy diferente en los países desarrollados. Los brotes epidémicos solo se producen en países en desarrollo y se asocian con la contaminación de aguas por el VHE. Las tasas de morbilidad durante los brotes 1 2 Sesión 1 - HEPATITIS VIRALES van del 1 al 15%, afectan, sobre todo, a adultos jóvenes y son más frecuentes en varones que en mujeres. Es remarcable que los brotes de hepatitis E se asocian a una alta morbilidad y mortalidad en mujeres gestantes (el 19% en embarazadas frente al 2,1% en no embarazadas o el 2,8% en varones), con un alto riesgo obstétrico de prematuridad y riesgo de mortalidad perinatal. En regiones sin brotes epidémicos, la hepatitis E representa solo una minoría de las hepatitis virales agudas y hasta hace unos años la mayoría de los casos estaban relacionados con viajes a zonas donde la infección por VHE es endémica. Sin embargo, en los últimos años se han reportado casos de hepatitis E de transmisión autóctona en Estados Unidos, Europa y en países desarrollados de Asia-Pacífico (Japón, Taiwán, Hong Kong, Australia). Estos casos esporádicos se trata de zoonosis y el mecanismo de transmisión es a través de la ingesta o manipulación de carne de cerdo u otros animales como el jabalí, contaminados por el VHE: el “verdadero” número de infecciones por el VHE es difícil de conocer, ya que la presencia de infecciones subclínicas en humanos es frecuente y probablemente superior a la de las infecciones clínicas. En países no endémicos, la infección por VHE suele estar causada por los genotipos 3 y 4, y suele detectarse mediante pruebas serológicas en casos con hepatitis inexplicables, sobre todo en pacientes de más de 60 años. La enfermedad clínica en estos casos es similar a la observada en las regiones endémicas, aunque con una proporción mayor de hepatitis ictéricas. La mayoría de los pacientes son varones de mediana edad o mayores, a menudo con patologías previas que parece justificar su peor pronóstico en comparación con los casos de zonas endémicas. En España, la prevalencia de anticuerpos antiVHE de tipo IgG oscila entre el 0,6 y el 7,3%, y es más baja del 1% en individuos jóvenes y más elevada del 3,6% en sujetos de mayor edad. También se han reportados casos de hepatitis aguda clínica, la mayoría casos autóctonos, que correspondían a genotipo 3 y que han presentado una evolución favorable hacia la curación. La edad media de estos casos es elevada, entre 50-70 años, a diferencia de lo que ocurre en las epidemias de hepatitis aguda que afectan a sujetos más jóvenes. El VHE se ha asociado excepcionalmente con manifestaciones extrahepáticas como manifestaciones neurológicas (encefalitis, ataxia, neuritis braquial y síndrome de Guillain-Barré), manifestaciones hematológicas (aplasia medular y trombocitopenia), manifestaciones musculares (miopatía proximal y miositis necrosante) y manifestaciones renales (nefropatía IgA y glomerulonefritis membranoproliferativa). Hasta hace poco se creía que el VHE, al igual que el VHA, solo causaba cuadros de hepatitis aguda autolimitada y casos de fallo hepático fulminante, y no se asociaba a procesos de cronificación. Sin embargo, recientemente se han descrito en pacientes inmunodeprimidos, como receptores de trasplante de órganos sólidos, pacientes hematológicos o en quimioterapia o infectados por el VIH, casos de infección por VHE con enfermedad hepática crónica, que incluso pueden progresar a cirrosis. Todos estos casos de infección crónica por VHE en personas inmunodeprimidas se deben al genotipo 3, que es el genotipo mayoritario en las zonas en que se han descrito estos casos. En un estudio retrospectivo, Kamar et al observaron que el 60% de 85 casos de receptores de órganos sólidos infectados por VHE desarrollaron una hepatitis crónica por este agente, y sugieren que la inmunosupresión con tacrolimus podría ser un factor fuertemente relacionado con la cronificación. Asimismo se ha constatado que la reducción de la dosis de tacrolimus podría asociarse con el aclaramiento del virus. Estas observaciones han modificado la visión sobre la historia natural de la infección por VHE y sugieren la posibilidad de una forma de infección persistente por VHE con daño hepático crónico y progresivo que puede evolucionar a cirrosis. DIAGNÓSTICO DE LA INFECCIÓN POR EL VIRUS DE LA HEPATITIS E El diagnóstico de la infección por el VHE se realiza mediante la determinación de anticuerpos específicos (anti-VHE) de tipo y/o la presencia de ARN-VHE. Los anticuerpos IgM anti-VHE aparecen durante la fase aguda de la enfermedad, de forma muy precoz durante el final del período de incubación o al inicio de la ictericia, y permanecen detectables 4-5 meses y constituyen un marcador muy adecuado para el diagnóstico de la infección aguda. Sin embargo, hay que tener en cuenta que la detección de anticuerpos IgM VHE presenta problemas tanto de especificidad (del 78 al 98%) como de sensibilidad (del 72 al 98%), por lo que a veces es necesario determinar el ARN-VHE. El ARN-VHE es el marcador virológico más importante, aunque tiene un valor limitado por lo breves que son los períodos de viremia (unas 2 semanas en suero y unas 4 en heces, con casos de 8 a 12 semanas). Por ello, su ausencia no permite descartar el diagnóstico de infección aguda por VHE. Además, la mayoría de las técnicas de PCR para la determinación del ARN-VHE no están comercializadas ni estandarizadas, lo que dificulta su interpretación y comparación entre las distintas técnicas. Por lo tanto, el diagnóstico de laboratorio de la hepatitis E aguda se basa en la presencia de IgM antiVHE en suero y/o la detección de ARN del VHE en suero o heces. Desde el punto de vista serológico, en la infección aguda por el VHE se detectan anticuerpos anti-VHE de tipo IgM e incluso IgG anti-VHE, mientras que la detección aislada de este último indica infección pasada y es útil en los estudios de prevalencia. En pacientes inmunodeprimidos es básica la detección de ARN-VHE, ya que los anticuerpos pueden aparecer tardíamente o incluso pueden estar ausentes. Hepatitis E 3 ción. Existe una vacuna eficaz para prevenir la hepatitis E comercializada en China. No hay datos sobre la utilización de gammaglobulina para prevenir la postexposición a la hepatitis E. Bibliografía recomendada TRATAMIENTO No existe un tratamiento específico de la hepatitis aguda E. La ribavirina y el interferón pegilado se han mostrado eficaces en la hepatitis crónica por VHE en sujetos inmunodeprimidos. La ribavirina es el fármaco más utilizado, aunque la dosis y la duración del tratamiento no están bien estandarizadas. La mayoría de los estudios utilizan 600 mg/día durante 6-12 semanas. La eliminación del VHE se consigue en el 70-80% de los casos; los fracasos del tratamiento están relacionados con recaídas. Es aconsejable remitir a los pacientes con hepatitis aguda grave y/o de evolución fulminante, independientemente del origen etiológico, a un centro que disponga de trasplante hepático para su evalua- Aggarwal R. Hepatitis E: Historical, contemporary and future perspectives. J Gastroenterol Hepatol. 2011;26 Suppl 1:72-82 . Balayan MS, Andjaparidze AG, Savinskaya SS, Ketiladze ES, Braginsky DM, Savinov AP, et al. Evidence for a virus in non-A, non-B hepatitis transmitted via the fecal-oral route. Intervirology. 1983;20:23-31. Buti M, Clemente-Casares P, Jardi R, Formiga-Cruz M, Schaper M, Valdes A, et al. Sporadic cases of acute autochthonous hepatitis E in Spain. J Hepatol. 2004;41:126-31. Gerolami R, Moal V, Colson P. Chronic hepatitis E with cirrhosis in a kidney-transplant recipient. N Engl J Med. 2008;358: 859-60. Khudyakov Y, Kamili S. Serological dianostics of hepatitis E virus infection. Virus Res. 2011;161:84-92. Purcell RH, Emerson SU. Hepatitis E: an emerging awareness of an old disease. J Hepatol. 2008;48:494-503. SESIÓN 1 HEPATITIS VIRALES Temas no resueltos en hepatitis B Marta Hernández Conde, Christie Perelló y José Luis Calleja Servicio de Gastroenterología y Hepatología, Hospital Universitario Puerta de Hierro, Universidad Autónoma de Madrid, Madrid, España INTRODUCCIÓN La Organización Mundial de la Salud estima en 2.000 millones el número de personas infectadas por el virus de la hepatitis B (VHB), con 350-400 millones infectados crónicamente1. Hemos seleccionado 4 temas controvertidos que creemos de interés; para ello nos hemos basado en estudios relevantes publicados recientemente. MÉTODOS NO INVASIVOS PARA EVALUAR LA FIBROSIS Se precisa una evaluación adecuada del grado de fibrosis para poder determinar la indicación de tratamiento. Por ello, en los últimos años han surgido diversos métodos de evaluación clínicos y de imagen, que se han postulado como alternativas a la biopsia hepática (BH), un método invasivo con error de muestreo y elevada variabilidad inter e intraobservador. Además, estos métodos no invasivos aportan un rango continuo de valores, mientras que la BH tiene un número limitado de categorías. Con el VHB nos enfrentamos a 2 escenarios clínicos diferentes: a) la presencia de fibrosis significativa, que es una indicación de tratamiento en estos pacientes, y b) la presencia de cirrosis hepática (CH), que es una indicación para realizar seguimiento, prevención y tratamiento de las posibles complicaciones relacionadas con la CH. La exactitud y la aplicabilidad de los estudios sobre biomarcadores séricos y elastografía de transición (ET) son diferentes para los pacientes con hepatitis B o con hepatitis C2. Los métodos no invasivos para medir el grado de fibrosis hepática son menos utilizados en pacientes con VHB que en pacientes con virus de la hepatitis C, debido a la existencia inicial de un menor número de estudios y porque la inflamación hepática y la replicación del virus pueden confundir en la interpretación de los resultados de los tests. 4 La última guía de la EASL3 recomienda la BH como método de evaluación de la fibrosis en la cirrosis por VHB (CHB). Sin embargo, el papel de los marcadores no invasivos se clasifica como “asignatura pendiente”, con la sugerencia para su posible aplicación en los pacientes que inician el tratamiento sin biopsia activa. La AEEH, sin embargo, refiere que en la práctica clínica habitual la BH no es imprescindible en la mayoría de los casos, y su realización queda para casos en que los exámenes no invasivos no proporcionen la información necesaria para adoptar decisiones clínicas4. Por el contrario, en la guía de la AEEH se acepta la utilización de métodos no invasivos. La medición de la elasticidad hepática mediante ET o Fibroscan® (Echosens, Paris, France) es un método no invasivo para el diagnóstico de la fibrosis hepática, que en la hepatitis por virus B permite discriminar 3 estadios de fibrosis: ausencia o fibrosis leve, fibrosis avanzada o “zona gris” (que se encuentra entre las anteriores)5. Viganò et al6 aportaron un algoritmo basado en 2 valores de corte (“dual cut-off”) para clasificar a los pacientes con hepatitis crónica, pudiendo identificar a los pacientes con fibrosis significativa (≥ F2) y cirrosis (F4), aunque este algoritmo depende de la prevalencia de fibrosis significativa y cirrosis en la población en la que se aplica. Utilizando el corte predictivo negativo < 6,2 kPa junto con el corte predictivo positivo > 9,4 kPa se diagnosticaba a los pacientes ≥ F2 en dos tercios de los casos. Ocurría similar en el caso de los pacientes F4, para ello se utilizaba un corte predictivo negativo < 9,4 kPa junto con el corte predictivo positivo > 13,1 kPa. En el resto de pacientes se indicaba la realización de BH. Para evitar el riesgo de falsos positivos, algunos autores han propuesto adaptar los puntos de corte de la ET en función de los valores de aminotransferasas (ALT), una estrategia que no se debería utilizar en pacientes con valores fluctuantes de estas7. Sin embargo, en pacientes con antígeno e de la hepatitis B (HBeAg) negativo y ALT en rango de la normalidad, los métodos no invasivos, en particular la ET, se podrían utilizar como herramienta complementaria a la medición del ADN-VHB en el seguimiento de estos pacientes portadores inactivos y así poder identificar a los pacientes que precisen BH. Hay una relación entre el valor de elasticidad hepática y las diversas complicaciones de la CH8. Además parece que la elasticidad hepática disminuye con el tratamiento antiviral, lo que sugiere que probablemente también mejore la supervivencia9. En la actualidad, la ET puede identificar a los pacientes con mayor probabilidad de desarrollar hipertensión portal (HTP) clínicamente significativa, pero no resulta efectiva en la identificación de los pacientes con varices esofágicas, por lo que no puede sustituir a la gastroscopia en pacientes con CHB. No obstante se puede utilizar para identificar a los pacientes que presentan mayor probabilidad de progresión de su enfermedad10. La estrategia de combinar ET, el diámetro esplénico y el número de plaquetas (denominado LSPS) aumenta la exactitud del diagnóstico en la detección de varices esofágicas con elevado riesgo de hemorragia y el riesgo de hepatocarcinoma (CHC) en pacientes con CHB11-13. Estos hallazgos concuerdan con los del estudio que determinaba que los valores de elasticidad hepática pueden ser tan efectivos como la medida del gradiente de hipertensión portal a la hora de predecir los pacientes que desarrollarán descompensaciones clínicas y complicaciones relacionadas con la HTP. Si estos hallazgos se confirman, la ET podría utilizarse como herramienta de pronóstico14. ARFI® (Acuson S20000; Siemens Medical Solu­tions, Mountain View, CA, USA), en comparación con la ET, mide la elasticidad hepática en un área menor, pero esta puede ser elegida por el operador. La ventaja de este es que se puede añadir al ecógrafo, aunque los valores se presentan en un rango estrecho, por lo que resulta difícil establecer puntos de corte a la hora de tomar decisiones. Park el at15 compararon ARFI® frente a BH y demostraron que es un buen predictor de fibrosis avanzada (F3-4), y que su resultado se puede ver influenciado por el índice de masa corporal. Además propusieron nuevos valores de corte (1,31 m/s para ≥ F2; 1,81 m/s para ≥ F3, y 1,98 m/s para F4) que precisan ser validados. Los marcadores serológicos presentan una elevada aplicabilidad (95%), una reproducibilidad entre laboratorios y una amplia disponibilidad. Sin embargo, no son específicos del hígado, por lo que su resultado se puede ver influenciado por las condiciones externas, y son menos eficaces a la hora de detectar estadios intermedios de fibrosis que en detectar o no cirrosis2. Existen varios algoritmos disponibles; cabe destacar el Fibrotest® (fórmula patentada; Biopredictive, Paris, France), ya que fue el primer algoritmo que se creó, muy útil para excluir CH, pero tiene una exactitud Temas no resueltos en hepatitis B 5 subóptima a la hora de detectar fibrosis significativa y cirrosis, por lo que precisa combinarse con otros métodos no invasivos16. Debido a su elevado coste, estos métodos no se utilizan habitualmente en países en desarrollo, por lo que están en desarrollo alternativas más baratas. Salkic et al17 publicaron recientemente un estudio en el que creaban un algoritmo basado en 4 scores para seleccionar a los pacientes VHB con mayor probabilidad de tener presencia o ausencia de fibrosis significativa y cirrosis (http://www.chb-lfc.com). Para ello evaluaron 6 marcadores no invasivos de fibrosis en VHB y los compararon con la BH. Llegaron a la conclusión de que APRI y FIB-4 son los métodos alternativos no invasivos más adecuados para clasificar el grado de fibrosis, con una precisión del 93,5%. Ocurría de forma similar a la hora de combinar los scores GUCI y LoK para diagnosticar a pacientes con cirrosis, ya que alcanzaban un grado de precisión del 97,8%. En conclusión, se recomienda evaluar el grado de fibrosis en todos los pacientes con VHB. Esta evaluación puede hacerse inicialmente utilizando métodos no invasivos (ET, marcadores serológicos o ambos), realizando BH solo en los casos donde no se obtenga la información necesaria para adoptar una decisión sobre el manejo del paciente. TRATAMIENTO COMBINADO INTERFERÓN PEGILADO + ANÁLOGOS Existen 2 tipos de fármacos aprobados para el tratamiento de la hepatitis crónica B: interferón pegilado (interferón pegilado alfa-2a) y los 5 análogos de nucleós(t)ido (AN), lamivudina, adefovir, entecavir, telbivudina y tenofovir. El objetivo del tratamiento es la supresión persistente de la replicación viral para evitar la progresión de la enfermedad hepática. Sin embargo, el objetivo ideal es el aclaramiento del antígeno de superficie de la hepatitis B (HBsAg) y/o seroconversión a anti-HBs, algo infrecuente actualmente. Los AN se han convertido en la estrategia de tratamiento más utilizada, pero la administración durante un período tan prolongado se puede relacionar con un elevado coste, valores de seguridad desconocidos, falta de adherencia al tratamiento, riesgo por las resistencias al fármaco o disminución en el aclaramiento de HBsAg. Por otro lado, PEG-IFN alcanza la respuesta viral sostenida (RVS) en casi un cuarto de los pacientes tratados y el aclaramiento de HBsAg en el 30-50% de los pacientes respondedores18. Para aumentar la respuesta de los pacientes tratados con PEG-IFN se debe realizar una selección de los pacientes teniendo en cuenta la edad, las ALT y el ADN-VHB, así como utilizar las reglas de parada basadas en la cinética del ADN-VHB y del HBsAg. 6 Sesión 1 - HEPATITIS VIRALES Tabla 1 Estrategia Objetivo Duración Eficacia PEG-IFN Control inmune (respuesta sostenida tras el tratamiento) ADN-VHB < 2.000 UI/ml y ALT normal Finita Respuesta sostenida en el 30% de los pacientes tras 48 semanas de tratamiento, puede elevarse al 50% si factores de buen pronóstico previo al tratamiento AN Control viral (respuesta mantenida durante el tratamiento) ADN-VHB indetectable y ALT normal Prolongada o indefinida Supresión profunda del AND-VHB con tratamiento mantenido, sin producir resistencias ALT: aminotransferasas; AN: análogos de nucleós(t)ido; PEG-INF: interferón pegilado; VHB: virus de la hepatitis B. En los últimos años se ha postulado sobre si el tratamiento combinado de PEG-IFN con AN puede mejorar la respuesta al tratamiento y el porcentaje de seroconversión de HBsAg para acortar la duración de los tratamientos19. En la combinación de PEG-IFN con lamivudina se llegó a la conclusión de que, a pesar de que la inhibición de la replicación viral es más intensa con el tratamiento combinado, esto no se traduce en un incremento en la respuesta20,21. Estos estudios estaban limitados por 2 aspectos. Primero, la mayoría de los estudios utilizaban lamivudina, reconocida como una opción subóptima de tratamiento (modesto efecto antiviral y baja barrera genética). Segundo, IFN y lamivudina se administraban con el mismo tiempo finito, por lo que lamivudina finalizaba antes de alcanzar el objetivo22. Actualmente se está investigando con la combinación PEG-IFN y entecavir o tenofovir23. Además, no hay datos sobre la ventaja de esta combinación en pacientes naïve para AN y se precisan más estudios en pacientes con viremia basal elevada (> 108 UI/ml). En el congreso americano (AASLD) de 2014 se publicó un estudio llevado a cabo por Marcellin et al, en el que 740 pacientes con hepatitis B crónica HBeAg positivo y negativo recibieron PEG-IFNa y tenofovir o PEG-IFNa o tenofovir. Los resultados preliminares demostraron que la incidencia de pérdida del HBsAg era mayor en los pacientes tratados con PEG-IFNa + tenofovir durante 48 semanas que en los tratamientos administrados por separado. Debido a que los AN no eliminan el cccADN de las células, hablamos de “cura funcional o clínica del VHB”, ya que al seroconvertir el HBsAg en anti-HBs, las células que quedan infectadas están controladas por el propio sistema inmune del paciente. Se está investigando in vitro e in vivo sobre nuevos fármacos que actúen sobre nuevas dianas. COHORTES DE VIDA REAL Tabernero et al publicaron en el congreso europeo (EASL) de 2014 los datos del estudio de cohorte de vida real (CIBERHEP-AEEH) sobre el tratamiento con tenofovir en 383 pacientes con VHB (el 47% naïve, el 53% tratados previamente) de 24 centros españoles, que evidenciaron que los pacientes tratados con tenofovir alcanzaban la supresión viral en una proporción similar entre los pacientes naïve y los previamente tratados (el 78 frente al 84%; p = no significativa). Sin embargo, la pérdida de HBeAg ocurría más frecuentemente en los pacientes naïve que en los previamente tratados (el 26,5 frente al 4,5%; p = 0,004). Recientemente se ha publicado un estudio de cohorte de vida real24, en el que se analizan los cambios en la infección por VHB entre la población de Taiwán vacunada y no vacunada. Taiwán fue el primer país en realizar programas nacionales de inmunización al VHB, que comenzaron en julio de 1984. La prevalencia global de la infección crónica por el VHB fue del 13,7% y cerca de dos tercios han tenido exposición (anti-HBc: 68,46%) en el año 2002. La cohorte vacunada tendía a tener menor prevalencia de HBsAg y anti-HBc, y una mayor proporción de anti-HBs y HBeAg positividad, genotipo C y elevada carga viral. La mayoría (85,42%) fue consistentemente HBsAg–, mientras que el 12,65% fue positivo y el 8,98% logró aclaramiento en un período de 5 años. En la cohorte vacunada, ningún sujeto adquirió nueva exposición y se convirtió a HBsAg+, y solo 1 sujeto (0,54%) aclaró el HBsAg, lo que demuestra la durabilidad de la vacunación hasta la edad adulta joven y adolescente. HEPATOCARCINOMA EN PACIENTES TRATADOS Se estima que la incidencia anual de CHC es del 0-1,4% en pacientes asiáticos no cirróticos, del 0,1-1,0% en caucásicos no cirróticos, del 0,9-5,4% en asiáticos con CHB y del 1,5-5,2% en pacientes caucásicos con CHB25. Numerosos estudios sugieren que el VHB está asociado al proceso de carcinogénesis, mientras que la replicación persistente del VHB y la progresión hacia CH son factores de riesgo independientes de CHC. No hay datos que evalúen el efecto del tratamiento con PEG-IFNa sobre el riesgo de CHC en hepatitis B crónica. Los datos que se disponen provienen de estudios realizados sobre IFNa, que parece tener un efecto moderado sobre el riesgo de CHC; este efecto es mayor en pacientes procedentes de Asia y en casos con elevado riesgo basal de CHC26. El efecto sobre el CHC de los pacientes con RVS tras tratamiento con IFNa permanece en debate, por lo que actualmente se recomienda que los pacientes con RVS tras PEGIFNa y riesgo basal elevado de CHC mantengan el cribado. Respecto a los AN, a pesar de que la remisión virológica se acompaña de mejora de la histología de la CH en la mayoría de los casos, su efecto sobre la prevención del CHC es controvertido, ya que no eliminan la carcinogénesis producida por la infección. Un estudio retrospectivo llevado a cabo por Wu et al27 demostró que el efecto de disminuir el riesgo de CHC de los AN se presentaba en pacientes con y sin CH. El tratamiento prolongado con AN de elevada barrera genética (entecavir o tenofovir) ofrece una reducción moderada del riesgo de CHC, en comparación con el no tratamiento, al menos durante los primeros 5 años de tratamiento (el seguimiento de los estudios presentados hasta ahora no excede de los 4-6 años)25. El beneficio en la reducción del riesgo de CHC es más evidente en pacientes con cirrosis con elevado riesgo basal de CHC y no es tan evidente en pacientes no cirróticos con bajo riesgo basal de CHC25. Además, en la revisión llevada a cabo por Papatheodoridis et al25 se demostró que no había diferencia respecto a la reducción del riesgo de CHC entre comenzar el tratamiento con entecavir o tenofovir y comenzar el tratamiento con lamivudina y, posteriormente, utilizar tratamiento de rescate en pacientes que creaban resistencias. Se ha estudiado la utilización de algoritmos que calculen el riesgo de CHC en población asiática (GAGHCC, CU-HCC y REACH-B). En Europa disponemos del algoritmo PAGE-B, que se estudió en pacientes con VHB que recibieron tratamiento con entecavir o tenofovir. Este algoritmo incluye las variables que no se afectan por el tratamiento antiviral (edad y sexo) y otra que sí se afecta (recuento plaquetario)25,28. Se precisan estudios con mayor número de pacientes y períodos de seguimiento mayores para poder determinar el beneficio potencial de estos agentes sobre el riesgo de CHC en pacientes no cirróticos. Mientras tanto, todos los pacientes con riesgo basal elevado de CHC deben continuar con el cribado de acuerdo con las recomendaciones existentes, incluso si se ha conseguido la respuesta virológica de forma mantenida, la normalización bioquímica y ha mejorado la histología hepática. Temas no resueltos en hepatitis B 7 REACTIVACIÓN EN PACIENTES INMUNODEPRIMIDOS La inmunosupresión debida a los fármacos utilizados como inmunosupresores o a la quimioterapia puede inducir la reactivación de VHB en 2 casos: el primero, en pacientes portadores de VHB (p. ej., HBsAg+) y el segundo, en pacientes con resolución de la infección por VHB (p. ej., HBsAg–, anti-HBc+ con o sin anti-HBs–), siendo el riesgo de reactivación del VHB más común en pacientes que reciben tratamiento quimioterápico por un tumor hematológico y en los sometidos a trasplante de médula ósea29. La AASLD define como reactivación del VHB al aumento de la replicación viral (aumento ≥ 2 log en comparación con los valores basales o aparición del ADN VHB en valores superiores a ≥ 100 UI/ml) en un individuo con valores previos estables o indetectables. El cribado recomendado debería incluir la determinación del HBsAg e IgG anti-HBc, y el estudio debería completarse mediante cuantificación del ADN-VHB y anti-HBs en los casos en los que el anti-HBc sea positivo. Se ha demostrado que el mejor predictor de reactivación de VHB es el valor basal de ADN-VHB30. Perrillo et al30 recomiendan realizar el cribado en pacientes con riesgo basal de reactivación elevado o moderado, mientras que en los pacientes con bajo riesgo de reactivación queda a indicación del servicio de medicina preventiva de cada centro. Entre las variables que se han descrito como factores de riesgo asociados a la reactivación del VHB se encuentran: características basales de los pacientes (varón joven), estado serológico previo (HBsAg+ y HBeAg+, ADN-VHB detectable y anti-HBs < 10 UI/ml), tipo de tumor (linfoma), tratamiento inmunosupresor (rituximab) y complicaciones asociadas al proceso (enfermedad injerto contra huésped)31. Aquellos pacientes sin marcadores serológicos de infección por VHB deberían ser vacunados. Se ha sugerido que la presencia de anti-HBs podría proporcionar una protección adicional frente a la reactivación, aunque faltan estudios que determinen qué valores de anti-HBs son necesarios para prevenir esta reactivación32. Por lo tanto, actualmente la decisión de realizar profilaxis no se puede basar en los valores de anti-HBs cuando anti-HBc es positivo30. Los pacientes HBsAg+, independientemente del ADN-VHB, que vayan a recibir tratamiento inmunosupresor o quimioterapia deben recibir profilaxis con AN durante el tratamiento y hasta 12-18 meses después de finalizar este. Respecto al AN de elección, el fármaco con el que se posee mayor experiencia es lamivudina, que podría ser suficiente en pacientes con baja carga viral y corta exposición al tratamiento inmunosupresor; sin embargo, en pacientes con carga viral elevada, con 8 Sesión 1 - HEPATITIS VIRALES Tabla 2. Grupos de riesgo Grupos de riesgo Fármaco de riesgo Enfermedad de tratamiento Riesgo elevado (> 10%) Rituximab y ofatumumab Linfoma/leucemia, AR, PTI, crioglobulinemia HBsAg+/anti-HBc+: 30-36% HBsAg–/anti-HBc+: > 10% Derivados de las antraciclinas HBsAg+/anti-HBc+: 15-30% Corticoides > 4 semanas (dosis moderada/alta) HBsAg+/anti-HBc+: > 10% Riesgo moderado (1-10%) Anti-TNFa Tumores sólidos. TACE, linfoma/leucemia EII, vasculitis, sarcoidosis, enfermedades autoinmunes EII, AR, EA HBsAg+/anti-HBc+: 1-10% HbsAg–/anti-HBc+: 1% Inhibidores de citoquinas e integrinas EII, psoriasis HBsAg+/anti-HBc+: 1-10% HBsAg–/anti-HBc+: 1% Inhibidores de la tirosincinasa HBsAg+/anti-HBc+: 1-10% LMC, tumores estromales gastrointestinales HBsAg–/anti-HBc+: 1% Corticoides > 4 semanas HBsAg+/anti-HBc+: 1-10% (baja dosis) EII, vasculitis, sarcoidosis, enfermedades autoinmunes HBsAg–/anti-HBc+: 1-10% (dosis moderada/alta) Derivados de las antraciclinas Riesgo bajo (< 1%) Tumores sólidos. TACE, linfoma/leucemia HBsAg+/anti-HBc+: 1-10% HBsAg–/anti-HBc+: 1-10% AZT, 6 MCP, metotrexato EII, vasculitis, sarcoidosis HBsAg+/anti-HBc+: < 1% HBsAg–/anti-HBc+: < 1% Corticoides intraarticulares Artritis HBsAg+/anti-HBc+: < 1% HBsAg–/anti-HBc+: < 1% Corticoides < 1 semana Asma, dermatitis HBsAg+/anti-HBc+: < 1% HBsAg–/anti-HBc+: < 1% Corticoides > 4 semanas (baja dosis) EII, vasculitis, sarcoidosis, enfermedades autoinmunes HBsAg–/anti-HBc+: < 1% AR: artritis reumatoide; AZP: azatioprina; EA: espondilitis anquilosante; EII: enfermedad inflamatoria intestinal; HBc: anticuerpo del núcleo de la hepatitis B; HBsAg: antígeno de superficie de la hepatitis B; LMC: leucemia mieloide crónica; TNFa: factor de necrosis tumoral alfa. Modificada de referencia 30. Temas no resueltos en hepatitis B enfermedad hematológica o que vayan a requerir tratamiento inmunosupresor a largo plazo o combinación de varios inmunosupresores, se recomienda el uso de un AN más potente y con alta barrera genética (entecavir o tenofovir). Todos los pacientes HBsAg+ o anti-HBc+ con ADNVHB detectable deben recibir profilaxis previamente al inicio del tratamiento inmunosupresor con rituximab, que se debería continuar hasta 12 meses después de la finalización del tratamiento inmunosupresor. Esta recomendación se afianza con los datos publicados en el trabajo de Seto et al, donde se detectaron reactivaciones del VHB después del año de seguimiento. Sin embargo, para los pacientes anti-HBc+ con ADNVHB indetectable, a día de hoy no hay consenso sobre qué estrategia seguir, por lo que tanto el tratamiento con AN como la monitorización trimestral de los valores de ALT y ADN-VHB son actitudes correctas. En los pacientes HBsAg+ que van a recibir un injerto hepático se recomienda tratamiento con AN de alta barrera genética, con el fin de lograr un ADN-VHB indetectable previo al trasplante. Tras el procedimiento, el tratamiento con AN en combinación con la administración de IGHB ha reducido el riesgo de infección del injerto a menos del 10%. A día de hoy, no existe ni una dosis o frecuencia de administración de la IGHB estandarizada, aunque sí datos a favor de que dosis reducidas (1.000-2.000 UI/mes) son suficientes en pacientes con ADN-VHB indetectable en el momento del trasplante hepático. Asimismo, actualmente tampoco hay datos suficientes para recomendar la profilaxis inicial sin IGHB, basadas únicamente en la administración de AN. Por otra parte, debido a la disparidad entre el número de pacientes en lista de trasplante y el número de donaciones, cada día es más frecuente el empleo de órganos de pacientes anti-HBc+ y, en ese caso, se recomienda la profilaxis indefinida con lamivudina31. Bibliografía 1.Lavanchy D. Hepatitis B virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J Viral Hepat. 2004;11:97-107. 2.Castera L. Noninvasive methods to assess liver disease in patients with hepatitis B or C. Gastroenterology. 2012;142:12931302.e4. 3.European Association For The Study Of The Liver. EASL clinical practice guidelines: Management of chronic hepatitis B virus infection. J Hepatol. 2012;57:167-85. 4.Buti M, García-Samaniego J, Prieto M, Rodríguez M, SánchezTapias JM, Suárez E, et al. [Consensus document of the Spanish Association for the Study of the Liver on the treatment of hepatitis B infection (2012)]. Gastroenterol Hepatol. 2012;35:512-28. 5.Castera L, Pinzani M. Biopsy and non-invasive methods for the diagnosis of liver fibrosis: does it take two to tango? Gut. 2010;59:861-6. 9 6.Viganò M, Paggi S, Lampertico P, Fraquelli M, Massironi S, Ronchi G, et al. Dual cut-off transient elastography to assess liver fibrosis in chronic hepatitis B: a cohort study with internal validation. Aliment Pharmacol Ther. 2011;34:353-62. 7.Chan HL, Wong GL, Choi PC, Chan AW, Chim AM, Yiu KK, et al. Alanine aminotransferase-based algorithms of liver stiffness measurement by transient elastography (Fibroscan) for liver fibrosis in chronic hepatitis B. J Viral Hepat. 2009;16:36-44. 8.Foucher J, Chanteloup E, Vergniol J, Castéra L, Le Bail B, Adhoute X, et al. Diagnosis of cirrhosis by transient elastography (FibroScan): a prospective study. Gut. 2006;55:403-8. 9.Poynard T, Ngo Y, Marcellin P, Hadziyannis S, Ratziu V, Benhamou Y; Adefovir Dipivoxil 437 and 438 Study Groups. Impact of adefovir dipivoxil on liver fibrosis and activity assessed with biochemical markers (FibroTest-ActiTest) in patients infected by hepatitis B virus. J Viral Hepat. 2009;16:203-13. 10.De Ledinghen V, Vergniol J, Barthe C, Foucher J, Chermak F, Le Bail B, et al. Non-invasive tests for fibrosis and liver stiffness predict 5-year survival of patients chronically infected with hepatitis B virus. Aliment Pharmacol Ther. 2013;37:97988. 11.Kim BK, Kim do Y, Han KH, Park JY, Kim JK, Paik YH, et al. Risk assessment of esophageal variceal bleeding in B-viral liver cirrhosis by a liver stiffness measurement-based model. Am J Gastroenterol. 2011;106:1654-62, 1730. 12.Kim BK, Han KH, Park JY, Ahn SH, Kim JK, Paik YH, et al. A liver stiffness measurement-based, noninvasive prediction model for high-risk esophageal varices in B-viral liver cirrhosis. Am J Gastroenterol. 2010;105:1382-90. 13.Shin SH, Kim SU, Park JY, Kim do Y, Ahn SH, Han KH, et al. Liver stiffness-based model for prediction of hepatocellular carcinoma in chronic hepatitis B virus infection: comparison with histological fibrosis. Liver Int. 2015;35:1054-62. 14.Bosch J. Soft technique, hard end-points. J Hepatol. 2011; 55:955-6. 15.Park MS, Kim SW, Yoon KT, Kim SU, Park SY, Tak WY, et al. Factors influencing the diagnostic accuracy of acoustic radiation force impulse elastography in patients with chronic hepatitis B. Gut and Liver. 2015; doi:10.5009/gnl14391. [Epub ahead of print]. 16.Salkic NN, Jovanovic P, Hauser G, Brcic M. FibroTest/Fibrosure for significant liver fibrosis and cirrhosis in chronic hepatitis B: a meta-analysis. Am J Gastroenter. 2014;109:796-809. 17.Salkic NN, Cickusic E, Jovanovic P, Denjagic MB, IljazovicTopcic S, Bevanda M, et al. Online combination algorithm for non-invasive assessment of chronic hepatitis B related liver fibrosis and cirrhosis in resource-limited settings. Eur J Int Med. 2015; pii:S0953-6205(15)00231-9. [Epub ahead of print]. 18.Zoulim F. Are novel combination therapies needed for chronic hepatitis B? Antiviral Res. 2012;96:256-9. 19.Kim V, Abreu RM, Nakagawa DM, Baldassare RM, Carrilho FJ, Ono SK. Pegylated interferon alfa for chronic hepatitis B: systematic review and meta-analysis. J Viral Hepat. 2015; doi:10.1111/jvh.12418. [Epub ahead of print]. 20.Marcellin P, Lau GK, Bonino F, Farci P, Hadziyannis S, Jin R, et al; Peginterferon Alfa-2a HBeAg-Negative Chronic Hepatitis B Study Group. Peginterferon alfa-2a alone, lamivudine alone, and the two in combination in patients with HBeAg-negative chronic hepatitis B. N Engl J Med. 2004;351:1206-17. 21.Lau GK, Piratvisuth T, Luo KX, Marcellin P, Thongsawat S, Cooksley G, et al; Peginterferon Alfa-2a HBeAg-Positive Chronic 10 Sesión 1 - HEPATITIS VIRALES Hepatitis B Study Group. Peginterferon Alfa-2a, lamivudine, and the combination for HBeAg-positive chronic hepatitis B. N Engl J Med. 2005;352:2682-95. hepatocellular carcinoma in patients with chronic hepatitis B: a nationwide cohort study. Gastroenterology. 2014;147:143151.e5. 22.Wi CI, Kim WR, Gross JB, Stadheim LM, Poterucha JJ. Potential efficacy of pegylated interferon-alpha and a nucleos(t) ide analogue as combination therapy for hbeag-positive chronic hepatitis B. Gut Liver. 2015; doi:10.5009/gnl14256. [Epub ahead of print]. 28.Papatheodoridis GV, Dalekos GN, Yurdaydin C, Buti M, Goulis J, Arends P, et al. Incidence and predictors of hepatocellular carcinoma in Caucasian chronic hepatitis B patients receiving entecavir or tenofovir. J Hepatol. 2015;62:363-70. 23.Kao JH. HBeAg-positive chronic hepatitis B: why do I treat my patients with pegylated interferon? Liver Int. 2014;34 Suppl 1:112-9. 29.Hwang JP, Lok AS. Management of patients with hepatitis B who require immunosuppressive therapy. Nat Rev Gastroenterol Hepatol. 2014;11:209-19. 24.Chen CL, Yang JY, Lin SF, Sun CA, Bai CH, You SL, et al. Slow decline of hepatitis B burden in general population: Results from a population-based survey and longitudinal follow-up study in Taiwan. J Hepatol. 2015;63:354-63. 30.Perrillo RP, Gish R, Falck-Ytter YT. American Gastroenterological Association Institute technical review on prevention and treatment of hepatitis B virus reactivation during immunosuppressive drug therapy. Gastroenterology. 2015;148:221-244 e3. 25.Papatheodoridis GV, Chan HL, Hansen BE, Janssen HL, Lampertico P. Risk of hepatocellular carcinoma in chronic hepatitis B: assessment and modification with current antiviral therapy. J Hepatol. 2015;62:956-67. 31.Riveiro-Barciela M, Buti M. [Hepatitis B virus infection in pregnancy and the immunosuppressed patient]. Gastroenterol Hepatol. 2015;38:31-9. 26.Vlachogiannakos J, Papatheodoridis G. Hepatocellular carcinoma in chronic hepatitis B patients under antiviral therapy. World J Gastroenterol. 2013;19:8822-30. 27.Wu CY, Lin JT, Ho HJ, Su CW, Lee TY, Wang SY, et al. Association of nucleos(t)ide analogue therapy with reduced risk of 32.Reddy KR, Beavers KL, Hammond SP, Lim JK, Falck-Ytter YT, American Gastroenterological Association I. American Gastroenterological Association Institute guideline on the prevention and treatment of hepatitis B virus reactivation during immunosuppressive drug therapy. Gastroenterology. 2015;148:215-9; quiz e216-7. SESIÓN 1 HEPATITIS VIRALES Tratamiento de la hepatitis C: genotipos 3, 5 y 6 Raquel Souto Rodríguez y Juan Turnes Servicio de Aparato Digestivo, Complejo Hospitalario Universitario de Pontevedra, Pontevedra, España INTRODUCCIÓN Los genotipos 3, 5 y 6 del virus de la hepatitis C (VHC) tienen algunas características diferenciales epidemiológicas, virológicas y relativas al tratamiento con respecto a los otros genotipos. En el caso de los genotipos 5 y 6 su baja prevalencia, particularmente en España, limita la información disponible relativa a la eficacia de los nuevos tratamientos antivirales orales. El genotipo 3 es el segundo más frecuente en nuestro medio y se ha convertido, en esta nueva era de antivirales de acción directa (AAD) de elevada efectividad, en el genotipo más difícil de curar, especialmente en pacientes con cirrosis, y en el que tan solo disponemos de 3 AAD activos aprobados: sofosbuvir (inhibidor de NS5B), daclatasvir y ledipasvir (inhibidores de NS5A). GENOTIPO 3 Epidemiología El genotipo 3 del VHC es común en el sudeste de Asia, y es el genotipo predominante en países como India y Pakistán1. En algunos países de Europa como Grecia, Polonia y Países Bajos representa hasta el 30% de todos los casos2. En España, la proporción de pacientes infectados por el genotipo 3 del VHC se sitúa en el 19,6% (fig. 1)3. La expansión del genotipo 3 se atribuye a la asociación del subtipo 3a al uso de drogas por vía parenteral y a la migración de la población de países donde originariamente era endémico, como el Sudeste Asiático4. Además de los rasgos epidemiológicos descritos posee algunas características que le confieren ciertas diferencias en la historia natural de la enfermedad hepática en comparación con otros genotipos5. El genotipo 3 se relaciona con una progresión de la fibrosis más rápida y un mayor riesgo de hepatocarcinoma en comparación con otros genotipos, características que Figura 1. Distribución del virus de la hepatitis C por genotipos en España. son independientes de otros factores de riesgo como la edad, el tiempo de evolución, ser varón, el consumo de alcohol, el índice de masa corporal elevado y la coinfección por el virus de la inmunodeficiencia humana6. Como una posible causa de este comportamiento biológico más agresivo, se postula el impacto de este genotipo sobre el metabolismo lipídico y la resistencia a la insulina por interferencia de las proteínas del core en el efecto metabólico de esta hormona, lo que conduce a una mayor incidencia de esteatosis2. Estándar de tratamiento antes de la introducción de los antivirales de acción directa Durante la última década, el tratamiento estándar de la infección por VHC genotipo 3 ha sido la combinación de interferón (INF) pegilado (pegINF) más ribavirina (RBV) a dosis fija de 800 mg/día durante 24 semanas. En algunas circunstancias se podía plantear acortar el tratamiento a 16 semanas en pacientes con factores 11 12 Sesión 1 - HEPATITIS VIRALES pronósticos de buena respuesta al tratamiento, como aquellos con respuesta viral rápida y carga viral basal < 600.000 UI/ml en pacientes sin fibrosis avanzada. Por otra parte, en pacientes con cinética viral lenta y ARN indetectable a la semana 24 de tratamiento se podía considerar prolongar el tratamiento hasta 48 o 72 semanas7. Globalmente, con esta línea de tratamiento se obtenía una tasa de respuesta viral sostenida (RVS) en torno al 70%. Recomendaciones de tratamiento actuales La introducción de los regímenes de tratamiento basados en AAD que combinan diferentes dianas moleculares del ciclo de replicación del VHC, con sus elevadas tasas de eficacia y excelente perfil de seguridad, ha revolucionado el tratamiento de la hepatitis C. Sin embargo, en comparación con otros genotipos, las ganancias de eficacia en el genotipo 3 son relativamente menores en comparación con el estándar previo de tratamiento (INF/RBV), y determinados subgrupos de pacientes con factores de mal pronóstico de respuesta al tratamiento tienen tasas de RVS subóptimas. De los AAD aprobados, únicamente el inhibidor de la polimerasa (NS5B) sofosbuvir y los inhibidores de NS5A daclatasvir y, en menor medida, ledipasvir son activos frente al genotipo 3. Esta situación es notablemente diferente en los genotipos 1 y 4, en donde disponemos de un mayor número de moléculas activas que permiten múltiples combinaciones posibles de elevada eficacia. Los regímenes actuales de tratamiento se basan en la combinación de estos AAD con o sin RBV, y en algunos casos se contempla la utilización de una triple terapia de sofosbuvir, INF y RBV. La selección del régimen de tratamiento y su duración van a depender de la presencia o no de cirrosis hepática y de la respuesta a un trata- miento previo, disponiendo para cada perfil de paciente de diversas opciones de tratamiento (tabla 1). Pacientes naïve sin cirrosis • PegIFN-a más RBV ajustada por peso (1.000 mg o 1.200 mg/día en pacientes < 75 o ≥ 75 kg, respectivamente) y sofosbuvir 400 mg/día durante 12 semanas. Los primeros datos de eficacia en este grupo de pacientes proceden de un ensayo fase II (estudio PROTON)8, en el que 9 de 10 pacientes tratados alcanzaron RVS, mientras que del otro paciente se perdió el seguimiento. Recientemente, en un estudio fase III (BOSON), en el que los pacientes con genotipo 3 fueron aleatorizados a recibir triple terapia con sofosbuvir o bien sofosbuvir y RBV ajustada a peso durante 16 o 24 semanas, se observó que el 96% (68/71) de los pacientes naïve sin cirrosis presentaban RVS con esta combinación9. • Sofosbuvir 400 mg/día más RBV ajustada por peso (1.000 mg o 1.200 mg/día en pacientes < 75 o ≥ 75 kg, respectivamente) durante 24 semanas. En el estudio FISSION se evaluó esta pauta durante 12 semanas, con resultados subóptimos (RVS del 61% en pacientes sin cirrosis)10. Para superar esta limitación se evaluó la prolongación del tratamiento a 24 semanas en el estudio VALENCE11, y se observó una tasa de RVS del 92% en pacientes naïve sin cirrosis. Sin embargo, en pacientes considerados difíciles de curar, aquellos con fracaso a un tratamiento previo con pegIFN y RBV y cirrosis, la tasa de RVS se reducía hasta un 60%, resultados claramente subóptimos. • Sofosbuvir 400 mg/día más daclatasvir 60 mg durante 12 semanas. La combinación de sofosbuvir y daclatasvir sin RBV durante 12 semanas ha demos- Tabla 1. Datos de eficacia de las distintas combinaciones según subgrupos de pacientes con infección por el virus de la hepatitis C genotipo 3 Pacientes naïve Sin cirrosis Con cirrosis Sin cirrosis Con cirrosis Peginterferón + RBV + sofosbuvir 96%9 91%9 94%9 86%9 Sofosbuvir + RBV 92%22 82%9 87%22 No se recomienda Sofosbuvir + daclatasvir 97% (12 semanas sin RBV)12 58% (12 semanas sin RBV)12 94% (12 semanas sin RBV)12 69% (12 semanas sin RBV)12 Sofosbuvir + ledipasvir 100% (12 semanas con RBV)13 No es posible extraer conclusiones 89-100% (12 semanas con RBV)13 No es posible extraer conclusiones RBV: ribavirina. Pacientes con fracaso a tratamiento previo Tratamiento de la hepatitis C: genotipos 3, 5 y 6 13 Tabla 2. Recomendaciones de tratamiento de pacientes con hepatitis por el virus de la hepatitis C genotipo 3 Pacientes sin cirrosis Pacientes con cirrosis Naïve Fracaso a tratamiento previo Naïve Fracaso a tratamiento previo Peginterferón + RBV + sofosbuvir 12 semanas 12 semanas 12 semanas 12 semanas Sofosbuvir + RBV 24 semanas 24 semanas 24 semanas No se recomienda Sofosbuvir + daclatasvir 12 semanas sin RBV 12 semanas sin RBV 24 semanas con RBV* 24 semanas con RBV* Sofosbuvir + ledipasvir 12 semanas con RBV 12 semanas con RBV No se recomienda No se recomienda RBV: ribavirina. *La European Association for the Study of the Liver (EASL), sobre la base de los resultados subóptimos en pacientes cirróticos en el estudio ALLY-312, recomienda de forma “empírica” prolongar el tratamiento a 24 semanas y añadir RBV. trado excelentes resultados en estudios fase II y en un reciente ensayo clínico fase III (ALLY-3)12, en el que se observó una tasa de RVS del 97% (73/75) en el subgrupo de pacientes naïve sin cirrosis, con un excelente perfil de seguridad. • Sofosbuvir 400 mg/día más ledipasvir 90 mg/día más RBV ajustada por peso (1.000 mg o 1.200 mg/día en pacientes < 75 o ≥ 75 kg, respectivamente) durante 12 semanas. Existen escasos datos sobre la eficacia de esta combinación. Únicamente se dispone de un estudio fase II, el ELECTRON-2, en el que el 100% de los pacientes naïve tratados, de un total de 26 pacientes, alcanzaron RVS, el número de pacientes cirróticos fue 5, por lo que no se pueden extrapolar resultados a esta subpoblación. Este mismo régimen sin RBV presentó resultados subóptimos (RVS del 64% en este grupo de pacientes naïve)13. En resumen, hay varias opciones de tratamiento eficaces en este grupo de pacientes que, dentro de los pacientes con genotipo 3, representan el subgrupo más “fácil de curar” (tabla 2). La triple terapia con INF proporciona unas tasas de RVS excelentes, y probablemente es el único escenario en el que su uso podría estar justificado en la actualidad. Sin embargo, las contraindicaciones que presentan algunos pacientes al INF y la propia resistencia de los enfermos para su administración debido a la mala tolerancia limitan en gran medida su utilización. La combinación de AAD sin INF, con o sin RBV, permite alcanzar tasas de RVS igualmente elevadas, con un excelente perfil de seguridad y tolerancia. La actividad in vitro de daclatasvir es notablemente mayor que la de ledipasvir y, por otra parte, disponemos de un nivel de evidencia más sólido de la eficacia de la combinación sofosbuvir/daclatasvir, aunque posiblemente sofosbuvir/ledipasvir y RBV sea también una opción de tratamiento adecuada. Pacientes naïve con cirrosis • PegIFN-a más RBV ajustada por peso (1.000 mg o 1.200 mg/día en pacientes < 75 o ≥ 75 kg, respectivamente) y sofosbuvir 400 mg/día durante 12 semanas. Los resultados más recientes sobre esta combinación en este subgrupo de pacientes proceden del ensayo clínico fase III BOSON, en el que en los pacientes tratados con triple terapia se observó una tasa de RVS del 91% (21/23), en comparación con el 57% (12/21) y el 82% (18/22) en pacientes tratados con sofosbuvir y RBV 16 o 24 semanas, respectivamente9. • Sofosbuvir 400 mg/día más RBV ajustada por peso (1.000 mg o 1.200 mg/día en pacientes < 75 o ≥ 75 kg, respectivamente) durante 24 semanas. El estudio FISSION evaluó esta pauta durante 12 semanas con resultados subóptimos: RVS 34% en pacientes con cirrosis10. La prolongación del tratamiento a 24 semanas se evaluó en el estudio VALENCE, observando un incremento de la RVS en hasta el 92% (12/13) de pacientes naïve con cirrosis, si bien el número de pacientes incluidos con estas características fue reducido11. • Sofosbuvir 400 mg/día más daclatasvir 60 mg/día más RBV durante 24 semanas. El ensayo ALLY-312, con una RVS del 58%, mostró unos resultados subóptimos en términos de eficacia de esta combinación durante 12 semanas sin RBV en pacientes con cirrosis. La utilización de RBV y/o la prolongación del tratamiento en estos pacientes se está evaluando en la actualidad en un estudio complementario al ALLY-3 (NCT02319031), cuyos resultados se comunicarán en los próximos meses. Mientras tanto, la recomendación de la European Association for the Study of the Liver (EASL) es prolongar el tratamiento a 24 semanas y añadir RBV en los pacientes con cirrosis. 14 Sesión 1 - HEPATITIS VIRALES • Sofosbuvir 400 mg/día más ledipasvir 90 mg/día más RBV ajustada por peso (1.000 mg o 1.200 mg/ día en pacientes < 75 o ≥ 75 kg, respectivamente) durante 12 semanas. El único estudio que evalúa esta combinación es el ELECTRON-2, con solo 5 pacientes cirróticos incluidos13, lo que limita en gran medida la extracción de recomendaciones para su utilización en la práctica clínica. El tratamiento con triple terapia con sofosbuvir en pacientes naïve con cirrosis ofrece unos resultados excelentes en términos de RVS. Sin embargo, es frecuente que los pacientes cirróticos presenten alguna contraindicación para recibir tratamiento con INF (descompensación clínica, plaquetas < 100.000 × 109, albúmina < 3,5, etc.), por lo que habitualmente tendremos que elegir un régimen que no lo contenga. A pesar de que no hay datos de eficacia de la combinación de sofosbuvir, daclatasvir y RBV durante 24 semanas, en el momento actual probablemente sea la opción más prudente, con objeto de garantizar las mayores probabilidades de curación en este grupo de pacientes (tabla 2). Pacientes con fracaso a tratamiento previo sin cirrosis • PegIFN-a más RBV ajustada por peso (1.000 mg o 1.200 mg/día en pacientes < 75 o ≥ 75 kg, respectivamente) y sofosbuvir 400 mg/día durante 12 semanas. El ensayo fase IIb LONESTAR-2 evaluó la eficacia de este régimen en pacientes que habían fracasado a tratamiento previo con INF y RBV. El 83% de los pacientes en este subgrupo alcanzó respuesta viral sostenida, manteniéndose este porcentaje de respuesta entre los pacientes cirróticos (10/12 pacientes)14. Más recientemente, el ensayo clínico BOSON confirmó los buenos resultados de esta combinación con un 94% de RVS en un mayor número de pacientes (49/52) con cirrosis y fracaso al tratamiento previo9. Este régimen también se ha mostrado útil en pacientes con fracaso a tratamiento previo con sofosbuvir y RBV, con una RVS del 91% (20 de 22 pacientes tratados) tras 12 semanas de tratamiento15. • Sofosbuvir 400 mg/día más RBV ajustada por peso (1.000 mg o 1.200 mg/día en pacientes < 75 o ≥ 75 kg, respectivamente) durante 24 semanas. El estudio FUSION comparó la eficacia de este régimen si se administraba 12 frente a 16 semanas en pacientes con fracaso previo a INF y RBV, con resultados subóptimos incluso en el grupo de 16 semanas, en el que únicamente un 62 y un 61% sin cirrosis y con cirrosis, respectivamente, alcanzaban RVS16. Esta limitación se puede superar prolongando el tratamiento a 24 semanas, estrategia evaluada en el estudio VALENCE, lo que permite alcanzar una RVS del 87% (87/100) en este subgrupo de pacientes11. Resultados similares se observaron en el estudio BOSON, con una eficacia en el brazo tratado con sofosbuvir y RBV durante 24 semanas del 82%9. • Sofosbuvir 400 mg/día más daclatasvir 60 mg durante 12 semanas. El estudio ALLY-3 mostró unos resultados excelentes de esta combinación en pacientes pretratados sin cirrosis, con un tasa de RVS del 94%12. • Sofosbuvir 400 mg/día más ledipasvir 90 mg/día más RBV ajustada por peso (1.000 mg o 1.200 mg/ día en pacientes < 75 o ≥ 75 kg, respectivamente) durante 12 semanas. En el estudio ELECTRON-2 se observó que el 89% (25/28) de pacientes presentaban RVS tras 12 semanas de este régimen. Cabe destacar que la misma combinación de antivirales sin RBV tan solo obtuvo un 64% de eficacia13. En resumen, los diferentes regímenes disponibles muestran unos resultados excelentes en pacientes sin cirrosis, independientemente del antecedente de haber fracasado a un tratamiento previo con INF y RBV. En consecuencia, en la selección del tratamiento van a influir otras variables como la tolerancia/efectos adversos, la duración (siendo preferibles tratamientos de 12 semanas) o la simplificación del régimen excluyendo la RBV (tabla 2). Pacientes con fracaso a tratamiento previo y cirrosis • PegIFN-a más RBV ajustada por peso (1.000 mg o 1.200 mg/día en pacientes < 75 o ≥ 75 kg, respectivamente) y sofosbuvir 400 mg/día durante 12 semanas. Esta combinación se ha mostrado eficaz en el estudio LONESTAR-2, en el que el 83% de los pacientes cirróticos con fracaso a tratamiento previo alcanzaron RVS14. Estos resultados han sido confirmados posteriormente en el estudio BOSON, con una RVS del 86% (30/35 pacientes) en pacientes tratados con triple terapia frente al 77% (28/34) con sofosbuvir más RBV durante 24 semanas. • Sofosbuvir 400 mg/día más daclatasvir 60 mg/día más RBV ajustada por peso (1.000 mg o 1.200 mg/ día en pacientes < 75 o ≥ 75 kg, respectivamente) durante 24 semanas. Al igual que en pacientes naïve con cirrosis, la combinación de sofosbuvir y daclatasvir sin RBV en este subgrupo de pacientes ofrece unos resultados subóptimos, con una tasa de RVS del 69% en el estudio ALLY-312, por lo que se recomienda prolongar el tratamiento a 24 semanas y añadir RBV en pacientes cirróticos en espera de disponer de más datos, como se ha mencionado anteriormente. • Sofosbuvir 400 mg/día más ledipasvir 90 mg/día más RBV ajustada por peso (1.000 mg o 1.200 mg/ día en pacientes < 75 o ≥ 75 kg, respectivamente) durante 12 semanas. Disponemos únicamente de datos procedentes del estudio ELECTRON-2, en el que en una muestra reducida de pacientes con estas características se observó una RVS del 73% (16/22)13. En este grupo de pacientes con un perfil que condiciona unos resultados más pobres, la opción que cuenta con un nivel de evidencia más consistente es la triple terapia con INF + RBV y sofosbuvir. Sin embargo, es probable que una proporción significativa de pacientes presente algún tipo de circunstancia clínica que desaconseje el tratamiento con INF; en estos casos es preferible la combinación de sofosbuvir, daclatasvir y RBV durante 24 semanas, con objeto de garantizar las mayores probabilidades de curación en este subgrupo de pacientes. No obstante, en los próximos meses dispondremos de información adicional que podría permitir refinar la utilización de sofosbuvir y daclatasvir con/sin RBV en estos pacientes, lo que potencialmente evitaría sobretratar a los pacientes y reducir la incidencia de efectos adversos (tabla 2). Tratamiento en situaciones especiales Cirrosis descompensada. En el estudio ALLY-1, 5 de 6 pacientes con cirrosis descompensada tratados con sofosbuvir/daclatasvir/RBV durante 12 semanas alcanzaron RVS17. Por otro lado, disponemos de algunos datos procedentes de los estudios de práctica clínica real, como los datos del uso compasivo en el Reino Unido18, con 189 pacientes con cirrosis descompensada (el 64% Child-Pugh B y el 12,7% Child Pugh C) con una media en la puntuación MELD de 12,6 (rango, 6-36). En total, 114 pacientes fueron tratados con sofosbuvir/daclatasvir/RBV, 61 con sofosbuvir/ledipasvir/RBV y 14 con los regímenes anteriores sin RBV. Los resultados fueron mejores en el grupo de sofosbuvir/daclatasvir, sin diferencias entre la adición o no de RBV (el 70% de RVS en el grupo con RBV y el 71% de pacientes en el grupo sin RBV). Tan solo el 59% de los pacientes tratados con sofosbuvir/ledipasvir/RBV y el 43% de los que recibieron sofosbuvir/ledipasvir alcanzaron RVS. Estos resultados de eficacia deben valorarse con cautela, ya que la selección del tratamiento y su duración se realizaron exclusivamente bajo criterio del médico responsable del paciente y, en consecuencia, no se pueden realizar comparaciones directas entre regímenes de tratamiento debido a los sesgos de selección inherentes. El 25% de pacientes sufrió algún efecto adverso, la mayoría en relación con su enfermedad hepática, dato que no Tratamiento de la hepatitis C: genotipos 3, 5 y 6 15 resulta sorprendente si tenemos en cuenta el perfil de pacientes tratados. En resumen, aunque es evidente que son necesarios más estudios parece que las combinaciones de sofosbuvir, daclatasvir y ledipasvir son razonablemente seguras en este escenario y permiten alcanzar tasas de RVS próximas a las observadas en pacientes con cirrosis compensada, que se deberán confirmar en posteriores ensayos clínicos y estudios de práctica clínica real. Retratamiento tras fracasos con regímenes que contienen antivirales de acción directa En el momento actual, no disponemos de estudios que evalúen alternativas terapéuticas eficaces en pacientes que fracasen a la combinación de sofosbuvir y un inhibidor de NS5A. En cambio, hay algunos datos en pacientes con genotipo 3 con fracaso a un tratamiento previo con sofosbuvir y RBV y que hayan sido rescatados con triple terapia con sofosbuvir o la combinación de sofosbuvir y daclatasvir con RBV. • PegIFN-a más RBV ajustada por peso (1.000 mg o 1.200 mg/día en pacientes < 75 kg o ≥ 75 kg, respectivamente) y sofosbuvir 400 mg/día durante 12 semanas. En un estudio presentado recientemente, el 91% (22/24 pacientes) que había recaído tras un tratamiento previo con sofosbuvir y RBV en los ensayos clínicos alcanzó RVS con este régimen15. • Sofosbuvir 400 mg/día más daclatasvir 60 mg/día más RBV ajustada por peso (1.000 mg o 1.200 mg/ día en pacientes < 75 o ≥ 75 kg, respectivamente) durante 12 semanas con RBV si ≤ F2 o 24 semanas si F3 o cirrosis. En el estudio ALLY-312, 7 pacientes que habían fracasado a regímenes que contenían sofosbuvir fueron tratados con la combinación de sofosbuvir más daclatasvir, 5 de los cuales (71%) alcanzaron RVS. Algunas consideraciones sobre el tratamiento del genotipo 3 La EASL, en sus últimas guías clínicas, no recomienda la asociación sofosbuvir/ledipasvir en pacientes infectados por VHC genotipo 3. Ello podría explicarse básicamente por 3 factores: la actividad in vitro significativamente inferior frente al genotipo 3 del inhibidor NS5A ledipasvir frente a daclatasvir; la escasez de información procedente de ensayos clínicos, y los resultados aparentemente inferiores respecto a sofosbuvir/daclatasvir observados en el programa de acceso precoz del Reino Unido en pacientes con cirrosis avanzada. El inhibidor de NS5A daclatasvir no está aprobado por la Food and Drug Administration; por lo tanto, la 16 Sesión 1 - HEPATITIS VIRALES guía para el manejo de la infección por el VHC de la American Association for the Study of Liver Diseases no contempla regímenes que contengan daclatasvir. Opciones de tratamiento en desarrollo Se están llevando a cabo estudios con nuevos fármacos para pacientes infectados por el genotipo 3 del VHC, concretamente nuevos inhibidores de la proteína NS5A como elbasvir y grazoprevir, un inhibidor de la proteasa de nueva generación, que tiene actividad in vitro frente al genotipo 3, aunque esta combinación ha mostrado unos resultados pobres19. Más prometedora es la combinación de grazoprevir, elbasvir y sofosbuvir, con RVS del 100% (14/14) en pacientes naïve sin cirrosis y del 91% (10/11) en pacientes naïve con cirrosis20. Adicionalmente se están evaluando otras posibles combinaciones de fármacos antivirales con actividad pangenotípica como inhibidores de proteasa con alta barrera genética, inhibidores de NS5A e inhibidores de la polimerasa no nucleósidos. GENOTIPOS 5 Y 6 Epidemiología El genotipo 5 es el responsable de menos casos de infección por el VHC (< 1% de casos globalmente), la mayoría de los cuales se concentra en el sudeste de África Subsahariana. El genotipo 6 alcanza su mayor prevalencia en el Sudeste Asiático (16% de todos los casos) y es endémico en Laos. El mecanismo fundamental por el que este genotipo se ha amplificado en estos países ha sido a través de los usuarios de drogas por vía parenteral4. Su presencia en nuestro país es marginal. Recomendaciones de tratamiento actuales La mayoría de la información de que disponemos sobre la eficacia de los tratamientos con AAD en estos genotipos se basa en la actividad in vitro de los fármacos antivirales, y no en estudios de registro, dada la escasa prevalencia de estos genotipos en muchas de las áreas donde se han llevado a cabo los estudios. Además, los escasos estudios en los que se han incluido pacientes con estos genotipos no tienen una representación de todos los subgrupos de pacientes (con cirrosis, con fracaso a tratamiento previo, etc.), por lo que en la mayoría de los casos, la base de las recomendaciones se fundamenta en la actividad in vitro e inferencias del comportamiento en genotipo 1. • PegIFN-a más RBV ajustada por peso (1.000 mg o 1.200 mg/día en pacientes < 75 o ≥ 75 kg, respectivamente) y sofosbuvir 400 mg/día durante 12 semanas. Esta combinación se ha evaluado en el estudio NEUTRINO10 en pacientes naïve. Se trató a 1 paciente con genotipo 5 y a 6 con genotipo 6, de los cuales todos alcanzaron RVS. No hay datos de este régimen en pacientes con fracaso a tratamiento previo. Por otra parte, debido al escaso número de pacientes tratados, no disponemos de datos de eficacia que diferencien pacientes con o sin cirrosis. • Sofosbuvir 400 mg/día más ledipasvir 90 mg/día. Sofosbuvir tiene una acción pangenotípica que incluye a los genotipos 5 y 6. Ledipasvir es activo frente a los genotipos 5 y 6 in vitro, pero no disponemos de datos sobre la eficacia de esta combinación en pacientes con hepatitis C infectados por el genotipo 5. En genotipo 6, la combinación de sofosbuvir y ledipasvir sin RBV durante 12 semanas fue eficaz en 24 de 25 pacientes tratados (96%), entre los que había pacientes naïve y con fracaso a tratamiento previo21. Las pautas y recomendaciones de tratamiento son extrapoladas de los datos disponibles para genotipo 1: – En pacientes sin cirrosis (tanto naïve como con fracaso a tratamiento previo) se recomienda esta combinación sin añadir RBV durante 12 semanas. – En pacientes con cirrosis (incluyendo pacientes naïve y pacientes con fracaso a tratamiento previo) se recomienda esta combinación más RBV ajustada por peso (1.000 mg o 1.200 mg/día en pacientes < 75 o ≥ 75 kg, respectivamente). En pacientes con cirrosis que presentan contraindicación para recibir RBV se debe prolongar el tratamiento con sofosbuvir más ledipasvir a 24 semanas. – En pacientes cirróticos con factores de mal pronóstico con menos de 75.000 plaquetas × 109/dl se recomienda asociar RBV ajustada por peso al tratamiento de 24 semanas. • Sofosbuvir 400 mg/día más daclatasvir 60 mg/día durante 12 semanas. El inhibidor de NS5A daclatasvir es activo in vitro frente a los genotipos 5 y 6 del VHC. No existen datos en los ensayos clínicos llevados a cabo para evaluar la eficacia de esta combinación en estos genotipos; por lo tanto, las pautas son extrapoladas de los datos de otros genotipos. En pacientes con cirrosis se recomienda esta combinación más RBV ajustada por peso (1.000 mg o 1.200 mg/día en pacientes < 75 o ≥ 75 kg, respectivamente) durante 12 semanas. En pacientes con cirrosis que presentan contraindicación para recibir RBV se debe prolongar el tratamiento con sofosbuvir más ledipasvir a 24 semanas. Tratamiento de la hepatitis C: genotipos 3, 5 y 6 CONCLUSIONES A diferencia de lo que sucedía en la era del pegINF y la RBV, los pacientes con hepatitis C crónica infectados por el genotipo 3 se han convertido en el grupo de pacientes más difícil de curar. A pesar de que el arsenal de tratamiento se ha incrementado significativamente respecto a la era de la biterapia, y de que las combinaciones de AAD sin INF ofrecen excelentes resultados en pacientes sin cirrosis, en los pacientes con cirrosis los resultados todavía son subóptimos, tanto en naïve como en pretratados con INF y RBV. Probablemente existan múltiples factores, todavía no bien conocidos, que contribuyan a la mayor tasa de recaída en estos pacientes y que justifican la utilidad de la RBV o la prolongación del tratamiento en pacientes con factores predictivos de mal pronóstico. Es en estos pacientes difíciles de curar en los que la triple terapia con sofosbuvir todavía puede ofrecer ventajas adicionales en términos de RVS, aunque en muchos casos va a estar limitada por la elegibilidad de unos pacientes que pueden presentar contraindicaciones para la utilización de INF debido al deterioro de su función hepática. Los pacientes con genotipos 5 y 6 representan un grupo marginal en nuestro medio, en el que, si bien disponemos de un cuerpo de evidencia muy pobre generada por ensayos clínicos, los regímenes basados en combinaciones de inhibidores de NS5B y NS5A o bien la triple terapia con sofosbuvir parecen ofrecer resultados similares a los observados en genotipo 1. Bibliografía 1.Shepard CW, Finelli L, Alter MJ. Global epidemiology of hepatitis C virus infection. Lancet Infect Dis. 2005;5:558-67. 2.Ampuero J, Romero-Gómez M, Reddy KR. Review article: HCV genotype 3 - the new treatment challenge. Aliment Pharmacol Ther. 2014;39:686-98. 3.Gower E, Estes C, Blach S, Razavi-Shearer K, Razavi H. Global epidemiology and genotype distribution of the hepatitis C virus infection. J Hepatol. 2014;61 Suppl:S45-57. 4.Messina JP, Humphreys I, Flaxman A, Brown A, Cooke GS, Pybus OG, et al. Global distribution and prevalence of hepatitis C virus genotypes. Hepatology. 2015;61:77-87. 5.Pol S, Vallet-Pichard A, Corouge M. Treatment of hepatitis C virus genotype 3-infection. Liver Int. 2013;34:18-23. 6.Kanwal F, Kramer JR, Ilyas J, Duan Z, El-Serag HB. HCV genotype 3 is associated with an increased risk of cirrhosis and hepatocellular cancer in a national sample of U.S. Veterans with HCV. Hepatology. 2014;60:98-105. 17 patients with genotypes 1, 2, and 3 hepatitis C infection: a randomised, double-blind, phase 2 trial. Lancet Infect Dis. 2013;13:401-8. 9.Foster GR, Pianko S, Cooper C, Brown A, Forton D, Nahass RG, et al. LO5: sofosbuvir + peginterferon/ribavirin for 12 weeks vs sofosbuvir + ribavirin for 16 or 24 weeks in genotype 3 HCV infected patients and treatment-experienced cirrhotic patients with genotype 2 HCV: The boson study. J Hepatol. 2015;62:S259-60. 10.Lawitz E, Mangia A, Wyles D, Rodríguez-Torres M, Hassanein T, Gordon SC, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med. 2013;368:1878-87. 11.Zeuzem, Dusheiko GM, Salupere R, Mangia A, Flisiak R, Hyland RH, et al. Sofosbuvir and ribavirin in HCV genotypes 2 and 3. N Engl J Med. 2014;370:1993-2001. 12.Nelson DR, Cooper JN, Lalezari JP, Lawitz E, Pockros PJ, Gitlin N, et al. All-oral 12-week treatment with daclatasvir plus sofosbuvir in patients with hepatitis C virus genotype 3 infection: ALLY-3 phase III study. Hepatology. 2015;61:1127-35. 13.Gane EJ, Hyland RH, An D, et al. Sofosbuvir/ledipasvir fixed dose combination is safe and effective in difficult-to-treat populations including genotype-3 patients, decompensated genotype-1 patients, and genotype-1 patients with prior sofosbuvir treatment experience. J Hepatol. 2014; Suppl EASL 2014. 14.Lawitz E, Poordad F, Brainard DM, Hyland RH, WT S. Sofosbuvir in combination with PegIFN and ribavirin for 12 weeks provides high SVR rates in HCV-infected genotype 2 or 3 treatment-experienced pataients with and without compensated cirrhosis: results from the LONESTAR-2 study. Hepatology. 15.Esteban R, Nyberg L, Lalezar J, Ni L, Doehle B, Kanwar B, et al. Succesful retreatment with sofosbuvir-containing regimens for HCV genotype 2 or 3 infected patients who failed prior sofosbuvir plus ribavirin therapy. J Hepatol. 2014;60:S4. 16.Jacobson IM, Gordon SC, Kowdley KV, Yoshida EM, RodríguezTorres M, Sulkowski MS, et al. Sofosbuvir for hepatitis C genotype 2 or 3 in patients without treatment options. N Engl J Med. 2013;368:1867-77. 17.Poordad F, Schiff ER, Vierling JM, Landis C, Fontana RJ, Yang R, et al. LO8 : daclatasvir, sofosbuvir, and ribavirin combination for HCV patients with advanced cirrhosis or posttransplant recurrence: Phase 3 ALLY-1 study. J Hepatol. 2015;62: S261-2. 18.Foster GR, Irving W, Cheung M, Hudson B, Verma S, Agarwal K. Treatment of decompensated HCV cirrhosis in patients with diverse genotypes: 12 weeks sofosbuvir and NS5A inhibitors with/without ribavirin is effective in HCV genotypes 1 and 3. J Hepatol. 2015;62:S190-1. 19.Gane E, Nahass R, Luketic V, Hwang P, Robertson M, Wahl J, et al. P0776: Efficacy of 12 or 18 weeks of grazoprevir plus elbasvir with ribavirin in treatment-naive, noncirrhotic HCV genotype 3-infected patients. J Hepatol. 2015;62:S621. 7. European Association for the Study of the Liver. EASL Clinical Practice Guidelines: management of hepatitis C virus infection. J Hepat. 2011;55:245-64. 20.Poordad F, Lawitz E, Gutierrez JA, Evans B, Howe A, Feng HP, et al. C-swift: grazoprevir/elbasvir + sofosbuvir in cirrhotic and noncirrhotic, treatment-naive patients with hepatitis C virus genotype 1 infection, for durations of 4, 6 or 8 weeks and genotype 3 infection for durations of 8 or 12 weeks. J Hepatol. 2015;62:S192-3. 8.Lawitz E, Lalezari JP, Hassanein T, Kowdley KV, Poordad FF, Sheikh AM, et al. Sofosbuvir in combination with peginterferon alfa-2a and ribavirin for non-cirrhotic, treatment-naive 21.Gane EJ, Hyland RH, An D, et al. High efficacy of LDV/SOF regimens for 12 weeks for patients with HCV genotype 3 or 6 infection. Hepatology. 2015; Supp AASLD 2014. SESIÓN 1 HEPATITIS VIRALES Hepatitis colestásica fibrosante María-Carlota Londoño Servicio de Hepatología, Hospital Clínic, Barcelona, España INTRODUCCIÓN La hepatitis C es la principal indicación de trasplante hepático (TH) en el mundo occidental. La recidiva de la hepatitis C tras el TH es universal en pacientes que llegan al momento del TH con un ARN del virus de la hepatitis C (VHC) detectable. La historia natural de la hepatitis C después del trasplante es acelerada cuando se compara con la de los pacientes inmunocompetentes. Aproximadamente un 20-30% de los pacientes desarrollan una cirrosis a los 5 años del trasplante1. El riesgo de descompensación en pacientes con cirrosis es un 40% al año y un 60% a los 3 años del diagnóstico2. La presencia de descompensación hepática en estos pacientes es un indicador de mortalidad precoz, ya que el 60% de estos fallece en el primer año tras el primer episodio3. Un pequeño porcentaje de pacientes (5-10%) desarrolla una forma grave de recidiva de la hepatitis C conocida como hepatitis colestásica fibrosante (HCF), que puede llevar a una pérdida rápida del injerto y a la muerte del paciente4. Aquí se revisarán las principales características de la HCF y las opciones actuales de tratamiento. DEFINICIÓN La primera definición de HCF fue realizada por un panel internacional de expertos en 2003 con los siguientes criterios analíticos e histológicos: a) aparición después del primer mes postrasplante; b) bilirrubina sérica ≥ 6 mg/dl; c) fosfatasa alcalina y gamma-glutamil transpeptidasa 5 veces el valor de la normalidad; d) valores de ARN-VHC muy elevados (generalmente > 6 log10 UI/ml; e) presencia en la biopsia hepática de balonización de los hepatocitos generalmente perivenular, escasa inflamación portal y lobulillar, proliferación colangiolar sin pérdida de conductos biliares, y f) ausencia de complicaciones biliares o vasculares4. Más recientemente, Verna et al propusieron nuevos criterios para el diagnóstico de HCF basados en los 18 Tabla 1. Nuevos criterios diagnósticos de la hepatitis colestásica fibrosante Criterios específicos • > 1 mes tras el TH • Al menos 3 de 4 hallazgos histológicos: – Proliferación colangiolar – Colestasis –B alonización de los hepatocitos –F ibrosis perisinusoidal o periportal Criterios de soporte • Bilirrubina total > 2 mg/dl • GGT > 150 UI/l • AST > 70 UI/l • Donante añoso • Biopsia reciente con signos de rechazo Criterios de exclusión • Ausencia de obstrucción biliar • Ausencia de trombosis de la arteria hepática AST: aspartato aminotransferasa; GGT: gamma-glutamil transpeptidasa; TH: trasplante hepático. hallazgos de un estudio en el que se evaluaron las biopsias de pacientes con recidiva del VHC tras el TH. Estos criterios son más sensibles que los anteriores y permiten detectar un mayor número de casos con la enfermedad (tabla 1). FACTORES DE RIESGO Se han descrito una serie de factores de riesgo que contribuyen al desarrollo de una recidiva grave de Hepatitis colestásica fibrosante Tabla 2. Factores de riesgo para una rápida progresión de la recidiva del virus de la hepatitis C 1. Virus Genotipo (?) Carga viral (?) 2. Donante Edad del donante Esteatosis Donante vivo (?) Corazón parado (?) 3. Receptor Sexo Diabetes mellitus 4. Trasplante Complicaciones biliares Inmunosupresión Infección por CMV 5. Polimorfismo IL28B CC frente a TT o CT CMV: citomegalovirus. la hepatitis C y a una rápida progresión de la fibrosis. Estos son: factores virales, factores relacionados con el donante, factores relacionados con el receptor y los intrínsecamente relacionados con el trasplante (tabla 2). 19 donantes de edad avanzada en receptores VHC positivo9. Donante vivo Existe controversia sobre la asociación entre la utilización de donantes vivos y la gravedad de la recidiva del VHC. Los primeros informes señalaban que en el TH de donante vivo los signos de recidiva de la hepatitis C aparecían antes, la progresión de la fibrosis era más rápida y las formas graves de la enfermedad, como la hepatitis colestásica, eran más frecuentes en comparación con los pacientes trasplantados con hígados procedentes de donante de cadáver10,11. Estudios más recientes analizando biopsias de protocolo no observaron diferencias significativas en la progresión de la enfermedad. Un estudio multicéntrico realizado en 9 centros de Estados Unidos demostró la importancia de la experiencia del cirujano. Así, cuando se excluyeron los primeros 20 trasplantes de donante vivo realizados en cada centro, no se observaron diferencias significativas en la supervivencia del injerto entre donantes vivos y donantes cadavéricos12. Esteatosis del donante Factores relacionados con el donante Mientras que en el paciente inmunocompetente, la asociación entre esteatosis y progresión de la fibrosis en pacientes con VHC es clara, en el TH los datos son menos concluyentes. Burra et al13 no encontraron ninguna asociación entre la progresión de la fibrosis y la esteatosis del donante. Por el contrario, Briceño et al14 encontraron que la esteatosis del donante (> 30%) se asocia con una recidiva precoz, rápida progresión de la fibrosis y peor supervivencia del injerto. Estos resultados, sin embargo, deben interpretarse con cautela, ya que las biopsias de hígado solo se realizaron cuando los valores de transaminasas aumentaban 1,5 veces el valor normal. Ello puede ocasionar un sesgo en el diagnóstico de la recidiva, ya que los pacientes con pruebas hepáticas normales no fueron sometidos a biopsia hepática. Edad Donación después de la muerte cardíaca La edad del donante es probablemente el factor que se ha asociado de manera más consistente con peores resultados en pacientes trasplantados por VHC6-8. Los mecanismos por los que la edad del donante está asociada con la gravedad de la fibrosis son poco conocidos y merecen más investigación, pero podría ser debido a la presencia de algún grado de lesión histológica en el injerto. Estos resultados podrían tener implicaciones importantes en la asignación de los donantes, ya que algunos centros recomiendan evitar Los injertos de donación después de la muerte cardíaca (DCD) presentan varios trastornos que se asocian con una mayor gravedad de la recidiva: tiempo de isquemia prolongada, lesión de la preservación y mayor riesgo de complicaciones biliares. Hernández-Alejandro et al15 compararon los resultados del TH con hígados procedentes de DCD en receptores VHC positivos y negativos. Los autores encontraron una tasa significativamente mayor de recidiva grave del VHC (fibrosis ≥ 2) y pérdida del injerto. Factores virales Algunos estudios han observado una forma más grave de progresión de la recidiva de la hepatitis C en pacientes infectados con el genotipo 1, aunque este hallazgo no se ha confirmado en todos los estudios. Por otro lado, algunos datos sugieren que una carga viral del VHC elevada pre y post-TH podría estar relacionada con mayor pérdida del injerto y mayor mortalidad. De hecho, la HCF se caracteriza por cargas virales muy elevadas (> 6 log10 UI/ml)2,5. 20 Sesión 1 - HEPATITIS VIRALES Factores relacionados con el receptor Sexo femenino El sexo femenino del receptor parece ser un factor predictivo independiente para el desarrollo de fibrosis en puente o cirrosis y una menor supervivencia, especialmente cuando se combina con la edad de los donantes16. La explicación para esta asociación es actualmente desconocida, pero algunos estudios en mujeres posmenopáusicas sugieren una tendencia a un estado proinflamatorio, como se indica por un aumento de los valores del TNF e IL-617. Esto podría ser responsable de la gravedad de la fibrosis y la ausencia de respuesta al tratamiento con interferón en mujeres receptoras de TH. Diabetes mellitus postrasplante La asociación entre la hepatitis C y la diabetes mellitus (DM) es bien conocida. La incidencia de DM de novo después del trasplante es significativamente mayor en receptores VHC. Además, en los pacientes VHC positivos, aquellos con DM o un estado prediabético, que se caracteriza por resistencia a la insulina, desarrollan fibrosis grave significativamente más rápido que los pacientes sin DM. Se han propuesto varias teorías para explicar esta asociación: a) la resistencia a la insulina aumenta la progresión de la fibrosis; b) la leptina sérica activa las células de Kupffer y aumenta la producción de citocinas proinflamatorias, y c) la esteatosis hepática, que se asocia con la resistencia a la insulina, podría ser la responsable del desarrollo de fibrosis grave18. Factores propios del trasplante Inmunosupresión El estado neto de inmunosupresión en el receptor es un poderoso determinante del pronóstico de la recidiva del VHC. De hecho, la HCF solo se ha descrito en pacientes inmunodeprimidos. Además, es bien sabido que la tasa de progresión de la fibrosis es mayor en pacientes inmunodeprimidos y coinfectados con el virus de la inmunodeficiencia humana (VIH). Berenguer et al19 observaron que al evitar el exceso de inmunosupresión (bolos de esteroides y terapias triples o cuádruples), la tasa de recurrencia de la hepatitis C grave se redujo del 54 al 33%. Los estudios que evalúan agentes inmunosupresores individuales tienen varias limitaciones: la mayoría son retrospectivos, tienen un seguimiento corto, se centran en la supervivencia del paciente y del injerto y no en la enfermedad del injerto, y carecen de biopsias protocolo. Se ha demostrado claramente que el uso de bolos de esteroides para tratar el rechazo se asocia con una carga viral más elevada, una recidiva precoz y más grave y una mayor mortalidad20. Los datos sobre los inhibidores de la calcineurina y la recurrencia de la hepatitis C son controvertidos. Los primeros estudios que compararon el efecto de la ciclosporina (CsA) y el tacrolimus (TAC) en la recurrencia de la hepatitis C no encontraron diferencias significativas en la supervivencia del paciente y del injerto y la gravedad de la recidiva21. Sin embargo, un estudio reciente con datos de 8.809 receptores de TH del registro de la UNOS encontró que los pacientes tratados con CsA presentaron un mayor riesgo de fracaso del injerto y de muerte en comparación con los pacientes tratados con TAC22. La explicación de esta diferencia es que los pacientes tratados con CsA presentan una mayor tasa de rechazo agudo y rechazo resistente a esteroides, ambos factores de riesgo bien conocidos para una peor evolución de la enfermedad en pacientes VHC positivos. Estos resultados, sin embargo, deben interpretarse con precaución debido a las limitaciones del análisis de grandes bases de datos, falta de información sobre las dosis y valores valle, el cambio de un fármaco a otro, el cambio histológico de seguimiento y otros posibles factores de confusión. También es importante tener en cuenta que se ha encontrado que la CsA tiene un efecto in vitro frente a la replicación del VHC23. Aunque este efecto no se ha replicado en la clínica, algunos estudios retrospectivos han sugerido que la terapia antiviral con interferón y ribavirina es más eficaz en pacientes que reciben CsA24. Por lo tanto, un enfoque racional de la inmunosupresión en la hepatitis C recurrente sería reducir al mínimo el uso de la terapia triple/cuádruple, bolos de esteroides y altos valores de inmunosupresión. Esto es particularmente importante durante el primer año del TH, ya que parece que los primeros eventos que podrían activar la fibrogénesis son cruciales para determinar el pronóstico del injerto. Otros factores Otros factores de riesgo implicados en la gravedad de la recidiva son la infección por citomegalovirus25 (que quizás refleja un mayor estado de inmunosupresión), la presencia de complicaciones biliares26 (que se ha implicado como factor fibrogénico) y el polimorfismo de la IL-28B en donante y receptor27. TRATAMIENTO El tratamiento de la hepatitis C en pacientes con HCF es el mismo que en pacientes con cualquier tipo de recidiva (fig. 1A y B)28-34. Sin embargo, en los estu- Hepatitis colestásica fibrosante A SOLAR-1 (28) 55 26 26 SATURN (30) SOLAR-1 (29) 5 56 25 18 3 21 14 21 CORAL-I (31) 34 B ALLY-1 CULPIT CULPIT Mayo 5 6 5 6 Figura 1. A y B) Eficacia del tratamiento antiviral en pacientes con trasplante hepático. dios realizados hasta ahora parece que el tratamiento en pacientes con esta forma grave de recidiva de la hepatitis es más eficaz que en pacientes con fibrosis establecida. Ello se evidenció en el estudio publicado por Forns et al35, en el que se analizaron los datos del uso compasivo de sofosbuvir en pacientes con recidiva grave del VHC. Es este estudio retrospectivo se incluyeron 104 pacientes divididos en 2 grupos: a) pacientes con recidiva grave precoz (en el pri- mer año postrasplante) incluyendo HCF (n = 52), y b) pacientes con cirrosis diagnosticada después del primer año postrasplante (n = 52). Todos los pacientes recibieron tratamiento con sofosbuvir + ribavirina (± interferón pegilado a criterio del investigador). La tasa de respuesta viral sostenida (RVS) fue mayor en pacientes con recidiva grave precoz (el 73 frente al 43%). Aunque esta combinación actualmente se considera subóptima en pacientes con genotipo 1, los 22 Sesión 1 - HEPATITIS VIRALES resultados son alentadores para esta población de pacientes que anteriormente tenía pocas posibilidades de tratamiento. Recientemente se ha publicado la única serie específicamente de 23 pacientes con HCF tratados con sofosbuvir y daclatasvir33. La tasa de RVS fue del 96% (solo un fallo virológico en un paciente VIH positivo) y la tolerancia fue excelente, ya que no se reportaron eventos adversos grado 3 o 4. La elección del tratamiento, pues, se basa en el genotipo del virus, el grado de disfunción hepática y la presencia de interacciones farmacológicas con los inmunosupresores. CONCLUSIONES La HCF es la forma más grave de la recidiva de la hepatitis C tras el trasplante hepático y se presenta en un 5-10% de los pacientes que se trasplantan por esta indicación. Se caracteriza por la presencia de colestasis analítica e histológica, carga viral elevada, lesión de los hepatocitos y se asocia con una elevada tasa de pérdida del injerto y del paciente. Afortunadamente, la aparición de nuevos fármacos antivirales administrados en regímenes sin interferón ha cambiado completamente el pronóstico de esta patología. Muchos pacientes reciben el tratamiento antes del TH, con lo que se evita su aparición, y también los pacientes no tratados que desarrollen HCF, cuyas probabilidades de curación y mejoría de la función hepática son excelentes. Bibliografía 1.Garcia-Retortillo M, Forns X, Feliu A, Moitinho E, Costa J, Navasa M, et al. Hepatitis C virus kinetics during and immediately after liver transplantation. Hepatology. 2002;35:680-7. 2.Berenguer M, Ferrell L, Watson J, Prieto M, Kim M, Rayon M, et al. HCV-related fibrosis progression following liver transplantation: increase in recent years. J Hepatol. 2000;32:673-84. 3.Berenguer M, Prieto M, Rayon JM, Mora J, Pastor M, Ortiz V, et al. Natural history of clinically compensated hepatitis C virus-related graft cirrhosis after liver transplantation. Hepatology. 2000;32(4 Pt 1):852-8. 4.Wiesner RH, Sorrell M, Villamil F. Report of the first International Liver Transplantation Society expert panel consensus conference on liver transplantation and hepatitis C. Liver Transpl. 2003;9:S1-9. 5.Feray C, Gigou M, Samuel D, Paradis V, Mishiro S, Maertens G, et al. Influence of the genotypes of hepatitis C virus on the severity of recurrent liver disease after liver transplantation. Gastroenterology. 1995;108:1088-96. 6.Berenguer M, Prieto M, San Juan F, Rayón JM, Martínez F, Carrasco D, et al. Contribution of donor age to the recent decrease in patient survival among HCV-infected liver transplant recipients. Hepatology. 2002;36:202-10. 7.Gallegos-Orozco JF, Yosephy A, Noble B, Aqel BA, Byrne TJ, Carey EJ, et al. Natural history of post-liver transplantation hepatitis C: A review of factors that may influence its course. Liver Transpl. 2009;15:1872-81. 8.Wali M, Harrison RF, Gow PJ, Mutimer D. Advancing donor liver age and rapid fibrosis progression following transplantation for hepatitis C. Gut. 2002;51:248-52. 9.Valadao RM, Terrault NA. Older donors: mounting risks for the hepatitis C-infected liver transplant recipient? Liver Transpl. 2009;15:677-81. 10.García-Retortillo M, Forns X, Llovet JM, Navasa M, Feliu A, Massaguer A, et al. Hepatitis C recurrence is more severe after living donor compared to cadaveric liver transplantation. Hepatology. 2004;40:699-707. 11.Thuluvath PJ, Yoo HY. Graft and patient survival after adult live donor liver transplantation compared to a matched cohort who received a deceased donor transplantation. Liver Transpl. 2004;10:1263-8. 12.Terrault NA, Shiffman ML, Lok AS, Saab S, Tong L, Brown RS Jr, et al. Outcomes in hepatitis C virus-infected recipients of living donor vs. deceased donor liver transplantation. Liver Transpl. 2007;13:122-9. 13.Burra P, Loreno M, Russo FP, Germani G, Galligioni A, Senzolo M, et al. Donor livers with steatosis are safe to use in hepatitis C virus-positive recipients. Liver Transpl. 2009;15: 619-28. 14.Briceno J, Ciria R, Pleguezuelo M, De la Mata M, Muntané J, Naranjo A, et al. Impact of donor graft steatosis on overall outcome and viral recurrence after liver transplantation for hepatitis C virus cirrhosis. Liver Transpl. 2009;15:37-48. 15.Hernández-Alejandro R, Croome KP, Quan D, Mawardi M, Chandok N, Dale C, et al. Increased risk of severe recurrence of hepatitis C virus in liver transplant recipients of donation after cardiac death allografts. Transplantation. 2011;92: 686-9. 16.Belli LS, Burroughs AK, Burra P, Alberti AB, Samonakis D, Camma C, et al. Liver transplantation for HCV cirrhosis: improved survival in recent years and increased severity of recurrent disease in female recipients: results of a long term retrospective study. Liver Transpl. 2007;13:733-40. 17.Villa E, Karampatou A, Camma C, Di Leo A, Luongo M, Ferrari A, et al. Early menopause is associated with lack of response to antiviral therapy in women with chronic hepatitis C. Gastroenterology. 2011;140:818-29. 18.Veldt BJ, Poterucha JJ, Watt KD, Wiesner RH, Hay JE, Rosen CB, et al. Insulin resistance, serum adipokines and risk of fibrosis progression in patients transplanted for hepatitis C. Am J Transplant. 2009;9:1406-13. 19.Berenguer M, Aguilera V, Prieto M, San Juan F, Rayón JM, Benlloch S, et al. Significant improvement in the outcome of HCV-infected transplant recipients by avoiding rapid steroid tapering and potent induction immunosuppression. J Hepatol. 2006;44:717-22. 20.Roche B, Samuel D. Risk factors for hepatitis C recurrence after liver transplantation. J Viral Hepat. 2007;14 Suppl 1: 89-96. 21.Berenguer M, Royuela A, Zamora J. Immunosuppression with calcineurin inhibitors with respect to the outcome of HCV recurrence after liver transplantation: results of a metaanalysis. Liver Transpl. 2007;13:21-9. 22.Irish WD, Arcona S, Bowers D, Trotter JF. Cyclosporine versus tacrolimus treated liver transplant recipients with chronic hepatitis C: outcomes analysis of the UNOS/OPTN database. Am J Transplant. 2011;11:1676-85. 23.Watashi K, Hijikata M, Hosaka M, Yamaji M, Shimotohno K. Cyclosporin A suppresses replication of hepatitis C virus genome in cultured hepatocytes. Hepatology. 2003;38:1282-8. 24.ReViS-TC Study Group. Cyclosporine a-based immunosuppression reduces relapse rate after antiviral therapy in transplanted patients with hepatitis C virus infection: a large multicenter cohort study. Transplantation. 2011;92:334-40. 25.Rosen HR, Chou S, Corless CL, Gretch DR, Flora KD, Boudousquie A, et al. Cytomegalovirus viremia: risk factor for allograft cirrhosis after liver transplantation for hepatitis C. Transplantation. 1997;64:721-6. 26.Katz LH, Mor E, Brown M, Bar-Nathan N, Shaharabani E, Sulkes J, et al. Recurrent hepatitis C virus disease after liver transplantation and concurrent biliary tract complications: poor outcome. Clin Transplant. 2006;20:465-70. 27.Charlton MR, Thompson A, Veldt BJ, Watt K, Tillmann H, Poterucha JJ, et al. Interleukin-28B polymorphisms are associated with histological recurrence and treatment response following liver transplantation in patients with hepatitis C virus infection. Hepatology. 2011;53:317-24. 28.Flamm S, Everson G, Charlton M, Denning J, Arterburn S, Brandt-Sarif T, et al. Ledipasvir/sofosbuvir with ribavirin for the treatment of HCV in patients with decompensated cirrhosis: preliminary results of a prospective, multicenter study. Hepatology. 2014;60:320A. 29.Manns M, Forns X, Samuel D, Denning J, Arterburn S, BrandtSarif T, et al. Ledipasvir/sofosbuvir with ribavirin is safe and efficacious in decompensated and post-liver transplantation patients with HCV infection: Preliminary results of the SOLAR-2 trial. J Hepatol. 2015;62:S187. Hepatitis colestásica fibrosante 23 30.Forns X, Berenguer M, Herzer K, Sterneck M, Donato MF, Andreone P, et al. On-treatment virologic responde and tolerability of Simeprevir, Daclatasvir and Ribavirin in patients with recurrent hepatitis C virus genotype 1b infection after orthotopic liver transplantation: interim data from the phase II SATURN study. J Hepatol. 2015;62:S191. 31.Kwo P, Gitlin N, Nahass R, Bernstein D, Rojter S, Schiff E, et al. A phase 3, randomised, open-label study to evaluate the efficacy and safety of 12 and 8 weeks of simeprevir (SMV) plus sofosbuvir (SOF) in treatment-naïve and -experienced patients with chronic HCV genotype 1 infection without cirrhosis: OPTIMIST-1. J Hepatol. 2015;62:S270. 32.Poordad F, Schiff E, Vierling J, Landis C, Fontana R, Yang R, et al. Daclatasvir, sofosbuvir, and ribavirin combination for HCV patients with advanced cirrhosis or post-transplant recurrence: ALLY-1 Phase 3 Study. J Hepatol. 2015;62:S261. 33.Leroy V, Dumortier J, Coilly A, Sebagh M, Fougerou-Leurent C, Radenne S, et al. Efficacy of sofosbuvir and daclatasvir in patients with fibrosing cholestatic hepatitis C after liver transplantation. Clin Gastroenterol Hepatol. 2015; pii:S15423565(15)00765-X. [Epub ahead of print]. 34.Pungpapong S, Aqel B, Leise M, Werner KT, Murphy JL, Henry TM, et al. Multicenter experience using simeprevir and sofosbuvir with or without ribavirin to treat hepatitis C genotype 1 after liver transplant. Hepatology. 2015;61:1880-6. 35.Forns X, Charlton M, Denning J, McHutchison JG, Symonds WT, Brainard D, et al. Sofosbuvir compassionate use program for patients with severe recurrent hepatitis C following liver transplantation. Hepatology. 2015; doi:10.1002/hep.27681. [Epub ahead of print]. SESIÓN 2 CIRROSIS E HIPERTENSIÓN PORTAL Profilaxis y tratamiento de la peritonitis bacteriana espontánea: implicaciones de los cambios en el espectro microbiano Germán Soriano Hospital de la Santa Creu i Sant Pau, Barcelona, España INTRODUCCIÓN La peritonitis bacteriana espontánea (PBE) es la infección del líquido ascítico (LA) sin evidencia de un foco séptico intraabdominal. Aunque se puede observar en otras situaciones, como la ascitis por insuficiencia cardíaca, el síndrome nefrótico o la ascitis neoplásica, es una complicación característica de los pacientes con cirrosis y ascitis. Se considera que hasta una cuarta parte de los pacientes cirróticos hospitalizados pueden presentar una PBE al ingreso o desarrollarla durante este1-3. En su etiopatogenia desempeñan un papel importante las alteraciones en la microbiota y la barrera intestinales, la traslocación bacteriana y las alteraciones en los mecanismos de defensa inmunológica, tanto a nivel del LA como sistémico3,4 (fig. 1). Como consecuencia de estas alteraciones, la mayoría de episodios de PBE son causados por bacterias procedentes de la propia flora intestinal del paciente, principalmente bacilos gramnegativos, aunque en los últimos años han aumentado los episodios producidos por cocos grampositivos1-4. DIAGNÓSTICO La clínica de la PBE puede consistir en los síntomas clásicos de fiebre y dolor abdominal, aunque con frecuencia es inespecífica. Por ello, el clínico que atiende a pacientes con cirrosis debe tener en cuenta que se les debe realizar una paracentesis diagnóstica para descartar esta complicación al ingreso en el hospital y ante cualquier deterioro en la situación clínica o analítica del paciente, por leve que sea. Hallazgos tan variados como vómitos, diarrea, encefalopatía, hemorragia digestiva, insuficiencia renal, acidosis, hiponatremia, aumento de la proteína C reactiva o neutrofilia en sangre pueden ser la manifestación de una PBE1-3. 24 Cuanto antes se diagnostique, antes se iniciará el tratamiento antibiótico, y el tratamiento precoz es un factor pronóstico fundamental en estos pacientes1-3. Recientemente se ha estimado que cada hora de retraso en la paracentesis diagnóstica aumenta un 3,3% la mortalidad en los pacientes con PBE5. Para el diagnóstico de PBE se precisa una paracentesis con determinación de neutrófilos, proteínas totales (PT), lactatodeshidrogenasa (LDH), glucosa, tinción de Gram y cultivo con inoculación del LA en frascos de hemocultivo (10 ml en cada frasco, aerobio y anaerobio)1-3. Un recuento de neutrófilos en LA ≥ 250/µl en ausencia de otras causas como hemoperitoneo, carcinomatosis o pancreatitis es diagnóstico de PBE. Las tiras reactivas no se consideran de utilidad para diagnosticar una PBE2. El cultivo puede ser positivo (PBE con cultivo positivo) o negativo (PBE con cultivo negativo), siendo negativo en más del 50% de los casos. Es importante la toma de hemocultivos, ya que pueden ser positivos aunque el cultivo del LA sea negativo2. Las determinaciones de PT, LDH, glucosa y la tinción de Gram en LA son útiles en el diagnóstico diferencial entre PBE y peritonitis bacteriana secundaria (PBS)6,7. La PBS es consecuencia de un foco séptico intraabdominal y en su tratamiento debe considerarse la cirugía o el drenaje percutáneo guiado radiológicamente del foco infeccioso, además del tratamiento antibiótico de amplio espectro sobre gérmenes aerobios y anaerobios. Aunque representa menos del 10% de las peritonitis en pacientes cirróticos, tiene una especial gravedad, con una mortalidad superior al 60%. La presencia de 2 de los 3 criterios de Runyon en LA (PT > 10 g/l, LDH > al límite superior en sangre, glucosa < 2,7 mmol/l) nos debe hacer sospechar una PBS y realizar una tomografía computarizada abdominal, que tiene una sensibilidad superior al 90% para detectar el foco infeccioso. La tinción de Gram en la PBS es positiva en el 40% de los casos, frente a solo el 10% en la PBE, y el cultivo suele ser polimicrobiano mientras que en la PBE es monomi- Profilaxis y tratamiento de la peritonitis bacteriana espontánea: implicaciones de los cambios en el espectro microbiano 25 Figura 1. Etiopatogenia de la peritonitis bacteriana espontánea en la cirrosis. IgA: inmunoglobulina A; IL-6: interleucina 6; TNFα: factor de necrosis tumoral alfa. crobiano. En la PBS, el recuento de neutrófilos en LA no desciende más de un 25% a las 48 h de tratamiento antibiótico, lo cual sí ocurre en la PBE cuando se utiliza el antibiótico adecuado6,7. como ofloxacino también se habían mostrado eficaces en el tratamiento de la PBE10. TRATAMIENTO CLÁSICO Sin embargo, la situación ha cambiado en los últimos años con la aparición de episodios de PBE causados por bacterias multirresistentes, y específicamente resistentes al tratamiento antibiótico empírico más utilizado en la PBE, las cefalosporinas de tercera generación. Se consideran bacterias multirresistentes las que son resistentes a 3 o más de las principales familias de antibióticos, incluyendo los betalactámicos. Las más frecuentes aparecen en la tabla 1. Sin embargo, la aparición de bacterias multirresistentes es muy diferente según si las infecciones son extrahospitalarias o adquiridas en la comunidad, asociadas al sistema sanitario, o nosocomiales o intrahospitalarias (tabla 2)4,11,12. Efectivamente, 2 estudios recientes en nuestro medio han demostrado el aumento de la incidencia El tratamiento de la PBE es antibiótico, no debiendo plantearse la cirugía, a diferencia de la PBS. La duración del tratamiento debe ser entre 5 y 10 días, habitualmente 1 semana, y debe comprobarse la normalización del recuento de neutrófilos y la negativización del cultivo (si fue positivo) en el LA con el tratamiento1,2. Después de que en la década de los ochenta del siglo pasado se demostrara que las cefalosporinas de tercera generación eran más eficaces y seguras que las combinaciones de un betalactámico y un aminoglucósido en la PBE8, las cefalosporinas han sido el tratamiento de elección durante los últimos 30 años1-3. Otros antibióticos como amoxicilina/ácido clavulánico9 o quinolonas RESISTENCIAS BACTERIANAS 26 Sesión 2 - CIRROSIS E HIPERTENSIÓN PORTAL Tabla 1. Principales bacterias multirresistentes • Enterobacterias productoras de BLEA, principalmente Escherichia coli y Klebsiella pneumoniae • Bacilos gramnegativos no fermentadores como Pseudomonas aeruginosa, Stenotrophomonas maltophilia y Acinetobacter baumanii • Staphylococcus aureus resistente a meticilina • Enterococos sensibles o resistentes a vancomicina BLEA: betalactamasas de espectro ampliado. Tabla 2. Tipos de infecciones según el lugar de adquisición • Infecciones adquiridas en la comunidad: diagnosticadas en el momento del ingreso o en las primeras 48 h de este • Intrahospitalarias o nosocomiales: diagnosticadas después de las primeras 48 h del ingreso hospitalario • Asociadas al sistema sanitario: diagnosticadas en el momento del ingreso o en las primeras 48 h de este, aunque el paciente ha tenido un contacto reciente con el sistema sanitario: – Estancia hospitalaria durante al menos 2 días en los 90 días previos – Residencia en centro sociosanitario – Hemodiálisis crónica de infecciones por bacterias multirresistentes en los pacientes con cirrosis en la última década11,12. En el estudio de Fernández et al11, las bacterias multirresistentes se aislaron en el 39% de las infecciones nosocomiales, el 20% de las infecciones asociadas al sistema sanitario y en el 0% de las extrahospitalarias. En este estudio, los antibióticos empíricos recomendados por las guías vigentes (fundamentalmente cefalosporinas de tercera generación) fueron capaces de resolver la infección en solo el 26% de los episodios de PBE nosocomiales, frente al 78% en las extrahospitalarias y el 71% en las PBE asociadas al sistema sanitario. Ariza et al12 han observado resultados similares. En su estudio, la incidencia de resistencias a cefalosporinas de tercera generación fue del 41% en las PBE nosocomiales, el 21% en las PBE asociadas al sistema sanitario y el 7% en las extrahospitalarias. CAUSAS DE LAS MULTIRRESISTENCIAS BACTERIANAS Además de causas comunes a la población general, como es el uso creciente y en ocasiones indiscriminado de antibióticos, tanto en humanos como en animales de granja, se han identificado factores específicos que han favorecido la aparición de bacterias multirresisten- tes en los pacientes cirróticos. Efectivamente, diversos estudios han relacionado el desarrollo de infecciones por bacterias multirresistentes en estos pacientes con la profilaxis con norfloxacino, la administración de betalactámicos —ampliamente utilizados no solo como tratamiento de infecciones bacterianas, sino también como profilaxis de estas durante una hemorragia digestiva— y con la creciente instrumentalización a que son sometidos los pacientes con cirrosis11-13. CONSECUENCIAS DE LA APARICIÓN DE RESISTENCIAS BACTERIANAS La aparición de infecciones por bacterias resistentes a las quinolonas en los pacientes con cirrosis durante la década de los noventa, unos años después de la instauración de la profilaxis con norfloxacino, no tuvo gran trascendencia clínica debido a que la mayoría de estas bacterias seguían siendo sensibles a las cefalosporinas14. Sin embargo, el aumento de bacterias resistentes a las cefalosporinas de tercera generación sí ha tenido consecuencias graves sobre el pronóstico de los pacientes con PBE, debido a 2 motivos principales. En primer lugar, como ya se ha comentado, las cefalosporinas eran el tratamiento empírico de elección y, por tanto, el más utilizado en los pacientes con PBE. Por Profilaxis y tratamiento de la peritonitis bacteriana espontánea: implicaciones de los cambios en el espectro microbiano 27 Tabla 3. Indicaciones para la profilaxis de PBE (peritonitis bacteriana espontánea) en los pacientes con cirrosis • Hemorragia digestiva: – Función hepática preservada: 400 mg/12 h de norfloxacino v.o. durante 7 días –C irrosis avanzada (al menos 2 de ascitis, ictericia, encefalopatía o malnutrición): 1 g/día de ceftriaxona i.v. durante 7 días • Profilaxis primaria en pacientes con ascitis y PT < 15 g/l e insuficiencia hepática avanzada (puntuación de Child-Pugh ≥ 9 con bilirrubina ≥ 3 mg/dl) y/o disfunción renal (creatinina sérica ≥ 1,2 mg/dl, BUN ≥ 25 mg/dl y/o sodio sérico ≤ 130 mEq/l): 400 mg/día de norfloxacino o 500 mg/día de ciprofloxacino durante un período indefinido • Profilaxis secundaria en pacientes con PBE previa: 400 mg/día de norfloxacino durante un período indefinido o hasta mejoría de la función hepática con resolución de la ascitis de forma persistente BUN: nitrógeno ureico en sangre; i.v.: vía intravenosa; PT: proteínas totales; v.o.: vía oral. otra parte, en más del 50% de los episodios de PBE no se identifica la bacteria responsable, lo cual implica que el tratamiento es empírico y no puede ser guiado por datos microbiológicos, sino solo por la evolución clínica y analítica. Dado que el tratamiento antibiótico adecuado precoz es un factor fundamental en el pronóstico de la PBE1-3, no es sorprendente que la mortalidad sea mayor en los pacientes en los que se debe cambiar el tratamiento antibiótico que en aquellos en los que no es preciso cambiarlo15, y que la presencia de bacterias multirresistentes sea un destacado factor predictivo independiente de la mortalidad12. CAMBIOS EN LAS GUÍAS CLÍNICAS La escasez de datos, las variaciones geográficas y entre hospitales, la necesidad del uso de antibióticos de amplio espectro, con elevado coste y con efectos secundarios, y la dificultad para realizar estudios clínicos comparativos aleatorizados han complicado los cambios en las guías clínicas. Sin embargo, al final se han publicado recientemente los cambios en las recomendaciones de la EASL (European Association for the Study of the Liver) para el manejo de los pacientes con PBE4. Según este documento, el tratamiento empírico actual de un episodio de PBE debería ser diferente si se trata de una PBE extrahospitalaria o nosocomial. En las extrahospitalarias se recomienda el tratamiento “clásico” con cefalosporinas de tercera generación (ceftriaxona o cefotaxima) o amoxicilina/ácido clavulánico. En las nosocomiales, sin embargo, se recomienda un carbapenémico (meropenem o imipenem) o piperacilina/tazobactam (en áreas con baja prevalencia de bacterias multirresistentes) asociado o no a un glucopéptido (vancomicina o teicoplanina), dependiendo de si la prevalencia de Staphylococcus aureus resistente a meticilina y enterococos sensibles a la vancomicina es alta o baja. Los glucopéptidos deberían sustituirse por linezolid en áreas de alta prevalencia de enterococos resistentes a la vancomicina. Con respecto a las PBE asociadas al sistema sanitario, el documento aconseja tratarlas como nosocomiales si se presentan con sepsis grave o si se trata de un área con elevada prevalencia de bacterias multirresistentes en las infecciones asociadas al sistema sanitario. En todo caso, se destaca la importancia de conocer en cada centro el perfil actualizado de las resistencias bacterianas para el adecuado manejo de los pacientes con PBE. Desde luego, si se aísla la bacteria responsable, el tratamiento antibiótico empírico inicial se debe cambiar de acuerdo al resultado del cultivo y el antibiograma1-3. PROFILAXIS La profilaxis antibiótica con norfloxacino o ceftriaxona es eficaz para prevenir las infecciones en general, y la PBE en particular, y mejorar el pronóstico en los pacientes con cirrosis1-3,16-20. Sin embargo, ya se ha comentado que el uso de estos antibióticos es un factor fundamental para el desarrollo de infecciones por bacterias multirresistentes9,10. Por ello, la EASL recomienda restringir estrictamente la profilaxis a aquellos pacientes con mayor riesgo de infección4. Las indicaciones actuales de profilaxis se pueden consultar en la tabla 3. Recientemente se ha cuestionado la necesidad de profilaxis antibiótica en los pacientes Child-Pugh A con hemorragia digestiva21 y si los valores bajos de proteínas en LA realmente predisponen al desarrollo de una PBE22. Ante el problema de la aparición de las resistencias bacterianas se están investigando alternativas a los antibióticos utilizados actualmente. Entre estas posibles alternativas destacan la rifaximina, el trimetoprim-sulfametoxazol, los probióticos, los bloqueadores 28 Sesión 2 - CIRROSIS E HIPERTENSIÓN PORTAL beta, los procinéticos y los ácidos biliares. Sin embargo, hasta el momento ningún estudio aleatorizado ha demostrado que estos tratamientos puedan sustituir a norfloxacino en la prevención de la PBE4. Bibliografía 1.Runyon BA; AASLD Practice Guidelines Committee. Management of adult patients with ascites due to cirrhosis: an update. Hepatology. 2009;49:2087-107. 2.European Association for the Study of the Liver. EASL clinical practice guidelines on the management of ascites, spontaneous bacterial peritonitis, and hepatorenal syndrome in cirrhosis. J Hepatol. 2010;53:397-417. 3.Guarner C, Soriano G. Spontaneous bacterial peritonitis. Semin Liver Dis. 1997;17:203-17. 4.Jalan R, Fernandez J, Wiest R, Schnabl B, Moreau R, Angeli P, et al. Bacterial infections in cirrhosis: a position statement based on the EASL Special Conference 2013. J Hepatol. 2014;60:1310-24. 5.Kim JJ, Tsukamoto MM, Mathur AK, Ghomri YM, Hou LA, Sheibani S, et al. Delayed paracentesis is associated with increased in-hospital mortality in patients with spontaneous bacterial peritonitis. Am J Gastroenterol. 2014;109:1436-42. 6.Runyon BA, Hoefs JC. Ascitic fluid analysis in the differentiation of spontaneous bacterial peritonitis from gastrointestinal tract perforation into ascitic fluid. Hepatology. 1984;4: 447-50. 7.Soriano G, Castellote J, Alvarez C, Girbau A, Gordillo J, Baliellas C, et al. Secondary bacterial peritonitis in cirrhosis: a retrospective study of clinical and analytical characteristics, diagnosis and management. J Hepatol. 2010;52:39-44. 8.Felisart J, Rimola A, Arroyo V, Perez-Ayuso RM, Quintero E, Gines P, et al. Cefotaxime is more effective than is ampicillin-tobramycin in cirrhotics with severe infections. Hepatology. 1985;5:457-62. 9.Ricart E, Soriano G, Novella MT, Ortiz J, Sàbat M, Kolle L, et al. Amoxicillin-clavulanic acid versus cefotaxime in the therapy of bacterial infections in cirrhotic patients. J Hepatol. 2000;32:596-602. 10.Navasa M, Follo A, Llovet JM, Clemente G, Vargas V, Rimola A, et al. Randomized, comparative study of oral ofloxacin versus intravenous cefotaxime in spontaneous bacterial peritonitis. Gastroenterology. 1996;111:1011-7. 11.Fernández J, Acevedo J, Castro M, Garcia O, De Lope CR, Roca D, et al. Prevalence and risk factors of infections by multiresistant bacteria in cirrhosis: a prospective study. Hepatology. 2012;55:1551-61. 12.Ariza X, Castellote J, Lora-Tamayo J, Girbau A, Salord S, Rota R, et al. Risk factors for resistance to ceftriaxone and its impact on mortality in community, healthcare and nosocomial spontaneous bacterial peritonitis. J Hepatol. 2012;56:825-32. 13.Fernández J, Navasa M, Gómez J, Colmenero J, Vila J, Arroyo V, et al. Bacterial infections in cirrhosis: epidemiological changes with invasive procedures and norfloxacin prophylaxis. Hepatology. 2002;35:140-8. 14.Ortiz J, Vila MC, Soriano G, Miñana J, Gana J, Mirelis B, et al. Infections caused by Escherichia coli resistant to norfloxacin in hospitalized cirrhotic patients. Hepatology. 1999;29: 1064-9. 15.Umgelter A, Reindl W, Miedaner M, Schmid RM, Huber W. Failure of current antibiotic first-line regimens and mortality in hospitalized patients with spontaneous bacterial peritonitis. Infection. 2009;37:2-8. 16.Ginés P, Rimola A, Planas R, Vargas V, Marco F, Almela M, et al. Norfloxacin prevents spontaneous bacterial peritonitis recurrence in cirrhosis: results of a double-blind, placebocontrolled trial. Hepatology. 1990;12:716-24. 17.Soriano G, Guarner C, Tomás A, Villanueva C, Torras X, González D, et al. Norfloxacin prevents bacterial infection in cirrhotics with gastrointestinal hemorrhage. Gastroenterology. 1992;103:1267-72. 18.Fernández J, Ruiz del Arbol L, Gómez C, Durandez R, Serradilla R, Guarner C, et al. Norfloxacin vs ceftriaxone in the prophylaxis of infections in patients with advanced cirrhosis and hemorrhage. Gastroenterology. 2006;131:1049-56. 19.Fernández J, Navasa M, Planas R, Montoliu S, Monfort D, Soriano G, et al. Primary prophylaxis of spontaneous bacterial peritonitis delays hepatorenal syndrome and improves survival in cirrhosis. Gastroenterology. 2007;133:818-24. 20.Chavez-Tapia NC, Barrientos-Gutierrez T, Tellez-Avila F, Soares-Weiser K, Mendez-Sanchez N, Gluud C, et al. Metaanalysis: antibiotic prophylaxis for cirrhotic patients with upper gastrointestinal bleeding—an updated Cochrane review. Aliment Pharmacol Ther. 2011;34:509-18. 21.Tandon P, Abraldes JG, Keough A, Bastiampillai R, Jayakumar S, Carbonneau M, et al. Risk of bacterial infection in patients with cirrhosis and acute variceal hemorrhage, based on Child-Pugh class, and effects of antibiotics. Clin Gastroenterol Hepatol. 2015;13:1189-96. 22.Bruns T, Lutz P, Stallmach A, Nischalke HD. Low ascitic fluid protein does not indicate an increased risk for spontaneous bacterial peritonitis in current cohorts. J Hepatol. 2015;63:527-8. SESIÓN 2 CIRROSIS E HIPERTENSIÓN PORTAL TIPS: indicaciones y resultados Juan Carlos García-Pagán Laboratorio de Hemodinámica Hepática, Servicio de Hepatología, Hospital Clínic, Barcelona, IDIBAPS y CIBERehd, Universidad de Barcelona, Barcelona, España Introducción La derivación portosistémica percutánea intrahepática (DPPI), más conocida como TIPS, acrónimo de las iniciales de su denominación anglosajona (transjugular intrahepatic portosystemic shunt), ha sustituido prácticamente a la cirugía derivativa, tanto en situaciones de urgencia como electivas. El TIPS consiste en la creación de una derivación portosistémica intrahepática de diámetro prefijado (“calibrada”) por métodos de radiología intervencionista. Una vez realizado el TIPS ocasiona un rápido descenso en el gradiente de presión portal y esta es la base para su utilización en diferentes complicaciones de la hipertensión portal. El procedimiento comienza por la cateterización de la vena suprahepática a través de la vena yugular interna derecha. Por medio de un equipo de punción transvenosa se perfora el parénquima hepático bajo control ecográfico hasta alcanzar una rama portal intrahepática. Una vez cateterizada la vena porta, y tras la medición de presiones y determinación de gradientes, se procede a dilatar el tracto parenquimatoso mediante un balón de angioplastia. Finalmente se coloca una prótesis metálica de 8-10 mm de diámetro, que evita que se cierre la comunicación portosistémica establecida a través del parénquima hepático. El uso de prótesis recubiertas con PTFE (politetrafluoroetileno) se asocia a una tasa muy baja de oclusión/estenosis. En este capítulo se revisarán los resultados de las indicaciones en las que el TIPS se ha mostrado claramente eficaz y de las que existen más datos en la bibliografía: la hemorragia por varices esofágicas, el tratamiento de la ascitis refractaria y en el síndrome de Budd-Chiari (SBC). El TIPS en la hemorragia digestiva por rotura de varices esofágicas Actualmente, las indicaciones para la realización de un TIPS en el contexto de la hemorragia por varices esofágicas son: a) el tratamiento de episodios agudos de hemorragia por varices esofágicas que no pueden ser controlados con tratamiento farmacológico y endoscópico; b) en los pacientes en los que la hemorragia recidiva en 2 o más ocasiones, a pesar de haber sido instaurado un tratamiento farmacológico y endoscópico para la prevención de la recidiva, y c) en pacientes con un episodio de hemorragia por varices y alto riesgo de fracaso terapéutico utilizando el tratamiento médico farmacológico y endoscópico (denominado TIPS precoz). Uso de TIPS en el fracaso del tratamiento farmacológico y endoscópico En el contexto del uso del TIPS como tratamiento de rescate, un estudio aleatorizado comparó la eficacia del TIPS frente a la cirugía derivativa mediante la anastomosis esplenorrenal distal en pacientes con buena reserva hepática1. En este estudio, ambas alternativas terapéuticas fueron igual de eficaces en la prevención de la hemorragia, en el riesgo de presentar encefalopatía hepática en el seguimiento o en la supervivencia. No obstante, los pacientes tratados con TIPS requerían frecuentes revisiones y reintervenciones, dado el elevado numero de episodios de disfunción. Cuando se diseñó el estudio, todavía no se había implementado la utilización de prótesis recubiertas y actualmente sabemos que su utilización disminuye de forma drástica el número de disfunciones2. Es por ello que el TIPS es actualmente la primera opción derivativa en el tratamiento de rescate de la hemorragia variceal y la cirugía derivativa se reserva para los pacientes con buena función hepatocelular donde el TIPS no sea técnicamente factible (menos del 5% de casos). Dado el acumulativo riesgo de muerte en pacientes con episodios repetidos de hemorragia variceal, el TIPS siempre debería considerarse cuando un paciente ha presentado más de un episodio de hemorragia. 29 30 Sesión 2 - CIRROSIS E HIPERTENSIÓN PORTAL Uso de TIPS en pacientes con hemorragia variceal y alto riesgo de fracaso terapéutico A pesar de los avances en el tratamiento farmacológico y endoscópico de la hemorragia variceal, todavía en un 10-20% de pacientes estos tratamientos fracasan. Es precisamente en este grupo de pacientes donde la mortalidad es mayor, alcanzando en algunas series hasta el 50% de estos. Por ello, con la identificación de estos pacientes con alto riesgo de fracaso al tratamiento estándar se podría plantear una estrategia más agresiva de forma precoz, para evitar el fracaso terapéutico y potencialmente mejorar la sobrevida. En este sentido, el uso precoz del TIPS en pacientes que tienen un elevado riesgo de fracaso del tratamiento médico/endoscópico convencional ha mostrado mejorar el pronóstico de estos pacientes. Así, la utilización del TIPS durante las primeras horas de la hemorragia logra el control rápido de esta, evita al mismo tiempo la recidiva precoz y mejora la supervivencia de estos pacientes. Así, en pacientes con hemorragia variceal aguda y criterios de alto riesgo para fallo terapéutico con el manejo estándar, la colocación de un TIPS precoz en las primeras 72 h disminuye la tasa de recidiva y aumenta la supervivencia, sin incrementar la tasa de encefalopatía hepática, con una incidencia de eventos adversos graves que no mostró diferencias significativas con el grupo control. TIPS en la prevención de la recidiva hemorrágica Dada la elevada eficacia del TIPS en el control de la hemorragia, numerosos estudios han evaluado la posibilidad de utilizar el TIPS como tratamiento inicial a todo paciente con cirrosis hepática, independientemente del riesgo de que tengan fracaso terapéutico a la terapia convencional. Así, el TIPS se ha comparado frente a las terapias endoscópicas como tratamiento inicial en la prevención de la recidiva hemorrágica. Estos estudios muestran claramente que el TIPS tiene una eficacia mayor que las técnicas endoscópicas. No obstante, el TIPS se asocia a una mayor incidencia de encefalopatía sin ningún beneficio en la supervivencia3. De igual forma, el TIPS se ha comparado frente al tratamiento farmacológico combinado (propranolol más mononitrato de isosorbida) y nuevamente se ha demostrado una mayor eficacia del TIPS, que logra el control de la hemorragia pero con más encefalopatía e igual supervivencia4. Es por ello que actualmente no se recomienda el TIPS como tratamiento inicial para prevenir la recidiva hemorrágica. No obstante, es posible que EL TIPS pudiera ser una buena alternativa para prevenir la recidiva hemorrágica en pacientes que tengan un elevado riesgo de recidiva, a pesar de recibir tratamiento farmacológico y endoscópico. El TIPS en la ascitis refractaria El objetivo tras la colocación del TIPS en la ascitis refractaria, como en el caso de la hemorragia digestiva por varices, es el descenso del gradiente de presión portal por debajo de 12 mmHg, lo que disminuirá la presión en los sinusoides hepáticos. Ello se asocia a una disminución de la actividad de la renina plasmática y de los valores de aldosterona y a un aumento en la excreción renal de sodio. Diversos estudios prospectivos controlados y aleatorizados han comparado la colocación del TIPS frente a la paracentesis evacuadora con reposición de albúmina en el manejo de la ascitis refractaria. En estos estudios se demostró que la colocación de un TIPS era más efectiva que la paracentesis evacuadora con reposición de albúmina en la recurrencia de la ascitis, siendo la incidencia de encefalopatía mayor en el TIPS y existiendo controversias sobre el efecto sobre la supervivencia6,7. Un metaanálisis basado en los datos individuales de 4 estudios aleatorizados que comparan TIPS frente a paracentesis evacuadoras con reposición de seroalbumina ha mostrado que el TIPS logra una mejoría significativa de la supervivencia libre de trasplante en pacientes con ascitis refractaria8. En algunos de estos estudios se detectaron como factores de mayor riesgo de presentar encefalopatía hepática: bilirrubina > 3 mg/dl, edad superior a 60 años y una clasificación C de la escala Child-Pugh. En el análisis univariante, el predictor de mortalidad independiente más importante fue el valor de bilirrubina sérica. Los estudios previamente referenciados han utilizado prótesis no recubiertas para la realización del TIPS. El uso actual de prótesis recubiertas de politetrafluoretilo (e-PTFE), con un índice mucho menor de disfunción, puede incrementar la indicación del TIPS en la ascitis refractaria en los próximos años. El TIPS en el síndrome de Budd-Chiari El TIPS con prótesis recubiertas de e-PTFE es la técnica derivativa de elección en los pacientes con SBC que no responden al tratamiento médico. Se requiere un entrenamiento especial para la realización del TIPS en estos pacientes, ya que la técnica es habitualmente más compleja que en pacientes con cirrosis hepática, al no poder, en ocasiones, canular ninguna vena suprahepática, siendo necesario realizar una punción directa desde la vena cava intrahepática hacia la rama portal derecha. No obstante, en centros con experiencia el TIPS es técnica- mente realizable y descomprime eficazmente el sistema venoso portal en más del 90% de los casos. La trombosis precoz del stent en las primeras horas no es infrecuente en los pacientes en los que se retrasa el inicio de la anticoagulación, por lo que se recomienda iniciar una perfusión de heparina sódica inmediatamente después de lograr la punción de la vena porta9. Un reciente estudio multicéntrico internacional, con participación de centros de referencia en esta patología, ha evaluado el impacto sobre la supervivencia y factores pronósticos de mala evolución en una serie amplia de 124 pacientes con SBC tratados con TIPS en los que había fracasado el tratamiento médico previo. Este estudio ha puesto de manifiesto los excelentes resultados que se obtienen mediante la utilización del TIPS. Así, la supervivencia libre de trasplante a 1 y 5 años fue, respectivamente, del 88 y el 78%. Esta excelente supervivencia fue especialmente evidente en los pacientes con mal pronóstico. En este subgrupo, el TIPS logró obtener una supervivencia libre de trasplante a los 5 años del 71%, muy superior a la supervivencia estimada del 42% en este grupo de alto riesgo. Por todo ello consideramos que el TIPS realizado en centros con experiencia deberá ser el tratamiento de elección en pacientes con SBC que no responden al tratamiento médico, reservando el trasplante para los enfermos en los que el TIPS no logre controlar los síntomas. En el estudio previamente mencionado un pequeño grupo de pacientes (menos del 10% de la población estudiada) presentaron una mala evolución a pesar del TIPS. Datos preliminares sugieren que estos enfermos pueden ser identificados mediante un modelo pronóstico basado en la edad, valores de bilirrubina y cociente internacional normalizado. Si estos datos se confirman, en este subgrupo de pacientes debería plantearse el trasplante hepático como primera opción terapéutica. TIPS: indicaciones y resultados 31 Bibliografía 1.Henderson JM, Boyer TD, Kutner MH, Galloway JR, Rikkers LF, Jeffers LJ, et al. Distal splenorenal shunt versus transjugular intrahepatic portal systematic shunt for variceal bleeding: a randomized trial. Gastroenterology. 2006;130:1643-51. 2.Bureau C, García-Pagán JC, Otal P, Pomier-Layrargues G, Chabbert V, Cortez C, et al. Improved clinical outcome using polytetrafluoroethylene-coated stents for tips: Results of a randomized study. Gastroenterology. 2004;126:469-75. 3.Luca A, D’Amico G, La Galla R, Midiri M, Morabito A, Pagliaro L. TIPS for prevention of recurrent bleeding in patients with cirrhosis: meta-analysis of randomized clinical trials. Radiology. 1999;212:411-21. 4.Escorsell A, Banares R, García-Pagán JC, Gilabert R, Moitinho E, Piqueras B, et al. TIPS versus drug therapy in preventing variceal rebleeding in advanced cirrhosis: a randomized controlled trial. Hepatology. 2002;35:385-92. 5.Bureau C, Pagán JC, Layrargues GP, Metivier S, Bellot P, Perreault P, et al. Patency of stents covered with polytetrafluoroethylene in patients treated by transjugular intrahepatic portosystemic shunts: long-term results of a randomized multicentre study. Liver Int. 2007;27:742-7. 6.D’Amico G, Luca A, Morabito A, Miraglia R, D’Amico M. Uncovered transjugular intrahepatic portosystemic shunt for refractory ascites: a meta-analysis. Gastroenterology. 2005;129:1282-93. 7.Albillos A, Banares R, González M, Catalina MV, Molinero LM. A meta-analysis of transjugular intrahepatic portosystemic shunt versus paracentesis for refractory ascites. J Hepatol. 2005;43:990-6. 8.Salerno F, Camma C, Enea M, Rossle M, Wong F. Transjugular intrahepatic portosystemic shunt for refractory ascites: a meta-analysis of individual patient data. Gastroenterology. 2007;133:825-34. 9.Perello A, García-Pagán JC, Gilabert R, Suárez Y, Moitinho E, Cervantes F, et al. TIPS is a useful long-term derivative therapy for patients with Budd-Chiari syndrome uncontrolled by medical therapy. Hepatology. 2002;;35:132-9. SESIÓN 2 CIRROSIS E HIPERTENSIÓN PORTAL Insuficiencia renal en la cirrosis Xavier Ariza y Pere Ginès Servicio de Hepatología, Hospital Clínic, Barcelona, España Universitat de Barcelona, Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), Barcelona, España CIBERehd (Centro de Investigación Biomédica en Red de enfermedades hepáticas y digestivas) INTRODUCCIÓN La insuficiencia renal es una complicación frecuente de los pacientes con cirrosis descompensada, que se produce en un 20% de los pacientes hospitalizados y que condiciona su pronóstico1-4. Se estima que dos terceras partes de los casos de insuficiencia renal de estos pacientes son producidas por alteraciones de la función circulatoria secundarias a los cambios hemodinámicos, sin coexistencia de daño renal estructural5. El desarrollo progresivo de este tipo de insuficiencia renal asociado a una alteración de la función circulatoria en ausencia de lesiones histológicas renales es conocido como síndrome hepatorrenal (SHR)1,2. El SHR es la única causa de insuficiencia renal con una fisiopatología específica que tan solo ocurre en el contexto de la cirrosis hepática evolucionada. Además del SHR, los pacientes con cirrosis pueden desarrollar insuficiencia renal secundaria a otras etiologías, como hipovolemia, infecciones bacterianas, administración de fármacos nefrotóxicos o enfermedades renales parenquimatosas1,2. Dada la existencia de este estado de disfunción circulatoria, junto con la alteración del volumen arterial efectivo y el aumento de la actividad de los sistemas vasoconstrictores endógenos que afectan la circulación intrarrenal, la función renal de los pacientes con cirrosis es lábil y es relativamente frecuente que desarrollen insuficiencia renal asociada a otras complicaciones de la enfermedad, como las infecciones bacterianas o la hemorragia digestiva. FISIOPATOLOGÍA La insuficiencia renal en los pacientes con cirrosis se relaciona principalmente con la presencia de alteraciones de la circulación sistémica, que se caracteri32 zan por una intensa vasodilatación arterial esplácnica, una reducción del volumen arterial efectivo y una activación de los sistemas vasoconstrictores endógenos secundarios a la hipertensión portal2,4-8. Las alteraciones funcionales renales más comunes son el trastorno en la excreción de sodio y de agua libre, responsables del desarrollo de ascitis e hiponatremia dilucional, respectivamente. En estadios más avanzados existe una reducción del flujo sanguíneo renal debido a una intensa vasoconstricción renal que conduce al desarrollo del SHR. Esta vasoconstricción renal es la última alteración funcional renal que ocurre, cronológicamente, en los pacientes con cirrosis y ascitis6,8. El origen funcional del SHR queda demostrado por la ausencia de alteraciones histológicas renales y la normalización de la función renal tras el trasplante hepático o el tratamiento farmacológico1. En la fisiopatología están implicados distintos mecanismos, que se comentan a continuación. Alteraciones de la circulación sistémica Los pacientes con cirrosis y ascitis muestran una marcada alteración de la función circulatoria, que se caracteriza por una baja presión arterial, un descenso de las resistencias vasculares periféricas y un alto gasto cardíaco, como consecuencia de la intensa vasodilatación arterial esplácnica2,4-6. Esta vasodilatación arterial esplácnica es producto de un aumento de la producción o actividad de factores vasodilatadores, particularmente el óxido nítrico (NO), el monóxido de carbono y los cannabinoides endógenos, incrementados debido a la hipertensión portal. La existencia de estas alteraciones circulatorias precede a la retención renal de sodio y agua, el acúmulo de ascitis y el desarrollo de insuficiencia renal2,6. En estadios iniciales, cuan- Insuficiencia renal en la cirrosis 33 do los pacientes están asintomáticos, la hipertensión portal es moderada y tan solo existe un descenso leve de las resistencias vasculares sistémicas, que se consigue compensar gracias al desarrollo de una circulación hiperdinámica, que se caracteriza por un aumento del gasto cardíaco, manteniendo unos valores de presión arterial y de volumen arterial efectivo normales2,4,6. Sin embargo, en estadios más avanzados, la reducción de las resistencias vasculares periféricas es progresiva, lo que conlleva una hipovolemia arterial efectiva que no se consigue compensar mediante un mayor aumento del gasto cardíaco, lo que condiciona una activación de los sistemas vasoconstrictores endógenos. la administración de antiinflamatorios no esteroideos (AINE), fármacos que inhiben su síntesis, es una causa de insuficiencia renal en los pacientes con cirrosis16,17. Esto sugiere que la función renal de los pacientes con cirrosis depende en gran parte de la producción renal de PG. Otra sustancia que podría estar implicada en la regulación de la función renal es el NO18,19. El NO tiene un papel en la regulación de la microcirculación glomerular y facilita la natriuresis en respuesta a los cambios de perfusión renal18,19. Activación de los sistemas neurohormonales Estudios experimentales y clínicos sugieren que la traslocación bacteriana puede tener un papel importante en la disfunción circulatoria, pudiendo conllevar el desarrollo de SHR20,21. La traslocación bacteriana induce una respuesta inflamatoria, con la consiguiente producción de citocinas proinflamatorias (especialmente IL-6 y TNFa) y factores vasoactivos (NO) en la circulación esplácnica, conduciendo a la vasodilatación de esta8. Los pacientes con cirrosis y valores elevados de LPS o ADN-bacteriano tienen aumentados los valores de citocinas proinflamatorias, reducidas las resistencias vasculares periféricas y aumentado el gasto cardíaco, en comparación con los pacientes sin signos de traslocación bacteriana22,23. Además, la administración de norfloxacino, un antibiótico que reduce la traslocación bacteriana, mejora las alteraciones circulatorias y reduce el riesgo de desarrollo de SHR24,25. La actividad de estos sistemas vasoconstrictores, como el sistema renina-angiotensina-aldosterona, el sistema nervioso simpático y la hipersecreción no osmótica de vasopresina, está aumentada en una gran proporción de pacientes con cirrosis, particularmente en aquellos con una enfermedad más evolucionada, para mantener la presión arterial dentro de unos valores normales2,4,6. La activación de estos sistemas vasoconstrictores tiene efectos homeostáticos en relación con el mantenimiento del volumen arterial efectivo, pero también conlleva efectos negativos en la función renal, produciendo una retención marcada de sodio y agua libre, que conduce al desarrollo de ascitis y edemas, así como hiponatremia hipervolémica. En los estados más avanzados, el aumento de actividad de los sistemas vasoconstrictores induce una intensa vasoconstricción renal que condiciona un descenso de la filtración glomerular y el desarrollo del SHR. Otras sustancias potencialmente implicadas en las alteraciones hemodinámicas son la endotelina y las hormonas natriuréticas, como los péptidos natriuréticos atrial y cerebral. A pesar de que sus valores están aumentados en los pacientes con cirrosis, su papel en la disfunción circulatoria no está claramente establecido9-12. Los datos que provienen de estudios experimentales sugieren que tienen un papel en el mantenimiento de la perfusión renal y la modulación de la actividad del sistema renina-angiotensina-aldosterona13. Factores renales Las prostaglandinas (PG), metabolitos del ácido araquidónico, se producen en el riñón vía ciclooxigenasa y tienen efectos protectores, compensando los efectos vasoconstrictores de los sistemas neurohormonales14. Las PG renales (PGI2 y PGE2) tienen efectos vasodilatadores en el riñón y sus valores están aumentados en los pacientes con cirrosis y ascitis2,14,15. Además, Inflamación sistémica DIAGNÓSTICO Los métodos más precisos para medir la tasa de filtración glomerular en pacientes con cirrosis se basan en técnicas de aclaramiento de inulina o sustancias radioisotópicas. No obstante, estos métodos son caros y complicados y no están disponibles de modo generalizado en el medio sanitario. Por eso, en la práctica clínica se sigue aceptando que la creatinina sérica (sCr) es el método más útil para estimar la función renal en los pacientes con cirrosis7,26. A pesar de esto, está ampliamente reconocido que el valor de sCr no es un marcador óptimo de la función renal, ya que puede sobrestimar la tasa de filtrado glomerular, especialmente en los pacientes con cirrosis, debido a la menor producción de esta. Además, la síntesis de sCr está condicionada por el peso corporal, los estados de malnutrición, la edad o el sexo2,27-30. Clásicamente, la insuficiencia renal en los pacientes con cirrosis se ha definido como un incremento de la sCr con un valor > 1,5 mg/dl (133 µmol/l)2,7,26. Sin embargo, esta definición tiene diversos inconvenien- 34 Sesión 2 - CIRROSIS E HIPERTENSIÓN PORTAL Tabla 1. Criterios diagnósticos de acute kidney injury (AKI) en pacientes con cirrosis hepática según el International Club of Ascites39 Variable Definición sCr basal • Valor de sCr disponible en los 3 meses previos al ingreso hospitalario • En caso de disponer de más de un valor de sCr en los 3 meses previos, usar el valor más cercano a la hospitalización • En caso de ausencia de valores previos usar la sCr obtenida al ingreso AKI • Aumento sCr ≥ 0,3 mg/dl (≥ 26,5 mmol/l) durante un período de 48 h respecto a sCr basal o • Aumento sCr ≥ 50% respecto a sCr basal en los 7 días previos Estadios de AKI • Estadio 1: aumento sCr ≥ 0,3 mg/dl (≥ 26,5 mmol/l) o aumento sCr ≥ 1,5-2 veces respecto a sCr basal • Estadio 2: aumento sCr > 2-3 veces respecto a sCr basal • Estadio 3: aumento sCr > 3 veces respecto a sCr basal o sCr ≥ 4,0 mg/dl (353,6 mmol/l) con un aumento agudo ≥ 0,3 mg/dl (≥ 26,5 mmol/l) o inicio de tratamiento renal sustitutivo Progresión de AKI • Progresión a un estadio superior de AKI y/o necesidad de tratamiento renal sustitutivo Regresión de AKI • Descenso a un estadio inferior de AKI Respuesta al tratamiento • No respuesta: no regresión de AKI • Respuesta parcial: regresión del estadio de AKI con una reducción sCr ≥ 0,3 mg/dl ≥ 26,5 mmol/l), pero manteniendo un valor de sCr superior al basal • Respuesta completa: descenso del valor sCr al valor basal ± 0,3 mg/dl (≥ 26,5 mmol/l) sCr: creatinina sérica. tes: en primer lugar, el nivel de sCr > 1,5 mg/dl identifica a pacientes con una reducción marcada de la tasa de filtrado glomerular (en general, < 30 ml/min), lo que puede comportar un retraso en el diagnóstico y tratamiento de la insuficiencia renal; en segundo lugar, el uso de un valor fijo de sCr no tiene en cuenta los cambios respecto a los valores previos, lo cual es fundamental para distinguir entre los casos de insuficiencia renal aguda y crónica5,31. En la última década se han propuesto nuevas definiciones de insuficiencia renal, actualmente denominada acute kidney injury (AKI), que se han validado en la población general32-34. La última de estas, propuesta por un panel de expertos, la Kidney Disease Improving Global Outcome (KDIGO) criteria, define la presencia de AKI como un incremento de sCr > 0,3 mg/dl en 48 h o un aumento ≥ 50% respecto a la sCr basal34. En este sentido, estudios recientes han evaluado la utilidad de los criterios de AKI en pacientes con cirrosis, y han demostrado que la utilización de esta definición identifica a pacientes con un mal pronóstico, en términos de estancia hospitalaria y mortalidad35-38. Con todos estos condicionantes, recientemente, el International Club of Ascites (ICA) ha validado y aplicado estos criterios para el diagnóstico de AKI en pacientes con cirrosis hepática (tabla 1)39. CAUSAS, TRATAMIENTO Y PRONÓSTICO El tratamiento inicial de los pacientes con cirrosis e insuficiencia renal depende de la causa, la gravedad del cuadro clínico y sus posibles complicaciones asociadas. Es fundamental identificar la etiología, ya que condiciona el pronóstico a corto plazo40. No existen pruebas específicas que ayuden a diferenciar la causa de la insuficiencia renal. Para el enfoque diagnóstico es necesario realizar una historia clínica detallada, una exploración física y una evaluación de la función renal con un estudio de electrólitos en suero y orina41. La realización de una ecografía renal es importante para descartar alteraciones estructurales sugestivas de enfermedad renal crónica o patología obstructiva del tracto urinario. Por otro lado, estudios recientes sugie- ren que los valores urinarios de neutrophil gelatinaseassociated lipocalin (NGAL) son útiles en el diagnóstico diferencial entre el SHR y la necrosis tubular aguda (NTA)42-44. La insuficiencia renal en los pacientes con cirrosis puede tener diversas causas: • Infecciones bacterianas. Son la causa más frecuente de insuficiencia renal en los pacientes con cirrosis. Su patogenia se relaciona con un aumento de la vasodilatación arterial sistémica ya existente producida por la respuesta inflamatoria, que aparece en este contexto clínico. Este efecto hemodinámico es característico de los pacientes que desarrollan peritonitis bacteriana espontánea o bacteriemia espontánea, aunque puede ocurrir en cualquier tipo de infección bacteriana, a pesar de que la gravedad de la respuesta inflamatoria y la disfunción renal no sea tan marcada45-47. De este modo debe evaluarse cuidadosamente la posibilidad de la existencia de una infección bacteriana en cualquier paciente con cirrosis e insuficiencia renal, así como iniciar el tratamiento antibiótico adecuado de forma precoz para conseguir la resolución del cuadro clínico y evitar la progresión de la disfunción renal. • SHR. Tal como se ha indicado con anterioridad, el SHR es un tipo de insuficiencia renal específica de los pacientes con cirrosis, que se caracteriza por una vasoconstricción renal funcional con una reducción grave del filtrado glomerular y la ausencia de alteraciones histológicas1,2,7,26. Debido a la ausencia de marcadores diagnósticos específicos, el diagnóstico del SHR es clínico y se realiza mediante unos criterios aceptados que excluyen otras causas de insuficiencia renal (tabla 2)7. Las infecciones bacterianas pueden precipitarlo, y puede ser reversible después de la resolución de la infección, pero en otros puede persistir o progresar47,48. Para su diagnóstico se debe descartar la causa hipovolémica mediante la reposición de volumen con albúmina vía intravenosa. Existen 2 tipos de SHR en función de las características clínicas y pronósticas7,26. El tipo 1 se caracteriza por un deterioro brusco de la función renal, con un aumento del valor de sCr > 2,5 mg/dl en menos de 2 semanas. Estos pacientes padecen disfunción multiorgánica grave y sin tratamiento este tipo de SHR tiene muy mal pronóstico, con una mediana de supervivencia de unas 2 semanas49. En el tipo 2 existe una alteración estable de la función renal, con valores de sCr que oscilan entre 1,5 y 2,5 mg/ dl. El curso clínico de estos pacientes se caracteriza por ascitis refractaria y sin trasplante la mediana de supervivencia es de unos 6 meses49. Los pacientes con SHR tipo 2 pueden evolucionar hacia el tipo 1 debido a la progresión de la enfermedad u otros factores desencadenantes, como infecciones bacte- Insuficiencia renal en la cirrosis 35 Tabla 2. Criterios diagnósticos del síndrome hepatorrenal (SHR) en pacientes con cirrosis hepática7 • Cirrosis hepática con ascitis • sCr > 1,5 mg/dl • Ausencia de mejoría en el valor de sCr (reducción hasta un nivel < 1,5 mg/dl) después de 2 días sin diuréticos y con expansión de volumen mediante albúmina (1 g/kg de peso corporal, hasta un máximo de 100 g/día) • Ausencia de shock • Ausencia de tratamiento actual o reciente con fármacos nefrotóxicos • Ausencia de signos de enfermedad renal parenquimatosa: – Proteinuria (> 500 mg/día) – Hematuria (> 50 hematíes/campo) – Hallazgos ecográficos renales anómalos sCr: creatinina sérica. rianas o paracentesis de gran volumen. Los pacientes sometidos a paracentesis de gran volumen presentan mayor riesgo de desarrollar disfunción circulatoria posparacentesis, un desajuste circulatorio que se acompaña de la activación del sistema renina-angiotensina consecuencia de la evacuación de grandes cantidades de líquido ascítico50. Aproximadamente el 20% de los pacientes tratados con paracentesis de gran volumen sin reposición de albúmina desarrollan SHR y/o una retención de agua sin solutos que provoca hiponatremia51. La administración de albúmina intravenosa es eficaz para prevenir la disfunción circulatoria posparacentesis y tiene mayor eficacia que otros expansores plasmáticos, especialmente en los casos de paracentesis > 5 l52. El diagnóstico diferencial entre el SHR y la NTA puede resultar difícil. La presencia de células epiteliales tubulares en el sedimento urinario o la existencia previa de hipovolemia/shock favorecen el diagnóstico de NTA. Por otro lado, una fracción de excreción de sodio urinaria < 1% (en ausencia de diuréticos) indica integridad de la reabsorción tubular y apoya el diagnóstico de SHR. Por otra parte, tal y como se ha indicado con anterioridad, estudios recientes sugieren la utilidad en la determinación de los valores urinarios de NGAL, un biomarcador de lesión tubular, para el diagnóstico diferencial42-44. Unos valores urinarios altos de este biomarcador traducirían lesión tubular y favorecerían el diagnóstico de NTA, mientras que unos valores bajos indicarían ausencia de daño tubular y el posible diagnóstico de SHR. 36 Sesión 2 - CIRROSIS E HIPERTENSIÓN PORTAL El tratamiento del SHR se realiza mediante la administración de fármacos vasoconstrictores (fundamentalmente terlipresina) junto con expansión con albúmina intravenosa2,7,26,53. Este tratamiento es efectivo en el 40-50% de los pacientes con SHR tipo 1. La información en relación con la eficacia del tratamiento farmacológico en el SHR tipo 2 es limitada y no se recomienda su utilización. En este sentido, un estudio reciente no ha encontrado diferencias por lo que respecta al pronóstico en pacientes en lista de espera de trasplante hepático con SHR tipo 2 tratados con terlipresina y albúmina54. • Hipovolemia. En la insuficiencia renal inducida por hipovolemia, el principal mecanismo responsable de la hipoperfusión renal es la reducción del volumen intravascular, que en los casos graves puede producir el desarrollo de NTA. Las principales causas son la hemorragia digestiva, la pérdida de líquidos por vómitos o diarreas y un exceso de administración de tratamiento diurético55,56. La función renal suele mejorar en estos casos al eliminar la causa precipitante y con la expansión del volumen plasmático, a no ser que exista shock hipovolémico grave. • Enfermedades renales parenquimatosas. Deben sospecharse en caso de presencia de proteinuria > 500 mg/24 h, un sedimento urinario con > 50 hematíes/campo o hallazgos ecográficos patológicos, en ausencia de otras causas de insuficiencia renal7,53. La mayor parte de estas patologías guardan relación con marcadores de riesgo cardiovascular (como hipertensión, diabetes mellitus o sobrepeso) y factores etiológicos de la cirrosis, donde se produce un depósito de inmunocomplejos circulantes en los glomérulos57. Las causas más frecuentes se resumen en la tabla 3. En casos seleccionados, puede ser útil la realización de una biopsia renal en pacientes con etiología incierta, ya que los pacientes con enfermedad renal intrínseca concomitante pueden necesitar diálisis o trasplante doble de riñón e hígado. • Nefrotoxicidad farmacológica. Puede aparecer en tratamientos con AINE, antihipertensivos u otros fármacos que pueden causar NTA o nefritis intersticial aguda (tabla 3)16. La insuficiencia renal por AINE o por los inhibidores de la enzima de conversión de angiotensina se produce principalmente por una alteración del equilibrio entre los factores vasodilatadores y vasoconstrictores de la microcirculación renal, aunque se ha documentado que estos fármacos también pueden producir NTA17. Otros fármacos como los aminoglucósidos, la anfotericina B o el tenofovir también pueden ocasionar NTA. Algunos antibióticos pueden producir nefritis intersticial debido a una reacción de hipersensibilidad (tabla 3). En estos casos es necesaria la interrupción del fármaco para que la función renal vuelva a la normalidad. Tabla 3. Causas de insuficiencia renal en pacientes con cirrosis hepática • Infecciones bacterianas* – Peritonitis bacteriana espontánea – Bacteriemia espontánea – Infección del tracto urinario, neumonía, otras • SHR • Insuficiencia renal inducida por hipovolemia – Hemorragia digestiva – Diuréticos – Vómitos y/o diarreas • Enfermedades renales parenquimatosas –G lomerulopatías: IgA, nefropatía membranosa, membranoproliferativa, crioglobulinemia – IRC debida a diabetes, hipertensión u otras • Insuficiencia renal inducida por fármacos – Inducida hemodinámicamente: AINE, bloqueadores del receptor de angiotensina – NTA: aminoglucósidos, anfotericina B, tenofovir –N efritis intersticial aguda: penicilina, rifampicina, sulfonamidas AINE: antiinflamatorios no esteroideos; IRC: insuficiencia renal crónica; NTA: necrosis tubular aguda; SHR: síndrome hepatorrenal. *La insuficiencia renal que ocurre en el contexto de infecciones bacterianas se considera un síndrome hepatorrenal si cumple los criterios diagnósticos de este. FINANCIACIÓN PG: Fondo de Investigación de Salud Carlos III (FIS PI12/0330) cofinanciado por los fondos europeos de desarrollo (FEDER). Agencia de Gestió d’Ajuts Universitaris i de Recerca (2014SGR 708). Fondos de Investigación: PG: Sequana Medical, Grifols S.A. Consultant/advisory board: PG: Ferring, IKARIA, Noorik. Bibliografía 1.Fagundes C, Ginès P. Hepatorenal syndrome: a severe, but treatable, cause of kidney failure in cirrhosis. Am J Kidney Dis. 2012;59:874-85. 2.Ginès P, Schrier RW. Renal failure in cirrhosis. N Engl J Med. 2009;361:1279-90. 3.Fede G, D’Amico G, Arvaniti V, Tsochatzis E, Germani G, Georgiadis D, et al. Renal failure and cirrhosis: a systematic review of mortality and prognosis. J Hepatol. 2012;56: 810-8. 4.García-Tsao G, Parikh CR, Viola A. Acute kidney injury in cirrhosis. Hepatology. 2008;48:2064-77. 5.Wong F. Definition and diagnosis of acute kidney injury in cirrhosis. Dig Dis. 2015;33:539-47. 6.Schrier RW, Arroyo V, Bernardi M, Epstein M, Henriksen JH, Rodés J. Peripheral arterial vasodilation hypothesis: a proposal for the initiation of renal sodium and water retention in cirrhosis. Hepatology. 1988;8:1151-7. 7.Salerno F, Gerbes A, Ginès P, Wong F, Arroyo V. Diagnosis, prevention and treatment of hepatorenal syndrome in cirrhosis. Gut. 2007;56:1310-8. 8.Epstein M, Berk DP, Hollenberg NK, Adams DF, Chalmers TC, Abrams HL, et al. Renal failure in the patient with cirrhosis. The role of active vasoconstriction. Am J Med. 1970;49: 175-85. 9.Leivas A, Jiménez W, Lamas S, Bosch-Marcé M, Oriola J, Clària J, et al. Endothelin 1 does not play a major role in the homeostasis of arterial pressure in cirrhotic rats with ascites. Gastroenterology. 1995;108:1842-8. 10.Sogni P, Moreau R, Gomola A, Gadano A, Cailmail S, Calmus Y, et al. Beneficial hemodynamic effects of bosentan, a mixed ET(A) and ET(B) receptor antagonist, in portal hypertensive rats. Hepatology. 1998;28:655-9. Insuficiencia renal en la cirrosis 37 22.Albillos A, De la Hera A, González M, Moya JL, Calleja JL, Monserrat J, et al. Increased lipopolysaccharide binding protein in cirrhotic patients with marked immune and hemodynamic derangement. Hepatology. 2003;37:208-17. 23.Francés R, Zapater P, González-Navajas JM, Muñoz C, Caño R, Moreu R, et al. Bacterial DNA in patients with cirrhosis and noninfected ascites mimics the soluble immune response established in patients with spontaneous bacterial peritonitis. Hepatology. 2008;47:978-85. 24.Rasaratnam B, Kaye D, Jennings G, Dudley F, Chin-Dusting J. The effect of selective intestinal decontamination on the hyperdynamic circulatory state in cirrhosis. A randomized trial. Ann Intern Med. 2003;139:186-93. 25.Fernández J, Navasa M, Planas R, Montoliu S, Monfort D, Soriano G, et al. Primary prophylaxis of spontaneous bacterial peritonitis delays hepatorenal syndrome and improves survival in cirrhosis. Gastroenterology. 2007;133:818-24. 26.Arroyo V, Ginès P, Gerbes AL, Dudley FJ, Gentilini P, Laffi G, et al. Definition and diagnostic criteria of refractory ascites and hepatorenal syndrome in cirrhosis. International Ascites Club. Hepatology. 1996;23:164-76. 27.Bataller R, Ginès P, Guevara M, Arroyo V. Hepatorenal syndrome. Semin Liver Dis. 1997;17:233-47. 11.Ginès P, Jiménez W, Arroyo V, Navasa M, López C, Titó L, et al. Atrial natriuretic factor in cirrhosis with ascites: plasma levels, cardiac release and splanchnic extraction. Hepatology. 1988;8:636-42. 28.Caregaro L, Menon F, Angeli P, Amodio P, Merkel C, Bortoluzzi A, et al. Limitations of serum creatinine level and creatinine clearance as filtration markers in cirrhosis. Arch Intern Med. 1994;154:201-5. 12.La Villa G, Romanelli RG, Casini Raggi V, Tosti-Guerra C, De Feo ML, Marra F, et al. Plasma levels of brain natriuretic peptide in patients with cirrhosis. Hepatology. 1992;16:156-61. 29.Sherman DS, Fish DN, Teitelbaum I. Assessing renal function in cirrhotic patients: problems and pitfalls. Am J Kidney Dis. 2003;41:269-78. 13.Angeli P, Jiménez W, Arroyo V, Mackenzie HS, Zhang PL, Clària J, et al. Renal effects of natriuretic peptide receptor blockade in cirrhotic rats with ascites. Hepatology. 1994;20: 948-54. 30.Cholongitas E, Shusang V, Marelli L, Nair D, Thomas M, Patch D, et al. Review article: renal function assessment in cirrhosis - difficulties and alternative measurements. Aliment Pharmacol Ther. 2007;26:969-78. 14.Laffi G, La Villa G, Pinzani M, Marra F, Gentilini P. Arachidonic acid derivatives and renal function in liver cirrhosis. Semin Nephrol. 1997;17:530-48. 31.Solà E, Ginès P. Assessment of acute kidney injury at hospital admission in cirrhosis: estimating baseline serum creatinine is not the answer. Liver Int. 2015;35:2079-81. 15.Pérez-Ayuso RM, Arroyo V, Camps J, Rimola A, Gaya J, Costa J, et al. Evidence that renal prostaglandins are involved in renal water metabolism in cirrhosis. Kidney Int. 1984;26: 72-80. 32.Bellomo R, Ronco C, Kellum JA, Mehta RL, Palevsky P; Acute Dialysis Quality Initiative workgroup. Acute renal failure - definition, outcome measures, animal models, fluid therapy and information technology needs: the Second International Consensus Conference of the Acute Dialysis Quality Initiative (ADQI) Group. Crit Care. 2004;8:R204-12. 16.Salerno F, Badalamenti S. Drug-induced renal failure in cirrhosis. En: Ginès P, Arroyo V, Rodes J, Schrier RW, editors. Ascites and Renal Dysfunction in Liver Disease. 2nd ed. Malden: Blackwell; 2005. p. 372-82. 17.Elia C, Graupera I, Barreto R, Solà E, Moreira R, Huelin P, et al. Severe acute kidney injury associated with non-steroidal anti-inflammatory drugs in cirrhosis: A case-control study. J Hepatol. 2015;63:593-600. 18.Blantz RC, Deng A, Lortie M, Munger K, Vallon V, Gabbai FB, et al. The complex role of nitric oxide in the regulation of glomerular ultrafiltration. Kidney Int. 2002;61:782-5. 19.Clària J, Jiménez W, Ros J, Asbert M, Castro A, Arroyo V, et al. Pathogenesis of arterial hypotension in cirrhotic rats with ascites: role of endogenous nitric oxide. Hepatology. 1992;15:343-9. 20.Wiest R, Das S, Cadelina G, García-Tsao G, Milstien S, Groszmann RJ. Bacterial translocation in cirrhotic rats stimulates eNOS-derived NO production and impairs mesenteric vascular contractility. J Clin Invest. 1999;104:1223-3. 21.Wiest R, Lawson M, Geuking M. Pathological bacterial translocation in liver cirrhosis. J Hepatol. 2014;60:197-209. 33.Mehta RL, Kellum JA, Shah SV, Molitoris BA, Ronco C, Warnock DG, et al. Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney injury. Crit Care. 2007;11:R31. 34.Kidney Disease: Improving Global Outcomes (KDIGO) Acute Kidney Injury Work Group. KDIGO Clinical Practice Guideline for Acute Kidney Injury. Kidney Int. 2012;2 Suppl:1-138. 35.De Carvalho JR, Villela-Nogueira CA, Luiz RR, Guzzo PL, Da Silva Rosa JM, Rocha E, et al. Acute kidney injury network criteria as a predictor of hospital mortality in cirrhotic patients with ascites. J Clin Gastroenterol. 2012;46:e21-6. 36.Belcher JM, García-Tsao G, Sanyal AJ, Bhogal H, Lim JK, Ansari N, et al. Association of AKI with mortality and complications in hospitalized patients with cirrhosis. Hepatology. 2013;57:753-62. 37.Fagundes C, Barreto R, Guevara M, García E, Solà E, Rodríguez E, et al. A modified acute kidney injury classification for diagnosis and risk stratification of impairment of kidney function in cirrhosis. J Hepatol. 2013;59:474-81. 38 Sesión 2 - CIRROSIS E HIPERTENSIÓN PORTAL 38.Piano S, Rosi S, Maresio G, Fasolato S, Cavallin M, Romano A, et al. Evaluation of the Acute Kidney Injury Network criteria in hospitalized patients with cirrhosis and ascites. J Hepatol. 2013;59:482-9. 48.Barreto R, Fagundes C, Guevara M, Solà E, Pereira G, Rodríguez E, et al. Type-1 hepatorenal syndrome associated with infections in cirrhosis: natural history, outcome of kidney function, and survival. Hepatology. 2014;59:1505-13. 39.Angeli P, Ginès P, Wong F, Bernardi M, Boyer TD, Gerbes A, et al. Diagnosis and management of acute kidney injury in patients with cirrhosis: revised consensus recommendations of the International Club of Ascites. J Hepatol. 2015;62: 968-74. 49.Alessandria C, Ozdogan O, Guevara M, Restuccia T, Jiménez W, Arroyo V, et al. MELD score and clinical type predict prognosis in hepatorenal syndrome: relevance to liver transplantation. Hepatology. 2005;41:1282-9. 40.Martín-Llahí M, Guevara M, Torre A, Fagundes C, Restuccia T, Gilabert R, et al. Prognostic importance of the cause of renal failure in patients with cirrhosis. Gastroenterology. 2011;140:488-96. 41.Cárdenas A, Ginès P. A patient with cirrhosis and increasing creatinine level: what is it and what to do? Clin Gastroenterol Hepatol. 2009;7:1287-91. 42.Fagundes C, Pépin MN, Guevara M, Barreto R, Casals G, Solà E, et al. Urinary neutrophil gelatinase-associated lipocalin as biomarker in the differential diagnosis of impairment of kidney function in cirrhosis. J Hepatol. 2012;57:267-73. 43.Verna EC, Brown RS, Farrand E, Pichardo EM, Forster CS, Soladel Valle DA, et al. Urinary neutrophil gelatinase-associated lipocalin predicts mortality and identifies acute kidney injury in cirrhosis. Dig Dis Sci. 2012;57:2362-70. 44.Belcher JM, Sanyal AJ, Peixoto AJ, Perazella MA, Lim J, Thiessen-Philbrook H, et al. Kidney biomarkers and differential diagnosis of patients with cirrhosis and acute kidney injury. Hepatology. 2014;60:622-32. 45.Terra C, Guevara M, Torre A, Gilabert R, Fernández J, MartínLlahí M, et al. Renal failure in patients with cirrhosis and sepsis unrelated to spontaneous bacterial peritonitis: value of MELD score. Gastroenterology. 2005;129:1944-53. 46.Fasolato S, Angeli P, Dallagnese L, Maresio G, Zola E, Mazza E, et al. Renal failure and bacterial infections in patients with cirrhosis: epidemiology and clinical features. Hepatology. 2007;45:223-9. 47.Terg R, Gadano A, Cartier M, Casciato P, Lucero R, Muñoz A, et al. Serum creatinine and bilirubin predict renal failure and mortality in patients with spontaneous bacterial peritonitis: a retrospective study. Liver Int. 2009;29:415-9. 50.Ruiz-del-Árbol L, Monescillo A, Jimenéz W, García-Plaza A, Arroyo V, Rodés J. Paracentesis-induced circulatory dysfunction: mechanism and effect on hepatic hemodynamics in cirrhosis. Gastroenterology. 1997;113:579-86. 51.Ginès P, Titó L, Arroyo V, Planas R, Panés J, Viver J, et al. Randomized comparative study of therapeutic paracentesis with and without intravenous albumin in cirrhosis. Gastroenterology. 1988;94:1493-502. 52.Bernardi M, Caraceni P, Navickis RJ, Wilkes MM. Albumin infusion in patients undergoing large-volume paracentesis: a metaanalysis of randomized trials. Hepatology. 2012;55:1172-81. 53.European Association for the Study of the Liver. EASL clinical practice guidelines on the management of ascites, spontaneous bacterial peritonitis, and hepatorenal syndrome in cirrhosis. J Hepatol. 2010;53:397-417. 54.Rodríguez E, Pereira G, Solà E, Elia C, Barreto R, Pose E, et al. Treatment of type-2 hepatorenal syndrome in patients awaiting transplantation. Effects on kidney function and transplantation outcomes. Liver Transpl. 2015.[Epub ahead of print]. 55.Thabut D, Massard J, Gangloff A, Carbonell N, Francoz C, Nguyen-Khac E, et al. Model for end-stage liver disease score and systemic inflammatory response are major prognostic factors in patients with cirrhosis and acute functional renal failure. Hepatology. 2007;46:1872-82. 56.Cárdenas A, Ginès P, Uriz J, Bessa X, Salmerón JM, Mas A, et al. Renal failure after upper gastrointestinal bleeding in cirrhosis: incidence, clinical course, predictive factors, and short-term prognosis. Hepatology. 2001;34:671-6. 57.Meyers CM, Seeff LB, Stehman-Breen CO, Hoofnagle JH. Hepatitis C and renal disease: an update. Am J Kidney Dis. 2003;42:631-57. SESIÓN 2 CIRROSIS E HIPERTENSIÓN PORTAL Bloqueadores beta: indicaciones, contraindicaciones y riesgos en cirrosis Agustín Albillos Facultad de Medicina, Universidad de Alcalá, Alcalá de Henares, Madrid, España Servicio de Gastroenterología y Hepatología, Hospital Universitario Ramón y Cajal, Madrid, España introducción Desde hace algunos años se ha abierto un intenso debate sobre el riesgo/beneficio respecto al uso de los bloqueadores beta en los pacientes con cirrosis hepática, sobre todo en los estadios más avanzados de la enfermedad. Sin embargo existe una evidencia robusta y contrastada de su efecto beneficioso en la reducción de la presión portal. Es por ello que los bloqueadores beta se consideran la piedra angular del tratamiento médico de la hipertensión portal. Aprovechando la actual controversia sobre los bloqueadores beta, a continuación se repasan los conceptos fisiopatológicos más relevantes, sus indicaciones y los aspectos relacionados con su seguridad en las fases más avanzadas de la cirrosis. BENEFICIOS HEMODINÁMICOS Y NO HEMODINÁMICOS DE LOS BLOQUEADORES BETA Los bloqueadores beta son el tratamiento médico más importante de la hipertensión portal1. Sus efectos beneficiosos en los pacientes con cirrosis hepática se deben, principalmente, a sus efectos sobre la hemodinámica del sistema venoso portal hepático. Sin embargo, estos fármacos tienen acciones no hemodinámicas que también son de interés en los pacientes con cirrosis hepática. Los bloqueadores beta utilizados en la cirrosis hepática son los no cardioselectivos. Sus efectos hemodinámicos se deben al bloqueo de receptores beta-adrenérgicos a nivel cardíaco y renal (B1) y a nivel esplácnico (B2). El bloqueo B1 a nivel cardíaco tiene efectos cronotrópicos e inotrópicos negativos, y a nivel renal disminuye la actividad del sistema renina-angiotensina-aldosterona, que se encuentra hiperactivado en los pacientes con hipertensión portal clínicamente significativa. El bloqueo B2 produce una vasoconstricción en el territorio esplácnico, lo que disminuye el flujo y la presión portal. Los beneficios hemodinámicos de los bloqueadores beta son evidentes en la prevención de la hemorragia por varices esofágicas (v. apartado indicaciones) y en la prevención del incremento progresivo de la hipertensión portal2,3. Sin embargo, los bloqueadores beta también son beneficiosos por sus acciones no hemodinámicas. La diana de esta acción asienta principalmente a nivel. Es importante destacar que los efectos no hemodinámicos de los bloqueadores beta están presentes en los pacientes no respondedores desde un punto de vista puramente hemodinámico3. En varios estudios se ha observado que el uso de bloqueadores beta previene el desarrollo de peritonitis bacteriana espontánea y otras infecciones, así como episodios de encefalopatía hepática. Asimismo, gracias al efecto sobre el sistema ácigos incluso los pacientes sin una respuesta hemodinámica a los bloqueadores beta presentan un menor riesgo de hemorragia variceal que los pacientes no tratados con bloqueadores beta4. En las fases avanzadas de la hipertensión portal, el sistema nervioso simpático (SNS) presenta un aumento de su actividad intestinal. Como consecuencia del aumento de noradrenalina en las terminaciones nerviosas intestinales se produce un enlentecimiento del tránsito intestinal, sobrecrecimiento bacteriano, deterioro de la barrera defensiva de la mucosa intestinal y alteración de las funciones de las células del sistema inmune entérico. Por otro lado, la hipertensión portal produce congestión venosa, edema y alteración en la microcirculación intestinal. Todo ello origina un 39 40 Sesión 2 - CIRROSIS E HIPERTENSIÓN PORTAL aumento de la permeabilidad intestinal, lo que favorece la traslocación bacteriana, que es el fenómeno que precede a la peritonitis bacteriana espontánea y a gran parte de las infecciones en los pacientes cirróticos5. Los bloqueadores beta, por su efecto hemodinámico sobre la hipertensión portal, mejoran la vascularización y perfusión intestinal. Por otro lado, el bloqueo del SNS (acción no hemodinámica) mejora la motilidad intestinal disminuyendo la estasis luminal y el sobrecrecimiento bacteriano y evita el deterioro de la barrera mucosa intestinal. De esta forma, los bloqueadores beta previenen la traslocación bacteriana y las infecciones en el paciente cirrótico. Este mismo mecanismo de acción explica la disminución del riesgo de desarrollar episodios de encefalopatía hepática3,5. el riesgo de recidiva hemorrágica es del 60% a los 2 años, con una mortalidad del 30%1. El pilar del tratamiento más importante en la profilaxis secundaria son los bloqueadores beta7. No se debe utilizar la ligadura endoscópica en monoterapia, y el tratamiento con bloqueadores beta en monoterapia solamente está aceptado en los pacientes en los que la ligadura endoscópica no es posible o la rechazan persistentemente. De nuevo, en este escenario, los pacientes respondedores presentan un riesgo de recidiva del 15% en 2 años, frente al 37-55% en los pacientes no respondedores. Sin embargo, gracias a los efectos no hemodinámicos de los bloqueadores beta se consigue un menor riesgo de hemorragia en los pacientes tratados no respondedores respecto a los pacientes no tratados (el 37-55% frente al 55-67%, a los 2 años)1,4. INDICACIONES DE LOS BLOQUEADORES BETA EN LA CIRROSIS Tanto para la prevención primaria como secundaria, los bloqueadores beta disponibles son propranolol, nadolol, timolol y carvedilol. El más utilizado es el propranolol. La dosis inicial es de 10-20 mg/12 h. La dosis se debe ir incrementando cada 2-3 días. Inicialmente se aceptaba como un objetivo del tratamiento obtener una frecuencia cardíaca de 55-60 lpm, o un descenso de las pulsaciones del 20-25% respecto a la frecuencia cardíaca basal del paciente o conseguir un gradiente de presión portal < 12 mmHg. Dado que el estudio hemodinámico no es accesible en la práctica clínica diaria y que no se ha demostrado correlación entre la reducción de la frecuencia cardíaca y la reducción de la presión portal, actualmente se acepta que se debe titular la dosis de bloqueadores beta hasta la dosis máxima tolerada (propranolol 320 mg/día o nadolol 160 mg/día) siempre y cuando se tolere, la presión arterial sistólica (PAS) no sea inferior a 100 mmHg y la frecuencia cardíaca no sea menor de 50-55 lpm1. Lamentablemente, no hay ningún dato clínico fiable que nos permita saber si realmente el paciente presenta respuesta hemodinámica o no a los bloqueadores beta. Se ha observado que los pacientes respondedores tienen valores más elevados de albúmina y más bajos de bilirrubina respecto a los no respondedores8,9. Sin embargo, no se puede extrapolar estos resultados para tomar decisiones individuales. El único método para evaluar la respuesta hemodinámica es el cateterismo hepático con determinación del gradiente de presión portal. Solamente un 30% de los pacientes en tratamiento con propranolol presentan una respuesta hemodinámica1. Sin embargo, los beneficios no hemodinámicos de los bloqueadores beta son independientes de la respuesta hemodinámica2,3. Entre todos los bloqueadores beta, el carvedilol es un nuevo bloqueador beta que ha mostrado resultados interesantes en varios estudios publicados recientemente. Desde un punto de vista fisiopatológico, además del efecto bloqueador beta presenta un efecto En los pacientes con cirrosis hepática e hipertensión portal los bloqueadores beta están indicados en las siguientes situaciones6: • Profilaxis primaria de hemorragia por varices esofágicas en pacientes con: – Varices pequeñas con puntos rojos o con clase funcional Child C. – Varices grandes, independientemente del grado de función hepática (Child). La profilaxis primaria en pacientes con estadio funcional Child B con varices pequeñas sin estigmas de riesgo hemorrágico no está completamente aclarada sobre la base de la evidencia científica actual. Sin embargo, en el consenso internacional sobre hipertensión portal (Baveno VI) se recomienda firmemente la utilización de los bloqueadores beta en este escenario clínico6. Beneficios de la profilaxis primaria1. La profilaxis primaria reduce el riesgo de la primera hemorragia de un 25% sin tratamiento a un 15% en los pacientes tratados. Sin embargo, los pacientes respondedores son los que presentan unos mejores resultados, con una reducción del riesgo de hemorragia de un 40-50%. Aunque el beneficio es menor en los no respondedores, están igualmente indicados los bloqueadores beta. Por tanto, en conclusión, todo paciente con indicación de profilaxis primaria debe ser tratado con bloqueadores beta. • Profilaxis secundaria de hemorragia por varices. Los pacientes que hayan sobrevivido a un episodio de hemorragia variceal deben ser tratados con bloqueadores beta en combinación con un programa de ligadura endoscópica. En ausencia de este tratamiento, Bloqueadores beta: indicaciones, contraindicaciones y riesgos en cirrosis bloqueador alfa, lo que produce vasodilatación intrahepática y disminución de la presión portal. Este efecto aditivo de los bloqueadores alfa explica por qué el carvedilol consigue un mayor descenso de la presión portal respecto al propranolol1,5,10,11. Por otro lado, con el carvedilol se consigue una mayor proporción de pacientes respondedores y además rescata al 40% de los no respondedores a propranolol12. Sin embargo, no se han realizado ensayos clínicos comparando ambos bloqueadores beta en los que se hayan analizado resultados clínicos, y la evidencia existente es aún débil y escasa como para generalizar su uso. Por otro lado, el efecto alfa-1-bloqueador se relaciona con frecuentes efectos adversos en relación con la hipotensión que produce, lo que puede limitar su uso, especialmente en pacientes cirróticos, los cuales suelen presentar cierto grado de hipotensión por la vasodilatación esplácnica secundaria a la disfunción circulatoria10. Un escenario potencial donde el carvedilol podría ser de utilidad es en pacientes cirróticos con hipertensión arterial. Los bloqueadores beta no están indicados en la profilaxis preprimaria, ya que se ha demostrado que no evitan el desarrollo de las varices esofágicas. Este efecto fútil de los bloqueadores beta cuando el gradiente de presión portal es < 10 mmHg se debe a que en las fases iniciales de la enfermedad el aumento de la presión portal se debe al aumento de la resistencia vascular intrahepática, donde los bloqueadores beta no tienen efecto alguno1,4,6. Una vez que el gradiente supera los 10 mmHg, la activación del SNS origina vasodilatación esplácnica e hiperaflujo portal. En este momento se abre la ventana y los bloqueadores beta comienzan a ser eficaces por su acción vasoconstrictora esplácnica, reduciendo el flujo y la presión portal13,14. Un 15% de los pacientes tratados con bloqueadores beta presenta contraindicaciones, las más frecuentes son la hiperreactividad bronquial, la enfermedad pulmonar obstructiva crónica, el bloqueo auriculoventricular de segundo y tercer grados, la valvulopatía aórtica, la claudicación intermitente y la psicosis grave. La bradicardia sinusal y la diabetes insulinodependiente son contraindicaciones relativas. Excluyendo estas contraindicaciones, la incidencia de efectos adversos es del 15%, la mayoría de ellos leves: fatiga muscular, disnea de esfuerzo, insomnio, cansancio muscular, impotencia y apatía. Estos efectos secundarios desaparecen con la disminución de la dosis. Solo en un 5% de los casos los efectos adversos obligan a la retirada del tratamiento1. BLOQUEADORES BETA EN LOS PACIENTES CON CIRROSIS AVANZADA (ASCITIS) Este es un apartado de gran debate en los últimos años dadas las conclusiones contradictorias de varias publicaciones. 41 El estudio inicial de Serste et al15 abrió la caja de Pandora con la publicación de un estudio retrospectivo donde se demostraba que los bloqueadores beta eran un factor de riesgo independiente de mortalidad en los pacientes con ascitis refractaria. Como se muestra en la tabla 1, los bloqueadores beta han demostrado ser perjudiciales en los pacientes con ascitis refractaria o peritonitis bacteriana espontánea en los estudios de Serste et al, Mandofer et al y Kimer et al. Por el contrario han demostrado ser beneficiosos en los pacientes con ascitis y ascitis refractaria en el estudio de Leithead et al, y neutrales en los pacientes con ascitis en los estudios de Galbois et al, Mandofer et al, Robbins et al y Bossen et al16-21. En general, los estudios adolecen de ciertas limitaciones metodológicas que impiden extraer conclusiones firmes y generalizables: a) los estudios son heterogéneos entre sí (el estudio de Leithead et al está realizado en un grupo muy seleccionado de pacientes en lista de trasplante hepático); b) distribución desigual de los pacientes más graves entre los distintos grupos de tratamiento15; c) la definición de ascitis refractaria es variable; d) la proporción de pacientes con ascitis refractaria o con antecedente de hemorragia variceal previa es desigual, lo que puede afectar a la interpretación de los datos, ya que los bloqueadores beta mejoran la supervivencia en los pacientes en profilaxis secundaria y dichos beneficios superan los riesgos potenciales en los pacientes con ascitis refractaria15,18; e) el grado de función hepática (MELD o Child-Pugh) tampoco es homogéneo entre grupos15-19; f) existe heterogeneidad en el grado de disfunción hemodinámica (valorada con el sodio y la presión arterial media)16-20; g) dificultad para establecer una posible relación causa/efecto entre el uso de bloqueadores beta y la causa de la muerte (p. ej., hepatocarcinoma)15; h) causas de muerte no bien tipificadas en algunos estudios. A pesar de toda la información disponible, los datos actuales no se pueden considerar robustos para establecer que los bloqueadores beta son beneficiosos o deletéreos en los pacientes con ascitis refractaria. El mecanismo fisiopatológico que subyace al efecto deletéreo de los bloqueadores beta en pacientes con ascitis y ascitis refractaria es el deterioro que producen estos fármacos en la disfunción circulatoria, que ya está presente en esta fase de la enfermedad2. La ascitis refractaria representa el extremo de gravedad de la cirrosis avanzada. En esta fase se produce un aumento de la vasodilatación arterial periférica, una activación del sistema renina-angiotensina-aldosterona y del SNS, y una disminución progresiva de la reserva cardíaca para mantener el gasto cardíaco. Bajo estas circunstancias, la acción de los bloqueadores beta sobre los receptores B1 cardíacos origina una disminución del gasto cardíaco que es crítica para mantener la perfusión tisular. Como consecuencia de ello se produce hipotensión, deterioro 42 Sesión 2 - CIRROSIS E HIPERTENSIÓN PORTAL Tabla 1. Series publicadas donde se ha estudiado la influencia de los bloqueadores beta (BB) en los pacientes con cirrosis y ascitis Autor, año (referencia) Serste et al, 20102 Galbois et al, 20113 Mandorfer et al, 20144 Robins et al, 20145 Leithead et al, 20141 Kimer et al, 20156 Bossen et al, 20157 BB No BB BB No BB BB No BB BB No BB BB No BB BB No BB BB No BB Número de pacientes 77 74 26 42 245 362 36 78 159 163 23 38 562 636 Población Ascitis refractaria Cirrosis y sepsis ingresados en UCI (62% con ascitis) Ascitis ± PBE Ascitis resistente a diuréticosa Ascitis en pacientes en lista de TH Ascitis refractariab Cirróticos con ascitis incluidos en 3 ensayos clínicos con satavaptán Tipo de publicación/ análisis Publicación completa/ análisis retrospectivo de una base de datos prospectiva Carta/análisis retrospectivo Publicación completa/ análsis retrospectivo Carta/ análisis retrospectivo Publicación completa/ análisis retrospectivo Publicación completa/ análisis retrospectivo Abstract/ análisis retrospectivo de una base de datos prospectiva Dosis de propranolol/ carvedilol (rango), mg 80 (40-160) ? 40 (20-120)/ 12,5 (6,25-25,0) 48,9 (40-80) 80 (10-240)/ 6,25 (3,12-12,5) 80 (40-200) ? Ascitis refractaria (%) 100 100 54 41 32 40 ? 35 100 100 46 Presencia de varices (%) 100 4 92 38 90 62 100 54 ? 95c 46 ? Hemorragia variceal previa (%) ? 18 15 69 32 40 25 ? MELD (mediana) 18,8 18,9 23 24 17,2 17,8 ? 16 17 15 Sodio sérico, media (mmol/l) 125 133 135 131 134 134 ? 136 134 133 PAS, media (mmHg) 103 123 120 106 114 117 116 116 115d 122 ? PBE (%) ? 23 17 35 27 50 44 ? 35 29 ? Seguimiento (meses) 8 Estancia en UCI 12,9 10,6 10 9 Hasta el trasplante 36,6 46,6 12,2 Muerte (%) 82 58 Ascitis 36 50 25 19 22e 65 68 24 Tras SBP 53 27 Efecto de los bloqueadores beta ? 46 Perjudicial 62 Neutral Neutral en ascitis Neutral 37 29 Beneficioso en ascitis y en ascitis refractaria Neutral 52 30 13 15,5 12 11 135 137 136 ? 27 Neutral en ascitis y en ascitis refractaria Perjudicial después de un episodio de PBE PAS: presión arterial sistólica; PBE: peritonitis bacteriana espontánea; TH: trasplante hepático; UCI: unidad de cuidados intensivos. aPacientes con paracentesis regulares. bPacientes con paracentesis al menos 2 veces al año a pesar de tratamiento con diuréticos. cEndoscopia no disponible en todos los pacientes. dPresión arterial solamente disponible en 81 pacientes. eSin diferencias en mortalidad entre la presencia de ascitis refractaria o PBE. Bloqueadores beta: indicaciones, contraindicaciones y riesgos en cirrosis de la función renal e hiperactivación sostenida del resto de sistemas presores, que por vasoconstricción de la arteria aferente empeoran aún más la función renal con riesgo de desarrollar síndrome hepatorrenal y muerte del paciente3,5. Se ha observado que un gasto cardíaco bajo y la hipotensión (presión arterial media [PAM] < 80 mmHg) son 2 factores que predicen el desarrollo de síndrome hepatorrenal y mortalidad en los pacientes con cirrosis y ascitis13,14. En el consenso internacional de Baveno VI se destacó la falta de evidencia robusta en este campo. Las recomendaciones de Baveno VI se basan en el metaanálisis de datos individuales (sobre todo de los pacientes incluidos en el estudio de Serste et al y Mandofer et al) y en la opinión de expertos6. En los pacientes con ascitis refractaria en profilaxis primaria de hemorragia variceal, se recomienda reducir la dosis o suspender el tratamiento con bloqueadores beta si el paciente desarrolla hipotensión (PAS < 90 mmHg), hiponatremia (Na < 130) o daño renal agudo. Aunque esto está basado en series de pacientes con ascitis refractaria, estas recomendaciones podrían ser extensibles a los pacientes con ascitis tratada con diuréticos que desarrollen cualquier complicación aguda de su enfermedad6. Desde un punto de vista práctico, sería recomendable identificar a los pacientes con una enfermedad grave mediante factores relacionados con la hemodinámica (hipotensión, hiponatremia, PAM), en los que existe riesgo de producirse un efecto adverso grave por parte de los bloqueadores beta, más que intentar definir entidades clínicas específicas, como la ascitis refractaria. Es importante destacar que la decisión de suspender los bloqueadores beta debe ser cuidadosamente valorada en los pacientes que se encuentran en profilaxis secundaria de hemorragia por varices esofágicas, donde los bloqueadores beta proporcionan un beneficio claro de la supervivencia (Albillos et al, datos no publicados). HIPÓTESIS DE LA VENTANA Con todo lo expuesto anteriormente se observa cómo los bloqueadores beta son útiles durante una fase de la historia natural de la cirrosis, lo que ha dado lugar al desarrollo de la hipótesis de la ventana13. En la fase inicial de la cirrosis hepática, cuando el gradiente de presión portal es < 10 mmHg (sin varices y sin ascitis), el SNS se encuentra mínimamente activado o casi normal, el flujo esplácnico es normal (no hay vasodilatación esplécnica) y la reserva funcional cardíaca está intacta2,5,13. En esta fase, el aumento de presión portal se debe fundamentalmente al aumento de resistencias intrahepáticas, por lo que los bloqueadores beta (que actúan sobre la vasculatura esplácnica) no han mostrado efectos beneficiosos clínicos. En esta fase, la ventana de los bloqueadores beta permanece cerrada13. 43 Conforme la enfermedad va progresando, la presión portal va aumentando (gradiente de presión portal > 10 mmHg). En este momento comienza la activación del SNS, que produce vasodilatación esplácnica, la cual, a través del aumento del flujo portal, contribuye al aumento de la presión portal. En esta fase, la reserva cardíaca permite mantener aún la hemodinámica y la perfusión tisular del paciente. En esta fase pueden aparecen varices esofágicas y la cirrosis se puede descompensar (hemorragia por varices y/o ascitis). Es en este momento cuando la ventana de los bloqueadores beta se abre y su uso está indicado según todas las recomendaciones de las guías clínicas para la profilaxis primaria y secundaria de hemorragia por varices esofágicas13,14. Los efectos no hemodinámicos de los bloqueadores beta sobre la traslocación bacteriana se hacen evidentes en esta fase, aunque en modelos experimentales solamente se han demostrado en presencia de ascitis (no se ha demostrado traslocación bacteriana patológica en modelos sin ascitis). Desde un punto de vista clínico, la traslocación bacteriana se ha demostrado en pacientes con cirrosis hepática y grado funcional Child C, pero no en los pacientes con Child A/B13. En las fases finales de la cirrosis hepática, la activación del SNS y el grado de vasodilatación esplácnica del paciente son máximos. La reserva cardíaca se encuentra limitada para seguir compensando la disfunción circulatoria tan acusada. El resultado es un deterioro progresivo de dicha reserva cardíaca, lo que conlleva el desarrollo de síndrome hepatorrenal5,13,14. En este momento, el riesgo de traslocación bacteriana e infecciones es máximo. En esta fase, la ventana de los bloqueadores beta se cierra, ya que sus acciones sobre los receptores B1 cardíacos tienen efectos deletéreos sobre las funciones inotrópicas y cronotrópicas. Su uso en esta fase aceleraría el fracaso de la bomba cardíaca para mantener la perfusión renal13, desencadenándose el síndrome hepatorrenal y la muerte del paciente. CONCLUSIÓN Los bloqueadores beta son el tratamiento médico más importante de la hipertensión portal. Su acción principal es la reducción de la presión portal (efecto hemodinámico), que comienza a ser evidente cuando existe activación del SNS y vasodilatación esplácnica. Sus acciones no hemodinámicas sobre la función de barrera intestinal previenen la traslocación bacteriana y el desarrollo de infecciones, incluso en los pacientes no respondedores. Los bloqueadores beta no están indicados en las fases iniciales de la enfermedad, donde el aumento de la presión portal se debe al aumento de resistencias intrahepáticas y no al hiperaflujo secundario a la vaso- 44 Sesión 2 - CIRROSIS E HIPERTENSIÓN PORTAL dilatación esplácnica. Los bloqueadores beta no están indicados en la profilaxis preprimaria. Están indicados en la prevención primaria y secundaria de la hemorragia por varices esofágicas. En la fase final de la enfermedad, dado el grado máximo de vasodilatación esplácnica, la función cardíaca es fundamental para mantener la perfusión tisular. El efecto inotrópico y cronotrópico negativo de los bloqueadores beta disminuiría aún más la reserva funcional cardíaca, lo que agravaría la ya crítica hemodinámica del paciente cirrótico. Por ello se recomienda disminuir la dosis/suspender los bloqueadores beta en esta fase de la enfermedad, que clínicamente está definida por la ascitis refractaria, la hiponatremia (Na < 130), la hipotensión (PAS < 90 mmHg) o la insuficiencia renal aguda. Bibliografía 1.Bosch J, Abraldes JG, Albillos A, Aracil C, Bañares R, Berzigotti A, et al. Portal hypertension: recommendations for evaluation and treatment: consensus document sponsored by the AEEH and the CIBERehd. Gastroenterol Hepatol. 2012;35:421-50. 2.Giannelli V, Lattanzi B, Thalheimer U, Merli M. Beta-blockers in liver cirrhosis. Ann Gastroenterol. 2014;27:20-6. 3.Ge PS, Runyon BA. The changing role of beta-blocker therapy in patients with cirrhosis. J Hepatol. 2014;60:643-53. 4.Albillos A, Bañares R, González M, Ripoll C, González R, Catalina MV, et al. Value of the HVPG to monitor drug therapy for portal hypertension: a meta-analysis. Am J Gastroenterol. 2007;102:1116-26. 5.Iwakiri Y. Pathophysiology of portal hypertension. Clin Liver Dis. 2014;28:281-91. 6.De Franchis R; Baveno VI Faculty. Expanding consensus in portal hypertension: Report of the Baveno VI Consensus Workshop: Stratifying risk and individualizing care for portal hypertension. J Hepatol. 2015;63:743-52. 7.González R, Zamora J, Gómez-Camarero J, Molinero LM, Bañares R, Albillos A. Meta-analysis: Combination endoscopic and drug therapy to prevent variceal rebleeding in cirrhosis. Ann Intern Med. 2008;149:109-22. 8.Abraldes JG, Tarantino I, Turnes J, García-Pagán JC, Rodés J, Bosch J. Hemodynamic response to pharmacological treatment of portal hypertension and long-term prognosis of cirrhosis. Hepatology. 2003;37:902-8. 9.Augustin S, González A, Badia L, Millán L, Gelabert A, Romero A, et al. Long-term follow-up of hemodynamic responders to pharmacological therapy after variceal bleeding. Hepatology. 2012;56:706-14. 10.Bañares R, Moitinho E, Matilla A, García-Pagán JC, Lampreave JL, Piera C, et al. Randomized comparison of long-term carvedilol and propranolol administration in the treatment of portal hypertension in cirrhosis. Hepatology. 2002;36: 1367-73. 11.Tripathi D, Therapondos G, Lui HF, Stanley AJ, Hayes PC. Haemodynamic effects of acute and chronic administration of low-dose carvedilol, a vasodilating beta-blocker, in patients with cirrhosis and PHT. Aliment Pharmacol Ther. 2002;16: 373-80. 12.Reiberger T, Ulbrich G, Ferlitsch A, Payer BA, Schwabl P, Pinter M, et al. Carvedilol for primary prophylaxis of variceal bleeding in cirrhotic patients with haemodynamic non-response to propranolol. Gut. 2013;62:1634-41. 13.Krag A, Wiest R, Albillos A, Gluud LL. The window hypothesis: haemodynamic and non-haemodynamic effects of b-blockers improve survival of patients with cirrhosis during a window in the disease. Gut. 2012;61:967-9. 14.Ge PS, Runyon BA. When should the B-blocker window in cirrhosis close? Gastroenterology. 2014;146:1597-9. 15.Sersté T, Melot C, Francoz C, Durand F, Rautou PE, Valla D, et al. Deleterious effects of beta-blockers on survival in patients with cirrhosis and refractory ascites. Hepatology. 2010;52:1017-22. 16.Mandorfer M, Bota S, Schwabl P, Bucsics T, Pfisterer N, Kruzik M, et al. Nonselective b blockers increase risk for hepatorenal syndrome and death in patients with cirrhosis and spontaneous bacterial peritonitis. Gastroenterology. 2014;146: 1680-90. 17.Kimer N, Feineis M, Møller S, Bendtsen F. Beta-blockers in cirrhosis and refractory ascites: a retrospective study and review of the literature. Scand J Gastroenterol. 2015;50: 129-37. 18.Leithead JA, Rajoriya N, Tehami N, Hodson J, Gunson BK, Tripathi D, et al. Nonselective b blockers are associated with improved survival in patients with ascites listed for liver transplantation. Gut. 2015;64:1111-9. 19.Galbois A, Das V, Thabut D, Maury E, Ait-Oufella H, Housset C, et al. Beta-blockers have no effect on outcomes in patients with cirrhosis and severe infections. Hepatology. 2011;53:1412-3. 20.Robins A, Bowden A, Watson W, Smith F, Gelson W, Griffiths W. Beta-blockers in cirrhosis patients with refractory ascites. Hepatology. 2014;59:2054-5. 21.Bossen L, Krag A, Vilstrup H, Watson H, Jepsen P. Non-selective beta-blockers and mortality in cirrhosis patients with or without refractory ascites: post-hoc analysis on three large RCT’S with 1 198 patients. J Hepatol. 2015;62;S213. SESIÓN 2 CIRROSIS E HIPERTENSIÓN PORTAL Fracaso hepático agudo sobre crónico Rafael Bañares Servicio de Aparato Digestivo, Hospital General Universitario Gregorio Marañón, Madrid, España Instituto de Investigación Sanitaria Gregorio Marañón, Madrid, España Facultad de Medicina, Universidad Complutense, Madrid, España CIBERehd INTRODUCCIÓN La historia natural de la cirrosis está marcada por diferentes fases evolutivas, que van desde la enfermedad compensada, en la cual no existen manifestaciones clínicas relacionadas con la cirrosis y que se caracteriza por un pronóstico favorable, hasta la enfermedad descompensada grave, que requiere trasplante u ocasiona la muerte del paciente. Esta evolución está bien aceptada actualmente y ha permitido modificar el modo de entender la cirrosis. Sin embargo, el curso de la enfermedad no es lineal y puede verse alterado por cambios bruscos en su presentación y en su gravedad. En este sentido, hace unos años se acuñó el concepto de fracaso hepático agudo sobre crónico (ACLF, acute on chronic liver failure), intentando describir un síndrome que se caracteriza por la aparición súbita de deterioro grave de la función hepática en pacientes con enfermedad compensada, asociado a fallo de otros órganos y a un marcado deterioro del pronóstico. No obstante, hasta muy recientemente no se han precisado unos criterios diagnósticos y no se ha profundizado en los mecanismos fisiopatológicos subyacentes de este síndrome. El objetivo de este capítulo es describir el concepto actual y las características más relevantes del ACLF, a partir de la información acumulada en el estudio CANONIC (CLIF Acute-on-Chronic Liver Failure in Cirrhosis), que trató esencialmente de desarrollar una definición de fracaso hepático agudo sobre crónico capaz de identificar a los pacientes con alto riesgo de mortalidad a corto plazo, además de precisar otras características hasta ahora desconocidas como su prevalencia, los factores precipitantes y los principales mecanismos patogénicos. BASES CONCEPTUALES PARA LA DEFINICIÓN DEL SÍNDROME El objetivo esencial del estudio CANONIC fue establecer unos criterios diagnósticos operativos a partir de una definición conceptual del síndrome asumida a priori y que incluyó 3 condiciones: presencia de descompensación aguda de la cirrosis, fallo de al menos un órgano (riñón, cerebro, pulmón, etc.) y mortalidad a los 28 días desde la admisión > 15%. De acuerdo con estas consideraciones, se reclutó en un corto espacio de tiempo (9 meses) a una amplia cohorte multicéntrica e internacional de pacientes cirróticos que ingresaron en un hospital por descompensación de su enfermedad hepática (ascitis, hemorragia digestiva, encefalopatía, infección bacteriana). Otro elemento novedoso y de importancia es la forma de valorar el fracaso de un determinado órgano. Clásicamente, la valoración de la gravedad de los pacientes con cirrosis se realiza mediante la puntuación MELD o la escala de Child-Pugh. Sin embargo, estas clasificaciones no reflejan con precisión el impacto del fracaso de otros órganos en el pronóstico. Por otra parte, las clasificaciones habitualmente empleadas en el entorno de las unidades de cuidados intensivos (UCI) no tienen en cuenta aspectos característicos de la enfermedad hepática. Por este motivo, se adaptó la escala SOFA a las características propias de la enfermedad hepática (tabla 1). Finalmente se analizó la mortalidad de los pacientes de la cohorte en función de qué órganos presentaban fracaso y el número de estos para intentar estratificar de forma operativa la gravedad del síndrome. De esta forma se pudo establecer una gradación específica del síndrome (tabla 2). 45 46 Sesión 2 - CIRROSIS E HIPERTENSIÓN PORTAL Tabla 1. Adaptación de la escala SOFA (Sepsis-related-Organ Failure Assessment) a las características de la enfermedad hepática Órgano/sistema Parámetro Puntuación = 1 Puntuación = 2 Puntuación Hígado Bilirrubina (mg/dl) <6 6-12 > 12 Riñón Creatinina (mg/dl) <2 2-3,5 > 3,5 o diálisis Cerebro Grado de encefalopatía (West-Haven) 0 1-2 3-4 Coagulación INR < 2,0 2,0-2,5 > 2,5 Circulación PAM (mmHg) > 70 < 70 Necesidad de vasopresores Respiratorio (1 de los 2 parámetros) PaO2/FiO2 > 300 < 300 y > 200 < 200 SatO2/FiO2 > 357 > 214 y < 357 < 214 FiO2: fracción inspiratoria de oxígeno; INR: índice normalizado internacional; PaO2: presión arterial de oxígeno; PAM: presión arterial media; SatO2: saturación de oxígeno. Los números en negrita reflejan la definición de cada fracaso orgánico. Tabla 2. Distribución de los pacientes del estudio CANONIC. Porcentaje de mortalidad de pacientes en función del número y tipo de fracaso de órganos Número y tipo de fracaso de órganos No disfunción renal ni EH leve-moderada Disfunción renal con o sin EH leve-moderada Sin fracaso orgánico 3,6 6,2 Un órgano 9,2 26,5 Hígado 5,9 30,3 Cerebro 8 20 Coagulación 5,3 22,2 Circulación 6,7 28,6 Riñón 15,8 24,1 Dos órganos 28,8 38,7 Tres o más órganos 86,2 61,5 EH: encefalopatía hepática. Los números en negrita representan los grupos en los que apareció fracaso de algún órgano y mortalidad superior al 15%. CARACTERÍSTICAS CLÍNICAS DE LOS PACIENTES CON FRACASO HEPÁTICO AGUDO SOBRE CRÓNICO Las características basales de los pacientes reproducen las conocidas características epidemiológicas de la cirrosis descompensada en nuestro medio; así, la edad media fue de 57,2 ± 12,2 años, el 63,3% eran varones, más frecuentemente alcohólicos (51,9%) y con una puntuación MELD media de 18,8 ± 7,5 puntos. Un 26,8% no había tenido descompensaciones agudas (DA) previas, siendo la ascitis y la encefalopatía hepática (EH) las descompensaciones más frecuentemente responsables de la hospitalización. El 46,5% de los pacientes había estado hospitalizado en los 3 meses previos y un 14,6% ingresó directamente en la UCI. El primer resultado relevante del estudio procede de la constatación, lógica por otra parte, de que la gravedad conferida por cada tipo de fracaso orgánico (FO) no fue similar; así, la mortalidad a los 28 días de la inclusión en el estudio fue de un 14,6% en los pacientes con fallo de un solo órgano, excepto en el caso del fracaso renal aislado (18,6%). Igualmente, la combinación de fracaso de un órgano aislado acompañado de deterioros intermedios de otros órganos se asoció fuertemente a la mortalidad. De este modo, los pacientes con fallo de un único órgano que presentaban además deterioro moderado de la función renal (creatinina sérica 1,5-1,9 mg/dl) y/o EH I-II tuvieron una mortalidad de prácticamente el doble que en ausencia de estos datos. Teniendo en cuenta todas estas consideraciones, el estudio permitió establecer 4 categorías en los pacientes cirróticos ingresados en el hospital como consecuencia de una DA de la enfermedad hepática. 1. Pacientes sin ACLF (77,4% de la población). Definido por la ausencia de FO, o con FO aislado de un órgano distinto del riñón (con creatinina sérica < 1,5 mg/dl y sin EH) o FO cerebral aislado con creatinina sérica < 1,5 mg/dl. 2. ACLF grado 1 (11% de la población). Definido por la presencia de FO renal aislado o FO hepático, de coagulación, circulatorio o respiratorio aislados con creatinina sérica entre 1,5-1,9 mg/dl y/o EH I-II o FO cerebral con creatinina sérica entre 1,5-1,9 mg/ dl. 3. ACLF grado 2 (8% de la población). Dos FO. 4. ACLF grado 3 (3,5% de la población). Tres o más FO. Esta clasificación permitió definir una clara diferencia en la supervivencia de los grupos; así, la mortalidad a los 28 días del ingreso en el hospital fue del 4,7, 22,1, 32 y 76,7%, para cada uno de los 4 grupos. Este diferente comportamiento clínico entre los distintos tipos de ACLF permitiría, hipotéticamente, actuar de forma precoz, estratificar la agresividad del tratamiento y adecuar futuras estrategias terapéuticas y nuevos estudios clínicos en este campo. Un dato epidemiológico de gran valor procedente del estudio CANONIC es la elevada incidencia de ACLF. De los 1.343 pacientes ingresados en el hospital y reclutados en el estudio, 303 (22,6%) cumplían criterios de ACLF en el momento de la hospitalización y 112 (8,3%) lo desarrollaron en los primeros 28 días del ingreso. Así, este síndrome está presente en el momento del ingreso o aparece durante la hospitalización en 1 de cada 3 pacientes cirróticos descompensados que ingresan en el hospital. No hubo diferencias en la mortalidad a los 28 y 90 días entre los pacientes con ACLF al ingreso y los que lo desarrollaron durante el ingreso (el 33,9 frente al 29,7% y el 51,2 frente al 51,1%, respectivamente). Sin embargo, la mortalidad de los pacientes que desarrollaron ACLF durante el ingreso hospitalario fue muy superior a la de los pacientes que no lo desarrollaron, tanto a los 28 como a los 90 días Fracaso hepático agudo sobre crónico 47 (el 29,7 frente al 1,9% y el 51,1 frente al 9,7%), confirmando la marcada influencia pronóstica del diagnóstico de ACLF. Los enfermos con ACLF fueron más jóvenes, más frecuentemente alcohólicos y tuvieron más infecciones bacterianas asociadas; por otra parte, los pacientes con ACLF tuvieron un mayor grado de respuesta inflamatoria evidenciada por un significativo aumento en el número de leucocitos y un valor más elevado de proteína C reactiva (PCR). Desde el punto de vista de los factores asociados a la mortalidad, la presencia de una mayor puntuación CLIF-SOFA y el número de leucocitos fueron factores predictivos independientes de mortalidad en pacientes con ACLF. Inesperadamente, los pacientes sin historia previa de DA que desarrollaron ACLF presentaron un mayor número de FO, mayor número de leucocitos y PCR y mayor mortalidad comparados con los pacientes con historia previa de descompensación aguda (mortalidad a los 28 días del 42,2 frente al 29,6%; p = 0,03). Los factores precipitantes más frecuentes fueron el alcoholismo activo y la presencia de infección bacteriana; sin embargo, es muy interesante el hecho de que hasta en el 43,6% de los pacientes con ACLF en el momento del ingreso no se identificó ningún potencial factor precipitante. Este dato debe ser interpretado con cautela, porque la metodología del estudio no permitió obtener con precisión todos los potenciales factores predisponentes; así, no es posible descartar que la prevalencia de infecciones bacterianas hubiera sido más elevada si se hubieran utilizado técnicas más sensibles para su diagnóstico. Otra característica relevante de este síndrome es su marcado carácter dinámico, de forma que solamente un tercio de los pacientes no experimentó cambios en lo que respecta a la evaluación inicial de la gravedad. De hecho, aunque la gravedad inicial se asoció a la supervivencia, el curso clínico del paciente durante los 2 primeros días fue la variable más fuertemente asociada al pronóstico. Este dato puede ayudar de forma clara a la toma de decisiones de cara a la intensidad de la terapia a efectuar y a la eficacia o futilidad de esta. CARACTERÍSTICAS PATOGÉNICAS DEL FRACASO HEPÁTICO AGUDO SOBRE CRÓNICO (fig. 1) Un elemento predominante en la patogenia del ACLF es su asociación a infección bacteriana. De hecho, el 30% de los casos del estudio CANONIC se asociaron a infección bacteriana. La presencia de bacterias supone la expresión de diferentes patrones moleculares asociados a patógenos capaces de estimular receptores específicos celulares de reconocimiento de estos 48 Sesión 2 - CIRROSIS E HIPERTENSIÓN PORTAL ACLT Liberación aguda de PAMP (sepsis) Liberación aguda de DAMP (ACL injury) en cirrosis compensada o descompensada Reacción inflamatoria excesiva Inflamación sistémica grave Estrés oxidativo Extensión del estado inflamatorio y del estrés oxidativo a los órganos; descenso de la tolerancia orgánica Vasodilatación arterial Disfunción ventricular izquierda Descenso del gasto cardíaco Hipoperfusión de órganos Disfunción celular Fracaso de órganos ACLF I Renal Fracaso cerebral Fracaso circulatorio Fracaso respiratorio Coagulopatía Figura 1. Propuesta patogénica para el fracaso hepático agudo sobre crónico (ACLF). La exposición del sistema inmune a patrones moleculares asociados a infección bacteriana o a daño tisular genera mecanismos patogénicos capaces de producir daño tisular y, eventualmente, fracaso de órganos. DAMP: moléculas endógenas generadas por el daño y muerte celular; PAMP: patrones moleculares asociados a patógenos. patrones y que se asocian a la inducción de una gran cantidad de genes, incluyendo los que codifican para proteínas proinflamatorias. Se considera que la sobreproducción precoz de estos elementos resulta en daño tisular inmunomediado y fallo de órganos. Por otra parte, es evidente la posibilidad de producción de daño directamente mediado por patógenos cuya intensidad puede ser modulada por el mayor o menor grado de tolerancia a la agresión por parte del hospedador. Además, en un número significativo de pacientes que no presentan infección bacteriana evidente, la exposición del sistema inmune a productos bacterianos procedentes de la traslocación bacteriana puede promover igualmente respuestas inmunomediadas con daño tisular. Alternativamente, esta respuesta excesiva puede estar vinculada a la estimulación de los receptores de reconocimiento de patrones por parte de moléculas endógenas generadas por el daño y muerte celular. CONCLUSIONES La definición conceptual y patogénica del nuevo síndrome de ACLF puede suponer realmente un cambio relevante en la forma de entender la enfermedad hepática crónica descompensada. Sin embargo son precisos nuevos estudios encaminados a mejorar el conocimiento patogénico de la enfermedad y sus implicaciones clínicas y pronósticas. Bibliografía recomendada Arroyo V, Moreau R, Jalan R, Gines P. Acute-on-chronic liver failure: A new syndrome that will re-classify cirrhosis. J Hepatol. 2015;62:S131-43. Das V, Boelle PY, Galbois A, Guidet B, Maury E, Carbonell M, et al. Cirrhotic patients in the medical intensive care unit: early prognosis and logn term survival. Crit Care Med. 2010;38:2108-16. Jalan R, Gines P, Olson JC, Mookerjee R, Moreau R, García-Tsao G, et al. Acute on chronic liver failure. J Hepatol. 2012;57:1336-48. Jalan R, Williams R. Acute on chronic liver failure: pathophysiological basis of therapeutics options. Blood Purif. 2002;20:252-61. Medzhitov R, Schneider DS, Soares MP. Disease tolerance as a defense strategy. Science. 2012;335:936-41. Moreau R, Jalan R, Gines P, Pavesi M, Angeli P, Cordoba J, et al. Acute-on-chronic liver failure is a distinct syndrome that develops in patients with acute decompensation of cirrhosis. Gastroenterology. 2013;144:1426-37, 1437.e 1-9. Fracaso hepático agudo sobre crónico 49 Olson JC, Kamath PS. Acute on chronic liver failure: concept, natural history and prognosis. Curr Opin Crit Care. 2011;17: 165-9. Vincent JL, Moreno R, Takala J, Willatts S, De Mendonça A, Bruining H, et al. The SOFA (Sepsis-related-Organ Failure Assessment) score to describe organ dysfunction/failure. Intensive Care Med. 1996;22:707-10. Wehler M, Kokoska J, Reulbach U, Hahn EG, Strauss R. Short term prognosis in critically ill patients with cirrosis assessed by prognostic scoring systems. Hepatology. 2001;34:255-61. SESIÓN 2 CIRROSIS E HIPERTENSIÓN PORTAL Impacto de la encefalopatía hepática mínima en la morbimortalidad del paciente con cirrosis hepática Javier Ampuero Herrojo y Manuel Romero Gómez UGC Aparato Digestivo Intercentros, Hospitales Universitarios Virgen Macarena-Virgen del Rocío, Sevilla, España Instituto de Biomedicina de Sevilla, Sevilla, España ESPECTRO CLÍNICO DE LA ENCEFALOPATÍA HEPÁTICA La encefalopatía hepática (EH) se considera una alteración neuropsiquiátrica que tiene lugar en pacientes con disfunción hepática, tanto aguda como crónica, que es potencialmente reversible. La patogenia de la EH se relaciona principalmente con la hiperamoniemia, aunque han emergido factores coexistentes en su desarrollo, como son el incremento de la respuesta inflamatoria sistémica, la diabetes mellitus1 o la hiponatremia2. La EH se clasifica en distintos estadios (I-IV), según los criterios de West Haven3. En un estadio inicial (0), se situaría la EH mínima (EHM), que se encuentra presente en el 30-50% de los pacientes cirróticos (que no manifiesten evidencia clínica de EH) (tabla 1)4. La EHM se define como la presencia de alteraciones cognitivas, en pacientes con disfunción hepática, que no pueden ser detectadas en la exploración habitual. En esta enti- dad, además de déficit cognitivo, podemos encontrar enlentecimiento motor, alteraciones en la atención y la percepción visual y disminución de la destreza manual por una alteración de los movimientos finos. La presencia de EHM se asocia a una alteración de la calidad de vida y a una disminución de la supervivencia, como se desarrollará a continuación. DIAGNÓSTICO DE ENCEFALOPATÍA HEPÁTICA MÍNIMA El diagnóstico de la EH se realiza mediante la exploración clínica, sin necesidad de otros tests diagnósticos. En cambio, la EHM requiere la realización de algún test especial, ya que sus manifestaciones son imperceptibles en la simple exploración. La mayoría de los métodos diagnósticos se basan en el empleo de pruebas psicométricas y neurofisiológicas, por su sencillez y coste5. Tabla 1 Nivel de conciencia Síntomas neuropsiquiátricos Síntomas neurológicos Grado 0 (EHM) Normal Alteración solo advertida por tests psicométricos Ninguno Grado 1 Lentitud mental Distrofia, irritabilidad, ansiedad Alteración de la destreza manual Grado 2 Fatiga, apatía, letargia Trastorno de personalidad, leve desorientación temporoespacial Flapping, ataxia, bradilalia Grado 3 Somnolencia Agresividad, marcada desorientación temporoespacial Clonus, asterixis Grado 4 Coma No valorable Signos de hipertensión craneal EHM: encefalopatía hepática mínima. 50 Impacto de la encefalopatía hepática mínima en la morbimortalidad del paciente con cirrosis hepática Pruebas psicométricas Dentro de las pruebas psicométricas, el Psychometric Hepatic Encephalopathy Score (PHES) es capaz de detectar la mayoría de las alteraciones neuropsicológicas de la EHM6. Se trata de un test sencillo y con escaso coste. Evalúa la velocidad y precisión del movimiento, la concentración, la atención, la memoria, la orientación espacial y la percepción visual7. El PHES combina 5 tests: a) test de símbolos y números (DST); b) test de conexión numérica A (NCT-A); c) test de conexión numérica B (NCT-B); d) test de puntos seriados (SD), y e) test de la línea quebrada (LT) (fig. 1). La puntuación total oscila entre –15 y +3, siendo –4 puntos el límite que define la existencia de EHM8. Sin embargo, los resultados del PHES (y de los demás tests psicométricos existentes) se ven influidos por factores como la edad y el sexo o el nivel educativo de los pacientes, por lo que los resultados de estos pueden variar según la población sobre la que se aplican. se va ralentizando. El punto de corte es 39 Hz, de manera que valores superiores se consideran normales y valores inferiores patológicos (diagnósticos de EHM)11. La FCP, cuando es patológica, es capaz de predecir de forma independiente el riesgo de desarrollar EH clínica y la supervivencia de los pacientes con enfermedad hepática en estadio de cirrosis12. La ventaja de este test diagnóstico es que, a diferencia de los tests psicométricos, no se ve influenciado por la edad, el sexo o el nivel educativo de los pacientes. Tampoco está influido por el aprendizaje, por lo que es reproducible en el tiempo. Otro test neurofisiológico útil es la electroencefalografía, ya que los pacientes con EH muestran una actividad eléctrica cerebral disminuida. Sin embargo, este test presenta una sensibilidad muy variable (43-100%)13. Por último, los potenciales evocados (P300) también han mostrado cierta utilidad en el diagnóstico de la EHM, con una gran sensibilidad14. Pruebas de imagen Pruebas neurofisiológicas Además de los tests psicométricos, existen pruebas neurofisiológicas que aportan cierta objetividad al diagnóstico de la EHM9. Sin embargo, este tipo de pruebas precisa de aparataje específico, que no siempre es accesible o está disponible. La medición de la frecuencia crítica de parpadeo (FCP) se ha mostrado útil en el diagnóstico de la EHM y es una prueba fácil y rápida de realizar10. Esta prueba se basa en la fisiología de las células presentes en la retina, que muestran una alteración similar a la de los astrocitos cerebrales. Para la realización de la prueba se coloca el aparato en la cabeza del paciente y este debe pulsar un botón cuando nota un parpadeo en un punto luminoso en el interior de este. El parpadeo comienza a una frecuencia muy rápida y con el paso del tiempo Puntos seriados 51 Línea quebrada Se recomienda el uso de una prueba de imagen para descartar otras causas de alteración neuropsicológica en pacientes con disfunción hepática, si existen dudas acerca del diagnóstico. En cuanto a la detección de EHM, la resonancia magnética muestra cambios significativos en los pacientes con esta entidad. Mediante esta técnica se detecta una señal hiperintensa bilateral y simétrica a la altura de los ganglios basales (globo pálido y sustancia negra) en las imágenes potenciadas en T1. Los hallazgos encontrados en la resonancia magnética están en relación con depósitos de manganeso15. Con la espectroscopia por resonancia se puede objetivar el incremento de amonio cerebral y el estrés osmótico derivado de este. Esta técnica muestra un aumento de la glutamina y una disminución del mioinositol16. NCT-A NCT-B Figura 1. NCT-A: test de conexión numérica A; NCT-B: test de conexión numérica B. Clave de números 52 Sesión 2 - CIRROSIS E HIPERTENSIÓN PORTAL Biomarcadores Se ha estudiado poco acerca de la utilidad de los biomarcadores en el diagnóstico de la EHM. Un biomarcador periférico, la 3-nitrotirosina, parece que puede ser un buen indicador de la presencia de EHM. Este biomarcador se eleva ostensiblemente en los pacientes afectados de EHM, mostrando una sensibilidad del 89% y una especificidad del 93%17. Por tanto, la determinación de este biomarcador podría permitir el diagnóstico de esta enfermedad, aunque necesita ser validado en cohortes externas. MORBILIDAD ASOCIADA A LA ENCEFALOPATÍA HEPÁTICA MÍNIMA La EHM influye directamente en la calidad de vida de los pacientes18. La alteración neuropsicológica presente en estos pacientes impide incluso que, en ocasiones, completen los cuestionarios sobre calidad de vida. Las caídas accidentales y los accidentes de tráfico son 2 de las principales preocupaciones en estos enfermos. En los pacientes con EHM se ha observado un 40% de caídas frente a un 6-12% en los pacientes sin EHM19. Además de tener más caídas, los pacientes con EHM sufren caídas más graves y con mayor consumo de recursos sanitarios20. Este hecho se ve agravado en los pacientes cirróticos, ya que el riesgo de fracturas en ellos es superior al de la población general debido a que presentan mayor prevalencia de osteoporosis (por malnutrición, hipogonadismo e insuficiencia hepática)21. Por tanto, las caídas en estos pacientes son muy relevantes desde el punto de vista de la morbimortalidad. Por otra parte, los accidentes de tráfico también representan una causa de alta morbimortalidad con un alto coste socioeconómico en los pacientes que sufren EHM22. Se debe, principalmente, a la importante alteración en la atención, la concentración y los déficits motores y visuales presentes en esta entidad. Se ha documentado que los pacientes sin EHM presentan un 4% de accidentes de tráfico, mientras que los que sí la presentan tienen hasta 4 veces más riesgo (un 16%, aproximadamente)23. Se ha publicado, incluso, que el diagnóstico precoz de la EHM mediante un test computarizado y su tratamiento precoz con lactulosa representa una estrategia coste-efectiva, comparada con los costes directos e indirectos estimados de los accidentes de tráfico24. MORTALIDAD ASOCIADA A LA ENCEFALOPATÍA HEPÁTICA MÍNIMA Hay pocos estudios que hayan evaluado el impacto de la EHM en la supervivencia. Además, generalmente no han tenido en cuenta esta variable como objetivo primario y su diagnóstico ha sido heterogéneo. El primer estudio fue realizado por Amodio et al, que incluyeron 94 pacientes y evaluaron el deterioro cognitivo mediante NCT y tests psicométricos computarizados. Encontraron que Scan y Choice2 influían en el pronóstico de pacientes con cirrosis hepática (seguimiento medio de 14 meses)25. Un año después, Hartmann et al incluyeron 116 pacientes con cirrosis hepática (seguimiento medio de 29 meses) y evaluaron la EHM mediante NCT-A, DST y electroencefalograma. Los pacientes con EHM tuvieron más episodios de EH clínica, pero no presentaron peor tasa de supervivencia durante el seguimiento26. Posteriormente, nuestro grupo realizó un estudio prospectivo en el que se incluyeron 126 pacientes cirróticos; se evaluó la EHM mediante NCT, DST y BDT. Tras un seguimiento medio de 25 meses, los pacientes con EHM no presentaron un incremento de la mortalidad. Sin embargo, cuando el diagnóstico de EHM se combinó con la alteración de la sobrecarga oral de glutamina se observó un incremento significativo de mal pronóstico27. En 2010, Dhiman et al incluyeron 104 pacientes cirróticos, que fueron seguidos durante un período medio de 22 meses. Los autores evaluaron la EHM mediante PHES < –4 puntos. En el análisis univariante, la EHM diagnosticada mediante PHES se asoció a un incremento de la mortalidad (el 39 frente al 22%), aunque este hecho no fue confirmado en el análisis multivariante. Sin embargo, los autores seleccionaron otro punto de corte (PHES < –6 puntos) mediante un análisis ROC (receiver operating characteristic). Con este nuevo valor, la EHM sí se asoció a mortalidad en el análisis multivariante28. Finalmente, nuestro grupo realizó un nuevo estudio multicéntrico sobre el impacto de la EHM en la supervivencia de pacientes cirróticos. Se incluyeron 231 pacientes (117 cohorte de estimación y 114 de validación), cuyo seguimiento fue durante más de 5 años y el diagnóstico de EHM se realizó mediante PHES y FCP. Observamos que la FCP era capaz de predecir la supervivencia en ambas cohortes de manera independiente a otros factores. Además, observamos que los pacientes con FCP patológica (respecto a los que tenían FCP > 39 Hz) tenían menor supervivencia, tanto en pacientes con MELD 10-14 como con MELD > 1529. Las razones por las que pudimos encontrar una asociación relevante entre la EHM y la supervivencia radican en el mayor número de pacientes incluidos y en el mayor tiempo de seguimiento de los pacientes. CONCLUSIÓN La EHM es una entidad que se sitúa al inicio del espectro de la EH. En los últimos años ha crecido su importancia debido a su íntima relación con el mayor riesgo de caídas, el mayor número de accidentes y la bien conocida Impacto de la encefalopatía hepática mínima en la morbimortalidad del paciente con cirrosis hepática relación con episodios de EH clínica. Sin embargo, el impacto de la EHM en la supervivencia no ha sido bien estudiado y casi siempre se han obtenido resultados negativos. Recientemente se ha demostrado que esta entidad se asocia a una clara disminución de la supervivencia, condicionando claramente el pronóstico de la cirrosis hepática. Estos hallazgos hacen que la EHM sea una complicación derivada de la cirrosis hepática de obligada evaluación, ya que su aparición debe condicionar el manejo de estos pacientes, estableciendo una monitorización más cercana. Bibliografía 1.Ampuero J, Ranchal I, Del Mar Díaz-Herrero M, Del Campo JA, Bautista JD, Romero-Gómez M. Role of diabetes mellitus on hepatic encephalopathy. Metab Brain Dis. 2013;28:277-9. 2.Frederick RT. Current concepts in the pathophysiology and management of hepatic encephalopathy. Gastroenterol Hepatol. 2011;7:222-33. 3.Romero-Gómez M, Ampuero J. Deciphering the spectrum of lowgrade hepatic encephalopaty in clinical practice. Gastroenterology. 2014;146:887-90. 4.Maldonado-Garza HJ, Vázquez-Elizondo G, Gaytán-Torres JO, Flores-Rendón AR, Cárdenas-Sandoval MG, Bosques-Padilla FJ. Prevalence of minimal hepatic encephalopathy in cirrhotic patients. Ann Hepatol. 2011;10:40-4. 5.Dhiman RK, Chawla YK. Minimal hepatic encephalpathy: time to recognise and treat. Trop Gastroenterol. 2008;29:6-12. 6.Seo YS, Yim SY, Jung JY, Kim CH, Kim JD, Keum B, et al. Psychometric hepatic encephalopathy score for the detection of minimal hepatic encephalopathy in Korean patients with liver cirrhosis. J Gastroenterol Hepatol. 2012;27:1695-704. 7.Li SW, Wang K, Yu YQ, Wang HB, Li YH, Xu JM. Psychometric hepatic encephalopathy score for diagnosis of minimal hepatic encephalopathy in China. World J Gastroenterol. 2013;19:8745-51. 8.Romero Gómez M, Córdoba J, Jover R, Del Olmo J, Fernández A, Flavià M, et al; Red Nacional de Investigación en Encefalopatía Hepática. [Normality tables in the Spanish population for psychometric tests used in the diagnosis of minimal hepatic encephalopathy]. Med Clin (Barc). 2006; 127:246-9. 9.Randolph C, Hilsabeck R, Kato A, Kharbanda P, Li YY, Mapelli D, et al; International Society for Hepatic Encephalopathy and Nitrogen Metabolism (ISHEN). Neuropsychological assessment of hepatic encephalopathy: ISHEN practice guidelines. Liver Int. 2009;29:629-35. 10.Sharma P, Sharma BC, Sarin SK. Critical flicker frequency for diagnosis and assessment of recovery from minimal hepatic encephalopathy in patients with cirrhosis. Hepatobiliary Pancreat Dis Int. 2010;9:27-32. 11.Torlot FJ, McPhail MJ, Taylor-Robinson SD. Meta-analysis: The diagnostic accuracy of critical flicker frequency in minimal hepatic encephalopathy. Aliment Pharmacol Ther. 2013;37:527-36. 12.Romero-Gómez M, Córdoba J, Jover R, Del Olmo JA, Ramírez M, Rey R, et al. Value of the critical flicker frequency in patients with minimal hepatic encephalopathy. Hepatology. 2007;45:879-85. 53 13.Montagnese S, Amodio P, Morgan MY. Methods for diagnosing hepatic encephalopathy in patients with cirrhosis: a multidimensional approach. Metab Brain Dis. 2004;19:281-312. 14.Ciecko-Michalska I, Wojcik J, Wyczesany M, Binder M, Szewczyk J, Senderecka M, et al. Cognitive evoked response potentials in patients with liver cirrosis without diagnosis of minimal or overt hepatic encephalopathy. A pilot study. J Physiol Pharmacol. 2012;63:271-6. 15.Qi R, Zhang LH, Zhong J, Zhang Z, Ni L, Zheng G, et al. Disrupted thalamic resting-state funcional connectivity in patients with minimal hepatic encephalopathy. Eur J Radiol. 2013;82:850-6. 16.Sarma MK, Huda A, Nagarajan R, Hinkin CH, Wilson N, Gupta RK, et al. Multi-dimensional MR spectroscopy: towards a better understanding of hepatic encephalopathy. Metab Brain Dis. 2011;26:173-84. 17.Montoliu C, Cauli O, Urios A, ElMlili N, Serra MA, Giner-Duran R, et al. 3-nitro-tyrosine as a peripheral biomarker of minimal hepatic encephalopathy in patients with liver cirrhosis. Am J Gastroenterol. 2011;106:1629-37. 18.Bianchi G, Giovagnoli M, Sasdelli AS, Marchesini G. Hepatic encephalopathy and health-related quality of life. Clin Liver Dis. 2012;16:159-70. 19.Román E, Córdoba J, Torrens M, Torras X, Villanueva C, Vargas V, et al. Minimal hepatic encephalopathy is associated with falls. Am J Gastroenterol. 2011;106:476-82. 20.Soriano G, Román E, Córdoba J, Torrens M, Poca M, Torras X, et al. Cognitive dysfunction in cirrhosis is associated with falls: a prospective study. Hepatology. 2012;55:1922-30. 21.Savic Z, Damjanov D, Curic N, Kovacev-Zavisic B, Hadnadjev L, Novakovic-Paro J, et al. Vitamin D status, bone metabolism and bone mass in patients with alcoholic liver cirrhosis. Bratisl Lek Listy. 2014;115:573-8. 22.Bajaj JS, Hafeezullah M, Hoffmann RG, Saeian K. Minimal hepatic encephalopathy: a vehicle for accidents and traffic violations. Am J Gastroenterol. 2007;102:1903-9. 23.Bajaj JS, Saeian K, Schubert CM, Hafeezullah M, Franco J, Varma RR, et al. Minimal hepatic encephalopathy is associated with motor vehicle crashes: the reality beyond the driving test. Hepatology. 2009;50:1175-83. 24.Bajaj JS, Pinkerton SD, Sanyal AJ, Heuman DM. Diagnosis and treatment of minimal hepatic encephalopathy to prevent motor vehicle accidents: a cost-effectiveness analysis. Hepatology. 2012;55:1164-71. 25.Amodio P, Del Piccolo F, Marchetti P, Angeli P, Iemmolo R, Caregaro L, et al. Clinical features and survivial of cirrhotic patients with subclinical cognitive alterations detected by the number connection test and computerized psychometric tests. Hepatology. 1999;29:1662-7. 26.Hartmann IJ, Groeneweg M, Quero JC, Beijeman SJ, De Man RA, Hop WC, et al. The prognostic significance of subclinical hepatic encephalopathy. Am J Gastroenterol. 2000;95:2029-34. 27.Romero-Gómez M, Grande L, Camacho I. Prognostic value of altered oral glutamine challenge in patients with minimal hepatic encephalopathy. Hepatology. 2004;39:939-43. 28.Dhiman R, Kurmi R, Thumburu K, Venkataramarao SH, Agarwal R, Duseja A, et al. Diagnosis and prognostic significance of minimal hepatic encephalopaty in patients with cirrhosis of liver. Dig Dis Sci. 2010;55:2381-90. 29.Ampuero J, Simón M, Montoliú C, et al. minimal hepatic encephalopathy and critical flicker frequency are associated with survival of patients with cirrhosis. Gastroenterology. 2015 Aug 20. pii:S0016-5085(15)01176-2. SESIÓN 3 ENFERMEDADES METABÓLICAS Y SISTÉMICAS Diagnóstico no invasivo de cirrosis e hipertensión portal Salvador Augustin, Mónica Pons, Begoña Santos, Meritxell Ventura y Joan Genescà Medicina-Interna-Hepatología, Hospital Universitari Vall d’Hebron, Vall d’Hebron Institut de Recerca (VHIR), Universitat Autònoma de Barcelona, Barcelona, España Centro de Investigación Biomédica en Red de enfermedades hepáticas y digestivas (CIBERehd), Instituto de Salud Carlos III, Madrid, España Introducción El diagnóstico de cirrosis representa un hito de consecuencias prácticas determinantes en el manejo del paciente con enfermedad hepática crónica. Por un lado implica la búsqueda mediante endoscopia de varices esofágicas, cuya presencia condiciona la indicación de profilaxis primaria de hemorragia1,2. Así, las guías clínicas internacionales establecen que todos los pacientes cirróticos deben ser estudiados mediante endoscopia en el momento del diagnóstico para evaluar la presencia de varices2,3. Por otro lado, dichas guías también aconsejan medir el gradiente de presión venosa hepática (GPVH) en los centros donde sea posible dado su valor pronóstico y para evaluar la respuesta al tratamiento bloqueador beta. Finalmente, el diagnóstico de cirrosis implica iniciar el cribado periódico y permanente del desarrollo de hepatocarcinoma4. El diagnóstico clásico de cirrosis es histológico y viene definido por la presencia de nódulos de regeneración rodeados de tejido fibroso, dando lugar a una distorsión de la angioestructura hepática. La biopsia hepática ha sido, consecuentemente, el “estándar de oro” en la evaluación de la gravedad de la fibrosis y en el diagnóstico de la cirrosis. Sin embargo, las limitaciones del procedimiento son ampliamente conocidas (invasividad, complicaciones, error de muestreo, etc.)5,6. Por los mismos motivos, la biopsia hepática no es adecuada para la monitorización continua y dinámica de la progresión de la enfermedad hepática. Además de la biopsia hepática, y desde un punto de vista práctico, la cirrosis puede diagnosticarse también desde un punto de vista clínico por la presencia de una combinación de signos físicos (ascitis, ictericia, arañas 54 vasculares, bazo palpable, etc.), bioquímicos (plaquetas bajas, alteración de pruebas hepáticas), de imagen (hígado nodular, esplenomegalia, circulación colateral) y/o endoscópicos (varices). Esta definición práctica es de uso común por los hepatólogos, pero la sensibilidad y especificidad de estos criterios clínicos es variable y en ocasiones son subóptimos. Así, no existe un consenso claro sobre cómo integrar estos criterios en una definición precisa y útil. Finalmente, la cirrosis puede diagnosticarse indirectamente a través del diagnóstico de hipertensión portal. El desarrollo de la hipertensión portal es un evento crucial en la evolución de la cirrosis. La hipertensión portal puede cuantificarse mediante el GPVH. La medición del GPVH es una técnica que aporta una valiosa información pronóstica en los pacientes con cirrosis. Para valores de GPVH < 10 mmHg, el riesgo de desarrollo de varices o descompensación clínica es muy bajo. Una vez el GPVH pasa a ser > 10 mmHg, el paciente con cirrosis entra en riesgo de desarrollar varices y descompensación clínica7-9. Por ello, valores de GPVH ≥ 10 definen lo que se conoce como hipertensión portal clínicamente significativa (HPCS). La técnica de medición del GPVH es precisa y reproducible, pero nuevamente tiene el problema de que es invasiva y, dados sus requerimientos técnicos, solo está disponible en unos pocos centros especializados. A pesar del valor de esta prueba, la realidad es que el GPVH no se ha incorporado a la práctica clínica habitual en la gran mayoría de hospitales del mundo. Por las limitaciones expuestas anteriormente, en los últimos años se han desarrollado y evaluado intensivamente nuevas alternativas diagnósticas para determinar de forma no invasiva la presencia de fibrosis hepática, cirrosis, HPCS y varices. La aparición, la evaluación y posterior generalización del uso de algunos de estos métodos diagnósticos están cambiando a pasos acelerados el escenario clínico de la enfermedad hepática crónica. En particular han proporcionado una nueva perspectiva sobre la historia natural de la enfermedad en las primeras fases del proceso. Este cambio de perspectiva ha venido dado por la capacidad de estas técnicas de identificar pacientes con enfermedad significativa entre aquellos en los que por criterios clínicos no se sospechaba. Estos pacientes previamente no detectados están entrando ahora en la categoría operacional de pacientes cirróticos compensados, ya que se les asocian los riesgos propios de la cirrosis (a pesar de que se estima que al menos un 10-15% de ellos no tiene cirrosis por histología)10. El diagnóstico de “cirrosis” en este nuevo y numeroso grupo de pacientes implica, por tanto, la decisión de iniciar la detección de varices, HPCS y hepatocarcinoma, lo que puede conllevar un importante aumento en el uso de procedimientos innecesarios11. Para enmarcar adecuadamente y describir esta nueva situación clínica en la enfermedad hepática crónica y proporcionar recomendaciones, se ha propuesto utilizar el término operativo de enfermedad hepática crónica avanzada compensada (cACLD, de sus siglas en inglés compensated advanced chronic liver disease). Dicho término engloba los pacientes con cirrosis hepática diagnosticada mediante técnicas clásicas (biopsia, signos evidentes de hipertensión portal en pruebas de imagen, varices, descompensación clínica), así como aquellos con enfermedad significativa detectada por técnicas no invasivas y antes de la aparición de los signos clásicos. La incorporación de la información proporcionada por pruebas no invasivas en la definición de cACLD podría ser útil para la optimización del manejo práctico del paciente con cirrosis compensada, y así se ha recogido en la reciente conferencia internacional de consenso en hipertensión portal de Baveno9. Papel de la elastografía en el cambio epidemiológico en enfermedad hepática crónica avanzada compensada En los últimos años se han explorado diferentes estrategias para evaluar la utilidad de diferentes métodos no invasivos en la evaluación del grado de fibrosis hepática y, en consecuencia, el reconocimiento de la cirrosis, las varices o la HPCS. Entre las diferentes modalidades se incluyen biomarcadores séricos (directos e indirectos) de fibrosis, así como métodos físicos. Del conjunto de todos ellos, la que ha logrado sin duda una mayor aceptación y de la que se va a hablar más Diagnóstico no invasivo de cirrosis e hipertensión portal 55 extensamente en el presente capítulo es la elastografía de transición (ET) hepática mediante FibroScan® (Echosens, París, Francia). Esto se debe, por una parte, a su uso relativamente sencillo y con resultados cuantificables directos y rápidos y, por otra, porque ha demostrado proporcionar un excelente rendimiento en la detección de la cirrosis y la exclusión de la fibrosis significativa, así como en la monitorización en el tiempo de la progresión de la enfermedad10,12. Por todo ello, la ET se ha incorporado como una herramienta valiosa en la evaluación de la enfermedad hepática crónica en muchos centros, sobre todo en Europa. Esto ha hecho que el número de biopsias hepáticas con fines de estadificación se haya reducido sustancialmente en muchos hospitales. El impacto de la ET en el cambio de la epidemiología de las enfermedades crónicas del hígado se puede ilustrar de diferentes maneras. En este sentido, cabe destacar el hecho de que la ET es capaz de revelar la presencia de cACLD en pacientes con enfermedad hepática crónica en los que el médico no sospechaba de ella por criterios clásicos. Esta circunstancia ha implicado un aumento sustancial en el número de pacientes que necesita un seguimiento más estrecho (fig. 1). El análisis de estudios de cribado disponibles realizados con ET en poblaciones sanas no seleccionadas puede ayudar a precisar la prevalencia de cACLD en la población general. En un estudio francés realizado en más de 1.000 individuos sanos, la prevalencia de mediciones de la rigidez hepática (MRH) ≥ 8 kPa (compatibles con una sospecha de fibrosis avanzada) fue del 7,5%. De estos, el 10% (el 0,8% de la población total) tenía una MRH > 13 kPa, sugestiva ya de la presencia de cirrosis13. En la cohorte de Rotterdam, con 1.324 participantes mayores de 65 años, la prevalencia de MRH > 9,5 kPa y > 13 kPa fue del 4,2 y el 1,1%, respectivamente14. Estudios similares realizados en poblaciones de Asia, incluyendo a más de 3.000 personas, muestran MRH indicativas de fibrosis avanzada o cirrosis en el 1-2% de los individuos15-17. La enfermedad por hígado graso no alcohólico fue la etiología predominante de la enfermedad hepática en todos estos estudios. Una manera diferente de analizar la importancia de la ET para revelar cACLD oculta es mediante el estudio sistemático de series de pacientes con enfermedad hepática crónica sin signos clínicos que hicieran sospechar la presencia de cirrosis (plaquetas normales y ecografía abdominal). Resultados de Barcelona18, Montreal19 y Seúl20 indican una prevalencia del 8 al 14% de los pacientes con MRH ≥ 13-13,6 kPa en estas cohortes (fig. 2A). Estos pacientes con cACLD oculta llegan a representar el 24-37% del número total de pacientes con MRH ≥ 13-13,6 kPa cuando se analizan conjuntamente datos de pacientes incluidos en las cohortes de Montreal y del estudio ANTICIPATE21 (fig. 2B). Este último es un estudio cooperativo (Edmonton, Barcelona, 56 Sesión 3 - ENFERMEDADES METABÓLICAS Y SISTÉMICAS ELASTOGRAFÍA DE TRANSICIÓN BIOPSIA HEPÁTICA DIAGNÓSTICO CLÍNICO Figura 1. Representación del impacto epidemiológico de la elastografía de transición a través de la identificación de pacientes con enfermedad hepática crónica avanzada o cirrosis, comparado con el de la biopsia hepática y el del diagnóstico clínico de cirrosis. Toulouse, Cluj-Napoca) dirigido a evaluar estrategias diagnósticas no invasivas para identificar el riesgo de HPCS y varices en pacientes con cirrosis hepática compensada confirmada o sospechada por criterios clásicos. Los datos disponibles de este estudio solo han sido comunicados hasta la fecha en forma de comunicación a congresos internacionales. En cualquier caso, considerando el conjunto de todos estos datos, los pacientes con cACLD oculta representan una parte sustancial de los pacientes en los diferentes estudios. Por último cabe mencionar que en la cohorte de Montreal19, los pacientes con cACLD habían recibido significativamente menor vigilancia (cribado de varices o hepatocarcinoma) que los pacientes con cirrosis hepática clínicamente evidente. Esto dio lugar a una mayor tasa de diagnóstico tardío (carcinoma hepatocelular avanzado, hemorragia por varices). Los resultados de este estudio observacional avisan sobre la posibilidad de que los pacientes con cACLD oculta estén infradiagnosticados y peor monitorizados que aquellos con cirrosis hepática evidente. ¿Cuál es la utilidad de considerar el concepto de enfermedad hepática crónica avanzada compensada? Cuando un paciente con enfermedad hepática crónica presenta varices esofágicas o desarrolla una descompensación clínica (ascitis, hemorragia varicosa, encefalopatía, ictericia) puede asumirse que el paciente tiene una cirrosis y HPCS. Sin embargo, la presencia o no de la cirrosis en sus etapas iniciales puede no resultar fácil, ya que el espectro de la fibrosis avanzada y cirrosis es en realidad un proceso continuo. El nuevo término de lo que hemos llamado cACLD podría ser útil en este contexto. Este nuevo concepto clínico sería útil por varias razones: a) para enmarcar adecuadamente una situación clínica en función del pronóstico (riesgo de eventos clínicos) esperado; b) para seleccionar a los pacientes para estudios clínicos y terapéuticos, y c) para proporcionar recomendaciones para el cribado de hepatocarcinoma, varices y HPCS. Diagnóstico no invasivo de cirrosis e hipertensión portal 57 A. ENFERMEDAD HEPÁTICA CRÓNICA SIN SIGNOS APARENTES DE CIRROSIS *Pacientes con elastografía hepática ≥ 13-13,6 kPa n = 173 Augustin, et al n = 702 Chen, et al n = 2.876 Kim, et al B. PACIENTES CON ELASTOGRAFÍA HEPÁTICA ≥ 13-13,6 kPa *Pacientes con cACLD oculta (sin signos aparentes de cirrosis) n = 54 Augustin, et al n = 270 Chen, et al n = 221 ANTICIPATE Figura 2. A) Prevalencia de pacientes con elastografía hepática ≥ 13-13,6 kPa en 3 series de pacientes con enfermedad hepática crónica sin sospecha de cirrosis (plaquetas normales y la ecografía abdominal). B) Prevalencia de enfermedad hepática crónica avanzada compensada (cACLD) oculta (plaquetas y ecografía normales) en 3 series de pacientes con elastografía hepática ≥ 13-13,6 kPa. El fundamento del uso de cACLD, que incluiría tanto a los pacientes con fibrosis grave como a aquellos con cirrosis compensada, especialmente en las primeras etapas, podría ser el siguiente: • La cirrosis hepática tal y como la conocemos es un diagnóstico histológico. • La cirrosis no está presente histológicamente en todos los pacientes clasificados como portadores de fibrosis avanzada por métodos no invasivos. • No existe un consenso preciso explícito sobre la definición clínica de la cirrosis hepática. • Los pacientes en etapas precirróticas pueden tener hipertensión portal22,23. • Las pruebas no invasivas han cambiado la situación clínica de la enfermedad hepática crónica. • El cribado de hepatocarcinoma puede estar indicado en las etapas precirróticas4,24. Un paciente con cACLD sería un paciente con enfermedad hepática crónica con signos de fibrosis hepática avanzada o un paciente con cirrosis hepática compensada (con o sin signos de hipertensión portal). La identificación de un paciente con cACLD implicaría la derivación por médicos de atención primaria a un especialista en enfermedades del hígado para el seguimiento y decidir sobre cribado de HPCS, varices y hepatocarcinoma. En la última reunión de consenso de Baveno se propusieron valores de ET de referencia orientativos9. En esencia, un MRH < 10 kPa (alto valor predictivo negativo) excluirá cACLD, y valores > 15 kPa (alto valor predictivo positivo) serán altamen- 58 Sesión 3 - ENFERMEDADES METABÓLICAS Y SISTÉMICAS te indicativos de cACLD. Para los valores de ET entre estos 2 puntos (“zona gris”) se necesitarían pruebas adicionales. ¿En qué pacientes con enfermedad hepática crónica avanzada compensada se puede evitar la endoscopia de cribado de varices? Uno de los principales retos que conlleva la detección cACLD por métodos no invasivos es la gran cantidad de endoscopias innecesarias que podrían llegar a realizarse en pacientes con un riesgo muy bajo de presentar varices11. El uso de ET como predictor no invasivo de varices se ha evaluado en varios estudios. En general, los estudios muestran que la ET es más fiable para descartar (alta sensibilidad y valor predictivo negativo) que para confirmar la presencia de varices12,18,25,26. Sin embargo, la heterogeneidad en los resultados y puntos de corte propuestos han dificultado hasta ahora un uso extensivo en la práctica clínica. Ya que la ET es más útil para descartar varices, resulta crucial consensuar cuál sería un riesgo “aceptable” de omisión del diagnóstico de varices cuando se utiliza esta técnica a efectos de cribado. Para varices que implican necesidad de iniciar tratamiento (VNT: medianas o grandes o de cualquier tamaño con puntos rojos), el riesgo de omisión ha de ser lo más cercano al 0%, aceptando una variabilidad de un 5% como máximo. En el caso de analizar la presencia de varices de cualquier tamaño (incluyendo las que no requieren tratamiento), un riesgo del 20% de omisiones diagnósticas podría ser aceptable (ya que el riesgo real que se quiere evitar, el de una hemorragia varicosa, sería mucho menor). El rendimiento diagnóstico parece mejorar cuando la elastografía se combina con otros parámetros clínicos sencillos, incluyendo principalmente las plaquetas y el tamaño del bazo. El índice LSPS (que incluye MRH, diámetro esplénico y plaquetas)27,28 o el VRS (Varices Risk Score)29 son muy buenos ejemplos de esta estrategia, y ambos se comportan mejor que la MRH en detección de varices. También en el estudio de ANTICIPATE, el LSPS fue el mejor predictor de varices en un análisis de modelos de predicción de riesgo21. Sin embargo, estos índices basados en combinación de pruebas no invasivas requieren un cálculo más o menos complejo y la memorización de dinteles adimensionales y poco intuitivos para aplicar a la práctica clínica diaria. Así, se han propuesto como alternativa práctica el uso de reglas de clasificación algorítmicas simples y visuales, que podrían ser más fácilmente implementables. En la tabla 1 se resumen 3 estudios que utilizan una combinación secuencial de dinteles de ET y plaquetas18,30,31. Además, en la tabla también se muestra la validación de las reglas de clasificación de estos estudios en la cohorte del estudio ANTICIPATE. Los resultados indican que el uso de una combinación de MRH (con un punto de corte de 25 kPa) y recuento de plaquetas (con puntos de corte entre 100 y 150 × 103/µl) permitiría evitar un 20-40% de endoscopias en estos pacientes, con un riesgo aceptable de omisión de VNT bajo (5% en el peor de los casos). La simplicidad de uso de esta regla de clasificación podría permitir a los médicos diferir la realización de endoscopias durante la visita al paciente y, por lo tanto, facilitar la incorporación de la clasificación a la práctica clínica. ¿En qué pacientes con enfermedad hepática crónica avanzada compensada podría asumirse la presencia de hipertensión portal clínicamente significativa? De manera similar a la detección de varices, la ET también se ha utilizado para predecir HPCS. La detección de pacientes de muy alto riesgo de tener HPCS podría ser de utilidad para evaluar el riesgo de descompensación clínica, para seleccionar pacientes para estudios clínicos y/o para indicar la terapia profiláctica empírica de descompensación (si diferentes estudios en marcha y previstos al respecto demuestran finalmente la utilidad de esta profilaxis). En este sentido, parece bastante claro que la ET no es capaz de predecir con suficiente precisión el valor numérico de GPVH y, por tanto, sería poco adecuada para la monitorización de cambios de GPVH. La ET ha demostrado consistentemente obtener excelentes resultados a la hora de confirmar (alta especificidad y valor predictivo positivo) la presencia de HPCS, aunque es menos fiable a la hora de descartarla12,18,25,26. En estos estudios, puntos de corte MRH de 25 kPa se asociaban a valores predictivos positivos de HPCS > 90%. Una vez más, los datos del estudio ANTICIPATE21 confirman que en la subcohorte de pacientes con una MRH ≥ 25 kPa (un 37% de la cohorte total), el 96% de estos pacientes tenían HPCS (fig. 3). Conclusiones La reciente introducción del término cACLD puede ayudar a clarificar el escenario clínico cambiante de la cirrosis en sus fases más tempranas. El término cACLD englobaría tanto a los pacientes con cirrosis hepática compensada confirmada mediante técnicas clásicas, como a aquellos con enfermedad significativa (fibrosis avanzada o cirrosis) detectada por técnicas no invasivas y antes de la aparición de los signos clásicos. El uso de reglas de clasificación clínicas sencillas podría Diagnóstico no invasivo de cirrosis e hipertensión portal 59 Tabla 1. Resumen de los resultados para descartar presencia de varices de los estudios que utilizan una combinación de medición de la rigidez hepática (MRH) y plaquetas N Augustin et al18 49 Montes et al30 85 Varices (todas) 10 45* VNT (%) Regla de clasificación 0 20 Varices (todas) (%) VNT (%) Varices (%) VNT (%) Endoscopias evitables (%) VPN (%) VPN (%) No detectadas (%) No detectadas (%) MRH < 25 93 100 7 0 61 MRH < 25 + plaquetas ≥ 150 100 100 0 0 20 MRH < 20 90 – 9,5 – 25 MRH < 20 y/o plaquetas > 120 100 100 0 0 15 Ding et al31 272 – – MRH < 25 + plaquetas ≥ 100 – 100 – 0 42 ANTICIPATE20 379 42 15 MRH < 25 + plaquetas ≥ 100 79 95 21 5 45 MRH < 25 + plaquetas ≥ 150 86 96,5 14 3,5 23 VNT: varices que necesitan tratamiento; VPN: valor predictivo negativo. MRH en kPa; plaquetas: × 103/µl. *Incluye varices y gastropatía portal. 393 pacientes 182 GPVH HPCS 109 (60%) MRH < 25 kPa MRH ≥ 25 kPa N = 115 (63%) HPCS = 45 (39%) N = 67 (37%) HPCS = 64 (96%) Plq ≥ 150.000 Plq < 150.000 Plq ≥ 150.000 Plq < 150.000 N = 46 (25%) HPCS = 8 (17%) N = 69 (38%) HPCS = 37 (54%) N = 17 (9%) HPCS = 16 (94%) N = 50 (28%) HPCS = 48 (96%) Figura 3. Evaluación de la presencia de hipertensión portal clínicamente significativa (HPCS) en la cohorte del estudio ANTICIPATE utilizando mediciones de rigidez hepática (MRH) mediante elastografía de transición y recuento plaquetario (Plq). GPVH: gradiente de presión venosa hepática. 60 Sesión 3 - ENFERMEDADES METABÓLICAS Y SISTÉMICAS ser útil para evitar endoscopias y determinaciones de GPVH innecesarias en estos pacientes. Con la combinación de MRH < 25 kPa y recuento de plaquetas ≥ 100 × 103/µl podría evitarse hasta un 40-45% de las endoscopias de cribado de varices, asumiendo un riesgo relativamente bajo (5%) de omitir el diagnóstico de varices tributarias de tratamiento. Del mismo modo, en los pacientes con MRH ≥ 25 kPa se puede asumir una HPCS y en aquellos con MRH < 25 kPa y plaquetas normales se puede asumir su ausencia. Con estas reglas clínicas podría evitarse un número sustancial de procedimientos invasivos innecesarios en estos pacientes. Bibliografía international consensus workshop. Oxford: Wiley-Blackwell; 2011. p. 18-27. 13.Roulot D, Costes JL, Buyck JF, Warzocha U, Gambier N, Czernichow S, et al. Transient elastography as a screening tool for liver fibrosis and cirrhosis in a community-based population aged over 45 years. Gut. 2011;60:977-84. 14.Koehler EM, Schouten JNL, Hansen BE, Stricker BH, Castera L, Janssen HLA. Prevalence and risk factors of severe fibrosis in the elderly: transient elastography in a population-based study. Autumn meeting of the Dutch Society of Hepatology, Zeist. Abstract book. Oct 4-5 2012; p. 13. 15.Wong VW, Chu WC, Wong GL, Chan RS, Chim AM, Ong A, et al. Prevalence of non-alcoholic fatty liver disease and advanced fibrosis in Hong Kong Chinese: a population study using proton-magnetic resonance spectroscopy and transient elastography. Gut. 2012;61:409-15. 1.D’Amico G, Garcia-Tsao G, Pagliaro L. Natural history and prognostic indicators of survival in cirrhosis: a systematic review of 118 studies. J Hepatol. 2006;44:217-31. 16.Fung J, Lee CK, Chan M, Seto WK, Lai CL, Yuen MF; Hong Kong Liver Health Census Study Group. High prevalence of nonalcoholic fatty liver disease in the Chinese – results from the Hong Kong liver health census. Liver Int. 2015;35:542-9. 2.De Franchis R; Baveno V Faculty. Revising consensus in portal hypertension: report of the Baveno V consensus workshop on methodology of diagnosis and therapy in portal hypertension. J Hepatol. 20101;53:762-8. 17.You SC, Kim KJ, Kim SU, Kim BK, Park JY, Kim do Y, et al. Factors associated with significant liver fibrosis assessed using transient elastography in general population. World J Gastroenterol. 2015;21:1158-66. 3.Garcia-Tsao G, Sanyal AJ, Grace ND, Carey W; Practice Guidelines Committee of the American Association for the Study of Liver Diseases; Practice Parameters Committee of the American College of Gastroenterology. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Hepatology. 2007;46:922-38. 18.Augustin S, Millán L, González A, Martell M, Gelabert A, Segarra A, et al. Detection of early portal hypertension with routine data and liver stiffness in patients with asymptomatic liver disease: a prospective study. J Hepatol. 2014;60:561-9. 4.European Association For The Study Of The Liver; European Organisation For Research And Treatment Of Cancer. EASLEORTC clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol. 2012;56:908-43. 5.Bedossa P, Dargère D, Paradis V. Sampling variability of liver fibrosis in chronic hepatitis C. Hepatology. 2003;38:1449-57. 6.Bedossa P, Carrat F. Liver biopsy: the best, not the gold standard. J Hepatol. 2008;50:1-3. 7.Groszmann RJ, Garcia-Tsao G, Bosch J, Grace ND, Burroughs AK, Planas R, et al; Portal Hypertension Collaborative Group. Beta-blockers to prevent gastroesophageal varices in patients with cirrhosis. N Engl J Med. 2005;353:2254-61. 8.Ripoll C, Groszmann R, Garcia-Tsao G, Grace N, Burroughs A, Planas R, et al; Portal Hypertension Collaborative Group. Hepatic venous pressure gradient predicts clinical decompensation in patients with compensated cirrhosis. Gastroenterology. 2007;133:481-8. 9.De Franchis R; Baveno VI Faculty. Updating consensus in portal hypertension: report of the Baveno III. Expanding consensus in portal hypertension: Report of the Baveno VI Consensus Workshop: Stratifying risk and individualizing care for portal hypertension. J Hepatol. 2015;63:743-52. 10.Castera L. Noninvasive methods to assess liver disease in patients with hepatitis B or C. Gastroenterology. 2012;142:1293302. 11.Rudler M, Benosman H, Lebray P, Nghiem D, Ngo Y, Munteanu M, et al. Screening for esophageal varices in patients newly diagnosed in 2011: 84% of upper gastrointestinal endoscopies are futile. Hepatology. 2011;54 Suppl:935A. 12.Castera L. Elastography in the non-invasive evaluation of the extent of fibrosis and in the diagnosis of portal hypertension. En: De Franchis R, editor. Proceedings of the fifth Baveno 19.Chen T, Wong R, Wong P, Rollet-Kurhajec KC, Alshaalan R, Deschenes M, et al. Occult cirrhosis diagnosed by transient elastography is a frequent and under-monitored clinical entity. Liver Int. 2015; doi:10.1111/liv.12802. [Epub ahead of print]. 20.Kim MN, Kim SU, Kim BK, Park JY, Kim DY, Ahn SH, et al. Increased risk of hepatocellular carcinoma in chronic hepatitis B patients with transient elastography defined subclinical cirrhosis. Hepatology. 2015; doi:10.1002/hep.27735. [Epub ahead of print]. 21.Abraldes J, Bureau C, Stefanescu H, Augustin S, Ney M, Procopet B, et al. Non-invasive tools and risk of varices and clinically significant portal hypertension in compensated cirrhosis: the “ANTICIPATE” Study. J Hepatol. 2015;62 Suppl 2:S198-9. 22.Krogsgaard K, Gluud C, Henriksen JH, Christoffersen P. Correlation between liver morphology and portal pressure in alcoholic liver disease. Hepatology. 1984;4:699-703. 23.Van Leeuwen DJ, Howe SC, Scheuer PJ, Sherlock S. Portal hypertension in chronic hepatitis: relationship to morphological changes. Gut. 1990;31:339-43. 24.Rinella ME, Sanyal AJ. NAFLD in 2014: genetics, diagnostics and therapeutic advances in NAFLD. Nat Rev Gastroenterol Hepatol. 2015;12:65-6. 25.Castera L, Pinzani M, Bosch J. Non invasive evaluation of portal hypertension using transient elastography. J Hepatol. 2012;56:696-703. 26.Shi KQ, Fan YC, Pan ZZ, Lin XF, Liu WY, Chen YP, et al. Transient elastography: a meta-analysis of diagnostic accuracy in evaluation of portal hypertension in chronic liver disease. Liver Int. 2013;33:62-71. 27.Kim BK, Han KH, Park JY, Ahn SH, Kim JK, Paik YH, et al. A liver stiffness measurement based, noninvasive prediction model for high-risk esophageal varices in B-viral liver cirrhosis. Am J Gastroenterol. 2010;105:1382-90. 28.Colecchia A, Montrone L, Scaioli E, Bacchi-Reggiani ML, Colli A, Casazza G, et al. Measurement of spleen stiffness to evaluate portal hypertension and the presence of esophageal varices in patients with HCV-related cirrhosis. Gastroenterology. 2012;143:646-54. 29.Berzigotti A, Seijo S, Arena U, Abraldes JG, Vizzutti F, García-Pagán JC, . Elastography, spleen size, and platelet count identify portal hypertension in patients with compensated cirrhosis. Gastroenterology. 2013;144:102-11. 30.Montes Ramírez ML, Pascual-Pareja JF, Sánchez-Conde M, Bernardino de la Serna JI, Zamora Vargas FX, Miralles P, Diagnóstico no invasivo de cirrosis e hipertensión portal 61 et al. Transient elastography to rule out esophageal varices and portal hypertensive gastropathy in HIV-infected individuals with liver cirrhosis. AIDS. 2012;26:1807-12. 31.Ding NS, Nguyen T, Iser DM, Hong T, Flanagan E, Wong A, et al. Liver stiffness plus platelet count can be used to exclude high risk oesophageal varices. Liver Int. 2015; doi:10.1111/ liv.12916. [Epub ahead of print]. 32.Kitson MT, Roberts SK, Colman JC, Paul E, Button P, Kemp W. Liver stiffness and the prediction of clinically significant portal hypertension and portal hypertensive complications. Scand J Gastroenterol. 2015;50:462-9. SESIÓN 3 ENFERMEDADES METABÓLICAS Y SISTÉMICAS Patogenia de la enfermedad hepática por depósito de grasa no alcohólica María Teresa Arias Loste y Javier Crespo Servicio de Digestivo, Hospital Universitario Marqués de Valdecilla, Instituto de Investigación Marqués de Valdecilla (IDIVAL), Santander, España INTRODUCCIÓN La enfermedad hepática por depósito de grasa engloba un amplio espectro de lesiones hepáticas cuyo denominador común es la presencia de un acúmulo ectópico de grasa en el hígado conocido como esteatosis. Aunque la asociación existente entre obesidad y diabetes mellitus con la presencia de esteatosis es bien conocida desde principios del siglo pasado, no es hasta 1980 cuando aparecen las primeras referencias a una forma de esteatohepatitis que, aunque remeda a la presente en el paciente alcohólico, aparece en sujetos sin este antecedente. La prevalencia de la enfermedad hepática por depósito de grasa (EHDG) ha experimentado un crecimiento exponencial, convirtiéndose en la primera causa de hepatopatía en los países desarrollados, aumento paralelo a la epidemia de obesidad y síndrome metabólico. La Organización Mundial de la Salud estima que, en el año 2014, más de 1.900 millones de adultos de 18 o más años tenían sobrepeso, de los cuales más de 600 millones eran obesos. Estos datos ponen de manifiesto un problema global de salud pública que se va a traducir en los próximos años en un aumento muy importante en la incidencia de la patología crónica asociada a la obesidad, siendo la EHDG el reflejo de esta en el hígado. La EHDG presenta una fuerte asociación con la obesidad, la resistencia a la insulina (RI) y el síndrome metabólico. Además, la presencia de RI se relaciona con la gravedad de la lesión hepática, la presencia de esteatohepatitis, cirrosis e incluso el desarrollo de complicaciones como el hepatocarcinoma. Por otro lado, en los pacientes con EHDG es frecuente la presencia de dislipemia, fundamentalmente en forma de hipertrigliceridemia y bajas concentraciones de colesterol unido a lipoproteínas de alta densidad. De manera global, un 80% de los casos de EHDG presenta alguno de los factores de riesgo cardiovascular 62 que constituyen el síndrome metabólico (RI, obesidad, dislipemia e hipertensión arterial), y la prevalencia de EHDG aumenta de manera directa según el número de estos factores que estén presentes. Además, cabe destacar la asociación entre EHDG y el síndrome de apneas-hipopneas del sueño, el síndrome del ovario poliquístico y endocrinopatías como el hipotiroidismo, el hipopituitarismo y el hipogonadismo. ETIOPATOGENIA El acúmulo de grasa intrahepatocitario —en forma predominantemente de triglicéridos— es la piedra angular de la EHDG y, por lo general, tiene un curso benigno. En menos de un 25% de los casos, este depósito se acompaña de un grado variable de inflamación, con el consiguiente riesgo de progresión de la enfermedad hepática con desarrollo de fibrosis, cirrosis e incluso hepatocarcinoma. Por qué unos casos progresan a formas más graves y otros permanecen estables en lo que se denomina una esteatosis simple es objeto de estudio en la actualidad. En 1998, Day desarrolló la teoría del “doble impacto” en la patogenia de la EHDG. Según esta teoría, el desarrollo de la enfermedad seguiría 2 etapas; una inicial con un primer impacto, con un acúmulo excesivo de triglicéridos en el hepatocito, que supondría el desarrollo de esteatosis. Tras este primer hecho, una segunda noxa, como podría ser la presencia de una endotoxemia de origen intestinal, sería la responsable del desarrollo de inflamación y daño celular. Una década más tarde, Herbert Tilg desarrolló una teoría patogénica diferente en la cual se postula que la inflamación y el daño hepatocelular pueden llegar a preceder al desarrollo de esteatosis en el tiempo, respondiendo este hecho a la aparición de múltiples noxas que incidirían sobre el hígado de manera paralela. Patogenia de la enfermedad hepática por depósito de grasa no alcohólica Estas noxas incluirían diferentes mediadores procedentes fundamentalmente de 2 lugares: el intestino y el tejido adiposo. Esta teoría, conocida como de “impactos múltiples paralelos”, es ampliamente aceptada en la actualidad y abre el debate acerca de la posibilidad de considerar la esteatosis simple y la esteatohepatitis no alcohólica (EHNA) no como un espectro continuo de la misma enfermedad, sino como 2 enfermedades diferentes. DESARROLLO DE ESTEATOSIS HEPÁTICA La forma principal de almacenamiento de lípidos en el hepatocito es en forma de triglicéridos, que se sintetizan a partir de ácidos grasos libres (AGL). Sin embargo, la esteatosis hepática puede deberse también al acúmulo de otros lípidos, como es el caso de los AGL, los ésteres de colesterol, el colesterol libre, las ceramidas, los fosfolípidos o el diacilglicerol. La acumulación de triglicéridos depende de la disponibilidad de AGL en el hepatocito, que procede de 3 fuentes principales. La primera y mayoritaria proviene de ácidos grasos plasmáticos no esterificados, que a su vez derivan de la lipólisis del tejido adiposo. Cuando la capacidad de almacenamiento del tejido adiposo se sobrepasa —situación que se observa frecuentemente en la obesidad por un exceso de aporte energético— se produce un aumento en la movilización de AGL. Esta situación se puede ver agravada en los casos de RI, pues es esta hormona la encargada de inhibir la lipólisis en el adipocito. La segunda fuente de AGL proviene de la lipogénesis de novo intrahepatocitaria, de donde deriva aproximadamente una cuarta parte de estos ácidos grasos. Recientemente se ha demostrado que los fenómenos de lipogénesis están aumentados de manera significativa en los pacientes con un diagnóstico de EHDG, lo que probablemente responde a un fenómeno derivado del hiperinsulinismo. Por último, un 15% de los ácidos grasos procede directamente de la dieta y, por lo tanto, se ven incrementados en dietas en las que más del 30% del aporte calórico es en forma de grasas. Estos AGL van a seguir 3 rutas a nivel del hepatocito: a) oxidación mitocondrial; b) síntesis de triglicéridos y almacenamiento en forma de vacuolas lipídicas, y c) acoplamiento y eliminación como lipoproteínas de muy baja densidad (VLDL). Por lo tanto, los mecanismos subyacentes al desarrollo de hígado graso pueden responder no solo a un aumento en las diferentes fuentes de aporte de AGL en el hepatocito, sino también a una disminución en su oxidación mitocondrial o en su eliminación en forma de VLDL. La insulina disminuye la síntesis y estabilidad de la apolipoproteína B, lo que explica que en situaciones de hiperinsulinismo la eliminación de ácidos grasos en forma de VLDL esté disminuida. Este fenómeno también explica la presencia de 63 EHDG y EHNA de manera independiente a la obesidad o la RI en casos de hipolipoproteinemia familiar. A pesar de representar la forma predominante de almacenamiento de grasa, los triglicéridos han demostrado tener un efecto citoprotector, al contrario de otros lípidos como el colesterol o los AGL, que sí presentan un efecto lipotóxico. Este efecto protector se ha comprobado mediante el estudio de las enzimas diacilglicerol-aciltransferasa 1 y 2, responsables del último paso en la síntesis de triglicéridos. La sobreexpresión de estas protege frente a la activación macrofágica, previene la respuesta inflamatoria sistémica y el desarrollo de RI, mientras que su inhibición —aunque disminuye el grado de esteatosis— empeora el daño hepático. El concepto de lipotoxicidad se ha introducido recientemente y permite explicar la presencia, en algunos casos, de inflamación hepática previa al desarrollo de esteatosis. Los AGL y el colesterol acumulados en el interior de los hepatocitos pueden condicionar daño a nivel mitocondrial y estrés del retículo endoplásmico, daño celular y apoptosis. Estos fenómenos son capaces de provocar una respuesta inflamatoria y fibrogénica. MEDIADORES DE DAÑO HEPÁTICO DE ORIGEN INTESTINAL Existe una evidencia creciente en la bibliografía acerca del papel clave ejercido por la microbiota intestinal tanto en la obesidad como en el síndrome metabólico y, por tanto, en el desarrollo de EHDG. Se ha comprobado en modelos animales de esterilidad intestinal cómo, al restablecer una flora normal, estos animales ganan peso y desarrollan obesidad, RI y EHNA. Este fenómeno sugiere que la microbiota y sus interacciones con el sistema inmune a nivel intestinal son clave en el metabolismo energético, el almacenamiento de grasa y la adiposidad, así como en los fenómenos de inflamación sistémica asociados a la obesidad que conllevan el desarrollo de RI. De este modo, se ha comprobado que los cambios en la composición habitual del microbioma intestinal, conocidos como disbiosis, se relacionan con el desarrollo de EHNA en humanos. Esta disbiosis va a ser capaz no solo de modificar la absorción de algunos nutrientes y su metabolismo, sino que, además, se relaciona también con cambios a nivel del epitelio intestinal que aumentan su permeabilidad y la traslocación de diferentes componentes bacterianos. De este modo, se produce lo que se conoce clásicamente como endotoxemia y que hace referencia al aumento en la concentración plasmática de lipopolisacárido (LPS) de origen endoluminal. El LPS es un componente de la pared celular de bacterias Gram-negativas. Tanto las dietas ricas en grasa como en fructosa se han relacionado con aumento en la concentración plasmá- 64 Sesión 3 - ENFERMEDADES METABÓLICAS Y SISTÉMICAS tica de LPS, que es reconocido por el sistema inmune innato a través de sus receptores de tipo Toll (TLR), concretamente el TLR4. En condiciones normales, el hígado presenta una baja expresión y actividad de TLR, generándose así una inmunotolerancia que es necesaria para mantener el equilibrio en un órgano tan ampliamente expuesto a diferentes estímulos antigénicos. Sin embargo, este equilibrio parece estar alterado en el caso de la EHNA. La activación de la vía de señalización dependiente de TLR4 conlleva una respuesta inflamatoria que se asocia tanto al desarrollo de RI como de EHNA. Pero no solo el TLR4 está implicado en la patogenia de la EHDG. El TLR9, que reconoce característicamente fragmentos de ADN de origen bacteriano, tiene una expresión aumentada en el tejido hepático afectado de EHNA, y su activación se relaciona con la progresión de la enfermedad y el desarrollo de RI. El TLR2 detecta característicamente el peptidoglicano de la pared celular de las bacterias Gram-positivas. Entre las bacterias Gram-positivas que componen el microbioma intestinal destacan la familia de las firmicutes, que están muy aumentadas en pacientes obesos, probablemente por efecto de dietas ricas en grasa, lo que determina que la flora de estos pacientes sea rica en ligandos de TLR2. En este sentido, en diferentes modelos animales se ha comprobado que al bloquear la señalización dependiente de TLR2 se previene el desarrollo de RI y EHNA. Por último, el TLR5, que detecta específicamente la flagelina bacteriana, tiene un papel clave a nivel del epitelio intestinal. Los modelos animales carentes de este receptor que se han estudiado desarrollan un síndrome metabólico florido, con hiperfagia, obesidad, RI, inflamación pancreática y esteatosis hepática. La deficiencia en este receptor afecta a la composición de la flora intestinal, que si se transfiere a animales sanos será capaz de desarrollar un síndrome metabólico per se. Estos estudios ponen de manifiesto la importancia de la microbiota y su interacción con el epitelio intestinal como desencadenantes y perpetuadores de la inflamación de origen metabólico. MEDIADORES DE DAÑO HEPÁTICO ORIGINADOS EN EL TEJIDO ADIPOSO Como se ha descrito anteriormente, la RI es un elemento clave en la patogenia de la esteatosis, pero el aumento en los depósitos de grasa también puede promover RI, generándose de este modo un círculo vicioso que perpetuará la situación. Así, la expansión del tejido adiposo, fundamentalmente el visceral, promueve un estado proinflamatorio crónico de bajo grado en este tejido que responde a varios mecanismos fisiopatológicos, entre los que destacan la hipoxia y el estrés oxidativo, y que conllevan la secreción de citocinas proinflamatorias entre las que destacan la interleu- cina-6 y el factor de necrosis tumoral-alfa. El tejido adiposo, fundamentalmente visceral, se comporta —al contrario de lo que se pensaba tradicionalmente— como un órgano metabólicamente muy activo con gran actividad endocrina y paracrina. Sin embargo, en el paciente obeso se produce —además del estado inflamatorio descrito— una disregulación en la liberación de diferentes péptidos bioactivos, conocidos como adipocinas, que regulan la homeostasis dentro de este tejido. Así, se produce una disminución en adipocinas con un efecto fundamentalmente antiinflamatorio, como la adiponectina, mientras que se produce un aumento en la secreción de leptina directamente proporcional a la expansión del tejido adiposo. La leptina desencadena una respuesta proinflamatoria y profibrogénica a través de su interacción con diferentes elementos, tanto de la inmunidad innata como adaptativa, además de ejercer un efecto anorexígeno. Sin embargo, en el paciente obeso los valores permanentemente elevados de la leptina acaban condicionando una resistencia a esta hormona a nivel del sistema nervioso central, perdiéndose este efecto sobre el apetito. La liberación de todos estos mediadores solubles a la circulación general tendrá, entre otros, como órgano diana el hígado. ESTRÉS OXIDATIVO El desequilibrio energético y los fenómenos de lipotoxicidad que aparecen en la obesidad generan determinadas respuestas celulares de estrés. Esto, unido a la disfunción mitocondrial demostrada en diversos estudios, conlleva una situación de estrés oxidativo que se ha asociado con el desarrollo de EHDG. El aporte excesivo de AGL al hígado conlleva un aumento excesivo en los procesos de beta-oxidación que desencadenan una acumulación de especies reactivas de oxígeno (ROS) e inflamación. Las ROS se comportan como agentes oxidantes muy potentes, generando un daño indiscriminado a nivel de diversas estructuras celulares, entre las que se incluyen ADN, membranas lipídicas y proteínas. De este modo, se activan diversas vías proapoptóticas que inician la muerte celular programada, dando lugar a un fenómeno que se conoce como lipoapoptosis. FACTORES GENÉTICOS A pesar de que los factores ambientales como la dieta y la actividad física son determinantes en el desarrollo de la EHDG, se ha descrito también una susceptibilidad genética a presentar esta enfermedad. Esta predisposición genética se sustenta en la observación de una mayor agregación familiar no explicada por hábitos de vida comunes dentro de esas familias, así como las diferencias que ya se han mencionado en cuanto a la Patogenia de la enfermedad hepática por depósito de grasa no alcohólica prevalencia de la enfermedad entre diferentes grupos raciales. La asociación entre el gen PNPLA3 (patatinlike phospholipase domain containing 3) y la EHDG fue descrita inicialmente por Romeo mediante un GWAS (genome-wide association study) realizado en más de 2.100 pacientes de diferentes razas procedentes del Dallas Heart Study. En este estudio se describió la presencia de un SNP (polimorfismo de un solo nucleótido) en la posición rs738409 que se traduce en una sustitución de isoleucina por metionina en el residuo 148 (I148M) de la proteína codificada, la adiponutrina. La adiponutrina se expresa fundamentalmente a nivel hepático y se localiza en el retículo endoplásmico y las vacuolas lipídicas, pero su función en el metabolismo lipídico no es totalmente conocida. Esta mutación se asocia con la presencia de EHDG y su gravedad de manera independiente a la coexistencia de obesidad o síndrome metabólico. Esta asociación ha sido ampliamente corroborada y validada en diversos estudios de genes candidatos y GWAS realizados en diferentes cohortes independientes de pacientes. Además, este polimorfismo se encuentra más frecuentemente representado en la población hispánica y es raro en la afroamericana, lo que explicaría, al menos en parte, las diferencias descritas en cuanto a prevalencia de la EHDG entre estas 2 poblaciones. En este mismo sentido, se ha descrito otro polimorfismo dentro de este mismo gen, en este caso con una sustitución de isoleucina por metionina en el residuo 48 (I48M), que se asocia en esta ocasión con menores depósitos de grasa en el hígado y que, por el contrario, es más frecuente entre los afroamericanos. Otros estudios de asociación genética en EHDG realizados mediante GWAS y confirmados mediante estudios de genes candidatos apuntan a otras asociaciones relevantes que implicarían genes asociados con los diferentes mecanismos patogénicos 65 como genes implicados con RI (IRS-1 y ENPP-1), con estrés oxidativo (SOD2), ciclo celular (CDKN1A), fibrosis (KLF6) y metabolismo de ácidos grasos (FADS1). Por último, otros estudios genéticos han demostrado que la obesidad tiene un importante componente genético, siendo la asociación más fuerte y consistente la descrita en los intrones 1 y 2 del gen FTO, que alberga al menos 89 variantes comunes. Sin embargo, la interpretación fisiopatogénica de esta asociación es a menudo compleja y no del todo conocida. En este sentido, recientemente se ha descrito la repercusión funcional de una de las variantes conocidas dentro de este gen, el polimorfismo rs1421085, que explicaría el aumento en los depósitos de grasa asociados a la obesidad en los portadores. Brevemente, la variante rs1421085 condiciona un polimorfismo T/C que altera el represor ARID5B, lo que conlleva una potenciación de los preadipocitos y un aumento en la expresión de IRX3 e IRX5 durante las primeras etapas de la diferenciación de los adipocitos. Esto resulta en un cambio en el desarrollo de los adipocitos, que pasan de un fenotipo beige, con una mayor capacidad de disipación de energía, a un fenotipo blanco, donde predomina el almacenamiento de la grasa debido a la disminución en la termogénesis de origen mitocondrial. Desde nuestro punto de vista, este hallazgo es clave en la patogenia de la obesidad y, quizás, en la patogenia de la EHNA, aunque este hecho todavía no se ha demostrado. Por otro lado, se sugiere que en el tratamiento de la obesidad, además de una mayor actividad física y de los imprescindibles cambios nutricionales, la manipulación de la termogénesis mitocondrial ofrece una potencial tercera vía para facilitar la transición entre el acúmulo de grasas en el adipocito hacia el aumento del gasto energético mediante el incremento de la termogénesis. SESIÓN 3 ENFERMEDADES METABÓLICAS Y SISTÉMICAS Toxicidad hepática inducida por fármacos Raúl J. Andrade Unidad de Gestión Clínica de Aparato Digestivo, Hospital Universitario Virgen de la Victoria, Instituto de Investigación Biomédica de Málaga (IBIMA), Málaga, España Departamento de Medicina, Universidad de Málaga, Málaga, España Centro de Investigación Biomédica en Red de enfermedades hepáticas y digestivas (CIBERehd), Madrid, España Introducción La toxicidad hepática idiosincrásica o impredecible causada por fármacos (DILI de su acrónimo en inglés, drug-induced liver injury) es actualmente un problema de salud de importancia creciente y un desafío de la hepatología moderna. Al ocurrir con una frecuencia muy baja, las reacciones adversas hepáticas a fármacos no son habitualmente detectadas durante el proceso de desarrollo y, por ello, típicamente se hacen visibles tras la exposición de decenas de miles de pacientes una vez autorizado el medicamento. La DILI es capaz de remedar todas las variedades de enfermedad hepática aguda y crónica y aún se carece de marcadores específicos de toxicidad hepática aplicables a la práctica clínica, por lo que el diagnóstico es difícil y suele retrasarse, requiriendo la cuidadosa exclusión de causas alternativas de enfermedad hepática. Por otra parte, una proporción no despreciable de las reacciones hepáticas a fármacos son graves e incluso fatales. En Occidente, los fármacos (incluyendo el paracetamol) son responsables de más del 50% de los casos de hepatitis fulminante, aunque en este capítulo, por razones de espacio, nos centraremos en la DILI idiosincrásica, excluyendo el paracetamol. Definiciones y fenotipos La DILI tiene un espectro de expresión fenotípica muy amplio, que abarca desde alteraciones asintomáticas del perfil hepático hasta fallo hepático agudo, pero la presentación más común es un cuadro clínico indistinguible de la hepatitis viral. Debido al hecho de que 66 en sujetos que ingieren fármacos, las alteraciones del perfil hepático de escasa magnitud tienen un significado incierto (pueden ser transitorias producidas por el propio fármaco y/o deberse a otras causas tales como hígado graso), un grupo de consenso ha propuesto que para considerar lesión hepática tóxica, los valores de ALT deben ser mayores de 5 veces el límite superior de la normalidad (LSN) y/o los valores de fosfatasas alcalinas (FA) > de 2 × LSN o bien la combinación de ALT > 3 × LSN y bilirrubina total > 2 × LSN. Por otra parte, debido a que en muchos casos no se dispone de material histológico, los fenotipos de toxicidad hepática se caracterizan habitualmente mediante criterios bioquímicos sobre la base de la actividad de la ALT y de la FA séricas (cuando se elevan de forma aislada) y la relación entre ambas (cuando la elevación es simultánea) en hepatocelular (R ≥ 5), colestásica (R ≤ 2) y mixta (R 2-5). La clasificación de la lesión hepática utilizando este criterio exhibe una buena correlación, en general, con la lesión hepática subyacente y es excelente a efectos pronósticos cuando se combina con un parámetro específico de función hepática, la bilirrubina. El valor pronóstico de los valores de ALT combinados con los de bilirrubina total fue anunciado hace más de 4 décadas por Hyman Zimmerman, quien observó que los pacientes que sufrían una lesión hepatocelular con ictericia atribuible a un fármaco (una vez excluidas otras causas de hepatitis e hiperbilirrubinemia) tenían un riesgo de muerte por fallo hepático no < 10%. Dicha observación es conocida como la “ley o regla de Hy”. Esto es debido a que una elevación de bilirrubina con un patrón de citólisis y sin elevación significativa de fosfatasa alcalina, en este contexto, es indicativa de disfunción hepatocitaria grave. Toxicidad hepática inducida por fármacos Epidemiología La incidencia real de DILI es muy poco conocida. En estudios retrospectivos que conectan bases de datos de prescripción con eventos clínicos se calculó una incidencia anual de 2,4 casos por 100.000 habitantes. Muy probablemente, solo una minoría de los casos se comunica a las agencias reguladoras por el sistema de tarjeta amarilla o se publica en revistas científicas. De hecho, en un estudio poblacional prospectivo llevado a cabo en Francia, la incidencia cruda anual de reacciones hepáticas a fármacos fue de 13,9 casos por 100.000 habitantes (16 veces mayor que las notificadas por el sistema de tarjeta amarilla). Recientemente, en la población de Islandia (250.000 habitantes) —cuyo avanzado sistema de salud posibilita acceso a información precisa de cada paciente, médica y farmacológica, y proporciona el número total de sujetos expuestos a cada fármaco— se estimó que la incidencia cruda anual de DILI era de 19,1 casos por 100.000 habitantes y se pudo calcular el riesgo relativo para cada uno de los fármacos implicados. La elevada incidencia comunicada en estos estudios poblacionales, superior a la de otras enfermedades hepáticas como la hepatitis autoinmune o la cirrosis biliar primaria e incluso a la hepatitis C, puede explicarse, en parte, por la inclusión de una proporción numerosa de casos asintomáticos detectados durante el seguimiento sistemático de los sujetos expuestos a fármacos, a diferencia de los casos incluidos en registros, en los que, probablemente por un sesgo de selección, se capturan las incidencias más graves. En la práctica clínica, el diagnóstico de DILI es poco frecuente. Entre los casos de ictericia ingresados en un hospital general, la causa tóxica supuso el 4,7%, pero la mayoría se debían a paracetamol y solo un 0,7% se atribuyó a mecanismo idiosincrásico. La ocurrencia de DILI idiosincrásica entre pacientes hospitalizados fue del 1,4% en un estudio. Los agentes antibacterianos, los antiinflamatorios no esteroideos y anticonvulsionantes ocupan los primeros lugares en la lista de grupos terapéuticos incri- 67 minados en DILI en cohortes prospectivas (registros), y la amoxicilina/ácido clavulánico es, en términos absolutos, la molécula involucrada en un mayor número de incidencias. Existe una marcada variabilidad geográfica en cuanto al agente responsable del daño hepático. En los países occidentales, la mayor parte de los casos se asocian a antibióticos, anticonvulsionantes y a fármacos psicotrópicos, correspondiendo menos del 10% a productos de herboristería y suplementos dietéticos, aunque esta proporción tiende a incrementarse en los últimos años­. En Asia, estos últimos son una causa relevante de DILI. Patogenia y factores de riesgo La DILI idiosincrásica se debe, básicamente, a la interacción de 3 circunstancias: un fármaco que tiene potencial de generar radicales tóxicos para el hígado, un sujeto genéticamente susceptible y la intervención de otros factores del huésped y ambientales. Los estudios genéticos, ya sea con un diseño de genes candidatos por su implicación en el metabolismo de los fármacos (de la fase I o bioactivadora, de la fase II o depuradora y de la fase III o de transporte) o bien sin hipótesis a priori (análisis amplios del genoma [genome-wide association, GWA]), han encontrado asociaciones significativas en casos de DILI. Los GWA realizados en grupos de casos de DILI producidas por fármacos específicos como flucoxacilina, amoxicilina-clavulánico, ximelagatrán o lumiracoxib han identificado sistemáticamente señales en el “Manhattan plot” correspondientes a la región HLA del cromosoma 6, tanto de clase I como de clase II, y específicos para algunos de ellos o compartido en algún caso (tabla 1). Estos estudios apuntan al carácter inmune de las reacciones hepatotóxicas y han cambiado la concepción mecanística de la DILI idiosincrásica. En el momento actual se cree que la predisposición a la DILI con una amplia gama de fármacos estaría vinculada a ser portador del alelo HLA de clase de ries- Tabla 1. Alelos HLA asociados a hepatotoxicidad Principio activo Número de casos Alelo HLA OR (IC del 95%) p Flucloxacilina 51 B*57:01 80,6 (22,8-284,9) 9 × 10–19 Amoxicilina-clavulánico 201 A*02:01 2,3 (1,8-2,9) 1,8 × 10–10 DRB1*15:01-DQB1*06:02 2,8 (2,1-2,8) 3,5 × 10–11 Lumiracoxib 41 DRB1*15:01-DQB1*0,6:02 5,0 (3,6-7,0) 6,8 × 10–25 Lapatinib 35 DRB1*07:01-DQA1*02:01 2,9 (1,3-6,6) 0,007 Ximelagatrán 74 DRB1*07:01-DQA1*02:01 4,4 (2,2-8,9) 6 × 10–6 Ticlopidina 22 A*33:03 13,0 (4,4-38,6) 1,2 × 10–5 IC: intervalo de confianza; OR: odds ratio. 68 Sesión 3 - ENFERMEDADES METABÓLICAS Y SISTÉMICAS Figura 1. Mecanismos de toxicidad hepática inducida por fármacos (DILI) y vías implicadas. BSEP: bomba transportadora de sales biliares; DAMP: daño asociado a patrones moleculares; FHF: fallo hepático fulminante; GSH: glutatión; TNFa: factor de necrosis tumoral alfa. go, que determinaría la presentación del metabolito reactivo hapetenizado (o la molécula nativa) por las células presentadoras de antígenos al sistema inmune adaptativo (hipótesis del hapteno) (fig. 1). En numerosas ocasiones es necesario un coestímulo iniciado por la liberación de DAMP (daño asociado a patrones moleculares) producido por los metabolitos reactivos que dañan el hepatocito y que, a través de los “toll-like receptors” de las células presentadoras de antígeno, inician una inflamación estéril que estimula el sistema inmune innato, el cual libera citocinas proinflamatorias que estimulan al sistema inmune adaptativo, el cual produce una respuesta citotóxica directa o medida por anticuerpos frente a los hepatocitos (hipótesis de la señal de peligro). El sistema inmune innato puede tener un efecto dual coestimulando la respuesta del sistema inmune adaptativo y modulando el grado de inflamación y regeneración. Para numerosos fármacos en los que el riesgo de DILI se asocia a determinados alelos HLA, solo se produciría toxicidad en los sujetos portadores de dichos alelos, pero estarían exentos de riesgo los que carecen de ese perfil genético (fig. 2). Así pues, la carencia de alelos de riesgo en un sujeto que desarrolla una hepatitis cuando se expone al fármaco sospechoso tiene un alto valor predictivo negativo y podría usarse en un test farmacogenético con fines diagnósticos. Por el contrario, solo una minoría de sujetos con los alelos de riesgo expuestos al fármaco desarrollaría DILI, debido a que algunos otros factores, tanto del huésped como del fármaco, son acompañantes necesarios. Es también evidente que la DILI asociada a algunos fármacos no depende de la susceptibilidad genética vinculada a alelos HLA; en estos casos la toxicidad mitocondrial que algunas estructuras químicas muestran, favorecida por polimorfismos de genes mitocondriales (SOD2 y GPX1), podría jugar un papel fundamental. Otros polimorfismos en genes de los sistemas antioxidantes como los genotipos doble nulo del gen de la glutatión transferasa mitocondrial (GST) o de transporte, afectando al ABCB11, el gen que codifica a la bomba exportadora de sales biliares (BSEP), se han encontrado todos ellos con más frecuencia en sujetos Toxicidad hepática inducida por fármacos 69 Figura 2. Susceptibilidad genética a la toxicidad hepática inducida por fármacos (DILI) y probabilidad de lesión clínicamente expresiva. ALF: fallo hepático fulminante. Tomado de Dara L, et al. Liver Int. 2015 [Epub ahead of print]. con DILI idiosincrásica que en controles. Entre los factores no genéticos del huésped se ha sugerido que la edad avanzada, el sexo femenino y las comorbilidades, entre otros, incrementarían el riesgo. Sin embargo, a este respecto hay algunos mitos y pocos hechos contrastados. En el reciente estudio poblacional islandés se evidenció un incremento lineal de la incidencia con la edad (41 casos por 100.000 habitantes en el grupo > 80 años), que iba paralelo al aumento del número de fármacos consumidos con la edad. En los registros español y americano (DILIN), en cambio, la distribución de casos por edad y sexo fue similar. Respecto a la enfermedad subyacente, aunque la diabetes no incrementó el riesgo de sufrir hepatotoxicidad, se asoció a un mayor riesgo de mortalidad en el DILIN. Estudios que conectan bases de datos farmacéuticas con eventos clínicos indican que hay una relación lineal entre dosis diaria (> 50 mg) y metabolismo hepático > 50% y riesgo de hepatotoxicidad idiosincrásica. La combinación en un fármaco determinado de ambos factores (dosis diaria > 100 mg/día) y elevada lipofilidad (que requiere de metabolismo hepático) resulta en el máximo perfil de riesgo, se ha denominado “regla de 2” y se encuentra entre una mayoría de fármacos con toxicidad hepática demostrada. Asimismo, fármacos que in vitro muestran una capacidad inhibitoria sobre la BSEP, en su mayoría han sufrido restricciones de uso o retiradas del mercado por hepatotoxicidad. Cuando concurren todas estas circunstancias se producirá la lesión hepática que, en una mayoría de casos, es de escasa entidad y que se resuelve pese a la continuación del tratamiento con el fármaco. Este fenómeno se conoce como adaptación, y su fracaso conduce a la progresión del daño y la aparición de una lesión hepática manifiesta e incluso fallo hepático fulminante. Entre las causas de fracaso de adaptación podría haber causas inmunológicas como polimorfismos genéticos de citocinas inmunorreguladoras, como la IL-10, o alteraciones del microbioma (inducidos por el propio fármaco; p. ej., antibióticos) o no inmunológicas, como alteración de los sistemas antioxidantes, de la mitofagia, etc. Presentación clinicopatológica y factores pronósticos La DILI se presenta habitualmente simulando a la hepatitis viral aguda, con ictericia, náuseas, astenia y malestar o dolor abdominal, pero son posibles otras 70 Sesión 3 - ENFERMEDADES METABÓLICAS Y SISTÉMICAS presentaciones, que incluyen la hepatitis o colestasis crónica, la cirrosis hepática, la enfermedad venooclusiva e incluso las neoplasias. Aunque, en teoría, la histología hepática sería la herramienta más apropiada para definir el patrón de lesión, rara vez está disponible, y en el momento actual existe un consenso amplio respecto a que la mejor forma de definir la lesión hepática tóxica es utilizando los valores de las aminotransferasas séricas y de la bilirrubina plasmática. El tipo de lesión resultante se clasificará en función de la relación (R) entre el incremento de ALT/fosfatasa alcalina en hepatocelular, colestásica o mixta. En la práctica clínica, la toxicidad hepatocelular representa aproximadamente un 55%, la colestásica un 21% y la mixta un 25% de los casos. La lesión hepatocelular (citolítica, citotóxica) aguda se define, siguiendo estos criterios, como una ALT > 5 LSN o R > 5. En ocasiones coexisten manifestaciones de hipersensibilidad como fiebre, exantema cutáneo o eosinofilia periférica, que sugieren alergia farmacológica. La expresión hepatocelular de la hepatotoxicidad es más frecuente en mujeres jóvenes y este perfil (lesión hepatocelular en mujeres) comporta un mayor riesgo de evolución grave en presencia de ictericia. Histológicamente pueden encontrarse grados variables de inflamación y necrosis, pero el predominio centrolobulillar de las lesiones y la presencia de un infiltrado inflamatorio rico en eosinófilos sugieren una etiología tóxica. La necrosis centrolobulillar es especialmente prominente en casos de intoxicación con algunos hepatotóxicos intrínsecos como el paracetamol o la cocaína. En estas circunstancias, los valores séricos de transaminasas suelen estar extraordinariamente elevados (100 veces o más el LSN), situándose en el rango de la hepatitis isquémica. La lesión colestásica aguda se define como un incremento en los valores séricos de FA > 2 N o R < 2, y se clasifica en 2 subtipos: colestasis pura, “blanda” o canalicular y hepatitis aguda colestásica o hepatocanalicular. La probabilidad de una expresión colestásica de la hepatotoxicidad se incrementa con la edad avanzada y es más frecuente en varones. La presentación habitual de la colestasis aguda es con ictericia y prurito. La variedad canalicular se caracteriza por un incremento en la bilirrubina conjugada, FA y G-glutamil transpeptidasa con alteración mínima o nula de las transaminasas. La biopsia hepática muestra colestasis hepatocitaria y canalículos biliares dilatados con trombos de bilis, sin evidencia de necrosis o inflamación, y el curso es benigno con recuperación completa y sin secuelas. Este tipo de lesión es característica de los anticonceptivos y esteroides anabólicos. En el daño hepatocanalicular puede existir dolor abdominal y fiebre, que simula la obstrucción biliar aguda, y con frecuencia manifestaciones asociadas de hipersensibilidad. Los hallazgos histológicos incluyen inflamación portal y ductal y necrosis hepatoci- taria junto a marcada colestasis de predominio centrolobulillar. Entre los fármacos que se han involucrado en este subtipo de toxicidad hepática merece destacarse la amoxicilina-clavulánico, los antibióticos macrólidos y las fenotiazinas. En algunos casos puede instaurarse un síndrome de desaparición de conductos biliares, con colestasis persistente e incluso cirrosis biliar. De hecho, la resolución de las lesiones colestásicas es más lenta que en las lesiones hepatocelulares y con mayor frecuencia evolucionan a la cronicidad. En realidad, el tiempo de resolución de las reacciones hepatotóxicas agudas en su conjunto es más prolongado que el de la hepatitis viral, por lo que se ha determinado que un punto de corte de 1 año sería el idóneo para establecer cronicidad. Se utiliza la nomenclatura de daño hepático mixto cuando las alteraciones clínicas y de laboratorio se sitúan en un rango intermedio entre las de tipo hepatocelular y las de tipo colestásico (R > 2 y < 5), aunque pueden predominar las de uno u otro. En esta variedad son más frecuentes las manifestaciones de alergia farmacológica, y el médico debe conocer que el daño hepático mixto es más probablemente causado por tóxicos que por agentes virales. Casi todos los fármacos que inducen hepatitis colestásica pueden causar, asimismo, una lesión mixta. Algunos casos de hepatotoxicidad se asocian a lesiones en otros órganos, fundamentalmente la piel, el riñón y el páncreas. Este contexto refuerza la naturaleza tóxica de la lesión hepática frente a otras alternativas diagnósticas. Diagnóstico La hepatotoxicidad idiosincrásica es muy compleja y los agentes implicados muy numerosos, por lo que la búsqueda de biomarcadores mecanísticos y diagnósticos es aún incipiente. Están en proceso de exploración en hepatotoxicidad 3 tecnologías: la metabolómica, la transcriptómica y la proteómica, con la finalidad de descubrir biomarcadores más precoces, sensibles y específicos que la ALT, basándose en el hecho de que el hígado libera alguna clase de señal, como los recientemente identificados DAMP, antes de que ocurra lesión hepática manifiesta. Los tests de proliferación linfocitaria al exponer suero de pacientes afectados al fármaco no han conseguido estandarización suficiente para ser incorporados a la práctica clínica. Muy recientemente, un nuevo test in vitro, que expone al fármaco sospechoso monocitos modificados con fenotipo de hepatocitos del paciente y que mide la citotoxicidad, ha mostrado una elevada sensibilidad y especificidad, pero requiere aún validación. Los tests genéticos basados en alelos HLA de riesgo por su alto valor predictivo negativo (su ausencia disminuye la probabilidad de hepatotoxicidad), podrían usarse con la finalidad de Toxicidad hepática inducida por fármacos 71 Figura 3. Algoritmo diagnóstico de una sospecha de toxicidad hepática inducida por fármacos (DILI). excluir el diagnóstico de DILI si se carece de estos. No obstante, este abordaje es inaccesible por el momento en la mayoría de los escenarios clínicos. El diagnóstico de DILI requiere en primera instancia la demostración de la existencia de lesión hepática, caracterizada por actividad de la ALT, AST y fosfatasa alcalina séricas en un sujeto expuesto a fármacos. Estas enzimas reflejan necrosis hepatocitaria y colestasis y su valor en sangre depende del balance neto entre su liberación desde el citoplasma de las células hepáticas (y de otros tejidos como músculo esquelético y cardíaco, intestino, etc.) y su degradación por el sistema reticuloendotelial. La ALT se considera más específica del hígado que la AST, debido a que su concentración en el citoplasma hepatocitario es más elevada que en el músculo esquelético, pero el hecho de que la masa muscular supere en 15-20 veces la masa hepática determina que una lesión muscular pueda ser una fuente considerable de incrementos de ALT. Además, las transaminasas son muy volátiles y sus valores pueden elevarse y disminuir en horas o días. Por ello, una única determinación no establece un valor “pico”. Por otra parte, erróneamente se denomina a las transaminasas parámetros de “función hepática”, pero no existe una relación lineal entre los valores de transaminasas y la intensidad de la lesión hepática ni el grado de disfunción hepática, por lo que, en sí mismos, no tienen valor pronóstico. A este efecto debe determinarse simultáneamente la concentración sérica de bilirrubina total y directa, cuya elevación indica una disminución de la reserva funcional hepática. Una vez establecida la lesión hepática, el diagnóstico de DILI idiosincrásica es de exclusión y se basa en un alto grado de sospecha clínica y la existencia de una cronología compatible de exposición a un fármaco o tóxico (que es muy variable en su latencia), conjuntamente con una batería de pruebas diagnósticas que sean capaces de eliminar una serie de causas alternativas (fig. 3). Los estudios pertinentes se recogen en la tabla 2. Frecuentemente, la sospecha diagnóstica emerge tras un cribado negativo, más o menos exhaustivo, para otras causas específicas. Entre los criterios favorables al diagnóstico se encuentran las manifestaciones de hipersensibilidad y una evidente mejoría clinicobiológica tras la retirada de los fármacos. El patrón oro para el diagnóstico de hepatotoxicidad es la demostración de una recrudescencia de las alteraciones clinicobiológicas tras la reexposición al agente causal. Sin embargo, por razones éticas tal práctica no está justificada salvo en circunstancias excepcionales. No obstante, un cuidadoso interrogatorio puede descu- 72 Sesión 3 - ENFERMEDADES METABÓLICAS Y SISTÉMICAS Tabla 2. Principales causas de enfermedad hepática que hay que excluir antes de considerar un diagnóstico de hepatotoxicidad Test/características clínicas Enfermedades • Serología viral IgM anti-HVA IgM anti-HBc Anti-VHC, ARN-VHC (RT-PCR) IgM anti-VHE, ARN-VHE IgM-CMV IgM-VEB Hepatitis virales • Serología bacteriana: si fiebre persistente, diarrea (Salmonella, Campylobacter, Listeria, Coxiella) Hepatitis bacterianas • Serología sífilis/FA muy elevada Sífilis secundaria • AST/ALT > 2 , VCM y GGT incrementados Hepatopatía alcohólica • Autoanticuerpos (ANA, ANCA, AMA, ASMA, anti-LKM-1) Hepatitis autoinmune, cirrosis biliar primaria • Ceruloplasmina, cobre en orina (pacientes < 40 años) Enfermedad de Wilson • Alfa-1 antitripsina Déficit de alfa-1 antitripsina • Saturación de transferrina (en daño hepatocelular anictérico) Hemocromatosis • Hipotensión, shock, insuficiencia cardíaca Hepatitis isquémica • Enfermedad vascular, ancianos Hepatopatía congestiva • Técnicas de imagen: radiológicas/endoscópicas, ecografía abdominal, TCA, colangiorresonancia, ERCP Obstrucción biliar AMA: anticuerpo antimitocondrial; ANA: anticuerpo antinuclear; ANCA: anticuerpo antineutrófilo citoplasmático perinuclear; anti-HBc: anticuerpo hepatitis B core; anti-LKM-1: anticuerpo microsomal tipo1 hígado-riñón; anti-VHA: anticuerpo virus de la hepatitis A; anti-VHC: anticuerpo virus de la hepatitis C; anti-VHE: anticuerpo virus de la hepatitis E; ASMA: anticuerpo antimúsculo liso; CMV: citomegalovirus; ERCP: colangiografía endoscópica retrógrada; GGT: gamma glutamil transpeptidasa; TC: tomografía computarizada abdominal; VCM: volumen corpuscular medio; VEB: virus de Epstein-Barr. brir en algunos casos evidencias sutiles de reexposición accidental que serían de gran utilidad para el diagnóstico. En tales casos, el primer episodio tras la administración del medicamento no se acompañó de ictericia y los síntomas habrían sido inespecíficos (malestar abdominal, fiebre, astenia), lo que habría dificultado su identificación por parte del médico no familiarizado con la hepatotoxicidad. Aunque prácticamente cualquier fármaco comercializado se ha involucrado en incidencias de hepatotoxicidad, la información acerca de la probabilidad de causar lesión hepática es escasa o ambigua para otros muchos fármacos. Puede recurrirse a las bases de datos de fármacos hepatotóxicos como LIVERTOX, una potente herramienta informática puesta a punto por los National Institutes of Health (http://www.livertox.nih.gov/) o la del Spanish DILI Registry (http://www.spanishdili.uma.es). No existen manifestaciones histológicas que se puedan considerar absolutamente específicas de DILI y, en consecuencia, no debe practicarse una biopsia hepática de forma rutinaria con esta indicación. La biopsia hepática es especialmente útil cuando otro diagnóstico es posible (p. ej., alcohólico en tratamiento aversivo, exacerbación de hepatitis crónica viral), para determinar el patrón de lesión con fármacos anteriormente no implicados en hepatotoxicidad y para caracterizar el sustrato de lesión en formas crónicas (cuya escasa expresividad clínica no permite estimar el verdadero alcance de la lesión) o infrecuentes (p. ej., sospecha de lesiones vasculares). Diversos grupos han intentado dar consistencia al diagnóstico de DILI desarrollando escalas para la evaluación de causalidad que proporcionen un enfoque homogéneo y, usualmente, una categoría de probabilidad basada en una puntuación numérica. En la actualidad se utilizan 2 escalas o algoritmos diagnósticos para la evaluación de causalidad en DILI: la escala de CIOMS/RUCAM y la escala de María y Victorino, también denominada escala diagnóstica clínica. Ambas escalas proporcionan un sistema de puntuación para 6 apartados en la estrategia de decisión. Las respuestas corresponden a valores ponderados que se suman Toxicidad hepática inducida por fármacos 73 Tabla 3. Criterios y puntuaciones para los apartados individuales de las escalas diagnósticas de CIOMS/ RUCAM y M & V CIOMS/RUCAM Criterios M&V Puntuación Criterios cronológicos Criterios Puntuación Criterios cronológicos Desde inicio del tratamiento hasta comienzo del evento +2 a +1 Desde inicio del tratamiento hasta comienzo del evento +1 a +3 Desde retirada del fármaco hasta comienzo del evento +1 a 0 Desde retirada del fármaco hasta comienzo del evento –3 a +3 Curso de la reacción –2 a +3 Curso de la reacción 0 a +3 Exclusión de causas alternativas –3 a +3 Factores de riesgo Edad (> 55 años) +1 a 0 Manifestaciones extrahepáticas 0 a +3 Alcohol +1 a 0 Datos en la bibliografía –3 a +2 Tratamiento concomitante –3 a 0 Reexposición 0 a +3 Exclusión de causas alternativas –3 a +2 Datos en la bibliografía 0 a +2 Reexposición –2 a +3 Con CIOMS la puntuación total puede ser clasificada en 5 categorías: ≤ 0, excluido; 1-2, improbable; 3-5, posible; 6-8, probable; > 8 altamente probable o definido. Con M & V las categorías de probabilidad son: > 17, definido; 14-17, probable; 10-13, posible; 6-9, dudoso; < 6, excluido. para proporcionar una puntuación total. Dichas puntuaciones se trasladan a categorías de sospecha (tabla 3). El clínico debe contestar las diferentes cuestiones pertinentes a la cronología de la reacción (tiempo de latencia y evolución de la bioquímica hepática tras la supresión del tratamiento), factores de riesgo (alcohol y embarazo, este último solo para las reacciones colestásicas/mixtas), comedicación, exclusión de causas alternativas, información previa de la toxicidad hepática del fármaco bajo sospecha y respuesta a una nueva exposición al compuesto (si ha existido), todo ello con una puntuación predeterminada en función de la información disponible. En muchos casos falta información para contestar a algunas de las preguntas. Esta circunstancia, aunque puede reducir la puntuación final obtenida, está contemplada en las escalas y no debe suponer un impedimento para su aplicación. El único requisito realmente limitante para el uso de la escala de CIOMS/RUCAM es conocer con certeza la cronología de la exposición al(los) fármaco(s) en relación con el inicio de la reacción. Estos instrumentos tienen fortalezas y debilidades, pero quizás su mayor inconveniente es no poder confrontarse con un parámetro objetivo de diagnóstico. En un estudio, la escala de CIOMS/RUCAM ha demostrado una mayor consistencia con el juicio hecho por clínicos y, en el momento actual, es la recomendada para la evaluación de causa- lidad, aunque dista de ser perfecta y existe un amplio consenso en que debe reducirse la ambigüedad de las cuestiones que plantea. Tratamiento La principal medida terapéutica en una sospecha de hepatotoxicidad es la inmediata supresión de cualquier tratamiento farmacológico no esencial, ya que la continuación del fármaco responsable podría determinar una mayor probabilidad de evolución fulminante o crónica. La hospitalización es aconsejable para los pacientes con sospecha de hepatotoxicidad que muestren un fenotipo de mayor riesgo de evolución fulminante (lesión hepatocelular con ictericia), aquellos con ictericia franca y/o con manifestaciones de hipersensibilidad prominentes y los que presentan afectación de otros órganos. Un algoritmo que utiliza la AST y la bilirrubina al inicio del cuadro clínico ha mostrado una elevada sensibilidad y especificidad para predecir evolución fulminante (fig. 4). Los pacientes con criterios de fallo hepático agudo en progresión (INR > 1,5, encefalopatía) deben ser trasladados a un centro de trasplante. Los resultados obtenidos con el trasplante hepático son, en términos de supervivencia, superponibles a los obtenidos con otras indicaciones. La N-acetilcisteína, 74 Sesión 3 - ENFERMEDADES METABÓLICAS Y SISTÉMICAS Figura 4. Algoritmo para estimar el riesgo de fallo hepático fulminante/trasplante (ALF/OLT) al inicio del episodio de hepatotoxicidad. Un nivel AST > 17,3 × límite superior de la normalidad (ULN), bilirrubina total (TBL) > 6,6 × ULN y AST/ALT > 1,5 ×. ULN identificó pacientes que desarrollaron ALF/OLT con especificidad del 82% y sensibilidad del 80%. Tomado de Robles-Díaz M, et al. Gastroenterology. 2014;147:109-18. probablemente por sus propiedades antioxidantes, ha mostrado una eficacia modesta en reducir la necesidad de trasplante en la insuficiencia hepática aguda no relacionada con intoxicación por paracetamol, de origen variado (incluyendo hepatitis tóxica idiosincrásica), a condición de que se administre en los grados iniciales de encefalopatía. Los corticoides, aunque ocasionalmente se utilizan y parecen justificados en pacientes con DILI y manifestaciones de hipersensibilidad y en aquellos con rasgos de autoinmunidad, han sido ineficaces en ensayos clínicos controlados. Debido a que no existe antídoto para la toxicidad hepática por fármacos, el tratamiento se circunscribe a las medidas terapéuticas sintomáticas que se emplean en otras variedades de enfermedad hepática. El prurito puede ser una manifestación prominente en casos de colestasis y se deben emplear como primera alternativa las resinas de intercambio (resincolestiramina) y la rifampicina como segunda línea de tratamiento. Los suplementos de vitaminas liposolubles estarían asimismo indicados en la colestasis crónica. El ácido ursodeoxicólico, en estudios no controlados parece ser beneficioso en casos de colestasis prolongada y ductopenia. Bibliografía recomendada Aithal GP, Watkins PB, Andrade RJ, Larrey D, Molokhia M, Takikawa H, et al. Case definition and phenotype standardiza- tion in drug-induced liver injury (DILI). Clin Pharmacol Ther. 2011;55:683-91. Aithal GP. Pharmacogenetic testing in idiosyncratic drug-induced liver injury: current role in clinical practice. Liver Int. 2015;35:1801-8. Andrade RJ, Camargo R, Lucena MI, González-Grande R. Causality assessment in drug-induced hepatotoxicity. Expert Opin Drug Saf. 2004;3:329-44. Andrade RJ, Lucena MI. Drug-Induced Liver Disease-Mechanism and Diagnosis. En: McDonald JWD, Burroughs AK, Feagan BG, editors. Evidenced Based Gastroenterology and Hepatology. 3rd ed. West Sussex: Wiley-Blackwell; 2010. p. 771-86. Andrade RJ, Lucena MI, Fernández MC, Peláez G, Pachkoria K, García-Ruiz E, et al. Drug-induced liver injury: An analysis of 461 incidences submitted to the Spanish Registry over a 10year period. Gastroenterology. 2005;129:512-21. Andrade RJ, Lucena MI, Kaplowitz N, García-Muñoz B, Borraz Y, Pachkoria K, et al. Outcome of acute idiosyncratic druginduced liver injury: Long-term follow-up in a hepatotoxicity registry. Hepatology. 2006;44:1581-8. Andrade RJ, Robles M, Lucena MI. Rechallenge in drug-induced liver injury: The attractive hazard. Expert Opin Drug Saf. 2009;8:709-71. Andrade RJ, Tulkens PM. Hepatic safety of antibiotics used in primary care. J Antimicrob Chemother. 2011;66:1431-46. Benesic A, Leitl A, Gerbes AL. Monocyte-derived hepatocyte like cells for the causality assessment of idiosyncratic druginduced liver injury. Gut. 2015 Jun 4. pii:gutjnl-2015-309528. [Epub ahead of print]. Bénichou C. Report of an International Consensus Meeting. Criteria of drug-induced liver disorders. J Hepatol, 1990;11: 272-6. Björnsson E, Olsson R. Outcome and prognostic markers in severe drug-induced liver disease. Hepatology. 2005;42:481-9. Björnsson ES, Bergmann OM, Björnsson HK, Kvaran RB, Olafsson S. Incidence, presentation, and outcomes in patients with drug-induced liver injury in the general population of Iceland. Gastroenterology. 2013;144:1419-25. Chalasani N, Björnsson E. Risk factors for idiosyncratic drug-induced liver injury. Gastroenterology. 2010;138:2246-59. Chalasani N, Bonkovsky Hl, Fontana R, Lee W, Stolz A, Talwalkar J, et al. Features and outcomes of 899 patients with druginduced liver injury: The DILIN Prospective Study. Gastroenterology. 2015;148:1340-52. Chen M, Borlak J, Tong W. High lipophilicity and high daily dose of oral medication are associated with signicant risk for druginduced liver injury. Hepatology. 2013;58:388-96. Chen M, Suzuki A, Borlak J, Andrade RJ, Lucena MI. Drug induced liver injury: interactions between drug properties and host factors. J Hepatol. 2015;63:503-14. Daly AK, Donaldson PT, Bhatnagar P, Shen Y, Pe’er I, Floratos A, et al; DILIGEN Study; International SAE Consortium. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat Genet. 2009;41:816-9. Danan G, Bénichou C. Causality assessment of adverse reactions to drugs I. A novel method based on the conclusions of international consensus meetings: application to drug-induced liver injuries. J Clin Epidemiol. 1993;46:1323-30. Dara L, Liu ZX, Kaplowitz N. Mechanisms of adaptation and progression in idiosyncratic drug induced liver injury, clinical implications. Liver Int. 2015. [Epub ahead of print]. De Abajo FJ, Montero D, Madurga M, García Rodríguez LA. Acute and clinically relevant drug-induced liver injury: a population based case-control study. Br J Clin Pharmacol. 2004;58:71-80. Devarbhavi H, Andrade RJ. Drug-induced liver injury due to antimicrobials, central nervous system agents, and nonsteroidal anti-inflammatory drugs. Semin Liver Dis. 2014;34:145-61. Fontana R, Seef L, Andrade RJ, Björnsson E, Day CP, Serrano J, et al. Standardization of nomenclature and causality assessment in drug-induced liver injury: summary of a clinical research workshop. Hepatology. 2010;52:730-42. García-Cortés M, Stephens C, Fernández-Castañer A, Lucena MI, Andrade RJ. Causality assessment methods in drug induced liver injury: strengths and weaknesses. J Hepatol. 2011:55:683-91. Kaplowitz N. Idiosyncratic drug hepatotoxicity. Nat Reviews Drug Discov. 2005;4:489-99. Lammert C, Einarsson S, Saha C, Niklasson A, Bjornsson E, Chalasani N. Relationship between daily dose of oral medications and idiosyncratic drug-induced liver injury: search for signals. Hepatology. 2008;47:2003-9. Lammert C, Niklasson A, Bjornsson E, Chalasani N. Oral medicationswith significant hepatic metabolism are at higher risk for hepaticadverse events. Hepatology. 2010;51:615-20. Lee WM, Hynan LS, Rossaro L, Fontana RJ, Stravitz RT, Larson AM, et al. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology. 2009;137:856-64. Lee WM, Squires RH Jr, Nyberg SL, Doo E, Hoofnagle JH. Acute liver failure: Summary of a workshop. Hepatology. 2008;47:1401-15. Toxicidad hepática inducida por fármacos 75 Lucena MI, Andrade RJ, Kaplowitz N, García-Cortes M, Fernández MC, Romero-Gómez M, et al. Phenotypic characterization of idiosyncratic drug-induced liver injury: the influence of age and sex. Hepatology. 2009;49:2001-9. Lucena MI, Andrade RJ, Martínez C, Ulzurrun E, García-Martín E, Borraz Y, et al; Spanish Group for the Study of Drug-Induced Liver Disease. Glutathione S-transferase M1 and T1 null genotypes increase susceptibility to idiosyncratic drug-induced liver injury. Hepatology. 2008;48:588-96. Lucena MI, Camargo R, Andrade RJ, Pérez-Sénchez C, Sánchez de la Cuesta F. Comparison of two clinical scales for causality assessment in hepatotoxicity. Hepatology. 2001;33:123-30. Lucena MI, García-Martín E, Andrade RJ, Martínez C, Stephens C, Ruiz JD, et al. Mitochondrial superoxide dismutase and glutathione peroxidase in idiosyncratic drug-induced liver injury. Hepatology. 2010;52:303-12. Lucena MI, Molokhia M, Shen Y, Urban TJ, Aithal GP, Andrade RJ, et al. Susceptibility to amoxicillin-clavulanate induced liver injury is influenced by multiple HLA class I and II alleles. Gastroenterology. 2011:141:338-47. Maria V, Victorino R. Development and validation of a clinical scale for the diagnosis of drug-induced hepatitis. Hepatology. 1997;26:664-9. Meier Y, Cavallaro M, Roos M, Pauli-Magnus C, Folkers G, Meier PJ, et al. Incidence of drug-induced liver injury in medical inpatients. Eur J Clin Pharmacol. 2005;61:135-43. Rakela J, Mosley JW, Edwards VM, Govindarajan S, Alpert E. A double-blinded, randomized trial of hydrocortisone in acute hepatic failure. The Acute Hepatic Failure Study Group. Dig Dis Sci. 1991;36:1223-8. Robles-Díaz M, Lucena MI, Kaplowitz N, Stephens C, Medina-Cáliz I, González-Jimenez A, et al. Use of Hy’s law and a new composite algorithm to predict acute liver failure in patients with drug-induced liver injury. Gastroenterology. 2014;147: 109-18. Sgro C, Clinard F, Ouazir K, Chanay H, Allard C, Guilleminet C, et al. Incidence of drug-induced hepatic injuries: a French population-based study. Hepatology. 2002;36:451-5. Spagnuolo MI, Iorio R, Vegnente A, Guarino A. Ursodeoxycholic acid for treatment of cholestasis in children on long-term total parenteral nutrition: a pilot study. Gastroenterology. 1996;111:716-9. Stephens C, Andrade RJ, Lucena MI. Mechanisms of drug-induced liver injury. Curr Opin Allergy Clin Immunol. 2014;14:286-92. Ulzurrun E, Stephens C, Crespo E, Ruiz-Cabello F, Ruiz-Núñez J, Sáenz-López P, et al. Role of chemical structures and the 1331T>C bile salt export pump polymorphismin idiosyncratic drug-induced liver injury. Liver Int. 2013;33:1378-85. Vuppalanchi R, Liangpunsakul S, Chalasani N. Etiology of newonset jaundice: how often is it caused by idiosyncratic druginduced liver injury in the United States? Am J Gastroenterol. 2007;102:558-62. Watkins PB, Seligman P, Pears JS, Avigan MI, Senior JR. Using controlled clinical trials to learn more about acute drug-induced liver injury. Hepatology. 2008;48:1680-9. Zimmerman HJ. Hepatotoxicity. The adverse effects of Drugs and Other Chemicals on the Liver. 2nd ed. Philadelphia: Lippincott Williams & Wilkins: 1999. SESIÓN 3 ENFERMEDADES METABÓLICAS Y SISTÉMICAS Riesgo vascular en el paciente con esteatosis hepática: implicaciones en el entorno del trasplante hepático Guillermo Ontanilla Clavijo y Manuel Romero Gómez Unidad de Aparato Digestivo Intercentros, Hospital Universitario Virgen Macarena-Virgen del Rocío, Universidad de Sevilla, Sevilla, España INTRODUCCIÓN Desde que a principios de la década de los ochenta Ludwig et al1 establecieran el término nonalcoholic steatohepatitis (NASH), la enfermedad hepática por depósito graso se ha convertido en una entidad con un espectro clínico muy amplio. La presencia de esteatosis hepática es común en la enfermedad hepática grasa, tanto de origen etílico como no alcohólica, pero también se ha observado en pacientes con hepatopatías secundarias a etiología viral, farmacológica, metabólica o autoinmune. La esteatosis hepática es especialmente relevante en pacientes con trastornos metabólicos y hepatitis viral. La presencia de esteatosis hepática junto a inflamación y fibrosis se ha asociado a un mayor riesgo de progresión de la enfermedad hepática, al tiempo que se incrementa el riesgo cardiovascular. En cambio, en los pacientes con cirrosis biliar primaria (CBP), que suelen presentar cifras elevadas de colesterol, tanto colesterol total como colesterol unido a lipoproteínas de baja densidad (cLDL) o colesterol unido a lipoproteínas de alta densidad (cHDL), así como de triglicéridos, la reducción de la actividad plasmática de la enzima lecitin-colesterol acil transferasa, así como el descenso en la secreción de ácidos biliares y la infraexpresión de receptores de LDL en los hepatocitos, junto al incremento de la síntesis hepática de colesterol, parecen estar detrás de la hiperlipidemia mixta característica de los pacientes con CBP. La hiperlipidemia está fuertemente asociada con el riesgo cardiovascular en la población general, pero no en pacientes con CBP. Los pacientes con CBP no muestran mayor riesgo cardiovascular cuando se monitorizan parámetros de aterosclerosis ni se ha descrito aumento de episodios cardiovasculares. La alta prevalencia en mujeres, con un perfil antiaterogénico (debido al aumento de las cifras de HDL), valores bajos 76 de lipoproteína A y de lipoproteína X —una LDL modificada—, así como la escasa prevalencia de obesidad visceral en estas mujeres y las elevadas concentraciones de adiponectina, pueden explicar esta protección cardiovascular en un escenario hiperlipidémico. Además, desde el punto de vista metabólico y hepático, la prevalencia de resistencia a la insulina y diabetes en pacientes con CBP es baja y la presencia de vacuolas grasas en los hepatocitos, excepcional. Por tanto, la sensibilidad normal a la insulina y la ausencia de esteatosis pueden proteger frente al riesgo cardiovascular2. Por último, agentes hipolipemiantes como las estatinas y los fibratos han demostrado efectos antiinflamatorios y en pacientes con CBP son seguros y ejercen un efecto beneficioso sobre la colestasis y la inflamación hepática cuando se administran a pacientes con respuesta subóptima al ácido ursodeoxicólico; por tanto, pueden ser otro factor asociado a una disminución del riesgo cardiovascular en pacientes hiperlipémicos. La esteatosis hepática aparece por el desequilibrio en el metabolismo lipídico, que a nivel hepático se traduce en un aumento de la lipogénesis frente a la lipólisis, lo que conlleva el acúmulo de triglicéridos. Esta situación está condicionada por un aumento de las concentraciones plasmáticas de ácidos grasos libres, secundario a una mayor lipólisis en el tejido graso, y por una captación de estos aumentada a nivel hepático, todo ello desencadenado por la resistencia insulínica a nivel del tejido adiposo3. Este daño hepático está modulado por factores ambientales, destacando el papel de la microbiota y, sobre todo, genéticos como el gen de la adiponutrina (PNPLA3) y el TM6SF2 que, junto a factores epigenéticos, impactan en el desarrollo de inflamación, necrosis y activación de los procesos de fibrogénesis. El síndrome metabólico se compone de diferentes factores de riesgo vascular y metabólico como la Riesgo vascular en el paciente con esteatosis hepática: implicaciones en el entorno del trasplante hepático hipertensión, la dislipemia, la obesidad visceral y el trastorno del metabolismo de la glucosa o la resistencia insulínica. Los pacientes con esteatosis hepática muestran alteraciones leves del valor de las transaminasas, con valores no superiores a 2 veces el límite superior de la normalidad, con un cociente AST/ALT < 1, y discretas alteraciones de la fosfatasa alcalina y la gammaglutamil transpeptidasa. Aun así, hasta en el 80% de los pacientes estos valores serán normales4, y lo que encontraremos son alteraciones metabólicas como: hipertrigliceridemia, disminución de los valores de HDL, hiperglucemia e hiperinsulinemia. Asimismo, en un 25% de los pacientes podremos encontrar la presencia de autoanticuerpos positivos, a títulos < 1:3205. La prueba inicial de imagen que solicitaremos será una ecografía abdominal, que mostrará un hígado con un aumento de la ecogenicidad y atenuación posterior del sonido, datos que con una sensibilidad del 100% y un valor predictivo positivo del 62% corresponderán con un porcentaje > 33% de grasa a nivel hepático6. En pacientes con alteraciones analíticas y ecográficas compatibles con esteatosis hepática hemos de descartar consumo de alcohol, hepatitis viral, hepatitis autoinmune, hemocromatosis y enfermedad de Wilson. Si todos los datos son negativos haremos el diagnóstico de exclusión de enfermedad hepática grasa no alcohólica (NAFLD). La presencia de esteatohepatitis y fibrosis incrementa el riesgo de progresión de la enfermedad hepática y la posibilidad de sufrir episodios cardiovasculares. Para ello, el único método fiable que teníamos disponible era la realización de una biopsia hepática. Recientemente se han desarrollado técnicas no invasivas capaces de detectar esteatohepatitis (NASH-MRi, mediante el análisis óptico de imágenes de resonancia magnética de hígado y Owl-Liver®, un análisis metabolómico que confirma la presencia de esteatohepatitis de acuerdo al índice de masa corporal), así como métodos para conocer el estadio de fibrosis como la elastografía transitoria o una combinación de determinaciones bioquímicas y antropométricas como el NAFLD fibrosis score (NFS). El uso combinado de estas 2 últimas técnicas nos ha permitido conocer más de cerca la historia natural de la enfermedad, cuya prevalencia se estima entre un 6,3-33% para NALFD y entre un 3-5% para NASH7. La prevalencia, no obstante, es especialmente elevada en pacientes con diabetes tipo 2 (DM2), donde un estudio demostró que el 87% de pacientes presentaba NAFLD en ecografía7, y en obesos mórbidos que se van a someter a cirugía bariátrica, donde la prevalencia es > 90%8. Un metaanálisis de 13 estudios puso de manifiesto una sensibilidad y una especificidad del NFS del 90 y el 60%, respectivamente, para descartar fibrosis avanzada, con un valor ≤ –1,455, y con un 67 y un 97% de sensibilidad y especificidad para identificarla, con un 77 valor ≥ 0,676, aunque la zona gris entre estos valores se detectó en casi la mitad de los pacientes. La elastografía transitoria alcanzó una sensibilidad del 94% y una especificidad del 95%, no obteniéndose mediciones válidas en un tercio de los pacientes9. En pacientes con NASH, la principal causa de muerte son las enfermedades cardiovasculares10, mientras que los pacientes con esteatosis simple muestran un curso benigno sin un aumento de la mortalidad11. Además, la detección de fibrosis avanzada mediante métodos no invasivos se asocia a un incremento del riesgo cardiovascular: en pacientes con NFS > 0,676 en el 7,7 frente al 2,3% en pacientes sin fibrosis avanzada (p < 0,002). En pacientes con FIB-4 > 3,25 (fibrosis avanzada) se detectaron complicaciones vasculares en el 9,0 frente al 2,3% (p < 0,001)12. ESTEATOSIS HEPÁTICA, ATEROSCLEROSIS SUBCLÍNICA Y MORTALIDAD CARDIOVASCULAR La evaluación del riesgo es fundamental cuando nos enfrentamos a pacientes con NALFD que han evolucionado a cirrosis hepática y que por insuficiencia hepática, hepatocarcinoma o alguna otra de las complicaciones (ascitis refractaria, encefalopatía o síndrome hepatorrenal) precisan trasplante hepático, dado que el 70% de los eventos clínicos adversos mayores en el primer año postrasplante son de origen cardiovascular13. Las características basales a evaluar tendrán, por tanto, implicaciones pronósticas pre y postrasplante, que se deben tener en cuenta en el proceso de evaluación pretrasplante. La obesidad per se supone un riesgo añadido de complicaciones posquirúrgicas14, aunque hay que corregirla por el volumen de ascitis, teniendo en cuenta que cada litro de ascitis supone un incremento del 7% de la mortalidad15. Los obesos también verán comprometida su supervivencia a largo plazo postrasplante14. Las complicaciones cardiovasculares tras el trasplante hepático son tan prevalentes como en la población general16. El estudio cardiovascular pretrasplante consiste en un electrocardiograma, una ecocardiografía y pruebas de esfuerzo para descartar enfermedad aterosclerótica coronaria. No obstante, el 90% de los infartos de miocardio se produce por la ruptura y trombosis de una placa de ateroma que no condicionaba una estenosis que limitara el flujo sanguíneo17; por ello, la evaluación del calcio arterial coronario (calcio-score) mediante CT es la prueba de elección, ya que evalúa la presencia de placas de ateroma calcificadas. En un estudio que evaluó mediante calcio-score a 101 pacientes candidatos a trasplante con diferentes etiologías (NALFD 5,94%), se encontró que el 19,8% presentaba valores de alto riesgo 78 Sesión 3 - ENFERMEDADES METABÓLICAS Y SISTÉMICAS de enfermedad arterial coronaria (> 400) y el 37,6% riesgo moderado18. La enfermedad aterosclerótica a nivel carotídeo es un factor independiente de accidentes cerebrovasculares19 y episodios cardiovasculares relacionados20, pudiendo determinarse mediante la medición del grosor de la íntima media carotídea u objetivando la presencia de placas ateromatosas en la arteria carótida. Un metaanálisis de nuestra Unidad21 analizó múltiples estudios (n = 14), que evaluaban la relación entre NAFLD y la enfermedad arteriosclerótica coronaria (n = 4) y la arteriosclerosis subclínica carotídea (n = 10). Con respecto a la primera entidad, el análisis de un total de 1.198 pacientes, en los que se había realizado angiografía coronaria diagnóstica, puso de manifiesto que la NALFD es un factor de riesgo independiente para la presencia de esta (odds ratio [OR]: 3,31; intervalo de confianza [IC] del 95%, 2,21-4,95). La relación de la esteatosis hepática y la arteriosclerosis subclínica se analizó en 10 estudios, 4 de ellos medían el grosor de la capa íntima media carotídea (CIMT) y 6 evaluaban la presencia de arteriosclerosis subclínica carotídea. Independientemente del método diagnóstico utilizado, en el análisis global se encontró una relación directa e independiente entre la esteatosis hepática y estos cambios subclínicos, con una OR global de 2,04 (IC del 95%, 1,65-2,51) y 4,41 (IC del 95%, 2,63-7,40), respectivamente. Mohammadi et al examinaron el engrosamiento de la capa íntima media de la arteria carótida y la vasodilatación mediada por flujo en la arteria braquial en 84 pacientes con diagnóstico de NAFLD (mediante ecografía) y 65 sujetos control, y concluyeron que las diferencias en los cambios morfológicos y funcionales eran significativas (p = 0,001). Los sujetos incluidos en el estudio eran menores de 45 años y no presentaban características propias de síndrome metabólico, por lo que los autores concluyeron que la influencia de NAFLD es una variable independiente asociada a la aterosclerosis subclínica en población de mediana edad sin síndrome metabólico22. En otro artículo publicado por el mismo autor, se compararon 250 pacientes con diagnóstico de NAFLD (mediante ecografía) con 85 sujetos control aleatorizados según edad y sexo, en relación con el grosor de la CIMT y la presencia de placas ateroscleróticas en dicha arteria. En este caso, sí se trata de individuos con síndrome metabólico y tras el análisis multivariante la presencia de NAFLD se relacionó con el engrosamiento de la CIMT (p = 0,001), independientemente de otros factores de riesgo aterogénicos asociados con el síndrome metabólico23. Por otro lado, Poanta et al publicaron un estudio de 56 sujetos con DM2 no complicada; los que presentaban esteatosis hepática (38/56; 67%) mostraban un índice mayor de factores de riesgo cardiovascular (índice de masa corporal, hipertensión arterial [HTA], valor de triglicéridos, disminución de cHDL), pero no un aumento significativo del grosor de la CIMT24. McKimmie et al estudiaron en pacientes diabéticos la relación entre el hígado graso (diagnosticado por tomografía computarizada) y el aumento de grosor de la CIMT, llegando a la conclusión de que se trataba más de un epifenómeno que de una verdadera asociación25. Sin embargo, Agarwal et al evaluaron a 124 individuos con DM2 en tratamiento, de los cuales 71 (57,2%) mostraban NAFLD (mediante ecografía). Los que presentaban NAFLD tuvieron una prevalencia más alta de factores de riesgo cardiovascular (HTA, índice de masa corporal, obesidad abdominal, dislipemia aterogénica) y un engrosamiento en la CIMT respecto a los que no presentaban hígado graso26. Estos hallazgos coinciden con los publicados por Sookoian et al27, donde ya se estableció relación entre valores patológicos de CIMT y arteriosclerosis carotídea y esteatosis hepática. A pesar de esta marcada relación, los estudios sobre la mortalidad cardiovascular son más escasos y menos clarificadores. Lazo et al incluyeron a 11.371 pacientes en un estudio para evaluar la mortalidad por causa cardiovascular, hepática o por cáncer. Los pacientes se clasificaron como NAFLD cuando presentaban esteatosis hepática (mediante ecografía) y normalidad en las transaminasas (n = 2.089; 16,4%); NASH (esteatohepatitis no alcohólica) cuando presentaban esteatosis hepática y elevación de las transaminasas (n = 426; 3,1%). No se detectó un aumento de mortalidad relacionada con la presencia de NAFLD o NASH28. Bhala et al compararon 247 pacientes con NAFLD y 264 pacientes con hepatitis C (näive o no respondedores). Ambas cohortes eran Child-Pugh A y tenían fibrosis avanzada o cirrosis (evaluada mediante biopsia hepática). No se detectaron diferencias en términos de mortalidad global ni de episodios cardiovasculares entre la cohorte NAFLD y hepatitis C; sin embargo, sí hubo una menor tasa de complicaciones hepáticas (incluyendo hepatocarcinoma) en la cohorte NAFLD29. En un estudio recientemente publicado por Wong et al se incluyeron 612 pacientes con indicación clínica de coronariografía. Los pacientes se clasificaron en NAFLD (n = 356; 58,2%) mediante ecografía o pacientes con ecografía hepática normal. Se consideró enfermedad coronaria significativa una estenosis > 50% en, al menos, una arteria coronaria, lo que se detectó en 456 sujetos (76%). Los pacientes con hígado graso mostraron una prevalencia de enfermedad coronaria significativa del 84,6% (301/356), mientras que esta cifra descendía hasta el 64,1% (164/256) si no mostraban NAFLD (p < 0,001). No obstante, NAFLD no pudo predecir la mortalidad cardiovascular en estos pacientes con enfermedad coronaria establecida30. La principal limitación de estos estudios se debe a la incapacidad para segregar a los pacientes con esteatosis simple y pacientes con esteatohepatitis mediante métodos no invasivos o biopsia hepática. Riesgo vascular en el paciente con esteatosis hepática: implicaciones en el entorno del trasplante hepático RIESGO CARDIOVASCULAR Y MORTALIDAD EN EL PACIENTE TRASPLANTADO Los pacientes trasplantados presentan una mayor incidencia de ECV, el 9% a los 5 años y el 25% a los 10 años31, comparado con la población general. Este aumento del riesgo es multifactorial y se asocia con alteraciones metabólicas, factores genéticos y medicación inmunosupresora. Wang et al32 han publicado un metaanálisis que incluía 9 estudios con 4.237 pacientes trasplantados, de los cuales 717 tenían NASH como causa de su enfermedad hepática. En este estudio se comprueba que no hay diferencias en la mortalidad a 1, 3 y 5 años entre pacientes con NASH u otras etiologías como motivo del trasplante. No obstante, sí se comprueba al analizar 6 de estos 9 estudios que la causa de muerte difiere entre los 2 grupos. Los pacientes con NASH tienen mayor riesgo de morir por episodios cardiovasculares (OR: 1,65; IC del 95%, 1,01-2,70; p = 0,05) y sepsis (OR: 1,71; IC del 95%, 1,17-2,50; p = 0,006). De hecho, la esteatohepatitis se ha identificado como una etiología de trasplante que está directamente relacionada con la aparición de isquemia miocárdica en el postrasplante 33. En cuanto a las infecciones, es conocido que la diabetes y la hipertensión predisponen a estas, siendo ambas entidades más prevalentes en pacientes con NASH, lo que puede explicar que los datos de mortalidad por sepsis en pacientes con NASH, 4-16%, sean superiores a los de los que no tienen NASH, 1-8%, de forma significativa32. Estos datos vienen a reafirmar las conclusiones publicadas en otros 2 estudios previos34,35. Sin embargo, los pacientes con NASH tienen un riesgo disminuido de morir por fallo del injerto (OR: 0,21; IC del 95%, 0,05-0,89; p = 0,03), en probable relación con una menor tasa de recurrencia de la enfermedad32. La recidiva de la NAFLD es prácticamente universal34 y la enfermedad esteatósica aparecerá de novo en un 25%36 de los pacientes. La obesidad constituye el factor de riesgo más importante en la recurrencia de la enfermedad37. De forma concomitante, tanto el uso de medicación inmunosupresora como los glucocorticoides se han implicado en la recurrencia o aparición de esteatosis de novo. La esteatosis en el postrasplante se asocia a biomarcadores de riesgo cardiovascular, como son ciertas lipoproteínas aterogénicas, y marcadores subclínicos de resistencia insulínica38. Y es que en estos pacientes existen concentraciones mayores de insulina en suero condicionado por un cierto grado de resistencia insulínica subclínica, que en el tejido adiposo conlleva la no supresión de la lipólisis y la liberación de ácidos grasos libres. Asimismo, también se ha demostrado, aunque no de forma significativa, la asociación entre la ciclosporina y las lipoproteínas aterogénicas y la del tacrolimus con parámetros indicativos de resistencia insulínica. 79 Bibliografía 1.Ludwig J, Viggiano T, McGill D, Oh BJ. Nonalcoholic steatohepatitis. Mayo clinic experiences with a hitherto unnamed disease. Mayo Clin Proc. 1980;55:434-8. 2.Loria P, Marchesini G, Nascimbeni F, Ballestri S, Maurantonio M, Carubbi F, et al. Cardiovascular risk, lipidemic phenotype and steatosis. A comparative analysis of cirrhotic and non-cirrhotic liver disease due to varying etiology. Atherosclerosis. 2014;232:99-109. 3.Cheung O, Sanyal AJ. Recent advances in nonalcoholic fatty liver disease. Curr Opin Gastroenterol. 2010;26:202-8. 4.Wilfred de Alwis NM, Day CP. Non-alcoholic fatty liver disease: the mist gradually clears. J Hepatol. 2008;48:S104-12. 5.Adams L, Lindor K, Angulo P. The prevalence of autoantibodies and autoimmune hepatitis in patients with nonalcoholic fatty liver disease. Am J Gastroenterol. 2004;99:1316-20. 6.Saadeh S, Younossi Z, Remer E, Gramlich T, Ong JP, Hurley M, et al. The utility of radiological imaging in nonalcoholic fatty liver disease. Gastroenterology. 2002;123:745-50. 7.Vernon G, Baranova A, Younossi ZM. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and nonalcoholic steatohepatitis in adults. Aliment Pharmacol Ther. 2011;34:274-85. 8.Leite NC, Salles GF, Araujo AL, Villela-Nogueira CA, Cardoso CR. Prevalence and associated factors of non-alcoholic fatty liver disease in patients with type-2 diabetes mellitus. Liver Int. 2009;29:113-9. 9.Musso G, Gambino R, Cassader M, Pagano G. Meta-analysis: Natural history of non-alcoholic fatty liver disease (NAFLD) and diagnostic accuracy of non-invasive tests for liver disease severity. Ann Med. 2011;43:617-49. 10.Fargion S, Porzio M, Fracanzani AL. Nonalcoholic fatty liver disease and vascular disease: state-of-the-art. World J Gastroenterol. 2014;20:13306-24. 11.Dam-Larsen S, Franzmann M, Andersen IB, Christoffersen P, Jensen LB, Sørensen TI, et al. Long term prognosis of fatty liver: risk of chronic liver disease and death. Gut. 2004;53:750-5. 12.Takahashi Y, Kurosaki M, Tamaki N, Yasui Y, Hosokawa T, Tsuchiya K, et al. Non-alcoholic fatty liver disease fibrosis score and FIB-4 scoring system could identify patients at risk of systemic complications. Hepatol Res. 2015;45:667-75. 13.Dec GW, Kondo N, Farrell ML, Dienstag J, Cosimi AB, Semigran MJ. Cardiovascular complications following liver transplantation. Clin Transplant. 1995;9:463-71. 14.Nair S, Verma S, Thuluvath PJ. Obesity and its effect on survival in patients undergoing orthotopic liver transplantation in the United States. Hepatology. 2002;35:105-9. 15.Zipprich A, Garcia-Tsao G, Rogowski S, et al Fleig WE, Seufferlein T, Dollinger MM. Prognostic indicators of survival in patients with compensated and decompensated cirrhosis. Liver Int. 2012;32:1407-14. 16.McAvoy NC, Kochar N, McKillop G, Newby DE, Hayes PC. Prevalence of coronary artery calcification in patients undergoing assessment for orthotopic liver transplantation. Liver Transpl. 2008;14:1725-31. 17.Giroud D, Li JM, Urban P, Meier B, Rutishauer W. Relation of the site of acute myocardial infarction to the most severe coronary arterial stenosis at prior angiography. Am J Cardiol. 1992;69:729-32. 18.McAvoy NC, Kochar N, McKillop G, Newby DE, Hayes PC. Prevalence of coronary artery calcification in patients under- 80 Sesión 3 - ENFERMEDADES METABÓLICAS Y SISTÉMICAS going assessment for orthotopic liver transplantation. Liver Transpl. 2008;14:1725-31. tality among US adults: prospective cohort study. BMJ. 2011;343:d6891. 19.Hermann DM, Gronewold J, Lehmann N, Seidel UK, Möhlenkamp S, Weimar C, et al. Intima-media thickness predicts stroke risk in the Heinz Nixdorf Recall study in association with vascular risk factors, age and gender. Atherosclerosis. 2012;224:84-9. 29.Bhala N, Angulo P, Van der Poorten D, Lee E, Hui JM, Saracco G, et al. The natural history of nonalcoholic fatty liver disease with advanced fibrosis or cirrhosis: An international collaborative study. Hepatology. 2011;54:1208-16. 20.Sillesen H, Muntendam P, Adourian A, Entrekin R, Garcia M, Falk E, et al. Carotid plaque burden as a measure of subclinical atherosclerosis: Comparison with other tests for subclinical arterial disease in the High Risk Plaque Bio Image study. JACC Cardiovasc Imaging. 2012;5:681-9. 21.Ampuero J, Gallego-Durán R, Romero-Gómez M. Association of NAFLD with subclinical atherosclerosis and coronary-artery disease: Meta-analysis. Rev Esp Enferm Dig. 2015;107:10-6. 22.Mohammadi A, Habibpour H, Ghasemi-Rad M. Evaluation of carotid intima-media thicknessand flow-mediated dilatation in middle-agedpatients with nonalcoholic fatty liver disease. Vasc Health Risk Manag. 2011;7:661-5. 23.Mohammadi A, Bazazi A, Ghasemi-Rad M. Evaluation of atherosclerotic findings in patients with nonalcoholic fatty liver disease. Int J Gen Med. 2011;4:717-22. 24.Poanta L, Albu A, Fodor D. Association between fatty liver disease and carotid atherosclerosis inpatients with uncomplicated type 2 diabetes mellitus. Med Ultrason. 2011;13:215-9. 25.McKimmie RL, Daniel KR, Carr JJ, Bowden DW, Freedman BI, Register TC, et al. Hepatic steatosis and subclinical cardiovascular disease in a cohort enriched for type 2 diabetes: the Diabetes Heart Study. Am J Gastroenterol. 2008;103: 3029-35. 26.Agarwal AK, Jain V, Singla S, Baruah BP, Arya V, Yadav R, et al. Prevalence of non-alcoholic fatty liver disease and its correlation with coronary risk factors in patients with type 2 diabetes. J Assoc Physicians India. 2011;59:351-4. 27.Sookoian S, Pirola CJ. Non-alcoholic fatty liver disease is strongly associated with carotid atherosclerosis: A systematic review. J Hepatol. 2008;49:600-7. 28.Lazo M, Hernaez R, Bonekamp S, Kamel IR, Brancati FL, Guallar E, et al. Non-alcoholic fatty liver disease and mor- 30.Wong V, Wong G, Yip G, Lo AO, Limquiaco J, Chu WC, et al. Coronary artery disease and cardiovascular outcomes in patients with non-alcoholic fatty liver disease. Gut. 2011;60:1721-7. 31.Ciccarelli O, Kaczmarek B, Roggen F, DeReyck C, Goffette P, Danse E, et al. Long-term medical complications and quality of life in adult recipients surviving 10 years or more after liver transplantation. Acta Gastroenterol Belg. 2005;68:323-30. 32.Wang X, Li J, Riaz DR, Shi G, Liu C, Dai Y. Outcomes of liver transplantation for nonalcoholic steatohepatitis: systematic review and meta-analysis. Clin Gastroenterol Hepatol. 2014;12:394-402. 33.Nicolau-Raducu R, Gitman M, Ganier D, Loss GE, Cohen AJ, Patel H, et al. Adverse cardiac events after orthotopic live transplantation: a cross-sectional study in 389 consecutive patients. Liver Transpl. 2015;21:13-21. 34.Charlton MR, Burns JM, Pedersen RA, Watt KD, Heimbach JK, Dierkhising RA. Frequency and outcomes of liver transplantation for nonalcoholic steatohepatitis in the United States. Gastroenterology. 2011;141:1249-53. 35.Afzali A, Berry K, Ioannou GN. Excellent posttransplant survival for patients with nonalcoholic steatohepatitis in the United States. Liver Transpl. 2012;18:29-37. 36.Contos MJ, Cales W, Sterling RK, Luketic VA, Shiffman ML, Mills AS, et al. Development of nonalcoholic fatty liver disease after orthotopic liver transplantation for cryptogenic cirrhosis. Liver Transpl. 2001;7:363-73. 37.Dureja P, Mellinger J, Agni R, Chang F, Avey G, Lucey M, et al. NAFLD recurrence in liver transplant recipients. Transplantation. 2011;91:684-9. 38.Idowu MO, Chhatrala R, Siddiqui MB, Driscoll C, Stravitz RT, Sanyal AJ, et al. De novo hepatic steatosis drives atherogenic risk in liver transplant recipients. Liver Transpl. 2015; doi:10.1002/lt.24223. [Epub ahead of print]. SESIÓN 3 ENFERMEDADES METABÓLICAS Y SISTÉMICAS Caso clínico: Enfermedad sistémica con manifestaciones hepáticas: fiebre, hepatoesplenomegalia y síndrome poliadenopático Joan Francesc Andrés Cordón y Joan Genescà Ferrer Servicio de Medicina Interna-Hepatología, Hospital Universitario Vall d’Hebron, Barcelona, España INTRODUCCIÓN Paciente de 74 años, que consultó por un cuadro clínico de 2 meses de evolución consistente en pérdida de aproximadamente 10 kg de peso y astenia, al que se añadió en los últimos días coluria y fiebre. Como antecedentes citar que se encontraba afectado de hipertensión arterial en tratamiento con hidroclorotiazida, asma en tratamiento con broncodilatadores inhalados, psoriasis palmoplantar e hiperuricemia. Como hábitos tóxicos destacar que era ex fumador, con un factor de exposición de 30 paquetes-año. A la exploración física se objetivó ictericia, hepatoesplenomegalia y adenopatías inguinales bilaterales. Analíticamente destacaba elevación de enzimas de colestasis (FA 350 UI/l, GGT 440 UI/l, bilirrubina 4,7 mg/dl) y elevación de reactantes de fase aguda (velocidad de sedimentación globular 120 mm/h). Las serologías de hepatitis virales (HBsAg, anti-VHC, VHE IgM), virus de la inmunodeficiencia humana y anticuerpos antimitocondriales fueron negativas. Una ecografía abdominal mostró hepatomegalia heterogénea con engrosamiento mural de colédoco y alguna ectasia segmentaria de la vía biliar intrahepática. Se realizó una colangiorresonancia, que evidenció hepatoesplenomegalia e irregularidades de vía biliar intra y extrahepática, así como múltiples adenopatías mesentéricas e hiliares hepáticas. Una tomografía por emisión de positrones-tomografía computarizada (PETTC) puso de manifiesto la presencia de una captación anormal de 18-FDG en adenopatías supra e infradiafragmáticas, bazo, hígado, hueso y peritoneo (fig. 1). La biopsia hepática y ganglionar resultó compatible con enfermedad por inmunoglobulina G4 (IgG4) (ratio células IgG4+/IgG+ del 74 y el 65%, respectivamente) (fig. 1). La biopsia ósea descartó malignidad y resultó también compatible con enfermedad por IgG4. La IgG4 en suero era de 1.900 mg/dl (rango normal: 5-156 mg/ dl). El recuento de plasmablastos en sangre periférica era del 9,7% del total de células B (rango normal: 0,11,5%). Tras iniciar corticoides a dosis de 1 mg/kg/día, el paciente experimentó una rápida mejoría del estado general, con desaparición de la fiebre en 24 h y mejoría progresiva de los parámetros de colestasis. ENFERMEDAD RELACIONADA CON IGG4 La enfermedad relacionada con IgG4 (ER-IgG4) es una entidad inmunomediada, que puede cursar con afectación multiorgánica y simular patologías malignas, infecciosas e inflamatorias. Su descripción y reconocimiento ha sido reciente (en 2001 como causa de pancreatitis autoinmune y en 2003 como enfermedad sistémica multiorgánica), por lo que muchos aspectos de su epidemiología, patogenia y tratamiento siguen siendo poco conocidos. Puede afectar a prácticamente cualquier órgano, aunque los más frecuentemente afectados son el páncreas y las glándulas exocrinas. Diversas patologías que se habían considerado previamente como entidades únicas (la tiroiditis de Riedel, el tumor de Küttner o la enfermedad de Ormond, entre otras) se consideran actualmente parte del espectro de la ER-IgG4. EPIDEMIOLOGÍA No se dispone de datos sobre la prevalencia o incidencia de la ER-IgG4 en la población general. En Japón se 81 82 Sesión 3 - ENFERMEDADES METABÓLICAS Y SISTÉMICAS Figura 1. A) Tomografía por emisión de positrones-tomografía computarizada (PET-TC) que muestra afectación hepática, esplénica, adenopática y ósea. B) Biopsia ganglionar con tinción inmunohistoquímica para IgG4. C) Biopsia hepática con tinción inmunohistoquímica para IgG4. estima que la prevalencia de pancreatitis autoinmune relacionada con IgG4 es de 2,2 casos por 100.000 habitantes. El paciente prototipo es un varón de edad mediana o avanzada; el predominio del sexo masculino es un importante factor que contrasta con otras enfermedades autoinmunes. FISIOPATOLOGÍA La fisiopatología de la enfermedad no está aclarada. Desde un punto de vista genético se ha descrito susceptibilidad de algunos subtipos de HLA. Se sugiere también una base de autoinmunidad o alergia, dada la frecuente positividad para anticuerpos antinucleares, elevación de IgE y eosinofilia periférica. También se considera la posibilidad de que infecciones bacterianas actúen como detonantes de la enfermedad y se ha propuesto el mimetismo molecular con Helycobacter pylori. Es importante conocer en más profundidad la molécula de IgG4, pues es un anticuerpo con función y estructura únicas. Representa menos del 5% del total de IgG en una persona sana y es la subclase menos abundante. A diferencia del resto de subclases, su concentración sérica varía en individuos sanos por un factor de 100, aunque las concentraciones individuales se suelen mantener estables. Las cadenas de IgG4 tienen pequeñas diferencias en cuanto a la composición de aminoácidos del segundo dominio constante, lo que les confiere una unión muy débil al C1q y al receptor Fcγ. Por lo tanto, se considera que la IgG4 no activa la vía clásica del complemento; consecuentemente es una Ig no inflamatoria. Otra característica única de la IgG4 es lo que se conoce como el intercambio del fragmento de unión al antígeno (antigen binding fragment), que se produce por la inestabilidad de los enlaces entre cadenas pesadas. Esta inestabilidad permite que las cadenas se separen y se recombinen aleatoriamente, formando anticuerpos asimétricos con 2 fragmentos de unión a antígenos distintos. Esto resulta en anticuerpos biespecíficos (aunque funcionalmente monovalentes) incapaces de formar inmunocomplejos. Se sabe también que las concentraciones de IgG4 se elevan tras el descenso de IgE en los trastornos alérgicos. Por lo tanto, una posible visión es que la IgG4 ha evolucionado como una Ig no inflamatoria que actúa como “limpiadora” de antígenos para atenuar procesos inflamatorios. Sin embargo, y aunque no hay evidencia al respecto, se ha descrito también la posibilidad de que la IgG4 fuera patogénica y actuara en colaboración con lectinas circulantes activando el complemento en las zonas de lesión por la enfermedad. Así pues, se teoriza que en un individuo genéticamente susceptible, un estímulo lesivo ambiental desencadena daño tisular y altera la inmunotolerancia. Esta alteración induciría una respuesta T CD4 helper inducida por antígenos propios, que provocaría un proceso fibrótico en una o diversas localizaciones. Los motivos de la afectación de órganos concretos se desconocen. En los órganos afectados, el incremento de células CD4 activaría células del sistema inmune innato, que a su vez secretarían otras citocinas que promueven la patología. Los linfocitos T CD4 de memoria que inician el proceso se perpetúan probablemente por la acción de células B presentadoras de antígeno (lo que explicaría la respuesta de la enfermedad a la depleción de células B). El mismo antígeno inicial, u otros procesos desencadenados por la fibrosis, producirían una respuesta paralela de células T, que induciría la formación de centros germinales en los ganglios linfáticos generando plasmablastos secretores de IgG4. Caso clínico: Enfermedad sistémica con manifestaciones hepáticas: fiebre, hepatoesplenomegalia y síndrome poliadenopático 83 Tabla 1. Criterios diagnósticos de la enfermedad relacionada con IgG4 Criterio clínico Presencia de masa localizada, difusa o engrosamiento que afecta a uno o más órganos Criterio hematológico IgG4 sérica > 135 mg/dl Criterio histopatológico Presencia de infiltrado linfoplasmocitario denso con fibrosis Presencia > 10 células IgG4+ por campo de gran aumento Ratio células IgG4+/células IgG+ > 40-50% HISTOLOGÍA DIAGNÓSTICO La histología es la clave para el diagnóstico de la ERIgG4. Hay 3 características centrales: infiltración linfoplasmocítica densa, flebitis obliterativa y fibrosis estoriforme. En los estadios iniciales, los tejidos afectados están difusa y densamente infiltrados por linfocitos T y células plasmáticas, con presencia aislada de macrófagos y eosinófilos. En segundo lugar, se produce fibrosis a partir de los fibroblastos y miofibroblastos presentes en el infiltrado inflamatorio, que característicamente se dispone en un patrón estoriforme. La lesión venosa característica es la flebitis obliterativa, que debe diferenciarse de la oclusión venosa fibrosa no inflamatoria que ocurre en otras patologías, como por ejemplo la colangitis esclerosante. La tinción inmunohistoquímica permite la detección de células plasmáticas IgG4+, que suelen infiltrar difusamente las lesiones. El número absoluto de células plasmáticas IgG4+ debe interpretarse según el órgano afectado. El ratio de células plasmáticas IgG4+/IgG+ debe ser > 40%. Con el curso de la enfermedad, la fibrosis se convierte en la característica predominante, cosa que puede dificultar el diagnóstico histológico en casos evolucionados. Las técnicas de imagen son frecuentemente la primera herramienta que identifica las lesiones tipo masa producidas por la ER-IgG4. En algunas ocasiones pueden llegar a permitir diferenciar entre masas inflamatorias o neoplásicas, pero el gold standard para el diagnóstico es el estudio histológico del tejido afectado. La PET-TC es especialmente útil en la valoración de la extensión de la enfermedad y en la monitorización de la actividad de la enfermedad en el seguimiento. Se han desarrollado unos criterios diagnósticos, que se resumen en la tabla 1. Si se cumplen los 3 criterios se considera un diagnóstico definitivo. Si se cumple el criterio clínico e histopatológico se considera probable y si solo se cumple el criterio clínico y hematológico se considera posible. No obstante, se recomienda la biopsia del órgano afectado siempre que sea técnicamente posible y no esté contraindicada. SEROLOGÍA Las concentraciones elevadas de IgG4 en suero no son lo suficientemente sensibles ni específicas para el diagnóstico de la enfermedad. Se ha descrito hasta un 20% de pacientes con pancreatitis autoinmune tipo I (relacionada con IgG4) con valores normales de IgG4 en suero. Asimismo, la elevación de IgG4 puede producirse en otras enfermedades e incluso en pacientes sanos. La identificación de plasmablastos en sangre mediante citometría de flujo es más sensible que la determinación de concentraciones de IgG4, pero no está disponible de forma generalizada. La monitorización de los valores de IgG4 puede ser útil en algunos pacientes, pues disminuyen significativamente tras la administración de corticoides. Sin embargo, se ha descrito que hasta en el 63% de los casos los valores no llegan a normalizarse. PRESENTACIÓN CLÍNICA Y DIAGNÓSTICO DIFERENCIAL Dada la afectación potencial de prácticamente todos los órganos, la ER-IgG4 plantea un diagnóstico diferencial amplio. La clínica aparece de forma subaguda y, además de síntomas específicos del órgano, se suele acompañar de astenia y pérdida de peso. La fiebre no es una manifestación frecuente. La tabla 2 resume las presentaciones clínicas y el diagnóstico diferencial según los órganos afectados de la ER-IgG4. AFECTACIÓN PANCREÁTICA, BILIAR Y HEPÁTICA La pancreatitis fue la primera afectación que se asoció a la elevación de IgG4 sérica. Se han descrito 2 tipos de pancreatitis autoinmune: tipo 1 (relacionada con IgG4) y tipo 2 (infiltración neutrofílica). La presentación clínica más frecuente de la pancreatitis autoinmune tipo 1 es la ictericia obstructiva inducida por una colangitis esclerosante concomitante relacionada con IgG4. La TC suele mostrar engrosamiento pancreático difuso con 84 Sesión 3 - ENFERMEDADES METABÓLICAS Y SISTÉMICAS Tabla 2. Presentación clínica y diagnóstico diferencial de la enfermedad relacionada con IgG4 (ER-IgG4) Órgano ER-IgG4 Diagnóstico diferencial Órbita Orbitis/miositis orbitaria Linfoma GPA Dacrioadenitis Oftalmopatía de Graves Sarcoidosis Área ORL Glándulas salivares Seudotumor orbitario Alergia Sarcoma Rinitis alérgica Faringitis Síndrome de Churg-Strauss Infecciones crónicas Poliposis nasal Traqueítis GPA Masas Inflamación de las cuerdas vocales Enfermedad de Mikulicz Linfoma Sarcoidosis Engrosamiento glándula submandibular Síndrome de Sjögren Sialolitiasis Xerostomía Meninges Paquimeningitis hipertrófica Paquimeningitis hipertrófica idiopática TMI Linfoma GPA Arteritits de células gigantes Histiocitosis de células de Langerhans Sarcoidosis Hipófisis Hipofisitis Neoplasias Histiocitosis Hipofisitis primaria Hipofisitis secundaria Adenopatías Tiroides Poliadenopatías generalizadas o en territorio de órgano afectado Tiroiditis de Riedel Enfermedad de Castleman Sarcoidosis Linfoma LES Linfoma Carcinoma papilar Pulmonar Engrosamiento broncovascular Neoplasias Granulomatosis linfomatoide Nódulos pulmonares TMI Neumonitis intersticial idiopática Opacidades en vidrio deslustrado Sarcoidosis Erdheim-Chester Engrosamiento pleural GPA Enfermedad intersticial Enfermedad de Castleman Continúa en página siguiente Caso clínico: Enfermedad sistémica con manifestaciones hepáticas: fiebre, hepatoesplenomegalia y síndrome poliadenopático 85 Tabla 2. Continuación Órgano ER-IgG4 Diagnóstico diferencial Vascular Aortitis Arteritis de Takayasu Histiocitosis Aneurismas aórticos Arteritis de células gigantes Linfoma Disección aórtica Sarcoidosis Aortitis infecciosa Afectación de vasos medianos Erdheim-Chester Periaortitis crónica Linfoma Retroperitoneo Sarcoma Fibrosis por metisergida Fibrosis idiopática Riñón Nefritis tubulointersticial Linfoma Glomerulopatía membranosa Carcinoma renal Nefritis tubulointersticial farmacológica Glomerulonefritis membranosa idiopática Glomerulonefritis necrosante pauciinmune Sarcoidosis Síndrome de Sjögren LES (nefropatía membranosa) Páncreas Pancreatitis autoinmune tipo I Neoplasia Hígado y vía biliar Colangitis esclerosante IgG4 Neoplasia pancreática Colecistitis Colangiocarcinoma Hepatopatía IgG4 Colangitis esclerosante primaria Seudotumor hepático Hepatocarcinoma Próstata Hipertrofia Hipertrofia benigna de próstata Piel Pápulas eritematosas o hiperpigmentadas Linfoma cutáneo GPA: granulomatosis con poliangeítis; LES: lupus eritematoso sistémico; ORL: otorrinolaringológica; TMI: tumor miofibroblástico inflamatorio. captación tardía de contraste y un borde hipodenso. El hallazgo de una estenosis difusa e irregular del conducto pancreático principal en la colangiopancreatografia endoscópica o en la colangiorresonancia es altamente específico de pancreatitis autoinmune. La PET-TC per- mite valorar la afectación de otros órganos, que puede dar soporte al diagnóstico de ER-IgG4. Para el diagnóstico histológico se recomienda la ecoendoscopia con punción. Es importante realizarla antes de plantear cualquier tratamiento, ya que se han descrito casos 86 Sesión 3 - ENFERMEDADES METABÓLICAS Y SISTÉMICAS de cáncer de páncreas en pacientes con pancreatitis tipo I. La pancreatitis tipo I se acompaña hasta en el 70% de los casos de colangitis esclerosante relacionada con IgG4, lo que la convierte en la manifestación extrapancreática más frecuente de la pancreatitis tipo I. Los principales diagnósticos diferenciales son la colangitis esclerosante primaria y el colangiocarcinoma. Ni las concentraciones de IgG4 sérica ni la morfología en la colangiografía permiten diferenciar claramente dichas patologías. Por lo tanto, se suele requerir una biopsia endoscópica transpapilar (o biopsia hepática en casos de afectación de la vía biliar intrahepática). Las biopsias endoscópicas permiten descartar la afectación neoplásica, pero raramente son lo suficientemente profundas para diferenciar claramente entre colangitis esclerosante primaria o por IgG4. Se ha descrito también colecistitis relacionada con IgG4, que suele cursar con engrosamiento mural asintomático. Sin tratamiento, la colangitis por IgG4 puede evolucionar a fallo hepático en meses. Aunque con menor frecuencia, también se ha descrito la afectación del parénquima hepático por infiltración de células plasmáticas IgG4+. Clínicamente, la presentación es similar a la de la hepatitis autoinmune. Por otra parte, se ha descrito también la afectación hepática en forma de seudotumor inflamatorio, con las características histológicas típicas de la ER-IgG4. TRATAMIENTO La ER-IgG4 responde de forma significativa al tratamiento inmunosupresor, aunque la respuesta está condicionada de forma importante por el grado de fibrosis. Actualmente, las recomendaciones de tratamiento se basan en estudios observacionales y consensos de expertos. El tratamiento de primera línea para la induc- ción a la remisión son los glucocorticoides. La duración de dicho tratamiento no está definida. Algunos grupos son partidarios de suspender el tratamiento a los 2-3 meses, mientras que otros defienden mantener dosis bajas de corticoides durante 2-3 años. Un estudio japonés mostró remisión completa del 61% con tratamiento con corticoides durante 1 año. El uso de agentes ahorradores de corticoides, como azatioprina o micofenolato, está extendido, pero no ha sido evaluado con estudios prospectivos. En los pacientes refractarios o con brotes recurrentes, pese a corticoides o inmunosupresores, se ha usado rituximab con buenos resultados. La eficacia del rituximab se podría explicar porque la depleción de células B elimina los precursores de las células plasmáticas productoras de IgG4 que están infiltrando los tejidos afectados. Bibliografía recomendada Islam AD, Selmi C, Datta-Mitra A, Sonu R, Chen M, Gershwin ME, et al. The changing faces of IgG4-related disease: Clinical manifestations and pathogenesis. Autoimmun Rev. 2015;14 914-22. Kamisawa T, Zen Y, Pillai S, Stone JH. IgG4-related disease. Lancet. 2015;385:1460-71. Khosroshahi A, Wallace ZS, Crowe JL, Akamizu T, Azumi A, Carruthers MN, et al; Second International Symposium on IgG4-Related Disease. International Consensus Guidance Statement on the Management and Treatment of IgG4-Related Disease. Arthritis Rheumatol. 2015;67:1688-99. Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med. 2012;366:539-61. Umemura T, Zen Y, Hamano H, Ichijo T, Kawa S, Nakanuma Y, et al. IgG4 associated autoimmune hepatitis: a differential diagnosis for classical autoimmune hepatitis. Gut. 2007;56:1471-2. Zen Y, Fujii T, Sato Y, Masuda S, Nakanuma Y. Pathological classification of hepatic inflammatory pseudotumor with respect to IgG4-related disease. Mod Pathol. 2007;20:884-94. SESIÓN 4 OTRAS ENFERMEDADES HEPÁTICAS Inclusión de candidatos en lista de espera de trasplante hepático: evaluación e indicación Manuel de la Mata Unidad Clínica de Aparato Digestivo, Unidad de Hepatología y Trasplante Hepático, Hospital Universitario Reina Sofía, Córdoba, España Instituto de Investigación Biomédica de Córdoba (IMIBIC), CIBERehd INTRODUCCIÓN El trasplante hepático (TH) es el tratamiento de elección para los pacientes con enfermedad hepática avanzada. En España se realizan más de 1.000 trasplantes al año gracias a una tasa de donantes por millón de población, que es la más elevada del mundo. Sin embargo, las indicaciones de TH se han visto incrementadas a lo largo de las 2 últimas décadas, lo que ha hecho necesario un procedimiento de evaluación multidisciplinar cuidadoso que permita incluir en lista de espera a los pacientes con una probabilidad mayor de supervivencia. INDICACIONES DEL TRASPLANTE HEPÁTICO Una gran variedad de enfermedades del hígado deben ser consideradas como indicación de trasplante, ya sea porque condicionan complicaciones derivadas de insuficiencia hepatocelular o bien por su naturaleza tumoral. Con menor frecuencia, la causa de una enfermedad grave que solo puede ser tratada mediante el trasplante es un trastorno genético o metabólico que reside en el hígado (tabla 1). Insuficiencia hepatocelular Las indicaciones generales por insuficiencia hepatocelular, en su mayor proporción por complicaciones derivadas de una cirrosis hepática en fase avanzada, suponen aproximadamente el 60% del total de trasplantes. En estos casos, el modelo MELD (model end-stage liver disease) predice la mortalidad a corto plazo sin trasplante y permite determinar el momento óptimo para la inclusión de los pacientes en lista de espera (cuando la puntuación es > 14), así como su priorización en lista. Con mucha menor frecuencia, el deterioro de la función hepática es brusco y ocurre sobre un hígado previamente sano (fallo hepático agudo). Las principales causas de fallo hepático agudo son hepatitis virales, fármacos y tóxicos, mientras que las causas más frecuentes de cirrosis hepática en nuestro medio son el consumo de alcohol y el virus de la hepatitis C. Mención especial requiere la hepatitis aguda alcohólica, que solo recientemente se contempla como posible indicación de trasplante. Desde hace muchos años se ha venido aplicando como requisito indispensable para poder ser trasplantado una abstinencia etílica superior a 6 meses. Este criterio, meramente convencional, se maneja en la actualidad con flexibilidad y se tienen en cuenta elementos clave de índole sociofamiliar y psicológicos que se estiman determinantes. Los resultados de un estudio reciente en Francia han mostrado un beneficio significativo, en términos de supervivencia, para casos muy seleccionados de pacientes con hepatitis aguda alcohólica refractaria a corticoides. La recidiva del consumo etílico fue poco frecuente y sin compromiso sobre la función del injerto. Indicaciones especiales o excepciones al MELD En un grupo de pacientes, la puntuación MELD no predice de forma adecuada la mortalidad a corto plazo, o bien la indicación de trasplante proviene de un deterioro significativo de la calidad de vida. El paradigma de este grupo de pacientes lo constituye el hepatocarcinoma, que por sí mismo justifica el 25% del total de los trasplantes hepáticos realizados en España. La mayoría de los centros de trasplante utilizan los criterios de Milán para seleccionar a estos pacientes (un nódulo único < 5 cm o con hasta 3 nódulos, todos < 3 cm, en ausencia de macroinvasión vascular o enfermedad extrahepática). La expansión de estos criterios, incrementando 87 88 Sesión 4 - OTRAS ENFERMEDADES HEPÁTICAS Tabla 1. Indicaciones de trasplante hepático Insuficiencia hepatocelular • Fallo hepático agudo • Cirrosis hepática avanzada – Virus de la Hepatitis B y C – Etílica – Esteatohepatitis no alcohólica – Autoinmune – Criptogenética • Hepatopatías colestásicas – Cirrosis biliar primaria – Colangitis esclerosante primaria • Retrasplante Indicaciones especiales Tumores hepáticos • Primarios – Carcinoma hepatocelular – Hemangioendotelioma – Colangiocarcinoma intrahepático • Secundarios – Metástasis de tumores neuroendocrinos – Ascitis refractaria – Encefalopatía crónica/recurrente – Colangitis recurrente – Prurito intratable – Hidrotórax recurrente Indicaciones inusuales • Enfermedades hereditarias/sistémicas – Hemocromatosis hereditaria – Enfermedad de Wilson – Poliquistosis hepática/hepatorrenal – Déficit de alfa-1-antitripsina – Polineuropatía amiloidótica familiar – Enfermedad de Caroli – Hipercolesterolemia familiar – Fibrosis quística – Hiperoxaluria primaria • Enfermedades vasculares hepáticas – Síndrome de Budd-Chiari – Enfermedad venooclusiva hepática Inclusión de candidatos en lista de espera de trasplante hepático: evaluación e indicación el número de nódulos permitidos o el tamaño máximo de dichos nódulos, se traduce en un aumento variable en la tasa de recidiva tumoral postrasplante, especialmente cuando en la pieza del explante se confirma la existencia de microinvasión vascular. Otro subgrupo relevante de indicaciones especiales lo conforman los pacientes con ascitis refractaria. En ellos, a pesar de su mal pronóstico, la puntuación MELD puede ser baja y no alcanzan prioridad en las listas de espera. Se ha propuesto aplicar en ellos una modificación del baremo MELD, que añade a su fórmula la concentración sérica de sodio. Con menor frecuencia, pero en un contexto similar, se encuentran los pacientes con hidrotórax refractario o los cirróticos con encefalopatía recurrente. La aparición de colangitis de repetición en el caso de la colangitis esclerosante primaria, de prurito invalidante o de osteopenia grave en el caso de la cirrosis biliar primaria, determina la indicación de TH, al margen del deterioro de la función hepática subyacente. Indicaciones inusuales Se trata de indicaciones de trasplante poco frecuentes (< 5%), cuya gravedad no está relacionada con el deterioro de la función hepática en todos los casos. En este grupo se sitúan una serie de enfermedades hereditarias con afectación sistémica, cuyo pronóstico está determinado por el hígado de uno u otro modo. En las enfermedades metabólicas, como la enfermedad de Wilson, y en la hemocromatosis, el depósito de cobre y de hierro, respectivamente, en el parénquima hepático produce un daño crónico que puede acabar en cirrosis y, sobre todo, en el caso de la hemocromatosis puede incrementar el riesgo de hepatocarcinoma. En estos casos, la presencia de enfermedad cardíaca (hemocromatosis) o neurológica (enfermedad de Wilson) significativa puede limitar la opción del trasplante. En otras enfermedades hereditarias, el trastorno primario se encuentra en el hígado, debido a la incapacidad de producir o aclarar ciertos mediadores que, a su vez, condicionan un daño extrahepático. Es el caso de la hipercolesterolemia familiar homocigota, en la que el déficit del receptor hepático de lipoproteínas de baja densidad hace que estas lipoproteínas se encuentren en concentraciones muy elevadas en sangre desde la infancia y ocasionen una ateromatosis acelerada y la aparición de episodios cardiovasculares en la adolescencia. En el déficit de alfa-1-antitripsina también se produce un daño, en este caso pulmonar, que favorece el desarrollo precoz de enfisema. En la hiperoxaluria primaria hay un déficit de ciertas enzimas encargadas de metabolizar el oxalato, en ausencia de las cuales este se acumula produciendo litiasis renal de repetición. La indicación del TH en estas enfermedades debe 89 ser precoz, antes de que se desarrollen complicaciones extrahepáticas que a menudo son irreversibles. CONTRAINDICACIONES PARA EL TRASPLANTE HEPÁTICO Algunas situaciones clínicas o anatómicas limitan el acceso al TH por reducir drásticamente la probabilidad de superar este procedimiento con éxito. No obstante, la relación de contraindicaciones para el trasplante de hígado se ha reducido a lo largo de las 2 últimas décadas debido a los notables avances medicoquirúrgicos que se han producido en las áreas de conocimiento de la hepatología y la cirugía hepatobiliar (tabla 2). En la actualidad, la mayoría de los programas de trasplante admiten pacientes entre 65 y 70 años, e incluso mayores. Por tanto, la edad per se no debe suponer una contraindicación absoluta en ausencia de comorbilidades. Es importante evaluar el riesgo cardiovascular del paciente. Desde el punto de vista cardiológico, las contraindicaciones más frecuentes son la miocardiopatía con disfunción ventricular grave, la cardiopatía isquémica avanzada, la hipertensión pulmonar grave (presión arteria pulmonar > 50 mmHg) y las valvulopatías con repercusión funcional significativa. Las contraindicaciones pulmonares incluyen cualquier patología irreversible que condicione insuficiencia respiratoria crónica, excluido el síndrome portopulmonar. La presencia o los antecedentes recientes de enfermedad neoplásica suponen una contraindicación formal para el trasplante, debido a que la inmunosupresión aceleraría la progresión de la enfermedad tumoral y agravaría su pronóstico. Se debe contraindicar el TH en todo paciente con infección activa no controlada a pesar de tratamiento Tabla 2. Contraindicaciones para el trasplante hepático • Enfermedad cardíaca o pulmonar grave • Sida • Abuso de alcohol o drogas • Hepatocarcinoma con extensión extrahepática • Sepsis no controlada • Trombosis portal masiva • Neoplasia extrahepática • Enfermedad psiquiátrica grave • Falta de adherencia o soporte social mínimo 90 Sesión 4 - OTRAS ENFERMEDADES HEPÁTICAS antibiótico. La infección por virus de la inmunodeficiencia humana ha dejado de ser una contraindicación absoluta cuando se encuentra bien controlada con medicación antirretroviral. En la mayoría de los casos, el trasplante se indica por cirrosis asociada a virus C y debe contarse con que la recidiva viral reduce de modo significativo la supervivencia a los 5 años. No obstante, en el escenario de los nuevos antivirales es muy probable que se amplíe el acceso al trasplante de este subgrupo de pacientes. La evaluación psicológica debe identificar a los pacientes incapaces de comprender el procedimiento del TH, la importancia de los cuidados y de las revisiones tras este. Es importante contar con apoyo familiar para facilitar la adherencia al tratamiento y a las revisiones. En ausencia de estas condiciones se debe contraindicar el trasplante. El alcoholismo activo y el consumo activo de drogas son una contraindicación formal para el TH. La existencia de ciertas variantes anatómicas vasculares puede dificultar el procedimiento quirúrgico. La trombosis extensa del eje esplenoportal es para la mayoría de los grupos una contraindicación para el TH. Cuando la trombosis se limita a la vena porta principal puede hacerse una trombectomía durante el trasplante, aunque el riesgo de trombosis portal postrasplante aumenta de forma significativa. PRIORIZACIÓN EN LISTA DE ESPERA A lo largo de la última década, las unidades de TH han implantado en sus programas un modelo de gestión de la lista de espera basado en la gravedad de los receptores. Desde el año 2002 se ha generalizado el uso del baremo MELD para seleccionar de modo objetivo a los pacientes con mayor riesgo de mortalidad pretrasplante. Sin embargo, el sistema MELD no está exento de limitaciones, ya que como se ha citado previamente, en un considerable número de pacientes no refleja adecuadamente la gravedad de la insuficiencia hepática. Asimismo se ha cuestionado el impacto que sobre las variables que lo determinan pueden tener situaciones transitorias como la deshidratación o el uso de fármacos diuréticos o anticoagulantes orales. Otra limitación importante es que no predice la mortalidad postrasplante. Para afrontar estas debilidades del modelo MELD se han introducido modificaciones empíricas para las denominadas indicaciones especiales o excepciones al MELD. Entre ellas se incluye el hepatocarcinoma, que supone aproximadamente el 25% de los trasplantes anuales en España, en el que el principal riesgo es la exclusión por progresión de la enfermedad, que se ha estimado entre el 10-25% a los 6 y 12 meses, respectivamente. Emparejamiento donante-receptor: matching En la gestión de la lista de espera es clave el concepto de emparejamiento donante-receptor (matching), que se ha desarrollado de modo paralelo a los intentos de definición de donante subóptimo, es decir, aquel donante que por tener ciertas características, como edad avanzada, esteatosis hepática moderada-grave, estancia prolongada en unidad de cuidados intensivos o un tiempo prolongado de isquemia, puede aumentar el riesgo de complicaciones postrasplante, particularmente de fallo primario del injerto. Por otra parte, también en el receptor confluyen factores de riesgo que influyen negativamente sobre el resultado del trasplante: edad avanzada, sodio sérico bajo y elevación rápida de la puntuación MELD pretrasplante. Sin embargo, es la combinación de factores del donante y del receptor lo que compromete de forma significativa la viabilidad del TH. El sistema de matching ideal debería incluir una valoración multivariante basada en parámetros objetivos y fáciles de medir, que identificasen la pareja donante-receptor que ofreciese una mayor supervivencia postrasplante al paciente (principio de utilidad). Los modelos publicados hasta el momento actual, entre los que destacan el donor risk index (DRI), el sistema D-MELD y el sistema BAR, están lejos de lo que debería ser un matching real en la práctica clínica, bien por su excesiva simplicidad o bien por la ausencia de validación externa prospectiva, entre otras limitaciones. Recientemente se ha propuesto un modelo basado en redes neuronales y modelos de análisis computacional denominado sistema MADRE, capaz de combinar un elevado número de variables que permitiría un matching donantereceptor objetivo, con resultados preliminares muy prometedores. Bibliografía recomendada Berenguer J, Parrilla P. Trasplante hepático. Madrid: Elsevier; 2008. Briceño J, Ciria R, De la Mata M. Donor-recipient matching: Myths and realities. J Hepatol. 2013;58:811-20. Briceño J, Cruz-Ramírez M, Prieto M, Navasa M, Ortiz de Urbina J, Orti R, et al. Use of artificial intelligence as an innovative donor-recipient matching model for liver transplantation: Results from a multicenter Spanish study. J Hepatol. 2014;61:1020-8. Cuervas-Mons V. Receptores de trasplante hepático de alto riesgo. Gastroenterol Hepatol. 2006;29:65-9. Martin P, Dimartini A, Feng S, Brown R, Fallon M. Evaluation for liver transplantation in adults: 2013 Practice guideline by the American Association or the study of Liver Diseases and the American Society of Transplantation. Hepatology. 2014;59:1144-65. Registro Español de Trasplante Hepático. Disponible en: http:// www.sethepatico.org SESIÓN 4 OTRAS ENFERMEDADES HEPÁTICAS Cirrosis biliar primaria: nuevas opciones de tratamiento Albert Parés Unidad de Hepatología, Hospital Clínic, IDIBAPS, CIBERehd, Universidad de Barcelona, Barcelona, España INTRODUCCIÓN La cirrosis biliar primaria (CBP) es una enfermedad colestática crónica de presunta patogenia autoinmune, que se caracteriza por la inflamación y lesión de conductos intrahepáticos de mediano calibre, que finalmente produce una cirrosis. La presentación clínica ha cambiado en gran medida en las últimas décadas, y ahora la mayoría de los pacientes son mujeres de mediana edad, asintomáticas, con alteraciones bioquímicas que se caracterizan por aumentos poco importantes de la fosfatasa alcalina (FA), gammaglutamil transferasa (GGT) y transaminasas y con lesiones iniciales en la biopsia hepática1,2. Estos cambios están asociados con una supervivencia prolongada y, en consecuencia, el objetivo principal del tratamiento es mejorar la colestasis y evitar la progresión del daño hepático, que finalmente dará lugar a una cirrosis, y en consecuencia alargar la supervivencia libre de trasplante. Por lo tanto, el tratamiento de la CBP está orientado a corregir el daño hepático debido a la inflamación de las vías biliares mediante tratamientos específicos y también atenuar la sintomatología y las consecuencias de la colestasis crónica, particularmente el prurito, la fatiga, la osteoporosis y las deficiencias de vitaminas liposolubles. TRATAMIENTOS ESPECÍFICOS Debido a la supuesta patogenia autoinmune de la enfermedad se han realizado una serie de estudios aleatorizados, observacionales y piloto, que han evaluado el efecto de agentes como la azatioprina, el clorambucil, el metotrexato, el micofenolato mofetilo, la prednisona y la ciclosporina. También se han evaluado otros fármacos como la penicilamina, la colchicina, el malotilato, la talidomida, la silimarina, la atorvastatina y la simvastatina1,2. No hay resultados concluyentes de estos estudios e incluso en algunos casos estos fármacos han sido perjudiciales. El ácido ursodesoxicólico (AUDCl) se ha convertido en el único fármaco aprobado para el tratamiento de la CBP, basado en evidencias experimentales y clínicas, incluyendo la estimulación hepatocelular y la secreción ductular, la citoprotección frente a la lesión inducida por el ácido biliar, el aumento del índice de hidrofilicidad del conjunto de ácidos biliares circulante y más inmunomodulador, antiinflamatorio y efecto3,4 antiapoptótico. Ácido ursodesoxicólico y respuesta terapéutica Durante las últimas 2 décadas, varios estudios han indicado que el AUDCl (13-16 mg/kg/día) es el tratamiento de elección en pacientes con CBP, basado en algunos ensayos controlados con placebo y otras evidencias más recientes. El primer estudio piloto demostró que el AUDCl disminuyó marcadamente las FA y las transaminasas con pocos efectos adversos5. Estos resultados sobre la mejoría bioquímica hepática se confirmaron en ensayos controlados de AUDCl comparados con placebo, en los que se observó una disminución significativa de los valores de FA, GGT, colesterol, bilirrubina sérica e inmunoglobulina M6-10, así como la falta de progresión del estadio histológico y la mejoría de algunas lesiones histológicas10, especialmente en los pacientes con estadios iniciales de CBP11. A pesar de estos efectos favorables, el uso de AUDCl en la CBP se ha cuestionado porque no hay resultados claros respecto a la supervivencia o el tiempo para el trasplante de hígado. De hecho, los efectos del AUDCl sobre la supervivencia solo se han demostrado en un análisis que combina los datos originales de 3 ensayos clínicos frente a placebo. El tratamiento con AUDCl se asoció con una reducción significativa de la probabilidad de un trasplante o fallecimiento. Este beneficio se 91 92 Sesión 4 - OTRAS ENFERMEDADES HEPÁTICAS Probabilidad de supervivencia libre de trasplante 1,0 0,8 0,6 0,4 0,2 0,0 0 4 8 12 16 20 Años Figura 1. Supervivencia libre de trasplante en pacientes con cirrosis biliar primaria y respuesta óptima al tratamiento con ácido ursodesoxicólico según el criterio de Barcelona, en comparación con la población estándar de edad y sexo similares. observó en los pacientes con enfermedad moderada y grave, pero no en aquellos con enfermedad leve (concentración de bilirrubina sérica < 1,4 mg/dl, en estadio histológico I o II en quienes la progresión a la etapa terminal de la enfermedad no se produjo durante este corto intervalo de tiempo)12. Los resultados de la terapia AUDCl sobre la supervivencia se han cuestionado por algunos metaanálisis13,14 y el análisis Cochrane15. Sin embargo es conveniente señalar que en la mayoría de los estudios incluidos en el metaanálisis, el tratamiento duró 2 años, que probablemente es demasiado corto para determinar la eficacia, en términos de supervivencia, de cualquier tratamiento de una enfermedad como la CBP, que tiene una supervivencia estimada mucho más larga. Otro metaanálisis, en el cual se excluyeron los estudios de corta duración (menos de 24 meses) y los que utilizaron una dosis baja de AUDCl (< 10 mg/kg/día) concluyó que el AUDCl mejoraba significativamente la supervivencia libre de trasplante y retrasaba la progresión histológica en pacientes con enfermedad inicial16. Por tanto, la dosis apropiada de AUDCl es crítica, como se ha demostrado en los ensayos controlados frente a placebo y en otro estudio que comparaba 3 dosis diferentes y que mostró que la dosis óptima de AUDCl era de 13 a 15 mg/kg de peso17. Estudios más recientes han demostrado los efectos favorables del AUDCl sobre la supervivencia a largo plazo en pacientes que recibieron dosis estándar (13 a 16 mg/kg/día)18-23. El primer estudio, que incluyó a pacientes tratados durante una mediana de 7,5 años, indica claramente que el AUDCl mejora la supervivencia en comparación con la supervivencia esperada según el índice pronóstico de la Clínica Mayo18, pero significativamente más corta que la predicha para la población estándar de similares edad y sexo. Estas diferencias pueden explicarse por la respuesta bioquímica desigual al AUDCl, tal como se define por una normalización o descenso > 40% en los valores de FA al cabo de 1 año de tratamiento18. Posteriormente, otros análisis han demostrado que la respuesta bioquímica a AUDCl evaluada al cabo de 6 meses o 1 año predice claramente el pronóstico a largo plazo, ya que en los respondedores al AUDCl, la supervivencia fue similar a la estimada para la población control (fig. 1). Se han propuesto varias definiciones de respuesta bioquímica óptima al AUDCl, y aproximadamente el 40% de los pacientes cumplen al menos 1 de los criterios de respuesta bioquímica incompleta (tabla 1)19-23. Estos pacientes tienen un mayor riesgo de presentar manifestaciones de progresión de la enfermedad y menor supervivencia. Por lo tanto, los pacientes con respuesta bioquímica subóptima al AUDCl definen el grupo de pacientes que precisan otros tratamientos, solos o asociados a AUDCl. El AUDCl también previene el desarrollo de carcinoma hepatocelular en la CBP, ya que disminuye la progresión de la enfermedad y, como consecuencia, reduce la probabilidad de desarrollar cirrosis, que es el principal factor de riesgo para cáncer primario de hígado24. Ciertamente, varios estudios apoyan este efecto del AUDCl para reducir el riesgo de desarrollo de un carcinoma hepatocelular25,26. Cirrosis biliar primaria: nuevas opciones de tratamiento 93 Tabla 1. Criterios bioquímicos de respuesta óptima al ácido ursodesoxicólico Criterio Tiempo Respuesta bioquímica insuficiente al tratamiento con ácido ursodesoxicólico Barcelona18 1 año FA > 1 × LAN o descenso de FA ≤ 40% I19 1 año FA ≥ 3 × LAN o AST ≥ 2 × LAN o bilirrubina > 1 mg/dl Rotterdam20 1 año Bilirrubina y/o albúmina anormal Toronto21 2 año A FA > 1,67 × LAN Paris II22 1 año FA ≥ 1,5 × LAN o AST ≥ 1,5 × LAN o bilirrubina ≥ 1 mg/dl Beijing23 6 meses FA > 3 LAN, bilirrubina aumentada, albúmina baja Paris AST: aspartato aminotransferasa; FA: fosfatasa alcalina; LAN: límite alto de normalidad. TRATAMIENTOS ALTERNATIVOS EN PACIENTES CON RESPUESTA BIOQUÍMICA SUBÓPTIMA A ÁCIDO URSODESOXICÓLICO Fibratos En los últimos 15 años se han publicado los efectos potencialmente beneficiosos de los fibratos en la CBP. Las características de estos estudios se relacionan en la tabla 2. Los primeros estudios de Japón describían una mejoría de la colestasis al administrar fibratos (bezafibrato o fenofibrato). Posteriormente se llevaron a cabo algunos estudios prospectivos para evaluar la eficacia de añadir fibratos a AUDCl en pacientes con insuficiente respuesta al tratamiento convencional. Estos estudios, sin embargo, tienen algunas limitaciones como un corto período de tratamiento, el escaso número de pacientes, la falta de un grupo control y el uso de dosis bajas de AUDCl27-32. Más recientemente, el tratamiento de la CBP con fibratos se ha evaluado en países occidentales. Un estudio realizado en el Reino Unido en 16 pacientes sin respuesta bioquímica observó una disminución significativa de FA después de añadir fenofibrato (160 mg/día) al tratamiento con AUDCl en el 90% de pacientes33. También se han descrito efectos favorables de los fibratos en otros 2 estudios de 834 y 6 pacientes35. Otro estudio realizado en 20 pacientes sin respuesta bioquímica suficiente observó una disminución significativa en la colestasis (FA) y la citólisis, así como de algunos marcadores de inflamación como la IL-1 y la IL-6. Estos datos son parecidos a los de una observación de nuestro centro, que ha evaluado a 30 pacientes sin respuesta bioquímica a AUDCl o con FA > 1,5 veces la normalidad37. La adición de bezafibrato (400 mg/día) tiene unos claros efectos favorables, que se caracterizan por la normalización o una rápida y profunda disminución de los indicadores de colestasis (fig. 2) y citólisis al cabo de 1 año, así como una disminución notable del prurito. La interrup- ción del bezafibrato durante 3 meses se asoció con un aumento significativo de los marcadores de colestasis y la reaparición del prurito. El efecto del bezafibrato fue más notorio en los pacientes con enfermedad más leve. Otros estudios han constatado este efecto38. Los mecanismos anticolestáticos de los fibratos no están completamente dilucidados, pero la acción principal se puede deber a que es un agonista del PPAR que tiene una acción muy variada sobre transportadores biliares y receptores nucleares que regulan la excreción biliar y la inflamación38-40; también se puede deber a un efecto agonista sobre el receptor nuclear del pregnano X. No se observaron efectos adversos graves asociados al tratamiento combinado con fibratos, pero algunos pacientes tienen dispepsia o náuseas. En resumen, los datos de los diferentes estudios de los fibratos en CBP son comparables. En última instancia concluyen que el tratamiento combinado con fibratos en pacientes sin respuesta bioquímica óptima a AUDCl podría ser útil, ya que mejoraría el grado de colestasis. Sin embargo, todavía se necesitan más estudios con una cohorte de pacientes más grande y con seguimiento a largo plazo para determinar la eficacia y la seguridad a largo plazo de este tratamiento, y el efecto sobre la historia natural y la supervivencia. Un ensayo en fase III está en curso en Europa para evaluar el bezafibrato como terapia adyuvante al AUDCl. Nuestra experiencia reciente indica que el tratamiento es seguro, especialmente en pacientes con enfermedad inicial, pero otros estudios contemplan la posibilidad de ausencia de efectos sobre la supervivencia a largo plazo con aumento de la concentración de creatinina. Este estudio solo incluyó 13 pacientes que recibieron tratamiento combinado con bezafibrato (Am J Gastro 2015). Budesonida La budesonida es un glucocorticoide no halogenado que se absorbe en el intestino delgado y con la acti- 94 Sesión 4 - OTRAS ENFERMEDADES HEPÁTICAS Tabla 2. Estudios que han evaluado el tratamiento con fibratos en la cirrosis biliar primaria Autor País Año Dosis (mg/día) Dosis AUDCl (mg/día) Número de pacientes Duración (meses) Resultados mejoría Efectos adversos Nakai et al27 Japón 2000 400 600 10 12 FA, ALT, GGT, IgM No Kurihara et al28 Japón 2000 400 12 12 FA, ALT, GGT, IgM No Kanda et al30 Japón 2002 400 600 11 6 FA No Akbar et al31 Japón 2005 400 600 10 12 FA, ALT, GGT, IgM, colesterol No Kita et al32 Japón 2006 400 600 22 6 FA, GGT No Hazzan y Tur-Kaspa34 Israel 2010 400 9001.500 8 4 a 12 FA, GGT No Honda et al38 Japón 2013 400 600 19 3 FA, GGT, ALT, colesterol, TGR, IgM No Lens et al37 España 2014 400 13-15/kg 30 12 FA, GGT, ALT, colesterol, TGR Pirosis Ohira et al29 Japón 2002 150 a 200 600-900 7 6 FA, GGT, IgM No Walker et al33 UK 2009 134 a 200 16 3a4 FA, IgM No Liberopoulus et al35 Grecia 2010 200 600 6 2 FA, GGT No Levy et al36 USA 2011 160 13-15/kg 20 12 FA, AST, IgM, IL-1, IL-6 Esofagitis, aumento ALT Bezafibrato Fenofibrato ALT: alanina aminotransferasa; AST: aspartato aminotransferasa; AUDCl: ácido ursodesoxicólico; FA: fosfatasa alcalina; GGT: gammaglutamil transferasa; IgM: inmunoglobulina M; IL-1: interleucina-1; IL-6: interleucina-6; TGR: triglicéridos. vidad de unión al receptor de glucocorticoides de 15 a 20 veces mayor que la prednisolona. Dos ensayos aleatorios mostraron que la combinación de budesonida (6 a 9 mg/día) con AUDCl es más eficaz para mejorar las alteraciones bioquímicas hepáticas y la histología que la monoterapia con AUDCl en pacientes no cirróticos41,42. El estudio más reciente, prospectivo, aleatorizado, abierto y multicéntrico, que incluyó a 77 pacientes con CBP, mostró que el tratamiento con budesonida durante 3 años se asoció a una disminución significativa de las alteraciones bioquímicas hepáticas en los 2 brazos de tratamiento, pero el estadio histológico y la fibrosis mejoraron en el grupo de tratamiento combinado y se deterioraron en el grupo de monoterapia con AUDCl41. El mismo efecto positivo de la terapia combinada se ha descrito sobre la mejoría de la histología hepática (estadio, fibrosis e inflamación) y los valores de laboratorio, pero con una dosis más alta de budesonida (9 mg/día) y durante 2 años42. Otro estudio reportó beneficios mínimos al tratamiento combinado43. Los efectos secundarios relacionados con la budesonida fueron leves y claramente menos notables que los producidos por la prednisona. Sin embargo, la budesonida no debe administrase a pacientes con cirrosis, ya que puede suponer un mayor riesgo de trombosis portal44. En resumen, estos resultados son importantes, pero el efecto del tratamiento combinado con budesonida deberá establecerse en un ensayo aleatorizado de gran tamaño, que se está realizando actualmente en Europa (Número EudraCT: 2007004040-70). Mientras tanto, este tratamiento se puede ensayar en pacientes sin óptima respuesta bioquímica Cirrosis biliar primaria: nuevas opciones de tratamiento ticas49,50. Los ensayos clínicos de este nuevo agente indican que es claramente eficaz en la CBP51,52. La administración de OCA en pacientes que reciben AUDCl reduce en aproximadamente el 25% los valores de FA, de las transaminasas y la GGT, así como de la IGM y de C4, un marcador de la síntesis de ácidos biliares. En otros ensayos también se ha constatado este efecto anticolestático del OCA. Bezafibrato 400 mg/día 2.700 2.400 Fosfatasa alcalina (µ/l) 95 2.100 1.800 1.500 1.200 900 Otros tratamientos 600 300 0 0 3 6 9 12 15 18 Meses Figura 2. Cambios en los valores de fosfatasa alcalina en los pacientes con cirrosis biliar primaria tratados con ácido ursodesoxicólico que recibieron bezafibrato. La parada del tratamiento se siguió de un repunte de la fosfatasa alcalina que volvió a bajar al reiniciar bezafibrato. *p < 0,001 frente a basal; **p < 0,001 frente a 12 y 18 meses. El área sombreada indica los valores normales de fosfatasa alcalina. Tomada de referencia 37. Debido a las mejoras recientes en la comprensión de la patogenia de la enfermedad, actualmente se están ensayando un número variado de nuevas moléculas frente al antígeno CD2054 o frente a IL-1255. También se está evaluando un inhibidor del transportador de ácidos biliares dependiente del sodio, localizado en los enterocitos, y una nueva proteína recombinante derivada del FGF19. Bibliografía 1.European Association for the Study of the Liver. EASL Clinical Practice Guidelines: management of cholestatic liver diseases. J Hepatol. 2009;51:237-67. 2.Lindor KD, Gershwin ME, Poupon R, Kaplan M, Bergasa NV, Heathcote EJ; American Association for Study of Liver Diseases. Primary biliary cirrhosis. Hepatology. 2009;50:291-308. a AUDCl y probablemente en pacientes con mayores índices de inflamación. Ácido obeticólico El ácido obeticólico (ácido 6-alfa-etil-quenodesoxicólico, OCA) es un nuevo derivado sintético del ácido quenodesoxicólico45-47 y es 100 veces más potente como agonista del receptor farnesoide X (FXR). El FXR se expresa en el hígado, el intestino, las glándulas suprarrenales y los riñones y tiene un papel importante en la circulación enterohepática de los ácidos biliares. Entre otras funciones, incluyendo la regulación de los aspectos clave del metabolismo de carbohidratos y lípidos, reduce la síntesis de ácidos biliares hidroxilasa, actuando sobre la enzima 7 alfa-colesterol hidroxilasa y disminuyendo la entrada de ácidos biliares en el hepatocito. Además, la activación de FXR aumenta la expresión del transportador MDR3 que exporta bilirrubina y fosfolípidos. Asimismo, el FXR estimula el factor de crecimiento de fibroblastos 19 (FGF19) en el intestino, que está implicado en un mecanismo de paso de sales biliares desde el intestino a los hepatocitos y que impide la síntesis de ácidos biliares48. Estudios preclínicos en diversos modelos animales han demostrado que el OCA tiene propiedades coleréticas y antibió- 3.Beuers U. Drug insight: Mechanisms and sites of action of ursodeoxycholic acid in cholestasis. Nat Clin Pract Gastroenterol Hepatol. 2006;3:318-28. 4.Poupon R. Ursodeoxycholic acid and bile-acid mimetics as therapeutic agents for cholestatic liver diseases: an overview of their mechanisms of action. Clin Res Hepatol Gastroenterol. 2012;36:S3-12. 5.Poupon R, Chretien Y, Poupon RE, Ballet F, Calmus Y, Darnis F. Is ursodeoxycholic acid an effective treatment for primary biliary cirrhosis? Lancet. 1987;1:834-6. 6.Poupon RE, Balkau B, Eschwege E, Poupon R. A multicenter, controlled trial of ursodiol for the treatment of primary biliary cirrhosis. UDCA-PBC Study Group. N Engl J Med. 1991;324:1548-54. 7.Heathcote EJ, Cauch-Dudek K, Walker V, Bailey RJ, Blendis LM, Ghent CN, et al. The Canadian Multicenter Double-blind Randomized Controlled Trial of ursodeoxycholic acid in primary biliary cirrhosis. Hepatology. 1994;19:1149-56. 8.Lindor KD, Dickson ER, Baldus WP, Jorgensen RA, Ludwig J, Murtaugh PA, et al. Ursodeoxycholic acid in the treatment of primary biliary cirrhosis. Gastroenterology. 1994;106: 1284-90. 9.Combes B, Carithers RL Jr, Maddrey WC, Lin D, McDonald MF, Wheeler DE, et al. A randomized, double-blind, placebo-controlled trial of ursodeoxycholic acid in primary biliary cirrhosis. Hepatology. 1995;22:759-66. 10.Parés A, Caballeria L, Rodés J, Bruguera M, Rodrigo L, GarcíaPlaza A, et al. Long-term effects of ursodeoxycholic acid in primary biliary cirrhosis: results of a double-blind controlled multicentric trial. UDCA- Cooperative Group from the Spanish 96 Sesión 4 - OTRAS ENFERMEDADES HEPÁTICAS Association for the Study of the Liver. J Hepatol. 2000;32: 561-6. 11.Poupon RE, Lindor KD, Pares A, Chazouillères O, Poupon R, Heathcote EJ. Combined analysis of the effect of treatment with ursodeoxycholic acid on histologic progression in primary biliary cirrhosis. J Hepatol. 2003;39:12-6. 12.Poupon RE, Lindor KD, Cauch-Dudek K, Dickson ER, Poupon R, Heathcote EJ. Combined analysis of randomized controlled trials of ursodeoxycholic acid in primary biliary cirrhosis. Gastroenterology. 1997;113:884-90. 13.Goulis J, Leandro G, Burroughs A. Randomised controlled trials of ursodeoxycholic-acid therapy for primary biliary cirrhosis: a meta-analysis. Lancet. 1999;354:1053-60. 14.Gong Y, Huang Z, Christensen E, Gluud C. Ursodeoxycholic acid for patients with primary biliary cirrhosis: an updated systematic review and meta-analysis of randomized clinical trials using Bayesian approach as sensitivity analyses. Am J Gastroenterol. 2007;102:1799-807. 15.Rudic JS, Poropat G, Krstic MN, Bjelakovic G, Gluud C. Ursodeoxycholic acid for primary biliary cirrhosis. Cochrane Database Syst Rev. 2012;CD:000551. 16.Shi J, Wu C, Lin Y, Chen YX, Zhu L, Xie WF. Long-term effects of mid-dose ursodeoxycholic acid in primary biliary cirrhosis: a meta-analysis of randomized controlled trials. Am J Gastroenterol. 2006;101:1529-38. 17.Angulo P, Dickson ER, Therneau TM, Jorgensen RA, Smith C, DeSotel CK, et al. Comparison of three doses of ursodeoxycholic acid in the treatment of primary biliary cirrhosis: a randomized trial. J Hepatol. 1999;30:830-5. 18.Parés A, Caballeria L, Rodés J. Excellent long-term survival in patients with primary biliary cirrhosis and biochemical response to ursodeoxycholic acid. Gastroenterology. 2006;130:715-20. 19.Corpechot C, Abenavoli L, Rabahi N, Chrétien Y, Andréani T, Johanet C, et al. Biochemical response to ursodeoxycholic acid and long-term prognosis in primary biliary cirrhosis. Hepatology. 2008;48:871-7. 20.Kuiper EM, Hansen BE, De Vries RA, Den Ouden-Muller JW, Van Ditzhuijsen TJ, Haagsma EB, et al; Dutch PBC Study Group. Improved prognosis of patients with primary biliary cirrhosis that have a biochemical response to ursodeoxycholic acid. Gastroenterology. 2009;136:1281-7. 21.Kumagi T, Guindi M, Fischer SE, Arenovich T, Abdalian R, Coltescu C, et al. Baseline ductopenia and treatment response predict long-term histological progression in primary biliary cirrhosis. Am J Gastroenterol. 2010;105:2186-94. 22.Corpechot C, Chazouillères O, Poupon R. Early primary biliary cirrhosis: biochemical response to treatment and prediction of long- term outcome. J Hepatol. 2011;55:1361-7. 23.Zhang LN, Shi TY, Shi XH, Wang L, Yang YJ, Liu B, et al. Early biochemical response to ursodeoxycholic acid and long-term prognosis of primary biliary cirrhosis: results of a 14-year cohort study. Hepatology. 2013;58:264-72. 24.Cavazza A, Caballería L, Floreani A, Farinati F, Bruguera M, Caroli D, et al. Incidence, risk factors, and survival of hepatocellular carcinoma in primary biliary cirrhosis: comparative analysis from two centers. Hepatology. 2009;50:1162-8. 25.Kuiper EM, Hansen BE, Adang RP, Van Nieuwkerk CM, Timmer R, Drenth JP, et al; Dutch PBC Study Group. Relatively high risk for hepatocellular carcinoma in patients with primary biliary cirrhosis not responding to ursodeoxycholic acid. Eur J Gastroenterol Hepatol. 2010;22:1495-502. 26.Jackson H, Solaymani-Dodaran M, Card TR, Aithal GP, Logan R, West J. Influence of ursodeoxycholic acid on the mortality and malignancy associated with primary biliary cirrhosis: a population-based cohort study. Hepatology. 2007;46:1131-7. 27.Nakai S, Masaki T, Kurokohchi K, Deguchi A, Nishioka M. Combination therapy of bezafibrate and ursodeoxycholic acid in primary biliary cirrhosis: a preliminary study. Am J Gastroenterol. 2000;95:326-7. 28.Kurihara T, Niimi A, Maeda A, Shigemoto M, Yamashita K. Bezafibrate in the treatment of primary biliary cirrhosis: comparison with ursodeoxycholic acid. Am J Gastroenterol. 2000;95:2990-2. 29.Ohira H, Sato Y, Ueno T, Sata M. Fenofibrate treatment in patients with primary biliary cirrhosis. Am J Gastroenterol. 2002;97:2147-9. 30.Kanda T, Yokosuka O, Imazeki F, Saisho H. Bezafibrate treatment: a new medical approach for PBC patients? J Gastroenterol. 2003;38:573-8. 31.Akbar SM, Furukawa S, Nakanishi S, Abe M, Horiike N, Onji M. Therapeutic efficacy of decreased nitrite production by bezafibrate in patients with primary biliary cirrhosis. J Gastroenterol. 2005;40:157-63. 32.Kita R, Takamatsu S, Kimura T, Kokuryu H, Osaki Y, Tomono N. Bezafibrate may attenuate biliary damage associated with chronic liver diseases accompanied by high serum biliary enzyme levels. J Gastroenterol. 2006;41:686-92. 33.Walker LJ, Newton J, Jones DE, Bassendine MF. Comment on biochemical response to ursodeoxycholic acid and longterm prognosis in primary biliary cirrhosis. Hepatology. 2009;49:337-8. 34.Hazzan R, Tur-Kaspa R. Bezafibrate treatment of primary biliary cirrhosis following incomplete response to ursodeoxycholic acid. J Clin Gastroenterol. 2010;44:371-3. 35.Liberopoulos EN, Florentin M, Elisaf MS, Mikhailidis DP, Tsianos E. Fenofibrate in primary biliary cirrhosis: a pilot study. Open Cardiovasc Med J. 2010;4:120-6. 36.Levy C, Peter JA, Nelson DR, Keach J, Petz J, Cabrera R, et al. Pilot study: fenofibrate for patients with primary biliary cirrhosis and an incomplete response to ursodeoxycholic acid. Aliment Pharmacol Ther. 2011;33:235-42. 37.Lens S, Leoz M, Nazal L, Bruguera M, Parés A. Bezafibrate normalizes alkaline phosphatase in primary biliary cirrhosis patients with incomplete response to ursodeoxycholic acid. Liver Int. 2014;34:197-203. 38.Honda A, Ikegami T, Nakamuta M, Miyazaki T, Iwamoto J, Hirayama T, et al. Anticholestatic effects of bezafibrate in patients with primary biliary cirrhosis treated with ursodeoxycholic acid. Hepatology. 2013;57:1931-41. 39.Matsumoto T, Miyazaki H, Nakahashi Y, Hirohara J, Seki T, Inoue K, et al. Multidrug resistance is in situ detected in the liver of patients with primary biliary cirrhosis, and induced in human hepatoma cells by bezafibrate. Hepatol Res. 2004;30:125-36. 40.Chianale J, Vollrath V, Wielandt AM, Amigo L, Rigotti A, Nervi F, et al. Fibrates induce mdr2 gene expression biliary phospholipid secretion tion in the mouse Biochem J. 1996;314:781-6. 41.Leuschner M, Maier KP, Schlichting J, Strahl S, Herrmann G, Dahm HH, et al. Oral budesonide and ursodeoxycholic acid for treatment of primary biliary cirrhosis: results of a prospective double-blind trial. Gastroenterology. 1999;117:918-25. 42.Rautiainen H, Kärkkäinen P, Karvonen AL, Nurmi H, Pikkarainen P, Nuutinen H, et al. Budesonide combined with UDCA to Cirrosis biliar primaria: nuevas opciones de tratamiento improve liver histology in primary biliary cirrhosis: a threeyear randomized trial. Hepatology. 2005;41:747-52. 43.Angulo P, Jorgensen RA, Keach JC, Dickson ER, Smith C, Lindor KD. Oral budesonide in the treatment of patients with primary biliary cirrhosis with a suboptimal response to ursodeoxycholic acid. Hepatology. 2000;31;318-23. 44.Hempfling W, Grunhage F, Dilger K, Reichel C, Beuers U, Sauerbruch T. Pharmacokinetics and pharmacodynamic action of budesonide in early- and late-stage primary biliary cirrhosis. Hepatology. 2003;38:196-202. 45.Pellicciari R, Fiorucci S, Camaioni E, Clerici C, Costantino G, Maloney PR, et al. 6alpha-ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J Med Chem. 2002;45:3569-72. 46.Fiorucci S, Rizzo G, Donini A, Distrutti E, Santucci L. Targeting farnesoid X receptor for liver and metabolic disorders. Trends Mol Med. 2007;13:298-309. 47.Pellicciari R, Costantino G, Fiorucci S. Farnesoid X receptor: from structure to potential clinical applications. J Med Chem. 2005;48:5383-403. 48.Trauner M, Halilbasic E. Nuclear receptors as a new perspective for the management of liver diseases. Gastroenterology. 2011;140:1120-25. 49.Fiorucci S, Antonelli E, Rizzo G, Renga B, Mencarelli A, Riccardi L, et al. The nuclear receptor SHP mediates inhibition of hepatic stellate cells by FXR and protects against liver fibrosis. Gastroenterology. 2004;127:1497-512. 97 50.Fiorucci S, Rizzo G, Antonelli E, Renga B, Mencarelli A, Riccardi L, et al. Cross- talk between farnesoid- X- receptor (FXR) and peroxisome proliferator- activated receptor gamma contributes to the antifibrotic activity of FXR ligands in rodent models of liver cirrhosis. J Pharmacol Exp Ther. 2005;315: 58-68. 51.Mason A, Luketic VA, Lindor KD, Hirschfield G, Gordon SC, Mayo ML, et al. Farnesoid-X receptor agonists: a new class of drugs for the treatment of PBC? An international study evaluating the addition of obeticholic acid (int-747) to ursodeoxycholic acid. Hepatology. 2010;52:375A. 52.Marschall HU, Luketic V, Lövgren-Sandblom A, Benthin L, Kowdley K, Hirschfield G, et al. The farnesoid X receptor (FXR) agonist obeticholic acid (OCA) increases plasma FGF-19 concen- trations and decreases bile acid synthesis in primary biliary cirrhosis (PBC). J Hepatol. 2012;5 Suppl 2:S377. 53.Kowdley KV, Jones D, Luketic V, Chapman R, Burroughs A, Hirschfield G, et al. An international study evaluating the farnesoid X receptor agonist obeticholic acid as monotherapy in PBC. J Hepatol. 2011;54:S13. 54.Myers RP, Swain MG, Lee SS, Shaheen AAM, Burak, KW. B-cell depletion with rituximab in patients with primary biliary cirrhosis refractory to ursodeoxycholic acid. Am J Gastroenterol. 2013;108,933-41. 55.Hirschfield GM, Liu X, Xu C, Lu Y, Xie G, Lu Y, et al. Primary biliary cirrhosis associated with HLA, IL12A, and IL12RB2 variants. N Engl J Med. 2009;360:2544-55. SESIÓN 4 OTRAS ENFERMEDADES HEPÁTICAS Hepatocarcinoma: manejo del estadio intermedio María Varela Sección de Hepatología, Servicio de Aparato Digestivo, Hospital Universitario Central de Asturias, Oviedo, Asturias, España El carcinoma hepatocelular es una de las principales causas de muerte en los pacientes con cirrosis. Su incidencia está creciendo en todo el mundo, sobre todo debido a la expansión de la enfermedad por depósito de grasa en los países desarrollados y a la mayor supervivencia de los pacientes con infección crónica por virus de la hepatitis C y B. El estadio intermedio se define, según la clasificación BCLC, como aquel tumor no quirúrgico, asintomático, sin invasión vascular ni enfermedad extrahepática. Las guías clínicas recomiendan la quimioembolización transarterial hepática (TACE) como tratamiento de primera elección, pero solamente es aplicable a los pacientes con cirrosis compensada. Los pacientes con cirrosis descompensada no pueden recibir TACE y han de quedar en historia natural, puesto que la aplicación del tratamiento agrava la cirrosis y empeora el pronóstico debido a las complicaciones asociadas. Si por alguna razón técnica, la TACE no puede aplicarse o se ha llegado a una situación de fracaso de la TACE y el tumor sigue en estadio intermedio, el paciente debe ser evaluado para recibir sorafenib. A caballo entre la TACE y el sorafenib emerge la radioembolización con itrio-90, un tratamiento intraarterial que es muy eficaz y que ofrece en estudios de cohortes la misma supervivencia que la TACE, con menos efectos secundarios y mejor tolerancia. INTRODUCCIÓN El carcinoma hepatocelular (CHC) es el tumor primario hepático más frecuente y es el tercero a nivel mundial en cuanto a muertes relacionadas con cáncer, tras el de pulmón y el de estómago1. En Europa occidental, la incidencia anual es de 10-12 casos/100.000 habitantesaño. Asienta fundamentalmente en hígados con enfermedad hepática crónica (fibrosis avanzada/cirrosis) y es una de las principales causas de mortalidad más 98 allá de las complicaciones de la cirrosis (hemorragia digestiva, insuficiencia renal, infecciones, etc.). El sistema de clasificación pronóstica más extendido, adoptado por las guías españolas, americanas y europeas de enfermedades hepáticas y reconocido por las guías europeas de oncología es el sistema BCLC (BarcelonaClinic-Liver-Cancer), presentado por primera vez en 1999 con la última modificación realizada en 20142 (fig. 1). Este sistema ha sido validado de forma externa por numerosos grupos de trabajo y tiene la virtud de vincular cada estadio tumoral con su opción terapéutica de elección. Así, en el estadio BCLC-0 (muy inicial), que comprende los tumores únicos < 2 cm, el tratamiento ideal es la ablación con radiofrecuencia. Si el paciente es candidato a trasplante y no presenta datos de hipertensión portal clínicamente significativa, la opción preferida es la resección, porque el análisis del espécimen puede detectar invasión vascular o satelitosis, que predicen alto riesgo de recidiva, y permitir la posibilidad de inclusión en lista de espera de trasplante antes de que se produzca dicha recidiva. En el estadio BCLC-A (inicial) se consideran todos los tumores aptos para cirugía, idealmente los tumores únicos sin hipertensión portal clínicamente significativa y con bilirrubina normal3, y aquellos en estadio Child-Pugh A-B, con tumores únicos hasta 5 cm o en un número menor de 3 hasta 3 cm de diámetro cada uno. Si estos pacientes no son candidatos óptimos a resección deben evaluarse para trasplante y si no son candidatos a trasplante por edad avanzada o comorbilidad pueden recibir tratamiento con ablación percutánea, generalmente radiofrecuencia4-6. La expectativa vital mediana de estos pacientes compensados con tumores iniciales supera los 36 meses y con tratamiento se estima una supervivencia > 50-70% a 5 años. La ablación con radiofrecuencia, la resección y el trasplante se consideran tratamientos curativos. El problema principal de los pacientes con tumores iniciales es la recidiva tumoral, de hasta el 70% a 5 años, salvo en los trasplantados, en los que está en torno al 15% postrasplante. Hepatocarcinoma: manejo del estadio intermedio 99 Figura 1. Esquema BCLC-2014. 100 Sesión 4 - OTRAS ENFERMEDADES HEPÁTICAS Los pacientes Child-Pugh C se consideran terminales (BCLC-D), a menos que sean candidatos a trasplante, así como aquellos con tumores avanzados con mala función hepática y síntomas invalidantes (ECOG Performance Status 3-4) (fig. 1). El pronóstico de los pacientes en estadio terminal no supera los 3-6 meses de vida y el único tratamiento posible es el control de síntomas en historia natural. Entre ambos extremos están los estadios intermedio (BCLC-B) y avanzado (BCLC-C). La mayoría de los pacientes en nuestro medio se diagnostica en estadio intermedio, que lo constituyen los pacientes con tumores únicos > 5 cm no quirúrgicos y aquellos con enfermedad multinodular que excede el “3 < 3” (BCLC-B). Los pacientes en estadio intermedio son candidatos a quimioembolización transarterial hepática (TACE) y su expectativa vital mediana pasa de 16 a 20 meses con este tratamiento (fig. 1). Si los pacientes presentan síntomas leves o moderados asociados al cáncer (ECOG PS 1-2), invasión vascular o enfermedad extrahepática se consideran avanzados (BCLC-C). Los pacientes en estadio avanzado son candidatos a sorafenib (SOR) y su expectativa vital mediana pasa de 6 a 9 meses con este tratamiento (fig. 1). Nódulos Cirrosis hepática HEPATOCARCINOMA EN ESTADIO INTERMEDIO. CARCINOMA HEPATOCELULAR ESTADIO BCLC-B Tal y como se muestra en la figura 2, el espectro de enfermedad que abarca el estadio intermedio es amplísimo y es difícil comparar estudios, puesto que el pronóstico de los pacientes depende de la presencia de comorbilidad, de la función hepática, del tamaño y número de nódulos tumorales y del tratamiento aplicado. La mayoría de los nódulos de CHC reciben su vascularización por vía arterial, mientras que el hígado se nutre sobre todo de la sangre que llega por la vía portal. Esta peculiaridad anatómica es la base racional para el uso de terapias intraarteriales en el CHC. La TACE se inició en Japón hace más de 30 años7. Consiste en la inyección por vía arterial, generalmente accediendo a través de la arteria femoral derecha mediante técnica de Seldinger, de un agente quimioterápico (los más utilizados son cisplatino y adriamicina) emulsionado en un agente oleoso llamado lipiodol, con el objetivo de que la medicación se quede retenida en Nódulos Cirrosis hepática Figura 2. Espectro del BCLC-B. TACE: quimioembolización transarterial hepática. Nódulos Cirrosis hepática Hepatocarcinoma: manejo del estadio intermedio 101 Figura 3. TACE (quimioembolización transarterial hepática). Procedimiento técnico. PVA: alcohol polivinílico. el lecho vascular tumoral y peritumoral. Seguidamente se inyecta el agente embolizante: gelfoam, coils, coágulos, esferas de polivinilo, etc., para conseguir que los nódulos tumorales se queden hipóxicos y los agentes quimioterápicos tengan mayor efecto (fig. 3). La evidencia de que la TACE aumenta la supervivencia se basa en un metanálisis de ensayos clínicos aleatorizados8. En la última década se ha refinado la técnica, se ha pasado de una embolización lobar o segmentaria a una embolización supraselectiva a través de las arterias nutricias tumorales, con el uso de microcatéteres, y se han diseñado unas partículas que cargan la medicación en su interior (drug eluting beads) no reabsorbibles y que a la vez que embolizan liberan la quimioterapia de forma lenta y sostenida, consiguiendo un procedimiento menos doloroso y con menor toxicidad sistémica. El 80-85% de los pacientes sometidos a TACE desarrollan dolor, fiebre y malestar los siguientes días al procedimiento, es el llamado síndrome postembolización, y suele resolverse sin complicaciones en casa en 10-15 días. En la tabla 1 se muestran las contraindicaciones técnicas al procedimiento y en la tabla 2 se muestra la tasa ideal de las complicaciones que pueden seguir a la TACE. Hay estudios que sugieren que solamente el 12% de los pacientes en estadio intermedio son candidatos ideales a TACE, bien por flujo portal invertido (hepa- tofugal), porque presentan mala función hepática con bilirrubina > 2, ascitis o encefalopatía, o bien porque presentan un tumor multinodular bilobar que precisa una embolización de prácticamente todos los segmentos hepáticos. Es importante hacer una cuidadosa selección de los pacientes candidatos a TACE y su evaluación en comités multidisciplinares en que intervengan radiólogos, hepatólogos, oncólogos, cirujanos, etc. El manejo multidisciplinar de los pacientes con CHC aumenta su supervivencia9. Auque la expectativa vital mediana con la TACE es superar los 20 meses de supervivencia, recientes series publicadas superan los 40 meses10. Una vez realizada la TACE se realiza la evaluación de la respuesta al tratamiento mediante examen físico, analítica y prueba de imagen a las 4-6 semanas del procedimiento. Si el paciente está compensado asintomático y se objetiva respuesta parcial (necrosis > 30%), enfermedad estable o progresión tratable (progresión < 20%) se indica una segunda sesión de TACE. Tras la evaluación de la segunda sesión de TACE se decide seguir con TACE si aparece una nueva progresión tratable (p. ej., un nódulo nuevo en el lóbulo contralateral al tratado). Por el contrario, si con la segunda TACE no se ha conseguido una respuesta significativa o hay una progresión intratable es el momento de cambiar a SOR. 102 Sesión 4 - OTRAS ENFERMEDADES HEPÁTICAS Tabla 1. Selección de candidatos para la quimioembolización transarterial hepática (TACE) Tabla 2. Tasa ideal de posibles complicaciones de la quimioembolización transarterial hepática (TACE) BCLC-B Complicaciones de la TACE (%) • Asintomáticos Fallo hepático 2-3 Absceso hepático 1-2 Síndrome postembolización con hospitalización prolongada 4-6 Absceso con vía biliar alterada (post-CPRE) 0-15 • Leucopenia (< 1.000 leucocitos/ ml) Colecistitis quirúrgica <1 • Insuficiencia cardíaca Biloma que requiera drenaje percutáneo <1 • Insuficiencia renal (creatinina sérica > 2 mg/dl) Embolia de lipiodol a arterias pulmonares <1 Úlcera gastrointestinal o hemorragia <1 Disección iatrogénica que impida el tratamiento <1 Mortalidad en 30 días 2-4 • Cirrosis compensada • Ausencia de invasión vascular • Ausencia de enfermedad extrahepática Contraindicaciones • Infección sistémica intratable • Flujo portal hepatofugal • Obstrucción biliar El SOR fue aprobado en 2008 como tratamiento de elección para el CHC en estadio avanzado tras la publicación de los resultados de 2 ensayos multicéntricos aleatorizados doble ciego en Europa-América (el ensayo SHARP)11 y en Asia (el ensayo Asia-Pacific), que demostraron un aumento significativo de la supervivencia frente a placebo en pacientes con estadio intermedio y avanzado. SOR es un inhibidor multicinasa que se administra por vía oral, con propiedades antiangiogénicas y antiproliferativas. Actúa inhibiendo la proliferación tumoral, aumentando la apoptosis y disminuyendo la angiogénesis. Asimismo hay evidencias preclínicas que indican que el SOR modula el microambiente tumoral actuando sobre los macrófagos asociados al tumor y sobre las células naturalkiller estimulando la respuesta inmune oncolítica del organismo. La dosis recomendada es 800 mg/día en forma de comprimidos de 200 mg (2-0-2). Se metaboliza a través del citocromo P-450 CYP3A4 y es preciso conocer que deben evitarse los fármacos que utilizan esta misma vía metabólica, en especial los antibióticos macrólidos, el hipérico, la carbamaze- CPRE: colangiopancreatografía retrógrada endoscópica. pina, etc. Puede producir crisis hipertensivas, por lo que es necesario vigilar frecuentemente la presión arterial, es recomendable intensificar los controles del índice internacional normalizado en aquellos que toman dicumarínicos y aumenta los valores séricos de digoxina. Los efectos secundarios dermatológicos (queilitis, estomatitis, alopecia, exantema y síndrome mano-pie) se asocian a mayor supervivencia12. Por último, cabe hablar de la radioembolización con itrio-90 (RE-Y90), un tratamiento intraarterial no embolizante que consiste en la inyección selectiva tumoral de partículas de resina o de cristal que llevan en su constitución partículas de Y90. Estas partículas emiten radiación ionizante beta, que se caracteriza por ser de alta intensidad y de baja penetrancia (tabla 3). Si es importante la coordinación multidisciplinar en el tratamiento del CHC, a la hora de aplicar la RE-Y90 esta es esencial, ya que es preciso realizar primero una arteriografía de planificación, Tabla 3. Características de las partículas de itrio-90 TheraSphere® SIR-Spheres® Material Vidrio Resina Tamaño (µm) 25 (20-30) 32,5 (20-60) Radiación Beta Profundidad 2,5 mm Semivida 64,1 h Número de partículas por tratamiento (en millones) 1,2-8 Hasta 30 Hepatocarcinoma: manejo del estadio intermedio Tabla 4. Supervivencia de los pacientes en estadio intermedio (BCLC-B) tratados con RE-Y90 (en negrita) y con sorafenib (SOR) BCLC-B Autor Child n OS (IC del 95%) Hilgard et al (n = 108) A-B 51 16,4 (12,1-NC) Salem et al (n = 291) A 48 17,3 (13,7-32,5) Sangro et al (n = 325) A 82 18,4 (13,6-23,2) Mazzaferro et al (n = 52) A 15 18 (13-38) Bruix et al (SHARP), SOR A-B 54 14,5 Bruix et al (SHARP), placebo A-B 51 11,4 IC: intervalo de confianza; OS: supervivencia global. Hilgard P, et al. Hepatology. 2010;52:1741-9. Salem R, et al. Gastroenterology. 2010;138:52-64. Sangro B, et al. Hepatology. 2011;54:868-78. Mazzaferro V, et al. Hepatology. 2013:57:1826-37. Bruix J, et al. J Hepatol. 2012;57:821-9. inyectar macroagregados de albúmina marcados con tecnecio-99 para evaluar la distribución de las partículas radiactivas a nivel tumoral, medir el shunt pulmonar y realizar las embolizaciones necesarias de las ramas extrahepáticas aberrantes que llevan sangre al tumor, para evitar que en el segundo tiempo, cuando se inyecten las partículas radiactivas con Y90, estas no lleguen a zonas extrahepáticas no deseables como el territorio gastrointestinal. La tolerancia de la RE-Y90 es excelente. Hay que destacar que la correcta selección de candidatos en cuanto a función hepatocelular (bilirrubina sérica < 1,5 mg/dl, idealmente), shunting pulmonar (< 50 Gy), volumen tumoral (< 70% del volumen hepático) y apropiada embolización de las arterias aberrantes, hacen de esta técnica intraarterial una alternativa muy interesante a la TACE en los casos en que está contraindicada y al SOR en los pacientes con perfil cardiovascular adverso13,14 (tabla 4). 103 Bibliografía 1.Ferlay J et al. Int J Cancer. 2014. doi: 10.1002/ijc.29210. 2.Reig M, Darnell A, Forner A, Rimola J, Ayuso C, Bruix J. Systemic therapy for hepatocellular carcinoma: the issue of treatment stage migration and registration of progression using the BCLC-refined RECIST. Sem Liv Dis. 2014;34:444-55. 3.Berzigotti AL, Reig M et al. Hepatology. 2014. doi: 10.1002/ hep.27431. 4.Orlando A, Leandro G, Olivo M, Andriulli A, Cottone M. Radiofrequency thermal ablation vs. percutaneous ethanol injection for small hepatocellular carcinoma in cirrhosis: metaanalysis of randomized controlled trials. Am J Gastroenterol. 2009;104:514-24. 5.Cho YK, Kim JK, Kim MY, Rhim H, Han JK. Systematic review of randomized trials for hepatocellular carcinoma treated with percutaneous ablation therapies. Hepatology. 2009;49:453-9. 6.Germani G, Pleguezuelo M, Gurusamy K, Meyer T, Isgrò G, Burroughs AK. Clinical outcomes of radiofrequency ablation, percutaneous alcohol and acetic acid injection for hepatocelullar carcinoma: a meta-analysis. J Hepatol. 2010;52:380-8. 7.Yamada R, Sato M, Kawabata M, Nakatsuka H, Nakamura K, Takashima S. Hepatic artery embolization in 120 patients with unresectable hepatoma. Radiology. 1983;148:397-401. 8.Llovet JM, Bruix J. Systematic review of randomized trials for unresectable hepatocellular carcinoma: Chemoembolization improves survival. Hepatology. 2003;37:429-42. 9.Barone C, Koeberle D, Metselaar H, Parisi G, Sansonno D, Spinzi G. Multidisciplinary approach for HCC patients: hepatology for the oncologists. Ann Oncol. 2013;24 Suppl 2:ii15-23. 10.Burrel M, Reig M, Forner A, Barrufet M, De Lope CR, Tremosini S, et al. Survival of patients with hepatocellular carcinoma treated by transarterial chemoembolisation (TACE) using Drug Eluting Beads. Implications for clinical practice and trial design. J Hepatol. 2012;56:1330-5. 11.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al; SHARP Investigators Study Group. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378-90. 12.Reig M, Torres F, Rodriguez-Lope C, Forner A, LLarch N, Rimola J, et al. Early dermatologic adverse events predict better outcome in HCC patients treated with sorafenib. J Hepatol. 2014;61:318-24. 13.Salem R, Lewandowski RJ, Mulcahy MF, Riaz A, Ryu RK, Ibrahim S, et al. Radioembolization for hepatocellular carcinoma using Yttrium-90 microspheres: a comprehensive report of long-term outcomes. Gastroenterology. 2010;138:52-64. 14.Sangro B, Carpanese L, Cianni R, Golfieri R, Gasparini D, Ezziddin S, et al; European Network on Radioembolization with Yttrium-90 Resin Microspheres (ENRY). Survival after yttrium-90 resin microsphere radioembolization of hepatocellular carcinoma across Barcelona clinic liver cancer stages: a European evaluation. Hepatology. 2011;54:868-78. SESIÓN 4 OTRAS ENFERMEDADES HEPÁTICAS Colangiocarcinoma intrahepático Bruno Sangro Unidad de Hepatología, Clínica Universidad de Navarra, Pamplona, España IDISNA, CIBERehd, Pamplona, España INTRODUCCIÓN El término colangiocarcinoma (CC) engloba un grupo heterogéneo de tumores con características histológicas de diferenciación a tracto biliar. Pueden surgir de la vía biliar extrahepática o del hígado, presumiblemente a partir de los conductos biliares intrahepáticos, grandes o pequeños, aunque es posible que también lo hagan directamente por transdiferenciación de hepatocitos1. En función de su origen anatómico se diferencian en CC intrahepático (CCI), perihiliar (también llamado tumor de Klatskin) y distal2. El objeto de esta revisión es exclusivamente el CCI, que tiene peculiaridades etiológicas, patológicas, clínicas y terapéuticas. Puede aparecer en personas con hígado sano o con hepatopatías crónicas, se manifiesta habitualmente por masas hepáticas que solo infrecuentemente obstruyen la vía biliar produciendo ictericia y desde el punto de vista histológico tiene el aspecto de un adenocarcinoma, aunque en ocasiones puede verse en un mismo tumor doble diferenciación en los llamados tumores mixtos o hepatocolangiocarcinomas3. EPIDEMIOLOGÍA Y CAUSAS La incidencia de CCI varía mucho de unos países a otros y, en general, los datos no son muy fiables, ya que los registros rara vez distinguen al CCI de las otras variantes anatómicas y puede quedar englobado dentro de los tumores del hígado o de la vía biliar. La incidencia es máxima en el Lejano Oriente (más de 80 casos por 100.000 habitantes en Tailandia) y en países occidentales puede alcanzar los 2,1 casos por 100.000 habitantes4. Es, después del hepatocarcinoma, el segundo cáncer más frecuente del hígado. Caben pocas dudas de que la incidencia va en claro ascenso en los países europeos y norteamericanos. En Estados Unidos se ha descrito un aumento anual del 9%, tanto en la incidencia como en la mortalidad por CCI entre 1973 y 19975, en Alemania la mortalidad se ha triplicado entre 104 1998 y 2008 y en el Reino Unido el CCI ha sobrepasado al hepatocarcinoma como causa de muerte por tumor hepático primario. El proceso de colangiocarcinogénesis es probablemente multifactorial y las diferencias geográficas razonablemente reflejan distinta exposición a las causas. La gran mayoría de los casos son de etiología desconocida. Entre las causas reconocidas (tanto para CCI como para las otras formas anatómicas de CC) figuran los trematodos hepatobiliares, la colangitis esclerosante primaria, la hepatolitiasis, los quistes biliares, determinadas toxinas y, más recientemente reconocidos como factores de riesgo, la cirrosis, las hepatitis crónicas B y C, la obesidad, la diabetes y el alcohol6,7. La circunstancia de que CCI y hepatocarcinoma compartan factores de riesgo apunta a la existencia de vías patobiológicas comunes para los tumores hepáticos primarios. El hecho de que solo una minoría de los pacientes con factores de riesgo desarrolle la enfermedad hace probable que los factores genéticos del huésped desempeñen también un papel importante. Aunque pequeños, varios estudios de casos-control han sugerido que variaciones en los genes que codifican para distintos sistemas enzimáticos pueden asociarse a un mayor riesgo de desarrollar CCI. Entre ellos están los genes de la 5,10-metilentetrahidrofolato reductasa, implicados en el metabolismo de los folatos y la metilación del ADN; el de la timidilato sintetasa, asociado a la reparación del ADN; el de la proteína asociada a resistencia múltiple a drogas tipo 2, transportador celular implicado en la depuración de toxinas, o el de la ciclooxigenasa 2, un mediador de inflamación8. En cuanto a las alteraciones genéticas, se han descrito una serie de mutaciones, aberraciones cromosómicas y cambios epigenéticos. Entre ellas las mutaciones activadoras de k-ras y las mutaciones de p53, que no solo se han descrito en porcentajes altos de los pacientes con CCI sino que tienen un papel carcinogénico en modelos animales. Aunque no hay una clasificación molecular universalmente aceptada, se ha avanzado en la definición molecular de subclases CCI basadas en el análisis del transcriptoma y otros parámetros biológicos9,10. En pacientes con CC de cualquier subtipo se han descrito 2 subclases, una de ellas asociada a mal pronóstico y que se caracteriza por la activación de receptores de tirosincinasas como EGFR, ErbB2 y MET. Específicamente, en pacientes con CCI se ha confirmado la existencia de 2 subgrupos con perfiles genómicos y clínicos diferentes. Uno inflamatorio, enriquecido en la activación de vías de inflamación y citocinas, y otro proliferativo, enriquecido en la activación de vías oncogénicas como RAS/MAPK y MET. Aunque importantes, ya que suponen la primera aproximación a una clasificación molecular que permita distinguir a los tipos de CCI en función de su pronóstico y, lo que es más importante, respuesta a tratamiento, estas u otras clasificaciones deben ser validadas en el futuro antes de adoptarse en clínica. CLÍNICA, DIAGNÓSTICO Y ESTADIFICACIÓN La presentación clínica del CCI es muy variable e inespecífica y depende en parte del subtipo morfológico: nodular (que supone más del 85% de los casos), infiltrante periductal y de crecimiento intraductal. Los pacientes con enfermedad en estadio temprano suelen estar asintomáticos y el tumor se detecta incidentalmente al hacer análisis o pruebas de imagen por otro motivo. En estadios más avanzados pueden presentar pérdida de peso, malestar general, dolor abdominal, ictericia, hepatomegalia o una masa abdominal palpable. La obstrucción del tracto biliar es poco frecuente salvo en los subtipos no nodulares. Desde el punto de vista analítico es frecuente hallar colostasis disociada en la enfermedad precoz y colostasis franca, con o sin alteración de la función hepática, en las fases más avanzadas. En los pacientes con hepatolitiasis o colangitis esclerosante primaria se debe sospechar CCI en aquellos con afectación del estado general o empeoramiento analítico. El diagnóstico firme de CCI es histológico y se basa en el hallazgo de un adenocarcinoma o carcinoma mucinoso, con estructuras tubulares o papilares y estroma fibrosa habitualmente prominente11. Cabe excepcionalmente hacer un diagnóstico clínico radiológico de presunción cuando por la situación clínica únicamente se plantea un tratamiento paliativo. La sensibilidad de la biopsia hepática depende del tipo de ubicación y tamaño del tumor, la experiencia del operador y el componente fibroso. Una biopsia negativa no excluye necesariamente el diagnóstico. Además de entidades benignas, como los microhamartomas biliares (complejos von Meyenburg) o los adenomas biliares, el diagnóstico diferencial puede ser muy difícil con las metástasis de adenocarcinoma extrahepático, especialmente el de origen en pulmón, páncreas, esófago y estómago. Colangiocarcinoma intrahepático 105 En la diferenciación entre CCI y hepatocolangiocarcinoma pueden ayudar los marcadores específicos de hepatocitos o células progenitoras como Hep-Par-1, GPC3, HSP70, glutamina sintetasa, EpCAM y citoqueratinas 7, 19 y 2012,13. En los estudios de imagen es frecuente hallar características que sugieren intensamente el diagnóstico de CCI, aunque no sean cien por cien específicas (fig. 1). En la tomografía computarizada (TC), el aspecto típico del subtipo nodular es el de una masa hepática hipodensa en la fase sin contraste, con márgenes irregulares, realce periférico en la fase arterial y progresivamente más intenso y central en las fases venosa y tardía, frecuentemente con retracción capsular14. En la resonancia magnética (RM), el CCI aparece hiperintenso en T2, con el mismo comportamiento ante el contraste que en la TC y con restricción en las secuencias de difusión15. La tomografía por emisión de positrones (PET) con fluorodesoxiglucosa (FDG) es útil en el diagnóstico y estadificación del CCI, ya que puede detectar lesiones de hasta 1 cm con una sensibilidad del 85-95%16. Los marcadores tumorales en bilis o suero no son específicos, pero pueden ayudar al diagnóstico. El antígeno CA19.9 y el antígeno carcinoembrionario tienen una baja sensibilidad y especificidad (el 62 y el 63%, respectivamente), sobre todo en la enfermedad precoz o en presencia de obstrucción de la vía biliar. El CA19.9 tiene, sin embargo, utilidad pronóstica y valores > 100 U/ml se asocian a una peor supervivencia libre de recidiva tras resección quirúrgica17. Los estudios de imagen deben determinar el grado de extensión locorregional y distante, incluyendo la invasión vascular y biliar, y permitir la evaluación volumétrica de hígado no afectado, clave para establecer la resecabilidad. En los tumores de crecimiento periductal o intraductal, la utilidad es muy limitada. La ecografía con Doppler color puede identificar la afectación vascular con valores predictivos > 95%18, comparables a la angiografía por TC. El papel de la PET-FDG es controvertido, pero diversos estudios indican que posee una gran capacidad para detectar metástasis ocultas, ganglionares o viscerales, especialmente en el CCI nodular, que capta más ávidamente la FDG. Por ello es una herramienta que debe considerarse siempre a la hora de valorar la resecabilidad, ya que permite detectar también adenocarcinomas primarios ocultos de otros órganos. De hecho, puede cambiar la indicación quirúrgica en hasta un 30% de los pacientes19. Aunque hay distintos sistemas de estadificación, existe consenso en utilizar la séptima edición del sistema TNM de la AJCC/UICCA20. La determinación del componente T se basa en el número de lesiones y la presencia de invasión vascular, metástasis intrahepáticas e invasión de estructuras adyacentes (T1: tumores solitarios y sin invasión vascular; T2: tumores múltiples 106 Sesión 4 - OTRAS ENFERMEDADES HEPÁTICAS A) B) C) D) E) F) G) H) Figura 1. Aspecto característico del colangiocarcinoma intrahepático en las pruebas de imagen. Tomografía computarizada en fase arterial (A) y tardía (B). Resonancia magnética en fase arterial (C) y tardía (D) y en secuencias ponderadas en T2 (E) y de difusión (F). G y H) Tomografía por emisión de positrones con fluorodesoxiglucosa. [enfermedad multifocal, satelitosis, metástasis intrahepáticas] y tumores con cualquier tipo de invasión vascular, microscópica o macroscópica; T3: tumores que invaden estructuras adyacentes; T4: tumores con cualquier componente infiltrante periductal). La calificación N1 se basa en la afectación de ganglios hiliares, periduodenales o peripancreáticos y la enfermedad a distancia se considera M1. TRATAMIENTO Cirugía La resección quirúrgica es la base del tratamiento del CCI. Hay gran controversia sobre la utilidad de la laparoscopia de estadificación, pero con los datos actualmente disponibles no hay pruebas suficientes para recomendar esta prueba de forma rutinaria. La resección está indicada en todos los pacientes con enfermedad potencialmente resecable (fig. 2), aunque solo se ofrece a un 20-40% de ellos21. Esto se debe en parte al hecho de que a menudo se trata de tumores grandes, localmente avanzados, que precisan operaciones técnicamente complejas. El objetivo de la resección es eliminar toda la enfermedad con márgenes negativos (resección oncológica o R0) preservando un volumen de hígado remanente adecuado. Esto puede requerir hepatectomías extensas, que incluyan estructuras adyacentes como la vía biliar extrahepática. Como ejemplo, el 73% de los pacientes de una gran serie requirió una hemihepatectomía o hepatectomía extendida, a menudo con reconstrucción biliar22. Y esto debe hacerse con tasas de mortalidad perioperatoria < 5%. Por ello, la indicación quirúrgica se debe evaluar y realizar por equipos muy expertos en cirugía hepática, y se debe referir al paciente precozmente a estos centros de referencia. Colangiocarcinoma intrahepático 107 Colangiocarcicoma intrahepático Estadio I Estadio II Estadio III Estadio IV Tumor único Único o multiple IV Perforación peritoneovisceral Invasión hepática local Invasión periductal, N1M1 Resecable (30-40%) No resecable (60-70%) Ressección curativa Ressección no curativa Observación Ensayos clínicos Supervivencia a 5 años RO: 40% Supervivencia a 5 años N1 e IV: 20% Enfermedad solo intrtahepática Enfermedad extrahepática Terapia locorregional Terapia sistémica RF/TACE: mediana supervivencia 15 meses Quimioterapia: mediana supervivencia 12 meses Figura 2. Algoritmo de tratamiento propuesto por la Asociación Internacional de Cáncer Hepático (ILCA). Define recomendaciones basadas en el estándar de práctica, ya que no existen pruebas suficientemente sólidas para hablar de estándar de tratamiento en la mayoría de las indicaciones. IV: invasión vascular; RF/TACE: quimioembolización transarterial hepática. La resección de la enfermedad ganglionar clínicamente sospechosa es obligatoria, pero el papel de la linfadenectomía de rutina está mal definido. Aunque el hallazgo de enfermedad ganglionar no sospechada marca un mal pronóstico, no hay tratamientos adyuvantes que hayan demostrado mejorar la supervivencia, por lo que la linfadenectomía reglada debe hacerse solo en el marco de estudios prospectivos. El 50-60% de los pacientes resecados presenta recidiva de la enfermedad con una mediana de supervivencia libre de enfermedad de 26 meses23,24. La recurrencia suele ser en el hígado (50-60%) seguido de los ganglios linfáticos regionales o el peritoneo (20-25%)23. Los factores asociados con un mayor riesgo de recidiva son los que forman la base del sistema de estadificación: tumores múltiples, invasión vascular o linfática y metástasis. La supervivencia a 5 años de la cirugía es del 15-40%25. El trasplante hepático está contraindicado sobre la base de la alta tasa de recidiva, aunque recientemente se ha descrito que en pacientes con tumores muy pequeños y únicos la supervivencia puede ser similar a la de los pacientes con hepatocarcinoma26,27. Antes de extender de forma indiscriminada esta indicación, los buenos resultados deben confirmarse en series prospectivas, ya que en los estudios publicados hay un importante sesgo de selección, pues muchos de los tumores son hallazgos incidentales o errores del diagnóstico por imagen. Procedimientos locorregionales Muchos pacientes con CCI se presentan con enfermedad irresecable. En ellos, la evidencia que apoya el uso de cualquier tratamiento es científicamente ende- 108 Sesión 4 - OTRAS ENFERMEDADES HEPÁTICAS ble. Algunos estudios han sugerido que los tratamientos locorregionales pueden tener un efecto favorable en la supervivencia28. Estos tratamientos incluyen fundamentalmente la radioterapia externa, la quimioembolización transarterial y la radioembolización. La irradiación externa, fundamentalmente en forma de técnicas modernas que limitan la irradiación del tejido hepático no tumoral como la radioterapia de intensidad modulada, la radioterapia corporal estereotáctica, la braquiterapia con agujas de iridio o la irradiación con protones se ha empleado en pacientes con tumores únicos o paucinodulares. Las tasas de supervivencia a 1 año descritas van del 36 al 73%. Aunque el CCI no es un tumor francamente hipervascular en la TC o RM, su irrigación es preferentemente arterial. En los escasos estudios publicados, sin duda con un fuerte sesgo de selección de pacientes, tanto la quimioembolización como la radioembolización han mostrado altas tasas de respuesta. En un reciente metaanálisis, la mediana de supervivencia global ponderada fue de 15,6 ± 1,1 meses y casi el 50% de los pacientes tenía enfermedad estable como mejor respuesta29. Tratamiento sistémico Hasta la fecha no hay ensayos clínicos con tratamientos sistémicos en la población exclusiva de pacientes con CCI, sino que estos pacientes se han incluido, de forma minoritaria, junto con pacientes con otros tumores de la vía biliar. Dos estudios aleatorizados han comparado la quimioterapia sistémica con el mejor tratamiento de soporte en pacientes con cáncer biliar avanzado e inoperable. Aunque con baja potencia estadística, ambos sugieren un beneficio para los pacientes tratados con quimioterapia30,31. Además, un análisis de 112 ensayos clínicos en cáncer biliar avanzado sugiere que la gemcitabina y las fluoropirimidinas, solas o en combinación con cisplatino, pueden ser beneficiosas32. Con esta base, un reciente estudio aleatorizado que comparó la combinación de cisplatino y gemcitabina (cisplatino 25 mg/m2 seguido de gemcitabina 1.000 mg/m2, días 1 y 8 cada 21 días), con gemcitabina en monoterapia (gemcitabina 1.000 mg/m2, días 1, 8 y 15 de un ciclo de 28 días)33,34 mostró un incremento significativo de la supervivencia global en el brazo de la combinación frente a la monoterapia (mediana: 11,7 frente a 8,1 meses). Este beneficio se mantuvo en el análisis del subgrupo de 80 pacientes con CCI. A partir de aquí, este régimen se ha convertido en un estándar de práctica internacional y la base de estudios posteriores. Sin embargo, la extrapolación de los resultados de estos estudios a la población específica de CCI es comprometida por la falta de potencia estadística y, por lo tanto, los datos pueden orientar la práctica clínica pero no deben considerarse como el aval suficiente para establecer un estándar de tratamiento. Finalmente, no hay ninguna evidencia de que la quimioterapia de segunda línea mejore la supervivencia. Bibliografía 1.Fan B, Malato Y, Calvisi DF, Naqvi S, Razumilava N, Ribback S, et al. Cholangiocarcinomas can originate from hepatocytes in mice. J Clin Invest. 2012;122:2911-5. 2.Blechacz B, Komuta M, Roskams T, Gores GJ. Clinical diagnosis and staging of cholangiocarcinoma. Nat Rev Gastroenterol Hepatol. 2011;8:512-22. 3.Razumilava N, Gores GJ. Classification, diagnosis, and management of cholangiocarcinoma. Clin Gastroenterol Hepatol. 2013;11:13-21 e1; quiz e3-4. 4.Yang JD, Kim B, Sanderson SO, Sauver JS, Yawn BP, Larson JJ, et al. Biliary tract cancers in Olmsted County, Minnesota, 1976-2008. Am J Gastroenterol. 2012;107:1256-62. 5.Patel T. Increasing incidence and mortality of primary intrahepatic cholangiocarcinoma in the United States. Hepatology. 2001;33:1353-7. 6.Khan SA, Thomas HC, Davidson BR, Taylor-Robinson SD. Cholangiocarcinoma. Lancet. 2005;366:1303-14. 7.Palmer WC, Patel T. Are common factors involved in the pathogenesis of primary liver cancers? A meta-analysis of risk factors for intrahepatic cholangiocarcinoma. J Hepatol. 2012; 57:69-76. 8.Bridgewater J, Galle PR, Khan SA, Llovet JM, Park JW, Patel T, et al. Guidelines for the diagnosis and management of intrahepatic cholangiocarcinoma J Hepatol. 2014;60:1268-89. 9.Sia D, Hoshida Y, Villanueva A, Roayaie S, Ferrer J, Tabak B, et al. Integrative molecular analysis of intrahepatic cholangiocarcinoma reveals 2 classes that have different outcomes. Gastroenterology. 2013;144:829-40. 10.Andersen JB, Thorgeirsson SS. Genetic profiling of intrahepatic cholangiocarcinoma. Curr Opin Gastroenterol. 2012;28: 266-72. 11.Nakanuma Y, Sato Y, Harada K, Sasaki M, Xu J, Ikeda H. Pathological classification of intrahepatic cholangiocarcinoma based on a new concept. World J Hepatol. 2010;2:419-27. 12.Nakanuma Y, Sasaki M, Ikeda H, Sato Y, Zen Y, Kosaka K, et al. Pathology of peripheral intrahepatic cholangiocarcinoma with reference to tumorigenesis. Hepatol Res. 2008;38:325-34. 13.Goodman ZD. Neoplasms of the liver. Mod Pathol. 2007;20: S49-60. 14.Valls C, Guma A, Puig I, Sánchez A, Andia E, Serrano T, et al. Intrahepatic peripheral cholangiocarcinoma: CT evaluation. Abdom Imaging. 2000;25:490-6. 15.Murakami T, Nakamura H, Tsuda K, Ishida T, Tomoda K, Hori S, et al. Contrast-enhanced MR imaging of intrahepatic cholangiocarcinoma: pathologic correlation study. J Magn Reson Imaging. 1995;5:165-70. 16.Lan BY, Kwee SA, Wong LL. Positron emission tomography in hepatobiliary and pancreatic malignancies: a review. Am J Surg. 2012;204:232-41. 17.Tamandl D, Herberger B, Gruenberger B, Puhalla H, Klinger M, Gruenberger T. Influence of hepatic resection margin on recurrence and survival in intrahepatic cholangiocarcinoma. Ann Surg Oncol. 2008;15:2787-94. 18.Bach AM, Hann LE, Brown KT, Getrajdman GI, Herman SK, Fong Y, et al. Portal vein evaluation with US: comparison to angiography combined with CT arterial portography. Radiology. 1996;201:149-54. 19.Corvera CU, Blumgart LH, Akhurst T, DeMatteo RP, D’Angelica M, Fong Y, et al. 18F-fluorodeoxyglucose positron emission tomography influences management decisions in patients with biliary cancer. J Am Coll Surg. 2008;206:57-65. 20.Edge SB, Compton CC. The American Joint Committee on Cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol. 2010;17:1471-4. 21.Tan JC, Coburn NG, Baxter NN, Kiss A, Law CH. Surgical management of intrahepatic cholangiocarcinoma - a populationbased study. Ann Surg Oncol. 2008;15:600-8. 22.De Jong MC, Nathan H, Sotiropoulos GC, Paul A, Alexandrescu S, Marques H, et al. Intrahepatic cholangiocarcinoma: an international multi-institutional analysis of prognostic factors and lymph node assessment. J Clin Oncol. 2011;29:3140-5. 23.Choi SB, Kim KS, Choi JY, Park SW, Choi JS, Lee WJ, et al. The prognosis and survival outcome of intrahepatic cholangiocarcinoma following surgical resection: association of lymph node metastasis and lymph node dissection with survival. Ann Surg Oncol. 2009;16:3048-56. 24.Endo I, Gonen M, Yopp AC, Dalal KM, Zhou Q, Klimstra D, et al. Intrahepatic cholangiocarcinoma: rising frequency, improved survival, and determinants of outcome after resection. Ann Surg. 2008;248:84-96. 25.Paik KY, Jung JC, Heo JS, Choi SH, Choi DW, Kim YI. What prognostic factors are important for resected intrahepatic cholangiocarcinoma? J Gastroenterol Hepatol. 2008;23: 766-70. Colangiocarcinoma intrahepático 109 26.Robles R, Figueras J, Turrión VS, Margarit C, Moya A, Varo E, et al. Spanish experience in liver transplantation for hilar and peripheral cholangiocarcinoma. Ann Surg. 2004;239:265-71. 27.Sapisochin G, Fidelman N, Roberts JP, Yao FY. Mixed hepatocellular cholangiocarcinoma and intrahepatic cholangiocarcinoma in patients undergoing transplantation for hepatocellular carcinoma. Liver Transpl. 2011;17:934-42. 28.Lazaridis KN, Gores GJ. Cholangiocarcinoma. En: Boyer T, Manns MP, Sanyal A, editores. Elservier; 2011. p. 1032-44. 29.Edwards A, Ray JC. Transarterial therapies for unresectable cholangiocarcinoma: a meta-analysis. J Vasc Interv Radiol. 2012;23:S101. 30.Glimelius B, Hoffman K, Sjoden PO, Jacobsson G, Sellstrom H, Enander LK, et al. Chemotherapy improves survival and quality of life in advanced pancreatic and biliary cancer. Ann Oncol. 1996;7:593-600. 31.Sharma A, Dwary AD, Mohanti BK, Deo SV, Pal S, Sreenivas V, et al. Best supportive care compared with chemotherapy for unresectable gallbladder cancer: a randomized controlled study. J Clin Oncol. 2010;28:4581-6. 32.Eckel F, Schmid RM. Chemotherapy in advanced biliary tract carcinoma: a pooled analysis of clinical trials. Br J Cancer. 2007;96:896-902. 33.Valle JW, Wasan H, Johnson P, Jones E, Dixon L, Swindell R, et al. Gemcitabine alone or in combination with cisplatin in patients with advanced or metastatic cholangiocarcinomas or other biliary tract tumours: a multicentre randomised phase II study – The UK ABC-01 Study. Br J Cancer. 2009;101:621-7. 34.Valle J, Wasan H, Palmer DH, Cunningham D, Anthoney A, Maraveyas A, et al. Cisplatin plus gemcitabine vs. gemcitabine for biliary tract cancer. N Engl J Med. 2010;362:1273-81. SESIÓN 4 OTRAS ENFERMEDADES HEPÁTICAS Hipertensión portal idiopática Virginia Hernández-Gea Laboratorio de Hemodinámica Hepática, Servicio de Hepatología, Hospital Clínic-IDIBAPS, Barcelona, España CIBERehd (Centro de Investigación Biomédica en Red de enfermedades hepáticas y digestivas) INTRODUCCIÓN La hipertensión portal idiopática (HPI) es una causa rara de hipertensión portal (HTP) intrahepática de etiología incierta. Esta entidad ha recibido otros nombres como esclerosis hepatoportal, fibrosis portal no cirrótica, cirrosis septal incompleta, hiperplasia nodular regenerativa, transformación nodular parcial del hígado o venopatía portal obliterativa1-8. Para el diagnóstico de la HPI se requiere la presencia de signos inequívocos de HTP con permeabilidad de las venas suprahepáticas y del eje esplenoportal en ausencia de cirrosis hepática y de otras causas conocidas de HTP9-11. Las alteraciones arquitecturales hepáticas abarcan un amplio espectro de lesiones, que van desde cambios mínimos hasta alteraciones histológicas más relevantes, como la hiperplasia nodular regenerativa, pasando por una leve o moderada fibrosis portal, pero no hay ninguna de estas alteraciones histológicas que sea patognomónica. Típicamente, los pacientes con HPI tienen un curso más benigno que la HTP cirrótica y la función hepática suele estar conservada. EPIDEMIOLOGÍA La HPI tiene una distribución mundial, pero su prevalencia es variable. Se ha descrito una mayor prevalencia en países subdesarrollados y afecta fundamentalmente a personas de un nivel socioeconómico bajo1,10-13. Es particularmente frecuente en Asia: en India, la HPI constituye el 7,9-48% de los pacientes con HTP8,14,15 y en Japón es responsable del 30%9,16. No existe una clara predominancia en cuanto al sexo y edad; sin embargo, pueden observarse discretas diferencias entre estudios de diferentes países9,17,18. Aunque no está claramente establecido, las diferencias en el nivel socioeconómico, las condiciones de vida, la exposición a diferentes patógenos o la etnia podrían jugar un papel. En Japón, la incidencia de HPI ha disminuido en las últimas déca110 das, probablemente debido a una mejoría en la higiene y en los estándares de vida18. ETIOPATOGENIA La etiología de la HPI es desconocida. Se han postulado diferentes mecanismos en la etiopatogenia de esta entidad, pero hasta el momento ninguno de ellos ha sido demostrado. • Infecciones crónicas o recurrentes. Las infecciones repetidas del tracto digestivo con émbolos sépticos y la sepsis umbilical se han propuesto como una de las causas de HPI, ya que ocasionarían pileflebitis y, consecuentemente, daño endotelial, microtrombosis, esclerosis y obstrucción de las ramas portales de pequeño y mediano tamaño19. • Tóxicos y drogas. La exposición a algunos tóxicos y drogas se ha relacionado con la inducción de fibrosis en el espacio de Disse y desarrollo de HPI7. Entre los tóxicos asociados a HPI se encuentra la azatioprina y otros citotóxicos20,21. • Infección por el virus de la inmunodeficiencia humana (VIH). Se ha relacionado la infección por VIH en la patogenia de la HPI, fundamentalmente en los pacientes que habían recibido tratamiento con terapia antiviral (en especial larga exposición a didanosina y/o estavudina)22 o por efecto directo del VIH23-27. La discontinuación del tratamiento con didanosina se ha relacionado, en algunos estudios, con una reducción de la progresión de la HPI28,29. • Predisposición genética. Hasta la fecha no se ha identificado una base genética de la enfermedad. Sin embargo se ha visto agregación familiar30 y una mayor prevalencia de HLA-DR3 en pacientes indios31. • Alteraciones inmunológicas. Se ha descrito una mayor prevalencia de trastornos de la respuesta inmune y disminución en la respuesta inmune celular en pacientes con HPI32,33. En la mayoría de series de pacientes con HPI se constata una asociación más frecuente de la esperada con enfermedades autoinmunes (enfermedades del tejido conectivo, tiroiditis)34-36 y se ha descrito la presencia de anticuerpos anti-ADN hasta en un 65% de pacientes japoneses con HPI. Estudios recientes han descrito la presencia de HPI en pacientes con deficiencias inmunes primarias graves37. • Hipercoagulabilidad. Varios estudios muestran una mayor prevalencia de alteraciones protrombóticas en pacientes con HPI. Estas se hallaron hasta en un 50% de pacientes en un estudio europeo incluyendo 28 pacientes, de los cuales 13 desarrollaron trombosis portal durante el seguimiento10. En cambio, otro estudio más reciente38 objetivó alteraciones protrombóticas en tan solo un 7% de pacientes. Por ello, el papel de estas alteraciones requiere más estudios. • Miscelánea. Otros estudios han descrito el papel de la endotelina-1, el óxido nítrico y el factor de crecimiento del tejido conectivo en el desarrollo de HPI39-43. PRESENTACIÓN La HPI se caracteriza por la presencia de complicaciones derivadas de la HTP. Habitualmente, la función hepática está conservada10,11,44. Las varices gastroesofágicas están presentes en un 85-95% de los pacientes en el momento del diagnóstico de la enfermedad45. La gastropatía de la HTP está presente en menor frecuencia que en los pacientes con cirrosis hepática; sin embargo, las varices rectales son más comunes que en la cirrosis45,46. Es característico hallar una gran esplenomegalia10,11. La hemorragia digestiva por HTP es la manifestación clínica más frecuente, pero tiene mejor pronóstico que en la cirrosis hepática8,10,11,47,48. La causa principal son las varices esofágicas13. Se ha descrito la presencia de ascitis en aproximadamente el 50% de los pacientes con HPI10 y su presencia se ha relacionado con un peor pronóstico49. Suele ocurrir de forma transitoria, igual que el empeoramiento de las pruebas de función hepática, en el contexto de un episodio de hemorragia digestiva, cirugía o infección8,10,11. La encefalopatía hepática es una complicación rara, pero que en ocasiones puede ser grave50-52. Dos estudios prospectivos han descrito una prevalencia de síndrome hepatopulmonar del 10%53,54. No hay una relación evidente entre la HPI y el desarrollo de hepatocarcinoma55-58. Hasta en un 45,6% de los casos puede evidenciarse anemia, leucopenia y trombocitopenia como manifestaciones del hiperesplenismo11. La trombosis de la vena porta tiene una incidencia superior a la observada en los pacientes con cirrosis hepática, de Hipertensión portal idiopática 111 aproximadamente el 15% en 1 año10,22,49,59, llegando al 75% en pacientes con HPI asociada al VIH22,59,60. En una cohorte de 69 pacientes con HPI, la infección por VIH y la hemorragia variceal como manifestación al diagnóstico de la enfermedad fueron los 2 únicos factores independientes asociados al desarrollo de trombosis portal durante el seguimiento38. DIAGNÓSTICO Frecuentemente, los pacientes con HPI son diagnosticados erróneamente de cirrosis hepática. En un estudio patológico de hígados explantados se halló que de los pacientes que tenían diagnóstico histológico compatible con HTI, cerca del 80% llegaron al trasplante con el diagnóstico previo de cirrosis hepática criptogénica52. Ello probablemente refleja el hecho de que en la actualidad no hay ninguna prueba que permita el diagnóstico definitivo de HPI y este es un diagnóstico de exclusión. Sin embargo, existen datos que nos pueden hacer sospechar que estamos ante un caso de HPI. Las alteraciones histológicas son variables y, aunque pueden ser sugestivas de HPI, no son patognomónicas. Son hallazgos comunes la esclerosis portal, con engrosamiento de la capa íntima e hipertrofia de la muscular, que ocasionan un estrechamiento y la obliteración del lumen de las vénulas portales, la dilatación sinusoidal, la presencia de vasos portales aberrantes, la fibrosis perisinusoidal, la microtrombosis o la presencia de hiperplasia nodular regenerativa10,44,61,62. Sin embargo, la biopsia hepática es fundamental para descartar cirrosis y otras causas de HTP. Las exploraciones de imagen pueden mostrar un hígado de tamaño normal y ecoestructura homogénea61. Sin embargo, más frecuentemente las alteraciones ecográficas sugieren una enfermedad hepática crónica: superficie hepática nodular, engrosamiento de las paredes de la vena porta > 3 mm (principalmente en las ramas intrahepáticas) y afilamiento repentino de segunda división de las radículas intrahepáticas de la vena porta5,11,63. Como ya hemos mencionado, con mucha frecuencia el bazo es de gran tamaño y si se realiza una resonancia magnética pueden visualizarse los nódulos de Gamma Gandy (núcleos sideróticos que se encuentran frecuentemente en el bazo congestivo)61. Otros hallazgos en las pruebas de imagen son la existencia de frecuentes colaterales portosistémicas. El estudio ecográfico y la tomografía computarizada o la resonancia magnética son indispensables para evaluar el eje esplenoportal y las venas suprahepáticas, que por definición deben ser permeables para el diagnóstico de esta entidad. Sin embargo, no es infrecuente que estos pacientes desarrollen a lo largo de su historia natural trombosis del eje esplenoportal, y pueden ser erróneamente diagnosticados de trombosis portal pri- 112 Sesión 4 - OTRAS ENFERMEDADES HEPÁTICAS maria si son estudiados por primera vez en el momento en que la trombosis ya está presente. La práctica de un estudio hemodinámico hepático y/o la elastografía hepática pueden dar datos muy útiles de sospecha de HPI. Así, los pacientes con HPI presentan una alta incidencia de comunicantes vena-vena en el cateterismo de venas suprahepáticas (que en muchas ocasiones dificultan lograr una correcta oclusión de la vena suprahepática) e, incluso si se logra una adecuada oclusión, el gradiente de presión venosa hepática (GPVH) es habitualmente normal o está solo discretamente elevado, muy por debajo de lo esperado en un paciente con cirrosis hepática y signos semejantes de HTP, infraestimando la verdadera presión portal que presentan10,22,64,65. Esta infraestimación de la verdadera presión portal se debe fundamentalmente al aumento de la resistencia intrahepática a nivel presinusoidal66,67. De forma semejante, los pacientes con HPI presentan valores usualmente elevados en la elastografía hepática de transición (Fibroscan®), pero claramente inferiores a los valores descritos en pacientes con cirrosis hepática y signos semejantes de HTP22,68. Raramente, los pacientes con HPI presentan valores > 12 kPa22,65. Ante un paciente con signos claros de HTP, estos hallazgos en el GPVH y en el Fibroscan® nos deberían hacer sospechar la presencia de una HPI. Un estudio reciente utilizando ARFI (acoustic radiation force impulse, una técnica ecográfica que también mide la rigidez hepática de forma no invasiva) sugiere que el hallazgo de una razón rigidez del bazo/hígado ≥ 1,71 m/s tendría una gran capacidad diagnóstica para diferenciar a los pacientes con HPI de los pacientes con cirrosis hepática y hepatitis crónica69. Se están evaluando otros métodos no invasivos para ayudar a establecer el diagnóstico de HPI. Datos preliminares sugieren que los pacientes con HPI presentan un perfil metabolómico sérico diferencial (que incluye entre 3 y 5 metabolitos), cuya determinación podría permitir el diagnóstico específico positivo de esta entidad70. También se ha sugerido que el estudio del patrón de perfusión hepática mediante ultrasonografía con contraste71 o el hallazgo de valores séricos de vitamina B12 ≤ 250 pg/ml72 podrían permitir la diferenciación entre HPI y cirrosis hepática criptogénica. No hay estudios en la actualidad que hayan validado estos hallazgos en muestras amplias de pacientes. En resumen, el diagnóstico de HPI se basa en la presencia de signos inequívocos de HTP (varices gastroesofágicas, esplenomegalia, ascitis, colaterales), la exclusión de cirrosis hepática mediante biopsia hepática o de otras enfermedades que ocasionen HTP y, además, la evidencia de permeabilidad de las venas suprahepáticas y del eje esplenoportal. En ocasiones, nos encontraremos con pacientes en los que hay una alta sospecha de que tengan HPI, pero que no cumplen todos estos criterios (p. ej., presencia de trombosis portal en el momento del diagnóstico) y, hasta el descubrimiento de una prueba diagnóstica especifica de esta entidad, deberemos catalogarlos como de “posible” HPI. TRATAMIENTO No existen guías clínicas de manejo de esta enfermedad ni un tratamiento específico. Por lo tanto, se aplican las mismas medidas terapéuticas y preventivas que en pacientes con cirrosis hepática y HTP, que parecen tener la misma aplicabilidad y efectividad que en estos73. Hemorragia aguda Hay escasos estudios que evalúen de forma específica el manejo de la hemorragia aguda en estos pacientes74-76. Por lo tanto, el tratamiento del episodio agudo de la hemorragia no difiere del manejo en un paciente con cirrosis hepática77. La terapia combinada con fármacos vasoactivos y el uso de la profilaxis antibiótica en pacientes con HPI son temas de controversia, ya que el comportamiento hemodinámico y la función hepática de estos pacientes son diferentes de los pacientes con cirrosis. No obstante, no hay datos que contraindiquen el uso de estos tratamientos en pacientes con HPI, por lo que se recomienda la aplicación de estos. La terapia endoscópica se ha mostrado efectiva en el control de la hemorragia aguda variceal en el 95% de los pacientes con HPI78. La TIPS (derivación portosistémica intrahepática transyugular) o el shunt portosistémico quirúrgico son alternativas para los pacientes en los que falla el tratamiento médico y endoscópico. Bissonnette et al reportaron recientemente una muy buena supervivencia en pacientes con HPI cirrótica y hemorragia variceal no controlada que fueron tratados con la colocación de TIPS79. La encefalopatía post-TIPS puede aparecer, pero usualmente es de fácil control, aunque ocasionalmente requiere reducir el calibre de la TIPS79. En referencia a las profilaxis primaria y secundaria de la hemorragia variceal, actualmente se recomienda seguir las mismas guías clínicas que se aplican en los pacientes cirróticos. Tratamiento anticoagulante Debe considerarse el uso de anticoagulación en pacientes con HPI que tengan una condición protrombótica asociada o en los que han desarrollado trombosis del eje portal7. No obstante, no hay estudios que hayan demostrado la efectividad de esta actitud y, por ello, la indicación o no del tratamiento anticoagulante deberá de individualizarse. Hipertensión portal idiopática Trasplante hepático Hay varios casos publicados de trasplante hepático en pacientes con HPI10,44,52,80-83. Las indicaciones han sido el desarrollo de complicaciones progresivas y potencialmente mortales de la HTP unidas a la imposibilidad de realización de un shunt quirúrgico o una TIPS. El pronóstico postrasplante a largo plazo es bueno y probablemente sin recurrencia de la enfermedad52,81,82, si bien, como hemos indicado, la experiencia no es amplia. Bibliografía 1.Mikkelsen WP, Edmondson HA, Peters RL, Redeker AG, Reynolds TB. Extra- and intrahepatic portal hypertension without cirrhosis (hepatoportal sclerosis). Ann Surg. 1965;162: 602-20. 2.Sherlock S, Feldman CA, Moran B, Scheuer PJ. Partial nodular transformation of the liver with portal hypertension. Am J Med. 1966;40:195-203. 3.Sama SK, Bhargava S, Nath NG, Talwar JR, Nayak NC, Tandon BN, et al. Noncirrhotic portal fibrosis. Am J Med. 1971;51: 160-9. 4.Wanless IR, Mawdsley C, Adams R. On the pathogenesis of focal nodular hyperplasia of the liver. Hepatology. 1985;5: 1194-200. 5.Wanless IR. Micronodular transformation (nodular regenerative hyperplasia) of the liver: a report of 64 cases among 2,500 autopsies and a new classification of benign hepatocellular nodules. Hepatology. 1990;11:787-97. 6.Sciot R, Staessen D, Van Damme B, Van Steenbergen W, Fevery J, De Groote J, et al. Incomplete septal cirrhosis: histopathological aspects. Histopathology. 1988;13:593-603. 7.Schouten JN, García-Pagán JC, Valla DC, Janssen HL. Idiopathic noncirrhotic portal hypertension. Hepatology. 2011;54: 1071-81. 8.Madhu K, Avinash B, Ramakrishna B, Eapen CE, Shyamkumar NK, Zachariah U, et al. Idiopathic non-cirrhotic intrahepatic portal hypertension: Common cause of cryptogenic intrahepatic portal hypertension in a Southern Indian tertiary hospital. Indian J Gastroenterol. 2009;28:83-7. 9.Okudaira M, Ohbu M, Okuda K. Idiopathic portal hypertension and its pathology. Semin Liver Dis. 2002;22:59-72. 10.Hillaire S, Bonte E, Denninger M-H, Casadevall N, Cadranel J-F, Lebrec D, et al. Idiopathic non-cirrhotic intrahepatic portal hypertension in the West: a re-evaluation in 28 patients. Gut. 2002;51:275-80. 11.Dhiman RK, Chawla Y, Vasishta RK, Kakkar N, Dilawari JB, Trehan MS, et al. Non-cirrhotic portal fibrosis (idiopathic portal hypertension): experience with 151 patients and a review of the literature. J Gastroenterol Hepatol. 2002;17:6-16. 12.Vakili C, Farahvash MJ, Bynum TE. “Endemic” idiopathic portal hypertension: report on 32 patients with non-cirrhotic portal fibrosis. World J Surg. 1992;16:118-24; discussion 124-5. 13.Sarin SK, Kumar A, Chawla YK, Baijal SS, Dhiman RK, Jafri W, et al. Noncirrhotic portal fibrosis/idiopathic portal hypertension: APASL recommendations for diagnosis and treatment. Hepatol Int. 2007;1:398-413. 113 14.Boyer JL, Sen Gupta KP, Biswas SK, Pal NC, Basu Mallick KC, Iber FL, et al. Idiopathic portal hypertension. Comparison with the portal hypertension of cirrhosis and extrahepatic portal vein obstruction. Ann Intern Med. 1967;66:41-68. 15.Datta D V. Non-cirrhotic portal fibrosis (‘idiopathic’ portal hypertension in India). J Assoc Physicians India. 1976;24: 511-27. 16.Okuda K, Nakashima T, Okudaira M, Kage M, Aida Y, Omata M, et al. Liver pathology of idiopathic portal hypertension. Comparison with non-cirrhotic portal fibrosis of India. The Japan idiopathic portal hypertension study. Liver. 1982;2:176-92. 17.Sarin SK, Kapoor D. Non-cirrhotic portal fibrosis: current concepts and management. J Gastroenterol Hepatol. 2002;17: 526-34. 18.Okuda K. Non-cirrhotic portal hypertension versus idiopathic portal hypertension. J Gastroenterol Hepatol. 2002;17 Suppl 3:S204-13. 19.Sarin SK, Aggarwal SR. Idiopathic portal hypertension. Digestion. 1998;59:420-3. 20.Nataf C, Feldmann G, Lebrec D, Degott C, Descamps JM, Rueff B, et al. Idiopathic portal hypertension (perisinusoidal fibrosis) after renal transplantation. Gut. 1979;20:531-7. 21.Barnard JA, Marshall GS, Neblett WW, Gray G, Ghishan FK. Noncirrhotic portal fibrosis after Wilms’ tumor therapy. Gastroenterology. 1986;90:1054-6. 22.Chang P-E, Miquel R, Blanco J-L, Laguno M, Bruguera M, Abraldes J-G, et al. Idiopathic portal hypertension in patients with HIV infection treated with highly active antiretroviral therapy. Am J Gastroenterol. 2009;104:1707-14. 23.Schiano TD, Kotler DP, Ferran E, Fiel MI. Hepatoportal sclerosis as a cause of noncirrhotic portal hypertension in patients with HIV. Am J Gastroenterol. 2007;102:2536-40. 24.Maida I, García-Gasco P, Sotgiu G, Ríos MJ, Vispo ME, MartínCarbonero L, et al. Antiretroviral-associated portal hypertension: a new clinical condition? Prevalence, predictors and outcome. Antivir Ther. 2008;13:103-7. 25.Saifee S, Joelson D, Braude J, Shrestha R, Johnson M, Sellers M, et al. Noncirrhotic portal hypertension in patients with human immunodeficiency virus-1 infection. Clin Gastroenterol Hepatol. 2008;6:1167-9. 26.Mallet VO, Varthaman A, Lasne D, Viard J-P, Gouya H, Borgel D, et al. Acquired protein S deficiency leads to obliterative portal venopathy and to compensatory nodular regenerative hyperplasia in HIV-infected patients. AIDS. 2009;23:1511-8. 27.Kovari H, Ledergerber B, Peter U, Flepp M, Jost J, Schmid P, et al. Association of noncirrhotic portal hypertension in HIVinfected persons and antiretroviral therapy with didanosine: a nested case-control study. Clin Infect Dis. 2009;49:626-35. 28.Cachay ER, Peterson MR, Goicoechea M, Mathews WC. Didanosine Exposure and noncirrhotic portal hypertension in a HIV clinic in North America: a Follow-up study. Br J Med Med Res. 2011;1:346-55. 29.Vispo E, Morello J, Rodríguez-Novoa S, Soriano V. Noncirrhotic portal hypertension in HIV infection. Curr Opin Infect Dis. 2011;24:12-8. 30.Dumortier J, Boillot O, Chevallier M, Berger F, Potier P, Valette PJ, et al. Familial occurrence of nodular regenerative hyperplasia of the liver: a report on three families. Gut. 1999; 45:289-94. 31.Sarin SK, Mehra NK, Agarwal A, Malhotra V, Anand BS, Taneja V. Familial aggregation in noncirrhotic portal fibrosis: a report of four families. Am J Gastroenterol. 1987;82:1130-3. 114 Sesión 4 - OTRAS ENFERMEDADES HEPÁTICAS 32.Nayyar AK, Sharma BK, Sarin SK, Malhotra P, Broor SL, Sachdev G. Characterization of peripheral blood lymphocytes in patients with non-cirrhotic portal fibrosis: a comparison with cirrhotics and healthy controls. J Gastroenterol Hepatol. 1990;5:554-9. 33.Tokushige K, Komatsu T, Ohzu K, Yamauchi K, Obata H. A defective autologous mixed lymphocyte reaction in patients with idiopathic portal hypertension. J Gastroenterol Hepatol. 1992;7:270-3. 34.Saito K, Nakanuma Y, Takegoshi K, Ohta G, Obata Y, Okuda K, et al. Non-specific immunological abnormalities and association of autoimmune diseases in idiopathic portal hypertension. A study by questionnaire. Hepatogastroenterology. 1993;40:163-6. 35.Inagaki H, Nonami T, Kawagoe T, Miwa T, Hosono J, Kurokawa T, et al. Idiopathic portal hypertension associated with systemic lupus erythematosus. J Gastroenterol. 2000;35:235-9. 36.Tsuneyama K, Harada K, Katayanagi K, Watanabe K, Kurumaya H, Minato H, et al. Overlap of idiopathic portal hypertension and scleroderma: report of two autopsy cases and a review of literature. J Gastroenterol Hepatol. 2002;17:217-23. 37.Pulvirenti F, Pentassuglio I, Milito C, Valente M, De Santis A, Conti V, et al. Idiopathic non cirrhotic portal hypertension and spleno-portal axis abnormalities in patients with severe primary antibody deficiencies. J Immunol Res. 2014;2014:672458. 38.Siramolpiwat S, Seijo S, Miquel R, Berzigotti A, García-Criado A, Darnell A, et al. Idiopathic portal hypertension: natural history and long-term outcome. Hepatology. 2014;59: 2276-85. 39.Kamath PS, Tyce GM, Miller VM, Edwards BS, Rorie DK. Endothelin-1 modulates intrahepatic resistance in a rat model of noncirrhotic portal hypertension. Hepatology. 1999;30: 401-7. 40.Kamath P. Hepatic localization of endothelin-1 in patients with idiopathic portal hypertension and cirrhosis of the liver. Liver Transplant. 2000;6:596-602. 41.Yamaguchi E, Yamanoi A, Ono T, Nagasue N. Experimental investigation of the role of endothelin-1 in idiopathic portal hypertension. J Gastroenterol Hepatol. 2007;22:1134-40. 42.Sato Y, Sawada S, Kozaka K, Harada K, Sasaki M, Matsui O, et al. Significance of enhanced expression of nitric oxide syntheses in splenic sinus lining cells in altered portal hemodynamics of idiopathic portal hypertension. Dig Dis Sci. 2007;52:1987-94. 43.Morikawa H, Tamori A, Nishiguchi S, Enomoto M, Habu D, Kawada N, et al. Expression of connective tissue growth factor in the human liver with idiopathic portal hypertension. Mol Med. 2007;13:240-5. 44.Schouten JNL, Nevens F, Hansen B, Laleman W, Van den Born M, Komuta M, et al. Idiopathic noncirrhotic portal hypertension is associated with poor survival: results of a long-term cohort study. Aliment Pharmacol Ther. 2012;35:1424-33. 45.Rangari M, Sinha S, Kapoor D, Mohan JC, Sarin SK. Prevalence of autonomic dysfunction in cirrhotic and noncirrhotic portal hypertension. Am J Gastroenterol. 2002;97:707-13. 46.Chawla Y, Dilawari JB. Anorectal varices--their frequency in cirrhotic and non-cirrhotic portal hypertension. Gut. 1991;32:309-11. 47.Okuda K, Kono K, Ohnishi K, Kimura K, Omata M, Koen H, et al. Clinical study of eighty-six cases of idiopathic portal hypertension and comparison with cirrhosis with splenomegaly. Gastroenterology. 1984;86:600-10. 48.Nevens F, Staessen D, Sciot R, Van Damme B, Desmet V, Fevery J, et al. Clinical aspects of incomplete septal cirrhosis in comparison with macronodular cirrhosis. Gastroenterology. 1994;106:459-63. 49.Matsutani S, Maruyama H, Akiike T, Kobayashi S, Yoshizumi H, Okugawa H, et al. Study of portal vein thrombosis in patients with idiopathic portal hypertension in Japan. Liver Int. 2005;25:978-83. 50.McDonald JA, Painter DM, Gallagher ND, McCaughan GW. Nodular regenerative hyperplasia mimicking cirrhosis of the liver. Gut. 1990;31:725-7. 51.Radomski JS, Chojnacki KA, Moritz MJ, Rubin R, Armenti VT, Wilson GA, et al. Results of liver transplantation for nodular regenerative hyperplasia. Am Surg. 2000;66:1067-70. 52.Krasinskas AM, Eghtesad B, Kamath PS, Demetris AJ, Abraham SC. Liver transplantation for severe intrahepatic noncirrhotic portal hypertension. Liver Transpl. 2005;11:627-34; discussion 610-1. 53.De BK, Sen S, Sanyal R. Hepatopulmonary syndrome in noncirrhotic portal hypertension. Ann Intern Med. 2000;132:924. 54.Kaymakoglu S, Kahraman T, Kudat H, Demir K, Cakaloglu Y, Adalet I, et al. Hepatopulmonary syndrome in noncirrhotic portal hypertensive patients. Dig Dis Sci. 2003;48:556-60. 55.Stromeyer FW, Ishak KG. Nodular transformation (nodular “regenerative” hyperplasia) of the liver. A clinicopathologic study of 30 cases. Hum Pathol. 1981;12:60-71. 56.Colina F, Alberti N, Solís JA, Martínez-Tello FJ. Diffuse nodular regenerative hyperplasia of the liver (DNRH). A clinicopathologic study of 24 cases. Liver. 1989;9:253-65. 57.Nzeako UC, Goodman ZD, Ishak KG. Hepatocellular carcinoma in cirrhotic and noncirrhotic livers. A clinico-histopathologic study of 804 North American patients. Am J Clin Pathol. 1996;105:65-75. 58.Nzeako UC, Goodman ZD, Ishak KG. Hepatocellular carcinoma and nodular regenerative hyperplasia: possible pathogenetic relationship. Am J Gastroenterol. 1996;91:879-84. 59.Zocco MA, Di Stasio E, De Cristofaro R, Novi M, Ainora ME, Ponziani F, et al. Thrombotic risk factors in patients with liver cirrhosis: correlation with MELD scoring system and portal vein thrombosis development. J Hepatol. 2009;51:682-9. 60.Amitrano L, Brancaccio V, Guardascione MA, Margaglione M, Sacco M, Martino R, et al. Portal vein thrombosis after variceal endoscopic sclerotherapy in cirrhotic patients: role of genetic thrombophilia. Endoscopy. 2002;34:535-8. 61.Chawla Y, Dhiman RK. Intrahepatic portal venopathy and related disorders of the liver. Semin Liver Dis. 2008;28:270-81. 62.Verheij J, Schouten JNL, Komuta M, Nevens F, Hansen BE, Janssen HLA, et al. Histological features in western patients with idiopathic non-cirrhotic portal hypertension. Histopathology. 2013;62:1083-91. 63.Ibarrola C, Colina F. Clinicopathological features of nine cases of non-cirrhotic portal hypertension: current definitions and criteria are inadequate. Histopathology. 2003;42:251-64. 64.García-Tsao G, Groszmann RJ, Fisher RL, Conn HO, Atterbury CE, Glickman M. Portal pressure, presence of gastroesophageal varices and variceal bleeding. Hepatology. 1985;5:419-24. 65.Seijo S, Reverter E, Miquel R, Berzigotti A, Abraldes JG, Bosch J, et al. Role of hepatic vein catheterisation and transient elastography in the diagnosis of idiopathic portal hypertension. Dig Liver Dis. 2012;44:855-60. 66.Sarin SK, Sethi KK, Nanda R. Measurement and correlation of wedged hepatic, intrahepatic, intrasplenic and intravariceal Hipertensión portal idiopática 115 pressures in patients with cirrhosis of liver and non-cirrhotic portal fibrosis. Gut. 1987;28:260-6. non-cirrhotic portal fibrosis, and extrahepatic portal venous obstruction. Gastrointest Endosc. 1991;37:460-4. 67.Ohnishi K, Chin N, Tanaka H, Iida S, Sato S, Terabayashi H, et al. Differences in portal hemodynamics in cirrhosis and idiopathic portal hypertension. Am J Gastroenterol. 1989;84: 409-12. 76.Sarin SK, Govil A, Jain AK, Guptan RC, Issar SK, Jain M, et al. Prospective randomized trial of endoscopic sclerotherapy versus variceal band ligation for esophageal varices: influence on gastropathy, gastric varices and variceal recurrence. J Hepatol. 1997;26:826-32. 68.Vizzutti F, Arena U, Romanelli RG, Rega L, Foschi M, Colagrande S, et al. Liver stiffness measurement predicts severe portal hypertension in patients with HCV-related cirrhosis. Hepatology. 2007;45:1290-7. 69.Furuichi Y, Moriyasu F, Taira J, Sugimoto K, Sano T, Ichimura S, et al. Noninvasive diagnostic method for idiopathic portal hypertension based on measurements of liver and spleen stiffness by ARFI elastography. J Gastroenterol. 2013;48:1061-8. 70.Seijo S, Lozano JJ, Alonso C, Reverter E, Miquel R, Abraldes JG, et al. Metabolomics discloses potential biomarkers for the noninvasive diagnosis of idiopathic portal hypertension. Am J Gastroenterol. 2013;108:926-32. 71.Maruyama H, Ishibashi H, Takahashi M, Imazeki F, Yokosuka O. Effect of signal intensity from the accumulated microbubbles in the liver for differentiation of idiopathic portal hypertension from liver cirrhosis 1. Radiology. 2009;252:587-94. 72.Goel A, Ramakrishna B, Muliyil J, Madhu K, Sajith KG, Zachariah U, et al. Use of serum vitamin B12 level as a marker to differentiate idiopathic noncirrhotic intrahepatic portal hypertension from cryptogenic cirrhosis. Dig Dis Sci. 2013;58:179-87. 73.Siramolpiwat S, Seijo S, Miquel R, Berzigotti A, García-Criado A, Darnell A, et al. Idiopathic portal hypertension: Natural history and long-term outcome. Hepatology. 2014;59:2276-85. 74.Kiire CF. Controlled trial of propranolol to prevent recurrent variceal bleeding in patients with non-cirrhotic portal fibrosis. BMJ. 1989;298:1363-5. 75.Kochhar R, Goenka MK, Mehta SK. Outcome of injection sclerotherapy using absolute alcohol in patients with cirrhosis, 77.De Franchis R. Evolving consensus in portal hypertension. Report of the Baveno IV consensus workshop on methodology of diagnosis and therapy in portal hypertension. J Hepatol. 2005;43:167-76. 78.Chawla YK, Dilawari JB, Dhiman RK, Goenka MK, Bhasin DK, Kochhar R, et al. Sclerotherapy in noncirrhotic portal fibrosis. Dig Dis Sci. 1997;42:1449-53. 79.Bissonnette J, García-Pagán J-C, Albillos A, Ferreira C, Turón F, Téllez L, et al. Assessment of the transjugular portosystemic shunt (TIPSS) in the management of complications of noncirrhotic portal hypertension. J Hepatol. 2015;62 Suppl 2:S228. 80.Bernard PH, Le Bail B, Cransac M, Barcina MG, Carles J, Balabaud C, et al. Progression from idiopathic portal hypertension to incomplete septal cirrhosis with liver failure requiring liver transplantation. J Hepatol. 1995;22:495-9. 81.Loinaz C, Colina F, Musella M, López-Ríos F, Gómez R, Jiménez C, et al. Orthotopic liver transplantation in 4 patients with portal hypertension and non-cirrhotic nodular liver. Hepatogastroenterology. 1998;45:1787-94. 82.Dumortier J, Bizollon T, Scoazec JY, Chevallier M, Bancel B, Berger F, et al. Orthotopic liver transplantation for idiopathic portal hypertension: indications and outcome. Scand J Gastroenterol. 2001;36:417-22. 83.Inokuma T, Eguchi S, Tomonaga T, Miyazaki K, Hamasaki K, Tokai H, et al. Acute deterioration of idiopathic portal hypertension requiring living donor liver transplantation: a case report. Dig Dis Sci. 2009;54:1597-601.