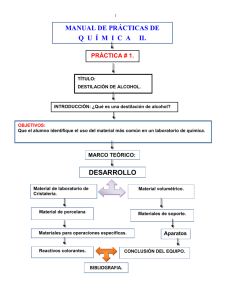
universidad de san carlos de guatemala
Anuncio

UNIVERSIDAD DE SAN CARLOS DE GUATEMALA FACULTAD DE AGRONOMÍA AREA DE CIENCIAS SUBAREA DE CIENCIAS QUÍMICAS LABORATORIO DE QUÍMICA ORGÁNICA MANUAL DEL LABORATORIO DE QUÍMICA ORGÁNICA Compilado por: Lic. Enrique Flores Corrección 2013 por: Br. Yorik Tenes Guatemala, Mayo 2013 INTRODUCCIÓN Las prácticas contenidas en este manual, servirán de apoyo al laboratorio de Química Orgánica. Está distribuido de manera que la dificultad de las prácticas vaya en aumento a medida que el estudiante obtenga nuevas destrezas y habilidades. Se presentan doce prácticas, en donde cada práctica está dividida en seis partes. La primera es una breve introducción en la cual se orienta al estudiante sobre las aplicaciones prácticas del experimento, en la segunda se presentan los objetivos que se pretenden alcanzar con el desarrollo de la práctica, la tercera parte es un marco teórico, donde se orienta al usuario con nociones breves sobre los compuestos y reacciones que se usan durante el experimento, en la cuarta parte se detalla una lista de materiales, equipo y cristalería que son necesarios para completar adecuadamente el experimento, en la quinta parte se detalla la metodología que se empleará para desarrollar los experimentos y finalmente, en la sexta parte, se citan algunas referencias bibliográficas que serán útiles para ampliar los conocimientos sobre el tema tratado. Al final de las prácticas se presenta un cuestionario que ayudará a orientar el estudio de los experimentos realizados en el laboratorio por el estudiante. De las nueve prácticas contenidas en el presente manual, la primera trata sobre análisis empírico de sustancias orgánicas, la segunda hace énfasis en la calibración del termómetro, mediante el punto de fusión, la tercera se relaciona con el uso de la ampolla de decantación en la separación de mezclas, la cuarta, sexta y séptima practicas son experimentos de síntesis de compuestos orgánicos. En el resto de las prácticas se manejan temas sobre destilación simple, reacciones típicas de los aldehídos, extracción de compuestos orgánicos a partir de material vegetal, empleando el sistema Soxhlet y saponificación de una grasa como última práctica. Con estas prácticas, se espera que el estudiante de Química Orgánica comprenda las reacciones y principios básicos que se requiere que conozcan para desarrollarse en el campo de la Agronomía. 2 UNIVERSIDAD DE SAN CARLOS DE GUATEMALA FACULTAD DE AGRONOMIA AREA DE CIENCIAS SUBAREA DE CIENCIAS QUÍMICAS. NORMAS GENERALES Y CUIDADOS DE MATERIALES DE LOS LABORATORIOS DE QUÍMICA En los laboratorios de química se hace indispensable cumplir con ciertas normas de conducta, pues de su cumplimiento dependen, el orden y la eficiencia del trabajo. Así mismo, es importante señalar que para que usted y sus compañeros trabajen con seguridad, deben seguirse determinados métodos de trabajo ya establecidos, por lo que es de carácter obligatorio el cumplimiento de las siguientes medidas de seguridad, para realizar las prácticas de laboratorio. 1. Cualquier cambio de jornada sólo podrá ser autorizado por el Coordinador de la Subárea de Química. 2. No se permitirá la permanencia en el laboratorio de ninguna persona ajena a la actividad docente que se desarrollará en cada práctica. 3. Se da un periodo de cinco minutos después de la hora establecida de inicio del laboratorio, para poder ingresar al mismo, luego se cierra la puerta y se realiza en un tiempo de 10 minutos la evaluación respectiva de la práctica; pasado este tiempo se abre nuevamente la puerta y se permite la entrada, sin reposición de la evaluación inicial. 4. Sólo se acepta un 20 por ciento máximo de ausencias, calculándose del número total de prácticas. 5. Previamente a la realización de la práctica, el estudiante deberá leer el instructivo de la misma, esto con el fin de prepararse para el examen corto inicial. 6. Para tener derecho a ingresar al laboratorio y realizar las prácticas, obligatoriamente deben cumplirse los siguientes requisitos: a. Presentar el diagrama de flujo y tener el instructivo de laboratorio así como un cuaderno que servirá para hacer las anotaciones respectivas de la práctica. b. Vestir bata de laboratorio larga, de mangas largas y de algodón, de color blanco. c. Contar con anteojos de seguridad para utilizarse a lo largo de la práctica. d. Entregar el reporte de la práctica anterior y cualquier otro tipo de materiales que se le solicite. 7. Cada día de práctica se formarán grupos de trabajo y a cada uno de ellos, se les asignará un locker con candado, ambos debidamente identificados. 8. Al iniciar cada una de las prácticas, un representante del grupo deberá solicitar al instructor de laboratorio la llave que corresponda a su locker de trabajo. Se deberá evitar perder la llave, pues queda bajo responsabilidad del grupo. 9. Cada estudiante es responsable de la cristalería y equipo que utilice. 10. Antes de utilizar el material para la práctica de trabajo, se debe revisar que dicho material esté completo. Para ello es indispensable el listado de materiales que aparecerá en el instructivo de la práctica, o lo que indique el instructor de laboratorio. 3 11. si al inicio de la práctica se encontrara algún daño en el material, el grupo de trabajo deberá informarlo de inmediato al instructor (éste habrá de informarlo al coordinador de laboratorios o al preparador del laboratorio). En este caso, se hará responsable del daño al último grupo que trabajó con ese material. 12. Si durante el desarrollo de la práctica de laboratorio se perdiese o dañase algún material, el estudiante responsable deberá dirigirse a la bodega de reactivos y cristalería para llenar un vale interno de cristalería rota, de modo que el grupo responsable reponga el material en un lapso de tiempo que no supere los cinco días hábiles. 13. Debido a lo indicado en el inciso 11, se recomienda a cada grupo de trabajo cuidar su material durante la práctica y revisar al final de la misma que no exista daño alguno. Una vez hecho esto se devuelve la llave del candado al instructor, quien velará por entregarlas al preparador del laboratorio. 14. El trabajo de laboratorio se debe realizar en silencio, con orden y disciplina. 15. Sólo deben estar sobre las mesas de trabajo los materiales que se estén utilizando. Los frascos de reactivos deben colocarse en su sitio inmediatamente después de utilizarlos. 16. Deben extraerse únicamente un excedente mínimo a la cantidad de reactivo que se va a utilizar, de modo que NO deben verterse de nuevo dentro de los frascos originales, pues el contenido puede contaminarse. 17. Los materiales sólidos inservibles, reactivos y mezclas ya utilizados deben descartarse en los recipientes destinados para ello, y nunca en el lavadero. 18. La cristalería debe lavarse cuidadosamente antes y después de efectuada la práctica. 19. Fumar e ingerir alimentos en el laboratorio está prohibido. 20. Para ausentarse temporalmente del laboratorio el estudiante deberá solicitar y obtener autorización de su profesor de laboratorio. 21. Al concluir la práctica las llaves de agua y gas deben dejarse cerradas, el equipo y área de trabajo perfectamente limpios. ______________________________________ Lic. Romeo Alfonso Pérez Morales, Q.B., M.A. Coordinador de la Subárea de Ciencias Químicas 4 Práctica 1. Análisis empírico de sustancias orgánicas I Introducción Uno de los primeros pasos cuando se tiene una muestra desconocida es determinar su estado físico: sólido, líquido o gaseoso. Luego se anota el color de la muestra original así como cualquier cambio que pueda ocurrir. El color de algunas sustancias se debe a las impurezas, éstas ocurren frecuentemente debido a la oxidación lenta del compuesto por el oxígeno del aire. (Por ejemplo la anilina recién destilada es incolora). Muchos líquidos y sólidos son realmente coloridos debido a la presencia de grupos cromóforos en la molécula, entre ellos compuestos nitrados, quinonas, azocompuestas y compuestos con sistemas conjugados extensos. Si un compuesto desconocido es un líquido estable incoloro o un sólido cristalino blanco, ésta información es valiosa porque excluye los grupos funcionales cromóforos, así como muchos otros grupos que por oxidación podrían convertirse en cromóforos. No es posible describir los olores pero el estudiante debe familiarizarse con los olores de los compuestos usuales. Los alcoholes tienen olores diferentes a los ésteres, los fenoles de las aminas, los aldehídos de las cetonas. El olor es más pronunciado en un grupo determinado mientras más corta es la cadena, dado su bajo peso molecular, debido a que son más volátiles. II Objetivos Al finalizar la práctica el estudiante estará en capacidad de: Realizar un análisis empírico preliminar a sustancias orgánicas desconocidas. Conocer distintos reactivos orgánicos y su aplicación en el laboratorio de química orgánica. Manipular cristalería y equipo para la identificación de sustancias desconocidas. III Marco Teórico 3.1 Compuestos Orgánicos Los compuestos orgánicos son todas las especies químicas que en su composición contienen el elemento carbono y, usualmente, elementos tales como el Oxígeno (O), Hidrógeno (H), Fósforo (F), Cloro (CL), Yodo (I) y nitrógeno (N), con la excepción del anhídrido carbónico, los carbonatos y los cianuros. Características Son Combustibles Poco Densos Todos contienen el elemento Carbono Poco Hidrosolubles Pueden ser de origen natural u origen sintético 5 3.1.1 Hidrocarburos Son los compuestos orgánicos más sencillos y se caracterizan por estar formados únicamente por hidrogeno y carbono. Algunos poseen una estructura molecular constituida por largas cadenas lineales que se denominan polímetros: otras cadenas son ramificadas. Son insolubles en agua, pero pueden disolver en disolventes orgánicos, como éter, benceno. Tetracloruro, cloroformos y otros. 3.2.1 Características: Están constituidos únicamente por átomos de carbonos o hidrógenos. Su fuente principal es el petróleo, gas natural, la hulla. En condiciones ambientales se encuentra en estado gaseoso (C1 al C4). En estado liquido desde el carbono 15 en estado (C5 al C15) En estado sólido el C16. Por condición completa origen el dióxido de carbono y agua. 3.2.2 Tipos de hidrocarburos Alcanos: se denominan también parafinas y constituyen un grupo de hidrocarburos cuyo carbono se une a través de un enlace covalente sencillo. Se denomina de acuerdo con el numero de carbono que poseen de la siguiente manera: con un carbono, metano; con dos etano; con tres propano; y así sucesivamente, butano, pentano, hexano, heptano, octano, nonato, decano, undecano, etc. Alquenos: se denominan también olefinas y constituyen un grupo de hidrocarburos cuya cadena carbonada uno o más enlaces dobles. El alqueno mas sencillo posee dos carbonos y se denomina eteno o etileno (CH2 = CH2). Otros son propeno, butano, centeno y así sucesivamente. La terminación usa para sus nombres es - eno. Se pueden generar diferentes tipos de alquenos a partir de un hidrocarburo con igual número de carbono y un solo enlace doble, ya que la posición del doble enlace puede variar ; por otra parte puede haber más de un enlace doble en un alqueno, y en tal caso reciben el nombre general de dienos, trienos y así sucesivamente. Alquinos: poseen cadenas carbonadas con uno o más enlaces triples. El alqueno mas sencillo es el etino o el acetileno (CH = CH), gas usado en soldaduras y en fabricación de plásticos. La terminación de sus nombres es - ino. Hidrocarburos aromáticos: estos compuestos se caracterizan por tener un olor fragante asociado, en un principio, a sustancias de origen vegetal El nombre de aromáticos, en la actualidad, no tiene nada que ver con el olor, La nominación de aromáticos data de los primeros compuestos de este tipo que fueron descubiertos, que se caracterizaban porque las fuentes de donde se obtenían tenían olores agradables en unos casos y en otros era el propio hidrocarburo el que poseía el aroma agradable. La estructura de estos compuestos revela que son derivados del benceno, compuesto cíclico con un anillo central que representa tres dobles enlaces. Algunos compuestos aromáticos son los siguientes: tolueno, xileno, estireno, antraceno, fenantreno, naftaleno entre otros. 6 3.1.2 Identificación de los compuestos orgánicos: Un compuesto orgánico se reconoce porque al arder deja un residuo negro de carbón. Al comparar el estado físico y la solubilidad de diferentes compuestos orgánicos, nos percatamos de que pueden existir en estado sólido, liquido o gaseoso y de que la solubilidad en el agua varia, desde los que son totalmente insolubles hasta los componentes solubles. Los compuestos orgánicos, esencialmente no polares, son insolubles. Ello se debe a que las moléculas de agua, fuertemente polares, se adhieren entre si y no permiten a las moléculas no polares introducirse en ellas. El grato sabor de un fruto o una flor se debe fundamentalmente a los ingredientes activos que forman parte de su composición. En su mayoría estos compuestos del carbono conocidos con el nombre de ésteres. 3.1.3 Algunos Compuestos Orgánicos que Utilizamos Diariamente: La mayoría de los alimento ( frutas, harinas, aceites comestibles, carnes) Medicamentos (tranquilizantes, antibióticos, aspirinas) Fibras naturales (algodón, lana, seda) Fibras artificiales(dacrón, nylon, orlón y rayón, utilizados para la fabricación de telas para vestir) Bebidas alcohólica (vinos, cidras) Insecticidas Detergentes Desinfectantes Colorantes Recipientes plásticos Gas de cocina Combustible para motores ( gasolina, kerosén, gas-oil) 3.1.4 Prueba de Ignición Muchos líquidos arden con llama característica que ayuda a determinar la naturaleza del compuesto. Así un hidrocarburo aromático (que tiene un contenido de carbono relativamente alto) arde con una llama fuliginosa. Los hidrocarburos alifáticos arden con llamas amarillas pero menos fuliginosas. A medida que aumenta el contenido de oxígeno en el compuesto, la flama se vuelve más clara (azul). Si la sustancia es inflamable deberá tomarse las precauciones necesarias para la manipulación subsecuente del compuesto. Esta prueba también muestra si deberá determinarse el punto de fusión de un sólido (mucho compuestos no tienen puntos de fusión sino que se descomponen definitivamente sin pasar por la fusión), también indica si el sólido es explosivo. Por último se debe observar si después de la combustión deja un residuo, el cual indicaría presencia de heteroátomos (probablemente metales). 3.1.5 Prueba de Baeyer (con permanganato de potasio): Una solución de permanganato de potasio se decolora con compuestos que tienen enlaces etilénicos y/o acetilénicos. Este ensayo se conoce como la prueba de Baeyer para insaturaciones. 7 3.1.6 Yodo en Hexano Este reactivo se utilizar para determinar la presencia de dobles enlaces en el compuesto a examinar. 3.1.7 Acetileno Etino o Acetileno, gas inflamable, inodoro e incoloro, algo más ligero que el aire, de fórmula HC:CH. En su forma más frecuente tiene un olor desagradable debido a sus impurezas. Puede obtenerse a partir de diversos compuestos orgánicos calentándolos en ausencia de aire, pero comercialmente se prepara por reacción del bicarburo de calcio con el agua. Aunque el etino puede licuarse a temperatura ambiente y alta presión, resulta violentamente explosivo en estado líquido. El etino arde al aire con una llama caliente y brillante. Antiguamente se utilizaba como fuente de iluminación, pero hoy en día se emplea principalmente en la soldadura oxiacetilénica, en la que el etino se quema con oxígeno produciendo una llama muy caliente que se usa para soldar y cortar metales. El acetileno también se utiliza en los procesos de síntesis química, y en especial en la fabricación del cloro etileno (cloruro de vinilo) para plásticos, del etanal (acetaldehído) y de los neoprenos de caucho sintético. El etino tiene un punto de fusión de -81 °C y un punto de ebullición de -57 °C. En la presente práctica se obtendrá acetileno a partir de carburo de calcio. La reacción es la siguiente: CaC2 + 2H2O → H - C ≡ C - C + Ca(OH)2 IV Marco Experimental 4.1 Materiales: Cant. Equipo y Cristalería 6 Tubos de ensayo 1 Gradilla de metal 1 Pipeta de 5 mL 1 Balón de destilación 1 Tapón de hule monohoradado 1 Mechero y su manguera 2 Pinzas Fisher 1 Soporte universal 1 Placa de porcelana Goteros 1 Beacker de 100 mL 1 Probeta de 5 Ml 1 Anillo de hierro 1 Manguera 1 Ampolla de decantación 1 Espátula 1 Micro espátula Benzaldehído Reactivos Isopropanol Acetona Acetato de etilo Naftol Difenilamina Heptanal Tolueno Hexano Naftaleno Ciclo hexano Sal Común Hidróxido de calcio Propano Parafina Acetato cúprico Glucosa Tetracloruro de carbono Carburo de calcio Yodo en Hexano 8 Fenol Acido Acético Permanganato de K al 2% Éter de petróleo Etanol Ciclo hexeno 4.2 Metodología: 4.2.1 PRUEBA DE IGNICIÓN En una de las celdas de la placa de porcelana, se coloca la muestra desconocida y se acerca la llama para determinar su inflamabilidad. Calentar suavemente sobre una llama débil Incrementar la intensidad del calentamiento hasta incinerar. Anotar los resultados en la siguiente tabla No. Muestra Distancia (1-4) Color de la llama Consistencia Color de Conclusión de la llama los humos 1 Hexano 2 Hidróxido de Calcio 3 Propano 4 Éter de Petróleo 5 Etanol 6 Acetona 7 Ciclo hexano 8 Tolueno 9 Acetato cúprico 10 Acetato de etilo 11 Acetona 12 Ciclo hexeno * La casilla de distancia es únicamente para las muestras sólidas y hace referencia a la distancia a la cual el sólido comienza a fundirse. La escala oscila entre 1 y 4. El 1 es cuando el sólido se funde muy cerca de la llama y el 4 cuando se funde lejos. ** En la columna de consistencia de la llama indicar si ésta es tenue o fuerte ***Color de humos: blanco, negro o no produce humos **** En conclusión indicar según los resultados de la prueba si la muestra es un compuesto orgánico o inorgánico. Si se trata de un compuesto orgánico indicar si es saturado, insaturado o aromático. 4.2.2 PRUEBA DE pH Determinación de pH con papel indicador En una de las celdas de la placa de porcelana, se colocan las siguientes muestras en solución: Acido Acético, Urea, Fenol. Cortas tiras de papel Indicador y observar el cambio de color Determinar el pH que indique el papel Indicador 9 4.2.3 PRUEBA DE INSATURACIÓN Prueba de Baeyer (con permanganato de potasio): Enumerar 5 tubos de ensayo de 1 a 5. Agregar 0.1 g (0.2 mL) en los tubos 1 a 4 respectivamente del compuesto que se va a examinar, y al tubo 5 colocar 2 mL de agua destilada. En el tubo 5 colocar una microespatula de Glucosa y diluir con el agua contenida en el tubo de ensayo. Añadir una solución de KMnO4 al 2% gota a gota con agitación hasta que persista el color púrpura de la solución de permanganato de potasio. La prueba es positiva cuando la solución de permanganato se decolora hasta desaparecer y se forma un precipitado de color marrón, entonces la muestra probablemente sea un compuesto insaturado. NOTA: Si el color no cambia entre 0.5 y 1 minuto dejar reposar los tubos durante 5 minutos con ocasional agitación vigorosa. No se engañe con una reacción débil, pues ésta puede deberse a la presencia de impurezas (el producto de reducción del Mn es un precipitado marrón del MnO2). La decoloración de una o dos gotas de la solución de permanganato no siempre puede aceptarse como prueba positiva. Anotar los resultados en el siguiente cuadro: No Compuesto Resultado de la Conclusión prueba de Baeyer 1 Ciclo hexeno 2 Hexano 3 Tolueno 4 Benzaldehído 5 Glucosa (*) Anotar “+” cuando la prueba sea positiva o “-“cuando la prueba sea negativa. (**) En la conclusión indicar si el compuesto es saturado o insaturado dependiendo del resultado de la prueba. 4.2.3 YODO EN HEXANO Se tienen tres muestras desconocidas y se desea saber si son saturadas, insaturadas o aromáticas. Prepare una serie de tres tubos identificados como A, B y C. Agregar 2 mL de las muestras que se le presentan en el cuadro a continuación en el tubo respectivo. Agregar 1 mL de Yodo en Hexano a cada uno de los tubos y observar los resultados 10 Muestra Yodo en Hexano Conclusión A Tolueno B Ciclo Hexano C Ciclo Hexeno (*) En los resultados escribir si la reacción es positiva o negativa. Es positiva cuando el bromo decolora y no tiñe la muestra. (**) En la conclusión indicar si la muestra es saturada, insaturada o aromática 4.2.4 OBTENCIÓN DE ACETILETINO (ETINO) Armar un sistema generador de gases (ver figura). Para ello debe utilizar un balón de destilación, un tapón de hule monohoradado, una ampolla de decantación, una pinza fisher, un anillo de hierro, un soporte universal, una manguera y una pipeta de 5 mL IMPORTANTE: REVISAR Y CORREGIR CUALQUIER POSIBLE FUGA DE GAS EN EL SISTEMA QUE NO SEA LA BOCA DE LA PIPETA Agregar de 10 a 20 g de carburo de calcio en el balón de destilación (evitar que el balón contenga agua). Con la ampolla de decantación se deja caer gotas de agua sobre el carburo de calcio. Se forma un gas incoloro. Encender el gas con el mechero o fósforo. El gas arde con llama blanca produciendo hollín. Por su seguridad apagar inmediatamente la llama, luego de haberla observado. Sistema Generador de Gases ‘V Cuestionario 1. ¿Cuál es el principio químico de la prueba de Bayer para instauración? 2. ¿Describa otros métodos prácticos que existan, para el examen preliminar de sustancias orgánicas. 3. ¿Qué otros métodos existen para la obtención de acetileno y cuál es el uso industrial de este compuesto? 11 VI Bibliografía 1 Compuestos orgánicos (en línea). Consultado el 2 de noviembre de 2007. Disponible en: http://html.rincondelvago.com/compuestos-organicos_3.html 2 GRAHAM SOLOMONS, T.W. 1,990. Fundamentos de Química Orgânica. Editorial Limusa. México, D.F. 903 p. 3 PINAGEL, D.; SLOWING, I.; DE LA ROCA, W. 2,003. Manual de prácticas de laboratorio de Química Orgánica 1. Facultad de Ciencias Químicas y Farmacia, USAC. Guatemala, Gua. 53 p. 12 Práctica 2. Determinación de propiedades físicas: Puntos de ebullición y Puntos de fusión para la calibración del termómetro I Introducción Cuando usted comienza a trabajar en un lugar donde no ha estado antes, lo primero que trata de averiguar, es qué tareas va a desempeñar, quiénes son sus compañeros de trabajo y cuáles son sus responsabilidades, para identificarse con su medio. De la misma manera, en el laboratorio de Química Orgánica, cuando usted trabaja con un compuesto que nunca ha manipulado, lo primero que deberá determinar son sus propiedades físicas. La importancia de determinar las propiedades físicas reside en que estas son específicas de un compuesto o familia de compuestos. En este sentido, la calibración del termómetro para tomar lecturas más exactas sería el principio, antes de determinar el resto de propiedades físicas para identificar una muestra desconocida, lo cual será particularmente útil, si usted trabaja con compuestos desconocidos o que han sido extraídos de plantas o animales. II Objetivos Elaborar la curva de calibración para un termómetro particular, utilizando puntos de fusión de diferentes sustancias orgánicas. Desarrollar habilidades en el uso de cristalería y equipo para la determinación del punto de fusión de diferentes sustancias orgánicas. Determinar experimentalmente el punto de fusión de cinco sustancias sólidas orgánicas. III Marco teórico 3.1 Calibración del termómetro: En trabajos prácticos de laboratorio de química con frecuencia se utiliza el termómetro para la medición de temperaturas. Un termómetro corriente de mercurio sirve para medir temperaturas comprendidas entre -38°C y 350°C aproximadamente, ya que el mercurio hierve a 359°C a una atmósfera de presión; este intervalo de temperatura suele bastar para la mayoría de trabajos de laboratorio de química orgánica. Sin embargo, la exactitud de los termómetros que se obtienen en el mercado varía de unos a otros, es decir, la lectura con uno de ellos puede ser completamente exacta o presentar un error de hasta 54°C, especialmente a temperaturas altas. Es por eso que el termómetro que debe utilizarse debe ser calibrado por lo menos una vez al año. Desde el punto de vista práctico, lo mejor es calibrarlo en condiciones lo más parecidas a aquellas en las que el termómetro va a ser utilizado, y después hacer las correcciones oportunas a las lecturas obtenidas del termómetro. Siembre debe anotarse la temperatura obtenida por lectura directa del termómetro, y la temperatura corregida debe indicarse claramente en las anotaciones de informes, o reportes de práctica. 13 En la calibración del termómetro se determinan los puntos de fusión de por lo menos cinco sustancias puras, de puntos de fusión conocidos. Para elaborar la curva de calibración se grafican las lecturas obtenidas del termómetro en el eje X de un cuadrante contra los valores teóricos de puntos de fusión de las sustancias en el eje Y del mismo cuadrante; se plotean y se unen los diferente puntos de las diferentes sustancias. Asimismo, experimentalmente debe determinarse la temperatura 0°C con una mezcla de hielo-agua. 3.2 Evaporación y presión de vapor La energía cinética de las moléculas de un líquido está cambiando continuamente a medida que chocan con otras moléculas. En cualquier instante, algunas de las moléculas de la superficie adquieren la suficiente energía para superar las fuerzas atractivas y escapan a la fase gaseosa ocurriendo la evaporación. La velocidad de evaporación aumenta a medida que se eleva la temperatura del líquido. Si el líquido se encuentra en un recipiente cerrado, las moléculas del vapor quedarán confinadas en las vecindades del líquido, y durante el transcurso de su movimiento desordenado algunas de ellas pueden regresar de nuevo a la fase líquida. Al principio, la velocidad de condensación de las moléculas es lenta puesto que hay pocas moléculas en el vapor. Sin embargo, al aumentar la velocidad de evaporación, también aumenta la velocidad de condensación hasta que el sistema alcanza un estado en el que ambas velocidades son iguales (véase la figura 6.1). Figura 1 Evaporación de un líquido en un recipiente cerrado En este estado de equilibrio dinámico, la concentración de las moléculas en el vapor es constante y por lo tanto también es constante la presión. La presión ejercida por el vapor cuando se encuentra en equilibrio con el líquido, a una determinada temperatura, se denomina presión de vapor y su valor aumenta al aumentar la temperatura. 3.2.2 Temperatura de ebullición La temperatura de ebullición es aquella a la cual la presión de vapor del líquido es igual a la presión externa. En este punto, el vapor no solamente proviene de la superficie sino que también se forma en el interior del líquido produciendo burbujas y turbulencia que es característica de la ebullición. La temperatura de ebullición permanece constante hasta que todo el líquido se haya evaporado. El punto de ebullición que se mide cuando la presión externa es de 1 atm se denomina temperatura normal de ebullición y se sobreentiende que los valores que aparecen en las tablas son puntos normales de ebullición. 14 3.2.3 Corrección de la temperatura de ebullición En el caso de los líquidos, la temperatura de ebullición se ve afectada por los cambios en la presión atmosférica debidos a las variaciones en la altura. A medida que un sitio se encuentra más elevado sobre el nivel del mar, la temperatura de ebullición se hace menor. A una altura de 1500 m o 0.84 atm (Medellín, por ejemplo), el agua ebulle a 95 °C mientras que al nivel del mar el agua hierve a 100 °C. Con el propósito de realizar comparaciones con los valores reportados por la literatura, se hace necesario corregir la temperatura normal de ebullición en un factor proporcional a la diferencia de presiones. Los factores de corrección se muestran en la tabla 6.1 y dependen de la polaridad del líquido. Ejemplo 1 La temperatura normal de ebullición del agua es de 100 °C. ¿Cuál será el punto de ebullición del agua en Medellín (p = 640 torr) y Bogotá (p = 560 torr)? Para Medellín: p = 760 torr – 640 torr = 120 torr = 120 mm Hg Fc = 120 mm Hg x 0.370 °C/10 mm Hg = 4.4 °C Te = 100 °C – 4.4 °C = 95.6 °C Tabla 1 Factores de corrección del punto de ebullición por cambios en la presión Teb normal (°C) 50 60 70 80 90 100 110 120 130 Para Bogotá: Variación en T por p = 10 mm Hg Líquidos no polares Líquidos polares 0.380 0.320 0.392 0.330 0.404 0.340 0.416 0.350 0.428 0.360 0.440 0.370 0.452 0.380 0.464 0.390 0.476 0.400 p = 760 torr – 560 torr = 200 torr = 200 mm Hg Fc = 200 mm Hg x 0.370 °C/10 mm Hg = 7.4 °C Te = 100 °C – 7.4 °C = 92.6 °C 3.2 Puntos de fusión: El punto de fusión de una sustancia es el intervalo de temperatura en el cual la fase sólida cristalina ordenada y la fase líquida aleatoria se encuentran en equilibrio. El valor del punto de fusión de un sólido es una de las constantes físicas de una sustancia que tiene 15 una gran utilidad para confirmar la identidad del compuesto y también es utilizado para determinar el grado de pureza del mismo. El principal factor que afecta la temperatura de fusión de un compuesto es la naturaleza de las fuerzas que mantienen la estructura cristalina de un sólido. Si las fuerzas son iónicas, los puntos de fusión de esos sólidos serán muy elevados, como es el caso de las sales y los compuestos inorgánicos, que por lo general tienen puntos de fusión superiores a 300°C. En la mayoría de los compuestos orgánicos las fuerzas intermoleculares son covalentes y por lo tanto presentan puntos de fusión entre 30°C y 300°C. En la práctica, el punto de fusión no se determina experimentalmente como una temperatura puntual, sino más bien como un intervalo de temperatura entre las cuales el sólido se convierte en un líquido; en este sentido, el punto de fusión que se reporta es la temperatura final de intervalo de fusión. En el caso de sustancias puras, generalmente presentan un intervalo de fusión que no excede de 2°C entre la temperatura a la cual el sólido empieza a fundir y la temperatura a la cual todo el sólido ha fundido. Cuando la muestra contiene impureza el intervalo de temperaturas es más grande. Se ha reportado que las impurezas disminuyen el punto de fusión de un sólido, y que esta disminución es tanto mayor en tanto más impura es la sustancia, disminuyendo el punto de fusión en 30°C. IV Marco Experimental 4.1 Materiales: Cant. Equipo y Cristalería 1 Mechero y su manguera Tubos capilares 1 Mortero y su pistilo 1 Termómetro 1 Tapón de hule monohoradado 2 Pinza Fisher 1 Tubo de thiele 1 Soporte universal Hilo 1 Espátula Reactivos Acido Benzoico Acido Salicílico Hidroquinona Acido succínico Di fenilamina Hexano 2-Propanol 1-butanol Tubos de Contención 4.2 Metodología Utilizando la llama del mechero Bunsen, selle 12 tubos capilares por un extremo. Llene cada tubo capilar hasta una altura de 1 cm. de muestra previamente pulverizada. Compacte la muestra dentro del tubo capilar. Sujete, utilizando hilo, los tubos capilares al termómetro, de tal modo que el nivel inferior de los tubos y del bulbo del termómetro queden alineados. Inserte el termómetro de un tapón de hule horadado, y sujete el tapón con una pinza Fisher. 16 Verifique que su tubo de thiele no contenga agua, llene el mismo con glicerina hasta cubrir justamente la entrada superior del brazo lateral. Sujete el tubo de thiele con una pinza de fisher, e instálelo al soporte universal. Coloque el termómetro y las muestras dentro del tubo de thiele de la manera que indique su instructor. Caliente suavemente el tubo de thiele, utilizando el mechero de Bunsen, flameando el codo del brazo lateral de tubo. Puede utilizar un tubo largo de vidrio para compactar la muestra que está dentro del tubo capilar. Observe atentamente las muestras, y anote la temperatura a la cual empieza a licuarse y a adherirse a la pared del tubo capilar la primera muestra sólida. En este momento debe suspender inmediatamente el calentamiento. Enseguida anote la temperatura a la cual la muestra queda completamente fundida. Siga calentado hasta determinar los intervalos de temperatura en que funden el resto de las muestras. Repita el ensayo hasta que los resultados sean constantes, eliminando los datos atípicos, utilizando nuevas porciones de las muestras. Determinación de Puntos de ebullición Selle 6 tubos capilares igual que en el experimento anterior. Arme un sistema similar al empleando en el experimento anterior. Amarrar 2 tubos de contención a un termómetro, de la misma manera que lo hizo en el experimento anterior con los tubos capilares. Agregar suficiente muestra dentro del tubo de contención. Coloque el tubo capilar con el extremo sellado en la parte de afuera del tubo de contención y la parte sin sellar dentro del líquido que se encuentra en el tubo de contención. Calentar el sistema, flameando el brazo del tubo de Thiele. A medida que se el sistema de caliente, observará como comienzan a salir burbujas por el extremo inferior del tubo capilar. Cuando el burbujeo sea uniforme y continuo, formando lo que se conoce como el “rosario de burbujas”, se suspenderá el calentamiento. Mantener en observación el sistema. Notará que a medida que va descendiendo la columna de mercurio dentro del termómetro, la velocidad con que salen las burbujas 17 del capilar va disminuyendo. Esperar hasta que salga la última burbuja del capilar. En este momento anotar la temperatura. Continuar todo el procedimiento hasta completar todas las sustancias. Repetir todo el procedimiento para corroborar sus resultados. V Cuestionario 1. ¿Por qué es aconsejable usar un baño de aceite para la determinación de la temperatura de fusión? ¿Por qué debe ser muy lento el calentamiento del baño de aceite? 2. ¿Además del aceite es posible utilizar otros líquidos para esta misma práctica? ¿Cuáles? ¿Qué criterios deben tenerse en cuenta para su selección? 3. ¿Por qué la temperatura de fusión de muchos sólidos se reporta como un rango? (Ej. acetanilida: 113-114 °C) 4. ¿Cuál es la influencia de una impureza insoluble en la temperatura de fusión de un sólido? 5. ¿Cómo son, comparativamente, las temperaturas de fusión de sólidos iónicos, covalentes, polares y no polares? 6. Analice el por qué de la forma tan particular del tubo de Thiele. VI. Bibliografía 1. GRAHAM SOLOMONS, T.W. 1,990. Fundamentos de Química Orgânica. Editorial Limusa. México, D.F. 903 p. 2. PINAGEL, D.; SLOWING, I.; DE LA ROCA, W. 2,003. Manual de prácticas de laboratorio de Química Orgánica 1. Facultad de Ciencias Químicas y Farmacia, USAC. Guatemala, Gua. 53 p 3. Puntos de fusión. (en línea). Consultado el 2 de noviembre de 2007. Disponible en: http://docencia..edu.co/cudeaen/tecnicaslabquimico/02practicas/practica04.htm 18 Práctica No. 3 Uso de la ampolla de decantación, extracción simple. Separación en mezclas I. Introducción La ampolla de decantación es una pieza de cristalería que se emplea en los laboratorios de química. Al tratarse de mezclas heterogéneas, es un instrumento sumamente útil. La separación de mezclas empleando la ampolla de decantación, se basa en la solubilidad relativa de la sustancia que se desea separar de la mezcla. Una aplicación práctica del uso de la ampolla de decantación es la separación de diferentes pigmentos, presentes en una muestra vegetal. La manera de llevar a cabo esto es emplear solventes polares y apolares para que cada uno extraiga diferentes tipos de pigmento. II. Objetivos Que el estudiante aprenda el uso adecuado de la ampolla de decantación. Que el estudiante aprenda y aplique experimentalmente la técnica de extracción simple, empleando un solvente orgánico. Que el estudiante aplique técnicas para purificar y filtrar una sustancia orgánica III. Marco teórico 3.1 Purificación con carbón activado Cuando se tiene un compuesto líquido o una solución la cual está contaminada por impurezas solubles (las que le dan una apariencia turbia a la solución) o está coloreada, se le debe aplicar un proceso que permita eliminar las impurezas y aclarar o decolorar la solución. En estos casos normalmente se emplea carbón activado, esta es una sustancia de color negro que tiene la habilidad de adsorber partículas finamente dispersas, como las de las suspensiones coloidales, y precipitarlas para que sean eliminadas posteriormente, normalmente haciendo uso de filtración (2). Es un material que se caracteriza por poseer una cantidad muy grande de microporos (poros menores que 2 nanómetros). A causa de su alta microporosidad, un solo gramo de carbón activado posee un área superficial de aproximadamente unos 500 m². El uso de carbón activado también permite eliminar o disminuir considerablemente el olor de una sustancia, cuando éste es causado por la presencia de contaminantes, dentro de la sustancia. (4) Carbón activado 19 3.2 Filtración La filtración es un proceso en el cual se hace pasar una mezcla heterogénea o una solución sobresaturada, a través de una membrana porosa con la finalidad de que esta retenga las partículas sólidas y permita pasar la solución (1). La eficiencia del proceso de filtración dependerá directamente del diámetro del poro. Los poros con diámetros grandes, permiten que la filtración se lleve a cabo rápidamente, pero también dejarán pasar los contaminantes insolubles que sean de menor tamaño que el poro de la membrana. Al contrario, cuando se tiene un poro de diámetro reducido, la velocidad del proceso de filtración será menor, pero impedirá el paso de los contaminantes de tamaño pequeño. Proceso de Filtración 3.3 Extracción La extracción es un proceso de laboratorio empleado para la separación y aislamiento de sustancias que están en forma de mezclas, usando un disolvente (2). Generalmente, la extracción se usa para separar un compuesto orgánico que está en disolución o suspensión acuosa, para lo que se emplea un solvente orgánico inmiscible con el agua. La separación se basa en la solubilidad relativa, a una temperatura dada, de la sustancia a separar, en cada uno de los solventes empleados (2). Para elegir el solvente orgánico se deben los que reúnan características como: alta afinidad por la sustancia que se quiere extraer, inmiscibilidad en agua, alta capacidad de solvatación, bajo punto de ebullición para que sea fácilmente eliminado por evaporación (2). Los solventes orgánicos más empleados son: Éter de petróleo Tetracloruro de carbono Benceno Éter diisopropil Éter etílico Cloroformo Cloruro de metileno Keroseno 20 El Éter etílico tiene un alto poder disolvente de la mayoría de compuestos orgánicos, además su punto de ebullición es bastante bajo (35°C), pero tiene el inconveniente de ser muy volátil e inflamable (2). Estos solventes pueden sustituirse en muchos casos por el Keroseno o gas corriente, el cual es más barato que estos. Presenta el inconveniente de estar coloreado, por lo que antes de su utilización es conveniente purificarlo. IV Marco Experimental 4.1 Materiales: Cant. 4 1 1 1 1 1 1 1 1 Equipo y Cristalería Beackers de 250 mL Espátula Varilla de vidrio Papel filtro Reactivos Carbón activado Yodo en solución con agua Hexano Agua Gaseosa de sabor de UVA, ROJA, TIKY ** Soporte Universal Anillo de hierro Embudo Ampolla de decantación Probeta de 10 mL Probeta de 100 mL **Este material debe proporcionarlo el estudiante 4.2 Metodología 4.2.1 EXTRACCIÓN DEL YODO Medir 50 mL de yodo en solución con agua, trasvasarlos hacia la ampolla de decantación. Medir 50 mL de hexano, agregar una alícuota de 10 ml a la ampolla de decantación. Tapar la ampolla. Agitar con movimientos suaves y circulares la ampolla de decantación. Liberar la presión acumulada dentro de la ampolla, abriendo la llave, cerrando la llave y agitando nuevamente, liberar nuevamente la presión. 21 Colocar la ampolla de decantación en el anillo de hierro. Previamente colocar pedazos de masking tape en las protuberancias del anillo, para proteger la ampolla. Dejar que la mezcla repose un minuto, decantar. Recoger en un beacker la solución de yodo. Destapar la ampolla y verter el extracto por la boca de la ampolla en otro beacker. Repetir la anterior operación hasta que la solución de yodo se aclare. Normalmente bastarán cinco extracciones. Tenga cuidado de que la cantidad acumulada de hexano y de solución de yodo, no supere la capacidad de la ampolla de decantación. Agregar 50 mL de solución de yodo y 50 mL de hexano al mismo tiempo en la ampolla, realizar la extracción. Comparar la apariencia de los extractos. Lavar cuidadosamente la ampolla de decantación. 4.2.2 PURIFICACIÓN Y FILTRACIÓN CON CARBON ACTIVADO Trasvasar 200 mL de la gaseosa hacia un beacker de 250 mL. Agregar una espátula de carbón activado dentro de la muestra, agitar vigorosamente con la varilla. Deje reposar durante 10 minutos. Durante el tiempo que está reposando la mezcla de gas y carbón, preparar un papel filtro no. 4. Preparar el sistema de filtración. Pasados los diez minutos, filtrar cuidadosamente 22 V. Cuestionario 1. ¿Por qué cree que es posible que el gas extraiga el yodo de la solución? 2. ¿Por qué es más eficiente hacer la extracción haciendo uso de varias alícuotas, en vez de una sola? 3. De los solventes presentados en este manual, ¿cuál es su polaridad en orden decreciente? 4. ¿Qué es una emulsión?, ¿cómo puede romperse una emulsión? 5. ¿Qué pigmentos vegetales cree usted que sean solubles en solventes polares?. ¿Cuáles cree que son solubles en solventes apolares? VI. Bibliografía 1. GRAHAM SOLOMONS, T.W. 1,990. Fundamentos de Química Orgânica. Editorial Limusa. México, D.F. 903 p. 2. PINAGEL, D.; SLOWING, I.; DE LA ROCA, W. 2,003. Manual de prácticas de laboratorio de Química Orgánica 1. Facultad de Ciencias Químicas y Farmacia, USAC. Guatemala, Gua. 53 p. 3. Separación de Mezclas (en línea). Consultado el 4 de noviembre de 2007. disponibleen:http://www2.uah.es/quimica_organica/docencia/practicas/practicas_qu imica_organica_farmacia.pdf 4. Carbón Activado. (en línea) 2009. A Wikimedia Project. Consultado el 27 de octubre de 2009. Disponible en: http://es.wikipedia.org/wiki/Carb%C3%B3n_activado. 23 Práctica 4. Síntesis de un compuesto orgánico: alcohol I. Introducción Los alcoholes son un grupo de compuestos orgánicos que se caracterizan por la presencia del grupo funcional Hidroxilo (-OH). Actualmente poseen gran cantidad de usos, desde productos para consumo humano, hasta productos para desinfección y limpieza. Una de las formas más empleadas para obtener alcoholes es fermentar frutas o partes vegetales. La presencia de ciertos microorganismos anaeróbicos facultativos es necesaria para obtener alcoholes aptos para consumo humano. Dentro de estos organismos, tradicionalmente se han empleado levaduras pertenecientes a las especies Saccharomyces cerevisiae y Kluyveromyces fragilis, los cuales degradan enzimáticamente los carbohidratos para producir dióxido de carbono y alcohol etílico. II. Objetivos Que el estudiante aprenda la forma correcta de determinar la viabilidad de inóculos de levaduras. Que el estudiante comprenda y aplique correctamente las técnicas necesarias para lograr una fermentación anaeróbica. III. Marco teórico 3.1 Alcoholes Los alcoholes son una familia de compuestos orgánicos que poseen como grupo funcional característico al Hidroxilo (OH) fijo a un átomo de carbono tetraédrico. El mas simple de los alcoholes es el alcohol metílico (metanol) cuya fórmula estructural es CH3 OH, otro alcohol ampliamente conocido es el etílico (etanol, apto para consumo humano), su fórmula estructural es CH3CH2OH (2). En general los alcoholes se pueden clasificar como primarios, secundarios o terciarios, dependiendo de la condición del carbono al que se fija directamente el grupo hidroxilo. Si el carbono enlazado al grupo hidroxilo está fijo únicamente a otro carbono, se considera al alcohol como primario. En el caso de que el carbono enlazado al grupo hidroxilo se encuentre ligado a otros dos átomos de carbono, se identifica a este como un alcohol secundario. Será un alcohol terciario, cuando el carbono enlazado con el grupo hidroxilo, se encuentre ligado con otros tres átomos de carbono (2). 3.1.2 Propiedades físicas de los alcoholes Los alcoholes son compuestos cuyas moléculas tienen un grupo hidroxilo fijo a un carbono saturado. Existen, sin embargo, otros compuestos orgánicos que poseen el grupo hidroxilo, pero este está enlazado con un anillo aromático, estos compuestos se conocen como fenoles (2). 24 Si se comparan los puntos de ebullición (Pe) de los alcoholes y los fenoles con los puntos de ebullición de hidrocarburos que posean aproximadamente el mismo peso molecular, se observará que los alcoholes y fenoles poseen Pe mas elevados (2). El Pe del metanol es mas de 150°C superior al del etano, esto sucede a pesar de que ambos compuestos poseen casi el mismo peso molecular. El etanol ebulle 123°C por encima del propano (2). Si se comparan los Pe de los hidrocarburos con otras familias de compuestos orgánicos con pesos moleculares semejantes, se encontrará que las diferencias no son tan marcadas. Por ejemplo el éter dietílico y el pentano poseen casi el mismo Pe (2). Este comportamiento se puede explicar únicamente al considerar la presencia o ausencia de puentes de hidrógeno fuertes. En los alcoholes y fenoles un átomo de hidrógeno está fijo a un átomo de oxígeno, debido a que el oxígeno es muy electronegativo, las moléculas de alcoholes y fenoles son capaces de formar puentes de hidrógeno fuertes entre si. Debido a ello, los alcoholes y fenoles poseen Pe mayores que otros compuestos orgánicos de peso molecular similar, ya que para lograr la volatilización, se deben romper estos puentes de hidrógeno (2). Tabla1. Propiedades físicas de algunos alcoholes Compuesto: Nombre: Pf (°C) Pe (°C, 1 Densidad atm) (g*mL-1) Solubilidad en agua (g/100g) - CH3 OH Metanol -97 64.7 0.792 CH3CH2OH Etanol -114 78.3 0.789 CH3CH2CH2OH n-propanol -126 97.2 0.804 CH3 CH(OH)CH3 Isopropanol -88 82.3 0.786 (CH3 )3COH Tert-butanol 25 82.3 0.789 CH3(CH2)7CH2OH n-nonanol 212 0.829 CH2 OHCH2 OH Etilenglicol -16 197 1.113 CH2OHCHOHCH2O Glicerol 18 290 1.261 H Fuente: GRAHAM SOLOMONS, T.W. 1,990. Fundamentos de Química Orgánica (2) Los alcoholes como el etanol, que se emplean a nivel industrial, se elaboran mediante procesos sintéticos, como la hidratación de Etileno, pero el etanol que se destina para consumo humano se prepara por procesos de fermentación de azúcares (3). 3.2 Levaduras Las levaduras son organismos unicelulares eucariotas, situándose evolutivamente entre los mohos y las bacterias. Son organismos anaeróbicos facultativos, es decir que son capaces de vivir en presencia o ausencia de oxígeno. Cuando las levaduras encuentran oxígeno disponible en su medio, respiran, es decir oxidan glucosa y obtienen ATP. En condiciones anaeróbicas, las cepas de Saccharomyces cerevisae (levaduras de la 25 panificación) y otras especies de levaduras transforman la glucosa en ácido pirúvico, siguiendo la secuencia de reacciones de la pirólisis. La mayoría de seres vivientes pueden seguir este proceso, pero las levaduras consiguen degradar el ácido pirúvico hasta etanol. Esta ventaja adaptativa, permite a las levaduras sobrevivir en condiciones anaeróbicas. Pero emplean este sistema únicamente cuando no hay oxígeno disponible, debido al bajo rendimiento energético de este sistema al compararlo con la degradación oxidativa de la glucosa (1). La capacidad de las levaduras de fermentar algunos azúcares, se debe principalmente a la actividad catalítica de una enzima llamada Cimasa, la que a su vez es un complejo de por lo menos 22 enzimas separadas. Cada una de estas enzimas cataliza un paso específico en la secuencia de la fermentación. Las enzimas se caracterizan por ser altamente específicas, es decir una enzima actuará solamente sobre un compuesto o grupo de compuestos altamente relacionados entre si (3). La Cimasa actúa sobre azúcares sencillos. Estos azúcares son polihidroxialdehidos o polihidroxiacetonas de cinco o seis carbonos, existen algunos azúcares sencillos de cuatro carbonos. Los azúcares complejos se clasifican en función de la cantidad de monosacáridos que producen al ser hidrolizados. De acuerdo a esto pueden haber disacáridos como el azúcar común o sacarosa, trisacáridos como la rafinosa y polisacáridos como la celulosa o el almidón (3). Levadura en polvo Microfotografía de la levadura 3.3 Fermentación alcohólica La fermentación alcohólica está ligada directamente a la cantidad de levaduras presentes durante el proceso. El tamaño de la población de levaduras dentro de un fermento sigue cuatro etapas principales: adaptación, durante esta etapa las levaduras no se reproducen. Posteriormente se da una etapa de crecimiento exponencial, durante el cual las levaduras se reproducen asexualmente, durante esta etapa se desprende una gran cantidad de dióxido de carbono. En la tercera etapa, se da un crecimiento poblacional nulo, es decir que la tasa de natalidad iguala a la tasa de defunción. Finalmente se produce una etapa de disminución de la población, en la cual la tasa de mortalidad supera a la tasa de natalidad. Durante esta última etapa, las levaduras se reproducen sexualmente, para tratar de adaptarse al medio que les es cada vez mas desfavorable (1) Durante la primera etapa, la de adaptación, las levaduras deben hidrolizar los di, tri y polisacáridos por medio de las Hidrolasas, para poder fermentarlas. La maltasa transforma la maltosa en glucosa. La Invertasa transforma la sacarosa en glucosa y fructosa. La Cimasa convierte la glucosa en dióxido de carbono y etanol (3). 3.3.1 Condiciones que afectan la fermentación alcohólica 26 Temperatura Durante la fermentación alcohólica existe liberación de energía térmica. Las levaduras son organismos mesófilos, es decir que están adaptadas a un rango de temperatura entre los 13 y 35°C. Si se supera este rango de temperaturas, las levaduras morirán. En el rango inferior de temperatura, la fermentación es lenta, pero produce un mayor grado alcohólico. En el rango superior de temperatura, la fermentación es mas veloz, pero el grado alcohólico es menor (1,3). Disponibilidad de oxígeno Las levaduras son organismos anaeróbicos facultativos, es decir que pueden sobrevivir en presencia o ausencia de oxígeno. En las primeras etapas de la fermentación alcohólica se necesita una gran cantidad de oxígeno, para lograr que las levaduras se reproduzcan adecuadamente. La producción de etanol se lleva a cabo en condiciones anaeróbicas. Las mismas levaduras producen dióxido de carbono como consecuencia de la actividad de la Cimasa, por lo que ellas mismas producen las condiciones necesarias para la fermentación alcohólica óptima (1,3). Si las levaduras se encuentran en un ambiente con oxigenación constante, se obtendrá únicamente dióxido de carbono y agua, esto debido a que bajo estas condiciones las levaduras realizan la fermentación oxidativa de donde obtienen mas energía (1,3). pH Las levaduras se desarrollan mejor en un medio ácido ( 3.5 –5). La fermentación tiende a acidificar el medio (1). Nutrientes y activadores Las levaduras necesitan azúcares para su catabolismo. Sin embargo la concentración de azúcares no debe ser tan elevada que provoque la plasmólisis de las mismas. Además de azúcares necesitan elementos como: N, P, C, S, K, Mg, Ca y vitaminas, especialmente vitamina B1 (1). De estos elementos el mas importante es el nitrógeno. El sustrato que se desea fermentar debe poseer concentraciones de N, en forma amoniacal y de aminoácidos, de por lo menos 130-150 ppm. La deficiencia de este nutriente provocará que las levaduras ataquen a las proteínas del sustrato y liberen sulfuro de hidrógeno (olor a huevos podridos) (1). Inhibidores Los principales inhibidores de la actividad enzimática de las levaduras son los restos de productos fitosanitarios y ácidos grasos saturados de cadena corta (1). 27 IV Marco Experimental 4.1 Materiales: Cant. Equipo y Cristalería 4 Tubos de ensayo 1 Gradilla de metal 1 Beacker de 600 mL 1 1 1 2 1 Beacker de 250 mL Varilla de agitación Termómetro Beacker de 1000 mL Pizeta 1 Espátula Reactivos Nitrato de Plata al 1% Sulfato de cobre al 10% 15 gramos de Levadura (Para panaderia)* 5 lbs. de azúcar* 1 garrafón con agua pura* 1 galón de plástico* 1 bolsa negra extragrande* Dulce de panela* (Se diluye en agua caliente con la menor cantidad de agua posible.) Fruta picada (aprox. 1/3 del garrafón)* 1 Estufa eléctrica 1 Tapón de hule No. 10 monohoradado* 1 Sifón* * Este material deberá proporcionarlo el alumno. 4.2 Metodología 4.2.1 DETERMINACIÓN DE LA ACTIVIDAD ENZIMÁTICA DE LA LEVADURA Lavar, secar, enumerar y colocar los tres tubos de ensayo en la gradilla. Agregar en cada tubo de ensayo 1g de levadura y 1g de azúcar. En el primer tubo de ensayo agregar 5 mL de agua destilada. En los tubos de ensayo dos y tres agregar seis gotas de CuSO4 al 10% y seis gotas de AgNO3 al 1% respectivamente. Calentar los tres tubos de ensayo en baño María a 37°C por un máximo de 10 minutos. Observar y anotar los resultados. En el primer tubo de ensayo, deberá observarse la formación de espuma. En este caso, se considera a la levadura activa. En los tubos de ensayo dos y tres no se deberá formar espuma. Esto debido a que los iones Cu2+ y Ag+ destruyen la enzima Cimasa de las levaduras. 4.2.2 PREPARACIÓN DE LA MEZCLA A FERMENTAR Seguir las indicaciones del Instructor respecto al tratamiento que deberá darle a su muestra. En algunos casos será necesario cocerla y/o hidrolizarla. Pesar 15 g de levadura, a la cual previamente le determinó la viabilidad. Disolver en 200 mL de agua destilada. Apartar y conservar esta mezcla. Usando del galón de plástico, disolver las siete libras de azúcar. Agregar la mitad de esta solución al recipiente de cinco galones. Agregar la mitad de la muestra vegetal previamente tratada. Agitar vigorosamente. 28 En la mitad restante de su muestra vegetal tratada, disuelva la mezcla de agua y levadura. Agite vigorosamente por cinco minutos. Agregue la mezcla al recipiente de cinco galones. Agite nuevamente. Verter en el recipiente de cinco galones el resto de la solución de agua y azúcar. Aforar con agua destilada el recipiente hasta donde indique el Instructor, no sobrepasar la mitad de la altura del recipiente. Sifón Este es el límite máximo hasta donde debe dejarse la mezcla a fermentar, de lo contrario puede reventarse el tambo por la acumulación de CO2 Colocar el sifón con el tapón horadado en la boca del recipiente. Sellar con parafilm, o bien con maskin tape. Identificar los recipientes. Tapar el recipiente con la bolsa negra extragrande y guardar en un lugar fresco y seco, alejado de la luz directa. Almacenar por lo menos una semana. Verificar cada dos días el estado del fermento, de ser necesario agregar mas agua al sifón. El sifón siempre debe de mantener el nivel de agua para evitar la entrada de oxigeno. V Cuestionario 1. ¿Qué otros organismos son capaces de producir etanol al llevar a cabo una fermentación anaeróbica de su sustrato? 2. ¿Cuál es la función del sifón durante la fermentación? 29 VI. Bibliografía 1. GRAHAM SOLOMONS, T.W. 1,990. Fundamentos de Química Orgânica. Editorial Limusa. México, D.F. 903 p. 2. PINAGEL, D.; SLOWING, I.; DE LA ROCA, W. 2,003. Manual de prácticas de laboratorio de Química Orgánica 1. Facultad de Ciencias Químicas y Farmacia, USAC. Guatemala, Gua. 53 p. 30 Práctica 5 Extracción de pigmentos vegetales, (extracción por cromatografía). I. Introducción Uno de los procesos metabólicos más importantes de las células vegetales lo constituye la Fotosíntesis. Durante la fase luminosa de la misma es imprescindible la participación de diferentes pigmentos fotosintéticos que se encuentran dentro de los plástidos, y que le confieren la coloración característica de las hojas de las plantas. Los pigmentos de mayor importancia son las clorofilas: la clorofila A que tiene un color verde azulado, y la clorofila B de color verde amarillento. También son importantes los carotinoides, de color anaranjado-rojizo, y las xantofilas de color amarillo. Las técnicas cromatográficas son muy variadas, pero en todas ellas hay una fase móvil que consiste en un fluido (gas, líquido o fluido supercrítico) que arrastra a la muestra a través de una fase estacionaria que se trata de un sólido o un líquido fijado en un sólido. Los componentes de la mezcla interaccionan en distinta forma con la fase estacionaria. De este modo, los componentes atraviesan la fase estacionaria a distintas velocidades y se van separando. Después de que los componentes hayan pasado por la fase estacionaria, separándose, pasan por un detector que genera una señal que puede depender de la concentración y del tipo de compuesto. Diferencias sutiles en el coeficiente de partición de los compuestos da como resultado una retención diferencial sobre la fase estacionaria y por tanto una separación efectiva en función de los tiempos de retención de cada componente de la mezcla. II. Objetivos Que el estudiante reconozca y aprenda a manipular correctamente el equipo y cristalería necesarios para extraer pigmentos. Que el estudiante comprenda y realice el proceso necesario para obtener pigmentos por medio de vegetales. III. Marco teórico Entre todos los caracteres más externos de los vegetales, el más notable y característico es probablemente el color. El color no es únicamente un carácter llamativo de la vegetación, sino que, además, algunos de los pigmentos que lo condicionan están estrechamente ligados a las actividades fisiológicas del propio vegetal. Por consiguiente, el estudio de cómo las plantas viven y se desarrollan requiere el previo conocimiento de los pigmentos vegetales. 3.1 ¿Qué son los pigmentos? Si es posible encontrar en el reino vegetal todos los matices y combinaciones de colores del espectro, existe un predominio general de los colores primarios: verde, amarillo, rojo y azul. Estos colores son conferidos a los vegetales por determinados compuestos químicos denominados pigmentos. El color particular que presenta un determinado órgano vegetal 31 depende generalmente del predominio de uno u otro o la combinación de ellos. Se debe tener claro que cuando un vegetal presenta un color blanco, es debido a la falta de pigmentos. La luz solar que incide sobre ellas no es absorbida selectivamente como ocurre en las partes coloreadas, sino que es transmitida o reflejada prácticamente sin sufrir modificación. 3.2 ¿Dónde están los pigmentos? Estos pigmentos se encuentran en el interior de las células vegetales específicamente en una organelo llamada cloroplasto. Los cloroplastos son simplemente plástidos que contienen pigmentos clorofílicos. Los compuestos clorofílicos están ligados químicamente con las estructuras internas del cloroplasto (membrana tilacoides) y se hallan retenidos en estado coloidal. Asociados con las clorofilas, existen también en los cloroplastos dos clases de pigmentos amarillos y amarillo-anaranjados que son las xantofilas y carotenides. Las Clorofilas. El color verde tan uniformemente presente en los vegetales es debido a la presencia de dos pigmentos estrechamente emparentados llamados clorofila "A" y clorofila "B". Se encuentran prácticamente en todas las plantas con semilla, helechos, musgos y algas. Pueden formarse en las raíces, tallos, hojas y frutos a condición de que estos órganos estén situados por encima del suelo y queden expuestos a la luz. También aunque aparentemente falten en algunas hojas de color rojo o amarillo, cuando se extraen las otras sustancias colorantes de estas, puede comprobarse incluso allí la presencia de las clorofilas, que estaban enmascaradas por los demás pigmentos. 3.3 ¿Cómo se dividen los solventes? Los pigmentos clorofílicos son insolubles en el solvente universal llamado agua. Pero sí son solubles en solventes orgánicos (afinidad química) como por ejemplo alcohol etílico y acetona. A los solventes que extraen simultáneamente todos los pigmentos de la hoja se los suele llamar extractantes. Existen otros solventes que presentan afinidad cor algunos pigmentos y se llaman separadores, como por ejemplo el Tetracloruro de carbono y el éter de petróleo. Las distintas técnicas cromatográficas se pueden dividir según cómo esté dispuesta la fase estacionaria: Cromatografía plana. La fase estacionaria se sitúa sobre una placa plana o sobre un papel. Las principales técnicas son: o Cromatografía en papel o Cromatografía en capa fina Cromatografía en columna. La fase estacionaria se sitúa dentro de una columna. Según el fluido empleado como fase móvil se distinguen: o Cromatografía de líquidos o Cromatografía de gases o Cromatografía de fluidos supercríticos 32 La cromatografía de gases es útil para gases o para compuestos relativamente volátiles, lo que incluye a numerosos compuestos orgánicos. En el caso de compuestos no volátiles se recurre a procesos denominados de "derivatización", a fin de convertirlos en otros compuestos que se volatilizen en las condiciones de análisis. Dentro de la cromatografía líquida destaca la cromatografía líquida de alta resolución (HPLC, del inglés High Performance Liquid Chromatography), que es la técnica cromatográfica más empleada en la actualidad, normalmente en su modalidad de fase reversa, en la que la fase estacionaria tiene carácter no polar, y la fase móvil posee carácter polar (generalmente agua o mezclas con elevada proporción de la misma, o de otros disolvente polares, como por ejemplo metanol). El nombre de "reversa" viene dado porque tradicionalmente la fase estacionaria estaba compuesta de sílice o alúmina, de carácter polar, y por tanto la fase móvil era un disolvente orgánico poco polar. IV Marco Experimental 4.1 Materiales: Cant. Equipo y Cristalería 1 Tubo de ensayo grande 1 Soporte universal 1 Pinza Fisher 2 Beackers 250 ml. 1 Probetas 100 ml. 1 Mortero con pistilo 1 Embudo plástico 1 Pizeta 1 Balanza monoplato *Nota: Lo debe llevar el estudiante a la práctica Reactivos Arena fina Lavada y Tamizada * 1 lb de Espinaca * Acetona Solución extractora de Eter de petróleo, Tolueno y Isopropanol Tapón de Hule # 3 Estufa Electrica 4.2 Metodología Procedimiento 1: a. Pesar 50 grs. de material vegetal (fresco), y macerarlas en un mortero con un poco de arena fina bien lavada. Triturar. b. Agregar 50 ml. de acetona y continuar macerando hasta que las hojas parezcan decoloradas y la acetona de color muy oscuro. (Si se evapora la acetona agregar nuevamente 50 ml.) 33 c. Filtrar con la gasa, en el embudo plástico. Dentro de un Beacker de 100 mL. d. Colocar 3-5 mL de solución extractora dentro del tubo de ensayo y taparlo con el tapón de hule e. Cortar el papel filtro, en forma de Flecha, procurando de colocar una línea que tocara el solvente, y realizar una línea arriba del punto de contacto del papel con el solvente f. Con lo filtrado, colocarlo en una estufa y evaporar con poco calor hasta formar una pasta verde. g. Con un tubo capilar, colocar puntos de extracto en la base del papel filtro en el centro de la línea superior en el papel filtro h. Tapar el sistema y observar el movimiento del de los colores. V. Cuestionario 1. Mencione que pigmentos se identifican al momento de la separación de las capas a lo largo de la práctica, basándose en la coloración de los extractos. 2. Por que se le agrega agua al extracto de coloración roja? 3. Investigue que otros pigmentos fotosintéticos existen? 4. Indique las estructuras moleculares de la clorofila (A y B), carotenos y xantofilas. VI. Bibliografía 1. GRAHAM SOLOMONS, T.W. 1,990. Fundamentos de Química Orgânica. Editorial Limusa. México, D.F. 903 p. 2. PINAGEL, D.; SLOWING, I.; DE LA ROCA, W. 2,003. Manual de prácticas de laboratorio de Química Orgánica 1. Facultad de Ciencias Químicas y Farmacia, USAC. Guatemala, Gua. 53 p. 3. CATALAN R. 1999. Manual de prácticas de laboratorio de Química Orgánica. Facultad de Agronomía 4. Pigmentos fotosintéticos (en línea). Ing. Agr. Carlos Gonzalez. Consultado el 14 de Mayo de 2008. disponible en: www.botanica.cnba.uba.ar/Trabprac 34 Práctica 6. Obtención de un plástico a partir de urea y formaldehído, resistencia de polímeros a ácidos y bases, comprobación del Cloro en PVC I. Introducción Polímeros sintéticos como Orlón®, Plexiglás®, Lucite®, polietileno, Teflón®, Dacrón®, Mylar® y nylón son en la actualidad bastante comunes y conocidos. Estos plásticos o polímeros se utilizan para elaborar muchos de objetos de uso cotidiano. La mayoría de estos compuestos se desconocían antes de la Segunda Guerra Mundial. El desarrollo de los procesos de síntesis de polímeros sintéticos ha sido la principal causa del notable crecimiento de la industria química del siglo XX. Hoy día los plásticos representan una alternativa barata y duradera de construir estructuras y almacenar sustancias. Sin embargo también son una fuente importante de contaminación, al haber una gran cantidad de plásticos que no son biodegradables. II. Objetivos Desarrollar en el estudiante de Química Orgánica algunas destrezas básicas necesarias para el manejo adecuado de la cristalería y reactivos empleados en la práctica. Obtener un polímero sintético partiendo de urea y formaldehído. Observar la resistencia del PVC y el polímero sintetizado a ácidos y bases. Comprobar la presencia de Cloro en PVC. III. Marco teórico 3.1 Polímeros Los polímeros son compuestos que constan de moléculas muy grandes, las cuales a su vez están formadas por muchas subunidades que se repiten. Estas subunidades moleculares de los polímeros se denominan monómeros y las reacciones mediante las cuales se unen los monómeros se denominan reacciones de polimerización (1). 3.1.1 Polímeros de adición La polimerización de algunos polímeros se da gracias a reacciones de adición. Los alquenos son muy convenientes para obtener polímeros de adición. Las reacciones de adición ocurren mediante mecanismos catiónicos o aniónicos de radicales libres, lo que depende de la manera en que se inician (1). 35 Las reacciones de polimerización ocurren en cadena. El etileno, por ejemplo se polimeriza a través de un mecanismo de radicales libres, cuando se le calienta a una presión de 1.00 atm con una pequeña cantidad de peróxido orgánico. El peróxido se disocia para producir radicales libres que, a su vez, inician las cadenas. De estas cadenas se obtiene el polietileno (1). El polietileno producido por la polimerización de radicales libres en general no es útil, a menos que su peso molecular sea de casi 1*106 daltons. El polietileno de muy elevado peso molecular se obtiene con el uso de una concentración muy baja de iniciador (1). El polietileno se produce a escala comercial desde 1,943. Se emplea para fabricar envases flexibles, películas, láminas y aislamientos para cables. El polietileno producido por polimerización de radicales libres tiene un punto de ablandamiento de aproximadamente 110° C. La polimerización de radicales libres del cloroetano (cloruro de vinilo) produce un polímero denominado poli cloruro de vinilo (PVC). CH2=CH l Cl -CH2-CHl Cl n Cloruro de Vinilo Policloruro de vinilo Esta reacción produce un polímero que tiene peso molecular de aproximadamente 1.5 *106 daltons y que es duro, quebradizo y rígido. En esta forma se le emplea a menudo para construir tubos y varillas (1). El Orlón®, se obtiene de la polimerización del acrilonitrilo (CH2=CHCN). El Teflón®, se obtiene de la polimerización del tetrafluoroetileno (nCF2=CF2) en suspensión acuosa, posee un punto de fusión de 372° C, que es inusualmente alto para un polímero de adición. Otros polímeros de adición son el Lucite®, Plexiglás® y Perspex®, que se obtienen de la polimerización de los radicales libres de metilmetacrilato (1). 3.1.2 Polímeros por condensación Estos polímeros de preparan por reacciones de condensación, es decir, reacciones en las que se ligan las subunidades monoméricas mediante eliminaciones intermoleculares de pequeñas moléculas, como agua o alcoholes. Los polímeros de condensación más importantes son las poliamidas y los poliésteres (1). 3.1.2.1 Poliamidas La seda y la lana son dos polímeros naturales que pertenecen a una familia de compuestos llamados proteínas. Debido a que las subunidades de las proteínas se derivan de los α-aminoácidos y están ligadas por medio de enlaces amida, son poliamidas (1). 36 La búsqueda de un material sintético cuyas propiedades fueran semejantes a las de la seda llevó al descubrimiento de una familia de poliamidas sintéticas llamadas nylones. Uno de los nylones mas importantes, llamado nylon 6,6®, se sintetiza a partir del ácido dicarboxílico de seis carbonos llamado adípico y de la diamina de seis carbonos llamada hexametilendiamina (1). 3.1.2.2 Poliésteres Uno de los poliésteres de mayor importancia es el politereftalato de etileno, polímero comercializado con los nombres de Dacrón®, Teryleno® y Mylar®. El método más empleado para sintetizar este polímero se basa en reacciones de transesterificación, es decir, reacciones en las que un éster se convierte en otro. En la síntesis comercial de este compuesto se emplean dos transesterificaciones (1). 3.1.2.3 Poliuretanos Un uretano es el producto de la reacción de un alcohol y un isocianato: R-OH + O=C=N-R´ Alcohol Isocianato O H Ħ R-O-C-N-R´ un uretano (carbamato) Los uretanos también reciben el nombre de carbamatos ya que en el sentido estricto son ésteres de un alcohol y un ácido carbámico. 3.2 Urea La urea es una sustancia nitrogenada producida por los seres vivos ureotélicos (mamíferos) como producto de alimentación del amoníaco. Se halla en la sangre, orina, bilis, sudor, etc. Fue la primera molécula orgánica sintetizada por el hombre (Wöhler, 1,828) con lo que se demostró que no es una sustancia exclusivamente biogena. Por la acción de la ureasa, se descompone en anhídrido carbónico y agua por lo que se utiliza como diurético (2). 3.3 Formaldehído Gas de olor picante, fácilmente soluble en agua, generalmente se emplea en solución acuosa. Se polimeriza fácilmente dando como resultado paraformaldehido o metaformaldehido, tiene gran importancia en la fabricación de materias plásticas (3). Se produce por oxidación del alcohol metílico o del etileno en presencia de un catalizador. Es un desinfectante y endurece las sustancias albuminoides, además es un necrosante del tejido orgánico. 37 Existen dos tipos de resina de formaldehído. Urea formaldehído y fenol formaldehído. Los productos elaborados con el primer tipo de sustancias liberan formaldehído. La emisión de este gas es menor en las resinas del segundo tipo (2). IV Marco Experimental 4.1 Materiales: Cant. Equipo y Cristalería 1 Estufa eléctrica 5 Tubos de ensayo 1 Gradilla de metal 1 Rejilla de asbesto 1 Beacker de 600 mL 1 Varilla de vidrio 1 Pinza para tubos de ensayo 1 Mechero y su manguera 1 Frasco de polietileno 2 Probeta de 10 mL 1 Beacker de 100 mL 1 Termómetro 1 Frasco de compota* 1 Microespátula * Este material lo debe proporcionar el estudiante Reactivos Glicerina Resorcinol Hidróxido de potasio al 20% Sulfito de sodio Formaldehído Acido Clorhídrico conc. Ácido sulfúrico al 50% Hidróxido de sodio al 40% Urea molida* Lana y algodón natural* Alambre de cobre* Tiritas de PVC* 4.2 Metodología 4.2.1 OBTENCIÓN DE PLÁSTICO A PARTIR DE UREA Pesar 4g de urea y agregarlo a un tubo de ensayo, agregar 10 mL de formaldehído. Calentar en baño María, durante cinco minutos o hasta la disolución de la urea, ocasionalmente agitar con la varilla de vidrio. Trasvasarlo a un frasco de compota, cuando la solución se comience a tornar lechosa. Agregar cuidadosamente de tres a cinco gotas de HCl ac. Observar la formación del plástico. Determinar el rendimiento de la reacción, comparando la masa de los reactivos con la masa de los productos. 4.2.2 RESISTENCIA DEL PVC A ÁCIDOS Y BASES Colocar en cada uno de tres tubos de ensayo: una tirita de PVC, un pedazo del polímero sintetizado, una hebra de lana, una muestra de algodón. Numerarlos del uno al tres. 38 Agregar 6 mL de H2SO4 al 50% en el tubo 1, 6 mL de HCl concentrado en el tubo 2, y 6 mL de NaOH al 40% en el tubo 3. Observar las reacciones. Transcurridos diez minutos, colocar los tres tubos en baño María, caliéntelos cuidadosamente por diez minutos. Observar las reacciones. 4.2.3 COMPROBACIÓN DEL CLORO EN EL PVC Con ayuda del alicate, cortar un pedazo de alambre de cobre de 10 cm de longitud. Sostener el alambre con la pinza para crisoles, calentar con la llama del mechero durante cinco minutos, o hasta que no se distinga una coloración verde en la llama. Preparar una tira de PVC sobrante del experimento anterior, mientras el alambre de cobre esté caliente, atravesar la tira con este. Calentar el alambre de cobre en la llama del mechero, esperar a que aparezca una coloración verde en la llama. Esta coloración es causada por la formación de CuCl2. Durante el calentamiento del PVC, este se descompone, dando lugar a HCl, el ácido reacciona con el cobre, formando la sal. NOTA: Los productos sintetizados antes de tocarse deben lavarse con suficiente agua, esto con el fin de eliminar el exceso de formaldehído que pueda tener el polímero ya que al haber contacto con la piel la destruye (necrosa el área). 4.2.4 RESINA ROJO RUBÍ En un tubo de ensayo de 16 * 150 mm, agregar 1 gramo de resorcinol con 1.5 mL de agua, agitar para disolver el resorcinol. Este será el tubo de ensayo A. En otro tubo de ensayo disolver 1 microespatulada de sulfito de sodio en 1 mL de hidróxido de potasio al 20%. Agitar para disolver el bisulfito. Este será el tubo de ensayo B. Agregar el contenido del tubo A en el tubo B y homogenizar la mezcla, se debe agregar inmediatamente 2 mL de formaldehído y seguir homogenizando. El líquido se hace viscoso y poco después solidifica repentinamente, dando una masa de resina artificial de color rojo rubí. Para poder expulsar el producto del tubo, caliente el fondo y vera que la resina avanza progresivamente. El producto primeramente es elástico quebradizo, pero debe dejarse durante unas horas para que se endurezca. 39 Resina sintetizada V. Cuestionario 1. ¿En la comprobación de Cu en el PVC, que otros colores podrían indicar diferentes elementos? 2. ¿Cuál es la función del HCl en la síntesis de la resina urea formaldehído? VI. Bibliografía 1. GRAHAM SOLOMONS, T.W. 1,990. Fundamentos de Química Orgánica. Limusa. México, Mex. 903 p. 2. Uretano (en línea). Consultado el 5 de www.sandretto.it/museo/SPAGNOLO/splati.htm-24k noviembre. Disponible en: 40 Práctica 7. Destilación simple de un compuesto orgánico: alcohol etílico I. Introducción En la práctica anterior se ha obtenido un compuesto orgánico por medio de la fermentación, el alcohol. Sin embargo, la fermentación no produce únicamente este compuesto, sino que también produce aldehídos y cetonas, las cuales no son aprovechables por el organismo y también pueden ser tóxicas. La destilación se lleva a cabo para obtener el mayor porcentaje de alcohol puro, a partir del fermento que se elaboró. La destilación simple se basa en la diferencia de los puntos de ebullición de diferentes sustancias. La destilación también es empleada para obtener agua pura a partir de agua de mar o para obtener agua potable a partir de agua contaminada. II. Objetivos Que el estudiante se familiarice con la cristalería necesaria para llevar a cabo una destilación simple. Que el estudiante aprenda y aplique la metodología para determinar el grado alcohólico de un fermento. Que el estudiante lleve a cabo la destilación simple de una alícuota del fermento obtenido previamente. III. Marco Teórico 3.1 Purificación de sustancias miscibles El proceso de destilación de un fermento, concentra el alcohol y elimina una gran cantidad de impurezas de sabor desagradable. Si se sobrepasa la rectificación se eliminan también los componentes saborizantes y se obtiene alcohol puro. En el proceso de envejecimiento, que generalmente tiene lugar en barriles de madera quemada, las impurezas, normalmente una mezcla de alcoholes superiores, se oxidan parcialmente a ácidos, que reaccionan con los alcoholes remanentes, formándose ésteres de sabor agradable. Los saborizantes que quedan en el producto final representan menos de 0.5% de éste, el resto es agua y entre un 38 a 45% de alcohol. Los licores, bebidas alcohólicas endulzadas y aromatizadas, tienen un contenido alcohólico entre 20 a 40% (1). La destilación es un proceso que consiste en calentar una mezcla líquida hasta que sus componentes mas volátiles pasan a la fase de vapor. El vapor se enfría para recuperar estos componentes en forma líquida, esto es se condensa el vapor. La finalidad de la destilación es separar una mezcla de varios componentes de acuerdo a su Pe o bien, separar componentes volátiles de los no volátiles. Normalmente el componente que se obtiene con la destilación no se desecha (1). 41 Si la diferencia entre los Pe de las sustancias es grande, es muy fácil separar estas por medio de destilación simple (1). En el caso del laboratorio de Química Orgánica las sustancias que se quieren separar son principalmente alcohol del agua, los cuales poseen una diferencia entre sus puntos de ebullición de 20°C (Pe agua 100°C, Pe etanol 78.3°C, 1 atm de presión) (2). En la destilación del compuesto orgánico se relaciona un volumen inicial de fermento contra un volumen final de etanol. Se debe tomar en cuenta que parte de este alcohol se debió a la adición de azúcar al fermento. El contenido de alcohol es directamente proporcional a la cantidad de azúcares presentes en la sustancia, si las condiciones son adecuadas (1). IV Marco Experimental 4.1 Materiales: Cant. 1 1 1 1 1 2 1 1 Equipo y Cristalería Cant. Balón de destilación 1 Condensador Liebig 2 Enlermeyer de 100 mL 2 Probeta de 100 mL 1 Probeta de 10 mL 1 Beacker de 250 mL 2 Termómetro Tapón monohoradado Mechero y su manguera Mangueras de hule Soportes universales Anillo de hierro Rejilla de asbesto Pinzas fisher Perlas de ebullición Fermento. 4.2 Metodología Destapar cuidadosamente el fermento que se ha almacenado. Tener cuidado de no agitarlo. Extraer una alícuota de 100 mL del fermento. Armar el sistema de destilación simple. Se deben atender las indicaciones del Instructor. Se debe tener la precaución de conectar las mangueras al condensador Liebig antes de armar el sistema. 42 Vertir la alícuota del fermento dentro del balón de destilación. Colocar de seis a diez perlas de ebullición dentro del balón. Cerrar cuidadosamente y colocar el termómetro de 0-400°C. Antes de que se comience la destilación se debe revisar que el condensador Liebig esté lleno de agua fría y que esta esté circulando. Una vez que se ha revisado el sistema, se comenzará la destilación. Se deberá tener presente que la temperatura del fermento no sobrepase los 70-78°C. El destilado se recibirá en un Enlermeyer. Para asegurar que el destilado es etanol, se humedecerá un pedazo de papel en el destilado y se acercará al mechero, si este prende fuego fácilmente, es etanol. Para determinar el contenido de etanol del fermento, se usará esta fórmula: % de alcohol = mL de destilado *100 100 mL V. Cuestionario 1. ¿Qué consecuencias traería al organismo el consumo de los subproductos de la fermentación anaeróbica de la levadura? 2. ¿Cuáles son estos subproductos? 3. ¿Qué procedimiento o metodología se deberá aplicar en el caso de que se quiera separar un compuesto soluble en agua, si éste tiene un Pe muy cercano al del agua? 4. ¿Cuál es el consumo de energía (Kcal), de la destilación simple, si se mantienen 200 ml de agua a 78°C, durante 30 minutos? VI. Bibliografía 1. CANCINOS, M.R.; SOLÍS, J. A. Orgánica. USAC, FAUSAC. 36 p. 2,003. Manual de prácticas de Química 2. GRAHAM SOLOMONS, T.W. 1,990. Fundamentos de Química Orgânica. Editorial Limusa. México, D.F. 903 p. 43 Práctica 8. Extracción de aceites de semillas I. Introducción Los aceites fijos son ésteres de ácidos grasos de cadena larga y glicerol o derivados estrechamente relacionados. Estos se obtiene a partir de las semillas de diversas plantas, tales como: aceitunas, algodón, manía, girasol, etc. Estos aceites son insolubles en agua, y otros solventes polares. En cambio son solubles en solventes apolares (éter, benceno, Tetracloruro de carbono, hexano). Su densidad es menor a la del agua. Los aceites son importantes en la solubilización de algunas vitaminas en el organismo. También son causantes indirectos de algunos problemas de salud. II. Objetivos Que el estudiante de Química Orgánica reconozca y aprenda a manipular correctamente el equipo y cristalería necesarios para extraer aceites de semillas. Que el estudiante determine el contenido de aceite presente en el material designado. III. Marco teórico 3.1 Naturaleza y aplicación de los lípidos Los lípidos son compuestos ternarios (compuestos de carbono, oxígeno y hidrógeno) insolubles en agua y solubles en solventes apolares. Provienen tanto de vegetales como animales. Sus constituyentes específicos son los llamados ácidos grasos (3). Las grasas se dividen en saturadas e insaturadas, dependiendo de que los enlaces químicos entre los átomos de carbono de las moléculas estén ocupados con todos los átomos de hidrógeno que estos puedan contener. Las grasas insaturadas se conocen como aceites, la naturaleza cis o trans de estas no permite el comportamiento propio de un sólido (3). Como norma general, las grasas saturadas son sólidas a temperatura ambiente, mientras que las insaturadas son líquidas. Estas últimas pueden convertirse en grasas saturadas por hidrogenación catalítica, al añadir átomos de hidrógeno (de esta manera se obtiene comercialmente la margarina) (3). Los ácidos grasos no saturados cumplen una trascendental misión en la alimentación humana, ya que no pueden ser sintetizados en el organismo. Dentro de este tipo de ácidos, los más conocidos son el linoleico y linolénico. Estos se encuentran en los aceites vegetales de oliva, maíz, soya y girasol (2). 44 Los aceites fijos son extraídos en su mayoría de plantas. Se obtienen por diferentes métodos físicos y químicos. Los aceites de oliva y maní, que son típicamente comestibles son llamados aceites secantes, porque al ser expuestos al aire se recubren con una capa elástica y poco a poco se solidifican hasta transformarse en una masa de aspecto resinoso. Este secado se obtiene debido a la absorción de oxígeno atmosférico, formándose peróxidos, luego se polimerizan los ácidos no saturados del aceite que se unen mediante enlaces oleofínicos (3). 3.2 Funciones de los lípidos Los lípidos que no se utilizan dentro de los organismos, se almacenan en el tejido adiposo de los animales o en forma de aceite, generalmente dentro de las semillas, en los vegetales. Los lípidos desempeñan cuatro tipos de funciones: Función transportadora: el transporte de lípidos desde el intestino hasta su lugar de destino se realiza mediante su emulsión, gracias a los ácidos biliares y los proteolípidos. Función de reserva: son la principal reserva energética del organismo. Un gramo de grasa produce 9.4 kilocalorías en las reacciones metabólicas de oxidación. En contraste las proteínas y los glúcidos solo producen 4.1 kilocalorías por gramo. Función estructural: los lípidos forman las bicapas lipídicas de las membranas. Recubren órganos y le dan consistencia. También protegen mecánicamente como el tejido adiposo de pies y manos. Función biocatalizadora: en este papel los lípidos favorecen o facilitan las reacciones químicas que se producen en los seres vivos. Cumplen esta función las vitaminas lipídicas, las hormonas esteroideas y las prostaglandinas (3). 3.3 Extracción de lípidos Existen tres métodos ampliamente usados a nivel industrial para la extracción de lípidos a partir de semillas, estos son: Extracción por solvente: Este tipo de extracción permite obtener los máximos rendimientos en la extracción de aceite, pero el aceite asi obtenido es a menudo de baja calidad. El solvente actúa a la vez sobre otras sustancias orgánicas (proteínas, clorofila, etc.) por lo que es necesario recurrir a la filtración al final del proceso. Extracción por prensado en frío o caliente: El aceite que se obtiene por este método es el de mejor calidad de los tres. Sin embargo su rendimiento es muy deficiente. Las prensas empleadas pueden ser hidráulicas, de vapor o molinos. Extracción por centrifugación: Este método se emplea principalmente para la separación de aceites que están en suspensión. A nivel industrial se someten las semillas oleaginosas a dos extracciones por prensado, posteriormente se somete el residuo a extracción con solventes. De esta manera 45 se obtienen tres tipos de aceite, siendo el de la proveniente de la primera extracción por prensado, el de mejor calidad y mayor precio (2). 3.3.1 Extracción continúa En este tipo de extracciones, se aprovecha la capacidad de los solventes apolares de disolver los lípidos que están presentes en una mezcla sólida. La extracción será mas eficiente si se calienta la mezcla de donde se desea extraer el lípido. También es recomendable emplear, a nivel de laboratorio, el sistema Soxhlet. Tabla 1. Contenido de lípidos en diferentes semillas Nombre común % de aceite Características Maíz 14.4 Aceite amarillo o amarillo claro. Corozo 62.8 Grasa sólida, blanco o ligeramente amarillento. Almendra 45.2 Grasa semifluida, amarilla. Pulpa aguacate 28.1 Grasa semifluida, amarillo verdoso. Cacao 55.1 Grasa sólida, blanco amarillento. Coco 60.3 Grasa semifluida, blanco amarillento. Nogal 76.5 Aceite color blanco amarillento. Durazno 33.2 Aceite amarillo claro, aromático. Manía 48.8 Aceite denso, amarillo Jocote marañon 47.9 Aceite color amarillo pálido. Aceituno 58.3 Grasa sólida, blanco verdosa, aromática. Cerezo 36.5 Aceite amarillo, aromático Zapote 50.6 Grasa blanca, consistencia de vaselina. Girasol 32.7 Aceite color amarillo verdoso. Fuente: Aguilar, 1,966 (1). IV Marco Experimental 4.1 Materiales: Cant. Equipo y Cristalería 1 Mortero y su pistilo Hilo 1 Sistema soxhlet 1 Rejilla de asbesto 1 Estufa eléctrica 1 Beacker de 600 mL y 1 de 100 mL 1 Soporte universal 1 2 pinzas fisher 1 Probeta de 100 mL y 1 de 10 mL 1 Balanza *Este material será proporcionado por el alumno Reactivos Hexano 15 gms. de Semillas oleaginosas* 46 4.2 Metodología Pesar de 10 a 12 g de la muestra vegetal requerida. Con ayuda del mortero y el pistilo, moler la muestra o bien picarla, según sea el caso. Elaborar un dedal con el papel filtro, introducir en el la muestra. Introducir el dedal en la cámara del Soxhlet. Agregar 150 mL de hexano en el balón Soxhlet. Armar el sistema Soxhlet, colocar la rejilla de asbesto sobre la estufa y colocar el sistema 1 cm arriba de la estufa. Hacer circular el solvente por 45 minutos. Transcurrido este tiempo, apagar la estufa y retirarla del sistema. Dedal Tarar un beaker de 250 mL. Transferir a este beaker el extracto del sistema Soxhlet. Evaporar el hexano con un suave calentamiento en baño María. La temperatura debe ser baja. Determinar el porcentaje de aceite extraído. Emplee la fórmula: % aceite = masa del aceite (g) * 100 masa de la muestra (g) 47 V. Cuestionario 1. ¿Cuál es el rendimiento de su extracción? 2. ¿Qué otros métodos de extracción, aparte de la extracción continua, se pueden emplear a nivel de laboratorio? 3. ¿Qué tratamiento previo se debe aplicar a las muestras de aguacate y zapote? 4. ¿Cuáles son las principales funciones de los lípidos dentro de los organismos vegetales y animales? VI. Bibliografía 1. AGUILAR, J. 1,966. Relación de unos aspectos de la flora útil de Guatemala. 2 ed. . Guatemala, Gua. 382 p. 2. CANCINOS, M.R.; SOLIS GOMEZ, J. A.. 2,002. Manual de prácticas de Química Orgánica. USAC. Guatemala, Gua. 56 p. 3. LEHNINGER, A.; NELSON, D.; COX, M.. 1,995. Principios de Bioquímica. 2ed. Barcelona, Esp. 1,013 p. 48 Práctica 9. Saponificación de una grasa I. Introducción La saponificación consiste en una hidrólisis alcalina de un lípido, normalmente empleando hidróxido de sodio o potasio. Los lípidos derivados de ácidos grasos al ser neutralizados con hidróxidos, dan lugar a sales de ácido o carboxílicas de Na y K (jabones) y alcoholes que son fácilmente extraíbles en medio acuoso (1). Las grasas, son compuestos de origen animal o vegetal, y son ésteres de un ácido carboxílico de 4 a 22 carbonos y glicerol. Pueden ser sólidas o semisólidas a temperatura ambiente y sus enlaces son más sobresaturados que los de un aceite (1). Los jabones y detergentes son compuestos ampliamente usados en la actualidad, especialmente por sus propiedades limpiadoras (formadores de micelas) y, en algunos casos, como emulsificantes de soluciones acuosas de plaguicidas hidrofóbicos para las plantas. II. Objetivos Que el estudiante aprenda a manipular adecuadamente las bases fuertes. Que el estudiante comprenda y compruebe el proceso de saponificación de una grasa. III. Marco teórico 3.1 Saponificación La saponificación es la hidrólisis alcalina de una preparación lipídica. Los lípidos derivados de ácidos grasos (ácidos monocarboxílicos de cadena larga) dan lugar a sales de ácido o carboxilatos de Sodio o Potasio (jabones) y alcoholes (glicerol), los cuales son fácilmente extraíbles del medio acuoso. La reacción es la siguiente: No todos los lípidos presentes en una muestra biológica dan lugar a la saponificación, por lo tanto se distinguen dos tipos de lípidos (1, 3). 3.1.1 Lípidos saponificables Dentro de este tipo de lípidos se agrupan a los derivados por esterificación u otras modificaciones de ácidos grasos. Estos se sintetizan en los organismos a partir de la aposición sucesiva de unidades de dos átomos de carbono, en este grupo se incluyen: 49 Ácidos grasos y sus derivados eicosianoides1: prostaglandinas, tromboxanos y leucotrienos. Lípidos neutros: acilgliceroles y ceras. Lípidos anfipáticos: glicerolípidos y esfingolípidos (1). 3.1.2 Lípidos insaponificables Estos lípidos se derivan por la aposición de varias unidades isoprénicas. La unidad básica de estas unidades es el Isopreno, un hidrocarburo no saturado, de cinco carbonos, con dos dobles enlaces. Este además es un componente de los cauchos naturales (1, 3). En este grupo se incluyen: Terpenos: retinoides, carotenoides, tocoferoles, naftoquinonas, dolicoles. Esteroides: esteroles, sales y ácidos biliares, hormonas esteroideas. Existen otros tipos de lípidos no saponificables, los cuales no están relacionados estructuralmente con el Isopreno: Hidrocarburos. Lípidos pirrólicos (1). 3.2 Jabones Los jabones son sales carboxílicas de cadena larga, derivados de la saponificación un ácido graso (2). Los jabones son casi totalmente miscibles en agua. Sin embargo no se disuelven a nivel de iones. Con excepción de soluciones muy diluidas, los jabones siempre forman micelas. Estas suelen ser agrupaciones, normalmente esféricas, de iones carboxilato que se dispersan en la fase acuosa. Los iones carboxilato se agrupan con sus grupos carboxilato negativamente cargados (polares) hacia la superficie de la micela, mientras que sus cadenas alifáticas apolares se orientan hacia adentro. Los iones sodio se distribuyen en la fase acuosa como iones individuales solvatados2 (2). 3.2.1 Acción limpiadora de los jabones La formación de micelas hace posible que los jabones se disuelvan en agua. Las cadenas alquílicas apolares, e hidrófobas, del jabón permanecen en un medio apolar en el interior de la micela. Mientras tanto los grupos carboxilato polares, e hidrofílicos, están expuestos a un ambiente polar en la fase acuosa. Al estar las micelas negativamente cargadas, las micelas individuales se repelen entre sí y permanecen dispersas en la fase acuosa (2). 1 2 Derivado del Eicosano, un n-alcano de 20 carbonos, sólido a temperatura ambiente (2). Las micelas son las estructuras que justifican el mecanismo aislante de la membrana celular. 50 De manera similar, todas las partículas de polvo (por ejemplo sobre la piel) se cubren con una capa de aceite o grasa. Las moléculas de agua son incapaces de dispersar estos glóbulos grasosos por si mismas. Esto debido a que no son capaces de penetrar en la capa aceitosa para separar las partículas individuales y desprenderlas de la superficie en la que están adheridas (2). Por el contrario, las soluciones jabonosas sí son capaces de separar las partículas individuales gracias a que sus cadenas alifáticas se disuelven en la capa aceitosa. Cuando esto sucede, cada partícula individual adquiere una capa externa de iones carboxilato. Gracias a esto ofrece a la fase acuosa un exterior mas compatible, que es una superficie polar. En este momento los glóbulos individuales se repelen entre si y se dispersan en la fase acuosa (2). 3.3 Detergentes sintéticos Los detergentes funcionan de manera similar a los jabones, tienen largas cadenas alifáticas apolares y grupos polares en un extremo. Los grupos polares de casi todos los detergentes sintéticos son sulfonatos de sodio o sulfatos de sodio (2). Los detergentes sintéticos poseen la capacidad de conservar su capacidad limpiadora (formando micelas), incluso cuando están disueltos en aguas duras. Estas son las que contienen iones Ca++, Fe++, Fe+++ y Mg++. Son empleados como emulsificantes de mezclas inmiscibles de plaguicidas. La desventaja de éstos, es que muchos no son biodegradables (2). IV Marco Experimental 4.1 Materiales: Cant. Equipo y Cristalería Reactivos 1 Balanza Grasa* 2 Beacker de 600 mL Hidróxido de Sodio al 40% 1 Probeta de 10 mL Etanol al 80% 1 Varilla de vidrio Hidróxido de Potasio al 20% 1 Estufa eléctrica 1 Espátula 1 Pinzas para capsulas de porcelana 1 Rejilla de asbesto *Este material será asignado por el Instructor y deberá proporcionarlo el alumno 4.2 Metodología 4.2.1 SAPONIFICACIÓN DE UNA GRASA Trasvasar a un beacker la muestra de grasa con 100 mL de su capacidad. Agregar NaOH o KOH 100 mL. Agregar Alcohol 15 mL. Calentar suavemente, la proporción entre el volumen del lípido y el volumen de hidróxido debe ser aproximadamente de 1:1. 51 Calentar en la estufa eléctrica hasta que la mezcla comience a ebullir. Agitar fuertemente durante todo el proceso. Alícuotas de NaOH Se deberá marcar en la parte exterior del beacker el nivel que tiene la mezcla. Calentar nuevamente. Agitar fuertemente. Cuando la mezcla comience a ebullir se deberá observar que no disminuya el nivel previamente marcado. En caso de que ocurra, se deberá ajustar el nivel con el etanol. ! No vertir etanol sobre una llama abierta¡. Se deberá ajustar el nivel de la mezcla cada vez que sea necesario. La saponificación se considera completa cuando la mezcla no tenga olor a grasa y su pH sea neutro. En caso de que el pH sea alcalino, se deberá neutralizar con HCl. Tomar una pequeña alícuota de lo saponificado, comprobar si produce espuma al entrar en contacto con agua. No deberá dejar las manos grasosas. V. Cuestionario 1. ¿Con que sustancias de uso común se puede llevar a cabo la hidrólisis alcalina de un lípido? 2. ¿Qué es la lejía?, ¿qué es la soda cáustica? 3. ¿Qué características físicas y químicas debe presentar el producto de la saponificación para considerarse jabón? VI. Bibliografía 1. FESSENDEN, R. J. 1,988. Química Orgânica. México, Me.. Grupo Editorial Iberoamericana. 2. GRAHAM SOLOMONS, T.W. 1,990. Fundamentos de Química Orgânica. Limusa. México, Mex. 903 p. 3. RIODUERO. 1,975. Química. 2 ed. Madrid, Esp. 269 p. 52 Práctica 10 Reacciones de aldehídos I. Introducción Los aldehídos son un grupo de compuestos orgánicos, nombrados así en el siglo XIX por Justus Liebieg a los productos procedentes de alcoholes deshidrogenados, es decir que son el primer producto de la oxidación de los alcoholes primarios (2). Los aldehídos a su vez pueden oxidarse fácilmente, son capaces de reducir las disoluciones de plata. Son muy reactivos y sirven para obtener sustancias aromáticas y pigmentadas, productos medicinales, sintetizar resinas artificiales y espejos de plata. En la naturaleza es común encontrarlos en las frutas, flores y aceites esenciales (2). II. Objetivos Que el estudiante aprenda a manipular correctamente la cristalería y reactivos necesarios para llevar a cabo esta práctica. Que el estudiante comprenda y realice el proceso necesario para obtener un espejo de plata III. Marco teórico 3.1 Aldehídos y cetonas Los aldehídos y las cetonas, son compuestos orgánicos que contienen el grupo carbonilo, en este un átomo de carbono forma un doble enlace con el oxígeno. C=Ö: El grupo carbonilo El grupo carbonilo de los aldehídos está unido por lo menos a un átomo de carbono y a uno de hidrógeno, mientras que en las cetonas, este se fija a dos átomos de carbono (1). Ö: R–C–H fórmula general de aldehído Ö: R–C–R fórmula general de cetona 3.1.1 Propiedades físicas Debido a que el grupo carbonilo es polar, los aldehídos y las cetonas poseen puntos de ebullición más elevados que los hidrocarburos con el mismo peso molecular. Los aldehídos y las cetonas no forman puentes de hidrógeno fuertes entre sus moléculas, por lo tanto sus puntos de ebullición son menores que los correspondientes a los alcoholes del mismo peso molecular (1). 53 El oxígeno carbonílico permite que las moléculas de aldehídos y cetonas formen puentes de hidrógeno fuertes con las moléculas de agua. Por esto, los aldehídos y cetonas de bajo peso molecular son considerablemente solubles en agua. La acetona (dos grupos alquilo unidos a un grupo carbonilo) y el acetaldehído ( o etanal, un grupo carbonilo unido a un alquilo y a un hidrógeno) son infinitamente solubles en agua (1). Tabla 1. Algunas propiedades físicas de aldehídos y cetonas Fórmula: Nombre: Pf °C Pe °C HCHO Formaldehído -92 -21 CH3CHO Acetaldehído -125 -21 CH3CH2CHO Propanal 81 49 CH3(CH2)2CHO Butanal -99 76 CH3(CH2)3CHO Pentanal -91.5 102 CH3(CH2)4CHO Hexanal -56 128 C6H5CHO Benzaldehído -57 178 C6H5CH2CHO Fenilacetaldehído 33 193 CH3COCH3 Acetona -95 56.1 CH3COCH2CH3 Butanona -86 79.6 CH3COCH2CH2CH3 2-Pentanona -78 102 CH3CH2COCH2CH3 3-Pentanona -42 102 C6H5COCH3 Acetofenona 21 202 C6H5COC6H5 Benzofenona 48 306 Fuente: GRAHAM, SOLOMONS, 1990 (1) Solubilidad en agua Muy soluble ∞ Muy soluble Soluble Poco soluble Poco soluble Poco soluble Poco soluble ∞ Muy soluble Soluble Soluble Insoluble Insoluble 3.1.3 Preparación de aldehídos Los aldehídos se preparan con métodos en los que hay oxidorreducción. Sin embargo, como se oxidan o reducen fácilmente, se deben emplear técnicas y reactivos especiales. 3.1.3.1 Preparación de aldehídos por oxidación de alcoholes primarios con cromato Los alcoholes primarios se oxidan a aldehídos. Un reactivo comúnmente usado para esos fines un complejo que se forma cuando el óxido crómico reacciona con la piridina (1). CH2CL2 C6H13CH2OH + CrO3* 2C5H5N C6H13CHO 1-Heptanol Óxido crómico piridina Heptanal (93%) 3.1.3.2 Preparación de aldehídos por reducción de derivados ácidos Se emplean varios métodos especiales para convertir derivados de ácidos en aldehídos. Los cloruros de acilo, por ejemplo, se pueden reducir a aldehídos mediante el uso de hidrógeno y un catalizador de paladio parcialmente desactivado por tratamiento con azufre. Esta técnica, llamada reducción de Rosenmund, suele producir excelentes cantidades del aldehído (1). O O SOCl2 R– C– OH R – C – Cl O H2 R–C–H Pd(S) (80-90%) 54 IV Marco Experimental 4.1 Materiales: Cant. Equipo y Cristalería Reactivos 5 Tubos de ensayo Nitrato de plata al 8% 1 Gradilla de metal Hidróxido de Sodio al 40% 1 Beacker de 100 mL Amoniaco 1 Probeta de 10 mL Acetona 1 Varilla de vidrio Benzaldehido, Formaldehido 1 Mechero con su manguera 2,4-Dinitrofenilhidracina (FDNH)* 1 Pinza para tubos de ensayo Etanol al 95% 1 Espátula Glucosa diluida (debe de prepararla el estudiante.) 1 Pizeta *Nota: El reactivo se prepara disolviendo 3 gramos de 2,4-dinitrofenilhidracina en 15 mL de ácido sulfurico concentrado. Entonces se añade esta solución, agitando a 20 mL de agua y 70 mL de etanol al 95%. Se mezcla perfectamente la solución. 4.2 Metodología 4.2.1 PRUEBA DE CLASIFICACIÓN: Lave 2 tubos de ensayo. Agregue a cada tubo 2 mL de etanol al 95%. en cada tubo de ensayo agregar 1 o 2 gotas del compuesto que se va a investigar. Añada a cada tubo de ensayo 10 gotas del reactivo 2,4-dinitrofenilhidracina. Debe observar la formación de un precipitado, lo que indica que el compuesto tiene un grupo carbonilo en su estructura. Ver ejemplo en figura Precipitado formado 4.2.1 “ENSAYO DE TOLLENS” PARA CARBONILOS Prueba del espejo de plata: Lave cuidadosamente 3 tubos de ensayo Posteriormente lavar 3 veces, aplicando sucesivamente NaOH y alcohol al 95%. Finalmente lavar con agua destilada En uno de los tubos de ensayo agregar 30 gotas de solución de nitrato de plata y 1 mL de agua destilada. Distribuir esta solución en partes iguales en tres tubos de ensayo ( tubos 1, 2 y 3) 55 En el tubo de ensayo 1, 2 y 3 una o dos gotas de NaOH al 40%, posteriormente agrege amoniaco (NH3) hasta que desaparezca el precipitado formado y se torne traslucida la solución. Agregar amoniaco hasta que desaparezca el precipitado En el tubo de ensayo 1 agregar tres gotas de Glucosa diluida, al tubo 2 agregar tres gostas de Formaldehido y al tubo 3 agregar tres gotas de acetona. Flamear cada uno de los tubos en el orden en el que se presentan, evitando que la llama sea directa y observar la reacción. Después de flamear debe observarse la formación de un espejo de plata. *Este reactivo debe prepararlo el estudiante disolviendo 1 espatulada de glucosa en 50 mL de agua destilada V. Cuestionario 1. ¿Qué se persigue al calentar la mezcla? 2. ¿Qué cuidados se deben tener con el manejo del amoníaco? 3. Indique cuales de las sustancias empleadas en la práctica son corrosivas y cuales tóxicas. 4. ¿A que atribuye usted la diferencia de velocidad en la formación de productos al emplear hidróxido de sodio comparándolo con el amoníaco? VI. Bibliografía 1. GRAHAM SOLOMONS, T.W. 1,990. Fundamentos de Química Orgânica. Limusa. México, Mex. 903 p. 2. RIODUERO. 1,975. Química. 2 ed. Madrid, Esp. 269 p. 56 Práctica 11. Obtención de aceites esenciales por arrastre de vapor I. Introducción Los aceites esenciales son los primeros odoríferos que se encuentran en las plantas. Debido a que se evaporan con facilidad al exponerse al aire a temperatura ambiente, se les llama también aceites volátiles o etéreos. La obtención de los aceites esenciales se efectúa mediante la destilación simple y por arrastre de vapor. De ellos el último es el método que se emplea con mayor frecuencia. Para ello se utiliza un generador de vapores del aceite esencial, un refrigerante y un recipiente para recibir el destilado. Un líquido hierve cuando la presión de su vapor iguala a la presión atmosférica. Por otro lado, cuando se hallan en presencia dos sustancias inmiscibles entre si, la presión de vapor de la mezcla es igual a la suma de las dos presiones individuales. Así entonces una destilación por corriente de vapor no es más que una destilación a presión reducida, lo cual implica a su vez, reducción en la temperatura normal de ebullición. Esto último es de suma importancia para la obtención de los aceites esenciales ya que muchos de ellos se ven afectados en sus propiedades al aplicárseles temperaturas elevadas. II. Objetivos Aplicar los fundamentos, procedimientos y cristalería que están involucrados con la técnica de destilación por arrastre con vapor de agua. Obtener aceite esencial a partir de una muestra vegetal utilizando destilación por arrastre con vapor de agua. Conocer el uso de los aceites esenciales en la industria. III. Marco teórico La extracción por arrastre de vapor de agua es uno de los principales procesos utilizados para la extracción de aceites esenciales. Los aceites esenciales están constituidos químicamente por terpenoides (monoterpenos, sesquiterpenos, diterpenos, etc.) y fenilpropanoides, compuestos que son volátiles y por lo tanto arrastrables por vapor de agua. Están formados principalmente por terpenoides volátiles formados por unidades de isopreno unidas en estructuras de 10 carbonos (monoterpenoides) y 15 carbonos (sesquiterpenoides). Las sustancias responsables del olor suelen poseer en su estructura química grupos funcionales característicos: aldehídos, cetonas, ésteres, etc. 3.1 Origen Las plantas elaboran los aceites esenciales con el fin de protegerse de las enfermedades, ahuyentar insectos depredadores, atraer insectos benéficos que producen la polinización. Los aceites esenciales son característicos de los Magnoliales, los Laurales, los Austrobaileyales, y los Piperales, y también de algunas familias no emparentadas con estos órdenes, como Myrtaceae, Rutaceae, las familias de Apiales, Lamiaceae, Verbenaceae y Asteraceae. 57 Están presentes en distintas partes de la planta: En las flores como lavanda, jazmín y rosa. En todo el árbol como sucede con el eucalipto. En las hojas como citronela. En la madera como en el sándalo. En la raíz como en el vetiver. En la resina que exhudan como en el incienso, la mirra y el benjuí. En la cáscara de los frutos como el limón, la naranja y la bergamota. Dentro de los tejidos vegetativos, se encuentran en células esféricas o diferentes cavidades o canales en el parénquima, y cuando dan el olor a las flores, se encuentran en las glándulas odoríferas desde donde son liberados. 3.2 Nomenclatura Se denominan con el mismo nombre de la planta de origen: aceite esencial de lavanda, aceite esencial de limón, etc. De algunas plantas se extrae más de un aceite esencial, en ese caso el nombre varía. Por ejemplo de las flores del naranjo se extrae por destilación el nerolí o azahar, por destilación de los frutos recién formados el petit grain y de la cáscara o corteza de los frutos el aceite esencial de naranjo. 3.3 Clasificación Los aceites esenciales se clasifican con base en diferentes criterios: consistencia, origen y naturaleza química de los componentes mayoritarios. De acuerdo con su consistencia los aceites esenciales se clasifican en esencias fluídas, bálsamos y oleorresinas. Las Esencias fluídas son líquidos volátiles a temperatura ambiente. Los Bálsamos son de consistencia más espesa, son poco volátiles y propensos a sufrir reacciones de polimerización. Las Oleorresinas tienen el aroma de las plantas en forma concentrada y son típicamente líquidos muy viscosos o sustancias semisólidas (caucho, chicle, oleorresina de paprika, de pimienta negra, de clavero, etc.). De acuerdo a su origen los aceites esenciales se clasifican como naturales, artificiales y sintéticos. Los naturales se obtienen directamente de la planta y no sufren modificaciones físicas ni químicas posteriores, debido a su rendimiento tan bajo son muy costosas. Los artificiales se obtienen a través de procesos de enriquecimiento de la misma esencia con uno o varios de sus componentes, por ejemplo, la mezcla de esencias de rosa, geranio y jazmín enriquecidas con linalool, o la esencia de anís enriquecida con anetol. Los aceites esenciales sintéticos como su nombre lo indica son los producidos por la combinación de sus componentes los cuales son la mayoría de las veces producidos por procesos de síntesis química. Estos son más económicos y por lo tanto son mucho más utilizados como aromatizantes y saborizantes (esencias de vainilla, limón, fresa, etc.). 58 Desde el punto de vista químico y a pesar de su composición compleja con diferentes tipos de sustancias, los aceites esenciales se pueden clasificar de acuerdo con el tipo se sustancias que son los componentes mayoritarios. Según esto los aceites esenciales ricos en monoterpenos se denominan aceites esenciales monoterpenoides (p.ej. hierbabuena, albahaca, salvia, etc.). Los ricos en sesquiterpenos son los aceites esenciales sesquiterpenoides (p.ej. copaiba, pino, junípero, etc.). Los ricos en fenilpropanos son los aceites esenciales fenilpropanoides (p.ej. clavo, canela, anís, etc.). Aunque esta clasificación es muy general nos resultará útil para propósitos de estudiar algunos aspectos fitoquímicos de los monoterpenos, los sesquiterpenos y los fenilpropanos. IV Marco Experimental 4.1 Materiales: Cant. Equipo y Cristalería Reactivos 4 Soportes universales Nacl 3 Pinzas Fisher Eter 2 Mecheros Bunsen Material vegetal* 4 Mangueras de hule 2 Balones de fondo plano de 500 ml. 1 Condensador de Liebig 1 Earlemeyer de 250 ml. 1 Beacker de 150 ml. 1 Varilla de 7 ó 10 mm de diámetro 3 Tapones horadados 2 Varillas conectoras 1 Ampolla de decantación 1 Espátula 1 Balanza monoplato 1 Baño de María 1 Estufa Perlas de ebullición **Este material deberá proporcionarlo el alumno. 4.2 Metodología Arme el sistema como se muestra en la figura 1. Agregue 300 ml. de agua en el matraz generador de vapor. Coloque las perlas de ebullición dentro del matraz generador de vapor. Coloque la mayor cantidad posible de la muestra vegetal a trabajar, con un poco de agua (más o menos 200 ml.) en el matraz de destilación. Tape los matraces con los tapones horadados y conéctelos a la tubería. Envuelva la tubería doblada que conecta a los dos matraces y al condensador Liebig con papel aluminio. Caliente el agua del matraz generador de vapor y cuando empiece a pasar vapor al matraz de destilación, caliente el matraz de destilación para evitar la condensación 59 del vapor de agua. Puede colocar otro mechero en el matraz de destilación para calentarlo y no perder el calor en el cambio del mechero del matraz generador de vapor hacia el matraz de destilación o viceversa. (Recuerde que el calor no debe de suspenderse, si no, dejará de pasar vapor y por consiguiente no recogerá el aceite esencial). Pase vapor hasta que se hayan recogido alrededor de 100 ml de destilado. Vierta el destilado en una ampolla de decantación. Agregue pequeñas cantidades de cloruro de sodio a la ampolla de decantación, agite un poco y observe si se rompe la emulsión, de no romperse agregue otra cantidad de cloruro de sodio y deje reposar la mayor cantidad de tiempo posible (24 hrs.), luego quedará el agua en la parte inferior y en la superior encontrará una mínima cantidad de aceite, que por lo general quedará adherido a las paredes de la ampolla. También puede agregar 25 ml de eter, agitar bien la ampolla, dejar reposar un momento y descartar el agua de la ampolla y posterior evaporar el eter en baño de María. Figura 1. Sistema de extracción de aceites esenciales por arrastre de vapor V. Cuestionario 1. Al disminuir uno de los componentes en la destilación de arrastre con vapor, ¿cómo evolucionará la temperatura de la destilación?. 2. El punto de ebullición de un compuesto para destilarlo en arrastre de vapor debe ser ¿inferior o superior al del agua?. 3. ¿Cómo se ven afectadas las tensiones de vapor de los componentes de una mezcla en función de las cantidades relativas existentes en la misma?. 4. Aplicaciones de la destilación en corriente de vapor de agua. 5. ¿Qué desventajas se podrían citar de la destilación en corriente de vapor, como método de separación y purificación?. 60 VI. Bibliografía 1. GRAHAM SOLOMONS, T.W. 1,990. Fundamentos de Química Orgânica. Editorial Limusa. México, D.F. 903 p. 2. PINAGEL, D.; SLOWING, I.; DE LA ROCA, W. 2,003. Manual de prácticas de laboratorio de Química Orgánica 1. Facultad de Ciencias Químicas y Farmacia, USAC. Guatemala, Gua. 53 p. 3. CATALAN R. 1999. Manual de prácticas de laboratorio de Química Orgánica. Facultad de Agronomía 61