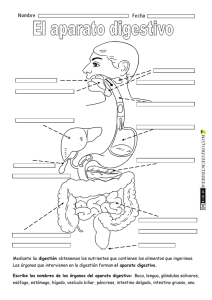
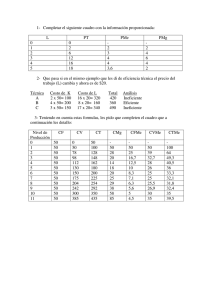
diagnóstico
Anuncio

CMG MÉDICA DE APARATO DIGESTIVO CMG TEMA 1 ENFERMEDAD POR REFLUJO GASTROESOFÁGICO DEFINICIÓN Y VARIANTES “La ERGE es una condición que aparece cuando el reflujo del contenido del estómago produce síntomas molestos y/o complicaciones.”(Montreal 2006). La definición de Montreal (definición actualmente vigente para la enfermedad por reflujo gastroesofágico) fue realizada por un grupo de consenso multinacional compuesto por 44 expertos de 18 países diferentes que decidieron establecer la enfermedad por reflujo gastroesofágico como, tal y como se dijo anteriormente: “La afección que se desarrolla cuando el reflujo del contenido del estómago causa síntomas molestos y/o complicaciones”. Con esta nueva definición, la clave del diagnóstico se basa en la severidad y frecuencia de los síntomas y es el propio paciente quien debe determinar si son lo suficientemente molestos como para ver afectada su calidad de vida. Como podemos observar, la definición hace referencia a “manifestaciones clínicas” (que son valoradas como tales por el paciente) y “complicaciones” (ya que hay un porcentaje de pacientes de que presentan complicaciones pero no tienen síntomas). importante ya que dependiendo de que haya esofagitis o no el tratamiento variará. En la ERGE CON ESOFAGITIS el tratamiento será un tratamiento de mantenimiento, mientras que en la ERGE SIN ESOFAGITIS el tratamiento es, generalmente, a demanda1. SIGNOS Y SÍNTOMAS Desde el punto de vista clínico, esta entidad puede ser responsable de un amplio abanico de manifestaciones clínicas, tanto a nivel esofágico como extraesofágico (Tabla 1). Síndromes Esofágicos Sintomáticos Con lesiones Síndrome de Esofagitis por Reflujo Típi- Reflujo co Estenosis EsofáSíndrome de gica por Reflujo Dolor Torácico por Esófago de Reflujo Barret Síndrome de Tos por Reflujo Faringitis Síndrome de Asma por Reflujo Fibrosis Pulmonar Sinusitis Otitis Media Recurrente Síndrome de Erosiones Dentales por Reflujo Es la enfermedad esofágica más frecuente. La ERGE se puede manifestar con síntomas, con lesiones de la mucosa esofágica o con la presencia simultánea de síntomas y lesiones. Tabla 1 Según los resultados de la endoscopia, la ERGE se divide en ERGE con esofagitis (ERGE erosiva) o ERGE con endoscopia negativa (ERGE no erosiva). Esta diferencia es muy 1 Prof. Fuenzalida Establecidos Propuestos Síndrome de Adenocarcinoma Laringitis por Reflujo de Esófago Con respecto a la epidemiología, indicar que es una entidad muy prevalente pero que, del tercio de la población que la presenta, sólo un 10% consulta al médico. TEMA 1 Síndromes Extraesofágicos La mayoría de los pacientes con síntomas de ERGE no presentan lesiones esofágicas y la clínica no guarda una buena correlación con las lesiones. Comisionista: María Lozano 1-1 CMG MÉDICA DE APARATO DIGESTIVO Además de factores genéticos asociados, la obesidad, la ganancia reciente de peso independientemente del IMC, el consumo de tabaco, la presencia de una hernia hiatal por deslizamiento y dormir en posición de decúbito lateral derecho son factores de riesgo para la ERGE. El ejercicio físico intenso, así como la toma de diversos fármacos (antagonistas del calcio, anticolinérgicos, aminofilinas, nitratos, opioides, esteroides,...) pueden empeorar los síntomas de ERGE. El ejercicio físico moderado, elevar la cabecera de la cama unos centímetros o la presencia de infección por H. Pylori podrían actuar como factores protectores. A continuación desarrollaremos una de las etiologías más importantes de la ERG: la hernia de hiato por deslizamiento (que es la que se puede asociar a reflujo gastroesófágico). HERNIA DE HIATO POR DESLIZAMIENTO (HHD) CMG sencia de una HHD y se correlaciona bastante bien con la tasa de RGE patológico. Esta hipotonía podría deberse a que está situado en el tórax en un ambiente de presión negativa o a alteraciones intrínsecas del mismo. FISIOPATOLOGÍA La enfermedad por RGE surge cuando se desequilibra el balance entre los factores agresores (reflujo ácido, potencia del reflujo) y los factores defensivos de la mucosa esofágica (aclaración del ácido esofágico, resistencia de la mucosa). Contenido Ampliado Para comprender lo relacionado a las alteraciones en los factores defensivos debemos recordar el importante papel antirreflujo o de barrera que juegan el esfínter esofágico inferior, el aclaramiento esofágico y la resistencia de la mucosa esofágica. Al ascender al tórax la unión esofagogástrica (UEG) ocurren dos hechos que facilitan el RGE, ya que producen incrementos bruscos de la presión intrabdominal: El esfínter esofágico inferior (EEI) más que una estructura anatómica debe ser considerado una unidad funcional donde se define un área con características especificas de altas presiones que hacen de la misma una zona que impide el paso retroactivo hacia el esófago del contenido gástrico, por tanto cualquier detalle que incida negativamente en esta adecuada funcionalidad, provocará dificultades en este mecanismo. Por ello se pueden producir determinadas características que alteren el normal estado del mecanismo esfinteriano. Ellas son: • El mecanismo esfinteriano del músculo estriado de los pilares diafragmáticos (esfínter extrínseco) está debilitado. - Presión en reposo del EEI menor de 8 mmHg. • - Longitud del EEI menor de 2 cms. La desaparición del EEI intraabdominal supone que los aumentos de presión en esta cavidad no se transmitan al EEI, con lo que la presión gástrica superaría la presión esfinteriana posibilitando el RGE. - Situación inadecuada del EEI, como cuando se ubica en la posición intratorácica. Es la causa más frecuente de ERGE y está presente en el 75 – 90 % de los casos. Pero hay que tener en cuenta que no siempre la ERGE es causada por hernia de hiato por deslizamiento, ni todas las hernias de hiato por deslizamiento producen ERGE2. Muchas HHD no se acompañan de ERGE. La relación causa-efecto entre HHD y ERGE no está totalmente aclarada, ya que hay reflujo sin hernia (15- 20%) y sobretodo muchas hernias que no se acompañan de ERGE. La hipotonía del EEI es un hecho frecuente en pre2 El Dr. Fuenzalida dijo que era muy importante que esto quedara claro. TEMA 1 Prof. Fuenzalida El aclaramiento esofágico, que no es más que la capacidad del esófago para vaciar de forma rápida y completa el contenido gástrico refluido, está condicionado por la acción en primer lugar de la saliva como medio de arrastre y tamponamiento, lo cual juega un papel fundamental. A esto se une la acción de la gravedad y la normal peristalsis o actividad motora esofágica. Cuando uno de estos factores fallan se pone en peligro el mecanismo de aclaramiento. Comisionista: María Lozano 1-2 CMG MÉDICA DE APARATO DIGESTIVO La resistencia de la mucosa esofágica estará condicionada por el balance entre diversos factores: Pre-epiteliales: característico de mas de 50 episodios al día con tiempo total de pH<4 mayor de una hora en el día, comprobado por pHmetria. Zollinger Ellison: Entidad en la cual a pesar de que el mecanismo o barrera antirreflujo permanece intacto, el estado de hiperacidez mantenido favorece el daño tisular. - Moco, Bicarbonato. - Acuosa. - Epiteliales: - Membranas celulares. - Complejos intercelulares. - Uniones densas y matriz intercelular. Patología Bronquial: Se destacan dos factores uno es la micro aspiración continua de contenido gástrico hacia el árbol bronquial y el otro es un mecanismo indirecto de bronconstricción inducido por la presencia de ácido en esófago y mediado por vía vagal. Post epiteliales: - CMG Flujo sanguíneo. En cuanto a los mecanismos agresivos encontramos: la acción del ácido clorhídrico, la pepsina, los ácidos biliares, así como el llamado enlentecimiento gástrico dado por un vaciamiento retardado. En la fisiopatología de esta enfermedad se deben considerar tres aspectos: la patogénesis del episodio de RGE, la cantidad de reflujo y la patogénesis de la esofagitis. 1. Esclerodermia: Patología en la cual el EEI pierde su existencia al igual que la peristalsis esofágica se ve dañada provocando un trastorno motor secundario definido, que favorece la ERGE. Episodio de reflujo gastroesofágico: Se deben dar dos condiciones para que ocurra. La primera que el contenido gástrico esté preparado para refluir y esto puede verse en situaciones en las que aumenta el volumen del contenido gástrico (postpandrial, obstrucción pilórica, gastroparesia, estados hipersecre-tores), situaciones en las que aumente la presión intragástrica (obesidad, embarazo, ascitis, o vestir ropas apretadas). La segunda, que haya una alteración de los mecanismos antirreflujo, cuya integridad funcional depende de la presión intrínseca del EEI, compresión extrínseca del EEI por las curas diafrag-máticas, la localización intraabdominal del EEI, la integridad del ligamento frenoesofágico y el mantenimiento de un ángulo agudo de His. 2. Embarazo: La acción de la progesterona sobre el EEI también está demostrada así como el aumento de presión intraabdominal por la masa en abdomen que conlleva el embarazo. La cantidad de reflujo: Depende de la cantidad de material refluido y la frecuencia, del aclarado esofágico por gravedad y por la peristalsis, y de la neutralización por la secreción salival. 3. Patogénesis de la esofagitis: Es una inflamación de la mucosa esofágica que puede ser debida a distintas causas como son: bacterias, virus, etc., pero sin lugar a duda, la causa más común es el RGE patológico. La esofagitis por RGE se diferencia de las demás en que las lesiones son máximas en la porción distal del esófago, Existen muchos agentes externos que pueden inducir al fallo de los diferentes mecanismos entre ellos se citan la ingestión de algunos medicamentos como las benzodiacepinas, xantinas, nitratos, anticálcicos, los cuales (al igual que las grasas y el alcohol) pueden incidir negativamente en la función barrera del EEI ya que lo relajan. Algunas situaciones clínicas pueden además alterar la fisiología de estos mecanismos de defensa como son: Cirugía esofágica: Donde se pierde el mecanismo antirreflujo. Infancia: El EEI aún no ha alcanzado su madurez y complejidad lo cual provoca una tendencia al reflujo aunque para que este se considere patológico como tal debe cumplir un patrón TEMA 1 Prof. Fuenzalida Comisionista: María Lozano 1-3 CMG MÉDICA DE APARATO DIGESTIVO zona que mayor tiempo permanece en contacto con el contenido ácido procedente del estómago. El contacto del contenido refluido con la mucosa esofágica da lugar a una destrucción epitelial seguida de una reepitelización. Entre las alteraciones micros-cópicas que se observan encontramos el alargamiento de las crestas e hipertrofia de la capa basal y metaplasia de epitelio esofágico que se transforma en columnar3. Macroscópicamente destacamos la visión de úlceras y estenosis. MANIFESTACIONES CLÍNICAS Y COMPLICACIONES Los síntomas típicos de ERGE son la pirosis y la regurgitación. El término de pirosis describe la sensación de ardor o quemazón en el área retroesternal y el de regurgitación, la sensación de retorno del contenido gástrico a la boca y a la hipofaringe. Este binomio es prácticamente diagnóstico de ERGE en su presentación típica. No obstante, la ausencia de pirosis y regurgitación no excluye el diagnóstico. Otros síntomas que se pueden presentar en esta enfermedad son dolor epigástrico y alteraciones del sueño. La ERGE se considera potencialmente responsable de una gran variedad de otros síntomas y entidades clínicas, algunas de origen esofágico (dolor torácico por reflujo), y otras extraesofágicas (tos crónica, asma, laringitis, erosiones dentales, faringitis, sinusitis, fibrosis pulmonar idiopática y otitis media). El dolor torácico, la tos crónica y el asma son los procesos que presentan datos más concluyentes sobre su asociación con la ERGE. Se consideran síntomas de alarma la disfagia persistente y/o progresiva, vómito persistente, hemorragia gastrointestinal, anemia ferropénica, pérdida de peso no intencionada y/o una tumoración epigástrica palpable. La complicación esofágica más frecuente de la ERGE es la esofagitis. La estenosis, la hemorragia, el esófago de Barrett y el adenocarcinoma son complicaciones más raras. 3 Hablaremos de Esófago de Barrett cuando esa metaplasia columnar sea de tipo intestinal TEMA 1 Prof. Fuenzalida CMG DIAGNÓSTICO El diagnóstico de la ERGE se basa en la combinación de una adecuada anamnesis y exploración física, con el empleo racional de las pruebas complementarias. La pirosis y la regurgitación son los síntomas característicos de la ERGE y, basándose en ellos, se puede establecer el diagnóstico clínico de ERGE en su forma de Reflujo Típico, e iniciar el tratamiento, sin necesidad de realizar otras pruebas comple-mentarias para confirmar el diagnóstico. La sensibilidad y la especificidad de la pirosis para el diagnóstico del síndrome de reflujo típico se estima que es del 75-83% y el 5563%, respectivamente. Estos porcentajes aumentan significativamente si se asocia regurgitación. La respuesta terapéutica a los inhibidores de la bomba de protones (IBP) se utiliza para validar el diagnóstico de sospecha de ERGE, aunque no lo establece o descarta de forma totalmente concluyente. Se debe realizar una endoscopia ante la presencia de signos y/o síntomas de alarma de complicación de una ERGE (disfagia, vómito persistente, hemorragia gastro-intestinal, anemia ferropénica, pérdida de peso no intencionada y/o tumoración epigástrica). Asimismo está indicada cuando la respuesta al tratamiento no es adecuada. No obstante, no existe una buena correlación entre las manifestaciones clínicas y los hallazgos endoscópicos. En pacientes que no responden al tratamiento empírico y en los que la endoscopia no demuestra lesiones de esofagitis, deberían someterse a una pH-metría para investigar la presencia de reflujo. TRATAMIENTO El tratamiento de la ERGE tiene como objetivo evitar o reducir la exposición ácida del esófago y la sintomatología asociada al mismo. Los tratamientos abarcan los consejos sobre estilos de vida y medidas higiénico-dietéticas, fármacos y ocasionalmente cirugía. (La ERGE tiene principalmente un tratamiento médico y no quirúrgico. Comisionista: María Lozano 1-4 CMG MÉDICA DE APARATO DIGESTIVO Los consejos sobre estilos de vida y medidas higiénico-dietéticas deben individualizarse e ir orientados a los factores de riesgo que presente cada paciente, valorando la respuesta clínica a los cambios introducidos. Incluyen: - La pérdida de peso en pacientes obesos. - El abandono del tabaquismo. - Evitar comidas copiosas, ricas en grasas y aquellas que desencadenen síntomas. - La práctica de ejercicio físico moderado. - Evitar en lo posible medicamentos favorecedores de reflujo. - En algunos casos, medidas posturales como dormir sobre el lado izquierdo o elevar la cabecera de la cama 15-30 cms. - Algunos pacientes también pueden beneficiarse de no acostarse hasta transcurrida una hora después de comer. Los fármacos utilizados en el tratamiento de la ERGE son los antiácidos (solos o asociados a alginatos), los antagonistas H2 (antiH2), los IBP y los procinéticos. - Los antiácidos han demostrado ser superiores a placebo en el control de los síntomas de ERGE, y la combinación antiácidos-alginatos es más eficaz que los antiácidos solos. Su uso actualmente queda restringido al tratamiento de algunos episodios leves de pirosis. - Los antagonistas H2 han demostrado ser más eficaces que placebo en el control de los síntomas de ERGE y en el tratamiento de la esofagitis por reflujo. Se pueden considerar una opción para el tratamiento del síndrome típico de la ERGE y de la esofagitis por reflujo en su fase aguda (dosis estándar) y de la de mantenimiento (dosis estándar y/o mitad de dosis). Los antagonistas H2 a demanda o pautados de forma intermitente se pueden utilizar para el control de los síntomas crónicos de reflujo4. En la práctica han sido sustituidos por los inhibidores de la bomba de protones (IBP). - CMG •Los inhibidores de la bomba de protones son los fármacos más efectivos en el tratamiento de la ERGE. Constituyen actualmente la mejor opción terapéutica (pregunta MIR) tanto para la terapia continua como a demanda. Han demostrado su eficacia en el control de los síntomas, en la curación de la esofagitis y en la prevención de las recurrencias. Son más eficaces que los antiH2 (pregunta MIR) en todos estos supuestos. No existen diferencias clínicas significativas entre los distintos IBP a dosis estándar. En caso de fracaso terapéutico es eficaz doblar la dosis en el caso de Omeprazol y Lansoprazol. No se han descrito efectos secundarios importantes de los IBP utilizados a las dosis habituales5, incluso en tratamientos prolon-gados. La mayor preocupación actualmente se centra en su relación con la malabsorción de minerales y el riesgo de fractura de cadera, efecto que aumenta en relación a los años de tratamiento. - Procinéticos: Tienen un papel muy limitado en el tratamiento de la ERGE. El uso de la cisaprida queda restringido actualmente al tratamiento de algunos pacientes concretos en el ámbito hospitalario. El resto de procinéticos (cinitaprida, metoclopramida, domperidona, cleboprida) no tienen suficientes evidencias en el tratamiento de la ERGE, aunque se sugiere que pueden tener algún efecto beneficioso en pacientes en los que el síntoma predominante es la regurgitación. Teóricamente deberían ser los más eficaces (ya que su acción consiste en el aumento del tono del esfínter esofágico inferior) pero en la práctica clínica no se han obtenidos buenos resultados. En el caso de llevar a cabo un TRATAMIENTO QUIRÚRGICO (por ejemplo en pacientes jóvenes que prefieren “atajar” definitivamente el problema) se lleva a cabo una funduplicatura de Nissen, preferentemente por vía laparoscópica (porque es menos agresiva). 4 El Dr. Fuenzalida me comentó que en pacientes con reflujo importante y que llevan tratamiento crónico con IBP se les puede recomendar que, además, tomen antes de dormir un anti-H2, y funciona muy bien. TEMA 1 Prof. Fuenzalida 5 Esto es muy importante ya que a diferencia de los IBP, algunos anti-H2 sí que tenían efectos secundarios tales como la impotencia. Comisionista: María Lozano 1-5 CMG MÉDICA DE APARATO DIGESTIVO CMG En el tratamiento del esófago de Barrett si se hacen tratamientos por endoscopia, vuelve a regenerarse el epitelio cilíndrico. En caso de que el enfermo tuviese una displasia o una metaplasia de alto grado (es decir, alterándose todo el epitelio) su indicación sería quirúrgica. Por tanto debemos señalar la importancia del seguimiento en aquellos pacientes con Esófago de Barrett, por el riesgo asociado de adenocarcinoma. Sin embargo no se ha demostrado que este seguimiento aumente la supervivencia. No debemos olvidar que también nos podemos encontrar, como complicación a la ERG, una estenosis esofágica. En este caso el tratamiento indicado es la dilatación con balón. TEMA 1 Prof. Fuenzalida Comisionista: María Lozano 1-6 CMG MÉDICA DE APARATO DIGESTIVO CMG TEMA 2 DISPEPSIA FUNCIONAL CONCEPTO Causas: La dispepsia funcional es una de las patologías más importantes a la que se debe enfrentar diariamente el especialista digestivo. De hecho, el 5 % de las consultas médicas en Atención Primaria y el 50% de los pacientes que consulta a un especialista digestivo presentan alteraciones funcionales. Enfermedad ulcerosa péptica. Además, su importancia también es debida a: Enfermedades metabólicas: Diabetes mellitus, uremia, hipercalcemia, IRC, enfermedad de Adisson, hipertiroidismo, hipotiroidismo. Cáncer gástrico. Las enfermedades bilio-pancreáticas (Litiasis biliar, pancreatitis crónica, cáncer de páncreas…). Enfermedad inflamatoria intestinal. a) Alta morbilidad. b) La alta cantidad de recursos que se consumen. Relacionada con fármacos: AINE, antibióticos, L-Dopa, digital, antiarrítmicos, antineoplásicos, estrógenos… c) Su diagnóstico no siempre es fácil, debido al gran desconocimiento de los mecanismos fisiopatológicos de muchas de las dispepsias. Alcohol. El comité de Roma III recomienda la siguiente definición de dispepsia: Por tanto, como podéis observar, las causas de dispepsia orgánica son múltiples. “Síntoma o conjunto de síntomas que la mayoría de médicos consideran tienen su origen en la región gastroduodenal” teniendo los siguientes síntomas: DISPEPSIA F UNCIONAL • Pesadez postpandrial • Saciedad precoz • Dolor o ardor epigástrico Es aquélla en la que no existe una explicación identificable de los síntomas. Más adelante comentaremos los criterios diagnósticos. La dispepsia funcional también ha recibido otros nombres como dispepsia no-orgánica, dispepsia idiopática o dispepsia esencial. CLASIFICACIÓN A su vez, la dispepsia funcional se clasifica en: - Síndrome del distress post- pandrial. Puede clasificarse en tres tipos: - Síndrome del dolor epigástrico. DISPEPSIA ORGÁNICA Es aquélla con una causa orgánica o metabólica identificada en la que si la enfermedad mejora o se elimina, los síntomas dispépticos también mejoran o desaparecen. TEMA 2 Prof. Fuenzalida DISPEPSIA NO INVESTIGADA Es aquélla en la que no se ha realizado un estudio (básicamente endoscópico) que permita establecer fehacientemente si existe o no una causa orgánica de la dispepsia. Esta ca- Comisionista: María Lozano 2-1 CMG MÉDICA DE APARATO DIGESTIVO CMG tegoría no es infrecuente, ya que en muchos pacientes no será absolutamente necesario realizar exploraciones complementarias si no cumplen los criterios de edad ni existen síntomas ni signos de alarma. 2. Las características de los síntomas no son suficientemente específicas para diferenciar la dispepsia orgánica de la funcional y, por tanto, es preferible etiquetarla como lo que es: dispepsia no investigada. • Pueden estar presentes la hinchazón en el abdomen superior, o náuseas pospandriales, o eructos excesivos. • Puede coexistir el SDE (Síndrome del Dolor Epigástrico). DISPEPSIA FUNCIONAL • Síntomas dispépticos inducidos por la comida (Síndrome del Distrés Postpandrial o SDP) antiguamente denominado “dispepsia tipo dismotilidad” debido a que en la mayoría de los pacientes se encontraban ciertas alteraciones de la motilidad gastroduodenal. Dolor epigástrico (Síndrome del Dolor Epigástrico o SDE) Esta subdivisión se debe al hecho de que, aunque los síntomas en muchos pacientes con dispepsias se inician o agravan con la ingesta, también los hay en los que las molestias aparecen en ayunas. El SDE antes era denominado “dispepsia no ulcerosa” DEFINICIÓN DE DISPEPSIA FUNCIONAL Sensación molesta de plenitud postpandrial, saciedad precoz, dolor epigástrico o ardor epigástrico, SIN evidencias de enfermedades estructurales (incluida la realización de una endoscopia digestiva alta) que puedan explicar los síntomas. DEFINICIÓN DE SDP 1. Sensación molesta de plenitud postpandrial que ocurre después de las comidas de volumen normal, al menos varias veces por semana, o TEMA 2 Prof. Fuenzalida Criterios de apoyo: DEFINICIÓN DE SDE En su reciente publicación, el comité de Roma III ha propuesto definir la dispepsia funcional a dos niveles. Uno, más general para uso fundamentalmente clínico y otro más específico, para estudios fisiopatológicos y ensayos terapéuticos, en el que se definen dos entidades nuevas: • Sensación precoz que impide la terminación de una comida normal, al menos varias veces por semana. 1. Dolor o ardor localizado en el epigastrio, de intensidad al menos moderada, y con una frecuencia mínima de una vez por semana. 2. El dolor es intermitente. 3. No generalizado o localizado a otras regiones abdominales o torácicas. 4. No se alivia con la defecación ni con el ventoseo. 5. No cumple criterios de dolor biliar. 6. El dolor puede ser de tipo quemante (ardor) pero sin ser retroesternal. 7. El dolor frecuentemente se induce o alivia con la ingesta de comidas, pero puede ocurrir en ayunas. 8. Puede coexistir el SDP. ESTOS CRITERIOS TIENEN QUE ESTAR PRESENTES DURANTE LOS ÚLTIMOS 3 MESES Y HABER COMENZADO UN MÍNIMO DE 6 MESES ANTES DEL DIAGNÓSTICO ETIOPATOGENIA DE LA D ISPEPSIA FUNCIONAL La etiopatogenia de la dispepsia funcional no se conoce con certeza. Antiguamente se relacionaba la dispepsia funcional con la gastritis crónica o con una hipersecreción de ácido clorhídrico, pero actualmente los estudios no avalan estas hipótesis. Comisionista: María Lozano 2-2 CMG MÉDICA DE APARATO DIGESTIVO También ha sido descartada la hipótesis que relacionaba la infección por E.coli con la etiología de la dispepsia funcional Y ¿Es el H. pylori una causa de dispepsia funcional? Éste es uno de los interrogantes que ha generado mayor controversia en los últimos 10 años. Pero actualmente, considerando en conjunto los numerosos ensayos terapéuticos realizados no avalan la hipótesis que postula que H. pylori desempeñaría un papel etiológico en la dispepsia funcional. Al igual que otros trastornos funcionales del tracto gastrointestinal, la dispepsia funcional puede ser mejor comprendida en el contexto de un modelo biopsicosocial de enfermedad según el cual los síntomas son consecuencia de una interacción compleja de rasgos fisiológicos gastrointestinales anormales y factores psicosociales. A día de hoy, se consideran como posibles causas de dispepsia funcional. 1. 2. 3. Alteraciones de la motilidad: En algunos pacientes se ha observado retraso del vaciamiento gástrico, hipomotilidad astral y relajación del fundus gástrico durante la ingesta( y que ésta se correlaciona con la saciedad precoz) Alteraciones de la sensibilidad visceral: Se demostró el aumento de la sensibilidad visceral( hiperestesia visceral) en pacientes con dispepsia funcional y este hallazgo no se relaciona con la presencia de alteraciones psicológicas Alteraciones psicosociales: Se ha observado una cierta relación con la ansiedad y/ o depresión. DIAGNÓSTICO La prevalencia de la dispepsia es enorme, y por tanto, es necesaria una sistemática diagnóstica racional e individualizada que permita seleccionar a los pacientes que deban ser estudiados con profundidad. En primer lugar, el diagnóstico de dispepsia ha de ser clínico; ello exige una minuciosa historia clínica y una cuidadosa exploración física. En la historia clínica debe constatarse cuándo se produjo el inicio de los síntomas, la duración, localización e irradiación del dolor, así como la sintomatología asociada, hábitos tóxicos y uso de medicación. Debemos tener presente que TEMA 2 Prof. Fuenzalida CMG las patologías que con mayor frecuencia producen síntomas dispépticos son la úlcera gástrica o duodenal, el cáncer gástrico y la dispepsia funcional. Una historia previa de síndrome constitucional, masa abdominal o anemia nos orientará más hacia el cáncer gástrico mientras que una historia larga de dolores episódicos hacen más probable el diagnóstico de dispepsia funcional o úlcera péptica, siendo más frecuente esta última cuando el dolor aumenta por la noche y se alivia con la ingesta. Cuando la clínica y la exploración física no permiten un diagnóstico relativamente preciso, el siguiente paso en el diagnóstico es la visualización del tracto digestivo superior. La endoscopia es mucho más sensible y específica que la radiología baritada y, además, permite la toma de biopsias. Pero: ¿A qué pacientes debemos una endoscopia? realizarle Edad >45-55 años Inicio de los síntomas en edad adulta Signos de alarma: o Clínica: cuadro tóxico, disfagia, vómitos, pérdida de peso, pérdida de apetito. o Exploración física: masa abdominal. o Datos de laboratorio: Los pacientes que presenten anemia o den positivo en una prueba de Sangre Oculta en Heces( SOH). o Ausencia de respuesta al tratamiento empírico (que son los casos que más se dan). DIAGNÓSTICO D IFERENCIAL Es importantísimo llevar a cabo un diagnóstico diferencial con otras posibles etiologías. La mayoría de pacientes acuden a la consulta por “dolor”. Y nosotros, mediante la realización de una buena anamnesis debemos contestar a diversas preguntas tales como cómo es ese dolor, su duración, cuándo se presenta, cómo se alivia, cómo se induce… para descartar otras patologías. Comisionista: María Lozano 2-3 CMG MÉDICA DE APARATO DIGESTIVO CMG Características del dolor ulceroso Tratamiento farmacológico: • Es rítmico y periódico. • Calma con la ingesta. Actualmente se están utilizando los siguientes fármacos: • Intensidad leve o moderada. • Suele presentarse por la noche. • Metoclopramida (antagonista dopaminérgico) Primperan®. • Domperidona (un antagonista dopaminérgico D2) Motilium®. • Cinitrapida (antidopaminérgico y agonista 5-HT4) Cidine®. Características del dolor biliar • Dolor en epigastrio que irradia a hipocondrio derecho. • Pasa de ser un dolor de intensidad baja a una intensidad muy alta en un periodo muy pequeño de tiempo. • Desciende y desaparece hasta la próxima “crisis”. • Es un dolor tipo “todo o nada”. Características del dolor dispéptico • Dolor moderado/ leve. • Se presenta varias veces a la semana. • La mayoría empeoran con la ingesta (aunque hay unos pocos casos donde mejora con la ingesta). • Hemos de señalar que mientras el dolor ulceroso es un dolor que el paciente te señala a “punta de dedo”, el dolor dispéptico es un dolor “a mano desplegada”. TRATAMIENTO Cuando el diagnóstico es de dispepsia funcional suele plantearse un serio problema ya que, actualmente, no existe un tratamiento que sea verdaderamente eficaz en todos los casos. Es importante indicar que simultánea-mente al diagnóstico empieza el tratamiento. Tratamiento dietético: • Es recomendable evitar el alcohol, el tabaco y los fármacos antiinflamatorios. • También debe aconsejarse comer despacio y masticar adecuadamente para favorecer el procesamiento gástrico de los alimentos. • Tomar una dieta pobre en grasas y no excederse con las legumbres. TEMA 2 Prof. Fuenzalida Debemos señalar que algunos de estos fármacos presentan efectos secundarios, entre los que destacamos alteraciones menstruales y galactorrea en mujeres jóvenes y temblor (tipo Parkinson) en ancianos. La Metoclopramida puede producir crisis extrapiramidales, sobre todo si se administra IV en bolo rápido, y más aún en niños. En caso de aparición de esta última reacción adversa o temblor Parkinsoniano, se tratará con Biperideno (Akinetón®). Elección del tratamiento: Dado que la dispepsia funcional es un síndrome heterogéneo con diferentes posibilidades patogénicas, es improbable que un único tratamiento beneficie a todos los pacientes. El dilema se plantea en el momento de escoger un fármaco en concreto para un determinado paciente. Desde un punto de vista práctico, y sin apoyos conceptuales, puede seguirse la siguiente estrategia: en caso de dispepsia funcional tipo ulceroso (dolor epigástrico), con antecedentes de enfermedad ulcerosa (una vez erradicado el Helicobacter pylori) o síntomas asociados de reflujo gastroesofágico, puede iniciarse tratamiento con inhibidores de la bomba de protones. Por el contrario, si la dispepsia es de tipo dismotilidad, se comenzará con fármacos procinéticos (Metoclopramida, domperidona y cinitrapida). Es muy importante recordar que muchos pacientes con dispepsia funcional no precisan de ningún tratamiento farmacológico. Lo que necesitan es que se excluya la existencia de enfermedades orgánicas graves y se les tranquilice. Aquí, EL APOYO PSICOLÓGICO POR PARTE DEL MÉDICO ES FUNDAMENTAL. Dedicar un cierto tiempo a explicar el origen de las molestias y el buen pronóstico de la enfermedad es una inversión que se verá recompensada para el paciente y para el médico. Comisionista: María Lozano 2-4 CMG MÉDICA DE APARATO DIGESTIVO CMG TEMA 3 (I) GASTRITIS Y GASTROPATÍA CONCEPTOS BÁSICOS Se define como gastropatía toda lesión gástrica sin inflamación de la mucosa. En el momento en que se aprecie inflamación, se denominará gastritis, debiéndose reservar este término a la inflamación histológicamente demostrada, no siendo aplicable al eritema de la mucosa apreciable por endoscopia, ni tampoco siendo intercambiable por el término dispepsia. La lesión gástrica no es más que un daño epitelial acompañado de procesos de regeneración en la mucosa gástrica. GASTROPATÍA AGUDA EROSIVA HEMORRÁGICA Este cuadro se caracteriza por la presencia de lesiones erosivas sangrantes (hemorrágicas) en la mucosa, inducidos por: 1. Irritantes 2. Hipovolemia - Úlcera de Curling: en politraumatizado, grandes quemados… - Sepsis. La gastropatía se debe a agentes irritantes de la mucosa, entre los que destacan los AINEs, alcohol, reflujo biliar, estados de hipovolemia o congestión crónica. 3. Mecanismos mixtos: Quimioterapia. La gastritis, por el contrario, se clasifica en gastritis por agentes infecciosos (Helicobacter pylori, Treponema palidum, Micobacterias,…), por autoinmunidad o por hipersensibilidad. El agente agresor de la mucosa es el ácido del estómago y la pepsina. Cuando se rompe la barrera de moco y bicarbonato, el ácido entra en contacto con la mucosa y lesiona los vasos, lo que induce un estímulo neural cuyo resultado es la liberación de histamina y otros mediadores de la inflamación. DIAGNÓSTICO DIFERENCIAL GASTROPATÍA VS GASTRITIS DE 4. Úlcera de Cushing: Grandes daños en el sistema nervioso central. Los hallazgos radiológicos y endoscópicos pueden ser similares, y con la historia clínica no podemos predecir la presentación histológica de la mucosa. Debido a que la diferencia entre ambas entidades diagnósticas reside en la existencia o no de inflamación, la Biopsia se hace imprescindible. TEMA 3 (I) Prof. Carballo Comisionista: Jose 3 (I)-1 CMG MÉDICA DE APARATO DIGESTIVO CMG Los AINEs inhiben la producción de prostaglandinas, sobre todo de tipo PGE, cuya presencia favorece la producción de bicarbonato y moco, agentes protectores de la mucosa, así como también estimula el flujo sanguíneo a la misma. Al realizar la endoscopia, podemos observar la presencia de múltiples petequias hemorrágicas, erosiones (rojas o negras, Fig.1), y es frecuente hallar también las lesiones en el duodeno de forma simultánea (gastroduodenitis, Fig. 2). Fig. 3: Lesión por AINE´s Fig. 4: Lesión por alcohol En estas úlceras, la prevención y tratamiento pasan por evitar la presencia del agente agresor (AINEs, alcohol….) e inhibir la producción de ácido mediante inhibidores de la bomba de protones (IBP’s), que son mucho más efectivos que la Ranitidina (Zantac®). Entre los IBP’s tenemos el Omeprazol y toda la familia de fármacos derivados del mismo (Lansoprazol, Pantoprazol, Desomeprazol…), con diferentes cualidades farmacocinéticas y farmacodinámicas básicamente. Fig. 1 GASTROPATÍA QUÍMICA CRÓNICA Fig. 2 En las úlceras de Curling podemos encontrar lesiones de estrés en fundus y cuerpo, y en las producidas por irritantes (AINEs y alcohol fundamentalmente) las veremos de forma generalizada en todo el estómago, aunque de mayor intensidad en el antro. Histológicamente, las lesiones de estrés no presentan inflamación, con lo que el término gastritis es incorrecto, siendo los mecanismos responsables de su aparición la isquemia de la mucosa, rotura de la barrera protectora e hipersecreción ácida. TEMA 3 (I) Prof. Carballo Se caracteriza por presentar una escasa inflamación, hiperplasia foveolar variable, edema de la mucosa, proliferación de fibras musculares en la lámina propia y dilatación vascular con congestión. Su causa más frecuente es el reflujo biliar intenso crónico, consumo mantenido de AINEs, Alendronato (en el tratamiento de la osteoporosis),… Comisionista: Jose 3 (I)-2 CMG MÉDICA DE APARATO DIGESTIVO CMG Por consumo crónico de AINE´s Los AINEs inhiben la atividad de la ciclooxigenasa, bloqueando la síntesis de prostaglandinas, estando algunas de ellas implicadas en el mantenimiento de la integridad de la protección de la mucosa gástrica (PGE2 sobre todo), con lo cual, ésta se ve debilitada y puede ser dañada por la acción de los jugos gástricos. Con el consumo prolongado ocurre un efecto de adaptación, con el que se reducen las lesiones hemorrágicas erosivas en la mucosa, pero este mecanismo puede fallar en un 15% de los pacientes. Por reflujo biliar duodeno-gástrico Puede deberse a anastomosis quirúrgicas, incompetencia pilórica o a una alteración de la motilidad duodenal. La lesión la producen las sales biliares, estando favorecidas por la acción bacteriana (desconjugación), pH gástrico y la acción conjunta de las enzimas pancreáticas. La sintomatología es muy variable, pudiendo cursar asintomático, o presentar dolor, vómitos o pérdida de peso. Si ha habido una cirugía previa de vías biliares o sobre el duodeno, el diagnóstico se hace fácil, pues ya tenemos la sospecha, pero si por el contrario no la ha habido, debemos hacer una endoscopia y evidenciar el reflujo biliar, analizar el contenido gástrico (para demostrar la presencia de sales biliares) o realizar estudios isotópicos de medicina nuclear. El tratamiento médico se basa en dos pilares fundamentales, conseguir la mejoría clínica y la mejoría histológica. Para mejorar la clínica utilizamos el ácido ursodeoxicólico, que al entrar en la circulación enterohepática y recircular, desplaza a las otras sales biliares (que son excretadas con las heces) y es éste el que será excretado vía biliar, con la característica de que es menos agresivo que los otros. Para mejorar la histología, se utiliza el sucralfato, que actúa como protector de la mucosa gástrica. Los tratamientos con PGE y colesteramina han demostrado que no son eficaces. En casos graves en los que el tratamiento médico no sea eficaz, se puede plantear el tratamiento quirúrgico, implantando una Y de Roux. TEMA 3 (I) Prof. Carballo Comisionista: Jose 3 (I)-3 CMG MÉDICA DE APARATO DIGESTIVO CMG TEMA 3 (II) GASTRITIS AGUDA Y CRÓNICA POR HELICOBACTER PYLORI CARACTERÍSTICAS DE LA GASTRITIS AGUDA Infección Helicobacter Esta enfermedad tiene una alta prevalencia, siendo su inicio paucisintomático, lo que hace difícil su diagnóstico salvo si se sufre alguna complicación. Los hallazgos macroscópicos son muy variados, desde una mucosa de aspecto normal hasta una mucosa sugerente de carcinoma o linfoma. Normalmente se cronifica, a no ser que sea tratada con antibióticos. CARACTERÍSTICAS DE LA GASTRITIS CRÓNICA Se asocia a úlcera péptica, cáncer gástrico y linfoma MALT. La bacteria, que tiene una alta afinidad por las células mucosas del estómago es identificable en biopsias. En un 80% de los casos, se localiza en antro y cuerpo, en un 8% sólo en antro y en un 10% sólo en cuerpo, por eso, la biopsia ha de hacerse de ambas zonas. Respecto a su asociación con el cáncer gástrico, se acepta la hipótesis de Pelayo Correa, que describe la secuencia de eventos para el desarrollo de adenocarcinoma según el siguiente esquema: TEMA 3 (II) Prof. Carballo Respuesta huésped Gastritis aguda T I E M Atrofia gástrica Metaplasia PR EM I O! ! P O Displasia Adenocarcinoma Gastritis crónica La infección por Helicobacter pylori (Hp) desencadena inflamación como respuesta por parte del huésped (gastritis aguda), que suele cronificarse (gastritis crónica) y causar atrofia gástrica, metaplasia intestinal, displasia y, finalmente, adenocarcinoma. Obviamente, la infección por Hp no es suficiente para causar cáncer gástrico, pero crea un estado de susceptibilidad donde los factores genéticos y ambientales pueden favorecer su aparición. La relación entre la infección por Hp y el desarrollo del linfoma MALT ha sido demostrada en diferentes estudios: La inflamación aguda evoluciona a crónica, con aumento de linfocitos, células plasmáticas y eosinófilos. Inicialmente, puede existir una gastritis antral difusa donde los linfocitos emigran al territorio gástrico, hasta los capilares de la lámina propia. En el curso de la gastritis crónica, pueden aparecer folículos linfoides y agregados linfáticos en la base de la mucosa gástrica, lo que constituye el llamado tejido MALT. Este es el sustrato anatómico necesario para que se desarrolle un linfoma MALT. Comisionista: Mariló 3 (II)-1 CMG DIAGNÓSTICO MÉDICA DE APARATO DIGESTIVO DE LA GAS- TRITIS CRÓNICA CMG primir el tratamiento quince días antes de la prueba. Histología TÉCNICAS NO INVASIVAS Requiere el procesamiento y la tinción anatomopatológicos. Serología Determinación de anticuerpos IgM o IgG contra Hp en suero. Esta prueba es barata y cómoda. Tiene una sensibilidad de alrededor del 90%, pero después de tratar la infección, los títulos de anticuerpos siguen altos por tiempo prolongado y, por tanto, no es útil para comprobar su erradicación. Cultivo Lleva tiempo, es cara, y depende de la experiencia. Permite determinar la sensibilidad a los AB. GASTRITIS CRÓNICA: HALLAZGOS HISTOLÓGICOS Prueba de la urea en aliento EN LA MUCOSA Consiste en suministrar urea marcada con C13 y medir a continuación, el CO2 marcado con el isótopo radiactivo en el aire espirado. Hp tiene actividad ureasa, y degrada la urea dejando libre el C13, que llega al aliento. Es una prueba simple y rápida y con una sensibilidad del 90%. A diferencia de la anterior, sí es útil para el seguimiento precoz, pero puede dar falsos negativos con tratamiento reciente con inhibidores de la bomba de protones (IBP). En general, hay un aumento de la actividad antral, gastritis superficial aguda y crónica y folículos linfoides, que están relacionados con el desarrollo del linfoma. Determinación de Antígeno fecal De elevada sensibilidad también, son apropiadas para el seguimiento siempre que se realice con un intervalo de ocho semanas después de realizado el tratamiento, aunque su uso no está muy extendido. PRUEBAS INVASIVAS Prueba de la ureasa rápida Introducción de la biopsia en un medio rico en urea, que permite detectar la presencia de la enzima ureasa en la muestra. Es el método de elección de entre los invasivos y posee sensibilidad y especificidad altas. Con el empleo reciente de IBP, antibióticos (AB) o compuestos de bismuto puede dar falsos negativos. Es importante destacar que tras la erradicación de Hp, desaparece el componente agudo, pero no el crónico. Es decir, se detiene el progreso de la infección aguda y los neutrófilos desaparecen rápidamente. La desaparición de linfocitos y eosinófilos es más lenta, y aún más la de los folículos linfoides. La mucosa con metaplasia y atrofia no se resuelven tras la erradicación. CITOTOXINA ASOCIADA GEN A (CAGA) A La citotoxina más conocida de las producidas por las cepas de Hp es la CagA. Se correlaciona con la intensidad de la gastritis: su presencia implica más Hp, más inflamación aguda y crónica, más daño epitelial, mayor probabilidad de desarrollar una úlcera y de desarrollar atrofia, metaplasia y adenocarcinoma. Está presente en el 60% de las cepas. Los IBP debilitan a la bacteria haciéndola menos visible en biopsias y reduciendo su capacidad de metabolizar urea y romper la unión con el C13. Es por esto que puedan dar falsos negativos estos métodos. La solución es su- TEMA 3 (II) Prof. Carballo Comisionista: Mariló 3 (II)-2 CMG GASTRITIS MÉDICA DE APARATO DIGESTIVO CRÓNICA ME- TAPLÁSICA Y ATRÓFICA Este tipo de gastritis está asociada a adelgazamiento mucoso, pérdida glandular y cambios en el tipo de las células epiteliales (metaplasia). Es un factor de riesgo para el desarrollo de enfermedad. G ASTRITIS ATRÓFICA METAPLÁ- CMG G ASTRITIS ATRÓFICA METAPLÁSICA “ AMBIENTAL” Está asociada a factores como la dieta (componentes nitrosos) y a Hp. No hay anticuerpos frente a células parietales y factor intrínseco. No existe hipoclorhidria tan marcada como en la gastritis autoinmune y la gastrina conserva sus valores normales. Pueden existir úlceras y cáncer gástrico (metaplasia intestinal). SICA AUTOINMUNE De carácter autosómico dominante, está asociada a los haplotipos HLA b-8 y DR-3. En cuanto a la epidemiología, afecta a la población del Norte de Europa, siendo más frecuentemente en mujeres (3:1). Puede asociarse a tiroiditis autoinmune (Hasimoto). La inflamación, la metaplasia y la atrofia afectan al cuerpo y al fundus gástrico. La autoinmunidad se produce frente a las células parietales y el factor intrínseco mediante la producción de anticuerpos contra la ATPasa de H+-K+, lo que produce una disminución de la secreción y de la capacidad de absorción de la vitamina B12, generando hipoclorhidria y un alto riesgo de anemia perniciosa. G ASTRITIS GRANULOMATOSA Un granuloma es una agrupación de células gigantes y macrófagos (ver AP). La gastritis granulomatosa puede darse de forma idiopática o secundaria a otras enfermedades como la de Crohn y la sarcoidosis. Otras causas de este tipo de gastritis son la tuberculosis, la histoplasmosis, la enfermedad de Whipple, la sífilis, la enfermedad de Anisaki y Hp. También puede generarse por tumores (linfoma o adenocarcinoma), cuerpos extraños y vasculitis (enfermedad de Wegener). La mala absorción de la vitamina B12 por déficit de factor intrínseco produce neuropatía. Esta afecta más a piernas que a brazos y se caracteriza por parestesias, ataxia, espasticidad, y en fases más avanzadas, paraplejia e incontinencia. La hipoclorhidria produce una hiperplasia de las células G. En consecuencia, hay hipergastrinemia, se generan nódulos hiperplásicos y aumenta el riesgo de padecer tumor carcinoide. El riesgo de adenocarcinoma gástrico también aumenta de 3 a 18 veces. Por eso, se debe realizar endoscopia con resección de nódulos y valoración de los mismos (también aplicable para carcinoide). Si los nódulos no se extirpan o si la displasia es significativa, ha de hacerse un seguimiento estrecho. En caso contrario, la revisión se hará cada cinco años. TEMA 3 (II) Prof. Carballo Comisionista: Mariló 3 (II)-3 CMG MÉDICA DE APARATO DIGESTIVO CMG TEMA 4 ÚLCERA PÉPTICA CONCEPTO CLASIFICACIÓN SEGÚN LOCALI- Párrafo texto normal. Enfermedad de origen multifactorial que se caracteriza por la existencia de una o más lesiones situadas en el estómago y/o duodeno, que alcanzan al menos la muscular de la mucosa. Está asociada a Hp y a los AINEs. ZACIÓN: EPIDEMIOLOGÍA Y FACTORES DE RIESGO La epidemiología ha sufrido cambios significativos en las últimas décadas. El riesgo de infección por Hp ha disminuido considerablemente en los países desarrollados. Sin embargo, el consumo de AINEs ha aumentado progresivamente y, en consecuencia, también las úlceras causadas por este tipo de fármacos. Otros factores que influyen en el desarrollo de la úlcera péptica son la agregación familiar, el tabaco y el alcohol. Dieta, café, estrés o personalidad no quedan aún demostrados como factores de riesgo. ANATOMÍA P ATOLÓGICA CONCEPTOS PREVIOS “Erosión” es la afectación de la mucosa exclusivamente. “Úlcera aguda” es también una afectación superficial, pero que llega a alcanzar la muscular de la mucosa. “Úlcera crónica” alcanza la submucosa y la muscular. TEMA 4 Prof. Carballo 1. ÚLCERA GÁSTRICA: de tipo I (cuerpo gástrico), II (cuerpo gástrico asociada a úlcera duodenal), III (prepilórica). Las úlceras gástricas pueden ser malignas o benignas. Las benignas suelen localizarse en el antro, don raras en el fundus e histológicamente, guardan semejanza con las duodenales 2. ÚLCERA DUODENAL : más relacionada con Hp. Asienta sobre todo en la primera porción del duodeno en los primeros 3 cm. siguientes al píloro. Habitualmente, es menor de 1 cm. de diámetro. Está claramente delimitada y tiene una profundidad que en ocasiones alcanza la muscular propia. La base de la úlcera suele estar formada por necrosis eosinófila con fibrosis circundante. Las úlceras duodenales malignas son extraordinariamente raras. ETIOPATOGENIA La úlcera se produce por desequilibrio entre los mecanismos de agresión y de defensa. La agresión la produce la secreción ácidopéptica y las sales biliares. La secreción de ácido tiene lugar en condiciones basales y estimuladas, cuando ingerimos alimentos. La producción basal sigue un patrón circadiano, con niveles máximos durante la noche y primeras horas de la mañana. Se estimula a través del vago (estímulo colinérgico) y a partir de fuentes locales gástricas (histaminérgico). Comisionista: Mariló 4-1 CMG MÉDICA DE APARATO DIGESTIVO ¿C UÁLES SON LOS COMPONENTES DEFENSIVOS? CMG ¿CUÁLES SON LOS AGENTES PATOGÉNICOS ? Helicobacter pylori Barrera Pre-epitelial Formada por la capa de moco y bicarbonato que producen las células epiteliales de la superficie gastroduodenal. El moco es un compuesto de agua, lípidos y glucoproteínas que forma una capa hidrófoba con los ácidos grasos que impide la difusión de iones y moléculas como la pepsina. El bicarbonato forma un gradiente que oscila entre 1 y 2 en la superficie gástrica y 6 y 7 en la superficie celular epitelial; su secreción es estimulada por el calcio, las prostaglandinas, la acción colinérgica y la acidificación de la luz. Barrera Epitelial Compuesta por las células epiteliales de la superficie que se encargan de la producción de moco, transportadores iónicos que mantienen el pH intracelular y la producción de bicarbonato y poseen estrechas uniones intracelulares. Si la barrera preepitelial es superada, las células epiteliales que bordean la lesión pueden migrar para restituir una región dañada. Este proceso tiene lugar independientemente de la división celular, requiere un flujo ininterrumpido y un pH alcalino en el medio circundante, y es regulado por diversos factores de crecimiento. Cuando los defectos son de gran tamaño y no pueden repararse mediante restitución, exigen proliferación celular, que es regulada por prostaglandinas y factores de crecimiento. Produce gastritis antral fundamentalmente por la citotoxina CagA o VacA. Aumenta la secreción mediada por la hipergastrinemia y esto favorece la acidificación duodenal y la metaplasia gástrica en duodeno. Además, su actividad ureasa genera NH3, y es capaz de lesionar las células epiteliales; produce factores quimiotácticos de neutrófilos y monocitos que contribuyen a la lesión; proteasas y lipasas que rompen el componente mucoso; y adhesinas, que facilitan la fijación de la bacteria a las células epiteliales del estómago. La reacción inflamatoria del huésped frente a la infección por Hp contribuye a la lesión. La respuesta de neutrófilos es intensa tanto en la infección aguda como en la crónica. Los linfocitos T y las células plasmáticas están presentes en la inflamación crónica, lo que apoya la implicación de las respuestas celular de Ag y humoral. Diversas citocinas y quimiocinas son producidas por las células epiteliales y las inmunorreguladoras (TNF, IL, IFN, etc.) en respuesta a Hp. AINE´s Inhiben la producción de prostaglandinas, especialmente las de tipo E. Estas son protectoras de la mucosa gástrica mediante la estimulación de la producción de bicarbonato y moco y manteniendo el flujo sanguíneo de la mucosa. Barrera Subepitelial CLÍNICA Proporcionada por el sistema microvascular de la submucosa gástrica. Suministra HCO3-, neutraliza la secreción ácida de las células parietales, proporciona el aporte adecuado de oxígeno y nutrientes y elimina los productos tóxicos metabólicos. Lo más característico es el dolor epigástrico quemante, que cursa con ritmo horario y estacional (recidivante), exacerbado por el ayuno y que mejora con la ingesta. TEMA 4 Prof. Carballo También disminuye el apetito, hay pérdida de peso, náuseas y vómitos y síntomas dispépticos. Comisionista: Mariló 4-2 CMG MÉDICA DE APARATO DIGESTIVO DIAGNÓSTICO CMG creatitis, mientras que las gástricas tienden a hacerlo hacia el lóbulo hepático izquierdo. También se han descrito fístulas gastrocólicas asociadas con úlceras gástricas. ENDOSCOPIA Además de permitir la visualización directa de la mucosa, permite hacer biopsias de los tejidos para descartar lesiones malignas o infección Hp. El estudio de una úlcera es iniciado con radiografía con bario habitualmente, pero hasta el 8% de las úlceras gástricas de aspecto radiológico benigno son en realidad malignas en endoscopia o cirugía. Por ello, a continuación de los estudios radiológicos que demuestran la presencia de una úlcera gástrica es necesario realizar una endoscopia con biopsia. También es útil para identificar lesiones demasiado pequeñas como para ser detectadas en la exploración radiológica o para determinar si una úlcera es el origen de una hemorragia. QUIMISMO GÁSTRICO Y DETERMINACIONES DE GASTRINA Y PEPSINÓGENO Actualmente en desuso ESTENOSIS PILÓRICA Es la complicación menos frecuente. Puede ser secundaria a la inflamación y el edema provocados por la úlcera y resolverse cuando la esta cicatrice. También es posible que exista una obstrucción mecánica fija secundaria a la cicatrización en las áreas peripilóricas. En este caso, es necesario realizar una endoscopia para proceder a una dilatación con globo o resolverla quirúrgicamente. TRATAMIENTO Se basa fundamentalmente en la inhibición ácida potente: Inhibidores de la bomba de protones (IBP´s) DIAGNÓSTICO DE HP Omeprazol, pantoprazol, lansoprazol, rabeprazol, esomeprazol. (Ver tema anterior) Es la complicación más frecuente de la úlcera péptica. Se producen en aproximadamente el 15% de los pacientes, y con mayor frecuencia en mayores de 60 años, por ser este grupo de edad el que más consume AINE. Son los fármacos inhibidores de ácido más potentes conocidos y los más utilizados en el tratamiento de las úlceras. Se unen de forma covalente e inhiben irreversiblemente la bomba de protones. Los pacientes que toman IBP presentan una hipergastrinemia leve o moderada, que se resuelve entre una y dos semanas después de interrumpir el tratamiento. Como sucede con cualquier fármaco que produce hipoclorhidria significativa, pueden interferir en la absorción de fármacos como el ketoconazol, la ampicilina, el hierro y la digoxina. PERFORACIÓN Sucralfato Es la segunda complicación más frecuente de la úlcera y, como la hemorragia, su incidencia en ancianos es más elevada a causa del empleo de AINE. Una forma de perforación es la penetración, en la que el lecho ulceroso tuneliza hasta un órgano adyacente. Las úlceras duodenales tienden a penetrar en dirección posterior, hacia el páncreas, provocando pan- Fármaco citoprotector que contiene hidróxido de aluminio y sulfato. Es insoluble en agua y se convierte en una pasta viscosa en el estómago y duodeno, uniéndose principalmente a los lugares de ulceración activa. El hidróxido de aluminio se disocia en medio gástrico, liberando el anión sulfato, que se puede unir a las proteínas cargadas positivamente que están COMPLICACIONES HEMORRAGIA DIGESTIVA TEMA 4 Prof. Carballo Comisionista: Mariló 4-3 CMG MÉDICA DE APARATO DIGESTIVO en el lecho ulceroso, proporcionando una barrera fisicoquímica que impide que prosiga la lesión del tejido por los ácidos y la pepsina. También puede inducir un efecto trófico al unirse a factores de crecimiento, estimulando la síntesis de prostaglandinas, provocando la secreción de moco y bicarbonato, y favoreciendo la defensa y reparación de la mucosa. La toxicidad por este fármaco es rara, y el efecto secundario más frecuente es el estreñimiento. CMG (400mg./6h.), tetraciclina o amoxicilina (500mg./6h.). Esta terapia no es menos eficaz pero sí más compleja y difícil de cumplir. Por eso, suele empezarse con la anterior. Prostaglandinas Los análogos de las prostaglandinas potencian la secreción de bicarbonato en el moco, estimulan el flujo sanguíneo de la mucosa y disminuyen el recambio celular. Pero provocan diarrea frecuentemente y otros efectos tóxicos importantes como las hemorragias y las contracciones uterinas. Antagonistas de los receptores H 2 Cimetidina, ranitidina, famotidina y nizatidina. Inhiben significativamente la secreción ácida basal y estimulada. Actualmente, se utilizan para el tratamiento de úlceras activas (durante 4 a 6 semanas) en combinación con los antibióticos para la erradicación de Hp. La cimetidina puede tener efectos secundarios antiandrogénicos leves que causan ginecomastia e impotencia, principalmente en pacientes tratados con dosis altas durante periodos prolongados. Tratamiento antibiótico contra Hp 1. PRIMERA LÍNEA : OCAX7 DÍAS Omeprazol u otro IBP, claritromicina (500mg.) y amoxicilina (1g.) cada 12 horas (1-0-1). Es posible sustituir uno de los dos antibióticos por metronidazol (400mg./12 h.) o tinidazol (500mg./12h.). 2. SEGUNDA LÍNEA : CUÁDRUPLE TERAPIA X 7 DÍAS Omeprazol u otro IBP (1-0-1), subcitrato de bismuto11(120mg./6h.), metronidazol 1 Los preparados con bismuto inducen la cicatrización de la úlcera por mecan ismos que no están claros. Se ha TEMA 4 Prof. Carballo dicho que la recubren, evitan que prosiga la lesión ind ucida por la pepsina y el ácido, se unen a la pepsina y estimulan la secreción de prostaglandinas, bicarbonato y moco. Los efectos secundarios a largo plazo son raros, mientras que cuando se utilizan a largo plazo pueden producir neurotoxicidad. Comisionista: Mariló 4-4 CMG MÉDICA DE APARATO DIGESTIVO CMG TEMA 4 ÚLCERA PÉPTICA CONCEPTO Enfermedad de origen multifactorial que se caracteriza por la existencia de una o más lesiones situadas en el estómago y/o duodeno, que alcanzan al menos la muscular de la mucosa. Está asociada a Hp y a los AINEs. EPIDEMIOLOGÍA Y FACTORES DE RIESGO La epidemiología ha sufrido cambios significativos en las últimas décadas. El riesgo de infección por Hp ha disminuido considerablemente en los países desarrollados. Sin embargo, el consumo de AINEs ha aumentado progresivamente y, en consecuencia, también las úlceras causadas por este tipo de fármacos. Otros factores que influyen en el desarrollo de la úlcera péptica son la agregación familiar, el tabaco y el alcohol (gastritis). Dieta, café, estrés o personalidad no quedan aún demostrados como factores de riesgo. Clasificación según localización: 1. ÚLCERA GÁSTRICA: de tipo I (cuerpo gástrico), II (úlcera antral asociada a úlcera duodenal), III (prepilórica). Las úlceras gástricas pueden ser malignas o benignas. Las benignas suelen localizarse en el antro, son raras en el fundus e histológicamente guardan semejanza con las duodenales 2. ÚLCERA DUODENAL: más relacionada con Hp. Asienta sobre todo en la primera porción del duodeno, en los primeros 3 cm siguientes al píloro. Habitualmente, es menor de 1 cm de diámetro. Está claramente delimitada y tiene una profundidad que en ocasiones alcanza la muscular propia. La base de la úlcera suele estar formada por necrosis eosinófila con fibrosis circundante. Las úlceras duodenales malignas son extraordinariamente raras. ANATOMÍA PATOLÓGICA ETIOPATOGENIA CONCEPTOS PREVIOS La úlcera se produce por desequilibrio entre los mecanismos de agresión y de defensa. “Erosión” es la afectación de la mucosa exclusivamente. “Úlcera aguda” es también una afectación superficial, pero que llega a alcanzar la muscular de la mucosa. “Úlcera crónica” alcanza la submucosa y la muscular, e incluso puede penetrar órganos vecinos. TEMA 4 Prof. Carballo La agresión la produce la secreción ácido péptica y las sales biliares. La defensa es a cargo del moco y el bicarbonato. La secreción de ácido tiene lugar en condiciones basales, y estimuladas cuando ingerimos alimentos. La producción basal Comisionista: Mariló 4-1 CMG MÉDICA DE APARATO DIGESTIVO sigue un patrón circadiano, con niveles máximos durante la noche y primeras horas de la mañana. Se estimula a través del vago (estímulo colinérgico) y a partir de fuentes locales gástricas (estímulo histaminérgico). CMG parietales, proporciona el aporte adecuado de oxígeno y nutrientes y elimina los productos tóxicos metabólicos. ¿CUÁLES SON LOS AGENTES PATOGÉNICOS? ¿CUÁLES SON LOS COMPONENTES Helicobacter pylori DEFENSIVOS? Barrera Pre-epitelial Formada por la capa de moco y bicarbonato que producen las células epiteliales de la superficie gastroduodenal. El moco es un compuesto de agua, lípidos y glucoproteínas que forma una capa hidrófoba con los ácidos grasos que impide la difusión de iones y moléculas como la pepsina. El bicarbonato forma un gradiente que oscila entre 1 - 2 en la superficie gástrica y 6 - 7 en la superficie celular epitelial; su secreción es estimulada por el calcio, las prostaglandinas, la acción colinérgica y la acidificación de la luz. Barrera Epitelial Compuesta por las células epiteliales de la superficie que se encargan de la producción de moco, de transportadores iónicos que mantienen el pH intracelular, de la producción de bicarbonato y poseen estrechas uniones intracelulares. Si la barrera preepitelial es superada, las células epiteliales que bordean la lesión pueden migrar para restituir una región dañada. Este proceso tiene lugar independientemente de la división celular, requiere un flujo ininterrumpido y un pH alcalino en el medio circundante, y es regulado por diversos factores de crecimiento. Cuando los defectos son de gran tamaño y no pueden repararse mediante restitución, exigen proliferación celular, que es regulada por prostaglandinas y factores de crecimiento. Produce gastritis antral fundamentalmente por la citotoxina CagA o VacA. Aumenta la secreción mediada por la hipergastrinemia y esto favorece la acidificación duodenal y la metaplasia gástrica en duodeno. Además, su actividad ureasa genera NH3, y es capaz de lesionar las células epiteliales; produce factores quimiotácticos de neutrófilos y monocitos que contribuyen a la lesión; proteasas y lipasas que rompen el componente mucoso; y adhesinas, que facilitan la fijación de la bacteria a las células epiteliales del estómago. La reacción inflamatoria del huésped frente a la infección por Hp contribuye a la lesión. La respuesta de neutrófilos es intensa tanto en la infección aguda como en la crónica. Los linfocitos T y las células plasmáticas están presentes en la inflamación crónica, lo que apoya la implicación de las respuestas celular y humoral. Diversas citocinas y quimiocinas son producidas por las células epiteliales y las inmunorreguladoras (TNF, IL, IFN, etc.) en respuesta a Hp. AINE´s Inhiben la producción de prostaglandinas, especialmente las de tipo E. Éstas son protectoras de la mucosa gástrica mediante la estimulación de la producción de bicarbonato y moco y el mantenimiento del flujo sanguíneo de la mucosa. CLÍNICA Barrera Subepitelial Proporcionada por el sistema microvascular de la submucosa gástrica. Suministra HCO3-, neutraliza la secreción ácida de las células TEMA 4 Prof. Carballo Lo más característico es el dolor epigástrico quemante, que cursa con ritmo horario y estacional (recidivante), exacerbado por el ayuno y que mejora con la ingesta. Comisionista: Mariló 4-2 CMG MÉDICA DE APARATO DIGESTIVO CMG PERFORACIÓN También disminuye el apetito, hay pérdida de peso, náuseas y vómitos, y síntomas dispépticos. DIAGNÓSTICO ENDOSCOPIA Además de permitir la visualización directa de la mucosa, permite hacer biopsias de los tejidos para descartar lesiones malignas o infección por Hp. El estudio de una úlcera es iniciado con radiografía con bario habitualmente, pero hasta el 8% de las úlceras gástricas de aspecto radiológico benigno son en realidad malignas en endoscopia o cirugía. Por ello, a continuación de los estudios radiológicos que demuestran la presencia de una úlcera gástrica es necesario realizar una endoscopia con biopsia. También es útil para identificar lesiones demasiado pequeñas como para ser detectadas en la exploración radiológica o para determinar si una úlcera es el origen de una hemorragia. Es la segunda complicación más frecuente de la úlcera y, como la hemorragia, su incidencia en ancianos es más elevada a causa del empleo de AINE. Una forma de perforación es la penetración, en la que el lecho ulceroso tuneliza hasta un órgano adyacente. Las úlceras duodenales tienden a penetrar en dirección posterior, hacia el páncreas, provocando pancreatitis, mientras que las gástricas tienden a hacerlo hacia el lóbulo hepático izquierdo. También se han descrito fístulas gastrocólicas asociadas con úlceras gástricas. ESTENOSIS PILÓRICA Es la complicación menos frecuente. Puede ser secundaria a la inflamación y el edema provocados por la úlcera y resolverse cuando ésta cicatrice. También es posible que exista una obstrucción mecánica fija secundaria a la cicatrización en las áreas peripilóricas. En este caso, es necesario realizar una endoscopia para proceder a una dilatación con globo o resolverla quirúrgicamente. TRATAMIENTO QUIMISMO GÁSTRICO Y DETERMINACIONES DE GASTRINA Y PEPSINÓGENO Se basa fundamentalmente en la inhibición ácida potente: Actualmente en desuso Inhibidores de la bomba de protones (IBP´s) DIAGNÓSTICO DE HP Omeprazol, pantoprazol, rabeprazol, esomeprazol. (Ver tema anterior) COMPLICACIONES HEMORRAGIA DIGESTIVA Es la complicación más frecuente de la úlcera péptica. Se produce en aproximadamente el 15% de los pacientes, y con mayor frecuencia en mayores de 60 años, por ser este grupo de edad el que más consume AINE. TEMA 4 Prof. Carballo lansoprazol, Son los fármacos inhibidores de ácido más potentes conocidos y los más utilizados en el tratamiento de las úlceras. Se unen de forma covalente e inhiben irreversiblemente la bomba de protones. Los pacientes que toman IBP presentan una hipergastrinemia leve o moderada, que se resuelve entre una y dos semanas después de interrumpir el tratamiento. Como sucede con cualquier fármaco que produce hipoclorhidria significativa, pueden interferir en la absorción de fármacos como el ketoconazol, la ampicilina, el hierro y la digoxina. Comisionista: Mariló 4-3 CMG MÉDICA DE APARATO DIGESTIVO Sucralfato CMG Tratamiento antibiótico contra Hp Fármaco citoprotector que contiene hidróxido de aluminio y sulfato. Es insoluble en agua y se convierte en una pasta viscosa en el estómago y duodeno, uniéndose principalmente a los lugares de ulceración activa. El hidróxido de aluminio se disocia en medio gástrico, liberando el anión sulfato, que se puede unir a las proteínas cargadas positivamente que están en el lecho ulceroso, proporcionando una barrera fisicoquímica que impide que prosiga la lesión del tejido por los ácidos y la pepsina. También puede inducir un efecto trófico al unirse a factores de crecimiento, estimulando la síntesis de prostaglandinas, provocando la secreción de moco y bicarbonato, y favoreciendo la defensa y reparación de la mucosa. La toxicidad por este fármaco es rara, y el efecto secundario más frecuente es el estreñimiento. 1. PRIMERA LÍNEA: CAOX72 DÍAS Claritromicina (500mg.), omeprazol u otro IBP y amoxicilina (1g.) cada 12 horas (1-0-1). Es posible sustituir uno de los dos antibióticos por metronidazol (400mg./12 h.) o tinidazol (500mg./12h.). 2. SEGUNDA LÍNEA: CUÁDRUPLE TERAPIA X 7 DÍAS Omeprazol u otro IBP (1-0-1), subcitrato de bismuto3(120mg./6h.), metronidazol (400mg./6h.), tetraciclina o amoxicilina (500mg./6h.). Esta terapia no es menos eficaz pero sí más compleja y difícil de cumplir. Por eso, suele empezarse con la anterior. Prostaglandinas Los análogos de las prostaglandinas potencian la secreción de bicarbonato en el moco, estimulan el flujo sanguíneo de la mucosa y disminuyen el recambio celular. Pero provocan diarrea frecuentemente y otros efectos tóxicos importantes como las hemorragias y las contracciones uterinas. Antagonistas de los receptores H2 Cimetidina, ranitidina1, famotidina y nizatidina. Inhiben significativamente la secreción ácida basal y estimulada. Actualmente, se utilizan para el tratamiento de úlceras activas (durante 4 a 6 semanas) en combinación con los antibióticos para la erradicación de Hp. La cimetidina puede tener efectos secundarios antiandrogénicos leves que causan ginecomastia e impotencia, principalmente en pacientes tratados con dosis altas durante periodos prolongados. 2 1 El profesor dijo que nos olvidáramos de la Ranitidina porque tiene bajísima eficacia, por lo que casi no se utiliza ya TEMA 4 Prof. Carballo RECORDAR QUE AL HP HAY QUE DEJARLO K.O!!!! (Muy bueno Cristina!! ;)) 3 Los preparados con bismuto inducen la cicatrización de la úlcera por mecanismos que no están claros. Se ha dicho que la recubren, evitan que prosiga la lesión inducida por la pepsina y el ácido, se unen a la pepsina y estimulan la secreción de prostaglandinas, bicarbonato y moco. Los efectos secundarios a corto plazo son raros, mientras que cuando se utilizan a largo plazo pueden producir neurotoxicidad. Comisionista: Mariló 4-4 CMG MÉDICA DE APARATO DIGESTIVO CMG TEMA 5 SÍNDROME DE MALABSORCIÓN SIGNOS DE SOSPECHA 2) Una fase absortiva de captación mucosa y reesterificación, y Diarrea Pérdida de peso o desnutrición Signos o síntomas de déficits específicos, como la anemia. Aunque son datos que nos pueden orientar en la sospecha, no siempre se presentan con tanta claridad que nos permita hacer un diagnóstico fácil de malabsorción. CONCEPTO En sentido amplio, la malabsorción es la dificultad para incorporar los nutrientes desde el tubo digestivo. Puede deberse a mala digestión intraluminal, a un defecto en la absorción a nivel de la célula intestinal o a dificultades en el transporte a la circulación. La malabsorción puede ser global o parcial, dependiendo de la localización del problema y del tipo de defecto. La global se da cuando no se absorbe ningún tipo de nutrientes, y la parcial cuando no se absorbe un determinado nutriente (ej.: defecto en el tramo donde se absorbe vitamina B12 y sales biliares). CLASIFICACIÓN 3) Una fase postabsortiva que comprende la formación de quilomicrones y su salida desde las células epiteliales intestinales a través de los linfáticos. Dicho esto, lo importante es tener en cuenta que un defecto en cualquiera de estas fases es causa de malabsorción de grasas y que su consecuencia es la esteatorrea (como todos sabemos, presencia de grasas en exceso en las heces). La esteatorrea se asocia a menudo a un déficit de vitaminas liposolubles, lo que puede hacer necesaria una suplementación con preparados hidrosolubles. ¿CUÁLES SON ESOS DEFECTOS? 1) En la fase digestiva. La alteración de la lipólisis se produce por una disminución de la secreción de lipasa, por ejemplo, por una pancreatitis crónica. Y la alteración de la formación de micelas responde a una disminución de ácidos biliares infraduodenales. 2) En la fase absortiva. La captación y reesterificación en la mucosa es alterada por una disfunción en la misma, como la que aparece en el esprue celíaco. MALABSORCIÓN DE GRASAS 3) En la fase postabsortiva. En primer lugar, un repaso al proceso global de digestión y absorción de lípidos. Puede dividirse en tres fases: La formación de quilomicrones se ve comprometida si hay ausencia de lipoproteínas beta (ej.: abetalipoproteinemia); y la salida del intestino, por anormalidades de los linfáticos (ej.: linfangiectasia intestinal). 1) Una fase digestiva que comprende la lipólisis y la formación de micelas, y que precisa de la presencia de lipasa pancreática y de ácidos biliares conjugados en el duodeno; TEMA # Prof. Miras Comisionista: Diego 5-1 CMG MÉDICA DE APARATO DIGESTIVO MALABSORCIÓN DE G LÚCIDOS Los carbohidratos en la dieta están presentes en forma de almidón, disacáridos (sacarosa, lactosa,…) y glucosa. Únicamente son absorbidos en el intestino delgado, y sólo en forma de monosacáridos. Por tanto, antes de su absorción, el almidón y los disacáridos deben ser digeridos por la amilasa pancreática y las disacaridasas del borde en cepillo intestinal a monosacáridos. Entendido esto, una malabsorción de hidratos de carbono puede producirse, pues, por una insuficiencia pancreática, una lesión de la célula intestinal o un déficit de disacaridasas. Las consecuencias son diarrea y malnutrición calórica. MALABSORCIÓN DE PROTEÍNAS Las proteínas están presentes en los alimentos casi exclusivamente como polipéptidos, por lo que es necesario que se sometan a una extensa hidrólisis hasta dipéptidos, tripéptidos y aminoácidos antes de poder absorberlos. La proteólisis ocurre tanto en el estómago como en el intestino delgado; es mediada por la pepsina secretada en forma de pepsinógeno por las células principales gástricas, y por la tripsina, secretada en forma de tripsinógeno y otras peptidasas por las células acinares pancreáticas. Los trastornos de la digestión y absorción de proteínas o aminoácidos son raros en clínica, incluso en presencia de extensa inflamación de la mucosa del intestino delgado. Pueden deberse a insuficiencia pancreática y lesión de la célula intestinal. Las consecuencias son diarrea y malnutrición. CMG mento del tiempo de protrombina), por déficit de vitamina K; enfermedad ósea metabólica, por déficit de vit.D; etc. PRUEBAS PARA DE LA DIAGNÓSTICAS MALABSORCIÓN GRASAS TEST DE VAN K AMER: Grasa cuantitativa en heces. Se da una dieta con un contenido conocido de grasas (70-100g.) y se recogen las heces de 3 días para ver qué cantidad se ha absorbido y qué se ha eliminado. En condiciones normales, se eliminan menos de 6g. en heces al día, es decir, no se absorbe menos del 94% de las grasas ingeridas. GRASA CUALITATIVA EN HECES Tinción de Sudán, Esteatocrito,… OTROS MÉTODOS Test NIRA: (near infrared reflectance analysis), mediante infrarrojos. Prueba del aliento 14C-Trioleína. Prueba del aliento 13C-Triglicéridos: Es una prueba emergente en los últimos años. Se hace ingerir al paciente triglicéridos unidos a Carbono 13, y se mide en el aliento la eliminación de 13C, de forma que obtenemos una curva indirecta de absorción de los triglicéridos. Cuanto mayor sea la curva, mayor será la absorción. MALABSORCIÓN DE VITAMINAS, MINERALES Y O LIGOELEMENTOS Este tipo de malabsorción también puede ser causada por una insuficiencia pancreática y por una lesión de la célula intestinal, global o parcial. Las consecuencias dependerán de la vitamina u oligoelemento que no se absorba: anemia macrocítica, por malabsorción de cobalamina o ácido fólico; trastornos coagulativos (incre- TEMA # Prof. Miras Comisionista: Diego 5-2 CMG MÉDICA DE APARATO DIGESTIVO PRUEBAS PARA DE LA DIAGNÓSTICAS MALABSORCIÓN CARBOHIDRATOS SOBRECRECIMIENTO BACTERIANO Permite valorar la función de la mucosa del intestino delgado proximal. La absorción de la D-xilosa, una pentosa, tiene lugar casi exclusivamente en el intestino delgado proximal, sin requerir la secreción exocrina pancreática. Por tanto, una prueba anormal de la D-xilosa nos permite localizar la causa de la malabsorción. Se realiza mediante la ingesta de 25g de Dxilosa y la cuantificación en suero y orina a las 5 horas. Lo normal es que haya más de 6g en orina y más de 20mg/dl en suero de D-xilosa. PRUEBA DE TOLERANCIA A LA L ACTOSA Sólo se realiza para confirmar un diagnóstico de déficit selectivo de disacaridasas. Se hace ingerir este disacárido y se mide la cantidad de glucosa en suero y en aliento (la lactosa es digerida por la lactasa del borde en cepillo a sus dos monosacáridos constituyentes, glucosa y galactosa). En suero, si la lactosa se absorbe correctamente, obtendremos una curva de glucosa que refleja la presencia de esta en la sangre. Si la lactosa no se absorbe, no habrá glucosa en sangre y la línea será plana. En aliento, ocurre lo contrario. Si obtenemos una curva de aumento en la medición de la glucosa querrá decir que la lactosa no ha sido absorbida y difunde más de lo normal. Si no hay alteraciones de la absorción, esta medición nos dará una línea plana. Esta prueba también puede hacerse para la fructosa y la sucrosa (disacárido artificial presente en chicles y caramelos sin azúcar), aunque son menos frecuentes. Prof. Miras OTRAS PRUEBAS PARA EL DIAGNÓSTICO DE LA MALABSORCIÓN PRUEBA DE LA D-X ILOSA TEMA # CMG Prueba del aliento de Hidrógeno (habitualmente con glucosa y lactulosa). La lactulosa no es absorbible. Por tanto, cuando se metaboliza y aparece hidrógeno en el aliento, es por sobrecrecimiento bacteriano en el colon. La glucosa se absorbe muy bien. Pero un aumento precoz de hidrógeno en aliento después de su ingesta indica una capacidad de metabolización aumentada por sobrecrecimiento bacteriano. El aumento de hidrógeno más tardío en aliento es debido a malabsorción. En cualquier caso, los resultados de esta prueba no deben ser suficientes porque genera muchos falsos positivos y negativos. Prueba del aliento 13 C-D-Xilosa: Se usa menos. TEST DE SCIHLLING Se realiza para determinar la causa de la malabsorción de cobalamina, administrándola marcada y recogiendo la orina de 24 h. Ampliación La cobalamina de la dieta se une en el estómago a la proteína captadora de R, que es sintetizada tanto en el estómago como en las glándulas salivales. Para la absorción de cobalamina es imprescindible la presencia de factor intrínseco, sintetizado y liberado por las células parietales gástricas, que permite su captación por los enterocitos del íleon. Las proteasas pancreáticas rompen el complejo cobalamina-proteína transportadora de R para liberar la cobalamina en el intestino delgado proximal, donde esta se une al factor intrínseco. Comisionista: Diego 5-3 CMG MÉDICA DE APARATO DIGESTIVO Como consecuencia, la absorción de cobalamina puede ser anormal en los siguientes procesos: 1) Anemia perniciosa, por ausencia de secreción de factor intrínseco; 2) pancreatitis crónica, como consecuencia de déficit de proteasas pancreáticas que rompan el complejo cobalamina-proteína captadora de R; 3) Aclorhidria o ausencia de otro factor secretado con el ácido que se encargue de separar la cobalamina de las proteínas alimentarias en que se encuentra; 4) Síndromes de sobrecrecimiento bacteriano, que conlleva déficit de cobalamina por la utilización que de ella hacen las bacterias; 5) Disfunción ileal (como consecuencia de inflamación o resección intestinal previa), debida a un trastorno del proceso de captación de cobalamina-factor intrínseco por las células epiteliales del íleon. PRUEBAS DE INSUFICIENCIA PAN- CMG Ampliación La cápsula endoscópica es un dispositivo de reducidas dimensiones que, a través de la ingestión oral, permite la obtención de imágenes de todo el tubo digestivo durante su recorrido fisiológico a través de éste. El sistema completo consta del dispositivo endoscópico en forma de cápsula, un videograbador que el paciente porta durante el tránsito intestinal de la cápsula y un ordenador externo que procesa las imágenes obtenidas. El enteroscopio de doble balón (EDB) está compuesto por un enteroscopio de calibre fino, que se acopla a un sobretubo especial flexible, ambos con un balón de presión monitorizada en el extremo distal. El sobretubo (ST) y el endoscopio (E) avanzan mediante movimientos alternativos de pulsión y tracción, ayudados por el inflado y desinflado secuencial de sus respectivos balones que permite que el intestino delgado quede replegado por fuera de ST y E. Es una técnica diagnóstica y terapéutica que permite acceder a la totalidad del intestino delgado y realizar toma de biopsias y otras acciones terapéuticas. CREÁTICA Directa: Mide las enzimas pancreáticas que hay en el duodeno directamente desde su interior. Indirecta en sangre y/o orina: En suero, se mide la isoamilasa pancreática y la tripsina inmunorreactiva. Indirecta en heces: Se mide la elastasa fecal o quimiotripsina fecal. Una disminución de estas enzimas indica una lesión pancreática. PRUEBAS RADIOLÓGICAS Las pruebas radiológicas sólo son sugerentes si hay una afectación extensa. En cualquier caso, no son concluyentes. BIOPSIA INTESTINAL La forma más habitual de tomar una muestra intestinal es la endoscopia. Pero en enfermedades más raras donde el diagnóstico se complica se realizan otras pruebas como la cápsula endoscópica y la enteroscopia de doble balón que, si bien muy invasiva, está dando muy buenos resultados. TEMA # Prof. Miras Comisionista: Diego 5-4 CMG MÉDICA DE APARATO DIGESTIVO CMG AMPLIACIÓN ESQUEMA DE LAS CAUSAS MÁS FRECUENTES DE LA MALABSORCIÓN Fibrosis quística (la causa número uno en los Estados Unidos). Pancreatitis crónica. Intolerancia a la lactosa. Enfermedad celíaca. Enfermedad de Whipple. Síndrome de Shwachman-Diamond (una enfermedad genética que afecta al páncreas y la médula ósea). Intolerancia a la proteína de la leche de vaca. Intolerancia a la proteína de la leche de soja. Atresia biliar. Abetalipoproteinemia. Malabsorción de vitamina B-12 debida a: o Infestación por Diphyllobothrium latum o Anemia perniciosa juvenil o Parásitos Giardia lamblia Strongyloides stercoralis Necator americanus (anquilostoma) TEMA # Prof. Miras Comisionista: Diego 5-5 CMG MÉDICA DE APARATO DIGESTIVO ENFERMEDAD CELIACA CMG ANATOMÍA P ATOLÓGICA La celiaquía es una enteropatía autoinmune inducida por la ingestión de gluten, en individuos genéticamente predispuestos. Se caracteriza por una atrofia vellositaria de intensidad variable y predominantemente en el intestino proximal, y por una regresión histológica una vez que se retira el gluten. OJO: Si bien se da en individuos genéticamente predispuestos, NO es una enfermedad hereditaria. Para confirmar el diagnóstico de la enfermedad, YA NO se exige la comprobación de recaída, basta con que las lesiones preatróficas desaparezcan tras retirar el gluten. La lesión característica, aunque no patognomónica, de la celiaquía es la siguiente: Atrofia vellositaria total o subtotal, con localización referentemente proximal. Hipertrofia críptica. Alteraciones epiteliales: enterocitos aplanados y cuboides, a menudo descamados, e hiperlinfocitosis intraepitelial. Hipercelularidad de la lámina propia (linfocitos, cel. plasmáticas, eosinófilos). Según cómo esté de avanzada la lesión, se distinguen 4 tipos histológicos: Enfermedad celíaca: ANATOMÍA PATOLÓGICA ETIOPATOGENIA Factores extrínsecos: alimentos que contienen gluten (harinas de trigo, centeno, avena y cebada). Tipos histológicos Factores intrínsecos o genéticos: HLA II (se ha encontrado DQ2 en el 95% de los celíacos y DQ8 en el 5% restante) y antecedentes familiares de primer grado (mayor prevalencia). La transglutaminasa tisular (TGt) es el autoantígeno más importante en la enfermedad celíaca. Produce determinantes antigénicos (neoepítopos) al reaccionar con la gliadina del trigo en sujetos genéticamente predispuestos (esto es, presentan HLA DQ2 y HLA DQ8). TGt es procesada por las células presentadoras de antígenos (linfocitos B, macrófagos y células dendríticas) y desencadena la activación de los linfocitos T CD+, que son los causantes del daño tisular. EPIDEMIOLOGÍA Como se ha dicho, es más prevalente en personas con predisposición genética y con antecedentes familiares de primer grado. Hay entre 5 y 10 casos no diagnosticados por cada caso diagnosticado. En España se ha pasado de una cifra de 14/100000 a 1/300. Es más frecuente en mujeres respecto a hombres (2:1), y tiene dos picos de incidencia: de 9 meses a 3 años y en la 3ª o 4ª década de la vida. TEMA # Prof. Miras Tipo 0 o Preinfiltrativo Tipo 1 o Infiltrativo Tipo 2 o Hiperplásico Tipo 3 o Destructivo Tipo 4 o Hipoplásico MANIFESTACIONES C LÍNICAS En la infancia suele presentarse con retraso del crecimiento; palidez, apatía, anorexia; atrofia muscular, hipotonía; distensión abdominal; heces blandas, voluminosas y claras; dolor abdominal; vómitos. En el adulto tenemos dos posibles presentaciones: Hay formas establecidas con afectación extensa de la mucosa: esteatorrea, debilidad, astenia, pérdida de peso, intolerancia a la lactosa, lesiones bucales, tetania, lesiones cutáneas y hemorrágicas, anemia… Formas con afectación menos extensa: anemia, osteopenia e infertilidad. Comisionista: Diego 5-6 CMG MÉDICA DE APARATO DIGESTIVO CMG Presencia de Ac + infiltración linfocitaria, pero ausencia de atrofia: se retira el gluten de la dieta y se sigue la evolución del paciente. Si mejora, podremos confirmar la existencia de enfermedad. ENFERMEDADES A SOCIADAS Dermatitis herpetiforme: erupción cutánea pruriginosa y ampollosa en la que existe depósito de IgA anular en la unión dermoepidérmica. Responde a la retirada de gluten. Si no hay anticuerpos, pero hemos hecho una biopsia por los motivos expuestos y encontramos una pequeña infiltración de linfocitos: actuaremos de la misma forma que en el caso anterior, retirando el gluten y siguiendo la evolución. Diabetes Mellitus. Afectación hepática: hepatitis C, cirrosis en vía biliar primaria, colangitis esclerosante primaria. Alveolitis esclerosante primaria. OTROS MÉTODOS DIAGNÓSTICOS Colitis (ulcerosa o microscópica). - Radiología: tránsito intestinal. Sólo es útil en fases tardías de la enfermedad. DIAGNÓSTICO - Videocapsuloendoscopia. Los pacientes con enfermedad celíaca pueden ser asintomáticos (en este sentido, aún no queda demostrada la eficacia de un screening); oligosintomáticos o sintomáticos atípicos, con dispepsia, anemia, etc; y sintomáticos típicos, que cursan con síndrome de malabsorción. - Enteroscopia. - Lesiones malignas: linfoma T, carcinomas epiteliales, adenocarcinomas. Cuando hay síndrome de malabsorción, el diagnóstico no es específico y se basa en observar la evolución de la malabsorción y sus consecuencias. La principal diana de diagnóstico específico son los pacientes con síntomas atípicos, pues el objetivo es evitar su evolución a un síndrome de malabsorción. - Yeyuno-ileítis ulcerativa. - Esprue colágeno. El estudio comienza con la serología. Podemos medir anticuerpos antigliadina (Ig G o Ig A), anticuerpos antiendomisio (Ig A) y anticuerpos antitransglutaminasa (Ig A), pero son éstos últimos los que presentan mayor sensibilidad y especificidad, y por tanto, los que habitualmente se miden. Si no hay Ac, no se requiere seguir con el estudio de la morfología, a excepción de que se trate de un paciente de elevado riesgo y/o tenga síntomas que nos hagan seguir sospechando de la enfermedad. COMPLICACIONES TRATAMIENTO El tratamiento es la exclusión completa y definitiva del gluten de la dieta. Puede tolerarse arroz y maíz. Si no hay respuesta, es probable el incumplimiento. Pero hay casos refractarios: falta de respuesta clínica a los seis meses o falta de respuesta histológica a los 12 meses. En este caso, valorar tratamiento corticoideo o inmunosupresor. Tratamiento de sostén. Si los Ac son positivos, se pasa al estudio de la morfología mediante biopsia intestinal. En ella podemos encontrar atrofia vellositaria y/o infiltración de linfocitos. Presencia de Ac + atrofia: confirmación de enfermedad celíaca. TEMA # Prof. Miras Comisionista: Diego 5-7 CMG MÉDICA DE APARATO DIGESTIVO CMG TEMA 5 (II) SD DE MALABSORCIÓN HIPOGAMMAGLOBULINEMIAS EN EL ADULTO Las células T presentan alteraciones fenotípicas y funcionales de la subpoblación CD4+, probablemente secundarias a las de los linfocitos B. CONCEPTO Clínicamente los pacientes presentan infecciones bacterianas de repetición de inicio precoz (en los primeros 6-9 meses de vida), que en muchas ocasiones determinan el ingreso hospitalario por su gravedad; el curso de la enfermedad puede complicarse seriamente debido a la infección por Mycoplasma, cuya manifestación más grave es la artritis, que es dolorosa, de difícil resolución y de la que se han conseguido algunas remisiones parciales con dosis altas de gammaglobulina. Consiste en un déficit selectivo de Ig A. Es relativamente frecuente, de 1/500 a 1/1000 individuos afectados. Generalmente es silente y la histología intestinal suele ser normal. AGAMMAGLOBULINEMIA INFANTIL LIGADA AL CROMOSOMA X Su herencia está ligada al cromosoma X, afecta por lo tanto a varones; las mujeres de la familia pueden ser portadoras sanas de la enfermedad y suelen existir antecedentes familiares de procesos infecciosos de repetición y muertes prematuras. Los niveles de IgG son siempre bajos, inferiores a 250 mg/dL, y los de IgA e IgM, ausentes o muy disminuidos. La respuesta de anticuerpos es nula y los linfocitos B maduros (CD19, CD20) periféricos están ausentes, rasgo característico de la enfermedad. Por el contrario, en la médula ósea1 se observa un número normal de linfocitos pre-B, lo cual indica que el defecto radica en un bloqueo selectivo del desarrollo de los linfocitos B, ya que las demás poblaciones linfocitarias son normales. El gen causante de la enfermedad, pertenece a la familia de los protooncogenes src y codifica una tirosincinasa expresada en células B. 1 Las infecciones del aparato digestivo son frecuentes y habitualmente están causadas por G. lamblia, Campylobacter, Yersinia y Cryptosporidium. La respuesta a antibióticos es muy pobre. El diagnóstico precoz y el tratamiento con dosis elevadas de gammaglobulina intravenosa han mejorado mucho la calidad y expectativa de vida de estos pacientes, que han empezado a superar la segunda y la tercera décadas de la vida. INMUNODEFICIENCIA COMÚN VARIABLE La inmunodeficiencia común variable, conocida también como hipogammaglobulinemia adquirida, es un síndrome heterogéneo caracterizado por niveles bajos de inmunoglobulinas del suero (anticuerpos) y una mayor susceptibilidad a infecciones. Prevalece en la tercera y cuarta década de la vida y su incidencia es similar en ambos sexos. Por lo general, la suma de IgA, IgG e IgM es inferior a 500 mg/dL. Tienen mayor riesgo de presentar linfomas y leucemias. TEMA 5 (II) Dr. Carballo 5(II)-1 CMG MÉDICA DE APARATO DIGESTIVO CMG Existe una pobre o nula respuesta de los anticuerpos frente a las infecciones bacterianas de repetición que se producen, especialmente respiratorias y digestivas. Se observa de forma predominante en varones de raza blanca, entre la cuarta y la séptima décadas de la vida. La presencia de autoanticuerpos sin traducción clínica es frecuente, así como una mayor incidencia de procesos neoplásicos, en especial, carcinoma gástrico y linfomas. HISTOLOGÍA Algunos pacientes presentan trastornos gastrointestinales tales como dolor abdominal, indigestión, náuseas, vómito, diarrea y pérdida de peso. Una evaluación cuidadosa de los órganos digestivos puede revelar una pobre absorción de grasas y ciertos azúcares. Si se obtiene una biopsia de mucosa intestinal, se podrán observar atrofia vellositaria intestinal. Estos cambios son de gran ayuda para diagnosticar y tratar el problema. La histología demuestra un infiltrado de la lámina propia del intestino delgado por macrófagos, que se colorean de rojo con la técnica del PAS. Estos gránulos PAS-positivos son glicoproteínas de los restos de la pared celular de los bacilos fagocitados por los macrófagos. Estos macrófagos, con gránulos PAS-positivos en su interior, se observan en la mayoría de los tejidos afectos, como corazón, hígado, bazo, pulmones, riñones, ganglios linfáticos y SNC. En algunos pacientes con problemas digestivos, se ha identificado Giardia lamblia, en las biopsias y muestras fecales. La erradicación de estos parásitos por medio de medicamentos puede eliminar los síntomas gastrointestinales. Puede existir también, Desde el punto de vista anatomopatológico se observan unas paredes intestinales edematosas y engrosadas. Los vasos linfáticos de la mucosa y de la submucosa están dilatados y traducen la obstrucción linfática causada por los nódulos linfáticos mesentéricos engrosados. El tratamiento consiste en: 1. Antibióticos (entre otros, el metronidazol). 2. Gammaglobulina intravenosa. 3. Dieta sin gluten. 4. Corticoides. ENFERMEDAD DE WHIPLLE CONCEPTO Es una rara enfermedad sistémica de origen infeccioso, que puede involucrar a cualquier órgano del cuerpo, pero que afecta fundamentalmente al intestino delgado (cursando con malabsorción), al sistema nervioso central y a las articulaciones. CLÍNICA 1. Síntomas generales: El agente causal es la bacteria Tropheryma whippelii, perteneciente al grupo de las Actinobacterias2. 2 Las actinobacterias son bacterias Gram +, no esporuladas, anaerobias, con distribución amplia, que forman parte de la flora habitual de los aparatos respiratorio, digestivo y genital femenino. TEMA 5 (II) Dr. Carballo a. Astenia. b. Anorexia. c. Fiebre: en ocasiones, la fiebre es el único síntoma durante cierto tiempo, por lo que debe de hacerse un diagnóstico diferencial de la fiebre de origen desconocido. d. Adelgazamiento. 5(II)-2 CMG MÉDICA DE APARATO DIGESTIVO 2. Afectación articular: alrededor de dos tercios de los pacientes presentan síntomas articulares. La artritis no es deformante, suele ser migratoria, y afecta al mismo tiempo articulaciones pequeñas y grandes, es especial de miembros inferiores y se acompaña de signos inflamatorios. 3. Diarrea con esteatorrea (80%): es el síntoma más frecuente. Suele acompañarse de dolor cólico abdominal. 4. Afectación: a. Neurológica: destacan las parestesias debidas a neuropatía periférica y la tetania causada por hipocalcemia o hipomagnesemia. CMG DIAGNÓSTICO 1. ANÁLISIS DE LABORATORIO Está relacionado con la malabsorción, hay: a. Hipolipemia. b. Hipocalcemia. c. Hipoproteinemia. d. Hipoprotrombinemia. e. Anemia ferropénica y, en ocasiones, macrocítica. f. Esteatorrea. g. Absorción de la D-xilosa está disminuida. b. Pulmonar. 2. RADIOLOGÍA c. Ocular. d. Hematológica: aparece la púrpura secundaria a alteraciones de la coagulación e. Cardiaca. EXPLORACIÓN FÍSICA Revela los hallazgos relacionados con malabsorción pronunciada: 1. Disminución de peso. 2. Hipotensión. 3. Linfadenopatías. 4. Hiperpigmentación cutánea. 5. Fiebre. 6. Edemas periféricos. 7. Glositis. 8. Sensibilidad abdominal incrementada. 9. Abdomen distendido y sensible a la palpación. TRATAMIENTO Penicilina I.V (intravenosa) + Estreptomicina I.V durante 10-14 días, seguido de Trimetropin – Sufametoxazol oral durante 1 año. 10. Pueden palparse masas abdominales mal definidas debidas a linfadenopatías mesentéricas. 11. Ascitis. 12. Esplenomegalia. TEMA 5 (II) Se realiza una endoscopia en la que se ve la mucosa tapizada con un punteado fino, irregular y concluyente, de color blanco amarillento. La confirmación de esta enfermedad se realiza mediante biopsia intestinal, en el que comprobamos la presencia de gránulos PASpositivos en los macrófagos de la lámina propia de la mucosa intestinal. Idénticas lesiones pueden encontrarse en cualquiera de los tejidos afectados. Los bacilos intracelulares pueden demostrarse mediante microscopia electrónica. El tratamiento utilizado hasta la actualidad proporciona una rápida mejoría clínica, lográndose la curación al cabo de 1-3 meses de iniciado. En todos los casos se obtiene la pronta desaparición de los síntomas digestivos y extradigestivos en esta enfermedad, que era siempre mortal antes de descubrirse la eficacia de los antibióticos. Una vez interrumpido el tratamiento los pacientes deben controlarse periódicamente, pues puede haber recidivas y es posible que aparezcan síntomas neurológicos aún en ausencia de enfermedad intestinal o articular. Dr. Carballo 5(II)-3 CMG MÉDICA DE APARATO DIGESTIVO GASTROENTERITIS EOSINOFÍLICA b. c. d. e. f. g. h. CONCEPTO CMG Vómitos. Diarreas. Pérdida de peso. Dolor abdominal cólico. Síndrome de malabsorción. Anemia. Enteropatía perdedora de proteínas. Es una enfermedad rara, caracterizada por la infiltración por leucocitos eosinófilos maduros de la mucosa del tracto digestivo, fundamentalmente de estómago e intestino delgado, asociada por lo general a hipereosinofilia sanguínea periférica. 2. La afectación de la muscular se manifiesta como: a. Estenosis pilórica intermitente. b. Obstrucción intestinal incompleta. FISIOPATOLOGÍA 3. Aunque la patogenía es desconocida, se piensa que puede tener alguna relación con reacciones alérgicas alimentarias, a parásitos o a fármacos, puesto que se observa: La afectación predominantemente serosa puede presentarse como una ascitis exudativa con eosinofilia. AMPLIACIÓN: 1. Asociación con síntomas alérgicos sistémicos. DIAGNÓSTICO: 1. En la forma de predominio mucoso el diagnóstico se establece mediante biopsia de la zona afecta. En estos casos el diagnóstico diferencial debe hacerse con: a. Infecciones parasitarias. b. Linfangiectasia intestinal. c. Enfermedad de Crohn. d. Síndrome de hipereosinofilia. e. Enfermedades del tejido conjuntivo. 2. La forma de predominio muscular se diagnostica radiológicamente con contraste baritado, al poner de manifiesto la zona de estenosis. El diagnóstico histológico suele efectuarse con una biopsia quirúrgica de toda la pared del intestino. El diagnóstico diferencial se plantea, sobre todo, con la enfermedad de Crohn. 3. El diagnóstico de la forma serosa se realiza por el hallazgo de proteínas elevadas y eosinofilia importante (12-95% de eosinófilos) en el líquido ascítico. En la laparotomía se aprecia una serosa engrosada con infiltración eosinófila subserosa y ganglios mesentéricos con eosinófilos. El diagnóstico diferencial se plantea principalmente con el linfoma y la panarteritis nudosa. 2. Concentraciones elevadas de IgE. 3. Respuesta al tratamiento con glucocorticoides. FORMAS ANATOMOPATOLÓGICAS Y CLÍNICA Se han descrito tres formas anatomopatológicas, según la localización preferente del infiltrado: 1. La forma más común es la localización mucosa (60%). 2. Con menor frecuencia, el infiltrado eosinófilo se localiza en la capa muscular (30%), ocasionando engrosamiento del intestino. 3. La forma más rara de presentación (10%) es la infiltración eosinófila de la serosa. Sea cual fuere la forma de presentación, en el 40-50% de los casos los pacientes refieren una historia de alergia o intolerancia alimentaria, aunque no hay que descartar la alergia a parásitos o a fármacos. Sin embargo, la sintomatología depende de la forma de afectación predominante, así tenemos: 1. La forma mucosa se caracteriza por la aparición de: a. Náuseas. TEMA 5 (II) PRONÓSTICO Y TRATAMIENTO El pronóstico es generalmente favorable, con tendencia a las remisiones, pero la enfermedad es crónica y de larga duración. La mortalidad es rara y suele ser consecuencia de las complicaciones. Dr. Carballo 5(II)-4 CMG MÉDICA DE APARATO DIGESTIVO En cuanto al tratamiento: - La eliminación de determinados antígenos de la dieta sólo consigue una mejoría transitoria de los síntomas. - Si hay sospecha de que la enfermedad está producida por un parásito, hay que dar el correspondiente tratamiento antiparasitario: Mebendazol 100 mg, dos veces al día durante 3 días. - También se pueden dar corticoides y azatioprina. LINFANGIECTASIA INTESTINAL 5. Edemas, en general miembros inferiores. 6. En fases avanzadas: a. Anasarca.3 b. Ascitis quilosa en la mitad de los casos. En estadios posteriores las adherencias creadas por las ascitis pueden provocar una oclusión intestinal. 2. Cualquier entidad que determine una oclusión de los linfáticos intestinales o una hipertensión de la vía principal de drenaje linfático (conducto torácico) o una dificultad crónica para el retorno venoso (Insuficiencia Cardiaca Congestiva) puede causar una linfangiectasia intestinal adquirida o secundaria, como TBC intestinal, linfoma abdominal, enfermedad de Crohn, pancreatitis crónica y cáncer pancreático, pericarditis constrictiva, fibrosis o tumores retroperitoneales y sarcoidosis. en ANÁLISIS DE LABORATORIO: 1. Moderada esteatorrea, aunque en ocasiones puede ser muy intensa. 2. El hallazgo característico es la hipoproteinemia con niveles bajos de albúmina, debido a un aumento de pérdidas proteicas intestinales, secundario a hiperpresión en el interior de los vasos linfáticos intestinales que provoca su disrupción y el paso de su contenido a la luz intestinal. 3. Niveles séricos disminuidos de: - Inmunoglobulinas. - Transferían. - Ceruloplasmina. - Calcio. - Vitamina B12. 4. Linfocitopenia. Existen dos tipos de linfangiectasia intestinal: La linfangiectasia intestinal primaria, es una enfermedad de tipo genético caracterizada por producir malabsorción de grasa y pérdida intestinal de proteínas, debido a un bloqueo del sistema linfático intestinal por dilatación. El cuadro clínico aparece inmediatamente después del nacimiento y existe con frecuencia linfedema periférico y el antecedente familiar de diarreas e hipoproteinemia. asimétricos, AMPLIACIÓN: CONCEPTO 1. CMG RADIOLOGÍA: La radiología del intestino delgado muestra un engrosamiento de los pliegues, por el edema existente, y signos de dilución de la papilla, dada la hipersecreción en la luz intestinal. Puede observarse dilatación de las asas y nodularidad discreta. La linfografía pone de manifiesto estasis, distorsión y reflujo en los linfáticos abdominales. DIAGNÓSTICO Se llega a través de un diagnóstico de sospecha, al excluir otras causas de enteropatía exudativa y tras la realización de una biopsia intestinal, que muestra dilatación de los vasos linfáticos de la lámina propia con macrófagos cargados de lípidos. CLÍNICA Se manifiesta con: 1. 2. 3. 4. Diarrea esteatorreica. Debilidad. Náuseas. Malestar abdominal. TEMA 5 (II) 3 Edema generalizado que se caracteriza por una excesiva colección líquida en el espacio extravascular (intersticial). Dr. Carballo 5(II)-5 CMG MÉDICA DE APARATO DIGESTIVO TRATAMIENTO No es específico, sino que se limita a una dieta pobre en lípidos, con suplementos de triglicéridos de cadena media y aporte de vitaminas liposolubles. Cuando la linfangiectasia es secundaria a una enfermedad inflamatoria o neoplásica, su tratamiento específico puede restablecer el flujo linfático y limitar la pérdida intestinal de proteínas. SOBRECRECIMIENTO BACTERIANO medio alcalino intestinal y ya no pueden participar de forma eficaz en la solubilización micelar de los lípidos. Además, las bacterias fijan el complejo vitamina B12-factor intrínseco, impidiendo así la absorción de vitamina B124. La flora anormal del intestino delgado puede también metabolizar los azúcares de la dieta (y por tanto, la D-xilosa). De este modo, el sobrecrecimiento bacteriano intestinal puede determinar esteatorrea, por lo general leve, malabsorción acentuada de vitamina B12 y una prueba de la D-xilosa en el límite de la normalidad o definitivamente anormal. AMPLIACIÓN: CLÍNICA: PATOGENÍA La flora del intestino delgado, en condiciones normales, es la siguiente: 1. Intestino delgado proximal: a. Es estéril en 1/3 de las personas. b. Los 2/3 restantes de personas, presentan una baja concentración de lactobacilos, enterococos y anaerobios Gram +. 2. Intestino delgado distal: zona de transición entre ileon (baja concentración de flora microbiana) y colon (alta concentración de flora microbiana). Los factores que condicionan el sobrecrecimiento bacteriano son: 1. Secreción ácida gástrica deficitaria. 2. Secreción de inmunoglobulinas. 3. Peristaltismo: cualquier anomalía estructural o funcional del intestino delgado que determina estasis local o recirculación del contenido intestinal puede acompañarse de una proliferación intraluminal de microrganismos, con flora variada similar a la colónica en la que predominan las bacterias y lactobacilos anaerobios. La patogenía de la malabsorción está relacionada con las propiedades metabólicas de la flora bacteriana. Los gérmenes son capaces de desconjugar y deshidroxilar los ácidos biliares; los ácidos biliares libres precipitan en el TEMA 5 (II) CMG Las manifestaciones clínicas varían considerablemente y dependen, en parte, de la enfermedad de base que causa el sobrecrecimiento bacteriano (como la enfermedad de Crohn, enfermedades de la pared intestinal, diverticulosis yeyunal, lesiones postquirúrgicas, entre otras). Sea cual fuere la causa de sobrecrecimiento bacteriano, las consecuencias para el paciente son las mismas: 1. Diarrea con esteatorrea. 2. Pérdida de peso. 3. Anemia megaloblástica, es secundaria a la deficiencia de vitamina B12 y puede cursar con síntomas neurológicos. 4. Puede observarse osteomalacia, deficiencia de vitamina K e incluso tetania hipocalcémica. 5. Hipoproteinemia, es una manifestación frecuente, y a veces, suficientemente importante para causar edemas. Su etiología es multifactorial, pudiendo resultar de la hipocaptación de aminoácidos por la mucosa intestinal alterada secundariamente, la digestión intraluminal de péptidos por las bacterias o una enteropatía perdedora de proteínas. DIAGNÓSTICO Esta entidad debe considerarse en el diagnóstico diferencial del paciente que presenta diarrea, esteatorrea, pérdida de peso o anemia macrocítica, en particular si se trata de un anciano o existen antecedentes de cirugía abdominal. 4 Los niveles séricos de vitamina B12 están por debajo de lo normal, mientras que los de ácido fólico tienden a estar elevados. Dr. Carballo 5(II)-6 CMG 1. 2. 3. MÉDICA DE APARATO DIGESTIVO Métodos directos: a. El cultivo de aspirado yeyunal es diagnóstico si revela concentraciones de bacterias superiores a 105/mL; sin embargo, la técnica para el cultivo intestinal debe ser muy cuidadosa y es poco factible en la práctica habitual. Aunque se utilice poco, la detección de ácidos biliares libres en el jugo yeyunal tiene un gran valor diagnóstico. Métodos indirectos: las exploraciones más comúnmente utilizadas para confirmar el diagnóstico son: a. Prueba del aliento de D-xilosa5 o lactulosa C14. b. Prueba de aliento de hidrógeno con D-xilosa, lactulosa o glucosa. Empírico con tratamiento antibiótico. TRATAMIENTO El objetivo del tratamiento es la reducción o la eliminación de la flora bacteriana del intestino delgado y consiste en la administración oral de antibióticos o, si es posible, la corrección quirúrgica de la anomalía responsable de la estasis intestinal. El antibiótico principal que se utiliza es: Amoxicilina clavulánico, 875 mg / 12 h, durante 10 días. También hay otras alternativas como las cefalosporinas, metronidazol, tetraciclinas o ciprofloxacino. El soporte nutricional es importante y puede ser necesario a pesar de la eficacia de los antibióticos. La supresión de la lactosa de la dieta, los triglicéridos de cadena media y la administración parenteral de vitamina B12 y otros nutrientes son medidas útiles. CMG SÍNDROME DE INTESTINO CORTO CONCEPTO El intestino delgado posee una extensa superficie de absorción, muy superior a la necesaria para mantener una nutrición suficiente. Sin embargo, cuando se realizan resecciones quirúrgicas extensas o existen lesiones inflamatorias amplias se reduce de forma notable la superficie absortiva y puede aparecer un cuadro clínico de malabsorción, cuya gravedad dependerá de dos factores: 1. La longitud de la resección (mayor del 80%). 2. La zona anatómica resecada. Ésta es importante si se tiene en cuenta la especialización funcional de los distintos segmentos intestinales (absorción de hierro, calcio y folatos en el duodeno y el yeyuno proximal, y de ácidos biliares y vitamina B12 en el íleon). ETIOLOGÍA 1. Enfermedad de Crohn. 2. Enfermedades que comprometen la vascularización mesentérica, como: infarto intestinal, trombosis venosa mesentérica, isquemia mesentérica no oclusiva, trombosis y embolias de la arteria mesentérica superior, entre otras. 3. Cáncer. 4. Enteritis post radiación. 5. Vólvulos y hernias estranguladas del intestino delgado. ETIOPATOGENÍA Y CLÍNICA: Mayor gravedad de la enfermedad si: 1. La diarrea y las consecuencias metabólicas de las resecciones ileales amplias entrañan mayor gravedad que las resecciones yeyunales. 2. Se agravan cuando además se extirpan la válvula ileocecal y el colon derecho, pues éste pierde la capacidad absortiva, 5 Prueba del aliento con xilosa-14C: la aparición de 14CO2 en el aire espirado dentro de los primeros 60 min. de la ingesta de 1 g de xilosa-14C, es diagnóstica de sobrecrecimiento bacteriano. TEMA 5 (II) Dr. Carballo 5(II)-7 CMG MÉDICA DE APARATO DIGESTIVO disminuye el tiempo de tránsito intestinal y puede aparecer sobrecrecimiento bacteriano en el intestino delgado restante. Cuando se efectúa una resección ileal se rompe el círculo enterohepático de los ácidos biliares. Se pueden dar 2 situaciones: 1. 2. Si la resección ileal es pequeña (menos de 100 cm), la síntesis hepática de ácidos biliares experimenta un aumento compensatorio de las pérdidas fecales excesivas, que permite una concentración y una absorción prácticamente normales de los lípidos. Como contrapartida, en el colon se alcanza una elevada concentración de ácidos biliares, cuyas propiedades catárticas determinan una diarrea coleriforme, que puede eliminarse con colestiramina. Si la resección ileal es superior a 100 cm, existe malabsorción de ácidos biliares y el aumento de la síntesis hepática no es suficiente para compensar las pérdidas fecales, por lo que disminuyen sus niveles intraluminales y se compromete la solubilización micelar de las grasas. En estas condiciones predomina la esteatorrea y no es tan importante el efecto catártico de los ácidos biliares sobre el colon por estar diluidos en un gran volumen intraluminal de bajo pH. Por otra parte, la malabsorción de grasa determina que los ácidos grasos que entran en el colon sean hidroxilados por la flora bacteriana, con lo cual tiene el efecto de inhibir la absorción y estimular la secreción colónica, con el consiguiente empeoramiento de la esteatorrea. Por lo explicado, deducimos que la clínica que presenta es de malabsorción. parados predigeridos de nutrición entera, triglicéridos de cadena media, abundante aporte vitamínico y mineral y/o nutrición parenteral total prolongada, y a veces permanente, en régimen ambulatorio. 3. La diarrea coleriforme responde uniformemente a la administración oral de colestiramina, 4 g con las comidas. 4. Triglicéridos de cadena media. 5. Transplante intestinal (en estudio). ABETALIPOPROTEINEMIA CONCEPTO Es una rara afección que se hereda con carácter autosómico recesivo, caracterizada por la incapacidad para sintetizar la apoproteína B, necesaria para la formación de los quilomicrones (lipoproteínas ricas en triglicéridos), por parte de intestino delgado e hígado. CLÍNICA 1. Malabsorción exclusiva de grasa con estatorrea (que generalmente es leve), esto es debido a que los triglicéridos se acumulan en las células epiteliales intestinales y, al no formarse quilomicrones, pasan con dificultad a la linfa. A pesar de un ayuno prolongado, la biopsia intestinal revela grandes cantidades de grasa intraepitelial, como si se acabara de ingerir una comida rica en triglicéridos. 2. Existe una acusada hipolipemia, con cifras de colesterol inferiores a 100 mg/dL y de triglicéridos por debajo de 30 mg/dL. 3. El suero de estos pacientes carece de: a. Quilomicrones. b. Lipoproteínas de baja densidad (LDL). c. Lipoproteínas de muy baja densidad (VLDL). d. No pueden detectarse apoproteínas B. 4. La dislipemia determina alteraciones de la función de las membranas celulares, que se manifiestan con síntomas neuromusculares secundarios a la desmielinización TRATAMIENTO 1. 2. Los pacientes que han sido sometidos a bypass del intestino delgado para tratamiento de la obesidad o han sufrido anastomosis quirúrgicas defectuosas son candidatos a la cirugía con el objetivo de restablecer la integridad del tubo digestivo. En las resecciones intestinales amplias e irreversibles es básico el soporte nutricional que comprende dietas selectivas, pre- TEMA 5 (II) CMG Dr. Carballo 5(II)-8 CMG MÉDICA DE APARATO DIGESTIVO CMG selectiva, en particular de los haces posteriores de la médula espinal. Esto causa pérdida de los reflejos osteotendinosos y ataxia sensorial cerebelosa. cosagalactosa, o ser secundaria a una enfermedad intestinal, la cual puede motivar disfunción de más de una disacaridasa, además del mecanismo de transporte. 5. Retinitis pigmentaria atípica, por la disfunción de las membranas oculares, con poca pérdida visual. 6. La alteración de la membrana lipídica de los hematíes conduce a acantocitosis y anemia hemolítica por destrucción prematura de los hematíes. La enfermedad se manifiesta dentro del primer año de vida en un niño nacido presuntamente sano, pero que no se desarrolla con normalidad y presenta: anorexia, diarrea y distensión abdominal. Los hidratos de carbono no absorbidos se acumulan en la luz intestinal y determinan la secreción de agua y electrólitos por un mecanismo osmótico, lo que causa distensión de asas y dolor cólico asociado a borborigmos. El tránsito se acelera y el contenido intestinal pasa rápidamente al colon, donde los hidratos de carbono son fermentados por las bacterias con producción de ácidos grasos de cadena corta, que disminuyen el pH, y gases (H2, CO2 y metano). El bajo pH intracolónico altera la absorción de agua y electrólitos, y esta secuencia de acontecimientos se traduce en diarrea hídrica y flatulencia. DIAGNÓSTICO INTOLERANCIA A LA LACTOSA Durante la niñez aparecen alteraciones neurológicas, y en la adolescencia, retinitis pigmentaria atípica. Es la causa más frecuente de malabsorción de los hidratos de carbono. Puede ser: 1. Primaria: a. Congénita: manifestándose en el recién nacido. b. Adquirida: se presenta en el adulto por un mecanismo hereditario. 2. Secundaria: debido a enteropatías. Las pruebas de laboratorio demuestran malabsorción selectiva de las grasas (la prueba de la D-xilosa es normal) e hipolipemia. El diagnóstico se confirma mediante la biopsia intestinal, que muestra unas vellosidades normales con enterocitos repletos de gotas de grasa, la cual está ausente de la lámina propia de la submucosa. TRATAMIENTO El tratamiento es empírico, con una dieta de bajo contenido graso y suplementos de triglicéridos de cadena media y vitaminas liposolubles como la vitamina E (que sirve para el control de la retinitis y la ataxia). Existen importantes diferencias raciales en cuanto a su incidencia (en adultos de raza blanca es alrededor del 5%, mientras que la raza negra, bantúes y razas orientales es del 60 al 90%). CLÍNICA: 1. Distensión abdominal. 2. Borborigmos6. 3. Retortijones. 4. Diarrea osmótica acuosa que aparece de 30 minutos a 2 horas después de la ingesta de leche o derivados. INTOLERANCIA A HIDRATOS DE CARBONO La malabsorción o intolerancia a los hidratos de carbono puede deberse a una anomalía constitucional, que altera una disacaridasa específica o el mecanismo de transporte glu- TEMA 5 (II) 6 Ruidos intestinales. Dr. Carballo 5(II)-9 CMG MÉDICA DE APARATO DIGESTIVO CMG DIAGNÓSTICO: Estos síntomas no se deben a fenómenos alérgicos producidos por las proteínas de la leche, lo cual se demuestra: 1. Prueba de lactosa en sangre: administrando al paciente 0,75-1,5 g de lactosa/kg de peso corporal y practicando una prueba de tolerancia con determinación de las concentraciones de glucosa en sangre. Normalmente deben aumentar más de 20 mg/100 mL (un 20%) respecto a los valores de la glucemia en ayunas. Los valores inferiores sugieren la existencia de una deficiencia de lactasa. 2. Prueba del aliento de H2 tras la ingesta de lactosa. 3. Provocación de los síntomas tras dieta. 4. En la práctica, por lo general es suficiente con comprobar el efecto sobre los síntomas de la eliminación de leche y derivados. TRATAMIENTO: Consiste en excluir de la dieta la leche y los productos lácteos con excepción del yogur, que es una fuente de lactosa autodigerida. Los pacientes con deficiencia de lactasa secundaria a otras enteropatías (por ejemplo en la enfermedad celíaca) pueden reintroducir la leche en la dieta una vez resuelta la enfermedad primaria. TEMA 5 (II) Dr. Carballo 5(II)-10 CMG MÉDICA DE APARATO DIGESTIVO CMG TEMA 6 DIARREAS AGUDAS Y CRÓNICAS INTRODUCCIÓN CLASIFICACIÓN La diarrea afecta a todas las edades y etnias, constituyendo una causa importante de morbilidad y mortalidad mundial. En España, la diarrea es el síntoma más frecuente que se consulta en Atención Primaria, hasta un 20% de las personas que acuden al médico lo hacen por esta patología. 1. Desde un punto de vista cronológico, es decir, en función del tiempo que dure la diarrea. Es la clasificación más importante ya que nos orientará hacia la etiología de la enfermedad, puesto que no será la misma causa la que produzca una diarrea de 7 días de evolución que otra de 30 días. Las podemos dividir en: En este tema analizaremos el concepto y clasificación de diarrea, los aspectos clínicos más relevantes en la evaluación del paciente con diarrea, las pruebas diagnósticas disponibles y los principios generales del tratamiento de las diarreas. DIARREAS Aguda: evolución de menos de 14 días o 2 semanas. b. Persistente: dura entre 14 y 30 días. c. Crónica: dura más de 30 días o 4 semanas. 2. Desde un punto de vista clínico: se fundamenta en las características de las heces: CONCEPTO La diarrea es un signo que revela una alteración fisiopatológica de una o varias funciones del intestino (secreción, digestión, absorción o motilidad) y que, en último término, indica un trastorno del transporte intestinal de agua y electrólitos. La diarrea consiste en una disminución de la consistencia de las heces con más de 3 deposiciones/día. Consideramos normal: Hasta 3 deposiciones/día de consistencia normal. Hasta 1 deposición cada 3 días. Cuando se superan estos límites habrá: a. Diarrea: si hay más de 3 deposiciones/día con disminución de la consistencia de las heces. b. Estreñimiento: si el periodo de tiempo entre dos deposiciones es superior a 3 días. TEMA 6 a. a. Acuosa: son heces líquidas sin productos patológicos. b. Inflamatoria: cuando la diarrea se acompaña de moco o sangre. Este punto de vista nos da una aproximación subjetiva, puesto que depende de cómo pregunte el médico, las preguntas que realice al paciente y de las características del mismo. Por ejemplo: puede ocurrir que un anciano que no vea bien, refiera tener heces negras (melenas) cuando en realidad son normales; o bien, que tenga heces negras porque toma hierro, pero el médico no le pregunte sobre eso y lo asocie a hemorragia digestiva. Otras veces, puede pasar que el paciente diga que tiene hebras de sangre en heces debido a su alimentación (porque ha comido tomate o pimiento) y nosotros podemos pensar que tiene una diarrea inflamatoria y que hay que dar antibióticos. Dr. Pons 1 CMG 2. MÉDICA DE APARATO DIGESTIVO Desde un punto de vista fisiopatológico (se aleja más de lo clínico), se clasifican en: a. Osmótica: producida por un aumento de sustancias hiperosmolares de las heces que arrastran agua a la luz intestinal. Ocurre en la enfermedad celiaca, laxantes osmóticos con magnesio y en el SD de malabsorción. 4. AMPLIACIÓN: Las CAUSAS más frecuentes de diarrea osmótica son: a. b. c. La ingestión excesiva de hidratos de carbono poco absorbibles (tratamiento con lactulosa, ingesta de fructosa, sorbitol o manitol utilizados como sustitutos del azúcar en alimentos de régimen). La ingestión de antiácidos o laxantes ricos en sulfato de magnesio o laxantes que contienen aniones poco absorbibles (sulfato sódico o fosfato sódico). La malabsorción de hidratos de carbono (síndrome de malabsorción, déficit de dis acaridasas, malabsorción congénita de fructosa y glucosa-galactosa). 5. 6. CMG de receptores en la membrana de las células epiteliales. Otros mediadores celulares, como neurotensina, serotonina, sustancia P y secretina, ejercen un efecto paracrino (la hormona actúa sobre células vecinas) estimulando receptores de membrana que aumentan la secreción de líquidos y electrólitos. Finalmente, mediadores celulares, como prostaglandinas, histamina, factor activador de las plaquetas, bradicinina e interleucina 1, actúan estimulando directamente la secreción de células epiteliales o, de forma secundaria, estimulando neuronas sensoriales y motoras del intestino. Sustancias que actúan desde la luz intestinal, como enterotoxinas bacterianas, laxantes y ácidos grasos o sales biliares mal absorbidos. Adenoma velloso gigante (> 4 cm.) localizado en la región rectosigmoidea. La elevada secreción del tumor y la escasa superficie de mucosa disponible distalmente para reabsorber líquidos y electrólitos hacen que el volumen de las heces alcance hasta 3.000mL al día en algunos pacientes. En estos casos se asocian deshidratación, hiponatremia, hipocloremia, hipopotas emia y acidosis metabólica. Enfermedades hereditarias raras. Desde el PUNTO DE VISTA CLÍNICO la diarrea osmótica se caracteriza por: Las principales CARACTERÍSTICAS CLÍNIC AS de la diarrea secretora son: a) Las heces suelen ser voluminosas (> 1 L/día). a) b) b) c) d) Cesar con el ayuno o cuando se suspende la ingesta del soluto no absorbible. El volumen de las heces es generalmente inferior a 1 L/día. El pH fecal suele ser bajo debido a la fermentación bacteriana de los hidratos de carbono no absorbidos. c. Existe tendencia a la hipernatremia. b. Secretora: sin que haya sustancias hiperosmolares hay un aumento de la secreción de agua y electrolitos. Está provocada por bacterias enterotoxígenas. AMPLIACIÓN: Las principales CAUSAS de diarrea secretora son: 1. Enfermedad intestinal difusa con destrucción importante de células epiteliales (por ejemplo: enferm edad celíaca, enfermedad inflamatoria crónica intestinal, gastroenteritis eosinófila, etc.). 2. Resección intestinal. La diarrea que sigue a la resección del íleon terminal o hem icolectomía derecha es multifactorial. 3. Factores humorales con efecto endocrino o paracrino. Tumores productores de hormonas circulantes (serotonina, polipéptido intes tinal vasoactivo, calcitonina) pueden inducir diarrea mediante el estímulo TEMA 6 La diarrea persiste tras un ayuno de 48-72 h. Aunque la mayoría de las diarreas secretoras cumplen estos criterios, existen algunas excepciones. Ciertas diarreas secretoras ceden con el ayuno. Ello s ucede en la diarrea por malabsorción de ácidos grasos o por ingesta subrepticia de laxantes. Grasa: esteatorrea, producida en los SD de malabsorción. d. Inflamatoria: debida a infecciones enteroinvasivas o por procesos inflamatorios intestinales como la colitis ulcerosa y la enfermedad de Crohn. AMPLIACIÓN: Las CAUSAS más frecuentes de diarrea exudativa son: a) La enfermedad inflamatoria crónica intestinal (colitis ulcerosa y enfermedad de Crohn). b) Infecciones por microorganismos invasivos o citotoxinas (Shigella, Salmonella, Campylobacter, Yersinia, Mycobacterium tuberculosis, Entamoeba histolytica, Clostridium difficile). c) d) Dr. Pons Colitis isquémica. Enteritis actínica. 2 CMG MÉDICA DE APARATO DIGESTIVO DIARREAS AGUDAS CONCEPTO Rotavirus. Adenovirus. c. Parásitos: Giargia lamblia. Criptosporidium. Amebas. Disminución de la consistencia de las heces con más de 3 deposiciones/día durante menos de 14 días o dos semanas. 2. No infecciosa: producida por: a. Fármacos, como el uso de antibióticos. b. Tóxicos como el alcohol. c. Enfermedad inflamatoria intestinal: colitis ulcerosa y enfermedad de Crohn que pueden debutar con un cuadro de diarrea. d. Diarrea crónica. ETIOLOGÍA 1. CMG Infecciosa: producida por: a. Bacterias: las más frecuentes son: Staphylococcus. Salmonella. Shigella. Campilobacter. E. coli. b. Virus: Norwalk 1 (es un norovirus y es el más frecuente) 1 El virus Norwalk pertenece a la familia de los Calciviridae y provocan cuadros de gastroenteritis. Su transmisión es fecooral o a través de alimentos contaminados. Sus síntomas se presentan de manera brusca, y son: náuseas, vómitos, diarrea, cólicos, cefaleas, fiebre, escalofríos y dolores musculares. Estas manifestaciones suelen aparecer 1 a 2 días después de la exposición al virus y, en la mayoría de los casos, a las 48 TEMA 6 horas remiten. No existe un tratamiento específico para la infección por NLV. El NLV no puede tratarse con antibióticos pero las personas suelen mejorar sin atención médica en 1 ó 2 días. Implica riesgo de deshidratación debido a los vómitos y la diarrea, por lo que es importante beber abundante cantidad de líquidos). Dr. Pons 3 CMG MÉDICA DE APARATO DIGESTIVO De las infecciosas, la causa más frecuente de diarrea aguda se produce por el virus Norwalk en adultos y Rotavirus en niños. Seguido de las bacterias, como segunda causa más frecuente. Los parásitos constituyen la causa etiológica más frecuente en países en vías de desarrollo o subdesarrollados. De las no infecciosas, la causa más habitual es por fármacos como los antibióticos. Estas diarreas se producen en un 10% de los pacientes que los han consumido, y de éstos, en un 99% se presentan como diarreas autolimitadas y leves, y el 1% restante son colitis pseudomembranosas producidas por la toxina de Clostridium difícile. CAUSA Diarrea infecciosa Infección viral Infección bacteriana Parásitos (NOTA: La enterotoxina A (atrae leucocitos al foco y produce inflamación) es la que induce principalmente la clínica, siendo la B (efecto lítico contra la actina de las células) un elemento que actúa sinérgicamente con la anterior. Las manifestaciones clínicas oscilan desde una diarrea leve a una colitis pseudomembranosa. Este clostridio es causante de una proporción significativa de las diarreas asociadas a antibióticos, ya que aprovecha el desplazamiento de la flora normal afectada por los antibióticos para colonizar el tubo digestivo. El diagnóstico se lleva a cabo demostrando la citotoxina en un infiltrado de heces. Los antibióticos para luchar contra el clostridios son el metronidazol y la vancomicina.) CARACTERÍSTICAS TRATAMIENTO Disminuyen la consistencia de las Ninguno, por lo heces, fiebre de bajo grado. general, se resuelve en 48 horas. Fiebre (temperatura> 38,4 ° C), Ninguna. puede haber sangre en heces. No es común en los países des- Antibióticos. arrollados. Diarrea no infecciosa Antibióticos Disminuyen la consistencia de las heces, comienza al iniciar tratamiento con antibióticos y remite pocos días después de su detención. Intolerancia alimentaria Diarrea, dolor abdominal, y / o de (por ejemplo, intoleran- gas después de consumir alimencia a la lactosa) tos Enfermedad inflamatoria Llagas en la boca, diarrea, dolor intestinal abdominal, pérdida de peso y fiebre. Síndrome de colon irri- Dolor abdominal crónico, diarrea y table / o estreñimiento La enfermedad celíaca Diarrea, pérdida de peso, dolor (sensibilidad al gluten) abdominal. TEMA 6 CMG Dr. Pons Ninguno. Determinar si la comida es la causa de intolerancia. Evaluación de la gravedad y tratamiento. Tratamiento sintomático Evitar la ingesta de trigo, centeno, cebada 4 CMG MÉDICA DE APARATO DIGESTIVO PRESENTACIÓN CLÍNICA DE LA DIARREA. E SCENARIOS MÁS 3. Diarrea del viajero: diarrea que se produce cuando se viaja a países en vías de desarrollo. Está producida por: a. E.coli enterotoxígena (la más frecuente). b. Shigella. c. Campilobacter. 4. Diarrea del inmunocomprometido: se produce en pacientes que son VIH positivos o trasplantados. La primera causa de diarrea es la bacteriana, pero también se ha de pensar en Criptosporidium, Isosporas y Mycobacterium avium intracelular (MAI) 3. 5. Diarrea nosocomial: es la producida en hospitales con un 20% de frecuencia. En estos pacientes, prevalece la enfermedad de base y a la diarrea no se le da mucha importancia. Se produce por (en orden de importancia): a. Antibióticos. b. Nutriciones generalmente enterales. c. Por infecciones asociadas al uso de antibióticos, en estos pacientes, hay que descartar la colitis pseudomembranosa. FRECUENTES Hay una serie de características de la diarrea que nos van a orientar hacia la etiología de la misma: 1. Diarrea disenteriforme: diarrea que se presenta con dolor abdominal + fiebre + moco o sangre en las heces. Lo suele producir: a. E. coli. b. Shigella. c. Salmonella. d. Campilobacter. Siendo las dos primeras las más frecuentes. Claramente, es una diarrea infecciosa, bacteriana, producida, principalmente, por estas dos bacterias y podemos poner tratamiento empírico. 2. Toxiinfección alimentaria2: suele producirse por: a. Staphylococcus aureus: presente en cremas y tartas…el periodo de incubación es menor de 6 horas. b. Salmonella: periodo de incubación de más de 6 horas y en general mayor a 14 horas. c. Otras: Campilobacter, Shigella y E.coli. CMG ACTITUD FRENTE A ANA DIARREA AGUDA No hay que estudiar todas las diarreas que nos lleguen. Un 20% de pacientes acuden al médico por diarrea; de éstos, en un 80% no se investigará la causa de la diarrea, a excepción de aquellas que presenten mayor gravedad. 3 2 El S. Aureus provoca intoxicación alimentaria, un alimento se contamina por la bacteria, ésta se multiplica en el alimento y libera las toxinas en él. Aunque se cocine el alimento, en él persisten las enterotoxinas pues son termoestables, por lo que son ingeridas y siguen produciendo la intoxicación. No suele haber fiebre directa ya que no hay pirógenos bacterianos, pero puede haberla a causa de la deshidratación. La salmonella, entre otros, produce toxiinfección alimentaria, que se da cuando una persona ingiere un alimento con una bacteria y ésta se multiplica dentro de ella; mientras que en la intoxicación alimentaria la multiplicación se produce en el alimento y no se ingiere la bacteria, sino la toxina. El efecto de la intoxicación se produce en el momento, mientras que la toxiinfección necesita un tiempo para la multiplicación bacteriana y además puede dar fiebre. TEMA 6 M AI es un patógeno oportunista presente en inmunocomprometidos. Esta bacteria se encuentra en el aire, polvo, agua, tierra y en los alimentos que ingerimos. La bacteria M AI puede afectar todos los órganos del cuerpo como también localizarse solamente en uno, siendo el intestino el más afectado. Es difícil de detectar y tiene el poder de causar otras infecciones, es uno de los principales causantes del "Síndrome de des gaste relacionado con el VIH". Para su diagnóstico es necesario su aislamiento en sangre o biopsia de ganglios, médula ósea. Síntomas: Fiebre continua, diarrea, pérdida de peso, escalofríos, sudoración nocturna, Adenopatía, dolor estomacal y Anemia, entre otros. Prevención y tratamiento: Se utilizan: la Claritromicina o Azitromicina son los agentes profilácticos preferidos. El tratamiento no está perfectamente establecido dándose Claritromicina sola o combinada con Etambutol y Rifabutina. Dr. Pons 5 CMG MÉDICA DE APARATO DIGESTIVO CMG Algoritmo a seguir: F ÁRMACOS QUE SE USAN EN EL 1. TRATAMIENTO ANTIBIÓTICOS 2. Historia clínica: preguntar por el número de deposiciones, si hay sangre o no en heces, si ha viajado al extranjero, si está tomando antibióticos, qué ha cenado el día anterior… Tratamiento. Ha de empezar siempre con el paciente sintomático: a. Dieta astringente: que en función de la gravedad puede empezar siendo líquida. Dar limonada alcalina 4. b. Espasmolíticos para el dolor. c. Sólo se usan fármacos que disminuyan la motilidad intestinal como el Fortasec que es Clorhidrato de loperamida o el Tanagel que es Tanato de gelatina. Estos fármacos se usan en pacientes con diarreas leves no disenteriformes y que no ceden con tratamiento dietético, pero no se pueden usar siempre, puesto que, en el caso de que el paciente tenga una diarrea infecciosa va a empeorar. d. También se pueden dar bebidas como el Acuarius sin sabor y la isotónica de Mercadona, ya que están bastante equilibradas. 3. Tratamiento empírico con antibióticos: a. Diarrea disenteriforme con sangre y moco. b. Fiebre mantenida y dolor abdominal. c. Diarrea que dura + de 6 días. d. Diarrea con + de 6 deposiciones/día. e. Personas ancianas. EMPÍRICO CON Se usan quinolonas como el ciprofloxacino, ya que cubre todas las bacterias. La única que no podría cubrir es Campilobacter y por ello usamos macrólidos como eritromicina o azitromicina. En adultos, como Campilobacter no es la causa más frecuente, no se usan como primera opción los macrólidos, dándose quinolonas. Sin embargo en niños sí se dan macrólidos, puesto que Campilobacter es la causa de diarrea más habitual. Si no hay mejoría, se pueden usar métodos endoscópicos como la colonoscopia para encontrar el proceso inflamatorio y tomar muestras para biopsiarlas. Pero lo más frecuente, si no cesa, es que la diarrea sea de causas inflamatorias y se trate de colitis ulcerosa o la enfermedad de Crohn. DIARREAS CRÓNICAS CONCEPTO Disminución de la consistencia de las heces con más de 3 deposiciones/día durante más de 30 días o 4 semanas. Se produce en un 5% de la población que consulta en Atención Primaria. Sus causas son muy diversas. Este tratamiento hay que utilizarlo después de pedir un coprocultivo y parásitos en heces. 4 La limonada alcalina se hace con: 30 gr. de cloruro sódico, 30 gr. de bicarbonato, 1gr. de cloruro potásico, 30-40 gr. de azúcar y limón. TEMA 6 Dr. Pons 6 CMG MÉDICA DE APARATO DIGESTIVO CMG DIARREA CRÓNICA DESDE UN PUNTO DE VISTA FISIOLÓGICO DIARREA OSMÓTICA 1. Ingestión de Mg, fosfatos, sulfatos 2. SD malabsorción de hidratos de carbono DIARREA GRASA 1. SD Malabsorción a. Enfermedades de la mucosa b. SD de intestino corto c. Isquemia mesentérica d. Postresection diarrea e. Small bowel bacterial overgrowth 2. Maladigestión a. Insuficiencia pancreática exocrina b. Secreción inadecuada de ácidos biliares DIARREA INFLAMATORIA 1. Enfermedad Inflamatoria Intestinal a. Colitis ulcerosa DIARREA SECRETORA 1. Abusos de laxantes b. Enfermedad de Crohn c. Diverticulitis 3. SD Congénitos 4. Toxinas Bacterianas a. Malabsorción de ácidos biliares b. Enfermedad Inflamatoria Intestinal 5. Vasculitis d. Yeyunoileitis ulcerativa 2. Enfermedades Infecciosas a. Colitis pseudomembranosa b. Infecciones bacte- 6. Drogas y Venenos rianas c. Infecciones Virales 7. Alteraciones en la Motilidad d. Infecciones Parasitarias 3. Colitis Isquémica 4. Colitis por radiación 5. Neoplasias a. Cáncer de colón a. Diarrea postvagal b. Diarrea postsimpática c. Neuropatía diabética d. Hipertiroidismo e. SD de intestino irritable 8. Tumores Neuroendocrinos a. Gastrinoma b. VIPoma c. Somatostinoma d. Mastocitosis e. SD Carcinoide f. Cáncer de tiroides 9. Neoplasias a. Carcinoma de colón b. Linfoma c. Adenoma velloso 10. Enfermedad de Addison b. Linfoma TEMA 6 2. Colecistectomia Dr. Pons 7 CMG MÉDICA DE APARATO DIGESTIVO C AUSAS DE DIARREA CRÓNICA Varían mucho en función del país en el que se presentan: 1. Países en vías de desarrollo: la causa más frecuente es la infección por: a. Bacterias. b. Tuberculosis. c. Parásitos: lo más frecuente, y entre ellos las Amebas (lo más frecuente según la zona), Giargia lamblia y Criptosporidium. 2. Países desarrollados: las causas más frecuentes son: (Cada enfermedad la veremos en su tema correspondiente) a. Síndrome de intestino o colon irritable (ya no se llama colon irritable). Es frecuente en estudiantes. Sus síntomas principales son el dolor abdominal y una alteración en los hábitos intestinales (diarrea o estreñimiento), pero no hay una causa identificable. ya que la clínica de las enfermedades digestivas es muy variada. Hay que ver la diarrea según el contexto del paciente, es decir, si se trata de una chica joven con anemia sin más síntomas asociados, no pensaremos en una diarrea crónica, pero si es un paciente que está bien, y de repente empieza con una diarrea aguda que se le cronifica, acompañada de estos síntomas, lo deberemos sospechar. DIAGNÓSTICO DE LA DIARREA AGUDA 1. Historia clínica: síntomas acompañantes, exploración física anómala (artritis, adenopatias, etc.) dirigida a descartar estas enfermedades. 2. Mucho menos valor tienen los análisis de heces, pero sí las analíticas de sangre, en las que veremos: a. Hemograma: para ver si hay anemia. b. Bioquímica: para ver si hay hipoproteinemia, ya que la disminución de albúmina significa SD de malabsorción. b. Enfermedad inflamatoria intestinal: - Colitis ulcerosa. - Enfermedad de Crohn. c. SD de malabsorción: lo más frecuente es la intolerancia a la lactosa y la enfermedad celiaca, la cual es muy sutil en el adulto y los síntomas que presentan son: personas que no engordan a pesar de comer mucho, con anemias leves, caída de pelo, uñas frágiles, etc…No se caracteriza por los síntomas típicos de los SD de malabsorción CLÍNICA 1. Diarrea de más de 30 días. 2. Síntomas de organicidad o de alarma: más allá de un trastorno funcional es un trastorno orgánico. Se ha de pensar cuando hay pérdidas de peso, anemia, fiebre y hemorragias en heces oculta o manifiesta (rectorragia). CMG c. Anticuerpos: - Antiendomisio. - Antitransglutaminasa. - Antigliadina. Estos anticuerpos van a ser muy sensibles para el diagnóstico de la enfermedad celiaca. Cuando no son suficientes las pruebas anteriores porque son normales, podemos asumir que es un intestino irritable siempre que cumpla los criterios. Pero si tiene síntomas acompañantes o de alarma, ha de hacerse una colonoscopia para descartar colitis ulcerosa o enfermedad de Crohn. Si tiene SD de malabsorción, hay que ver la grasa en heces y estudiar el intestino delgado por radiología o biopsia. Ante estos síntomas, se ha de pensar en una enfermedad orgánica, pero no siempre, TEMA 6 Dr. Pons 8 CMG MÉDICA DE APARATO DIGESTIVO CMG TRATAMIENTO Aquí el tratamiento ya no es empírico, porque la diarrea crónica no es amenazante para la vida y es de larga evolución, y hay que esperar el diagnóstico para que el tratamiento sea específico de la enfermedad. TEMA 6 Dr. Pons 9 CMG MÉDICA DE APARATO DIGESTIVO CMG TEMA 7A ENFERMEDAD INFLAMATORIA INTESTINAL La Enfermedad Inflamatoria Intestinal (EII) agrupa básicamente a dos enfermedades, la enfermedad de Crohn y la colitis ulcerosa, que cursa con inflamación intestinal crónica, de causa desconocida, presentando brotes y remisiones. EPIDEMIOLOGÍA La EII es más frecuente en el norte de Europa y EEUU que en los países meridionales (aunque debemos señalar que en nuestro medio está aumentando la incidencia de EII). Es más frecuente en blancos y en raza judía. La edad para el comienzo de la enfermedad está situada entre los 15-30 años. Se produce un segundo pico entre los 60 y 80 años (concretamente las edades más frecuentes de aparición son entre los 15 y 34 años para la enfermedad de Crohn y de 25 a 54 años para la colitis ulcerosa). Se ha observado que hay cierto agrupamiento familiar para EII con mayores concordancias en Crohn que en colitis ulcerosas. De hecho, en un porcentaje elevado de pacientes con Enfermedad de Crohn (10-20 %) existen antencedentes familiares de EII. La colitis ulcerosa es más frecuente que la enfermedad de Cronh (2:1) La incidencia es de 8 casos nuevos por cada 100.000 habitantes en la colitis ulcerosa frente a los 5.5 casos nuevos por cada 100.000 habitantes en la enfermedad de Cronh. Prof. Fuenzalida Se ha descrito la EEI como entidad clínica pero no se han definido sus causas ni su patogenia. Sin embargo, se han barajado como posibles causas: Factores genéticos: es una enfermedad donde hay agregación familiar. La incidencia de la enfermedad inflamatoria intestinal varía entre las diferentes zonas geográficas. TEMA 7A ETIOPATOGENIA Factores infecciosos: no demostrados. Factores inmunológicos: Se ha visto que guarda relación con enfermedades donde hay alteración de la inmunidad. Factores psicológicos: fueron relacionados con esta enfermedad pero actualmente no se acepta dicha hipótesis. ANATOMÍA PATOLÓGICA ENFERMEDAD DE C ROHN En la enfermedad de Crohn la afectación es DISCONTINUA, es decir, puede afectar a cualquier segmento o combinación de ellos del tracto digestivo desde la boca hasta el ano, aunque la más frecuente es la afectación del íleon terminal y colon derecho. A diferencia de lo que sucede en la colitis ulcerosa, en la que siempre está implicado el recto, esta región puede estar indemne en la enfermedad de Crohn. La mucosa ileal o colónica puede aparecer normal o mostrar erosiones redondeadas superficiales, pequeñas y puntiformes. Estas úlceras se denominan “úlceras aftoides o aftosas”. La combinación de úlceras longitudinales y transversales en una mucosa edematosa es la responsable del aspecto macroscópico característico en empedrado. Comisionista: María Lozano 7A-1 CMG MÉDICA DE APARATO DIGESTIVO Las úlceras aftosas, el patrón en empedrado y la presencia de úlceras irregulares desde pequeñas a grandes en una mucosa por otro lado de aspecto normal son característicos de la Enfermedad de Crohn (aunque las úlceras aftosas se pueden ver también en algunas infecciones). Siendo una enfermedad transmural, la pared del intestino se encuentra engrosada con afectación de la submucosa, la muscular propia, la subserosa y la masa mesentérica. Los senos y las úlceras profundas que generan fisura (que alcanzan la muscular de la mucosa) pueden dar lugar a abscesos o fístulas (comunicaciones anormales) entre los segmentos afectos y los órganos adyacentes o las asas cercanas no afectadas. La enfermedad de Crohn activa se caracteriza por inflamación focal y trayectos fistulosos que se resuelven con fibrosis y estenosis del intestino. La pared intestinal aumenta de grosor y su luz se vuelve estrecha y fibrótica, provocando obstrucciones intestinales cróni-cas y recurrentes. Las proyecciones del mesenterio engrosado encapsulan el intestino y la inflamación de la serosa y el mesenterio favorecen las adherencias y la formación de fístulas. Los cambios histológicos consisten en una inflamación de las criptas, formando microabscesos de neutrófilos con las consiguientes ulceraciones, pero a diferencia de la colitis ulcerosa, la inflamación es más profunda, invade la lámina propia por agregados linfoides y macrofágicos que producen una inflamación transmural inespecífica, aunque en un 50% de los casos conducen a la formación, en cualquier capa de la pared, en el mesenterio o en los ganglios linfáticos, de granulomas no caseificantes muy característicos de la enfermedad. Los granulomas no caseificantes (que pueden verse en ganglios linfáticos, mesenterio, peritoneo, hígado y páncreas) constituyen una manifestación patognomónica de Crohn, pero rara vez se identifica en las biopsias de mucosa. La infamación puede extenderse por todo el espesor de la pared, provocando fístulas. CMG un 20% de los pacientes con colitis, los hallazgos no permiten una clara diferenciación entre las dos enfermedades, considerándose como colitis indeterminada, aunque generalmente con la evolución van marcándose las diferencias. COLITIS ULCEROSA La lesión en la colitis ulcerosa es siempre CONTINUA, de forma que no hay zonas sanas dentro del área afectada, aunque la intensidad de la inflamación no tiene por qué ser homogénea. La colitis ulcerosa siempre afecta al recto y se extiende en sentido proximal hasta abarcar todo o parte del colon. La extensión proximal se produce en contigüidad, sin dejar áreas de mucosa indemne (como hemos dicho anteriormente). Ampliación Cuando todo el colon está afectado, la inflamación se extiende 1 a 2 cm. en el íleon Terminal en 10-20% de los pacientes. Es la denominada ileítis por reflujo. En los casos de inflamación leve se observa una mucosa hiperémica, edematosa y granular. A medida que la inflamación se torna más severa, la mucosa pasa a ser hemo-rrágica y aparecen úlceras puntiformes y pequeñas que a menudo son irregulares y socavadas y son las responsables de la imagen en “botón de camisa” observada en los estudios baritados. En la enfermedad de larga evolución, puede observarse pólipos inflamatorios (pseudospólipos) que se encuentran en colon, y no en recto. A diferencia de lo observado en la Enfermedad de Crohn, la fibrosis es un hallazgo infrecuente y la enfermedad crónica de larga evolución rara vez se asocia con la formación de estrecheces regulares. Generalmente se puede distinguir entre colitis ulcerosa y enfermedad de Crohn , en base a que en el Crohn se halla afectación segmentaria o discontinual, transmural, aftas o úlceras lineales, fisuras, fístulas, afectación perianal y en intestino delgado. Sin embargo, hasta en TEMA 7A Prof. Fuenzalida Comisionista: María Lozano 7A-2 CMG MÉDICA DE APARATO DIGESTIVO Diferencias Macroscópicas entre Crohn y Colitis Ulcerosa Colitis Ulcerosa Crohn Recto afectado Recto puede estar indemne Ileon rara vez afectado (Ileitis por reflujo) Afectación íleon Lesión contínua Lesión discontínua Pseudopólipos frecuentes Pseudopólipos infrecuentes Estenosis luminar rara Estenosis luminar frecuente común del CMG La intensidad de los síntomas depende en gran medida de la extensión de la enfermedad. Como es de esperar, en aquellos pacientes que presentan afectación de todo el marco cólico (pancolitis) el estado general será peor. Según el perfil evolutivo, la colitis ulcerosa se puede clasificar en: Leve: menos de cuatro deposiciones diarias, con escasa sangre en heces y sin alteraciones sistémicas. Moderada: más de cuatro deposiciones diarias pero con alteraciones sistémicas mínimas. Ésta es la forma de presentación más frecuente. Grave: más de seis deposiciones diarias con sangre y evidencias de trastornos sistémicos que pueden manifestarse con fiebre, taquicardia o anemia. CLÍNICA COLITIS ULCEROSA Los principales síntomas de la colitis ulcerosa son: Diarrea en general sanguinolenta. Según la extensión de la enfermedad nos podemos encontrar con: a) Rectitis b) Proctosigmoiditis Rectorragia, que es muy habitual ya que la fragilidad de la mucosa provoca que sangre con facilidad. c) Colitis izquierda d) Pancolitis Tenesmo rectal. Secreción de moco. ENFERMEDAD DE C ROHN Dolor abdominal, que suele ser leve, tipo cólico o “retortijón” y se alivia con la defecación. En la enfermedad de Crohn (EC), la sintomatología depende del lugar de afectación. Astenia. Pérdida de peso. Dolor abdominal: El dolor abdominal es más importante en la enfermedad de Crohn que en la colitis ulcerosa y es bastante invalidante. Puede tener un origen multifactorial, siendo la causa y la localización anatómica de la enfermedad de Crohn la que determinará las características del mismo. Fiebre. Anemia. La anemia es secundaria a la pérdida de sangre, pero también puede contribuir con supresión medular ósea resultante de la inflamación crónica; en los que reciben tratamiento también deben tenerse presentes posibles efectos relacionados con los fármacos. En general el síntoma o signo más frecuente es la diarrea sanguinolenta. Cuando aumenta la severidad de la inflamación es más probable la aparición de síntomas sistémicos como fiebre, malestar, náuseas y vómitos. TEMA 7A Prof. Fuenzalida Los síntomas ante los que nos podemos encontrar son: Diarrea: En la afectación de Crohn, la emisión de sangre en heces es más habitual en la afectación cólica, si bien en estos casos la rectorragia es menos frecuente que en la colitis ulcerosa. Pérdida de peso: La pérdida de peso es mucho más frecuente en la EC que en la colitis ulcerosa. Es debida a la propia acti- Comisionista: María Lozano 7A-3 CMG MÉDICA DE APARATO DIGESTIVO vidad inflamatoria de la enfermedad (incremento del catabolismo), a fenómenos de malabsorción cuando existe afección de intestino delgado, a la anorexia coexistente en una importante proporción de pacientes, a la diarrea y al temor a comer presente en estos pacientes. Estos tres síntomas (diarrea, dolor abdominal y pérdida de peso) son los síntomas clásicos de la Enfermedad de Crohn. Sin embargo, esta triada clásica sólo se presenta en el 25 % de los casos en el momento del diagnóstico. También es frecuente que estos pacientes presenten fiebre (de bajo grado). Constituye otro de los síntomas de la enfermedad de Crohn, aunque sólo suele presentarse en casos de afección inflamatoria grave o como consecuencia de complicaciones (abscesos, perforación,…), de forma que va acompañada habitualmente de otros síntomas. A veces nos encontramos ante FOD (fiebre de origen desconocido) siendo la enfermedad de Crohn una entidad incluida en el diagnóstico diferencial de FOD. Desnutrición A veces, cuando hay afectación ileal (ileitis de Crohn) se presenta como dolor en fosa iliaca derecha con una masa a ese nivel y puede cursar con un cuadro típico de apendicitis aguda. La presencia de masa o plastrones es propia de esta entidad, como reflejo de la inflamación transmural, que se pueden abscesificar. La enfermedad perianal en forma de abscesos y fístulas perianales o fisuras anales de localización atípica (fuera de la línea media) están presentes en un 5-10% en el debut de la enfermedad y puede preceder a la aparición de los síntomas digestivos. La enfermedad perianal aparece en aproximadamente el 20% de los pacientes con enfermedad de Crohn a lo largo de su evolución y es más frecuente cuando existe afección de colon. TEMA 7A Prof. Fuenzalida CMG COMPLICACIONES ENFERMEDAD DE C ROHN Las complicaciones típicas son nutricionales, por disminución de la ingesta (debido al dolor que conlleva) y por malabsorción. Son pacientes que presentan pérdida de peso y estado carencial de vitaminas. Más adelante pueden desarrollarse fístulas y abscesos, que conllevan complicaciones inflamatorias, pudiendo llegar a producir sepsis. En tal caso se hace necesaria la intervención quirúrgica. También se producen estenosis, que pueden deberse a reacciones inflamatorias o cicatriciales, siendo éstas complicaciones típicas de la enfermedad de Crohn. Éstas pueden llegar a afectar completamente el tránsito intestinal, ocasionando un cuadro completo de íleo mecánico. Los pacientes con enfermedad de Crohn presentan, a su vez, mayor incidencia de litiasis biliar (por la interrupción del circuito enterohepático) y de litiasis renal (por la mayor absorción de oxalatos). Como complicación tardía, puede aparecer un carcinoma de colon, pero es poco frecuente. COLITIS ULCEROSA Entre las complicaciones de la colitis ulcerosa figuran, en primer término, las hemorragias gastrointestinales bajas y el llamado “megacolon tóxico”. El megacolón tóxico se diagnostica con la presencia de dilatación mayor de 6 cm en colon transverso (radiografía simple de abdomen). Finalmente aparece fiebre séptica, fuertes dolores abdominales y, en la radiografía simple de abdomen, un colon lleno de gas, con peligro agudo de perforación. Existe un aumento del riesgo de presentar adenocarcinoma colorrectal en los pacientes con colitis ulcerosa de años de evolución (sobre todo a partir de 10 años). El riesgo se relaciona con el tiempo y con la extensión de las lesiones (mientras que en la enfermedad de Crohn, si bien puede aparecer carcinoma colónico, lo más frecuente es que la neoplasia aparezca en segmentos aislados por cirugía resectiva parcial o por fístulas). Comisionista: María Lozano 7A-4 CMG MÉDICA DE APARATO DIGESTIVO Una característica importante del cáncer asociado a la colitis es su aparición a una edad temprana, con una media de aproximadamente 25-30 años menos que la del cáncer colorrectal en la población sin colitis. Los cánceres colorrectales de entre 20 y 30 años, que prácticamente no se dan en la población general, no son raros en el marco de la colitis ulcerosa. MANIFESTACIONES EXTRAINTESTINALES DE LA EII Espondilitis que no se relaciona con la extensión ni con la localización de la enfermedad inflamatoria intestinal. Evoluciona de forma independiente a la enfermedad. Sacroileitis. MANIFESTACIONES D ERMATOLÓGICAS Eritema nodoso: El eritema nodoso es la causa más habitual de nódulos inflamatorios en las extremidades inferiores. Se caracteriza por la aparición brusca de nódulos subcutáneos múltiples, bilaterales, simétricos, calientes, dolorosos y de color rojoviolácea. Estas lesiones aparecen más a menudo en la cara anterior de la pierna, pero también pueden verse en las pantorrillas, el tronco o en la cara. Aunque la enfermedad inflamatoria intestinal afecta sobre todo al tracto gastrointestinal, tanto la colitis ulcerosa como la enfermedad de Crohn deben considerarse como trastornos sistémicos, ya que con frecuencia aparecen síntomas extraintestinales que no siempre coinciden con la actividad de la enfermedad intestinal de base. Los órganos afectados con mayor frecuencia son las articulaciones, la piel, los ojos y la vía biliar. La afectación extraintestinal en la enfermedad inflamatoria intestinal depende de varios factores y, a veces, es difícil distinguir las verdaderas manifestaciones extraintestinales de la enfermedad (afectación sistémica primaria), de las complicaciones extraintes-tinales de la misma, secundarias a inflamación crónica, malnutrición o efectos adversos. Algunas manifestaciones extraintestinales (por ejemplo, colangitis esclerosante primaria, espondilitis anquilosante) tienen un curso independiente de la enfermedad, pero en general, las manifestaciones extraintestinales siguen el curso de la enfermedad de base y pueden tener un impacto importante en la calidad de vida, la morbilidad y la mortalidad de estos pacientes. MANIFESTACIONES ARTICULARES Pioderma gangrenoso: es una dermatosis neutrofílica grave de carácter no infeccioso. Es más frecuente en la colitis ulcerosa. Aftas bucales: Sobre todo se observan en enfermedad de Crohn y pueden preceder al diagnóstico de esta última. MANIFESTACIONES O CULARES Las manifestaciones oculares de la enfermedad inflamatoria intestinal son infrecuentes (se encuentran en menos del 10% de los casos), pero con frecuencia son ignoradas o confundidas con trastornos banales frecuentes, a pesar que pueden comportar problemas graves, incluida la ceguera. Los síntomas oculares más frecuentes, que suelen ir paralelos a la afectación intestinal, en la enfermedad inflamatoria intestinal son: a) Artralgias Artritis periféricas de grandes articulaciones (rodillas, codos , tobillos) que suele ir paralela a la inflamación intestinal; se trata de una artritis asimétrica, poliarticular y migratoria, no deformante y seronegativa, respondiendo al tratamiento de la inflamación. CMG Conjuntivitis. b) Uveítis. La uveítis anterior es la manifestación extraintestinal ocular más frecuente. Se presenta con dolor ocular, visión borrosa, y fotofobia, aunque también puede ser asintomática y preceder al diagnóstico de EII. Su curso clínico suele coincidir con la actividad de la EII. c) Episcleritis. d) Iritis. TEMA 7A Prof. Fuenzalida Comisionista: María Lozano 7A-5 CMG MÉDICA DE APARATO DIGESTIVO MANIFESTACIONES HEPATOBILIARES Colangitis esclerosante primaria: caracterizada por inflamación y fibrosis de los conductos biliares intrahepáticos y extrahepáticos, y con frecuencia desemboca en cirrosis biliar e insuficiencia hepática . La complicación más temible de la colangitis esclerosante primaria es el colangiocarcinoma. No se conocen bien los factores de riesgo para la presentación del colangio-carcinoma. Se ha sugerido una relación estrecha con el tabaquismo y algunos estudios apuntan a una cierta coincidencia entre colangiocarcinoma y displasia o cáncer colorrectal en pacientes con colitis ulcerosa. Pericolangitis: es una forma particular de colangitis esclerosante primaria. Está confinada a los conductos biliares pequeños y habitualmente es benigna. Otras alteraciones hepáticas descritas en la EII son la esteatosis hepática, la hepatomegalia y la colelitiasis. DIAGNÓSTICO PROCEDIMIENTOS ENDOSCÓPICOS Es necesario realizar una colonoscopia. Con esta prueba no sólo podremos diagnosticar la colitis ulcerosa, sino que también podremos valorar su extensión y, en algunos casos, facilitar la diferenciación con la enfermedad de Crohn. Es preciso señalar que la colonoscopia debe realizarse con muchas precauciones en un brote agudo de la enfermedad debido al riesgo de perforación intestinal. La colonoscopia también se debe realizar en aquellos pacientes con sospecha de ileitis de Crohn o colitis de Crohn. El cuadro endoscópico típico en la enfermedad de Crohn es el de una inflamación segmentaria con lesiones aftosas, extensas ulceraciones geográficas y, a veces, estenosis y fístulas. La mucosa presenta el característico aspecto en “empredado”. En la colitis ulcerosa se aprecia una afectación uniforme desde la zona distal a la proximal con enrojecimiento difuso, mucosa friable (al menor roce la mucosa sangra), hemorragias difusas y ulceraciones mal delimi- TEMA 7A Prof. Fuenzalida CMG tadas. Más tarde se forman los denominados pseudopólipos; las haustras pueden haber desaparecido por completo. Como alternativa a la colonoscopia podríamos hacer un enema opaco con el inconveniente de que no nos dejaría realizar biopsias. PROCEDIMIENTOS R ADIOLÓGICOS Clásicamente se ha realizado el tránsito intestinal para valorar el intestino. De hecho, la radiología con contraste del intestino delgado está indicada para comprobar signos de afectación del intestino delgado, de una estenosis o para poner de manifiesto la existencia de fístula. En la enfermedad de Crohn aparece el denominado relieve en empedrado en la mucosa. El enema opaco de colon actualmente sólo se realiza en caso de que la colonoscopia sea impracticable, a efectos de descartar extensas alteraciones de carácter grosero. En caso de una colitis ulcerosa de larga evolución, el colon toma la forma de un neumático rígido desapareciendo las haustras (forma en “neumático de bicicleta”). El tránsito intestinal en algunos casos proporciona un diagnóstico de alta certeza de la enfermedad de Crohn pero a veces no es suficiente por lo que disponemos de otras alternativas: a) La enteroscopia. b) La cápsula endoscópica, siendo ésta una prueba sencilla para el paciente. Antes de realizarla debemos saber con seguridad que el paciente no tiene una estenosis a nivel intestinal y ésta es la razón por la que, previamente a la cápsula endoscópica, se realiza un tránsito. OTRAS PRUEBAS ECO y TC. La TC es más útil en la enfermedad de Crohn que en la colitis ulcerosa. La TC no revela detalles de la mucosa pero es una prueba muy valiosa para la definición de los detalles extramurales, por lo que puede ser útil para detectar la presencia de abscesos, fístulas y tractos sinusales. Por tanto, la TC es el estudio Comisionista: María Lozano 7A-6 CMG MÉDICA DE APARATO DIGESTIVO diagnóstico de elección para identificar las complicaciones piógenas de la enfermedad de Crohn (como abscesos intraabdominales y retroperitoneales). En los pacientes con una crisis grave de colitis ulcerosa es necesaria la radiología simple de abdomen en bipedestación. Cuando la enfermedad es grave, la pared del colon se vuelve edematosa e irregular. El engrosa-miento del colon y la dilatación tóxica (megacolón tóxico) pueden observarse con esta sencilla imagen radiológica. DIAGNÓSTICO DIFERENCIAL Ante un paciente con diarrea, fiebre y rectorragia del que sospechamos una EII debemos hacer diagnóstico diferencial con una gastroenteritis enteroinvasiva. Si el paciente sólo presenta rectorragia, debemos descartar hemorroides y/ o cáncer de recto. En el caso de la enfermedad de Crohn el diagnóstico diferencial es mucho más amplio y más variado: pérdida de peso por otras etiologías, paciente con obstrucción intestinal por otras causas, FOD (fiebre de origen desconocido) o apendicitis aguda (señalar que la apendicitis aguda presenta un curso clínico más agudo) entre otras. Muchas veces el diagnóstico de EC se confunde con el de colon irritable. TRATAMIENTO El tratamiento inicial es el tratamiento médico. El tratamiento quirúrgico se reserva para ciertas situaciones. Mientras que el tratamiento conservador medicamentoso de la enfermedad de Crohn y el de la colitis ulcerosa no se diferencian considerablemente, el tratamiento quirúrgico es distinto. En la enfermedad de Crohn el tratamiento quirúrgico NO ES DEFINITIVO; la cirugía en la enfermedad de Crohn NO ES CURATIVA y sólo se utiliza en el caso de tratar complicaciones que no responden a tratamiento médico; son frecuentes las recidivas en las zonas de resección. Por el contrario, en la colitis ulcerosa, se dispone de un método curativo mediante una colectomía TEMA 7A Prof. Fuenzalida CMG completa con conservación del esfínter mediante anastomosis ileoanal. Una vez hechas estas aclaraciones, desarrollaremos el tratamiento médico. TRATAMIENTO F ARMACOLÓGICO Preparados 5-ASA El tratamiento medica-mentoso está basado en los preparados del ácido 5-aminosalicílico (5-ASA) o MESALAZINA, que han desplazado a los antiguos preparados de compuestos de sulfonamidas (que presentaban toxicidad a largo plazo). Se presentan en distintas fórmulas farmacológicas en función del lugar en que se liberan las sustancias en el intestino. Ejercen una buena acción antiinflamatoria. Pueden presentar complicaciones como el fracaso renal. La dosis es de 2-3 gramos por día vía oral o de 0,5-1 gramos por vía rectal (en forma de supositorio o espuma). Corticoides En el brote agudo grave (tanto en la colitis ulcerosa como en la enfermedad de Crohn) es necesario un tratamiento con corticoides que se instaura al principio a dosis elevadas para ir reduciéndose en las semanas siguientes. Es importante señalar que los corticoides no son eficaces en el tratamiento de sostén de la colitis ulcerosa ni en la enfermedad de Crohn; una vez que se ha inducido la remisión clínica se deben retirar de manera gradual ya que presentan numerosos efectos secundarios tales como aumento de peso (por la retención de líquidos), hiperglucemmia, osteonecrosis (osteoporosis), miopatías, estrías abdominales y alteraciones cutáneas, trastornos emocionales y síntomas de supresión ( hay pacientes que son corticodependientes). Inmunosupresores Que están desplazando a los corticoides en muchos casos. AZATIOPRINA (Inmurel®) se ha utilizado con éxito en lugar de los glucocorticoides hasta en Comisionista: María Lozano 7A-7 CMG MÉDICA DE APARATO DIGESTIVO dos tercios de los pacientes con enfermedad de Crohn y colitis ulcerosa en los que no había sido posible suprimir los esteroides. Existe entusiasmo por el uso de estos fármacos inmunorreguladores en el tratamiento de sostén tanto de la EC como en la CU y para tratar la enfermedad perianal activa y las fístulas en la enfermedad de Crohn. Debemos señalar que este inmunosupresor tarda en actuar: la eficacia del tratamiento se observa a las tres o cuatro semanas. Aunque la azatioprina suele tolerarse bien, en un porcentaje bajo de pacientes (3 o 4%) ocurre pancreatitis. Otros efectos secundarios son náuseas, fiebre, exantema y hepatitis. Además uno de cada 300 individuos carece de la metiltransferasa de tiopurina (TPMT), la enzima responsable de la metabolismo del fármaco y otro 11% de la población es heterocigoto, con actividad intermedia de la enzima. Ambas poblaciones tienen mayor riesgo de toxicosis a causa de la acumulación de los metabolitos de la tioguanina. Por lo que antes de instaurar la azatioprina pediremos análisis del nivel de esta enzima del paciente para así poder adecuar la dosis. CMG TRAT AMIENTO DE COLITIS ULCEROSA Exige un tratamiento indefinido con 5-ASA, que disminuye el número de recaídas. La excepción sería el paciente con rectitis que responde bien y se decide quitar el tratamiento indefinido. En las recaídas añadimos esteroides según la intensidad del brote. Cuando la afectación es severa o extensa los usamos vía parenteral; si por el contrario la afectación es distal y leve utilizaremos esteroides vía local. TRAT AMIENTO DEL CROHN Tratamiento de mantenimiento con 5-ASA, pero no está tan clara como en la colitis ulcerosa la evidencia de que reduzca las recaídas. En los brotes: cada día va quedando más clara la utilización de inmunosupresores que de corticoides (como se ha comentado anteriormente). Es la llamada “inversión de la pirámide”. TRAT AMIENTO DE COMPLICACIONES COLITIS ULCEROS A: tratamiento con inmuno- supresores del megacolon tóxico. ENFERMEDAD DE CROHN: en la afectación CICLOSPORINA Se utiliza en el “rescate” de pacientes con colitis ulcerosa grave. Actúa como un potente inhibidor de las respuestas mediadas por las células T. A diferencia de la azatioprina no se ha demostrado su eficacia como tratamiento de sostén, sin embargo tiene un comienzo de acción más rápido que ésta. Terapia biológica El infliximab es un anticuerpo monoclonal antiTNF muy útil en la enfermedad de Crohn con patrón inflamatorio o fistuloso refractario a inmunosupresores. Sin embargo no es útil para la enfermedad de Crohn estenosada. La terapia biológica también es útil en la colitis ulcerosa refractaria Antibióticos Se utilizan en la afectación perianal de la enfermedad de Crohn. Los antibióticos que se utilizan son el metronidazol y el ciprofloxacino y se deben utilizar durante periodos prolongados. TEMA 7A Prof. Fuenzalida perianal utilizaremos antibióticos (también utilizados como tratamiento de base). En caso de estenosis, normalmente nutrición enteral, dilatación con tratamiento endoscópico o finalmente cirugía (como última opción). Decir que tanto en Crohn como en colitis ulcerosa en caso de necesitar nutrición artificial (en un ingreso hospitalario) es preferible utilizar la nutrición enteral que la parenteral por el riesgo de tromboflebitis de esta última. EII Y GESTACIÓN Las mujeres con colitis ulcerosa o Crohn tienen índices normales de fecundidad. En casos leves o quiescentes de EC y CU el pronóstico fetal es casi normal. Aumentan los índices de abortos espontáneos, óbitos fetales y defectos al intensificarse la actividad de la enfermedad pero no por acción de fármacos. La evolución de EC y CU durante el embarazo más bien se relaciona con la actividad de la enfermedad en la fecha de la concepción. Las pacientes deben estar en fase de remisión durante 6 meses antes de concebir. Comisionista: María Lozano 7A-8 CMG MÉDICA DE APARATO DIGESTIVO CMG TEMA 8 SÍNDROME DEL INTESTINO IRRITABLE (SII) CONCEPTOS BÁSICOS DEFINICIÓN DEL SII Conocido comúnmente con el clásico nombre de “colon irritable” o “colon espástico” (estas definiciones no deben emplearse actualmente, al considerar que no sólo se afecta el colon, sino también el intestino delgado), el síndrome del intestino irritable o SII es una anomalía funcional del aparato digestivo que se caracteriza por una asociación de dolor abdominal con alteración del hábito intestinal normal, con ausencia de anormalidades estructurales. La prevalencia de esta entidad en España es del 10% aproximadamente, y presenta una frecuencia de 2-4 veces mayor en el sexo femenino. Cabría destacar que presenta similar frecuencia a cualquier edad, quizás ligeramente inferior en ancianos. El síndrome del intestino irritable es una patología con una clínica similar a la dispepsia funcional (en el 50% de los casos), de carácter crónico, con baja capacidad terapéutica, asociado con frecuencia a enfermedades subyacentes, y con un alto consumo de recursos sanitarios. Requiere una gran habilidad por parte del profesional médico tanto en su diagnóstico como en su seguimiento, así como una estrecha relación de comprensión y empatía con el paciente al acarrear problemas psicológicos tales como desmotivación, además de limitaciones importantes de la vida diaria, y un tratamiento sintomático, no curativo. Como “punto a su favor” podríamos afirmar que consta de una morbimortalidad nula en todos los casos. CRITERIOS DIAGNÓSTICOS El SII consta de una serie de criterios o pautas básicos que permiten elaborar un diagnóstico, basado en aspectos clínicos de la patología. De esta forma, actualmente se emplean los criterios de Roma III, teniendo como precursor primigenio los criterios de Manning (Ampliación: criterios de Roma: simposio internacional en la 7a Semana Europea Unida de Gastroenterología, Roma, Italia, noviembre 1999 se establecen una serie de pautas básicas a la hora de diagnosticar determinadas enfermedades como el SII, enfermedad de Crohn o colitis ulcerosa). Por tanto, el diagnóstico del SII se apoya en: 1. Dolor abdominal que debuta al menos 3 días/mes, en asociación con al menos dos de los tres aspectos siguientes: - Alivio con la defecación. - Comienzo del dolor con cambio de consistencia en las heces. - Comienzo del dolor con cambio en la frecuencia de las deposiciones. 2. Historia con inicio de los síntomas al menos desde los últimos 6 meses, con presencia clínica destacable durante al menos los 3 últimos meses. TIPOS DE SII Podemos encontrar varios subtipos de esta entidad, también aceptados en el tercer simposio de Roma III: 1. Con predominancia de diarrea 2. Con predominancia de estreñimiento 3. Combinado (combinación de diarrea y estreñimiento 4. De patrón alternante 5. Inclasificable TEMA 8 Prof. Pons Comisionista: Diego 8-1 CMG MÉDICA DE APARATO DIGESTIVO Para diferenciar los subtipos combinado y alternante podríamos considerar que el SII combinado se caracteriza por diarreas y estreñimiento que se manifiestan al mismo tiempo, mientras que el segundo se determina por épocas transitorias con sólo diarreas seguidas de otras sólo de estreñimiento, o viceversa. tos factores que explicarían el origen de esta enfermedad, como son: - Alteración funcional del tracto intestinal (delgado y grueso) sin anomalía estructural alguna (se acepta actualmente como causa más que probable del SII) - Trastornos psicológicos o psiquiátricos: clásicamente se ha considerado al SII como una entidad psicosomática asociada a individuos con problemática psiquiátrica o psicológica (ansiedad, trastornos obesivo-compulsivos, stress...), aunque actualmente existe cierta controversia en este aspecto. Hoy en día hay una tendencia a considerar que tales trastornos pueden desencadenar un SII cuando existe combinación con otros factores de marcado carácter etiológico. Cabría destacar que sí se ha observado una amplia prevalencia de estos trastornos en individuos que padecen SII. Por su parte, ya fuera de la etiopatogenia, el SII por sí mismo puede dar lugar a trastornos psicológicos tales como fobias a determinadas situaciones (viajes largos, atascos, etc...). - Alteración de la motilidad, que por si sola no explica suficient ement e la patología. - Sensibilidad visceral alterada: aumento de la sensibilidad visceral, sobretodo a nivel de recto y sigma. Es muy común. - Infecciones: cabe considerar la existencia de un intestino irritable postinfeccioso que tiene su origen en infecciones severas, tales como gastroenteritis provocadas por Shigella, Campylobacter o Salmonella. Se ha observado que pacientes que padecen infecciones intestinales tienen más probabilidad de desarrollar intestino irritable. El estudio anatomopatológico microscópico revela la existencia de microinflamación mucosa que altera la motilidad y flora normal intestinales. - Factores ambientales: a pesar de que se tratan de simples suposiciones y no hay estudio alguno que lo avale, el SII podría tener origen en la dieta o en patologías intestinales tales como celiaquía, alergias intestinales o intolerancia a fructosa/lactosa. CONSISTENCIA DE LAS HECES La escala de Bristol permite determinar la consistencia de las heces en siete grados de orden decreciente: 1. Bolitas duras y separadas (tránsito lento) 2. Bolitas duras y agrupadas 3. Heces como salchichas con la superficie cuarteada 4. Heces como salchicha blanda y lisa 5. Heces semipastosas 6. Heces pastosas 7. Heces líquidas (tránsito rápido) 1 2 3 4 5 6 7 ETIOPATOGENIA El origen del SII no es bien conocido en la actualidad, de hecho se podría considerar como una entidad idiopática. Podemos barajar cier- TEMA 8 CMG Prof. Pons Comisionista: Diego 8-2 CMG MÉDICA DE APARATO DIGESTIVO CMG CLÍNICA - Comienzo de intestino irritable en mayores de 50 años. Debemos incidir en el aspecto de que el SII es una patología de carácter crónico cuyos síntomas presentan una historia clínica de larga evolución. - Hematoquecia: sangre viva y de color rojo en heces, proveniente de cualquier punto del tracto intestinal, lo que la diferencia de la rectorragia (origen rectal). - - Síntomas nocturnos. - Diarrea grave y persistente combinada con pérdida de peso no explicada (destacar en este punto la importancia de la anamnesis, puesto que la pérdida de peso, hecho nunca asociado a esta patología, puede deberse a la autolimitación alimentaria del propio paciente con la intención de aliviar la sintomatología). - Antecedentes de patología digestiva: como celiaquía, cáncer colorrectal... - Dolor cólico: síntoma fundamental y básico de esta enfermedad. Puede debutar frecuentemente como sensación difusa de malestar, pesadez o distensión, en hemiabdomen inferior normalmente, más comúnmente en la mitad izquierda. Se trata de un dolor leve/moderado, siempre diurno, con cronicidad y que alivia con ventoseo/defecación. Alteración del hábito intestinal normal: bien diarrea, bien estreñimiento o una combinación de ambos en patrones combinado o alternante. En el caso de la diarrea, suele ser explosiva, diurna, asociada comúnmente a tenesmo rectal y evacuación de moco con las heces, en ningún caso sangre (nunca rectorragia o hematoquecia). - Polaquiuria, nicturia. - Dolor de espalda y cefaleas como síntomas acompañantes. - Destaca especialmente la ausencia de síntomas tales como malestar general, malnutrición, pérdida de peso... que normalmente suelen ocurrir como consecuencia de la dierrea, debido a la pérdida extrema de nutrientes. DIAGNÓSTICO El diagnóstico del intestino irritable se basa en la clínica fundamentalmente (cambio de hábitos intestinales + síntomas prolongados en el tiempo + dolor) y en la anamnesis al paciente (carácter crónico de la patología). En ocasiones el intestino irritable puede enmascarar una patología subyacente que es necesario abordar, y cuya sospecha se iniciará en caso de que el paciente presente alguno de los síntomas siguientes (“síntomas de alarma”), los cuales nunca se asocirían a la manifestación clásica de esta enfermedad: - Por otra parte, es importante establecer un diagnóstico diferencial con diversas patologías en función de las características del paciente y sus síntomas: celiaquía (que cursa con diarreas), hipotiroidismo (cursa con diarreas), hipertiroidismo (cursa con estreñimiento), CCR (cursa con estreñimiento+rectorragia), diversas parasitosis... También tener en cuenta la edad del paciente a la hora de diferenciar el SII con otras patologías que debutan en determinados momentos de la vida como: - Cáncer colorrectal: suele aparecer en ancianos - Enfermedad de Crohn y celiaquía: más común en jóvenes TRATAMIENTO El tratamiento del SII no es en ningún caso curativo, únicamente sintomático. Dicho tratamiento se fundamenta en dos puntos esenciales: 1. Dieta: no está demostrada su utilidad. El tratamiento mediante una dieta específica está especialmente enfocado a individuos con trastornos metabólicos de índole digestiva tales como intolerancia a la lactosa, al sorbitol o a la fructosa. Síntomas análogos a la dispepsia. TEMA 8 Prof. Pons Comisionista: Diego 8-3 CMG MÉDICA DE APARATO DIGESTIVO CMG 2. Tratamiento farmacológico: se centra en tres grandes aspectos: - Dolor: se emplean antiespasmódicos (anticolinérgicos) y antidepresivos tricíclicos (los cuales administrados a dosis muy bajas de 10-25 mg alivian el dolor abdominal además de disminuir la motilidad intestinal en pacientes con diarrea). - Diarrea: Antidiarreicos (Aloperamida, p. ej.). - Estreñimiento: con los laxantes sintéticos no irritantes o fibra vegetal (esta última provoca meteorismo). TEMA 8 Prof. Pons Comisionista: Diego 8-4 CMG MÉDICA DE APARATO DIGESTIVO CMG TEMA 8 SÍNDROME DEL INTESTINO IRRITABLE (SII) CONCEPTOS BÁSICOS DEFINICIÓN DEL SII Conocido comúnmente con el clásico nombre de “colon irritable” o “colon espástico” (estas definiciones no deben emplearse actualmente, al considerar que no sólo se afecta el colon, sino también el intestino delgado), el síndrome del intestino irritable o SII es una anomalía funcional del aparato digestivo que se caracteriza por una asociación de dolor abdominal con alteración del hábito intestinal normal, y ausencia de anormalidades estructurales. La prevalencia de esta entidad en España es del 10% aproximadamente, y presenta una frecuencia de 2-4 veces mayor en el sexo femenino. Cabría destacar que presenta similar frecuencia a cualquier edad, quizás ligeramente inferior en ancianos. CRITERIOS DIAGNÓSTICOS El SII consta de una serie de criterios básicos que permiten elaborar un diagnóstico, basado en aspectos clínicos de la patología. De esta forma, actualmente se emplean los criterios de Roma III, teniendo como precursor primigenio los criterios de Manning, y posteriormente Roma I y Roma II (Ampliación: criterios de Roma: simposio internacional en la 7a Semana Europea Unida de Gastroenterología, Roma, Italia, noviembre 1999 se establecen una serie de pautas básicas a la hora de diagnosticar determinadas enfermedades como el SII, enfermedad de Crohn o colitis ulcerosa). Por tanto, el diagnóstico del SII se apoya en: El síndrome del intestino irritable es una patología con una clínica similar a la dispepsia funcional (en el 50% de los casos), de carácter crónico, con baja capacidad terapéutica, asociado con frecuencia a enfermedades subyacentes, y con un alto consumo de recursos sanitarios. 1. Dolor1 o molestia abdominal que debuta al menos 3 días/mes, en asociación con al menos dos de los tres aspectos siguientes: Requiere una gran habilidad por parte del profesional médico tanto en su diagnóstico como en su seguimiento, así como una estrecha relación de comprensión y empatía con el paciente al acarrear problemas psicológicos tales como desmotivación, además de limitaciones importantes de la vida diaria, y un tratamiento sintomático, no curativo (al menos hasta hoy en día). - Alivio con la defecación. - Comienzo del dolor con cambio de consistencia en las heces. - Comienzo del dolor con cambio en la frecuencia de las deposiciones. 2. Historia con inicio de los síntomas al menos desde los últimos 6 meses, con presencia clínica destacable durante al menos los 3 últimos meses2. Como “punto a su favor” podríamos afirmar que consta de una morbimortalidad nula en todos los casos. 1 2 TEMA 8 IMPORTANTE IMPORTANTE Prof. Pons Comisionista: Diego 8-1 CMG MÉDICA DE APARATO DIGESTIVO CMG 1 TIPOS DE SII Podemos encontrar varios tipos de esta entidad, también aceptados en el tercer simposio de Roma III: 2 3 1. Con predominancia de diarrea 2. Con predominancia de estreñimiento 4 3. Combinado (combinación de diarrea y estreñimiento) 5 4. De patrón alternante 5. Inclasificable 6 Para diferenciar los tipos combinado y alternante podríamos considerar que el SII combinado se caracteriza por diarreas y estreñimiento que se manifiestan al mismo tiempo, mientras que el segundo se determina por épocas transitorias con sólo diarreas seguidas de otras sólo de estreñimiento, o viceversa. 7 CONSISTENCIA DE LAS HECES La escala de Bristol permite clasificar la consistencia de las heces en siete grados de orden decreciente: 1. Bolitas duras y separadas (tránsito lento); heces caprinas 2. Bolitas duras y agrupadas 3. Heces como salchichas con la superficie cuarteada 4. Heces como salchicha blanda y lisa (heces normales) 5. Heces semipastosas 6. Heces pastosas 7. Heces líquidas (tránsito rápido) ETIOPATOGENIA Actualmente se acepta como causa más probable del SII, un trastorno funcional del I. Delgado y Grueso SIN alteración estructural detectable (hasta ahora). El origen del SII no es bien conocido en la actualidad, de hecho se podría considerar como una entidad idiopática. Podemos barajar ciertos factores que explicarían el origen de esta enfermedad, como son: - Trastornos psicológicos o psiquiátricos: clásicamente se ha considerado al SII como una entidad psicosomática asociada a individuos con problemática psiquiátrica o psicológica (ansiedad, trastornos obesivocompulsivos, estrés...), aunque actualmente existe cierta controversia en este aspecto. Hoy en día hay una tendencia a considerar que tales trastornos pueden desencadenar un SII cuando existe combinación con otros factores de marcado carácter etiológico3. Cabría destacar que sí se ha observado una amplia prevalencia de estos trastornos en individuos que padecen SII. Por su parte, ya fuera de la 3 Nota de la revisora: Yo entendí en clase que se consideraba un sesgo en la muestra de pacientes que consultan y que en realidad la prevalencia es igual que en la población general. Cristina. TEMA 8 Prof. Pons Comisionista: Diego 8-2 CMG MÉDICA DE APARATO DIGESTIVO etiopatogenia, el SII por sí mismo puede dar lugar a trastornos psicológicos tales como fobias a determinadas situaciones (viajes largos, atascos, etc...) - Alteración de la motilidad intestinal, pero por si sola no explica suficientemente la patología. - Sensibilidad visceral alterada: aumento de la sensibilidad visceral, sobretodo a nivel de recto y sigma. Es muy común4. - Infecciones: cabe considerar la existencia de un intestino irritable postinfeccioso que tiene su origen en infecciones más o menos severas, tales como gastroenteritis provocadas por Shigella, Campylobacter o Salmonella. Se ha observado que pacientes que padecen infecciones intestinales tienen más probabilidades de desarrollar intestino irritable. El estudio anatomopatológico microscópico de una pieza de biopsia revela la existencia de microinflamación mucosa que altera la motilidad y flora normal intestinales. - Factores ambientales y genéticos: a pesar de que se trata de simples suposiciones y no hay estudio alguno que lo avale, el SII podría tener origen en la dieta o en patologías intestinales tales como celiaquía, alergias alimentarias o intolerancia a fructosa/lactosa5. CLÍNICA Es necesario incidir en el carácter crónico de esta patología, cuyos síntomas presentan una historia clínica de larga evolución. - Dolor: síntoma fundamental y básico de esta enfermedad. Puede ser cólico o sordo. Puede debutar frecuentemente como sensación difusa de malestar, pesadez o distensión, en hemiabdomen inferior normalmente, más comúnmente en la mitad izquierda. Se trata de un dolor leve/moderado, siempre diurno, con cronicidad y que alivia con ventoseo/defecación6. - Alteración del hábito intestinal normal: bien diarrea, bien estreñimiento o una combinación 4 Ej: hipersensibilidad a la distensión visceral. En general hay muy poca correlación con la ingesta. Por supuesto habría que descartar celiaquía e intolerancia a fructosa antes de diagnosticar SII. 6 El profesor dijo que no es un dolor tan fuerte como para despertarte de noche, y no debería ser motivo para ir a urgencias. 5 TEMA 8 CMG de ambos en patrones combinado o alternante. En el caso de la diarrea, suele ser explosiva, diurna, asociada comúnmente a tenesmo rectal y evacuación de moco con las heces, en ningún caso sangre (nunca rectorragia o hematoquecia7) - Polaquiuria, nicturia - Dolor de espalda, cefaleas y dismenorrea en mujeres, como síntomas acompañantes - Destaca especialmente la ausencia de síntomas tales como malestar general, malnutrición, defectos de absorción, pérdida de peso o defectos carenciales, que normalmente suelen ocurrir como consecuencia de la diarrea puntual y ocasional (fuera del SII), consecuencia de la pérdida extrema de nutrientes. DIAGNÓSTICO El diagnóstico del intestino irritable se basa en la clínica fundamentalmente (cambio de hábitos intestinales + síntomas prolongados en el tiempo + dolor) y en la anamnesis al paciente (carácter crónico de la patología). En ocasiones el intestino irritable puede enmascarar una patología subyacente que es necesario abordar, y cuya sospecha se iniciará en caso de que el paciente presente alguno de los síntomas siguientes (“síntomas de alarma”), los cuales nunca se asociarían a la manifestación clásica del SII. Los síntomas de alarma que nos permiten sospechar patología orgánica son: - Comienzo de síntomas en mayores de 50 años - Hematoquecia: sangre viva y de color rojo en heces, proveniente de cualquier punto del tracto intestinal, lo que la diferencia de la rectorragia (origen rectal) - Síntomas nocturnos que te despiertan - Diarrea grave y persistente combinada con pérdida de peso no explicada (destacar en este punto la importancia de la anamnesis, puesto que la pérdida de peso, hecho nunca asociado a esta patología, puede deberse a la 7 Deposición intestinal sanguinolenta, rojo menos intenso, que NO te asegura que provenga del recto. Prof. Pons Comisionista: Diego 8-3 CMG MÉDICA DE APARATO DIGESTIVO autolimitación alimentaria del propio paciente con la intención de aliviar la sintomatología) - Antecedentes personales y/o familiares de patología digestiva: como celiaquía, cáncer colorrectal... Por otra parte, es importante establecer un diagnóstico diferencial con diversas patologías en función de las características del paciente y sus síntomas: celiaquía (que cursa con diarreas), hipotiroidismo (cursa con estreñimiento), hipertiroidismo (cursa con diarreas), CCR8 (cursa con pancreatitis estreñimiento+rectorragia), crónica con malabsorción (diarrea+ alcoholismo), diversas parasitosis... También tener en cuenta la edad del paciente a la hora de diferenciar el SII con otras patologías que debutan en determinados momentos de la vida como: - Cáncer ancianos colorrectal: suele aparecer CMG antidepresivos tricíclicos (los cuales administrados a dosis muy bajas de 1025 mg alivian el dolor abdominal además de disminuir la motilidad intestinal en pacientes con diarrea) - Diarrea: Antidiarreicos (Aloperamida, p. ej.); ADT en dosis bajas en pacientes de predominio diarreico - Estreñimiento: con los laxantes sintéticos no irritantes (lactulosa) o fibra vegetal (esta última provoca meteorismo intestinal, por lo que se debe dar en dosis graduales para evitar la intolerancia) en - Enfermedad de Crohn y celiaquía: más común en jóvenes TRATAMIENTO El tratamiento del SII no es en ningún caso curativo, únicamente sintomático. Dicho tratamiento se fundamenta en dos puntos esenciales: 1. Dieta: no está demostrada su utilidad. El tratamiento mediante una dieta específica está especialmente enfocado a individuos con trastornos metabólicos de índole digestiva tales como intolerancia a la lactosa, al sorbitol o a la fructosa9. 2. Tratamiento farmacológico: se centra en tres grandes aspectos: - Dolor: se emplean antiespasmódicos a demanda (anticolinérgicos) y 8 Cáncer Colo-Rectal. En la mayoría no existe correlación síntomasalimentación; y en los que sí parece haberlas seguramente se trata de gente obsesiva. Se aconseja dieta normal. 9 TEMA 8 Prof. Pons Comisionista: Diego 8-4 CMG MÉDICA DE APARATO DIGESTIVO CMG TEMA 9 TUMORES DE INTESTINO DELGADO ESTE TEMA (JUNTO CON LA SEGUNDA PARTE DE LOS SÍNDROMES DE MALABSORCIÓN Y EL CÁNCER DE COLON) FUE DADO POR EL DR . CARBALLO EN UNA CLASE…COMO OS PODEIS IMAGINAR LO DIO SUPER RÁPIDO…EN CLASE SÓLO SE EXPLICÓ LOS TIPOS MÁS FRECUENTES, PERO HE HECHO UNA INTRODUCCIÓN GENERAL (BASADA EN LA COMISION DE CX DIGESTIVA ) SOBRE EL TEMA PARA QUE SE ENTIENDA MEJOR CADA TIPO DE TUMOR DESPUÉS. 3. De la localización en éste órgano. 4. De la tendencia a la necrosis o ulceración. Hay que pensar en la posibilidad diagnóstica de que exista un tumor de intestino delgado en las siguientes situaciones: 1. Episodios recidivantes e inexplicables de dolores cólicos abdominales. 2. Brotes intermitentes de obstrucción intestinal, sobre todo en ausencia de enfermedad inflamatoria intestinal o de cirugía abdominal previa. MIRIAM 3. Invaginación o cuadros de perforación intestinal. INTRODUCCIÓN 4. Signos de hemorragia intestinal crónica. El intestino delgado ocupa el 75% de la longitud del tubo digestivo, sin embargo, los tumores que en él se originan no suponen más del 5% de toda la patología neoplásica. Los tumores benignos del intestino delgado suelen ser asintomáticos y su diagnóstico se realiza en la mayoría de las ocasiones de forma accidental a través de laparotomía, endoscopia o estudio necrópsico. Cuando ocasionan síntomas, se deberán a tumores malignos en más del 80% de los casos. Su incidencia es similar en varones y mujeres y pueden diagnosticarse a cualquier edad, aunque es más frecuente que se detecten a edades avanzadas, probablemente porque estos pacientes son sometidos más a menudo a exploraciones diagnósticas por otros motivos. 5. Presentar un efecto de masa abdominal palpable. 6. Síndrome general neoplásico: astenia, anorexia, pérdida de peso… Estos tumores suelen ser de difícil diagnóstico, y pueden pasar inadvertidos en la exploración radiológica convencional, sobre todo si no son de gran tamaño; si bien, ésta nos puede orientar a la existencia de un tumor del ID. Las exploraciones radiológicas que podemos usar son: 1. El tránsito intestinal con bario constituye un buen examen de detección para las lesiones mucosas (ulceración, irregularidad mucosa o estenosis); pero a menudo, omite los tumores benignos o aquellos que se encuentran en un estadio temprano. Por este motivo, ha sido reemplazado en parte por el estudio de enteroclisis, que permite evaluar con más precisión los pliegues intestinales. 2. Mediante gastrosduodenoscopia podemos llegar hasta duodeno y localizar tumores en el duodeno. Los síntomas que originan, van a depender: 1. Del tamaño del tumor. 2. De su situación en la pared intestinal (endoluminales o exoluminales). TEMA 9 Dr. Carballo 1 CMG MÉDICA DE APARATO DIGESTIVO 3. La colonoscopia no suele ofrecer buenos resultados. 4. La TC abdominal suele ser de ayuda para estudiar molestias abdominales inespecíficas e intermitentes, pero a veces no localiza bien la lesión. 5. 6. La capsuloendoscopia o capsulografía tiene como gran inconveniente que no se pueden realizar biopsias. TUMORES DERIVADOS DEL EPITELIO, ADENOMAS : Los adenomas forman entre el 25-40% de los tumores benignos intestinales. Pueden ser únicos o múltiples y se suelen encontrar en duodeno e íleon. Se pueden diferenciar en 3 tipos: 1. Pólipo adenomatoso: Tiene origen en las células epiteliales de la mucosa, tanto de la superficie como de las glándulas intestinales. Clínicamente, tienen un comportamiento relativamente uniforme, presentando síntomas como ictericia, anemia11 y obstrucción e invaginación intestinal. El diagnóstico se realiza mediante: endoscopia, radiografía de abdomen y cápsula endoscópica. El tratamiento es endoscópico y quirúrgico. Un método más novedoso es la enteroscopia de doble balón, que permite ver desde la boca a la válvula ileocecal o desde el ano al duodeno. Al contrario que la capsuloendoscopia si se pueden tomar muestras, incluso realizar algunas acciones terapéuticas. TUMORES DE INTESTINO DELGADO 2. Adenoma Velloso: Constituye un hallazgo excepcional, pero tienen mayor potencial malignizante. A. Benignos 3. Adenoma de las Glándulas de Brunner: Son los más frecuentes hallados en intestino y son formas localizadas de hiperplasia 2. 1. Tumores derivados de epitelio: adenomas. 2. Tumores no epiteliales o mesenquimatosos: a. b. c. d. e. f. Leiomioma. Lipoma. Neurofibroma. Angioma. Hamartoma. Fibroma. B. Malignos 1. 1 El examen hematológico permite confirmar anemia ferropénica consecutiv a a hemorragias (ocultas o vis ibles). En caso de darse éstas, la ausencia de lesiones gástric o cólicas nos hará sospechar una lesión en el intestino delgado. Primarios: a. b. c. d. Adenocarcinomas. Tumores carcinoides. Linfomas. Tumores estromales. 2 2. Secundarios. TIPOS FRECUENTES Los adenomas, leiomiomas y lipomas son los tumores benignos de intestino delgado más frecuentes. TEMA 9 CMG Las glándulas de Brunner se concentran fundamentalmente en la submucosa y la mucosa duodenal, disminuyendo gradualmente en número desde el píloro hacia la ampolla de Vater, no observándose posterior al ligamento de Treitz. Sus funciones son: Secreción de un fluido alcalino rico en mucina que neutraliza la acidez del contenido gástric o, de manera que protege el epitelio duodenal. La producción de factores de crecimiento epidérmico. Las teorías señalan que la hiperestimulación (acidez gástrica) o el aumento de la actividad vagal o irritación local, serían las responsables de desencadenar un crecimiento desmedido del número de glándulas, del tejido conectivo y del músculo liso, determinando de esta manera la formación de estructuras lobuladas pedunculadas y sésiles, que serán llamados comúnmente como hiperplasias de glándulas de Brunner 1 Dr. Carballo 2 CMG MÉDICA DE APARATO DIGESTIVO TUMORES NO EPITELIALES O MESENQUIMATOSOS: 1. LEIOMIOMA. Los leiomiomas son los tumores benignos más frecuentes del intestino delgado (2030%). Estos tumores derivan de la capa de músculo liso del intestino y crecen de forma intramural erosionando a veces la mucosa y provocando hemorragias digestivas. Se encuentran en yeyuno e íleon, de manera exoluminal o endoluminal. Es frecuente en estos tumores la necrosis o la ulceración, lo que justifica uno de sus síntomas más constantes, la hemorragia digestiva reiterada, que da lugar a anemia. Otro rasgo clínico frecuente es la presencia de molestias abdominales pero de características inespecíficas. El diagnostico se realiza con cirugía y cápsula. El tratamiento es quirúrgico. CMG Los tumores vasculares pueden también adoptar la forma de hemangiomas aislados, sobre toso en el yeyuno. La angiografía, especialmente durante un episodio de hemorragia, es el procedimiento de elección para diagnosticar este tipo de lesiones. 5. H AMARTOMA. Los hamartomas suelen estar asociados a la poliposis de Peutz – Jeghers 4 y son más frecuentes en la porción distal. 6. FIBROMA. Los fibromas se encuentran en íleon, pueden ser exo o endoluminales y pueden provocar obstrucción o invaginación. 2. LIPOMA. Los lipomas representan el 8-20% de todos los tumores benignos del intestino delgado, y se encuentran con mayor frecuencia en la porción distal del íleon y en la válvula ileocecal. Tienen un característico aspecto transparente, son intramurales y, en general, asintomáticos aunque a veces pueden producir obstrucción, invaginación o hemorragias. 3. NEUROFIBROMA. El neurofibroma se encuentra en yeyuno o íleon, puede ser único o múltiple, presentarse de manera exoluminal o endoluminal, y provocar hemorragias. De todos ellos, un 10% puede degenerar a formas malignas. 4. ANGIOMA. Aunque no son verdaderas neoplasias, estas lesiones son importantes debido a la frecuencia con que provocan hemorragias intestinales. Se encuentran múltiples telagientasias intestinales en una forma no hereditaria limitada al tubo digestivo o formando parte del síndrome hereditario de Osler – Rendu Weber3 3 El s índrome de Osler - Weber-Rendu se caracteriza por telangiectasias vasculares y malformaciones arterio venosas en la nariz, la piel, los pulmones, el cerebro y el tracto digestivo. Las vénulas y los capilares de la piel y las membranas mucosas están dilatados y enrevesados y pueden sangrar espontáneamente o como resultado de un pequeño trauma. TEMA 9 La manifestación más frecuente de esta enfermedad es la epistaxis y ocurre en la mayoría de los pacientes antes de los 21 años. Las hemorragias digestiv as recurrentes del estómago, intestino delgado y colon se manifiestan, sobre todo a partir de los 50 años y solo en una minoría de los pacientes. Las hemorragias se pueden detectar endoscópicamente en diversas partes del tracto digestiv o, desde el paladar, las encías y los labios hasta el estómago, el intestino delgado y el colon. Ta mbién pueden encontrarse telangiectasias en los pulmones y, en el 30% de los pacientes también el hígado está afectado aunque este último no causa por regla general ningún problema serio. Durante la adolescencia, suelen aumentar de tamaño as emejándose a las telangiectasias de la cirrosis hepátic a. Estas lesiones ocasionan molestias por su tendencia a sangrar. En la edad adulta, las epistaxis o las hemorragias gástric as pueden llegar a producir anemia. 4 El s índrome de Peutz -Jeghers es un proceso hereditario autosómico dominante. Consiste en el desarrollo de pólipos hamartomatosos que afectan fundamentalmente al intestino delgado y pueden extenderse a colon y estómago. Se asocia con lesiones mucocutáneas de pigmentación melánica (en labios, mucosa oral, etc.…). La his toria natural de la enfermedad demuestra que no es una enfermedad benigna. Se estima que el riesgo de estos pacientes de padecer cáncer es 18 veces mayor que el resto de la población, por lo que el screening con gastro y colonoscopia se recomienda a partir de los 20 años, y posteriormente de forma periódica cada tres años. Dr. Carballo 3 CMG MÉDICA DE APARATO DIGESTIVO CMG TEMA 10 CÁNCER DE COLON PÓLIPOS INTESTINALES TIPOS HISTOLÓGICOS CONCEPTO Los pólipos intestinales se dividen en dos grandes grupos: Se denomina pólipo intestinal a todo tumor circunscrito que protruye desde la pared a la luz intestinal. 1. Neoplásicos: pueden clasificarse en: a. Según la superficie de fijación a la pared intestinal, los pólipos pueden ser: plano, sesil, semipediculados o pediculados. Benignos: adenomas: se trata de tumores de epitelio neoplásico benigno, pero con degeneración maligna potencial, raros en el intestino delgado y muy frecuentes en el colon. Los adenomas colónicos pueden ser únicos o múltiples; cuando se localizan en el rectosigma suelen ser más grandes y con mayor tendencia a la malignidad. Existen tres tipos histológicos de adenoma: En el tracto digestivo pueden hallarse pólipos únicos a cualquier nivel, desde el estómago hasta el recto, o bien pólipos múltiples1 afectando el estómago, el intestino delgado y/o el colon en forma difusa. Prevalencia de pólipos por edades en la población general: - Los adenomas tubulares suelen tener un tamaño inferior a 1 cm y la probabilidad de hallar focos de carcinoma en ellos es baja. - Los adenomas tubulovellosos tienen un tamaño algo mayor y una incidencia de transformación cancerosa de aproximadamente el 10%. - El 50-60% de los adenomas vellosos tienen un tamaño superior a los 2 cm y alrededor del 40% presenta transformación maligna. b. Malignos: adenocarcinoma in situ o invasivo. 2. 1 No neoplásicos: incluyen pólipos mucosos (Peutz-Jeghers), hiperplásicos (10-30%), juveniles e inflamatorios (seudopólipo). Cuando hay más de 100 se denomina poliposis. TEMA 10 Dr. Carballo 1 CMG MÉDICA DE APARATO DIGESTIVO CMG 2. Tipo histológico: los adenomas vellosos son los que tienen mayor probabilidad de transformación maligna. Seguido de los túbulos-vellosos y el tubular. 3. Grado de displasia: se distingue en alto, moderado y bajo grado. 4. Número de pólipos: mayor probabilidad cuantos más pólipos existen. 5. Pólipo plano. La mayor parte de los cánceres se desarrollan de pólipos: esta afirmación tiene evidencias escasas directas, sin embargo indirectas si que las tiene, puesto que se ha visto: 1. Pólipo y cáncer colorrectal tienen igual distribución anatómica. 2. Raramente se ven cánceres en ausencia de pólipos. 3. Claros factores de riesgo en dependencia del tipo de pólipo. 4. Pacientes con poliposis hereditaria desarrollan cánceres. 5. Extirpación de pólipos se asocia a reducción de mortalidad por cáncer colorrectal. Como contrapartida, pocos pólipos progresan a cáncer: 1. 2,5 pólipos por cada 1000 y año. 2. El tiempo de evolución es largo: a. Diferencia de edad desde que se descubren los pólipos, hasta que se descubren los cánceres es diferente, de 18 años. b. Observaciones directas sugieren 10-15 años. c. Experiencias directas sugieren que un pólipo rara vez es maligno en un intervalo de menos de 3 años. d. Los programas de screening sugieren un efecto protector en 10 años. e. Estimación de inicio de síntomas desde que se hace maligno es de 4,8 años. La probabilidad de que un pólipo desarrolle cáncer, depende de varios factores: 1. Tamaño: a. Menos del 1% de los pólipos menores de 1 cm son malignos. b. Más del 10% de los pólipos de más de 1 cm son malignos. TEMA 10 Dr. Carballo 2 CMG MÉDICA DE APARATO DIGESTIVO CMG Variantes de la PAF son el SD de Turcot (es una enfermedad hereditaria autosómica recesiva, que consiste en la asociación de poliposis adenomatosa cólica y tumores del SNC. El número de adenomas suele ser menor que en los casos de poliposis cólica famiPOLIPO MALIGNO liar) y el SD de Gardner (se trata de una entidad familiar que se manifiesta por la presencia •Escision •Escision difusa de pólipos adenomatosos en el tracto Completa Incompleta gastrointestinal, tumoraciones de tejidos blan•Criterios •Criterios dos y osteomas en diferentes localizaciones. Favorables DesfavorablesLas lesiones extraintestinales más frecuentes son osteomas de mandíbula, cráneo y huesos largos, tumores desmoides, fibromatosis mesentérica y cavidades quísticas subdentarias). ESQUEMA SEGUIMIENTO TRAS POLIPECTOMIA + COLONOSCOPIA POLIPO BENIGNO MULTIPLE ó > 1CM UNICO REVISION AÑ AÑO REVISION 3 AÑ AÑOS CIRUGIA Variantes del PAF POLIPOSIS ADENOMATOSA FAMILIAR (PAF) Forma atenuada de PAF (Mutaciones en 5’-codon 158, 3’-codon 1596) se asocian a formas menos severas: Menos pólipos Inicio más tardío Desarrollo de cáncer 12 años más tarde Más lesiones en tracto digestivo superior Mutación APC I1307K que lleva a mutaciones somáticas del otro alelo y a un riesgo aumentado de CCR. En población judía con alta incidencia de CCR. (6% población judía de origen europeo). Se trata de una enfermedad hereditaria autosómica dominante (consiste en una mutación germinal del gen APC situado en el cromosoma 5) que se caracteriza por la presencia de numerosos pólipos adenomatosos (más de 100) a lo largo de todo el intestino grueso2. El diagnóstico suele hacerse entre la segunda y la cuarta décadas de la vida. Esta enfermedad presenta un 100% de riesgo de cáncer a los 40 años, y la esperanza de vida en aquellos pacientes no tratados es de 42 años. La poliposis adenomatosa familiar es una enfermedad con un alto potencial de malignización, de manera que si el diagnóstico se ha realizado a partir de síntomas causados por la presencia de los pólipos en el intestino, el porcentaje de pacientes con carcinoma en el momento inicial del diagnóstico es de alrededor del 75%. Alrededor del 40% de los pacientes presentan lesiones gastroduodenales asociadas, cáncer gástrico, duodenal, pancreático, de la vía biliar, del tiroides y del SNCentral, y es muy frecuente la existencia de lesiones hiperpigmentadas de la retina, así como lesiones no malignas de la piel. 2 Constituye un 1% de nuevos casos de cáncer colorrectal, 1 caso cada 8300-14025 nacimientos. TEMA 10 OTRAS POLIPOSIS FAMILIARES 1. SD de Peutz - Jeghers. El síndrome de Peutz - Jeghers es un proceso hereditario autosómico dominante (cromosoma 19q). Consiste en el desarrollo de pólipos hamartomatosos que afectan fundamentalmente al intestino delgado y pueden extenderse a colon y estómago. Se asocia con lesiones mucocutáneas de pigmentación melánica (en labios, mucosa oral, etc.…). La historia natural de la enfermedad demuestra que no es una enfermedad benigna. Se estima que el riesgo de estos pacientes de padecer cáncer es 18 veces mayor que el resto de la población, por lo que el screening con gastro y colonoscopia se recomienda a partir de los 20 años, y posteriormente de forma periódica cada tres años. Dr. Carballo 3 CMG MÉDICA DE APARATO DIGESTIVO 2. SD de Cowden: Es un síndrome de poliposis hereditario (herencia autosómica dominante, con mutación del gen PTEN situado en el cromosoma 10q) caracterizado por la presencia de múltiples hamartomas en piel y mucosas. Los pólipos intestinales (lesiones digestivas en un 70%) son de tipo hamartomatoso y no sufren transformación maligna, pero la enfermedad se asocia con relativa frecuencia a cáncer de tiroides y enfermedad fibroquística y cáncer de mama. 3. Poliposis juvenil: consiste en una mutación del gen DPC4 40% situado en el cromosoma 18q, que se caracteriza por la presencia de pólipos de probable origen hamartomatoso que afectan invariablemente el recto y a veces el colon de los niños. Los pólipos presentan un tamaño en general superior a 2 cm y se caracterizan por contener glándulas con dilatación quística y por cierto grado de infiltración inflamatoria de la lámina propia. Los síntomas, que pueden deberse a invaginación, hemorragia o prolapso del pólipo por el ano, suelen manifestarse a partir de los 5 años de edad. CMG CÁNCER COLORRECTAL EPIDEMIOLOGÍA Aunque las neoplasias malignas de pulmón y mama sean las más frecuentes en los sexos masculino y femenino, respectivamente, el adenocarcinoma de colon y recto es la neoplasia interna más frecuente que afecta a ambos sexos y, como tal, la primera causa de muerte por cáncer en los países occidentales. CÁNCER COLORRECTAL:INCIDENCIA MAXIMA 4. Neurofibromatosis o SD de los nevos de células basales: esta enfermedad sistémica puede afectar el tracto gastrointestinal con presencia de neurofibromas submucosos susceptibles de degenerar. OTRAS POLIPOSIS NO FAMILIARES 1. SD de Cronkhite – Canada: se trata de una enfermedad rara, no familiar, caracterizada por poliposis gastrointestinal difusa, hiperpigmentación cutánea, onicodistrofia y alopecia. Se manifiesta en la edad media de la vida con anorexia, pérdida de peso, dolor abdominal, diarrea, que puede contener moco y sangre, y un cuadro de malabsorción con esteatorrea y pérdida proteica por heces que determina hipoalbuminemia y edemas. 2. Poliposis linfoide. 3. Poliposis hiperplásica. 4. Poliposis lipomatosa. TEMA 10 Dr. Carballo 4 CMG MÉDICA DE APARATO DIGESTIVO CMG Los factores genéticos son: 1. Adenoma colónicos esporádicos. 2. Síndromes familiares hereditarios: a. Poliposis familiar adenomatosa (AFP). b. Síndrome hereditario del cáncer colorrectal no ligado a poliposis (HNPCC). 3. Colitis ulcerosa (UCAN). 4. Cáncer de ovario y de endometrio. 5. Portador de cáncer colorrectal previo. 6. Antecedentes familiares de cáncer colorrectal. ADENOMAS ESPORÁDICOS En España la incidencia de cáncer colorrectal parece ir en aumento en los últimos años, como sucede en general en todos los países industrializados. Alto riesgo de degeneración: 1. si el tamaño es mayor de 1,5 cm. 2. sesil o plano. 3. displasia grave (severa). 4. metaplasia escamosa. 5. velloso (alrededor del 40). 6. pólipos múltiples. Bajo riesgo de degeneración: 1. si el tamaño es menor de 1 cm. 2. pediculado. 3. displasia leve/moderada. 4. sin áreas de metaplasia. 5. tubular. 6. pólipo único. FACTORES DE RIESGO3 Existen múltiples evidencias que apoyan la participación de factores tanto genéticos como ambientales en la patogenia del cáncer colorrectal. 3 Otro factor de riesgo es la edad. Antes de los 40 años el riesgo de padecer cáncer colorrectal en personas sin predisposición para esta neoplasia es bajo. Después de los 40 años el riesgo aumenta progresivamente, duplicándose la incidencia con cada década y alcanzando un pico entre los 75 y los 80 años. En grupos de elevado riesgo (los que explicamos a continuación) es alta la posibilidad de desarrollar cáncer en edades más tempranas. TEMA 10 Dr. Carballo 5 CMG MÉDICA DE APARATO DIGESTIVO SÍNDROMES FAMILIARES HEREDITARIOS 1. Poliposis familiar adenomatosa (AFP): a. Herencia dominante. b. Penetrancia casi completa. c. Historia familiar previa de cáncer colorrectal. d. Mutación del gen APC Cr 5. e. Cientos y miles de pólipos. f. CMG En general, todo el tema el profesor lo dio a modo de “recordatorio”, y fue bastante rápido (por si os tranquiliza, Andrés G.L tiene copiado una cara de folio). A partir de aquí, el profesor fue pasando diapositivas, en la única que se detuvo un poco fue en la del cribado del cáncer de colon…Pongo las diapositivas para que os las miréis, por si acaso preguntara algo…Miriam Cáncer alrededor de los 35 años. 2. Síndrome hereditario del cáncer colorrectal no ligado a poliposis (HNPCC): a. Herencia dominante. b. Aparece, en especial, en colon derecho. c. Historia familiar de cáncer colorrectal. CANCER COLORRECTAL RAZONES PARA EL DIAGNÓSTICO PRECOZ Notable incidencia y mortalidad Estadios tardíos en el diagnóstico Lesiones premalignas detectables Curable en estadios precoces d. Mutación: inestabilidad de microsatélites. e. No obligado pólipos. f. Cáncer antes de los 50 años. COLITIS ULCEROSA CONCEPTO DE SCREENING EN CÁNCER Alto riesgo de degeneración: 1. Duración de la enfermedad superior a 10 años. Uso de té écnicas de ttécnicas intervenció ón para descubrir intervenci intervención c á nceres así íntomá ntomáticos o cánceres as asíntomáticos estados precancerosos 2. Gran extensión. 3. Displasia. 4. Colangitis esclerosante primaria. 5. Historia familiar de cáncer. Spratt, Spratt, 1984 1984 6. Presencia de adenomas. Bajo riesgo de degeneración: 1. Corta duración. 2. Sólo proctitis. 3. No displasia. 4. No colangitis esclerosante. 5. No historia familiar de cáncer colorrectal. 6. No presencia de adenoma. TEMA 10 Dr. Carballo 6 CMG MÉDICA DE APARATO DIGESTIVO CMG CÁNCER COLORRECTAL ALTERNATIVAS EN TÉCNICAS DE SCREENING II CONDICIONES DEL SCREENING TACTO RECTAL Problema de salud pú pública Tratamiento aceptable Historia natural y “punto crí crítico” tico” Costes screening / costes tratamiento Amplia disponibilidad Cáncer de Próstata Hasta 13% de lesiones Dudosa aceptabilidad tardí tardío Test aceptable CÁNCER COLORRECTAL ALTERNATIVAS EN TÉCNICAS DE SCREENING III 4 HEMORRAGIAS OCULTAS EN HECES Detectable Terminal Letal a b c d e Ventana Tiempo Test más utilizado Hemoccult II Variables Sensibilidad, Especificidad y Valor Predictivo Apto para screening de masas Eficacia / Reducción de mortalidad de control Cambio maligno CÁNCER COLORRECTAL ALTERNATIVAS EN TÉCNICAS DE SCREENING I TACTO TACTO RECTAL RECTAL HEMORRAGIAS HEMORRAGIAS OCULTAS OCULTAS EN EN HECES HECES RX RX ENEMA ENEMA OPACO OPACO ULTRASONOGRAFÍA ULTRASONOGRAFÍA ENDOSCÓPICA ENDOSCÓPICA TAC TAC HELICOIDAL HELICOIDAL CON CON RECONSTRUCCIÓN RECONSTRUCCIÓN EN EN 3D 3D ENDOSCOPIA ENDOSCOPIA 4 Cribado del cáncer de colon: a. totalmente negativo. b. Cambio maligno que no lo puedo detectar. c. Se detecta. d. Terminal. e. Letal. Ventana de control: se tiene que tratar de diagnosticar y es vital. Los mejores screening: endoscopia y HOS. TEMA 10 Dr. Carballo 7 CMG MÉDICA DE APARATO DIGESTIVO CMG CÁNCER COLORRECTAL ALTERNATIVAS EN TÉCNICAS DE SCREENING V ULTRASONOGRAFÍA INTESTINAL Identifica pólipos >7 mm en el 90% de los casos Sensibilidad 94% / Especificidad 100% ¿Aplicable en el futuro al screening? •RECONSTRUCCIÓN EN 3D TAC HELICOIDAL –Sensibilidad 80% con baja Especificidad – Escala de grises TEMA 10 Dr. Carballo 8 CMG MÉDICA DE APARATO DIGESTIVO CMG TEMA 11 HEPATITIS AGUDA DEFINICIÓN La hepatitis vírica es una inflamación aguda, ocasionada por virus y otras causas, en personas con hígado sano. ANATOMÍA PATOLÓGICA 1. Inflamación, pero sin polimorfonucleares. La inflamación suele disponerse en los espacios porta y en los sinusoides: luz y espacio de Disse (subendotelial). Hay predominio de mononucleares (linfocitos y monolitos), no obstante, puede haber eosinófilos y polimorfonucleares de manera secundaria. 2. Necrosis hepatocitaria: el agente lesivo atrae a los linfocitos hacia los hepatocitos, y junto a la inflamación aparece también necrosis. 3. Colestasis: alteración de la excreción biliar de bilirrubina y sales biliares, fosfatasa alcalina y gamma GT. Es debida a la acumulación de pigmento biliar en el hepatocito, no se debe confundir con la ictericia. MANIFESTACIONES TRAHEPÁTICAS EX- 1. Son frecuentes las linfadenomegalias. 2. Pueden aparecer también linfopenia y leucopenia, por la emigración hacia los hepatocitos que linfocitos y leucocitos llevan a cabo, y porque estos pueden ser destruidos directamente por los agentes lesivos. En especial en hepatitis C, en la fase tardía de la enfermedad, puede surgir anemia aplásica (ausencia de producción de hematíes en la médula ósea). TEMA 11 Dr. Pons CAUSAS AGUDA DE HEPATITIS 1. VÍRICA: puede estar causada por los llamados virus hepatotropos: VHA, VHB, VHC, VHD, VHE, VHG; o por virus no hepatotropos: virus de Ebstein Baar (mononucleosis infecciosa), virus del Herpes Simple y Varicela Zoster, Adenovirus, Parotiditis, etc. 2. POR FÁRMACOS: los medicamentos pueden inducir hepatitis aguda por dos mecanismos: a. Toxicidad directa del fármaco: se manifiesta de manera predecible en los individuos expuestos al agente nocivo y depende de la dosis. El periodo de latencia entre la exposición al agente tóxico y la lesión hepática suele ser corto (horas), aunque las manifestaciones clínicas tardan a veces hasta 24 o 48 h. El medicamento que con más frecuencia aparece como responsable de hepatitis por este mecanismo es el paracetamol (se estima que se requieren entre 6 y 7g. para inducir hepatitis aguda, aunque en alcohólicos bastaría con 3 o 4g.), que es la primera causa de hepatitis fulminante, normalmente por suicidio. También pueden causar hepatitis aguda el halotano y el tetracloruro de carbono. b. Toxicidad inmunoalérgica (reacción idiosincrásica): entre las reacciones idiosincrásicas por medicamentos, la hepatitis suele ser infrecuente e impredecible, la respuesta no guarda relación con la dosis y puede suceder en cualquier momento durante la exposición, o al poco tiempo después de ella. Comisionistas: Mariló y Miriam 1 CMG MÉDICA DE APARATO DIGESTIVO Además de la dificultad de predecir o identificar la hepatotoxicidad idiosincrásica a fármacos, está la aparición de incrementos leves y transitorios no progresivos de aminotransferasas séricas, que muestran resolución si se continúa el uso del fármaco (“adaptación”). En alrededor de la cuarta parte de los pacientes con reacciones hepatotóxicas idiosincrásicas a medicamentos se producen manifestaciones extrahepáticas de hipersensibilidad, como erupción cutánea, fiebre, leucocitosis y eosinofilia; este hecho, junto con la imposibilidad de predecir la hepatotoxicidad idiosincrásica de los medicamentos, refuerza la hipótesis de que este tipo de reacciones inducidas por fármacos son mediadas inmunitariamente. Los medicamentos que destacan en este grupo son los antibióticos (amoxicilina+clavulánico), los AINEs, las estatinas y los tuberculostáticos. 3. POR TÓXICOS: el alcohol es el más frecuente. No sólo puede ser víctima de hepatitis aguda el alcohólico crónico, sino también en el alcohólico agudo, “el del fin de semana”. 4. TRASTORNO HEMODINÁMICO (ISQUEMIA HEPÁTICA): para que se produzca hepatitis aguda por esta causa debe tener lugar un proceso dramático como un shock cardiogénico o hipovolémico. 5. ENFERMEDAD BILIAR: por ejemplo, una obstrucción que cause reflujo biliar al hepatocito puede causar hepatitis aguda. 6. METABÓLICA Y OTRAS CAUSAS MÁS RARAS. FORMAS CLÍNICAS DE LA HEPATITIS AGUDA 1. FORMA CLÁSICA: ICTÉRICA O ANICTÉRICA: en la forma ictérica se distinguen tres fases: a. La primera es la fase de pródromos, que se caracteriza por un cuadro pseudogripal, con febrícula, malestar general, etc., aunque también pueden apa- TEMA 11 Dr. Pons CMG recer también síntomas digestivos como náuseas, vómitos o diarrea. Comienza entre 2 y 4 semanas antes. b. La siguiente es la fase clínica, en la que aparecen de forma secuencial coluria, heces hipocólicas e ictericia. Cuando aparece la ictericia, que es síntoma de alarma, suelen disminuir los síntomas generales prodrómicos. c. La última fase es la de convalecencia, en la que aparece astenia y una recuperación progresiva. La forma anictérica es asintomática y se diagnostica de forma casual. Se caracteriza por un aumento de las transaminasas y aparece en un poco menos de la mitad de las hepatitis agudas. Las crónicas, en una amplia mayoría, son precedidas, aunque muchos años atrás, por una hepatitis aguda anictérica. 2. FORMA COLESTÁSICA: empieza con ictericia y prurito. Es la forma clínica más frecuente de la hepatitis A. 3. FORMA RECURRENTE: debuta con la forma clásica de hepatitis, y tras la recuperación vuelve a aparecer la enfermedad, con un cuadro de astenia que puede durar muchos meses. Está relacionada con el ejercicio, el incumplimiento del reposo que se prescribe. Aparece más frecuentemente en la hepatitis A. 4. FORMA FULMINANTE: sólo un 1% de las hepatitis agudas son de esta forma. Sus características son trastorno grave de la coagulación y/o encefalopatía hepática, que aparecen entre la primera y la octava semana después de la hepatitis. Con frecuencia se confunde con la clásica; para distinguirlas, debemos echar mano del estudio de la coagulación. En la forma clásica, la colestasis puede causar una disminución de los niveles de vitamina K y una alteración de la coagulación, alteración que remite sin problemas con la reposición de la vitamina K. En la forma fulminante, en cambio, el trastorno de la coagulación no responde a la vitamina K. Comisionistas: Mariló y Miriam 2 CMG MÉDICA DE APARATO DIGESTIVO CMG La clasificación de hepatitis fulminante más usada se basa en el tiempo ictericiaencefalopatía: a. Hiperaguda: 1-7 días. b. Aguda: 8-28 días. c. Subaguda: 5-26 semanas. DIAGNÓSTICO EXPLORACIÓN FÍSICA: En el 70% de los casos encontraremos hepatomegalia, esplenomegalia en un 30% e ictericia en un 50%. Es frecuente palpar adenopatías. EXPLORACIONES COMPLEMENTARIAS: (medición de transaminasas, fosfatasa alcalina y bilirrubina en suero). Un aumento de las transaminasas de tres veces su valor normal nos permite diagnosticar una hepatitis aguda. La fosfatasa alcalina no suele aumentar más de tres veces salvo en la colestasis. En cuanto a la bilirrubina, aumenta de forma variable en un 50% de los casos. La ictericia se hace visible cuando la bilirrubina supera la cifra de 1’6 mg/dl. En orina se determina la bilirrubina directa y el urobilinógeno. VIRUS DE LA HEPATITIS A (VHA) VIROLOGÍA Y ETIOLOGÍA El virus de la hepatitis A es un virus RNA que pertenece al género hepadnavirus, con 27 nm de diámetro, sin cubierta. El ARN codifica una poliproteina de 2227 aas, de las que derivan las 4 proteínas de la nucleocápside (VP1, VP2, VP3, VP4, estas proteínas son contra las que se dirigen los anticuerpos cuando el virus entra en el organismo) y varias proteínas estructurales. TEMA 11 Dr. Pons El virus de la hepatitis A sólo se reproduce en el hígado, pero está presente en hígado, bilis, heces y sangre durante la fase final del período de incubación, que dura cuatro semanas, y en la fase aguda preictérica de la enfermedad. La presencia del virus en heces se puede detectar desde dos semanas antes de la aparición de la clínica y hasta diez días después de la misma. El aislamiento del paciente no debe durar más de estos diez días, porque con la aparición de la clínica desaparece el riesgo de contagio (las personas que han estado expuestas al enfermo durante ese periodo deberán someterse a profilaxis). A pesar de la persistencia del virus en el hígado, su paso a las heces y a la sangre disminuye rápidamente una vez que la ictericia se hace evidente. MARCADORES SEROLÓGICOS Se pueden detectar IgG anti-VHA durante la fase aguda, cuando está elevada la actividad de las aminotransferasas séricas y aún hay virus en las heces. Esta respuesta precoz se mantiene durante varios meses, normalmente entre 6 y 12 meses. Durante la convalecencia, sin embargo, los anticuerpos anti-HVA que predominan son de tipo IgG. Por tanto, el diagnóstico de hepatitis A durante la fase aguda de la enfermedad se basa en la demostración de antiHVA de tipo IgM. Una vez superada la enfermedad aguda, los anti-HVA de tipo IgG se detectan toda la vida y los pacientes con antiHVA son inmunes a la reinfección. Comisionistas: Mariló y Miriam 3 CMG MÉDICA DE APARATO DIGESTIVO CMG La mayoría de las hepatitis se contagian en la edad infantil, de manera que el 50% de la población adulta está inmunizado frente a la hepatitis A. EPIDEMIOLOGÍA Se transmite casi exclusivamente por la vía fecal-oral. La diseminación del HVA de una persona a otra aumenta con la higiene personal deficiente y el hacinamiento, y se han detectado brotes epidémicos así como casos individuales en relación con alimentos, agua, leche, frambuesas, fresas congeladas y mariscos. También es frecuente la diseminación intrafamiliar e intrainstitucional (cárceles, residencias de ancianos, colegios, militares, etc.). La prevalencia de anticuerpos en la población, es distinta según sean países desarrollados o no. Esta gráfica nos va a medir de manera indirecta el grado de desarrollo de un país, este seria el caso de España. En la población general, la prevalencia del anti-HVA, un marcador excelente de infección previa por el HAV se incrementa con la edad y la precariedad socioeconómica. En los países en vías de desarrollo, la infección y la consiguiente inmunidad son casi universales en la infancia. Conforme disminuye en los países desarrollados la frecuencia de infecciones subclínicas en la infancia, emerge una cohorte de adultos predispuestos. PROFILAXIS Se dispone de inmunización pasiva con inmunoglobulina (Ig) y de inmunización activa con una vacuna de virus muertos. Cuando la Ig se administra antes de la exposición o durante la fase inicial del período de incubación, previene la hepatitis A clínicamente manifiesta. Para la profilaxis postexposición de contactos íntimos (en hogar, instituciones o relaciones sexuales) de personas con hepatitis A, la administración de Ig debe hacerse tan pronto como sea posible, pues es eficaz hasta dos semanas después de la exposición. La vacuna contra la hepatitis A confiere protección al cabo de cuatro semanas de la primera dosis. Si puede administrarse al menos cuatro semanas antes de que se vaya a producir una posible exposición, como un viaje a una zona endémica, la vacuna constituye el método de elección para la inmunoprofilaxis preexposición. TEMA 11 Dr. Pons Comisionistas: Mariló y Miriam 4 CMG MÉDICA DE APARATO DIGESTIVO CMG VIRUS DE LA HEPATITIS B (VHB) VIROLOGÍA El virus de la hepatitis B es un virus DNA bicatenario con una estructura genómica muy compacta basada en cuatro genes superpuestos: S, C, P y X; que le permite codificar la síntesis de múltiples proteínas.Tiene forma redondeada, de 42 nm. Existen diferentes partículas virales: grandes y esféricas (partículas de DANE), con forma tubular, es el virus completo. Tiene envoltura externa de carácter lipídico, Partículas cilíndricas con trozos de Ag Australia1, que es el Ag de superficie (HBs Ag). Cápside icosaédrica (nucleocápside), HBcAg (anti core). En su interior tiene ADNpolimerasas. El gen S codifica la proteína “mayor” de la envoltura, el antígeno de superficie (HBsAg). Antes del gen S se encuentran los genes preS, que codifican los productos génicos pre-S, entre los que figuran los receptores de superficie del VHB para la albúmina sérica humana polimerizada y para las proteínas de la membrana del hepatocito. La región pre-S está constituida por dos componentes, pre-S1 y pre-S2. Dependiendo de dónde se inicie el proceso de traducción del gen pueden sintetizarse tres productos diferentes del gen HBsAg. El producto proteínico del gen S es el HBsAg (proteína mayor), el producto de la región S más la región adyacente pre-S2 es la proteína intermedia, y el producto de las regiones pre-S1 más pre-S2 más S es la proteína grande. El gen C codifica la síntesis de dos proteínas de la nucleocápside: HBcAg y HBeAg. El antígeno central del virus de la hepatitis B (HBcAg) es el antígeno que se expresa en la superficie de la nucleocápside. El antígeno e del virus de la hepatitis B es una proteína soluble de la nucleocápside que no forma partículas y que difiere inmunológicamente del HBcAg. 1 Fue el primero que se descubrió y lo hizo Blumberg en Filadelfia en 1965, con el suero de dos hemofílicos politransfundidos, lo mezcló con suero de un aborigen australiano y había precipitación. Había un Ag Australia (por el aborigen) que luego se vio que era un Ag de superficie. TEMA 11 Dr. Pons Mientras las partículas de HBcAg permanecen el hepatocito y no pasan al suero, las de HBeAg sí se secretan y constituyen un marcador cualitativo conveniente y fácilmente detectable de multiplicación del VHB y del grado de infecciosidad relativa. Comisionistas: Mariló y Miriam 5 CMG MÉDICA DE APARATO DIGESTIVO El gen P es el más grande de los genes del VHB y codifica la síntesis de la polimerasa de DNA. Esta enzima posee actividades de la polimerasa de DNA dependiente de DNA y de transcriptasa inversa dependiente de RNA. El cuarto gen es el X, que codifica la síntesis de una proteína pequeña que no forma partículas, antígeno x de la hepatitis B, y que ha mostrado su capacidad de estimular la transcripción inversa de VHB y la réplica de DNA de VHB. MARCADORES SEROLÓGICOS Tras la infección por el VHB, aproximadamente dos semanas después, HBsAg es el primer marcador vírico detectable en el suero. La presencia de HBsAg circulante precede a las elevaciones de la actividad de aminotransferasas séricas y a los síntomas clínicos, y se sigue detectando durante toda la fase ictérica o sintomática de la hepatitis B aguda e incluso después. En los casos típicos, el HbsAg (IgM) deja de detectarse al cabo de uno o dos meses (puede llegar a tres) de la aparición de la ictericia y raras veces persiste más de seis meses (en tal caso, hablaríamos de hepatitis crónica). Una vez que desaparece el HBsAg, se detecta en el suero el anticuerpo contra HBsAg (antiHBs), que a partir de entonces persiste indefinidamente y es el anticuerpo (IgG) protector contra la reinfección. CMG ca de infección actual o reciente por VHB. No obstante, debido en parte a que la sensibilidad de los inmunoanálisis para HBsAg y anti-HBs ha mejorado, este período ventana es raro en la actualidad. La diferenciación entre infección reciente o antigua por el VHB puede lograrse determinando el tipo de inmunoglobulina que constituye el anti-HBc. La IgM anti-HBc predomina más o menos los seis primeros meses después de producida la infección aguda, mientras que la IgG antiHBc es el tipo predominante de anti-HBc transcurrido este período. Por tanto, los pacientes con hepatitis B actual o reciente, incluidos los que están en periodo ventana, tienen IgM anti-HBc en el suero. En aquellos pacientes que se recuperaron de hepatitis B en un pasado lejano y en los portadores de infección crónica por el VHB, el anti-HBc es sobre todo de tipo IgG. El HBeAg es detectable al mismo tiempo o poco después que el HBsAg. Su aparición coincide en el tiempo con tasas elevadas de multiplicación vírica y refleja la presencia de viriones íntegros y de DNA de VHB circulantes. En la infección aguda deja de detectarse antes de 10 semanas, de lo contrario nos encontraríamos ante una infección crónica, y a continuación aparece el anti-HBe, coincidiendo con un periodo de infecciosidad relativamente más baja. Dado que el HbcAg está enclaustrado dentro de una cubierta de HBsAg, no se detecta con los métodos habituales en el suero de los pacientes con infección por el VHB. Sin embargo, el anti-HBc sí es fácilmente detectable en el suero al cabo de una o dos semanas de la aparición de HBsAg, y semanas o meses antes de que existan concentraciones detectables de anti-HBs. Dado que existe variabilidad en el momento de aparición de anti-HBs tras la infección por el VHB, en ocasiones hay un espacio de varias semanas, o mayor, entre la desaparición del HBsAg y la aparición de anti-HBs. Durante este periodo, llamado “ventana”, el anti-HBc puede constituir una prueba serológi- TEMA 11 Dr. Pons Comisionistas: Mariló y Miriam 6 CMG MÉDICA DE APARATO DIGESTIVO MANIFESTACIONES HEPÁTICAS EXTRA- El síndrome prodrómico que se observa en la hepatitis B aguda responde, al parecer, al depósito de inmunocomplejos circulantes de HBsAg-anti-HBs en las paredes de los vasos sanguíneos de los tejidos, lo cual activa el sistema del complemento y disminuye sus valores séricos. Los pacientes con hepatitis B crónica pueden sufrir otras enfermedades por inmunocomplejos. Ocasionalmente se detecta glomerulonefritis. En un porcentaje más bajo (menor del 1%), también aparece poliarteritis. Otras manifestaciones extrahepáticas son la crioglobulinemia mixta esencial2, la mialgia reumática y la polineuropatía de Guillain-Barré. EPIDEMIOLOGÍA La prevalencia de la infección, sus modos de transmisión y la conducta humana conforman patrones geográficos diferentes desde el punto de vista epidemiológico de la infección por VHB. Para entender la magnitud del problema, es muy prevalente en países subsaharianos (10-20% de prevalencia de portadores) y en países del Este (6% de prevalencia en rumanos, rusos, ucranianos por transmisión perinatal). En China hay mucha incidencia, pero con la vacuna está disminuyendo. En los países desarrollados, Estados Unidos y Europa occidental, la hepatitis B es fundamentalmente una enfermedad de la adolescencia y de las primeras etapas de la vida adulta, periodos de la vida en que suelen ser más frecuentes los contactos sexuales y la exposición percutánea recreacional o laboral. En España, los principales mecanismos de transmisión son: transfusión y vía sexual y en drogadictos. CMG PROFILAXIS Y TRATAMIENTO La profilaxis contra la hepatitis B preexposición se basa en la vacuna recombinante de forma universal. Se realizan tres inyecciones intramusculares de vacuna en los meses cero, uno y seis. Para la profilaxis postexposición se recomienda una combinación de gammaglobulina (para obtener rápidamente títulos elevados de anti-HBs circulante) y de vacuna (para conseguir inmunidad duradera y aprovechar su eficacia frente a la enfermedad clínica en caso de administrarse tras el contagio).3 En cuanto al tratamiento de la fase aguda de la enfermedad, no es necesario aplicarlo. Esto es debido a que el 99% de los adultos previamente sanos que presentan un cuadro clínico evidente de hepatitis aguda se recuperan del todo, y es poco probable que el tratamiento antivírico mejore las tasas de recuperación. No está definido que se haya que poner interferon, hay que esperar y si a los 6 meses no ha desaparecido pone. Hay que administrar una dosis recuerdo a los 5 años. Para saber la efectividad de la vacuna se hace una serología y si tenemos mas de 100 UI de antiS o 10% UI/mg de anti S, estamos bien vacunados. Es efectiva en el 90% de la población. La hepatitis B se puede cronificar: - Si se adquiere por vía perinatal, la probabilidad de cronificación es del 90%. - Si se adquiere a la edad adulta, es del 10%. Embarazos: no se transmite por vía placentaria en los primeros meses de gestación, pero conforme se acerca el momento del parto hay más riesgo de infección, por lo que al niño hay que vacunarlo nada mas nacer y poner globulina en los primeros 15 días. 3 2 Se caracteriza por fiebre, artritis y erupción cutánea. TEMA 11 Dr. Pons La vacuna se pone una dosis basal, otra al mes y la última a los 6 meses. Comisionistas: Mariló y Miriam 7 CMG MÉDICA DE APARATO DIGESTIVO CMG VIRUS DE LA HEPATITIS C (VHC) GENERALIDADES El virus de la hepatitis C es un virus RNA. Se trata de un virus de la familia flaviridae de 60 nm de diámetro, envuelto. El ARN codifica a una poliproteina precursora de la que se derivan por fragmentación enzimática 2 proteínas estructurales y 5 no estructurales. Su diagnóstico en fase aguda se basa en la detección del RNA del virus y del anticuerpo anti-VHC en suero. El RNA desaparece antes de los seis meses, si no, hablaríamos de una hepatitis crónica. Hay un 10% de pacientes que presenta anti-VHC pero no tiene VHC. La causa es que estos pacientes han sufrido una hepatitis C aguda en el pasado y el anti-VHC ha permanecido. La hepatitis C cronifica entre un 50 y 80%. Si a partir de los tres meses no desaparece el RNA, debe aplicarse tratamiento con interferón alfa, pues reduce considerablemente la tasa de cronicidad. DIAGNÓSTICO Para el diagnóstico del RNA y del anti-VHC se usan dos métodos: - Método ELISA: contiene una mezcla de péptidos sintéticos o recombinantes, o una combinación de ambos, frente a los que se miden los anticuerpos IgG que tiene el suero de la muestra. Cuando se indica que un suero es reactivo con este método se está afirmando que tiene anticuerpos frente a alguno o todos los antígenos empleados en la prueba, pero no sabemos frente a cuál o cuáles. - Método RIBA (radio inmuno blooth): se emplea para conocer qué antígenos virales son los responsables de la reactividad obtenida mediante una prueba de ELISA convencional, es decir, pone de manifiesto frente a qué péptidos existen anticuerpos. TEMA 11 Dr. Pons Comisionistas: Mariló y Miriam 8 CHC Profilaxis Portador Hep. fulminante Infec. Crónica No Ig vacuna No 0,1% Picornavirus 15-45 (2sem.-6sem.) +++ Rara ? Leve generalmente No Familia P. Incubación (días) Si Ig vacuna 1-10% (90% neonatos) Si 1% 42 DNA 3,2 Orthohepadnavir us Hepadnavirus 30-180 (1m- 6m) +++ ++ +++ Moderada 27 RNA 7,5 Hepatovirus Transmisión Feco-oral Percutánea Sexual Parenteral Clínica Gravedad VHB VHA Tamaño Ác. Nucleico Longitud genoma Clasif. Género VHC Si No Raro Raro 40-80% Like Flavivirus 15-160 (2sem.-5m) +++ Poco frecuente Poco frecuente Leve 55 (40-60) RNA 9,4 Hepacivirus ? Vacuna VHB Si Si sobrinfec. 20% Satélite 21-140 (2sem.-5m) +++ ++ + Posiblemente grave Frecuente 36 RNA 1,7 Deltavirus VHD No Embarazada si 10-20% No No Calcivirus 14-63 (2sem.-2m) +++ Leve generalmente No 32 RNA 7,5 Sefavirus VHE ? No Si ? Si ? ++ ? ? Leve ? ? RNA 9,4 Flavivirus VHG CMG MÉDICA DE APARATO DIGESTIVO CMG TEMA 12 HEPATITIS CRÓNICA CONCEPTO DE HEPATITIS CRÓNICA El concepto de hepatitis crónica incluye diversas alteraciones hepáticas de etiología y gravedad variables caracterizadas en su conjunto por inflamación y necrosis hepática que tienen una persistencia superior a seis meses. Algunas formas leves no experimentan progresión o lo hacen lentamente, en tanto que las formas graves suponen una cicatrización y reorganización estructural que culminan en cirrosis. Encontramos tres grandes tipos de hepatitis crónica según su etiología1: víricas, inducidas por fármacos y autoinmunitarias. Seguidamente se procederá a explicar en este tema las hepatitis crónicas de origen viral, que incluyen las hepatitis crónicas de los tipos B y C. mecanismo de transmisión es tanto vertical (durante el parto) como sexual y percutáneo. Este virus presenta numerosos genotipos (A, B, C, D, E, F, G, H) y serotipos con importancia a la hora del tratamiento y de la seroconversión. Es importante mencionar la existencia de una distribución geográfica de los distintos genotipos del HBV, que se puede observar en la siguiente tabla: A ByC HEPATITIS CRÓNICAS VÍRICAS Los datos clínicos y serológicos permiten diferenciar entre dos grandes tipos de hepatitis vírica crónica, como son las hepatitis crónicas B y C. Cabe destacar no obstante la existencia de un tercer tipo de hepatitis crónica, D (delta), que no se desarrollará en esta lección. Como dato informativo debemos conocer la existencia de dos formas de hepatitis víricas, de transmisión enteral, que presentan sin embargo un carácter agudo, como son las hepatitis agudas A y E, que curan de manera espontánea y que en ningún caso se cronifican. HEPATITIS B CRÓNICA (HBC) Se trata de un tipo de hepatitis crónica vírica causada por el “virus de la hepatitis B” (VHB) de la familia Hepadnaviridae, virus ADN cuyo 1 Las hepatitis crónicas se pueden clasificar también en función de criterios histológicos o grados y de su nivel de progresión o estadio. TEMA 12 Prof. Miras D E, F, G, H Noroeste de Europa y América del Norte Asia Lejano Oriente No definida Si consideramos su epidemiología, esta patología afecta a unos 400 millones de personas en todo el mundo; presenta una alta prevalencia en latitudes en vías de desarrollo tales como África, el Sudeste Asiático o Sudamérica, de alrededor del 8%, (afectando fundamentalmente a recién nacidos y niños pequeños), lugares donde la forma de transmisión más común es la perinatal. La prevalencia en Europa, concretamente en el Sur del continente, aunque menor (1-7%, con afectación más frecuente en adolescentes y jóvenes), está en aumento debido a la inmigración creciente. La prevalencia de la hepatitis crónica B es baja (<1%) en países desarrollados como EE UU, Canadá o Suecia. Haciendo referencia a España, la prevalencia es relativamente baja, cerca del 2%. Comisionista: Diego 12-1 CMG MÉDICA DE APARATO DIGESTIVO Anatomía Patológica Las lesiones morfológicas de todos los tipos de hepatitis víricas son similares, y consisten en un infiltrado panlobulillar de células mononucleares, necrosis hepatocitaria, fibrosis, hiperplasia de las células de Kupffer y grados variables de colestasis, junto con regeneración hepatocitaria e infiltrado mononuclear de predominio linfocitario. Concretamente, en la infección crónica por VHB se pueden observar hepatocitos grandes con citoplasma de aspecto similar al vidrio esmerilado. A través de una biopsia hepática podemos realizar el diagnóstico diferencial de la HBC, la cual permite determinar simultáneamente la magnitud de la necrosis y de la fibrosis, dos de las lesiones más características de esta entidad. Actualmente contamos con diversos sistemas de gradación de la lesión anatomopatológica de la hepatitis crónica B (Knodell, Metavir, Ishak, planimetría) que cuantifican la dimensión de necrosisfibrosis en una escala 010 en base a la biopsia hepática. No obstante, dichos sistemas están en detrimento, siendo el “fibroscan”, técnica de imagen no-invasiva y fiable (el aparatejo de la imagen de la derecha), un procedimiento empleado en nuestros días y que ha sustituido a la biopsia a la hora de cuantificar el grado de fibrosis. Clínica El espectro de las manifestaciones clínicas de esta patología es amplio y va desde la infección asintomática hasta una enfermedad debilitante que puede terminar incluso en una insuficiencia hepática terminal y fatal. El comienzo de la enfermedad suele ser insidioso, con excepciones de hepatitis crónica derivada de una hepatitis aguda debido a un fracaso en la curación. La probabilidad de que una hepatitis B aguda se vuelva crónica depende de la edad y del estado inmune, de tal forma que la infección se cronificará en un 90% de los casos de RN infectados por el HBV, pero sólo en un 1% en jóvenes inmunocompetentes. TEMA 12 Prof. Miras CMG Esta patología suele ser asintomática en un 95% de los casos, en otros suele ocasionar molestias vagas en hipocondrio derecho. Los síntomas más frecuentes son astenia e ictericia (continua o intermitente). Los síntomas de una HBC avanzada son complicaciones de la cirrosis, como ascitis, edema, varices gastroesofágicas, coagulopatías... y suelen ser motivo de consulta del paciente por vez primera. Aunque en casos excepcionales, la HBC puede asociarse a depósitos de inmunocomplejos en articulaciones, riñón y vasos, complicando hacia artralgias y artritis, glomerulonefritis membranosa y vasculitis generalizada, respectivamente. Historia Natural de la Enfermedad La hepatitis B crónica evoluciona hacia una cirrosis hepática en alrededor del 2-5% de los enfermos al año; el avance de la enfermedad depende de muchos factores, entre los cuales destacan el SIDA, alcoholismo, toxicidad hepática... A su vez, la cirrosis hepática puede progresar mediante dos vías diferentes: a) Hepatocarcinoma, CHC2 o carcinoma hepatocelular: aparece en el 3-5% de los enfermos por cirrosis/año. La cirrosis es un factor de riesgo para el desarrollo del tumor. b) Otras complicaciones: tales como insuficiencia hepática y descompen-sación, ascitis, hemorragias, encefalo-patía... en el 3-5% de los enfermos por cirrosis/año. Diagnóstico Para el diagnóstico de la HBC contamos con diversas pruebas, de entre las cuales cabe destacar la analítica de marcadores; esta prueba consiste en la detección sérica de marcadores víricos del VHB y de la carga viral, y permite distinguir las diferentes cepas del virus (cepas salvaje, mutante o precore, seroconversión e inactiva) en los enfermos por HBC y en los portadores del VHB (la cepa es inactiva en portadores). A continuación se adjunta un cuadro que relaciona las distintas cepas del VHB con la detección o no de los marcadores víricos y con la carga viral (cuadro MUY IMPORTANTE, posible pregunta de examen!!!): 2 No confundir las siglas CHC de carcinoma hepatocelular con HCC de hepatitis C crónica Comisionista: Diego 12-2 CMG MÉDICA DE APARATO DIGESTIVO CMG b) En pacientes HbeAg -: ALT normal ADN – Podemos categorizar la respuesta del paciente al tratamiento mediante el empleo de diversas técnicas que se comentan a continuación; de esta forma, la respuesta al tratamiento será buena en caso de que: a) Bioquímica: ALT normal. b) Histológica: mejoría de lesiones necroinflamatorias. Es importante señalar que la cepa del VHB mutante o precore es la más frecuente en la Región de Murcia. Otros métodos diagnósticos de la HBC son: - Serología - Alfa-fetoproteína: su utilidad fundamental es descartar hepatocarcinoma - ECO - Biopsia: su realización no está exenta de controversia Tratamiento Para el tratamiento de la HBC se suele utilizar una combinación de interferón pegilado con análogos de nucleósidos, que son antivirales puros cuyo mecanismo de actuación evita la replicación viral (como lamivudina, adefovir, entecavir y telbivudina). El tratamiento de esta entidad patológica se aplicará siempre que exista una hepatitis grave o evolución hacia cirrosis, pero no será necesario en los casos en que el individuo sea portador del HBV o en hepatitis leves. En casos de HBC moderadas, el tratamiento será individualizado. La definición de una buena respuesta al tratamiento por parte del paciente debe ser: a) En pacientes HBeAg +: ALT3 normal ADN – Anti-HBe + 3 c) Virológica: el ADN disminuye progresivamente hasta hacerse negativo Existirá curación completa si se cumplen y mantienen los resultados anteriores junto con el cumplimiento y mantenimiento de un resultado negativo en la detección del HbsAg. HEPATITIS C CRÓNICA (HCC) Es una enfermedad infecciosa que afecta al hígado, causada por la infección del “virus de la hepatitis C” o HCV, virus ARN + perteneciente a la familia Flaviviridae, cuyos genotipos más comunes son 1, 2, 3 y 4. Alrededor del 50-85% de las hepatitis C agudas cronifican hacia una HCC. Epidemiología El VHC presenta una distribución mundial, afectando en mayor medida al continente africano. El periodo de incubación es siempre de aproximadamente 6 meses. Si nos referimos a los mecanismos de transmisión: 1. Transmisión sanguínea: es la forma de transmisión clásica, y la más importante. El VHC se transmite mediante el contacto con la sangre de individuos infectados. Explicaría la mayor prevalencia de HCC en UDVP (usuarios de drogas por vía parenteral), en hemodializados, trasfundidos (bien por trasfusión sanguínea o de factores de coagulación) o trasplantados. Otras situaciones que se relacionan con este mecanismo de transmisión son determinadas prácticas tales como la ablación, entre otras, o la realización de body piercing. Las enzimas ALT son más conocidas clásicamente como SGOT y SGPT TEMA 12 Prof. Miras Comisionista: Diego 12-3 CMG MÉDICA DE APARATO DIGESTIVO CMG 2. Transmisión sexual: actualmente existe cierta controversia respecto a la validez de este mecanismo de transmisión para la infección por el VHC al no haberse podido demostrar con certeza. La posibilidad de transmisión tanto sexual como perinatal es del 5%, muy inferior a las que se observan en infecciones por VIH o VHB. 3. Transmisión perinatal: coincidiendo con la transmisión sexual, aún no se conoce con exactitud la posibilidad de la transmisión perinatal del agente. En España aprox. unas 800.000 personas padecen esta enfermedad, de las cuales únicamente un 20% están diagnos-ticadas. Como dato interesante cabe señalar que en nuestro país le corresponden, a cada médico de atención primaria, 40 enfermos por HCC. Anatomía Patológica Las lesiones anatomopatológicas de la HCC son muy similares a las ya anteriormente comentadas lesiones generales de las hepatitis víricas. Si bien la HCC se caracteriza a menudo por la escasa presencia de inflamación, el marcado incremento de la activación de las células del revestimiento sinusoidal, la presencia de cúmulos linfoides y grasos, y, en ocasiones, lesiones de los conductos biliares. La HCC se caracteriza por una velocidad de evolución, bien lenta o bien rápida, que puede variar en función de determinados agentes conocidos como “factores de progresión”. Dichos factores pueden ser: a) Propios del virus: carga viral en la infección, genotipo viral... b) Propios del huésped: sexo (masculino o femenino), edad de infección... c) Externos. Historia Natural de la Enfermedad Como se ha indicado anteriormente, la aparición de HCC viene condicionada casi en todos los casos (50-85%) por una hepatitis C aguda previa de 6 meses de evolución (y sin resolución de la infección, evidentemente), independientemente del mecanismo de adquisición de la infección. La HCC, a lo largo del tiempo, puede progresar hacia cirrosis hepática en el 2-20% de los afectados. De hecho, la complicación hacia cirrosis es común incluso en pacientes leves desde el punto de vista clínico o en aquellos que tienen HCC compensada. A pesar de ello, el pronóstico a largo plazo suele ser benigno en la mayoría de los enfermos. Sin tratamiento eficaz, esta cirrosis puede descompensarse (6% de los casos), y desembocar en un hepatocarcinoma o ser fatal. A continuación se expone un cuadro que explica la historia natural de la HCC desde el momento de la infección por el agente vírico hasta los tres finales posibles: infección autolimitada, hepatocarcinoma o muerte. TEMA 12 Prof. Miras Continuando con lo comentado, hay factores que favorecen la evolución rápida de la patología (menos de 20 años de desarrollo) tales como coinfección por VIH, alcoholismo, fibrosis hepática, o infección por VHC después de los 40; como factores de progresión lentos (más de 30 años de desarrollo) podemos señalar infección en edad temprana o el hecho de pertenecer al sexo femenino. Comisionista: Diego 12-4 CMG MÉDICA DE APARATO DIGESTIVO CMG del VHC (1, 2, 3 ó 4) y en coinfección VHC+VIH: Clínica Las manifestaciones clínicas de la HCC son similares a las descritas para la HBC. En general, la astenia es el síntoma más frecuente, siendo rara la ictericia. Las com-plicaciones extrahepáticas de la HCC son menos comunes que las de la HBC, aunque podemos destacar vasculitis generalizada, liquen plano, porfiria cutánea tarda, glomerulo-nefritis por inmunocomplejos o sialoadenitis, entre otras. Diagnóstico Para establecer un diagnóstico correcto de la HCC debemos seguir los pasos siguientes: Analítica: determinación de transminasas (pueden ser normales), ALT (suelen ser normales), pruebas de función hepática, anticuerpos frente al VHC, carga viral, genotipo y viremia (suele ser positiva). Técnicas de imagen: destacando el empleo de la ecografía. Biopsia hepática o fibroscan. Por último, para confirmar el diagnóstico de HCC, hemos de establecer un diagnóstico diferencial con otras patologías hepáticas, fundamentalmente HBC y hepatitis autoinmune. Tratamiento El tratamiento actual de la HCC consiste en una combinación de interferón pegilado y ribavirina (RBV), y suele tener una duración de entre 6 y 12 meses. A continuación se adjunta un cuadro con el tratamiento a aplicar (dosis, duración del tratamiento y duración del seguimiento) dependiendo de los genotipos TEMA 12 Prof. Miras Una de las principales características del tratamiento de la HCC es su baja aceptabilidad, al ser ciertamente radical, con aparición de complicaciones severas. Su objetivo primordial es la eliminación total del virus, o, en el caso de no lograrlo, intentar detener/impedir su progresión para evitar la aparición de cirrosis o de hepatocarcinoma. Actualmente el tratamiento para la enfermedad se aplica a la totalidad de los pacientes afectos, incluso si tienen ALT normales o cirrosis compensada. El PCR debe ser negativo en dos determinaciones consecutivas realizadas en los últimos 6 meses para considerar curación completa. La tasa de curación de la HCC, considerando los cuatro genotipos VHC en conjunto, es del 60%. La curación de los genotipos 1 y 4 es del 52% en un tratamiento de 12 meses de duración, y del 84% en los genotipos 2 y 3 en un tratamiento de 6 meses de duración. En la actualidad está en auge la posibilidad de recurrir a nuevos tratamientos contra la infección por el virus VHC: a) Fármacos que potencian el tratamiento actual: terapias basadas en el empleo de interferón, incluyendo así: Albuferon. ANA 245. Omega. b) Análogos de la ribavirina o res de monofosfato genasa. inhibidodeshidro- c) Fármacos con dianas moleculares: Inhibidores de la proteasa. Inhibidores de la polimerasa. Fármacos que minimizan la fibrosis hepática. Comisionista: Diego 12-5 CMG MÉDICA DE APARATO DIGESTIVO CMG TEMA 13 HEPATOPATÍAS METABÓLICAS Y AUTOINMUNES INTRODUCCIÓN Dentro de la patología hepática general, las dos entidades patológicas más frecuentes en la población son la cirrosis hepática y la hepatitis. Seguidamente, son las hepatopatías metabólicas y autoinmunes las que ocupan el segundo lugar en frecuencia. A continuación se explicarán brevemente las enfermedades más importantes englobadas en los dos conjuntos anteriores: 1. Hepatopatías metabólicas: - Enfermedad de Wilson (degeneración hepatolenticular progresiva) - Hemocromatosis hereditaria 2. Hepatopatías autoinmunes: - Cirrosis biliar primaria (CBP) - Hepatitis autoinmune - Colangitis esclerosante primaria (CEP) --- no explicada HEPATOPATÍAS METABÓLICAS ENFERMEDAD DE WILSON También conocida con el nombre de “degeneración hepatolenticular progresiva”, se trata de una patología hereditaria de carácter autosómico recesivo definida por un trastorno en el metabolismo del cobre, concretamente en su excreción biliar intrahepática., con una prevalencia de 1/30000 habitantes. Patogenia La enfermedad de Wilson se caracteriza por una acumulación excesiva de cobre en el hígado y otros tejidos (principalmente SNC y córnea), como consecuencia de la ausencia de función metabólica de una enzima de mem- TEMA 13 brana transportadora de cobre anómala del tipo P-ATPasa, encargada de movilizar dicho metal desde el interior del hepatocito hasta la vía biliar. Dicha enzima transportadora se encuentra codificada por el gen ATP7B del brazo largo del cromosoma 13, cuyas múltiples mutaciones pueden derivar en multitud de enzimas ATPasas alteradas diferentes, siendo todas ellas completamente afuncionales (destacar la mutación H1069Q del gen). Cabe enfatizar en el importante papel patogénico de una cuproproteína del tipo alfa2-globulina, la ceruloplasmina (con misión de transporte del cobre en la circulación sanguínea), cuyo déficit actúa de forma conjunta con la alteración de la ATPasa en el desarrollo de la enfermedad. (Ampliación de interés: metabolismo del cobre: el cobre es un metal que se halla en cantidades ínfimas en el organismo humano, aunque es imprescindible para la correcta funcionalidad de determinados sistemas enzimáticos. Tras su ingesta, es transportado vía portal hasta el hígado unido a la proteína albúmina, lugar donde puede alcanzar dos vías diferentes: bien regresar al torrente sanguíneo unido a ceruloplasmina, o bien ser metabolizado por enzimas lisosomales en el interior del hepatocito para luego ser eliminado vía biliar. Actualmente se explica la fisiopatología de la enfermedad de Wilson como un error de transporte del cobre desde el interior del lisosoma hepatocitario hacia la vía biliar intrahepática, con la consecuente acumulación del metal en el interior de la célula hepática) De esta forma, la ausencia de excreción biliar del cobre y la disminución de los niveles plasmáticos de ceruloplasmina determinan la acumulación progresiva y lesiva de este metal a nivel del hígado en primera instancia. Cuando el hepatocito se encuentre saturado, el cobre escapará a la circulación sanguínea, acu- Prof. Pons Comisionista: Diego 13-1 CMG MÉDICA DE APARATO DIGESTIVO mulándose en el resto de los tejidos periféricos, pero con especial tropismo por el SNC, hematíes, riñón y membrana de Descemet corneal. tectable a simple vista, sino que requiere el uso de la lámpara de hendidura. (Fig. 1) 4. Riñón: nefrolitiasis, hematuria microscópica, y quizás fosfaturia, aminoaciduria y glucosuria por alteración de la reabsorción tubular renal. Clínica dependiente de acidosis tubular renal. 5. Sangre: es común la anemia hemolítica por toxicidad de cobre, así como hiperuricemia. 6. Hueso: la alteración del recambio óseo condiciona una desmineralización progresiva que puede desencadenar osteopenia y osteoporosis. Clínica Los síntomas de la enfermedad de Wilson se deben a la toxicidad del cobre derivada de su acumulación, y varían en función del estadio de evolución en el que se encuentre, así como de los territorios dañados por el depósito del metal. A continuación se exponen los principales órganos afectados, recordando la predominancia de sintomatologías hepática, nerviosa y oftalmológica: 1. 2. Hígado: los síntomas hepáticos son los más importantes y predominantes. La lesión hepática siempre está presente a pesar de que no ocurra manifestación clínica inicial de la enfermedad, o de que ésta debute con síntomas por lesión de otros órganos distintos. La acumulación de cobre condiciona cuadros muy variados de patología hepática secundaria, siendo el más común la hepatitis crónica asintomática con transaminasas elevadas, o bien hepatitis aguda y fulminante o cirrosis hepática, estos últimos menos frecuentes. SNC: la sintomatología puede ser de carácter neurológico y neuropsiquiátrico. - - 3. Manifestaciones neurológicas: ataxia por afectación cerebelosa, y distonía (rigidez), incoordinación, atetosis y temblor por afectación de los núcleos de la base. Temblor y rigidez son las más típicas. Existe gran afectación del SNC debido a la gran sensibilidad del mismo. En un Wilson no suele haber enfermedad neurológica sin enfermedad hepática. Es más, cuando llega el daño al SNC ya tiene normalmente Cirrosis hepática. Manifestaciones neuropsiquiátricas: retraso escolar, depresión, trastornos de la personalidad, hiperactividad, pérdida de inhibición sexual, demencia… Córnea: el depósito cúprico en la membrana de Descemet da lugar a una imagen anular de color amarillo-azul denominada “anillo de Kayser-Fleischer”, que no es de- TEMA 13 CMG Fig. 1: en la imagen se aprecia el anillo de Kayser-Fleischer, señalado por las flechas, en el limbo esclero-corneal. Diagnóstico El diagnóstico de esta entidad patológica se fundamenta casi totalmente en las pruebas analíticas de laboratorio, aunque hay susceptibilidad de realizar una biopsia hepática para complementar o confirmar dicho diagnóstico. - Analítica hepática: se observará siempre un aumento de transaminasas séricas, acompañado de elevación de fosfatasa alcalina y bilirrubina únicamente cuando se acompañe de cuadros de cirrosis o de hepatitis agudas. - Metabolismo del cobre: conectando directamente con la patogenia de la enfermedad, los niveles séricos de cobre libre o no ligado a ceruloplasmina se encontrarán aumentados; por otra parte, los niveles de cobre unidos a ceruloplasmina estarán disminuidos por disminución de los niveles plasmáticos de ceruloplasmina. Estos hechos determinan una disminución de los niveles totales de cobre. Las pruebas más Prof. Pons Comisionista: Diego 13-2 CMG MÉDICA DE APARATO DIGESTIVO importantes dentro de este grupo son el cobre total y la ceruloplasmina sérica, cuya disminución por debajo de 20 mg/dl es de sospecha diagnóstica, aunque puede ser normal en un pequeño porcentaje de la población (el 90 % de la población puede tenerlo por encima de 20mg/dl y el otro 10 % aproximadamente aún teniendo niveles inferiores al 20mg/dl y resulta ser un hallazgo totalmente normal). - - Análisis de orina: es otra de las pruebas más importantes en el diagnóstico de la enfermedad de Wilson. Habrá una sospecha muy alta de esta entidad en el caso de que los niveles de cobre en 24 horas en orina sean superiores a 100 microgramos, con sospecha si son mayores de 50 microgramos (casi certeza). Biopsia hepática: se realizará en último lugar para confirmar el diagnóstico. La muestra de parénquima hepático se analiza mediante un espectrómetro de masas para determinar la cantidad de cobre por gramo de tejido hepático, que debe ser superior a 250 microgramos por gramo de tejido hepático para el diagnóstico de certeza. Tratamiento Al no poder actuar aún sobre el gen, el tratamiento de la enfermedad de Wilson se basa en la eliminación del cobre depositado, a través de diversas estrategias: - Dieta: se debe restringir de forma considerable la ingesta de determinados alimentos ricos en cobre tales como frutos secos, mariscos, setas o carne roja. - Empleo de sales de zinc: estos compuestos constituyen el tratamiento de elección en la enfermedad de Wilson asintomática, pues si misión es disminuir la absorción cúprica intestinal. Suele emplearse el acetato de zinc, el cual, en el borde en cepillo del enterocito, induce a una mayor actividad de la proteína metalotioneína, que se une sólidamente a metales entre ellos el cobre, el cual quedará depositado en el interior celular. La continua descamación de la mucosa intestinal asegura la eliminación del cobre ligado a la proteína junto con las heces. TEMA 13 CMG - Quelantes de cobre: se trata de sustancias que ligan el cobre para aumentar su eliminación renal por la orina. Los más comúnmente utilizados son la trientina (dosis de 750 mgr-1 gramo/día) y la Dpenicilamina (dosis de 1-1-„5 gramos/día). La D-penicilamina es de elección por su rápido efecto, aunque puede producir manifestaciones clínicas no deseadas semejantes a las de la enfermedad del suero (exantema, artritis, fiebre...). - Trasplante hepático: sólo indicado en casos de cirrosis hepática evolucionada o afectación neurológica en estadios tempranos con presencia de inflamación (para así minimizar las lesiones nerviosas por depósito de cobre, que pueden ser muy graves y limitantes en estadios avanzados, donde ya existe necrosis irreversible). También se indica en casos de hepatitis fulminante. Los dos últimos puntos, sólo se utilizan en caso de grave enfermedad hepática y/o neurológica. HEMOCROMATOSIS HEREDITARIA Supone un trastorno hereditario de carácter autosómico recesivo con alteración severa del metabolismo del hierro, en la que tiene lugar una excesiva absorción de este metal en intestino delgado. La génesis de esta entidad patológica se halla en la mutación génica más frecuente en la población general (mutación C282Y del gen HFE), aunque dicha mutación no implica el desarrollo de la enfermedad, al ser la hemocromatosis una patología multifactorial y no unifactorial. Aproximadamente 1/400 personas tienen esta mutación, y aunque se trate de un homocigoto (2 alelos mutados) la aparición de la enfermedad depende además de otros factores. Patogenia La hemocromatosis hereditaria se caracteriza por un depósito progresivo, masivo y tóxico de hierro en el parénquima visceral, provocado por un aumento anormal de la absorción de este metal en el intestino delgado. Esta acumulación excesiva de hierro condiciona lesión hística de las células parenquimatosas y trastornos funcionales viscerales. Prof. Pons Comisionista: Diego 13-3 CMG MÉDICA DE APARATO DIGESTIVO El aumento de la capacidad de absorción intestinal se puede producir por multitud de mutaciones del gen HFE, localizado en el brazo corto del cromosoma 6. La mutación más común de este gen, presente en el 90% de los individuos afectados, es la C282Y (además de ser la mutación general más común de la población), seguida de la H63D. - Afectación cardiaca: miocardiopatía hipertró-fica, IC congestiva, arritmias cardiacas - Afectación hepática: el hígado suele ser el primer órgano afectado, detectándose hepatomegalia en el 95% de pacientes sintomáticos. Como patologías derivadas cabe mencionar la hepatitis crónica y la cirrosis. El desarrollo de cirrosis hepática secundaria a hemocromatosis primaria es un factor de riesgo que predispone en alto grado a la aparición de hepatocarcinoma. Normalmente el diagnóstico se realiza en fase de inflamación, en la que aún no hay fibrosis. Si el diagnóstico se retrasa o no se hace, es cuando se hace más frecuente encontrar ya cirrosis hepática. Los depósitos de hierro en los órganos ocurren en forma de hemosiderina, molécula formada por la unión de cuatros proteínas ferritina, conocido almacén de este metal. Cabe diferenciar la “hemocromatosis hereditaria o primaria” de las formas secundarias de hemocromatosis, las cuales se deben a patologías varias tales como anemias hemolíticas, talasemias, anemias sideroblásticas, o incluso aumento de ingesta de hierro, pero nunca con etiología genética. Clínica El hierro se suele depositar en hígado fundamentalmente, y también en la piel, órganos endocrinos (testículos, ovarios, páncreas, tiroides, timo...), articulaciones y miocardio. Las manifestaciones generales más comunes de esta enfermedad son: - Hiperpigmentación cutánea: debido al depósito del metal en los macrófagos, y secundariamente por la estimulación de la síntesis de melanina, provocada por la estimulación de la ACTH, indirectamente en su síntesis conjunta con la MSH, debido a una insuficiencia de la corteza suprarrenal. Además, la propia cirrosis puede estimular la hiperpigmentación debido al aumento de los estrógenos (diabetes bronceada, ¿recordáis de la AP?) - Hipogonadismo: que desencadena pérdida de la líbido, impotencia, infertilidad, amenorrea. - Diabetes mellitus: el depósito pancreático de hierro conlleva la destrucción de los islotes de Langerhans. El desarrollo de este cuadro junto con la hiperpigmentación cutánea ha provocado que se conozca a la hemocromatosis hereditaria como “diabetes bronceada”. - Artropatías degenerativas, y artritis. TEMA 13 CMG Diagnóstico Encontramos diversos métodos analíticos válidos para cuantificar el depósito parenquimatoso de hierro que, junto con los datos clínicos, permiten confirmar el diagnóstico de hemocromatosis primaria: - Indice de saturación de la transferrina (IST): la transferrina es una proteína del tipo globulina encargada del transporte y almacenamiento del hierro circulante. En individuos sanos, el porcentaje de transferrina plasmática que se satura al fijarse con este metal es de 22-46 %, rango por encima del cual se establecen los niveles patológicos. Se considera patológico si IST > 45%). El IST es el test más sensible para el diagnóstico de HH, al ser la anomalía fenotípica más precoz de esta enfermedad. - Concentración sérica de ferritina: los niveles plasmáticos de ferritina se encontrarán elevados en esta enfermedad, por encima de 1000 microgramos/dl. Se trata de un indicador fiable de hemocromatosis, aunque puede encontrarse elevada en otras patologías hepáticas (como hepatopatía alcohólica). (Nota a pie de página: en la práctica clínica, la medición combinada del porcentaje de saturación de transferrina y de la concentración sérica de ferritina constituyen el método más sencillo para la detección de hemocromatosis). Prof. Pons Comisionista: Diego 13-4 CMG - MÉDICA DE APARATO DIGESTIVO Índice hepático de hierro (IHH): el cálculo de los niveles de hierro hepáticos se realiza en una muestra de biopsia. Los enfermos de hemocromatosis hereditaria presentan un nivel superior de 1‟9 de hierro hepático, cuya fórmula es: [Fehepático] Edad x Pm Tratamiento El tratamiento de esta patología consiste en eliminar el exceso de hierro corporal y en el soporte de los órganos lesionados. La manera óptima de eliminar hierro consiste en realización de flebotom ías (sangrías) de 300-500 ml una o dos veces por semana, pudiendo observar una disminución progresiva de los niveles plasmáticos de ferritina hasta niveles normales de menos de 50 microgramos/dl. El empleo de agentes quelantes, como la desferrioxamina, permite una disminución reducida de los niveles de hierro corporales en comparación con la fracción de hierro eliminada en una flebotomía, constituyendo un método poco eficaz aunque de elección en casos de anemia o hipoproteinemia graves que impiden proceder mediante sangrías. Por último destacar que la dieta restrictiva no consigue disminuir en ningún caso los niveles de hierro. HEPATOPATIAS AUTOINMUNES HEPATITIS AUTOINMUNE Esta entidad patológica constituye la enfermedad hepática inmune más frecuente, y tiende a evolucionar hacia hepatitis crónica, cirrosis e insuficiencia hepática. Se trata de un trastorno crónico (y por tanto una forma de hepatitis crónica según la clasificación clínica y serológica) caracterizado por necrosis hepatocelular sostenida e inflamación, con frecuencia de fibrosis, y más frecuente en el sexo femenino. Podemos encontrar tres tipos analíticos de hepatitis autoinmune diferentes según el tipo de marcador (anticuerpo plasmático con valor diagnóstico) que presenten cada uno de ellos: - H. autoinmune tipo I: es la forma más frecuente de esta enfermedad. Sus marcadores fundamentales son ANA (anti- TEMA 13 CMG cuerpos antinucleares) y SMA (anticuerpos anti músculo liso). Este último es imprescindible que esté. - H. autoinmune tipo II: presenta como marcador fundamental el AcLKM (anticuerpo antimembrana de hígado y riñón). - H. autoinmune tipo III: es la forma más infrecuente de esta patología, cuyo marcador principal es AcSLA (anticuerpo contra el antígeno específico hepático). En todas hay una elevación crónica de las transaminasas. Clínica Destacar como manifestaciones fundamentales astenia, malestar general, artritis frecuente, ictericia o edemas por complicación de cirrosis, encefalopatía y hepatoesplenomegalia. Tratamiento La base fundamental del tratamiento de la hepatitis autoinmune son los inmunosupresores esteroideos con respuesta terapéutica en un 80% de los pacientes, aunque no suele evitar la evolución final hasta la cirrosis u otra patología hepática grave. El tratamiento esteroideo puede combinarse con azatioprina, cuya ventaja es la reducción de la incidencia de las complicaciones derivadas del uso de esteroides. Cabe destacar la ausencia de patrones de duración del tratamiento, lo cual significa que puede mantenerse de por vida, aunque se aconseja retirarlo a los dos años con la subsiguiente observación para descartar recidivas (que se presentan en el 60% de los pacientes); en el caso de que se produzcan, no se volverá a retirar el tratamiento. CIRROSIS BILIAR PRIMARIA Se conoce con el nombre de “cirrosis biliar primaria” a una forma autoinmune de cirrosis hepática caracterizada por lesión del parénquima hepático como consecuencia de la inflamación crónica, destrucción epitelial y obliteración fibrosa de los conductillos biliares intralobulares. El 90% de los casos de CBP ocurren en mujeres, y es una entidad patológica de etiología desconocida. Prof. Pons Comisionista: Diego 13-5 CMG MÉDICA DE APARATO DIGESTIVO CMG Diagnóstico Clínica La CBP se caracteriza por la presencia del marcador específico AMA (anticuerpo antimitocondrial) como dato fundamental de laboratorio, aunque cabe señalar que su detección no es patogénica de la enfermedad, sólo presenta valor diagnóstico (puede dar falsos positivos o verdaderos negativos). Más concretamente, suele determinarse la detección del marcador AMA M2, que reconoce la subunidad E2 de la piruvatodeshidrogenasa de la membrana mitocondrial interna. Parece ser que en el epitelio biliar se expresan Ag similares a E2 y el organismo genera Ac. La mayor parte de los enfermos afectados por esta patología se mantienen asintomáticos durante largos periodos de tiempo, si bien la mayoría terminan por presentar lesión hepática progresiva. Las manifestaciones clínicas más frecuentes son: 1. PRURITO : es a menudo el síntoma inicial, y puede ser bien generalizado o bien localizado a nivel de plantas y palmas de las extremidades. Debido a la ictericia. 2. OSTEOPENIA : con predisposición a frácturas atraumáticas. (De hecho, la preexistencia de osteopenia al momento del transplante hepático se ha convertido en el factor de riesgo más importante). El equilibrio formación/reabsorción ósea está alterado. Aunque se desconoce el mecanismo que conduce a estos pacientes a la osteoporosis, se sabe que participan múltiples factores de riesgo generales: malnutrición, malabsorción, cambios hormonales… También ocurre en otras enfermedades hepáticas crónicas. 3. ESTEATORREA Y MALABSORCIÓN GRASA : manifestaciones tempranas de la CBP. 4. XANTOMAS , XANTELASMAS: estos signos característicos se deben a la elevación prolongada de los lípidos séricos, que se acumulan alrededor de los ojos (xantelasma) o sobre articulaciones y tendones (xantoma). 5. ICTERICIA : se trata de una manifestación que aparece en estadios intermedios. 6. COLESTASIS : definida como la disminución o carencia del flujo biliar normal, se produce por la lesión y obliteración de los conductillos biliares, acompañada de disminución de la excreción biliar. Lo más común es que no muestre síntomas, acompañándose de elevación sérica de la enzima fosfatasa alcalina. Lo más habitual, mujer asintomática con resultado en analítica casual de aumento de la fosfatasa alcalina 7. CIRROSIS : tal y como se comentó en los estadios anatomopatológicos de la CBP, la evolución final e inevitable de esta enfermedad es una cirrosis hepática grave con trasplante hepático como solución única. Fig. 2: Representación de un hígado cirrótico. Pregunta de examen: Los AMA no tienen valor patogénico (no causan la enfermedad) aunque sí diagnóstico. Anatomía Patológica La CBP se clasifica en cuatro estadios evolutivos según los hallazgos morfológicos: 1. ESTADIO I: denominada “colangitis destructiva no-supurativa”, se caracteriza por inflamación y necrosis de los espacios porta. 2. ESTADIO II: esta fase viene definida por inflamación y necrosis tanto portal como periportal. 3. ESTADIO III: además de las lesiones anteriores, encontramos fibrosis periportal con red cicatricial de tejido conjuntivo. 4. ESTADIO IV: etapa final de la evolución de la CBP que se identifica por el establecimiento de una cirrosis hepática, bien macronodular, bien micronodular. El profesor dijo que era importante la presencia de granulomas en los espacios Porta, ya que es característico (¿¿¿posible pregunta de examen???). TEMA 13 Prof. Pons Comisionista: Diego 13-6 CMG MÉDICA DE APARATO DIGESTIVO CMG Tratamiento Aunque no existe ningún tratamiento específico para la CBP, se ha comprobado que el empleo de UDCA (ursodiol o ácido ursodesoxicólico) alivia los síntomas de la patología y puede prolongar la supervivencia de los enfermos, en particular la supervivencia sin trasplante hepático; no obstante, el empleo de UDCA no evita la evolución final de la CBP hacia cirrosis, cuya solución única es dicho trasplante. El ursodiol se administra con alimentos, y sólo se precisa de una dosis diaria. Su mecanismo de actuación consiste en disminuir la capacidad lesiva de las sales biliares, aumentando también su excreción. CUADRO RESUMEN DIAGNÓSTICO TEMA 13 DEL Prof. Pons Comisionista: Diego 13-7 CMG MÉDICA DE APARATO DIGESTIVO CMG TEMA 14 CIRROSIS HEPÁTICA DEFINICIÓN La cirrosis hepática fue descubierta por Laennec en 1826 en una autopsia. La llamó “scirrhus” (naranja o ámbar oscuro). Se define como un proceso difuso caracterizado por la fibrosis y conversión de la arquitectura hepática normal en nódulos de estructura anómalos. Este proceso refleja un daño crónico e irreversible en el parénquima hepático. La cirrosis hepática puede clasificarse por características morfológicas, etiología, estado evolutivo, actividad de la enfermedad y complicaciones. Clasificación morfológica: - Cirrosis micronodular - Cirrosis macronodular - Cirrosis mixta Clasificación etiológica de cirrosis TEMA 14 El grado de actividad de la enfermedad hace referencia a la destrucción hepatocelular y a la infiltración inflamatoria. No obstante, la infiltración inflamatoria puede no existir y la analítica de un cirrótico puede ser normal. Por otro lado, la alteración de las enzimas hepáticas puede constituir un marcador, pero suele ser baja. Al hablar de evolución, se distingue entre la clínica, la bioquímica y la histológica. CLASIFICACIÓN Alcohol Hepatitis víricas Obstrucción biliar • Cirrosis biliar primaria • Cirrosis biliar secundaria Enfermedad metabólica congénita • Hemocromatosis • Enfermedad de Wilson • Deficit de alfa-1-antitripsina • Fibrosis quistica • Galactosemia • Glucogenosis tipo III y IV • Tirosinemia • Intolerancia hereditaria a la fructosa • Telangiectasia hereditaria familiar Enfermedad metabolica adquirida • Esteatohepatitis no alcoholica Causa vascular • Higado de estasis crónico • Enfermedad veno-oclusiva del hígado Hepatitis autoinmune Drogas y toxinas Actividad y evolución: Sifilis Sarcoidosis Bypass yeyuno-ileal Hipervitaminosis A Criptogenetica EPIDEMIOLOGÍA Se desconoce con exactitud la prevalencia de la cirrosis, aunque se sabe que la causa más frecuente es la alcohólica. Se estima que hay un 30% de cirróticos asintomáticos y no diagnosticados, en los que la enfermedad se descubre por casualidad en una intervención quirúrgica o en una autopsia. Es tres veces más frecuente en hombres que en mujeres. CLÍNICA CIRROSIS COMPENSADA (30/40%) Se caracteriza por febrícula o deterioro general con astenia, anorexia o pérdida de peso, o por hiperesplenismo. CIRROSIS NO COMPENSADA Es la cirrosis avanzada, que presenta insuficiencia hepática e hipertensión portal y sus consecuencias principales son la ascitis, ictericia, encefalopatía y hemorragia digestiva. También suele cursar con complicaciones Prof. Miras Comisionista: Mariló Jiménez 14-1 CMG MÉDICA DE APARATO DIGESTIVO hematológicas tales como la anemia y alteraciones de la coagulación. Complicaciones endocrinas como la feminización, hipogonadismo, diabetes, hipoglucemias. Complicaciones pulmonares como el síndrome hepatopulmonar, hipertensión pulmonar. Cálculos biliares y ulcus péptico. PRINCIPALES COMPLICA- CIONES En clase se habló muy por encima. La mayor parte del texto es contenido ampliado. ASCITIS CMG expresarse con manifestaciones dispépticas, seguidas de distensión o sensación abdominal de peso. Mediante la palpación, con una mano colocada sobre un hemiabdomen, puede percibirse la ola líquida al golpear con la otra mano el lado opuesto. La percusión descubre matidez en los flancos que cambia según la posición del enfermo y ocupa las porciones más declives. Si hay organomegalias, la presión con una mano sobre ese órgano hace descender y rebotar el órgano aumentando el tamaño (signo del témpano). La eco y la TC completan el diagnóstico. Por último, los derrames pleurales asociados a ascitis se producen por movimiento directo del líquido a través de los pequeños orificios del diafragma hacia el espacio pleural. Suelen producirse en el lado derecho y frecuentemente son lo bastante grandes como para producir disnea grave. Parece que es una manifestación de la hipertensión portal. El diagnóstico diferencial debe hacerse con neoplasias, como el cáncer de ovario, y con infecciones como la tuberculosis. Se asocia a derrame pleural en un 5-10%. Contenido Ampliado Como sabemos, la ascitis es la acumulación excesiva de líquido libre dentro de la cavidad peritoneal. Representa una situación de exceso corporal total de sodio y agua, pero no se conoce con exactitud el acontecimiento que pone en marcha este desequilibrio. Parece que la hipertensión portal origina una vasodilatación arteriolar esplácnica, mediada por el óxido nítrico, produciéndose un hiperaflujo de sangre al territorio venoso portal que contribuye al mantenimiento de la hipertensión portal a pesar de que escape sangre por las colaterales. Esta vasodilatación arteriolar esplácnica determina un llenado insuficiente del espacio vascular arterial (hipotensión) y provoca un estímulo mediado por barorreceptores del sistema renina-angiotensina, del tono simpático y de la liberación de hormona antidiurética, con lo que aumenta la retención de líquidos. ICTERICIA Es una manifestación de insuficiencia hepatocelular grave. La bilirrubina que aumenta es predominantemente conjugada. El diagnóstico diferencial debe hacerse con: hemólisis, insuficiencia renal funcional, infecciones bacterianas, hepatitis alcohólica, litiasis biliar, neoplasia y pancreatitis crónica. Contenido Ampliado La ictericia por daño hepatocelular es causada por las hepatitis agudas y por la cirrosis en fase avanzada. En este tipo de ictericia, se producen trastornos en la captación y eliminación de la bilirrubina, mientras que la conjugación no se afecta de forma significativa en las enfermedades adquiridas. La bilirrubina conjugada que no puede ser eliminada a la vía biliar, pasa a la sangre, donde tiende a ser recaptada por el hepatocito, compitiendo con la bilirrubina no conjugada, que por tanto también aumenta en sangre. En cuanto al diagnóstico, son claves la historia clínica (antecedentes de alcoholismo u otros tóxicos hepáticos, de hepatitis o de tuberculosis), la exploración física y las pruebas complementarias. Clínicamente, la ascitis suele TEMA 14 Prof. Miras Comisionista: Mariló Jiménez 14-2 CMG MÉDICA DE APARATO DIGESTIVO ENCEFALOPATÍA HEPÁTICA HEMORRAGIA DIGESTIVA Es una manifestación de insuficiencia hepática e hipertensión portal. Es manifestación de hipertensión portal. Puede ser espontánea, o como consecuencia de un factor desencadenante como por ejemplo una hemorragia digestiva, infecciones bacterianas, medicamentos (diuréticos, benzodiacepinas), proteínas (aminoácidos aromáticos),... Contenido Ampliado La encefalopatía hepática es un síndrome neuropsiquiátrico caracterizado por altera-ciones de la consciencia y de la conducta, cambios de la personalidad, signos neuro-lógicos fluctuantes, asterixis y alteraciones electroencefalográficas características. Es secundario a la pérdida de la función hepática, ya sea porque el parénquima hepático esté gravemente dañado o porque existan comunicaciones portosistémicas intra o extrahepáticas. En ambos casos, deter-minadas sustancias tóxicas de procedencia sobre todo intestinal, escapan del paso por el hígado y por tanto, eluden su desintoxicación, actuando a nivel sistémico, penetrando en el SNC a través de la barrera hematoencefálica. El amonio es la sustancia que más se ha visto implicada en la patogenia de la encefalopatía hepática. Puede ser espontánea o, como suele ocurrir en cirróticos, consecuencia de un factor desencadenante. El más común es la hemorragia gastrointestinal, que produce un aumento de amoniaco y otras sustancias nitrogenadas que se absorben a continuación (por un aumento de las proteínas en la luz del tubo digestivo, que pasa a contener gran cantidad de sangre). El aumento de las proteínas de la dieta también puede desencadenar encefalopatía por aumento de la generación de productos nitrogenados por las bacterias del colon. El uso excesivo de diuréticos puede causar encefalopatía por alcalosis hipopotasémica; y las benzo-diacepinas, por depresión del SNC. CMG Puede producirse por tres causas: rotura de varices esofágicas o gástricas, gastropatía hipertensiva y ulcus duodenal. Contenido Ampliado La causa más frecuente de hemorragia digestiva es la rotura de varices esofágicas o gástricas. En los pacientes con hipertensión portal, el bloqueo de la circulación portal condiciona la apertura de comunicaciones llamadas portocavas o porto-sistémica. Las de mayor interés clínico, por las graves consecuencias que pueden desencadenar, son las denominadas comunicaciones porta-ácigos, que se crean en la unión esofagogástrica originando unas dilataciones venosas varicosas en la submucosa. Estas son las denominadas varices esofagogástricas que, en función de su tamaño y del grado de hipertensión portal, pueden romperse y ocasionar una grave hemorragia digestiva alta. La gastropatía hipertensiva se trata de una congestión de la mucosa gástrica motivada por la dificultad del retorno venoso gástrico, ya que la vena coronaria estomáquica es tributaria de la porta. Dicha congestión produce pequeñas dilataciones venulares, que también pueden ser causa de hemorragia. Por último, en pacientes con comunicaciones porto-cava, parece que hay una tendencia a la hiperclorhidria como consecuencia del déficit de inactivación hepática de la histamina circulante. Esto favorece la aparición de ulcus péptico, y una de sus complicaciones es la hemorragia digestiva. INFECCIONES BACTERIANAS Aparecen en el 30-40% de los casos de cirrosis hepática y conllevan una alta mortalidad (30%). La más grave es la peritonitis bacteriana primaria (PBP). Siempre debemos tenerla en mente para procurar que no aparezca, y para ello, es útil evitar es uso de antibióticos nefro y hepatotóxicos en estos pacientes. Otras infecciones que pueden aparecer son las urinarias, respiratorias y dermatológicas. TEMA 14 Prof. Miras Comisionista: Mariló Jiménez 14-3 CMG MÉDICA DE APARATO DIGESTIVO Contenido Ampliado La PBP (sin origen manifiesto de contaminación) se relaciona con mayor frecuencia con cirrosis hepática y afecta prácticamente a todos los pacientes con ascitis. Parece que consiste en una disemi-nación hematógena de microorganismos que, junto con un hígado enfermo y una circulación portal alterada, provoca una alteración de la función de filtro normal de este órgano. Además, el líquido ascítico es un buen medio de cultivo para la multiplicación de micro-organismos. La manifestación más frecuente es la fiebre, por eso, en cualquier cirrótico con ascitis y fiebre, es esencial tomar una muestra de líquido peritoneal. Otros síntomas claves son el dolor abdominal, el comienzo agudo de los mismos y la irritación peritoneal. Pero el diagnóstico de PBP no es fácil y se basa en la exclusión de una fuente intraabdominal de infección. COMPLICACIONES HEMATOLÓGICAS Básicamente, la anemia y alteraciones de la coagulación. Contenido Ampliado El hiperesplenismo (consecuencia de la esplenomegalia congestiva, que es otra complicación frecuente en los pacientes con hipertensión portal grave) contribuye a la trombocitopenia o pancitopenia. En alcohólicos, el etanol ejerce a veces efecto depresor de la médula ósea. Y por otro lado, la disminución de la síntesis proteínica es capaz de reducir la producción de fibrinógeno, protrombina y otros factores de la coagulación. COMPLICACIONES ENDOCRINAS Son el hipogonadismo, que se caracteriza por impotencia, atrofia testicular, amenorrea e infertilidad; y la diabetes (a veces existe intolerancia a la glucosa por resistencia a la insulina endógena, sin embargo, la diabetes clínicamente manifiesta es rara). TEMA 14 Prof. Miras CMG COMPLICACIONES PULMONARES Síndrome hepatopulmonar. Se caracteriza por hipoxemia secundaria a vasodilatación microvascular pulmonar. Hay alteración de la relación V/Q, disminución de la difusión y desaturación de oxígeno. Es indicación de trasplante hepático, siempre en casos que todavía no se han complicado con hipertensión pulmonar avanzada, con el que se consigue restaurar la pO2. Hipertensión portopulmonar. Es el aumento de la presión en la arteria pulmonar en el contexto de una hipertensión portal. La obstrucción del flujo sanguíneo produce un aumento de las resistencias vasculares pulmonares. También puede requerir trasplante. CARCINOMA HEPATOCELULAR Aparece en un 10-25% de los pacientes con cirrosis. Los factores de riesgo que predisponen al desarrollo de carcinoma hepatocelular son la infección crónica por VHB y VHC, la hemocromatosis y el etanol, y en definitiva, todos los factores que contribuyen al daño crónico de la célula hepática y a la mitosis, pues hacen que el DNA del hepatocito sea más susceptible a las alteraciones genéticas. DIAGNÓSTICO SIGNOS Y SÍNTOMAS A menudo los síntomas tienen un comienzo gradual y son lentamente progresivos. La anorexia y la malnutrición originan pérdida de peso y decremento de la masa muscular. El paciente notará debilidad creciente y astenia y aparición de equimosis con facilidad. En un momento dado, se presentan las manifestaciones propias de la disfunción celular y la hipertensión portal: ictericia progresiva, hemorragia digestiva, ascitis y encefalopatía. Un signo precoz de la enfermedad suele ser la palpación no dolorosa de un hígado duro y nodular, cuyo tamaño puede ser normal o estar disminuido. Comisionista: Mariló Jiménez 14-4 CMG MÉDICA DE APARATO DIGESTIVO Otros signos frecuentes son ictericia, eritema palmar, angiomas en araña, aumento de tamaño de las glándulas parótidas y lagrimales, acropaquias y esplenomegalia. Los varones pueden presentar disminución del vello corporal, o ginecomastia y atrofia testicular, debidas, lo mismo que las alteraciones cutáneas, a trastornos del metabolismo hormonal. En las mujeres hay ocasionalmente signos de virilización o irregularidades menstruales. La contractura de Dupuytren, debida fibrosis de la fascia palmar y que origina contractura en flexión de los dedos, es consecuencia del alcoholismo, pero no es específica de la cirrosis. CMG DIAGNÓSTICO DIFERENCIAL De la cirrosis compensada: hepatitis crónica, fibrosis hepática congénita, esquistosomiasis hepática e hiperplasia nodular regenerativa. De la cirrosis no compensada: síndrome de Budd Chiari, que presenta obstrucción de las venas suprahepáticas que desencadena hipertensión portal y ascitis; y enfermedad venooclusiva. HALLAZGOS ANALÍTICOS CLASIFICACIÓN DE CHILEPUGH Anemia, como consecuencia de pérdidas gastrointestinales, hiperesplenismo o por el efecto mielosupresor del alcohol sobre la médula ósea. Permite obtener información sobre la gravedad y el pronóstico de un paciente con cirrosis. La clase A es una cirrosis compensada; la clase B es indicación de trasplante hepático. Hiperbilirrubinemia, generalmente asociada con aumentos variables de la fosfatasa alcali5 na sérica. Concentraciones elevadas de AST (aminotransferasa de aspartato). Tiempo de protrombina aumentado, reflejo de la menor síntesis hepática de proteínas de la coagulación. Albúmina disminuida, normalmente, debido también a la afección de la síntesis de proteínas. Actualmente, hay otra clasificación (MELD) que se usa para priorizar a los pacientes en lista de espera para trasplante. Tiene en cuenta la evolución de los enfermos a lo largo de un mes basándose en tres parámetros: IRN, bilirrubina y creatinina. Child-Pugh Puntaje 1 Ascitis Ausente Encefalopatía TÉCNICAS DE IMAGEN Albúmina (g/L) Ecografía, TC, RMN, arteriografía, endoscopia y laparoscopia. BIOPSIA HEPÁTICA La biopsia hepática con aguja percutánea puede ser útil para diferenciar a los pacientes con hepatopatía menos avanzada de aquellos que ya tienen cirrosis, para excluir otros tipos de lesión hepática, como la hepatitis vírica, y para valorar a los pacientes con clínica sugerente de hepatopatía alcohólica que niegan el consumo de alcohol. TEMA 14 Prof. Miras Bilirrubina (mg/dL) No 2 3 Leve Moderada Grado 1 a Grado 3 a 4 2 >3.5 2.8 - 3.5 <2.8 <2 2-3 >3 (En enf. colestásicas) (<4) (4-10) (>10) T. Protrombina % ó INR >50 <1.7 30 - 50 1.8-2.3 <30 >2.3 Clase Puntaje Sobrevida 1 año Sobrevida 2 años A 5-6 100 85 B 7-9 80 60 C 10-15 45 35 Comisionista: Mariló Jiménez 14-5 CMG MÉDICA DE APARATO DIGESTIVO CMG TRATAMIENTO Cuando la cirrosis es compensada, basta con medidas generales como evitar el alcohol y los fármacos con hepatotoxicidad. Cuando aparecen las complicaciones, éstas se tratan en el hospital. Estos enfermos deben llevar un seguimiento por parte del médico para evitar que empeoren. El tratamiento definitivo es el trasplante ortotópico hepático. TEMA 14 Prof. Miras Comisionista: Mariló Jiménez 14-6 CMG MÉDICA DE APARATO DIGESTIVO CMG TEMA 15 (I) HIPERTENSIÓN PORTAL CONCEPTO La hipertensión portal, clínicamente significativa y con potencial para producir complicaciones, se define como el incremento de la presión hidrostática en el interior del sistema venoso portal. Este incremento determina que el gradiente de presión entre la vena porta y la vena cava se eleve por encima del rango normal (2-5 mmHg), sobreviniendo las complicaciones cuando el rango es mayor de 10 mmHg. El flujo sanguíneo hepático, en condiciones normales, es de alrededor 1500 ml/ minuto (25% del Gasto Cardiaco). La arteria hepática (procedente del tronco celiaco), aporta aproximadamente el 30% del flujo sanguíneo hepático y el 50% de oxígeno utilizable por el hígado; el resto lo suministra la vena porta. La arteria hepática se ramifica en el interior del hígado. La vena porta es el tronco final formado por la unión de las venas esplénica y la mesentérica, y conduce al hígado toda la sangre procedente del bazo, el páncreas, el estómago, el duodeno, el intestino y el mesenterio. Este aumento sostenido de la presión portal, ocasiona el desarrollo de circulación colateral, que deriva una parte del flujo sanguíneo portal a la circulación sistémica sin pasar por el hígado (shunt portosistémico), y que incluye las varices esofágicas. La trascendencia de este síndrome, muy frecuente en el curso de las enfermedades crónicas del hígado, está determinada por sus graves consecuencias: hemorragia digestiva por rotura de varices esofagogástricas, ascitis y encefalopatía hipertensiva, entre otras. Para comprender las consecuencias fisiopatológicas y la clínica es necesario conocer un poco la anatomía y fisiología esplácnica, que iremos viendo a continuación. ANATOMIA HEPÁTICA El hígado es el único órgano de la cavidad abdominal que tiene un doble sistema de aporte de sangre (vena porta y arteria hepática). La sangre procedente de ambos sistemas se mezcla en los sinusoides hepáticos. El retorno venoso del hígado es recogido por las venas suprahepáticas que desembocan en la vena cava inferior, cerca del diafragma. TEMA 15 (I) Dr. Miras 1 CMG MÉDICA DE APARATO DIGESTIVO AMPLIACIÓN FORMACIÓN DE LAS COLATERALES PORTOSITÉMICAS Cuando existe hipertensión portal (“cuando hay un obstáculo en sentido literal o figurado”), una proporción sustancial del flujo portal no llega al hígado puesto que es derivado a la circulación sistémica a través de una extensa red de colaterales. El aumento de la presión portal promueve la form ación de esta circulación colateral porto-sistémica, por la dilatación de las comunicaciones preexistentes pero funcionalmente cerradas . Hay varios sistemas anastomóticos entre el territorio portal y el de la vena cava. Los más importantes son: 1. CMG Luego, un aumento del flujo portal o de la resistencia vascular en cualquiera de estos niveles provocará un incremento de la presión de perfusión efectiva en el sistema portal (el gradiente de presión entre la vena porta y cava) y podrá dar lugar a hipertensión portal. La resistencia, a su vez, depende proporcionalmente de diferentes factores como la viscosidad de la sangre, la longitud del vaso e inversamente proporcional del radio del vaso. Los radiculares, constituidos por: a. El pedículo porto-cava superior, colaterales ascendentes que a partir de la vena coronaria estomática y venas gástricas cortas originan varices esofágicas y drenan a la vena ácigos. b. El pedículo porto-cava inferior, colaterales descendentes que, a través de la vena mesentérica inferior y plexo hemorroidal, drenan en la vena cava inferior. c. El pedículo posterior o sistema de Retzius, colaterales posteriores, que a través de venas retroperitoneales, drenan en las venas renales y ca va inferior. 2. Derivado de los remanentes de la circulación fetal. 3. Venas portas accesorias o venas de Sappey, que pueden conducir sangre portal al hígado cuando existe un bloqueo mecánico en el tronco de la vena porta. Estás últimas se acompañan con frecuencia del desarrollo de varices esofágicas. FISIOPATOLOGÍA HEPÁTICA La circulación portal se rige, básicamente, por los principios que determinan el flujo de fluidos en cualquier sistema hidrodinámico, es decir, P=QxR P = Gradiente de presión entre los dos extremos de un vaso, es decir, sería el gradiente de presión entre la vena porta y las venas suprahepáticas. Q = Flujo sanguíneo que circula a través de un vaso, en este caso, sería el flujo sanguíneo portal. R = Resistencia que se opone a este flujo, es decir, la resistencia vascular al flujo portal ejercida por la vena porta, las vénulas hepáticas terminales y las venas suprahepáticas. TEMA 15 (I) Una pequeña disminución del calibre de los vasos puede provocar un aumento acusado de la resistencia vascular y del gradiente de presión si no se acompaña de una reducción simultánea del flujo sanguíneo. En condiciones normales, la circulación hepática tiene una elevada distensibilidad, por lo que amplios cambios en el flujo sanguíneo sólo originan pequeñas variaciones en la presión de perfusión, pero un aumento de la resistencia vascular, es el factor inicial que provoca la aparición de hipertensión portal en la mayor parte de los casos. Una vez que se ha producido la elevación de la presión portal y el desarrollo de la circulación colateral secundaria, se desarrolla un aumento del flujo sanguíneo por el sistema venoso portal, que impide que la presión portal disminuya, a pesar del mecanismo compensatorio, que es la circulación colateral. Ahora, a su vez el aumento del flujo sanguíneo, se puede deber, a una vasodilatación esplácnica, probablemente promovida por factores endógenos, de origen endotelial y humorales, como la excesiva producción de: 1. Óxido nítrico que origina vasodilatación esplácnica que eleva el flujo sanguíneo y la presión portal y también sistémica que disminuye la presión arterial y promueve la retención de sodio. 2. Prostaciclina. 3. Glucagón. 4. Factor natriurético auricular que también originan una vasodilatación. Dr. Miras 2 CMG MÉDICA DE APARATO DIGESTIVO La excesiva producción de vasodilatadores endógenos explica la reducción de la sensibilidad vascular a vasoconstrictores endógenos observada en la hipertensión portal. En la hipertensión portal se asocia constantemente la presencia de hipovolemia, esta- blecida por la retención de sodio y agua ocasionada por la vasodilatación periférica. Esta hipovolemia contribuye a elevar la presión portal, que disminuye de forma significativa con dieta hiposódica y administración de espironolactona. a. AMPLIACIÓN ETIOLOGÍA 1 Numerosas enfermedades pueden causar hipertensión portal. Esta se clasifica según la localización de la lesión que la origina y los resultados del cateterismo de las venas suprahepáticas. Las causas más importantes son: 1. 1 HT presinusoidal: es aquella que cursa con una PSE (presión suprahepática enclavada) normal. A su vez se distinguen dos subtipos: Esta ampliación, la pongo “a modo de curiosidad”… b. Prehepáticas: la lesión asienta en el eje esplenoportal (aumento de presión en el bazo y en el segmento de la vena porta anterior a la lesión). Ejemplos: - Malformaciones portales como la hipoplasia portal. - Trombosis esplenoportal, que suele ser secundaria a casos de pancreatitis, neoplasias pancreáticas y de otros órganos intraabdominales. Intrahepática: originada por enfermedades hepáticas que afectan las ramificaciones intrahepáticas de la vena porta, sin distorsionar la circulación por los sinusoides. Ejemplos: - TEMA 15 (I) CMG Dr. Miras Esquistomatosis. 3 CMG MÉDICA DE APARATO DIGESTIVO - Enfermedades difusas del hígado en las que la lesión afecta casi exclusivamente a los espacios portales. - Hipertensión portal idiopática o esencial. - mmHg. La presión portal es idéntica o sólo ligeramente superior (1 mmHg) a la PSE. Este tipo de hipertensión portal es el más frecuente, puesto que es el que se observa en la cirrosis hepática, sobre todo en la de etiología alcohólica. también la originan, como la hepatitis alcohólica y algunas hepatopatías por fármacos. Hiperplasia nodular regenerativa. - Intoxicación por arsénico, sulfato de cobre y cloruro de vinilo. - Fibrosis hepática congénita. Enfermedades venoclusivas. - Cirrosis no alcohólicas. Hepatits alcohólicas y no alcohólicas. - Hepatocarcinomas. Tumores metastáticos. - Esteatosis hepática del embarazo. 2. HT mixta (sinusoidal y presinusoidal). 3. HT sinusosoidal: ésta es siempre de origen intrahepático y cursa con una PSE elevada y una PSL normal, de forma que existe un gradiente de presión en las venas suprahepáticas superior a 6 TEMA 15 (I) CMG 4. Dr. Miras Ht postsinusoidal: el aumento de la presión portal se debe a una obstrucción a la salida de sangre del hígado. En estos casos, la PSL está elevada, al igual que la PSE y la presión portal, y todos ellos cursan con un cuadro histológico de congestión pasiva del hígado. Según la localización del obstáculo puede ser intrahepática, cuyo paradigma es la trombosis de las venas suprahepáticas (síndrome de Budd-Chiari), o posthepática, cuyo paradigma lo constituyen la pericarditis constrictiva y los casos de malformaciones congénitas y de trombosis de la vena cava inferior en su segmento torácico. Estos dos tipos de hipertensión portal postsinusoidal (intrahepática o posthepática) cursan con una presión sinusoidal elevada, y producen una ascitis importante. 4 CMG TEMA 15 (I) MÉDICA DE APARATO DIGESTIVO Dr. Miras CMG 5 CMG MÉDICA DE APARATO DIGESTIVO CONSECUENCIAS DE LA HIPERTENSIÓN PORTAL 4. Alteraciones de la hemodinámica sistémica: consiste en un aumento del gasto cardiaco y una disminución de la presión arterial, con una reducción muy acentuada de la resistencia vascular sistémica e hipervolemia. Estos pacientes presentan, una circulación hiperdinámica, con una intensa vasodilatación arteriolar, responsables de algunas manifestaciones clínicas de la enfermedad como taquicardia o la piel caliente. Estas manifestaciones desencadenan una serie de respuestas humorales dirigidas a mantener la presión arterial en niveles normales, como la activación del SNSimpático y del sistema ReninaAngiotensina. 5. Esplenomegalia e hiperesplenismo: el aumento de la presión portal determina que con mucha frecuencia aumente el tamaño del bazo y se asocie a menudo a trastornos hematológicos (trombocitopenia y leucopenia). 6. Circulación colateral abdominal: en ocasiones la formación de colaterales se efectúa también por la pared anterior del abdomen, dando lugar a venas subcutáneas dilatadas, que constituyen la denominada circulación colateral abdominal. La existencia de hipertensión portal determina la aparición de alteraciones anatómicas, fisiológicas y hemodinámicas, que condicionan las manifestaciones clínicas de la enfermedad. Las manifestaciones de la hipertensión portal dependen en parte del nivel del territorio portal en el que se localice la lesión que ocasiona la hipertensión. En general, son más numerosas cuanto más alta sea la lesión. 1. Varices esofágicas y hemorragia digestiva: la hipertensión portal promueve la apertura de vasos, que comunican el sistema porta con las venas cavas superior e inferior. Las varices esofágicas son las colaterales más relevantes, puesto que la hemorragia digestiva por rotura de varices, es la principal complicación de la hipertensión portal. 2. Ascitis: la hipertensión portal es un factor sine qua non para la formación de ascitis. Sin embargo, sólo aparece cuando la hipertensión portal es de origen intrahepático o posthepático. 3. Derivación porto-sistémica2: la derivación de la sangre portal a la circulación sistémica (shunt porto-sistémico) tiene importantes consecuencias fisiológicas y hemodinámicas, no sólo por la disminución de la cantidad de sangre portal que perfunde el hígado que puede ocasionar atrofia hepática, sino que al no existir el filtro por parte del hígado, numerosas sustancias que se metabolizan en este órgano pasan a la circulación sistémica. Esto último repercute en la fisiopatología de la encefalopatía hepática, en la aparición de bacteriemias y en el aumento de las concentraciones en sangre periférica de algunas hormonas (como la insulina) y de fármacos administrados por vía oral. MÉTODOS DE EXPLORACIÓN DE LA HIPERTENSIÓN PORTAL En general, la exploración de la hipertensión portal exige utilizar una combinación de varias técnicas: 1. 2 El cortocircuito porto-sistémico interviene directa o indirectamente, en la patogenía del aumento del flujo sanguíneo esplácnico, que se observa en la hipertensión portal avanzada. TEMA 15 (I) CMG Dr. Miras Medición de la presión portal: a. Presión esplénica: consiste en medir la presión de los sinusoides esplénicos mediante la punción percutánea del bazo. b. Cateterismo de la vena porta: dentro de las variantes que tiene esta técnica, la que más se utiliza es la punción transhepática de la vena portal. 6 CMG c. d. 2. MÉDICA DE APARATO DIGESTIVO Cateterismo de las venas suprahepáicas: es la técnica más simple y con menor riesgo en la evaluación hemodinámica de la hipertensión portal. Consiste en la cateterización bajo control fluoroscópico de una vena suprahepática, mediante punción de la vena yugular interna o de la vena femoral. Con la punta del catéter libre dentro de la vena suprahepática se registra la presión suprahepática libre (PSL); cuando se hace avanzar el catéter hasta enclavarlo, se registra la presión suprahepática enclavada o PSE. La diferencia entre ambas es el gradiente de presión en las venas suprahepáticas. La PSE refleja la presión en los sinusoides hepáticos, por lo que también se denomina presión sinusoidal. Por su simplicidad, ausencia de riesgo y la riqueza de la información que aporta, el cateterismo de las veas suprahepáticas debe ser siempre la primera técnica empleada en el estudio hemodinámico de un paciente con hipertensión portal. También permite expresar los resultados en forma de gradiente de presión. 3. CMG Determinación del flujo sanguíneo hepático, portal y colateral. a. Flujo sanguíneo hepático. b. Flujo sanguíneo portal. c. Flujo sanguíneo de la vena ácigos. d. Medición el grado de cortocircuitos portosistémicos. Medición de la presión de las varices esofágicas. Visualización del sistema porto-colateral. a. Angiografía. b. Endoscopia: es esencial la práctica de una esofagogastroscopia, ya que permite además efectuar una valoración cuantitativa de la extensión y el volumen de las varices esofagogástricas. c. Ecografía: la ecografía es la primera técnica que se ha de utilizar para el estudio de la hipertensión portal, pues permite observar con un alto grado de precisión si existen o no malformaciones o trombosis en el eje esplenoportal; al mismo tiempo permite obtener información sobre el tamaño del bazo, la presencia de ascitis y de colaterales y, si existe o no una hepatopatía asociada. TEMA 15 (I) Dr. Miras 7 CMG MÉDICA DE APARATO DIGESTIVO CMG TEMA 15 (II) COMPLICACIONES DE LA HTP HEMORRAGÍA DIGESTIVA La causa más frecuente de hemorragia digestiva en pacientes con hipertensión portal es la rotura de varices esofágicas, siendo por otra parte la complicación más grave de la misma. Además, es una de las causas de muerte más comunes en pacientes cirróticos. HISTORIA NATURAL DE LAS VARICES ESOFÁGICAS 1. Otras causas de hemorragia digestiva en pacientes con hipertensión portal son: 1. 2. 3. Suelen formarse en la unión gastroesofágica (trayecto submucoso) y dicha formación se ve favorecida por los siguientes factores: Gastropatía de la HTP: se caracteriza por una ectasia difusa de los vasos de la mucosa gástrica que no presenta signos inflamatorios. En la patogenia de esta lesión se hallan implicados los trastornos humorales y de la hemodinámica esplácnica provocados por la hipertensión portal. Erosiones agudas de la mucosa gástrica: los pacientes cirróticos descompensados, en particular los que presenta ascitis e insuficiencia renal, peritonitis bacteriana espontánea e insuficiencia respiratoria aguda, padecen con frecuencia hemorragias por erosiones agudas de la mucosa gástrica (gastritis erosiva hemorrágica). Rara vez es masiva. a. b. c. d. 2. Varices ectópicas, es decir, aunque la localización habitual es en región esófagogástrica, pueden desarrollarla en otros lugares inusuales Ej. Vaginales, vesicales, bronquiales, coledocales y vesiculares. En este tema, desarrollaremos la hemorragia digestiva a consecuencia de varices esofagogástricas, por su mayor importancia. TEMA 15 (II) Formación de las varices esofágicas: el factor responsable del desarrollo de varices esofágicas es el aumento de la presión portal por encima de 10-12 mmHg1 (aunque no todos los pacientes con un gradiente de presión portal superior a 12 mmHg tienen varices). Ausencia de tejido de sostén. Presión negativa intratorácica. El efecto de succión durante la inspiración. La existencia de venas perforantes que comunican las varices con las colaterales periesofágicas y que actúan en forma similar a las venas comunicantes. Dilatación de las varices esofágicas: una vez formadas las varices, éstas crecen por dos motivos: un aumento de la presión en su interior y por el mantenimiento de un elevado flujo sanguíneo porto-colateral. 1 Lo subrayado, tengo puesto en los apuntes que es ¡¡básico!! También es básico: 1. Si, por efecto del tratamiento o espontáneamente, el GPVH se reduce por debajo de 12 mmHg el riesgo de hemorragia es prácticamente nulo. 2. Si, por efecto del tratamiento o espontáneamente, el GPVH se reduce >20% respecto al valor basal, el riesgo de recidiva disminuye en forma muy acentuada. Dr. Miras 1 CMG 3. MÉDICA DE APARATO DIGESTIVO Mecanismo de rotura de las varices. Aquí existen dos teorías: CMG TRATAMIENTO DE LA HEMORRAGÍA ACTIVA a. Teoría de la erosión: las varices se rompen por erosión de su frágil pared, promovida por la existencia de reflujo gastroesofágico y por el traumatismo mecánico que acompaña la deglución de alimentos sólidos. HDA por varices esofá esofágicas SMT/terlipresina Al ingreso Esclerosis o ligadura (continuar durante 5 días Con tratamiento farmacológico) b. Teoría de la explosión: como consecuencia de una excesiva presión hidrostática en su interior. Aunque hay que tener en cuenta que el factor más importante en esta teoría no es el aumento de la presión de la variz per se, sino la tensión excesiva a la que se halla sometida su pared. De acuerdo con la ley de Laplace, la tensión de la pared de las varices se expresa por la ecuación: Fracaso Control Considerar Esclerosis o Ligadura de rescate Tratamiento electivo Gradiente de presión transmural x Radio Control Fracaso TIPS/Cirugía derivativa de rescate Tensión = Grosor de la pared Esta ecuación destaca un hecho importante: el aumento de tamaño (y el adelgazamiento de la pared) multiplica el efecto deletéreo del incremento de la presión de la variz acercando la tensión de la pared al punto de rotura. Ello explica el hecho de que las varices grandes sangran con mayor frecuencia que las pequeñas y de que la presencia en la pared de las varices de signos endoscópicos que traducen un menor grosor de la pared (signos rojos) se acompaña de un mayor riesgo de hemorragia. El tratamiento debe perseguir: 1. La reposición de la volemia se efectúa con transfusiones (hasta que el hematocrito esté entre el 25-30%) y con la perfusión simultánea de expansores plasmáticos, para mantener una presión arterial sistólica superior a 90 mmHg, una frecuencia cardíaca inferior a 100 lat/min y una presión venosa central superior a 5 cm H2O. DIAGNÓSTICO El mejor método de diagnóstico en la hemorragia digestiva es la gastroscopia de urgencia. Con ésta prueba se podrá confirmar si la hemorragia digestiva corresponde a varices (sangrado activo, coágulo sangre o fibrina sobre variz) o descartarlo, así como determinar el punto sangrante. la reposición de la volemia: requiere un catéter para medir la presión venosa central y una cánula intravenosa de grueso calibre, que permita la transfusión rápida si ésta es necesaria. 2. la profilaxis de las complicaciones secundarias a la hemorragia: las más importantes son: a. Las relacionadas con el shock hipovolémico (insuficiencia renal y respiratoria). b. La encefalopatía hepática en los pacientes cirróticos (lactulosa lactilol por vía oral, eliminación de sangre del tubo digestivo, enemas de limpieza). TRATAMIENTO En el tratamiento de la hipertensión portal deben distinguirse varias fases: 1. Tratamiento de la hemorragia activa. 2. Prevención de la recidiva hemorrágica. 3. Tratamiento profiláctico. TEMA 15 (II) Dr. Miras 2 CMG c. 3. MÉDICA DE APARATO DIGESTIVO La aparición de infecciones por gérmenes entéricos, que pueden prevenirse con antibióticos. Técnicas complementarias de la HDA: 1. Escleroterapia endoscópica de urgencias: el uso de esta técnica unida o no a otros procedimientos hemostáticos, permite reducir los episodios de recidiva y la necesidad de cirugía de urgencia. Debido a las complicaciones graves suele reservarse para cuando falla el tratamiento farmacológico. 2. Ligadura endoscópica de las varices mediante bandas elásticas, con la idea de reducir el riesgo asociado a la escleroterapia. 3. Derivación porto-sistémica percutánea intrahepática (TIPS): por vía yugular se crea una comunicación entre un brazo de la vena porta y una vena suprahepática, colocando una prótesis metálica que se expande en el trayecto parenquimatoso creado. La hemostasia de la lesión sangrante. Tratamiento Farmacológico en la HDA2: La vasopresina y sus derivados sintéticos es la sustancia que más se ha utilizado. La vasopresina es un potente vasoconstrictor, sobre todo en el territorio vascular esplácnico, por lo que ocasiona reducción del flujo sanguíneo de las colaterales gastroesofágicas, puesto en evidencia por la medición del flujo sanguíneo de la vena ácigos. La vasopresina reduce significativamente la presión de las varices esofágicas. Este efecto vasoconstrictor se manifiesta también en el territorio sistémico, ocasionando un aumento de la presión arterial y un descenso del gasto cardiaco, del flujo sanguíneo coronario y de la frecuencia cardiaca. El principal inconveniente de este tratamiento se da en paciente cardiacos, contraindicado en hipertensos. Junto con la vasopresina, administramos nitroglicerina, fármaco con acción vasodilatadora sobre todo venosa, permite reducir los efectos adversos de la vasopresina. La terlipresina, es un derivado sintético de la vasopresina con acción prolongada y tiene menos efectos secundarios que la combinación anteriormente expuesta. Es el único fármaco con el que se ha demostrado una disminución de la mortalidad asociada a la hemorragia por varices. La somatostatina sustancia que inhibe la secreción de hormonas del crecimiento y de la mayoría de hormonas gastrointestinales, tiene la propiedad de disminuir el flujo sanguíneo y la presión portal sin presentar los efectos adversos de la vasopresina; luego, se puede administrar durante periodos más largos. 2 CMG Permite un mayor descenso de la presión portal y es más eficaz para detener la hemorragia y para prevenir nuevos episodios de sangrado, aunque la mortalidad quirúrgica asociada y el riesgo de encefalopatía y de empeorar la evolución de la cirrosis son sus grandes complicaciones. Las indicaciones aceptadas para la utilización de esta técnica son: a. Hemorragia por varices esofagogástricas que no ha cesado con tratamiento médico y endoscópico. b. Fracaso del tratamiento para prevenir la recidiva hemorragica y presentan dos o más recidivas significativas a pesar del tratamiento farmacológico o endoscópico. c. Recidiva hemorrágica por varices esofagogástricas en pacientes portadores de una derivación portosistémica. d. Prevención de la recidiva hemorrágica por varices esofagogástricas. e. Ascitis refractaria o recidivante. Hemorragía Digestiva Aguda TEMA 15 (II) Dr. Miras 3 CMG MÉDICA DE APARATO DIGESTIVO Esta técnica la realiza el radiólogo intervencionista, y el enfermo ha de estar en buen estado general, estadio A de Child-Pugh, con una hipertensión portal muy evolucionada y con poca probabilidad de presentar encefalopatía hepática (ya que ésta es una contraindicación del tratamiento). CMG TRATAMIENTO PROFILÁCTICO El objetivo del tratamiento profiláctico es el de prolongar la supervivencia de los pacientes eliminando la mortalidad asociada al primer episodio hemorrágico. Profilaxis del primer episodio de hemorragia Derivació Derivación portosistemica percutanea intrahepatica (TIPS) Gastroscopia Control 1-2 años Control 2-3 años Varices esofágicas Pequeñas < 5 mm Sin varices Grandes > 5 mm y/o signos rojos Tratamiento farmacologico Con beta-bloqueantes PREVENCIÓN DE LA RECIDIVA HEMORRÁGICA Ligadura endoscopica Prevenció Prevención recidiva hemorrá hemorrágica Los mejores resultados del tratamiento se obtienen en los pacientes con elevado riesgo de sangrado, que se correlaciona con: ¿Tratamiento previo con beta-bloqueantes Para profilaxis primaria? SI Añadir tratamiento Endoscópico (ligaduras) Mantener beta-bloqueantes NO Beta-bloqueantes y/o nitritos 1. El tamaño de las varices. 2. La presencia de “signos rojos”. 3. El grado de insuficiencia hepática3. 4. La presión y el índice de tensión de la pared de las varices esofágicas. Recidiva A efectos prácticos, los pacientes con varices mayores y menores de 5 mm, deben considerarse candidatos para el tratamiento profiláctico4. Recidiva Menor Seguir ligadura y fármacos Mayor TIPS/cirugía 3 Recidiva Derivación portosistemica percutanea intrahepatica TIPS El riesgo de hemorragia es variable de acuerdo con la presencia de factores de riesgo. Los principales factores de riesgo de presentar la primera hemorragia por varices son el tamaño de las varices, la presencia de signos rojos y el grado de insuficiencia hepática. 4 El tamaño de las varices es el mejor parámetro pronóstico, con un riesgo de hemorragia de alrededor del 10% a los 2 años en los pacientes con varices de pequeño tamaño y superior al 30% en los pacientes con varices de gran tamaño. TEMA 15 (II) Dr. Miras 4 CMG MÉDICA DE APARATO DIGESTIVO Tratamiento farmacológico en la profilaxis: CMG Se utilizan beta bloqueantes (propranolol) para prevenir el primer episodio de hemorragia por rotura de varices esofágicas. MEDIDAS COMPLEMENTARIAS QUE CONTRIBUYEN A DISMINUIR LA MORBILIDAD ASOCIADA A LA HDA POR HTP El tratamiento reduce el riesgo de sangrado y de fallecer por hemorragia en cualquier subgrupo de pacientes: alcohólicos y no alcohólicos, con ascitis o sin ésta, etc. 1. Evitar una sobreexpansión excesiva de la volemia. 2. Disminuir el riesgo de broncoaspiración. 3. Profilaxis de la infección entérica, para evitar bacteriemias. 4. Profilaxis de la encefalopatía. 5. Evitar el riesgo de descompensación hidrópica, ya que puede provocar aumento de la ascitis, reteniendo agua y sodio. 6. Disminuir el riesgo de fallo renal. 7. Profilaxis de la deprivación alcohólica en alcohólico activo. El único dato que permite predecir la eficacia del tratamiento es la medida del gradiente de presión en las venas suprahepáticas: los estudios han demostrado que ninguno de los pacientes cuyo gradiente se había reducido a menos de 12 mmHg sangraron; asimismo, se comprobó una disminución significativa del tamaño de las varices esofágicas y una prolongación de la supervivencia en dichos pacientes. Estos datos sugieren que es conveniente efectuar controles hemodinámicos para valorar la respuesta de la presión portal al tratamiento con beta bloqueantes; cuanto más se reduce la presión portal, más eficaz es el tratamiento, por lo que si se consigue potenciar la reducción de la presión portal, por ejemplo, mediante la terapéutica combinada, es posible que se pueda mejorar aún más la eficacia del tratamiento. Conducta frente a pacientes sin varices o con varices poco evolucionadas: Estos pacientes no tienen un riesgo significativo de presentar complicaciones por su hipertensión portal y, por lo tanto, no requieren un tratamiento inmediato. Sin embargo, ello no significa que estén libres de desarrollar varices voluminosas y de sangrar en el futuro, lo cual obliga a instaurar un seguimiento. ENCEFALOPATÍA HEPÁTICA CONCEPTO El shunt portosistémico y la insuficiencia hepática5 producen una disfunción reversible de la función cerebral, responsable de síntomas neuropsiquiátricos. Es importante destacar que si el gradiente de presión portal es inferior a 12 mmHg, el paciente no desarrollará varices a corto plazo, por lo que es posible diferir una nueva evaluación hemodinámica-endoscópica hasta 2-3 años. En caso de que existan varices pequeñas ya en el examen inicial, el riesgo de que se desarrollen y sangren es mucho más alto: alrededor del 40% tiene varices grandes al año, y el 70% a los 2 años. Por consiguiente, en estos pacientes es prudente efectuar estudios endoscópicos-hemodinámicos de seguimiento con intervalos más cortos. TEMA 15 (II) 5 La aparición de encefalopatía hepática depende, como factor imprescindible, de la existencia de insuficiencia hepática, aunque con esto no se excluyen el resto de factores. Dr. Miras 5 CMG MÉDICA DE APARATO DIGESTIVO de la tirosina (por hiperconsumo de tirosina-hidroxilasa) desviándolo de su vía normal (tirosina-dopa-dopaminanoradrenalina), para la cual aquella enzima es imprescindible, hacia la síntesis de tiramina y octopamina. Esta última sustancia se comporta como un falso neurotransmisor, bloqueando las sinapsis e interfiriendo así en la acción de los neurotransmisores fisiológicos (dopamina y noradrenalina). Pueden considerarse: 1. Factores predisponentes. 2. Factores determinantes. 3. Factores desencadenantes. FACTORES PREDISPONENTES La existencia de un grado más o menos avanzado de insuficiencia hepática, es un factor prácticamente imprescindible para que se produzca encefalopatía hepática, junto con la existencia de shunt portosistémico. El hígado insuficiente6 no puede depurar sustancias con potencial tóxico para el cerebro o dejar de sintetizar otras necesarias para el metabolismo normal de Sistema Nervioso Central. FACTORES DETERMINANTES Y PATOGENIA7 CMG 3. Benzodiacepinas. 4. GABA. En resumen, cualquiera de estas sustancias provoca las alteraciones del metabolismo energético cerebral y de la neurotransmisión y el incremento de la permeabilidad de la membrana hematoencefálica (a través de una inhibición de la actividad Na/ K ATPasa). Patogenia de la encefalopatía hepática Varias son las sustancias nitrogenadas potencialmente tóxicas para el SNC incriminadas en la patogenia de la encefalopatía hepática. Muchas de ellas tienen su origen principal en el colon, por acción de la flora proteolítica sobre las proteínas de la alimentación y, en condiciones normales, son metabolizadas por el hígado. En presencia de los factores predisponentes, se las halla en concentraciones elevadas en suero y en SNC. Incremento de la permeabilidad De la barrrera hematoencefalica Shunting Hiperamoniemia Aminoacidos aromaticos Benzodiacepinas GABA Alteración de neurotransmisores Acumulo de manganeso Estas sustancias son: 1. 2. Amoniaco, que se produce en el estómago por hidrólisis de la urea gracias a las ureasas gástricas y en el riñón (amoniogénesis renal), es normalmente utilizado por el hígado para la síntesis de urea. La toxicidad cerebral del amoniaco es bien conocida, aunque su mecanismo permanece oscuro. Aumento de aminoácidos aromáticos (fenilalanina y tirosina particularmente) en detrimento de aas de cadena ramificada (valina, leucina, isoleucina). Este desequilibrio alteraría el metabolismo intracerebral 6 Cuando la función hepática es normal, aún existiendo hipertensión portal y anastomosis portosistémicas, la encefalopatía es excepcional. Trastorno del ciclo de la urea Degradación de productos nitrogenados en el intestino FACTORES DESENCADENANTES O PRECIPI- TANTES En gran proporción de casos de encefalopatía hepática es posible identificar una situación clínica previa que ha actuado como factor desencadenante. Pueden actuar como factor desencadenante todas las circunstancias que, de alguna manera, aumentan la producción intestinal de sus- 7 Aquí pongo la patogenia para no repetir lo mismo dos veces… TEMA 15 (II) Dr. Miras 6 CMG MÉDICA DE APARATO DIGESTIVO tancias nitrogenadas. Las causas más frecuentes son: 1. Hemorragia digestiva. 2. Estreñimiento, por la prolongada permanencia de las heces en el colon y la ingesta abundante de proteínas animales provocan un aumento en la producción intestinal de sustancias nitrogenadas y actúan como factor desencadenante de encefalopatía. 3. Uso de sedantes, que requieren ser transformados en el hígado. 4. Uso de diuréticos en pacientes cirróticos con ascitis sin el debido control pueden producir trastornos electrolíticos ácido– base e insuficiencia renal funcional. 5. Hipopotasemia. 6. Infecciones. 7. Disfunción hepática aguda e insuficiencia renal. 8. Alcalosis metabólica. 9. Uso de antihistamínicos. 10. Derivación portosistémica TIPS o espontánea. CUADRO CLÍNICO Es un síndrome complejo en el que no hay un síntoma concreto, sino que el diagnóstico se realiza en base al conjunto de síntomas y signos. Vamos a diferenciar: 1. Encefalopatía aguda. 2. Encefalopatía crónica. A su vez y en cada una de ellas se aprecian: a. b. quirúrgica / Para todos estos síntomas puede establecerse una gradación. La clasificación de Trey es la más utilizada: Grado I. Euforia-depresión. Desorientación temporo-espacial. Dificultad en el habla. Dificultad en el habla. Insomnio nocturno- somnolencia diurna. Irritabilidad. Déficit de atención. Grado II. Acentuación de los signos del Grado I. Trastornos del comportamiento. Somnolencia intensa. Cambio marcado de personalidad. Gran dificultad para realizar tareas mentales. Grado III. Pérdida de conciencia (somnolencia constante y profunda, respondiendo sólo a estímulos intensos). Lenguaje incomprensible. Agitación psicomotriz. Amnesia. Desorientación temporo-espacial. AMPLIACIÓN8 ENCEFALOPATÍA AGUDA Puede aparecer en el curso de una necrosis hepática masiva o, con mayor frecuencia, en forma de brotes agudos en la cirrosis hepática. Suele iniciarse con cambios de personalidad a los que siguen trastornos del comportamiento con cambios bruscos en el humor (iras repentinas y euforias injustificadas), con frecuencia se asocian trastornos del sueño, con somnolencia diurna e insomnio nocturno. En otras ocasiones aparece una sintomatología esquizoide (síndrome paranoico, hipomaniaco, alucinatorio o megalomaniaco). Entre los signos neurológicos, el flapping tremor o temblor aleteante es el más frecuente. Puede observarse además de en las manos, en los pies, en la lengua, y en casos extremos en otros músculos sometidos a tensión. Este signo aunque muy característico no es patognomónico de encefalopatía hepática, ya que puede observarse también en las encefalopatías anóxica (cor pulmonare, policitemia) y urémica. La hiperreflexia y el signo de Babinski unilateral o bilateral son también frecuentes, sobre todo este último, en la fase de coma. Signos muy característicos son la disgrafía y la apraxia de construcción. ENCEFALOPATÍA CRÓNICA Los síntomas neuropsiquiátricos de la encefalopatía se instauran de manera lenta y progresiva con posibles exacerbaciones a menudo puramente psiquiátricas, y su curso es crónico y rebelde. Pueden desarrollarse cuadros neurológicos orgánicos irreversibles: paraplejía espástica sin afección sensitiva por desmielinización de los cordones laterales de la médula, síndrome Parkinsoniano sintomático por afectación de los ganglios basales, síndrome de focalidad cerebral y ataques epilépticos. Este tipo de encefalopatía se presenta en enfermos con función hepatocelular conservada relativamente pero con importantes comunicaciones portosistémicas espontáneas o quirúrgicas. 8 TEMA 15 (II) Síntomas neuropsiquiátricos. Síntomas neuromusculares. Disminución de la capacidad intelectual (memoria y concentración), y desorientación temporo-espacial. ESTADIOS CLÍNICOS DE LA ENCEFALOPATÍA HEPÁTICA Grado IV. Coma profundo. CMG Esta ampliación es a modo de curiosidad… Dr. Miras 7 CMG MÉDICA DE APARATO DIGESTIVO En anatomía patológica en el caso de la encefalopatía aguda y dado que inicialmente es un trastorno funcional, no suelen existir lesiones orgánicas cerebrales. En los casos crónicos, o agudos recidivantes hay proliferación difusa y agrandamiento de los astrocitos protoplásmicos, en la sustancia gris del cerebro y del cerebelo, así como en el putamen y el núcleo pálido, sin grandes alteraciones visibles en las neuronas. EXPLORACIONES COMPLEMENTARIAS 1. 2. 3. Análisis de Laboratorio: proporcionan datos pocos específicos. La amoniemia arterial o venosa es un parámetro poco fidedigno, ya que sus variaciones no siempre son paralelas al grado de encefalopatía. El LCR suele mostrar un aumento de las proteínas, del ácido glutámico y de la glutamina. Las pruebas funcionales hepáticas estarán alteradas en relación a la hepatopatía de base. Lo más importante de la analítica es para administrar si existen alteraciones electrolíticas, que hay que corregir para el control de la terapéutica de reanimación. PRONÓSTICO La encefalopatía hepática es siempre un índice de insuficiencia hepática grave. El significado pronóstico a corto plazo no es, sin embargo, el mismo en todos los casos, dependiendo del factor patogénico predominante. TRATAMIENTO De todas las terapéuticas propuestas, las que han sido verdaderamente útiles según la experiencia son las encaminadas a disminuir la producción intestinal de sustancias nitrogenadas y aquellos tratamientos de mantenimiento. Ambos los podemos englobar en: EEG: constituye el parámetro más fiel para detectar una encefalopatía hepática y valorar su curso. Sus alteraciones, aunque inespecíficas por ser comunes a otros sufrimientos cerebrales metabólicos son constantes y precoces. Provocan un enlentecimiento de la frecuencia (pasa del ritmo alfa normal, a un ritmo delta no modificable por estímulos). En la fase de coma, el trazado tiene las mismas características, con un mayor enlentecimiento. Índice de encefalopatía: combinando datos clínicos, biológicos y electroencefalográficos se ha confeccionado este parámetro para cuantificar la encefalopatía con fines de investigación clínica. Para ello se puntúan de 0 a 4 los resultados de las pruebas de conexión numérica, las diferentes intensidades de flapping tremor, la frecuencia de las alteraciones EEG, la cifra de amoniemia plasmática y, por último, el estado mental según la clasificación de Trey; con la peculiaridad de que los valores correspondientes a este último dato se multiplican por tres. La puntuación máxima es de 28 puntos. DIAGNÓSTICO DIFERENCIAL A. Control de los factores precipitantes. 1. Monitorizar los efectos de los diuréticos. 2. En caso de hemorragia digestiva, aspiración nasogástricas, enemas de limpieza, profilaxis de las infecciones. 3. Evitar la deshidratación. 4. Reposición adecuada de electrolitos. 5. Cierre o reducción de shunt portosistémico. B. Reducción de la producción intestinal de amonio. 1. Dieta. En el caso de E. Aguda 2000 cal/ día, suspender proteínas; en el caso de E. Crónica, reducir proteínas de 1.2 -1.5 g/ día, proteínas vegetales /aas ramificados. 2. Fármacos. Disacáridos no absorbibles (lactulosa o cactitol), antibióticos no absorbibles, enemas de limpieza. Diagnostico diferencial Anamnesis Semiologia Laboratorio Alcohol Hepatopatia previa Shunt portosistemico Uso de sicotropos Uso de diureticos Transgresion dietetica Cefalea intensa previa Traumatismo cerebral Fiebre Focalidad neurológica Rigidez de nuca Insuficiencia respiratoria Signos de cirrosis Signos septicos Signos de hemorragia Signos deshidratación Asterixis Foetor hepático Hemograma: Trombopenia, anemia TEMA 15 (II) CMG Tiempo de protrombina Iones Bioquimica Liquido ascitico Dr. Miras C. Efectos de las sustancias nitrogenadas sobre el cerebro. Flumazemil (eficaz en la fase aguda y en la encefalopatía precipitada por sedantes e hipnóticos). D. Transplante. 8 CMG MÉDICA DE APARATO DIGESTIVO CMG TEMA 16 ASCITIS INTRODUCCIÓN 3. La ascitis es la complicación más frecuente de la cirrosis. Hasta un 50% de los pacientes cirróticos la desarrollan en un periodo de 10 años. En general, las complicaciones más frecuentes de las cirrosis son: PATOGENIA 1. Ascitis. 2. Encefalopatía hepática. 3. Hemorragia digestiva. Las causas más frecuente de ascitis son: a. En un 75% de los casos, es debida a la cirrosis. b. En el 25% restante, se debe a otras causas como son: Insuficiencia cardiaca. Pancreatitis. Neoplasias. El que aparezca la ascitis en un paciente cirrótico tiene consecuencias desde un punto de vista pronóstico, ya que a partir de su presencia la mediana de vida de estos pacientes es de 2 años. La aparición de ascitis es una indicación de transplante hepático. La cirrosis determina cambios importantes en la microcirculación hepática (alteración de la arquitectura hepática con formación de nódulos de regeneración, capilarización de los sinusoides hepáticos con depósito de colágeno en el espacio de Disse, etc.) y esplácnica (vasodilatación con pérdida de los mecanismos de autorregulación de la presión capilar), que favorecen la formación de líquido intersticial que, al no ser drenado en su totalidad por los vasos linfáticos (conducto torácico) se acumula en la cavidad peritoneal. Al mismo tiempo los pacientes con cirrosis hepática e hipertensión portal presentan cambios importantes en la circulación sistémica, que consisten en: 1. Aumento del volumen plasmático y del gasto cardíaco. 2. Reducción de las resistencias vasculares sistémicas. 3. Activación de los sistemas vasoconstrictores que tienden a mantener constante la presión arterial. ASCITIS Se denomina ascitis a la acumulación de líquido en la cavidad peritoneal. CLASIFICACIÓN 1. Grado I ó mínima, aquella que se detecta porque en la ecografía se ve un poco de líquido. 2. Grado II ó moderada, se puede detectar por la exploración física, debido a la distensión del abdomen del paciente. TEMA 16 Grado III ó severa, gran distensión o que el abdomen, que se encuentra a tensión (barriga como en el embarazo). Esta circulación hiperdinámica se desarrolla cronológicamente antes de que aparezca la ascitis, empeora con la progresión de la enfermedad y desempeña un papel clave en la patogenia de los trastornos de la función renal que se observan en los pacientes con cirrosis y ascitis. La causa de la disminución de las resistencias vasculares se debe a la marcada vasodilatación esplácnica que se observa en pacientes cirróticos con hipertensión portal. Dr. Pons 1 CMG MÉDICA DE APARATO DIGESTIVO Transplante Hepático CMG CIRROSIS Resistencia vascular intrahepática DPPI o TIPS HTPortal VD esplácnica VC VD esplácnica Hipervolemia arterial Incapacidad cardiaca para compensar la de la precarga Presión arterial periférica Estímulos sistemas vasoconstricción (SNSimpático, SRAA, ADH) DIURÉTI COS Retención Na + Retención H2O VC Renal PG E2 Ascitis y Edemas Hiponatremia dilucional En pacientes cirróticos hay un aumento de los niveles plasmáticos de las sustancias vasodilatadoras, como: glucagón, péptido intestinal vasoactivo, prostaglandinas, sustancia P, péptido relacionado con el gen de la calcitonina y el factor activador de las plaquetas. Pero, el mediador más importante de la vasodilatación arterial en la cirrosis es el óxido nítrico (NO). El NO es una sustancia vasodilatadora muy potente, que es sintetizada en el endotelio vascular por la enzima NO-sintetasa a partir del aminoácido L-arginina. En pacientes cirróticos con ascitis: 1. Está aumentada la síntesis vascular de NO. 2. La concentración de NO en el plasma está aumentada, comparado con sujetos sanos. 3. La concentración de NO es mayor en sangre portal que en sangre venosa periférica, lo que sugiere que la producción de NO está aumentada sobre todo en la circulación esplácnica. TEMA 16 4. VC SD. HR Por último, los pacientes cirróticos tienen niveles séricos elevados de nitratos y nitritos, que son los metabolitos del NO, y aumento de la concentración de NO en el aire exhalado. En la patogenia de los trastornos de la función renal1 en la cirrosis, participan diversos sistemas neurohumorales y sustancias endógenas con propiedades vasoactivas y actividad sobre el transporte renal de sodio y agua. SISTEMAS EXTRARRENALES2: El sistema renina-angiotensinaaldosterona, que estimula la reabsorción de sodio en el túbulo distal y colector, y el sistema nervioso simpático, que activa la reabsorción de sodio en el túbulo proximal, el asa de Henle y los túbulos distal y colector y la hormona natriurética (ADH) están estimulados en la cirrosis con ascitis y, en particular, en la insuficiencia renal funcional. 1 Ahora, vamos a empezar a hablar de la “segunda parte” del esquema. 2 Que, su actuación, da lugar a la ascitis y edemas e hiponatremia dilucional. Dr. Pons 2 CMG MÉDICA DE APARATO DIGESTIVO El trastorno de la función renal más característico de los pacientes cirróticos con ascitis es la retención de sodio. La importancia de esta alteración, queda demostrada por el hecho de que la ascitis puede desaparecer en algunos pacientes reduciendo la cantidad de sodio en la dieta o bien, en otros, aumentando la excreción renal de sodio mediante la administración de diuréticos. La intensidad de la retención de sodio en la cirrosis con ascitis varía de unos pacientes a otros. Estas diferencias en la retención de sodio tienen trascendencia terapéutica y pronóstica. Los pacientes con intensa retención de sodio requieren en general dosis elevadas de diuréticos, presentan con relativa frecuencia una ascitis resistente al tratamiento diurético y tienen una supervivencia corta. Por el contrario, los pacientes con retención moderada de sodio suelen responder muy fácilmente al tratamiento diurético y tienen una supervivencia prolongada. En la mayoría de los casos la retención renal de sodio en la cirrosis se debe a un aumento de la reabsorción tubular de este ion, puesto que ocurre en presencia de un filtrado glomerular normal. En los pacientes cirróticos con insuficiencia renal, la intensidad de la retención de sodio suele ser mayor que en los enfermos sin insuficiencia renal puesto que, además de estar aumentada la reabsorción tubular, existe una disminución de la cantidad de sodio filtrada. En la cirrosis hepática con ascitis suele existir también una alteración de la capacidad renal de excretar agua. Las consecuencias clínicas de esta anomalía son un aumento de la cantidad total de agua corporal y, en casos extremos, hiponatremia dilucional. La intensidad del trastorno de excreción de agua en la cirrosis es variable de unos pacientes a otros. En algunos pacientes el trastorno es tan intenso que son incapaces de eliminar el agua que ingieren de forma normal con la dieta y presentan hiponatremia e hiposmolaridad espontáneas. La hiponatremia se denomina dilucional puesto que ocurre en el contexto de un aumento de la cantidad de agua corporal. La hiponatremia también es paradójica, dado que se asocia a retención de sodio y aumento de la cantidad total de sodio existente en el organismo. Por estos motivos, la hiponatremia TEMA 16 CMG dilucional de la cirrosis debe tratarse mediante restricción acuosa. La administración de sodio no sólo no mejora la hiponatremia sino que aumenta la formación de ascitis. La teoría fisiopatológica que mejor explica la secuencia de hechos que ocurren en los pacientes cirróticos con ascitis es la denominada “teoría de la vasodilatación arteriolar periférica”. Según esta teoría, la hipertensión portal, produce una vasodilatación esplácnica, que produciría hipotensión arterial. Automáticamente, se estimulan los sistemas reninaangiotensina-aldosterona, sistema nervioso simpático y la secreción de ADH, los cuales retornarían la presión arterial a sus límites normales o casi normales. Estos sistemas mantienen la presión arterial mediante su acción vasoconstrictora y también aumentando la reabsorción tubular renal de sodio y agua, lo cual conduce a una expansión del volumen circulante. En los pacientes con hipertensión portal moderada, el mantenimiento de la presión arterial se produciría sobre todo a expensas de la expansión del volumen circulante, el cual, al rellenar el árbol vascular dilatado, suprimiría los estímulos que activan a los sistemas vasoactivos endógenos y normalizaría el metabolismo renal de sodio y agua. Esto sería lo que ocurriría en los pacientes cirróticos sin ascitis, en los que existen hipertensión portal moderada, volumen plasmático e índice cardíaco altos, resistencias periféricas disminuidas, concentraciones plasmáticas de renina, noradrenalina y ADH normales y capacidad renal de excretar sodio y agua libre también normales. Sin embargo, a medida que la enfermedad hepática avanza y la hipertensión portal se hace más acusada, la retención renal de sodio y agua no es capaz de corregir el trastorno hemodinámico sistémico, porque la vasodilatación esplácnica es más importante, pero, sobre todo, porque casi todo el líquido retenido por el riñón se extravasa desde los sinusoides hepáticos hasta la cavidad peritoneal produciendo ascitis. En estos pacientes, por lo tanto, la actividad nerviosa simpática y la secreción de renina y ADH permanecerán elevadas para mantener la presión arterial, perpetuando así la retención renal de sodio y agua, y la formación de ascitis. Dr. Pons 3 CMG SISTEMAS TORRENAL MÉDICA DE APARATO DIGESTIVO INTRARRENALES: SÍNDROME HEPA3 : VALORACIÓN DE LA ASCITIS: CLÍNICA Y DIAGNÓSTICO4 El trastorno de la función renal más grave de la cirrosis con ascitis es la insuficiencia renal funcional, también denominada síndrome hepatorrenal. Este síndrome aparece en general en pacientes con enfermedad hepática avanzada y se caracteriza por el aumento de la concentración sérica de BUN y creatinina, y oliguria. CIRROSIS CON ASCITIS Evaluación ANAMNESIS EXPLORACIÓN FÍSICA Presión arterial La causa de este síndrome es una vasoconstricción renal que determina un descenso del flujo sanguíneo renal y del filtrado glomerular. Prostaglandinas (PGE2 y PGI2): vasodilatadoras, que participarían en el mantenimiento del flujo sanguíneo renal y del filtrado glomerular (en especial PGI2) y en la excreción de agua (PGE2). Los pacientes cirróticos con síndrome hepatorrenal tienen una disminución de la producción renal de PG. Estos hallazgos sugieren que la regulación de la función renal de los pacientes cirróticos con ascitis depende de un balance adecuado entre los sistemas vasoconstrictores y la producción intrarrenal de PG. 2. NO: la inhibición simultánea de la síntesis de NO y PG se asocia a una marcada vasoconstricción renal, lo que sugiere que el NO interacciona con las PG para mantener la hemodinámica renal de la cirrosis. 3. Sistema calicreína-bradicina: es natriurético y actúa sobre todo en el túbulo distal. Parte de su actividad se debe a su capacidad de estimular la síntesis de PG. Pero en pacientes con SD hepatorrenal está disminuida. 4. Adenosina: es un potente vasodilatador en la mayoría de los territorios vasculares excepto la circulación pulmonar y renal, donde produce vasoconstricción intensa. Función hepática Urea o BUN Creatinina sérica Sodio sérico Sodio urinario DETERMINACIONES ANALÍTICAS Primera ascitis Ingreso hospitalario Sospecha de PBE En la cirrosis hepática se han demostrado alteraciones en distintos sistemas vasoactivos intrarrenales: 1. CMG PARACENTESIS DIAGNÓSTICA Proteínas totales Recuento celular y cultivo ECOGRAFÍA ABDOMINAL Lo primero que hay que ver en todo paciente con ascitis es: 1. Situación general hepática mediante analítica. 2. Situación renal, para ver si ha desarrollado un SD hepatorrenal. (ClCr, creatinina). 3. Situación hidroelectrolítica (Na + en orina y en sangre, el sodio más bajo de lo normal es un dato de muy mal pronóstico). 4. Tener en cuenta que, cuando un paciente excreta menos de 10 mEq/24h, tiene gran retención de sodio y no le sirven los diuréticos. Aunque el diagnóstico etiológico de la ascitis es relativamente simple en la mayoría de las ocasiones por los datos clínicos y analíticos, el análisis del líquido ascítico obtenido por paracentesis constituye una maniobra obligada, porque suministra una información importante, tanto desde el punto de vista diagnóstico como pronóstico. La paracentesis se hace en el punto de McBurney izquierdo, con agujas largas mediante aspiración, con a la presión que se genera, el líquido va saliendo, y esta medida se puede usar como terapia o diagnóstico. 4 3 Que, su actuación, da lugar al SD. Hepatorrenal. TEMA 16 He encontrado unas diapositivas en Internet que me han gustado y que están bastante bien… Dr. Pons 4 CMG MÉDICA DE APARATO DIGESTIVO Medimos: 1. proteínas. 2. albúmina. 3. leucocitos. 4. LDH. 5. Amilasa (sospecha de pancreatitis). 6. ADA (sospecha de tuberculosis). que como efecto secundario puede producir ginecomastia dolorosa), si no lo toleran se puede usar amilorida o furosemida (Seguril). Primero se empieza con la espironolactona y luego se continúa con la furosemida. Otra cosa importante, es distinguir si el paciente tiene edemas o no. Si los hay, el paciente ha de perder más de 500 g. (8001Kg/ día) y si no hay edemas ha de perder entre 300-800 g. (hay que perder menos puesto que hay insuficiencia renal). La ascitis de origen cirrótico es un líquido de color cetrino con una concentración de proteínas por lo general inferior a 2,5 g/ml (trasudado)5. El contenido celular del líquido ascítico oscila entre 20 y 100 células/ml, la mayoría de ellas de origen endotelial. El contenido de neutrófilos es inferior a 250 x cl. Albúmina: cociente seroascítico: Albúmina en suero – albúmina líquido ascítico III. Diuréticos de mantenimiento. El volumen medio de la ascitis en estos pacientes es de alrededor de 10 L, oscilando entre 6 y más de 15 litros, por ello hay que realizar paracentesis evacuadora asociada a la infusión intravenosa de albúmina (8 g por litro de ascitis extraída). Si no hacemos esto, se puede producir una disfunción circulatoria (empeoramiento de la enfermedad). Si se extrae una cantidad menor a 5 litros, se puede usar expansores de volumen sistémicos como la dextrano poligelina, aunque son menos eficaces que la albúmina en prevenir la disfunción circulatoria postparacentesis Si es mayor 1,1g. /dl es un trasudado (cirrosis) más albúmina en la sangre. Si es menor de 1,1 g. /dl exudado. Por ejemplo: 3 Alb, suero - 2,8 Alb. LA = 0,2, es un exudado, echo muchas proteínas y hay una gran diferencia seroascíticas. La paracentesis evacuadora hay que hacerla las veces que necesite el paciente. A veces tiene poco valor porque hay pacientes malnutridos que tienen un cociente mayor a 2,5 debido a que tienen pocas proteínas en sangre. ASCITIS GRADO 3. Tratamiento TRATAMIENTO ASCITIS GRADO 3 Tratamiento inicial Se realiza en función de los distintos grados: I. II. PARACENTESIS TOTAL Solo se trata con restricción de sodio. Entre 50-90 mEq/Na+ ó 1-2 gr sal/dia. < 5 litros Restricción de sodio y diuréticos, como espirololactona6 (que es el Aldactone 5 Hay un 5% de pacientes, con concentración 2,5 g/mL, ya que excretan linfa. 6 La espironolactona actúa en el túbulo contorneado (TC) distal. La furosemida actúa en el asa ascendente de + Henle y elimina Na . NO hay que usar diuréticos de asa larga (furosemida) como monoterapia, ya que elimino el + Na en el asa ascendente de Henle y cuando llego al TC distal no excreto casi nada, produciendo deshidratación e Insuficiencia Renal. TEMA 16 CMG Expansores sintéticos (8 g/L de ascitis) (No paracentesis parciales) > 5 litros Albúmina (8 g/L de ascitis) Tratamiento de mantenimiento Dieta hiposódica (<80mEq/día) Tratamiento diurético Contraindicaciones relativas de la paracentesis - Coagulopatía: Protrombina < 40% y/o plaquetas < 40.000 - Peritonitis bacteriana espontánea Dr. Pons 5 CMG MÉDICA DE APARATO DIGESTIVO ASCITIS REFRACTARIA TRATAMIENTO ASCITIS REFRACTARIA. Tratamiento ASCITIS REFRACTARIA Falta de respuesta a : Esp 400mg/día + Fur 160 mg/día o complicaciones con dosis menores PARACENTESIS TOTAL MÁS ALBÚMINA ( 8 g/L) Como tratamiento farmacológico, se pueden dar Acuaréticos, que son inhibidores de los receptores de la vasopresina. Hay más fármacos en ensayo clínico y no están comercializados. COMPLICACIONES Recidiva de la ascitis TIPS Se basa en la restricción hídrica, menos de 1 litro de agua al día. Si el sodio suero fuera menor 120 mEq/l, hay que suspender los diuréticos, le puedes provocar sino insuficiencia renal. Dieta hiposódica (<80mEq/día) Tratamiento diurético si sodio urinario > 30mEq/día Paracentesis repetidas más albúmina CMG Indicaciones 1. Recidiva muy frecuente Ascitis tabicada Contraindicaciones Child Pugh >13 Encefalopatía crónica SD hepatorrenal: IRenal funcional sin lesión orgánica del riñón, en pacientes con cirrosis hepática y a hepatopatia fulminante. 2. Peritonitis bacteriana espontánea. Es aquella que no se puede eliminar a pesar del tratamiento médico. Incluye las siguientes situaciones: a. b. Cuando no puedo controlar la ascitis con tratamiento con diuréticos o existe resistencia a los mismos: alcanzamos dosis plenas de diurético y no hay mejoría. (160 g. de furosemida y 400 g. de aldactone en 4 semanas). Intratable, aunque podemos aumentar los diuréticos, aparecen efectos secundarios como la encefalopatía, hiperpotasemia, insuficiencia renal e hiponatremia. TRATAMIENTO DEL SD HEPATORRENAL Los criterios diagnósticos del SD hepatorrenal son: 1. Más de 1,5 mg/ dl de creatinina. 2. No presencia de factores que puedan provocar IRenal: deshidratación, hemorragias digestivas… 3. Mala respuesta a la supresión de diuréticos y administración de albúmina. ¿CÓMO SE TRATA LA ASCITIS REFRACTARIA? Valor pronóstico: Con paracentesis. Hay que tener cuidado con los diuréticos y no se deben dar si, el Na+LA del paciente es menor de 30 mg/día y el Na+ suero menor de 120 mg/día (esto indica disminución de capacidad de excreción de sodio). 1. deterioro rápido de la función renal, alcanzando más de 2,5 mg/dL de creatinina en menos de 2 semanas. HIPONATREMIA DILUCIONAL En estos casos los niveles de Na menores a 130 mEq/l. TEMA 16 + 2. deterioro más estable de la función renal, con valores de creatinina entre 1,5 mg/dL y 2,5 mg/dL durante varias semanas. Se da en el marco de ascitis refractaria. son Dr. Pons 6 CMG MÉDICA DE APARATO DIGESTIVO Tratamiento: El 100% de estos pacientes, se mueren en menos de un mes sin tratamiento. Como el SD hepatorrenal se produce por vasoconstricción, hay que dar vasodilatadores como la terlipresina (vasodilatador esplácnico). También se puede administrar: Sodio y la midotrina, 0,2-2 mg/dl IV + albúmina. Los AINEs están contraindicados, se usan lo menos posible Si el tratamiento médico fracasa, hay que poner un TIPS, para disminuir la presión portal y disminuir la vasodilatación hepática y si esto vuelve a fracasar, nos planteamos el transplante7. TRATAMIENTO DE LA PERITONITIS BACTERIANA ESPONTÁNEA (PBE) Normal Per. Be PBE cultivo ascitis neutrofílica Bacteriascitis CMG CULTIVO Liq. Ascítico + - + PMN/ l LA 250 250 criterio PBE tratar 250 tratar 250 no respuesta inflamatoria En las bacteriascitis, hay que repetir paracentesis. Si el cultivo vuelve a dar de nuevo positivo, tratar y si es negativo es una bacteriascitis transitoria. En general, el tratamiento es empírico: cefalosporinas de 3ª generación (cefotaxima, ceftriaxona) y después según venga el cultivo (Para cultivar, lo que se hace es que hay que coger 10 cm de líquido ascético e inocularlo en botellas de cultivo y aumenta mucho la eficacia). Consiste en la infección del líquido ascítico sin lesión dentro del tubo digestivo. (Peritonitis primaria). Se produce debido a que en la cirrosis hay más sobrecrecimiento bacteriano (hay más estasis por la HTPortal, hemorragia digestiva, disminución de defensa sistémica, etc.). Ello condiciona, que aumente la concentración de las bacterias ya presentes en la flora intestinal (G-), que tienen tendencia a translocarse (paso de bacterias de la luz intestinal a los ganglios mesentéricos) pasando así a la sangre produciendo bacteriemias y en el líquido da lugar a bacteriascitis y PBE. La infección más común es la urinaria, por eso la causa más frecuente de infección del líquido ascético es por E.Coli. 7 Hay una clasificación para priorizar la lista de espera del transplante que se llama MELD (Model for End Liver Disease), que se basa en una fórmula matemática que comprende: BT, INR y creatinina. TEMA 16 Dr. Pons 7 CMG MÉDICA DE APARATO DIGESTIVO CMG Flora Intestinal CIRROSIS Sobrecrecimiento Bacteriano Alteraciones Permeabilidad Intestinal Hemorragia Digestiva Maniobras Invasivas Translocación bacteriana (Gánglios mesentér.) Alteración mecánica defensas sistémicas Sangre Alteración de las defensas del líq. Ascítico Bacteriemia Bacteriascitis PBE TEMA 16 Dr. Pons 8 CMG MÉDICA DE APARATO DIGESTIVO CMG TEMA 17 PATOLOGÍA BILIAR II COLEDOCOLITIASIS La coledocolitiasis, tal y como su nombre indica, es una patología caracterizada por la presencia de cálculos biliares, únicos o múltiples, y de tamaño variable, en el conducto colédoco. La inmensa mayoría de estos cálculos son de colesterol, los cuales una vez formados en la vesícula migran a la vía biliar extrahepática a través del conducto cístico; por tanto, su origen suele ser vesicular (80-90% de los casos), y raramente se forman en la misma vía biliar extrahepática. CLÍNICA Una de las principales manifestaciones clínicas de esta patología es la presencia de dolor en hipocondrio derecho en la casi totalidad de los casos. Como signo acompañante podemos encontrar ictericia intermitente (el cálculo permite el paso de la bilis de manera discontinua, alternando periodos de excreción biliar normal con otros ictéricos). impactado distalmente, pudiendo quedar solapado por burbujas de aire intestinal. 2. COLANGIORRESONANCIA : a pesar de su valor diagnóstico, tiene capital importancia en el tratamiento endoscópico de esta patología en algunos casos. 3. TC 4. ECOENDOSCOPIA 5. COLANGIOPANCREATOGRAFÍA RETRÓGRADA ENDOSCÓPICA (ERCP O CPRE): esta técnica se suele emplear en pacientes no-colecistectomizados (a los que no se les ha extirpado la vesícula biliar) con el fin de determinar si la coledocolitiasis tiene o no resolución. En este último caso, se debe valorar el riesgo quirúrgico para decidir la intervención a realizar, que será quirúrgica si el riesgo es bajo, o tratamiento percutáneo (técnica transparietohepática) si el riesgo es alto. Normalmente, la coledocolitiasis suele afiliarse a colestasis disociada (diferenciada de la colestasis asociada en el aumento de los niveles de gamma GT y fosfatasas alcalinas junto con bilirrubinemia normal), y muy frecuentemente a colelitiasis (70-85% de los casos). Considerándolo de manera inversa, no son raros, por otra parte, los casos de colelitiasis que se complican con una coledocolitiasis derivada. DIAGNÓSTICO El diagnóstico de coledocolitiasis se puede establecer mediante diversas técnicas: 1. ECOGRAFÍA : se trata de una prueba que otorga valiosa información y que, en este caso, consta de altas sensibilidad y especificidad. No obstante, existe dificultad diagnóstica de coledocolitiasis mediante ecografía cuando el cálculo se encuentra TEMA 17 Imagen 1: CPRE a la altura de la ampolla de Vater con extracción de un cálculo de colesterol. Prof. Miras Comisionista: Diego 17-1 CMG MÉDICA DE APARATO DIGESTIVO CMG son tanto CA supurativa como CA no supurativa (más habitual). TRATAMIENTO El tratamiento de la coledocolitiasis es variable según el enfermo. De manera muy general, las prácticas más habituales son: Esfinterotomía endoscópica Litotricia mecánica: actualmente en desuso Endoprótesis Técnica transparietohepática: esta intervención únicamente se realiza en casos graves de coledocolitiasis, y requiere además de la existencia de dilatación previa de la vía biliar intrahepática, pues se procede a su canalización con el exterior mediante un catéter transcutáneo que llega hasta el conducto colédoco. Este procedimiento permite la extracción del cálculo. COLANGITIS AGUDA Es un cuadro inflamatorio de la vía biliar generalmente causado por una infección bacteriana. Su frecuencia es más alta en la población anciana, más aún cuando el enfermo presenta patologías de base tales como DM, HTA, miocardiopatías, etc... La mayor parte de las colangitis agudas suelen estar desencadenadas por los géneros bacterianos Enterococcus, Proteus y Klebsiella, y además por la especie Escherichia Coli. CLÍNICA Los síntomas de la colangitis aguda (CA) son debidos a la inflamación, que generalmente requiere que exista una obstrucción al menos parcial del flujo biliar normal. La presentación o debut característico de la colangitis aguda consiste en la tríada de Charcot, constituida por dolor cólico, ictericia y fiebre séptica (ondulante y con picos) con escalofríos. Además, otros síntomas acompañantes pueden ser malestar general, tiritona, e incluso bacteriemias y sepsis en casos graves. Con menor frecuencia se desencadenan cuadros de coledocolitiasis o de carcinoma de la vía biliar. DIAGNÓSTICO En alrededor del 75% de los cultivos de bilis se recuperan bacterias poco tiempo después de la aparición de síntomas de la enfermedad. Al mismo tiempo, podemos observar leucocitosis con desviación izquierda. Las dos pruebas que aportan una mayor información diagnóstica de la colangitis aguda son tanto la ecografía como la ERCP. TRATAMIENTO La respuesta a antibióticos como tratamiento exclusivo y de elección es relativamente escasa, y con frecuencia suelen aparecer abscesos hepáticos múltiples con una mortalidad próxima al 100% salvo en aquellos casos en los que mediante endoscopia o cirugía se elimina la causa de la obstrucción y se drene la bilis infectada. Por otra parte, el tratamiento endoscópico suele tener una eficacia similar a la cirugía. De esta forma, será la ERCP con esfinterotomía endoscópica la técnica de elección de la colangitis aguda, además de su validez como método diagnóstico definitivo. TUMORES DE VÍA B ILIAR A continuación se explicará de manera muy superficial uno de los tumores más frecuentes de la vía biliar junto con el carcinoma in-situ, el colangiocarcinoma. El colangiocarcinoma es un tumor maligno que afecta al sistema de conductos que transportan la bilis desde el hígado hasta el duodeno, agresivo pero poco frecuente (10-15% de los tumores hepatobiliares). Se puede originar en cualquier punto de la vía biliar, tanto intrahepática como extrahepática. La evolución más frecuente de un colangiocarcinoma es la tendencia a la invasión del parénquima hepático, lo que podría conducir a un diagnóstico erróneo al confundirlo con un hepatocarcinoma. Clásicamente se han considerado dos formas anatomopatológicas de colangitis aguda, que TEMA 17 Prof. Miras Comisionista: Diego 17-2 CMG MÉDICA DE APARATO DIGESTIVO CMG presión, por lo que el tratamiento de estos tumores suele ser exclusivamente paliativo. Factores de Riesgo Algunos de los factores de riesgo más importantes para el desarrollo de colangiocarcinoma son la enfermedad inflamatoria intestinal, colangitis esclerosante proliferativa, coledocolitiasis o quiste de colédoco. Imagen 2: Múltiples metástasis hepáticas derivadas de un colangiocarcinoma primario Clasificación Anatómica El colangiocarcinoma puede clasificarse generalmente en intrahepático (10%), perihiliar (6080%) y distal (20%). Seguidamente atenderemos a la tipificación de Bismuth-Corlette para clasificar y diferenciar los tipos anatómicos de colangiocarcinoma en función de la zona de la vía biliar a la que afecten y su relación con el porcentaje de supervivencia: CLÍNICA Como en la mayor parte de los tumores, la clínica suele ser inespecífica, aunque algunas de las manifestaciones más destacadas son: ictericia, prurito, alteración de la función hepática, colestasis no-dolorosa (el dolor aparece en pocos casos), síndrome paraneoplásico o síndrome constitucional (caracterizado por anorexia, malestar general, etc...). DIAGNÓSTICO El diagnóstico pasa primero por una analítica, seguida de ecografía, RMN, ERCP... Tipo I: afectación del conducto hepático común. Tipo II: este tipo anatómico se conoce con el nombre de “tumor de Klatskin”, colangiocarcinoma que compromete el conducto hepático común y su bifurcación. Tipo IIIa: afectación del conducto hepático común, de la bifurcación de éste y del conducto hepático derecho. Tipo IIIb: afectación del conducto hepático común, de su bifurcación y del conducto hepático izquierdo Tipo IV: afectación del conducto hepático común, de su bifurcación y del conducto hepático derecho junto con compromiso del conducto colédoco. Los colangiocarcinomas del tipo I y II presentan una supervivencia del 20% a los 5 años si se diagnostican con precocidad en estadios iniciales, mientras que los tipo III y IV tienen un pronóstico nefasto, con supervivencia del 20% en un año incluso si se decide el trasplante. Es más, el hecho de proceder al trasplante de la zona comprometida puede llevar a la muerte temprana del enfermo debido a la inmunosu- TEMA 17 Prof. Miras Comisionista: Diego 17-3 ANEXO DEL TEMA 17: PATOLOGÍA BILIAR II Con el objetivo de mejorar la comprensión y facilitaros el estudio, a continuación se expone un esquema gráfico de los tipos anatómicos de colangiocarcinoma (según la clasificación de Bismuth-Corlette) que nos muestra la localización particular de la lesión en la vía biliar extrahepática. Al final se incluye también un recuerdo anatómico de dicha vía para evitar búsquedas innecesarias de atlas de anatomía, léase Netter ®, Sobota ®, etc. - Tipo I: afectación del conducto hepático común. - Tipo II (tumor de Klatskin): compromiso del conducto hepático común y su bifurcación. - Tipo IIIa: afectación del conducto hepático común, de la bifurcación de éste y del conducto hepático derecho. - Tipo IIIb: afectación del conducto hepático común, de su bifurcación y del conducto hepático izquierdo - Tipo IV: afectación del conducto hepático común, de su bifurcación y del conducto hepático derecho junto con compromiso del conducto colédoco Conducto hepático dcho. Conducto hepático izq. Conducto hepático común Bifurcación CMG MÉDICA DE APARATO DIGESTIVO CMG TEMA 18 LITIASIS BILIAR Aviso a los compis ;) Parte de este tema ha sido elaborado gracias a la colaboración de l@s comisionistas de Cirugía. Es por ello por lo que me gustaría agradecer su participación a est@s compañer@s, en especial a Rocío y a Elena, sin cuya aportación el tema no habría sido el mismo. Muchas gracias chicas. María Lozano INTRODUCCIÓN La litiasis biliar es la presencia de cálculos en el árbol biliar. Es una patología muy frecuente. Se calcula que aproximadamente el 20% de la población la desarrollará en algún momento de su vida. Es más frecuente en mujeres (dos veces más frecuente) sobre todo en las que han tenido varios embarazos (ser multípara es un factor predisponente). desembocará en el hepático común. A partir de la recepción del cístico, el hepático común pasa a denominarse colédoco, el cual sigue su trayecto hacia el duodeno, recibiendo en su camino al conducto de Wirsung (conductor excretor del páncreas). Ambos conductos, colédoco y Wirsung, terminarán desembocando en un orificio común, la papila duodenal, o ampolla de Váter2. Estos conductos en sus segmentos terminales se encuentran rodeados por el aparato muscular esfinteriano denominado Esfinter de Oddi3. 2 RECUERDO ANATÓMICO DEL ÁRBOL BILIAR ¿Qué habríamos hecho sin las lecciones de Pepe Edmundo? Ahora estaríamos perdidos!!! 3 Que asegura el control del flujo de la bilis y jugo pancreático a la luz del tubo digestivo. Se extiende desde el parénquima hepático a la segunda porción del duodeno. En el hígado, el hepatocito produce bilis por el polo biliar, pasando a los conductos biliares de calibre creciente, que acabarán confluyendo entre sí para formar los conductos hepáticos derecho e izquierdo. Ambos se reunirán, ya fuera del hígado, a 1-2cm1 por debajo del hilio hepático, para formar el conducto hepático común. Este conducto recibirá al cístico, que 1 Esto es una ampliación, para ponernos en situación, luego no es necesario aprendérselo ;) TEMA 18 Prof. Miras Comisionista: María Lozano 18-1 CMG MÉDICA DE APARATO DIGESTIVO VÍA BILIAR PRINCIPAL Y ACCESORIA Vía Principal: Formada por todos los conductos intrahepáticos, conducto hepático común y colédoco (hasta la ampolla). Vía Accesoria: Conducto cístico y vesícula. VÍA BILIAR INTRAHEPÁTICA Y EXTRAHEPÁTICA Vía Intrahepática: Incluye desde los conductos biliares de últimas generaciones hasta los hepáticos derecho e izquierdo. Vía Extrahepática: Conducto hepático común, colédoco, cístico y vesícula. Ampliación del Contenido: Porciones y trayecto del colédoco: La primera porción o supraduodenal, desciende manteniendo sus relaciones con el hilio hepático (arteria hepática y vena porta). Posteriormente cruza por detrás de la 1º porción duodenal, aproximándose a la 2º porción del duodeno. A esta parte del colédoco se le conoce como colédoco retroduodenal. En esta aproximación al duodeno, tunelizará la cabeza del páncreas (colédoco intrapancreático). Así alcanzará su pared posteromedial, la atravesará en un trayecto de unos pocos mm, para después confluir con el Wirsung, dando lugar a una dilatación común, la ampolla de Vater. Estos conductos se abren a la luz a través de un pequeño orificio o papila Colédoco supraduodenal → retroduodenal → intrapancreático → en interior del páncreas: Recepción del Wirsung → desembocadura en 2º porción duodeno Cuando comemos, especialmente alimentos grasos, la mucosa duodenal segrega colecistoquinina (CCK), que provoca la contracción vesicular y la relajación paralela del esfinter de Oddi. ¿Y la vesícula? Es el órgano encargado de modificar el flujo constante de la bilis que sale del hígado, de forma que llegue al tubo digestivo en su momento adecuado para la digestión de los alimentos. Propiedades → reservorio. → absorción de agua, para concentrar la bilis. → llenado vesicular → pasivo. → vaciado → mecanismos hormonales y neurogénicos. Composición de la Bilis La bilis está compuesta por: a) agua. b) pigmentos biliares, fundamentalmente bilirrubina. c) colesterol. d) lecitina. e) colesterina4 ( colesterina = colesterol) Las sales biliares junto con la lecitina y el colesterol, forman micelas para asegurar la solubilidad de estos lípidos en la bilis. Vesicular: más espesa, filante y color verdosonegro. La función de la vía biliar es asegurar el paso de la bilis, producida por el hígado, al tubo digestivo a nivel del duodeno. El hígado está produciendo bilis de forma constante, pero ésta sólo hace falta en el duodeno tras las comidas. Prof. Miras En los periodos interdigestivos, la bilis desciende, procedente del hígado, por el colédoco, pero se encuentra con el esfinter de Oddi cerrado, no pudiendo salir al duodeno. Lo que va a ocurrir es que esa bilis ascenderá de forma retrógrada hasta la vesícula, que está relajada, donde se almacenará. Diferencias entre Bilis Hepática y Vesicular FUNCIÓN DE LA VÍA BILIAR TEMA 18 CMG Hepática: amarilla y más clara (menos espesa). Esto nos sirve para entender por qué los cálculos se van a formar fundamentalmente en la vesícula, como consecuencia de las características de la bilis vesicular. 4 colesterina = colesterol Comisionista: María Lozano 18-2 CMG MÉDICA DE APARATO DIGESTIVO da esta situación va a favorecer que la bilis se infecte y se formen cálculos5. CONCEPTO DE LITIASIS BILIAR La litiasis biliar es una de las patologías más frecuentes del aparato digestivo y la colecistectomía, una de las intervenciones quirúrgicas abdominales más habituales llevadas a cabo (Everhart JE, 1999). Morfología y composición de los cálculos vesiculares: Encontramos dos tipos de cálculos: 1) 2) Cálculos de colesterol. Es el tipo más común de cálculos (75% ) Están compuestos puramente por colesterol o tienen al colesterol como su constituyente químico principal. Pueden ser identificados frecuentemente mediante la inspección; los que están constituidos enteramente por colesterol generalmente son grandes y tienen aspecto blancoamarillento. Los cálculos mixtos de colesterol están compuestos por más de 50% de colesterol y son levemente más comunes que los cálculos puros. Los cálculos mixtos tienden a ser más pequeños que los cálculos puros y frecuentemente son múltiples. Cálculos pigmentados negros. Se hallan compuestos por bilirrubinato cálcico puro o complejos poliméricos de calcio, cobre y grandes cantidades de glucoproteínas de mucina. Los cálculos negros son más comunes en pacientes que padecen cirrosis y enfermedades hemolíticas crónicas. Localización de los Cálculos: Cuando hablamos de litiasis biliar estamos haciendo referencia a la litiasis vesicular (85%) Además de en la vesícula, también podemos encontrar cálculos en la vía biliar principal (hepatocolédoco), pero es poco frecuente en relación a los de la vesícula (15%) y tienen una clínica especial. Los cálculos que nos encontramos aquí, pueden tener dos orígenes: 1. Que sea una litiasis que procede de la vesícula pero ha salido de ella y pasado al colédoco. 2. Litiasis de la vía biliar principal primaria, que es muy raro, pero puede aparecer cuando hay estenosis u obstrucción de esta vía. Hay por tanto un mal drenaje de la bilis, dándose un estasis no fisiológico. To- TEMA 18 Prof. Miras CMG Así que, si hablamos de litiasis biliar sin más, nos estaremos refiriendo a la litiasis vesicular. Cuando la litiasis está en la vía biliar principal debemos especificarlo. FACTORES DE RIESGO6 FACTORES DE RIESGO ASOCIADOS CON LA FORMACIÓN DE CÁLCULOS DE COLESTEROL: 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. Edad ( FORTY) Sexo femenino ( FEMALE) Embarazo ( FERTILE) Obesidad ( FAT) Anticonceptivos orales y terapia hormona sustitutiva con estrógenos Pérdida rápida de peso Nutrición parenteral Diabetes Mellitas Enfermedad de Crohn Cirrosis hepática Predisposición Consumo de determinados fármacos( por ej. Clofibrato) Como podréis comprobar los cuatro primeros factores de riesgo empiezan en inglés por “F”; la regla nemotécnica “de las 4 efes” resume los factores de riesgo más importantes de la aparición de cálculos de colesterol: lo más frecuente es que sean mujeres ( female), de unos cuarenta años( forty), si bien todavía en edad fértil (fertile). Además las pacientes se caracterizan por tener exceso de peso (Fat)7. 5 Todo esto se explicará con mayor profundidad más adelante. 6 He decidido comenzar poniendo un cuadro resumen de los factores de riesgo, tipo esquemático, para que sea más fácil a la hora de estudiar. Posteriormente se desarrolla cada uno de los puntos. Cierto es que este apartado es un pelín largo, pero creo que merecía la pena explicar el defecto fisiológico que hacía que esos factores fueran considerados de riesgo para esta patología. María Lozano 7 Esta regla nemotécnica va dedicada a todos aquellos que disfrutan con mis reglas nemotécnicas y en especial a ella( Si RAfa fuera FAMOso de CINE se iría a NIZA) y a un japonés, digo xino( ja,ja) por compartir las suyas conmigo XDD) Comisionista: María Lozano 18-3 CMG MÉDICA DE APARATO DIGESTIVO 1. Edad: más frecuente a partir de los 40 años (FORTY) cerca del 20% de los adultos a partir de esta edad y del 30% en los mayores de 70 años. Esto es debido a que es raro que los cálculos vesiculares se disuelvan espon-táneamente por lo que la prevalencia acumu-lativa de litiasis vesicular aumenta con la edad. Además, la secreción de la bilis se incrementa con el tiempo mientras que la secreción de ácidos biliares puede disminuir. Como resultado, la bilis que se forma es más litogénica con los años. 2. Sexo femenino: (FEMALE) La mayoría de los trabajos informan de un riesgo de 2 ó 3 veces más alto en mujeres que en hombres. 3. Embarazo: (FERTILE) Normalmente son formas asintomáticas de litiasis biliar y tanto el barro biliar como los cálculos menores de 10mm habitualmente desaparecen tras el parto8. La bilis se forma más litogénica durante la gestación, posiblemente por el aumento de los niveles de estrógenos, que lleva al incremento de la secreción de colesterol y la sobresaturación de la bilis. Además, el volumen de la vesícula se duplica y ello provoca éstasis, lo cual promueve la formación de barro biliar y cálculos, probablemente como resultado de los niveles más altos de proges-terona, que alteran la motilidad vesicular. 4. Obesidad. ( FAT) La obesidad es un factor conocido de riesgo de colelitiasis. La asociación entre la obesidad y la formación de cálculos podría ocurrir por el incremento d la secreción de colesterol hacia la bilis como resultado de la mayor actividad de la 3hidroxi-3 metilglutaril - coenzima A (HMGCoA) reductasa (la enzima que constituye el paso limitante de la síntesis de colesterol), la cual podría llevar a altos niveles de síntesis de colesterol en el hígado. 5. Anticonceptivos orales y terapia hormonal sustitutiva con estrógenos: Los estrógenos son los fármacos u hormonas más estudiados esn relación con la formación de cálculos vesiculares. En las mujeres, los estrógenos exógenos mejoran la captación de lipoproteínas por el hígado, aumentan la secreción de colesterol hacia la bilis e inhiben la síntesis de ácidos biliares. El uso CMG de anticonceptivos orales también se ha rela-cionado con aumento en la incidencia y con un aumento en la incidencia, demostrándose una respuesta dependiente de la dosis de estró-genos. Sin embargo, un estudio de 1997 en el que se utilizaron antinconceptivos orales más recientes no mostró un aumento de tal riesgo, presumiblemente por su menor contenido de estrógenos. 6. Pérdida rápida de peso: Las alteraciones fisiológicas que llevan a la formación de cálculos vesiculares como resultado de la pérdida de peso son múltiples. Muchos investigadores han demostrado que la secreción hepática de colesterol aumenta durante la restricción calórica. Factores adicionales podrían ser la elevación de la producción de mucina (un estimulador potente de la nucleación de cristales de colesterol) y la disminución de la motilidad vesicular. 7. Nutrición parenteral: El defecto fisiológico primario es la hipomotilidad de la vesícula con estasis biliar como consecuencia de ayuno prolongado. 8. Diabetes Mellitus: Se cree que los diabéticos son más propensos que los no diabéticos a las complicaciones asociadas con la colelitiasis. Se ha considerado durante mucho tiempo que los pacientes con diabetes resistente a la insulina tienen mayor riesgo de presentar cálculos vesiculares porque este tipo de diabetes se asocia con hipertrigliceridemia, obesidad e hipomotilidad vesicular, factores de riesgo conocidos para la formación de cálculos. 9. Enfermedad de Crohn: Como ya hemos estudiado, la enfermedad de Crohn es la enfermedad sistémica más común que afecta al íleon Terminal9. Los pacientes con enfermedad de Crohn tienen un riesgo de 2 o 3 veces mayor de formación de cálculos. La explicación clásica para este aumento del riesgo es que se pierden los receptores específicos de los ácidos biliares en el íleon terminal, con el resultado de la excreción excesiva de sales biliares y la disminución del tamaño de la reserva de ácidos biliares. Estos cambios, finalmente llevan a la formación de bilis litogénica. 10. Cirrosis hepática. 9 8 Ver el tema 7 de esta comisión: ¿Qué no tiene el pobre enfermo de Crohn? Maringhini, A., 1993 TEMA 18 Prof. Miras Comisionista: María Lozano 18-4 CMG MÉDICA DE APARATO DIGESTIVO 11. Antecedentes familiares de litiasis biliar: (“cálculos en la familia”). 12. Consumo de algunos fármacos: como estatinas, clofibrato10 y ceftriaxona11. FISIOPATOLOGÍA. FORMAS CLÍNICAS CMG cólico hepático: Molestia sorda, típica, periódica y que aparece en determinadas situaciones13. El cólico biliar puede ceder espontáneamente cuando el cálculo deja de obstruir el cístico y vuelve a su lugar en la vesícula, o bien desaparecer al administrar tratamiento médico con espasmolíticos. En la litiasis biliar y de la VBP lo que ocurre es que existe un problema mecánico-obstructivo. SI EL DOLOR DURA MÁS DE SEIS HORAS SUGIERE UNA COLECISTITIS AGUDA EN LUGAR DE UN SIMPLE CÓLICO BILIAR. 1.- CÓLICO BILIAR SIMPLE 2.- COLECISTITIS AGUDA14. El cólico biliar es el síntoma más común de la colelitiasis. Se produce cuando un cálculo (que se encontraba en el fondo de la vesícula) queda encajado en el conducto cístico. Es un cólico biliar mantenido, ya que el cálculo se ha quedado encajado en el cístico (obstrucción del cístico prolongada). Clínica: Comienza con un dolor súbito e intenso en epigastrio o hipocondrio derecho que se irradia en hemicinturón (derecho) hacia la espalda, ascendiendo a veces a la escápula12. Suele ir asociado a náuseas y vómitos tras una ingesta importante de comida. El dolor es de tipo visceral e intenso, haciendo que el paciente no pueda parar de moverse y tenga que acudir a la puerta de urgencias. Clínica: Comienza con un dolor de tipo visceral como el del cólico biliar, pero cuando la obstrucción se mantiene 8-10 horas, pasa a ser un dolor somático, más soportable pero el paciente apenas puede moverse. Este cambio en el dolor se debe a la infección de la bilis como consecuencia del mal drenaje de la que está almacenada en la vesícula. 3.-COLECOLEDOCOLITIASIS. No se acompaña de fiebre ni de ictericia (MUY IMPORTANTE!!!), por eso se llama cólico biliar simple. Ocurre cuando un cálculo de la vesícula atraviesa el cístico y pasa al colédoco, obstruyéndolo. Pueden aparecer irradiaciones atípicas del dolor: Clínica: Inicialmente se manifiesta como un cuadro hepático pero cuando la piedra entra en el colédoco provoca una colédocolitiasis. Comienza a haber ictericia, coluria, prurito, acolia, fiebre, escalofríos y tiritonas. Además se mantiene un dolor biliar importante. → Retroesternal→ pudiendo simular un cuadro cardiaco. → Sólo dolor en epigastrio, sin irradiaciones hacia la espalda. En estos casos los médicos lo primero que hacen es mirar el estómago para ver cómo está. Un dolor en esta localización también podría estar causado por una úlcera o una gastritis, pero en ambas, las características del dolor son diferentes a las del 10 Hipolipemiante que actúa inhibiendo la síntesis de colesterol. Este fármaco está indicado en el tratamiento de la arteriosclerosis y de las dislipemias de distinto orden. Está contraindicado su uso en pacientes con alteraciones de la vesícula biliar y en la insuficiencia hepática o renal. 11 Antibiótico de amplio espectro, bactericida perteneciente al grupo de las cefalosporinas de tercera generación, de acción prolongada. 12 La “punta de la paletilla”. TEMA 18 Prof. Miras TRIADA DE CHARCOT: a) Prurito b) Fiebre con escalofríos c) Ictericia Si existe la triada podemos hacer el diagnóstico de coledocolitiasis y colangitis aguda. A veces sólo aparece dolor e ictericia. En este caso, sólo habrá coledocolitiasis, ya que, para que haya colangitis aguda, es necesario la 13 Este es el dolor típico de la persona que padece del estómago, siendo muy diferente del cólico hepático. 14 La desarrollaremos más adelante. Comisionista: María Lozano 18-5 CMG MÉDICA DE APARATO DIGESTIVO presencia de fiebre y escalofríos15. Menos frecuente, pero que ocurre a veces, es que nos encontremos con un colédoco en empedrado en un enfermo sin ninguno de los componentes de la triada, con sólo algo de dolor. 4.- OBSTRUCCIÓN PAPILAR. A veces el cálculo es pequeño y se queda impactado en la papila provocando una obstrucción a este nivel. Esta obstrucción pro-duce la forma más frecuente de pancreatitis aguda. En Murcia, las pancreatitis agudas son provocadas por cálculos (80%), alcohol (15%) y otras causas (5%). La gravedad de ellas, conocidas como colecistopancreatitis, es variable: la mayoría son leves, habiendo únicamente algo de edema, pero en el 10% de los casos hay necrosis pancreáticas, siendo una situación grave. El Wirsung conduce los jugos pancreáticos hacia la luz duodenal. Estos jugos tienen pro- fermentos, que permanecen inactivos hasta que llegan a la luz duodenal, donde se activan. Si la papila está obstruida, el Wirsung no va a drenar hacia la luz. Los profermentos se van a mantener inactivos en el conducto pancreático durante horas, pero pasado cierto tiempo se activarán, digiriendo el páncreas del paciente. 5.- ÍLEO BILIAR16 Una vesícula con un cálculo único de colesterol de grandes dimensiones, fre-cuentemente asintomático, pero a veces, el peso del cálculo produce inflamación de la pared vesicular. Esta inflamación puede llevar a la formación de adherencias entre la vesícula y el primer codo duodenal, e incluso a la aparición de una fístula colecisto-duodenal. Si se forma la fístula, el cálculo pasa al intestino delgado, que se puede obstruir a nivel del íleon terminal (ya que es la zona más estrecha de delgado, y por las grandes dimensiones de la piedra). 16 15 CMG MUY IMPORTANTE!!! Será desarrollado en otro apartado. DIAGNÓSTICO objetivos). Haciendo uso de estos datos, establecemos un diagnóstico de sospecha. Vamos a dividir el diagnóstico en dos partes: a) el diagnóstico de la litiasis vesicular, y b) el diagnóstico de la litiasis de la vía biliar principal (hepatocolédoco). Para establecer el diagnóstico de certeza la exploración fundamental es la ECOGRAFÍA17. a) Diagnóstico de la litiasis vesicular Para diagnosticar al paciente disponemos de la clínica y las exploraciones complementarias. Dentro de la clínica, ya descrita, encontramos síntomas (datos subjetivos) y signos (datos En el diagnóstico de la litiasis vesicular la ECO presenta muy pocos falsos positivos (cuando la ECO dice que hay cálculos, es que realmente los hay). Sin embargo, sí podemos encontrar en ocasiones falsos negativos (en un 5-10 % de los casos). Esto ocurre cuando los cálculos son muy pequeños o por la falta de 17 TEMA 18 Prof. Miras MUY IMPORTANTE!!! Comisionista: María Lozano 18-6 CMG MÉDICA DE APARATO DIGESTIVO experiencia del ecografista. Cuando obtenemos un resultado negativo en la ecografía pero la sospecha clínica es muy alta es aconsejable repetir el estudio ecográfico, a ser posible por otro ecografista. Muchos informes que comienzan siendo negativos, acaban siendo positivos tras la repetición del estudio. A la hora de realizar la ecografía, es ideal que la vesícula se encuentre lo más distendida posible. Para esto, bastará con realizar la prueba tras un periodo largo de ayuno (8-9 horas), puesto que en este caso la cantidad de bilis almacenada en la vesícula será alta. Sin embargo, en ocasiones los cálculos son tan grandes que pueden verse incluso en el periodo post-ingesta, cuando la vesícula se encuentra replegada. Es un error común pensar que la TC será capaz de diagnosticar cálculos que no han podido verse con la ecografía. Esto es falso18, en el diagnóstico de la litiasis vesicular (veremos cómo en la de vía biliar principal es diferente) la ecografía presenta un rendimiento mayor que el de la TC y mayor que el de cualquier otra prueba. Rx simple de abdomen: Puede detectar cálculos biliares si éstos contienen el suficiente calcio para ser radioopacos. Sólo el 50% de los cálculos pigmentados y el 20% de los cálculos de colesterol contienen suficiente calcio como para ser visibles en la radiografías simples de abdomen. Como el 85% de los cálculos vesiculares en las poblaciones occidentales son de colesterol, se deduce que únicamente el 25% puede ser detectado por este método. Su mayor utilidad es evaluar a los pacientes que tienen complicaiones infrecuentes de los cálculos vesiculares, como colecistitis enfisematosa, fístula colecisto-entérica y vesícula en porcelana. No debemos olvidar realizar diagnóstico diferencial con la litiasis renal. TAC: Puede ser útil para visualizar abscesos o complicaciones. b) Diagnóstico de la litiasis de la vía biliar principal o Ecografía: en este caso es poco fiable. Va a presentar muchos falsos negativos y también algunos falsos positivos. Esta diferencia con el diagnóstico de la litiasis vesicular se debe a que el hepatocolédoco presenta una posición más profunda que la vesícula, en el hilio hepático, y además suele interponerse entre el colédoco y la superficie corporal del paciente el aire del antro gástrico, lo que deteriora mucho la imagen ecográfica. o Colangio–resonancia: consiste en inyectar un contraste intravenoso que será captado por el hepatocito y eliminado por el mismo hacia la vía biliar. El contraste opacificará dicha vía, dando una imagen bastante clara de la misma. El rendimiento de la colangio RMN para diagnóstico de la litiasis de la vía biliar principal es mucho mayor que el de la ECO. Sin embargo, existe una técnica aún más eficaz, la ERCP. o ERCP, CPRE o Colangiopancreatografía retrógrada endoscópica: consiste en alcanzar, de forma endoscópica, la segunda porción de duodeno y buscar allí la papila. A pesar de la dificultad que entraña la localización de la papila en una cirugía abierta (por su localización oculta entre los pliegues intestinales), sí podemos localizarla mediante endosco- Por último, decir que están en total desuso la colecistografía oral e intravenosa, aunque hace algunos años eran las pruebas más utili- zadas. Fig. 1: ¿Recordáis el signo del faro? OTRAS PRUEBAS que podemos realizar son: 18 OJO!!! TEMA 18 Prof. Miras CMG Comisionista: María Lozano 18-7 CMG MÉDICA DE APARATO DIGESTIVO pia. Ésta se observa como un pequeño “botoncito” por el cual se puede ver la salida de “gotitas de bilis”. Una vez localizada la papila, se introduce por la misma una sonda por la que se inyecta el contraste. Gracias a esto podemos observar perfectamente la vía biliar. El rendimiento de esta técnica es mucho mayor que el de la colangio RMN. CMG En la obstrucción secundaria a una litiasis el extremo distal que se observa del colédoco presenta una forma cóncava hacia abajo que se llama en “pata de cangrejo”. El problema es que, en ocasiones, el endoscopista necesita realizar una papilotomía para introducir la sonda por la papila. Esto puede presentar las siguientes complica-ciones: Pancreatitis aguda, proceso de altísima gravedad que puede incluso llevar a la muerte del enfermo. Hemorragias digestivas Debido a las graves complicaciones, esta técnica no se usa de forma sistemática, sino que usaremos siempre como primera opción diagnóstica la colangio RMN (que aún teniendo menor rendimiento es más segura). Si aún así siguiéramos con dudas diagnósticas, pasaríamos a realizar la ERCP (ya que, aunque ésta entrañe riesgos, es mejor confirmar el diagnóstico que operar al paciente directamente ante la sospecha y encontrarnos con una cirugía blanca). Ampliación de Contenido Diagnóstico diferencial de las obstrucciones de la vía biliar principal mediante técnicas de imagen: Podríamos pensar que el diagnóstico diferencial empleando técnicas de imagen entre los tumores de la vía biliar principal, la compresión del colédoco secundaria a una pancreatitis crónica y los cálculos de la vía biliar sería difícil. Sin embargo, existen diferencias manifiestas entre los tres procesos. En el tumor de la vía biliar principal o en el cáncer de páncreas se observa un colédoco bastante dilatado (el proceso de obstrucción tumoral lleva un tiempo y permite una cierta dilatación) y un cierre abrupto de la vía. En el caso de una pancreatitis crónica que comprime la vía, se observa una estenosis concéntrica que afecta a un segmento largo del colédoco, permitiendo que cierta cantidad de contraste llegue hasta el duodeno. TEMA 18 Prof. Miras TRATAMIENTO Cirugía. Hoy día, el procedimiento de elección es colecistectomía laparoscópica. Tiene mortalidad casi nula y mínima morbilidad cuando se realiza de forma electiva, obteniendo mejores resultados que ningún otro tratamiento19. Vivir sin vesícula…… Se vive bien. Solo que al no haber vesícula, la presión en la vía biliar principal está un poco aumentada, quedando el esfínter de Oddi algo más relajado de lo normal. La bilis ya no se puede almacenar en la vesícula, pasando al duodeno, pero este flujo “continuo” de bilis al duodeno no provoca ningún problema. Tratamiento médico con ácidos biliares, utilizándose el ácido ursodesoxicólico, con o sin quenodesoxicólico. Son candidatos a este tratamiento los pacientes asintomáticos con cálculos de colesterol no calcificados menores de 15 mm de diámetro y vesícula funcionante. Litotricia biliar extracorpórea. Puede aplicarse este tratamiento en pacientes con litiasis biliar no complicada que presentan cálculos radiotransparentes de un tamaño máximo de 20mm y con una vesícula biliar funcionante. Presentan como complicación: pancreatits aguda (2%) y hematuria (1%). Existe una recidiva de un 10% a un 15% por lo que se necesita tomar ácidos biliares de forma prolongada posteriormente, lo cual hace que apenas se emplee. 19 El Dr. Miras dijo que, TEÓRICAMENTE, una persona con litiasis biliar sin sintomatología no se debería operar. Pero, en la práctica clínica, la mayoría se opera, por una medicina defensiva, pues en un futuro puede dar lugar a una litiasis sintomática en cualquiera de sus presentaciones Comisionista: María Lozano 18-8 CMG MÉDICA DE APARATO DIGESTIVO COLECISTITIS AGUDA La colecistitis aguda se considera la complicación más frecuente de la litiasis vesicular. La inflamación de la pared de la vesícula, asociada al cuadro de dolor abdominal, dolor a la palpación en el hipocondrio derecho, fiebre y leucocitosis, es el sello de la colecistitis aguda. Aprox. en el 90% de los casos, la causa subyacente es un cálculo que obstruye el conducto cístico. En el 10% restante, la colecistitis se produce en ausencia de cálculos vesiculares y se denomina colecistitis aguda alitiásica. La colecistitis aguda alitiásica se origina en pacientes hospitalizados por otras razones (quemados, traumatismos, hiponutrición, deshidratación, hipoxia….), en los carcinomas de vesícula con infiltración del cístico, en la infección biliar por Salmonella y por vasculitis. Es más frecuente en personas mayores. En la etiología de la colecistitis también es posible encontrar una forma especial, y rara, de colecistitis aguda: la colecistitis aguda enfisematosa. La colecistitis aguda enfisema-tosa es una entidad clínica poco frecuente (1%) que se desencadena al invadir la pared de la vesícula microorganismos productores de gas, generalmente, Clostridium perfringens. Afecta de forma especial a varones diabéticos y se asocia a colelitiasis en una proporción menor que la colecistitis aguda simple; otra diferencia con ésta es la tendencia a la gangrena y perforación que da origen a cuadros sépticos con una alta mortalidad. PATOGENIA Mientras el cólico biliar tiene como causa la obstrucción intermitente del conducto cístico por cálculos, la colecistitis aguda generalmente ocurre cuando un cálculo se impacta en el conducto cístico y provoca la obstrucción crónica. La éstasis de la bilis en la luz de la vesícula determina el daño de la mucosa vesicular con la liberación consecuente de enzimas intracelulares y la activación de una cascada de mediadores de la inflamación. El proceso inflamatorio es inicialmente aséptico, invirtiéndose el mecanismo fisiológico de reabsorción de agua, con lo que se incrementa la presión intravesicular. Este incremento de presión dificulta el retorno venoso y el aporte arterial, lo que favorece la isquemia. Si el proceso TEMA 18 Prof. Miras CMG no evoluciona se produce un hidrops vesicular de contenido transparente por la reabsorción de pigmentos biliares y la secreción intravesicular de agua y moco. La contaminación del contenido provoca un empiema vesicular. Si la distensión progresa, con compromiso de la viabilidad de la pared, o se produce la sobreinfección del contenido vesicular, el proceso puede evolucionar a la gangrena, la perforación y el coleperitoneo. MANIFESTACIONES CLÍNICAS Como se ha comentado anteriormente, mientras que en el cólico biliar el dolor rara vez excede de 6 horas, el dolor que dura más de 6 horas sugiere una colecistitis aguda. El dolor se localiza en el hipocondrio derecho. Las náuseas con algunos vómitos son característicos de la colecistitis aguda, pero estos síntomas casi invariablemente suceden al dolor en lugar de precederlo. En contraste con el cólico biliar no complicado, el examen físico sugiere la colecistitis aguda en muchos casos. La fiebre es común, debido a la inflamación activa de la mucosa vesicular, pero en general no supera los 39º C, a menos que ocurra gangrena o perforación de la vesícula. En la exploración es característica la hipersensibilidad en hipocondrio derecho con dolor que impide la inspiración profunda (signo de Murphy positivo). No es habitual la ictericia20 y sólo cuando se produce una colecistopancreatitis o síndrome de Mirizzi (compresión extrínseca del colédoco por vesícula severamente inflamada). Si el dolor se intensifica de forma súbita, y la reacción peritoneal aumenta junto con fiebre mayor de 39ºC y leucocitosis debe sospecharse una perforación vesicular. DIAGNÓSTICO El diagnóstico de colecistitis aguda suele basarse en una anamnesis característica y en la exploración física. Lo habitual es detectar leucocitosis con desviación a la izquierda. Puede aparecer alteración de las enzimas hepáticas21. 20 De hecho, sólo aparece ictericia leve en 20 % de los pacientes. 21 La bilirrubina sérica se eleva ligeramente en menos de la mitad de los pacientes, mientras que alrededor del Comisionista: María Lozano 18-9 CMG MÉDICA DE APARATO DIGESTIVO La ecografía es la técnica más utilizada; sin embargo debemos señalar que la técnica más específica es la gammagrafía con HIDA, salvo si hay colestasis asociada. En la ecografía podremos ver una pared de la vesícula biliar en la que se observan 2 ó 3 capas (“la doble pared vesicular”). Dicha imagen también aparece en las hepatitis agudas graves así que solicitaremos un TAC para hacer el diagnóstico diferencial22. Diagnóstico diferencial: Realizaremos el diagnóstico diferencial con todas aquellas patologías que puedan producir dolor en hemiabdomen derecho: • Pancreatitis aguda. • Apendicitis retrocecal. • Hepatitis aguda. • Neumonía basal derecha (sobre todo, en niños y en ancianos). • Ulcus péptico. • Pielonefritis. El tratamiento de la colecistitis aguda es en la mayoría de los casos quirúrgico. No se considera una urgencia quirúrgica inmediata, excepto si existe un cuadro clínico de peritonitis difusa. La necesidad de instaurar un tratamiento médico y retrasar el tratamiento quirúrgico 8-12 semanas está justificada en pacientes en los que el diagnóstico se hace varios días después de comenzar el cuadro clínico, ante la insistencia de un cuadro local o en enfermos de elevado riesgo quirúrgico. Si tras la instauración del tratamiento médico la vesícula se “enfría” (es decir, que disminuye la inflamación) esperaremos un tiempo (alrededor de 1 ó 2 meses) y se operará. La vía de abordaje más recomendable para efectuar la colecistectomía en la colecistitis aguda es la colecistectomía laparoscópica. En un reducido subgupo de pacientes en situación crítica en los que no se puede plantear la colecistectomía, la colecistostomía percutánea supone una opción menos agresiva con resultados razonables23. 23 TRATAMIENTO 25% muestran ligeras elevaciones de las aminotransferasas séricas. 22 MUY IMPORTANTE!!! TEMA 18 CMG Prof. Miras La colecistostomía percutánea consiste en el drenaje de la vesícula al exterior mediante un tubo con el que se punciona a través de la pared abdominal o de la pared costal. Comisionista: María Lozano 18-10 CMG MÉDICA DE APARATO DIGESTIVO ÍLEO BILIAR Una vesícula con un cálculo único de colesterol de grandes dimensiones, frecuente-mente es asintomático, pero a veces, el peso del cálculo produce inflamación de la pared vesicular. Esta inflamación puede llevar a la formación de adherencias entre la vesícula y el primer codo duodenal, e incluso a la aparición de una fístula colecistoduodenal. Si se forma la fístula, el cálculo pasa al intestino delgado, el cual puede obstruirse a nivel del íleon terminal (ya que es la zona más estrecha de delgado, y por las grandes dimensiones de la piedra). El íleo biliar ocurre con más frecuencia en personas con edad avanzada. En estas obstrucciones, la radiología simple de abdomen es muy importante y aporta muchos datos. En el íleo biliar la obstrucción suele ser generalmente de íleon terminal, por lo que en la radiografía observaremos: • Niveles hidroaéreos (en pila de monedas), que son asas de delgado. • No presencia de gas en el colon. • Cámara del estómago. • NEUMOBILIA, es el dato más importante. Se debe a que hay paso de aire a través de la fístula a la vía biliar. • A veces, cuando el cálculo es radioopaco, lo podremos ver. CMG c) Eliminación asintomática en heces: No se conoce su frecuencia pero puede pasar que los cálculos no tengan el tamaño necesario para obstruir, por lo que tras viajar por todo el intestino delgado pasan al colon, eliminándose finalmente por las heces. Todo esto sin que el paciente se dé cuenta. d) Fístula colecistocólica: Otras veces la vesícula no se pega al duodeno sino al ángulo hepático del colon, haciendo este tipo de fístula. La piedra pasa al colon y sale por las heces. Cuando el enfermo expulsa la piedra en las heces, se da cuenta (ya que ha echado una “pelota” grande, que por supuesto lleva al médico para que la vea25) Neumobilia……. En la actualidad no sólo la vamos a encontrar en pacientes con fístulas, sino también en aquéllos a los que se le ha hecho una ERCP, tras la cual la papila queda abierta, y en pacientes con cirugía biliar previa o con alguna derivación de la vía biliar al tubo digestivo. En estos casos, el tratamiento es abrir el asa de intestino, sacar el cálculo y cerrar el asa. La fístula no tenemos que tocarla. Posibilidades clínicas con una fístula: a) Íleo biliar. b) Por su gran tamaño, el cálculo se puede encajar en el duodeno produciendo obstrucción duodenal, vómitos y HDA. Este cuadro se conoce como Síndrome de Bouveret, y su diagnóstico lo hace el endoscopista. Si el paciente está con vómitos, se le pone una sonda nasogástrica y posteriormente se procede a hacer la endoscopia. Cuando se alcanza el duodeno, el endoscopista se encuentra con la piedra24 y la obstrucción. 24 Más que piedra, pedrusco, porque para obstruir el duodeno… debe tener una cantera ;) TEMA 18 Prof. Miras 25 Quizá hasta se asuste al oir el click al caer a la taza del “señor Roca”. Comisionista: María Lozano 18-11 CMG MÉDICA DE APARATO DIGESTIVO CMG Bueno, parecía que no iba a pasar, pero el final llegó (por lo menos, el de la Comisión, jeje). Como Coordinador de ésta, nuestra Comisión, en primer lugar quiero pedir perdón por todos los errores, retrasos y demás problemas que hayan podido surgir, pero debo decir que estoy muy contento y agradecido con mis compañer@s de Comisión, por el gran trabajo que han realizado, tanto redactando como revisando los temas. Espero que este equipo, incluído el “nuevo fichaje” de principios de temporada (sí, tú, Mariló), siga trabajando en nuevos proyectos. Habéis funcionado como unos campeones. Gracias a todos los que alguna vez elaboraron una Comisión, porque nos ayudaron (sobre todo a los que trabajamos y no podemos asistir a clase asíduamente) a ir sacando los cursos adelante. Gracias a todos los que alguna vez se plantearán seguir con esta labor, pues gracias a ellos, podremos continuar con este gesto de solidaridad y compañerismo. Gracias a todos aquellos que confían en nosotros, pues sin ellos, este esfuerzo no tendría razón de ser. Gracias a todos los que vinieron al viaje y lo conviritieron en una experiencia maravillosa, inolvidable, y aquellos que no pudieron acompañarnos, también gracias, por estar ahí en los buenos y en los malos momentos, que también los hay. Vosotros sois quienes nos animáis a seguir adelante, y hacéis que nos sintamos orgullosos de pertenecer a nuestra Promoción, a este grupo de futuros médicos. Parece que fue ayer cuando llegué desde tierras gaditanas para unirme al “Grupo de los Protestones”, que tan bien me acogieron desde el primer día todos, sin excepción, y ya han pasado tres años, caminando de la mano en esta “Carrera de obstáculos”. Al principio, hicimos peña todos los “trasladados” (Leandro, Carlos, Juan Pablo, Almudena, Mª José, Angi, Blanca, Mariaje, Maqui), unos amigos desde el primer día, tan especiales, a quienes debo tantos buenos momentos, tantas fiestas, la Aventura-Congreso de Granada,… y porque siempre estáis ahí. Pero la clase pronto se abrió a todos nosotros, y nos conquistó. También daré las gracias a los compañeros que se nos han ido uniendo por el camino de la misma forma que yo lo hice, y enriquecen día a día nuestro grupo: sois geniales!! Esta es una de las razones que me animaron para embarcarme en la tarea de participar en una Comisión de Apuntes, porque en parte, me siento en deuda con todos vosotros. Sois unos Cracks!! Aún nos quedan dos añitos más, que seguramente, pasarán igual de rápido que los que ya dejamos atrás, pero espero que sean tan buenos como aquellos, y que sigamos tan unidos como hasta ahora, tanto para disfrutar de lo bueno como para plantarnos ante los problemas y las dificultades con tanta fuerza, y que sigamos mereciendo el apodo que nos hemos ganado. También quiero dedicar unas palabras a aquellos compañeros que se quedaron en el camino: os deseo toda la suerte y la felicidad del mundo, daros las gracias por los momentos que compartimos y recordaros que aquí tenéis un amigo para lo que os haga falta. Alguno marchó lejos en busca de su luz, a quien espero volver a abrazar pronto; otros se quedaron más cerca, y confío en que nuestros caminos se vuelvan a cruzar en un futuro no muy lejano. Bueno, esto de escribir a altas horas de la madrugada es lo que tiene, que igual te pones ñoño, o igual sueltas barbaridades, así que, voy a ir cediendo la palabra a mis compañeros de Comisión, para que os expresen lo que les dé la gana (que se lo han ganado). Un abrazo a tod@s, y mucha suerte!! José Martínez Más TEMA 18 Prof. Miras Comisionista: María Lozano 18-12 CMG MÉDICA DE APARATO DIGESTIVO CMG Bueno, ha llegado la hora de la despedida final. Debo dar las GRACIAS a todos mis compañeros de comisión por el trabajo realizado. Para la mayoría (por no decir todos, je,je) ha sido nuestra primera Comisión y la hemos realizado con mucha ilusión y esfuerzo. Gracias Juani, por corregir TODOS mis temas y ser tan meticulosa. Ya sabía que eras un bellísima persona, pero lo que he aprendido también haciendo esta Comisión es que eres una trabajadora y luchadora nata. Mi más sincero agradecimiento a las comisionistas de Cirugía Digestiva, en especial a su coordinadora Carmen Hernández, a Rocío García Paredes y Elena Vera, por su aportación en la realización del tema de Patología Biliar I (Litiasis biliar). Me gustaría dedicar esta comisión a todas aquellas personas que hacen que día tras día tras día me apetezca ir a prácticas, a seminarios y a clase (y si hay que ir a una manifestación….pues se va XDD) que día tras día, me demuestren que no son sólo simples compañeros de clase, sino que se han convertido en mis verdaderos amigos: A Andrés García Lax: qué te voy a decir que ya no sepas!!! Gracias por ser como eres, por ser una persona tan especial y aportar tanta alegría y optimismo a mi vida. Y por ser mi agobioso preferido (porque mira que te pones insoportable, eh???menos mal que ya nos conocemos XDD). Por aguantarme cuando tengo mis brotes de “diarrea cardiogénica”( ja,ja,ja) y por demostrarme que estás ahí incluso cuando nos ponemos a jugar a … lerda o tonta 1,2 y 3!!!ja,ja. Gracias!! A Miriam por su sinceridad y por estar siempre ahí. Porque debe ser cierto eso de que los polos opuestos al final se atraen, o coinciden…porque, mira que somos diferentes! A Ana Rodríguez porque que estuvo, está y estará. A Mª José Mayor: conocerte ha sido una de las mejores cosas que me ha pasado en este curso. Tengo tantísimas cosas que agradecerte….muchísimas gracias por darme tan buenos momentos, por tu apoyo incondicional, por ser tan buena gente y tener un corazón tan grande. No sabes cúanto me alegro de haberte conocido!! A Tayo, María Ramos, Alicia y Jose Alberto por proporcionarme tan buenos momentos en el viaje de estudios. A Leandro y a Laura Novella, por su lealtad. Porque a pesar del paso de los años, siguen estando ahí y me siguen demostrando lo buena gente que son. Os quiero muchísimo. A María Cerdán, por todos aquellos momentos pre-conferencia pasados, por aquellas reflexiones tan profundas, por ….tantas cosas!!! Sabes que eres mi madrina preferida!!!XDD A mi empiema favorito!!! Jaja. Por todos los buenos momentos. Nunca una crema NIVEA dio parta tanto;)). A Josemi (el mejor compañero de prácticas que una puede tener) a digo xina, ( ja,ja) y a Irene García-Escribano: ha sido un placer conoceros MÁS este año!! Gracias por todo. En definitiva, gracias a TODOS y también a ti, querido lector. Gracias por confiar en esta comisión. Espero que te haya gustado y que te haya sido útil. Te deseo lo mejor en lo que queda de carrera y en la vida. No te desanimes, sigue adelante y no tires la toalla. Porque, aunque es cierto que hay momentos duros, éstos quedarán ahí y con el paso del tiempo se olvidarán, pero las satisfacciones, la sonrisas de los pacientes, el poder ayudar a personas, la alegría del trabajo bien hecho….eso nunca se olvida. Un abrazo TEMA 18 Prof. Miras Comisionista: María Lozano 18-13 CMG MÉDICA DE APARATO DIGESTIVO CMG Ya estamos llegando al final de esta Comisión, y qué menos que dedicar unas merecidas palabras, en primer lugar, a mis compañeros comisionistas (Jose, Jose, María, MiMiriam, Mariló, Cristina, Juani): Juani admiro y agradezco vuestro enorme esfuerzo y trabajo, habéis demostrado saber y poder transmitir los conceptos básicos de esta asignatura de una forma amena y didáctica, así como un alto grado de competencia y excelencia. Deciros que fue un placer trabajar con todos vosotros, no me importaría en absoluto reunirnos de nuevo en años sucesivos para realizar una nueva comisión (es más, os lo propongo aprovechando esta dedicatoria ;). ¡Enhorabuena! Es un buen momento, aunque a estas alturas sea ya bastante tópico, para animar en la participación de alguna Comisión a todos aquellos que aún no lo habéis hecho, no sólo por el alto grado de satisfacción que se consigue al ayudar a estudiar al resto de compañeros (que compensa sobradamente nuestro esfuerzo dedicado), sino también por ser una forma de agradecimiento a todos aquellos que ya se preocuparon de elaborar otras Comisiones y facilitarnos la labor en este largo camino espartano que es Medicina. Esta ayuda y colaboración mutuas parten de dos conceptos que, aunque también suene a tópico, ya conocemos: solidaridad y compañerismo. Si bien en general tales conceptos se practican gratuitamente y de forma desinteresada, no faltan compañeros que se jactan de promover y enarbolar casi cínicamente esos consabidos lemas, cuando en realidad denotan cierta hipocresía, envidia y tensiones silenciadas. A todos ellos quisiera recitarles una conocida expresión que cita “practica la moral que predicas”. Por último, no podía olvidarme de vosotros: Graciosas, Estupendas, Gigolós... nos hemos regalado momentos inolvidables que no han hecho sino afianzar más aún nuestra amistad (y los que nos quedan), y ante eso sólo me queda insistiros en que esos lazos no se romperán. Grandes!! τ TEMA 18 Prof. Miras Comisionista: María Lozano 18-14 CMG MÉDICA DE APARATO DIGESTIVO CMG Hola!! Lo primero que quiero decir es que me ha gustado mucho participar en la comisión…la verdad es que da mucha alegría (a pesar de que algunas veces, hacerla se hace cuesta arriba), cuando ves los temas terminados y que a la gente le gustan y te felicitan…porque compensa todas las tardes y noches que te pasas delante del ordenador. Mi parte de comisión se la quiero dedicar a varias personas: a Juani, a la que le estoy muy agradecida por su minuciosa labor como revisora, ya que ha corregido TODOS los temas que se le enviaba. A Irene y Emilio (por su “falsa” puntualidad, jeje!, no es japonesa, digochina), a José Miguel por todo el follón que le he dí hasta que me enteré de cómo se daba formato, además del que le doy. A María Lozano porque a pesar de ser las dos como los dos polos de un imán, hay un punto en el que congeniamos (lo tengo que decir: ojala que todo el mundo que se compre un coche, le salga tan bueno como el tuyo jajaja!!), a la comisión de cirugía digestiva, es especial a Carmen, por enviarme aquellos temas que podían tener contenido en común con los míos, además por ser un y una estupendísima persona, y a Andrés G.L. por ser el más feo, el más tonto, el más egoísta, el más competitivo, el más agobioso (y esto si es verdad, que no hay quien te hable y quien te soporte cuando te pones así, pero en esos momentos, deberías decir lo mismo que dijo Lola Flores en la boda de Lolita “Si me queréis, irse!!!!!!!”Jajajajajajaja), el más antipático, el peor compañero…No te tengo que decir nada más, tú ya sabes lo especial que eres para mí. Un saludo Miriam En primer lugar, le dedico mi parte del trabajo a mis compañeros de Comisión, porque, aunque el temario de la asignatura y el encanto de la mayoría de los profesores nos han ayudado, creo que nos ha quedado un buen trabajo. En segundo lugar, mención especial a los delegados de la clase, a los claustrales, a los del CEUM, y a todos los idealistas que luchan por lo que creen que es justo, porque no es en vano. Por último, a Yaiza, Anica, Isa, María Ramos, María Cerdán, Ángel, Paola, Elena, Mariano... y a todos los que han hecho que me sienta aquí como si estuviera toda la vida. Mariló TEMA 18 Prof. Miras Comisionista: María Lozano 18-15 CMG MÉDICA DE APARATO DIGESTIVO CMG Hola a todo el que esté leyendo estas palabrillas...te darás cuenta de que no sé muy bien qué decir..así que mejor breve y sencillo. No sé si merezco dedicar lo poquito que haya podido ayudar en esta Comisión, pero si debiera hacerlo, considero que los que se llevan mi aplauso y enhorabuena son los chicos/as de esta Comisión...va por vosotros...oleeeeeeeee, oleeeeeeee y oleeeeeeeeeeeeeeee!!! Para los demás, gracias por cada palabra y cada sonrisa..sobre todo esta última..y si ha caido un abrazo..mejor que mejor! Gracias. Ah...todo aquel que crea que no puede...puedes!! Todo aquel con el que pude compartir al menos un minuto del viaje a la Repúbica Dominica consiguió algo extraordinario..y es cambiar mi desilusión..aseguro que no es nada fácil. Así que supongo que si no estamos solos en este mundo es por algo...y la próxima vez que creas que no puedes..pregúntate: y con la ayuda de otro/os podré?? Ánimo que va quedando menos para la graduación..y ya sabéis que los años de Universidad dicen que son los mejores! jeje. Oye, que ya paro que siempre que empiezo me engancho y jurf..ciao! PD1: Un montón de besicos y morreos para mis nenitas de Medicina..vosotras sabéis que os quiero...pero no me cansaré de repetirlo las veces que haga falta. PD2: Para el que aún no lo sepa..nuestro Pascuy ha aprobado Anatomía II!!!! ip hip hurra!! Otro que puede!! Que quede constancia para la eternidad!! Cris (Conte) Muy a mi pesar, y no pudiendo demorar más la publicación del último tema, por las muchas peticiones de los compañeros para que lo colgase cuanto antes, no hemos podido esperar a que Juani nos enviase su dedicatoria. Lo siento mucho, nos habría hecho muchísima ilusión contar con tu comentario tan merecido por tu aportación a la Comisión. Has hecho un trabajo genial, sin el que el temario no habría sido el mismo. Un beso, guapetona, y mucha suerte!! TEMA 18 Prof. Miras Comisionista: María Lozano 18-16 CMG MÉDICA DE APARATO DIGESTIVO CMG TEMA 19A PANCREATITIS AGUDA DEFINICIÓN Inflamación del páncreas1 con o sin compromiso de los órganos vecinos, así como la respuesta inflamatoria sistémica derivada de la lisis de células acinares y la liberación de su contenido enzimático (Consenso de Atlanta 1992). Otras definiciones asociadas de Atlanta 922: Pancreatitis aguda leve (80% de las pancreatitis agudas): edema intersticial con mínima repercusión sistémica. Pancreatitis aguda grave: se asocia a fallo multiorgánico y/o complicaciones locales como necrosis, pseudoquiste o absceso. Necrosis (estéril o infectada): zonas localizadas o difusas de tejido no viable con necrosis grasa peripancreática. (Menor densidad a la TC). Colecciones líquidas agudas: colecciones de densidad líquida que carecen de pared. Pseudoquiste: formación de densidad líquida con pared (4ª semana). Absceso pancreático: colección de pus en el páncreas o en su vecindad. Opie describe el origen biliar de la pancreatitis aguda (PA) por la existencia de cálculos que se enclavan en la papila y que generan, por un lado, aumento de la presión en el conducto biliar, que produce reflujo biliar, y por otro lado, fenómenos inflamatorios peripapilares. Efecto de la ingesta continuada de Alcohol sobre el páncreas El alcohol tiene dos importantes efectos sobre el páncreas: modifica la secreción pancreática favoreciendo la aparición de precipitados proteicos y dificulta la exocitosis en la célula acinar, permitiendo la activación intracelular de enzimas. Es un factor precipitante en las pancreatitis agudas y crónicas y un factor agravante en las crónicas. CAUSAS La causa más frecuente es la litiasis biliar, que aparece en un 60-70% de las PA. Va seguida del alcoholismo (20%) y de la pancreatitis idiopática (10%). Estas son todas las causas: 1. Obstructivas: coledocolitiasis, tumores periampulares, páncreas divisum3 , disfunción del esfínter de Oddi4 . 2. Tóxicos o drogas: alcohol, insec-ticidas organofosforados, azatioprina, ác. valproico, metronidazol. Componente Biliar en la Pancreatitis Aguda A diferencia de la pancreatitis crónica, en la aguda se observa con frecuencia afectación del colédoco: coledocolitiasis. La teoría de 1 Inflamación del páncreas hace referencia a afectación de grandes vasos y de la vía biliar, y activación de enzimas pancreáticas. 2 NOTA IMPORTANTE: Fue un congreso, las Olimpiadas de ese año fueron en Barcelona, y en Atlanta cuatro años después ;) TEMA 19A Prof. Carballo 3 El páncreas dividido ocurre cuando los primordios embriológicos dorsal y ventral del páncreas no se fusionan, de modo que el drenaje pancreático se efectúa principalmente a través de la papila accesoria. 4 Durante el ayuno, el esfínter de Oddi constituye una zona de resistencia de alta presión al paso de la bilis desde el conducto colédoco al duodeno. Esta contracción tónica sirve para impedir el reflujo del contenido duodenal a los conductos biliar y pancreático y para permitir que la vesícula se llene de bilis. Comisionista: Mariló Jiménez 19A-1 CMG MÉDICA DE APARATO DIGESTIVO 3. Traumáticas: trauma contuso o penetrante, CPRE (colangiopancreatografía retrógrada endoscópica), cirugía, manometría del Oddi. 4. Metabólicas: hipertrigliceridemia, hipercalcemia. 5. Infecciosas: ascaridiasis, urliana, virus de la hepatitis A, B y C, adenovirus, CMV, VIH. 6. Vascular: isquemia por hipoperfusión, vasculitis, microembolismo. 7. Hereditaria. 8. Miscelánea: úlcera péptica penetrante, enfermedad de Crohn, insuficiencia renal crónica terminal, trasplante de órgano, veneno de alacrán (escorpión). PATOGÉNESIS PATOGENESIS DE LA P.A . ENTEROQUINASA TRIPSINOGENO • TRIPSINA PROENZIMAS CALICREINA ELASTASA FOSFOLIPASA A Acido Biliar SHOCK 2/23/03 HEMORRAGIA CMG Con estos dos esquemas creemos que queda bien resumido el proceso. No obstante, os añadimos un... Contenido Ampliado La patogenia de la pancreatitis se puede dividir en tres fases. La primera se caracteriza por la activación intrapancreática de enzimas digestivas y por la lesión de células acinares como consecuencia de la activación del zimógeno. La segunda fase comprende la activación, quimioatracción y secuestro de neutrófilos en el páncreas, que origina una reacción inflamatoria intrapancreática de intensidad variable. Este secuestro de neutrófilos activa el tripsinógeno. La tercera fase se debe a los efectos de las enzimas proteolíticas y de mediadores activados, liberados por el páncreas inflamado, en órganos distantes. Las enzimas proteolíticas activadas y en particular la tripsina, además de digerir tejidos pancreáticos y peripancreáticos, también activan otras enzimas como la elastasa y la fosfolipasa. Como paso siguiente las enzimas activas digieren las membranas celulares y originan proteólisis, edema, hemorragia intersticial, daño vascular y necrosis. El daño y la muerte de las células hacen que se liberen péptidos de bradicinina, sustancias vasoactivas e histamina, que originarán vasodilatación, mayor permea-bilidad vascular y edema, con profundos efectos en muchos órganos, en particular el pulmón. Pueden ocurrir como consecuencia de la cascada de efectos locales y a distancia el síndrome de respuesta inflamatoria generalizada y el fallo multiorgánico. Lipasa Necrosis Grasa Necrosis Pancreática MANIFESTACIONES CLÍNICAS Principales: TEMA 19A Prof. Carballo Dolor abdominal. Es el síntoma principal de la PA. Está presente en el 90% de los casos. Normalmente es brusco, intenso, constante y localizado en hemiabdomen superior. Nauseas y vómitos. Son frecuentes y se deben a la hipomotilidad gástrica e intestinal. Síntomas de shock en casos graves. Comisionista: Mariló Jiménez 19A-2 CMG MÉDICA DE APARATO DIGESTIVO Otras manifestaciones: La TC es el mejor método diagnóstico de la gravedad de la PA. Con ella, podemos clasificar a los pacientes según los criterios de Balthazar: Alteraciones hemodinámicas: taquicardia, hipotensión, deshidratación, mala perfusión periférica, fiebre. Signos abdominales: palpación dolorosa, distensión abdominal, íleo, masa epigástrica, ascitis, equimosis parietales. Grado A: páncreas de aspecto normal. Score 0. Trastornos respiratorios: taquipnea, atelecta-sia, derrame pleural. Grado B: aumento de tamaño focal o difuso. Score 1. Manifestaciones hepatobiliares: signos de daño hepático crónico en pacientes alcohólicos e ictericia. Esta última es rara y suele deberse a edema de la cabeza del páncreas, que comprime la porción intrapancreática del conducto colédoco. Grado C: páncreas anormal con inflamación peripancreática y de la grasa peripancreática. Score 2. Alteraciones neurológicas: compromisos en el estado de consciencia, agitación psico-motora. Criterios de Balthazar Grado D: una colección intra o extrapancreática. Score 3. Grado E: dos o más colecciones y/o gas retroperitoneal. Score 4. DIAGNÓSTICO DE GRAVEDAD DIAGNÓSTICO En primer lugar, el médico debe tener un alto índice de sospecha en pacientes con antecedentes de litiasis biliar y de alcoholismo. Cuando se sospecha una PA, el diagnóstico es acertado en el 50% de los casos. El diagnóstico suele establecerse por la detección de un aumento en las concentraciones séricas de amilasa, lipasa e isoamilasa. La cifra normal de amilasa en sangre es de 4 a 25 mU/ml. Valores tres o más veces por encima de este, prácticamente aseguran el diagnóstico si se excluyen enfermedad manifiesta de las glándulas salivales y perforación o infarto intestinal. Puede presentarse una cifra más baja si el análisis se hace de forma precipitada, de manera que el aumento aún no es detectable, o si estamos ante una pancreatitis crónica. En orina, el aumento de enzimas se detecta con mayor sensibilidad. La ecografía suele ser el procedimiento inicial en la mayoría de los pacientes en quienes se sospecha enfermedad pancreática. En la PA, se puede ver un agrandamiento característico del páncreas y también descubrir la causa5. No obstante, factores como la obesidad o el exceso de gas intestinal dificultan la visualización de la glándula. 5 CMG Para evaluar el riesgo de fallecer en las personas con pancreatitis aguda hay diversos sistemas de puntuación de múltiples factores: Ranson, Imrie, Osborne, Balmey y APACHE II, que se usa en UCI. Pero tienen escasa capacidad predictiva y los clínicos no los han aceptado unánimemente. Los indicadores básicos de gravedad de pancreatitis son: 1. Disfunción/fallo multiorgánico: hipotensión o taquicardia, alteración de los valores normales de pO2 y pCO2, oliguria, hemorragia de las vías gastrointestinales. 2. Necrosis pancreática (véanse criterios de Balthazar). 3. Obesidad; edad > 70. 4. Hemoconcentración. 5. Proteína C reativa (>150mg/L). 6. Aumento de péptido activador del pepsinógeno. COMPLICACIONES Sistémicas: síndrome de respuesta inflamatoria sistémica o fallo multiorgánico. Locales: necrosis o necrosis infectada, colecciones, pseudoquistes, abscesos. Como puede ser una litiasis biliar, por ejemplo. TEMA 19A Prof. Carballo Comisionista: Mariló Jiménez 19A-3 CMG MÉDICA DE APARATO DIGESTIVO CMG TRATAMIENTO En la mayoría de los pacientes (85-90%) con PA, la enfermedad es de evolución limitada y cede espontáneamente, entre tres y siete días después de instaurado el tratamiento. Las medidas generales son: 1. Reposición de volumen con líquidos y coloides intravenosos. Si estamos ante una PA grave, tratamiento de sostén en UCI. 2. Alivio del dolor con analgésicos, incluso morfina si es necesario. 3. Reposo pancreático con ayuno. 4. Sonda nasogástrica sólo en presencia de íleo o dolor por distensión. Disminuye la liberación de gastrina en el estómago y evita que el contenido gástrico pase al duodeno. Otras medidas: 1. Ingreso en UCI. 2. Profilaxis antibiótica. Es una medida controvertida, que sólo se aplica cuando ay alta sospecha de infección. 3. Nutrición enteral. Se acompaña de una menor cifra de complicaciones que incluyen infecciones, cuando se compara con la nutrición parenteral. 4. CPRE si hay cálculos en papila. 5. Cirugía (extirpación de tejido necrótico) cuando hay necrosis infectada y cuando no se consigue frenar con las medidas ordinarias el deterioro de un paciente con necrosis estéril, pues también tiene un alto índice de mortalidad. TEMA 19A Prof. Carballo Comisionista: Mariló Jiménez 19A-4 CMG MÉDICA DE APARATO DIGESTIVO CMG TEMA 19B PANCREATITIS CRÓNICA DEFINICIÓN La pancreatitis crónica (PC) es una enfermedad inflamatoria del páncreas caracterizada por cambios morfológicos irreversibles que típicamente causan dolor y/o pérdida permanente de la función pancreática. CLASIFICACIÓN TIGAR-O Tóxico-metabólica: - Alcohólica - Tabáquica - Hipercalcemia - Hiperlipidemia - Insuficiencia renal crónica - Medicamentos: abuso de fenacetina - Tóxicos: (DBTC) Di-n-butyltin dichloride Idiopática: - Tropical: Pancreatitis tropical calcificante - Diabetes pancreática fibro-calculosa. Hay dos variantes: 1. Inicio temprano. Genética: - - Autosómica dominante: mutación de tripsinógeno catiónico (codón 29 y 122). Autosómica recesiva: o Mutación del CFTR o Mutación del SPINK 1 o Mutación de tripsinógeno catiónico (codón 16, 22 y 23) TEMA 19B Prof. Carballo - Pancreatitis crónica autoinmune aislada - Pancreatitis crónica autoinmune sindromática: - Síndrome de Sjögren - Enfermedad inflamatoria intestinal - Cirrosis biliar pancreática Recurrente: - Primaria - PA recurrente asociada a PC: o Postnecrótica o PA recurrente o Enfermedad sos/isquemia o Postirradiación de los va- Obstructiva: - Páncreas divisum - Obstrucción ductal - Cicatricial ductal postraumática PANCREATITIS ALCOHÓLICA El alcohol es el responsable del 70-80% de los casos de PC en los países desarrollados. 2. Inicio tardío. Autoinmune: Sólo el 10% de los alcohólicos desarrollan la enfermedad, desconociéndose si existen además factores genéticos o ambientales. Como ya sabemos, la ingesta crónica de alcohol tiene dos importantes efectos sobre el páncreas: modifica la secreción pancreática favoreciendo la aparición de precipitados proteicos, y dificulta la exocitosis en la célula acinar, permitiendo la activación intracelular de enzimas. Comisionista: Mariló Jiménez 19B-1 CMG MÉDICA DE APARATO DIGESTIVO Sobre la etiopatogenia de la PC de origen alcohólico hay dos hipótesis: 1. 2. Hipótesis de Sarles, según la cual en pacientes alcohólicos se produce un aumento de la concentración proteica en el jugo pancreático, aumentando su viscosidad, y una disminución de litostatina (PSP), que estabiliza el exceso de calcio en el jugo pancreático. Todo esto genera la precipitación de proteínas con formación de tapones proteínicos y atrofia de epitelio ductal. Hipótesis necrosis-fibrosis, que defiende que la pancreatitis crónica es sólo el estadio final de la pancreatitis aguda alcohólica. Reiterados ataques de pancreatitis aguda con múltiples focos de necrosis conducen a cambios fibróticos irreversibles, obstrucción de los conductos con precipitación proteica y formación de tapones ductales, precipitación de calcio y formación de cálculos. En cuanto al tabaco, este no causa por sí mismo PC pero agrava la PC alcohólica1. PANCREATITIS IDIOPÁTICA CRÓNICA Supone entre el 10-30% de las PC en países desarrollados. Tiene una distribución bimodal según la edad de aparición: PC idiopática de inicio temprano: Aparece en menores de 35 años (media de 25), cursa con un dolor severo de inicio, y los cambios morfológicos y funcionales ocurren lentamente. PC idiopática de inicio tardío: Aparece en mayores de 35 años (media de 52), es indolora en la mitad de los casos, y los cambios morfológicos y funcionales son rápidos. La PC idiopática tiene una fuerte asociación con antígenos HLA clase I y clase II, relación que no se ha encontrado en la PC alcohólica. Por tanto, podemos decir que en la PC idiopática están implicados factores genéticos y factores autoinmunes. CMG PANCREATITITS CRÓNICA GENÉTICA O HEREDITARIA Los portadores de la mutación pueden no llegar a desarrollar nunca la enfermedad. Los que sí la padecen pueden ser bebedores o no bebedores. El 80% de los pacientes que heredan el gen sufren reiterados ataques de PA, y el 50% de estos desarrolla PC. En la PC genética, la posibilidad de desarrollar cáncer de páncreas es del 50%. PANCREATITIS AUTOINMUNE CRÓNICA En ella se detectan autoanticuerpos en la sangre y valores aumentados de gamma globulina o Ig G sérica. Ocasionalmente, va asociada a enfermedades autoinmunes. Se caracteriza por un agrandamiento difuso del páncreas, estrechez segmentaria o difusa del Wirsung en la CPRE, compromiso del ducto biliar (colestasis) y ausencia de calcificaciones o pseudoquistes. Los síntomas suelen ser leves, sin ataques de recidiva agudos de la enfermedad. Los cambios fibróticos coexisten con infiltración linfocitaria. Responde bien al tratamiento inmunosupresor con corticoides. PANCREATITIS RECURRENTE CRÓNICA La teoría necrosis-fibrosis apoya que ataques reiterados de PA alcohólica conducen progresivamente a PC. No obstante, esta asociación ha sido motivo de controversias. La PA hereditaria resultaría una de las formas de la enfermedad que apoyaría esta teoría. 1 Otra razón más para dejar de fumar, ¿verdad Miguel? Jeje. TEMA 19B Prof. Carballo Comisionista: Mariló Jiménez 19B-2 CMG MÉDICA DE APARATO DIGESTIVO PANCREATITIS OBSTRUCTIVA CRÓNICA CMG tiene un coste elevado y su uso está restringido a centros especializados. 3.2. Test indirecto de Elastasa-1 fecal. Puede ser congénita: páncreas divisum. O adquirida: tumores, traumas, secuelas cicatrizales, pseudoquistes, disfunción del Oddi. DIAGNÓSTICO 1. Sospecha clínica. La triada clásica de dolor abdominal en paciente alcohólico, esteatorrea y diabetes prácticamente establece el diagnóstico de PC, pero sólo ocurre en menos de un tercio de los enfermos de PC. La elastasa fecal tiene dos funciones: actividad proteolítica y transporte del colesterol y sus metabolitos a través del tracto intestinal. Es muy estable en su pasaje por el tubo digestivo, por eso la concentración en heces refleja la cantidad de enzima secretada desde el páncreas, y no es interferida por la administración de enzimas exógenas. Con los datos obtenidos, podremos clasificar la función pancreática en: Normal: >200 μg/g de elastasa fecal Insuficiencia pancreática moderada: 100-200 μg/g Insuficiencia pancreática severa: <100 μg/g 2. Cambios morfológicos pancreáticos. La ecografía y la TC pueden mostrar calcificaciones diseminadas o conductos dilatados, además de excluir los pseudoquistes y el cáncer de páncreas. La CPRE aporta información del estado del conducto de Wirsung y otros de menor calibre y permite medir la gravedad de la enfermedad mediante la clasificación de Cambridge: Grado I: Wirsung normal y menos de tres ramas secundarias. Grado II: Wirsung normal y más de tres ramas secundarias. Grado III: Wirsung alterado y más de tres ramas secundarias. Grado IV: lo anterior más cavidades mayores de 10mm, defectos de relleno o cálculos y severa dilatación del Wirsung. 3. Pruebas funcionales. 3.1. Test directo de Secretina-CCK. Consiste en la estimulación directa del páncreas mediante administración intravenosa de secretina más CCK, colectando y midiendo el contenido duodenal. Es el método diagnóstico “gold estándar”, con una sensibilidad y especificidad que supera el 90%. Sin embargo, es un método invasivo y molesto para el paciente, requiere largo tiempo de realización, TEMA 19B Prof. Carballo La sensibilidad de la prueba en la PC leve es el 0%, sin embargo en la PC moderada-grave es del 100%. La especificidad es de 83%. TRATAMIENTO 1. Tratamiento del dolor. El dolor en la PC puede presentarse de varias formas: como episodios de dolor agudo (PA) separados por periodos libres de dolor, como dolor persistente durante periodos prolongados o como dolor recurrente con periodos libres de dolor. Las medidas de sostén para el tratamiento del dolor consisten en la abstinencia de alcohol y de comidas copiosas ricas en grasa, y en la administración de analgésicos (AINES, si el dolor es leve; opioides si es moderadosevero). Fisiopatológicamente, hay dos teorías que explican el dolor. La teoría de la hipertensión ductal e intersticial establece que el dolor es consecuencia de la obstrucción ductal, bien por estenosis, cálculos o hiperviscosidad del jugo pancreático. Y la teoría de la inflamación neural explica que el dolor es debido a atrapamiento neural por fibrosis, a inflamación perineural y a un incremento de la inervación peptidérgica sensorial. Por tanto, el tratamiento del dolor va a consistir en disminuir la hipertensión ductal y la secre- Comisionista: Mariló Jiménez 19B-3 CMG MÉDICA DE APARATO DIGESTIVO ción pancreática, tanto de enzimas pancreáticas como de octreótido. La administración de enzimas pancreáticas disminuye el dolor en la mayoría de los pacientes con PC. Puede intentarse tratamiento endoscópico: esfinterotomía del esfínter pancreático mayor o menor, dilatación de las estenosis, extracción de cálculos o colocación de endoprótesis (stent) en el conducto pancreático ventral o dorsal; pero conlleva complicaciones significativas en un alto porcentaje de pacientes que son sometidos a él. La estrategia diagnóstica del dolor abdominal secundario a PC es la siguiente: después de excluir otras causas del dolor, aplicar las medidas de sostén y efectuar una ecografía pancreática. Si no aparece ninguna masa, se puede realizar una prueba de secretina. Si el resultado es anormal, se aconseja la administración de enzimas pancreáticas. Si no se produce una mejoría, se debe efectuar ERCP. Si se descubre un pseudoquiste o una obstrucción ductal localizada, deberá considerarse la cirugía (cuando el Wirsung es menor de 6mm, cirugía resectiva; y cuando es mayor de 6mm, cirugía derivativa). CMG 3. Tratamiento de insuficiencia endocrina: Tratamiento de la Diabetes. El tratamiento de la diabetes consiste en adecuación de la dieta, hipoglucemiantes orales e insulina. 4. Tratamiento de las complicaciones. Las complicaciones más comunes de la PC son los pseudoquistes, la obstrucción biliar y los pseudoaneurismas. Otras poco frecuentes son: obstrucción duodenal, ascitis pancreática, trombosis de la vena esplénica y cáncer. 2. Tratamiento de la insuficiencia exocrina: Tratamiento de la esteatorrea. La maldigestión de grasas es un proceso más rápido y severo que la maldigestión de carbohidratos y proteínas. El tratamiento de la esteatorrea consiste en abstinencia de alcohol, adecuación de la dieta y sustitución enzimática. Las propiedades ideales del preparado de sustitución enzimática son: alta actividad de la lipasa, protección contra el ácido gástrico para que no desactive la lipasa, y vaciamiento de los alimentos del estómago simultáneo con las enzimas pancreáticas. Para abolir la esteatorrea, la cantidad de enzimas liberadas en duodeno debe ser ±10% de la secreción normal. TEMA 19B Prof. Carballo Comisionista: Mariló Jiménez 19B-4