FÍSICA Y QUÍMICA Temario
Anuncio

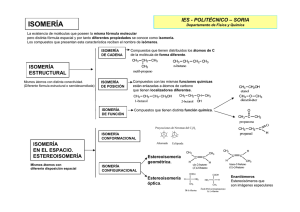
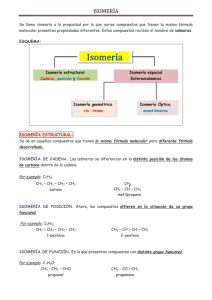
Temario FÍSICA Y QUÍMICA Estructura y enlaces del carbono. Nomenclatura. Isomería. Comprobación experimental de la actividad óptica. 25-15223-13 59 Química del carbono. FÍSICA Y QUÍMICA 59 1. ENLACES DEL CARBONO 2. NOMENCLATURA 2.1. ALCANOS, ALQUENOS Y ALQUINOS 2.2. HIDROCARBUROS AROMÁTICOS 2.3. FUNCIONES OXIGENADAS 2.3.1. Alcoholes, fenoles y éteres 2.3.2. Aldehídos, cetonas, ácidos carboxílicos y ésteres 2.4. FUNCIONES NITROGENADAS: AMINA, AMIDA, NITRILO Y NITRO 3. ISOMERÍA 3.1. CONCEPTO DE ISOMERÍA 3.2. ISOMERÍA ESTRUCTURAL O CONSTITUCIONAL 3.2.1. Isomería de cadena 3.2.2. Isomería de posición 3.2.3. Isomería de función 3.3. ESTEREOISOMERÍA 3.3.1. Isomería configuracional 3.3.2. Isomería conformacional 3 FÍSICA Y QUÍMICA 59 INTRODUCCIÓN En este tema se analiza el carbono y sus enlaces, y la nomenclatura y las características estructurales de los compuestos que forma, los compuestos orgánicos. La configuración electrónica del átomo de carbono es 1s2 2s2p2, con una capa de valencia semillena o semivacía, que le confiere la capacidad de compartir los electrones de la capa de valencia produciendo enlaces covalentes. Así, los átomos de carbono se unen a otros átomos, principalmente de hidrógeno, pero también de flúor, cloro, bromo, yodo, oxígeno, nitrógeno, azufre o fósforo, con facilidad y produciendo compuestos estables. Sin embargo, es su capacidad única de compartir los electrones con otros átomos de carbono lo que posibilita la existencia de millones de compuestos orgánicos. Pueden formar cadenas de miles de átomos o estructuras cíclicas de infinidad de tamaños; y estas cadenas y anillos pueden tener a su vez ramificaciones y/o uniones cruzadas, de modo que cada compuesto es diferente, y sus propiedades químicas y físicas, distintas. Por último, al tener una estructura electrónica tetravalente puede también formar enlaces múltiples (dobles o triples) consigo mismo C=C, C≡C, y con otros elementos, por ejemplo C=O o C≡N, lo que aumenta aún más la diversidad de las estructuras del carbono. 5 6 TEMARIO 1 ENLACES DEL CARBONO El átomo de carbono forma enlaces covalentes con otros átomos de carbono y con el hidrógeno. La naturaleza de este enlace determina la geometría de las moléculas que forma, su energía y su reactividad; de ahí la importancia de las teorías que explican la estructura del enlace. XX Enlaces simples: hibridación sp3. Metano El átomo de carbono en el metano tiene la capacidad de formar cuatro enlaces simples iguales con los átomos de hidrógeno, de una longitud aproximada de 1,09 Å y orientados hacia los vértices de un tetraedro, formando, por tanto, entre sí 109º. Esta estructura se explica por combinación del orbital atómico de valencia 2s con los tres orbitales atómicos de valencia 2p, para formar cuatro orbitales híbridos sp3 idénticos entre sí y cada uno con un electrón de valencia. 3 + 4 1 p s sp3 Cuando se une a cuatro átomos de hidrógeno, es la estructura del metano, CH4, molécula que experimentalmente demuestra ser simétrica, con cuatro enlaces iguales C-H de exactamente 1,094 Å de longitud y formando un ángulo de 109,5º. 109º H H H C H H sp3 XX Enlaces dobles: hibridación sp2. Eteno Cuando la hibridación se produce entre tres orbitales atómicos, uno s y dos p (dejando sin hibridar el orbital atómico pz), para dar tres orbitales híbridos idénticos denominados sp2, se obtiene la estructura de doble enlace C=C, en la que cada carbono tiene una estructura plana-trigonal, con los tres orbitales híbridos dirigidos hacia los vértices de un triángulo equilátero. El orbital pz sin hibridar queda perpendicular al plano formado por los híbridos sp2 y solapa con el otro orbital pz del átomo de carbono vecinal formando el denominado enlace π (enlace banana). Debido a que el solapamiento es menos efectivo entre estos dos orbitales que entre FÍSICA Y QUÍMICA 59 los sp2, el enlace π es más débil que el enlace σ. Esto explica que los alquenos sean más reactivos que los alcanos. π pz H H H H H sp2 σ XX Enlaces triples: hibridación sp. Etino El triple enlace C≡C del etino puede explicarse en función de la hibridación de dos orbitales atómicos, uno s y otro p (dejando sin hibridar dos orbitales atómicos, py y pz), para dar dos orbitales híbridos igualmente denominados sp. En este tipo de enlace los tres orbitales híbridos tienen los ejes a lo largo de una línea recta, con lo que los dos enlaces s que generan estarán dirigidos también a lo largo de una línea recta, y por tanto las moléculas son lineales. Los orbitales py y pz, sin hibridar, de cada átomo de carbono se solapan con los del otro formando dos enlaces π. El resultado es el triple enlace que une los dos átomos de carbono de la molécula: uno σ y dos π. π pz sp H H H py σ π XX Enlaces deslocalizados: nubes π. Benceno. En los tipos de moléculas estudiados hasta ahora, los enlaces están localizados, es decir, con la distribución electrónica situada en zonas muy específicas de la molécula, concretamente entre los núcleos de los átomos que enlazan. Existen, sin embargo, muchas otras moléculas orgánicas, denominadas conjugadas, cuyas fórmulas clásicas se representan en función de enlaces carbono-carbono simples y dobles alternados. Ejemplo de estas moléculas conjugadas son los polienos lineales, como el butadieno, moléculas aromáticas, como el benceno y el naftaleno, y heteromoléculas, como la anilina y la piridina. 7 8 TEMARIO Como ejemplo de molécula conjugada se estudiará el benceno, de la que se sabe experimentalmente que es plana y con ángulos de enlace de 120º, lo que indica para los enlaces σ una clara hibridación sp2. Benceno: formas resonantes Butadieno Los seis enlaces de la molécula de benceno son iguales, un intermedio entre simples y dobles, ya que su longitud entre carbonos es de 1,39 Å (el simple es de 1,54 Å, y el doble, de 1,33 Å). Esto se explica a partir de los seis orbitales pz (uno por cada átomo de carbono) que deja libres la hibridación sp2, que dan nubes π por encima y debajo del plano de la molécula, al estar deslocalizados los electrones. La deslocalización de los electrones en estos orbitales estabiliza de forma especial al benceno; esto es el carácter aromático. pz Nube π sp2 FÍSICA Y QUÍMICA 59 2 NOMENCLATURA1 2.1. ALCANOS, ALQUENOS Y ALQUINOS XX Alcanos Son hidrocarburos saturados. Los alcanos que presentan menor complicación son los de cadena lineal. A excepción de los cuatro primeros, que reciben los nombres de metano, etano, propano y butano, se nombran mediante un prefijo griego, que indica el número de carbonos, y la terminación -ano, que es genérica y se aplica a todos los hidrocarburos saturados (de ahí el nombre de alcanos). CH4 Metano H3C CH3 H3C CH2 CH3 H3C CH2 Propano Etano CH2 CH3 Butano Para nombrar los alcanos ramificados es preciso definir antes lo que se entiende, en nomenclatura, por radicales o grupos. Se llama así a los agregados de átomos que proceden de la pérdida de un átomo de hidrógeno en un hidrocarburo. Los radicales derivados de los alcanos (llamados radicales alquilo) se nombran sustituyendo la terminación -ano por -il o -ilo al principio o al final del nombre de la molécula. Para determinar la posición del radical se numera la cadena más larga, asignando el número más bajo a los carbonos con cadena lateral, y se escribe dicho número, llamado localizador, delante del nombre del radical. Si hay varias ramificaciones iguales, no se repite, sino que se usan prefijos indicando cuántas veces está repetido di-, tri-, etc. Y si hay algún sustituyente sobre la cadena, por ejemplo, un átomo de halógeno, se cita el nombre de este átomo y su posición (haluros de alquilo). Los hidrocarburos saturados cíclicos se nombran añadiendo el prefijo ciclo- al nombre del alcano equivalente de cadena abierta. Br 2,4-Dimetilpentano 2-Metilpentano Ciclopentano 1-Bromopropano 2-Ciclohexilhexano 1 Recuerda que debes repasar la nomenclatura que aquí desarrollamos cuando estudies los temas correspondientes de química orgánica. 9 10 TEMARIO XX Alquenos Los hidrocarburos que contienen uno o más dobles enlaces. Para su nomenclatura se siguen las mismas reglas que las ya vistas para alcanos (lo cual también ocurre con los demás compuestos); en este caso es la cadena principal la que contiene el doble enlace, y se cambian la terminación -ano, del alcano correspondiente, por -eno y se señala la posición del doble enlace con un número localizador. Cuando un hidrocarburo contiene más de un doble enlace, para nombrarlo se emplea la terminación -dieno, -trieno, etc., en lugar de -eno. 2-Buteno 1,4-Ciclohexadieno XX Alquinos Su nomenclatura es exactamente igual a la de los alquenos, pero en vez de acabar en -eno, acaban en -ino. Si hay varios triples enlaces, se usa, de forma análoga, -diino, -triino, etc. 2-Butino 1,4-Hexadiino 2.2. HIDROCARBUROS AROMÁTICOS El nombre genérico de los hidrocarburos aromáticos mono- y policíclicos es areno. Los radicales derivados de ellos se llaman radicales arilo. Junto a la nomenclatura sistemática de la IUPAC suelen presentar nombres propios también admitidos por la IUPAC, como benceno, naftaleno, fenantreno, etc. Benceno Naftaleno Fenantreno La presencia de sustituyentes se indica con un número localizador o, cuando hay dos sustituyentes, con los prefijos orto- (sustituyentes en las posiciones 1 y 2), meta- (en las 1 y 3), y para- (en las 1 y 4). FÍSICA Y QUÍMICA 59 NO2 I NO2 p-etiliodobenceno m-etilmetilbenceno o-Dinitrobenceno 2.3. FUNCIONES OXIGENADAS 2.3.1. Alcoholes, fenoles y éteres Los alcoholes se obtienen al sustituir uno o varios átomos de hidrógeno por uno o varios grupos hidroxilo (OH); serán alcoholes alifáticos cuando esta sustitución sea en un hidrocarburo no aromático, y un fenol cuando éste sea aromático. Se nombra añadiendo la terminación -ol al nombre del hidrocarburo de referencia e indicando su posición. Si hay varios grupos OH, también se usa di-, tri-, etc. Se puede utilizar la palabra hidroxi junto con la localización en la cadena para nombrarlos, o hidroxil para radicales. OH Propanol o 1-Hidroxipropano OH Fenol HO OH 1,3-Dihidroxipropano OH 2-Metilfenol o 1-Hidroxi-2-metilbenceno o 2-Hidroxitolueno Los éteres son los que presentan un átomo de oxígeno en lugar de un metileno (-CH2-). La nomenclatura más común es la que nombra los radicales en orden alfabético seguidos de la palabra éter. O Etil metil éter O Dietil éter 11 12 TEMARIO 2.3.2. Aldehídos, cetonas, ácidos carboxílicos y ésteres Todos estos compuestos se caracterizan por tener un doble enlace carbono-oxígeno, o grupo carbonilo (C=O). En el caso de los aldehídos su nomenclatura consiste en usar el sufijo -al (no es necesario especificar la posición, ya que se sabe que es en el extremo de su estructura o, lo que es lo mismo, en un carbono primario). En el caso de las cetonas el grupo carbonilo no está en el extremo de la cadena, sino que se encuentra en un carbono secundario. Se nombran usando el sufijo -ona. Si no es el grupo principal o está como radical, se usa el prefijo oxo-. O O O O OH H Pentanal 2-Pentanona ácido 3-oxo-butanoico Cuando el grupo carbonilo está unido a un grupo hidroxilo se denomina grupo carboxilo (-COOH), que evidentemente estará en el extremo de la cadena carbonada. Éstos son los ácidos carboxílicos. Se nombran usando la palabra ácido y la terminación -oico. Cuando se nombra como sustituyente, se usa la palabra carboxi-. Los ésteres son análogos a los ácidos carboxílicos, pero con la diferencia, que una cadena carbonada reemplaza al H del grupo ácido. Se nombran poniendo la terminación -ato a la cadena principal y la terminación -ilo a la cadena que sustituye al hidrógeno, uniendo las palabras con de. O O OH O OH Ácido etanoico Ácido propanoico O Acetato de etilo Otras funciones oxigenadas derivadas de ácidos carboxílicos son los haluros de ácido, anhídridos y sales orgánicas, sobre cuya nomenclatura se citan a continuación algunos ejemplos ilustrativos. Los anhídridos se nombran con la palabra anhídrido seguida del nombre del ácido del que provienen. Las sales y los haluros se nombran se nombran de forma similar, estos últimos como sales de los halógenos, cambiando la terminación -oico del ácido por -oilo. O Cl Cloruro de pentanoilo O O O Anhídrido acético O Na Butanoato sódico FÍSICA Y QUÍMICA 59 2.4. FUNCIONES NITROGENADAS: AMINA, AMIDA, NITRILO Y NITRO Las aminas se nombran usando la terminación amina y numerando la posición donde está. Según el número de sustituyentes del nitrógeno, podemos hablar de aminas primarias, secundarias o terciarias. Cuando la amina es secundaria (dos cadenas unidas al átomo de nitrógeno) o terciaria (tres cadenas unidas al átomo de nitrógeno) se nombran las cadenas como radicales y se escribe delante de cada una de ellas la letra N, salvo en la cadena principal, que lleva la terminación -amina. NH2 Propilamina H 2N NH2 1,3-Propanoamina N N-Etil-N-metil propilamina La amidas se consideran derivados de ácidos carboxílicos por sustitución del grupo OH por el grupo amina. Las amidas presentan el grupo funcional R’CONR2, donde CO es el grupo carbonilo, y R y R’ son sustituyentes carbonados o hidrógenos. Se nombran usando el nombre de la cadena principal de alcano, que contiene al grupo funcional amida, cambiando la «o» por la terminación -amida. No lleva localizador porque el grupo funcional siempre es terminal. O NH2 Pentanamida O O O NH2 NH2 4-Oxopentanamida (E)-Hept-2-enamida A los compuestos orgánicos que presentan un grupo funcional C≡N se les da el nombre genérico de nitrilos o cianuros. Se les puede nombrar de dos formas: a) añadiendo el sufijo -nitrilo al nombre del hidrocarburo de igual número de átomos de carbono, o b) considerándolo un derivado del ácido cianhídrico (HCN) e incluyendo las palabras cianuro delante del nombre de la cadena como radical (no cuenta en la nomenclatura el átomo de carbono del propio grupo). Cuando hay otras funciones que tienen prioridad sobre el grupo nitrilo, se cita éste mediante el prefijo ciano-. Los compuestos orgánicos con la función nitro en su estructura (-NO2) se nombran con el prefijo nitro- seguido del nombre del compuesto e indicando con un número la posición del grupo. NO2 C N Propanonitrilo o cianuro de etilo NO2 Nitrobutano NO2 m-dinitrobenceno o 1,3-dinitrobenceno 13 14 TEMARIO 3 ISOMERÍA 3.1. CONCEPTO DE ISOMERÍA La fórmula molecular indica el número de átomos de cada clase que hay en una molécula, pero no su distribución. En Química Orgánica se encuentra con frecuencia el caso de que una fórmula determinada representa a dos o más compuestos que difieren entre sí en sus propiedades físicas y químicas. Por ejemplo: existen por lo menos siete compuestos que tienen la fórmula molecular C4H10O, pero que difieren entre sí en cuanto a sus propiedades. Estos compuestos de idéntica fórmula molecular y propiedades diferentes se llaman isómeros, y al fenómeno correspondiente, isomería. Su existencia se explica admitiendo que los átomos se distribuyen en una forma definida en la molécula, habiendo una distribución diferente para cada isómero. En caso de que dos compuestos presenten la misma fórmula molecular pero un patrón de enlaces diferente, el tipo de isomería es constitucional o estructural y dentro de ésta, de cadena, de posición y de función. En caso de que los dos compuestos presenten una disposición espacial o en el plano diferente, se habla de estereoisomería. Dentro de la estereoisomería, si los compuestos son interconvertibles por rotación de enlaces simples, tenemos isomería conformacional, y si no son interconvertibles, tenemos la isomería configuracional: cis-trans y óptica. La interconversión entre estos últimos sólo se puede dar por rotura y formación sucesiva de enlaces. Estructura o constitucional Isomería Cadena Posición Función I. Configuracionales Cis-trans Ópticos Esteroisometría Confórmeros Los isómeros son, física y químicamente, compuestos distintos. Los confórmeros, sin embargo, son solamente compuestos distintos entre sí a bajas temperaturas, a las cuales y en algunos casos, se pueden aislar. 3.2. ISOMERÍA ESTRUCTURAL O CONSTITUCIONAL Cuando para una misma fórmula molecular se pueden escribir compuestos con distinta estructura carbonada y/o secuencia de heteroátomos en ella, así como cuando puede haber distintos tipos de enlace en la molécula, se habla de isomería estructural. FÍSICA Y QUÍMICA 59 Hay tres tipos: Isomería de cadena: varía la ordenación del esqueleto carbonado. Isomería de posición: varía la posición de uno o varios grupos funcionales en el esqueleto principal. Isomería de función: poseen grupos funcionales distintos. 3.2.1. Isomería de cadena Este tipo de isómeros presentan diferencias estructurales en cuanto a la ordenación del esqueleto carbonado. Puede ser considerada una isomería de posición, donde varía, por ejemplo, la posición de un grupo metilo. CH3 CH3 3-Metilpentano n-Hexano Al ir aumentando el número de átomos de carbono, el número de isómeros posibles crece enormemente. Las diferencias tanto químicas como físicas entre isómeros de cadena no son muy pronunciadas. Por ejemplo, los puntos de fusión del 2-metil hexano y del 3-metil hexano difieren solamente en un grado. En general, los isómeros más ramificados tienen puntos de ebullición algo más bajos que los isómeros menos ramificados, ya que al ser las fuerzas entre estas moléculas de Van der Waals, una menor ramificación implica un mejor empaquetamiento y una interacción más efectiva entre moléculas. 3.2.2. Isomería de posición Este tipo de isómeros viene caracterizado por el hecho de presentar el mismo esqueleto carbonado principal y los mismos grupos funcionales, pero la disposición relativa de éstos en el esqueleto principal varía de un isómero a otro. Por ejemplo: OH OH 3-Hidroxipentano o 3-Pentanol Br 1-Bromopropano 2-Hidroxipentano o 2-Pentanol Br 2-Bromopropano 15 16 TEMARIO También suelen considerarse isómeros de posición los compuestos heterocíclicos con dos o más átomos diferentes y en los que las posiciones de éstos varían. N N N Piridazina N Pirimidina Los isómeros de posición pueden presentar notables diferencias, tanto en sus propiedades físicas como químicas. Por ejemplo, el catecol (anillo de benceno con dos grupos OH en orto) tiene un punto de ebullición de 240 ºC, el resorcinol (con los grupos OH en meta) tiene un punto de ebullición de 277 ºC y la hidroquinona (con los grupos OH en para) tiene un punto de ebullición de 286 ºC. 3.2.3. Isomería de función Los isómeros de función poseen diferencias estructurales en cuanto que poseen grupos funcionales distintos. Aquí las diferencias químico-físicas son muy marcadas. O O 3-Pentanona H Pentanal Un caso especial de este tipo de isomería lo constituyen las cetonas y los enoles, ya que con ellos es posible pasar de un isómero a otro, debido a la existencia del denominado equilibrio ceto-enólico y que no se da en ningún otro grupo de isómeros funcionales. El equilibrio se representa: O R OH CH2R Forma ceto R CHR Forma enol El equilibrio suele estar desplazado en un sentido u otro, dependiendo de las características de los compuestos (R) entre los que se da dicho equilibrio. Entre los aldehídos y cetonas sencillos, el equilibrio está desplazado hacia la forma ceto. El fenómeno es un tipo tautomería. La separación de las dos formas tautómeras se puede conseguir a bajas temperaturas. FÍSICA Y QUÍMICA 59 3.3. ESTEREOISOMERÍA 3.3.1. Isomería configuracional XX Isomería cis-trans La imposibilidad de rotación alrededor de un enlace que tiene sustituyentes, debida a la existencia de un doble enlace o de un ciclo que haga inviable el proceso, determina la aparición de este tipo de isomería. Los isómeros se denominan en este caso cis-trans o geométricos. Cis-2-buteno o (Z)-Buteno Trans-2-buteno o (E)-Buteno Para su nomenclatura se utiliza el prefijo cis (o Z) si están los sustituyentes iguales en un mismo lado, y trans (o E) en el caso contrario. En el caso de isómeros cis-trans de compuestos cíclicos suele ser frecuente la nomenclatura ecuatorial-ecuatorial (ec-ec), axial-axial (ax-ax) y axial-ecuatorial (ax-ec), que hacen mención a la posición relativa de los sustituyentes entre ellos y respecto al anillo. De igual forma se pueden emplear los prefijos syn- y anti-. ax CH ec CH3 3 Cis-1,2-dimetilciclohexano CH3 CH3 CH3 CH3 Trans-1,2-dimetilciclohexano CH3 CH3 Cis-1,3-dimetilciclohexano Trans-1,3-dimetilciclohexano XX Isomería óptica Cuando en una molécula orgánica al menos uno de los átomos de carbono presenta cuatro sustituyentes distintos, la molécula presenta quiralidad, y el átomo de carbono se denomina carbono quiral o asimétrico (se representa mediante C*). Cuando la posición relativa de los sustituyentes sobre el carbono quiral cambia, cada una de las moléculas resultantes es la imagen especular de la otra, es decir, son enantiómeros. Los enantiómeros, por tanto, no son superponibles y cada uno es ópticamente activo, es decir, desvía el plano de la luz polarizada en sentidos 17 18 TEMARIO opuestos. (La luz polarizada es luz ordinaria en la que el vector eléctrico solamente vibra en un plano, ya que las demás direcciones de vibración se suprimen al hacerla pasar por un prisma de Nicol). Ésta es la única diferencia entre ambas moléculas. COOH H3C OH H COOH H OH CH3 Notación de Fisher COOH HO H3C COOH HO CH3 H H Notación de Fisher Enantiómeros entre sí. (R)-Ácido láctico (S)-Ácido láctico Los diastereoisómeros son los estereoisómeros que no son enantiómeros. Para ello es necesario que haya al menos dos carbonos quirales en la molécula. La diferencia entre enantiómeros y diastereoisómeros se observa en el caso del ácido tartárico. COOH H HO COOH OH HO H H COOH COOH H H OH OH H OH COOH Ácido (L)-t artárico Enantiómeros COOH Ácido (D)-tartárico Ácido (meso)-t artárico Diastereoisómeros La tercera posibilidad en moléculas con carbonos quirales son las formas meso. Éstas se observan cuando las moléculas que tienen al menos dos átomos de carbono quirales, tienen además un plano de simetría entre ellos, de forma que la mitad de la molécula es la imagen especular de la otra mitad. Y por ello esta forma es ópticamente inactiva. Formulación y nomenclatura de compuestos quirales. Para representar una molécula orgánica con carbonos quirales es necesario poder especificar la configuración de cada compuesto, es decir, la situación de sus átomos en el espacio. Fischer sugirió una representación abreviada para los carbonos quirales, en la que los trazos horizontales se dirigen hacia el exterior del papel y los verticales hacia dentro. Existen varias formas de especificar la disposición de los sustituyentes en torno al átomo de carbono asimétrico: a) Notación arbitraria. Viendo cómo desvían la luz polarizada las moléculas de gliceraldehído, las que la desvían a la derecha se denominan (+) gliceraldehído, y las que la desvían a la izquierda se llaman (–) gliceraldehído. FÍSICA Y QUÍMICA 59 b) Notación «D» y «L». El compuesto de referencia es el D-gliceraldehído y, atendiendo al convenio de Fischer, cuando el compuesto posee el grupo hidroxilo a la derecha, el compuesto es D, y su enantiómero será la forma L (hidroxilo a la izquierda). Inicialmente se asignó arbitrariamente a la forma (+) la forma D, y a la (–) la L. En 1949 se logró dilucidar las estructuras absolutas y se vio que la asignación arbitraria era cierta (+ → D, – → L). H O OH CH2OH (D)-Gliceraldehído H O HO CH2OH (L)-Gliceraldehído Aunque el sistema D-L es difícil de aplicar, se utiliza para designar la estereoquímica de la mayoría de los carbohidratos simples, aminoácidos, lípidos y ácidos nucleicos. c) Regla de la secuencia o sistema R-S. Es un método más general y fue ideado por Cahn, Ingold y Prelog. Se toma como centro el carbono asimétrico. Los sustituyentes del centro asimétrico se disponen en orden decreciente de número atómico, de los átomos unidos directamente al carbono asimétrico. Cuando dos o más de estos átomos poseen el mismo número atómico, se consideran sus sustituyentes. El grupo con sustituyentes de mayor número atómico, será el primero. Cuando se encuentra que algún sustituyente está unido mediante un enlace múltiple, este sustituyente se considera de modo arbitrario como si existieran dos, tres, etc. Una vez ordenados en sentido decreciente todos los sustituyentes, el más pequeño se coloca en la posición más alejada del observador (detrás del carbono asimétrico), proyectándose hacia él los otros tres. Si un giro en el sentido de las agujas del reloj nos permite encontrar a los sustituyentes ordenados de manera decreciente, el centro asimétrico se denota como R (rectus); si el giro para obtener lo mismo es en sentido contrario, se designa como S. La comprobación experimental de la actividad óptica2 se lleva a cabo utilizando el polarímetro, que mide el poder rotatorio [α] de la molécula. Esencialmente consta de dos prismas de Nicol: uno es el polarizador (P) y el otro el analizador (A), y entre ambos un tubo (T) que contiene la sustancia que se examina (un líquido o una solución); S es la fuente de luz monocromática. Si la sustancia gira el plano de polarización hacia la derecha, se dice que es dextrógira, y si lo desvía hacia la izquierda, levógira. Una mezcla equimolar de enantiómeros es inactiva ópticamente, y se la denomina racémica. La separación de los enantiómeros de dicha mezcla recibe el nombre de resolución. El ángulo de rotación producido por 2 Se incluye aquí el apartado de referencia en el título del tema. 19 20 TEMARIO una sustancia ópticamente activa depende también de factores como el espesor de la muestra, su concentración, el disolvente empleado, la temperatura y la longitud de onda de la luz utilizada. La rotación específica se representa como [α], y es la actividad óptica intrínseca de la sustancia en las condiciones de la medida; viene dada por: [α ]λ = T 100 α Lc En donde cada símbolo significa: α: ángulo de rotación observado (en grados). L: longitud del tubo de muestra (en dm). c: concentración de la sustancia (en g/100 mL). T: temperatura de la solución. λ: longitud de la onda de la luz utilizada. La rotación específica constituye una constante física única y característica para cada sustancia ópticamente activa3. 3.3.2. Isomería conformacional Las conformaciones son formas que adoptan las moléculas orgánicas cuando los grupos atómicos de éstas rotan alrededor de un enlace sencillo, o cuando una estructura compleja puede adoptar distintas disposiciones en el espacio. Cada conformación posee una determinada energía; esto hace que exista una barrera para la interconversión entre confórmeros y que la rotación deje de ser libre, por lo que se definen como «isómeros» conformacionales (confórmeros). Las barreras de rotación (diferencias energéticas) alrededor de un enlace sencillo son pequeñas en la mayoría de los casos, con lo cual, a temperatura ambiente, las moléculas tienen suficiente energía térmica para remontarlas. En referencia, por ejemplo, a la actividad biológica, al estar ésta definida por la estructura tridimensional de la molécula, ésta depende de las proporciones de los diferentes isómeros conformacionales presentes. Hidrocarburos saturados. En el etano (R=H) se pueden determinar dos ordenaciones extremas de un grupo metilo con respecto a otro, a consecuencia de la rotación alrededor del enlace carbono-carbono. Se puede obtener la fórmula de proyección mirando la molécula a lo largo de la línea de enlace de los dos átomos de carbono representa la forma «alternada» o trans o anti, en la que los átomos de hidrógeno están lo más separados posible (proyección de caballete). Para convertir esta forma en la cis o syn, hay que suministrar energía (superar la barrera energética) para vencer, entre otras cosas, la repulsión que existe entre los átomos de hidrógeno. Esta energía vale 2,75 kcal/ mol. Al sustituir los hidrógenos por grupos metilos (R=CH3), aumenta, por tanto, el volumen espacial, y con este los impedimentos estéricos a la rotación. 3 Puedes ampliar estos contenidos con los desarrollados en el tema 61. FÍSICA Y QUÍMICA 59 R H H H Rotación alrededor del enlace C-C R Anti R H H H H H R Syn Ciclos. En el caso de moléculas mayores y cíclicas, como en el ciclohexano, su estructura puede presentar diferentes disposiciones espaciales. En este caso concreto se conocen como formas de «bote» y «silla» resultado de la flexibilidad de la estructura. El confórmero más estable es la forma silla, debido a que no hay interacciones estéricas 1-4. Es un confórmero rígido en el sentido de que el cambio de un ángulo diedro alrededor de cualquier enlace requiere un cambio simultáneo en uno o más ángulos de la molécula. Interacción 1-4 H ax ax H H ax H ec ax H H ec Ciclohexano, silla Ciclohexano, bote Ciclohexano, silla Los enlaces que sujetan los hidrógenos que están en el plano del anillo se encuentran en un cinturón alrededor del «ecuador» de aquél, por lo que se llaman enlaces ecuatoriales, señalados con «ec». Las uniones de los hidrógenos, ubicados encima y debajo del plano, son paralelas a un eje perpendicular a éste y se denominan enlaces axiales, señalados con «ax». En la conformación silla, cada átomo de carbono posee un enlace ecuatorial y otro axial. En el ciclohexano las dos formas de silla no se diferencian; sin embargo, en los ciclohexanos sustituidos o en el caso de azúcares cíclicos, sí, ya que un isómero es axial y el otro ecuatorial; es más estable este último, ya que hay menos interacciones debidas a los volúmenes de los sustituyentes en una posición ecuatorial que en una axial. 21 22 TEMARIO CONCLUSIÓN En este tema se inicia el estudio de los compuestos orgánicos, que origina una rama de química, la Química Orgánica. En primer lugar se justifica, con base en la formación de enlaces muy estables con otros átomos de carbono, el número tan elevado de compuestos orgánicos conocidos. Y en segundo lugar se estudia la nomenclatura de los compuestos orgánicos, haciendo especial énfasis en las características estructurales, es decir, la isomería. FÍSICA Y QUÍMICA 59 BIBLIOGRAFÍA BIBLIOGRAFÍA COMENTADA CRAINE, L. E.; HART, H.; HART, D. J. (2007): Química orgánica. Madrid: McGraw-Hill. Desarrolla lo referente a la nomenclatura y formulación de los compuestos orgánicos, así como a su reactividad. Incluye, asimismo, la isomería y el análisis conformacional. En español. MCMURRAY, E. (2007): Organic chemistry. Brooks Cole. Este libro es similar al anterior, complementándose ambos. En inglés. SYKES, P. (1996): A quidebook to mechanism in organic chemistry. Nueva York: Longman. En inglés. Contenido mecanístico de las reacciones orgánicas. SYKES, P. (1981): Mecanismos de reacción en química orgánica. Barcelona: Reverté. El mismo contenido que el anterior, pero en español. VOLHARDT, K. P. C. y SCHORE, N. E. (2005): Organic chemistry. Structure and function. Freeman. Este es un libro más completo que el anterior, de contenido de un nivel superior, que de igual forma incluye nomenclatura y formulación de los compuestos orgánicos, y trata con detalle su reactividad. Incluye asimismo, la isomería y el análisis conformacional. En inglés. WEBGRAFÍA www.uned.es http://es.wikipedia.org/wiki/Portada http://www.quimicaorganica.net/nomenclatura_quimica_organica.htm 23 FÍSICA Y QUÍMICA 59 RESUMEN Química del carbono. Estructura y enlaces del carbono. Nomenclatura. Isomería. Comprobación experimental de la actividad óptica. 1. ENLACES DEL CARBONO Hibridaciones sp, sp2 y sp3. Enlaces simples, dobles y triples. Aromaticidad y la estructura del benceno. 2. NOMENCLATURA 2.4.FUNCIONES NITROGENADAS: AMINAS, AMIDAS, CIANUROS Y NITRO Aminas con la terminación -amina y las amidas con la terminación -amida. Los cianuros se suelen nombrar con el sufijo -nitrilo o con la palabra cianuro delante. El grupo nitro se nombra con esta palabra y especificando su posición en la cadena principal. 2.1.ALCANOS, ALQUENOS Y ALQUINOS A excepción de los cuatro primeros, que reciben los nombres de metano, etano, propano y butano, los restantes se nombran mediante un prefijo griego, que indica el número de carbonos, y la terminación -ano. Para nombrar los alcanos ramificados se numera la cadena más larga y se escribe el número donde se encuentra el sustituyente. Alquenos acabados en -eno y alquinos en -ino. 2.2.HIDROCARBUROS AROMÁTICOS En general se nombran arilo o arenos, y tienen nombres propios aceptados por la IUPAC. 2.3.FUNCIONES OXIGENADAS: 2.3.1. Alcoholes, fenoles y éteres Los alcoholes y fenoles se nombran añadiendo la terminación -ol al nombre del hidrocarburo de referencia e indicando la posición del grupo alcohol. La forma más común de nombrar los éteres es citando el nombre de los dos radicales en orden alfabético y añadiendo la palabra éter. 2.3.2. Aldehídos, cetonas, ácidos carboxílicos y ésteres Los aldehídos se nombran añadiendo el sufijo -al; las cetonas, -ona; los ácidos carboxílicos usando la palabra ácido y la terminación -oico, y los ésteres incluyendo la terminación -ato a la cadena principal y la terminación -ilo a la cadena que sustituye al hidrógeno del grupo carboxílico, uniendo las palabras con «de». 3. ISOMERÍA EN LOS COMPUESTOS ORGÁNICOS 3.1.CONCEPTO DE ISOMERÍA La misma fórmula molecular y distintas propiedades. 3.2.ISOMERÍA ESTRUCTURAL O CONSTITUCIONAL 3.2.1. Isomería de cadena Distinta forma de ordenar el esqueleto carbonado. 3.2.2. Isomería de posición Grupos funcionales en distinta posición del esqueleto carbonado. 3.2.3. Isomería de función Distintos grupos funcionales en el esqueleto carbonado. 3.3.ESTEREOISOMERÍA 3.3.1. Isomería configuracional Isomería cis-trans, y óptica: estereoisómeros o diastereoisómeros 3.3.2. Isomería conformacional Debida a las rotaciones a lo largo de enlaces simples, ya sea en moléculas lineales o cíclicas. 25