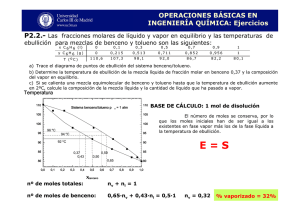
remoción de hidrocarburos en agua mediante la utilización de
Anuncio

UNIVERSIDAD SIMÓN BOLÍVAR DECANATO DE ESTUDIOS PROFESIONALES COORDINACIÓN DE LICENCIATURA EN QUÍMICA REMOCIÓN DE HIDROCARBUROS EN AGUA MEDIANTE LA UTILIZACIÓN DE RESIDUOS DE POLÍMEROS COMERCIALES Por Marco Antonio Belandria Joseph PROYECTO DE GRADO Presentado ante la ilustre Universidad Simón Bolívar Como requisito final para optar por el título de Licenciado en Química Sartenejas, Mayo de 2010 UNIVERSIDAD SIMÓN BOLÍVAR DECANATO DE ESTUDIOS PROFESIONALES COORDINACIÓN DE LICENCIATURA EN QUÍMICA REMOCIÓN DE HIDROCARBUROS EN AGUA MEDIANTE LA UTILIZACIÓN DE RESIDUOS DE POLÍMEROS COMERCIALES Por Marco Antonio Belandria Joseph Realizado con la asesoría de Prof. Fernando Morales PROYECTO DE GRADO Presentado ante la ilustre Universidad Simón Bolívar Como requisito final para optar por el título de Licenciado en Química Sartenejas, Mayo de 2010 REMOCIÓN DE HIDROCARBUROS EN AGUA MEDIANTE LA UTILIZACIÓN DE RESIDUOS DE POLÍMEROS COMERCIALES RESUMEN El objetivo del siguiente trabajo fue evaluar la eficiencia de residuos de polietileno de baja densidad (PEBD), de poliestireno (PS) y de una polimezcla física de los polímeros polietileno de baja densidad y poliestireno (PEBD-PS) (1:1), para la remoción de benceno y tolueno en agua. Para lograr este objetivo se estudió la cinética de cada proceso de adsorción y seguidamente cada isoterma de adsorción en base a los modelos de Langmuir y de Freundlich. Los polímeros utilizados fueron caracterizados por las técnicas de Espectroscopia Infrarrojo por Transformada de Fourier (FT-IR), Calorimetría Diferencial de Barrido (CDB) y Microscopía Electrónica de Barrido (MEB). Adicionalmente se empleó el método de microextracción en fase líquida para la cuantificación de los hidrocarburos mencionados en agua; dicho método fue validado en cuanto al cálculo de parámetros como precisión, exactitud, porcentajes de recuperación, límite de detección y de cuantificación. En general todas las adsorciones se ajustaron mejor al modelo de Langmuir (0,991<R2<0,970) que al de Freundlich (0,986<R2<0,963); además se obtuvo una pobre adsorción para ambos compuestos según lo reportado en la literatura cuando se emplean otras resinas poliméricas. Los estudios de CDB reflejaron una reducción del grado de cristalinidad del PEBD de la polimezcla en casi 1/3 de su valor original; comprobado por la MEB de la polimezcla, que reveló una matriz de PEBD en una fase interna de PS. A pesar de los resultados obtenidos en la caracterización, la polimezcla PEBD-PS no presentó mejorías como adsorbente al ser comparado con los dos polímeros solos en el caso del benceno, pero si en el caso del tolueno; demostrado en los parámetros obtenidos de cada modelo de isotermas. Se obtuvo que la naturaleza de estos procesos de adsorción son del tipo físicos, regido principalmente por el área superficial de cada polímero. En general los polímeros empleados en esta investigación no son eficaces en la remoción de benceno y tolueno en agua. iv AGRADECIMIENTOS Esta investigación no pudo realizarse sin la colaboración de ciertas personas. Primero que todo agradezco a mis padres, Aide y Marco, que siempre estuvieron pendientes de mi transitar por la carrera, y me apoyan incondicionalmente en mis decisiones; son mi ejemplo a seguir a cuando a perseverancia, responsabilidad y cariño. Al Profesor Fernando Morales, por darme la oportunidad de ser parte del Laboratorio de Química Ambiental, por compartir conmigo un poco de su amplia experiencia y sobre todo por obligarme a enfrentar las dificultades y resolverlas. Al Profesor Marco Sabino a través del cual se realizó la polimezcla PEBD-PS, así como también la Microscopia Electrónica de Barrido (MEB) y la Calorimetría Diferencial de Barrido (CDB). A la Profesora Ursula Erhman, por ayudarme en innumerables ocasiones y enseñarme, entre muchas cosas, un poco de cromatografía, métodos analíticos, entre otras cosas. A mis amigos: Jacquelin, Tomas, Jose Luis, Gustavo, Gabo y Jonathan… siempre pendientes de mis avances, problemas, y siempre dándome ánimo. Al T.S.U. Gilberto Gedler y a la Ing. Lucianna Viele Ianni del Laboratorio de Química Ambiental, al igual que al Licenciado Andrés Oliveros del Laboratorio de Desechos Tóxicos, por facilitar material y herramientas para la realización de este trabajo. A la T.S.U. Yetzana Sanchez y a la T.S.U. Soraya De León del laboratorio de Orgánica/Inorgánica USB por facilitar los viales de vidrio sin los cuales no se hubiese podido realizar los experimentos. v INDICE GENERAL INTRODUCCIÓN………………………………………………………………............... 1 CAPITULO 1……………………………………………………………………………... 5 MARCO TEORICO……………………………………………………………………… 5 1.1. Hidrocarburos aromáticos monocíclicos (HAM)………..…………………………… 5 1.1.1. Benceno……………………………………………………………………………. 5 1.1.2. Tolueno……………………………………………………………………………... 7 1.2. Normativa relacionada con la concentración de BTEX en agua……………………... 8 1.3. Análisis de BTEX: características generales de la cuantificación de BTEX en agua……………………………………………………………………………………….. 9 1.4. Métodos de preconcentración de analito (BTEX) en matriz acuosa…………………. 10 1.4.1. Inyección directa de muestra acuosa (DAI)………………………………………... 11 1.4.2. Método de purga y trampa (P&T)………………………………………………….. 12 1.4.3. Método headspace (HS)……………………………………………………………. 13 1.4.3.1. Método HS estático………………………………………………………………. 13 1.4.3.2. Método HS dinámico…………………………………………………………….. 14 1.4.4. Microextracción en fase sólida (SPME)……………………………………………. 15 1.4.5. Métodos de extracción en fase líquida (LLE)……………………………………… 16 1.4.5.1. Microextracción en fase líquida………………………………………………….. 17 1.5. Tratamientos para eliminar BTEX de cuerpos acuosos……………………………… 17 1.5.1. Métodos químicos………………………………………………………………….. 18 1.5.2. Métodos biológicos………………………………………………………………… 21 1.5.3. Métodos físicos…………………………………………………………………….. 23 1.6. Polímeros……………………………………………………………………………... 29 1.6.1. Polietileno…………………………………………………………………………... 30 1.6.2. Poliestireno…………………………………………………………………………. 31 1.7. Procesos de adsorción………………………………………………………………... 32 1.7.1. Isotermas de adsorción……………………………………………………………... 33 1.7.1.1. Isotermas de adsorción de Langmuir…………………………………………….. 35 1.7.1.2. Isoterma de Freundlich…………………………………………………………… 38 1.8. Antecedentes de esta investigación…………………………………………………... 39 vi CAPITULO 2……………………………………………………………………………... 42 PROCEDIMIENTO EXPERIMENTAL…………………………………………………. 42 2.1. Materiales……………………..……..………………………………………………... 42 2.2. Procedimiento Experimental………………………………………………………….. 43 2.1.1. Preparación de la polimezcla PEBD-PS……………………………………………. 43 2.2.2. Caracterización del material polimérico……..……………………………………... 44 2.2.2.1 Espectroscopía infrarrojo por transformada de Fourier (FT-IR)………………….. 44 2.2.2.2. Calorimetría diferencial de barrido (CDB)………………………………………. 44 2.2.2.3. Microscopio Electrónico de Barrido……………………..………………………. 45 2.2.2.4. Método Brunauer-Emmett-Teller (Método BET)………………………………… 46 2.2.3. Microextracción en fase líquida……………………………………………………. 46 2.2.4. Elaboración de blancos……………………………………………..………………. 50 2.2.5. Experimentos de cinética de adsorción……….……………………………………. 50 2.3.3. Experimentos de isotermas de adsorción…………………………………………… 50 2.3.4. Tratamiento de los desechos generados en la experimentación……………………. 51 CAPITULO 3……………………………………………………………….…………….. 52 RESULTADOS Y DISCUSIÓN………………………………………….……………… 52 3.1. Espectroscopía infrarrojo por transformada de Fourier (FT-IR)…….……………….. 52 3.2. Calorimetría diferencial de barrido (CDB)…………………………………………… 54 3.3. Microscopia electrónica de barrido (MEB)………………….……….………………. 58 3.4. Método BET………………………………………………………….………………. 62 3.5. Validación para la microextracción en fase líquida…………………...……………... 63 3.6. Cinética de adsorción……………………..………………….………………………. 68 3.7. Isotermas de adsorción……………………………………………………...………… 70 3.8. Isotermas de adsorción. Modelo Langmuir...……………………………………..….. 72 3.9. Isotermas de adsorción. Modelo Freundlich…………………………………………. 78 CONCLUSIONES………………………………………………………………………… 85 BIBLIOGRAFÍA………………………………………………………………………….. 87 APÉNDICE A……………..……………………………………………………………... 93 APÉNDICE B……………………………………………………………………………... 95 APÉNDICE C……………………………………………………………………………... 96 vii INDICE DE TABLAS Tabla 1.1 Estructura y propiedades fisicoquímicas del benceno.…………………............ 6 Tabla 1.2 Estructura y propiedades fisicoquímicas del tolueno………………………….. 7 Tabla 1.3 Valores permitidos de BTEX en agua potable…………………………………. 9 Tabla 1.4 Tratamientos de efluentes contaminados con compuestos aromáticos..……….. 18 Tabla 1.5 Modelo de la isoterma de Langmuir y sus formas lineales…………………….. 37 Tabla 2.1 Reactivos empleados…………………………………………….………….….. 42 Tabla 2.2 Polímeros usados……………………................................................................... 43 Tabla 2.3 Patrones de benceno y tolueno…………………………………………………. 48 Tabla 2.4 Condiciones cromatográficas para benceno y tolueno…………………………. 47 Tabla 3.1 Bandas de absorción en el espectro FT-IR de los polímeros utilizados 53 Tabla 3.2 Grado de cristalinidad del PEBD utilizado……………………………………... 55 Tabla 3.3 Grado de cristalinidad del PEBD matriz de la polimezcla PEBD-PS………….. 57 Tabla 3.4 Tamaño de los volúmenes libres de la polimezcla PEBD-PS………………….. 62 Tabla 3.5 Área superficial BET…………………………………………………………… 62 Tabla 3.6 Componentes y señales de los patrones de benceno y tolueno…………………. 64 Tabla 3.7 Parámetros de calibración de benceno y tolueno.………………………………. 65 Tabla 3.8 Porcentajes de recuperación de benceno y tolueno………………………..…… 67 Tabla 3.9 Límite de detección del método……………………………………………….. 67 Tabla 3.10 Tiempos de equilibrio de adsorción de benceno y tolueno con cada polímero…………………………………………………………………………………… 70 Tabla 3.11 Parámetros de Langmuir para el benceno…………………….…...………….. 73 Tabla 3.12 Parámetros de Langmuir para el tolueno……….……….……………………. 76 Tabla 3.13 Comparación entre cantidad de moléculas adsorbidas y permitidas………….. 77 Tabla 3.14 Parámetros de Freundlich para el benceno……………………………………. 81 Tabla 3.15 Parámetros de Freundlich para el tolueno…………………………………….. 81 Tabla 3.16 Comparación de polímeros……………………………………………………. 84 viii INDICE DE FIGURAS Figura 1.1 Síntesis de benceno por Mitchrlich……………………………………………. 5 Figura 1.2 Esquema de un sistema P&T………………………………...…….………….. 12 Figura 1.3 Método HS estático……………………………………………………………. 14 Figura 1.4 Método HS estático. HS-SDM………………………………………………… 14 Figura 1.5 Partes de la fibra de SPME……………………………...…………………….. 15 Figura 1.6 Técnicas SPME. a) Inmersión de la fibra b) Fibra en la fase gaseosa c) Desorción en el inyector del cromatógrafo……………...………………………………... 16 Figura 1.7 Reacción directa del contaminante con ozono………………………………… 19 Figura 1.8 a) Producción de radicales por descomposición de ozono b) Reacción del contaminante con el radical OH……………...…………………………………………… 19 Figura 1.9 Descomposición del ozono en presencia de peróxido de hidrógeno……..…... 20 Figura 1.10 Factores que afectan la biorremediación……………………………………... 21 Figura 1.11 Esquema de la técnica de "air sparging"……………………………………... 25 Figura 1.12 Esquema de una torre con flujo de aire en contracorriente…………………... 26 Figura 1.13 Barrera permeable reactiva………………......………………………………. 27 Figura 1.14 Polímeros naturales. a) Caucho natural b) Quitina……..………………..….. 29 Figura 1.15 Polímeros sintéticos. a) Nylon 66 b) Neopreno.………………………..……. 29 Figura 1.16 Unidad repetitiva del polietileno……………...……………………………… 30 Figura 1.17 Preparación de polietileno………...………………………………………….. 30 Figura 1.18 Unidad repetitiva del poliestireno……………………………………………. 31 Figura 1.19 Síntesis del estireno………………………………………………………….. 31 Figura 1.20 Esquema de a) adsorción de monocapa b) adsorción de multicapas………… 33 Figura 1.21 a) Fisisorción de H2 sobre una superficie metálica b) Quimisorción de H2 sobre una superficie metálica…………………...………………………………………… 33 Figura 1.22 Tipos de isotermas de adsorción……………………………………………... 34 Figura 3.1 Termograma de fusión del PEBD……………………………………………... 54 Figura 3.2 Termograma de cristalización del PEBD……………………………………… 55 Figura 3.3 Termograma de fusión del PEBD-PS…………………………………………. 56 Figura 3.4 Termograma de cristalización del PEBD-PS………………………………….. 56 Figura 3.5 Esquema de a) PEBD semicristalino b) PEBD-PS, disminución del grado de 58 ix cristalinidad original………………………………………………………………………. Figura 3.6 PEBD MEB del PEBD……………...………………………………………… 59 Figura 3.7 MEB del PEBD-PS………………………………………………………….... 59 Figura 3.8 MEB de PEBD-PS tratado con cloroformo..………………………………….. 60 Figura 3.9 MEB del PEBD-PS (80/20) en THF…………………………………………... 61 Figura 3.10 MEB del PEAD-PS (80/20)…….……………..……………………………... 61 Figura 3.11 Curva de calibración de benceno…………………………………………….. 65 Figura 3.12 Curva de calibración de tolueno…..…………………………………………. 65 Figura 3.13 Curva de calibración de surrogate……………………………………………. 66 Figura 3.14 Cinéticas de adsorción del benceno con los distintos polímeros…………….. 69 Figura 3.15 Cinéticas de adsorción del tolueno con los distintos polímeros……………... 69 Figura 3.16 Isoterma de adsorción benceno-PEBD…………………………….………… 71 Figura 3.17 Isoterma de adsorción benceno-PS………………………………….……….. 71 Figura 3.18 Isoterma de adsorción benceno-PEBD-PS………………………….……….. 71 Figura 3.19 Linealización de la isoterma benceno-PEBD. Langmuir-1………….………. 72 Figura 3.20 Linealización de la isoterma benceno-PS. Langmuir-1……………………… 72 Figura 3.21 Linealización de la isoterma benceno-PEBD-PS. Langmuir-1………………. 73 Figura 3.22 Isoterma de adsorción tolueno-PEBD………………………………………... 74 Figura 3.23 Isoterma de adsorción tolueno-PS…………………………………………… 74 Figura 3.24 Isoterma de adsorción tolueno-PEBD-PS……………………………………. 74 Figura 3.25 Linealización de la isoterma tolueno-PEBD. Langmuir-1…………………… 75 Figura 3.26 Linealización de la isoterma tolueno-PS. Langmuir-1………………………. 75 Figura 3.27 Linealización de la isoterma tolueno-PEBD-PS. Langmuir-1……………….. 76 Figura 3.28 Isoterma de adsorción de benceno-PEBD. Modelo de Freundlich…………... 79 Figura 3.29 Isoterma de adsorción de benceno-PS. Modelo de Freundlich………………. 79 Figura 3.30 Isoterma de adsorción de benceno-PEBD-PS. Modelo Freundlich………….. 79 Figura 3.31 Isoterma de adsorción de tolueno-PEBD. Modelo Freundlich………………. 80 Figura 3.32 Isoterma de adsorción de tolueno-PS. Modelo Freundlich…………………... 80 Figura 3.33 Isoterma de adsorción de tolueno-PEBD-PS. Modelo Freundlich…………... 80 x Lista de Símbolos Símbolos en Castellano C: Concentración Ceq: Concentración en equilibrio k: Constante de capacidad de adsorción del adsorbente (Freundlich) KL: Constante de afinidad(Langmuir) n: Constante de la intensidad de adsorción (Freundlich) qm: Cantidad de adsorbato adsorbido en los sitios disponibles en la monocapa del adsorbente (Langmuir) Símbolos en Griego ϑ: relación entre la cantidad de material adsorbido por masa de adsorbente (Freundlich) θ: fracción de posiciones ocupadas en el adsorbato µ/L: microgramos por litro µm: micrómetros ∆H: entalpía asociada a un proceso xi Lista de Abreviaciones BTEX: benceno, tolueno, etilbenceno y xilenos cal/J: Calorías por Joule CDB: Calorímetro diferencial de barrido COV: Compuestos orgánicos volátiles COSV: Compuestos orgánicos semivolátiles DAI: Inyección directa de muestra acuosa DCM: Diclorometano ECD: Detector de captura de electrones E.I: Estándar interno FID: Detector de ionización a la llama FT-IR: Espectroscopía infrarrojo por transformada de Fourier GC: Cromatografía de gas GPUSB: Grupo de Polímeros de la Universidad Simón Bolívar HAM: Hidrocarburos aromáticos monocíclicos HAP: Hidrocarburos aromáticos policíclicos HS-SDME: “headspace microextracción en gota” HS: Headspace HS-LPME: “headspace microextracción en fase líquida” J/g: Joule por gramo kPa: kilopascal LLE: Métodos de extracción en fase líquida MEB: Microscopía electrónica de barrido Método BET: Método Brunauer-Emmett-Teller mg/L: miligramos por litro Min: minutos mL: mililítros mmHg: milímetros de mercurio MNC: Máximo nivel del contaminante MS: Espectrómetro de masas nm: nanómetros xii ONMC: Objetivo de nivel máximo del contaminante PEAD: Polietileno de alta densidad PE: Polietileno PEBD: Polietileno de baja densidad ppm: Partes por millón PS: Poliestireno Psi: libra por pulgada cuadrada PTFE: Politetrafluoroetileno PVC: Polivinil cloruro P&T: Purga y Trampa Tc: Temperatura de cristalización Tg: Temperatura de transición vítrea Tf: Temperatura de fusión USEPA: US Environmental Protection Agency Xc: Grado de cristalinidad xiii INTRODUCCION En los procesos de extracción de crudo, refinación y petroquímica, así como también en los procesos de manufactura metalúrgicos, se emplea abundante agua que, al cumplir su función, se desecha contaminada con sustancias de distinta índole (polvos, iones metálicos, aceites y otros compuestos orgánicos). En una buena parte de los casos, la complejidad química de los contaminantes presentes en estas aguas dificulta su tratamiento de forma económica; específicamente cuando se trata de mezclas crudo/agua, la separación se hace muy complicada si el crudo se encuentra emulsionado en el agua (químicamente estabilizado o no) y más aún si las gotas del mismo son sumamente pequeñas (Rubio, 2002). En todo caso los procesos de tratamiento son siempre de naturaleza física. La fracción de los hidrocarburos que se disuelve es siempre la más difícil de tratar. Entre los compuestos monoaromáticos que pueden contaminar cuerpos acuosos se encuentran los denominados BTEX (benceno, tolueno, etilbenceno e isómeros de xilenos), los cuales constituyen un porcentaje significativo en peso de la gasolina (entre 10-59%), y son además muy solubles en agua si se comparan con los componentes alifáticos presentes en la gasolina. De hecho, cuando se mezcla gasolina con agua, entre (50-60) % del contenido másico de los hidrocarburos que se disuelven en el agua corresponden a los BTEX (Larroche, 2008). Los compuestos BTEX pueden contaminar los cuerpos de agua debido a la descarga de desechos industriales, fugas, derrames, disposición inapropiada y accidentes en labores de transporte de crudo (en refinerías y petroquímica), almacenamiento deficiente de tanques o tambores con productos de refinación petrolera (gasolina, diesel y otros aceites lubricantes), industrias de gas, aeropuertos, actividades de industrias de pinturas, así como también otras industrias químicas que trabajan con fibras sintéticas, plásticos y pesticidas (Larroche, 2008). En muchos países se ha planteado la recuperación y el reciclado de productos como una estrategia seria de gestión de residuos sólidos. Los materiales plásticos son probablemente los que, en mayor medida, se transforman en productos manufacturados destinados a pequeños 2 períodos de uso. Esto origina que cada día sea mayor la cantidad de residuos de este tipo, creando serios problemas ambientales. La asociación no gubernamental VITALIS indica que en Venezuela sólo se recicla un 2% del plástico desechado. Desde hace un tiempo se ha propuesto el uso de resinas poliméricas como adsorbente de estos contaminantes en medio acuoso. A pesar de tener menor capacidad de adsorción que el carbón activado, las resinas poliméricas han demostrado tener mayor afinidad por compuestos orgánicos de bajo peso molecular. La gran variedad de grupos funcionales, área superficial y porosidad, hacen posible la síntesis de resinas poliméricas específicas para la remoción de diversos compuestos orgánicos; además la regeneración de estas resinas se puede lograr, en muchas ocasiones, con el uso de un solvente u otras condiciones poco agresivas (Cohen, 1993). Simko y col. (1999) emplearon PEBD para determinar el proceso de difusión de HAP (hidrocarburos poliaromáticos) seleccionados en agua. Ellos publicaron en la revista Food Chemistry que la concentración del analito decrece en el agua tan pronto como comienza el experimento y se puede medir la adsorción en la superficie del polímero a los 45 minutos y en un tiempo de 1 hora y 30 minutos ya se puede medir la difusión de los HAP hacia el interior del polímero. Pan y col. (2009) realizaron una revisión bibliográfica sobre el material polimérico más ampliamente usado en procesos de adsorción de contaminantes, indicando que las resinas más frecuentemente usadas poseen matrices de poliestireno o poliéster acrílico. Considerando lo anteriormente dicho, el objetivo general de esta investigación fue evaluar la eficiencia de residuos de polietileno de baja densidad (PEBD), de poliestireno (PS) y de una polimezcla física (1:1) de ambos polímeros, en la remoción de hidrocarburos monoaromáticos (benceno, tolueno) en agua. Para llevar a cabo el objetivo general, se plantearon los siguientes objetivos específicos: 3 • Caracterización fisicoquímica y morfológica de los homopolímeros PEBD y PS empleados en esta investigación. • Preparación de una polimezcla 1:1 de PEBD-PS creada a partir de residuos de estos homopolímeros PEBD y PS. • Caracterización fisicoquímica y morfológica de la polimezcla PEBD-PS, mediante las técnicas Espectroscopía infrarrojo por transformada de Fourier (FT.IR), Calorimetría diferencial de barrido (CDB) y Microscopía electrónica de barrido (MEB). • Determinación del área superficial del PEBD, PS y de la polimezcla PEBD-PS por medio del método Brunauer, Emmett y Teller (Método BET). • Cuantificación de benceno y tolueno en agua mediante microextracción en fase líquida y el posterior análisis por cromatografía de gases acoplada a detector de ionización a la llama (GC/FID). • Validación de la microextracción en fase líquida como método de cuantificación en base a los parámetros de linealidad, precisión, exactitud, límites de detección, límites de cuantificación y porcentajes de recuperación. • Realización de cinéticas de adsorción de benceno y tolueno en PEBD, PS y la polimezcla PEBD-PS. • Determinación de las isotermas de adsorción de benceno y tolueno en PEBD, PS y la polimezcla PEBD-PS. Para la presentación de los aspectos fundamentales y el análisis de los resultados obtenidos, este libro se divide en las siguientes secciones. En el Capítulo 1 se presenta el marco teórico que sirvió de base para realizar esta investigación; en el Capítulo 2 se describen, el procedimiento experimental, materiales y equipos y la metodología utilizada. 4 Los resultados obtenidos y la discusión de los mismos están en el Capítulo 3. A continuación se presentan las conclusiones a las que se pudo llegar en esta investigación en base a los objetivos planteados, y las recomendaciones que surgen del mismo. Finalmente se presentan las referencias que sirven de apoyo y antecedentes, y los apéndices que complementan la información dada en los resultados. CAPITULO 1 MARCO TEÓRICO 1.1. Hidrocarburos aromáticos monocíclicos (HAM). Se define como compuestos aromáticos a aquellos compuestos cíclicos que contienen dobles enlaces conjugados, con energías de resonancia inusualmente grandes. Fundamentalmente, un compuesto aromático posee las siguientes características: estructura cíclica con enlaces pi conjugados, cada átomo del anillo tiene un orbital p no hibridado, los orbitales p no hibridados se solapan para formar un anillo continuo de orbitales paralelos (en la mayoría de los casos la estructura ha de ser plana) y la deslocalización de los electrones pi en el anillo debe disminuir la energía electrónica (Wade, 2003). Entre estos compuestos se hallan benceno y tolueno; los cuales poseen las características antes mencionadas. 1.1.1. Benceno El compuesto que hoy se conoce como benceno fue aislado por primera vez en 1825 por Michael Faraday, quien extrajo dicho compuesto de un residuo líquido obtenido luego de calentar aceite de ballena a cierta presión con el fin de producir un gas usado para iluminar edificios en Londres (Bruice, 2004). Nueve años más tarde Eilhardt Mitscherlich de la Universidad de Berlin preparó la misma sustancia por medio del calentamiento de ácido benzoico con óxido de calcio, hallando su fórmula empírica C6H6 (Carey, 2000). Figura 1.1 Síntesis de benceno por Mitchrlich (Carey, 2000) 6 En 1845 August von Hofmann preparó benceno a partir de alquitrán, que fue por muchos años, el reactivo primario para la producción industrial de benceno hasta la aparición de las tecnologías petroleras en 1950. En esta década se producen alrededor de 6 millones de toneladas de benceno por año, siendo una porción sustancial empleada de la producción de estireno para la posterior preparación de poliestireno (Carey, 2000). El benceno tiene una estructura plana y su esqueleto de carbonos tiene la forma de un hexágono regular. En la tabla 1.1 se muestra la estructura molecular del benceno y algunas de sus propiedades fisicoquímicas relevantes: Tabla 1.1 Estructura y propiedades fisicoquímicas del benceno (Wade, 2004, Larroche, 2008) Propiedades Benceno Estructura Química Fórmula Química Peso Molecular (g/mol) Solubilidad en agua (mg/L a 25ºC) Algunas Aplicaciones Temperatura de Ebullición Normal (ºC) Presión de Vapor (mmHg a 20ºC) Temperatura de Fusión Normal (ºC) Gravedad Específica (ºC) Coef. De Partición Octanol-Agua a 25ºC (LogP) C6H6 78,11 1785,5 Combustible, solvente en industrias químicas 80 95,19 5,5 0,8765 20 2,13 A corto plazo, la “US Environmental Protection Agency” (USEPA) ha encontrado que la exposición prolongada al benceno puede causar trastornos del sistema nervioso, sistema inmunológico y anemia. A largo plazo se ha determinado que es un compuesto cancerígeno. (USEPA, 2009) 7 1.1.2. Tolueno Algunos compuestos relacionados con benceno fueron obtenidos, en un principio, por extractos de plantas. Por ejemplo, una resina de olor característico conocida como “bálsamo tolu” se obtuvo en un árbol en Suramérica llamado “árbol Tolu”. En 1840 se descubrió que la destilación del bálsamo “tolu” originaba un derivado metilado del benceno, que luego se llamó tolueno (Carey, 2000). El tolueno es un líquido incoloro que posee un olor característico dulce; a una concentración de 8 mg/L se puede detectar su olor en el aire y a concentraciones entre 0,04 mg/L y 1 mg/L se puede sentir su sabor en el agua. Hoy en día el tolueno se obtiene de la elaboración de la gasolina y del crudo en general, además de la producción de coque a partir de carbón, entre otras fuentes (Health Protection Agency, 2008). En la tabla 1.2 se muestra la estructura molecular del tolueno y algunas de sus propiedades fisicoquímicas relevantes: Tabla 1.2 Estructura y propiedades fisicoquímicas del tolueno (Wade, 2004, Larroche, 2008) Propiedades Tolueno Estructura Química Fórmula Química Peso Molecular (g/mol) Solubilidad en agua (mg/L a 25ºC) Algunas Aplicaciones Temperatura de Ebullición Normal (ºC) Presión de Vapor (mmHg a 20ºC) Temperatura de Fusión Normal (ºC) Gravedad Específica (ºC) Coef. De Partición Octanol-Agua a 25ºC (LogP) C7H8 92,14 532,6 Solventes, TNT, Uretanos 110,6 28,4 -94,9 0,8669 20 2,73 8 A corto plazo, la USEPA ha determinado que los efectos a de la exposición al tolueno pueden causar trastornos menores del sistema nervioso, fatiga, nauseas, debilitamiento físico, mareos, etcétera. La exposición a largo plazo pueden causar trastornos pronunciados del sistema nervioso, espasmos, dificultad para hablar, puede afectar la visión, a memoria y la coordinación motora; además afecta al hígado y a los riñones (USEPA, 2009). 1.2. Normativa relacionada con la concentración de BTEX en agua Desde 1974 el Congreso de los Estados Unidos promovió una ley que requería que la USEPA determinara los niveles de seguridad en los cuales algunos compuestos químicos no causaran daños al encontrarse en agua potable. La USEPA llamó a esta cifra “Objetivo de nivel máximo del contaminante” (ONMC). La USEPA considera que el ONMC del benceno es cero, ya que así no causará daños a la salud (USEPA, 2009). Sin embargo, basado en el ONMC, la USEPA ha determinado una cantidad estándar llamada “máximo nivel del contaminante” (MNC); dicho número es lo más cercano posible al valor de ONMC y su valor se fundamenta en la capacidad para detectar y remover estos contaminantes utilizando tratamientos rentables (USEPA, 2009). También en Venezuela y otros países existen reglamentos destinados a controlar el contenido de estos contaminantes tanto en el agua potable como en los efluentes. La tabla 1.3 presentan los valores permitidos de BTEX en algunos países: 9 Tabla 1.3 Valores máximos permitidos de BTEX en agua potable Nivel máximo permitido en agua potable (mg/L) Compuesto Benceno Tolueno Etilbenceno Xilenos USEPAa (2009) 0,005 1 0,7 10 Venezuelab (1998) 0,01 0,7 0,3 0,5 Brasilc (2005) 0,005 0,002 0,9 0,3 Reino Unidod (2008) 0,001 0,7 - - a USEPA. National Primary Drinking Water Regulations, bMinisterios de Sanidad y Asistencia Social. Normas Sanitarias de Calidad de Agua Potable, cConsejo Nacional de Medio Ambiente (CONAMA). Resolución Conama N°357, dHealth Protection Agency. “Exposure Standards, Guidelines or Regulations” 1.3. Análisis de BTEX en agua: características generales de la cuantificación de BTEX en agua Hoy en día los análisis ambientales de compuestos orgánicos (multicomponentes) en aguas son mucho más complicados debido a dos razones; la gran cantidad de sustancias que deben ser monitoreados al mismo tiempo y la demanda de métodos que ofrezcan una mayor sensibilidad. La cromatografía proporciona una herramienta única en estos tipos de análisis, que, en muchos casos, son extremadamente complejos ya que muchas veces se encuentran mezclados distintos tipos de compuestos en diferentes concentraciones (Sarzanini, 2000). Básicamente se enfrentan tres problemas: la separación de los compuestos a ser analizados de la matriz en la que se encuentran, la necesidad de lograr límites bajos de detección y la identificación de analitos desconocidos. Como los métodos convencionales de separación acoplados con técnicas de separación no son suficientes para lograr solventar estos tres inconvenientes, se debe realizar un proceso de separación de matriz y uno de enriquecimiento (Sarzanini, 2000). 10 El término “preconcentración” o “enriquecimiento” responde a un paso previo en el análisis, es decir, una etapa de preparación de la muestra que resuelve los problemas antes mencionados y adicionalmente puede servir como paso de limpieza de otros compuestos no tóxicos que pueden estar acompañando al analito. El enriquecimiento suele lograrse con las técnicas de extracción, ya sea en fase líquida o sólida, previos al análisis cromatrográfico (Sarzanini, 2000). 1.4. Métodos de preconcentración de analito (BTEX) en matriz acuosa Dependiendo de la naturaleza del analito existen diversos métodos para lograr una separación de la matriz acuosa y la preconcentración para su posterior análisis. Los compuestos BTEX pueden ser incluidos en la categoría que toma en cuenta su volatilidad como propiedad fundamental; esta clasificación se conoce como “compuestos orgánicos volátiles” (COV). Existen muchas definiciones oficiales de lo que es un COV, unas orientadas hacia la contaminación que producen y otras que toman en cuenta su presión de vapor. (Dewulf, 2007). La USEPA define COV como compuestos orgánicos que contribuyen a la creación de “smog” fotoquímico (USEPA, 2006). Kennes y col. (2001) definen COV como compuestos orgánicos (vapor) que tienen una temperatura de ebullición por debajo de 373,15 K a una presión de 101 kPa. La Asociación Americana de Pruebas y Materiales (ASTM por sus siglas en idioma inglés) define COV como compuestos que poseen una presión de vapor superior a 13,3 Pa a 25°C y la Unión Europea limita a los COV a una presión de vapor superior a 10 Pa a 20°C (Dewulf, 2007). Se necesitan técnicas analíticas de alta precisión para estudiar este tipo de compuesto en base a su comportamiento y su destino en el ambiente. En términos de propiedades fisicoquímicas, los métodos analíticos más usados incluyen a la separación por cromatografía de gas (GC) acoplada tanto a un detector de ionización a la llama (FID), a un espectrómetro de masas (MS) o a un detector de captura de electrones (ECD) (Dewulf, 2002, Santos, 2002, Mangani, 2003, Eiceman, 2004, Dewulf, 2006). Sin embargo, un procedimiento de separación analítica no implica la separación de la matriz; sobre todo en el caso de matrices ambientales, en donde las concentraciones de COV son muy 11 bajas (pg/L o µg/L). Por esto se necesitan técnicas apropiadas de preconcentración; que serán descritas a continuación (Dewulf, 2007). 1.4.1. Inyección directa de muestra acuosa (DAI) El método DAI es el más sencillo de los métodos de preconcentración de analito, debido a que, como su nombre lo indica, evita el paso de preparación de la muestra para el posterior análisis en el cromatógrafo ya que se inyecta directamente (Kubinec, 2005). Las ventajas de este método son muy claras: se evita el paso de pretratamiento de la muestra para preconcentrar los analitos (se desestima pérdidas de analitos volátiles y contaminación de la muestra) (Dewulf, 2007, Kubinec, 2005), además permite analizar muestras sin discriminar entre analitos más polares y no requiere una instrumentación especial (Aeppli, 2008). En cuanto a desventajas, se pueden citar tres: las posibles interferencias que pueda producir la matriz acuosa, la no compatibilidad del agua con la fase estacionaria de las columnas comunes GC así como del detector FID (no sólo se daña la columna sino que disminuye la sensibilidad del detector). Por otra parte, como en la GC sólo se puede inyectar hasta 10 µL, el análisis por DAI quedaría restringido a sólo unos poco COV que podrían encontrarse en el agua en altas concentraciones (Aeppli, 2008, Dewulf, 2007, Kubinec, 2005). Por último se tiene que el método DAI no está normalizado por agencias como la USEPA. Aeppli y col. (2008) realizaron análisis de COV polares y no-polares en matrices acuosas utilizando la técnica DAI-GC/MS para poder inyectar volúmenes superiores a las 100 µL; estos investigadores obtuvieron límites de detección por debajo de las normas establecidas por la USEPA. Por otra parte, Kubinec y col. (2005) lograron inyectar, por GC-FID, volúmenes de hasta 250 uL para el análisis de BTEX con la ayuda de una precolumna empacada con “Chromosorb P NAW” colocada en el inyector; obtuvieron límites de detección entre 0,6 y 1 µg/L. 12 1.4.2. Método de purga y trampa (P&T) El método de purga y trampa (P&T) consiste en el burbujeo de un gas inerte a través de una porción de la muestra acuosa, a temperatura ambiente o elevada, dependiendo de los analitos de interés; todo esto con el fin de transferir dichos analitos de la fase acuosa a la fase gaseosa. El flujo del gas arrastra el vapor a una columna adsorbente donde los COV son adsorbidos. Una vez completada la purga, la columna es calentada y se invierte el flujo del gas inerte para desorber los COV hacia el columna cromatográfica (USEPA, Método 5030C, 2003). Esta técnica ha sido aplicada a matrices acuosas, nieve, suelos, tejidos biológicos, bebidas, entre otras (Sanz-Medel, 2008). Figura 1.2 Esquema de un sistema P&T Las ventajas básicas de esta técnica son tres: es una técnica que no requiere el uso de solventes, está estandarizada por la USEPA (USEPA, Método 5030C, 2003), y se integra, en un solo paso, la extracción y preconcentración de los analitos, por lo cual se evita pérdidas de COV (SanzMedel, 2008). Como limitación se tiene la utilización de un sistema automatizado especial para la aplicación de este método. Según el método de la USEPA, esta técnica puede emplearse en el análisis de la mayoría de los COV con puntos de ebullición por debajo de los 200°C, insolubles o poco solubles en agua (USEPA, Método 5030C, 2003). 13 Zoccolillo y col. (2005) emplearon este tipo de análisis para la determinación de hidrocarburos halogenados como tetracloruro de carbono, tetracloro etileno, cloroformo, entre otros, en agua mineral y agua de chorro, obteniendo límites de detección en el orden de los ng/L (Zoccolillo, 2005). Sanz-Medel y col. (2008) realizaron una comparación crítica entre el método P&T y el método de microextracción en fase sólida (SPME), concluyendo que el método de P&T fue el más acertado, analíticamente hablando, pues arrojó límites de detección más bajos (aproximadamente 0,06 ng/mL) para los hidrocarburos halogenados y el benceno. 1.4.3. Método headspace (HS) El método HS es ampliamente utilizado en el análisis de COV en suelos, desechos sólidos, soluciones acuosas con analitos miscibles en ellas, etcétera (USEPA, Método 5021ª, 2003). Se mencionan cuatro ventajas de esta técnica: es un método estandarizado por la USEPA, la extracción de los analitos, ya sea en aire o en un gas inerte, es compatible con los instrumentos analíticos (columnas cromatográficas, detectores, etcétera), es una técnica rentable y los efectos de la matriz son minimizados. Se deben vigilar ciertos parámetros como la temperatura de extracción, la fuerza iónica, el volumen de muestra extraído, entre otros (Dewulf, 2007). Existen dos tipos de HS: los métodos HS estáticos, en donde se realiza la extracción luego de alcanzar el equilibrio de partición entre la fase acuosa y la fase gaseosa; en los métodos HS dinámicos, los analitos presentes en la matriz acuosa pueden ser extraídos mediante P&T (Dewulf, 2007). 1.4.3.1. Método HS estático En este tipo de técnica se analiza una alícuota de la fase de vapor, la cual se encuentra en equilibrio termodinámico con la fase condensada. La manera más simple consiste en una jeringa que extrae muestra de la fase de vapor, sin preconcentración aparente. 14 Figura 1.3 Método HS estático Recientemente se ha propuesto técnicas como “headspace-microextracción en gota” (HSSDME) y “headspace-microextracción” en fase líquida (HS-LPME) que consisten en colocar en el embolo de la microjeringa, o en la punta de la misma, una pequeña cantidad de solvente orgánico (Figura 1.4) para favorecer la extracción (Dewulf, 2007). Sapelnikova y col. (2004) obtuvieron límites de detección para BTEX entre 0,2 y 0,4 µg/L utilizando HS-LPME. Figura 1.4 Método HS estático. HS-SDM (Dewulf, 2007). 1.4.3.2. Método HS dinámico En este tipo de técnica se emplea la P&T, es decir, una corriente de gas inerte para volatilizar los compuestos COV de la matriz acuosa; hay algunos casos en donde la purga se realiza en la 15 zona de vapor para evitar interferencias de la matriz acuosa. El HS dinámico tienes muchas desventajas asociadas, entre las cuales se pueden mencionar la compleja instrumentación, posible contaminación e interferencia del vapor de agua, y el tiempo de duración de cada extracción que oscila entre 10 y 30 minutos por muestra (Dewulf, 2007). 1.4.4. Microextracción en fase sólida (SPME) La SPME es una técnica introducida por primera vez en 1990 (Ouyanga, 2008). Se basa en la extracción de los analitos de la matriz de la muestra mediante una fibra de sílice fundida recubierta con un sorbente, en la mayoría de los casos polimérico (Figura 1.5), seguida de un proceso de desorción térmica del analito (Peñalver, 2002). Esta técnica ha sido ampliamente usada en análisis ambientales, forenses, farmacéuticos, de alimentos, entre otros (Ouyanga, 2008). El análisis se puede realizar de dos maneras (Figura 1.6), con la fibra inmersa en la matriz acuosa o con la fibra sobre la fase acuosa, en la fase gaseosa del vial contenedor de la muestra (Tena, 2007). Figura 1.5 Partes de la fibra de SPME (Peñalver, 2002) 16 Figura 1.6 Técnicas SPME. a) Inmersión de la fibra b) Fibra en la fase gaseosa c) Desorción en el inyector del cromatógrafo (Peñalver, 2002). Esta técnica posee varias ventajas, entre las cuales se tienen: el tamaño pequeño de la fibra y su geometría cilíndrica permiten que la misma esté incorporada a la jeringa; esto permite su fácil manipulación y protección de la fibra (Peñalver, 2002). Asimismo, no utiliza solventes tóxicos, e integra en un solo paso el aislamiento y la preconcentración de los analitos, además que la fibra es totalmente reusable (Ouyanga, 2008). 1.4.5. Métodos de extracción en fase líquida (LLE) La extracción en fase líquida es el método más antiguo de preconcentración en química analítica (Dewulf, 2007). Esta técnica consiste en la partición de un analito por contacto de dos fases inmiscibles, desde una solución acuosa homogénea (inicial) hasta una fase orgánica (solvente orgánico inmiscible en agua) (Assadi, 2006). Este método, muy simple, tiene muchas desventajas, entre las cuales se pueden mencionar: el uso de solventes puede limitar el rango de COV que pueden ser medidos en los cromatógrafos, los COV podrían perderse al momento de ser extraídos por su volatilidad, los límites de detección pueden ser deficientes si se toma en cuenta la poca cantidad de solvente que puede ser inyectado en el cromatógrafo. Por último, esta técnica consume mucho tiempo y necesita grandes cantidades de solvente, lo cual no concuerda con la idea de “química verde” (Dewulf, 2007). 17 1.4.5.1. Microextracción en fase líquida En la extracción en fase líquida, se han dado varios avances. La microextracción en fase líquida emplea pequeñas cantidades de un solvente inmiscible en agua, originando un sistema de dos y hasta tres fases. Entre las ventajas se pueden señalar la reducción del uso de solventes y la integración de la extracción y preconcentración en un solo paso. Vale la pena acotar que este método se encuentra estandarizado por la USEPA y ha sido codificado como 3511 (USEPA, 2002). El método 3511 de la USEPA, microextracción de compuestos orgánicos en agua, se ha aplicado para compuestos COV con excelentes porcentajes de recuperación. Sin embargo no se reportan límites de detección estimados. 1.5. Tratamientos para eliminar BTEX de cuerpos acuosos En su revisión bibliográfica, Larroche y col. (2008) hacen una separación entre los distintos métodos para tratar aguas contaminadas con compuestos orgánicos. En primer lugar dividen los métodos en químicos, biológico y físicos; seguidamente hacen una separación entre lo que son tecnologías aplicadas en el sitio de origen que está contaminado, llamadas “tecnologías in-situ” y tecnologías aplicadas en sitios alternos al origen, llamadas “tecnologías ex-situ”. Específicamente las tecnologías ex–situ también son conocidas con la expresión “bombear y tratar” ya que aguas subterráneas contaminadas son bombeadas a la superficie mediante pozos de extracción, en donde son tratadas para ser reusadas o reinyectadas al sitio de origen (Larroche, 2008). En la Tabla 1.4 se presentan las principales tecnologías utilizadas para el tratamiento de aguas contaminadas con compuestos aromáticos. 18 Tabla 1.4 Tratamientos de efluentes contaminados con compuestos aromáticos (Larroche, 2008). Métodos Tecnologías in-situ Tecnologías ex-situ Inyección de aire (Air sparging) Burbujeo de aire (Air stripping) Contención hidráulica Adsorción: Filtración con membranas reactivas, carbón activado, polímeros Físicos Remediación Fotocatalítica (TiO2/UV) Químicos Inyección de compuestos oxidantes (O3, O2, H2O2, Cl2) Aplicación de compuestos oxidantes (O3, O2, H2O2, Cl2) Coagulación, floculación y precipitación Biológicos Biorremediación (natural o artificial) Biorremediación (aeróbica o anaeróbica) A continuación se detallan los aspectos más significativos de los métodos más frecuentemente empleados en la remediación de efluentes acuosos contaminados con compuestos aromáticos. 1.5.1. Métodos químicos En los métodos químicos de remediación de aguas contaminadas se aplican compuestos químicos para originar reacciones favorables a la degradación de los contaminantes o para facilitar la remoción de los mismos. Los tratamientos químicos in-situ pueden ser considerados sólo en situaciones donde los contaminantes son conocidos y sus concentraciones son definidas. Este tipo de técnicas incluye generalmente la instalación de pozos de inyección a la cabeza de las aguas subterráneas, en donde un agente químico es bombeado hacia el cuerpo de agua. Dicho agente químico debe ser específico para la clase de contaminante con que se trabaja (Knox, 1986). 19 En los últimos 20 años ha crecido notablemente la ozonización como uno de los procesos empleados para la degradación in-situ de compuestos contaminantes en cuerpos acuosos. El ozono (O3) es una forma alotrópica del oxígeno, descubierto en 1783 por Van Marum y nombrado en 1840 por Schonbein. El ozono es producido por una descarga eléctrica de alto voltaje en presencia de oxígeno; es un gas a temperatura y presión normal y su solubilidad depende de la temperatura, su presión parcial en la fase gaseosa y hasta del pH. El ozono es inestable y su tasa de descomposición se incrementa con la temperatura y el pH; dicha descomposición se inicia con iones OH presentes en el medio acuoso, formándose, después de una serie de reacciones en cadena, radicales OH., entre otros (Muñoz, 2006). El ozono puede reaccionar con materia orgánica presente en el agua mediante dos mecanismos posibles descritos a continuación (Figuras 1.7 y 1.8): Figura 1.7 Reacción directa del contaminante con ozono (Muñoz, 2006). (a) (b) Figura 1.8 a) Producción de radicales por descomposición de ozono b) Reacción del contaminante con el radical OH (Muñoz, 2006) Usualmente ambos mecanismos explican la degradación de contaminantes, prevaleciendo uno u otro dependiendo del pH del medio; en medio ácido predomina el primer mecanismo (figura 1.7) y en condiciones alcalinas predomina el segundo mecanismo (figura 1.8). Cuando se aplica el ozono a compuestos orgánicos con alto peso molecular, estos pueden no degradarse de forma completa. Por esto se emplean compuestos oxidantes menos selectivos que el ozono molecular. El peróxido de hidrógeno (H2O2) es adecuado para estos casos ya que estimula la producción de radicales OH (Figura 1.9): 20 Figura 1.9 Descomposición del ozono en presencia de peróxido de hidrógeno (Muñoz, 2006). Por otra parte, los tratamientos químicos ex-situ engloban los procesos de remediación fotocatalítica. Se puede definir fotocatálisis como una aceleración de una fotorreacción por la presencia de un catalizador semiconductor que puede activarse por la adsorción de luz (radiación UV por debajo de los 380 nanómetros) para liberar electrones de la banda de valencia del semiconductor. Los electrones fotogenerados y los huecos reaccionan con el agua, el oxígeno disuelto y compuestos orgánicos, para formar radicales y crear un ambiente altamente oxidante. Entre las ventajas se tiene su rapidez de acción (resultados más rápidos que en biorreactores), menores costos que otros métodos como la ozonización (estos costos se pueden reducir empleando la luz solar) (Lair, 2008, Muñoz, 2006). El semiconductor empleado por excelencia es el dióxido de titanio (TiO2) aunque también se emplea sulfuro de cadmio (CdS), sulfuro de zinc (ZnS), oxido de zinc (ZnO), entre otros. El titanio es el novena elemento más abundante en la corteza terrestre, y específicamente el TiO2 tiene una hueco de energía en aproximadamente 3,2 eV. Las ventajas de este semiconductor son su condición de químico inerte, fotoestable, relativo bajo costo, etcétera (Muñoz, 2006). Los procesos de coagulación, floculación y precipitación también se emplean con frecuencia. La coagulación consiste en añadir al medio acuoso un coagulador químico (sales de hierro, polímeros orgánicos, entre otros) para acondicionar la suspensión o coloide presente y disolver la materia orgánica y proceder a la floculación que consiste en agregar partículas desestabilizadas (partículas desde las cuales la carga eléctrica superficial ha sido reducida) para llevar a cabo la precipitación de los productos formados que pueden ser removidos por sedimentación o filtración. Se dice que la coagulación ocurre en unos 10 segundos, mientras que la floculación ocurre en un período de 20 a 45 minutos (Crittenden, 2005). La efectividad los procesos de coagulación/floculación/precipitación dependen de la naturaleza y concentración de los contaminantes y el diseño general del proceso. En el diseño se deben considerar los compuestos 21 químicos empleados y las dosis óptimas de los mismos, valores de pH, agitación, temperatura, producción de lodos, remoción de agua, etcétera (Ginn, 2004). Generalmente no se utiliza este proceso como tratamiento dedicado para los BTEX. 1.5.2. Métodos biológicos Se define “biorremediación” como la eliminación, atenuación o transformación de sustancias contaminantes mediante procesos biológicos (Lynch y Moffat, 2005). Los procesos de biorremediación emplean microorganismos para tratar los contaminantes orgánicos, con la finalidad de degradarlos a compuestos menos tóxicos como dióxido de carbono (CO2), metano, agua y sales inorgánicas. Como indicó la tabla 1.4, las tecnologías incluyen biorremediación insitu o ex-situ (Larroche, 2008). Los procesos de biorremediación se ven afectados por una serie de factores ampliamente estudiados (Figura 1.10): Figura 1.10 Factores que afectan la biorremediación (Larroche, 2008, Boopathy, 2000). 22 En el caso de la biorremediación in-situ se tiene la ventaja de que los compuestos indeseados se destruyen completamente y se evita la remoción del cuerpo acuoso y tratamiento de residuos posteriores. Hay dos aproximaciones básicas: la primera es la actividad natural biológica en la superficie del cuerpo acuoso, en donde la intervención es mínima y sólo consiste en medidas de contención para prevenir el movimiento de los contaminantes. En este sentido, como los contaminantes orgánicos tienden a crear condiciones anaeróbicas, este tipo de aproximación es más adecuado para ellos. La segunda aproximación se conoce como “biorestauración” e involucra la estimulación de los microorganismos añadiendo nutrientes y oxígeno para promocionar la degradación aeróbica de los contaminantes (Russell y Ginn, 2004, Knox, 1986). De igual forma se plantea también la adición de microorganismos no nativos del sitio que se adapten rápidamente al medio y puedan ejercer funciones de degradación de los contaminantes (Knox, 1986). Entre las técnicas más empleadas se puede hablar de fitorremediación, que es un término aplicado a una variedad de tecnologías que consisten en el uso de plantas para la transformación, degradación o extracción de los contaminantes. La USEPA posee información acerca de la evaluación de la fitorremediación como método apropiado, costos, mecanismos, criterios de diseño y casos estudiados. Entre los contaminantes que han sido evaluados por este método se hallan hidrocarburos poliaromáticos (HAP), bifenilos policlorados, fenoles, pesticidas, entre otros. Los mecanismos que se manejan incluyen la fitoextracción, rizofiltración, fitoestabilización, rizodegradación, entre muchas otras (Russell y Ginn, 2004). En el caso de la biorremediación ex-situ existen diversas alternativas de tratamiento de acuerdo con las condiciones de trabajo y del sitio en donde se realizará el tratamiento. Los lodos activados consisten en un sistema biológico de crecimiento suspendido en donde una solución contaminada es mezclada en un tanque con suministro de aire. La actividad microbiana es capaz de degradar biológicamente los compuestos orgánicos a material celular, dióxido de carbono y agua (Russell y Ginn, 2004). 23 Los embalses o humedales superficiales también son empleados como tratamientos pasivos pero la población microbiana es menor, por lo que requieren tiempos de residencia en el orden de las semanas. Esto trae como consecuencia que se necesite una superficie más grande para su emplazamiento. (Russell y Ginn, 2004). Los filtros de goteo o biopercoladores son sistemas biológicos en donde las aguas contaminadas se ponen en contacto con microorganismos que crecen adheridos a un medio sólido. Los microorganismos crecen en forma de colonias formando una película que recubre el medio sólido metabolizando aeróbicamente los compuestos orgánicos solubles en el agua (Russell y Ginn, 2004). Los discos biológicos rotatorios consisten de una serie de discos mecánicos conectados a un eje y colocados en un tanque; los discos giran y aproximadamente 40% de su área superficial queda sumergida en el agua. A medida que el agua contaminada pasa los microorganismos que se desarrollan en los discos metabolizan los contaminantes orgánicos en el agua (Russell y Ginn, 2004). La alternancia inmersión-aireación garantiza el suministro de oxígeno a las colonias de microorganismos. En líneas generales, los métodos de biorremediación son empleados con frecuencia por su bajo costo operacional, lo cual proporciona una ventaja interesante sobre otros métodos de remediación de aguas contaminadas. Los métodos de biorremediación son utilizados generalmente para el tratamiento de aguas subterráneas contaminadas con BTEX. 1.5.3. Métodos físicos En lo que a los métodos físicos in-situ existen varias alternativas entre las cuales se encuentran el confinamiento y la tecnología conocida como “air sparging”. En primer lugar, los métodos de contención hidráulica han sido usados de forma frecuente, no sólo para aislar las fuentes de contaminación sino para contener la posible contaminación “agua 24 abajo”. Las aguas contaminadas pueden ser físicamente aisladas con barreras de baja permeabilidad. Los materiales típicos para la construcción de estas barreras incluyen arcilla, plásticos sintéticos, geo-membranas, bentonita, bentonita mezclada con cemento, etcétera. Evidentemente, un emplazamiento que rodee las aguas contaminadas tendrá mucho mayor efecto que barreras que no encierren el efluente. Se dice también que las barreras rectas tienen menor efectividad pues el agua puede fluir hacia los bordes. Hay limitaciones para el uso de este tipo de metodología y no se sabe a ciencia cierta su efectividad a largo plazo. En cuanto a ventajas se tiene que esta técnica es útil para sitios en donde no se posea la tecnología adecuada para eliminar los contaminantes del efluente como tal (Committee on Ground Water Cleanup Alternatives, 1994). La técnica conocida como “air sparging” se puede traducir como una aireación in-situ, en donde la circulación de aire se emplea para remover los COV. La aireación se realice en las zonas saturadas con agua subterránea y consiste en inyectar aire directamente en el acuífero a cierta presión con el fin de desplazar los COV hacia zonas no saturadas. El flujo de aire debe ser capturado por un sistema de extracción adecuado y debe ser tratado previo a su liberación. Esta técnica promueve la remoción de contaminantes tanto física como biológicamente; la eficiencia del sistema depende en gran parte del coeficiente de partición de los COV en aire (ley de Henry) y de la biodegradación aeróbica por la gran cantidad de oxígeno que se incluye en el sistema (Committee on Ground Water Cleanup Alternatives, 1994). En la figura 1.11, se presenta un esquema simple del método de aireación in-situ: 25 Figura 1.11 Esquema de la técnica de "air sparging" (Committee on Ground Water Cleanup Alternatives, 1994). Se conoce que este tipo de técnicas es aplicable para COV cuya constante de ley de Henry es mayor a 10-5 atm-m3/mol, y la presión de vapor es mayor a 1 mm Hg (Committee on Ground Water Cleanup Alternatives, 1994). El método de remediación físico conocido como “air stripping”, se aplica en pozos alternos al sitio original de contaminación, por lo cual es la versión ex-situ del “air sparging”. En este tipo de tecnologías se promueve el contacto entre una fase gaseosa y una líquida para crear una transferencia de masas de los contaminantes del medio acuoso al medio gaseoso. Este método es ampliamente usado para la remoción de COV y consiste básicamente en dos modalidades: torres empacadas con un flujo de aire en contracorriente (gas continuo en contracorriente con un flujo de agua discontinuo) y aireadores (Crittenden, 2005). A continuación (Figura 1.12) se presenta un esquema de una torre empacada con flujo de aire en contracorriente: 26 Figura 1.12 Esquema de una torre con flujo de aire en contracorriente (Crittenden, 2005). Al igual que con la técnica “air sparging” el éxito de este tipo de tecnologías depende del grado de remoción que se desea lograr, de la afinidad del COV por el agua (solubilidad y constante de ley de Henry), entre otros factores (Crittenden, 2005). Otra técnica de separación o retención física es el uso de barreras permeables reactivas. En este proceso el cuerpo acuoso contaminado es tratado mientras fluye por las zonas reactivas ubicadas en el acuífero. Mientras los gradientes hidráulicos naturales o inducidos mueven el agua hacia las zonas reactivas, una especie de filtros son utilizados para la retención y/o degradación de los contaminantes disueltos (Committee on Ground Water Cleanup Alternatives, 1994). A continuación (Figura 1.13) se presenta un esquema de las barreras permeables reactivas: 27 Figura 1.13 Barrera permeable reactiva (Committee on Ground Water Cleanup Alternatives, 1994) Las zonas reactivas de las barreras pueden combinar materiales para obtener resultados satisfactorios mediante procesos químicos, biológicos o físicos. La primera posibilidad emplea reacciones de oxidación-reducción para precipitar metales o catalizadores metálicos para degradar compuestos orgánicos; la segunda posibilidad emplea nutrientes y/o bacterias para favorecer la biodegradación de los contaminantes. Los investigadores han tratado de crear zonas de reacciones biológicas para optimizar el proceso de biodegradación de diversos contaminantes. La tercera posibilidad emplea materiales como el carbón activado o polímeros de manera que actúen como adsorbentes de contaminantes orgánicos. Cuando dichas barreras emplean materiales que se desgastan (como el carbón activado y otros químicos), los mismos deben ser instalados en especies de módulos para facilitar su reemplazo de forma periódica (Committee on Ground Water Cleanup Alternatives, 1994). El carbón activado es el material más ampliamente usado para la adsorción de contaminantes orgánicos en agua. Se dice que la adsorción usando carbón activado es efectiva y confiable para remover compuestos orgánicos de baja solubilidad en agua, de alto peso molecular, algunos metales y otras especies inorgánicas; además puede emplearse cuando los contaminantes se hallan en un amplio rango de concentraciones. Este material se emplea en muchas ocasiones luego de tratamientos preliminares para reducir los niveles de contaminación al mismo tiempo de extender la vida útil del adsorbente. Existen distintos tipos de carbón activado con distinta 28 porosidad y área superficial que se adecuan a cualquier contaminante. En Norteamérica y Europa se han instalado un gran número de barreras permeables reactivas que emplean al carbón activado como adsorbente. Una limitación es la cantidad de agua que puede ser tratada por una cierta cantidad de carbón activado dependiendo además del tipo de adsorbente que se use (Ginn, 2004, Cortina, 2008). La mayor desventaja del uso de carbón activado como adsorbente es justamente su reemplazo cuando se agota, además de su disposición final (uso de altas temperaturas, y otras condiciones agresivas) (Larroche, 2008). Es importante mencionar que la adsorción en carbón activado se debe a un fenómeno meramente superficial, es decir, varía dependiendo del área superficial y su interacción con el contaminante. Desde hace más de dos décadas se ha estudiado el uso de resinas sintéticas o polímeros como alternativa al carbón activado para la adsorción de contaminantes orgánicos específicos. Las primeras investigaciones demostraron que, a nivel másico, los polímeros tienen menor capacidad de adsorción para la mayoría de compuestos orgánicos que el carbón activado, sin embargo, las resinas mostraron alta selectividad en lo que se refiere a compuestos orgánicos de bajo peso molecular (Cohen, 1993). Puntualmente, las resinas poliméricas han demostrado amplia capacidad de adsorber compuestos fenólicos, ácidos orgánicos, hidrocarburos aromáticos monocíclicos y policíclicos, alcanos y otros derivados (Pan, 2009). Las resinas poliméricas ofrecen vastas áreas superficiales, rigidez mecánica, una química superficial ajustable (tamaño y distribución de los poros), y fácil regeneración (Pan, 2009). De igual manera, los polímeros ofrecen la ventaja de la desorción de los contaminantes para su recuperación y uso posterior. Las síntesis poliméricas pueden determinar la afinidad del mismo hacia un determinado contaminante (ya sea altamente soluble en agua, un metal pesado, etcétera) ya que se puede modificar su superficie con grupos funcionales determinados unidos a la matriz polimérica para incentivar al interacción con dichos contaminantes (Pan, 2009). La regeneración de los polímeros contaminados se puede lograr de manera muy sencilla con el uso de solventes (Cohen, 1993). En un principio los polímeros presentaron problemas en la optimización del proceso de adsorción-desorción de los contaminantes no sólo por las condiciones de equilibrio sino también por la cinética por la cual se rige (Hradil, 1991). 29 1.6. Polímeros Los polímeros son moléculas compuestas por muchas unidades más pequeñas, denominadas monómeros, las cuales se enlazan entre si y se repiten (Wade, 2003). A diferencia de las moléculas orgánicas pequeñas que son de interés por sus propiedades químicas, los polímeros son de interés por sus propiedades físicas que los hacen útiles en la vida cotidiana (Bruice, 2004). Según su naturaleza, los polímeros se dividen en: naturales, que son los que se encuentran presentes en la naturaleza como celulosa, almidones, proteínas, entre otros (Figura 1.14); y sintéticos (Figura 1.15), que son los que se obtienen por vía puramente sintética a partir de monómeros, entre los cuales se pueden mencionar las poliamidas, PE, PS, PVC, entre otros (Wade, 2003). Figura 1.14 Polímeros naturales. a) Caucho natural b) Quitina (Wade, 2003, Bruice, 2004) Figura 1.15 Polímeros sintéticos. a) Nylon 66 b) Neopreno (Bruice, 2004) 30 1.6.1. Polietileno El polímero más sencillo en cuanto a estructura química, más estudiado y frecuentemente usado para la fabricación de productos plásticos es el polietileno (PE) (Figuras 1.16 y 1.17). Entre sus características más destacables se encuentran su bajo costo, buena capacidad de aislamiento eléctrico, resistencia química, flexibilidad, fuerza, fácil procesamiento y transparencia (en el caso de capas delgadas). Es un polímero de adición, siendo su precursor la molécula de etileno. A lo largo de la historia se ha sintetizado polietileno de distintas formas y con diversas unidades monoméricas; esto ocasionó que, dadas las variedades de propiedades obtenidas en relación a su estructura molecular se clasificara como polietilenos de baja densidad (PEBD), polietilenos lineales de baja densidad (PELBD) y polietilenos de alta densidad (PEAD) (Brydson, 1999). Figura 1.16 Unidad repetitiva estructural del polietileno (Brydson, 1999). Figura 1.17 Preparación de polietileno (Brydson, 1999). La flexibilidad de sus cadenas C-C son la causa de su baja temperatura de transición vítrea (Tg), el cual se ha reportado que puede ser -20°C o más bajo (según los valores reportados), mientras que su temperatura de fusión (Tf) puede oscilar entre 108-132 °C dependiendo de su estructura molecular. Estos valores se atribuyen a que en el caso del PE, su estructura le permite “empacarse” o disponerse de forma ordenada al momento de la cristalizar, por la inexistencia de fuerzas intermoleculares importantes que impidan este proceso. Esta alta cristalinidad origina el nivel de opalescencia de su estructura; además, por no interactuar con ningún líquido, no tiene solventes a temperatura ambiente. Además, cuando el PE es de alta pureza, es un excelente aislante de alta frecuencia, debido a su naturaleza no polar (Brydson, 1999). 31 El PE es un polímero fuerte, flexible, de alta resistencia eléctrica y químicamente inerte (por su falta de grupos funcionales reactivos con otras especies químicas); hace que sea muy útil en actividades de cortos períodos de uso y donde no se aplique una fuerza prolongada sobre ellos, es decir, como bolsas, contenedores de comida, etcétera; sin embargo su inestabilidad ante altas temperaturas hace que no sea la resina más idónea para usos en ingeniería automotriz, ente otros (Peacock, 2000). 1.6.2. Poliestireno Por otra parte, el PS y puntualmente los polímeros basados en estireno (Figuras 1.18 y 1.19), se reconocen entre los más importantes en el rubro de los termoplásticos. Para 1990 la producción mundial de poliestireno estuvo en el orden de las 10x106 toneladas. Los polímeros basados en PS se desarrollaron rápidamente a lo largo de la historia por sus beneficiosas características de procesabilidad, bajo costo, coloración o transparencia (según sea el caso), rigidez, aislamiento eléctrico y baja capacidad de absorción de agua. La misma rigidez del poliestireno le otorga fragilidad, por eso se ha considerado la unión de este polímero con otro que modifique sus propiedades. Entre estos compuestos se mencionan los copolímeros estireno-butadieno, estirenopropileno, estireno-acrilonitrilo, entre otros (Brydson, 1999). Figura 1.18 Unidad repetitiva estructural del poliestireno (Brydson, 1999). Figura 1.19 Síntesis de estireno (Brydson, 1999). 32 La rigidez de las cadenas de PS a causa de los anillos bencénicos hace que esta resina posea una Tg aproximada entre 90-100°C. Su valor de temperatura de transición vítrea combinado con su condición de polímero amorfo hace que esta resina sea fuerte y transparente a temperatura ambiente. Por otra parte, su Tf oscila entre 230-270°C. A causa del anillo bencénico, el PS es soluble en varios solventes (cloroformo, benceno, tolueno, tetracloruro de carbono) y es mucho más reactivo que el PE, sobre todo a reacciones de sulfonación, cloración, nitración e hidrogenación (Brydson, 1999). El PS es un excelente aislante eléctrico, además adsorbe poca cantidad de agua, por lo cual su condición de aislante se mantiene intacta en condiciones húmedas. En cuanto a sus propiedades ópticas, el PS tiene un índice de refracción 1,592; el cual origina que sea una resina brillante. Sin embargo es un mal conductor térmico (Brydson, 1999). 1.7. Procesos de adsorción La adsorción se puede definir como un proceso donde las moléculas en fase gaseosa o acuosa son acumuladas sobre una superficie. La sustancia que se concentra en la superficie se llama adsorbato y el material sobre el cual se acumula se llama adsorbente. Este proceso es de gran importancia a nivel industrial, ya que se aplica en áreas como catálisis, separación de compuestos, etcétera (Levine, 2004). Particularmente, los gases se adsorben en la superficie de un sólido formando una capa de partículas gaseosas que recibe el nombre de “monocapa”; dichas partículas están en contacto directo con la superficie sólida. Asimismo, existe la posibilidad de que varias capas del gas se coloquen una encima de la otra, recibiendo el nombre de “multicapas” (Ortega, 2008). En la Figura 1.20 se ilustra la adsorción en mono y multicapas. Existen dos tipos básicos de adsorción que se diferencian por el tipo de interacciones que se dan entre adsorbente y adsorbato: en la fisisorción predominan las interacciones de tipo Van der Waals, entre las cuales se pueden mencionar las fuerzas dipolo-dipolo y las fuerzas de dispersión; 33 en la quimisorción las interacciones se asemejan a enlaces químicos, pues los electrones de adsorbato y adsorbente son compartidos (Ortega, 2008, Levine, 2004). Figura 1.20 Esquema de a) adsorción de monocapa b) adsorción de multicapas (Ortega, 2008). A medida que se llevan a cabo los procesos de adsorción se libera una cantidad de calor denominada “calor de adsorción” (∆Hads). Para el caso de la fisisorción el calor liberado es aproximadamente igual al “calor de condensación” (∆Hc). En el caso de la quimisorción el calor liberado es mucho mayor y se encuentra en el mismo orden de magnitud que el “calor de la reacción” (∆HR) (Ortega, 2008). La Figura 1.21 muestra los procesos de fisisorción y quimisorción. Figura 1.21 a) Fisisorción de H2 sobre una superficie metálica b) Quimisorción de H2 sobre una superficie metálica (Levine, 2004) 1.7.1. Isotermas de adsorción Para el estudio de los procesos de adsorción se necesita relacionar de alguna forma la cantidad de adsorbato que se acumula en el adsorbente con la concentración del mismo en la fase fluída, 34 de manera de obtener una relación de la transferencia de masa que se lleva a cabo. En los estudios de adsorción se mide la cantidad de gas o líquido adsorbido a una temperatura dada (en función de la presión en el caso de los gases). En el caso de los gases, se realiza la experiencia en un baño termostático a diferentes presiones iniciales, mientras que en el caso de los líquidos, se repite la experiencia en un baño termostático, cambiando las concentraciones del adsorbato. En ambos casos, la representación de la relación de la masa de adsorbato y la masa de adsorbente frente a la presión del gas o concentración del líquido a temperatura constante, se conoce con el nombre de “isotermas de adsorción” (Levine, 2004). En el año 1945, Stephen Brunauer clasificó los diferentes tipos de isotermas de adsorción que podrían describir distintos procesos de adsorción (Masel, 1996), los cuales se muestran en la Figura 1.22. Figura 1.22 Tipos de isotermas de adsorción (Adamson, 1997). La isoterma TIPO 1 indica que la cantidad de moléculas adsorbidas aumenta a medida que aumenta la presión, hasta llegar a un límite máximo donde se mantiene constante. Este tipo de 35 isotermas aplican en sistemas donde el fenómeno predominante es la quimisorción, ya que el adsorbato forma una monocapa (Masel, 1996, Shaw, 1966). La isoterma TIPO 2 es característica en la adsorción con formación de multicapas, es decir, fisisorción. Inicialmente las moléculas son adsorbidas y se llega a una saturación momentánea (formación de la monocapa) para luego continuar adsorbiendo a medida que la presión aumenta (formación de multicapas) (Shaw, 1966). La isoterma TIPO 3 se asemeja mucho a la de TIPO 2, ya que inicialmente la adsorción es baja para luego aumentar de forma drástica; esto sugiere la formación de multicapas en el adsorbente (Shaw, 1966). Las isotermas TIPO 4 y TIPO 5, usualmente describen como un gas se adsorbe en multicapas sobre una superficie porosa. En etapas iniciales, estas isotermas se asemejan a las de TIPO 1 y TIPO 2, pero eventualmente el sistema alcanza la saturación (Shaw, 1966). 1.7.1.1. Isotermas de adsorción de Langmuir En el año 1918, el físico Irving Langmuir empleó un modelo simplificado de la superficie de un sólido haciendo las siguientes suposiciones: el sólido en cuestión posee una superficie uniforme, no existe interacción entre las moléculas de adsorbato y las moléculas del adsorbato se hallan en posiciones especificas del adsorbente creando única y exclusivamente una monocapa de adsorción. Se habla de un estado de equilibrio en donde las velocidades de adsorción y desorción de las moléculas en las superficies del sólido son iguales; estas se caracterizan por sus constantes de adsorción y desorción “Ka” y “Kd” respectivamente. Si se representa la fracción de posiciones ocupadas en el adsorbato “θ” y el número total de moléculas del adsorbato “N”, se igualan las velocidades y se despeja θ: Ecuación 1.1 Velocidad en el proceso de adsorción (Levine, 2004). 36 Ecuación 1.2 Velocidad en el proceso de desorción (Levine, 2004). Ecuación 2.3 Fórmula de la isoterma de Langmuir (Levine, 2004). Se maneja la constante KL como la relación de Ka y Kd y el término “C” responde a la concentración de adsorbato en la fase fluida (Levine, 2004). Para sitios de adsorción equivalentes, tanto la energía de adsorción como KL son independientes del grado de recubrimiento, por lo que se puede definir θ o qe como el número total de moléculas de adsorbato entre el numero de moléculas de adsorbato que forman una monocapa “Nm” o qm, es decir N/Nm (Levine, 2004, Ortega, 2008). Si se representa N en función de C se obtiene una curva ascendente que tiende a la saturación o capacidad de la monocapa Nm, es decir, la ecuación 1.3 indica que la adsorción de las moléculas del adsorbato disueltas en el fluido en la superficie del sólido se ajustarán a una isoterma de adsorción TIPO 1 (Ortega, 2008). De la ecuación 1.3, se desprende que para sistemas donde KLC>>1, el grado de recubrimiento (θ) tenderá a la unidad. Por el contrario para sistemas donde KLC<<1, la gráfica de la ecuación 1.3 tenderá a una recta que pasa por el origen, donde θ será igual al producto KLC (Ortega, 2008). La adsorción de compuestos orgánicos puede ser estudiada mediante cinco expresiones diferentes de la isoterma de Langmuir. Se señala que, la linealización de datos se realiza más frecuentemente mediante la isoterma de Langmuir-1 y Langmuir-2, debido a que minimiza los errores del ajuste de la ecuación (Hamdaoui, 2007). La Tabla 1.5 muestra las diferentes formas propuestas de representar el ajuste lineal para comportamientos que siguen la ecuación de Langmuir. 37 Tabla 1.5 Modelo de la isoterma de Langmuir y sus formas lineales (Hamdaoui, 2007) A pesar de que se ha encontrado que la isoterma de Langmuir se ajusta para sistemas con quimisorción, hay quienes rechazan este modelo de isoterma, ya que sus deducciones no concuerdan con la realidad; por ejemplo, las superficies de la mayoría de los sólidos no son uniformes y la velocidad de desorción depende de la posición de la molécula adsorbida, además, existen evidencias claras que muestran que las moléculas adsorbidas pueden moverse a lo largo de la superficie (esta movilidad es mayor para aquellas moléculas fisisorbidas que para aquellas quimisorbidas) (Levine, 2004). Entonces, se han desarrollado distintas deducciones mecano-estadísticas de la isoterma de Langmuir. 38 1.7.1.2. Isoterma de Freundlich El físico Erwin Freundlich desarrolló otro tipo de isoterma para explicar el comportamiento de los sistemas de adsorción. La diferencia más marcada entre la isoterma de Langmuir y la de Freundlich es que en el segundo caso, su creador consideró la existencia de diferentes tipos de adsorción sobre la superficie del adsorbente, cada una con distinto calor de adsorción (Levine, 2004). Según estas aseveraciones, la isoterma de Freundlich se puede definir en la siguiente ecuación: Ecuación 3.4 Fórmula general de la isoterma de Freundlich (Levine, 2004). En la ecuación 1.4 “ϑ” representa la relación entre la cantidad de material adsorbido por masa de adsorbente, el término “k” indica la capacidad de adsorción del adsorbente y el término “n” es una constante indicativa de la intensidad de adsorción, la cual está restringida a valores mayores que la unidad, mientras que “C” es la concentración del absorbato en la solución. En líneas generales, los valores de n en el rango 2-10 representan una alta intensidad de adsorción, entre 12 la intensidad de adsorción es moderada y valores menores a 1 son característicos de una adsorción pobre (Ortega, 2008, Treybal, 1981). Si se toman logaritmos en ambos lados de la igualdad de la ecuación 1.4, se obtiene la siguiente expresión: Ecuación 4.5 Forma lineal de la isoterma de Freundlich (Levine, 2004). En la ecuación 1.5 se puede representar gráficamente como log (υ) versus logC, para obtener una recta cuyo intercepto será log k y la pendiente 1/n. La isoterma de Freundlich se aplica normalmente a la adsorción de los solutos de disoluciones líquidas sobre sólidos; sin embargo también cumple un comportamiento similar a las isotermas TIPO 1 (Levine, 2004). 39 1.8. Antecedentes de esta investigación Los polímeros han mostrado gran versatilidad en actividades de remediación de aguas contaminadas. Sus numerosas ventajas han originado una vasta corriente de estudios e investigaciones con el fin de entender, no sólo su eficiencia en procesos de remoción de compuestos orgánicos, sino los procesos fisicoquímicos que determinan estos comportamientos. Una serie de trabajos se avocan a dilucidar la cinética de los procesos de adsorción mientras que otra serie de investigaciones ahonda más en los procesos de adsorción como tal. Los primeros polímeros empleados como adsorbentes fueron desarrollados en 1960, originalmente con la finalidad de emplearlos en cromatografía de permeación de gel, sin embargo sus propiedades físicas sobresalientes dieron cabida a otros usos. Por razones ambientales, los adsorbentes poliméricos sintéticos se basan en matrices de ésteres poliacrílicos y poliestirenos modificadas (Pan, 2009). Hradil y col. (1991) emplearon tres familias de polímeros sintéticos para adsorber compuestos como nitrobenceno, fenol, anilina, p-cloroanilina, entre otros. En primer lugar emplearon tres tipos de copolímeros macroporosos de metacrilato, en segundo lugar un copolímero de estireno, divinilbenceno y ácido acrílico metil-ester (Wofatit Y-59), y por último un copolímero de estireno y divinilbenceno (Polysorb 40/100). Entre sus resultados demostraron que la adsorción de compuestos orgánicos de bajo peso molecular no sólo depende de la composición química del polímero sino de su morfología y estructura porosa. Además probaron que mientras menor sea el grado de entrecruzamiento de los polímeros (lo cual origina una menor superficie interna), los compuestos orgánicos se disuelven en el seno del polímero. En líneas generales el Wofatit Y-59 ofreció una mayor capacidad máxima de adsorción sobre todo de nitrobenceno, p-cloroanilina y fenol con valores de 3,31; 2,14 y 5,19 mmol/g de adsorbente respectivamente. Cohen y col. (1993) emplearon resinas entrecruzadas de poliacrilato (XAD-8), una resina hecha con matriz de poliestireno nitroso entrecruzada con divinilbenceno (XAD-12), copolímeros de poliestireno y divinilbenceno (XAD-4), una resina compuesta por divinilbenceno (XAD-16) y un polímero compuesto por una matriz de polivinilpiridina entrecruzada con divinilbenceno 40 (Reillex-425). Los adsorbatos fueron fenol, tolueno, clorobenceno, entre otros. Entre sus resultados más resaltantes está el hecho de que el hinchamiento de las resinas poliméricas, ya sea por el solvente o por el soluto, debe ser tomado en cuenta conjuntamente con sus propiedades físicas, área superficial, porosidad, etcétera, a la hora de realizar valoraciones en experimentos de adsorción. Además se encontró que en un mismo proceso con polímeros se puede dar tanto adsorción como absorción del contaminante en medio acuoso. En el caso de la adsorción del tolueno, en términos de monocapa, esta fue mayor para las dos resinas polares XAD-12 y Reillex-425, excediendo la monocapa sencilla (de concentración 1 mmol/L) y formando hasta 3 monocapas para Reillex-425 y siete monocapas para XAD-12 de concentraciones 4 mmol/L. Simko y col. (1999) emplearon PEBD para determinar el proceso de difusión de HAP seleccionados en agua. Ellos publicaron en la revista Food Chemistry que la concentración del analito decrece en el agua tan pronto como comienza el experimento y se puede observar que la adsorción cesa en la superficie del polímero a los 45 minutos. Luego de 1 hora y 30 minutos ya se puede medir la difusión de los HAP hacia el interior del polímero. Es importante mencionar que el proceso de adsorción fue llevado a cabo sólo con la influencia de la difusión del adsorbato hacia el adsorbente, es decir, sin agitación. En su publicación en la revista Reactive & Funcional Polymers, Jarabek y col. (2004) estudiaron la adsorción de benceno, tolueno y etilbenceno en copolímeros de estirenodivinilbenceno modificados para formar distintos niveles de entrecruzamiento. Se comprobó que no sólo tiene lugar el proceso de adsorción sino también el de absorción y como era de esperarse, mientras mayor es el grado de entrecruzamiento se favorece más el proceso de adsorción por la abundancia de poros estrechos en la superficie de la resina. Chen y col. (2006) estudiaron la adsorción de HAP seleccionados en PEBD, determinando que más de un 50% de la adsorción ocurre en las primeras 24 horas en las que el polímero está en contacto con la solución contaminada, llegando a la condición de equilibrio de adsorción al tercer día del experimento. Por último señalan que la adsorción de HAP en PEBD se ve favorecida porque estos compuestos orgánicos van de un medio altamente polar como el agua a uno no polar como lo es el PEBD, por lo cual siente mayor afinidad por este último. 41 Zhang y col. (2006) emplearon los adsorbentes macro-reticulados XAD-4 y NDA100 en la adsorción de 1-naftol y naftilamina. La capacidad de adsorción de ambos adsorbentes de base poliestirénica, fue comprobada mediante los modelos de isotermas de Langmuir y Freundlich obteniendo excelentes mejores resultados, basados en los valores de parámetros de adsorción, para la resina NDA100 que posee, a nivel estructural, mayor área superficial y volumen de poros. En la revista Journal of Hazardous Materials, Long y col. (2006) observaron la adsorción de fenol en las resinas XAD-4 y NDA103, resinas de base poliestirénica y de base poliestirénica aminada respectivamente. Sus resultados indican que, el polímero NDA103, a pesar de tener una menor área superficial que el XAD-4, es más eficaz en la adsorción de fenol (basado en la obtención de parámetros de adsorción de Langmuir y Freundlich, además de cálculos de entalpía del proceso a distintas temperaturas) por tener una sinergia entre el anillo aromático (que atrae al anillo aromático del fenol) y el grupo amino (en nitrógeno forma puentes de hidrógeno con el hidrógeno del grupo OH) del adsorbente. El trabajo de investigación realizado por Thomas y col. (2007) arrojó interesantes resultados. En su estudio sobre la absorción y difusión de benceno y tolueno en polimezclas no compatibilizadas de polietileno de baja densidad lineal-acetato de etilenvinilo se observó que mientras mayor era la fase de acetato de etilenvinilo, mayor capacidad de absorción tenía la polimezcla, debido a que el solvente tenía mayor fluidez por la flexibilidad que otorgaba la fase de ese polímero (que se hallaba en fase dispersa) a las cadenas en el interior de la polimezcla. Ceylan y col. (2009) emplearon poli-isobutileno y polipropileno para la absorción de tolueno en medio acuoso, pero de forma mecánica, estudiando la capacidad de hinchamiento del polímero. Ellos encontraron que la adsorción de tolueno para el primer polímero fue 11,5 g de adsorbato por g de adsorbente y para el segundo polímero fue 5,8 g de adsorbato por gramo de adsorbente. CAPITULO 2 PROCEDIMIENTO EXPERIMENTAL 2.1. Materiales En la preparación de las soluciones se utilizó agua destilada, con pH 6,2 y una conductividad de 18 mmho/cm. Dicha agua fue almacenada en una botella ámbar de 2,5 L, nueva, y siempre se mantuvo cerrada, de manera de evitar su contaminación con algún compuesto. Para realizar las soluciones de benceno y tolueno (soluciones de trabajo y patrones), se empleó los reactivos que se presentan en la tabla 2.1 Tabla 2.1 Reactivos empleados Reactivo Diclorometano (DCM) Metanol Benceno Tolueno Fluorobenceno Trifluorotolueno Sulfato de sodio anhidro Cloruro de sodio anhidro Proveedor Sigma Aldrich Burdick & Jackson Riedel – de Haën Riedel – de Haën Chem Service (Lote 132-42A) Chem Service (Lote 151-18B) Riedel – de Haën Riedel – de Haën Características 99,9% de pureza 99,9% de pureza Grado analítico Grado analítico Patrones certificados Patrones certificados Grado analítico Grado analítico Los polímeros empleados en los experimentos de adsorción, que fueron concebidos como material de desecho, se presentan en la tabla 2.2. 43 Tabla 2.2 Polímeros usados 2.2. Polímero Abreviación Polietileno de baja densidad PEBD Poliestireno PS Polimezcla Polietileno de baja densidad/Poliestireno PEBD-PS Proveedor Material de desecho de Laboratorio GPUSB Cajas de “compact discs” de desecho Preparado en Laboratorio GPUSB Procedimiento Experimental A continuación se describen los procedimientos para preparación de la polimezcla PEBD-PS, la caracterización de los homopolímeros PEBD, PS y la polimezcla utilizada, la cuantificación de benceno y tolueno en solución acuosa y la posterior realización de las cinéticas e isotermas de adsorción de benceno y tolueno en los adsorbentes seleccionados. 2.2.1. Preparación de la polimezcla PEBD-PS En la preparación de la polimezcla PEBD-PS se pesó aproximadamente 25 g de cada homopolímero en un beaker de 100 mL. Se realizó un mezclado en el beaker para garantizar la homogeneidad. La mezcla se añadió lentamente a la alimentación del mezclador “Miniextrusora ATLAS LME” del Laboratorio del Grupo de Polímeros de la Universidad Simón Bolívar (GPUSB), que cuenta con un sistema de termocuplas calentadoras y flujo en contracorriente para lograr la fundición y mezcla adecuada de los polímeros en la proporción deseada. Las condiciones del proceso fueron una temperatura de 210°C (tanto en el cabezal como en los tornillos ubicados en la cámara de mezclado) y a una velocidad de 40 rpm. El proceso se realizó en dos partes, una con 10 g de de la mezcla en la alimentación y, luego que se retiró el producto por la boquilla de salida del mezclador, otra porción con 10 g de la mezcla, para obtener una polimezcla homogénea (1:1) de PEBD-PS. Los tres polímeros fueron moldeados en una prensa, a cierta temperatura, para obtener láminas uniformes de aproximadamente 0,01 milímetros de espesor. 44 2.2.2. Caracterización del material polimérico. A continuación se describen las técnicas y equipos empleados en la caracterización del material polimérico empleado en esta investigación. 2.2.2.1. Espectroscopía infrarrojo por transformada de Fourier (FT-IR) Es una herramienta útil para realizar el análisis y caracterización de polímeros. Considerando las vibraciones los grupos funcionales, la FT-IR permite elucidar la estructura química del polímero. La radiación infrarroja se encuentra en un intervalo de 4000-400 cm-1, está radiación será absorbida por las moléculas a la longitud de onda específica dependiendo de su masa relativa, la fuerza entre los enlaces y geometría de los átomos (Areiza, 2002). Para los análisis FT-IR del homopolímero PEBD se obtuvo una película delgada, transparente bajo fusión a cierta temperatura en un portaobjeto; mientras que el homopolímero PS se disolvió en cloroformo y se colocó en una cápsula de petri de forma tal que, al evaporarse el solvente, quedó la fase PS en forma de película muy delgada. Dichas muestras fueron analizadas en el espectrofotómetro infrarrojo FT-IR marca “Bruker FT-IR-27” ubicado en el Laboratorio de Química Instrumental de la Universidad Simón Bolívar. 2.2.2.2. Calorímetría diferencial de Barrido (CDB) La calorimetría diferencial de barrido es un método utilizado para medir la energía producida o absorbida, monitoreando la diferencia de energía suministrada entre una muestra con respecto a una referencia, en función de la temperatura. La absorción de energía es producida por una reacción endotérmica, mientras que la generación de energía es producto de una reacción exotérmica. La CDB ha sido el método más ampliamente usado de todos los métodos térmicos para estudios de fusión, cristalización, curado, transición vítrea, entre otros (Silverstein 1974, Rosato, 2000). Se conocen dos variantes: la CDB de potencia compensada, donde la muestra y el material de referencia se calientan de manera separada aunque sus temperaturas se mantienen iguales 45 mientras se van aumentando o disminuyendo linealmente. Por otra parte, en la CDB de flujo de calor se mide la diferencia en cantidad de calor que fluye hacia la muestra y hacia la referencia cuando la temperatura de la muestra se aumenta o disminuye linealmente (Skoog, 2001). Los ensayos de CDB se realizaron en un equipo marca “Perkin-Elmer Modelo DSC 7”. La preparación de las muestras se llevó a cabo pesando 5,00 mg del homopolímero PEBD y de la polimezcla PEBD-PS en una balanza marca “Mettler Toledo Modelo A0249”. Luego de calibrar el equipo y obtener la línea base correspondiente, se procedió a realizar un barrido de calentamiento desde e 0 a 170ºC a razón de 40ºC/min, permaneció por 3 minutos en 170ºC para luego pasar desde 170ºC a 25ºC (la disminución fue a razón de 20ºC/min.) y por último desde 25ºC a 170ºC. 2.2.2.3. Microscopía Electrónica de Barrido La microscopía electrónica de barrido (MEB) es una técnica empleada con frecuencia en el campo de los polímeros para el estudio de su morfológico. Esta técnica es importante para la caracterización de la superficie de los sólidos, estructuras porosas, etc. La información se obtiene con resoluciones mayores a la microscopía óptica. La MEB permite observar la morfológica y topografía sobre la superficie de sólidos que normalmente es necesario para entender su comportamiento y propiedades (Skoog, 2001). Las láminas obtenidas por moldeo del homopolímero PEBD y de la polimezcla PEBD-PS fueron fracturadas criogénicamente en nitrógeno líquido, y posteriormente se recubiertas con una fina capa de oro (para otorgarles densidad electrónica necesaria para poder ser observadas) en un equipo de metalización marca “Balzars-SCD 030”. El equipo utilizado fue un microscopio electrónico de barrido, marca “JEOL”, modelo “JSM6390”, que se encuentra en el laboratorio de Superficies de la USB. El voltaje del equipo fue regulado 15 y 25 kV para los análisis. 46 2.2.2.4. Método Brunauer-Emmett-Teller (Método BET) El método de adsorción de BET es el método más comúnmente utilizado para la determinación del área superficial de materiales porosos. En este caso se supone que un gas como el nitrógeno, al licuarse y adsorberse sobre superficies sólidas limpias llenará la misma en su totalidad formando capas múltiples. La isoterma de adsorción BET se indica en la siguiente ecuación: Ecuación 2.1 Cálculo de la isoterma BET El término “n” corresponde a la cantidad (volumen) de gas adsorbido a una presión de trabajo “P”; “P0” indica la presión de saturación o presión de vapor del gas licuado a la temperatura de adsorción y “c” es la constante relacionada exponencialmente con la energía de adsorción de la capa adsorbida (Brunauer, 1838). El procedimiento se basa en determinar el valor del área superficial correspondiente a la medida por adsorción e idéntica a la de desorción. Una vez se tiene este valor se divide por el peso de muestra para determinar el área superficial específica. El valor utilizado para la determinación de la velocidad corresponde a la media del área superficial específica a partir de distintas cantidades iniciales de sólido (Brunauer, 1938). Las medidas de fisiadsorción de nitrógeno en la superficie de los hompolímeros PEBD, PS y en la polimezcla PEBD-PS se realizó en un equipo marca “Micromeritics”, modelo “ASAP 2000”, ubicado en el Laboratorio de Carbón de La Universidad Simón Bolívar. Para desalojar cualquier especie adsorbida, los sólidos fueron sometidos a un tratamiento previo con vacío a 25ºC. 2.2.3. Microextracción en fase líquida. Se utilizó material de vidrio común de laboratorio: balones aforados de 5, 10, 25, 100, 250 y 500 mL, viales con tapas de rosca de 20 mL, viales con tapa a presión de 2 mL, jeringas “gas- 47 tight” de 500 µL y 5 mL, pipetas volumétricas 2 y 15 mL. Además se utilizaron equipos usuales de laboratorio tales como balanza, mufla, agitador mecánico “shaker”, taladro, etcétera. Este metodología de extracción se tomó de la USEPA (Método 3511: Microextracción de compuestos orgánicos en agua), con ligeras modificaciones. Se trabajó con viales de 20 mL de vidrio, con tapas de rosca de plástico perforadas en el centro y un septum de PTFE colocado en su interior para asegurar el hermetismo en el vial y que la muestra no estuviese en contacto con el plástico de la tapa. En un primer paso se tiene la muestra inicial acuosa con el vial lleno hasta el tope, de manera de no tener un espacio vacío entre el líquido y la tapa. En el segundo paso se añade 120 µL de fluorobenceno, para que en un tercer paso se añada aproximadamente 5 g de NaCl anhidro y exactamente 2,00 mL de DCM. Se agita el vial por espacio de 5 minutos hasta que la sal añadida se disuelve. En un cuarto paso se extrae del vial, con una jeringa “gas-tight”, aproximadamente 1,5 mL de la fase orgánica (previamente separada de la fase acuosa), y se coloca en un vial de 2 mL que contiene aproximadamente 50 mg de Na2SO4 anhidro, para secar las trazas de agua que pueda tener el extracto. El quinto y último paso consiste en extraer, del vial de 2 mL, exactamente 1 mL, con una jeringa “gas-tight” y añadir 300 µL de trifluorotolueno. Al cerrar el vial con el extracto final, el mismo debía estar correctamente identificado y ser refrigerado a unos 4°C. La validación de un método analítico es la confirmación, a través de un examen y el aporte de evidencias objetivas, del cumplimiento de los requisitos particulares para un uso específico previsto. Es un requisito importante en la práctica de análisis químico, sobre todo a nivel de laboratorios comerciales y de investigación. La microextracción en fase líquida se validó mediante los parámetros de linealidad, límite de detección, límite de cuantificación, porcentajes de recuperación, precisión, exactitud. Para evaluar la linealidad, el procedimiento consistió en realizar, a partir de una solución madre de 10000 ppm de benceno y tolueno en metanol, una dilución de la misma en agua hasta alcanzar 100 ppm. A partir la de solución de 100 ppm de benceno y tolueno, en balones aforados de 5 mL, se preparó cada patrón. El trifluorotolueno (estándar interno) se preparó diluyendo 0,840 mL a una concentración de 2000 ppm en un balón aforado de 25 mL en metanol para obtener una 48 solución (67,20 ± 0,03) ppm. Por último el fluorobenceno “surrogate”, también calibrado, se preparó diluyendo 1 mL a una concentración de 2000 ppm en un balón aforado de 25 mL en metanol para obtener una solución 80 ppm. El “surrogate” es un compuesto similar al analito en composición química y comportamiento en el proceso analítico, que normalmente no se halla en muestras naturales. A continuación, en la tabla 2.3, se muestra de forma detallada la formulación de cada solución patrón de benceno y tolueno: Tabla 2.3 Patrones de benceno y tolueno Patrón A1 A2 A3 A4 A5 BTEX Cantidad Sol. Madre 100 ppm (±0,0001) ppm (±10) µL 1,0000 50 2,0000 100 5,0000 250 7,0000 350 10,0000 500 Estándar Interno (±10) µL 350 Surrogate (±10) µL 130 190 250 315 450 La cromatografía es una técnica que permite al experimentador determinar y separar los componentes de una mezcla compleja a partir de sus propiedades físicas (volatilidad, punto de ebullición) y su interacción con una fase móvil y con una fase estacionaria. Se usan los tiempos de retención (tiempo en el cual un compuesto recorre la columna hasta que alcanza el detector del equipo) como medida para obtener las características del cromatógrafo que tienen que ver con la eficacia del análisis (Skoog, 2001). La cromatografía de gas consiste, a grosso modo, en un gas portador, un sistema de inyección de muestra, una columna empacada, horno termostato y detector (Skoog, 2001). Los análisis de contaminante en medio acuoso se realizaron el un cromatógrafo de gases con detector de ionización a la llama (GC-FID) marca Hewlett Packard 5890 Series II, con una columna RTX-5 de 30 metros de longitud (0,25 mm. i.d. y 0,25 µm de espesor), de composición 5% difenil/95% polidimetilsiloxano y que emplea nitrógeno como gas portador; ubicado en el Laboratorio de Química Ambiental de la Universidad Simón Bolívar. 49 Las condiciones cromatográficas para lograr la separación óptima de cada componente de los patrones se presenta en la tabla 2.4: Tabla 2.4 Condiciones cromatográficas para benceno y tolueno Condiciones Temperatura inicial (°C) Grado de calentamiento (°C/min) Temperatura del inyector (°C) Temperatura del detector (°C) Tiempo inicial (min) Flujo de gas portador (mL/min) Presión de la cabeza de la columna (psi) Valores 30 3,0 150 200 2,00 30 17 El procedimiento para evaluar los parámetros límite de detección, límite de cuantificación, precisión y exactitud del método consistió en preparar una solución de concentraciones de benceno y tolueno conocidas y realizar cinco extracciones para analizar los parámetros antes mencionados. Se calculó el promedio de las concentraciones obtenidas, la desviación estándar (desviación estándar relativa) y de acuerdo al número de extracciones realizadas se calculó el límite de detección del método con un intervalo de confianza del 99%; todo lo mencionado anteriormente con las ecuaciones que se presentan a continuación. Ecuación 2.2 Promedios de una serie de muestras Ecuación 2.3 Variancia Ecuación 2.4 Desviación estándar Ecuación 2.5 Desviación estándar relativa 50 Ecuación 2.6 Límite de detección del método Ecuación 2.7 Límite de cuantificación 2.2.4. Elaboración de blancos Para descartar las pérdidas por volatilización de benceno y tolueno en las muestras acuosas durante la realización de los experimentos (cinética e isoterma de adsorción), se realizó el siguiente procedimiento. En un balón aforado de 250 mL se preparó soluciones con concentraciones conocidas de benceno y tolueno en agua. Luego de llenar cada vial previamente identificado con cada solución (lleno hasta el tope), se midió la concentración del hidrocarburo correspondiente en agua, con la microextracción en fase líquida, en los siguientes tiempos: (0; 10; 60, 90) minutos, (24, 168) horas, con agitación constante. 2.2.5. Experimentos de cinética de adsorción En un balón de 250 mL se preparó soluciones de benceno y de tolueno en agua. Luego de llenar cada vial previamente identificado con cada solución (lleno hasta el tope sin espacio entre la fase líquida y el septum), a cada solución se le agregó 30 mg de cada polímero por separado. Se midió las concentraciones en agua, luego de retirar los polímeros, en los siguientes tiempos de contacto con agitación constante: (0; 45; 90) minutos, (3, 7, 24, 48 y 72) horas. 2.2.6. Experimentos de isotermas de adsorción En un balón de 250 mL se preparó soluciones de benceno y de tolueno en agua. Luego de llenar cada vial previamente identificado con cada solución (lleno hasta el tope sin espacio entre la fase líquida y el septum), a cada solución se le agregó 30 mg de cada polímero por separado. Se midió las concentraciones en agua, luego de retirar los polímeros, con agitación constante, en el tiempo aproximado de equilibrio hallado con los experimentos de cinética de adsorción. 51 2.2.7. Tratamiento de los desechos generados en la experimentación Como parte del procedimiento experimental, los desechos generados debían ser tratados para su disposición final adecuada. El procedimiento empleado para el tratamiento de desechos de benceno y tolueno generados fue la Oxidación Fenton, en la cual se añade peróxido de hidrógeno (H2O2) y sulfato de hierro (FeSO4) en una relación molar con el contaminante presente en el agua de 1:4:1 respectivamente. Esta solución se acidifica hasta pH 2-3 de manera de maximizar la formación de radicales OH. y se agita por cierto tiempo. Por último se añade cal hidratada para que la solución aumente su pH y precipite el oxido férrico, asegurando la total degradación del compuesto orgánico. Luego de verificar la presencia de los contaminantes en el agua y obtener una prueba negativa, los desechos podían ser dispuestos apropiadamente por la cañería, sin riesgo alguno. CAPITULO 3 RESULTADOS Y DISCUSIÓN Al ser completados los experimentos necesarios para la verificación de los objetivos planteados, y siguiendo la metodología mencionada anteriormente, a continuación se muestran los resultados obtenidos y su respectiva discusión. Primeramente se muestra la caracterización del material polimérico empleado en esta investigación, para luego discutir los resultados obtenidos en la calibración del método empleado en las mediciones de contaminantes en agua. Por último se hablará de la cinética de adsorción y de las isotermas de adsorción y el por qué del comportamiento obtenido. 3.1. Espectroscopía infrarrojo por transformada de Fourier (FT-IR) El análisis FT-IR de los polímeros usados se llevó a cabo con la finalidad de caracterizarlos y compararlos con FT-IR teóricos de dichos polímeros, de manera de determinar la presencia de algún grupo funcional en la estructura química de los polímeros empleados que pudiese influir en los procesos de adsorción. Los espectros FT-IR se presentan en el apéndice A. La tabla 3.1 muestra las bandas de absorción en los espectros FT-IR realizados de PEBD y de PS., entre 4000-400 cm-1. 53 Tabla 3.1 Bandas de absorción en el espectro FT-IR de los polímeros utilizados (Workman, 2000) Polímero PEBD PS Grupo Funcional Longitud de onda obtenida (cm-1) Longitud de onda teórica (cm-1) Estiramiento (simétrico y asimétrico) de enlaces C-H de metilos 2936, 730 (2935, 2909, 2854, 2846), 720 Vibraciones de deformación simétrica de enlaces C-H de metilos 1472 1464 Vibraciones de los enlaces C-H en las ramificaciones (1377, 1303) (1376-1304) Estiramiento de enlaces C-H de aromáticos 3082-3026 3100-3003 Estiramiento de enlaces C-H de metilos 2926-2850 2923-2849 Vibraciones de enlaces C=C aromáticos (multiplete) 1944-1750 1943-1746 Vibraciones de enlaces C-C aromáticos 1601-1453 1600-1450 Vibraciones de enlaces C-H aromáticos fuera del plano 755, 699 756, 699 En principio, las longitudes de onda de los espectros obtenidos y las longitudes de onda reportadas en la teoría son similares, lo cual indica que efectivamente se trabajó con PEBD y PS, a pesar de ser material de residuo. En el caso de PEBD es de notar que, para el estiramiento simétrico y asimétrico de los enlaces C-H de los metilos, no se nota las distintas bandas (2935, 2909, 2854, 2846) cm-1 reportadas en la literatura, y esto se debe a que la película realizada para la elaboración del espectro no era lo suficientemente delgada, lo cual se reflejó en una saturación de la señal. Cabe destacar también que el ancho de la banda en 720 cm-1 puede ser un indicador de desordenes en la región cristalina del polímero; en este caso esta banda es atribuida a la región amorfa (Takahashi, 2001). Para el caso del PS, las bandas obtenidas concuerdan con las bandas reportadas en la literatura (Workman, 2000). Los espectros teóricos de PEBD y PS se pueden apreciar en el Apéndice A. 54 3.2. Calorimetría diferencial de barrido (CDB) La CDB se realizó con la finalidad de estudiar la morfología en cuanto a la relación orden/desorden de las cadenas poliméricas por acción de gradientes de temperatura, al ocurrir los procesos de fusión y cristalización. En las figuras 3.1 y 3.2 se presentan los termogramas de fusión y cristalización del PEBD. Figura 3.1 Termograma de fusión del PEBD 55 Figura 3.2 Termograma de cristalización del PEBD En el termograma de fusión y de cristalización del PEBD puede servir como de punto de referencia para estudiar la cristalización y fusión del PEBD de la polimezcla, de manera de evidenciar algún cambio sobre dichos procesos. Esto puede servir de base para afirmar el nivel de desorden que pudo provocar la inclusión de las cadenas de PS en el PEBD. Con los datos presentados en las figuras 3.1 y 3.2 se puede calcular el grado de cristalinidad (Xc) del homopolímero PEBD usado en esta investigación. En la tabla 3.2 se presenta el cálculo de Xc del PEBD, de acuerdo a los parámetros obtenidos de fusión y cristalización del PEBD homopolímero y tomando como punto de referencia el valor de la entalpía de fusión del PEBD 100% cristalino (69 cal/J que equivalen a 288,69 J/g) (Peacock, 2000). Tabla 3.2 Grado de cristalinidad del PEBD utilizado PEBD Cristalización Fusión T(°C) 91,97 110,7 ∆H (J/g) 117,99 123,32 Xc (%) 41,88 En las figuras 3.3 y 3.4 se muestran los termogramas de fusión y cristalización del PEBD-PS. 56 Figura 3.3 Termograma de fusión del PEBD-PS Figura 3.4 Termograma de cristalización del PEBD-PS 57 De la misma manera, con los parámetros obtenidos del proceso de fusión y cristalización de la polimezcla, se puede obtener el valor de Xc. De nuevo se toma como referencia el valor mencionado anteriormente para PEBD 100% cristalino. La tabla 3.3 muestra el cálculo de Xc para el constituyente de la polimezcla PEBD-PS. Tabla 3.3 Grado de cristalinidad del PEBD matriz de la polimezcla PEBD-PS PEBD Cristalización Fusión T(°C) 96,63 110,03 ∆H (J/g) 43,17 43,34 Xc (%) 14,98 Comparando la fusión del PEBD y de la polimezcla, no se observan cambios significativos en lo que es la temperatura de fusión (Tf), sin embargo si se evidencian cambios en los valores de la temperatura de cristalización (Tc). Esta variación de la Tc se puede atribuir al hecho que la fase PS es más viscosa y por lo tanto tiende a segregarse como microesferas (por la acción del corte o cizallamiento) que se dispersan en la fase menos viscosa de PEBD. Esto hecho puede permitir que, durante el enfriamiento de la polimezcla en el CDB, las cadenas de PEBD aprovechan la superficie de las microesferas de PS (que en esa temperatura aproximada se encuentra en estado sólido por su temperatura de transición vítrea), conduciendo así a que el proceso de cristalización del PEBD inicie a una temperatura mayor. Esto indica que, en presencia de PS, el PEBD sufre un proceso de nucleación heterogénea (proceso de iniciación de la cristalización en presencia de cuerpos que se hallan en estado sólido a la temperatura de cristalización) (Peacock, 2000). Evidentemente, la reducción el grado de cristalinidad implica un menor costo energético de dicho proceso, por lo cual el valor de ∆Hc pasa de 117,99 J/g en el homopolímero PEBD a 43,17 J/g. En cuanto al grado de cristalinidad, los valores de ∆Hc y ∆Hf permiten observar que la cristalinidad del homopolímero PEBD se redujo exactamente en 2,79 veces su valor inicial. Esto indica que en la polimezcla se tiene una fase de PEBD más amorfa (aumento de la relación desorden/orden) que podría facilitar la difusión del adsorbato, como se verificará más adelante. La figura 3.5 muestra como cambió el homopolímero PEBD en el aumento de su relación desorden/orden de cadenas al cristalizar con la fase de PS incluida y no compatibilizada. 58 Figura 3.5 Esquema de a) PEBD semicristalino b) PEBD-PS, disminución del grado de cristalinidad original 3.3. Microscopia electrónica de barrido (MEB) En el campo de los polímeros, es necesario precisar técnicas capaces de ofrecer una visión detallada de la morfología de los polímeros, a nivel microscópico. Así pues, las técnicas de microscopía óptica y electrónica son ampliamente utilizadas para el estudio de la morfología y la estructura de material polimérico. Los procesos de adsorción en material polimérico, y más específicamente a nivel morfológico, se llevan a cabo gracias a la flexibilidad que poseen las cadenas poliméricas, que permiten que las moléculas de adsorbato penetren en la masa del polímero dando lugar a procesos de adsorción y absorción. La adsorción se produce en la interface entre la fase polimérica y el fluido, siendo energéticamente más eficiente que la absorción, debido a que parte de la energía de la interacción adsorbato-polímero se consume para expandir la red polimérica (Jarabek, 2004). En la figura 3.6 se presentan todas las imágenes MEB del homopolímero PEBD empleado en esta investigación. Se puede notar que, en el frente de fractura, su morfología poco porosa. Es sabido que el PEBD posee en su estructura química ramificaciones aleatorias y con cadenas alifáticas de distinta longitud, lo cual le otorgan un peso molecular promedio menor a 50000 (Brydson, 1999). Dichas ramificaciones aleatorias originan una pequeña condición amorfa del PEBD, sin embargo se considera un polímero semicristalino; esto no puede apreciarse mediante MEB, sin embargo si se nota que la superficie de fractura es continua y en un plano. 59 Figura 3.6 MEB del PEBD En la figura 3.7 se observan las distintas micrografías del PEBD-PS. En el caso de la polimezcla PEBD-PS, se distingue una matriz (PEBD) y fases dispersa (PS), a pesar de que ambos polímeros están en proporción 1:1. Es importante mencionar que no se debe hablar de microfases dispersas en una matriz primaria, debido a que la polimezcla se realizó en iguales proporciones. Figura 3.7 MEB del PEBD-PS En este caso particular se puede mencionar que la mezcla es co-continua, pero entre el PS y PEBD existe una gran diferencia de viscosidad (PS>PEBD) y por ende el polímero viscoso tiende a formar gotas que son rodeadas por el polímero menos viscoso. Por este hecho Se observa como el PS 60 provoca que la superficie del PEBD pierda su uniformidad, formando especies de volúmenes libres aleatorios y de distintos tamaños, con cierta forma circular. A pesar de este hecho, no se distinguen interconexiones entre dichos volúmenes libres formados en la polimezcla, lo cual sugiere que la interacción física entre ambos polímeros fue muy pobre, demostrando a su vez la incompatibilidad de las fases. En la figura 3.8 se observa una porción de la polimezcla tratada con cloroformo; solvente totalmente selectivo para solubilizar el PS. Figura 3.8 MEB de PEBD-PS tratado con cloroformo Se puede apreciar que la lámina de la polimezcla se fue debilitando (en cuanto a rigidez) hasta quedar prácticamente como una lámina de PEBD. Sabiendo que esta poliolefina no es soluble en cloroformo, se asegura que la fase dispersa observadas en la figura 3.2 es PS, pues dichas partículas fueron disueltas por el solvente usado. Este comportamiento puede evidenciarse con bases en la literatura. Harrats y colaboradores realizaron polimezclas de PEBD-PS (80:20), sin compatibilizar, obteniendo que, luego de un tratamiento con tetrahidrofurano como marcador de PS, esta fase destacó en las micrografías 61 mostrando partículas amorfas de distintos tamaños (Harrats, 2002). En la figura 3.9 se muestra la micrografía obtenida en dicha publicación. Figura 3.9 MEB del PEBD-PS (80/20) en THF (Harrats, 2002). También Chen y colaboradores en 2002 realizaron polimezclas de PEAD-PS (80/20) sin compatibilizantes. En la figura 3.10 se evidencia el mismo comportamiento, una matriz de PEAD y una fase dispersa de PS en forma de semiesferas, demostrando que no hay adhesión ya que esa fase dispersa produce cavidades en la matriz (Chen, 2002). Figura 3.10 MEB del PEAD-PS (80/20) (Chen, 2002) La tabla 3.4 presenta los valores promedio de los volúmenes libres en el PEBD de la polimezcla, calculados a partir de las micrografías presentadas en la figura 3.2 y suponiendo que 62 las cavidades o volúmenes libres son perfectamente circulares, se puede obtener una estimación del diámetro de las mismas. Tabla 3.4 Tamaño de los volúmenes libres de la polimezcla PEBD-PS Tamaño promedio de los volúmenes libres (µm) Desviación estándar (±) 28 12 El diámetro promedio de dichas cavidades indica que estamos tratando con macroporos, pues su longitud es superior a 200 nm (Crittenden, 2005). 3.4. Método BET El análisis del área superficial de los polímeros puede servir para explicar la capacidad de adsorción del material que se está usando. En este caso nos proporciona cierta información que podría explicar porque un polímero adsorbe más que otro. A continuación la tabla 3.5 muestra las áreas superficiales calculadas para cada polímero, mediante el método BET, considerando que dichos análisis fueron realizados a partir de láminas de los mismos (obtenidas por moldeo y compresión). Tabla 3.5 Área superficial BET Muestra PEBD PS PEBD-PS Área (m2/g) 7,0±0,3 4,3±0,6 5,1±0,2 Con estos valores y la cantidad de moléculas adsorbidas en la monocapa de cada polímero, se puede comparar cuantas moléculas se adsorbieron frente a la cantidad de moléculas que el área superficial permite. De este hecho hablaremos más adelante. 63 3.5. Validación para la microextracción en fase líquida En el caso de la evaluación de la linealidad, por ser análisis cromatográficos de inyección directa de muestra en un solvente, se realizó una curva de calibración que consiste en introducir en el instrumento de análisis varios patrones que contienen concentraciones del analito exactamente conocidas. Al registrarse las señales instrumentales se corrigen con un blanco y los datos obtenidos se representan en una gráfica con la finalidad de obtener un comportamiento lineal. El método de estándar interno (E.I) consiste en añadir una sustancia determinada a todas las muestras, blancos y soluciones de calibrado en una cantidad fija. En lo que a calibración se refiere, se representará gráficamente el cociente resultante entre la señal del analito y la señal de estándar interno en función de la concentración del analito en los patrones; para las muestras como tal, dicho cociente se emplea para determinar la concentración del analito a partir de la curva de calibración. Una elección adecuada del E.I puede compensar algunos errores aleatorios y sistemáticos, ya que la señal del analito y del E.I tendrá una respuesta proporcional al error aleatorio instrumental y a las fluctuaciones del método, debido a que la relación entre dichas señales es independiente. De igual manera, si ambas señales se modifican por efectos de la matriz, también se compensan en ambas dichos efectos (Skoog y Holler, 2001). La mayor dificultad para aplicar el método de E.I es encontrar la sustancia adecuada que sirva para determinados análisis (incorporación a la muestra y a los patrones de forma reproducible). Básicamente el E.I debe dar una señal similar a la del analito en la mayoría de los casos, pero lo suficientemente diferente como para que ambas señales sean diferenciables por el instrumento; además se debe asegurar la ausencia de E.I en la matriz de la muestra, de tal forma que su única procedencia sea la cantidad conocida que se añadida (Skoog y Holler, 2001). La tabla 3.6 con los componentes de los patrones de benceno y tolueno: 64 Tabla 3.6 Componentes y señales de los patrones de benceno y tolueno Patrón Compuesto A1 A2 A3 A4 A5 Tolueno Benceno Surrogate E.I Tolueno Benceno Surrogate E.I Tolueno Benceno Surrogate E.I Tolueno Benceno Surrogate E.I Tolueno Benceno Surrogate E.I Areas 1172,2 1032,5 1315,1 3690,6 2681,3 2179,5 2054,7 4113,8 6409,1 5393,1 3127,2 3972,7 8325,1 7364,4 4079,7 3978,6 11853,4 10993,1 5195,7 4091,1 Benceno y Tolueno (0,01± ppm) E.I (0,01± ppm) Surr (0,01± ppm) 1,00 2,08 2,00 3,04 5,00 4,70 4,00 7,00 5,12 10,00 7,20 En la Tabla 3.6 se puede ver como a cada patrón se le añadió el compuesto “surrogate” (fluorobenceno) para ser calibrado conjuntamente con el benceno y tolueno para poder ser cuantificado en las muestras. El “surrogate” es un compuesto que, en las muestras reales, es añadido en una concentración conocida para sufrir por todos los procesos de extracción (reconcentración) del analito y así cuantificar la recuperación del método como tal. A continuación se muestran la tabla 3.6 con los parámetros empleados para la realización de las curvas de calibración de benceno, tolueno y “surrogate” 65 Tabla 3.7 Parámetros de calibración de benceno y tolueno Patrón A0 A1 A2 A3 A4 A5 A benceno/A E.I 0 0,2798 0,5298 1,3576 1,8510 2,6871 C (bencenoTolueno)/C E.I 0 0,2126 0,4252 1,0629 1,4881 2,1259 A Tolueno/A ASurrogate/A C Surrogate/C E.I E.I E.I 0 0 0 0,3176 0,3563 0,4422 0,6518 0,4995 0,6463 1,6133 0,7872 0,8503 2,0925 1,0254 1,0884 2,8973 1,2700 1,5306 En las figuras 3.11, 3.12 y 3.13 se presentan las curvas de calibración para benceno, tolueno y “surrogate”. Figura 3.11 Curva de calibración de benceno Figura 3.12 Curva de calibración de tolueno 66 Figura 3.13 Curva de calibración de surrogate Se observa excelente linealidad en el rango de concentraciones trabajadas para las curvas de calibración de cada compuesto. Los cromatográmas de los patrones de benceno y tolueno se presentan en el Apéndice B En lo que es la evaluación de la precisión, exactitud, % de recuperación, limites de detección y cuantificación, se realizó, una extracción de cada analito por separado, a una concentración conocida para evaluar todos estos parámetros. Para calcular los límites de detección y cuantificación, además de la precisión (desviación estándar relativa) se empleó las ecuaciones 2.2 a 2.7. De esta forma se puede estimar la concentración inicial a emplear en cada experimento de manera que el error del método no sea significativo a la hora de expresar los resultados finales de concentraciones de analito en la matriz acuosa. Para el caso del benceno y del tolueno se trabajó con soluciones de (1,50 ± 0,01) ppm y (1,00 ± 0,01) ppm respectivamente. Se realizó cada extracción (5 veces para cada analito) para evaluar la repetitibilidad de los resultados. En la tabla 3.8 se muestran los porcentajes de recuperación de cada analito y del “surrogate” para cada extracto realizado: 67 Tabla 3.8 Porcentajes de recuperación de benceno y tolueno Extracto Benceno (ppm) Tolueno (ppm) Surrogate (ppm) % Benceno % Tolueno % Surrogate A 1,52800757 1,01475911 0,56071333 102 102 94 B 1,4859222 0,96023735 0,59304522 99 96 99 C 1,51353844 0,99742056 0,5932541 101 99 99 D 1,53259671 0,99931858 0,59362709 102 100 99 E 1,47554691 1,00467546 0,57703277 98 99 96 En la tabla 3.8 se observa la exactitud del método, ya que partiendo de 1,5 y 1 ppm exactos de solución de benceno y tolueno respectivamente, las concentraciones obtenidas experimentalmente fueron similares a la concentración teórica. Además se obtuvo porcentajes de recuperación, siempre superiores a 96%, lo cual indica porcentajes de recuperación de los analitos en la matriz acuosa similares a los reportados en la USEPA para estas concentraciones (93-101% para benceno y 106-109% para tolueno). Es importante destacar los altos porcentajes de recuperación del “surrogate”, indicativo del porcentaje de extracción del método y por ende de su eficacia para la cuantificación de COV. La tabla 3.9 presenta los valores de los parámetros de validación indicados en las ecuaciones 2.2, 2.3, 2.4 y 2.7 para obtener los siguientes valores: Tabla 3.9 Límite de detección del método Compuesto S2 S (ppm) RS (%) MDL (ppm) MQL (ppm) Benceno Tolueno 0,0006 0,0004 0,03 0,02 3 2 0,1 0,1 0,3 0,3 68 El límite de detección del método para benceno y tolueno, con un intervalo de confianza del 99% fue de 0,1 ppm y el límite de cuantificación fue 0,3 ppm. Este último valor sirve de punto de partida para conocer las concentraciones más apropiadas para realizar los experimentos. Por ejemplo, si se inicia a partir de 1 ppm de concentración de benceno, el error del método es el 30% de la medida (0,3 ppm), lo cual indica que al momento en que la concentración descienda como consecuencia de la sorción en el polímero nuestra medida será menor pero con un porcentaje de error mucho más alto. Además, en cuanto a precisión, la desviación estándar relativa arrojó un valor de 3% y 2% para 1 ppm, lo cual refleja que los resultados son sumamente repetibles y precisos; más aun si se aumenta la concentración inicial de trabajo. 3.6. Cinética de adsorción Para realizar las isotermas de adsorción primero se debe conocer cómo se comporta el adsorbato en presencia del material adsorbente en función del tiempo. En el caso de la adsorción-desorción en polímeros, el problema principal para su optimización requiere no sólo un conocimiento del equilibrio de sorción sino de la cinética de sorción, que es la base de todo el proceso conjunto (Hradil, 1991). Se hacen estudios de la cinética de adsorción con la finalidad de conseguir el tiempo aproximado que muestre una tendencia de equilibrio en el cual el adsorbente se satura de adsorbato presente en el medio. Una vez obtenido el tiempo aproximado de equilibrio de adsorción, se pueden realizar los experimentos de isotermas para estudiar el tipo de proceso que se lleva a cabo. Vale destacar que este estudio cinético es más cualitativo que cuantitativo, ya que se desea observar una tendencia del proceso de adsorción en ese preciso espacio de tiempo. En las figuras 3.14 y 3.15 se presentan las gráficas de cinética para la adsorción de benceno y tolueno en cada polímero: 69 Figura 3.14 Cinéticas de sorción del benceno con los distintos polímeros Figura 3.15 Cinéticas de sorción del tolueno con los distintos polímeros 70 De las figuras 3.14 y 3.15 se pueden deducir los intervalos de tiempo aproximado a las condiciones de equilibrio de adsorción de benceno y tolueno con cada polímero, mostrados en la tabla 3.10. Tabla 3.10 Tiempos de equilibrio de adsorción de benceno y tolueno con cada polímero Compuesto Polímero Tiempo de Equilibrio PEBD > 24 h Benceno PS PEBD-PS (3-24) h (3-24) h PEBD (3-24) h Tolueno PS PEBD-PS (3-24) h > 24 h Los estudios de la cinética indican, de forma muy aproximada, que para el caso del benceno con PS y PEBD-PS hay una saturación momentánea entre 3 y 24 horas de estar en contacto con dichos polímeros, para luego continuar adsorbiendo en un probable proceso de difusión hacia el interior del polímero. Este mismo comportamiento se observó para el tolueno con el PEBD y el PS. En cambio, para el benceno con PEBD se observó una saturación del sistema que perduró por varios días, siendo igual el caso para el tolueno con PEBD-PS. Es importante destacar que no se asume la condición de equilibrio de adsorción en tiempos alejados (que tiendan al infinito) debido a que las gráficas de cinética indican que además del proceso de adsorción, en tiempos más largos se pueden llevar acabo procesos de absorción en los polímeros. 3.7. Isotermas de adsorción. Con el objetivo de estudiar y cuantificar la sorción de los cuatro compuestos en los tres polímeros, se construyeron las isotermas de sorción a partir de soluciones de diferentes concentraciones de cada compuesto en 30 mg. de cada polímero. Cada isoterma se monitoreó en el tiempo de equilibrio determinado por el estudio de la cinética. En las figuras 3.16, 3.17 y 3.18 se muestran las isotermas (mg de compuesto sorbido/g de polímero frente a la concentración en equilibrio) de cada compuesto en cada polímero. 71 Figura 3.16 Isoterma de adsorción benceno-PEBD Figura 3.17 Isoterma de adsorción benceno-PS Figura 3.18 Isoterma de adsorción benceno-PEBD-PS La isoterma correspondiente a benceno-PEBD parece tener un comportamiento de TIPO 1, en donde se produce adsorción hasta que el sistema llegue a una saturación; esto puedo deberse a la formación de una monocapa en la superficie del PEBD. Las isotermas correspondientes a 72 benceno-PEBD-PS y benceno PS, parecen tener un comportamiento propio del TIPO 3, en donde también se sugiere la formación de multicapas en la superficie del adsorbente; esto puede ser posible, sobre todo en el caso de la polimezcla, gracias a la presencia del PS en el adsorbente, el cual podría poseer una mayor interacción con el adsorbato. Además se sabe que la adsorción física es el mecanismo más esperado en tratamientos de agua contaminada con adsorbentes (Crittenden, 2005). 3.8. Modelo Langmuir La información correspondiente a los parámetros de Langmuir para la adsorción del benceno en cada polímero se obtuvo ajustando cada isoterma a un tipo de linealización de Langmuir descrito en la Tabla 1.8. Las figuras 3.19, 3.20 y 3.21 presentan las tres gráficas correspondientes a las formas lineales de las isotermas de Langmuir: Figura 3.19 Linealización de la isoterma benceno-PEBD. Langmuir-1 Figura 3.20 Linealización de la isoterma benceno-PS. Langmuir-1 73 Figura 3.21 Linealización de la isoterma benceno-PEBD-PS. Langmuir-1 De la tabla 3.11 se desprenden los valores de las ecuaciones de linealización de las isotermas del benceno con los polímeros y los parámetros de Langmuir. Tabla 3.11 Parámetros de Langmuir para el benceno Polímero Ecuación R2 qm KL PEBD 1/X = 8,714*(1/C) + 9,237 0,980 0,108 1,060 PS PEBD-PS 1/X = 156,7*(1/C) + 6,993 1/X = 151,6*(1/C) + 8,489 0,988 0,970 0,143 0,118 0,045 0,055 En el caso de benceno, la tabla 3.11 refleja, en primer lugar, la linealidad y factores de correlación de cada ajuste mayor a 0,970; además, un valor mayor qm (mg de benceno adsorbido en los sitios disponibles en la monocapa del adsorbente) para la adsorción en PS que en los polímeros restantes. La constante de adsorción KL arrojó un mayor valor para la adsorción con PEBD; dicho valor fue mayor que 1, lo cual indica una moderada adsorción en el polímero, mientras que en los otros casos el valor obtenido fue menor a 1, denotando una pobre adsorción en estos polímeros. En las figuras 3.22, 3.23 y 3.24 se muestran las isotermas del tolueno con cada uno de los polímeros. 74 Figura 3.22 Isoterma de adsorción tolueno-PEBD Figura 3.23 Isoterma de adsorción tolueno-PS Figura 3.24 Isoterma de adsorción tolueno-PEBD-PS La isoterma correspondiente a tolueno-PEBD parece tener un comportamiento del TIPO 3, mientras que las isotermas de tolueno-PS y tolueno-PEBD-PS aparentan tener un 75 comportamiento propio del TIPO 1. Esto podría indicar la formación de multicapas para el caso de tolueno-PEBD y la formación de monocapas para el tolueno con los polímeros restantes. Crittenden y colaboradores señalan que hay casos de adsorción que pueden tener un poco de ambos procesos (fisisorción y quimisorción); esto debido a que el hecho de compartir electrones no se diferencia del alto grado de distorsión producido por la nube de electrones que forma la adsorción física (Crittenden, 2005). Este comportamiento debe ser respaldado por los valores de los parámetros de Langmuir calculados a partir de la linealización de cada isoterma que se presenta en las figuras 3.25, 3.26 y 3.27. Figura 3.25 Linealización de la isoterma tolueno-PEBD. Langmuir-1 Figura 3.26 Linealización de la isoterma tolueno-PS. Langmuir-1 76 Figura 3.27 Linealización de la isoterma tolueno-PEBD-PS. Langmuir-1 Con ayuda de las linealizaciones de las tres isotermas del tolueno con los tres polímeros, se obtuvo los parámetros de Langmuir para cada adsorción, presentados en la tabla 3.12. Tabla 3.12 Parámetros de Langmuir para el tolueno Polímero PEBD PS PEBD-PS Ecuación 1/X = 18,38*(1/C) + 0,107 1/X = 27,68*(1/C) + 1,647 1/C = 10,87*(1/C) + 0,510 R2 qm KL 0,991 9,346 0,006 0,982 0,607 0,059 0,985 1,961 0,047 A priori se puede observar que se obtiene una excelente linealidad en las isotermas del tolueno con los tres polímeros, con valores de R2 mayores a 0,980. Además el valor de qm es considerablemente mayor para el PEBD, seguido de la polimezcla y por último del PS. Los valores de KL son bastante bajos para los tres polímeros, denotando una adsorción pobre en los tres casos. Es necesario estudiar más a fondo las afirmaciones de los distintos tipos de isotermas descritos en esta investigación, según el modelo de Langmuir, por lo cual se comparará la cantidad de moléculas que admite el área superficial de cada polímero con el valor de cada qm obtenido, es decir, la cantidad de moléculas adsorbidas en la monocapa, de manera de observar la concordancia entre ambos valores. Mediante esta comparación se observará si el benceno y el 77 tolueno se adsorben en forma de monocapa (Cohen, 1993). Para realizar esta comparación es necesario suponer que la molécula de adsorbato (benceno y tolueno) tienen forma esférica, por lo cual, adsorbidas por efectos de la nube pi, su disposición sobre el adsorbente es de forma plana u horizontal, es decir, en forma de circunferencia cuyo radio es igual al de la esfera antes mencionada. Una vez aceptadas las condiciones, se calculó, en primer lugar, el volumen molar de una molécula de benceno y el de una molécula de tolueno, para luego obtener el área que ocupa dichas moléculas y, conociendo el área superficial de cada polímero, saber cuántas moléculas ocuparían dicha área superficial. Seguidamente, con el valor de qm, se calculó la cantidad de moléculas adsorbidas en la monocapa. En la tabla 3.13 se muestra la comparación de las moléculas adsorbidas en la monocapa teórica con la experimental, para benceno y tolueno con todos los polímeros. Tabla 3.13 Comparación entre cantidad de moléculas adsorbidas y permitidas Benceno Polímero PEBD PS PEBD-PS Tolueno Moléculas adsorbidas en la monocapa (teórica) Moléculas adsorbidas en la monocapa (experimental) Moléculas adsorbidas en la monocapa (teórica) Moléculas adsorbidas en la monocapa (experimental) 6,21E+17 3,81E+17 1,53E+17 2,50E+16 3,31E+16 2,73E+16 5,51E+17 3,38E+17 4,01E+17 1,83E+18 1,19E+17 3,84E+17 Comparando los valores obtenidos de moléculas adsorbidas teórica y experimentalmente en cada polímero y de cada compuesto, se observa que para el caso de benceno con los tres polímeros no llega a formarse la monocapa. En el caso del tolueno, su adsorción en PEBD arrojó el valor de 3,32 capas, con PS menos de una monocapa y con PEBD-PS 1 monocapa aproximadamente. Esto indica que sólo se podría verificar la formación de multicapas en la adsorción de tolueno en PEBD, lo cual concuerda con la forma de la isoterma tipo 3 78 Otra hipótesis indica que estos valores podrían atribuirse a otra disposición de las moléculas, tanto de benceno como tolueno, sobre el adsorbente, que pueden colocarse de forma vertical o perpendicular y no de forma plana (como se supuso en un principio), permitiendo que una mayor cantidad de moléculas se adhieran a la superficie del polímero sin la necesaria formación de más de una monocapa. Es importante señalar que se sabe que la adsorción física conlleva un proceso exotérmico, reversible, cuyo calor de adsorción oscila entre 4 y 40 kJ/mol, mientras que la quimisorción, es también un proceso exotérmico, típicamente no-reversible, cuyo calor de adsorción es mayor a los 200 kJ/mol (Crittenden, 2005). Por lo mencionado anteriormente, una manera de corroborar el tipo de proceso que se lleva a cabo podría consistir en la realización de isotermas a diversas temperaturas, de manera de estudiar las constantes de cada proceso y poder, no solo hallar la naturaleza endotérmica o exotérmica del proceso, sino el valor numérico del calor de adsorción (Long, 2006). Haciendo una conexión entre los valores obtenidos en la tabla 3.11, 3.12 y 3.13, a pesar de que los valores de KL son superiores para la adsorción de benceno, el tolueno posee mejores números en cuanto a moléculas adsorbidas en los tres polímeros. Este hecho podría ratificarse con el ajuste a otro modelo de adsorción. 3.9. Isotermas de adsorción. Modelo Freundlich Se realizó un ajuste al modelo de adsorción de Freundlich. A pesar de ser considerado un modelo de ajuste que ofrece parámetros capaces de explicar capacidad de adsorción (k) y capaces de explicar la fuerza de adsorción del adsorbente (n), otros piensan que simplemente es un arreglo matemático para ajustar casi cualquier proceso de adsorción y principalmente de procesos en donde se lleve a cabo una fisisorción. Las figuras 3.28 a 3.33 muestran los ajustes realizados para las isotermas de benceno y tolueno con los tres polímeros usados. 79 Figura 3.28 Isoterma de adsorción de benceno-PEBD. Modelo de Freundlich Figura 3.29 Isoterma de adsorción de benceno-PS. Modelo de Freundlich Figura 3.30 Isoterma de adsorción de benceno-PEBD-PS. Modelo Freundlich 80 Figura 3.31 Isoterma de adsorción de tolueno-PEBD. Modelo Freundlich Figura 3.32 Isoterma de adsorción de tolueno-PS. Modelo Freundlich Figura 3.33 Isoterma de adsorción de tolueno-PEBD-PS. Modelo Freundlich De las figuras 3.28 a 3.33 se desprenden los valores de la linealización y los parámetros del modelo Freundlich, presentados en las tablas 3.14 y 3.15. 81 Tabla 3.14 Parámetros de Freundlich para el benceno Compuesto Polímero Ecuación R2 k n Benceno PEBD Log (x/m) = 1,830* Log(Ceq) - 0,435 0,980 0,367 0,546 PS Log (x/m) = 2,096* Log(Ceq) - 1,396 0,962 0,040 0,480 PEBD-PS Log (x/m) = 1,953* Log(Ceq) - 1,285 0,986 0,050 0,512 Tabla 3.15 Parámetros de Freundlich para el tolueno Compuesto Polímero Ecuación R2 k n Tolueno PEBD Log (x/m) = 1,039* Log(Ceq) + 0,338 0,980 2,170 0,962 PS Log (x/m) = 1,370* Log(Ceq) + 0,203 0,984 1,590 0,730 PEBD-PS Log (x/m) = 0,820* Log(Ceq) + 0,435 0,963 2,720 1,210 De las tablas 3.14 y 3.15 se puede observar que los valores de k son mayores para la adsorción de tolueno, denotando a priori que los polímeros son más capaces de adsorber tolueno que benceno. Además los valores de n, se encuentran, en el caso del tolueno, más cercanos a la unidad significando una adsorción de moderada, mientras que en el caso del benceno significa una adsorción pobre. Cabe destacar que en el caso del tolueno, el hecho de que los valores de n sean cercanos a la unidad indica que todos los sitios son equivalentes energéticamente, lo cual podría indicar la formación de la monocapa como vía de adsorción, que a su vez podría indicar la disposición de las moléculas en el adsorbato de forma vertical y no plana como se había suponido. En el caso del benceno, el valor de k para la adsorción con PEBD fue mucho mayor a los obtenidos con los otros polímeros, corroborado además por el valor de n, que también fue mayor para PEBD. Estos resultados se corresponden con aquellos obtenidos para el modelo de 82 Langmuir, en donde el PEBD arroja valores más altos en las constantes de adsorción que los polímeros restantes. En cuanto al tolueno, los valores de k y n para la adsorción con PEBD-PS son mayores a los otros dos polímeros, seguido por el PEBD y por último el PS. Estos resultados se corresponden para los valores de los parámetros de Langmuir con el tolueno, ya que para la polimezcla se hallan valores altos en comparación con los polímeros restantes. Para la adsorción con tolueno si se observó una mejoría, en relación al aumento de los valores de los parámetros de Freundlich, entre el PEBD y el PEBD-PS, y probablemente se debe a ese aumento de zonas amorfas en la estructura de el PEBD y además un efecto químico del anillo bencénico en la fase de PS, que ayudó a que más moléculas de tolueno se adsorbieran en la superficie. Para corroborar que la intensidad de adsorción de Freundlich “n”, puede o no ser relacionado efectivamente con los valores obtenidos para cada polímero, se podría estudiar la espontaneidad del proceso de adsorción con cada polímero, es decir, la energía libre de Gibbs. Mientras más negativo sea el valor, mayor será la intensidad de adsorción para cada polímero (Long, 2006). Comparando los resultados obtenidos con resultados arrojados por otros polímeros empleados en la adsorción de este tipo de hidrocarburos, se puede evidenciar la poca eficacia de adsorción de las tres resinas empleadas en esta investigación. Ceylan y col. (2009) estudiaron la sorción de de tolueno en poli-isobutileno y polipropileno (Apéndice C), hallando valores de capacidad máxima en 15,5 y 5,8 respectivamente, en g de tolueno por g de adsorbente, sin embargo este proceso no es por mecanismo de adsorción sino por hinchamiento de los polímeros (absorción). Ambos polímeros son poliolefinas hidrofóbicas y en un mecanismo distinto de sorción, sobrepasan en valor de qm obtenido para PEBD (9,346 mg de tolueno por g de adsorbente), lo cual podría indicar que poliolefinas más voluminosas son más productivas en la sorción por hinchamiento, de acuerdo a sus propiedades mecánicas, que en la adsorción física a nivel molecular de estos contaminantes. 83 Cohen y col. (1993), emplearon dos polímeros de matriz poliestirénica con divinilbenceno llamados “XAD-12” y “XAD-4”, obteniendo que para la adsorción de tolueno en el caso del primer polímero se sobrepasó el valor de la monocapa en 7 capas, y para el segundo se registró solo una monocapa (empleando las mismas suposiciones en cuanto a las moléculas de tolueno empleadas en esta investigación), por lo cual se puede decir que los resultados obtenidos con PS y PEBD-PS no son comparables con los mencionados anteriormente, ya que no alcanzan el valor de la monocapa, pero la adsorción de tolueno en PEBD si alcanza un valor de 3 monocapas, por lo cual puede ser considerado como un adsorbente aceptable de tolueno, al ser comparado con los valores reportados en la literatura. En la misma tónica, los estudios realizados por Pan y col. (2009) revelan que polímeros de matriz poliestirénica “XAD-2” y el mencionado “XAD-4” presentan capacidades máximas de adsorción de benceno de 730 y 1400 mg de benceno por g de adsorbente; valores que sobrepasan los obtenidos en esta investigación que no sobrepasan 0,143 mg de benceno por g de adsorbente. Long y col. (2006) emplearon también “XAD-4” (Apéndice C) y un modificado de ese polímero aminado llamado “NDA103” (Apéndice C) en la adsorción de fenol. La idea fue funcionalizar dicho polímero con un grupo que permitiera una sinergia en la adsorción. Ellos encontraron valores de qm 0,926 y 1,436 (mmol de fenol por g de adsorbente) y KL 0,910 y 1,676 para ambos polímeros respectivamente, lo cual prueba que la inclusión de un grupo polar ayudó a la adsorción de fenol. En líneas generales, por ser la adsorción de contaminantes en medio acuoso un proceso en donde fundamentalmente se produce fisisorción, la característica más importante es el área superficial del polímero usado. En la tabla 3.16 podemos ver una comparación del área superficial BET de los polímeros utilizados en esta investigación con otros empleados en la literatura. 84 Tabla 3.16 Comparación de polímeros Polímero PEBD PS PEBD-PS XAD-4 NDA103 XAD-12 Tipo de Polimezcla no Matriz Poliestireno PoliestirenoPoliolefina Poliestireno estructura compatibilizada poliestirénica aminado divinilbenceno Area superficial 7 4,3 5,1 914 611 21 BET (m2/g) La tabla 3.16 indica como los polímeros comerciales sobrepasan en área superficial a los utilizados en esta investigación en un mínimo de un orden de magnitud, eso les proporciona mayor capacidad de adsorción para compuestos aromáticos de distintas estructuras. Esta característica aunada a la poca porosidad y poca capacidad de hinchamiento evita que estos polímeros sean eficaces en la adsorción o al menos retenimiento de contaminantes en medio acuoso. El caso ideal para mejorar sus capacidades sería funcionalizar los polímeros con grupos que puedan aumentar el área superficial, originar microporos en la morfología y tener mayor afinidad por los contaminantes no sólo física sino químicamente. CONCLUSIONES Una vez realizada la caracterización de los polímeros empleados, la validación del método de cuantificación de benceno y tolueno en agua y los experimentos de cinéticas e isotermas de adsorción, en función de los objetivos planteados, se puede concluir: • La técnica FT-IR permitió caracterizar los polímeros PEBD y PS adquiridos para esta experimentación, al determinar los grupos característicos de cada polímero. • La microscopía electrónica de barrido (MEB) permitió detallar la morfología de la polimezcla PEBD-PS, observándose una estructura con una fase fija de PEBD en una fase móvil de PS, en forma de partículas amorfas, originando la aparición de volúmenes libres de distintos tamaños en la fase fija de PEBD sin interconexión entre ellas. • La calorimetría diferencial de barrido (CDB) se empleó para observar cambios en la cristalización del PEBD sólo en comparación con el PEBD de la polimezcla PEBD-PS. Se encontró una disminución en el grado de cristalinidad del PEBD de la polimezcla en un valor de 2,79 veces su valor original. • El método de microextracción en fase líquida fue validado en cuanto a linealidad, precisión, exactitud, % de recuperación, limites de detección y cuantificación; obteniendo excelentes resultados, adecuados para esta investigación. • Se determinó el tiempo aproximado de condiciones de equilibrio de adsorción de cada compuesto en cada polímero. • Se realizó cada isoterma de adsorción variando la concentración de adsorbato y manteniendo fijo la cantidad de adsorbente, en agitación constante. Se obtuvo que para benceno con todos los polímeros hay indicios que apuntan hacia una fisisorción, y para el tolueno puede ocurrir fisisorción en el caso de PEBD y quimisorción en el caso de PS y PEBD-PS. • El ajuste de cada isoterma al modelo de Langmuir fue logrado con una buena linealidad (R2>0,970) Se encontró una afinidad similar en ambos contaminantes con los PS y PEBD-PS, poca afinidad del tolueno-PEBD y moderada afinidad del benceno-PEBD. 86 • La determinación del área superficial BET de cada polímero permitió comparar el número de moléculas permitidas en el área superficial del adsorbente con el valor de qm. Se encontró que para el caso del benceno en PEBD, PS y PEBD-PS, no se alcanzó el valor de la monocapa; mientras que, para el caso del tolueno, se superó el valor de la monocapa en la adsorción con PEBD (3,31 capas) y no se alcanzó el valor de la monocapa simple con los adsorbentes restantes. • El ajuste de cada isoterma al modelo de Freundlich fue logrado con relativa linealidad 2 (R >0,960). Se encontró una mayor capacidad y moderada intensidad de adsorción en el caso de tolueno-PEBD y tolueno-PEBD-PS, y poca capacidad y pobre intensidad de adsorción para las adsorciones restantes. • Dado los resultados se puede afirmar que los polímeros empleados en esta investigación son más capaces de adsorber tolueno que benceno, sin embargo no son muy eficaces en dicha tarea, dado los valores obtenidos de los parámetros de adsorción y su comparación con polímeros similares usados en la literatura. BIBLIOGRAFIA • Adamson, A., “Physical Chemistry of Surfaces”. 6ta Edición. Wiley- Interscience Publication, pp. 617 (1997). • Aeppli, C., Berg, M., Hofstetter, T., Kipfer, R., Schwarzenbach, R., Simultaneous quantification of polar and non-polar volatile organic compounds in water samples by direct aqueous injection-gas chromatography/mass spectrometry. Journal of Chromatography A. 2008, 1181, 116-124. • Areiza J. “Polímeros”. Editorial Síntesis, Madrid, pp. 60, (2002). • Boopathy, R., Factors limiting bioremediation technologies. Bioresourc. Technol. 2000, 74, 63–67. • Bruice, P., “Organic Chemistry”. 4ta Edición. Prentice Hall, pp. 594-596 (2004). • Brunauer, S., Emmett, P. H., Teller, E. Adsorption of gases in multimolecular layers. Journal of American Chemical Society. 1938, 60, 309-319. • Bruzzoniti, E., Corrado Sarzanini, C., Bruzzoniti, M.C., Preconcentration of contaminants in water analysis. J. Chromatogr. A. 2000, 902, 289-309. • Brydson, J.A., “Plastic Materials”. 7ma Edición. Butterworth-Heinemann, pp. 205-207, 425-427, 433 (1999). • Carey, F., “Organic Chemistry”. 4ta Edición. McGraw Hill, pp. 399 (2000). • Ceylan, D., Dogu, S., Karacik, B., Yakan, S., Okay, O., Okay, A., Evaluation of butyl rubber as sorbent material for the removal of oil and polycyclic aromatic hydrocarbons from seawater. Environmental. Science. Technolgy. 2009, 43, 10, 3846-3852. • Chen, S., Chen, J., Removal of polycyclic aromatic hydrocarbons by low density polyethylene from liquid model and roasted meat. Food Chemistry. 2005, 90, 461-469. • Chen, B., Lee, X., Xu, S., Tang, T., Zhou, B. Huang, B. Compatibilization effect of block copolymers in high density polyethylene/syndiotactic polystyrene blends. Polymer, 2002, 43, 953-961 88 • Cohen, Y., Gusler, G., Browne, T. Sorption of Organics from Aqueous Solution onto Polymeric Resins. Ind. Eng. Chem. Res. 1993, 32, 2727-2735. • Committee on Ground Water Cleanup Alternatives, National Research Council. “Alternatives for Ground Water Cleanup”, National Academy of Sciences, pp. 126-164, (1994). • Consejo Nacional de Medio Ambiente (CONAMA). Resolución N° 357 de Conama. Brasil, 2005. • Crittenden, J., Trussell, R., Hand, D., Howe, K., Tchobanoglous, G., “Water Treatment: Principles and Design”, John Wiley & Sons, pp. 643-645, (2005). • Demeestere, K.; Dewulf, J.; Witte, B.; Langenhove. H.V., Sample preparation for the analysis of volatile organic compounds in air and water matrices. J. Chromatogr. A. 2007, 1153, 130-144. • Dewulf, J., Huybrechts, T., Langenhove, H.V., Developments in the analysis of volatile halogenated compounds. Trends Anal. Chem. 2006, 25, 300-317. • Dewulf, J., Langenhove, H. V., Wittmann, G., Analysis of volatile organic compounds using gas chromatography. Trends Anal. Chem. 2002, 21, 637-646. • Eiceman, G.A., Gardea-Torresdey, J., Overton, E., Carney, K., Dorman, F., Gas chromatography. Anal. Chem. 2004, 76, 3387-3394. • Hamdaoui, O., Noffrechoux, E., Modeling of adsorption isotherms of phenol and chlorophenols onto granular activated carbon: Part I. Two-parameter models and equations allowing determination of thermodynamic parameters. Journal of Hazardous Materials. 2007, 147, 381-394. • Harrats, C., Fayt, R., Jerome, R. Efect of block copolymers of various molecular architecture on the phase morphology and tensile properties of LDPE rich (LDPE/PS) blends. Polymer. 2002, 43, 863-873. • Hradil, J.; Svec, F., Podlesnyuk, V., Marutovskij, R., Friedman, L., Klimenko, N. Sorption and Desorption of Organic Compounds by Synthetic Polymeric Sorbents. Ind. Eng. Chem. Res. 1991, 30, 1926-1931. • Health Protection Agency. “Toluene - Production and Uses”. Reino Unido, 2008. 89 • Health Protection Agency. “Benzene-Exposure Standards, Guidelines or Regulations”. Reino Unido, 2008. • Health Protection Agency. “Toluene-Exposure Standards, Guidelines or Regulations”. Reino Unido, 2008. • Jerabek, K., Veverka, P. Influence of hypercrosslinking on adsorption and absorption on or in styrenic polymers. Reactive & Functional Polymers. 2004, 59, 71-79. • Jurdáková, H., Kraus, A., Lorenz, W., Kubinec, R., Krkošová, Ž., Blaškoa, J., Ostrovský, I., Soják, L., Pacákovác, V., Determination of gasoline and BTEX in water samples by gas chromatography with direct aqueous injection. Petroleum & Coal. 2005, 47, 3, 49-53. • Jurdáková, H., Kraus, A., Lorenz, W., Kubinec, R., Krkošová, Ž., Blaškoa, J., Ostrovský, I., Soják. L., Gas chromatographic determination of benzene, toluene, ethylbenzene and xylenes using flame ionization detector in water samples with direct aqueous injection up to 250 µL. Journal of Chromatography A. 2005, 1084, 90-94. • Kennes, C., Veiga, M.C., Bioreactors for waste gas treatment, Kluwer Academic Publishers. 2001, p. 3. • Knox, R.C., Canter, L.W., Kincannon, D.F., Stover, E.L., Ward C.H., “Aquifer Restoration”, Noyes Publications, pp. 195-200, (1986). • Lair, A., Ferronato, C., Chovelon, J.M., Herrmann, J.M., Naphthalene degradation in water by heterogeneous photocatalysis: An investigation of the influence of inorganic anions. Journal of Photochemistry and Photobiology A: Chemistry. 2008, 193, 193-203. • Lara-Gonzalo, A., Sánchez-Uría, J.E., Segovia-García, E., Sanz-Medel, A., Critical comparison of automated purge and trap and solid-phase microextraction for routine determination of volatile organic compounds in drinking waters by GC–MS. Talanta. 2008, 74, 1455-1462. • Larroche, C., Farhadian, M., Duchez, M., Vachelard, C., Monoaromatics removal from polluted water through bioreactors-A review. Water Research. 2008, 42, 1325-1341. • Lee, H.K., “Modern techniques for the analysis of polycyclic aromatic hydrocarbons”. Handbook of Analytical Separations, Vol. 3, Cap 2. (2001). • Levine, I. “Fisicoquímica Vol. 1”. 5ta Edición. Mc Graw Hill, pp. 483-488 (2004). 90 • Lide, D., “CRC handbook of chemistry and physics”, Internet Version, (2005). • Long, C., Ming, Z.W., Cai, P., Xing, Z., Zhang, B. Synergistic adsorption of phenol from aqueous solution onto polymeric adsorbents. Journal of Hazardous Materials. 2006, B128, 123129. • Lynch, J.M., Moffat, A.J., Bioremediation-prospects for the future application of innovative applied biological research. Ann. Appl. Biol. 2005, 146, 2, 217-221. • Mangani, F., Maione, M., Palma, P., GC-MS analysis of halocarbons in the environment. Adv. Chromatogr. 2003, 42, 139-239. • Marcé, R.M., Borrull, F., Solid-phase Extraction of Polycyclic Aromatic Compounds. Journal of Chromatography A. 2000, 885, 273-290. • Masel, R. I., “Principles of adsorption and reaction on solid surfaces”. Wiley- Interscience Publication (1996). • Ministerios de Sanidad y Asistencia Social. “Normas Sanitarias de Calidad de Agua Potable”. República de Venezuela, 1998. • Muñoz, I. Life cycle assessment as a tool for green chemistry: Application to different advanced oxidation processes for wastewater treatment. Universidad Autónoma de Barcelona, Bellaterra, 2006. • Ortega, N. Degradación fotocatalítica de fenoles empleando luz UV y TiO2 modificado. Universidad Simón Bolívar, Sartenejas, 2008. • Ouyanga, G., Pawliszyn J., A critical review in calibration methods for solid-phase microextraction. Analytica. Chimica Acta. 2008, 627, 184-197. • Pan, B., Pan,B., Zhang, W., Lv, L., Zhang, Q., Zheng, S. Development of polymeric and polymer-based hybrid adsorbents for pollutants removal from waters. Chemical Engineering Journal. 2009, 151, 19-29. • Peacock, A. Handbook of polyethylene. Structures, properties and applications. Marcel Decker, Inc., pp. 123, 124, 177 (2000). • Peñalver, A. Aplicación de la microextracción en fase sólida al análisis medioambiental. Universidat Rovira I Virgili, Tarragona, 2002. 91 • Rezaee, M., Assadi, Y., Hosseini, M., Aghaee, E., Ahmadia, F., Berijani, S. Determination of organic compounds in water using dispersive liquid–liquid microextraction. Journal of Chromatography A, 2006, 1116, 1-9. • Rosato, D. Concise Encyclopedia of Plastics. Kluwer Academic Publishers: EEUU (2000). • Rubio, J., Souza, M.L., Smith, R.W., Overview of flotation as a wastewater treatment technique. Minerals Engineering 15. 2002, 139-155. • Russell, J., Ginn J., “Practical Handbook of Soil, Vadose Zone, and Ground-Water Contamination. Assessment, prevention, and Remediation”, Lewis Publishers, Capítulo 14 (2004). • Safarova, V.I., Sapelnikova, S.V., Djazhenko, E.V., Teplova, G.I., Shajdulina, G.F., Kudasheva, F., Gas chromatography–mass spectrometry with headspace for the analysis of volatile organic compounds in waste water. J. Chromatogr. B. 2004, 800, 325. • Santos, F.J., Galceran, M.T., The aplication of gas chromatography to environmental analysis. Trends Anal. Chem. 2002, 21, 672-685. • Shaw, D., “Colloid & Surface Chemistry”. Butterworth-Heinemann, pp. 121-123 (1966). • Simko, P.; Simon, P.; Khunov, V. Removal of Polycyclic Aromatic Hydrocarbons from Water by Migration into Polyethylene. Food Chemistry. 1999, 64, 157-161. • Skoog, D.; Holler; Nieman. Principios de Análisis Instrumental. 5ta Edición. McGraw Hill. Madrid. (2001). • Takahashi, Y. Infrared and Raman Spectra of Polyethylene and Polyethylene-d4 in the Temperature Region (5-300 K). Macromolecules 2001, 34, 7836-7840. • Tena, M.; Carrillo, J. Multiple solid-phase microextraction: Theory and applications. Trends in Analytical Chemistry, 2007, 26, No. 3. • Treybal, R.E., “Mass Transfer operations”, McGraw-Hill, pp. 581-582 (1981). • Moly, K., Bhagawan, S., George, S. Sorption and diffusion of aromatic solvents through linear low density polyethylene–ethylene vinyl acetate blend membranes. J. Mater. Sci. 2007, 42, 4552-4561. 92 • United States Environmental Protection Agency (USEPA). “Drinking Water Contaminants. List of Contaminants & their MCLs”. (2009) Disponible en http://www.epa.gov/. Consultada 01/05/10. • United States Environmental Protection Agency (USEPA). Método 3511 “Organic Compounds In Water By Microextraction”. (2002). • United States Environmental Protection Agency (USEPA). SW-846. “Quality Control”. Cap 1. (1992). • United States Environmental Protection Agency (USEPA). Método 5030c “Purge-And- Trap For Aqueous Samples”. (2003). • United States Environmental Protection Agency (USEPA). Método 5021a “Volatile Organic Compounds In Various Sample Matrices Using Equilibrium Headspace Analysis”. (2003). • Valderrama, C., Gamisans, X., de las Heras, X., Farrán, A., Cortina, J.L. Sorption kinetics of polycyclic aromatic hydrocarbons removal using granular activated carbon: Intraparticle diffusion coefficients. Journal of Hazardous Materials. 2008, 157, 386-396. • Wade, L.G., “Química Orgánica”. 5ta Edición. Prentice Hall, pp. 679, 702-704, 1182 (2004). • Workman, J. “Handbook of Organic Compounds: NIR, IR, Raman, and UV-VIS Spectra Featuring Polymers and Surfactants”, Argose Incorporated, pp. 1029, 1293, (2000). • Zhang, W., Chen, J., Pan, B., Zhang Q. Cooperative adsorption behaviours of 1-naphthol and 1-naphthylamine onto nonpolar macroreticular adsorbents. Reactive & Functional Polymers. 2006, 66, 485-493. • Zoccolillo, L., Amendola, L., Cafaro, C., Insogna, S., Improved analysis of volatile halogenated hydrocarbons in water by purge-and-trap with gas chromatography and mass spectrometric detection. Journal of Chromatography A. 2005, 1077, 181-187. 93 APÉNDICE A Figura A.1 FT-IR del PEBD Figura A.2 FT-IR teórico del PEBD (Workman, 2000) 94 Figura A.3 FT-IR del PS Figura A.4 FT-IR del teórico de PS (Workman, 2000) APÉNDICE B 95 Figura B.1 Cromatogramas de curva de calibración de benceno y tolueno. (De izquierda de derecha: Benceno, Surrogate, E.I., Tolueno) 96 APÉNDICE C Figura C.1 Poli-isobutileno Figura C.3 “XAD-4” Figura C.2 Polipropileno Figura C.4 “NDA103”