UNIVERSIDAD AUTONOMA METROPOLITANA
Anuncio

UNIVERSIDAD AUTONOMA METROPOLITANA
PLANTEL IZTAPALAPA
'tax
/j-,c
. Q L " ~ ~
M a . Laura Cano Orúaz
ACERODra. Judith Cardoso Martínez
M. en Q. Eloísa Anleu Avda
-/c.
1998.
i
Contenido
CAPíTULO 1 INTRODUCCIÓN.......................................................................................
I.I INTRODUCCI~N...................................................................................................
1.2 ANTECEDENTES...................................................................................................
1.3 OBJETIVOS ............................................................................................................
REFERENCIAS .............................................................................................................
1
1
3
4
5
CAPíTULO 2 FUNDAMENTOS TEÓRICOS ..................................................................
6
2.1 POLIMERO.............................................................................................................. 6
2.2 POLIMERIZACIÓN EN CADENA POR RADICALES LIBRES .............................. 7
2.3 POLIMERIZACIÓN EN SOLUCIÓN ....................................................................... 8
2.4 FUNCIONALIZACIÓN DE POLIMEROS............................................................... 9
2.4.1 POLISULFOBETAINAS ........................................................................................ 9
2.4.2 POLlCARBOXIBETAíNAS.................................................................................... 10
2.4.3 N-ÓXIDOS ................................................................................................... 10
2.4.4 COMPUESTOS CUATERNIZADOS DE AMONIO .................................................... 11
2.5 PROPUESTAS DE OBTENCIÓN DE POLíMEROS FUNCIONALIZADOS....... 12
2.5.1 SíNTESlS DE LA POLISULFOBETAINAPDEAEEM-S ............................................... 12
2.5.2 SíNTESlS DE LA POLICARBOXIBETAINA PDEAEEM-C ........................................... 13
2.5.3 SINTESIS DEL N-ÓXIDO PDEAEEM-NO ............................................................. 14
2.5.4 SINTESIS DE COMPUESTOS CUATERNIZADOS DE AMONIO ................................. 15
2.6 CARACTERIZACIÓN FlSlCOQUíMlCA.............................................................. 16
2.6.1 ESPECTROSCOPIADE INFRARROJO (FT-IR) ....................................................... 17
2.6.2 ESPECTROSCOPIADE RESONANCIA MAGNÉTICANUCLEAR (RMN) ..................... 18
2.6.3 DISPERSIÓN DE LUZ ................................................................................... ..I 9
REFERENCIAS ........................................................................................................... 21
CAPITULO 3 PURIFICACIÓN DE REACTIVOS.......................................................
3.1 . PURIFICACIÓN DE REACTIVOS .......................................................................
3.1.1 PURIFICACIÓN DE HEXANO.................................................................................
3.1.2 PURIFICACIÓN DE NOo ......................................................................................
3.1.3 PURIFICACIÓN DE BENCENO...............................................................................
22
22
22
23
24
11
3.1.4 PURIFICACIÓN DE 1,3-DINITROBENCENO ............................................................
3.1.5 PURIFICACIÓN DE DIETILAMINOETOXIETANOL(DEAEE) .........................................
3.1.6 PURIFICACIÓN DE CLORURO DE METACRILOILO ..................................................
3.1.7 PURIFICACIÓN DEL MONÓMERO DEAEEM.............................................................
CAPITULO 4 TÉCNICAEXPERIMENTAL ..................................................................
4.1 SíNTESlS DEL MONÓMERO METACRILATO DE N,NDIETILAMINOETOXlETILO...... (DEAEEM)..............................................................
4.2 SíNTESlS DEL POLíMERO PDEAEEM...............................................................
4.3 SINTESIS DEL N-OXIDO DEL PDEAEEM.........................................................
4.4 PURIFICACIÓN DEL N-OXIDO DEL PDEAEEM.................................................
4.5 CARACTERIZACI~N............................................................................................
4.5.1 ESPECTROSCOPIADE INFRARROJO (FT-IR) .........................................................
4.5.2 ESPECTROSCOPIADE RESONANCIA MAGNÉTICA NUCLEAR (RMN) .........................
4.5.3 DISPERSIÓN DE LUZ ..........................................................................................
REFERENCIAS................................................................................................................................................
CAPITULO 5 RESULTADOS .......................................................................................
5.1 FORMACIÓN DEL ALCÓXIDO DEAEE...............................................................
5.1.ICARACTERIZACIÓN POR FT-IR .............................................................................
5.1.2 CARACTERIZACIÓNPOR RMN ..............................................................................
5.2 SINTESIS DEL DEAEEM......................................................................................
5.2.1 CARACTERIZACIÓN POR FT-IR ............................................................................
5.3 SINTESIS DEL POLI(DEAEEM)...........................................................................
5.3.1 CARACTERIZACI~NPOR FT-IR .............................................................................
5.3.2 CARACTERIZACIÓN POR RMN ..............................................................................
5.3.3 DETERMINACI~NDE PESO MOLECULAR ................................................................
REFERENCIAS ...........................................................................................................
CAPíTULO 6 CONCLUSIONES .................................................................................
25
26
27
28
29
29
31
32
33
34
34
34
34
35
36
36
37
38
40
41
42
43
44
45
46
47
1
Capítulo I
Introducción y Objetivos
1.1 Introducción 192
Una necesidad vigente es eliminar los sólidos suspendidos y los materiales
catiónicos que contaminan las aguas. Las fuentes naturales de abasto de agua para
las grandes ciudades son los ríos, los manantiales y las lluvias, y cada una de ellas
contiene diferentes tipos de contaminantes. La calidad de las aguas residuales,
provenientes de usos domésticos, industriales, municipales y agrícolas, varía en
función del uso original y puede tener diversas cantidades y concentraciones de
materiales orgánicos, inorgánicos y bacteriológicos. Estas aguas son muy utilizadas,
sobre todo en la agricultura y en la industria, y deben cumplir con las especificaciones
de la Norma Oficial Mexicana (NOM. CG4.03Z-ECOLp6),que establece los límites
permitidos de contaminantes, como son: pH, los sólidos suspendidos totales,
incluyendo los catiónes metálicos y aniónes que generalmente tienen un agua
residual. Alguno de estos materiales, como el níquel, manganeso, plomo, cromo, zinc
y mercurio son tóxicos y, las aguas que los contienen requieren de un tratamiento de
purificación especial para su eliminación. Una de las tecnologías, que han sido
desarrolladas principalmente en la iniciativa privada desde los años 50, esta basada
en el uso de materiales poliméricos que floculan.
El proceso de floculación se emplea para extraer del agua los sólidos que en
ella se encuentran suspendidos, siempre que su rapidez natural de asentamiento sea
demasiado baja para proporcionar clarificación efectiva.
La floculación es un proceso de agregación de pequeñas masas, que
generalmente se encuentran en un medio líquido, en masas mayores llamados
flóculos. El proceso de floculación se determina por la fuerte interacción de fuerzas
entre las partículas dispersas. La floculación es estimulada por un mezclado lento
que junta poco a poco los flóculos; un mezclado demasiado intenso los rompe y
raramente se vuelven a formar en su tamaño y fuerza óptimos. La floculación no sólo
incrementa el tamaño de las partículas del flóculo sino que también afecta su
naturaleza física.
Los floculantes se pueden clasificar bajo el punto de vista del que se estudian,
según se considere su carga, naturaleza química, modo de acción y modo de empleo.
Los polimeros floculantes Sintéticos y naturales son solubles en agua debido a la
presencia de grupos iónicos o grupos polares, y consisten de macromoleculas
orgánicas capaces de desestabilizar y flocular a la materia suspendida, durante el
tratamiento de aguas.
Los floculantes naturales son compuestos orgánicos y están formados por
polisacáridos, cuyo principal representante es el almidón donde el mecanismo
principal es el enlace por nitrógeno.
Los floculantes sintéticos son polímeros lineales de elevado peso molecular,
solubles en agua, efectivos generalmente a concentraciones muy pequeñas y poseen
grupos activos, distribuidos a lo largo de sus cadenas, que tienen gran afinidad por
supeificies sólidas. El principal mecanismo de floculación de estos agentes es la
formación de puentes.
Los materiales floculantes también se distinguen por el tipo de carga
(aniónicos, catiónicos, no iónicos y zwitteriónicos), su densidad de carga (definida por
el porcentaje de unidades monoméricas que portan una carga) y su peso molecular
(considerado dentro del intervalo de lo4 a lo7).
Los polímeros floculantes sintéticos de nuestro estudio son los polímeros
iónicos o también llamados zwitteriónicos que son aquellos que poseen ambas cargas
unidas por enlaces covalentes (positiva y negativa). El efecto de floculación de estos
polímeros, es mas notable a pH<4; las dosis Óptimas son 1 ppm Los flóculos
formados no son muy grandes, sin embargo, la sedimentación al cabo de 5 min es
total, indicio de que el flóculo es más compacto.
Los polímeros zwitteriónicos son actualmente de gran interés debido a su
facilidad de síntesis y sus aplicaciones industriales como la recuperación terciaria del
petróleo y en el tratamiento de aguas residuales; son una clase especial de materiales
que presentan cargas positivas y negativas en la cadena polimerica, dentro de estos
podemos encontrar aquellos en los cuales las cargas positivas y negativas están
unidas por enlaces covalentes y se encuentran en el mismo grupo.
Algunos de estos tipos de estructuras se muestran a continuación:
>
Sulfobetaínas
- k + - ( C H),;
I
S O-,
;
Carboxibetaínas
S+-(CH,)ñSO-,
3
Las reacciones de cuaternización en aminas terciarias presentes en los
polímeros, son de gran interés debido a que éstas tienden a ser muy rápidas aun a
bajas temperaturas.
Una de estas estructuras tipo catiónica es:
Compuestos cuaternizados de amonio
R
I
R-N+-fi
I
Br
R
Los floculantes sinteticos comparados con los floculantes naturales, ofrecen la
ventaja de tener una mayor pureza, una mayor estabilidad de su calidad y una mayor
eficacia, ya que al ser los pesos moleculares de los sintéticos más elevados que
algunos de los naturales empleados en floculación, el alto grado de floculación
obtenido con los sintéticos no podría lograrse con los 'naturales, así como también los
polímeros sinteticos no añaden sustancias insolubles a los Iodos y no modifican las
propiedades fisicoquímicas del agua, así que no impiden el reciclado de los líquidos
clarificados.
1.2Antecedentes
Una amplia gama de investigaciones1 2 3 4 5 6 ponen en manifiesto la factibilidad de
sintetizar estructuras zwitteriónicas, especialmente polianfolitos del tipo Sulfobetaína,
Carboxibetaína y N-Óxidos. Por ejemplo la síntesis de sulfobetaínas y electrolítos
catiónicos derivados de acrilatos, acrilamida, acrilato piridina, acrilamida piridina y
vinil piridina fue reportada por Laschewky y Zerbe6 y por Galín, Monrroy-Soto y
colaboradores2', respectivamente, estudiando las propiedades en estado sólido y en
solución acuosa de tales polímeros. Cardoso' reportó la síntesis, caracterización y el
estudio de propiedades térmicas y de conducción iónica de una serie de metacrilatos
y vinil piridinas. La síntesis y el estudio de propiedades en solución de
polisulfobetaínas fue ampliamente examinado por Salamone y colaboradores'.
Tambien investigaron las propiedades en solución de copolímeros electrolíticos
catiónicos y aniónicos.
4
1.3 Objetivos
El objetivo principal es:
Sintetizar polímeros derivados del ácido metacrílico que
sirvan como precursores para su posterior funcionalización y
poder obtener polímeros funcionalizados.
Los objetivos específicos son:
0
Sintetizar monómeros de N,N-dietilaminoetilmetacrilato DEAEM mediante
reacciones de sustitución nucleofílica.
Llevar a cabo la reacción de polimerización del monómero precursor, vía
radicales libres en solución.
Caracterizar los polímeros precursores sintetizados por técnicas
espectroscópicas.
Proponer rutas de síntesis para la obtención de polímeros floculantes del
tipo zwitteriónicos como las polisulfobetaínas, Carboxibetaínas y los NÓxidos y del tipo catiónico como los compuestos cuaternarios de amonio que
sirvan en la clarificación de aguas residuales.
5
Referencias
.-Galin J.C. y Monrroy V.M Polymer 25, 121 (1984).
.- Galin J.C.
y Gingrean C., Polymer 35, 4669 (1994).
.- Hart, R. Y Timmerman D.J., J. Am. Chem. Soc. 76, 6280 (1954).
.-Laschewsky, Andre y Zerbe, I., Polymer 32, 2070 (1991).
.-Liaw, D. J. Y Lee, W.F., J. Appl. Polymer 36, 357 (1995).
.-Salamone, J.C., volksen, W., Olson, A.P. y Raia, D.C., Polymer 18, 1058 (1977).
.-Cardoso M. J,, Síntesis, caracterización y propiedades de polimeros y co-polímeros
zwitteriÓnicos,Tesisi Doctoral. FQ-UNAM (1990).
6
Capítulo 2
Fundamentos Teóricos
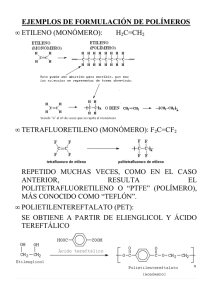
2.i PoZímero1*2
La materia esta constituida por moléculas que pueden ser de tamaño normal
o moléculas gigantes llamadas polímeros.
Una definición general de polímeros es: cualquier sustancia que posea un alto
peso molecular, comúnmente superior a 10,000 unidades.
Un polímero es un compuesto macromolecular, múltiplo de unidades químicas
más simples conocidada como monómeros y unidas químicamente, que forman
enormes cadenas de las formas mas diversas. Algunas parecen fideos, otra tienen
ramificaciones, algunas mas se asemejan a las escaleras de mano y otras son como
redes tridimensionales. La palabra proviene de las raices poli: muchos y mero:
unidad Las reacciones químicas por las cuales los monómeros se transforman en
polímeros de denomina pohmeriaciÓn. Por lo general, las macromoléculas tienen
enlaces covalentes (energías altas 35-150 Kcal/mol).
La enorme cantidad de interacciones entre las cadenas polimericas o
fuerzas intermoleculares, que aumentan en presencia de grupos polares y disminuye
al aumentar la distancia entre moléculas; tales interacciones consisten en diversas
clases de enlaces intermoleculares y entrecruzamientos físicos; la magnitud de dichas
interacciones depende de la naturaleza de las fuerzas intermoleculares, el peso
molecular, la forma en que las cadenas del polímero están empacadas entre sí y la
flexibilidad de las mismas. Por lo que la magnitud de las interacciones es distinta para
cada polímero.
7
2.2 Polimerización en Cadena por Radicales Libres 394
En la polimerización por reacción en cadena hay una serie de reacciones cada
una de las cuales consume una partícula reactiva y genera otra similar de modo que
cada reacción individual depende de otra previa. La presencia de un radical libre es
indispensable en la iniciación de la reacción de polimerización en cadena. Su
estabilidad dentro del medio reaccionante debe ser suficientemente grande para que
un cierto número de moléculas de monómero pueda reaccionar. Generalmente son
esenciales tres etapas para la formación de un polímero de alto peso molecular:
lniciación de la Cadena:
La iniciación, que es la creación de un centro "activo", tal como un radical
libre, se produce la fijación de una primera molécula de monómero.
Iniciador + RO
Ro +
CH2=CH
I
+
R4H24H0
I
X
Propagación de la Cadena:
X
La propagación, es la adición de mas moléculas de monómero en el extremo
de una cadena en crecimiento, generalmente muy rápida.
R 4 H 2A H o
I
+ CH2=CH
I
X
+ R 4 H 24
X
M H 2A H o
I
I
X
X
Terminación de la Cadena:
La terminación, es la desaparición de un centro "activo", es decir, el
crecimiento de los radicales se detiene por combinación de los radicales entre ellos
mismos.
Combinación.
DesproprciÓn.
2 R- (CH2 -CH)n-CH2-CH0 +R-(CH2-CH)n-CH2-CH2
I
X
I
X
I
X
I
X
+
R-(CH2-CH)n-CH=CH
I
X
I
X
8
Una desventaja importante que presenta este tipo de reacciones es la amplia
distribución de pesos moleculares en productos polimericos; esto se debe a que el
control sobre el crecimiento de las cadenas es limitado.
Iniciador
Un iniciador de polimerización es un compuesto capaz de producir radicales
libres generalmente por elevación de la temperatura. Su empleo asegura una
cantidad de radicales libres a una temperatura mucho mas baja que la que se
necesita para realizar una reacción de polimerización con iniciación térmica.
Los compuestos azoicos: R- N = N - R, se descomponen con eliminación de
una molécula de nitrógeno, uno de los mas empleados es el azobisisobutironitrilo
(AIBN); este es un iniciador que se usa frecuentemente en las reacciones de
polimerización por radicales libres. En el mecanismo la molécula se rompe
homolíticamente para dar lugar al radical libre que promueve la reacción en cadena:
20-100%
(CH,)TC-N=N-C-(CH,),
CN
CN
2(CH3),-C*
I
CN
+
N,
2.3 Polimerización en Solución 2,3
La reacción de polimerización en solución se efectúa en un disolvente que
tiene la finalidad de absorber el calor y reducir la viscosidad de la mezcla de reacción.
Permite realizar una reacción mas regular, el polímero puede ser soluble en el
disolvente o precipitar a partir de cierto grado de polimerización En este Último caso,
la polimerización en solución da polímeros con buena homogeneidad desde el punto
de vista de grados de polimerización; este proceso exige una cantidad considerable
de disolvente para limitar la viscosidad del medio.
Una vez que se obtiene el producto, el disolvente debe ser eliminado y
recuperado cuidadosamente. Este disolvente tiene la ventaja de que se limita la
temperatura de reacción con el punto de ebullición del disolvente y, operando con
disolventes de punto de ebullición bajo, se obtienen polímeros de pesos moleculares
muy elevados en el caso de una polimerización exotérmica.
Aunque la polimerización en solución tiene varias ventajas, presenta ciertos
problemas, por ejemplo, problemas de purificación. Con frecuencia es difícil eliminar
totalmente el disolvente del polímero, el polímero puede participar en reacciones de
transferencia de cadena favoreciendo la formación de polímeros de bajo peso
molecular. Sin embargo, con los iniciadores que se utilizan en la actualidad, en
concentraciones muy bajas de estos se obtienen pesos moleculares muy elevados
(210~).
9
2.4 Funcionaiización de Poiímeros
Debido al rápido crecimiento en la ciencia y en la tecnología de los polímeros,
se han desarrollar nuevos materiales polimericos que se ajusten a las necesidades
actuales. La introducción de un grupo funcional a la cadena polimerica permite
modificar sus propiedades físicas como la solubilidad, la cristalinidad, estabilidad
térmica.
La funcionalización de un polímero puede ser parcial o total, el porcentaje de
funcionalización puede ser controlado y se puede determinar por métodos químicos o
anaIíticos.
2.4.1 Polisulfobetaínas
Las sulfobetaínas que se pueden preparar a partir del polímero precursor
sintetizado se realizan mediante una reacción de apertura de anillo, como lo es la
1,3-propanosultona o 1,4-butanosultona cuaternizando el nitrógeno como se muestra
en la siguiente reacción:
R'
R= grupo arilo o alquilo
La amina terciaria del polímero precursor actúa como nucleófilo y ataca al
carbono electrofílico que esta unido directamente al oxígeno de la sultona.
Las polisulfobetaínas, pertenecientes a los llamados polizwitteriones, han sido
sujeto de escasos estudios, a pesar de sus diversas propiedades en estado sólido y en
solución; estas interesantes propiedades son debidas a que presentan una estructura
química Única, ya que poseen una naturaleza del tipo iónico, haciéndolas de gran
interés por su facilidad de síntesis y por sus aplicaciones en la recuperación terciaria
del petróleo, en el tratamiento de aguas residuales y en la elaboración de pilas secas.
2.4.2 Policarboxibetaínas
El polímero se cuaterniza para la obtención de la policarboxibetaína, con
reacciones de apertura de anillo con la 1,4-butirolactona:
La amina terciaria del polimero precursor actúa como nucleófilo y ataca al
carbón mas electrofnico que es el que esta unido dierectamente al oxígeno del ester
cíclico.
R-th
I
&+
O
R'
+
R"
R
Rd++CH2FOO-
I
R"
2.4.3 N-Óxidos
La amina terciaria puede ser oxidada por agentes oxidantes comunes como el
peróxido de hidrógeno o peroxiacidos aromáticos, produciendo grupos N-Óxidos
según la reacción siguiente:
I
R= grupo arilo o alquilo
RW
Las reacciones anteriores nos llevan a la formación de zwitteriónes que tiene
propiedades importantes, se incrementa la solubilidad en agua de los materiales
debido a que ahora se tiene una sal cuaternaria, que es una característica favorable
en materiales floculantes.
2.4.4 Compuestos Cuaternarios de Amonio
La reacción entre un halogenuro de alquilo y una amina terciaria alifática,
genera un producto catiónico llamado sal cuaternaria de amonio, donde la carga
positiva la posee el nitrógeno y la carga negativa el halogenuro. La conversión de
aminas terciarias a sales cuaternarias es llamada "Reacción de Menschutkin7.
Si el amoniaco es el nucleófilo, los tres o cuatro grupos alquilo sobre el
nitrógeno del producto final pueden ser idénticos. Si se usa una amina terciaria, los
diferentes grupos alquilo pueden ser colocados en el mismo átomo de nitrógeno
RX
+
NH, y
RNH+,X
amoniaco
Los compuestos que contienen cuatro grupos alquilo unidos al nitrógeno amino
son llamados compuestos cuaternarios de amonio; puesto que son iónicos,
generalmente son solubles en agua y tienen regularmente altos puntos de fusión.
12
2.5 Propuestas d e obtención de Polimeros Funcionalizados
A continuación se proponen rutas de síntesis de polímeros zwitteriónicos del
tipo polisulfobetaínico, policarboxibetaínico, N-Óxido y compuestos cuaternarios de
amonio.
2.5.1 Síntesis de la Polisulfobetaína PDEAEM-S
El polímero precursor poli(DEAEM) se obtiene por radicales libres en solución
como se menciona en la técnica experimental (Capítulo 4).
El polímero precursor se puede cuaternizar para obtener la polisulfobetaína
(PDEAEM-S) con reacciones de apertura de anillo con la 1,3-propanosultona.
El disolvente dimetilformamida (DMF) y la 1,3-propanosultona fueron
destilados a presión reducida.
En un matraz de tres bocas se disuelve el polímero precursor para obtener una
concentración al 2% en una corriente de nitrógeno seco se adicionan 1.1
equivalentes de 1,3-propanosultona, se mantiene en agitación utilizando
dimetilformamida como medio de reacción a una temperatura de reacción de 6OoC
por 24 horas.
En el transcurso de la reacción el producto final va precipitando hasta formar
una gran masa.
AI final de la reacción se decanta el disolvente y se adiciona 2,2,2-trifuuretanol
para disolver el producto, el cual se precipita en un exceso de acetato de etilo, se
filtra y se seca en un horno a vacío a 5OoC durante 24 horas.
Reacción de Polimerización de la polisulfobetaína
2.5.2 Síntesis de la policarboxibetaína PDEAEM-Clo
El polímero precursor poli(DEAEM) se obtiene por radicales libres en solución
como se menciona en la técnica experimental (Capítulo 4).
El polímero precursor se puede cuaternizar para obtener la policarboxibetaína
(PDEAEM-C) con reacciones de apertura de anillo con la 1,4-butiriIactona
En un matraz de tres bocas se disuelve el polímero precursor en 1,2dicloroetano (DCE) para obtener una solución al 2% y en una corriente de nitrógeno
seco se adiciona 1.1equivalentes de 1,4-butiroIactona, la cual fue destilada a presión
reducida. La reacción se lleva a una temperatura de 7OoC durante 72 horas.
El polímero ya cuaternizado precipita en el medio de reacción, se filtra y se
seca en una estufa con vacío a 6OoC durante 24 h.
CH3
I
-( C-CH2-)"
I
CH,
y 4 3
CH,
u
3 dias
Reacción de polimerización PDEAEM-C
14
2.5.3 Síntesis del N-Oxido del PDEAEMA-NOll
La reacción de oxidación del Poli[N-(-N-Dietilaminoetoxi)etilmetacrilato]
(PDEAEEM), la realizamos dentro de la campana de extracción en un sistema de
reflujo utilizando un baño de aceite. Se disuelve el polímero en la menor cantidad de
ácido acético g/acia/(2 mi) y cuando el sistema alcanza el equilibrio térmico a 6OoC
se adiciona lentamente el peróxido de hidrógeno (1.87 mi) en relación molar 1:1 por
unidad monomerica, después de 3 horas de reacción se adiciona un 30% de exceso
de peróxido. La reacción se llevó a cabo a una temperatura constante de 60OC por 18
horas con agitación magnética.
AI final de la reacción se elimina el exceso de ácido acético por destilación a
presión reducida, el polimero se lavó varias veces disolviéndolo en agua
tridesionizada, rotaevaporando el azeótropo de agua-ácido en cada lavado, hasta
obtener un pH de 4 en la solución del polímero (teniendo cuidado de que la
temperatura del baño del rotavapor no exceda de 60OC).
15
2.5.4 Síntesis de Compuestos Cuaternizados de Amonio12
Se utiliza como polímero precursor con aminas terciarias al PDEAEMA, el cual
se debe de secar en una estufa a 30 O C y con vacío por 24 h. Los halogenuros de
alquilo que se pueden emplear son el bromoetano y el bromometano, disueltos en
dicloroetano (DCE) y cloroformo (CHC13) respectivamente, sin purificar ya que es
difícil su manipulación. Para obtener el bromuro de poli(trietilaminoetiImetacrilato)
[(PTEAEMA)Br], en un matraz erlenmeyer con tapón esmerilado se hace la mezcla
DCE/bromoetano en la relación 10:1, y sobre ella se adiciona el PDEAEMA seco,
inmediatamente se tapa el matraz y se cubre perfectamente con papel aluminio para
protegerlo de la luz y del aire. Se agita manualmente para disolver el PDEAEMA,
después se deja reposar por 48 horas.
En la síntesis del bromuro de poi(dietiImetilaminoetiImetacrilato)
[(PDMEAEMA)Br], se disuelve primero el PDEAEMA en CHC13 en un matraz
erlenmeyer con tapón esmerilado, debido a que el bromometano se adiciona
inmediatamente después de haberlo disuelto en CHC13, se tapa el matraz
perfectamente y también se cubre con papel aluminio, se agita manualmente y se
deja reposar por 48 horas.
Los productos obtenidos son sólidos blancos a los que se les evaporó el
disolvente y el halogenuro de alquilo que no reacciona, después son secados en una
estufa a 30 O C y con vació por 24 horas. Se espera que ambos productos sean
solubles en agua.
2.6 Caracterización Fisicoquímica
La caracterización es el proceso mediante el cual se obtiene información
acerca de la estructura química de los polímeros, su forma, conformación y
movimientos moleculares causados por la acción del calor, etc.
La técnicas espectroscópicas utilizadas en la caracterización de los polímeros
sintetizados son espectroscopía de resonancia magnética nuclear (RMN) protónica y
de carbono trece y espectroscopia de infrarrojo con tansformada de Fourier.
Cuando se hace referencia al peso molecular de un polímero, no tenemos una
sustancia pura en el sentido estricto de la palabra, ya que en realidad tenemos una
mezcla de moléculas de diferente peso molecular; aún en el caso en que se sinteticen
estos materiales en ausencia de contaminantes e impurezas.
La mayoría de las propiedades mecánicas de estos materiales dependen del
peso molecular. Si la intensidad de las fuerzas electrostaticas por unidad de longitud
para una colección de moléculas es la misma, tal como en las series homólogas,
entonces la magnitud de las fuerzas atractivas se incrementa conforme crece el peso
molecular. Tal incremento conduce primero a cambios en el estado físico.
Se conocen varios métodos que determinan experimentalmente el peso
molecular promedio de un polímero. Dichos métodos se basan en propiedades
coligativas, refracción de la luz, viscosidad, ultracentrifugación y sedimentación. En el
presente trabajo nos enfocaremos Únicamente a la determinación de peso molecular
promedio por dispersión de luz.
17
2.6.1 Espectroscopia de Infrarrojo (FT-IR)
9 '
Para una irradiación electromagnética hay absorción cuando una molécula
pasa de un estado de energía El, a un estado de energía E2 superior. La frecuencia
de la irradiación adsorbida viene dada por la relación de Bohr.
En el estudio de un espectro de I R de una molécula, se trata primeramente de
determinar los tipos de vibración que provocan las bandas de absorción.
Los átomos que constituyen una molécula están en constante vibración.
Cuando el número de átomos en una molécula excede de 10 o mas, el número de
modos posibles de vibración se vuelve muy grande. Afortunadamente, muchas
frecuencias son características para enlaces localizados. Así la absorción de la luz por
el estiramiento del enlace C-H casi siempre ocurre a frecuencias entre 2880 y 2900
cm-'. La mayoría de los espectrofotómetros usan una fuente de luz incandescente
para proporcionar luz con longitudes de onda de 2.5 a unos 15 pm que corresponde
a una frecuencia de 4000 cm hasta 670 cm -'.Un prisma giratorio o una rejilla de
difracción descomponen la luz de la fuente en un espectro del que se aislan
progresivamente las diferentes longitudes de onda con la ayuda de filtros.
La muestra puede ser una película delgada sin soporte (generalmente
alrededor de 0.001 pulgadas de espesor), una película por evaporación sobre un
sustrato no absorbente como el NaCI, una oblea prensada transparente del material
mezclado con polvo de KBr, etc.
Además de usarse en la identificación de polímeros desconocidos, impurezas o
grupos en los extremos de la cadena de los polímeros; con algunas modificaciones,
el análisis de infrarrojo se usa para medir la cristalinidad y la orientación de grupos
específicos en los polímeros cristalinos. Las reacciones químicas de los polímeros
pueden seguirse por la aparición o desaparición de grupos funcionales tales como
grupos oxigenados en la oxidación o de grupos hoxidrilo libres que se originan por la
hidrólisis.
18
2.6.2 Espectroscopia de Resonancia Magnética Nuclear
(Rl")5
La resonancia magnética nuclear es una técnica espectroscópica que estudia
los núcleos en resonancia en presencia de un campo magnético, no desde el punto
de vista físico, sino en términos de la estructura molecular. Su fundamento
fisicoquímico se basa en el movimiento de precesión que presenta el espín de ciertos
átomos, específicamente los que cuentan número y/o masa impar, como el hidrógeno
y el isótopo de masa atómica 13 del carbono.
La resonancia magnética se sitúa en el espectro electromagnético en la zona
de la radio frecuencia. Los métodos espectroscópicos son clasificados de acuerdo con
la longitud de onda del espectro electromagnético con la que absorbe o emite energía
una muestra de material, o de acuerdo con el mecanismo involucrado en la absorción
de energía.
La frecuencia de precesión (Wo Ó vo)esta expresada por la ecuación de Larmor
como:
WO=-y.HO
y
vO=WO/2.n
donde y es la razón magnetogírica y se expresa como:
y = 2.n
pN/
h
Mi
donde pN = Momento magnético del protón.
h = constante de Plank.
M I = Número cuántico de espín.
En el espectro de RMN es posible medir el area bajo la curva o la integral de
cada uno de los picos de éstas señales, ya que la absorción es proporcional al
número de núcleos magnéticamente activos que la ocasionan. La integral se registra
electrónicamente como una señal separada, después de obtenido el espectro de
absorción. En un espectro de RMN protónico de un compuesto puro es posible
estimar el número de protones en la molécula.
Los equipos de RMN pueden ir de 60 Mhz a 500 MHz o mas. La muestra para
estos equipos es una solución que contenga una concentración mínima de 10 mg de
sustancia en 0.5 ml de disolvente. Los disolventes empleados no deben contener
protones, como el tetracloruro de carbono, o usar disolventes deuterados (uno de los
isótopos del hidrógeno), con el fin de evitar la interferencia de la señal del disolvente
con la señal de la muestra. La solución se coloca en tubo de vidrio de 5mm de
diámetro externo y se adiciona la sustancia de referencia (generalmente TMS en
solución del disolvente escogido). El tubo con la muestra se coloca en la sonda del
instrumento, y se hace girar para promediar las inhomogeneidades del campo
magnético y se determina el espectro de absorción. Generalmente el disolvente es el
cloroformo deuterado.
19
2.6.3 Dispersión de Luz2
Esta técnica proporciona basta información sobre las cadenas polimericas ya
que se puede obtener no solamente el peso molecular promedio, sino también el
radio de giro o distancia de extremo a extremo y el segundo coeficiente del virial
(Az). Además es posible tener idea de la forma de la macromolécula en solución
(esferas, ovillos, barritas). La limitación principal de esta técnica es el tiempo que
requiere en la preparación de las soluciones.
El fenómeno de dispersión de la luz por partículas fue estudiado por Raleigh. El
método consiste, esencialmente, en una medida de la intensidad de luz dispersada, a
diferentes ángulos, por soluciones de distinta concentración. Después se procesa esta
información para calcular los parámetros estadísticos.
Los cálculos teóricos muestran que la intensidad de la difusión de la luz se
puede poner en la forma:
I = KoMw OC
siendo Mw = Peso molecular promedio
C = Concentración de la solución
expresión en la cual:
K = (2~’n’/ho4N)(dn/dc)
’
en donde: :
n = índice de refracción del disolvente.
ho = longitud de onda en el vacío.
N = número de Avogadro
dn = variación del índice de la solución en función de la concentración.
dC
Esta fórmula es válida en el caso de moléculas pequeñas con respecto a la
longitud de onda (L < ho /20) y que no presenta interacciones.
De una manera mas general se puede escribir:
c= o
en donde:
k = 2x2n2 fdnI2
h$N \dcj
y
I‘ = (I - Io) a
20
Po = función sin dimensión cuyo máximo es 1 para 8 = O y cuya variación depende
de la forma y del tamaño de las moléculas estudiadas.
A2 = segundo coeficiente del virial
Por lo anterior se observa que la difusión de la luz aporta dos medidas
simultáneas: la primera en función de la concentración; la segunda en función del
ángulo de observación respecto al ángulo incidente, y las dos extrapolaciones, una a
8 = O otra a c = O serán necesarias para obtener el valor de Mw y A2.
De la representación gráfica de los valores de (KC / Re) en función de
[sen2@/2) + KC] se obtienen lo que se conoce como gráfica de Zmm como se
muestra en la figura 1. El valor de K es arbitrario para establecer las mejores
condiciones de la gráfica.
T
Sen28/2
+ KC
Figura 1. Representación de una gráfica de Zimm.
Referencias
1.- ANGULO SANCHEZ, Caracterización Fisicoquímica de los Polhneros,
LIMUSA, la. edición, México, 1994.
2.-RODRÍGUEZ F., Principios de Sistemas de Polimeros, Manual
Moderno, 2a. edición, México, 1984.
3 .- URIBE VELASCO M., Los Polimeros Síntesis y Caracterización,
LIMUSA, 2a. edición, Mexico, 1990.
4.- FLORY P., Principles of Polymer Chemistry, Cornel1 University Press,
9" edición, London 1975.
BOYD.Química Orgánica. 5" Edición. Pag 943 Editorial
Addison Wesley ( 1987).
5.-MORRISON AND
'.- J A N F. RABEK, Experimental Methods in Polymer Chemistry, John
Wiley and Sons, 1" edición, New York, 1980.
7.- JERRY MARCH. Advanced organic Chemistry. 4O Edición. Pag 411. Editorial
Wiley Interscience (1992).
8.- CARDOS0 M. J. Síntesis, caracterización y propiedades de polímeros y
co-polímeros zwitteriÓnicos.TesisiDoctoral. FQ-UNAM (1990).
9.- RAÚL G. MANRIQUE G. Síntesis y caracterización de poliesteres
zwitteriónicos del tipo sulfobetaínico. Tesis Profesional. Universidad Autónoma
de Tlaxcala. Depto de Ingeniería y Tecnología. (1995).
10.- ARMANDO CONTRERAS PINEDA. Síntesis de polímeros mitteriónicos de
alto peso molecular tipo sufobetaínicos y carboxibetaínicos. Tesis
Profecional. Universidad Nacional Autónoma De Mexico. Facultad de Química. (1991).
11.- V. M. MONROY SOTO and GAUN. 'Poly(sulphopropylbetaines: 1. Syntesis and
Characterization", Polymer 25, 1984 (January).
12.- ELOISA ANLEU A. Síntesis, Caracterización y Propiedades de FloculaciÓn
de Polímeros Cuaternizados. Investigación para el desarrollo de la Tesis Doctoral.
UAM-I (1998).
22
Capítulo 3
Desarrollo Experimental
3.1 Purificación de reactivos
Es necesario purificar los reactivos para tenerlos en estado anhídro y para
eliminar las impurezas que puedan contener, ya que al tener el mayor grado de
pureza podemos obtener un mayor peso molecular del polímero.
Los reactivos que fueron purificados son los siguientes: Hexano, Sodio
Metálico, Benceno, Acetona, 1,3-Dinitrobenceno, DEAEE, Cloruro de Metacriloilo y el
monómero DEAEEM.
3 . 1 . 1 Purificación de Hexano
Es necesario que el hexano este libre de agua, para lavar con el el sodio y para
precipitar los polímeros precursores. En un matraz bola de 1 L se adiciona cloruro de
calcio (5 g) y el hexano que se va a secar (500 mi) y se dejan reposar por 24 horas.
Para la destilación del hexano, se monta el sistema integrado de destilación
como de muestra la figura 3.1. El hexano se destila sobre CaC12 a 6OoC en un baño
de agua que se
Figura 3.1. Sistema integrado de destilación.
23
3.1.2 Purificación de Nao
Hay que tener en cuenta que el sodio metálico (Nao) es muy reactivo con el
agua y, que al contacto con el medio ambiente genera Óxidos.
Se toma una rodaja de Nao y rápidamente se limpia con papel para eliminar el
exceso de aceite, inmediatamente se coloca la rodaja en un vaso de p.p. chico que
contiene hexano anhídro y con la ayuda de la espátula se raspa la rodaja para
eliminar los residuos de cera, aceite y Óxidos de sodio que la protegen del medio
ambiente, cuando el hexano ya esta muy turbio cambiamos la rodaja a otro vaso de
p.p. con hexano limpio y seco, aplicando el procedimiento anterior, hasta que el
hexano ya no se vea turbio y que la rodaja de Nao presente un color gris metálico. AI
sodio metálico limpio rápidamente lo sacamos del hexano, lo secamos con un papel
absorbente para eliminar el exceso de disolvente, lo pesamos e inmediatamente lo
adicionamos al matraz que contiene el disolvente que se va a secar.
Para neutralizar los residuos de Nao se adiciona una muy pequeña cantidad
de estos en metano1 en la campana de extracción, ya que se produce una reacción
violenta con desprendimiento de gases. Si se adicionan cantidades grandes de
residuos de Nao se generara una explosión con formación de chispas y gases
inflamables (Hz).
Papel
+
-+
Papel
+
+
Disolvente
Figura 3.2 Purificación del Sodio Metálico
24
3.1.3 Purificación de Benceno
Montamos el sistema integrado como se muestra en la figura 3.3 de reflujodestilación. Adicionamos al matraz de reacción el benceno que vamos a secar (250
mi)/ benzofenona (1 g) y el sodio metálico limpio (1 g)/ ponemos en reflujo al
sistema donde la solución ira cambiando de color a medida que el benceno se va
secando. Éste cambiara de amarillo a verde y finalmente se pondrá azul intenso, lo
cual nos indicará que ya no hay agua presente en el benceno, de esta forma el sodio
ha reaccionado con toda el agua presente, formando un complejo con la benzofenona
dando una coloración azul intensa. Se procede a la destilación del benceno el cual
debe ser incoloro. Los residuos del Nao se neutralizan como se indica en la pagina
anterior.
O
Benceno
Benzofenona
Figura 3.3 Sistema de Reflujo-Destilación
Azu I
3.1.4 Purificación de 1,3-Dinitrobenceno
El 1,3-Dinitrobenceno (DNB) se usa como inhibidor en la formación de
radicales libres.
Se pesa el dinitrobenceno (5 g) y se disuelve en etanol caliente (150 ml); se
deja reposar a temperatura ambiente, cubriendo con papel el vaso. Después de que
la solución se haya enfriado se filtran con vacío las agujas y se lavan con 10 ml de
etanol frío. Se deja evaporar el etanol en un desecador. Se determina su p.f., y si
este es igual al reportado en la literatura (teórico 88-90 "C), se coloca en un frasco
ámbar y se guarda en el desecador. En caso de que el punto de fusión sea menor se
debe realizar el proceso anterior tantas veces como sea necesario hasta alcanzar el
p.f. teórico. Las aguas madres se pueden volver a concentrar y a filtrar, para obtener
más producto. Se puede seguir realizando lo anterior hasta que la producción de
cristales ya no se de, seguida de su purificación total. Colocar las agujas en un frasco
ámbar y guardarlas en el desecador.
Calentamiento
Filtrado
Secado
Figura 3.4 Cistalización del 1,3-dinitrobenceno
3.1.5 Purificación de Dietilaminoetoxietanol (DEAEE)
Se requiere utilizar presión reducida en la destilación ya que el punto de ebullición del 2-(2-Dimetilaminoetoxi)etanol es de 100-104°C a 10 mmHg y se recomendable utilizar
mínimo dos trampas de vacío con una mezcla frigorífica (hielo-sal 1:3, hielo seco- acetona
Ó N2 (I), para proteger la bomba.
Se monta el sistema de destilación integrado a presión reducida como se muestra en la
Figura 3.5. Se mide con una pipeta graduada el volumen que se va a destilar (50 mi) y se
adiciona a un matraz bola (100 mi) que contiene una pequeña cantidad de 1,3dinitrobenceno(0.5 g).
Se ayuda a dar calentamiento a la columna de destilación con franelas calientes para
que la destilación inicie rápidamente. Se descabeza con 3 gotas del alcohol, se recolecta el
cuerpo de la destilación. Cuando se observe que baja significativamente la temperatura de
destilación se cambia al matraz cola y se aumenta la temperatura para terminar la
destilación.
Las condiciones en las cuales se destilo el alcohol fueron a una presión de 8 mmHg y la
temperatura de destilación fue de 90°C, el reostato con el cual se trabajo se encontraba
en 40 volts. Obteniendo el destilado incoloro.
Los residuos que quedan en el matraz de reacción (mezcla chiclosa color cafe) se
pueden remover con un poco de benceno, los cuales se deben guardar en un frasco para
desechos.
Figura 3.5 Sistema de destilación integrado a presión reducida.
27
3.1.6 Purificación de Cloruro de Metacriloilo
Es necesario realizar esta destilación dentro de la campana de extracción y con
la debida protección personal ya que este reactivo es muy tóxico, flamable,
lacrimógeno e irritante de la piel.
Se monta el sistema de destilación integrado como se muestra en la Figura 3.6
Se mide con una pipeta graduada los 10 ml que se va a destilar, adicionándolos al
matraz de reacción de 50 ml que contiene 0.5 g de 1,3-dinitrobenceno, el cual evita
la polimerización del cloruro iniciada por calentamiento. Se cubre al matraz
recolector con papel aluminio y se pesa éste con el papel y su respectivo tapón. Se
da calentamiento a la columna de destilación para que la destilación inicie en menor
tiempo.
Es importante preparar una solución de hidróxido de sodio concentrada para
neutralizar los residuos del cloruro de metacriloilo, haciendo enjuagues con la
solución en el sistema de destilación-intregado, en los matraces recolectores, en el
matraz de reacción y, todo el material de vidrio que haya tenido contacto con el
reactivo; posteriormente sumergir el material de vidrio en una tina que contenga
agua con jabón y dejarlo reposar por un rato (todo esto se realiza dentro de la
campana de extracción).
Las condiciones en las cuales se obtuvo la destilación son: temperatura de
destilación 87OC a presión atmosférica, el reostato con el cual se trabajo se
encontraba en 40 volts, el recirculador de agua contenía una mezcla de agua-hielo y
se obtuvo un destilado incoloro (aprox. 6 mi).
Una vez destilado el cloruro de metacriloilo se debe usar inmediatamente.
Figura 3.6 Sistema de destilación simple.
3.1.7 Purificación del Monómero DEAEEM
La purificación del monómero 2-(2dietilaminoetoxi)etilmetacrilato)
(DEAEEM), se realizó en un sistema de destilación integrado a presión reducida,
modificando el sistema como se muestra en la figura 3.7, donde la entrada del vacío
se alejó de la parte donde condensan los vapores del monómero para que este no
fuera arrastrado por la bomba, se utilizaron dos trampas con N2 (I) para proteger la
bomba de vacío.
Se cubren con papel aluminio los matraces de destilación y recolector, se les
adiciona una pequeña cantidad de DNB para evitar la polimerización del monómero
por calentamiento, posteriormente se pesa cada uno de los matraz cubiertos con el
papel aluminio, con los sólidos y con su respectivo tapón, este procedimiento se
realiza para saber cuanto monómero se polimeriza, en caso de que suceda; una vez
montado el sistema se protege de la luz, ya sea con franelas o papel aluminio. Se
mide con una probeta los 6 ml que se van a destilar del monómero. Se adicionan al
matraz de destilación que contiene el DNB, y se le da calentamiento a la columna de
destilación con franelas calientes para que la destilación inicie rápidamente.
Descabezar con 2 Ó 3 gotas de monómero, girar el matraz "vaquita" para
recolectar el cuerpo y cuando se observe un cambio significativo en la temperatura
de destilación, se cambia de matraz y se aumenta el calentamiento para recolectar la
cola de destilación.
Las condiciones en las cuales se dio la la destilación son: Temperatura
de destilación 74% a presión reducida, el reostato con el cual se trabajo se
encontraba en 40 volts, el recirculador de agua contenía una mezcla de agua-hielo.
Se obtuvo aproximadamente 4 ml de destilado ligeramente amarillo transparente el
cual se protegió de la luz.
Figura 3.7 Sistema de destilación integrado con presión reducida modificado.
Capítulo 4
Técnica Experimental
4. Síntesis del MonÓmero N,N-dietilaminoetoxietiZmetacri1ato
(DEAEEM)
El N,N-dietilaminoetoxietilmetacrilato (DEAEEM) fue obtenido al realizar la
esterificación de la N,N-dietilamina (DEAEE) con el cloruro de metacriloilo, según la
técnica descrita por Monrroy y Galin'.
A un matraz bola de tres bocas de 100 ml se le coloca en una de las bocas un
adaptador con un globo con N2 (9) el cual se seco haciéndolo pasar a traves de
NaOH (s) y después por una solución de pirogalol, que nos permite tener una
atmósfera inerte y seca, en la segunda boca se le coloca un adaptador para
termómetro y en la tercer boca un adaptador con llave en la cual se le conecta una
trampa de aceite, esta trampa nos permite controlar el flujo de salida del H2 (9). La
agitación se realiza con un agitador magnético. Ver fig. 4.1.
Se adiciona al matraz de tres bocas 2 g de hídruro de s d í o y, 24 ml. de
benceno seco se observa una efervescencia debida a el desprendimiento de H2;
colocar el matraz en un baño con agua-hielo y bajo agitación se adicionan
lentamente 6.0 ml. de DEAEE N,N-díetí/amín~~xíe~no/
recién destilado, con
el baño y la adición lenta del DEAEE aseguramos que la temperatura se mantenga
por debajo de los 28OC, al final de la adición se retira el baño de hielo y se calienta
el sistema con baño María a 50OC hasta que deje de haber formación de H2(g)
(aprox. 4 horas). Simultáneamente se esta realizando la destilación del cloruro de
metacriloilo, ya que se requiere recién destilado.
CH
NaH
+
H-(-O-CH;CH;),-N
I
L
442
kNa+-(-O-CH;CHz)-2$
c H3
Formación del alcóxido
c H3
Cuando haya cesado la formación de H2(g) se retira el calentamiento y, se
adiciona al matraz de reacción 0.0117 g de 1,3-dínít.benceno.Se enfría la
mezcla a 100C en un baño de agua con hielo; gota a gota es adicionado 3.6 ml de el
c/oruro de me&crí/oí/o recién destilado, cuidando que en la adición la
temperatura de la solución no exceda los 20OC, es posible que pueda dar lugar la
gelación de la solución (amarilla) a pesar de la continua agitación. Se deja reposar
toda la noche.
CH3
y
3
C=CH,
c-CI-
+
II
O
kH,
I
Na+-(-O-CHiCH,)-,N
$ 4 2
Tolueno
N2
DNB
-NaCI
CH3
Cloruro de Metacriloilo
CH3
C=CH,
I
O=C(-O-CHiCH
CH3
CH2
-'$H2
CH3
Monómero DEAEEM
Reacción de Esterificación
Se separa el c/oruro de s d í o (NaCI) formado por centrifugación, los
residuos del cloruro de sodio se lavan con porciones de benceno seco, adicionando
los lavados al sobrenadante (monómero). Se elimina el benceno del monómero por
la rotaevaporación de la solución a temperatura ambiente y a presión reducida, con
lo que se obtiene el monómero (solución amarilla obscura ligeramente espesa).
Fig. 4.1 Sistema de Reacción.
31
4.2 S í n t e s i s d e l PolíDEAEEM
20-1 OOOC
(CH,),C-N=N-C-(CH,),
2(CH3),-C*
I
CN
I
CN
CN
AlBN
+ N,
Radical Libre
Reacción de Formación de Radicales
Reacción de Polimerización de DEAEEM
La reacción de polimerización en solución se llevó a cabo vía radicales libres,
usando azobisisobutironitrilo (AIBN) como iniciador.
La reacción de polimerización del [N-(N-dietilaminoetoxi)etilmetacrilato]
DEAEEM se llevó a cabo en un sistema similar al descrito en (4.1), con la diferencia
de que la fuente de calentamiento es un baño de aceite (glicerol o nujol) el cual debe
mantenerse a una temperatura de 50OC. Se le adiciona al matraz de reacción 5 ml
de benceno seco y 3 ml de monómero DEAEEM recién destilado; una vez alcanzado
el equilibrio térmico de 60OC son adicionados 0.00727 g. de AIBN en una
concentración de 4 ~ 1 0mol/l
- ~ previamente disuelto en benceno seco.
La reacción de polimerización se deja por 21 horas a una temperatura
constante de 6OoC, protegiendo al sistema de la luz para evitar la polimerización de
radicales libres iniciada por ésta.
El benceno se eliminó por rotaevaporación como se muestra en la figura 4.2, el
material resultante se precipitó en hexano frío y se dejó secar en la estufa con vacío
a 50OC por 24 horas.
,
I-
Figura 4.2. Rotavapor con vacío.
4.3 Síntesis del N-Oxido del PDEMAEEM
CH3
CH3
+Hi%
CH3
O=C - (O-CH2-C H 2 WI :
I
H A
CH,COOH
t{-CH2*
o=c - p
CH3
Reacción de N-oxidación de PDEAEEM
CH3
cH-c H2J+-CH3
La reacción de oxidación del Poli[N-(-N-Dietilaminoetoxi)etilmetacrilato]
(PDEAEEM), la realizamos dentro de la campana de extracción en un sistema de
reflujo (ver fig. 4.3) utilizando un baño de aceite. Se disuelve el polímero en la
menor cantidad de ácido acético glacial (2 mi) y cuando el sistema alcanzó el
equilibrio térmico a 6OoC se adiciona lentamente el peróxido de hidrógeno (1.87 mi)
en relación molar 1:l por unidad monomerica, después de 3 horas de reacción se
adicionó un 30% de exceso de peróxido. La reacción se llevó a cabo a una
temperatura constante de 60OC por 18 horas con agitación magnética.
Al final de la reacción se elimina el exceso de ácido acético por destilación a
presión reducida, el polímero se lavó varias veces disolviéndolo en agua
tridesionizada, rotaevaporando el azeótropo de agua-ácido en cada lavado, hasta
obtener un pH de 4 en la solución del polímero (teniendo cuidado de que la
temperatura del baño del rotavapor no exceda de 60%).
Fig. 4.3 Sistema de Oxidación.
33
4.4 Purificación del N-Oxido del PDEAEEM
Para purificar el polimero N-Óxido es necesario pasarlo por una resina de
intercambio iónico fuertertemente básica para eliminar a los iónes acetato del ácido
acético que contiene la solución, esta resina se necesita activar previamente de la
siguiente manera:
Para activar 200 ml de la resina de intercambio iónico se requieren
aproximadamente 30 ml de una solución de hidróxido de sodio 0.1 M y 70 ml de agua
tridesionizada con lo cual se obtiene un pH entre 12-13 y se deja reposar por 4
horas; después de transcurrido el tiempo la resina esta activada, es necesario lavarla
con agua tridesionizada hasta que el pH de los lavados sea de 7.
Se empaca una columna ancha con la resina activada, es recomendable
aplicarle vacío a la columna con un matraz kitazato para que el polímero baje más
rápido. La solución del polímero con pH 4 se adiciona a la columna poco a poco, la
cual va bajando de la columna con adiciones de agua tridesionizada, el polímero es
recolectado cuando el goteo en la salida de la columna presenta un pH 9, se sigue
adicionando agua tridesionizada hasta que el pH cambie a 6. Se hace otra adición de
la solución del polimero a la columna empacada con la resina activada y se procede
de manera análoga hasta terminar con la solución. El polímero que se recolectó con
pH 8-9 contiene agua, la cual se elimina rotaevaporando la solución, obteniendo al
final una solución viscosa amarilla, que se precipitó en acetona fría y se seco en la
estufa con vació a 50OC por 24 horas.
Figura 4.4. Sistema de Intercambio IÓnico.
34
4.5 Caracterización
Determinación de estructura química por espectroscopia de infrarrojo (FT-IR) y
resonancia magnética nuclear (RMN) y determinación de peso molecular promedio
por dispersión de luz.
4.5.1 Espectroscopia de Infrarrojo (FT-IR)
La caracterización de los polimeros precursores mediante esta técnica fue a
traves de películas de los materiales, las cuales se prepararon disolviendo los
polimeros en metano1 y colocándolos en unas celdas de cloruro de sodio (NaCI), se
dejaban secar a temperatura ambiente. El equipo en el cual se analizaron las
muestras es un espectrofotómetro F I I R marca PERKIN-ELMER, modelo 1600.
4.5.2 Espectroscopia de Resonancia Magnética Nuclear (RMN)
La caracterización de los polímeros precursores mediante esta técnica fue a
traves de RMN de 'H y de I3C. Las muestras se prepararon disolviendo los polimeros
precursores en cloroformo deuterado (CDC13) a 25OC. El equipo en el cual se
analizaron las muestras es un espectrofotómetro BRUKER modelo DMX500.
4.5.3 Dispersión de Luz
Primeramente hubo que determinar el dn/dc de los polimeros precursores se
preparaon 5 soluciones en cloroformo filtrado con una concentración del orden de
manteniendo estas en agitación constante hasta la disolución del polímero.
Para la técnica de dispersión de luz se preparan 5 soluciones en cloroformo
a
se mantienen en agitación hasta la
con una concentración del orden de
disolución el polímero. Las soluciones se prepararon con el disolvente desgasificado y
filtrado utilizando una membrana milipore VC de 0.10 micras para filtrar el disolvente
y después se filtraron las soluciones utilizando una membrana milipore HA de 0.45
micras. Las soluciones son filtradas directamente en las celdas de vidrio debidamente
lavadas en las cuales el equipo realiza la medición. Es muy importante que todo este
proceso de filtado se realice en un area aislada del polvo, ya que este afecta en las
mediciones. El equipo en el cual se analizaron las muestras es un DAWN, modelo F
W A l T TECHNOLOGY.
35
Una vez filtradas las muestras estas deben estar en agitación mínimo una hora
antes de realizar las corridas. Las muestras se introducen en el equipo iniciando por
la mas diluida y se corren las muestras en el programa DAWN. AI finalizar las corridas
de las muestras el programa AURORA muestra las gráficas de los datos colectados
(Zimm, Debye y Berry) en los cuales obtenemos el peso molecular (Mw), el segundo
coeficiente del virial (Az) y el radio promedio cuadratico (RMS).
LAVADO DEL MATERIAL DE VIDRIO
Las celdas de medición se lavan con una mezcla crómica durante 24 horas, se
enjuagan abundantemente con agua desionizada y después muchas veces con agua
desionizada previamente filtrada con membrana milipore VC 0.10 micras. Se colocan
boca abajo en un cristalizador se tapa éste con otro mas grande y se secan a la
estufa a 11OOC.
Referencias
1.- V. M. MONROY SOTO and GALIN. "Poly(sulphopropylbetaines:1. Syntesis and
Characterization", Polymer 25, 1984 (January).
Capítulo 5
Resultados
5.1 Formación del Aicóxido DEAEE
La formación del alcóxido del N,N-dietilaminoetoxietanol se obtiene de la
reacción de sustitución entre el alcohol N, N-dietilaminoetoxietanol (DEAEE) y el
hidruro de sodio como se muestra en la siguiente reacción:
Formación del alcóxido
donde el sodio del hidruro reemplaza a un átomo de hidrógeno del alcohol DEAEE,
dando origen a la formación del correspondiente alcóxido.
Antes de realizar cualquier reacción química es importante saber cual es la
confiabilidad de nuestros reactivos, para asegurarnos de la pureza del DEAEE con el
cual trabajamos fue necesario bidestilarlo y posteriormente caracterizarlo por técnicas
espectrofotométricas que verificaron la presencia y el acoplamiento de los grupos
funcionales en su estructura química; ya que el DEAEE puede degradarse o perderse
sino se mantienen las condiciones adecuadas en su destilación, como se mencionó
en la parte experimental.
I
&
H O-CH2-CH~0-CH~CH2-N
CH,
CHj
Estructura del DEAEE
37
5 . 1 . 1 Caracterización por FT-IR
La determinación de la estructura molecular del
2-(-2( N,Ndietilaminoetoxi)etanol)DEAEE recién destilado se realizó por FT-IR, obteniendo el
espectro que se muestra en la figura 1, en 3384 cm-' se observa la banda de una
vibración (O-H) asociada a un alcohol. La señal de 2970 cm-' corresponde a un
alargamiento del enlace (C-H) del metileno CH2; la señal en 2934 cm-' corresponde a
una tensión de la vibración (C-H) de un CH3 alifatico y la señal en 2872 cm-',
corresponde a una tensión de la vibración (C-H) de un CH2 alifatico. La señal en
1458 cm-' corresponde a la deformación simétrica del enlace (C-H) de un CH3 y la
banda en 1376 cm-' corresponde a la deformación simétrica de una vibración (C-H)
de un CH2. Las señales subsecuentes corresponden a flexiones simétricas y
asimétricas del tipo rocking de los metilenos. La banda de 1126 cm-' corresponde a
una vibración (O- CH2) del un ester y la banda en 1066 cm-' corresponde a la tensión
(C-O) de un alcohol primario. El análisis anterior nos muestra que el espectro de la
figura 1, corresponde a la estructura molecular del DEAEE.
CH,
CH2
H O-CH2-CH~0-CH~CH2-N
CH2
Figura 1. Espectro FT-IR de 2-(2-(N,N-dietilaminoetoxi)etanol)
(DEAEE).
38
5.1.2 Caracterización por R M N
Figura 2. Espectro d e RMN-IH de 2-(2-(N,N-dietilaminoetoxi)etanol)
(DEAEE).
Protón CH*3-CH2-N
a
ppm
1.o
CH3-CH*2-N
b
2.5
O-CH2-CH*2-N CH*2-O-CH*2
b'
C
2.7
3.6
H-O-C*Hz-CHz
d
3.7
39
La figura 3,
representa el espectro de 13C del 2-(2-(N,Ndietilaminoetoxi)etanol) DEAEE, este espectro nos proporciona información de
cuántos y de que tipo de átomos de carbonos hay presentes en la estructura del
DEAEE, el análisis se muestra en la tabla 2.
Tabla 2. Análisis del espectro de RMN de 13C del DEAEE
C"
ppm
C* H3-CH2- N
N-C"H2-CH3
O-CH2-C" H2-N
O-C"H2-CH2-N
O-CH2-C"H2-O
H-O-C"H2-CH2-O
11
47
53
62
69
73
Está caracterización espectroscópica nos permitió verificar que el compuesto
destilado si es el 2-(2-(N,N-dietilaminoetoxi)etanol)
(DEAEE), por lo cual pudimos
proseguir con la siguiente reacción para la obtención del monómero.
40
5.2 Síntesis del DEAEEM
Para la obtención del monómero N,N-dietilaminoetoxietilmetacrilato (DAEEM)
se realizó una reacción de esterificación entre el alcóxido del DEAEE y el Cloruro de
Metacriloilo, como se muestra en la siguientes reacción:
y43
C=CH2
I
CH3 Tolueno
¿H2
N*
+
Na+-(-O-CH,CH,)-,y
C-CIO
II
y
DNB
I
-NaCI
CH3
I
C=CH2
y 4 3
CH2
O=C- (-0-CHiC H2)-2-qi
2
y
CH3
2
CH3
Monómero DEAEEM
Cloruro de Metacriloilo
Reacción de Esterificación.
Para verificar que el producto obtenido de la reacción de esterificación fue el
monómero N,N-dietilaminoetoxietilmetacrilato DEAEEM fue necesario caracterizarlo
por técnicas espectroscópicas. La caracterización se realizó inmediatamente después
de que se destiló el DEAEEM y lo mas pronto posible, ya que este podía polimerizar
por la presencia de la luz.
CH3
CI X H ,
I
O=C -O-
I
cH*--c H?O- c H?cH~- N
&
CH,
CHJ
Estructura del Monómero DEAEEM)
41
5.2.1 Caracterización por FT-IR
Figura
4,
muestra
al
espectro
del
monómero
N,Ndietilaminoetoxietilmetacrilato DEAEEM, en el ya no se observa la señal de 3384
cm-' de la vibración (O-H) de un alcohol. Las señales en 2968 cm-' y en 2874 cm-'
corresponde a una tensión asimetrica y simétrica de las vibraciones (C-H) de los
grupos metilos (CH3) y metilenos (CH2) alifaticos. La señal en 2808 cm-' corresponde
a una tensión simétrica de las vibraciones (C-H) del un grupo N-CH2-CH3. Se observa
la presencia de una banda intensa en 1722 cm-l característica de la vibración de
tensión de un grupo carbonilo (C=O) de un ester y una señal en 1636 cm-'
característica de una tensión de un doble enlace carbono-carbono (C=C). La señal en
1452 cm-' corresponde a la deformación de la vibración (O-CH2) del ester. El intervalo
entre 1378 cm-' y 1296 cm-' corresponden a las deformaciones de la vibración (C-H)
de los grupos metil (CH3) y metileno (CH2). La señal en 1168 cm-' corresponde a
tensión (C-O) de un grupo ester. La banda en 1128 cm-' corresponde a la tensión
(O-CH2) del ester. Las bandas subsecuentes corresponden a vibraciones fuera del
plano y del tipo rocking del grupo metileno (CH2).
La
n
Figura 4. Espectro FT-IR del N,N-dietilaminoetoxietilmetacrilato(DEAEEM).
Este espectro nos permitió verificar que con la banda intensa de absorción del
grupo carbonilo y la banda del doble enlace carbono-carbono, se obtuvo el
monómero que se necesita para la poiimerización, por io que ya no fue conveniente
42
realizar una caracterización por RMN ya que el tiempo transcurrido para el
monómero destilado tenía que ser el menor posible para evitar que se polimerizara
inducido por la luz.
5.3 Síntesis del poli(DEAEEw
La polimerización del monómero se llevó a cabo mediante una reacción de
polimerización en solución vía radicales libres, usando azobisisobutironitrilo (AIBN)
como iniciador como se muestra en la siguiente reacción:
Reacción de Polimerización de DEAEEM
La polimerización se inicia en el radical libre que se forma cuando el AIBN
rompe el doble enlace carbono-carbono de la estructura molecular del monómero.
El material obtenido de la polimerización en solución es el
Poli(dietilaminoexotietiImetacrilato)(PoliDEAEEM) Ó polímero precursor.
El polímero precursor sintetizado puede ser funcionalizado para obtener un
polímero floculante del tipo zwitteriónico Ó catiónico como ya se mencionó
anteriormente.
La caracterización del polímero precursor se realizó por técnicas
espectroscópicas de FT-IR y RMN de 'H y su peso molecular promedio se determinó
por dispersión de luz.
CH3
CI H
,L
o=c-O-c H,-c H,O-c H ~ HC-, N
CH,
CH3
Estructura del Poli(DEAEEM)
43
5.3.1 Caracterización por FT-IR
La estructura molecular del polímero precursor se determinó por
espectroscopia de Infrarrojo con transformada de Fourier.
La Figura 5, muestra al espectro del polímero precursor en donde las señales
en 2946 cm-' y 2820 cm-' corresponde a una tensión asimétrica y simétrica de las
vibraciones (C-H) de los grupos CH2 y CH3 alifáticos. La señal en 2769 cm-'
corresponde a una tensión simétrica de las vibraciones (C-H) del un grupo N-CH2, se
observa una intensa banda de absorción en 1727 cm-' característica de la frecuencia
de tensión de la vibración (C=O) asociada a un ester y desaparece la señal de 1636
cm-' característica de una tensión de un doble enlace carbono carbono (C=C) como
esperábamos. La señal en 1456 cm-' corresponde a la deformación de la vibración
(O-CH2) de un grupo ester (CH2), en la señal en 1400 cm-' corresponden a la
deformación simétrica de una vibración (C-H) de los grupos CH2 y CH3. La señal en
1151 cm-' corresponde a tensión (C-O) de un grupo ester, las bandas subsecuentes
corresponden a vibraciones fuera del plano y del tipo rocking del grupo metileno
(CH2).
Poli(DEAEEM).
5.3.2 Caracterización por R M N de '
H
La figura 6 representa el espectro de RMN de 'H de PDEAEEM, y su análisis se
muestra en la tabla 3.
CH3
( C-CH2-)"
I
9
3
C
H,
i -L
o=C-o-c H~-CH?O-CH ~ H~-N
C
CH2
CH3
Estructura del Poli(DEAEEM)
Ta bl
Protón
ppm
O=C-O-CH *2-CH 2
O-C* H2-CH2-N
3.7
3.6
-I de PDEAEEM.
45
5.3.3 Determinación de Peso molecular
El peso molecular promedio para el polímero precursor se determinó por
Dispersión de Luz, disolviendo el polímero en cloroformo por 24 horas con agitación
constante. El procedimiento se explica en la parte experimental (capítulo 4).
La gráfica de Zimm nos proporciona información acerca del peso molecular
promedio Mw, radio cuadrático medio RMS y el segundo coeficiente del virial A2.
La tabla 4 muestra los valores obtenidos por dispersión de luz del polímero
precursor Poli(DEAEEMA) purificado, y en la figura 7 se ilustra su gráfico.
ZimmPlot -
7 . W
PDEAEMA
Figura 7 Gráfica de Zimm del polímero precursor Poli(DEAEEM).
dn/dc
,
Mw
RMS
A2
Zimm
O.1052
(2.19+/- 0.4) e4 g/mol
38.1 +/- 1.3 nm
(2.15+/- 0.4)e-4 mol ml/g
46
Referencias
1.- V. M. MONROY SOTO AND GALIN. "Poly(sulphopropylbetaines: I. Syntesis and
Characterization", Polymer 25, 1984 (January).
2. - MANRIQUE G. 'Síntesis y Caracterización de Poliesteres Zwitteriónicos
del tipo Sulfobetaínicos". Tesis de Licenciatura. Tlaxcala, Tlaxcala 1995.
3.- SILVERSTEIN, BASSLER AND MORRILL. Spectrometric Identification of
Organic Compounds. Fifth Edition. Published by John Wiley & Sons, INC. 1991.
4.- COLTHUP, DALY AND WILBERLEY. Introduction to Infrared and Raman
Spectroscopy. Third Edition. Published by Academic Press Limited. 1990.
5.- PREECH, CLERC, SEIBL AND SIMON. Tablas para la Elucidación Estructural
de Compuestos Orgánicos por Metodos Espectroscópicos. Editorial Alhambra
1980.
47
Capítulo 6
Conclusiones
La obtención del monómero (DEAEEM) a partir de la reacción
de sustitución entre el alcohol DEAEE y el cloruro de metacriloilo
procedió satisfactoriamente, generando el material para la obtención
del PDEAEEM.
La caracterización del monómero se realizó por espectroscopia
de FT-IR, en la cual pudimos identificar una intensa banda asociada al
grupo carbonilo del ester y otra banda asociada a un doble enlace
carbono carbono que son características de los grupos funcionales
presentes en el monómero.
La polimerización vía radicales libres en solución, sugerida en el
presente trabajo, nos conduce a la obtención del polímero precursor
poli(dietilaminoetoxietilmetacrilato)PDEAEEM .
La caracterización espectroscópica por FT-IR y RMN protónica
del polímero precursor nos permitió identificar a los grupos funcionales
que integran su estructura molecular y el acoplamiento químico de los
mismos grupos.
El peso molecular promedio del polímero precursor
poli(DEAEEM) fue calculado por dispersión de luz y es de 2 . 1 9 ~ 1 0 ~
g/mol.
El polímero precursor puede ser funcionalizado como un
polímero zwitteriónico del tipo N-Óxido, sulfobetaínico y
carboxibetaínico Ó como un polímero catiónico como los compuestos
cuaternarios de amonio.