DIAG diagnostico temprano 29
Anuncio

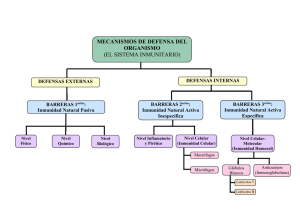
DIAGNÓSTICO Diagnóstico temprano de las inmunodeficiencias primarias F. Muñoz López Ex Jefe del Servicio de Inmunología y Alergia Pediátrica. Unidad Integrada Hospital Clínic-Sant Joan de Déu. Barcelona. Director de la revista Allergologia et Immunopathologia. L as inmunodeficiencias primarias (ID) son un grupo de enfermedades que se deben a diversos defectos congénitos del sistema inmunitario. Como consecuencia de ello, el niño está expuesto a sufrir con frecuencia infecciones graves, a veces por microorganismos “oportunistas”, que se comportan con inusitada virulencia. Bruton por primera vez, en 1952, describió un caso de deficiencia de la respuesta inmune, en un niño que desde los 4 años de edad había sufrido múltiples infecciones, y pudo demostrar en el suero sanguíneo la ausencia de anticuerpos frente a gérmenes a los que había sido vacunado (difteria, tifus) al mismo tiempo que en el trazado inmunoelectroforético no aparecía la banda correspondiente a la gammaglobulina. La administración repetida de globulina sérica consiguió mantener libre de infecciones al paciente. Este tipo de agammaglobulinemia se conoció por el nombre de su autor, observándose después que esta ausencia estaba ligada al sexo masculino, por lo que también de denominó agamma o hipogammaglobulinemia ligada al cromosoma X. Al identificarse en los años setenta dos poblaciones diferentes de linfocitos, B y T, se intuyó la posibilidad de que algunas variantes de inmunodeficiencias estuviesen relacionadas con ellas, y todavía más al comprobar que de los linfocitos B dependía la producción de anticuerpos. De ahí que se diferenciaran primeramente inmunodeficiencias en las que faltaban los anticuerpos circulantes, o ID humorales, dependientes de defectos de los linfocitos B, y otras de los linfocitos T. En el primer grupo cabía la mayoría de estos procesos, ya que la ausencia o disminución de alguna de las tres inmunoglobulinas identificadas (G, M y A) que soportan los anticuerpos, era la más frecuente, sobre todo, la ausencia de IgA sérica. Con posterioridad se comprobó que en algunos pacientes podían detectarse anomalías de ambas estirpes linfocitarias, por lo que se reconoció un grupo de ID combinadas. No tardó en conocerse que la producción de anticuerpos por los linfocitos B dependía de la acción de los linfocitos T, debido a la secreción de sustancias activadoras (linfocinas) sobre aquéllos, de lo que se dedujo que la disminución o ausencia de todas o alguna de las inmunoglobulinas no dependía exclusivamente de los linfocitos B, sino de un fallo de los T. De ahí que el grupo de ID humorales exclusivas se fue reduciendo, al mismo tiempo que se incrementaba el de las ID combinadas, en el que actualmente se incluyen la mayoría de las más graves, conocidas en general como ID combinadas graves. De este recuerdo cronológico, se deduce que la clasificación de las ID que periódicamente publica la OMS se ha ido modificando en función de estos conocimientos1,2. Además, cada vez se describen más casos de ID difíciles de catalogar en estos apartados, y el cuadro se ha ampliado con distintos subapartados. No sólo se detectan déficit de la inmunidad específica, sino que también puede estar alterada la defensa inespecífica, mediada por fagocitos, por lo que hay fallos de la fagocitosis y la lisis bacteriana o de alguno de las pasos intermedios, como la opsonización o la quimiotaxis, en lo que está implicado el sistema complemento. MECANISMOS INMUNITARIOS En síntesis, los mecanismos de defensa frente a los microorganismos con capacidad de originar enfermedades en la especie humana son los siguientes3: 1. Barrera cutaneomucosa: además de constituir una barrera física, la piel contiene diversos elementos que dificultan el paso de gérmenes, como los ácidos láctico, undecilénico y caprílico, la secreción sebácea y algunas células ligadas a la inmunidad, como linfocitos T (CD4 y CD8) y las de Langerhans, y también los queratocitos. Las mucosas disponen de un sistema físico, representado por los cilios en algunas de ellas y las glándulas mucosas. Pero en ellas, lo más representativo y eficaz es la presencia de IgA secretora, que es un dímero de esa inmunoglobulina, abundante sobre todo en el intestino, cuya función primordial es impedir el paso de gérmenes a través del epitelio. Otras inmunoglobulinas y diversas sustancias completan la función defensiva en este sistema. 2. Cuando un microorganismo pasa la primera barrera, encontrará un sistema inespecífico de defensa, representado por los fagocitos móviles (PMN) que tratarán de eliminarlo, en un proceso de acercamiento (quimiotaxis), unión celular (opsonización), ingestión (fagocitosis) y digestión (lisis), mecanismos para los que es fundamental la activación del sistema complemento, a partir de la fracción C3. 3. Cuando la fagocitosis no es efectiva, determinadas células presentadoras de antígenos (macrófagos) captan los microorganismos y tras su digestión intracelular aíslan determinadas fracciones con capacidad antigénica que son presentadas a los linfocitos T (CD4), previo reconocimiento del HLA, y se pone en marcha la activación de estas células, que van a estimular los linfocitos B, que activadas como células plasmáticas producirán anticuerpos específicos frente a esos antígenos. La activación del sistema linfocitario es compleja, y en ella intervienen múltiples sustancias, conocidas como citocinas, de ellas muy especialmente las interleucinas (IL). Esta fase de la defensa inmunitaria se conoce como inmunidad específica, al ser específicos los anticuerpos que se producen frente a los microorganismos. Se reconocen diversos tipos de linfocitos T, los cooperadores (helper), Th o CD4 y los supresores o citotóxicos (CD8). Los Th inactivos (Th0) pueden activarse como Th1, que por las IL-2, IL12 y el interferón gamma (INF-γ) darán lugar a la producción de los anticuerpos, por los linfocitos B. En la defensa frente a parásitos parece que intervienen los Th2 (procedentes de los Th0) que por la secreción de IL-4 activan las células B, dando lugar a la producción de IgE. En determinadas circunstancias y en individuos genéticamente predispuestos (atópicos) hay un predominio de linfocitos Th2, con producción de anticuerpos IgE específicos frente a alergenos, al mismo tiempo que la IL-5, de igual procedencia, activa los eosinófilos, células básicas en la inflamación alérgica. Las células natural killer (NK), también citotóxicas, son una línea celular, cuya relación con los linfocitos todavía no está bien establecida. DIAGNÓSTICO Diagnóstico temprano de las inmunodeficiencias primarias F. Muñoz López Los anticuerpos producidos por los linfocitos B (células plasmáticas) son inmunoglobulinas, en primera instancia IgM y posteriormente IgG (de la existen cuatro subclases), que se produce con rapidez frente a los estímulos antigénicos que puedan producirse con posterioridad al primer contacto con el antígeno. INMUNODEFICIENCIAS PRIMARIAS El defecto de cualquiera de los mecanismos inmunitarios citados puede originar enfermedades que se caracterizan sobre todo, por la incidencia de infecciones repetidas y graves. El fallo de la actividad de los fagocitos, bien por disminución en su número (neutropenias) o en su función (disfagocitosis) o también del complemento (falta o disfunción de alguno de sus componentes), es la base de las inmunodeficiencias inespecíficas. Las más frecuentes de estas ID se listan en la tabla I, y las tablas II y III muestran las características de las más destacadas. La escasa o nula capacidad de los linfocitos B para producir anticuerpos es la causa de las inmunodeficiencias predominantemente de anticuerpos (ID humorales) (tablas IV a VII). El defecto exclusivo de la acción de los linfocitos T, inmunodeficiencias celulares, sólo dudosamente está representado por el síndrome de Di George y la candidiasis mucocutánea. La mayoría de los procesos descritos corresponden a inmunodeficiencias combinadas, en las que una alteración de los linfocitos T da lugar a déficit de producción de anticuerpos por las células B (tablas VIII a XI). Algunas inmunodeficiencias son de difícil catalogación, siendo característica la presencia de ciertos defectos asociados, como ocurre en el síndrome de Wiskott-Aldrich, la ataxia-telangiectasia y el mismo síndrome de Di George citado, cuadros que la IUIS2 agrupa bajo la denominación de otros síndromes bien definidos (tablas XII a XIV), y en otro apartado incluye ID asociadas a otras enfermedades (tabla XV). La alteración de la respuesta inmunitaria puede residir en la propia célula o en los déficit de interleucinas u otras citocinas que participan en la maduración y estímulos celulares. Frecuencia En el Registro Español de ID de 1998, que recoge 1.420 diagnósticos, las inmunodeficiencias humorales representan el 69,5%, dado que el déficit selectivo de IgA es la más común de todas las ID, y comprende el 59% de esa cifra, seguida de la ID variable común, el 28,6% del total y la agammaglobulinemia ligada al cromosoma X (Bruton), el 6,2%. Las ID combinadas alcanzan el 14,8%. Mucho menos frecuentes son los demás procesos: defectos de fagocitos, el 6,1%, de complemento 6% y otras el 3,7%4. Signos de sospecha Ciertos hechos harán sospechar la existencia de alguna de estas enfermedades (tabla XVI). El diagnóstico temprano es fundamental, dado que en muchos casos es posible un tratamiento efectivo cuya prontitud en su aplicación puede ser fundamental para el futuro del paciente5, de ahí que sea preciso que el pediatra o el médico de familia conozca los signos de sospecha para poner en marcha las medidas diagnósticas oportunas, teniendo en cuenta que, además de las características clínicas, algunos análisis de fácil realización pueden confirmar la sospecha inicial. Después es obligada la colaboración del especialista, ya que en la mayoría de los casos el correcto diagnóstico se basará en un complejo estudio analítico, que en definitiva servirá para establecer el tratamiento oportuno. El antecedente familiar de otros casos de inmunodeficiencias o de muertes prematuras de niños que no medraban, con infecciones no controladas (neumonías, meningitis) o procesos diarreicos TABLA I Inmunodeficiencias inespecíficas 1. Neutropenia congénita grave 2. Neutropenia cíclica 3. Defecto de adhesión leucocitaria tipo I y tipo II 4. Síndrome de Chediak-Higashi 5. Síndrome de Schwachmann 6. Enfermedad granulomatosa crónica: a) Ligada al cromosoma X b) Autosómica recesiva TABLA II Enfermedad de Chediak-Higashi Características clínicas Características hematoinmunológicas Herencia autosómica recesiva: ambos sexos Infecciones bacterianas Adenopatías. Hepatosplenomegalia Fotofobia. Infecciones corneales. Nistagmo Albinismo parcial Cabellos color grisáceo, frágiles Neutropenia. Monocitopenia Disfunción de PMN Déficit enzimático endocelular Alteración de la quimiotaxis Alteración de las primeras etapas de la respuesta inmune Fagocitosis normal in vitro Trombopenia: hemorragias Inmunidad humoral normal Inmunidad celular: NK disminuidas NK: natural killer. TABLA III Enfermedad granulomatosa crónica Defecto de la lisis microbiana de los PMN Ligada al cromosoma X: defecto de la cadena gp91 kD del citocromo b, ligado a la activación de NADPH oxidasa (NADPH + O2 NADP + 2O2- + H+) Autosómica recesiva: defectos de la cadena de 22 kD del citocromo b o de los factores citosólicos p47 o p67 Clínica Infecciones repetidas de inicio temprano o no Microorganismos: estafilococo, Salmonella, serratia, Seudomonas, Aspergillus, Candida Localización: piel, pulmón, hígado, perianal, huesos Abscesos fríos y granulomas no caseosos Diagnóstico Test nitro azul tetrazolio: NAT (NBT) PMN: polimorfonucleares TABLA IV Inmunodeficiencias congénitas específicas. Deficiencias predominantes del número o de la función de los anticuerpos (Ig) Agammaglobulinemia ligada al cromosoma X Síndrome de hiper-IgM no ligado al cromosoma X Deficiencia selectiva de subclases de IgG con o sin déficit de IgA Deficiencia selectiva de IgA Síndrome variable común de inmunodeficiencia Deficiencia de anticuerpos con cifra normal de Ig Hipogammaglobulinemia transitoria de la infección TABLA V Agammaglobulinemia ligada al cromosoma X (agammaglobulinemia de Bruton) Causa: mutación del gen codificado de Btk (Xq21,3.22) Btk: tirosincinasa intracelular: interviene en la maduración de los linfocitos B Consecuencia: ausencia total o parcial de las tres Ig y de linfocitos B maduros (< 1.000/µl3 ≤ 5/1.000 linfocitos)-Pre-B en médula ósea Clínica dominante: infecciones sinupulmonares bacterianas, desde los 5-6 meses de edad Tratamiento: gammaglobulina intravenosa TABLA VI Déficit selectivo de IgA Criterio: IgA sérica < 5 mg/dl. Déficit parcial: 5-30 mg/dl Frecuencia: 1/1.000 de la población: sólo se manifiesta aproximadamente en 1/700 Clínica Infecciones respiratorias Alergopatías (50%) Posibilidad de enfermedades autoinmunes y neoplasias Formas clínicas Déficit sérico exclusivo Asociado a déficit de IgA-S Déficit de IgA-S exclusivo Secundaria: tratamiento hidantoínas, valproato, carbamacepina DIAGNÓSTICO Diagnóstico temprano de las inmunodeficiencias primarias F. Muñoz López TABLA VII Inmunodeficiencia variable común TABLA XIII Ataxia-telangiectasia Patogenia: diversos mecanismos. Casos esporádicos o relacionados con haplotipos HLA Edad de comienzo: infancia tardía o adulto joven Inmunidad humoral Linfocitos B: número normal o algo disminuidos Poco diferenciados pueden producir, pero no segregar anticuerpos IgG ⇓⇓ = a veces: ⇓ IgA e IgM Inmunidad celular A veces los linfocitos T están ⇓ o fallan al diferenciar o estimular a los linfocitos B: equiparable con la ID combinada Clínica Infecciones bacterianas recurrentes; giardiasis Enfermedad autoinmune: anemia hemolítica, trombopenia Neutropenia Neoplasias linforreticulares Mutación de un gen de la familia de la fosfatidil-inositol-3-cinasa: Roturas cromosómicas, inversiones, translocaciones: linfocitos T y sus receptores Ataxia progresiva Telangiectasias oculares y cutáneas. Discromias Disminución o ausencia de IgG2-IgA-IgE Disminución de la respuesta de los anticuerpos Disminución de los linfocitos T Neoplasias linforreticulares, tumores sólidos TABLA VIII Inmunodeficiencias (ID) combinadas ID combinada grave T (-) B (+) Ligada al cromosoma X Autosómica recesiva ID combinada grave T (-) B (-) Deficiencia de adenosisdeaminada (ADA) Disgenesia reticular Hiper-IgM ligada al X Deficiencia de purin nucleósido fosforilasa (PNF) Deficiencia de antígenos de HLA tipo II Otras: CDγ, CDε, Zap-70, TAO-2 TABLA IX Inmunodeficiencia combinada grave Inmunidad celular: dos patrones Ausencia linfocitos T y B Ausencia linf. T. B normales o ⇑: función anormal; más frecuente Inmunidad humoral Inmunoglobulinas con frecuencia ⇓ y no específicas frente antígenos Transmisión: ligada al cromosoma X. Autosómica recesiva Causas Defecto del gen codificador de la cadena γc de los receptores de IL-2, IL-4, IL-7, IL-9 e IL-15 Fallo en la diferenciación y proliferación de los linfocitos T y B Clínica: infecciones graves y repetidas, de inicio muy temprano Tratamiento: trasplante de médula ósea TABLA X Déficit de adenosindeaminasa (ADA) Se encuentra en todos los tejidos. Abunda en los linfocitos y es importante en el metabolismo de la purina Déficit de ADA: acumulación intracelular de tóxicos intermedios del metabolismo de la purina: deoxiadenosina, d-ATP, inosina Defecto de los linfocitos T y B: número y función Inmunoglobulinas: valores variables Clínica Infecciones graves Defectos óseos: Costillas cortas y cóncavas, escaso desarrollo de la cresta ilíaca, espículas en huesos largos TABLA XI Síndrome de hiper-IgM Distintas entidades con expresión clínica y fenotípica similar: Ligada al cromosoma X Autosómica recesiva En algunos grupos: déficit del ligando para el receptor CD40 de linfocitos B, que acopla IL-2, IL-4, IL-10 para sintetizar Ig Elevación de IgM, no siempre: respuesta primaria intacta. Déficit de IgAIgG Déficit celular: infecciones por Pneumocystis TABLA XII Síndrome de Wiskott-Aldrich: ID con plaquetopenia y eccema Mutación del gen WASP cromosoma Xp11 Plaquetas pequeñas, disfuncionales Sialoglicoproteínas CD43 y gpIb inestables en las membranas de los leucocitos y plaquetas Linfocitos de aspecto “liso” en el microscopio electrónico Ig normales: progresivo descenso de IgM Linfopenia progresiva Fenómenos autoinmunes: vasculitis, glomerulonefritis Neoplasias linforreticulares: muerte adultos TABLA XIV Síndrome de Di George Monosomía variable parcial del cr.22q11 o 10p Inmunodeficiencia T y B Embriogénesis anormal a las 4-6 semanas Múltiples anomalías: tercer-cuarto arco braquial: Hipoplasia o aplasia tímica Hipoparatiroidismo: convulsiones hipocalcémicas Atresia esofágica Cardiopatías Facies característica: hipertelorismo, hendidura antimongoloide de los ojos, malformación de las orejas TABLA XV ID asociadas o secundarias a otras enfermedades Síndrome de Bloom Displasia esquelética de miembros cortos Hipoplasia cartílago-pelo Albinismo parcial Displasia anhidrótica Acrodermatitis enteropática Hiper-IgE (síndrome de Job) Candidiasis mucocutánea crónica TABLA XVI Criterios de sospecha de inmunodeficiencias primarias Historia familiar de inmunodeficiencias primarias Infecciones bacterianas graves y repetidas: Neumonías recurrentes, meningitis, sepsis, otitis, mastoiditis Infecciones recurrentes por Neisseria (déficit del complemento) Evolución grave de infecciones víricas (varicela) Abscesos cutáneos graves y recurrentes Enfermedad pulmonar crónica, con alteraciones radiológicas y funcionales permanentes Diarrea persistente o recurrente Candidiasis orocutánea resistente, después de los 6 meses Reacciones vasculares inusuales o graves Dermatosis persistente: eccema, dermatitis seborreica, discromías Asociación de eccema y plaquetopenia Tetania en los primeros días de vida Ausencia de timo (RX) en período neonatal Presencia de ataxia y/o telangiectasia Retraso en el desarrollo pondoestatural prolongados, debe alertar sobre a posibilidad de que un recién nacido padezca alguno de estos procesos, dado que en su mayoría las inmunodeficiencias primarias son de transmisión genética. Algunos de estos procesos acaecen solamente en varones y algunos de ellos excepcionalmente también en mujeres (agammaglobulinemia X, ligada al cromosoma, Wiskott-Aldrich, hiper-IgM, enfermedad granulomatosa crónica, ID combinada grave). La repetición de infecciones, sobre todo por “gérmenes oportunistas” (Pseudomonas, Pneumocystis carinii, citomegalovirus, etc.), la escasa respuesta a los antibióticos y la cronificación de algunos procesos (neumonías, otitis, abscesos) son comunes a las ID. En cierta medida, pueden relacionarse algunos de los agentes infecciosos implicados, con el mecanismo inmunológico deficiente. Así, las bacterias son la causa común en los déficit de anticuerpos (linfocitos B) y las alteraciones de la fagocitosis, los virus y hongos cuando falla la respuesta de linfocitos T y diversas Neisserias en los defectos de la actividad complementaria (tabla XVII)6. También debe recaer la sospecha en aquellos niños que no se desarrollan correctamente, con escaso peso y talla, los que pade- DIAGNÓSTICO Diagnóstico temprano de las inmunodeficiencias primarias F. Muñoz López TABLA XVII Algunos agentes patógenos en los distintos déficit de la función inmunitaria Bacterias Déficit linfocitos T Sepsis bacterianas Déficit linfocitos B Estreptococos Estafilococos Haemofilus Estreptococos Estafilococos Pseudomonas Neisseria Otros piógenos Déficit fagocitos Déficit complemento Virus Citomegalovirus Epstein-Barr Varicela grave VRS, rotavirus Enterovirus (encefalitis) cen diarreas prolongadas, de difícil control, las dermatitis e infecciones cutáneas rebeldes al tratamiento o las candidiasis mucocutáneas que no ceden con antifúngicos. Algunas de estas enfermedades se asocian a otros procesos, como cardiopatías y atrófica esofágica (Di George), ataxia, telangiectasias en escleróticas, discromías (síndrome de ataxia-telangiectasia), albinismo (síndromes de Chediak-Higashi y de Griselli), malformaciones óseas (costales: déficit de adenosindeaminasa; miembros: hipoplasia cartílago-pelo). En todos estos casos es conveniente investigar la posibilidad de que exista una ID, aunque el niño no haya padecido aún infecciones importantes, siendo preciso hacerlo cuando se trate de una malformación, presente ya desde el nacimiento, como ocurre con la cardiopatía de Di George, que también puede acompañarse de otros signos (facies característica)7. La protección frente a las infecciones que confiere el paso de anticuerpos IgG de la madre al feto, con una persistencia entre 4 y 6 meses, hace que las primeras infecciones en los niños afectados de hipo o agammaglobulinemia ligada al cromosoma X no se presenten hasta pasado este tiempo, dada la incapacidad de producir anticuerpos propios. Por contra, como la inmunidad mediada por linfocitos T no se transmite, las infecciones suelen ser más tempranas y graves en las ID combinadas, en las que están implicados ambos mecanismos defensivos. Además de esta cronología en la aparición de los problemas, otros datos pueden ser igualmente orientadores. Entre ellos las mencionadas malformaciones o la presencia de convulsiones por hipocalcemia, por la asociación de hipoparatiroidismo en el síndrome de Di George. En esta misma enfermedad, puede comprobarse radiológicamente la ausencia de timo. Curiosamente, en otras ID en las que predomina el déficit de anticuerpos, el inicio de las infecciones es más tardío e incluso se manifiesta por signos distintos. Así, los primeros signos de la ID variable común, que es un síndrome incompletamente definido, suelen demorarse algunos años, incluso al adulto joven8. No es infrecuente encontrar déficit selectivo de IgA, en un estudio sistemático, en individuos que no padecen infecciones importantes (se encuentran a veces déficit parciales o ausencias totales en los familiares sanos de los enfermos), y en otras ocasiones la enfermedad más común en estos pacientes es la alergológica. Sin embargo, en edades posteriores no es infrecuente que aparezcan procesos autoinmunes e incluso neoplasias. Otras deficiencias de anticuerpos, como son los déficit de subclases de IgG, por lo general asociados entre sí (IgG1-IgG4) o con el de IgA, tienen una cronología variable, no definida. No es infrecuente la maduración tardía en el mecanismo productor de inmunoglobulinas, sobre todo de IgG, con una demora de entre 3 y 5 años, encontrándose cifras bajas de las mismas, sin que tengan lugar infecciones importantes. Por lo común, esta in- Hongos y parásitos Particularidades Candida albicans Pneumocystis carinii Enfermedades agresivas por patógenos oportunistas Giardiasis intestinal grave Infecciones sinopulmonares recurrentes, sepsis, meningitis crónica Candida albicans Nocardia Aspergillus sp. Impétigo, abscesos cutáneos y de órganos (hígado) munodeficiencia transitoria del lactante no requiere tratamiento específico, sino sólo de las infecciones intercurrentes, que no suelen cursar con mayor gravedad que en otros niños. Por otra parte, hay que recordar que es normal que durante los primeros 10 años, el niño padezca una media de una infección mensual, con predominio de las respiratorias, seguidas de otitis o gastroenteritis, con lo que se completa la madurez del sistema inmunitario. La ausencia total o casi total de IgE sérica se observa en algunas enfermedades, como la ataxia-telangiectasia, pero sobre todo tiene valor su gran aumento, que define el síndrome de hiper-IgE, en el que son características las infecciones cutáneas por Staphylococcus aureus, acompañándose a veces de un fallo de la quimiotaxis de los fagocitos (síndrome de Job). Cribado diagnóstico Al diagnóstico definitivo de muchas inmunodeficiencias sólo se llega tras una ardua labor en la que es preciso disponer de un laboratorio especializado. En los últimos años se han descrito cuadros complejos, cuya causa se debe a un defecto de alguno de los muchos elementos implicados en la respuesta inmunitaria, aparte los valores de anticuerpos o el número o función de los linfocitos B o T. Por fortuna, esas nuevas deficiencias sólo se han descrito en pocas familias, por lo que son cuadros poco habituales, en los que no obstante hay que pensar cuando la clínica o los hallazgos analíticos no son compatibles con las ID más frecuentes. Por esto, al médico generalista le basta con sospechar la presencia de una de estas enfermedades, apoyado en unos primeros datos analíticos, para después proseguir el estudio en un centro especializado9. Un simple hemograma puede evidenciar ya algunas alteraciones orientadoras. La neutropenia hará sospechar alguna de las variantes de ID inespecífica. La cifra de linfocitos está disminuida en la mayoría de los procesos con alteración de la inmunidad celular o retardada; cifras inferiores a 1.500/µl3 harán sospechar una ID, aunque son habituales cifras muy inferiores. No obstante, el contenido normal e incluso aumentado de linfocitos no descarta la ID celular, y debe valorarse siempre en función de la edad, dado el mayor contenido en los primeros meses de vida. La trombopenia, junto con el eccema, caracteriza el síndrome de Wiskott-Aldrich e igualmente acompaña al síndrome hiper-IgM. La presencia de anemia también requiere investigar la existencia de alguna ID. La actividad del complemento es de fácil valoración, tanto total (CH50-CH100) como de las fracciones clave, C’3 y C’4, con lo que puede orientarse un posible diagnóstico del déficit de alguno de los demás factores. Las deficiencias de la inmunidad humoral se detectan por el bajo contenido de inmunoglobulinas séricas (G, M, A y E), para lo que se tendrán en cuenta las variaciones que sufren desde el nacimiento, con un mayor contenido de IgG, nulo de IgA y escaso de IgM en el recién nacido, cifras que van cambiando hasta alcanzar DIAGNÓSTICO Diagnóstico temprano de las... F. Muñoz López valores próximos a las del adulto en la adolescencia. Sin embargo, el síndrome de déficit de anticuerpos cursa con cifras normales e incluso elevadas de IgG, aunque la actividad anticuerpo está ausente. Del mismo modo, la presencia de isoaglutininas (anti-A, anti-B) o de antiestreptolisinas (ASTO) informará sobre la inmunidad por anticuerpos. Aparte del recuento de linfocitos en la fórmula hemática, el fallo de la inmunidad celular se detecta mediante pruebas cutáneas de respuesta retardada, que están mediadas por linfocitos T: tuberculina, candidina, tricophyton, toxoides tetánico y diftérico. Hay que tener en cuenta que estas pruebas requieren el contacto previo o la vacunación con estos antígenos, por lo que en niños muy pequeños tienen escaso valor. Confirmada la sospecha a través de estas primeras pruebas de laboratorio, es necesario ampliar el estudio, dependiendo de la sospecha diagnóstica: estudio de la quimiotaxis (quimioluminiscencia, v. Rebuck); fagocitosis (NBT) y fracciones del complemento, para la inmunidad inespecífica. La humoral puede precisar la determinación de subclases de IgG, investigación de anticuerpos frente a antígenos conocidos (polio, sarampión, Salmonella, difteria), IgA secretora, linfocitos B (CD19-CD20) y la inmunidad celular se ampliará con el estudio de las poblaciones linfocitarias T (CD4-CD8), NK, actividad linfocitaria (estimulación linfoblástica con antígenos inespecíficos [CoA, PHA, PW], y específicos [candidina, tuberculina]), bioquímica (adenosindeaminasa, purina-nucleósido-fosforilasa, transcobalamina-2) y en estadios posteriores puede ser necesaria la determinación de diversas interleucinas u otros factores (ZAP-70, TAP-2, Jak 3, HLA). Bibliografía 1. World Health Organization Committee. Primary immunodeficiencies. Pediatrics 1971;47:927-46. 2. IUIS Scientific Group (WHO). Primary Immunodeficiency diseases. Cl Exp Immunol 1999;118:(Suppl 1):1-28. 3. Blanco Quirós, A. Fisiología y desarrollo de la inmunidad. En: Cruz M, editor. Tratado de Pediatría. 8.ª ed. Madrid: Ergon, 2001. 4. Milà J, Pons J, Raga S, Iglesias J, Matamoros N. Registro Español de inmunodeficiencias primarias. Informe de actualización Julio 1998. Inmunología 1999;18:87-90. 5. Blanco Quirós A, Muñoz López F, Martín Mateos MA. Tratamiento de las enfermedades inmunitarias, alérgicas y reumáticas en niños y adolescentes. Barcelona: Espaxs, 1999. 6. Puck JM. Primary immunodeficiency diseases. JAMA 1997;278:183541. 7. Martín Mateos MA, Pérez B, Iriondo M, Krauel, Gean E. Clinical and immunological spectrum of partial Di George síndrome. Invest Allergol Clin Immunol 2000;10:352-60. 8. Spickett GP, Farrant J, North ME. Common variable immunodeficiency: how many diseases? Immunol Today 1997;18:325-8. 9. Fontán Casariego G. Protocolo diagnóstico de las inmunodeficiencias. Pediat Integral 1999;4:13-26.