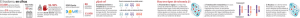Original - Fundació Catalana Síndrome de Down
Anuncio

SD 2001: vol. 5, núm. 2, pp. 19-24 REVISTA MÉDICA INTERNACIONAL SOBRE EL SÍNDROME DE DOWN 19 Original Correlaciones genotipo-fenotipo en casos de síndrome de Down con trisomía parcial del cromosoma 21 Marga Nadal y Xavier Estivill Centro de Genética Médica Molecular-IRO Correspondencia Dra. Marga Nadal Grupo de Investigación del Síndrome de Down Centro de Genética Médica Molecular-IRO Autovía de Castelldefels, Km. 2,7 08907 L’Hospitalet de Llobregat Barcelona e-mail: [email protected] Artículo recibido el: 14.06.01 Nota: Este artículo forma parte del estudio «Estudio citogenético-molecular del SD: Correlaciones genotipo-fenotipo en seis casos de trisomía parcial del cromosoma 21», que ha sido galardonado con el VII Premio Bienal de Investigación Ramon Trias Fargas, 1999-2001. Resumen El síndrome de Down (SD) es la causa principal de retraso mental y afecta a uno de cada 700 nacidos. En el 99 % de los casos el SD está asociado, a nivel genético, a una trisomía del cromosoma 21 (HSA21), pero también puede ser producido por una trisomía parcial de este cromosoma, tal y como se ha descrito en una cuarentena de casos en la literatura. Gracias al mapa genético del HSA21, que ha dado acceso a todo tipo de sondas, y al desarrollo concomitante de técnicas genéticas nuevas como la FISH, se ha podido ir identificando y caracterizando estos casos. Se estudian seis casos de SD debidos a trisomía parcial del HSA21. Dos de ellos se deben a la segregación familiar de una traslocación que implica un segmento del HSA21 y un segmento de otro cromosoma. Los otros cuatro casos recogidos sólo implican al HSA21. Uno presenta una traslocación (21; 21) no robertsoniana y los otros tres, una duplicación intersticial del HSA21. Todos estos casos se han podido estudiar a nivel citogenético-molecular, caracterizándose el punto de rotura de cada uno para determinar el alcance de la trisomía del HSA21 mediante FISH. La determinación del punto de rotura del HSA21 y los detallados datos clínicos obtenidos no sólo permiten el consejo genético, sino también el establecimiento de una correlación genotipo-fenotipo de los casos estudiados. Palabras clave: síndrome de Down, cromosoma 21, trisomía parcial, FISH, correlación genotipo-fenotipo. Genotype-phenotype correlations in cases of Down syndrome with partial trisomy of HSA21 Abstract Down syndrome (DS) is the chief cause of mental retardation, affecting 1 in 700 births. In 99% of cases, DS is genetically associated with trisomy of chromosome 21 (HSA21), but it may be caused by partial trisomy of this chromosome, as described in about 40 published cases. Thanks to genetic mapping of HSA21, which has made all kinds of new probes available, and to the concomitant development of new genetic techniques such as FISH, such cases have been identified and characterised. The present study addresses six cases of DS caused by partial trisomy of HSA21. Two are caused by familial segregation of a translocation involving a segment of HSA21 and a segment of a different chromosome. The other four cases only involve HSA21: one involves a non-Robertsonian (21; 21) translocation and the other three involve interstitial duplication of HSA21. All cases were studied at a cytogenetic-molecular level and the break point was identified to gauge the extent of HSA21 trisomy by FISH. By identifying the break point in HSA21 and gathering detailed clinical data, it became possible to provide genetic counseling and even to establish a genotype-phenotype correlation in the six cases studied. Keywords: Down syndrome, chromosome 21, partial trisomy, FISH, genotype-phenotype correlations. Introducción La trisomía del HSA21 que causa el síndrome de Down (SD) puede ser total o parcial. La trisomía total se produce por errores en la segregación equitativa de los cromosomas en la división meiótica. Dicha segregación desigual se llama «no disyunción» y hace que las células descendientes de esta célula germinal (materna o paterna) incorporen dos copias del HSA21 en lugar de una y que, en consecuencia, después de la fusión de los dos gametos para dar lugar al embrión, se produzca una trisomía y las células del nuevo individuo tengan 47 cromosomas en lugar de 46. Este fenómeno de la no-disyunción se da con mayor frecuencia en las células germinales femeninas que en las masculinas, de forma que la trisomía del HSA21 es, mayoritariamente, de origen materno [1]. SD 20 REVISTA MÉDICA INTERNACIONAL SOBRE EL SÍNDROME DE DOWN El SD abarca un abanico muy amplio de manifestaciones fenotípicas que comparten la mayoría de los casos estudiados, así como una serie de alteraciones clínicas que se encuentran sólo en algunos de los casos. El SD está asociado a retraso mental y a defectos cardíacos congénitos, pero también a alteraciones gastrointestinales, endocrinas, musculoesqueléticas, oculares, inmunológicas y auditivas. Estas manifestaciones clínicas son una preocupación constante en el SD ya que, aunque muchas pueden estar ausentes los primeros meses de vida, pueden ir apareciendo con el paso del tiempo. Dada la diversidad fenotípica, una de las tareas a las que los clínicos han dedicado más esfuerzos ha sido la elaboración de protocolos para caracterizar con detalle cada caso de SD. En este sentido, cabe mencionar la vigencia del protocolo elaborado por Epstein [2]. Ahora, gracias, por un lado, a los nuevos avances biomédicos y a los esfuerzos para caracterizar al máximo la patogenia de todas las anomalías clínicas y las malformaciones del SD y, por otro, al conocimiento cada vez más exhaustivo de los genes que se hallan en el HSA21 humano y de sus funciones, se puede empezar a establecer correlaciones entre el genotipo y el fenotipo, que deberán contribuir sustancialmente a un mejor conocimiento de este síndrome. Aspectos clínicos del síndrome de Down Los rasgos clínicos más evidentes del cuadro que caracteriza el SD incluyen varias malformaciones menores que se pueden reconocer durante el período neonatal y que, pese a no estar siempre presentes, constituyen de manera conjunta un fenotipo característico del síndrome. El retraso mental y la hipotonía están siempre presentes, y los defectos cardíacos congénitos, principalmente defectos del tabique interventricular, se dan en un 40% de los casos de SD. Las anomalías gastrointestinales (sobre todo la atresia duodenal y la enfermedad de Hirschsprung) se dan en un 5% de los casos. La tasa de mortalidad es más alta durante los primeros años de vida y la muerte suele ser debida a infecciones respiratorias, malformación cardíaca o leucemia [3]. De los pacientes con SD que superan la pubertad, muchos viven hasta la vida media adulta (35 ó 40 años) y casi todos sufren después una forma de degeneración cerebral o forma prematura de la enfermedad de Alzheimer. En el SD se da una gran variabilidad en cuanto a capacidades intelectuales y desarrollo psicomotor, que pueden verse influidos tanto por el entorno como por otros factores genéticos. No se conocen aún las causas concretas del retraso mental, pero se ha descrito una menor densidad de células nerviosas en regiones concretas del cerebro, cambios en la composición fosfolipídica de la mielina y alteraciones de determinadas propiedades electrofisiológicas de algunas neuronas. Los bebés con SD presentan un riesgo entre 15 y 20 veces superior al de la población general de desarrollar leucemia, sobre todo la leucemia megacarioblástica aguda [4]. Los hombres con SD casi siempre son infértiles, en tanto que las mujeres pueden ser fértiles [5]. Las causas principales de muerte en el SD son las infecciones, los defectos cardíacos congénitos y el cáncer. Se cree que la elevada susceptibilidad a las infecciones puede ser debida a alteraciones en el sistema inmunitario. En los últimos años ha aumentado la longevidad de tal forma que la esperanza de vida de las personas con SD sin defecto cardíaco congénito puede llegar a superar los 60 años. No hay ningún paciente con SD que presente todos los 2001: vol. 5, núm. 2, pp. 19-24 rasgos que se han descrito. Un caso notorio es el que publicaron Avramopoulos et al. en 1997 [6] de un paciente con trisomía regular del HSA21 con tan sólo dos rasgos (braquicefalia y puente nasal ausente). El caso inverso es el descrito por Ahlbom (1996) [7] de un paciente con un fenotipo indistinguible del del SD que no presentaba trisomía 21. Correlaciones genotipo-fenotipo En los úlimos años, todos los esfuerzos en el campo de la genética se han centrado en el estudio de la correlación entre el contenido genético del HSA21 y el fenotipo del SD. Por un lado, la genética molecular ha buscado la clonación de genes y la secuenciación completa del HSA21 [8]; por otro lado, la citogenética, mediante el desarrollo de técnicas más resolutivas que las bandas clásicas, ha perseguido la caracterización de casos de SD debidos a trisomía parcial del HSA21 de manera mucho más precisa. La complementariedad de ambas disciplinas se ha de sumar a los resultados de los estudios clínicos. El conjunto de toda la información debería resolver las correlaciones entre el genotipo y el fenotipo. El estudio de casos de SD debidos a la trisomía parcial del HSA21 se inició, estrictamente, en 1973, con la publicación del primer trabajo [9]. Las trisomías parciales del HSA21 incluyen las duplicaciones intersticiales, las traslocaciones que sólo implican al HSA21 y las traslocaciones entre el HSA21 y cualquiera de los demás cromosomas. Estos reordenamientos cromosómicos se traducen en la presencia por triplicado de determinados segmentos del HSA21, lo que permite la definición de regiones del HSA21 asociadas a determinados rasgos fenotípicos del SD. En la descripción de los primeros casos, Aula et al. [9] propusieron que la parte distal del HSA21 en trisomía era indispensable para producir el fenotipo del SD. Posteriormente, Niebuhr [10] propuso que la banda 21q22 en trisomía era «patogénica» para el SD i la denominó «región crítica del síndrome de Down» (Down Syndrome Critical Region o DSCR). En 1989, McCormick [11], mediante el uso de polimorfismos o el análisis de dosis de marcadores del HSA21 en 16 pacientes estudiados citogenéticamente, postuló que la región entre los marcadores D21S13 y D21S58 podía quedar excluida de la contribución al fenotipo final del SD, y que la región que realmente contribuía al fenotipo era la comprendida entre el marcador D21S55 y el telómero. Delabar et al. [12] describieron el primer mapa fenotípico del HSA21, construido a partir del estudio de 10 pacientes con SD y trisomía parcial. Este estudio reforzaba la hipótesis de la existencia y la importancia de la región crítica y la definía entre los marcadores D21S55 y MX1. A la inversa, excluía las regiones proximales (desde el pter hasta el marcador D21S54) y distal (desde el marcador PFKL hasta el S100B) de cualquier participación en el fenotipo final del SD. Un año más tarde, Korenberg [13] describió un mapa fenotípico del SD basado en 16 casos de SD debidos a diferentes tipos de trisomías parciales del HSA21. Este estudio, realizado mediante las técnicas de Southern blot y de hibridación in situ de fluorescencia (FISH) y mediante el análisis de 25 rasgos fenotípicos característicos de los pacientes, cuestionaba seriamente la hipótesis formulada por Niebuhr [10]. Estos autores determinaron que los rasgos fenotípicos –facies característica, microcefalia, baja estatura, hipotonía, dermatoglifos anómalos y retraso mental– quedaban fuera de la región crítica y que, por lo tanto, debía de haber una contribución importante al fenotipo de SD de genes de fuera de esta región. El mismo estudio también cuestionaba seriamente el trabajo publicado por De- SD 2001: vol. 5, núm. 2, pp. 19-24 REVISTA MÉDICA INTERNACIONAL SOBRE EL SÍNDROME DE DOWN 21 labar [12], argumentando que 9 de los 10 pacientes estudiados por Delabar et al. presentaban una trisomía parcial que incluía el marcador D21S55. El concepto de «región crítica del SD» sigue siendo objeto de controversia científica. El único punto sobre el que hay un acuerdo total es el de descartar que la trisomía parcial del brazo corto del HSA21 tenga ninguna consecuencia fenotípica. Por último, el estudio de las trisomías parciales y de las posibles correlaciones genotipo-fenotipo ha dividido la opinión de los científicos de este ámbito en dos grupos. El primero defiende que el fenotipo del SD no depende de la sobreexpresión de los genes del HSA21, sino del desequilibrio que produce la presencia de un exceso de material genético durante el desarrollo (hipótesis de la «amplified developmental instability», ampliamente defendida por Shapiro [14]). El segundo grupo defiende que la sobreexpresión de cada uno de los genes del HSA21 es directa e individualmente responsable de cada una de las manifestaciones clínicas que caracterizan, de forma conjunta, el fenotipo final del SD (hipótesis del «gene dosage effect»). empezar los estudios moleculares de búsqueda de genes en nuestro grupo. Debido a la presencia de quimerismo (presencia de señal en más de un cromosoma, es decir, contenido de secuencias diversas en un mismo YAC) en un número notable de estos YAC, la caracterización se extendió a un total de 49 YAC. En total se encontraron 12 quiméricos y 6 que no estaban en el HSA21, sino que pertenecían a otros cromosomas. Mediante la contratinción con DAPI se pudieron identificar los cromosomas en los que había señal. Los 6 YAC para los que no se vio señal de FISH en el HSA21 podían ser quiméricos, pero con una secuencia tan pequeña del HSA21 (probablemente inferior a 2 kb) que no se detectaron. Estos resultados indican que un 38% de los YAC son quiméricos, lo que se corresponde con el valor del 40% que da el CEPH y otorga a la FISH una fiabilidad del 92%. Para garantizar que los resultados fuesen unívocos se excluyeron del estudio todos esos YAC quiméricos. Materiales y métodos Se han identificado y estudiado a nivel clínico y molecular seis casos de SD debidos a trisomía parcial del HSA21 de procedencia diversa, nacional e internacional [15, 16, 17]. Los seis casos se han estudiado a nivel clínico siguiendo el protocolo de Epstein (1991). A nivel citogenético-molecular, la estrategia de estudio que se ha seguido es la misma en todos los casos: primero se ha hecho una hibridación de pintado cromosómico del HSA21 para detectar una posible traslocación de este cromosoma con otro cromosoma. A continuación, el proceso de estudio ha consistido en el análisis mediante FISH de los YAC del contiguo del HSA21, haciendo hibridaciones simultáneas con un YAC de la región centromérica y un YAC de la región telomérica del brazo largo, de forma que se fuese cubriendo el HSA21 hasta llegar al punto de rotura. Esta estrategia también ha permitido definir la orientación (directa o invertida) de los segmentos del HSA21 en trisomía. Los seis casos estudiados presentan los siguientes cariotipos: SD#1: 46,XX,-13,+der(13)t(21;13)(q32;q22.1) SD#2: 46,XY,-15,+der(15)t(21;13)(q26;q22.1) SD#3: 46,XY,-21,+der(21)dup(21)(q22.1-qter) SD#4: 46,XX,-21,+der(21)t(21;21)(p11;q22.1) SD#5: 46,XX,-21,+der(21)dup(21)(q22.1;qter) SD#6: 46,XX,-21,+der(21)dup(21)(q22.1) La Tabla 1 recoge los resultados de los estudios de fenotipo de los seis pacientes, ordenados según el alcance de su trisomía (de mayor a menor) para facilitar las comparaciones. En la figura 1B se recogen los resultados del análisis de los puntos de rotura en cada uno de los casos y se indican los marcadores de las regiones que contienen los puntos de rotura, junto a algunos genes comprendidos en estas regiones. Los casos SD#1 y SD#2 son de origen familiar (una traslocación en equilibrio, en ambos casos de origen paterno). La familia del caso SD#2 se estudió de la misma manera, siendo el padre, uno de los hermanos y la hermana (figura 1A) del caso, portadores de una traslocación equilibrada entre los cromosomas 15 y 21. De esta forma se pudo ofrecer consejo genético a la hermana, así como un diagnóstico prenatal de sus dos embarazos. El primer hijo resultó ser portador de la misma traslocación que la madre y, por lo tanto, también es totalmente asintomático. El segundo hijo tenía un cariotipo totalmente normal. Puesto que el estudio que se presenta está prácticamente basado en la técnica de la hibridación in situ de fluorescencia (FISH), se describe brevemente su principio. Hibridación in situ de fluorescencia La FISH es una técnica que consiste en marcar, con moléculas de biotina o digoxigenina (o directamente con un nucleótido conjugado a un fluorocromo), un fragmento de ADN de la región genómica que se quiere estudiar, para después hibridarlo sobre cromosomas, núcleos interfásicos o tejidos, aprovechando la afinidad entre las cadenas de ADN tras un proceso de desnaturalización por calor. Mediante incubaciones con anticuerpos o haptenos conjugados a fluorocromos que se unen con alta afinidad a las moléculas incorporadas en el ADN en el proceso de marcado, se llega a la observación en el microscopio de fluorescencia. De esta forma se puede localizar la señal fluorescente generada por la sonda marcada e hibridada. Esta técnica, además del mapaje cromosómico de cualquier tipo de sonda, permite detectar pérdidas o ganancias de ADN y reordenamientos cromosómicos (por pequeños que sean) y llegar hasta el punto de rotura de estos reordenamientos con una eficacia y fiabilidad realmente altas. Comparada con estrategias estrictamente moleculares, como la cuantificación de dosis de marcadores mediante Southern blot o la PCR cuantitativa, la FISH presenta la gran ventaja de ser siempre totalmente informativa y, al mismo tiempo, cubrir regiones genómicas mayores. Esta técnica ha supuesto un gran avance para el diagnóstico genético y ha establecido el vínculo entre dos disciplinas históricamente distantes, la citogenética y la genética molecular. Resultados Estudio del quimerismo de los YAC del cromosoma 21 De los YAC que CEPH/Généthon puso a disposición pública entre 1992 y 1993, se eligieron inicialmente 10 para que el HSA21 quedase cubierto en regiones grandes y para Estudio de casos de SD y trisomía parcial del cromosoma 21 SD 22 REVISTA MÉDICA INTERNACIONAL SOBRE EL SÍNDROME DE DOWN 2001: vol. 5, núm. 2, pp. 19-24 Figura 1. FISH en una metafase de uno de los casos estudiados. A. Pintado cromosómico de los cromosomas 15 y 21 de la hermana del caso SD#2, en la que se detecta una traslocación en equilibrio entre los dos cromosomas. B. Representación esquemática de todos los reordenamientos identificados en los pacientes con trisomía parcial 21 y SD. Al lado del ideograma del HSA21, se indican los marcadores de las regiones que contienen los puntos de rotura y las flechas indican el alcance de cada una de las trisomías. Discusión Mapa fenotípico del SD: Correlaciones genotipo-fenotipo Hay dos hechos fundamentales que dificultan la construcción de mapas fenotípicos en síndromes causados por aneuploidías. Por una parte, hay que tener en cuenta la penetrancia de cada uno de los rasgos clínicos que se quiere estudiar. La ausencia de un rasgo clínico determinado se puede deber, no tanto a la no trisomía del gen o genes como a la falta de penetrancia y expresividad del rasgo en ese paciente en concreto. Por esta razón, a la hora de intentar definir una región o grupo de genes responsables de una característica clínica del SD es indispensable basarse en fenotipos presentes, nunca en su ausencia. Sólo hay dos excepciones a esta regla: en primer lugar, cuando el grupo de pacientes es lo suficientemente grande y el rasgo no está presente en ninguno de ellos y, en segundo lugar, cuando la penetrancia del rasgo es del cien por cien. El otro aspecto muy importante que hay que tener en cuenta en este tipo de análisis es la implicación de otras cromosomopatías presentes en el paciente con SD y trisomía parcial del HSA21. Si la trisomía parcial se debe a una traslocación no recíproca entre el HSA21 y cualquier otro cromosoma, siempre hay implicada una monosomía parcial de este segundo cromosoma, que, entre otros rasgos clínicos, comportará con toda seguridad algún tipo de retraso mental. Muchos reordenamientos cromosómicos están asociados a manifestaciones clínicas que también se encuentran en el SD, como ocurre con el retraso mental; esto hace que no todas las manifestaciones clínicas del paciente se puedan atribuir de forma exclusiva a la trisomía del HSA21. Por este motivo, los casos de trisomía parcial de HSA21 más informativos son aquellos en los que no hay ninguna otra alteración cromosómica como, por ejemplo, los casos de traslocaciones 21; 21 o de duplicaciones intersticiales. Los casos SD#1 y SD#2 son ejemplos de los posibles efectos de una monosomía concomitante con la trisomía parcial del HSA21. SD#1 presenta un defecto de los factores VIII y X de la coagulación que se ubican en la región monosómica de 13q del paciente generada por la traslocación. Se han asociado defectos de estos factores hemostáticos a deleciones de q13-qter. SD#2 tiene unas extremidades muy cortas que se podrían relacionar con la monosomía de 15q26 debida también a la traslocación. En 15q26 se halla el gen IGFRI (Insulin-like Grow Factor Receptor I), que se ha descrito que puede estar asociado a un retraso del crecimiento [18]. Así pues, la hipotonía del recién nacido sería un fenotipo menos complicado de asignar a una determinada región del HSA21, ya que también tiene una penetrancia del cien por cien en el SD y no está descrita en las demás cromosomopatías. Según el mapa fenotípico del grupo de Korenberg [13], este rasgo se debe a la trisomía de las bandas 21q21.1q22.2. A partir de los estudios presentados en este trabajo se podría acotar más esta región excluyendo el segmento comprendido entre los marcadores D21S216 y D21S323, ya que el caso SD#6 tiene una duplicación intersticial que alcanza esta región y no presenta hipotonía. Las asignaciones de los fenotipos a las distintas regiones del HSA21 no están determinadas de forma indiscutible, porque, si nos basamos en este mismo caso (SD#6), los dos rasgos clínicos de SD que presenta son el surco palmar transverso y la comunicación interventricular. Estos dos rasgos fueron asignados por Korenberg [13] a la región más distal del brazo largo del HSA21, región que en SD#6 tampoco está triplicada. Esta discrepancia sólo se puede explicar de dos maneras: por la incidencia de los dos rasgos en la población general (en ambos casos, la incidencia es del 1% y podrían ser considerados acontecimientos independientes de la trisomía parcial), o bien porque hay más de una región del HSA21 cuya trisomía produce el mismo fenotipo. ¿Podría SD 2001: vol. 5, núm. 2, pp. 19-24 REVISTA MÉDICA INTERNACIONAL SOBRE EL SÍNDROME DE DOWN 23 Tabla 1. Resumen de los datos clínicos más relevantes de los casos de SD y trisomía parcial del HSA21 estudiados ser el gen GART, único gen conocido en la región trisómica de esta paciente, el responsable de sus rasgos clínicos? GART codifica por una proteína trifuncional implicada en la síntesis de purinas. Los nucleótidos purínicos funcionan como precursores de la síntesis de ARN y de ADN, de coenzimas, de moléculas transmisoras de energía y de factores de regulación. En particular, los niveles elevados de purinas que se detectan en el SD se han asociado al retraso mental [19]. Los seis casos de SD y trisomía parcial del HSA21 estudiados presentan cuatro regiones trisómicas diferentes (figura 1B, Tabla 1). SD#2 y SD#3 comparten la misma región en trisomía, desde el marcador D21S302 hasta el telómero; SD#4 y SD#5 presentan trisomía desde el marcador D21S226 también hasta el telómero; SD#1 tiene una región trisómica más pequeña, desde CBR hasta el telómero; y SD#6 presenta una duplicación intersticial desde el marcador D21S216 hasta el D21S323. Todos estos datos indican, por una parte, la posible existencia de puntos calientes de rotura (hot spots) en torno a los marcadores D21S302 y D21S226 y permiten, por otra parte, establecer comparaciones entre los fenotipos de los pacientes. SD#4 y SD#5, los dos pacientes con la trisomía parcial del HSA21 mayor, presentan casi todos los rasgos fenotípicos del SD excepto la lengua escrotal, las manchas de Brushfield, la boca permanentemente abierta y el paladar alto y estrecho. Por su parte, los casos SD#2 y SD#3 también presentan casi todos los rasgos clínicos del síndrome, salvo las manchas de Brushfield, la lengua escrotal y el paladar alto y estrecho. En consecuencia, las manchas de Brushfield y la lengua escrotal serían los dos rasgos clínicos ausentes en los cuatro casos y, según estos resultados, deberían situarse entre el centrómero y el marcador D21S226. Si se establece una correlación de estos cuatro casos con el caso SD#1, la lengua escrotal y las manchas de Brushfield siguen estando ausentes, lo cual corroboraría la hipótesis planteada. Estos resultados coinciden en parte con los de Delabar [12] y Korenberg [13], pero son más restrictivos, puesto que la región que aquí se define es muy pequeña. Por otra parte, los resultados presentados confirman que las regiones más proximales del brazo largo del HSA21 tienen una participación menos sustancial en el fenotipo final del SD. Si se comparan los rasgos clínicos estudiados en los seis pacientes y según el mismo protocolo, se ve que, conside- rando todos los casos en conjunto, se dan todos los rasgos clínicos del SD excepto los mencionados anteriormente, la estenosis duodenal y la leucemia, que no están presentes en ninguno de ellos. Ya se ha dicho que la incidencia de la estenosis duodenal en el SD es del 4-7%, lo que podría explicar la ausencia de esta alteración en los tres casos mencionados, ya que Korenberg et al. [20] situaron este rasgo entre los marcadores D21S8 i D21S15, en trisomía en los tres casos. En cuanto a las leucemias, hay que tener en cuenta que los pacientes SD#1, SD#3 y SD#4 aún no han cumplido los 16 años, por lo que presentan un riesgo entre 10 y 18 veces mayor que la población general de desarrollar esta enfermedad, a diferencia de los casos SD#2 y SD#5, que presentan un riesgo menor porque son mayores de 16 años. Por lo tanto, tampoco se puede afirmar que la ausencia de estas dos enfermedades en estos tres pacientes se deba a la diploidía de la región proximal al marcador D21S302. La enfermedad de Alzheimer en los casos de SD se debe a la trisomía del gen APP, que se encuentra cerca del marcador D21S386, fuera de la trisomía parcial en todos los casos. Los dos casos estudiados con edades superiores a los 30 años respaldarían esta hipótesis, ya que actualmente SD#2 tiene 30 años y SD#5, 34, y ninguno de los dos ha desarrollado rasgos clínicos de la enfermedad de Alzheimer. Sin embargo, para confirmar este extremo habrá que esperar a ver cómo evolucionan ambos en los próximos años. Para establecer las correlaciones genotipo-fenotipo hace falta disponer de muchos casos de trisomías parciales del HSA21 y tenerlos bien caracterizados desde el punto de vista clínico y molecular para poder hacer comparaciones válidas. Con esta finalidad, J. Delabar creó en 1996 una base de datos accesible a través de Internet que pretende recoger todos los casos de aneuploidías parciales del HSA21 (http://www.infobiogen.fr/services/aneu21). De esta base de datos se han extraído dos casos de trisomía parcial (DL y SOL) con puntos de rotura parecidos a los de los casos que se han estudiado aquí. Así se han establecido las comparaciones que se han presentado en la sección de Resultados. Posiblemente, si los estudios de marcadores fueran más completos se podría comparar un mayor número de casos. Sin embargo, bastaría con el número de trisomías que se han descrito hasta ahora si la información clínica, molecular y funcional del HSA21 fuese completa. SD 24 REVISTA MÉDICA INTERNACIONAL SOBRE EL SÍNDROME DE DOWN Perspectivas de futuro de los estudios de las trisomías parciales del cromosoma 21 Cuando se iniciaron los estudios y las caracterizaciones de las trisomías parciales en el SD, parecía que servirían para responder a preguntas que, de otra forma, no se podrían responder hasta que no se completase el proyecto de secuenciación del HSA21 y no se conociese la función de cada uno de los genes que lo componen. Tal y como se refleja en este trabajo, esto no ha ocurrido, sobre todo por el problema añadido de las penetrancias de cada una de las manifestaciones clínicas que componen este síndrome. Aunque a priori ya se sabía que en el grupo de personas con SD y trisomía regular del HSA21 había una gran variabilidad en la incidencia de determinadas manifestaciones clínicas y una diversidad interpersonal muy grande, se esperaba que los casos de trisomías parciales cubrirían muchas de las lagunas del conocimiento clínico y molecular de este síndrome. Se han hecho muchísimos progresos y quizá, a nivel genérico, la conclusión es que cada persona con SD es diferente debido a su carga genética particular. Con la publicación de la secuencia completa del HSA21, la clonación de todos los genes se acabará en un período de tiempo breve y habrá que estudiar entonces la función de cada uno de esos genes. Una clave para ver si una trisomía del HSA21 produce una sobreexpresión de otros genes en todo el genoma deberán darla los estudios de sobreexpresión de ARN mediante la técnica de análisis de expresión con microarrays. Agradecimientos Este trabajo se ha podido hacer gracias a la colaboración de los equipos clínicos del Hospital del Mar de Barcelona, del Servicio de Genética del Hospital Clínic de Barcelona, del Hospital de la Vall d’Hebron de Barcelona, del Hospital Virgen del Camino de Pamplona, del Hospital San Giovanni Rotondo de Foggia y de la Universidade Federal de Sâo Paulo. Asimismo, agradecemos al Dr. S. Antonarakis los cósmidos del YAC230e8. Este trabajo ha sido subvencionado en parte por la Fundació Catalana Síndrome de Down/Marató de TV3. 16. 17. 18. 19. 10. 11. 12. 13. 14. 15. 16. Bibliografía 11. Antonarakis SE, Petersen MB, McInnis MG, Adelsberger PA, Schinzel AA, Binkert F, Pangalos C, Raoul O, et al. The meiotic stage of nondisjunction in trisomy 21: determination by using DNA polymorphisms. Am J Hum Genet 1992; 50: 544-50. 12. Epstein CJ, Korenberg JR, Anneren G, Antonarakis SE, Ayme S, Courchesne E, Epstein LB, Fowler A, et al. Protocols to establish genotype-phenotype correlations in Down syndrome. Am J Hum Genet 1991; 49: 207-35. 13. Fryers T. Survival in Down’s syndrome. J Ment Defic Res 1986; 30: 101-10. 14. Robinson L, Neglia J. Epidemiology of Down syndrome and childhood acute leukaemia. En McCoy EE, ed. Oncology and Immunology of Down syndrome. New York: Liss; 1987: 19-32. 15. Johannisson R, Gropp A, Winking H, Coerdt W, Rehder 17. 18. 19. 20. 2001: vol. 5, núm. 2, pp. 19-24 H, Schwinger E. Down’s syndrome in the male: Reproductive pathology and meiotic studies. Hum Genet 1983; 63: 132. Avramopoulos D, Kennerknecht I, Barbi G, Eckert D, Delabar JM, Maunoury C, Hallberg A, Petersen MB. A case of apparent trisomy 21 without the Down’s syndrome phenotype. J Med Genet 1997; 34: 597-600. Ahlbom BE, Goetz P, Korenberg JR, Pettersson U, Seemanova E, Wadelius C, Zech L, Anneren G. Molecular analysis of chromosome 21 in a patient with a phenotype of Down syndrome and apparently normal karyotype. Am J Med Genet 1996; 63: 566-72. Hattori M, Fujiyama A, Taylor TD, Watanabe H, Yada T, Park HS, Toyoda A, Ishii K, et al. The DNA sequence of human chromosome 21. The chromosome 21 mapping and sequencing consortium. Nature 2000; 405: 311-9. Aula P, Leisti J, Koskull HV. Partial trisomy 21. Clin Genet 1973; 4: 241-51. Niebuhr E. Down’s syndrome. The possibility of a pathogenetic segment on chromosome 21. Hum Genet 1974; 21: 99-101. McCormick MK, Schinzel A, Petersen MB, Stetten G, Driscoll DJ, Cantu ES, Tranebjaerg L, Mikkelsen M, et al. Molecular genetic approach to the characterization of the «Down syndrome region» of chromosome 21. Genomics 1989; 5: 325-31. Delabar JM, Theophile D, Rahmani Z, Chettouh Z, Blouin JL, Prieur M, Noel B, Sinet PM. Molecular mapping of twenty-four features of Down syndrome on chromosome 21. Eur J Hum Genet 1993; 1: 114-24. Korenberg JR, Chen XN, Schipper R, Sun Z, Gonsky R, Gerwehr S, Carpenter N, Daumer C, et al. Down syndrome phenotypes: the consequences of chromosomal imbalance. Proc Natl Acad Sci USA 1994; 91: 49975001. Shapiro B. The Down syndrome critical region. En Lubec G. The molecular biology of Down Syndrome. Wien: Springer Medicine; 2000: 41-59. Nadal M, Mila M, Pritchard M, Mur A, Pujals J, Blouin JL, Antonarakis SE, Ballesta F, Estivill X. YAC and cosmid FISH mapping of an unbalanced chromosomal translocation causing partial trisomy 21 and Down syndrome. Hum Genet 1996; 98: 460-6. Nadal M, Moreno S, Pritchard M, Preciado MA, Estivill X, Ramos-Arroyo MA. Down syndrome: characterisation of a case with partial trisomy of chromosome 21 owing to a paternal balanced translocation (15;21) (q26;q22.1) by FISH. J Med Genet 1997; 34: 50-4. Nadal M, Guzmán-Vigo C, Melaragno M, Andrade J, García-Alonso L, Brunoni D, Pritchard M, Estivill X. Clinical and cytogenetic characterisation of a patient with Down syndrome due to a 22q22.1—>qter duplication. J Med Genet 2001; 38:73-76. Fryns JP, van den Berghe H. Possible excess of mental handicap and congenital malformations in autosomal reciprocal translocations. Ann Genet 1979; 22: 125-7. Jaeken J, Berghe GVD. An infantile autistic syndrome characterised by the presence of succinylpurines in body fluids. Lancet 1984; 2: 1058-61. Korenberg JR, Bradley C, Disteche CM. Down syndrome: molecular mapping of the congenital heart disease and duodenal stenosis. Am J Hum Genet 1992; 50, 294-302.