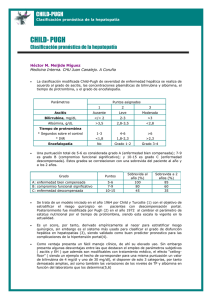
Continuación y bibliografía
Anuncio

Capítulo 352 Hepatopatía asociada con trastornos sistémicos & e352-1 © ELSEVIER. Fotocopiar sin autorización es un delito. ENFERMEDAD INTESTINAL INFLAMATORIA La colitis ulcerosa y la enfermedad de Crohn (cap. 328) se asocian con enfermedad hepatobiliar, que incluye procesos autoinmunitarios e inflamatorios relacionados con enfermedad intestinal inflamatoria (EII) (colangitis esclerosante, hepatitis autoinmunitaria), toxicidad medicamentosa (mercaptopurina, metotrexato, 6-tioguanina), malnutrición y trastornos fisiológicos (hígado graso, colelitiasis), translocación bacteriana e infecciones sistémicas (absceso hepático, trombosis de la vena porta), hipercoagulabilidad (infarto) y complicaciones a largo plazo de estas hepatopatías, como colangitis ascendente, cirrosis, hipertensión portal y carcinoma biliar. La colangitis esclerosante es una enfermedad hepatobiliar común asociada con EII, y se da en el 2-8% de los pacientes adultos con colitis ulcerosa, y con menor frecuencia en la enfermedad de Crohn. A la inversa, el 70-90% de los pacientes con colangitis esclerosante tiene colitis ulcerosa. En los pacientes pediátricos con EII el diagnóstico suele establecerse en la segunda década de la vida. La colangitis esclerosante se caracteriza por inflamación y fibrosis progresivas de segmentos de conductos biliares intra y extrahepáticos y puede progresar a una obliteración completa. Se ha demostrado susceptibilidad genética, con asociaciones con diversos antígenos leucocitarios humanos. Muchos pacientes están asintomáticos y se diagnostican inicialmente por las pruebas de función hepática que revelan una elevación de las funciones de fosfata alcalina (FA), 50 -nucleotidasa o g-glutamil transpeptidasa (GGT). También puede haber en el suero anticuerpos antinucleares o frente al músculo liso. El 10-15% de los pacientes adultos presenta síntomas consistentes en anorexia, pérdida de peso, prurito, fatiga, dolor en el hipocondrio derecho e ictericia; también puede haber colangitis aguda intermitente acompañada de fiebre, ictericia y dolor en el hipocondrio derecho. Puede desarrollarse hipertensión portal con la enfermedad progresiva. Estos síntomas son menos frecuentes en niños en los que la enfermedad hepatobiliar se reconoce con frecuencia por un cribado de rutina de las pruebas de función hepática. En ocasiones, los niños presentan inicialmente colangitis esclerosante y la EII acompañante se descubre sólo en una endoscopia posterior. La colangiografía por resonancia magnética (CRM) es una prueba diagnóstica de primera línea establecida para la colangitis esclerosante. Los hallazgos característicos consisten en un aspecto arrosariado e irregular de los conductos biliares intra y extrahepáticos. La biopsia hepática suele mostrar fibrosis e inflamación periductales, colangitis fibroobliterativa y fibrosis portal, pero puede no ser necesaria para el diagnóstico en pacientes con evidencia radiológica de colangitis esclerosante. La colangitis esclerosante presenta una estrecha asociación con neoplasias malignas hepatobiliares (colangiocarcinoma, carcinoma hepatocelular, carcinoma de la vesícula biliar), con una incidencia descrita que varía entre el 9% y el 14%. En una serie a gran escala, los pacientes con EII y colangitis esclerosante tenían un incremento de 10 veces del riesgo de carcinoma colorrectal y un aumento de 14 veces del riesgo de cáncer pancreático respecto a la población general. La serología tumoral (CA 19-9) y las pruebas de imagen en proyección transversal pueden constituir una estrategia de cribado útil para identificar a los pacientes con colangitis esclerosante que presenten un mayor riesgo de colangiocarcinoma. No hay tratamiento médico definitivo para la colangitis esclerosante; el trasplante hepático es la única opción a largo plazo para la cirrosis progresiva y la enfermedad autoinmunitaria puede recidivar en el aloinjerto. El tratamiento a corto plazo intenta mejorar el drenaje biliar y frenar el proceso obliterativo. El ácido ursodesoxicólico, en dosis de 15-30 mg/kg/24 horas, mejora el flujo de bilis y los parámetros de laboratorio, pero no se ha demostrado que mejore el pronóstico clínico. La vancomicina oral puede mejorar también los niveles bioquímicos séricos. Las estenosis biliares extrahepáticas dominantes pueden dilatarse o someterse a la colocación de endoprótesis por vía endoscópica. El tratamiento inmunosupresor con corticoides y/o azatioprina mejora los parámetros bioquímicos, pero ha sido decepcionante en la detención de la progresión histológica a largo plazo. El tratamiento sintomático debe iniciarse cuando haya prurito (rifampicina, ácido ursodesoxicólico, difenhidramina), malnutrición (suplementación enteral) y colangitis ascendente (antibióticos) según esté indicado. La colectomía total no ha sido beneficiosa en la prevención o tratamiento de las complicaciones hepatobiliares en pacientes con colitis ulcerosa. La hepatitis autoinmunitaria (HAI) asociada a EII se puede parecer mucho a la colangitis esclerosante asociada a la EII, afección denominada a menudo síndrome de solapamiento o colangitis esclerosante autoinmunitaria (CEA). Estos pacientes suelen tener hiperglobulinemia (aumento acusado de los niveles séricos de inmunoglobulina [Ig] G). Algunos niños son diagnosticados inicialmente de HAI y posteriormente se encuentra que tienen colangitis esclerosante después de la colangiografía; en otros casos, la HAI se presenta años después de haber diagnosticado colangitis esclerosante asociada a EII. La biopsia hepática en pacientes con síndrome de solapamiento de HAI/colangitis esclerosante (CEA) muestra hepatitis en interfase, además de lesión de los conductos biliares asociada con colangitis esclerosante. La medicación inmunosupresora (corticoides y/o azatioprina) es el pilar principal del tratamiento de la CEA; no parece que la respuesta a largo plazo sea tan favorable como en la HAI sola. La supervivencia a largo plazo en niños con CEA parece ser similar a la de los que tienen colangitis esclerosante, con una supervivencia mediana (50%) global sin trasplante hepático de 12,7 años. La esteatosis hepática puede ser también más prevalente en pacientes adultos con EII, oscilando entre un 25% y un 40% en una serie extensa. Los cálculos biliares son más prevalentes en los que tienen enfermedad de Crohn (11%) que en aquéllos con colitis ulcerosa (7,5%) y en personas sanas (5%). Sin embargo, la prevalencia verdadera de estas enfermedades hepáticas asociadas a la EII en los pacientes pediátricos es desconocida. SEPSIS BACTERIANA (CAPS. 103 Y 170) La sepsis puede simular una hepatopatía y debe descartarse en cualquier paciente en estado crítico que tenga colestasis en ausencia de una elevación acusada de los niveles séricos de aminotransferasas o FA, incluso cuando no sean manifiestos otros signos de infección. Los microorganismos gramnegativos son los que se aíslan con mayor frecuencia de los hemocultivos, en especial Escherichia coli, Klebsiella pneumoniae y Pseudomonas aeruginosa. Se cree que los lipopolisacáridos y otras endotoxinas bacterianas interfieren con la secreción biliar al alterar de modo directo la estructura o la función de las proteínas de transporte de la membrana de los canalículos biliares. El nivel sérico de la bilirrubina está aumentado, por lo general con predominio de la fracción conjugada. Las actividades séricas de la FA y las aminotransferasas no siempre están elevadas. La biopsia hepática muestra colestasis intrahepática con escasa o nula necrosis del hepatocito. También son comunes la hiperplasia de las células de Kupffer y un aumento de las células inflamatorias. Pueden observarse unos hallazgos parecidos en la sepsis urinaria. CARDIOPATÍA La lesión hepática puede producirse como complicación de una insuficiencia cardíaca congestiva aguda o crónica grave (cap. 436), cardiopatía congénita cianótica (caps. 423 y 424) y shock isquémico agudo. En todas las afecciones, tanto la congestión pasiva como un menor gasto cardíaco pueden contribuir al daño hepático. El aumento de la presión venosa central se transmite a las venas hepáticas, vénulas más pequeñas y, en último término, a los hepatocitos circundantes, lo que causa atrofia hepatocelular en la zona centrolobulillar del hígado. Debido a la disminución del gasto cardíaco hay una reducción del flujo de sangre arterial hepático, con lo que se produce hipoxia centrolobulillar. La necrosis hepática causa acidosis láctica, aumento de los niveles de aminotransferasas, colestasis, aumento del tiempo de tromboplastina parcial y, posiblemente, hipoglucemia debido a una alteración del metabolismo hepatocelular. Puede producirse ictericia, hepatomegalia dolorosa e352-2 & Parte XVIII Sistema digestivo y, en algunos casos, ascitis y esplenomegalia. En los adultos, las anomalías de las pruebas de función hepática se observan en el 3-18% de los pacientes con insuficiencia cardíaca crónica y la concentración sérica total de bilirrubina es un factor predictivo de mal pronóstico. Después de un shock hipovolémico agudo, los niveles séricos de aminotransferasas pueden elevarse espectacularmente, pero se normalizan con rapidez cuando mejoran la perfusión y la función cardíaca. En lactantes con síndrome de hipoplasia del corazón izquierdo y coartación de la aorta puede producirse necrosis hepática o insuficiencia hepática aguda. Unas presiones sistémicas venosas elevadas después de procedimientos de Fontan pueden casar también disfunción hepática, marcada por un tiempo de protrombina prolongado y cirrosis cardíaca. El objetivo del tratamiento en todas las causas de hepatopatía de origen cardíaco es mejorar el gasto cardíaco, reducir las presiones venosas sistémicas y monitorizar otros signos de hipoperfusión siguiendo muy estrechamente la diuresis y el nivel de consciencia. OBESIDAD La enfermedad del hígado graso no alcohólico (EHGNA) forma parte del espectro de hepatopatía fuertemente asociado con la obesidad. La EHGNA puede variar de hígado graso solo a una tríada de infiltración grasa, inflamación y fibrosis (esteatohepatitis no alcohólica [EHNA]), que se asemeja a la hepatopatía alcohólica pero que se produce con poca o nula exposición al etanol. Se pensaba que la EHGNA se daba principalmente en adultos obesos mayores (sobre todo mujeres) con obesidad central, resistencia a la insulina y diabetes mellitus de tipo II, pero cada vez se ha descrito con mayor frecuencia también en niños. Muchos pacientes están asintomáticos. La histología hepática en los datos de autopsia sugiere que el 10% de los niños y el 38% de los niños obesos de 2-19 años podrían tener EHGNA. El riesgo es menor en niños afroamericanos. La elevación del nivel sérico de las aminotransferasas no constituye un marcador sensible ni específico de la EHGNA. En el 21-23% de los pacientes pediátricos con EHGNA la concentración sérica de ALT es normal. Aunque la ecografía detecta la EHGNA, ninguna modalidad de imagen actual distingue entre la esteatosis y la EHNA. Puede requerirse una biopsia hepática para establecer el diagnóstico. Se cree que la prevalencia estimada en adultos es de hasta el 15-20% de EHGNA globalmente y del 2-4% de EHNA. Los factores de riesgo en las cohortes pediátricas son la obesidad, ser varón, etnia blanca o sudamericana, hipertrigliceridemia y resistencia a insulina. La esteatosis hepática sola puede ser benigna, pero hasta una cuarta parte de los pacientes con EHNA pueden desarrollar fibrosis progresiva con cirrosis resultante. El pronóstico a largo plazo de la EHNA que se ha desarrollado en la infancia es desconocido. Una pérdida de peso gradual es eficaz para lograr normalizar la ALT sérica y mejorar la EHGNA. Las vitaminas E y C no proporcionan beneficios adicionales a la eficacia de la modificación del estilo de vida (dieta y ejercicio) a la hora de mejorar la histología hepática y las anomalías bioquímicas en la EHGNA pediátrica. La metformina ha producido resultados mixtos en el tratamiento de la EHGNA. Las tiazolidinedionas (pioglitazona, rosiglitazona) mejoran la histología hepática en adultos con EHNA, pero no se han estudiado con detalle en niños. COLESTASIS ASOCIADA CON NUTRICIÓN PARENTERAL TOTAL La nutrición parental total (NPT) puede causar diversas hepatopatías, como la esteatosis hepática, daño en la vesícula biliar y conducto colédoco, y colestasis. Ésta es la complicación más grave y puede provocar una fibrosis progresiva y cirrosis, sobre todo en lactantes y niños pequeños sometidos a NPT prolongada. Es el principal factor limitante del empleo a largo plazo de la NPT tanto en niños como en adultos. Los factores de riesgo de la colestasis asociada a la NPT son la duración prolongada de la NPT, prematuridad, bajo peso al nacimiento, sepsis, enterocolitis necrosante y síndrome de intestino corto. En los lactantes de bajo peso al nacimiento se desarrolla colestasis asociada a la NPT en casi la mitad de aquéllos con un peso al nacimiento <1.000 g, en el 20% de los que pesaban 1.000-1.500 g y en el 5-10% de los que pesaban 1.5002.000 g. La patogenia de la colestasis asociada a la NPT es multifactorial. Sepsis, exceso de ingesta calórica, cantidades elevadas de proteína, grasa o carbohidratos, toxicidades específicas de aminoácidos, deficiencias de nutrientes y toxicidades relacionadas con componentes tales como manganeso, aluminio y cobre pueden contribuir a la lesión hepática. Un ayuno enteral prolongado compromete la integridad de la mucosa y aumenta la translocación mucosa bacteriana. El ayuno disminuye también las hormonas enterales tales como la colecistocinina, que estimula el flujo biliar. La sepsis, en especial debida a bacterias gramnegativas, y las toxinas asociadas pueden exacerbar el daño hepático. Los hallazgos histológicos precoces consisten en esteatosis macrovesicular, colestasis canalicular e inflamación periportal. Estos cambios pueden regresar después del cese de la NPT a corto plazo. La duración prolongada de la NPT se ve marcada por proliferación de conductos biliares, fibrosis portal y expansión de las tríadas portales y puede progresar a cirrosis y a hepatopatía terminal. El comienzo clínico se ve marcado típicamente por un inicio gradual de colestasis, que se desarrolla más de 2 semanas después de la NPT. En los lactantes de bajo peso al nacimiento, el comienzo de la ictericia puede superponerse con la fase de hiperbilirrubinemia (no conjugada) fisiológica. En cualquier lactante ictérico que haya recibido NPT durante más de 1 semana deben analizarse de forma fraccionada todas las determinaciones de bilirrubina que se le practiquen. Con una duración prolongada puede producirse hepatomegalia o esplenomegalia. Pueden aumentar las concentraciones séricas de ácidos biliares. Los aumentos de las actividades de las aminotransferasas séricas pueden constituir hallazgos tardíos. Una elevación de la actividad de FA sérica puede deberse a raquitismo, complicación común de la NPT en los lactantes de bajo peso al nacimiento. Además de la colestasis, las complicaciones biliares de la nutrición intravenosa consisten en colelitiasis y desarrollo de barro biliar, asociado con unos contenidos vesiculares densos y condensados. Pueden ser asintomáticos. También puede producirse una esteatosis hepática o una elevación de los niveles séricos de aminotransferasas en ausencia de colestasis, sobre todo en niños de mayor edad. Esto suele ser leve y se resuelve una vez que se suspende la NPT. Los niveles séricos de bilirrubina y de ácidos biliares permanecen dentro de la gama de la normalidad. También se deben considerar otras causas de hepatopatía, sobre todo si persisten datos de disfunción hepática a pesar de retirar la NPT y de dar comienzo a las alimentaciones enterales. El grupo de riesgo en el que es más común la colestasis asociada a la NPT recibe con frecuencia hemoderivados o fármacos. Por consiguiente, hay que considerar la hepatopatía inducida por virus o medicamentos. Si siguen elevados los niveles séricos de FA o de aminotransferasas puede requerirse una biopsia hepática para un diagnóstico exacto. El tratamiento de la colestasis asociada a la NPT se centra en evitar una lesión hepática progresiva limitando la duración cuando sea posible. La alimentación enteral se comenzará tan pronto como se tolere y se debe evitar un ayuno prolongado. Incluso volúmenes pequeños de nutrientes administrados por tomas orales intermitentes o por goteo nasogástrico continuo estimulan el flujo biliar, la recirculación enterohepática de ácidos biliares y la motilidad intestinal, y favorecen la función de barrera mucosa, lo que reduce el riesgo de translocación bacteriana. Unas mejores soluciones de NPT que satisfagan las necesidades específicas de los recién nacidos pueden prevenir las deficiencias y evitar toxicidades. Siempre se debe considerar el riesgo de una mayor lesión hepática cuando se sopesa la opción de continuar de modo indefinido con la NPT y se debe hacer todo lo posible para intentar avanzar las tomas enterales cuando sea posible. El tratamiento con ácido ursodesoxicólico puede ser beneficioso para mejorar la ictericia y la esplenomegalia. Otros tratamientos, como la administración de antibióticos para reducir el sobre- Capítulo 352 Hepatopatía asociada con trastornos sistémicos & e352-3 crecimiento bacteriano o la administración oral de taurina o colecistocinina, siguen siendo experimentales. FIBROSIS QUÍSTICA La fibrosis quística (FQ) está causada por mutaciones del gen regulador de la conductancia transmembrana de la FQ (CFTR), que alteran el transporte de cloruro a través de las membranas apicales de las células epiteliales en numerosos órganos (incluidos los colangiocitos) (cap. 395). La mayoría de los pacientes con FQ tienen algún signo de enfermedad hepatobiliar, pero menos de un tercio de éstos desarrollan una hepatopatía significativa desde el punto de vista clínico. Las complicaciones hepatobiliares suponen alrededor del 2,5% de la mortalidad global en los pacientes con FQ. La cirrosis biliar focal es la lesión hepática patognomónica de la FQ y se ha propuesto que se debe, en parte, a la alteración de la función secretora del epitelio de los conductos biliares. El bloqueo de los conductillos biliares debido a las secreciones viscosas provoca inflamación periductal, proliferación de los conductos biliares y aumento de la fibrosis en los tractos portales focales. Puede producirse una progresión gradual a cirrosis multilobulillar y dar lugar a hipertensión portal y hepatopatía terminal en el 1-8% de los pacientes. La hepatopatía tiende a ocurrir principalmente en pacientes con insuficiencia pancreática, asociación que no se comprende bien. El agrupamiento familiar sugiere una predisposición genética; sin embargo, no se ha observado asociación específica alguna entre genotipo-fenotipo y mutaciones específicas del CFTR. El análisis mutacional no es útil en este momento para predecir en qué pacientes con FQ se desarrollará la hepatopatía. Los factores de riesgo clínicos que pueden asociarse con hepatopatía son una mayor edad, insuficiencia pancreática, ser varón y, posiblemente, los antecedentes de íleo meconial. El tratamiento con ácido ursodesoxicólico (10-15 mg/kg/día) puede ser beneficioso para mejorar la función hepática, presumiblemente al mejorar el flujo biliar; se requieren nuevas investigaciones para determinar si existe un beneficio verdadero a largo plazo. Dado que es difícil predecir en qué pacientes se desarrollará hepatopatía, la profilaxis no es posible. La progresión de la hepatopatía suele ser lenta. Los pacientes en los que se desarrolla hepatopatía terminal pueden requerir trasplante hepático para su supervivencia. © ELSEVIER. Fotocopiar sin autorización es un delito. TRASPLANTE DE MÉDULA ÓSEA La hepatopatía es frecuente en pacientes que han recibido trasplante de células progenitoras (TCP) hematopoyéticas, tanto si las células se extraen de la médula ósea como de la sangre periférica (caps. 129133). La etiopatogenia es variada e incluye infecciones (víricas, bacterianas o micóticas), toxicidad por fármacos, nutrición parenteral, quimioterapia o radiación, enfermedad venooclusiva (EVO) o enfermedad del injerto contra el huésped (EICH), o bien hemosiderosis secundaria a sobrecarga de hierro por las frecuentes transfusiones de sangre. La EICH, la toxicidad medicamentosa y la sepsis son las causas más comunes de disfunción hepática después del trasplante de células progenitoras alogénicas. Con frecuencia, el diagnóstico es difícil debido a la coexistencia de múltiples factores de riesgo. Para efectuar el diagnóstico correcto hay que considerar la evolución clínica, los síntomas y los signos, así como las pruebas bioquímicas de función hepática y serológicas de virus. Puede requerirse una biopsia hepática percutánea; la histología puede revelar la presencia de una lesión extensa de los conductos biliares en la EICH, inclusiones víricas en la enfermedad por citomegalovirus o la lesión endotelial característica en la EVO. Es esencial diagnosticar la causa de modo exacto, ya que el tratamiento de la EICH difiere de modo acusado del de otras afecciones (p. ej., el tratamiento de la EICH implica iniciar una inmunosupresión) y puede empeorar una hepatitis secundaria a infecciones. La EICH del hígado puede ser aguda o crónica, pero a menudo se produce con la presencia de EICH en otros órganos diana tales como la piel y el intestino (cap. 131). La EICH hepática está causada por una reacción inmunológica contra el epitelio de los conductos biliares, lo que ocasiona colangitis no supurativa. Las características histológicas de la EICH son la pérdida de conductos biliares intralobulillares, lesión endotelial de las vénulas hepáticas y portales y necrosis hepatocelular. El comienzo suele producirse en el momento del prendimiento del donante (días 14-21 después del TCP). En la EICH aguda los niveles séricos de aminotransferasas pueden elevarse de modo acusado en ausencia de un aumento de los niveles de bilirrubina, FA y GGT, lo que simula una hepatitis vírica. La EICH aguda puede presentarse de forma precoz (días 14-21) y tardía (>70 días) después del TCP alogénico. En la EICH hepática crónica los niveles séricos de aminotransferasas no se hallan tan acusadamente elevados y la colestasis es más prominente, con elevaciones marcadas de los niveles séricos de bilirrubina conjugada, GGT y FA. Otros signos y síntomas pueden ser hipersensibilidad dolorosa hepática, coluria, heces acólicas, prurito y anorexia. La EVO hepática suele comenzar 1-3 semanas después del TCP. La incidencia es del 5-39% en pacientes pediátricos, con una mortalidad del 0-47% Los factores de riesgo son los traumatismos, coagulopatías, anemia drepanocítica, leucemia, policitemia vera, talasemia mayor, abscesos hepáticos, radiación, EICH, sobrecarga de hierro, pautas de quimioterapia de acondicionamiento y una edad más joven. La EVO está causada por obliteración fibrosa de las vénulas hepáticas terminales y de las venas lobulillares pequeñas, con lesión resultante de los hepatocitos y sinusoides circundantes. No se asocia con formación de trombos, a diferencia del síndrome de Budd-Chiari, que implica la oclusión de las venas hepáticas de mayor tamaño o de la vena cava inferior por una membrana, masa o trombo. La causa de la EVO después del trasplante de médula ósea no está clara; los factores de riesgo de EVO consisten en regímenes de acondicionamiento de dosis elevadas, radiación, leucemia, edad avanzada y hepatopatía preexistente. Los cambios anatomopatológicos en pacientes con EVO se muestran mejor con tinciones especiales (tricrómico) que resaltan las venas centrales. Una lesión precoz es el estrechamiento concéntrico de la luz de las pequeñas venas centrales, que provoca edema en la zona subendotelial. Hay una densa banda continua ondulada de colágeno en las venas centrales y necrosis hemorrágica centrolobulillar. Las lesiones pueden ser parcheadas. Más adelante en el curso de la enfermedad, las vénulas hepáticas pueden estar completamente obliteradas. Los síntomas suelen consistir en ictericia, hepatomegalia dolorosa, rápida ganancia de peso y ascitis. La EVO se resuelve en la mayoría de los pacientes pero puede causar insuficiencia multiorgánica, encefalopatía hepática e insuficiencia hepática fulminante. Las formas menos graves pueden caracterizarse por ictericia y ascitis con una lenta resolución; en los casos muy leves los cambios histológicos pueden ser la única manifestación. El diagnóstico se basa en la exclusión de otras enfermedades, tales como EICH, miocardiopatía congestiva, pericarditis constrictiva y síndrome de Budd-Chiari. No hay una profilaxis o tratamiento eficaz para la EVO. Los agentes profilácticos en investigación incluyen la heparina intravenosa o de bajo peso molecular, prostaglandina E1 y ácido ursodesoxicólico. El ácido ursodesoxicólico oral puede disminuir la incidencia de hepatopatía grave en pacientes sometidos a TCP y se ha demostrado que reduce la incidencia de EVO y de mortalidad relacionada con el trasplante en adultos. El tratamiento de soporte consiste en hidratación por vía intravenosa y perfusión renal. La EVO grave se ha tratado con defibrotida, agente experimental con propiedades antitrombóticas y trombolíticas, en pacientes adultos de alto riesgo. HEMOGLOBINOPATÍAS Los pacientes con anemia drepanocítica (cap. 456.1) o talasemia de células falciformes (cap. 456.1) pueden presentar disfunción hepática debido a asociación con hepatitis vírica crónica o aguda, hemosiderosis por tratamiento frecuente con transfusiones, crisis hepáticas por colestasis intrahepática grave, secuestro o necrosis isquémica. Además, la colelitiasis es común. e352-4 & Parte XVIII Sistema digestivo La crisis drepanocítica hepática o «hepatopatía falciforme» se da en alrededor del 10% de los pacientes con drepanocitosis. Se presenta con intenso dolor en el hipocondrio derecho, fiebre, leucocitosis, dolorimiento en hipocondrio derecho e ictericia. El nivel de bilirrubina puede estar muy elevado, mientras que los niveles séricos de FA pueden aumentar sólo de forma moderada. Puede ser difícil distinguirla de la hepatopatía drepanocítica debida a hepatitis vírica o de la colecistitis/coledocolitiasis aguda; por tanto, hay que excluir estas afecciones. Por lo general, la crisis drepanocítica hepática es autolimitada y los síntomas se resuelven en 1-3 semanas. La colestasis drepanocítica intrahepática se presenta con hepatomegalia, dolor abdominal, hiperbilirrubinemia y coagulopatía, y puede progresar a insuficiencia hepática aguda, dejando al trasplante hepático como única opción terapéutica. El trasplante conlleva un riesgo elevado de pérdida del injerto debido a complicaciones vasculares. Por fortuna, la colestasis intrahepática es infrecuente. A veces, los niños con drepanocitosis tienen niveles de bilirrubina >20 mg/dl, pero no acompañados de dolor intenso ni fiebre. No hay alteración del hematocrito ni del recuento de reticulocitos, ni asociación con crisis hemolítica. La evolución clínica es benigna. BIBLIOGRAFÍA Ahmad J, Slivka A: Hepatobiliary disease in inflammatory bowel disease, Gastroenterol Clin North Am 31:329-345, 2002. Alisi A, Manco M, Vania A, et al: Pediatric nonalcoholic fatty liver disease in 2009, J Pediatr 155:469-474, 2009. Allen LA, Felker GM, Pocock S, et al: Liver function abnormalities and outcome in patients with chronic heart failure: data from the Candesartan in Heart failure: Assessment of Reduction in Mortality and morbidity (CHARM) program, Euro J Heart Failure 11:170-177, 2009. Ardizzone S, Puttine PS, Cassinottie A, et al: Extraintestinal manifestations of inflammatory bowel disease, Digest Liver Dis 40S: S253-S259, 2008. Bargiggia S, Maconit G, Elli M, et al: Sonographic prevalence of liver steatosis and biliary tract stones in patients with inflammatory bowel disease: study of 511 subjects at a single center, J Clin Gastroenterol 36:417-420, 2003. Beale E, Nelson R, Bucciarelli R, et al: Intrahepatic cholestasis associated with parenteral nutrition in premature infants, Pediatrics 64:342-347, 1979. Belfort R, Harrison SA, Brown K, et al: A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis, N Engl J Med 355:2297-2307, 2006. Bergquist A, Ekbom A, Olsson R, et al: Hepatic and extrahepatic malignancies in primary sclerosing cholangitis, J Hepatol 36:321-327, 2002. Bhala N, Usherwood T, George J: Non-alcoholic fatty liver disease, BMJ 339:513-514, 2009. Black DD: What is the role of cystic fibrosis transmembrane conductance regulator dysfunction in primary sclerosing cholangitis? J Pediatr 151:230-232, 2007. Charatcharoenwitthaya P, Enders FB, Halling KC, et al: Utility of serum tumor markers, imaging, and biliary cytology for detecting cholangiocarcinoma in primary sclerosing cholangitis, Hepatology 48:1106-1117, 2008. Cheuk DKL, Wong P, Lee TL, et al: Risk factors and mortality predictors of hepatic veno-occlusive disease after pediatric hematopoietic stem cell transplantation, Bone Marrow Transplant 40:935-944, 2007. Claessen MM, Vleggaar FP, Tytgat KM, et al: High lifetime risk of cancer in primary sclerosing cholangitis, J Hepatol 50:158-164, 2009. Colombo C, Battezzati PM, Strazzabosco M, et al: Liver and biliary problems in cystic fibrosis, Semin Liver Dis 18:227-235, 1998. Cystic Fibrosis Foundation: Patient registry 2003 annual report to the Center Directors. Bethesda, Maryland, 2004, Cystic Fibrosis Foundation. Davies YK, Cox KM, Abdullah BA, et al: Long-term treatment of primary sclerosing cholangitis in children with oral vancomycin: an immunomodulating antibiotic, J Pediatr Gastroenterol Nutr 47 (1):61-67, 2008. Feldstein AE, Perrault J, El-youssif M, et al: Primary sclerosing cholangitis in children: a long-term follow-up study, Hepatology 38:210-217, 2003. Ferrara JL, Levine JE, Reddy P, et al: Graft-versus-host disease, Lancet 373(9674):1550-1561, 2009. Fracanzani AL, Valenti L, Bugianes E, et al: Risk of severe liver disease in nonalcoholic fatty liver disease with normal aminotransferase levels: a role for insulin resistance and diabetes, Hepatology 48:792-798, 2008. Giallourakis CC, Rosenberg P, Friedman LS: The liver in heart failure, Clin Liver Dis 6:947-967, 2002. Gregorio GV, Portmann B, Karani J, et al: Autoimmune hepatitis/sclerosing cholangitis overlap syndrome in childhood: A 16-year prospective study, Hepatology 33:544-553, 2001. Hong-Curtis J, Yeh MM, Jain D, et al: Rapid progression of autoimmune hepatitis in the background of primary sclerosing cholangitis, J Clin Gastroenterol 38:906-909, 2004. Kashi MR, Torees DM, Harrision SA: Current and emerging therapies in nonalcoholic fatty liver disease, Semin Liver Dis 28:396-406, 2008. Kaufman SS: Prevention of parenteral nutrition-associated liver disease in children, Pediatr Transplant 6:37-42, 2002. Kwan V, George J: Liver disease due to parenteral and enteral nutrition, Clin Liver Dis 8:893-913, 2004. Invernizzi P, Mackey IR: Clinical features and management of primary sclerosing cholangitis, World J Gastroenterol 14(21):3338-3349, 2008. Moseley RH: Sepsis and cholestasis, Clin Liver Dis 8:83-94, 2004. Moyer K, Balistreri W: Hepatobiliary disease in patients with cystic fibrosis, Curr Opin Gastroenterol 25:272-278, 2009. Narkewicz MR, Sondheimer HM, Ziegler JW, et al: Hepatic dysfunction following the Fontan procedure, J Pediatr Gastroenterol Nutr 36:352-357, 2003. Nobili V, Manco M, Devilo R, et al: Lifestyle intervention and antioxidant therapy in children with nonalcoholic fatty liver disease: a randomized, controlled trial, Hepatology 48:119-128, 2008. Pardi DS, Loftus EV, Kremers WK, et al: Ursodeoxycholic acid as a chemopreventive agent in patients with ulcerative colitis and primary sclerosing cholangitis, Gastroenterology 124:889-893, 2003. Patton HM, Lavine JE, Natta LV, et al: Clinical correlates of histopathology in pediatric nonalcoholic steatohepatitis, Gastroenterology 135:1961-1971, 2008. Quaglia A, Duarte R, Datch D, et al: Histopathology of graft versus host disease of the liver, Histopathology 50:727-738, 2007. Ribaud P, Gluckman E: Hepatic veno-occlusive disease, Pediatr Transplant 3:41-44, 1999. Schwimmer JB: Definitive diagnosis and assessment of risk for nonalcoholic fatty liver disease in children and adolescents, Semin Liver Dis 27:312-318, 2007. Schwimmer JB, Deutsch R, Kahen T, et al: Prevalence of fatty liver in children and adolescents, Pediatrics 118:1388-1393, 2006. Soden JS, Narkewicz MR, Haas JE, Sokol RJ: Hepatic veno-occlusive disease and human herpes virus 7 infection in primary agammaglobulinemia, J Pediatr 154:299-302, 2009. Zambrano E, El-Hennawy M, Ehrenkranz RA: Total parenteral nutrition induced liver pathology: an autopsy series of 24 newborn cases, Pediatr Development Pathol 7:425-432, 2004. Zakrzewski JL, Ballauff A, Wieland R: Differential diagnosis of cholestasis following allogeneic hematopoietic stem cell transplantation: the contribution of serum bile acid levels in relation to other liver tests, Pediatr Hematol Oncol 21:697-705, 2004.