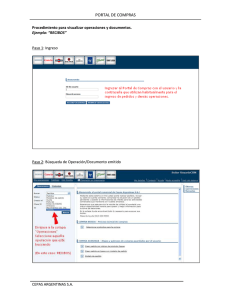
Tesis Electrónicas UACh - Universidad Austral de Chile
Anuncio

Universidad Austral de Chile Facultad de Ciencias Agrarias Escuela de Ingeniería en Alimentos Caracterización de Cepas de Bacillus Aisladas de Muestras de Miel y de Colmena Mediante la Secuenciación del Gen Ribosomal 16S Memoria presentada como parte de los requisitos para optar al Título de Ingeniero en Alimentos. Katherine Angélica Valderas Álvarez VALDIVIA – CHILE 2012 PROFESOR PATROCINANTE: ____________________________________ Renate Schöbitz Twele Tecnólogo Médico, M.Sc. Instituto de Ciencias y Tecnología de los Alimentos Facultad de Ciencias Agrarias PROFESORES INFORMANTES: ____________________________________ Alex Romero Zúñiga Bioquímico, Ph. D. Laboratorio de Biotecnología y Patología Acuática Facultad de Ciencias Veterinarias ____________________________________ Ociel Muñoz Fariña Bioquímico, Ph. D. Instituto de Ciencias y Tecnología de los Alimentos Facultad de Ciencias Agrarias ÍNDICE DE MATERIA Título Página RESUMEN 1 SUMMARY 2 1 INTRODUCCIÓN 3 2 REVISIÓN BIBLIOGRÁFICA 5 2.1 Las abejas y la apicultura 5 2.2 Enfermedades y enemigos de las abejas 6 2.2.1 Loque Americana (L.A). 8 2.2.2 Paenibacillus larvae. 10 2.3 Ribosoma bacteriano 11 2.3.1 Caracterización bacteriana utilizando el gen ribosomal 16S 13 2.3.2 Operones ribosómicos 13 2.4 Clonamiento del gen ribosomal 16S 15 3 MATERIAL Y MÉTODO 17 3.1 Materiales y equipos utilizados 17 3.1.1 Equipos 17 3.1.2 Medios de cultivo y reactivos 17 3.1.3 Material 17 3.1.4 Material biológico 17 3.2 Selección de la muestra a analizar 18 3.3 Método 18 2 3.3.1 Cultivo de las cepas de Bacillus 18 3.3.2 Extracción del ADN genómico de la cepas de Bacillus. 18 3.3.3 Amplificación del gen ribosomal 16S 19 3.3.4 Electroforesis en gel de agarosa al 1,5% 21 3.3.5 Purificación del gen ribosomal 16S amplificado por PCR desde el gel de 22 agarosa. 3.3.6 Clonamiento del gen ribosomal 16S 24 3.3.6.1 Ligación del producto PCR 24 3.3.6.2 Transformación de E. coli competentes con el ADN plasmidial 25 3.3.6.3 Extracción del ADN plasmidial por el Método de Ebullición 26 3.3.7 Análisis de restricción de ADN plasmidial purificado 26 3.3.8 Extracción de ADN plasmidial, para secuenciación 27 3.3.9 Lectura y análisis de secuencias 28 3.3.10 Prueba de antagonismo sobre césped 29 3.3.10.1 Antagonismo entre cepas de Bacillus 29 3.3.10.2 Antagonismo de cepas de Bacillus, frente a P. larvae 30 4 PRESENTACIÓN Y DISCUSIÓN DE RESULTADOS 32 4.1 Preparación de la muestra 32 4.2 Cuantificación del ADN genómico 33 4.3 Amplificación del gen ARNr 16S por medio de PCR 34 4.4 Purificación del gen ARNr 16S 35 4.5 Clonamiento del gen ARNr 16S de los aislados de Bacillus 36 4.5.1 Transformación bacteriana 37 4.6 Secuenciación e identificación de las cepas de Bacillus 41 4.7 Pruebas de antagonismo 43 3 4.7.1 Antagonismo entre las cepas de Bacillus identificadas 45 4.7.2 Antagonismo de las cepas de Bacillus frente a P. larvae. 47 5 CONCLUSIONES 50 6 BIBLIOGRAFÍA 51 ANEXO 58 ÍNDICE DE CUADROS Cuadro 1 Página Principales enfermedades apícolas en Chile, según naturaleza del agente, 7 etapa de la biología y ocurrencia. 2 Resumen de síntomas de la enfermedad de la Loque Americana. 9 3 Mezcla utilizada en la reacción de PCR para amplificar el gen ARNr 16S. 20 4 Componentes para agregar nucleótidos de adenina a los productos de PCR 24 purificados. 5 Componentes para la ligación del producto de PCR en el plasmidio 25 pGEMT-Easy. 6 Cuantificación del ADN genómico extraído de Bacillus. 33 7 Cuantificación del ADN enviado a secuenciar. 41 8 Resumen de la identificación bacteriana después de la secuenciación. 43 9 Densidad óptica (Do) a 620nm de las cepas incubadas en CST. 44 10 Recuento a las 12h de incubación en AST. 45 11 Halos de inhibición en las pruebas de antagonismo entre cepas del 46 género Bacillus. 12 Tamaño de los halos de inhibición de la prueba de antagonismo de las cepas 87, 95, 97 y mezclas de ellas, contra P. larvae ATCC 25745 y de Campo. 48 ÍNDICE DE FIGURAS Figura 1 Página Esporas de P. larvae de cadáveres de larvas de abejas (izquierda) y 11 esporas de P. larvae de cultivo (derecha). 2 El ribosoma bacteriano. Se muestra su estructura, las subunidades y 12 las macromoléculas que lo componen. 3 Representación esquemática de un operón ribosómico (rrn). 14 A) Genes estructurales de los tres tipos de ARNr (rrs, rrl y rrf), los promotores P1 y P2, y las regiones intergénicas (IG). B) Amplificación del gen rrs (ADNr 16S). Se indica la posición de los iniciadores I1 (directo), I2 e I3 (reversos) utilizados para la amplificación (y posterior secuenciación) del gen completo (I1-I3; amplicón de 1.500pb, aproximadamente) o de un fragmento menor (11-I2; 500pb correspondientes al extremo 59 del gen) C) Visualización de ambos fragmentos por electroforesis en gel de agarosa. 4 Esquema general del proceso de clonación. 16 5 Protocolo de extracción del ADN genómico de las cepas de Bacillus. 19 6 Protocolo de amplificación del ADN ribosomal 16S, método de la 21 polimerasa en cadena “PCR”. 7 Protocolo preparación del gel de agarosa al 1,5%. 22 8 Protocolo purificación del ADN amplificado por PCR desde el gel de agarosa. 23 9 Protocolo de extracción de ADN plasmidial. 28 10 Pasos para prueba de antagonismo entre cepas del género Bacillus. 30 11 Pasos para la prueba de antagonismo entre las cepas de P. larvae contra 31 las cepas del género Bacillus. 12 Colonias puras de las ocho cepas del género Bacillus en Agar Soya Tripticasa. 32 13 Electroforesis en gel de agarosa al 1,5%, que muestra la amplificación 35 del ADNr 16S mediante PCR de ocho aislados de Bacillus. Carriles: Est, marcador de peso molecular de 100-3000pb; 1g; 20; 22; 67; 87; 92; 95 y 97, aislados de Bacillus; (+), control positivo; (-), control negativo de F. psychrophilum. 14 Electroforesis del gen ARNr 16S purificado. Carriles: Est, marcador 36 de peso molecular de 100-3000pb; 1g; 20; 22; 67; 87; 92; 95 y 97, aislados de Bacillus. 15 Cultivo de E. coli transformantes en placas de agar LB/ampicilina/IPTG 37 y X-Gal. 16 Análisis electroforético del ADN plasmidial recombinante digerido 39 con EcoRI. Carriles: Est, marcador de peso molecular de 100-3.000pb; 921, 922, 923, 201, 202 y 203, aislados de Bacillus. 17 Análisis electroforético del ADN plasmidial conteniendo el gen 16S con 40 la enzima de restricción EcoRI. Carriles: Est, marcador de peso molecular de 100-3000pb; 97; 87; 67 y 95, aislados de Bacillus. 18 Análisis electroforético del ADN plasmidial conteniendo el gen 16S con 40 la enzima de restricción EcoRI. Carriles: Est, marcador de peso. 19 Pruebas de antagonismo sobre agar soya tripticasa entre cepas de 45 Bacillus, con inhibición de las cepas 95 y 97 por la cepa 87. 20 Pruebas de antagonismo sobre agar MYPGP entre cepas del género Bacillus frente a P. larvae ATCC 25745 y de Campo. 47 ÍNDICE DE ANEXOS Anexo Página 1 Medios de cultivos y buffer 58 2 Electroforesis en gel de garosa para la purificación de los productos de PCR 60 3 Análisis electroforético del ADN plasmidial recombinante digerido con EcoRI. 61 4 Secuencias de las cepas del genero Bacillus. 64 1 RESUMEN La Loque Americana (L.A), es una de las enfermedades más perjudiciales que afecta a las abejas de la especie Apis mellifera L, no sólo mata las larvas infectadas, también es potencialmente letal para las colonias. Esta enfermedad es altamente contagiosa, ya que su propagación se ve facilitada por el intercambio entre el material de la colmena y las colonias de abejas. El objetivo del presente trabajo fue aislar e identificar hasta el nivel de especie, cepas pertenecientes al género Bacillus, aisladas de muestras de miel y material de colmena para ser evaluadas como antagonistas frente a las cepas de Paenibacillus larvae (ATCC 25745 y de campo), agente causal de la L.A. La identificación se realizó mediante la secuenciación del gen ARNr 16S, para esto se amplificó y clonó el gen ribosomal 16S de las cepas en el plasmidio pGEMT-Easy y se envío a secuenciar el gen 16S contenido en el plasmidio de las ocho cepas de Bacillus. Las secuencias obtenidas se compararon con la base de datos para conocer su identidad. No se encontraron cepas de P. larvae entre los Bacillus identificados. Las especies correspondieron a B. cereus, B. subtilis y B. pumilus. De las ocho cepas identificadas, tres fueron chequeadas como mezcla e individualmente contra las cepas de P. larvae. Estas pruebas de antagonismo, revelaron inhibición de la cepa 87 (B. pumilus) al ser enfrentada a la cepa 95 (B. subtilis) y 97 (B. pumilus), de estas tres cepas las que presentaron mayor actividad antagónica contra P. larvae ATCC 25745 y de campo, fue la especie 95 (B. subtilis) y 97 (B. pumilus) respectivamente. Estas especies son inocuas para el apiario y para el ser humano, por lo tanto poseen un gran potencial como fuente de compuestos biocontroladores para la inhibición del patógeno. 2 SUMMARY American Foulbrood (A.F) is one of the most harmful disease that affects the bees of the Apis mellifera L. species. It not only kills the infected larvae, but also is potentially lethal for the colonies. This disease is highly contagious, because it is facilitated by the exchange between the material of the beehive and the bee colonies. The objective of the present study was, to isolate and identify to the species level, strains belonging to the genus Bacillus, isolated from honey samples and beehive material. The strains were evaluated as antagonists against Paenibacillus larvae ATCC 25745 and a field strain, causative agent of the A.F. The identification was performed by sequencing the 16S rRNA gene. For this purpose the gene of each strain was amplified and cloned in the plasmid pGEMT-Easy. The plasmid containing the gene was sent abroad for sequencing, wich was done for the eight Bacillus strains. The sequences obtained were compared with the database to find their identity. No strains belonging to the species P. larvae were identified among the Bacillus, the species were identified as B. cereus, B. subtilis and B. pumilus. Of the eight strains identified, three were checked against the two P. larvae strain, individually and as a mixture. Such antagonism tests revealed an inhibition of the strain 87 (B. pumilus) when faced with the strains 95 (B. subtilis) and 97 (B. pumilus). These two Bacillus strains showed the highest antagonistic activity against P. larvae ATCC 25745 and P. larvae field strain. These species are harmless to the apiary and humans, thus have a great potential like biocontroler compounds for the inhibition of the pathogen. 3 1 INTRODUCCIÓN La “Loque Americana” (LA) es una de las enfermedades que afecta más severamente a las crías de abejas del Apis mellifera L., según PICCINI et al., (2004). El agente causante de la L.A nombrado por HEYNDRICKX et al., (1996) es el Paenibacillus larvae, una bacteria Gram positiva, que produce esporas, las cuales resultan ser extremadamente resistentes a las altas temperaturas y agentes químicos (ANTÚNEZ et al., 2004). Esta enfermedad se encuentra presente en todo el mundo, reportándose casos en casi todas las regiones apícolas de los cinco continentes. En Chile se reportaron dos casos de P. larvae en el año 2001 fuera de la III región. Posteriormente el 15 de Octubre de 2005, en la provincia de Curicó, VII Región, se enfrentó un brote de “Loque Americana”, inmediatamente se presentaron casos en las Regiones III, V, RM, VI, VIII y X. Hoy en día esta enfermedad se considera establecida con una condición de endemismo, de presentación esporádica y localizada en ciertas regiones del país (SERVICIO AGRÍCOLA Y GANADERO (SAG), 2009). En el año 1980, un nuevo estándar para identificación de bacterias comenzó a ser desarrollado. Estudios mostraron que las relaciones filogenéticas de bacterias, y, en efecto, de todas las formas de vida, pueden ser determinadas por la comparación de una parte estable del código genético. La parte del ADN que ahora se utiliza comúnmente para propósitos taxonómicos en bacterias, es el gen ARNr 16S (CLARRIDGE, 2004). La elección de éste gen como herramienta óptima para tales efectos, se basa en las observaciones y las hipótesis de conservación ribosomal. La secuenciación del gen ARNr 16S proporciona lo medios precisos para la identificación de microorganismos. La calidad de esta identificación, depende principalmente de la base de datos que se utiliza (HARMSEN y KARCH, 2004). Es así como actualmente, los dos tratados fundamentales de bacteriología, el Bergey`s Manual of Systematic Bacteriology y The Prokaryotes, basan su estructuración del 4 mundo procariota, en las relaciones filogenéticas establecida por esta misma macromolécula. Como señala ANTÚNEZ et al., (2004), la confirmación específica de P. larvae, se puede realizar por medio del método de detección PCR (reacción en cadena de la polimerasa). Para esto se amplifica selectivamente por medio de partidores universales, un fragmento del gen ARNr 16S de P. larvae extraído de un cultivo puro (PICCINI et al., 2002). HIPOTESIS Cepas de bacterias pertenecientes al género Bacillus aisladas de miel y colmenas identificadas por medio de la secuenciación del gen ribosomal 16S, pueden utilizarse como controladores biológicos de Paenibacillus larvae. Objetivo general Caracterizar cepas del género Bacillus a partir de aislados de miel y colmena, por medio de la secuenciación del gen ribosomal 16S y evaluar el antagonismo de estas cepas frente a P. larvae mediante la prueba de antagonismo en placa. Objetivos específicos Confirmar la pureza de los cultivos conservados congelados de ocho cepas de Bacillus, aisladas a partir de miel y de material de colmenas. Amplificar y clonar el gen 16S de las cepas de Bacillus, en el plasmidio pGEMT- Easy. Secuenciar el gen 16S contenido en el plasmidio de las ocho cepas de Bacillus y compararlo con las secuencias de bases de datos, para conocer su identidad. Estudiar mediante pruebas de antagonismo en placa la interacción entre cepas de Bacillus. Realizar pruebas de antagonismo en placa de las cepas de Bacillus identificadas contra dos cepas de P. larvae. 5 2 REVISIÓN BIBLIOGRÁFICA 2.1 Las abejas y la apicultura La abeja es un insecto del orden de los himenópteros, la especie Apis mellifera L. es la más desarrollada en la apicultura y se ha domesticado para poder generar productos en cantidades importantes (TRHLIN et al., 2011). Es un insecto social cuya organización responde al de una colonia, en la cual existe una hembra fértil llamada reina, múltiples hembras infértiles llamadas obreras y una casta de machos con tareas principales de tipo reproductivas llamados zánganos, que viven sólo en época de primavera y verano (PADILLA y CUESTA, 2003). La apicultura es una actividad agropecuaria orientada a la crianza de abejas, para obtener beneficios derivados de la venta de una multiplicidad de productos apícolas (miel, polen, cera, jalea real, propóleo, veneno de abejas) y de la acción polinizadora en los cultivos, que contribuye a la preservación de la biodiversidad botánica, la actividad agrícola y más particularmente la fruticultura (ALVIS et al., 2009). En Chile, la apicultura es uno de los rubros que presenta mayor crecimiento, durante los últimos años ha alcanzado un gran desarrollo, por las características que presenta el país, principalmente por las condiciones ambientales y recursos naturales tales como un prodigioso clima templado, una gran diversidad de especies melíferas nativas y el resguardo sanitario dado por efectivas barreras naturales (GERDING y RODRIGUEZ, 2010; LESSER, 2004). El precio estable de las exportaciones de la miel y el trabajo de los apicultores, en su mayoría pequeños productores, sustentan el desarrollo de la actividad apícola nacional. La miel a granel lidera los envíos nacionales, con un 85% de participación. Sin embargo, la miel a granel con algún grado de diferenciación (monofloral u orgánica) ha ido ganando terreno, constituyendo más del 14% del mercado (ODEPA, 2011). Las estadísticas presentadas por ODEPA (2011) indican que las exportaciones chilenas totales de miel durante el año 2010, totalizaron 8.601 toneladas evaluadas en 6 US$28,9 millones, representando un descenso del 12,5% en volumen, pero manteniendo su valor respecto al año 2009. Por otra parte, hasta Junio de 2011, las exportaciones de miel totalizaron 6.337 toneladas, avaluadas en US$ 23,4 millones, lo que representa un aumento del 36,1% en valor y 22,2% en volumen, respecto al mismo período del año 2010. Estos resultados muestran una recuperación en las exportaciones a los niveles previos al terremoto del 27 de Febrero del año 2010, donde hubo pérdidas en las bodegas de las exportadoras y retrasos en el despacho de la miel en puertos nacionales. 2.2 Enfermedades y enemigos de las abejas Como ocurre con todas las especies vivientes, las abejas A. mellifera L. son atacadas por una variedad de enfermedades. Éstas generalmente se pueden clasificar en enfermedades que afectan a las abejas adultas y en enfermedades que afectan a las crías (larvas y pupa) (PIERRE y YVES, 2006). La mayoría de las enfermedades llegan a las colmenas, porque las abejas la transportan de una colmena a otra o por los manejos inadecuados por parte del apicultor, al no cumplir con los estándares de higiene al realizar trabajos e inspecciones en la colmena. Comúnmente los apicultores intercambian panales (vacíos, con cría, miel o polen) entre las colonias y ocasionalmente entre apiarios, lo que representa una vía importante en la diseminación de las enfermedades (MEDINA y MAY, 2005). Los factores determinantes de las enfermedades pueden ser bacterias, hongos, virus, parásitos, insectos “enemigos”, es decir, parásitos mayores, como la polilla o menores como el piojo (MENDIZABAL, 2004). Es de gran importancia conocer las enfermedades que afectan a las crías principalmente, para evitar o reducir el impacto negativo en las colonias de abejas, ya que de no tratarse a tiempo se puede incurrir en pérdidas del material vivo y en pérdidas económicas (CABALLERO, 2009). Algunas de las principales enfermedades que afectan a las A. melliferas L. se resumen a continuación en el CUADRO 1. 7 CUADRO 1 Principales enfermedades apícolas en Chile, según naturaleza del agente, etapa de la biología y ocurrencia. Nombre Nombre Naturaleza del Etapa de la Ocurrencia en común científico agente causal biología de la Chile abeja en que se presenta Varroa Varroa destructor Ácaro Crías y Adultos Presente Acariosis Acarapis woodi Ácaro Adultos Presente Nosema Nosema Apis Protozoo Adultos Presente Malpighamoaeba Protozoo Adultos Presente Bacteria Crías Presente Hongo Crías Presente Amebiasis mellificae Loque Paenibacillus americana larvae larvae Cría tiza Ascosphaera Apis Piojo Braula coeca Insecto Adultos Presente Loque Melissococcus Bacteria Crías Ausente europea plutonios Pequeño Aethina tumida Insecto Crías Ausente Tropilaelaps Ácaro Crías Ausente escarabajo de la colmena Ácaro asiático clareae FUENTE: NEIRA (2009). 8 Para el control de estas enfermedades es importante el manejo de las colmenas y su control periódico, más que los tratamientos (PADILLA y CUESTA, 2003). 2.2.1 Loque Americana (L.A). La “Loque Americana”, es una de las enfermedades más graves que ataca a las colonias de abejas, es infecciosa y altamente contagiosa. (SANAD y AL-BARRAK, 2010). Esta enfermedad provoca una pudrición de la cría operculada. La bacteria causante de ésta enfermedad es el P. larvae, la cual tiene efectos devastadores en apiarios infectados sufriendo grandes pérdidas económicas debido a una disminución significativa de las poblaciones de abejas y por consecuencia la producción de miel (LEE et al., 2009). Según CHANTAWANNAKUL y DANCER (2001), durante el proceso de infección de la larva, P. larvae secreta proteasas extracelulares, enzimas que probablemente ayudan en el proceso de descomposición de la cría después de la muerte. La miel, cera y propóleos son portadores de estas esporas, al igual que el polen, siendo estos productos contaminados una forma corriente de contagio. Las esporas son ingeridas por las larvas, se multiplican en el estómago y se extienden por todo el cuerpo, ocasionándoles la muerte, ataca a las larvas obreras, cuando ellas han sido operculadas (CHANTAWANNAKUL y DANCER, 2001). A modo de prevención, es importante mantener una vigilancia y monitoreo permanente, examinando todo el panal antes de seleccionar la muestra, buscando larvas o pupas con signos que sean característicos de la enfermedad de la Loque Americana. En caso de tener sospecha de la presencia de dicha enfermedad, deben enviarse inmediatamente muestras al laboratorio, para realizar un diagnóstico adecuado (USABIAGA et al., 2002). En el CUADRO 2, se describen algunos de los síntomas que se deben tener en cuenta, ya que son claros signos de presencia de la enfermedad de L.A. 9 CUADRO 2 Resumen de síntomas de la enfermedad de la Loque Americana. Características Síntomas de la Loque Americana. Apariencia general del panal - Cría salteada. - Opérculos hundidos, perforados, oscurecidos y aspecto grasoso. - Larvas muertas en celdas operculadas y celdas desoperculada por abejas. Edad de la cría muerta - Prepupa y pupas. Color de la cría muerta - Primero cremoso, luego café claro, café oscuro y finalmente negro. Consistencia de la cría muerta - Primero acuosa y luego viscosa. Olor de la cría muerta - Cola para pegar madera. Características de la escama - Se forma en el piso de la celda. - Quebradizas difícil de desprender. - Hilo pupal estirada hacia el techo, la cabeza plana y tendida - Color negro FUENTE: SHIMANUKI y KNOX (2000). 10 Debido a que es muy difícil de eliminar la L.A., se recomienda quemar todos los implementos y útiles, aunque también se puede tratar en forma menos drástica, eliminando el foco de infección, que son las crías y transfiriendo las abejas a una colmena limpia y desinfectada, cambiando la reina por una nueva, lo cual ayuda a la recuperación más rápida de la población (NEIRA, 2006). Diferentes técnicas son utilizadas por los apicultores para el tratamiento de las colonias infectadas con P. larvae. A menudo se utilizan agentes químicos, tales como el sulfatiazol sódico y la oxitetraciclina (CHANTAWANNAKUL y DANCER, 2001). Sin embargo, varios problemas han sido asociados al uso prolongado de antibióticos. El principal problema, es que estos residuos de antibióticos, pueden persistir en la miel afectando su calidad y seguridad para el consumo humano (GENERSCH, 2010). Por otro lado se sabe que la bacteria desarrolla gradualmente resistencia a los antibióticos que se utilizan para tratar la enfermedad, por lo tanto, se ha recomendado el uso de antibióticos sólo cuando la enfermedad ha sido identificada y no aplicar antibióticos a las colonias con fines preventivos (KILIC et al., 2010). Por todo lo anteriormente señalado, es que con el tiempo el control biológico a través de bacterias antagonistas representa un mecanismo de control promisorio. Diversos estudios han reportado que ciertos microorganismos asociadas normalmente a las abejas y la miel poseen la capacidad de inhibir hongos y bacterias, por la secreción de antibióticos con propiedades antifúngicas y bacteriocinas. Un estudio realizado por ALIPPI y REYNALDI (2006), tuvo por objetivo, investigar el antagonismo de bacterias formadoras de endosporas aisladas desde muestra de miel y de otras partes de la colmena, capaces de inhibir el P. larvae. Durante la investigación se encontró con cepas de B. pumilus, B. subtilis, B. cereus, B. circulans, B. megaterium B. licheniformis y B. laterosporus, capaces de inhibir el crecimiento de la cepa de P. larvae, corroborando así la producción de compuestos antibacterianos, que poseen algunas bacterias. 2.2.2 Paenibacillus larvae. Es la bacteria causante de la enfermedad Loque Americana. SHIMANUKI y KNOX (2000), señalan que su forma es de varilla delgada, con puntas ligeramente redondeadas y con tendencia a crecer en cadena. El largo de la bacteria es muy variable en longitud, de 2,5 a 5 µm de largo y 0.5 µm de ancho. La 11 espora es ovalada, dos veces más larga que ancha, entre 0.6 x 1.3 µm, como se puede observar en la FIGURA 1. Según HORNITZKY (1998), puede encontrarse en dos estadíos, en forma vegetativa y de espora, que es la que le asegura su sobrevivencia en el entorno (fuera del cuerpo de su huésped), siendo capaz de germinar cuando las condiciones son favorables. FIGURA 1 Esporas de P. larvae de cadáveres de larvas de abejas (izquierda) y esporas de P. larvae de cultivo (derecha). FUENTE: CHANTAWAWANNAKUL y DANCER (2001). Las esporas de P. larvae son muy resistentes a condiciones adversas, tales como, calor y frío extremo, desecación y desinfectantes químicos. Con estas características conserva su habilidad para germinar por muchos años. Se ha comprobado que esporas de 35 años presentes el medio ambiente, son capaces de causar la enfermedad (PICCINI y ZUNINO, 2001). 2.3 Ribosoma bacteriano Los ribosomas son orgánulos indispensables, altamente especializados, que utilizan los organismos para el proceso de síntesis de proteínas. 12 El ribosoma bacteriano tiene un coeficiente de sedimentación de 70S, que a su vez está formado por dos subunidades de diferentes tamaños (FIGURA 2). La subunidad más grande (50S), contiene una molécula de ARN ribosomal, el ARNr 5S, 23S y 34 proteínas. La subunidad menor (30S), está formada por una molécula de ARNr 16S y 21 proteínas (TRESGUERRES et al., 2003). Debido a su universalidad y la conservación estructural, el componente de ARN de la subunidad del ribosoma pequeño, ha demostrado ser un importante y útil cronómetro molecular para cuantificar las relaciones evolutivas entre los organismos (MEVARECH et al., 1989). FIGURA 2 El ribosoma bacteriano. Se muestra su estructura, las subunidades y las macromoléculas que lo componen. FUENTE: TRESGUERRES (2003). 13 2.3.1 Caracterización bacteriana utilizando el gen ribosomal 16S En la década de 1980, como lo señala CLARRIDGE (2004), un nuevo estándar para identificación de bacterias comenzó a desarrollarse en los laboratorios de Woese y de otros investigadores, demostrando que las relaciones filogenéticas de las bacterias y de todas las formas de vida, se pueden determinar mediante la comparación de una parte estable del código genético. La parte del ADN más utilizado para propósitos taxonómicos en bacterias es el gen ARNr 16S, siendo indiscutible que la información contenida en este gen, ha jugado un papel fundamental en la microbiología. El gen ARNr 16S es una secuencia de aproximadamente 1.500 nucleótidos, contiene regiones altamente conservadas, que permiten establecer la relación taxonómica profunda entre familias, clases y filos, así como regiones variables que posibilitan discriminar entre especies dentro del mismo género (TORTORI, 2003). Estas características permitieron utilizar el gen como marcador filogenético y herramienta de identificación (MAC-FADDIN, 2003). El análisis del los genes de ARNr, con el uso de las bases de datos de referencia de alta calidad, proporciona un método eficaz para la identificación de microorganismos (HARMSEN y KARCH, 2004). 2.3.2 Operones ribosómicos. Todas las bacterias contienen genes codificantes para diferentes ARNs ribosomales. Estos genes que codifican los ARN ribosomales están organizados en operones. En eubacterias, cada operón ribosómico (rrn) incluye genes para los ARNr: 23S (rrl), 16S (rrs) y 5S (rrf), separados por regiones espaciadoras o intergénicas (IG) y contiene además genes para uno o más ARN de transferencia (BARRY et al., 1991). El producto de la transcripción del operón a partir de dos promotores P1 y P2, situados en la región anterior a rrs, es procesado por la enzima ARNasa III, la que mediante cortes en sitios específicos da origen a las tres clases de ARNr, el o los ARNt y las dos regiones IG (GÜRTLER y STANISICH, 1996). A continuación en la FIGURA 3, se representa esquemáticamente la organización de un operón ribosómico. 14 FIGURA 3 Representación esquemática de un operón ribosómico (rrn). A) Genes estructurales de los tres tipos de ARNr (rrs, rrl y rrf), los promotores P1 y P2, y las regiones intergénicas (IG). B) Amplificación del gen rrs (ADNr 16S). Se indica la posición de los iniciadores I1 (directo), I2 e I3 (reversos) utilizados para la amplificación (y posterior secuenciación) del gen completo (I1-I3; amplicón de 1.500pb, aproximadamente) o de un fragmento menor (11-I2; 500pb correspondientes al extremo 59 del gen) C) Visualización de ambos fragmentos por electroforesis en gel de agarosa. FUENTE: RODICIO y MENDOZA (2004). 15 2.4 Clonamiento del gen ribosomal 16S El clonamiento de fragmentos de ADN es una técnica de biología molecular que surge gracias a las técnicas de aislamiento de ADN y al descubrimiento de las enzimas de restricción, que hacen posible cortar el ADN o lo que se desee amplificar, para posteriormente incorporar fragmentos a vectores plasmidiales, que a su vez se introducen en bacterias de Escherichia coli, también llamadas células anfitrionas, donde tiene lugar la propagación del plasmidio (LUQUE y HERRAEZ, 2008). El proceso de clonamiento, comienza con la ligación de un fragmento de ADN o gen al vector plasmidial en el sitio de múltiple clonamiento. Esta pequeña molécula de ADN circular es capaz de autoreplicarse en una bacteria y de esta forma propagar el fragmento de ADN clonado. Para ligar el ADN al plasmidio se utiliza la enzima ADN ligasa, formando lo que se conoce con el nombre de ADN recombinante. Además este vector plasmidial posee genes selectivos, como el gen de la resistencia a ampicilina, el cual permite que las bacterias conteniendo el plasmidio puedan desarrollarse de forma selectiva en medios de cultivo conteniendo el antibiótico. Además, estos plasmidios de clonamiento, contienen el gen de la beta-galactosidasa, lo que nos indica, cual de las colonias bacterianas posee el plasmidio con el inserto o fragmento (LEHNINGER et al., 2009). El proceso de transformación, consiste en ingresar el ADN recombinante hacia el interior de la célula bacteriana al modificar la membrana de esta, por medio de CaCl2 el cual cambia la permeabilidad de la membrana. Finalmente el cultivo bacteriano se realiza en medios sólidos suplementados con ampicilina junto al sustrato selectivo XGal, lo que determinan las colonias que realmente poseen el plasmidio y el inserto. (MATHEWS, 2002). En resumen las células transformadas se seleccionan por medio de un sistema de detección doble, que utiliza antibióticos y sustratos cromogénicos (que producen color) (VOET y VOET, 2004). Después las bacterias se lisan para extraer y purificar el ADN plasmidial recombinante del resto de los contenidos celulares. La preparación purificada de ADN plasmídico contendrá millones de copias del fragmento de ADN original (ALBERTS et al., 2004). En la FIGURA 4, se grafica un esquema general del proceso de clonación. 16 FIGURA 4 Esquema general del proceso de clonación. FUENTE: VOET y VOET (2004). 17 3 MATERIAL Y MÉTODO En esta sección se describirán los materiales y la metodología empleada en el desarrollo de la tesis. Las actividades prácticas del estudio fueron realizadas en el laboratorio de Biotecnología y Patología Acuática, de la facultad de ciencias veterinarias y el laboratorio Bioinsumos-Bacterialogía del Instituto de Producción y Sanidad Vegetal, de la Facultad de Ciencias Agrarias, pertenecientes a la Universidad Austral de Chile. Financiado por el Proyecto CAPI-IDO901. 3.1 Materiales y equipos utilizados A continuación se presentan los materiales y equipos utilizados durante el desarrollo de la investigación: 3.1.1 Equipos. Balanza digital (Shimadsu), agitador magnético, autoclave, baño termoregulado, estufa a 30+2ºC (Memmert), estufa a 37+2 (Shel lab), centrífuga (Equilab), refrigerador (Mademsa), pHímetro marca Hanna modelo 9321PH microscopio óptico (Zeiss), campana flujo laminar (ESCO, clase BSC II), vortex (Velp), espectrofotómetro (NANODROP 2000), espectrofotómetro (Multiskan FC, Thermo Scientific), micropipetas (Arquimed), termociclador (MULTIGENE, labnet), capturador fotos gel electroforesis Vilber lourmart (Arquimed), pie de metro (Mitutoyo). 3.1.2 Medios de cultivo y reactivos. Caldo Soya Tripticasa, Agar Soya Tripticasa, Agar MYPGP (DWORKIN et al., 2006), buffer fosfato salino como diluyente, gel agarosa, buffer TAE, (ANEXO 1). 3.1.3 Material. Tubos de vidrio con tapa, tubos falcon 50mL, placas petri, porta objetos, puntas de plástico, tubos Eppendorf, jarra anaerobiosis. 3.1.4 Material biológico. Cepas de Bacillus pertenecientes al cepario del laboratorio del ICYTAL, cepas de P. larvae otorgadas por el SAG de Chile, de tipo ATCC 25745 y otra identificada como de campo. 18 3.2 Selección de la muestra a analizar Se utilizaron ocho cepas aisladas de miel con código 20, 22, 67, 87, 92, 95, 97 y una aislada de colmena con código 1g. Estas cepas fueron aisladas e identificadas en trabajos de tesis previos (SANDOVAL, 2010; SANCHEZ 2010). Las cepas se encontraban congeladas en el cepario del Laboratorio del Instituto de Ciencias y Tecnología de los Alimentos, de la Universidad Austral de Chile. Las ocho cepas se seleccionaron por tener un halo de inhibición grande en investigaciones anteriores y por ser sospechosas de ser P. larvae. 3.3 Método A continuación se procede a describir la metodología utilizada durante la identificación de las cepas bacterianas y las pruebas de antagonismo. 3.3.1 Cultivo de las cepas de Bacillus. Las cepas congeladas a -20ºC se cultivaron en caldo soya tripticasa, durante 24 a 48h. Una vez cultivadas, se pasaron a placas con agar soya tripticasa. A partir de una colonia aislada se les realizó una tinción Gram, para verificar su pureza. De cada placa se tomó una colonia aislada pura con un asa estéril y se dejó crecer en caldo soya tripticasa, para el proceso de extracción de ADN. 3.3.2 Extracción del ADN genómico de la cepas de Bacillus. Para la extracción del ADN genómico se utilizó el kit “Wizard Genomic DNA Purification” de Promega, de acuerdo a las instrucciones del fabricante. En resumen el ADN genómico se extrajo a partir de 1mL del cultivo bacteriano puro (3.3.1), el que se centrifugó a 16.000 x g por 2min, obteniéndose un sedimento bacteriano. Una vez removido el sobrenadante, este sedimento fue resuspendido en 480µL de EDTA 50mM con 120µL de lisozima (10mg/mL), mezclándose lentamente, para luego colocarla a incubar a 37ºC por 60min. Luego la mezcla se centrifugó por 2min a 16.000 x g y se removió el sobrenadante. El propósito de este tratamiento previo es debilitar la pared celular para una lisis eficiente. Para la lisis bacteriana, el sedimento resultante fue resuspendido suavemente en 600µL de solución de lisis, para luego incubar a 80ºC por 5min. Finalmente, se dejó enfriar a temperatura ambiente y se agregó 3µL de la solución RNAasa, invirtiendo el tubo 2-5 veces para mezclar y se incubó a 37ºC por 60min y se enfrió la muestra a temperatura ambiente. El lisado celular obtenido fue incubado con 200µL de la solución de precipitación de proteínas mezclado en vortex por 20s e incubando la muestra en 19 hielo por 5min, para luego centrifugar a 16.000 x g por 3min. El sobrenadante que contenía el ADN se transfirió a un tubo microcentrífuga limpio de 1,5mL y se agregó 600µL de isopropanol a temperatura ambiente para precipitar el ADN, se mezcló lentamente invirtiendo el tubo hasta observar filamentos de ADN y se centrifugó a 16.000 x g por 2min. El sobrenadante se extrajo cuidadosamente y se secó el tubo en papel absorbente limpio. Una vez seco el pellet de ADN, éste se lavó con 600µL de etanol al 70% a temperatura ambiente, invirtiendo el tubo suavemente varias veces, posteriormente centrifugar 16.000 x g por 2min y descartar el sobrenadante. El sedimento de ADN se secó al aire por 15min. Al observar el sedimento de ADN seco, se rehidrató agregando 100µL de la solución de rehidratación al tubo, incubando durante la noche a temperatura ambiente o a 4ºC. Finalmente, el ADN resuspendido se almacenó a 4ºC. FIGURA 5 Protocolo de extracción del ADN genómico de las cepas de Bacillus. FUENTE: Elaboración propia. 3.3.3 Amplificación del gen ribosomal 16S. Las cepas de Bacillus se identificaron mediante la secuenciación del gen ARNr 16S, por medio de la amplificación por PCR. 20 Los partidores que se utilizaron para la amplificación fueron los siguientes: UN5´ACG GAT ACC TTG TTA CGA GTT 3´ y EB 5´AGA GTT TGA TCC TGG CTC AG 3´, capaces de amplificar el gen 16S de cualquier eubacteria. Los componentes, volúmenes y concentraciones para la mezcla de PCR utilizados, se detallan a continuación en el CUADRO 3. CUADRO 3 Mezcla utilizada en la reacción de PCR para amplificar el gen ARNr 16S. Componentes Concentración Volumen (µL) 10x 5,0 1x MgCl2 25mM 6,0 3,0mM dNTPs 10mM c/u 2,0 0,4mM c/u Primer 1 50pmol/µL 1,0 1,0µM Primer 2 50pmol/µL 1,0 1,0µM 5 U/µL 0,2 1,0 U/µL 100ng aprox. 5,0 - - 32,8 - - 50,0 - Buffer PCR ADN Taq polimerasa ADN templado Agua desionizada, Concentración final estéril Volumen final FUENTE: Elaboración propia. El programa de amplificación que se utilizó en el termociclador, es el que se detalla a continuación: Desnaturalización inicial 3min a 95ºC Posteriormente 35 Ciclos de PCR: 21 Desnaturalización 1min a 95ºC Elongación 1min a 45ºC Extensión 1min a 72ºC Extensión final 5min a 72ºC FIGURA 6 Protocolo de amplificación del ADN ribosomal 16S, método de la polimerasa en cadena “PCR”. FUENTE: Elaboración propia. 3.3.4 Electroforesis en gel de agarosa al 1,5%. Una vez finalizada la reacción de PCR, los fragmentos amplificados fueron visualizados por una electroforesis en gel de agarosa. Para ellos, se pesaron 0,6g de agarosa (Winkler) en un matraz y se disolvió en 40mL de buffer TAE 1X, la solución se calentó, para disolver por completo la agarosa. Posteriormente se agregaron 12µl de gel red (nucleic acid stain), cuidando que no se evapore. Esta solución fue vertida en molde de la cámara de electroforesis, cuidando de colocar las peinetas que dará origen a los pocillos del gel una vez 22 gelificada la agarosa. La corrida de electroforética se realizó en buffer TAE 1X a 100V por 1h, agregando 15µl de muestra mezclada con 3µl de buffer de carga 6X en los pocillos del gel de agarosa. Finalmente, terminada la electroforesis, se observó el gel en un transiluminador UV. FIGURA 7 Protocolo preparación del gel de agarosa al 1,5%. FUENTE: Elaboración propia. 3.3.5 Purificación del gen ribosomal 16S amplificado por PCR desde el gel de agarosa. Para tal efecto, se utilizó el kit “E.Z.N.A Gel extracción” de Omega Bio-Tek, según las indicaciones del fabricante. En breve se recortó desde el gel de agarosa, el producto de PCR amplificado con la ayuda de un bisturí. El trozo de agarosa conteniendo el ADN, se depositó en un tubo de 1,5mL y asumiendo que la densidad de la agarosa es 1mg/mL, se agregó un volumen igual de solución “binding buffer XP2”. La mezcla fue incubada a 60ºC por 7min, hasta que el gel se disolviera por completo (ir mezclando en vortex cada 2 o 3min). Una vez disuelto el trozo de gel conteniendo el ADN, se transfirieron 700µL de la solución ADN/agarosa a una columna de “ADN HiBinding” ensamblada en un tubo de 2mL (provisto por el kit). Se centrifugó a 10.000 x g por 1min a temperatura ambiente y descartar el líquido. Posteriormente se agregaron 23 300µL de solución Binding buffer, se centrifugó a 10.000 x g por 1min a temperatura ambiente. Para lavar la columna, se agregaron 700µL de “buffer SPW” diluido previamente en etanol, para luego incubar por 3min y centrifugar a 10.000 x g por 1min a temperatura ambiente. Enseguida se descartó el filtrado y se centrifugó la columna vacía por 1min a 10.000 x g para secar la matriz de la columna (paso crítico para obtención del ADN). Finalmente la columna se depositó en un tubo de 1,5mL, para luego agregar 50µL de “buffer de elución” sobre la matriz de la columna. Se centrifugó a 13.000 x g por 1min, con la finalidad de eluir el ADN (esta elución se repitió para mejorar el rendimiento del ADN). Para determinar la concentración del ADN obtenido, se procedió a determinar la cantidad de ADN en la solución por espectrofotometría, midiendo la absorbancia de la muestra obtenida a 260nm. FIGURA 8 Protocolo purificación del ADN amplificado por PCR desde el gel de agarosa. FUENTE: Elaboración propia. 24 3.3.6 Clonamiento del gen ribosomal 16S. El clonamiento del gen 16S se realizó utilizando el vector plasmidial pGEMT-Easy y se llevó a cabo a través de cuatro pasos, que se describen a continuación: 3.3.6.1 Ligación del producto PCR. Para la ligación de producto PCR se utilizó el plasmidio pGEMT-Easy de Promega. Antes de la ligación, los productos de PCR purificados fueron tratados con ADN Taq polimerasa para agregar nucleótidos de adenina, con el fin de facilitar la ligación con el plasmidio. Como se muestra en el CUADRO 4, se agregaron en un tubo de 1,5mL: 3,25 µl de agua, 5µl de buffer PCR 5X, 0,5µl de MgCl2, 1,0µl de dNTPs, 0,25µl de Taq ADN polimerasa y el producto PCR purificado. CUADRO 4 Componentes para agregar nucleótidos de adenina a los productos de PCR purificados. Componentes Componentes (ul) Producto PCR purificado 15 Buffer PCR 5x 5 Taq ADN polimerasa 0,25 dNTPs 1,0 Agua PCR 3,25 MgCl2 0,5 FUENTE: Elaboración propia. Realizada la mezcla, se incubó a 72ºC por 30min en el termociclador, así el ADN quedó listo para comenzar la ligación. 25 Para la ligación, se mezclaron en un tubo eppendorf las cantidades y volúmenes como se indica el CUADRO 5 en el siguiente orden: Agua estéril, buffer de ligación rápida 2X, plasmidio pGEMT- Easy, ADN ligasa T4 y el inserto. CUADRO 5 Componentes para la ligación del producto de PCR en el plasmidio pGEMT-Easy. Componentes Volumen (ul) Buffer rápida ligación 2x 5,0 Vector pGEM-T (50ng) 1,0 DNA ligasa T4 1,0 Inserto 2,5 Agua 2,5 FUENTE: Elaboración propia. La mezcla de ligación fue incubada a 4ºC durante toda una noche. 3.3.6.2 Transformación de E. coli competentes con el ADN plasmidial conteniendo el gen 16S de Bacillus. Este es un método realizado para incorporar ADN plasmidial recombinante en células de E. coli competentes. Primero se descongelaron las células competentes en hielo (almacenadas a -80ºC). Se transfirieron 100µL de células bacterianas a tubos de 1,5mL pre-enfriados en hielo y se añadió a las células bacterianas todo el volumen de la mezcla de ligación (vector/inserto) y se incubaron por 30min en hielo, agitando suavemente cada 5min. Posteriormente los tubos con la mezcla se incubaron en un baño de agua a 42ºC 1min 15s, este tiempo es crítico para la eficiencia de la transformación. Inmediatamente se incubó en hielo durante 2min. Después se trasfirió la mezcla de E. coli y ADN 26 plasmidial a un tubo de 15ml con 1mL de medio LB y se incubó a 37ºC en agitador orbital (200rpm) por 1h. Posteriormente, el cultivo de bacterias, se centrifugó a 3000rpm por 5min y se retiraron 900µL de sobrenadante y se resuspendieron las bacterias con 100µl de sobrante que quedó. Enseguida se sembró todo el volumen en placas de agar LB/ampicilina (50µg/mL), conteniendo además 20mg/mL de X-gal e IPTG 0,84M). Las placas sembradas fueron cultivadas a 37ºC durante 16h aproximadamente para la obtención de colonias recombinantes. 3.3.6.3 Extracción del ADN plasmidial por el Método de Ebullición. Para determinar la presencia de los plasmidios recombinantes en las colonias de E. coli, se procedió a realizar una extracción de ADN plasmidial por medio del método de ebullición para su posterior análisis. Para ello se sembró una colonia de E. coli recombinante (colonias blancas) en un tubo de 15mL conteniendo 4mL de medio LB fresco, con el antibiótico ampicilina y se incubó durante 16h a 37ºC. Posteriormente se colectaron 1,2mL de bacterias en un tubo microcentrífuga de 1,5mL y se centrifugó durante 3min a máxima velocidad. El sedimento bacteriano obtenido, se resuspendió en 110µL de solución STET (Solución STET que contiene: Sacarosa 8%; Tris-HCl 50mM pH 8,0; EDTA 50mM; Triton X-100 5%) y se mezcló en vortex por 2min. Enseguida la mezcla se incubó en baño maría por 1min y se centrifugó el lisado bacteriano a temperatura ambiente a 12.000 x g por 10min. El ADN plasmidial contenido en el sobrenadante se precipitó con 100µL de isopropanol (equivalente a la cantidad de sobrenadante que se obtiene) y luego se mezcló en vortex por algunos segundos, para luego ser centrifugado a temperatura ambiente a máxima velocidad durante 15min. El ADN precipitado se lavó con 0,5mL de etanol frío 70% (-20ºC) por la pared contraria al precipitado (lentamente) y se dejó reposar por 3min. Finalmente, el ADN plasmidial precipitado, se dejó secar a temperatura ambiente y se disolvió en 20µL de agua, agregando 0,7µL RNAasa a 2ng/mL. 3.3.7 Análisis de restricción de ADN plasmidial purificado. Para determinar la presencia del 16S insertado en los plasmidios purificados se realizó una digestión con la enzima de restricción EcoRI, la cual libera el fragmento de ADN insertado en el plasmidio. 27 Los componentes de la mezcla de digestión fueron las siguientes: 2µl buffer 10X, 0,5µl de la enzima EcoRI, ADN purificado y agua en cantidades suficientes para completar 20µl. La digestión se realizó a 37ºC por 3h y los productos de éstas se observaron en un gel de agarosa. 3.3.8 Extracción de ADN plasmidial, para secuenciación. Una vez identificados los clones de E. coli conteniendo los plasmidios recombinantes, se procedió a realizar una extracción de ADN plasmidial para su posterior secuenciación, utilizando el kit “E.Z.N.A miniprep para plasmidio” de Omega Bio-TeK. Primeramente una colonia de E. coli recombinante se inoculó en 10-15mL de caldo LB/ampicilina (50ug/mL), se incubó a 37ºC con agitación orbital (150rpm) por 16h y se centrifugó a 5.000 x g por 10min a temperatura ambiente. Se descartó el sobrenadante y el sedimento bacteriano se resuspendió en 500µL de solución I RNasa con agitación en vortex. Es vital resuspender completamente el sedimento bacteriano para obtener un buen rendimiento de ADN. Luego las células en suspensión se trasfirieron a un tubo de 2mL y se agregaron 500µL de la solución II, mezclando por inversión del tubo hasta obtener un lisado claro y se incubó por 5min a temperatura ambiente. Enseguida, se agregaron 700µL de la solución III, mezclando rápidamente por inversión del tubo hasta obtener un lisado blanco. Luego, se centrifugó a temperatura ambiente a 10.000 x g por 10min y el sobrenadante se aspiró cuidadosamente, cuidando de no tomar el sedimento. El sobrenadante obtenido se agregó a una columna HiBind™ miniprep ensamblada en un tubo colector de 2mL y se centrifugó a 10.000 x g por 1min, se descartó el filtrado obtenido y se lavó el ADN unido a la columna con 500µL de buffer HB. Se centrifugó a 10.000 x g por 1min, eliminando el sobrenadante y agregando luego 750µL de buffer de lavado diluido previamente con etanol. Nuevamente se centrifugó a 10.000 x g por 1min y se descartó el sobrenadante. Posteriormente, la columna se centrifugó seca a 10.000 x g por 1min, este paso es crítico para remover el etanol de la columna. Finalmente la columna fue puesta en un tubo microcentrífuga limpio de 1,5mL y se agregaron 80µL de agua desionizada estéril para eluir el ADN unido a la matriz de la columna. Se centrifugó a 10.000 x g por 1min, obteniéndose así al alrededor del 75- 28 80% de ADN plasmidial recombinante unido a la matriz, el cual se cuantificó en NANODROP 2000 para su posterior secuenciación. FIGURA 9 Protocolo de extracción de ADN plasmidial. FUENTE: Elaboración propia. 3.3.9 Lectura y análisis de secuencias. El ADN plasmidial recombinante, conteniendo el gen 16S, fue enviado a secuenciar a la empresa Macrogen (http://www.macrogen.com). Las secuencias obtenidas se compararon con las secuencias de ADNr 16S que se encuentran en la base de datos del GenBank mediante el software BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) del NCBI (National center for Biotechnology Information). La alineación de las secuencias se realizó mediante el software Clustal W de la EBI (http://www.ebi.ac.uk/Tools/msa/clustalw2/). ClustalW es un programa para la alineación de secuencias de ADN o proteínas. Produce alineaciones de secuencias múltiples biológicamente significativas de secuencias divergentes. Calcula el mejor 29 fósforo para seleccionar las secuencias que presentan el mayor porcentaje de identidad. 3.3.10 Prueba de antagonismo sobre césped. Se realizaron dos pruebas de antagonismo sobre césped, una con las cepas de Bacillus identificadas, seleccionando las que no causan daño a la salud, todo con el objetivo de evaluar como se comportan individualmente y estando juntas, para posteriormente ocuparlas frente a P. larvae. La segunda prueba de inhibición se hizo con las dos cepas de P. larvae en el césped, la cepa de campo (SAG) y la cepa ATCC 25745, frente a los antagonistas (cepas de Bacillus). 3.3.10.1 Antagonismo entre cepas de Bacillus. Antes de realizar las pruebas de antagonismo, se verificó su pureza y se hizo un recuento en placa de las cepas. Para esto, las cepas de Bacillus congeladas se inocularon en CST, se incubaron a durante 24h a 30+2ºC sembrándolas posteriormente en placas de AST en forma de estrías con la ayuda de un asa estéril, con el propósito de comprobar que el cultivo se encontrara completamente puro. A partir de una la colonia aislada, de cada cepa que creció en cultivo puro, con un asa estéril se inocularon en tubos con CST y se incubaron durante 24h a 30+2ºC. Terminada la incubación se les midió su densidad óptica a 620 nm en el espectrofotómetro Multiskan Fc. La muestra original se diluyó en forma seriada en buffer BPS, transfiriendo 1mL de cultivo en 9mL de buffer BPS y de este 1mL a 9mL BPS, siguiendo así sucesivamente hasta llegar a la dilución 10-6. Enseguida se sembraron 0,1mL en placas de AST, esparciéndolo sobre la superficie uniformemente con la ayuda de un rastrillo, se incubó durante 24h a 30+2ºC. Pasadas las 24h se realizó el recuento en placas, contando la dilución que presentó recuento de entre 25 a 250 colonias. De acuerdo a los recuentos obtenidos, se ajustó el inóculo de las cepas y se tomaron 300µL de la suspensión bacteriana en tubos con 10mL de medio semisólido de AST al 0,75% (dos tubos por placa) agitando en vortex y se depositó en placas estándar. Una vez gelificado el agar, con la ayuda de un sacabocado (Nº 4) se hicieron orificios en el césped, para inocular en ellos 50µL de un cultivo de 24-48h a 30+2ºC en CST, de las cepas de Bacillus mezcladas y por separadas. Después de inocular las placas, se incubaron durante 24h a 30ºC. 30 Transcurrida la incubación, se observó la presencia de halos de inhibición, producidos por los antagonistas. Aquellas en que había inhibición se tomaron tres mediciones al halo y se sacó un promedio. En la FIGURA 10 se muestra un esquema donde se resume la metodología utilizada en las pruebas de antagonismo entre las cepas de Bacillus. FIGURA 10 Pasos para prueba de antagonismo entre cepas del genero Bacillus. FUENTE: Elaboración propia. 3.3.10.2 Antagonismo de cepas de Bacillus, frente a P. larvae. Para las pruebas de inhibición frente a P. larvae, la preparación del césped se realizó para las dos cepas de P. larvae (previamente activadas en caldo MYPGP), esta cepa objetivo se inoculó en 2mL caldo MYPGP desde una colonia aislada de una placa con agar MYPGP, placa que fue incubada a 37+2ºC durante 72h en condiciones de anaerobiosis, obteniendo 31 de esta manera una suspensión bacteriana de 10 7 UFC/mL. Posteriormente se tomaron 300µL de esta suspensión y se introdujeron en 10mL de Agar MYPGP (dos tubos por placa) al 0,75% y luego se vertió en una placa estándar. Con la ayuda de un sacabocado se hicieron orificios en el césped de P. larvae para inocular en ellos 50µL de un cultivo de 24h a 30+2ºC (incubado en anaerobiosis) en CST de los posibles antagonistas, analizados en el punto anterior 3.3.10.1. En las placas que presentaron halos de inhibición frente a P. larvae, se tomaron tres mediciones al halo y se sacó un promedio. En la FIGURA 11 se presenta un esquema donde se resume los procedimientos utilizados en las pruebas de inhibición frente al P. larvae. FIGURA 11 Pasos para la prueba de antagonismo entre las cepas de P. larvae contra las cepas del género Bacillus. FUENTE: Elaboración propia. 32 4 PRESENTACIÓN Y DISCUSIÓN DE RESULTADOS Durante la investigación, se utilizaron ocho cepas pertenecientes al cepario del Proyecto CAPI IDO 901 (Laboratorio de Microbiología, ICYTAL, Facultad de Ciencias Agrarias, Universidad Austral de Chile). Las cepas fueron aisladas con anterioridad de muestras de miel y material de colmenas (trozos de panal). Las muestras de miel fueron facilitadas por la “Cooperativa Campesina Apícola Valdivia Limitada, APICOOP”, ubicada en Paillaco, Región de los Ríos. A continuación se presentan los resultados obtenidos, descritos en concordancia secuencial a los experimentos realizados. 4.1 Preparación de la muestra Para comenzar, se revivieron las cepas del género Bacillus que se encontraban congeladas en Caldo Soya Tripticasa (CST) y posteriormente se sembraron en (AST), con el objeto de verificar la pureza. FIGURA 12 Colonias puras de las ocho cepas del género Bacillus en Agar Soya Tripticasa. 33 En la FIGURA 12, se muestran los cultivos puros obtenidos en todas las cepas de Bacillus, con lo cual se asegura que todos los individuos del mismo, tengan la misma composición genética, para obtener una efectiva identificación. Esta etapa es esencial para tener mayor seguridad en la identificación de los microorganismos. 4.2 Cuantificación del ADN genómico Las cepas puras se inocularon en medio CST, desde el cual se extrajo el ADN genómico. En el CUADRO 6 se observan las concentraciones de ADN genómico aislado y medido por espectrofotometría, midiendo la absorbancia a 260 nm y utilizando la razón de absorbancias 260nm/280nm para verificar la calidad y pureza del ADN extraído. CUADRO 6 Cuantificación del ADN genómico extraído de Bacillus. Muestra Concentración (ng/µl) 260nm/280nm 1g 55,9 2,02 20 76,9 2,04 22 8 2,22 67 - - 87 59 1,75 92 98,1 1,7 95 - - 97 - - 34 De la medición obtenida en el equipo NANODROP 2000, se puede apreciar cuantificación de ADN genómico extraído en las muestras de las cepas 1g; 20; 22; 87 y 92, mientras que en las muestras 67; 95 y 97, el equipo no logró obtener una medición, lo cual pudo deberse al límite de detección que el equipo posee, el cual va de los 215.000ng/µl. Debido a los anillos aromáticos que poseen las bases nitrogenadas del ADN, éste tiene por absorbancia máxima 260nm, mientras que en las proteínas la absorbancia es medida a 280nm, por ende la determinación de la pureza y calidad del ADN, se determinó utilizando la razón de ambas absorbancias (260nm/280nm), con valores que deben estar por sobre 1,7, como sucede con las cepas 1g; 20; 22; 87 y 92. 4.3 Amplificación del gen ARNr 16S por medio de PCR El gen 16S fue amplificado por PCR a partir de ADN obtenido desde células vegetativas de cepas del género Bacillus. Para ello se utilizaron partidores universales UN y EB descritos por BARRY et al., (1990). Producto de la amplificación se obtuvo un fragmento de 1.500 pb, correspondiente al tamaño del gen ANRr 16S. Varias investigaciones han revelado que la secuencia del gen ARNr 16S presenta regiones conservadas y entre ellas se encuentran regiones que son variables, que han sido útiles para la diferenciación de género y especie en bacterias (JENSEN et al., 1993). En la FIGURA 13 se observa la separación en el gel de agarosa de los productos de la amplificación del gen de ADNr 16S de cada cepa, los que en todos los casos, al ser comparados con el estándar de ADN, corresponden a los 1.500 pb aproximadamente, descritos en la literatura por BARRY et al., (1990). Este producto PCR se obtuvo en todas las cepas en estudio, aún en las que no se había obtenido cuantificación de concentración de ADN genómico en el equipo NANODROP 2000, dando cuenta que no es necesario grandes cantidades de ADN para realizar la amplificación. La especificidad de la reacción fue determinada al no observar productos de amplificación inespecífica en el control negativo. 35 FIGURA 13 Electroforesis en gel de agarosa al 1,5%, que muestra la amplificación del ADNr 16S mediante PCR de ocho aislados de Bacillus. Carriles: Est, marcador de peso molecular de 1003000pb; 1g; 20; 22; 67; 87; 92; 95 y 97, aislados de Bacillus; (+), control positivo; (-), control negativo de F. psychrophilum. 4.4 Purificación del gen ARNr 16S Realizada y verificada la amplificación del gen 16S, se preparó un nuevo gel (ANEXO 2), con el objetivo de purificar los productos de PCR de cada uno de los aislados. Este procedimiento es indispensable para remover desde el producto de PCR obtenido el exceso de partidores y nucleótidos (CLARRIDGE, 2004). En la FIGURA 14 se muestran los resultados obtenidos y se aprecian los productos de 1.500pb, confirmando la obtención del gen 16S amplificado. 36 FIGURA 14 Electroforesis del gen ARNr 16S purificado. Carriles: Est, marcador de peso molecular de 100-3000pb; 1g; 20; 22; 67; 87; 92; 95 y 97, aislados de Bacillus. 4.5 Clonamiento del gen ARNr 16S de los aislados de Bacillus La reacción de PCR permite la obtención de una gran cantidad del gen que se desea clonar en vectores plasmidiales. Por esta razón se utilizó esta estrategia para clonar el gen 16S de los aislados de Bacillus. Este proceso consta de la purificación del gen amplificado por PCR, adición de nucleótidos de adenina en los extremos del producto amplificado y posterior ligación al vector de clonamiento pGEMT - Easy, utilizando la enzima T4 DNA ligasa. El clonamiento de productos de PCR se ve facilitado al utilizar ADN polimerasa termoestables como la Taq ADN polimerasa, la que dan lugar a productos con extremo 3´ conteniendo residuos de adenina, facilitando la inserción de los productos PCR en 37 vectores de clonamiento con extremos conteniendo timina (WILSON y WALKER, 2000). 4.5.1 Transformación bacteriana. El producto de la ligación 16S / pGEM-T Easy se utilizó para la transformación de E. coli competentes cepa JM109. El proceso de transformación bacteriana se refiere a la introducción de un ADN foráneo a una bacteria por medio de un “shok” térmico lo que provoca un cambio en la permeabilidad de la membrana de la E. coli, facilitando la entrada de este ADN, que en este caso correspondía a un ADN plasmidial conteniendo el gen 16S de aislados de Bacillus. Terminado el proceso de transformación, las bacterias son sembradas en placas con medio LB/amp, donde se obtienen finalmente las colonias recombinantes con un alto número de copias del vector con el inserto (LUQUE y HERRÁEZ, 2008). Posteriormente se seleccionaron tres colonias (clones) transformantes formadas por bacterias de E. coli que hayan incorporado el inserto, en este caso correspondiente a las colonias blancas, como se muestra en la FIGURA 15. FIGURA 15 Cultivo de E. coli transformantes LB/ampicilina/IPTG y X-Gal. en placas de agar 38 En la FIGURA 15 se aprecian colonias de E. coli conteniendo al vector sin inserto (colonias azules) y conteniendo el vector e inserto (colonias blancas). Esta coloración es debido a que el inserto ubicado en el sitio de múltiple clonamiento del vector, interrumpió la secuencia de parte del gen lacZ que codifica la proteína β-galactosidasa, originando una enzima no funcional. De esta manera, se seleccionan sólo las colonias que poseen el plasmidio e inserto, que en este caso corresponden a aquellas colonias blancas, que no son capaces de metabolizar el compuesto cromogénico X-Gal por parte de la enzima β-galactosidasa. Este proceso de selección es denominado α-complementación (VOET y VOET, 2007). Las tres colonias (clones) elegidos por placa, fueron cultivadas en medio líquido LB suplementado con ampicilina para extraer el ADN plasmidial recombinante, el cual fue digerido, en cada caso, con enzima de restricción EcoRI, para verificar la inserción de los productos de PCR. Los productos de digestión fueron analizados en un gel de agarosa al 1,5%, para finalmente seleccionar un clon plasmidial conteniendo el gen 16S por cepa de Bacillus. En la FIGURA, 16 se puede apreciar los productos de la digestión de los clones plasmidiales 92 y 20, donde se puede ver el plasmidio pGEM-T Easy sin inserto a las altura de 3.000pb y los fragmentos de ADN correspondiente al inserto, que en este caso son dos, ya que el gen 16S de Bacillus posee un sitio para la enzima EcoRI. Los clones plasmidiales seleccionados fueron 92 3 y 201. En las figuras del ANEXO 3 se aprecia el mismo análisis de restricción, donde los clones plasmidiales seleccionados fueron 1g 3, 221, 673, 871, 973 y 951. 39 FIGURA 16 Análisis electroforético del ADN plasmidial recombinante digerido con EcoRI. Carriles: Est, marcador de peso molecular de 1003.000pb; 921, 922, 923, 201, 202 y 203, aislados de Bacillus. 4.5.2 Purificación, cuantificación y secuenciación de ADN plasmidial seleccionado. Posteriormente a partir de los clones seleccionados, se realizó una segunda extracción de ADN plasmidial, esta vez con el kit comercial para obtener un ADN de mayor calidad para su secuenciación. Con el fin de visualizar el inserto de ADN incorporado al vector, el plásmido purificado fue sometido a una digestión con la enzima de restricción EcoRI y se procedió a visualizar el producto de la digestión en un gel de agarosa al 1,5%, que permitió visualizar los insertos buscados, como se muestra a continuación en las FIGURAS 17 y 18. En ambas figuras, se visualiza los 3.000pb del vector de clonamiento pGEM-T Easy, el inserto de 1.500pb y los fragmentos de los cortes de la enzima de restricción EcoRI para las muestras 97, 87, 67 y 95 (FIGURA 17) y las muestras 1g, 20, 22 y 92 respectivamente (FIGURA 18). 40 FIGURA 17 Análisis electroforético del ADN plasmidial conteniendo el gen 16S con la enzima de restricción EcoRI. Carriles: Est, marcador de peso molecular de 100-3000pb; 97; 87; 67 y 95, aislados de Bacillus. FIGURA 18 Análisis electroforético del ADN plasmidial conteniendo el gen 16S con la enzima de restricción EcoRI. Carriles: Est, marcador de peso. 41 La enzima de restricción EcoRI mostró 2 patrones de restricción, uno de los cuales liberó un fragmento de aproximadamente 1.500 pb en los ocho aislados, exponiendo así la existencia de la secuencia de reconocimiento de la enzima. El segundo patrón de restricción mostró dos fragmentos de restricción de 900 y 600pb aproximadamente, en promedio. El ADN purificado fue cuantificado (CUADRO 7), y enviado a secuenciar de acuerdo a las instrucciones señaladas en la página de la empresa Macrogen. CUADRO 7 Cuantificación del ADN enviado a secuenciar. Cepa Concentración ng/µl 260nm/280nm 1g 266,4 1,98 20 159,2 1,98 22 464,3 2,03 67 320,2 2,03 87 279,7 2,00 92 232,0 2,00 95 184,4 2,02 97 201,1 2,04 4.6 Secuenciación e identificación de las cepas de Bacillus La secuenciación se realizó por el método de didesoxinucleotido. Éste se llevó a cabo utilizando partidores complementarios a la secuencia de los promotores SP6 y T7 contenidos en el plasmidio, las que flanquean la región de múltiple clonamiento del vector. Las secuencias del gen 16S clonadas en el vector (ANEXO 4) se compararon 42 con las que se encuentran en la base de datos del GenBank mediante el software BLAST del NCBI (National Center for Biotechnology Information, EUA). El GenBank es parte de International Nucleotide Sequence Database Collaboration, que comprende the DNA DataBank of Japan (DDBJ), the European Molecular Biology Laboratory (EMBL), and GenBank at NCBI. Estas tres organizaciones intercambian datos diariamente, siendo el objetivo común dar a conocer abiertamente la información obtenida individualmente y formar un GenBank a nivel mundial cada día más amplio (http://www.ncbi.nlm.nih.gov/genbank/). Esta base de datos dispone de una colección de aproximadamente 126.551.501.141.135.440.924 bases en 135.440.924 expedientes de secuencias en las divisiones tradicionales de GenBank y 191.401.393.188 bases en 62.715.288 expedientes de secuencia registradas con fecha abril de 2011. En el CUADRO 8 se resume dicha comparación, donde se indican el origen bacteriano de los genes 16S que presentaron un mayor porcentaje de identidad con las secuencias 16S de los aislados de Bacillus de este estudio. La miel puede contener un número pequeño y una variedad limitada de microorganismos. Los microorganismos capaces de sobrevivir en la miel, son los que soportan la concentración de azúcar, la acidez y otras características antimicrobianas propias de la miel. Los microorganismos normalmente existentes en la miel se han investigado y se ha demostrado que se encuentran mohos, levaduras y bacterias formadoras de esporas. Esto se debe principalmente a las fuentes de contaminación microbiológica, que incluyen el polen, el tracto digestivo de las abejas, la suciedad, el polvo, el aire y las flores (LEE et al., 2008). Analizando los resultados obtenidos en el CUADRO 8, las especies del género Bacillus identificadas, resultan ser ubicuos de la miel. Estas bacterias son Gram positivas, formadoras de endosporas inocuos para el ser humano, con excepción del Bacillus cereus. Su hábitat principal es el suelo y debido a la formación de esporas, son extremadamente resistente al calor, pueden colonizar diferentes ambientes, como la miel, diversos alimentos, animales, insectos, etc. (MAHMOUND y ABOUWARDA, 2011). Cabe señalar que los microorganismos que se encuentran naturalmente en la miel del panal son incorporados por el néctar de las flores y por la abeja obrera. Las bacterias 43 (género Bacillus) provienen en gran parte solo de la abeja (SNOWDON y CLIVER, 1996). CUADRO 8 Resumen de la identificación bacteriana después de la secuenciación. Cepa Gen 16S % de identidad Código de acceso 1g Bacillus cereus 99% ID JQ435675.1 20 Bacillus cereus 79% ID EF513610.1 22 Bacillus cereus 79% ID EF428235.2 67 Bacillus pumilus 99% ID JN315777.1 87 Bacillus pumilus 99% ID EU620416.1 92 Bacillus subtilis 98% ID HQ727971.1 95 Bacillus subtilis 99% ID EU723210.1 97 Bacillus pumilus 99% ID EU620416.1 4.7 Pruebas de antagonismo Para realizar las pruebas de antagonismo, las cepas identificadas se volvieron a activar en CST, después de 24h se sembraron por estrías en AST. Luego de 24h a 48h se comprobó el desarrollo de colonias aisladas, verificando que el cultivo se encontrara puro. A partir de este cultivo, se inoculó en CST y se midió su densidad óptica (CUADRO 9). 44 CUADRO 9 Densidad óptica (Do) a 620nm de las cepas incubadas en CST. Cepa Do1 Do2 Do3 Promedio Do 1g 0,256 0,260 0,257 0,258 Dst 20 0,307 0,320 0,322 0,316 Dst 22 0,070 0,067 0,056 0,064 Dst 67 0,068 0,059 0,064 0,064 Dst 87 0,062 0,090 0,125 0,092 Dst 92 0,040 0,024 0,175 0,080 Dst 95 0,114 0,130 0,101 0,115 Dst 97 0,106 0,086 0,103 0,098 Dst Posteriormente se seleccionaron tres cepas del género Bacillus, descartando las identificadas como B. cereus, ya que esta especie es reconocida por ser causante de intoxicaciones alimentarias, aunque cabe señalar que para el desarrollo de un biocontrolador, se extrae la sustancias específica que actúa contra como antagonista dejando de lado los demás componentes y características propias de la célula bacteriana que son causante de las intoxicaciones (DWORKIN et al., 2006). Las tres cepas seleccionadas fueron las identificadas con código 87, 95 y 97, a las que se les realizaron diluciones seriadas en BPS (Buffer fosfato salino) a tres cepas previamente seleccionadas. Pasado 12h se realizó el recuento de las diluciones que presentaron entre 25-250 UFC/mL, como se muestra en el CUADRO 10. 45 CUADRO 10 Recuento a las 12h de incubación en AST. Cepa UFC/mL a las 12h Do 620nm a las 48h 87 9,8x107 0,098 95 8,0x107 0,115 97 1,3x107 0,098 Obtenidos los recuentos de los tres cultivos, se inoculó una colonia para realizar pruebas de antagonismo, entre las tres cepas y éstas frente a P. larvae. 4.7.1 Antagonismo entre las cepas de Bacillus identificadas. Previo a utilizar una mezcla de las tres cepas de Bacillus contra P. larvae, se realizaron pruebas de antagonismo entre las tres cepas de Bacillus, con el fin de observar su comportamiento, dada la posibilidad que estando juntas, estas potencien su capacidad para inhibir P. larvae, pero cuidando que no se inhiban entre ellas. Como se aprecia en la FIGURA 19, la prueba de antagonismo reveló que las cepas 97 y 95 resultaron ser inhibidas por la cepa 87, presentando un posible problema al ocuparla como mezcla en pruebas antagónicas posteriores. FIGURA 19 Pruebas de antagonismo sobre agar soya tripticasa entre cepas de Bacillus, con inhibición de las cepas 95 y 97 por la cepa 87. 46 A los halos obtenidos en las pruebas de inhibición, se les procedió a tomar tres mediciones, las que se detallan a continuación en el CUADRO 11. CUADRO 11 Halos de inhibición en las pruebas de antagonismo entre cepas del género Bacillus. Cepa césped Cepa antagonista Halo inhibición (mm) Promedio 87 2,09 2,21 97 2,26 2,27 95 95 - 87 2,24 2,15 2,12 2,10 87 97 - - 95 - - 97 - - Como se puede observar en el CUADRO 11, el halo de inhibición promedio que se obtuvo del antagonismo de la cepa 87 contra la 97, fue de 2,21mm y de 2,15mm contra la cepa 95. La cepa 87, identificada como B. pumilus, inhibió a las cepas 95 y 97, lo cual podría explicarse debido a que esta especie es reconocida por producir componentes antimicrobianos, que tienen una fuerte acción en contra de bacterias Gram positivas (LEIFERTL et al., 1995). 47 4.7.2 Antagonismo de las cepas de Bacillus frente a P. larvae. Analizado el comportamiento de antagonismo entre las cepas de Bacillus, se procedió a realizar pruebas de inhibición de éstas, frente a las cepas de P. larvae ATCC 25745 y de campo. La FIGURA 20, ilustra las fotografías de las pruebas de antagonismo, las tres cepas utilizadas, las 87, 95 y 97 inhibieron el P. larvae ATCC 25745, mientras que la cepa 87 no presento inhibición frente al P. larvae de campo. Por otro lado los mix (95+97), (87+97), (87+95) y (87+95+97) inhibieron el P. larvae ATCC 25745, mientras que las mezclas (87+97), (87+95), y (87+95+97) no presentaron halo de inhibición alguno frente al P. larvae de campo. FIGURA 20 Pruebas de antagonismo sobre agar MYPGP entre cepas del género Bacillus frente a P. larvae ATCC 25745 y de Campo. Observada la presencia de los halos, se procedió a tomar tres mediciones a cada halo. Los resultados obtenidos de las mediciones, se muestran a continuación en el CUADRO 12. 48 CUADRO 12 Tamaño de los halos de inhibición de la prueba de antagonismo de las cepas 87, 95, 97 y mezclas de ellas, contra P. larvae ATCC 25745 y de Campo. Cepa césped Cepa antagonista Halo inhibición (mm) ATCC 25745 87+97 1,80 87+95 1,17 87+97 - 87+95 - 95 7,78 97 8,63 87 1,74 95 1,78 97 1,39 87 - 95+97 5,72 87+95+97 2,38 95+97 1,34 87+95+97 - Campo ATCC 25745 Campo ATCC 25745 Campo Como se puede observar en el CUADRO 12, los mayores halos de inhibición promedio se obtuvieron en las cepas por individual, mientras que estando en mezcla no se mejoraron su capacidad antagónica, descartando la posibilidad de que pudieran potenciarse al estar en conjunto. 49 No es extraño que las cepas 87, 95 y 97, identificadas como B. pumilus, B. subtilis y B. pumilus respectivamente, hayan logrado inhibición en casi todas las cepas de P. larvae, pues estas son reconocidas por producir una gran variedad de antibióticos. Esto se corrobora con los resultados obtenidos en las pruebas de inhibición de P. larvae por ALIPPI y REYNALDI (2006), donde reconocen estas tres cepas como fuertes inhibidores del patógeno. El B. subtilis, es reconocida por producir una gran variedad de antibióticos con diferentes estructuras. La producción de componentes antimicrobianos activos que produce esta especie, incluye predominantemente péptidos, los que pueden o no ser sintetizados ribosomalmente, así como también algunos componentes no peptídico como fosfolípidos (LEIFERT et al., 1995; STEIN, 2005). 50 5 CONCLUSIONES Se seleccionaron ocho cepas del género Bacillus, en base a su capacidad para inhibir a P. larvae en pruebas de antagonismo en placa, las cuales fueron identificadas hasta el nivel de especie, mediante la secuenciación del gen ARNr 16S. Para la caracterización e identificación de las cepas se logró amplificar y clonar en el plasmidio pGEMT-Easy, el gen ribosomal 16S de las ocho cepas del género Bacillus, para su posterior secuenciación. Se obtuvieron las ocho secuencias del gen 16S, las cuales al ser comparadas con las secuenciaciones en la base de datos Gen Bank de NCBI, estas correspondieron a Bacillus subtilis, Bacillus pumilus y Bacillus cereus. Las pruebas de antagonismo entre las cepas de Bacillus, revelaron inhibición de la cepa 87 (B. pumilus), frente a la cepa 95 (B. subtilis) y 97 (B. pumilus), lo cual implica que al ser utilizadas en una mezcla frente a P. larvae, estas pueden inhibirse entre sí, disminuyendo su eficiencia contra el patógeno. Las cepas que presentaron mayor actividad antagónica contra la cepa de P. larvae ATCC 25745 y de campo, fueron B. pumilus (97) y B. subtilis (95) respectivamente. 51 6 BIBLIOGRAFÍA ALBERT, B; BRAY, D; HOPKIN, K; JOHNSON, A; LEWIS, J; RAFF, M; ROBERTS, K y WALTER, P. 2004. Editorial Panamericana. Segunda Edición. ALIPPI, A y REYNALDI, F. 2006. Inhibition of the growth of Paenibacillus larvae, the causal agente of American foulbrood of honeybees, by selected strains of aerobic spore-forming bacteria isolated from apiarian sources. Jouernal of Invertebrate Pathology 91: 141-146. ALVIS, V; CALLEJA, L; PEREIRA, M; RUIZ, L y CALAHORRA, F. 2009. Visión Actual de la Apicultura en España. Revista Complutense de Ciencias Veterinarias 3 (2): 139-148. ANTÚNEZ, K.; D`ALESSANDRO, B.; PICCINI, C.; CORBELLA, E. y ZUMINO, P. 2004. Paenibacillus larvae larvae Spores in Honey Samples from Uruguay: a Nationwide Survey. Journal of Invertebrate Pathology 86: 56-58. BARRY, T; POWELL, R y GANNON, F. 1990. A general method to generate DNA probes for microorganisms. Biotechnology 8: 233-236. BARRY, T; COLLERAN, G y GLENNON, M. 1991. The 16S/23S ribosomal spacer region as a target for DNA probes to identify aubacteria. Genome Res 1: 51-56 CABALLERO, D. 2009. Manual de Enfermedades Apícolas. Editorial Instituto Interamericano de Cooperación para la Agricultura. Honduras. 54p. 52 CHANTAWANNAKUL, P y DANCER. 2001. American Foulbrood in Honey. Iternational bee research association 82 (4): 168-180. CLARRIDGE, J. 2004. Impacto f 16S rRNA Gene Sequence Analysis for Identification of Bacterial on Clinical Microbiology and Infectious Diseases 17 (4): 840-862. DWORKIN, M.;FALKOW, S.; ROSENBERG, E.; SCHLEIFER, K y STACKEBRANDT, E. 2006. The prokaryots. A handbook on the biology of bacteria. Bacteria: Firmicutes, Cynobacteria. 3 th de. Estados Unidos. Springer. 572p. GENERSCH, E.; FORSGREN, E.;PETIKA, J.;ASHIRALIEVA,A.; RAUCH, S.; KILWINSKI, J. y FIES, I. 2006. Reclassification of Paenibacillus larvae subsp. pulvifaciens and Paenibacillus larvae subsp. Larvae as Paenibacillus larvae without subspecies differentiation. International Journal of Systematic and Evolutionary Microbiology 56; 506-511. GENERSCH, E. 2010. American Foulbrood in honeybees and its causative agent, Paenibacillus larvae. Journal of Invertebrate Pathology 103: S10-S19. GERDING, M y RODRIGUEZ, M. 2010. Valor de la apicultura y su relación con el control biológico. Revista Consorcio Apícola. Segunda edición: GÜRTLE, V y STANISICH, V. 1996. New approaches to typing and identification of bacteria using the 16S-23S rDNA spacer region. Microbiology 142: 3-16. HARMSEN, D y KARCH, H. 2004. 16S rDNA for Diagnosing Pathogens: a Living Tree. American Society for microbiology 70: 19-24. 53 HEYNDRICKX, M; VANDEMEULEBROECKE, K; HOSTE, B; JANSSEN, P; KERSTERS, K; DE VOS, P; LOGAN, N, ALI, N y BERKELEY, R. 1996. Reclassification of Paenibacillus (Formely Bacillus) pulvifaciens (Nakamura 1984) Ash et al. 1994, a Later Subjective Synonym of Paenibacillus (Formely Bacillus) larvae (While 1906) Ash et al. 1994, as a Subspecies of P. larvae as P. larvae subsp. larvae and P. larvae subsp. pulvifaciens 46: 270-279. HORNITZKY, M. 1998. The pathogenicity of Paenibacillus larave subsp. larave spores and vegetative cells to honey bee (Apis mellifera) colonies and their susceptibility to royal jelly. Journal of Apicultural Research 37 (4): 267-271. JENSEN, M; WEBSTER, J y STRAUS, N. 1993. Rapid Identification of Bacteria on the Basis of Polymerase Chain Reaction-Amplified Ribosomal DNA Spacer Polymorphisms. Applied and Environmental Microbiology 59 (4): 945-952. KILIK, A.; SIMSEK, H y KALENDER, H. 2010. Detection of American Foulbrood Disease (Paenibacillus larvae) By the PCR and Culture. Kafkas Univ Vet Fak Derg 16 (5): 841-845. LEE, H.; CHUREY, J y WOROBO, R. 2008. Antimicrobial activity of bacterial isolates from different floral sources of honey. International Journal of Food Microbiology 126; 240-244. LEE, H.; CHUREY, J y WOROBO, R. 2009. Isolation and characterization of a protective bacterial cultura isolated from honey active against American Foulbrood disease. FEMS Microbiology letters 296; 39-44. LEHNINGER, A.; NELSON, D y COX, M. 2009.Principios de Bioquímica. Editorial Omega. Quinta edición. Barcelona. 1124p. 54 LESSER, R. 2004. Manual de Apicultura Modera. Editorial Universitaria. Chile. 170p. LUQUE, J y HERRÁEZ, A. 2008. Biología molecular e Ingeniería genética. Editorial Elsevier S.A. Madrid, España. 468p. MAC-FADDIN, J. 2003. Pruebas Bioquímicas para la Identificación de Bacterias de Importancia Clínica. Editorial Médica Panamericana. España. Tercera Edición. 850p. MAHMOUND, H y ABOUWARDA, A. 2011. Antimicrobial Activity of Bacterial Isolates from Honey. International Journal of Microbiological Resarch 2: 82-85. MATHEWS, J. 2002. Bioquímica. Editorial Addison Wesley. Tercera edición. España. 1335p. MEDINA, L y MAY, W. 2005. Enfermedades de las abejas. Editorial Universidad Autónoma de Yucatán. México. 93p. MENDIZABAL, F. 2004. Abejas. Editorial Albatros. Aregentina. 259p. MEVARECH, M; HIRSCH, S; GOLDMAN, S; YAKOBSON, E; EISENBERG, H y DENNIS, P. 1989. Isolation and Characterization of the rRNA Gene Clusters of Halobacterium marismortui. Jouernal of Bacteriology 171 (6): 3479-3485. NEIRA, M. 2006. Sanidad apícola, principales enfermedades y enemigos de las abejas en Chile. 139p. 55 NEIRA, M. 2009. Almanaque Apícola. Editorial APICAP Chile. Chile. 150p. PADILLA, F y CUESTA, A. 2003. Zoología apicola. Editorial Díaz de Santos. España. 468p. PICCINI, C y ZUNINO, P. 2001. American foulbrood in Uruguay: Isolation of Paenibacillus larvae larvae from Larvae with Clinical Symptoms and Adult Honeybees and Susceptibility to Oxytetracycline. Journal of Invertebrate Pthology 78; 176-177. PICCINI, C; D’ ALESSANDRO, B; ANTÚNEZ, K y ZUNINO, P. 2002. Detection of Paenibacillus larvae subspecies larvae spores in naturally infected bee larvae and artificially contaminated contaminated honey by PCR. Journal of Microbiology y Biotechnology 18: 761-765. PICCINI, C; ANTÚNEZ, K y ZUNINO, 2004. An approach to the characterization of the Honey bee hive bacterial flora. Journal of Apicultural Research 43 (3): 101104. PIERRE, J y YVES, L. 2006. Apicultura. Editorial Mundi Prensa. Cuarta Edición. España. 790p. RODICIO, M y MENDOZA, M. 2004. Identificación bacteriana mediante la secuenciación del ARNr 16S: fundamento, metodología y aplicaciones en microbiología. Enferm Infecc Microbiol Clin 22 (4): 238-245. 56 SANAD, R y AL-BARRAK, A. 2010. Use of Clove and Watercress Oils for Controlling the American Foulbrood Disease Attacking Honeybee Colonies at El-Gharbia Governorate, Egypt 20: 33-36. SÁNCHEZ, C. 2010. Aislamiento de bacterias con propiedades antagonistas a Paenibacillus larvae subsp. Larvae a partir de fuentes apícolas. Valdivia. Universidad Austral de Chile. Facultad de Ciencias Agrarias. 78p. SANDOVAL, P. 2010. Aislamiento e identificación de especies del género Bacillus desde miel para su empleo como antagonistas a Paenibacillus larvae. Memoria de titulo Ing. en Alimentos. Valdivia. Universidad Austral de Chile, Facultad de Ciencias Agrarias. 124p. SHIMANUKI, H y KNOX, D. 2000. Diagnosis of Honey Bee Diseases. United States Departament of Agricultura. 61p. SNOWDON, J. y CLIVER, D. 1996. Microorganisms in honey. Journal International of Food Microbiology 31:1-26. STEIN, T. 2005. Bacillus subtilis antibiotics: Structures synthesis and specific functions. Molecular Mycrobiology 56(4):845-857. TORTORI, E. 2003. Impacto of Genotypic Studies on Mycobacterial Taxonomy: the New Mycobacteria of the 1990s. Journal Clinical Microbiology Reviews 16 (2): 319-354. TRESGUERRES, J; FERNAANDEZ, J y SERRANO, V. 2003. Biotecnología aplicada a la medicina. Editorial Diaz de Santos. España. 321p. 57 TRHLIN, M y RAJCHARD, J. 2011. Chemical communication in the honeybee (Apis mellifera L.): a review 56 (6): 265-273. USABIAGA, J.; GALLARDO, J. y CAJERO, S. 2002. Patología apícola. IICA. Costa rica. 80p.ç VOET, D y VOET, J. 2004. Bioquímica. Editorial Panamericana. Tercera Edición. 1763p. WILSON, K y WALKER, J. 2000. Principles and Techniques of Practical Biochemestry. Editorial Cambridge University Press. Fifth Edition. 792p. 58 1 ANEXO Medios de cultivos y buffer 1.1 Medios de cultivo Caldo soya tripticasa (CST) Ingrediente Cantidad Soya tripticasa 30g Agua destilada 1000mL Agar soya tripticasa (AST) Ingrediente Cantidad Soya tripticasa 30 Agar 15g Agua 1000mL Agar MYPGP Ingrediente Cantidad Extracto de lavadura 15g Caldo Muller-Hinton 10g Glucosa 2g 59 K2HPO4 anhidro 3g Piruvato de sodio 1g Agar-agar 15g Agua destilada 1000mL 1.2 Buffer Buffer fosfato salino 0,01M pH 7,2 (PBS) Ingredientes Cantidad NaCl 4g KCl 0,1g Na2HPO4 0,72 KH2PO4 0,12 Agua destilada 500ml * Ajustar pH 7,2 TAE 60 2 ANEXO Electroforesis en gel de garosa para la purificación de los productos de PCR A continuación se muestra la electroforesis en gel de garosa para la purificación de los productos de PCR. Carriles: Est, marcador de peso molecular de 100-3000pb; 1g; 20; 22; 67; 87; 92; 95 y 97, aislados de Bacillus. 61 3 ANEXO Análisis electroforético del ADN plasmidial recombinante digerido con EcoRI. 3.1 Análisis electroforético del ADN plasmidial recombinante digerido con EcoRI. Carriles: Est, marcador de peso molecular de 100-3000pb; 1g1; 1g2; 1g3; 221; 222 y 223, aislados de Bacillus. 62 3.2 Análisis electroforético del ADN plasmidial recombinante digerido con EcoRI. Carriles: Est, marcador de peso molecular de 100-3000pb; 671; 672; 673; 871; 872 y 873, cultivos. 63 3.3 Análisis electroforético del ADN plasmidial recombinante digerido con EcoRI. Carriles: Est, marcador de peso molecular de 100-3000pb; 971; 972; 973; 951; 952 y 953, cultivos. 64 4 ANEXO Secuencias de las cepas del genero Bacillus. A continuación se presentan las secuencias de las cepas del género Bacillus, estas se compararon la base de datos del GenBank de NCBI. 1g AGAGTTTGATCCTGGCTCAGGATGAACGCTGGCGGCGTGCCTAATACATGCAAGTCGAGCGAAT GGATTAAGAGCTTGCTCTTATGAAGTTAGCGGCGGACGGGTGAGTAACACGTGGGTAACCTGCC CATAAGACTGGGATAACTCCGGGAAACCGGGGCTAATACCGGATAACATTTTGAACTGCATGGT TCGAAATTGAAAGGCGGCTTCGGCTGTCACTTATGGATGGACCCGCGTCGCATTAGCTAGTTGG TGAGGTAACGGCTCACCAAGGCAACGATGCGTAGCCGACCTGAGAGGGTGATCGGCCACACTGG GACTGAGACACGGCCCAGACTCCTACGGGAGGCAGCAGTAGGGAATCTTCCGCAATGGACGAAA GTCTGACGGAGCAACGCCGCGTGAGTGATGAAGGCTTTCGGGTCGTAAAACTCTGTTGTTAGGG AAGAACAAGTGCTAGTTGAATAAGCTGGCACCTTGACGGTACCTAACCAGAAAGCCACGGCTAA CTACGTGCCAGCAGCCGCGGTAATACGTAGGTGGCAAGCGTTATCCGGAATTATTGGGCGTAAA GCGCGCGCAGGTGGTTTCTTAAGTCTGATGTGAAAGCCCAACGGCTCAACCGTGGAGGGTCATT GGAAACTGGGAGACTTGAGTGCAGAAGAGGAAAGTGGAATTCCATGTGTAGCGGTGAAATGCGT AGAGATATGGAGGAACACCAGTGGCGAAGGCGACTTTCTGGTCTGTAACTGACACTGAGGCGCG AAAGCGTGGGGAGCAAACAGGATTAGATACCCTGGTAGTCCACGCCGTAAACGATGAGTGCTAA GTGTTAGAGGGTTTCCGCCCTTTAGTGCTGAAGTTAACGCATTAAGCACTCCGCCTGGGGAGTA CGGCCGCAAGGCTGAAACTCAAAGGAATTGACGGGGGCCCGCACAAGCGGTGGAGCATGTGGTT TAATTCGAAGCAACGCGAAGAACCTTACCAGGTCTTGACATCCTCTGAAAACCCTAGAGATAGG GCTTCTCCTTCGGGAGCAGAGTGACAGGTGGTGCATGGTTGTCGTCAGCTCGTGTCGTGAGATG TTGGGTTAAGTCCCGCAACGAGCGCAACCCTTGATCTTAGTTGCCATCATTAAGTTGGGCACTC TAAGGTGACTGCCGGTGACAAACCGGAGGAAGGTGGGGATGACGTCAAATCATCATGCCCCTTA TGACCTGGGCTACACACGTGCTACAATGGACGGTACAAAGAGCTGCAAGACCGCGAGGTGGAGC TAATCTCATAAAACCGTTCTCAGTTCGGATTGTAGGCTGCAACTCGCCTACATGAAGCTGGAAT CGCTAGTAATCGCGGATCAGCATGCCGCGGTGAATACGTTCCCGGGCCTTGTACACACCGCCCG 65 TCACACCACGAGAGTTTGTAACACCCGAAGTCGGTGGGGTAACCTTTTTGGAGCCAGCCGCCTA AGGTGGGACAGATGATTGGGGTGAACTCGTAACAAGGTATCCGT 20 CCACGCGTTGGGAGCTCTCCCATATGGTCGACCTGCAGGCGGCCGCGAATTCACTAGTGATTAC AGATACCTTGTGACTAATGCTGAACGCCATCTGTCCCACCTTAGGTGGCTGGCTCCCGAATGTA ACCCCACGTTGAATGCTGGTGAGATTCAGCATGCAGAGGGCGGAAAGTCGTAATAACCACTGTA GCAGCTGATGAAATACGGACGTGATACTGGACGGAAGTACCATAATCCGAGTGTGGTCTCTCAC GCTTTGTAACCGACAGTGAGGGTTACAGAGCGTGAGAGATCACACTCGCCTAGATTGTTCTGGT AGTATCTCGTACGCATTTCATGAGTTACACAGTGGAATTCCACTTTCCTCTTCTGCACTCAAGT CTCCCAGTTTACAGCACTCCCTCCTGGGGATGAGCCGTGGGCTTTCACATCAGAATTAAGAAAC CACGGGGCGCCGCACAAGCGCCCAATCATTCCGGATAACGCGAAGCCAACGCGAAGAACCCGCG CAGGTTGGCACATCCTTAGAAAACCCTAGAGATAGGGCTTCTCGTCAAGGAGCCAGCTTATTCA ACTAGCAATGGTTGTTCCCTAACAACAGAGTGAGATGACCGGAAAACCTTCATCACTCGCGCGG CCTTGGTTCGTCAGACCCTTCTCCAATGTGGACGATTCCAGGTGGCTGCCTCCGGCAGGAGTGA GGAAGGTGTGTCAGTCGCAAAATGATCGATCACCCTATCAGGTGGGCTACGCATGGTTTCCAAG GGGAGCCGTTACGTCACCCAAGAGCTAGAGGGGGAGGGGTCCTTCCATAAGTGACATCAGAAGC GACCGTTCAATTTAACTCCATGCAGTTCAAAATATTATCCTAGATTAGCCGGATGTTTCCCGGA GTTATCCCAGTTTTATGGGCCGGTGTACACACGTGTTAGTCACCCCTCGAGAGTTAAAACACCC GAGCAAGGTGGGGATCCATTCGCTAGCCAGCCATGTATTGGGCGGACAGCCAGCGGGGTCATGA GTCACGATCAATCTGT 22 CGCCATGGCGGCCGCGGGATTCGATTACAGTTTGATCGTGGCTCATGACCCCGCTGGCTGTCCC CCTTATACATGGCTGGCTCCCAAGAAGNCGGAAAGTGGTAATACCACTGAGTAGCTGATGGATT ACGGACGTGGACTGGAGGGAGTATCAGAGGCGAATGTCGTCTTCACGCTCTGGAGCTGACAGTG TCACTCGAAGAGCGTGAGAGAGCGCACAGGACTAGATAGTTCTGGTAGTATCTCTACGCATCGA TGAGTTACTACAGTGTTAGAGGGTTTCCTCCTTTTCACTCAAGTGTTAACAGCATTACAGCACT CCCTCCTGGGGAGTAGCGGGTGCAAGGCACATCTCAAAGGAGATTGACGGGGGCGCCGCACAAG CGCTGGAGCATTCGGTTTAATTCGAAGCAACGCGAAGAACCTTACGCAGGTCTTGACATCCTTA GAAAACCCTAGAGATAGGGGTTCTCTTTCGGGAGCCAGAGTGACAGGTGGTGCATGGTTGTTGT 66 CAGACAGCAGAGTGAGATGACGGGAAAAGTCTCGTCACGCGCGCGACCCTGGTTCTTCGTACCC TTCTCCAAGTTGGACGATTTCAGGTGACTGCCTCCGACAAGCGTGAGGAAGGTGTGGCAGTCGC AAAATCATCAATCCCCTTATCAGGTGGGCTACGCATGGTTTCCAAGGGGAGGTACAACGACACG CAAGACCGAGAGGGGGAGGTAATCTTATAAAACCGTCATCAGTTCGGACTGTAGGATGCAACTC CATGCCATGAAGCTGGAATTCCTAGTAAAGTCGGATCATCCCGGAGTTATGAACAGTTTTATGG GCCGGTGTACACACGTGTTAGTCACACCTCGAGAGTTTGTAACAATAAGAGCAAGGTGGGGTAA CCTTTTTGGAGCCAGCCACCTAAGGTGGGACAGCCAGCGGGGAGCATTAGTCACAAGGTATCTG TAATCACTAGTGAATTCGCGGCCGCCTGCAGGTCGACCATATGGGAGAGCTCCCAACGCGT 67 AGAGTTTGATCCTGGCTCAGGACGAACGCTGGCGGCGTGCCTAATACATGCAAGTCGAGCGGAC AGAAGGGAGCTTGCTCCCGGATGTTAGCGGCGGACGGGTGAGTAACACGTGGGTAACCTGCCTG TAAGACTGGGATAACTCCGGGAAACCGGAGCTAATACCGGATAGTTCCTTGAACCGCATGGTTC AAGGATGAAAGACGGTTTCGGCTGTCACTTACAGATGGACCCGCGGCGCATTAGCTAGTTGGTG AGGTAACGGCTCACCAAGGCGACGATGCGTAGCCGACCTGAGAGGGTGATCGGCCACACTGGGA CTGAGACACGGCCCAGACTCCTACGGGAGGCAGCAGTAGGGAATCTTCCGCAATGGACGAAAGT CTGACGGAGCAACGCCGCGTGAGTGATGAAGGTTTTCGGATCGTAAAGCTCTGTTGTTAGGGAA GAACAAGTGCAAGAGTAACTGCTTGCACCTTGACGGTACCTAACCAGAAAGCCACGGCTAACTA CGTGCCAGCAGCCGCGGTAATACGTAGGTGGCAAGCGTTGTCCGGAATTATTGGGCGTAAAGGG CTCGCAGGCGGTTTCTTAAGTCTGATGTGAAAGCCCCCGGCTCAACCGGGGAGGGTCATTTGGA AACTGGGAAACTTGAGTGCAGAAGAGGAGAGTGGAATTCCACGTGTAGCGGTGAAATGCGTAGA GATGTGGAGGAACACCAGTGGCGAAGGCGACTCTCTGGTCTGTAACTGACGCTGAGGAGCGAAA GCGTGGGGAGCGAACAGGATTAGATACCCTGGTAGTCCACGCCGTAAACGATGAGTGCTAAGTG TTAGGGGGTTTCCGCCCCTTAGTGCTGCAGCTAACGCATTAAGCACTCCGCCTGGGGAGTACGG TCGCAAGACTGAAACTCAAAGGAATTGACGGGGGCCCGCACAAGCGGTGGAGCATGTGGTTTAA TTCGAAGCAACGCGAAGAACCTTACCAGGTCTTGACATCCTCTGACAACCCTAGAGATAGGGCT TTCCCTTCGGGGACAGAGTGACAGGTGGTGCATGGTTGTCGTCAGCTCGTGTCGTGAGATGTTG GGTTAAGTCCCGCAACGAGCGCAACCCTTGATCTTAGTTGCCAGCATTCAGTTGGGCACTCTAA GGTGACTGCCGGTGACAAACCGGAGGAAGGTGGGGATGACGTCAAATCATCATGCCCCTTATGA CCTGGGCTACACACGTGCTACAATGGACAGAACAAAGGGCTGCGAGACCGCAAGGTTTAGCCAA TCCCACAAATCTGTTCTCAGTTCGGATCGCAGTCTGCAACTCGACTGCGTGAAGCTGGAATCGC TAGTAATCGCGGATCAGCATGCCGCGGTGAATACGTTCCCGGGCCTTGTACACACCGCCCGTCA 67 CACCACGAGAGTTTGCAACACCCGAAGTCGGTGAGGTAACCTTTATGGAGCCAGCCGCCGAAGG TGGGGCAGATGATTGGGGTGAACTCGTAACAAGGTATCCGT 87 AGAGTTTGATCCTGGCTCAGGACGAACGCTGGCGGCGTGCCTAATACATGCAAGTCGAGCGGAC AGAAGGGAGCTTGCTCCCGGATGTTAGCGGCGGACGGGTGAGTAACACGTGGGTAACCTGCCTG TAAGACTGGGATAACTCCGGGAAACCGGAGCTAATACCGGATAGTTCCTTGAACCGCATGGTTC AAGGATGAAAGACGGTTTCGGCTGTCACTTGCAGATGGACCCGCGGCGCATTAGCTAGTTGGTG GGGTAATGGCTCACCAAGGCGACGATGCGTAGCCGACCTGAGAGGGTGATCGGCCACACTGGGA CTGAGACACGGCCCAGACTCCTACGGGAGGCAGCAGTAGGGAATCTTCCGCAATGGACGAAAGT CTGACGGAGCAACGCCGCGTGAGTGATGAAGGTTTTCGGATCGTAAAGCTCTGTTGTTAGGGAA GAACAAGTGCGAGAGTAACTGCTCGCACCTTGACGGTACCTAACCAGAAAGCCACGGCTAACTA CGTGCCAGCAGCCGCGGTAATACGTAGGTGGCAAGCGTTGTCCGGAATTATTGGGCGTAAAGGG CTCGCAGGCGGTTTCTTAAGTCTGATGTGAAAGCCCCCGGCTCAACCGGGGAGGGTCATTGGGA AACTGGGAAACTTGAGTGCAGAAGAGGAGAGTGGAATTCCACGTGTAGCGGTGAAATGCGTAGA GATGTGGAGGAACACCAGTGGCGAAGGCGACTCTCTGGTCTGTAACTGACGCTGAGGAGCGAAA GCGTGGGGAGCGAACAGGATTAGATACCCTGGTAGTCCACGCCGTAAACGATGAGTGCTAAGTG TTAGGGGGTTTCCGCCCCTTAGTGTTGCAGCTAACGCATTAAAGCACTCCGCCTGGGGAGTACG GTCGCAAGACTGAAACTCAAAGGAATTGACGGGGGCCCGCACAAGCGGTGGAGCATGTGGTTTA ATTCGAAGCAACGCGAAGAACCTTACCAGGTCTTGACATCCTCTGACAACCCTAGAGATAGGGC TTTCCCTTCGGGGACAGAGTGACAGGTGGTGCATGGTTGTCGTCAGCTCGTGTCGTGAGATGTT GGGTTAAGTCCCGCAACGAGCGCAACCCTTGATCTTAGTTGCCAGCATTCAGTTGGGCACTCTA AGGTGACTGCCGGTGACAAACCGGAGGAAGGTGGGGATGACGTCAAATCATCATGCCCCTTATG ACCTGGGCTACACACGTGCTACAATGGACAGAACAAAGGGCTGCAAGACCGCAAGGTTTAGCCA ATCCCATAAATCTGTTCTCAGTTCGGATCGCAGTCTGCAACTCGACTGCGTGAAGCTGGAATCG CTAGTAATCGCGGATCAGCATGCCGCGGTGAATACGTTCCCGGGCCTTGTACACACCGCCCGTC ACACCACGAGAGTTTGCAACACCCGAAGTCGGTGAGGTAACCTTTATGGAGCCAGCCGCCGAAG GTGGGGCAGATGATTGGGGTGAACTCGTAACAAGGTATCCGT 92 68 AGCTCTCCCATATGGTCGACCTGCAGGCGGCCGCGAATTCACTAGTGATTAGAGTTTGATCCTG GCTCAGGACGAACGCTGGCGGCGTGCCTAATACATGCAAGTCGAGCGGACAGATGGGAGCTTGC TCCCTGATGTTAGCGGCGGACGGGTGAGTAACACGTGGGTAACCTGCCTGTAAGACTGGGATAA CTCCGGGAAACCGGGGCTAATACCGGATGGTTGTCTGAACCGCATGGTTCAGACATAAAAGGTG GCTTCGGCTACCACTTACAGATGGACCCGCGGCGCATTAGCTAGTTGGTGAGGTAACGGCTCAC CAAGGCGACGATGCGTAGCCGACCTGAGAGGGTGATCGGCCACACTGGGACTGAGACACGGCCC AGACTCCTACGGGAGGCAGCAGTAGGGAATCTTCCGCAATGGACGAAAGTCTGACGGAGCAACG CCGCGTGAGTGATGAAGGTTTTCGGATCGTAAAGCTCTGTTGTTAGGGAAGAACAAGTGCCGTT CAAATAGGGCGGCACCTTGACGGTACCTAACCAGAAAGCCACGGCTAACTACGTGCCAGCAGCC GCGGTAATACGTAGGTGGCAAGCGTTGTCCGGAATTATTGGGCGTAAAGGGCTCGCAGGCGGTT TCTTAAGTCTGATGTGAAAGCCCCCGGCTGCTTCAACCGGGGAGTATCATTGGAAACTGGGGAT GTTGAGTGCAGAAGAGGAGAAGTGAAATTACACGTGTAGCGGTGAAATGCGTAGAGATGTGGAG GAACATCAGTGGCGAAGGCGACTCTGTGGTCTGTACCTGATGCTGAGGAGCGAAAGCGTGGAGA GCGAACAGGATTAGATACCTTGGTAGTCCACGCCGTAAACGATGAGTGATAAGTGTTAGGTGGT TTCCGCCCTCTTAGTGCTGCAGCTAACGCATTAAGCACTCCGCCTGGGGAGTACGGTCGCAAGA CTGAAACTCAAAGGAATTGACGGGGGCCCGCACAAGCGGTGGAGCATGTGGTTTAATTCGAAGC AACGCGAAGAACCTTACCAGGTCTTGACATCCTCTGACAATCCTAGAGATAGGACGTCCCCTTC GGGGGCAGAGTGACAGGTGGTGCATGGTTGTCGTCAGCTCGTGTCGTGAGATGTTGGGTTAAGT CCCGCAACGAGCGCAACCCTTGATCTTAGTTGCCAGCATTCAGTTGGGCACTCTAAGGTGACTG CCGGTGACAAACCGGAGGAAGGTGGGGATGACGTCAAATCATCATGCCCCTTATGACATGGGGT ACACACGTGCTACAATGGACAGAACAAAGGGCAGCGAGACCGCGAGGTTAAGCCAATCCCACAA ATCTGTTCTCAGTTCGGATCGCAGTCTGCAACTCGACTGCGTGAAGCTGGAATCGCTAGTAATC GCGGATCAGCATGCCGCGGTGAATACGTTCCCGGGCCTTGTACACACCGCCCGTCACACCACGA GAGTTTGTAACACCCGAAGTCGGTGAGGTAACCTTTATGGAGCCAGCCGCCGAAGGTGGGACAG ATGATTGGGGTGAACTAGTAACAGAGGTATCCGTT 95 AGAGTTTGATCCTGGCTCAGGACGAACGCTGGCGGCGTGCCTAATACATGCAAGTCGAGCGGAC AGATGGGAGCTTGCTCCCTGATGTTAGCGGCGGACGGGTGAGTAACACGTGGGTAACCTGCCTG TAAGACTGGGATAACTCCGGGAAACCGGGGCTAATACCGGATGGTTGTTTGAACCGCATGGTTC AGACATAAAAGGTGGCTTCGGCTACCACTTACAGATGGACCCGCGGCGCATTAGCTAGTTGGTG AGGTAACGGCTCACCAAGGCGACGATGCGTAGCCGACCTGAGAGGGTGATCGGCCACACTGGGA 69 CTGAGACACGGCCCAGACTCCTACGGGAGGCAGCAGTAGGGAATCTTCCGCAATGGACGAAAGT CTGACGGAGCAACGCCGCGTGAGTGATGAAGGTTTTCGGATCGTAAAGCTCTGTTGTTAGGGAA GAACAAGTGCCGTTCAAATAGGGCGGCACCTTGACGGTACCTAACCAGAAAGCCACGGCTAACT ACGTGCCAGCAGCCGCGGTAATACGTAGGTGGCAAGCGTTGTCCGGAATTATTGGGCGTAAAGG GCTCGCAGGCGGTTTCTTAAGTCTGATGTGAAAGCCCCCGGCTCAACCGGGGAGGGTCATTTGG AAACTGGGGAACTTGAGTGCAGAAGAGGAGAGTGGAATTCCACGTGTAGCGGTGAAATGCGTAG AGATGTGGAGGAACACCAGTGGCGAAGGCGACTCTCTGGTCTGTAACTGACGCTGAGGAGCGAA AGCGTGGGGAGCGAACAGGATTAGATACCCTGGTAGTCCACGCCGTAAACGATGAGTGCTAAGT GTTAGGGGGTTTCCGCCCCTTAGTGCTGCAGCTAACGCATTAAGCACTCCGCCTGGGGAGTACG GTCGCAAGACTGAAACTCAAAGGAATTGACGGGGGCCCGCACAAGCGGTGGAGCATGTGGTTTA ATTCGAAGCAACGCGAAGAACCTTACCAGGTCTTGACATCCTCTGACAATCCTAGAGATAGGAC GTCCCCTTCGGGGGCAGAGTGACAGGTGGTGCATGGTTGTCGTCAGCTCGTGTCGTGAGATGTT GGGTTAAGTCCCGCAACGAGCGCAACCCTTGATCTTAGTTGCCAGCATTCAGTTGGGCACTCTA AGGTGACTGCCGGTGACAAACCGGAGGAAGGTGGGGATGACGTCAAATCATCATGCCCCTTATG ACCTGGGCTACACACGTGCTACAATGGACAGAACAAAGGGCAGCGAAACCGCGAGGTTAAGCCA ATCCCACAAATCTGTTCTCAGTTCGGATCGCAGTCTGCAACTCGACTGCGTGAAGCTGGAATCG CTAGTAATCGCGGATCAGCATGCCGCGGTGAATACGTTCCCGGGCCTTGTACACACCGCCCGTC ACACCACGAGAGTTTGTAACACCCGAAGTCGGTGAGGTAACCTTTTAGGAGCCAGCCGCCGAAG GTGGGACAGATGACTGGGGTGAACTCGTAACAAGGTATCCGT 97 AGAGTTTGATCCTGGCTCAGGACGAACGCTGGCGGCGTGCCTAATACATGCAAGTCGAGCGGAC AGAAGGGAGCTTGCTCCCGGATGTTAGCGGCGGACGGGTGAGTAACACGTGGGTAACCTGCCTG TAAGACTGGGATAACTCCGGGAAACCGGAGCTAATACCGGATAGTTCCTTGAACCGCATGGTTC AAGGATGAAAGACGGTTTCGGCTGTCACTTACAGATGGACCCGCGGCGCATTAGCTAGTTGGTG GGGTAATGGCTCACCAAGGCGACGATGCGTAGCCGACCTGAGAGGGTGATCGGCCACACTGGGA CTGAGACACGGCCCAGACTCCTACGGGAGGCAGCAGTAGGGAATCTTCCGCAATGGACGAAAGT CTGACGGAGCAACGCCGCGTGAGTGATGAAGGTTTTCGGATCGTAAAGCTCTGTTGTTAGGGAA GAACAAGTGCGAGAGTAACTGCTCGCACCTTGACGGTACCTAACCAGAAAGCCACGGCTAACTA CGTGCCAGCAGCCGCGGTAATACGTAGGTGGCAAGCGTTGTCCGGAATTATTGGGCGTAAAGGG CTCGCAGGCGGTTTCTTAAGTCTGATGTGAAAGCCCCCGGCCTCAACCCGGGGAGGGTCATTGA AAACTGGGAAACTTGAGTGCAGAAGAGGAGAGTGGAATTCCACGTGTAGCGGTGAAATGCGTAG 70 AGATGTGGAGGAACACCAGTGGCGAAGGCGACTCTCTGGTCTGTAGCTGACACTGAGGAGCGAA AGCGTGGGGAGCGAACAGGATTAGATACCCTGGTAGTCCACGCCGTAAACGATGAGTGCTAAGT GTTAGGGGGTTTCCGCCCCTTAGTGCTGCAGCTAACGCATTAAGCACTCCGCCTGGGGAGTACG GTCGCAAGACTGAAACTCAAAGGAATTGACGGGGGCCCGCACAAGCGGTGGAGCATGTGGTTTA ATTCGAAGCAACGCGAAGAACCTTACCAGGTCTTGACATCCTCTGACAACCCTAGAGATAGGGC TTTCCCTTCGGGGACAGAGTGACAGGTGGTGCATGGTTGTCGTCAGCTCGTGTCGTGAGATGTT GGGTTAAGTCCCGCAACGAGCGCAACCCTTGATCTTAGTTGCCAGCATTCAGTTGGGCACTCTA AGGTGACTGCCGGTGACAAACCGGAGGAAGGTGGGGATGACGTCAAATCATCATGCCCCTTATG ACCTGGGCTACACACGTGCTACAATGGACAGAACAAAGGGCTGCAAGACCGCAAGGTTTAGCCA ATCCCATAAATCTGTTCTCAGTTCGGATCGCAGTCTGCAACTCGACTGCGTGAAGCTGGAATCG CTAGTAATCGCGGATCAGCATGCCGCGGTGAATACGTTCCCGGGCCTTGTACACACCGCCCGTC ACACCACGAGAGTTTGCAACACCCGAAGTCGGTGAGGTAACCTTTATGGAGCCAGCCGCCGAAG GTGGGGCAGATGATTGGGGTGAACTCGTAACAAGGTATCCGT