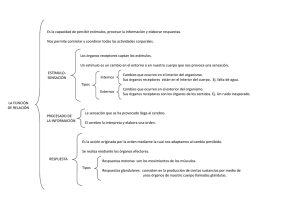
Módulo 1 Curso Pharma E Consultores
Anuncio

Derechos reservados de autor, prohibida su reproducción parcial o total sin permiso expreso de los autores PHARMA E CONSULTORES ÁREA DE CAPACITACIÓN CONTINUADA CURSO VIRTUAL DE CAPACITACIÓN CONTINUADA EN FARMACOTERAPIA MODULO 3: AVANCES EN FARMACOLOGÍA DEL SISTEMA NERVIOSO La fantasía nunca arrastra a la locura, lo que arrastra a la locura es precisamente la razón; los poetas no se vuelven locos, pero sí los jugadores de ajedrez. Chesterton 1. ANATOMÍA BÁSICA DEL SISTEMA NERVIOSO (SN) 1.1. ANATOMÍA Y FISIOLOGÍA Está formado aproximadamente por 300 x 109 células que pertenecen a dos tipos con morfología y funciones diferentes; neuronas y células gliales, siendo mayor el número de células gliales que el de neuronas. - LAS NEURONAS. Son células especializadas en conducir estímulos de una célula a otra mediante un código de señales electroquimicas en la cual hay una participación de la membrana celular. Ésta conformada por el cuerpo celular (soma), las dendritas y el axón terminal. - CÉLULAS GLIALES. Las células de gliales son mucho más numerosas que las neuronas. Sus funciones son muchas e importantes, entre ellas ejercer un papel estructural en el sistema nervioso, modificar la actividad eléctrica (comunicación) de las neuronas, intervenir en el reciclaje de los neurotransmisores, eliminar los fragmento de neurona después de su muerte y producir mielina, entre otras. Hay dos tipos de células gliales: la macroglia y la microglia. Algo muy importante de las células gliales, es la capacidad funcional de dividirse y por tanto reproducirse, gran diferencia con las neuronas. Los tumores del sistema nervioso, por ejemplo, se producen por una reproducción anómala de las células gliales. - ASTROSITOS. Tienen formas estrelladas y presentan largas prolongaciones que se extienden hacia las neuronas y hacia los láminas basales que rodean a los capilares sanguíneos (pies terminales), o que separan al tejido nervioso del conjuntivo laxo de la piamadre, constituyendo la glía limitante. - MIELINA. La mielina es una oligoproteina cuya naturaleza depende de su función y lugar de acción; la mielina de los nervios periféricos tiene su origen en la membrana de la célula de Schwann que envuelve al axón. La mielina de las fibras del sistema nervioso central procede de las membranas de los oligodendrocitos (una variedad de células gliales). Habitualmente, los oligodendrocitos mielinizan varios axones, mientras que la célula de Schwann se asocia a un solo axón. Microscópicamente en el tejido neural se identifican una “sustancia gris” y “sustancia blanca”, bioquímica y biológicamente diferentes. A nivel microscópico, la sustancia gris contiene los cuerpos de las células neuronales, mientras que la sustancia blanca es donde se encuentran las fibras neurales o axones. El aspecto “blanco” se debe a la mielina, que cubre estas fibras. 1.2. DIVISIÓN DEL SISTEMA NERVIOSO Funcionalmente el SN puede dividirse en dos compartimientos principales: el sistema nervioso somático transmite información sensitiva (tacto, temperatura, dolor, etc) desde los segmentos corporales y transporta las vías neurales que inervan y controlan el movimiento de los músculos esqueléticos; y el sistema nervioso visceral controla los órganos internos que no están normalmente bajo la influencia de los vasos sanguíneos (la dilatación y la contracción de las pupilas de los ojos, etc). Anatómica y morfológicamente el SN se diferencia en: sistema nervioso central (SNC) (cerebro y la médula espinal), sistema nervioso periférico (SNP) (formado por las neuronas situadas fuera del sistema nervioso central), sistema nervioso autónomo (SNA) (controla la actividad de los componentes viscerales del cuerpo humano. Anatómicamente el SNA tiene dos componentes principales: el sistema nervioso simpático o adrenérgico y el parasimpático o colinérgico) y el sistema neuroendocrino (formado por glándulas endocrinas que liberan mensajes químicos en el interior del organimos a través de hormonas). 2. BASES BIOLÓGICAS NEUROTRANSMISIÓN. Y FISIOLÓGICAS DEL SISTEMA NERVIOSO Y 2.1. SISTEMAS DE NEUROTRANSMISIÓN CEREBRAL El termino neurotransmisión es el proceso por el cual una neurona libera un neurotransmisor (NT) y éste se une a una proteína receptora de membrana de la neurona postináptica induciendo o generándose en ella, una serie de cambios eléctricos y bioquímicos. La neurotransmisión es el fenómeno esencial de comunicación entre neuronas y sobre él se basan todas las sinapsis, contacto especializados entre las neuronas. Estas sinapsis son sitios de unión donde el axón o alguna otra parte de una de las células presinápticas terminan sobre las dendritas, el soma o el axón de otra célula o en algunos casos, sobre una célula muscular o glandular (la célula o “target” posináptica). La transmisión en la mayor parte de las uniones sinápticas es de naturaleza química, a su vez la conducción nerviosa es un fenómeno eléctrico que causa en el axón presináptico la generación de un potencial de acción que se acompaña del evento bioquímico de síntesis, almacenamiento y secreción de un neurotransmisor - COMPONENTES DE LA SINAPSIS. El funcionamiento del SN se basa en un sistema de comunicación rápido y preciso. Una neurona establece un promedio de 1.000 sinapsis y recibe muchas más. Si el cerebro posee un número estimado de 1011 neuronas, el número de conexiones será de 1014, es decir, 100 billones de sinapsis. En la sinapsis el axón y la dendrita nunca se tocan. Siempre hay un pequeño vació llamado hendidura sináptica. Cuando la señal eléctrica llega a un terminal nervioso, hace que el nervio libere neurotransmisores. Los neurotransmisores son agentes químicos que viajan una corta distancia hasta las dendritas más próximas. A la neurona que libera el neurotransmisor se le llama neurona presináptica. A la neurona receptora de la señal se le llama neurona postsináptica. Los eventos de carácter físico y bioquímico que se presentan en una sinapsis son: • • • • • • • • • • • • Tabla 1. Eventos físicos y bioquímicos de una Sinapsis Despolarización de la neurona presináptica con la generación de un Potencial presináptico Ingreso de un precursor Síntesis del neurotransmisor Almacenamiento del Neurotransmisor en vesículas ó gránulos Liberación del neurotransmisor Interacción postsináptica con generación de un potencial postsináptico Transducción de señales en la célula postsináptica hasta la producción de un efecto biológico Modulación presináptica de la liberación del NT Difusión del NT Metabolismo sináptico o extraneuronal del NT Recaptación del NT Metabolismo intraneuronal del NT - PRECURSORES. Son sustancias precursoras de NT las siguientes: Fenilanina y tirosina: Colina: Triptofano: Acido Glutámico: para para para para Dopamina, Noradrenalina y Adrenalina Acetilcolina Serotonina el GABA - POTENCIALES PRESINÁPTICOS. La membrana en reposo mantiene un gradiente de potencial eléctrico de -70mv, el signo negativo se debe a que el citoplasma intracelular está cargado negativamente con respecto al exterior. Alcanzado un potencial crítico denominado "umbral", las cargas positivas como las generadas por el ingreso de sodio producen un efecto que obliga al potencial de membrana a cambiar de signo. Es decir, el interior de la célula se torna positivo con respecto al exterior, al cabo de 1 ms, la permeabilidad del sodio decae y el potencial de membrana retorna a -70mv, su valor de reposo. Los potenciales de acción, son señales de baja frecuencia conducidas en forma muy lenta, estos no pueden saltar de una célula a otra, la comunicación entre neuronas viene siempre mediada por transmisores químicos que son liberados en las sinapsis. - SÍNTESIS DE NEUROTRANSMISOR. Algunos NTs se sintetizan directamente en las terminaciones nerviosas a partir de sus precursores por acción de las enzimas que se han sintetizado en el soma y se han transportado a las terminaciones nerviosas. En función de la naturaleza del neurotransmisor se puede producir en el soma o en las terminaciones nerviosas. - ALMACENAMIENTO Y LIBERACIÓN DE NEUROTRANSMISOR. El NT puede almacenarse en vesículas sinápticas o gránulos, pequeños reservorios globulares que contienen sensores en su pared exterior y permiten que el neurotransmisor se libere en sitios específicos de la terminal presináptica debido a los movimientos iónicos generados por el potencial presináptico. Cuando un potencial de acción llega al terminal de un axón son liberados NTs alojados en las vesículas o gránulos y después son vertidos en la hendidura, la cual tiene unos 20 nanómetros de diámetro que separa la membrana presináptica de la postsináptica. Durante el desarrollo del potencial de acción presináptico, penetran iones de calcio en el terminal nervioso, su movimiento constituye una señal para la exocitosis sincronizada del NT. - INTERACCIÓN POSTSINÁPTICA. El NT liberado en la hendidura sináptica interacciona directamente con las moléculas del receptor en la membrana postsináptica. Mediante este tipo de interacción se abren un gran número de canales iónicos específicos que permiten el flujo de una corriente eléctrica, transportada por iones cargados a través de la membrana postsináptica lo que afecta al estado electroquímico de la membrana en el área inmediata al canal. De esta forma la excitabilidad eléctrica de esta pequeña porción de membrana puede aumentar o disminuir mediante despolarización o hiperpolarización de la misma. Las alteraciones eléctricas individuales de la membrana postsináptica ejercen un efecto en el potencial de membrana de la neurona, que puede llevar a la generación del impulso nervioso postsináptico. Cada sinapsis produce sólo un pequeño efecto, para determinar la intensidad (frecuencia de los potenciales de acción) de la respuesta cada neurona ha de integrar continuamente hasta unas 1000 señales sinápticas, que se suman en el soma o cuerpo de la célula. -POTENCIALES POSTSINÁPTICOS. La generación de un segundo potencial como consecuencia de la interacción del NT con los receptores postsinápticos puede producir eventos eléctricos (ingreso de iones) y bioquímicos (estímulo o inhibición de proteinas acopladoras de moléculas diana) con la subsecuente producción de segundos mensajeros y eventos biológicos que identifican una función especial. - RECEPTORES PRESINÁPTICOS Y POSTSINÁPTICOS. Los receptores son proteínas de membrana que se localizan en las membranas postsinápticas y presinápticas. El NT induce en su contacto con los receptores postsinápticos cambios estructurales en la proteína receptora (que puede estar unida a canales iónicos o a moléculas metabotrópicas acopladas a proteína G) que alteran la permeabilidad dando lugar al denominado potencial de placa o potencial sináptico, que cuando alcanza el voltaje umbral genera un potencial de acción. Es también trascendente la activación de los receptores específicos de membrana, situados en la neurona presináptica, que responden a la cantidad del neurotransmisor en la sinapsis con la misma afinidad que los receptores postsinápticos. Estos receptores, a diferencia de los postsinápticos, no controlan canales iónicos. Sin embargo, regulan la movilización de las vesículas, intervienen esencialmente en la disponibilidad del neurotransmisor y, por tanto, en su síntesis y en su liberación. - DIFUSIÓN. Al hablar de difusión del NT en la sinapsis permite incluir el concepto de producción de efectos a grandes distancias del lugar de secreción, y que afecta a grupos neuronales y otras áreas, moléculas y órganos. Este concepto es fundamental en la secreción de neuromoduladores y neuropéptidos más que de NTs clásicos, y aunque los propios neuropéptidos de secreción sináptica pueden actuar como auténticos neurotransmisores, la propia estructura de la molécula, así como el no metabolizarse “in situ” favorece su difusión, y alejamiento de su actuación local, por lo que pueden ser considerados como neuromoduladores. - METABOLISMO E INACTIVACIÓN. Esta biotransformación puede hacerse a nivel intraneuronal (célula presináptica) o a nivel extraneuronal (en el espacio sináptico o a distancia). El objetivo de este metabolismo es la inactivación del NT y la disipación de sus efectos biológicos. En algunos casos la inactivación no es metabólica, se realiza por la recaptación del NT por la neurona presináptica disminuyendo las concentraciones en la sinapsis y los impulsos postsinápticos. 2.2. NEUROTRANSMISORES Se distinguen tres tipos de neurotransmisores cerebrales: aminas biógenas, aminoácidos y polipéptidos. Entre ellos existen diferencias en su estructura molecular, distribución cerebral e implicación funcional, que viene determinada fundamentalmente por las proyecciones regionales de las neuronas sintetizadoras. Tabla 2. Clasificación de los Neurotransmisores Aminoácidos Aminas biógenas Polipeptidos Excitadores: Acetilcolina (Ach) Opiódes Ac. Glutámico Catecolaminas Sustancia P Ac. Aspartico Dopamina Colecistocinina Inhibidores: Noradrenalina Oxitocina Glicina Adrenalina Vasopresina Somastostaina GABA Indolamina Serotonina Otros Tomado y adaptado de: Consejo general de colegios oficiales de farmacéuticos de España. Actualización en Farmacoterapia. España. 2002. - Sistema adrenérgico. Este sistema neurotransmisor tiene como mediadores a la noradrenalina (el NT de tipo monoamina más abundante en el cerebro) y la adrenalina. Los núcleos noradrenérgicos más importantes se localizan en el tronco del encéfalo. Se reconocen dos grupos de receptores adrenérgicos con varios subtipos ∝ (alfa) y β (beta): (∝1a-c, ∝2a-c, ∝3) Figura 1. Proyecciones monoaminérgicas del SNC - Sistema dopaminérgico. El neurotransmisor que da nombre a este sistema es la dopamina. Se han clasificado dos grupos de receptores dopaminérgicos con una distribución y afinidad por la dopamina diferencial: D1 y D5; D2, D3, D4. Figura 2. Proyecciones dopaminérgicas del SNC Las principales vías dopaminérgicas son la vía nigroestriatal, tuberoinfundibular y corticomesolímbica con diferentes implicaciones biológicas y terapéuticas. La vía nigroestriada está relacionada con el control motor (tono, postura y movimiento) y desde el punto de vista clínico, está implicada en la presencia de síntomas extrapiramidales en la enfermedad de Parkinson y durante el tratamiento con antipsicóticos (APS). La segunda vía tuberoinfundibular se extiende desde los cuerpos celulares del hipotálamo hasta el infundíbulo y la hipófisis anterior. La acción de la dopamina es inhibidora de la liberación de prolactina a nivel de la hipófisis anterior. – Sistema Serotoninérgico. Los núcleos serotoninérgicos se localizan en el rafe y en locus ceruleus del troncoencéfalo proyectándose al sistema límbico, ganglios basales y córtex cerebral. Se han identificado siete tipos de receptores con afinidad para la serotonina con un total de 14 subtipos. – Sistema colinérgico. El neurotransmisor mediador es la acetilcolina. Las neuronas presinápticas se sitúan en el núcleo basal con proyecciones al córtex y sistema límbico, y en el sistema retícular cuyos axones se extienden hasta el córtex, sistema límbico, hipotálamo y tálamo. Diferenciamos dos tipos esenciales de receptores colinérgicos: muscarínicos y nicotínicos. – Sistema histaminérgico. Los cuerpos neuronales que liberan histamina se localizan en el sistema reticular mesencefálico con proyecciones ascendentes hasta el córtex, sistema límbico y tálamo. Los receptores histaminérgicos clasificados son tres: H1, H2 y H3, si bien es el receptor tipo H1 el que media la mayor parte de las acciones centrales (regulación de los ciclos vigiliasueño y del apetito). Las acciones de bloqueo histaminérgico de ciertos fármacos generan efectos colaterales como sedación, aumento de apetito y de peso. – Sistema gabaérgico. El ácido γ-aminobutírico (GABA) es el neurotransmisor inhibidor más abundante en el encéfalo. Las neuronas gabaérgicas se distribuyen de forma extensa en córtex, sistema límbico y en región troncoencefálica. El receptor GABAA es un receptor inhibidor sobre el que actúan las benzodiacepinas y otros fármacos con acción sedante. El GABAB también ejerce una acción inhibitoria a nivel presináptico. – Sistema glutamatérgico. De forma opuesta al sistema gabaérgico, el glutamatérgico es el sistema excitador central por excelencia. Ampliamente distribuido, se le relaciona con los procesos de degeneración y muerte neuronal presente en algunos trastornos neurológicos (pe, enfermedad de Huntington). En el ámbito psiquiátrico, la estimulación de los receptores NMDA (N-metil-D-aspartato) se asocia a un mecanismo de potenciación a largo plazo que se propone como fenómeno subyacente al proceso de aprendizaje asociativo. Los efectos psicomiméticos de sustancias como la fenciclidina están mediados por el bloqueo de los receptores NMDA del glutamato. – Neuropéptidos. Desde hace tiempo se conoce el papel como neurotransmisor de algunos péptidos o proteínas de bajo peso molecular. En la actualidad se conocen unos 300 péptidos que actúan en el cerebro humano. A diferencia de los neurotransmisores clásicos, cuya síntesis tiene lugar en las terminaciones sinápticas, los neuropéptidos se sintetizan en el citoplasma celular bajo la regulación de la síntesis proteica, es decir, existe una regulación genética de su síntesis. Estas moléculas son transportadas a la terminación presináptica en las vesículas derivadas del retículo endoplásmico y liberadas al espacio sináptico. La actividad de estos neuropéptidos termina con la acción de las peptidasas. La mayoría de las moléculas neuropeptídicas sintetizadas son precursores que se escinden (dividen) para dar lugar al péptido activo. Estas sustancias pueden ejercer una acción neurotransmisora típica o neuromoduladora sobre otros sistemas. Entre los neuropéptidos más estudiados se encuentran los opioides endógenos. Se han identificado tres tipos: endorfinas, encefalinas y dinorfinas. Otros neuropéptidos se han relacionado con la fisiopatología de la esquizofrenia (sustancia P, colecistocinina), la regulación del estado de ánimo (vasopresina, oxitocina, sustancia P) y la demencia tipo Alzheimer (sustancia P, somatostatina). 2.3. RECEPTORES Los receptores de NTs son proteínas de membrana sobre las que se fijan los neurotransmisores para ejercer su función. El rol de los receptores en la neurotransmisión es fundamental ya que más allá de las propiedades de las sustancias neurotransmisoras, la naturaleza de las respuestas neuronales a un determinado neurotransmisor depende de la presencia de un receptor. La principal función de un receptor postsináptico es alterar el potencial de membrana aumentando o disminuyendo la probabilidad de que se genere un potencial de acción. Básicamente, se diferencian dos tipos de receptores: unos que están unidos a proteínas G que abren canales (receptores dependientes ligados a proteínas G (pe, receptores adrenérgicos) y otros que son parte integrante de un complejo receptor-canal iónico (receptores dependientes de ligando; p. e., los receptores GABAA). Las proteínas G constituyen una familia de proteínas que median en la sensibilidad del receptor y la respuesta neuronal tras la unión del NT-receptor. Estas sustancias tienen tres subunidades (α, β, γ) y están vinculadas al guanosíntrifosfato. La unión NT-receptor provoca la desestibilización de la proteína G, una de cuyas subunidades se convierte en el fragmento activo que participa en la activación o inhibición de la molécula efectora (pe, la enzima adenilciclasa) y la actuación de los segundos mensajeros. De esta manera, la señal del neurotransmisor se traduce en una señal intraneuronal. 3. ANSIOLÍTICOS E HIPNÓTICOS. La ansiedad se define como la vivencia de un sentimiento o sensación de amenaza, tensión y alteraciones psicosomáticas, en ausencia de un peligro real o desproporcionado, en relación con el estímulo desencadenante. Dentro de la ansiedad se incluyen distintos cuadros patológicos, como: - Angustia: Los pacientes suelen estar ocupados en una actividad rutinaria y de repente, les invade un miedo sobrecogedor, con la sensación de que algo horrible les va a ocurrir. La mayor parte de las sensaciones físicas de una crisis de angustia son la manifestación de una hiperestimulación del sistema nervioso autónomo. Estas crisis duran de 5 a 20 minutos. Si el tiempo de la crisis es superior debería valorarse otra situación (ansiedad generalizada, trastorno obsesivo). - Agorafobia: temor patológico a espacios abiertos. La agorafobia es la más resistente de todas las fobias. - Fobia social: Se caracteriza por un miedo irracional a situaciones públicas (hablar en público, comer en público, utilizar baños públicos). Tiene una evolución crónica. Las fobias se pueden agravar o extender si no son tratadas. El pronóstico es bueno si se trata. - Fobia simple: a objetos o situaciones concretas. - Trastorno por estrés postraumático: revivir de forma parcial o total una situación que ha sido estresante. Este acontecimiento es revivido en sueños y pensamientos durante la vigila (flash back). Los síntomas de revivir la experiencia, de evitación y de hipervigilancia persisten más de 1 mes. Tiene evolución crónica. Durante años, el paciente vuelve a experimentar periódicamente el trauma. Tiene un peor pronóstico si existe psicopatología previa. - Trastorno de ansiedad generalizada: ansiedad y preocupaciones excesivas asociadas a una serie de acontecimientos y situaciones, que se observan casi a diario durante seis meses o más. Se presenta en niños y adultos. Su evolución es crónica. Los síntomas disminuyen con la edad - Trastorno Obsesivo Compulsivo (TOC): Se mezclan ideas, imágenes, reflexiones, impulsos, pensamientos intrusos y recurrentes (obsesiones), o patrones de comportamiento o acciones repetitivas (compulsiones). Tiene evolución crónica, con exacerbaciones y remisiones. Pronóstico regular con terapia, aunque algunos casos son intratables En términos generales, la ansiedad transcurre con tensión (temblor, dolor muscular, agitación motora, fatigabilidad), hiperactividad vegetativa (disnea, palpitaciones, hiperhidrosis, mareo inespecífico, náuseas), e hipervigilancia (dificultad de concentración y atención, insomnio, irritabilidad). Puede presentarse un tipo de ansiedad que está presente en la vida cotidiana; el cual, equivale a un adecuado estado de alerta y permite mejorar el rendimiento. Cuando no es excesiva, la ansiedad estimula el aprendizaje y el desempeño funcional de una persona. Sin embargo, cuando sobrepasa este margen, lleva a una sensación de malestar y disminución del rendimiento. No se conocen con exactitud las bases fisiopatológicas de la ansiedad, aunque se plantea que determinadas estructuras del sistema límbico sería activado por la percepción de situaciones amenazantes o de forma errónea evaluadas como amenazantes. Determinados sistemas neurotransmisores participarían en la regulación de tales estructuras límbicas y estarían involucrados en la génesis de la ansiedad (GABA, sistemas noradrenérgico y serotonérgico y determinados neuropéptidos). A la vez, se ven afectados el hipotálamo y su control sobre la médula (ocasionando alteraciones cardiovasculares y gastrointestinales), la hipófisis (alteraciones endocrinas), la corteza motora y la actividad muscular. Una gran mayoría de las investigaciones sobre la neuroquímica de la ansiedad se han centrado en las monoaminas serotonina (5-HT) y noradrenalina (NA), el complejo receptor GABA-benzodiazepinas y un número de compuestos no relacionados entre sí, de los cuales se sabe que provocan cambios en la ansiedad y/o el pánico. No obstante, la mayoría coinciden en la importancia que tienen los sistemas noradrenérgico, serotonérgico y GABAérgico. PROBLEMAS ORGÁNICOS QUE PUEDEN AGRAVAR O PRESENTAR SÍNTOMAS DE ANSIEDAD. 1. Endocrinológicos: hipo o hipertiroidismo, hipoglucemia, insuficiencia adrenal, hiperadrenocorticismo, feocromocitoma, menopausia 2. Cardiovasculares: ICC, insuficiencia coronaria, TEP, arritmias, prolapso mitral 3. Respiratorio: asma, EPOC, neumonías, trastornos de ventilación. 4. Alteraciones metabólicas: diabetes, porfiria, hipoxia, hipocapnia, hipoglucemia hiperpotasemia, hipercapnia, hiponatremia. 5. Neurológico: neoplasia, encefalitis, disfunción vestibular, epilepsia temporal, migrañas.. 6. Intoxicaciones: plomo 7. Hematológicas: anemia, déficit de B12. 8. Otras: infección urinaria en ancianos, síndrome de fatiga crónica, cáncer. 9. Yatrogenia: Antidepresivos (IRS), anticonvulsivantes (Carbamacepina, etosuximida), antimicrobianos (Cefalosporinas, Ofloxacino, Aciclovir, Isoniazida), broncodilatadores (teofilinas, beta 2 agonistas) intoxicación digitálica, estrógenos, insulina en hipoglucemias, AINES (Indometacina), Antihistamínicos, antagonistas del canal del calcio, dopamina, inotrópicos (adrenalina, noradrenalina), Levodopa, corticosteroides, Tiroxina. 10. Drogas: cafeína, anfetaminas, cocaína, alcohol 3.1. FARMACOTERAPIA ANSIOLITICA Tabla 3. Clasificación de fármacos ansiolíticos 1. Ansiolíticos que producen, además, un efecto sedante-hipnótico: a) Benzodiazepinas (BZD) b) Barbitúricos (BB) c) Meprobamato 2. Agonistas parciales de receptores 5-HT1A 3. Bloqueantes de algún componente vegetativo: a) Antihistamínicos b) Antidepresivos c) Beta-bloqueantes 3.1.1 BENZODIACEPINAS - Acciones farmacológicas de las benzodiacepinas Son sustancias potenciadoras de la acción del GABA, a través del cual se produce un influjo de iones cloro al interior de la neurona y su hiperpolarización, con bloqueo de la transmisión. • Acción ansiolítica - Disminuyen la ansiedad y la agresividad - Mejoran la tensión subjetiva - Mejoran los síntomas subjetivos • Acción sedante-hipnótica - Disminuyen el tiempo para conciliar el sueño - Aumentan la duración del sueño • Acción anticonvulsivante • Acción miorrelajante central - Aumentan la inhibición presináptica en médula espinal, FRAA, ganglios basales y cerebelo • Otras acciones - Sobre la agresividad y la memoria - Sobre el sistema cardiovascular - Sobre el sistema respiratorio - Características de Seguridad de las Benzodiacepinas Las sobredosis agudas de benzodiazepinas son mucho menos peligrosas que las de barbitúricos u otros fármacos, debido a que producen depresión del sistema nervioso central, que puede llegar a sueño prolongado e incluso al coma, pero que rara vez conduce a la muerte, ya que los efectos cardiovasculares y depresores respiratorios son poco importantes. El flumazenilo es un antagonista de BZD que contrarresta fácilmente una sobredosis de estos fármacos. El uso terapéutico y crónico produce como síntomas más frecuentes: sedación, somnolencia, ataxia, disartria, incoordinación motora e incapacidad para coordinar movimientos o para responder a estímulos de respuesta rápida. Alteran la capacidad de conducir vehículos y otras habilidades manuales, a veces incluso producen amnesia. En ocasiones, producen conducta agresiva y hostil por desinhibición, o un estado de nerviosismo previo al efecto ansiolítico o sedante. Las de acción corta pueden originar fenómenos ansiosos de rebote. Aunque no está bien establecido si inducen efectos teratógenos, deben ser evitadas durante el embarazo. Las benzodiazepinas tienen tendencia a producir dependencia psicológica y física con síndrome de abstinencia, como lo hacen los barbitúricos y otros hipnóticos. El cuadro que se produce es similar a la ansiedad inicial, aumentando con la dosis y duración del tratamiento. Las benzodiacepinas eliminan el recuerdo de los acontecimientos que tienen lugar mientras dura su acción, un efecto que no se observa con otros depresores del SNC. Esto permite practicar intervenciones quirúrgicas menores sin acumular recuerdos desagradables. 3.1.2. Agonistas 5-HT1A: buspirona Los agonistas parciales de receptores serotonérgicos 5-HT1A (buspirona, gepirona, ipsapirona), a nivel presináptico, inhiben o disminuyen la actividad de neuronas serotonérgicas centrales y a nivel postsináptico pueden actuar como agonistas o como antagonistas. Carecen de acción hipnótica, anticonvulsivante y miorrelajante y, en lugar de sedación, tienden a producir insomnio. No alteran la memoria ni producen trastornos cognitivos o psicomotores. A diferencia de las benzodiazepinas, no interaccionan con el alcohol ni con otros fármacos depresores centrales; tampoco desarrollan dependencia ni tolerancia, ni inducen tendencias al abuso. Como ansiolíticos son algo menos eficaces que las BZD, aunque su principal inconveniente es la lentitud de su acción y que el efecto ansiolítico tarda hasta dos semanas en aparecer, con el inconveniente añadido de resultar prácticamente ineficaces en pacientes que previamente han sido tratados con benzodiazepinas. Presentan menos efectos adversos que los ansiolíticos benzodiazepínicos, pueden producir cierta sensación de mareo, vértigo, cefaleas, sudor, inquietud, nerviosismo y náuseas, que tienden a remitir a lo largo del tratamiento.. 3.1.3. Barbitúricos Los barbituricos son depresores no selectivos del SNC con efectos que oscilan entre la sedación y la reducción de la ansiedad. Actúan en parte potenciando la acción del GABA, pero son menos especificos que las benzodiacepinas. Se ha abandonado su uso como sedantes e hipnóticos. Producen tolerancia y dependecia. 3.1.4. Meprobamato Está indicado en el tratamiento de la ansiedad generalizada, en el insomnio y en la espasticidad. Fue el primer fármaco no barbitúrico utilizado como ansiolítico, con una acción farmacológica similar a la de las benzodiazepinas y con ciertas desventajas: • La acción ansiolítica está más asociada a la hipnótica. • Es inductor del metabolismo de ciertos fármacos. • Produce farmacodependencia con más facilidad. • Tiene mayor capacidad de producir depresión completa, coma y muerte. Produce tolerancia y dependencia física y psicológica, con síndrome de abstinencia similar al de barbitúricos. La tolerancia y dependencia son cruzadas con barbitúricos y alcohol. Las reacciones adversas más importantes son reacciones hematológicas y cuadros de hipersensibilidad cutánea. El carisoprodol es un derivado de meprobamato, se utiliza como miorrelajante. 3.1.4. Ansiolíticos bloqueantes del sistema nervioso autónomo Se incluyen diversos fármacos desde el punto de vista químico y farmacológico. Tienen en común la capacidad de ejercer una acción ansiolítica y sedante y de bloquear las manifestaciones de determinados componentes del sistema nervioso vegetativo. Antidepresivos tricíclicos e inhibidores de la MAO. Son útiles en aquellos trastornos ansiosos cuyo principal síntoma sean los ataques de pánico; el más utilizado es imipramina. Antihistamínicos. De ellos se utilizan, además de doxilamina que posee una importante acción hipnótica y como tal es utilizado en el tratamiento del insomnio, hidroxizina, cuya acción es preferentemente ansiolítica. Estos fármacos se reservan a pacientes antecedentes de adicción, alcoholismo o a personas resistentes a otros tratamientos. β-bloqueantes. Útiles para controlar las manifestaciones somáticas adrenérgicas (palpitaciones, sudoración, temblor, etc.) de la ansiedad. A veces, se utilizan asociados a benzodiazepinas. EL mas utilizado es el propranolol 3.2. REVISIONES DE ENSAYOS CLÍNICOS CONTROLADOS Se reporta que los antidepresivos Imipramina, Paroxetina, Venlafaxina y Escitalopram mejoran los síntomas de ansiedad cuando se comparan con placebo. La Pregabalina y la hidroxicina también pueden ser eficaces. El Escitalopram parece mas eficaz y mejor tolerado que la Paroxetina. El Inhibidor Selectivo de la Recaptación de Serotonina y Noradrenalina –ISRSN- Venlafaxina también es efectivo, pero debido a cuestiones de tolerabilidad se recomienda con frecuencia para uso como segunda línea de tratamiento. Dado que la ansiedad generalizada en una condición con tendencia a la cronicidad, el tratamiento a largo plazo puede ser anticipado y preparado. Los ISRS Escitalopram y Paroxetina han demostrado su eficacia en la prevención de recaídas en pacientes con TAG. Las Benzodiacepinas pueden ser efectivas en 15-60 minutos, mientras que la buspirona tarda hasta 72 horas en actuar y su efecto está frecuentemente precedido de leve disforia. Los antidepresivos tardan hasta 4 semanas en mostrar efectividad, pero la mejoría clínica significativa puede percibirse a las 2 semanas. Como conclusión se puede decir que existe confusión sobre cual es el mejor método para tratar la ansiedad generalizada con psicofármacos. El tratamiento inicial probablemente debería ser una combinación de benzodiacepinas y antidepresivos. Las dosis de benzodicepinas se deberían reducir a las 2-3 semanas, al tiempo que los antidepresivos empiezan a ser efectivos. Esta pauta es de uso común en la práctica clínica, aunque no está respaldado por estudios controlados. Los pacientes que están siendo tratados sólo con benzodiacepinas, deberían ser tratados con benzodiacepinas y antidepresivos (ISRS), con el fin de retirar éstas en 2-3 semanas, aunque esta retirada no siempre es posible, según según algunos autores. En el tratamiento a largo plazo de la ansiedad crónica la Paroxetina, la Venlafaxina retard y el Escitalopram han demostrado su eficacia en tratamientos de más de 6 meses de duración. Tras la discontinuación del tratamiento, el 20-40% de los pacientes con ansiedad generalizada recae en un plazo de 6-12 meses, por lo que éste debe mantenerse al menos 12 meses. Algunos pacientes requieren tratamiento a más largo plazo para prevenir recaídas. 3.3. Otras alternativas como ansiolíticos Anestésicos como la metacuona y el hidrato de cloral se utilizan de manera ocasional para tratar el insomnio 3.4. NOVEDADES EN EL TRATAMIENTO DE LA ANSIEDAD Desde finales del siglo pasado se han sintetizado y evaluado nuevos fármacos como imidazenilo, abecarnilo, bretazenilo y divaplón, desprovistos de muchos de los efectos secundarios de las convencionales benzodiazepinas ansiolíticas. En general, son agonistas parciales de algún subtipo de receptor GABAA, aunque pueden ser agonistas totales de otros. De igual modo, se han conseguido otros compuestos desprovistos de actividad ansiolítica y que mantienen otras acciones distintas de las clásicas benzodiazepinas. Loreclezol, por ejemplo, es anticonvulsivante, con afinidad hacia receptores GABAA que carecen de subunidades α o γ. Los agonistas del receptor 5-HT3 como ondansetrón poseen actividad ansiolítica en modelos animales, pero no han resultado eficaces en estudios controlados en seres humanos 3.5. AGONISMO INVERSO Y ANTAGONISTAS DE LAS BENZODIAZEPINAS El término agonista inverso se aplica a los fármacos que se unen a los receptores de benzodiacepinas y ejercen un efecto opuesto al de éstas, con aumento de la ansiedad y las convulsiones. El DBI es un ejemplo y algunos análogos de las benzodiacepinas actúan de forma similar. Es posible explicar este complejo fenómeno según el modelo, admitiendo que los receptores de benzodiacepina adoptan dos conformaciones distintas, de las cuales sólo una (A) puede unirse a la molécula de GABA y abrir el canal de cloro. La otra conformación no puede unirse a GABA B. Parece que lo agonistas de las benzodiacepinas (p.ej., diacepam) se unen de manera preferente a la conformación A, desviando el equilibrio a su favor y desviando la sensibilidad al GABA. Los agonistas inversos se unen de manera selectiva a B y ejercen el efecto opuesto. 4. ANTIPSICÓTICOS. La esquizofrenia es una enfermedad crónica que no afecta por igual a todos los pacientes, generalmente se presenta mas en personas jóvenes. La sintomatología de la esquizofrenia se suele dividir en síntomas positivos (alucinaciones, delirios, conductas extravagantes, etc.) y síntomas negativos (alogia, falta de emotividad y afectos, etc.). 4.1. TIPOS DE ESQUIZOFRENIA El DSM-IV distingue cinco subtipos: paraniode, catatónica, hebefrénica, simple e indiferenciada, pero en la practica, mas específicamente en la evidencia clínica, la mayoría de los pacientes muestran una combinación de síntomas de los diferentes subtipos o cambian con el tiempo de un subtipo a otro, caracterizando la esquizofrenia por subtipos y por fases del problema de salud en el transcurso del tiempo y los síntomas. Subtipo Paranoide: En este grupo predominan los síntomas de tipo delirante y alucinatorio. Se le considera como la forma típica y la más diagnosticada en nuestro medio, aun cuando la conducta paranoide es compartida por otros tipos de psicosis. Las razones para sospechar de este subtipo del problema de salud serian la presencia de alucinaciones, ideas delirantes de influencia, persecución, referencia o alusión. Subtipo Catatónico: Se caracteriza por alteración de la conducta motora y puede presentarse bajo la forma de estupor o excitación. En la base de estupor aparece un notable retardo motor (disminución de los movimientos espontáneos o la actividad general), llegando a veces a la inmovilidad total, sin respuesta a estímulos ambientales. El subtipo de esquizofrenia catatónica es cada vez menos frecuente, al parecer por el diagnostico temprano de las crisis esquizofrenicas y los adelantos en los tratamientos farmacológicos. Subtipo Hebefrénico: Se caracteriza por la presencia de síntomas afectivos e ideativos. Desde el punto de vista emocional, lo más notable es la euforia, con risa fácil, sin motivo aparente o la incongruencia afectiva en su más clara expresión, las ideas delirantes son pobres, poco estructuradas y fragmentarias. El lenguaje esta profundamente alterado, presentando irrelevancia, disgregación y neologismos. La conducta es desorganizada, primitiva y pueril. Subtipo Simple: Su característica es la ausencia de síntomas perceptivos e ideativos, evolucionando de manera lenta e insidiosa hacia un estado de anhedonia, con pérdida de iniciativa cada vez mayor hacia el ambiente. Con frecuencia los enfermos consultan por quejas somáticas. Subtipo Indiferenciado: En estos pacientes se observan síntomas correspondientes a los diferentes subtipos, sin que los síntomas puedan ser clasificados en uno de ellos. Típicamente, los trastornos esquizofrénicos se preceden desde una fase premorbida con ninguna o pocas manifestaciones hacia una fase prodromal, caracterizado por muestras tempranas en la deterioración del funcionamiento personal. Las sintomatologías pueden incluir disturbios del sueño, ansiedad, irritabilidad, leve depresión, problemas de concentración, fatiga, además de retiro social, comportamiento peculiar y no característico, problemas en la comunicación, ideas y experiencias perceptivas inusuales, descuido de la higiene personal, e interés y poca motivación. Estos cambios pueden afectar la capacidad de la persona de mantener un trabajo o su vida académica, o su relación con los amigos y la familia. El período prodromal continua hasta el inicio de una fase psicotica aguda, esta fase se refiere a la presencia de características psicóticas mas marcadas, especialmente síntomas positivos. Los síntomas negativos llegan a ser a menudo más severos, y generalmente los pacientes no se pueden cuidar por sí mismos apropiadamente. La fase psicotica aguda transcurre luego hacia una fase de recuperación o de estabilización. La fase de estabilización o recuperación refiere un período de 6-18 meses después del tratamiento agudo. Durante la fase estable, los síntomas negativos y positivos residuales que pueden estar presentes son relativamente constantes en magnitud y generalmente menos severos que en la fase aguda. Algunos pacientes pueden ser asintomáticos mientras que otros experimentan síntomas no psicóticos tales como tensión, ansiedad, depresión, o insomnio 4.2. OTROS TRANSTORNOS PSICÓTICOS La esquizofrenia es el trastorno psicótico más común, sin embargo, existen otros síndromes psicóticos. Entre estos trastornos se incluyen: el trastorno esquizofreniforme, el trastorno esquizoafectivo, el trastorno delirante, el trastorno psicótico breve y los trastornos psicóticos inducidos por sustancias o debidos a una enfermedad médica. De forma esquemática, el trastorno esquizofreniforme se caracteriza por un cuadro clínico-psicopatológico similar a la esquizofrenia, su duración es mayor de 1 mes y menor de 6 meses. La característica esencial del trastorno psicótico breve viene determinada por la corta duración de los síntomas (al menos 1 día e inferior a un mes). 4.3. FARMACOTERAPÍA ANTIPSICOTICA 4.3.1. ANTIPSICOTICOS APs Los antipsicóticos son un grupo químicamente heterogéneo de sustancias que comparten sus indicaciones clínicas y algunos aspectos de su mecanismo de acción. Los antipsicóticos también se conocen como antagonistas del receptor de la dopamina, neurolépticos o tranquilizantes mayores. El descubrimiento de ciertos fármacos con acción antipsicótica pero con un mecanismo de acción que implica a otros sistemas neurotransmisores como la serotonina. - Clasificación de los APS Podemos establecer una primera clasificación de los antipsicóticos en dos grandes grupos: a. Antipsicóticos convencionales o típicos. Los APs Típicos son medicamentos utilizados para el tratamiento de las diversas formas de esquizofrenia. También se utilizan en la manía y cuadros de agitación, entre otros. Los APs Típicos se caracterizan por ser antagonistas competitivos de los receptores dopaminérgicos, aunque también actúan sobre receptores de otros neurotransmisores (adrenérgicos, muscarínicos, serotonérgicos, etc). Producen muchos efectos indeseables y en general no son muy bien aceptados por los pacientes, además de que son poco eficaces sobre la sintomatología negativa. Si consideramos su actividad sedativa e incisiva (acción antiesquizofrénica relacionada con la potencia), podemos clasificarlos de más sedativo a menos y de menos incisivo a más, de la siguiente manera: Levomepromacina > Tioridacina > Clorpromacina > Flufenacina > Haloperidol > Tioproperacina. Los de baja potencia precisan ser administrados a altas dosis para lograr el efecto terapéutico deseado, por lo que predomina su acción sedante. Los de alta potencia, en cambio, precisan dosis inferiores y muestran mayores y mejores efectos neurológicos. Existen diferencias en su perfil de acción clínica. El perfil de efectos adversos puede determinar la elección de un antipsicótico con un efecto predominantemente anticolinérgico (por ejemplo tioridazina) o extrapiramidal (por ejemplo haloperidol), según las características del paciente (edad, deterioro intelectual, estado físico). El efecto sedativo de los antipsicóticos clásicos puede ser interesante para otras indicaciones, como en pacientes agitados o de elevado nivel de ansiedad, los antipsicóticos de alto poder sedativo (clorpromacina, levopromacina, tioridacina, zuclopentixol) son los fármacos de elección. b. Antipsicóticos atípicos. Presentan un menor riesgo de efectos de tipo extrapiramidal o endocrino y poseen una mayor eficacia sobre los síntomas negativos de la esquizofrenia que los antipsicóticos típicos. Se incluyen como antipsicóticos atípicos a clozapina, risperidona, olanzapina, sertindol, quetiapina y ziprasidona. Igualmente se puede decir que el mecanismo de acción de estas sustancias se caracteriza por el bloqueo de receptores de dopamina y serotonina (5-HT2), hecho por el que también se les conoce como antagonistas de serotonina y dopamina. Existen diferencias entre los componentes de este grupo en cuanto a su afinidad por los distintos receptores de la dopamina, serotonina u otros tipos de receptores. Todos los fármacos de este grupo, cuyo mecanismo de acción consiste en el antagonismo de receptores dopaminérgicos, se asocian inevitablemente a la aparición de efectos secundarios extrapiramidales a las dosis efectivas. Entre las características que tienen los nuevos antipsicóticos podríamos enumerar: 1) Son eficaces tanto sobre los síntomas positivos como negativos. 2) Son eficaces en pacientes resistentes a tratamientos clásicos. 3) Son eficaces en las formas agudas y crónicas. 4) Producen menos efectos indeseables (afectan menos a la PRL (prolactina) y efectos extrapiramidales y, al parecer, poca incidencia de discinesia tardía). 5) Disminuye el riesgo de recaídas. 6) La aceptación por parte del paciente es mayor. 7) Disminuyen los días de hospitalización y es mejor la integración sociolaboral y familiar. 8) Son fármacos de primera elección y no únicamente para casos resistentes. 4.4. EFECTOS FARMACOLÓGICOS DE LOS AGENTES ANTIPSICÓTICOS. • Efecto antipsicótico • Efectos neurológicos • Efectos extrapiramidales: acatisia, reaciones distónicas, agudas y tardías, parkinsonismo; otros: efecto proconvulsivante, alteración de la temperatura corporal, síndrome neuroléptico maligno. • • • • • • • • Efecto sedante Efecto ansiolítico Efecto antiemético Efectos vegetativos Efectos cardíacos Efectos endocrinos y metabólicos Efectos psicofisiológicos y conductuales Tolerancia y dependencia física 4.4. EFECTOS ADVERSOS El perfil de efectos adversos del tratamiento difiere en gran medida entre los antipsicóticos de primera y segunda generación. Entre los más comunes asociados a los antipsicóticos convencionales se encuentran los efectos extrapiramidales, incluyendo parkinsonismo, distonías musculares, acatisia, discinesias tardías y síndrome neuroléptico maligno, potencialmente letal. Otros efectos adversos frecuentes son el incremento de peso moderado, los síntomas negativos secundarios y la prolongación del intervalo QT, con riesgo de arritmia cardiaca fatal. El perfil de efectos adversos frecuentes asociados a los antipsicóticos atípicos consiste fundamentalmente en ganancia de peso moderada o severa, diabetes mellitus tipo 2, dislipemia y efectos extrapiramidales moderados. Ambos grupos de antipsicóticos pueden diminuir el umbral convulsivo, producir sedación, hipotensión ortostática e hiperprolactinemia. Esta última puede originar alteraciones menstruales y de la función sexual, además de ginecomastia y galactorrea. 4.4.1. DISFUNCIONES SEXUALES ASOCIADAS A LOS ANTIPSICÓTICOS • En el varón - Galactorrea - Dificultad en la erección - Alteraciones de la eyaculación, sobre todo en aquellos fármacos con efecto anticolinérgico como la tioridazina • En la mujer - Anomalías en el ciclo menstrual - Disminución de la libido - Sequedad vaginal • Estos fenómenos son menos frecuentes con los antipsicóticos atípicos 4.4.2. AUMENTO DE PESO ASOCIADO AL TRATAMIENTO CON ANTIPSICÓTICOS • En el caso de la olanzapina, se ha constatado un incremento superior al 7% del peso corporal en el 40% de los pacientes tratados. • Con el resto de agentes (risperidona y quetiapina), este efecto parece menor, estimándose una ganancia de peso, a las 6-8 semanas, de 1-4 • La ganancia de peso parece menos manifiesta en el caso de la ziprasidona dada su escasa afinidad por los receptores H1. 4.5. UTILIDAD TERAPÉUTICA. En la actualidad los antipsicóticos atípicos se consideran el grupo de elección para el tratamiento de las fases agudas y el mantenimiento de la esquizofrenia, por su mejor tolerabilidad respecto a los convencionales. Poseen un nivel de eficacia similar o ligeramente superior a favor de los atípicos. El uso de los antipsicóticos convencionales se reserva para el tratamiento de las agitaciones psicomotoras agudas, así como para casos de resistencia a los fármacos atípicos. La clozapina es considerado el antipsicótico de mayor eficacia y se restringe a los casos de esquizofrenia fármaco-resistente, ya que se asocia a un perfil de efectos secundarios particularmente graves, incluyendo neutropenia en el 0,39%, miocarditis en el 0,032%. Existen en la actualidad diversas formulaciones de antipsicóticos de liberación retardada, de administración intramuscular con frecuencia bimensual a mensual, especialmente útiles en casos de incumplimiento terapéutico. Entre estas, disponemos en la actualidad de varias preparaciones de antipsicóticos de primera generación, incluyendo flufenazina y zuclopentixol, así como la preparación de risperidona de liberación prolongada. Se recomienda generalmente que el inicio del tratamiento antipsicótico sea a dosis bajas, en una o dos tomas diarias, aumentándose progresivamente hasta la dosis plena. Esta dependerá de la respuesta obtenida por cada paciente concreto, evaluada mediante la evolución de la semiología en las sucesivas entrevistas clínicas. 4.6. COMPARACIONES ENTRE LOS APS TÍPICOS Y ATÍPICOS Diferentes referencias describen revisiones basadas en la evidencia clínica comparando las alternativas de APs Típicos vs. APs Atípicos, de las cuales se rescatan las siguientes conclusiones: - Los APs Típicos son eficaces para reducir la mayor parte de los síntomas de la esquizofrenia, tanto los positivos (alucinaciones, delirios, conductas extrañas) como los negativos (apatía, embotamiento afectivo, alogia, abulia), siendo más eficaces en los positivos. Alrededor de 20% de los pacientes tienen remisión completa de sus síntomas. Diversos reportes de estudios de 2 años, muestran que aproximadamente 30% de los pacientes tienen recaídas durante el tratamiento con APs de primera generación (Típicos) comparado con 80% sin tratamiento. - Risperidona, quetiapina, sertindol, amisulpride, olanzapina y clozapina son tan o más eficaces que los APs convencionales para aliviar los síntomas globales de la esquizofrenia. - Clozapina y risperidona son tan o más eficaces que los APs convencionales para prevenir recaídas. - Sólo en pocos estudios se incluyeron pacientes con síntomas predominantemente negativos. La clozapina fue más eficaz que los APs convencionales en la mejoría de los síntomas negativos en pacientes resistentes al tratamiento. Del mismo modo, la clozapina es más eficaz que los APs convencionales para aquellos pacientes que no han respondido previamente al tratamiento. - La sedación puede aparecer más frecuentemente en los pacientes tratados con clozapina que en los tratados con los APs convencionales. La olanzapina, amisulpride, sertindol y quizá, risperidona pueden causar menos somnolencia. No hay suficiente evidencia científica que sugiera que los otros APs atípicos sean más o menos sedantes que los convencionales. - Los efectos secundarios anticolinérgicos y antiadrenérgicos pueden presentarse más frecuentemente en los pacientes tratados con clozapina y sertindol y menos frecuentemente en los tratados con quetiapina u olanzapina, que en los tratados con APs convencionales. - Se han descrito efectos secundarios cardíacos graves y potencialmente fatales al menos en dos APs atípicos (sertindol y clozapina) y en dos convencionales (pimozida y tioridazina). Se desconocen a largo plazo los efectos secundarios cardíacos, así como los hepáticos, de la mayoría de APs atípicos. - Parece que los APs atípicos pueden causar más aumento de peso que los convencionales. A excepción de risperidona, que se le atribuye alta ganancia de peso. - Los pacientes con esquizofrenia pueden percibir los APs atípicos más aceptables que los convencionales, debido a que menos pacientes tratados con atípicos (con excepción de ziprasidona) abandonaron precozmente los estudios. - A diferencia de la clozapina en los pacientes resistentes al tratamiento, ningún APs atípico se destaca por ser más eficaz que el resto de los otros. Estos fármacos tienen perfiles de efectos secundarios ligeramente diferentes entre sí cuya importancia puede variar en función del paciente y sus familias o cuidadores. - Si la esquizofrenia es crónica, hay poca diferencia en el tratamiento con un AP típico o uno atípico, a excepción de los casos en que los efectos secundarios, tales como parkinsonismo, obligaron a abandonar el estudio. - Se recomienda que los fármacos APs Típicos o Atípicos, no sean prescritos simultáneamente, excepto por cortos periodos para cubrir un cambio de fármaco. - Durante la fase aguda, antes de prescribir un AP deberá tenerse en cuenta la respuesta previa del paciente al tratamiento, el perfil de efectos secundarios del mismo y las preferencias por una determinada medicación en función de la experiencia previa y la vía de administración prevista. - En pacientes resistentes al tratamiento de la esquizofrenia, se recomienda el tratamiento con clozapina. - Se recomienda que las medicaciones coadyuvantes (litio, carbamacepina, ácido valproico y benzodiacepinas) se reserven para los casos en que la clozapina no sea apropiada en pacientes resistentes al tratamiento, bien por falta de eficacia, efectos adversos, preferencia del paciente o por probable falta de cumplimiento. - Dado el riesgo de recidiva rápida en la fase de postcrisis, se recomienda evitar la supresión o la reducción prematura del tratamiento farmacológico antipsicótico instaurado en la fase aguda; la continuación del tratamiento durante 1 o 2 años después de una crisis debe discutirse, cuando sea adecuado, con el paciente y su familia. - En pacientes estables que no presentan síntomas negativos puede estar indicada una reducción de dosis, que deberá ser reducida de forma gradual, hasta llegar como mínimo a una quinta parte de la dosis de mantenimiento habitual. - En un paciente que haya presentado sólo un episodio de sintomatología positiva y no haya sufrido ningún síntoma durante el año siguiente de tratamiento de mantenimiento puede plantearse un período de prueba sin medicación. - También se puede plantear una supresión de la medicación en pacientes con múltiples episodios previos que se hayan mantenido estables durante 5 años sin síntomas positivos y que cumplan bien el tratamiento. - Se recomienda tratamiento indefinido en pacientes con antecedentes de intentos de suicidio graves o de conductas violentas y agresivas. - Al tomar la decisión de suspender la medicación antipsicótica, se deben adoptar precauciones para reducir al mínimo el riesgo de recidiva: reducción gradual de la dosis durante varios meses, visitas más frecuentes y empleo de estrategias de intervención precoz. 4.7. NUEVOS ANTIPSICOTICOS Ritanserina, bloqueante selectivo 5-HT2A, utilizada de forma aislada, ha mostrado escasa eficacia como agente antipsicótico, aunque puede disminuir algunos síntomas negativos y aumentar la eficacia de los antipsicóticos clásicos cuando se asocia a éstos. 5. ANTIDEPRESIVOS Por definición, el concepto de depresión recoge la presencia de síntomas afectivos, de sentimientos o emociones: tristeza patológica, decaimiento, irritabilidad, sensación subjetiva de malestar e impotencia frente a las exigencias de la vida, aunque, en mayor o menor grado, siempre están también presentes síntomas de tipo cognitivo, volitivo (sentimientos de autoeliminación), o incluso somático como la disminución de la libido y del metabolismo corporal. Son muchas las formas de clasificar al depresión, la mas cercana a la fisiopatología y a la forma de tratamiento es: depresión primaria, mayor o endógena y depresión secundaria, menor o reactiva. La discusión generalmente ha girado en torno de cuales pacientes reciben un beneficio mayor del tratamiento con antidepresivos y cuales deben recibir un refuerzo psicoterapéutico más cercano y frecuente. Para el caso de la depresión secundaria a una enfermedad física como resultado de lo que se considera un SMO, síndrome mental orgánico, el tratamiento de la enfermedad de base es la base de la terapéutica; si es una depresión secundaria al empleo de sustancias o medicamentos, la suspensión o disminución de las dosis es el centro o eje de la intervención. Así mismo si la depresión es secundaria a una pérdida o a un duelo mal elaborado, el acompañamiento mediante la psicoterapia resulta siendo la mejor opción. Las Depresiones Secundarias son depresiones sintomáticas, de diversa etiología: a) De origen endocrino: Hiper e hipotiroidismo. Hiper o hipoaldosteronismo. Hiperfunción suprarenal. b) Tumorales: tumores del aparato digestivo. c) De origen neurológico: Enfermedad de Parkinson, esclerosis múltiple, Corea de Huntington. d) Enf. sistémicas o autoinmunitarias: Lupus eritematoso diseminado. e) Por agentes químicos y tóxicos: plomo, mercurio, monóxido de carbono, alucinógenos. f) Por fármacos: Antihipertensivos: Reserpina, alfa metildopa, clonidina, guanetidina, beta bloqueantes. Corticoides. Neurolépticos. Anticonceptivos hormonales. Analgésicos. Antineoplásicos. Depresores del SNC (alcohol). Estas formas de depresión, como su nombre lo indica, son secundarias a la enfermedad o problema de fondo, son de buen pronóstico ya que en general, desaparecida la etiología determinante el cuadro depresivo también desaparece. 5.6 CLASIFICACIÓN DE LOS ANTIDEPRESIVOS a. Tricíclicos o Clásicos Imipramina Clomipramina Desipramina Amitriptilina Nortriptilina Trimipramina Maprotilina Las sustancias de este grupo son todas derivadas de la dibenzoacepina o iminodibencilo, núcleo heterocíclico con dos anillos bencénicos, semejante a la fenotiazina en el que se reemplaza el átomo de azufre de ésta por un puente de etileno en la estructura cíclica central. Al igual que las fenotiazinas el iminodibencilo tiene una cadena lateral ligada al nitrógeno, con 3 átomos de carbono y un nitrógeno terminal. La imipramina tiene 2 grupos metilos en el N terminal. Huhn describió en Suiza en el año 1957, que la imipramina carece de efectos en la esquizofrenia, para la que originalmente fué sintetizada, pero en cambio resultó sumamente efectiva en el tratamiento de la depresión mayor, iniciando otra línea de avance terapéutico en psiquiatría de gran trascendencia. Pequeñas modificaciones en la cadena lateral de la imipramina, que determinan cambios de tipo farmacocinético principalmente, dan lugar al resto de los componentes del grupo. El agregado de un Cl en posición 7, origina la monoclorimipramina. La supresión de un metilo en la cadena lateral de la imipramina, demetilación, da origen a la desipramina. El agregado de un metilo en la cadena lateral da lugar a la trimipramina. El reemplazo en la molécula de la imipramina del puente de nitrógeno (N-CH2-...), en posición 5 por un puente de carbono (C=H2-....), origina la amitriptilina y la demetilación de ésta la nortriptilina. Estos agentes son inhibidores de la recaptación axonal de NA y 5-HT que constituye el principal mecanismo de inactivación fisiológica de los neurotransmisores, con distinta potencia y selectividad. También son antagonistas de los receptores muscarínicos, de los Alfa 1 y de los histaminérgicos H1 y H2, con potencia de moderada a elevada. Se absorben bien por vía oral. Debido a su elevada lipofilia se distribuyen ampliamente. La máxima concentración plasmática se alcanza en 8 a 12 horas. Poseen una vida media en general, prolongada, de 20 a 30 horas. La vida media de los derivados demetilados, como la desipramina o la nortriptilina es por lo menos el doble de los congéneres metilados. Los tricíclicos se metabolizan en hígado, con la participación de enzimas microsomales. El mecanismo más importante es la oxidación y posterior conjugación con ácido glucorónico. Un paso intermedio también muy importante es la demetilación ya que los metabolitos, como la desipramina por ejemplo, poseen acciones farmacológicas y adquieren mayor capacidad de inhibición de recaptación de NA, que los congéneres iniciales. b. De Segunda Generación Trazodone Amoxapina Mianserina c. ISRS (Inhibidores Selectivos de la Recaptación de Serotonina) Fluoxetina Paroxetina Sertralina Fluvoxamina Citalopram Escitalopram Estos agentes carecen casi por completo de actividad sobre la recaptación de otros neurotrasmisores aminérgicos. Tampoco tienen afinidad por adrenoceptores postsinápticos, o receptores muscarínicos, histaminérgicos, GABAérgicos o de 5-HT. La fluoxetina fué el primer agente utilizado y el prototipo del grupo. También es un potente inhibidor de una enzima citocromo P-450 hepática, conocida como la CYP2D6, que metaboliza a los antidepresivos tricíclicos, algunos neurolépticos, antiarrítmicos y betabloquentes. Ello explica las conocidas interacciones de la fluoxetina con los tricíclicos cuando se los administra conjuntamente (aumento de los niveles plasmáticos de éstos y potenciación de sus acciones) y sugiere la posibilidad de otras interacciones. La fluoxetina se absorbe completamente por vía oral pero su biodisponibilidad se reduce por su gran metabolismo de primer paso por el hígado (oxidación y conjugación posterior) La vida media de la fluoxetina y de su metabolito norfluoxetina es prolongada, de 40 a 70 horas. Se ligan firmemente a proteinas tisulares, por lo que después de suprimir su administración, la eliminación del organismo es muy prolongada. La sertralina, derivado 1.-aminotetrahidronaftaleno, diferente químicamente de la fluoxetina, es otro potente inhibidor de la recaptación de 5-HT. Se absorbe bien por vía oral, aunque mas lentamente que los otros agentes del grupo. Su metabolito demetilado pierde gran parte de su actividad farmacológica. La vida media plasmática es aproximadamente la mitad de la fluoxetina, es decir de 22 a 30 horas. La fluvoxamina es otro inhibidor de la recaptación de 5-HT cuya composición química es diferente de sertralina y fluoxetina. Es una arilcetona-2-aminoetil oxima. Se absorbe bien y rápidamente por vía oral. Se une firmemente a proteínas plasmáticas y tisulares. Se metaboliza en el hígado, en forma similar a los otros agentes pero no produce metabolitos activos. Su vida media es corta, 24 horas, aproximadamente. Todos los inhibidores de la recapatación de 5-HT pueden interaccionar con los antidepresivos tricíclicos y producir efectos adversos de sobredosis o intoxicación por tricíclicos de acuerdo al mecanismo metabólico explicado anteriormente. Lo mismo puede ocurrir, y a través del mismo mecanismo, con el uso simultáneo de neurolépticos. La administración conjunta de estos agentes con fármacos I-MAO pueden acusar importantes efectos adversos, siendo el principal el llamado sindrome de la serotonina, producido por un marcado incremento de serotonina en el espacio intersináptico, caracterizado por hipertermia, temblores, mioclonias, ansiedad, inquietud, incoordinación, ataxia, delirios, extrema agitación y coma. Este síndrome puede ser fatal. d. ISRD (Inhibidores Selectivos de la Recaptación de Dopamina) Amineptino e. ISRN (Inhibidores Selectivos de la Recaptación de Noradrenalina) Reboxetina Oxaprotilina f. ISRNS (Inhibidores Selectivos de la Recaptación de Noradrenalina y Serotonina) Venlafaxina Milnacipran Duloxetina g. Inhibidores Específicos de la Recaptación de Noradrenalina y Dopamina Bupropion Nomifesín h. Inhibidores Específicos de la Recaptación de Serotonina y Dopamina Banzinaprina i. IMAO (Inhibidores de la Monoaminoxidasa A y B) Tranilcipromina Fenelzina Estos fármacos inhiben la desaminación oxidativa de las monoaminas. Existen 2 isoenzimas de la MAO. La MAO-A es la forma más común en el intestino y desdobla selectivamente la 5-HT y la NA. La MAO-B es más común en el cerebro y sus substratos preferidos son la feniletilamina y la bencilamina. El resto de las aminas biógenas son metabolizxadas indistintamente por las 2 isoenzimas. Los primeros utilizados (fenelzina, nialamida yposteriormente tranilcipromina) son inhibidores irreversibles de ambos tipos de MAO, siendo éste su principal inconveniente, ya que la reactivación de la actividad enzimática depende prácticamente de nueva síntesis de la misma, proceso que necesita 2 - 3 semanas para alcanzar la situación inicial. Durante ese tiempo las monoaminas se acumulan por falta de metabolismo y pueden causar importantes efectos adversos. Los inhibidores reversibles de la MAO sólo lo hacen por unas horas, menos de 24, por lo que su uso es mas seguro. La fenelzina y nialamida son derivadas de la hidrazina, y como ella son hepatotóxicos. Por eso la utilización de estos agentes ha disminuido ampliamente. La tranilcipromina es un inhibidor de MAO, que resulta de la ciclización de la anfetamina. No es un derivado hidrazínico. Son agentes no selectivos que inhiben indistintamente a la MAO-A como a la MAO-B. j. RIMA (Reversibles Inhibidores de la Recaptación de la MAO A) Moclobemida Taloxotona Brofaromina k. IMAO-B (Inhibidor de la Monoaminoxidasa B) Selegilina La moclobemida es un derivado benzamídico, inhibidor selectivo de la MAO-A, de acción reversible. Inhibe selectivamente la desaminación de la 5-HT, NA y DA. El deprenil o selegilina es una fenilisopropil-N-metilpropinilamina, inhibidor altamente selectivo de la MAO-B. Esta isoenzima predomina en ciertas regiones del SNC, como los ganglios de la base. Por eso, la selegilina ha demostrado utilidad en la enfermedad de Parkinson ya que inhibe selectivamente la degradación de la dopamina intracerebral. l. Otros Mirtazapina Nefazodona Litoxetina Tianeptina Antidepresivos que se encuentran en fase de Investigación Clínica: 1. Inhibidores Triples de la Recaptación (de Serotonina, Noradenalina y Dopamina): - DOV - 21947 (nombre codificado) - NS - 2359 (nombre codificado) - DOV - 216303 (nombre codificado) 2. Melatoninérgico e Inhibidor de los receptores Serotoninérgicos 5-HT2C: - Agomelatina (Valdoxan) Algunas benzodiacepinas del grupo de las triazolobenzodiacepinas han demostrado poseer también acciones antidepresivas, aunque su uso en esa indicación es limitado. El rol del alprazolam ha sido evaluado en tal sentido Ha demostrando ciertas acciones antidepresivas y para el control de las crisis de pánico. Un compuesto relacionado, el adinazolam, ha demostrado en forma experimental sensibilizar a neuronas hipocampales a la serotonina, de tal manera que posiblemente sea un agente potencialmente útil como antidepresivo. Como pueden darse cuenta en la actualidad existen numerosos fármacos antidepresivos de eficacia terapéutica demostrada, aún para las formas mas graves de depresión. Se cuenta aún con agentes antidepresivos tricíclicos, los mas antiguos, pero aún no superados en eficacia específica, los de mas reciente aparición como los inhibidores de la recaptación de serotonina o 5-HT, cuya principal ventaja es la disminución de la incidencia de varios efectos adversos, También tiene aplicación terapéutica la terapia electroconvulsiva, que aunque muy limitada, posee indicaciones precisas. Finalmente debe considerarse especialmente a la psicoterapia como un arma terapéutica mas. En la casi totalidad de los pacientes con depresión, la psicoterapia actúa sinergísticamente con las drogas antidepresivas. 5.7 MECANISMOS DE ACCIÓN. El mecanismo de la acción timoléptica de los antidepresivos es aún un tema de discusión. Previamente se consideraba que la depresión se originaría en una desregulación del sistema noradrenérgico cerebral, sobre todo de las neuronas localizadas en el locus ceruleus, cerca del piso del IV ventrículo, neuronas que se proyectan ampliamente a centros cerebrales superiores. Posteriormente cobró vigencia la desregulación del sistema serotoninérgico, localizado principalmente en el cerebro medio y núcleo del rafe, que también se proyecta ampliamente en el cerebro. Sin embargo, como los sistemas monoaminérgicos cerebrales están interconectados, es posible que el origen del problema depresivo incluya a más de un sistema. Existen también hormonas como el cortisol, la T3, y sus factores hipotalámicos de liberación que juegan un rol en la génesis de las depresiones. De acuerdo con las acciones que producen los agentes antidepresivos, parece lógico relacionar dichas acciones con mecanismos de adaptación o regulación de receptores, sobre todo los de NA y 5-HT. Los antidepresivos que se utilizan actúan de acuerdo a uno de los siguientes mecanismos: reducen la degradación de neurotransmisores, como por ejemplo los inhibidores de la MAO, o bloquean la recaptación de los mismos en las sinapsis, como por ejemplo los tricíclicos o la fluoxetina. Otros como los tetracíclicos, además bloquean autoreceptores como los Alfa 2 adrenérgicos. En cualquier caso, la concentración de los neurotransmisores en el espacio intersináptico se incrementa marcadamente. Sin embargo, aunque esta acción ocurre inmediatamente, la respuesta clínica antidepresiva se observa recién después de varias semanas. Esto indica que se desarrollan mecanismos de adaptación, compensación o autoregulación de los receptores involucrados y que dichos mecanismos son mas importantes que la disponibilidad inmediata de los neurotransmisores en las sinapsis para la determinación del comienzo de la acción antidepresiva. Los siguientes efectos sobre los receptores de monoaminas se desarrollan por la acción de los agentes antidepresivos tricíclicos y por los inhibidores de la recaptación de 5-HT. 1. Bloqueo de la captación axonal de NA y de 5-HT, lo que produce aumento de la concentración y mayor disponibilidad de los neurotransmisores en la sinapsis. 2. Como consecuencia, se produce un estímulo desencadenado por la NA y 5-HT sobre los autoreceptores presinápticos Alfa 2 y de 5-HT. 3. Como el tratamiento es continuado, la actividad de las neuronas sufre una progresiva adaptación, que incluye una desensibilización de los receptores presinápticos, continuamente activados. 4. Ello da lugar a un retorno progresivo al estado previo y un aumento de la respuesta a los agonistas Alfa 1 y posiblemente a una aumento en la sensibilidad a la 5-HT. 5. En la administración crónica, por la mayor disponiblidad de NA, se reduce el número y la sensibilidad de los receptores Beta adrenérgicos, por down regulation. 6. También se observa un aumento de la respuesta de los receptores Alfa 1, por aumento del número y sensibilidad de los mismos. Ello ocurre porque los antidepresivos tricíclicos son también bloqueadores Alfa 1, lo que origina una regulación en ascenso de estos receptores, situación opuesta a lo que ocurre con los receptores Beta. 7. En tratamientos prolongados, la inhibición de la recaptación de 5-HT, sobre todo por los agentes inhibidores específicos, se traduce por un aumento de la disponibilidad de 5-HT y potenciación de la transmisión serotoninérgica postsináptica. También disminuye la sensibilidad de los autoreceptores presinápticos de 5-HT. 8. Los antidepresivos tricíclicos producen efectos similares sobre la transmisión serotoninérgica y los inhibidores de MAO disminuyen la sensibilidad de los receptores presináticos de la 5-HT y en consecuencia, tambien incrementan la actividad de 5-HT postsináptica. En resumen, el efecto neto de los agentes mencionados, es incrementar la transmisión serotoninérgica. Los agentes tetracíclicos no parecen interferir con la recaptación axonal de catecolaminas. La mayor disponibilidad de NA en la sinapsis se debería a un bloqueo de los receptores Alfa 2 presinápticos. Los tricíclicos también son antagonistas de los receptores muscarínicos y de los histaminérgicos H1 y H2. Como fuera especificado antes, también bloquean los receptores Alfa. 5.8 ACCIONES FARMACOLÓGICAS Sistema Nervioso Central: - Acción Antidepresiva: En personas normales los antidepresivos no producen efectos muy aparentes, salvo ligera euforia y discreta sedación o somnolencia inicial, cansancio, efectos anticolinérgicos y dificultad para la concentración, principalmente con los AD tricíclicos. En cambio en pacientes con depresión, luego de un periodo de latencia de 2 a 3 semanas, el estado de ánimo comienza a mejorar, el humor también mejora, y aparece paulatinamente una sensación de bienestar. Los trastornos del sueño tienden a desaparecer y disminuye el número de veces que el paciente se despierta. Disminuye el número de REM (movimientos oculares rápidos) y aumenta la latencia o tiempo de aparición de los REM, una vez iniciado el sueño. La amitriptilina, la clorimipramina y la trazadona son especialmente sedantes, en tal sentido. También los IMAO corrigen los trastornos del sueño, insomnio o hipersomnia. Por acción de los antidepresivos, el apetito mejora progresivamente, lo mismo que se eleva la autoestima y disminuyen las ideas de culpa. No todos los pacientes responden con una mejoría similar. Aparentemente la depresión mayor recurrente es la forma clínica que responde mejor. Un escaso número de pacientes no responde al tratamiento, aunque también es frecuente observar dosis insuficientes y abandono del tratamiento por no obtenerse respuesta rápidamente. En algunos pacientes, el efecto antidepresivo puede desencadenar un cuadro de excitación hipomaníaca o manía franca, sobre todo en pacientes con trastornos bipolares y con dosis altas. Debe considerarse en tal sentido, que la fluoxetina y congéneres tienen efectos estimulantes, y se presume (ya que no existen evidencias definitivas), que también incrementan en los pacientes la hostilidad y agresividad. Excluyendo estos pacientes, la incidencia de hipomanía o manía es de menos del 1 %. - Acciones sobre SNA: Los derivados tricíclicos poseen un potente efecto anticolinérgico, particularmente antimuscarínico de tipo atropínico. Los pacientes tratados con pueden presentar constipación, disminución de la secreciones exocrinas, xerostomia, midriasis, trastornos de la acomodación visual acompañados de visión borrosa, taquicardia, palpitaciones y retención urinaria. Este último efecto puede ser importante en pacientes de edad, sobre todo hombres con adenoma de benigno de próstata. La fluoxetina y los inhibidores específicos de la recaptación de 5-HT, prácticamente carecen de efectos anticolinérgicos. Los agentes tetracíclicos también ejercen escasas acciones antimuscarínicas pero como bloquen los receptores Alfa 2 presinápticos, los Alfa 1, los 5-HT2, y los H1, poseen acciones sedantes. Los IMAO, aunque no son bloqueadores de los receptores muscarínicos, producen con frecuencia xerostomía, visión borrosa, constipación y trastornos de la micción. También se ha observado Hiperhidrosis localizada por activación de los receptores Alfa 1 de las glándulas sudoríparas. - Acciones cardiovasculares. Los agentes AD tricíclicos pueden producir hipotensión ortostática, efecto relacionado con sus acciones bloqueadoras Alfa 1 y taquicardia sinusal por bloqueo muscarínico. Estas acciones son frecuentes (5 al 20 %). También pueden producir arritmias, particularmente en pacientes con patología preexistente como un bloqueo de rama por ejemplo, y desencadenar un bloqueo A-V completo. Los AD triciclicos son además cardiodepresores, efecto tipo quinidina, que es potencialmente peligroso por las interacciones con otras drogas, de acción similar. El ECG de los pacientes tratados con tricíclicos puede demostrar prolongación de los tiempos de conducción (P-R ; QRS y Q-T), e inversión o aplanamiento de la onda T. La mayor disponibilidad de catecolaminas a nivel miocárdico puede resultar en varias formas de cardiotoxicidad. Por las razones aludidas, el estado funcional cardiovascular de los pacientes tratados crónicamente con antidepresivos tricíclicos debe ser periódicamente evaluado. La fluoxetina y otros inhibidores de la recaptación de 5-HT producen pocos efectos cardíacos. La trazadona incluso produce una leve bradicardia. Los IMAO no selectivos, del tipo de la tranilcipromina o los hidrazínicos como la fenelzina, pueden producir hipotensión postural, invocándose varias razones como causa: inhibición dde la liberación de NA por activación de los receptores Alfa 2 presinápticos, formación de octopamina, inhibición de la tirosinhidroxilasa. La inhibición irreversible de MAO, determina que el efecto persiste por 2-3 semanas, ya que la recuperación depende de nueva síntesis de la MAO. Ello puede llevar a una acumulación de tiramina, precursor de las catecolaminas en el proceso biosintético y también presente en numerosos alimentos. El exceso de tiramina puede desencadenar uno de los efectos adversos mas serios de los IMAO, la crisis hipertensiva. Los IMAO-A, de acción reversible poseen menos efectos adversos cardiovasculares. La MAO-A metaboliza preferentemente a la tiramina y como la acción de inhibidores reversibles, como la meclobemida, dura sólo unas horas el efecto adverso mencionado es mucho mas difícil de ocurrir. Por otra parte, los inhibidores de la MAO-B, como el deprenil, a las dosis que son necesarias en terapéutica, dejan de ser selectivos y afectan preferentemente el metabolismo de la DA, por lo que son de utilidad en la enfermedad de Parkinson. Otras acciones: La fluoxetina y los inhibidores de la recaptación de 5-HT desarrollan efectos anorexígenos, que deben ser considerados en la institución del tratamiento. Su indicación sería más conveniente en pacientes obesos. Estos agentes también fueron involucrados en la regulación del consumo de alcohol en alcohólicos crónicos, reduciendo el consumo. 5. 9 EFECTOS ADVERSOS Antidrepresivos Tricíclicos: La incidencia de efectos adversos de los agentes AD tricíclicos es relativamente elevada, 5 % aproximadamente. Los mismos dependen de la extensión de las acciones que se generan por el bloqueo de los receptores sobre los que actúan. Pueden mencionarase los siguientes: 1. Trastornos de la acomodación visual, midriasis y posibilidad de desarrollo de glaucoma o complicación de un glaucoma de ángulo estrecho. 2. Retención urinaria, sobre todo en ancianos con hipertrofia prostática. Micción retardada. 3. Trastornos aparato digestivo: sequedad bucal, disartria, constipación, disminución de las secreciones, retardo del tiempo de vaciamiento gástrico, interacciones farmacocinéticas por la absorción de otros medicamentos. 4 Efectos sobre SNC: Trastornos de la memoria, confusión, debilidad, fatiga, delirio (tipo atropínico). Sedación. Además, potenciación de la acción depresora de otros agentes como el alcohol, barbitúricos o benzodiacepinas ansiolíticas. Reacciones extrapiramidales como el Temblor (que responde a los betabloqueantes), convulsiones tónico-clónicas en sobredosis, sobre todo en Niños. Hipomanía o manía franca. 5. Efectos sobre aparato cardiovascular: hipotensión ortostática, taquicardia, modificaciones del ECG (prolongación del P-R, QRS y Q-T), Bloqueo A-V, Depresión miocárdica, Precipitación de una insuficiencia cardiaca en pacientes predispuestos. 6. Trastornos de la función sexual: disminución de la líbido, anorgasmia, impotencia. 7. Erupciones cutáneas, agranulocitosis, ictericia colestásica, aumento de peso (efecto común de los antidepresivos, a excepción de la fluoxetina y congéneres). Sudoración excesiva, de causa no bien determinada. Inhibidores de MAO: 1. Hipotensión ortostática, por la desregulación noradrenérgica. Posibilidad de crisis hipertensiva, sindrome tiramínico o _sindrome del queso_, sobre todo con los IMAO irreversibles. La crisis hipertensiva por IMAO debe ser tratada con un bloqueador de los receptores Alfa 1, de acción rápida y corta vida media como la fentolamina (Regitina) , i.v., 5-10 mg. En su defecto prazocina. 2. Hepatotoxicidad: ictericia hepatocelular (más grave con los compuestos hidrazínicos). 3. Irritabilidad, hiperreflexia, trastornos de la eyaculación y de la líbido, agitación y alucinaciones, sobre todo en casos de sobredosificación. Inhibidores de la Recaptación de 5-HT. La fluoxetina y sus congéneres no producen efectos anticolinérgicos, ni neurotóxicos. Tampoco producen hipotensión ortostática o trastornos del ritmo cardíaco. Por ello son considerados agentes más seguros que los AD tricíclicos o los IMAO. 1. Sin embargo, son frecuentes otros efectos adversos menores como las nauseas, la anorexia y la diarrea. También ansiedad, nerviosismo, insomnio, mareos, fatiga o astenia, sudoración y disfunción sexual como disminución de la líbido y trastornos de la eyaculación. La frecuencia de estos los efectos adversos es de 5 a 30 %. Un pequeño porcentaje de pacientes debe suspender la administración de fluoxetina por los efectos adversos, pero debido a la vida media relativamente prolongada de este agente y su metabolito activo, norfluoxetina, la desaparición de los efectos adversos puede demorar en resolverse. Otros efectos frecuentes derivan de sus acciones estimulantes: ansiedad, nerviosismo, temblor e insomnio. 2. Pueden desencadenar hipomanía o manía franca, sobre todo en pacientes con trastornos bipolares. También reacciones psicóticas o paranoides. Se han registrado reacciones distónicas, convulsiones, disquinesia orolingual, acatisia y empeoramiento de reacciones extrapiramidales en pacientes tratados con neurolépticos. 3. Poco después de la comercialización de la fluoxetina, se describieron sospechas acerca que podría inducir ideación suicida y comportamiento violento. El primer efecto (ideación suicida), ha sido descartado en la actualidad pero se sigue dudando que no aumente la agresividad y la hostilidad. 5.10 INTERACCIONES MEDICAMENTOSAS MAYORES Los agentes antidepresivos pueden provocar graves interacciones cuando se administran conjuntamente y además interaccionan con otros fármacos o comidas por lo que su uso terapéutico debe ser cuidadoso en todos los casos. La interacción de AD tricíclicos con AD IMAO puede desencadenar graves efectos caracterizados por hiperpirexia, excitación del SNC, convulsiones, hipertensión arterial y coma. Su mecanismo no es bien conocido. Se aconseja un periodo libre de fármacos de por lo menos 14 días para prescribir el otro. En la actualidad se ha experimentado con la combinación de IMAO con tricíclicos, en dosis pequeñas, vigilando sus efectos. Los AD tricíclicos también potencian la acción del alcohol y otros depresores del SNC. Los efectos de los AD tricíclicos pueden potenciarse si se administran conjuntamente con fenitoina, fenilbutazona, aspirina, neurolépticos derivados de la fenotiazina, ya que los mismos desplazan a los tricíclicos de las proteínas transportadoras plasmáticas, como la albúmina, incrementando su fracción libre. De la misma manera también potencian sus efectos por inhibición del metabolismo los anticonceptivos hormonales y otros esteroides, el metilfenidato, otros neurolépticos, la cimetidina, el propranolol. Finalmente, los barbitúricos y el tabaquismo activan su metabolismo por inducción enzimática. Los IMAO producen un marcado incremento de las concentraciones de las aminas biógenas en el sistema nervioso. Por lo tanto la administración conjunta de agentes como las anfetaminas, efedrina, tiramina (presente en muchas comidas, vinos añejos, quesos estacionados), levodopa, aminas simpaticomiméticas, nafazolina, pseudoefedrina y otras, producen un desplazamiento de las catecolaminas almacenadas que pasan al espacio intersináptico y pueden desencadenar graves reacciones hipertensivas. Se han descrito accidentes cerebrovasculares y muerte por este mecanismo. Los inhibidores de la recaptación de 5-HT, fluoxetina y derivados, también pueden producir graves interacciones. La interacción con AD tricíclicos ya fué mencionada. La fluoxetina inhibe el CYP2D6, el resultado de la administración conjunta puede entonces producir un aumento de la concentración plasmática de los ADT de 4 a 5 veces la concentración inicial con efectos de intoxicación por estos últimos. La interacción con neurolépticos puede también ser de gravedad. Es un efecto de inhibición enzimática de la fluoxetina para los neurolépticos y viceversa. Por ejemplo la concentración de haloperidol puede incrementarse marcadamente si se lo administra en conjunto con fluoxetina. La administración conjunta de fluoxetina y AD IMAO, puede desencadenar el síndrome de la serotonina caracterizado por gran ansiedad, inquietud extrema, temblores, escalofríos, incoordinación, insomnio y efectos cardiovasculares. Este síndrome puede ser grave y fatal. Ha ocurrido incluso en pacientes que han suprimido la fluoxetina una o dos semanas antes de los IMAO. La combinación de fluoxetina y carbamazepina puede también desencadenar el síndrome de la serotonina. Otros agentes no psicotrópicos, como algunos betabloqueantes del tipo del metoprolol y varios antiarrítmicos son también metabolizados por la enzima CYP2D6 y por lo tanto su combinación con fluoxetina puede resultar en intoxicación por dichos fármacos. 6. PSICOESTIMULANTES Y ADICCIONES 6.1. TERMINOLOGÍA – Drogodependencia: se define como el estado físico y psíquico resultante de la interacción de una droga y el organismo. Se define adicción como la compulsión de consumir drogas con pérdida del control de lo que se consume y de la valoración de las consecuencias adversas del consumo. – Intoxicación: efectos psicológicos y físicos de la sustancia que desaparecen cuando es eliminada. – Abstinencia: síntomas y signos que surgen tras la suspensión o disminución del consumo de la sustancia. Son diferentes para cada sustancia. – Tolerancia: estado en el que la administración repetida de la sustancia conduce a un menor efecto. – Dependencia: se dice que un sujeto presenta un síndrome de dependencia si en el último año ha presentado tres o más de los siguientes puntos: a) deseo intenso o compulsión; b) problemas para dejar de tomar la sustancia o limitar la cantidad; c) cuadro fisiológico de abstinencia cuando se ha disminuido o suspendido el consumo; d) presencia de tolerancia, requiriendo dosis crecientes para obtener el mismo efecto que al principio; e) abandono progresivo de otras fuentes de placer o diversiones en favor del consumo de la sustancia, debido al tiempo que se emplea en conseguirla o recuperarse de sus efectos; y f) sigue consumiendo a pesar de los evidentes perjuicios que esto le ocasiona. Los criterios para el abuso de sustancias, a diferencia de la dependencia, no incluyen tolerancia, abstinencia ni el patrón de uso compulsivo, y en su lugar se citan las consecuencias dañinas del consumo repetido (p. ej. expulsión de la escuela en relación al uso de la sustancia) 6.2. CLASIFICACIÓN Drogas depresoras del sistema nervioso central: tienen en común su capacidad para entorpecer el funcionamiento habitual del cerebro, provocando reacciones que pueden ir desde la desinhibición hasta el coma, en un proceso progresivo de adormecimiento cerebral. Las más importantes son: a) alcohol; b) opiáceos: heroína, morfina, metadona, etc.; c) tranquilizantes: pastillas para calmar la ansiedad; y d) hipnóticos: pastillas para dormir. Drogas estimulantes del sistema nervioso central: aceleran el funcionamiento habitual del cerebro. Destacan: a) estimulantes mayores: (euforia, pupilas dilatadas, taquicardia) anfetaminas y cocaína; b) estimulantes menores: nicotina; y c) xantinas: cafeína, teobromina, etc. Drogas perturbadoras del sistema nervioso central: dan lugar a distorsiones perceptivas, alucinaciones, etc.: a) alucinógenos: LSD, etc.; b) derivados del cannabis: hachís, marihuana, etc.; y c) drogas de síntesis: éxtasis, etc. 6.3. EFECTOS PERJUDICIALES ASOCIADOS AL CONSUMO DE SUSTANCIAS – Físicos: trastornos gastrointestinales, trastornos del sistema nervioso. – Neuropsiquiátricos: amnesias, trastornos depresivos, ansiedad, cambios de la personalidad, conducta suicida, , disfunción sexual, alucinaciones. – Sociales: violencia familiar, problemas emocionales y de la conducta en los hijos, escaso rendimiento laboral. – Intoxicación: supone un efecto tóxico directo sobre ciertos tejidos, sobre todo hígado y cerebro. – Consumo crónico: puede conducir a una dieta insuficiente y déficit de proteínas y vitaminas de grupo B, susceptibilidad a infecciones. – Dependencia: graves síndromes de abstinencia. El síntoma más precoz de la abstinencia es el temblor, al que se le pueden añadir náuseas, sudoración, crisis epilépticas y, al cabo de 48 horas, puede desarrollarse el delirium tremens. – Propiedades tóxicas de la sustancia: como por ejemplo las drogas estimulantes pueden llegar a producir arritmias cardiacas, hipertermia, shock cardiovascular y paro cardiaco. – El método de administración: la administración intravenosa de drogas puede producir infecciones en el punto de inyección, trombosis venosas, transmisión de infecciones como el VIH y hepatitis. 6.4. TRATAMIENTO 6.4.1. ALCOHOL Tratamiento de la intoxicación alcohólica aguda. La intoxicación alcohólica requiere tratamiento urgente cuando cursa con agitación, agresividad o coma. Ante un paciente agitado suelen ser necesarias la sedación y la contención mecánica. Para la sedación se recomienda haloperidol por vía intramuscular. Síndrome de abstinencia alcohólica. En general se utilizan tratamientos sustitutivos, es decir, fármacos que presentan tolerancia cruzada con el etanol. Los tratamientos más utilizados son las benzodiacepinas. Síndrome de dependencia alcohólica. Se propone, en primer lugar, una pauta de desintoxicación comenzando con una benzodiacepina. Se han estudiado otros fármacos como la gabapentina y la carbamacepina. Para la posterior deshabituación, se ha utilizado en Europa fármacos que actúan a distintos niveles: los fármacos antidipsicotrópicos disulfiram y cianamida cálcica se han utilizado en las últimas décadas con resultados diversos. 6.4.1.1. ACTUALIDAD EN EL TRATAMIENTO – Disulfiram: interfiere en el metabolismo del alcohol bloqueando una de las enzimas implicadas. Se debe tener precaución ya que si se ingiere alcohol mientras se toma este fármaco, se acumula acetaldehído, provocándose enrojecimiento, cefalea, sensación de ahogo, taquicardia y ansiedad. – Acamprosato: se utiliza para disminuir los deseos de ingerir alcohol una vez desintoxicado, Ha demostrado en diversos ensayos clínicos controlados su superioridad frente al placebo para disminuir la tasa de recaídas a los 6 y 12 meses de tratamiento. La dosis habitual es de 2 comprimidos, tres veces al día. Efectos adversos: diarrea, prurito, mareo o cefalea. No se metaboliza por el hígado, por lo que es mejor tolerado por el paciente hepatópata. – Naltrexona: es un antagonista competitivo de los receptores opioides con capacidad para reducir el consumo excesivo de alcohol en estudios experimentales. Los estudios controlados en seres humanos han demostrado que la naltrexona reduce el consumo de alcohol y la tasa de recaídas, tanto en pacientes dependientes de alcohol como en los bebedores excesivos. – Otros: antidepresivos de perfil serotoninérgico; valproato, carbamacepina, gabapentina y topiramato. El valproato y la carbamazepina se pueden asociar con hepatotoxicidad, por lo que están contraindicados en alcohólicos con complicaciones hepáticas comórbidas. Se han realizado estudios observándose que los que tomaron topiramato consiguieron una mayor reducción del consumo, del deseo de tomar alcohol y de los niveles de GGT, que los que tomaron placebo. En la actualidad se están realizando estudios con fármacos como la oxcarbacepina, mirtazapina y vardenafilo entre otros. 6.4.2. Heroína Intoxicación aguda. La intoxicación aguda requiere tratamiento por los efectos depresores sobre la función respiratoria y el riesgo vital. – Naloxona: es antagonista opiáceo. La reversión del cuadro puede desencadenar un síndrome de abstinencia. Síndrome de abstinencia. Desde el punto de vista farmacológico puede tratarse de distintas maneras: – Sustituir el opiáceo de semivida corta por otro agonista opiáceo de semivida más larga (p. ej. metadona, dextropropoxifeno o brupenorfina). - Se puede administrar un agonista alfa-2-adrenérgico (p. ej. clonidina), y b) administrar de forma combinada clonidina y metadona. Síndrome de dependencia. Se puede sugerir la utilización de programas libres de drogas, programas de mantenimiento con sustitutivos y programas de mantenimiento con antagonistas. Dependiendo de diversos factores, los pacientes serán tributarios de uno u otro abordaje. Programas de mantenimiento con sustitutivos. La metadona y brupenorfina han demostrado su elevada eficacia y efectividad. 7. ANTIPARKINSONIANOS. La enfermedad de Parkinson (EP) es un proceso crónico y degeneración neuronal en la sustancia nigra, lo que conlleva una dopamina. Es una enfermedad clínicamente heterogénea, por lo puede variar de un paciente a otro y, si bien afecta a individuos aparece de manera más temprana (antes de los 40 años). progresivo provocado por la disminución en los niveles de que el grado de incapacidad de más de 45 años, también Su etiología es desconocida y probablemente multifactorial. Pueden estar implicados factores genéticos, ambientales, daño oxidativo y envejecimiento acelerado cerebral u apoptosis. Tiene la misma distribución de raza y sexo. Los síntomas guía son: - Temblor. Se caracteriza por ser de reposo, aunque a veces se presenta al mantener una postura. - Bradicinesia (enlentecimiento de los movimientos) acinesia (dificultad para el movimiento) e hipocinesia (reducción de la amplitud de los movimientos). Afecta principalmente a la cara y músculos axiales, por lo que se convierte en uno de los síntomas más incapacitantes. - Rigidez. Provocada por el aumento del tono, conlleva una mayor resistencia para la realización del movimiento pasivo. - Alteración de los reflejos posturales. - Afectación de la estabilidad y el equilibrio, provoca caídas frecuentes y la típica marcha en festinación. Trastornos cognitivos y neuropsiquiátricos: Deterioro cognitivo y demencia, depresión, ansiedad y ataques de pánico, alucinaciones y psicosis. - Trastornos del sueño. - Trastornos del habla y de la deglución. - Trastornos sensoriales, síndrome de las piernas inquietas, neuropatía periférica. - Alteraciones autonómicas, estreñimiento, trastornos genitourinarios, hipotensión ortostática, alteraciones de la termorregulación, olfato y sudor, dolor, seborrea y blefaritis. 7.1. TRATAMIENTOS Selegelina. Es un inhibidor selectivo de la monoaminooxidasa (IMAO). Produce náuseas, alucinaciones e hipotensión ortostática. Levodopa. Fármaco que sustituye la dopamina. Mejora la discapacidad y mortalidad. Su efecto mejora la rigidez y la bradiquinesia, pero tiene menos efecto en el temblor y en la inestabilidad postural. Se utiliza actualmente junto con inhibidores de la dopa descarboxilasa feriférica, como carbidopa y benseracida. La administración de levodopa exógena mejora la transmisión dopaminérgica estriatal y las manifestaciones clínicas. Esta opción terapéutica adolecía de un problema práctico importante. La levodopa presenta importantes efectos secundarios, debido a su acción fuera del sistema nervioso central. En la actualidad, las formulaciones que combinan levodopa más un inhibidor de la dopa-descarboxilasa, tanto carbidopa como benserazida, son la base del tratamiento de la EP. Carbidopa. Se suele combinar con levodopa. Impide la conversión de levodopa en dopamina en el sistema nervioso periférico. Agonistas dopaminérgicos. Los medicamentos de este grupo son bromocriptina, lisurida, pergolide, pramipexol, ropirinol. La bromocriptina y pergolide son derivados de la ergotamina, bien tolerados, aunque sus efectos secundarios incluyen alteraciones fibróticas (retroperitoneo, pleura y pulmón). – Pramipexol se ha asociado con somnolencia. Sus efectos secundarios incluyen alteraciones cardiovasculares (hipotensión ortostática), del SNC (somnolencia, alucinaciones, sueño alterado), gastrointestinales y neuromusculares. – Ropirinol se ha asociado con bradicardia en estados iniciales del tratamiento de EP (sin levodopa) y en pacientes con EP avanzada (con levodopa). Debe utilizarse con cuidado en pacientes con discinesia previa o enfermedad hepática o renal severa. Puede producir somnolencia, náuseas, síncope y fatiga. Anticolinérgicos. Los anticolinérgicos son útiles en el tratamiento del temblor. Los más habituales son trihexifenidilo y biperideno. Los efectos adversos son los típicos de los anticolinérgicos (boca seca, confusión, estreñimiento). Amantadina. Es un medicamento con acción antiviral que además tiene una acción serotoninérgica por bloqueo de la recaptación de serotonina en la neurona presináptica, o por estimulación de la liberación de serotonina en las neuronas presinápticas. No debe utilizarse en una reacción distónica aguda y, en general, la amantadina se considera menos eficaz que los agentes antiparkinsonianos tradicionales en el tratamiento de la acatisia, debido a su coste se reserva como segunda línea. La ventaja sobre los anticolinérgicos es que no produce deterioro cognitivo y su tolerancia en tratamientos a largo plazo es buena. La dosis en el tratamiento de la EP es de 100 mg cada 12 horas en monoterapia. La sobredosificación origina trastornos digestivos (náuseas y vómitos) y convulsiones que pueden controlarse con diacepam. Inhibidores de la COMT. La catecol-O-metil-transferasa (COMT) es otra enzima que metaboliza levodopa. Los inhibidores de este enzima tienen el poder de aumentar la dopamina extracelular y pueden ser utilizados en el tratamiento de la EP, en estadios precoces o asociarse a la levodopa en estadios avanzados. Sin embargo, el riesgo de toxicidad central es alto. Los dos medicamentos que representan a este grupo son: entacapona y tolcapona. – Tolcapona permite reducir la dosis de levodopa, mejorando tanto los fenómenos de picos de dosis. También mejora las funciones congnitivas (memoria, atención). Los efectos adversos incluyen discinesias, náuseas, vómitos, distonía, calambres, hipotensión postural, diarrea. Su efecto adverso más importante es la toxicidad hepática mortal. – Entacapona se absorbe rápidamente, es altamente selectivo de la COMT y no es tóxico. Permite la reducción de la dosis de levodopa en un 12%. No tiene toxicidad hepática que exija una vigilancia especial y sus efectos secundarios más frecuentes son: discinesia, náuseas, diarrea y mareos. 7.2. RECOMENDACIONES DE REVISIONES DE ESTUDIOS CLÍNICOS - Hasta el momento, ningún fármaco ha demostrado tener propiedades neuroprotectoras que pudieran detener el curso de la enfermedad. - Es recomendable iniciar el tratamiento cuando el paciente comience a notar que la sintomatología interfiere en sus actividades de la vida diaria y en su calidad de vida. Algunos autores recomiendan empezar el tratamiento con Levodopa en estadios iniciales para obtener el máximo beneficio en la función motora, otros prefieren instaurarlo con otros fármacos y esperar a usar la Levodopa cuando la clínica sea más severa, para retrasar la aparición de las complicaciones motoras. - En los pacientes con EP en los que se decide iniciar el tratamiento farmacológico se recomienda iniciarlo con Agonistas Dopaminergicos o con Levodopa individualizando la decisión en cada paciente, sopesando las necesidades de mejorar la disfunción motora, en cuyo caso sería mejor empezar con Levodopa o por el contrario si es preferible retrasar las complicaciones motoras, hacerlo con Agonistas Dopaminergicos. - No existen en la actualidad datos concluyentes acerca del mejor control de los síntomas motores en las fases iniciales de la EP si se utilizan los AD asociados desde el principio a la Levodopa, o esta última junto con la Selegilina frente a la Levodopa sola. 8. ANTIEPILÉPTICOS. La epilepsia es una afección crónica, de etiología diversa, caracterizada por crisis recurrentes debidas a una descarga excesiva de las neuronas cerebrales (crisis epiléptica) asociada eventualmente con diversas manifestaciones clínicas o paraclínicas. Las crisis epilépticas únicas, las ocasionales o las que aparecen durante una enfermedad aguda no constituyen una epilepsia. Es útil desde el punto de vista clínico y etiológico distinguir la epilepsia entre aquella que se origina como una descarga eléctrica generalizada de la que se genera en un lugar concreto del cerebro, para propagarse posteriormente y hacerse generalizada. Tabla 4. Etiología de la epilepsia según la edad. Hipoxia e isquemia perinatal. Infección (meningitis, encefalitis, abscesos cerebrales). Traumatismos craneoencefálicos (TCE). Trastornos metabólicos (hipoglucemia, hipocalcemia, hipomagnesemia, déficit de piridoxina). Malformaciones congénitas. Trastornos genéticos. Infancia (<12a) Crisis febriles. Infecciones. TCE. Tóxicos y defectos metabólicos. Enfermedades degenerativas cerebrales. Idiopáticas. Neonatos Adolescencia 18-35 años 35-50 años >50 años Idiopáticas. TCE. Infecciones. Enfermedades degenerativas cerebrales. Alcohol. TCE. Alcoholismo. Tumores cerebrales (primarios y secundarios). Tumores cerebrales (primarios y secundarios). Accidente vascular cerebral. Uremia, hepatopatía, hipoglucemia, alteraciones electrolíticas. Alcoholismo. Accidente vascular cerebral (como secuela). Tomado de: Zarranz JJ. Epilepsias. En: Farreras P, Rozman C editores. Medicina Interna. 145ª ed. Madrid: Elsevier; 2004. p. 1406-1423 8.1. Tipos de Epilepsias 8.1.1. Crisis generalizada. La descarga comienza en ambos hemisferios a la vez y se propaga de manera simétrica. – Ausencia (petit mal). Los típicos ataques de ausencia son breves y sorpresivos. Suelen producirse en complejos. Los ataques de ausencia pueden producir pérdida de consciencia, en otras ocasiones el paciente mira fijamente, con una total falta de fijación en su mirada. – Mioclonía. Breves y repentinos movimientos bruscos de la parte superior del tronco y ocasionalmente de las piernas. – Tónica-clónica (gran mal). Tradicionalmente, hay cinco fases en la epilepsia tónico-clónica: flexión, extensión, temblor, fase clónica y fase postictal. En la fase de flexión, el paciente mantendrá parcialmente abierta la boca, los ojos pueden sufrir una inversión y el paciente puede perder la consciencia. En la fase de extensión, el paciente sufre una extensión de su espalda y de su cuello, los músculos abdominales se ponen en tensión. Puede volverse apneico y sufrir flexión y extensión de los músculos de la pierna. El paciente puede gritar en esta fase, a causa de la expulsión de aire de sus pulmones. La fase de temblores ocurre mientras el paciente está realizando un tránsito entre la fase tónica, de enorme rigidez, hacia la clónica, de ligeras contracciones. La fase clónica se caracteriza por rítmicas contracciones similares a escalofríos. Después del ataque, el paciente se encuentra en estado postictal. La duración total suele ser de 3 minutos. – Clónica. Sería la epilepsia que sólo presenta ataques de tipo clónico (contracciones rítmicas y regulares seguidas de relajación). – Tónica. Sería la epilepsia que sólo presenta un estado de contracción continua con elevada rigidez (combina la fase de flexión y extensión de la tónica-clónica). – Atónica. Se caracteriza por la falta de tono muscular. El paciente pierde tensión en el músculo y suele caer al suelo. 8.1.2. Crisis parcial (epilepsia focal). La crisis se produce en uno de los hemisferios. – Simple. No existe pérdida de consciencia durante el ataque. Los síntomas son motores (afectan a cualquier parte del cuerpo), autonómicos (palidez, sudoración, vómitos, vértigo o taquicardia) o sensoriales (visuales, auditivos, olfativos y gustativos). – Compleja. Se produce pérdida de la consciencia. Suele precederse de un pródromo y comienzan con un aura. El aura que acompaña a la epilepsia parcial compleja suele ser un ataque parcial con sintomatología autonómica, el paciente suele sentir miedo y tiene la sensación de déjà vu. – Secundaria generalizada. Comienza como una crisis parcial, pero se propaga por los dos hemisferios y se convierte en generalizada. 8.1.3. Status epilepticus. Es una crisis generalizada que se mantiene durante 30 minutos o varias crisis cortas que se encadenan en el tiempo impidiendo que el paciente esté consciente. 8.1.4. Pseudocrisis. Son episodios que parecen crisis convulsivas epilépticas pero que tienen un origen orgánico o psicógeno. 8.5. TRATAMIENTO Tabla 5. Estrategias farmacológicas en terapia antiepiléptica Incrementar el umbral de excitación neuronal - Bloqueantes de canales iónicos de Na+ y Ca2+ Modulación de la neurotransmisión aminoacidérgica • Potenciación de la transmisión gabaérgica - Agonistas de los receptores del GABA - Inhibidores de la degradación del GABA - Inhibidores de la recaptación del GABA • Inhibición del sistema glutamatoaspartato - Inhibidores de la liberación de glutámico - Antagonistas de los receptores de glutámico 8.5.1. ANTICONVULSIVANTES PRINCIPALES - Carbamacepina. Bloqueante del canal de sodio. Es un potente inductor enzimático que autoinduce su metabolismo, lo cual exige que la dosificación en las primeras 4-8 semanas sea algo superior con el fin de contrarrestrar la autoinducción. Sus principales efectos adversos son rash, síndrome de liberación inapropiada de hormona antidiurética, anemia, anemia aplásica, trombocitopenia, leucopenia. No se recomienda combinarlo con otros antiepilépticos. - Etosuximida. Bloqueante del canal T del calcio. Sustituyo la Trimetadiona. Es de elección en las crisis de ausencia. Sus principales efectos secundarios son nauseas y anorexia. - Fenitoína. Bloquea los canales de sodio. Es un inductor enzimático. Su administración es i.v. y presenta como principales efectos adversos la hipotensión y la aparición de flebitis. La velocidad de administración debe de ser no superior a 50 mg/minuto, siempre diluido en solución fisiológica. Además pueden presentarse los siguientes efectos adversos relacionados con la dosis: nistagmus, ataxia, alteraciones cognitivas; y no relacionados con la dosis: hiperplasia gingival, hirsutismo, acné, rash, hepatotoxicidad. - Valproato. Aumenta la producción del GABA, bloquea los canales T del calcio y los canales del sodio. Es un inductor enzimático. En ocasiones la formulación parenteral se utiliza para controlar el status epilepticus. Sus efectos adversos son hepatotoxicidad, ganancia de peso, pancreatitis, alopecia, náuseas y vómitos e interferencia en la agregación plaquetaria. 8.5.2. FÁRMACOS DE SEGUNDA LÍNEA - Fenobarbital. Aumenta la afinidad del GABA y su acción sobre el ionóforo del cloro. Inductor enzimático. Es un barbitúrico, es ineficaz en el tratamiento de las ausencias al igual que la fenitoina. Primidona, hoy por hay en desuso, actúa tras su conversión a fenobarbital. Las aplicaciones clínicas de fenobarbital son prácticamente idénticas a la fenitoina, pero se prefiere esta última por que no produce sedación. Produce hiperactividad y alteración cognitiva. - Benzodiacepinas. Son sustancias no recomendables para esquemas terapéuticos a largo plazo por los problemas de tolerancia y dependencia. Las más habituales son clonacepam, diacepam y cloracepato. 8.5.3. FÁRMACOS MÁS RECIENTES - Vigabatrina. Actúa por inhibición de la GABA transaminasas. Eficaz en pacientes que no responden a los fármacos convencionales. Sus principales efectos secundarios son somnolencia, alteraciones del comportamiento y del estado de ánimo. - Lamotrigina. Bloquea los canales de sodio y reduce la liberación de glutamato y aspartato, evitando la descarga repetitiva de las neuronas. Es de amplio perfil terapéutico. El rash es un problema importante en el manejo de este medicamento. La dosis debe escalarse con cuidado para evitarlo. El ácido valproico reduce el metabolismo de lamotrigina, con lo que debe extremarse el escalado, situándose en dosis inferiores a las normales. - Felbamato. Análogo del meprobamato (un carbamato). No se conoce su mecanismo de acción. Tiene una acción saturable, por tanto, seguro en caso de sobredosis. Relativamente exento de efectos secundarios. - Gabapentina. No se conoce su mecanismo de acción. Es necesario el ajuste de dosis en caso de insuficiencia renal. La dosis debe escalarse para que su biodisponibilidad no se vea afectada. También tiene indicación en el dolor neuropático. - Tiagabina. Inhibidor de la captación del GABA Presenta un perfil toxicológico relativamente benigno. Sus efectos secundarios son mareos y confusión. Aún no esta completamente evaluado. - Topiramato. Bloquea los canales del sodio, potencia la acción del GABA y tiene débil acción como inhibidor de la anhidrasa carbónica. No se metaboliza de manera extensa y se elimina por el riñón. Similar a la fenitoian. Produce pérdida de peso, reducción de la motilidad, parestesias, acidosis metabólica e hipertermia (asociada a reducción de sudoración: oligohidrosis). Tener cuidado por su riesgo de teratogenia. 8.6. OTROS FÁRMACOS ANTIEPILEPTICOS DE ACTUALIDAD - Fosfenitoína. Es un profármaco de fenitoína. Bloquea los canales del sodio. Tiene una presentación parenteral para poder abordar con dosis de ataque o de mantenimiento aquellas situaciones susceptibles de ser tratadas con fenitoína (status epilepticus. Los efectos adversos son hipotensión y picor perianal. - Levetiracetam. Impide la sincronización en la propagación de la crisis, con lo que se detiene su desarrollo. No sufre metabolización y es necesario ajustar la dosis en caso de insuficiencia renal. No presenta interacciones importantes. - Oxcarbamacepina. Bloquea los canales de sodio. Es un inductor enzimático pero no presenta autoinducción. La hiponatremia es más frecuente que con carbamacepina, pero las discrasias sanguíneas son menos frecuentes que con carbamacepina. - Primidona. Aumenta la afinidad del GABA y su acción sobre el ionóforo del cloro. Se metaboliza a fenobarbital y feniletilmalonamida (PEMA). Inductor enzimático. También se utiliza en el temblor esencial. - Ozcarbazepina derivado de la carbamazepina, similar desde el punto de vista químico y farmacológico pero con diferencias farmacocinéticas, en especial en lo que se refiere a las vías de metabolización. Es un profármaco que es transformada rápidamente y casi por completo en el derivado monohidroxilado, conocido como MHD (10,11-dihidro- 10-hidroxicarbamazepina), complejo similar a la carbamacepina con acción muy similar a ésta. - Valpromida (amida del ácido valproico), tienen uno de los espectros antiepilépticos más amplios de todos los fármacos conocidos del grupo. TABLA 6. CLASIFICACIÓN DE LA EPILEPSIA Y TRATAMIENTO DE Tipo De elección De segunda Otros fármacos línea a tener en cuenta Focales o Carbamazepina a Clobazam Acetazolamida secundariamente Lamotrigina b Clobazam Clonazepam generalizadas Oxcarbazepinaa,b Gabapentina Fenobarbital Ácido Valproico Levetiracetam Fenobarbital Topiramato a Fenitoína a Primidona Tiagabina Generalizadas Carbamazepina a Clobazam Acetazolamida tónico-clónicas Lamotrigina b Levetiracetam Clonazepam Ácido Valproico Oxcarbazepina a Fenobarbital a a,b Topiramato Fenitoína a Primidona a Mioclónicas Ácido Valproico Clobazam Clonazepam ELECCIÓN Fármacos contraindicados Tiagabina Vigabatrina Carbamazepina a Gabapentina (Topiramato) a,d Tónicas Lamotrigina b Ácido Valproico Atónicas Lamotrigina b Ácido Valproico Ausencias Etosuximida Lamotrigina b Ácido Valproico Lamotrigina Levetiracetam Piracetam Topiramato a Clobazam Clonazepam Levetiracetam Topiramato a Clobazam Clonazepam Levetiracetam Topiramato a Clobazam Clonazepam Topiramato a Oxcarbazepina a Tiagabina Vigabatrina Acetazolamida Fenobarbital a Fenitoína Primidona a,c Acetazolamida Fenobarbital a Primidona a,c Carbamazepina a Oxcarbazepina a Carbamazepina a Oxcarbazepina a Fenitoína a Carbamazepina a Gabapentina Oxcarbazepina a Tiagabina Vigabatrina (a)Inductor de las enzimas hepáticas (b)Solo se deben usar como primera elección en las recomendaciones de NICE (c)Rara vez es utilizado, si se requiere, el fenobarbital es de elección (d)En la epilepsia mioclónica severa de la infancia Tomado de: http://www.fisterra.com/guias2/epilepsia.asp#Definiciones. 8.7. OTROS TIPOS DE TRATAMIENTO ANTICONVULSIVANTE 8.7.1. CIRUGÍA. Un 20-30% de los pacientes no responden al tratamiento farmacológico. La mayoría de los pacientes cuyas crisis se originan en un área focal cerebral con función anormal mejoran de forma significativa, e incluso se produce curación completa cuando se elimina o se desconecta dicha zona. Se calcula que tan solo en el 10-25% de los pacientes farmacorresistentes está indicada la cirugía de la epilepsia. Las modalidades quirúrgicas antiepilépticas actualmente disponibles son: Extirpación del área epileptógena. Reservada para casos en los que se ha identificado un foco único con presencia o no de lesión estructural. Es la técnica más utilizada, siendo la indicación típica de los casos de epilepsia temporal con esclerosis mesial del hipocampo. Las técnicas empleadas son: Desconexión del área epileptógena. La callosotomía es usada actualmente de forma parcial y limitada a los 2/3 anteriores del cuerpo calloso. Está indicada en casos de focos múltiples con crisis incontrolables. Las técnicas más aplicadas son: 8.7. PROFILAXIS. Las convulsiones febriles infantiles, incluso las complicadas, tienen un riesgo muy bajo de epilepsia o secuelas neurológicas permanentes. La profilaxis continuada con fenobarbital o ácido valproico sólo está justificada en casos muy especiales. La administración de diazepam en caso de fiebre alta (más de 38 ºC) reduce de forma muy significativa el riesgo de convulsión o las posibles complicaciones en caso de producirse. 9. ENFERMEDAD DE ALZHEIMER (EA) La demencia se define como un síndrome adquirido de alteración intelectual persistente que compromete la función de múltiples esferas de la actividad mental tales como la memoria, el lenguaje, las habilidades viso espaciales, la emoción o la personalidad y la cognición. La EA refiere la aparición de múltiples déficits cognitivos (trastornos de memoria, afasia, aprasia, agnosia o alteraciones de las funciones ejecutivas). La alteración no se debe a la situación del sistema nervioso central (ictus, tumor, enfermedad de Parkinson), condiciones sistémicas (infección VIH, hipotiroidismo, déficit vitamínico) o situaciones derivadas del consumo de sustancias adictivas. Los cambios en la capacidad cognitiva representan una disminución significativa de la autonomía social y ocupacional del paciente. Las alteraciones cognitivas no ocurren al mismo tiempo que el delirio. La historia familiar es el principal factor de riesgo: entre el 25 y 50% de los familiares de enfermos con EA puede padecer la enfermedad, frente al 10% entre aquellos que no poseen familiares. Los factores de riesgo mejor conocidos para EA son: edad elevada, parentesco, síndrome de Down y tener el alelo E4 para la lipoproteína. El rasgo patológico más característico es la presencia de placas de tejido fibroso (placas neuríticas o placas amieloides) en el hipocampo y córtex cerebral. Estas placas están formadas por proteína beta-amieloide, cuya agregación se favorece con elementos oxidativos como los radicales libres. 9.1. TRATAMIENTO Inhibidores de la colinesterasa (IAche). Actualmente, los inhibidores de la colinesterasa constituyen la primera línea de tratamiento en EA y no sólo mejoran las áreas cognitiva y del comportamiento, sino que parecen tener una influencia directa en la supervivencia y mantenimiento de la función neuronal. – Donepezilo: es el medicamento de elección. Se dosifica una vez al día y no presenta hepatotoxicidad. Los efectos adversos son de tipo digestivo (náuseas, vómitos, diarrea), junto con insomnio, calambres musculares y anorexia. No es hepatotoxico. – Rivastigmina: No presenta interferencias a nivel del sistema CYP y no altera la función hepática. Los efectos adversos típicos son de tipo gástrico. Dado que la inhibición de la acetilcolinesterasa es diferente a la que establecen tanto la tacrina como el donepezilo, los pacientes que hayan fracasado a estos medicamentos debieran intentar tratamiento con rivastigmin. Debe administrarse con algo de comida, en el desayuno y en la cena. Es de acción más prolongada que el donepezilo. – Galantamina: eleva la cantidad de acetilcolina por inhibición de la Ache (inhibición competitiva y reversible), actuando también como agonista en los receptores nicotínicos. Debe administrarse con el desayuno y la cena. En el caso de que se interrumpa el tratamiento durante 3 días, debe comenzarse de nuevo por la dosis más baja y escalar dosis. Los efectos adversos son: náuseas, vómitos, diarrea, mareo y anorexia. – Tacrina: está asociada a hepatotoxicidad y presenta múltiples interacciones clínicas. Es necesario dosificarla en varias tomas y los efectos adversos incluyen náuseas, vómitos, diarrea, anorexia, dispepsia, temblor, sudor, mialgia y acaloramiento. Los efectos gastrointestinales se minimizan administrándola con la comida, aunque la biodisponibilidad se reduce en un 30-40%, por lo que es conveniente administrarla una hora antes de las comidas. Otros fármacos – Memantina: Inhibidor del receptor N-metil-aspartato. El resultado terapéutico es un retraso en la evolución de la enfermedad más que una mejoría de la situación clínica. La memantina es bien tolerada, aunque pueden aparecer reacciones adversas de tipo neurológico (alucinación, confusión, cefalea, vértigo, ansiedad) y en menor frecuencia digestivo (náuseas, vómitos). – Vitamina E. Existe evidencia de que los compuestos con propiedades antioxidantes tienen efecto neuroprotector y pueden reducir el daño neuronal. Por otro lado, la acción de los radicales libres y sustancias oxidativas llevan a la agregación de la proteína amieloide. – Selegilina: es un potente inhibidor de la monoaminoxidasa (MAO) de tipo B. Su indicación aprobada es el tratamiento del Parkinson en estadios iniciales, como coadyuvante de la levodopa. Se puede utilizar como uso compasivo en el THDA, para tratar la sintomatología negativa en pacientes con esquizofrenia, en depresión y en EA. Los efectos adversos son hipotensión ortostática, ansiedad, irritabilidad y mareo. 9.2. MEDICAMENTOS ACTUALES PARA TRATAR LA DEMENCIA En la actualidad se vienen utilizando diversos medicamentos para tratar EA, entre ellos, posibles vasodilatadores, dihidroergotamina, agonistas muscarinicos; arecolina y pilocarpina, potenciadores de la cognición; piracetam y aniracetam. Además algunos antiinflamatorios (ibuprofeno e indometacina) y el clioquinol (quelantes de los metales) retrasan la neurodegeneración. 11. ANESTESIA GENERAL La anestesia general AG puede definirse de varias maneras: - Es un estado reversible de depresión del SNC, donde hay pérdida de la conciencia (hipnosis), de la sensibilidad (analgesia), de la actividad refleja (protección neurovegetativa) y de la motilidad (relajación muscular), situación que obtenemos con el uso de los anestésicos generales, cuando actúan sobre los diferentes órganos y sistemas del organismo, especialmente sobre el SNC. - Forma de anestesia en la que el paciente está inconsciente y no reacciona a estímulos dolorosos - Inducción farmacológica de la ausencia de percepción de todas las sensaciones Entonces, la anestesia general implica un estado de inconsciencia reversible en el cual el paciente ni percibe (ni recordará después) efectos nocivos de la cirugía. La progresión del efecto anestésico se realiza: 1. Corteza 2. Mesencéfalo y ganglios basales 3. Cerebelo 4. Médula espinal 5. Bulbo La recuperación se hace en sentido inverso: siendo lo último en recuperarse, las funciones corticales. No es un fin con AG llegar a la depresión bulbar dada la generación de paro cardio-respiratorio que puede hacer irreversible el proceso y llevar a la muerte cerebral. El tamaño de las pupilas (midriasis) y su poca respuesta a la luz, pueden ser indicadores de la intensidad de la depresión bulbar y de la hipoxia cerebral 11.1 ANESTESIA BALANCEADA: Es el resultado de combinar fármacos inhalados e intravenosos para lograr una mejor estabilidad de los signos vitales. CAM: concentración alveolar mínima Es la medida de potencia de los AG, expresada a nivel experimental como la dosis inhalada necesaria para producir analgesia en el 50% de la población expuesta. El CAM de los diversos AG es diferente 11.2. ETAPAS DE LA ANESTESIA Fueron definidas por Guedel y Artusio desde comienzos del siglo XX. Se sintetizan así: Etapa 1: Analgesia Disminución de la percepción del dolor y en ocasiones amnesia. Aún no se pierde la conciencia Etapa 2: Excitación Empieza con el ingreso a la inconsciencia, aunque existe un período de delirio y excitación: los reflejos se acentúan, puede aumentar la glicemia, la FC y la PA. Se experimenta una respiración irregular, náuseas e incontinencia. Etapa 3: Anestesia propiamente dicha Aunque a su vez se divide en 4 planos, esta etapa se caracteriza por la estabilidad de los Signos vitales, miosis, relajación muscular generalizada y parálisis de los músculos respiratorios. Es el momento ideal para las cirugías. Etapa 4: Depresión espinal completa y parálisis bulbar Depresión respiratoria y cardiovascular hasta el paro CR que puede llevar a la muerte Figura 3. Equipo de Inducción de Anestesia El período de inducción anestésica, farmacológicamente se compara con el periodo de latencia, es decir: el tiempo transcurrido entre la administración de un fármaco y la aparición de su efecto farmacológico básico: para el caso de los AG este efecto farmacológico es la pérdida de los reflejos superficiales (corneal, entre otros). Es muy frecuente que esta inducción se facilite con el empleo simultáneo de relajantes musculares intravenosos (succinil colina, galamina, dtubocurarina, pancuronio, atracurio, vecuronio, con lo cual el proceso de intubación endotraqueal y la conexión del paciente a una máquina de anestesia se haga más fácil. El período de inducción abarca las etapas 1 y 2 de la Anestesia general según Guedel y Artusio. Figura 4. Maquina de anestesia de circuito cerrado LA MÁQUINA DE ANESTESIA Toda máquina de anestesia debe realizar las siguientes funciones: a. Proporcionar cantidades medidas de gas anestésico, Oxígeno y Oxido Nitroso. b. Remover el CO2 exhalado. c. Proporcionar una trayectoria de baja resistencia que permita una fácil inhalación de la mezcla de gases. Un sistema anestésico consiste básicamente de 4 subsistemas: - Sistema de alimentación y control de gas. - Circuito de ventilación y respiración. - Sistema de purificación de gas. - Sistema de monitoreo. Normalmente las máquinas de anestesia utilizan un circuito cerrado, como el que se muestra en la Figura 1. Este tipo de sistema consiste de un vaporizador (VAP), la bolsa de inhalación, un arreglo de válvulas, un medidor múltiple de flujo ("manifold"), y válvulas de control del flujo para el oxígeno (y otros gases) que circula por el vaporizador y entran al circuito pasando por el absorbedor de CO2 (CO2A). El flujo de oxígeno mezclado con otros gases, como N2O, pasa a través del vaporizador tomando la concentración deseada del anestésico volátil. Luego la mezcla de gases pasa dentro del tubo de inspiración. El paciente inhala estos gases y exhala parte de ellos junto con CO2 hacia el absorbedor de CO2 (cal sodada). El gas espirado no fluye de regreso en el circuito, gracias a la válvula de inspiración (I). En el absorbedor, el CO2 es convertido a sólido. En esta reacción química, calor es liberado, y la cal sodada cambia de color en proporción a la cantidad de CO2 atrapado. La intensidad de color indica cuando el absorbedor pierde sus propiedades e indica cuando debería ser cambiado. Después de pasar por el absorbedor, la mezcla de gas es forzada a regresar al circuito a través de la válvula de espiración (ES). La bolsa de inhalación, en el lado de inspiración del circuito, realiza varias funciones importantes; por ejemplo, en el sistema de circuito cerrado, el flujo de oxígeno y de los otros gases debe ser igual a la cantidad absorbida por el paciente más una pequeña cantidad por cualquier fuga. El flujo de entrada del gas es constante y ocurre lentamente. Por otro lado, la inhalación requiere de un volumen de gas relativamente grande en un tiempo corto. Por lo que la bolsa de inhalación sirve como un reservorio elástico que acomoda la demanda en la inspiración. Sí a caso existe un excesivo flujo de entrada, la presión en el circuito se elevará y la válvula de exceso de presión (XP) se abrirá automáticamente para reducir la presión. La bolsa de inhalación actúa, a la vez, como un indicador de la respiración espontánea colapsando moderadamente durante la inhalación. Y lo que es más importante, proporciona los medios para suministrar una respiración artificial. Apretando la bolsa, se obliga al gas a entrar en los pulmones, y al soltarla se permite que los pulmones realicen la espiración. Mecánicamente, la respiración es lograda colocando la bolsa dentro de un ambiente en el que intermitentemente se proporcione presión positiva al gas. Algunas máquinas de anestesia cuentan con los medios para detectar el esfuerzo en la inhalación, y usar esta información para accionar el suministro de gas al sistema. 11.3. CLASIFICACIÓN DE LOS ANESTÉSICOS TABLA 7. CLASIFICACIÓN DE LOS ANESTÉSICOS GENERALES Líquidos Volátiles Gases Anestésicos Éteres. Hidrocarburos Halogenados. Oxido Nitroso - Simples: Éter Etílico - Simples: Cloroformo Xenón - Fluorados: - Fluorados: Halotano Isoflurano, Enfluorano, Metoxifluorano Dada su vía de administración y sus propias características físicas y químicas, existen determinantes particulares de la acción de los anestésicos por inhalación: a. Solubilidad del anestésico: – Coeficiente de Partición Sangre: Gas: Determina la solubilidad relativa del anestésico en sangre respecto a la que presenta en mezcla gaseosa, en otras palabras mide la tendencia del anestésico a pasar a la fase líquida en sangre. El anestésico en fase líquida (disuelto) no ejerce presión importante, por lo que el mismo no tiende a pasar significativamente al SNC desde la sangre. Por ejemplo, el Halotano tiene un coeficiente de partición Sangre:Gas de 2,3 y el Óxido Nitroso tiene un valor de 0,47 (tabla 1); esto implica que el Óxido Nitroso se disuelve menos que el Halotano en sangre y, por tanto tiende a permanecer en fase gaseosa, lo que le permite ejercer mayor presión parcial y, por tanto, inducir una anestesia más rápida. – Coeficiente de Partición Encéfalo:Sangre: Significado similar al anteriormente explicado b. Concentración del anestésico en el aire inspirado - CAM è Concentración Alveolar Mínima (CAM): “Dosis” que evita la respuesta a un determinado estímulo doloroso (incisión quirúrgica) en el 50% de los individuos. Por paralelismo con la curva dosis - efecto (“curva gradual”) Representa la “potencia” anestésica, comprendiéndose esta como que a menor CAM, mayor será la potencia. Este parámetro se utiliza en razón de que la “dosis real” es de más difícil determinación que la la fracción ocupada por un agente dado en una mezcla de gases. c. Gradiente de Concentración AV d. Flujo sanguíneo Pulmonar e. Ventilación Pulmonar 11.3.1. Inhalatorios Son sustancias que, administradas por vía inhalatoria, pueden inducir anestesia; por restricción de las propiedades de esa vía, estas sustancias deben ser necesariamente gases o líquidos volátiles. Los líquidos volátiles pueden definirse como sustancias que pueden pasar fácilmente a la fase gaseosa, por lo que, para efectos prácticos pueden considerarse como gases; una definición más precisa es la de que un agente volátil es una sustancia que a temperatura ambiente y a presión de una atmósfera permanece líquida, pero que puede pasar fácilmente a la fase gaseosa con cambios pequeños en su entorno (estos cambios son generados por las máquinas de anestesia). TABLA 8. CARACTERÍSTICAS DE ANESTÉSICOS INHALATORIOS TÍPICOS Óxido Xenón Halotano Enfluora Eter Isoflura Desflur Nitroso no no ano Sevoflur ano CAM Coeficientes de Partición - Sangre:Gas - Encéfalo:Sangre - Grasa:Sangre Inducción/Recu peración 100 70 0,8 0.7 1.9 1.2 6.1 2.1 0,47 1,10 2,30 Rápidas 0,47 2,5 1,9 51 Media 1,8 1,4 36 Rápidas 12 1.4 0.4 0.6 Lenta Media Rápidas Rápidas Profundidad Anestésica Baja Baja Moderada Completa Completa Analgesia Buena Buena Escasa Hipotensión Ligera (simpático) Frecuente y grave + Muy buena Moderada + Leve Moderada + Hepática (1/10.000 pacientes adultos) Irritación respiratori a, nauseas y vómitos. Riesgo de explosión Pocos efectos adversos. Isquemia coronaria Irritació n respirato ria, broncoe spasmo To0xicida d renal debida al fluor Depresión miocárdica Arritmias Toxicidad Muy Rápidas Casi Nula + + Hepática Tomado y adaptado de: Rang HP, Dale MM, Ritter JM et al. Farmacología. Quinta Edición. Elseiver España S.A. Madrid, España. 2004 y MARTINDALE. Guía Completa de Consulta Farmacoterapéutica. Primera Edición. Pharma Editores. España. 2003. 11.3.2. Intravenosos. Al usar la vía endovenosa, se evita el proceso de absorción, por lo que la inducción anestésica resulta más rápida que con los agentes inhalatorios; sin embargo, correlativamente, la eliminación tarda más, pues no depende de un factor controlable como la ventilación, sino de los propios procesos de eliminación del organismo. Algunos aspectos respecto a los anestésicos endovenosos son: • Ausencia de fase pulmonar: En algunos casos esto no es del todo cierto, pues los anestésicos intravenosos suelen combinarse con agentes Inhalatorios como el Óxido Nitroso para lograr “Anestesia Total Intravenosa”. • Farmacocinética más “habitual”: – Dependencia de la Unión a Proteínas Plasmáticas: la fracción unida a Proteínas no ejerce efecto – Influencia de la ionización: la fracción ionizada no difunde la barrera hematoencefálica – Fin de la acción: • Eliminación – Metabolismo, del cual, empero, pueden originarse metabolitos activos – Excreción • Redistribución El tiopental es un anestésico intravenoso particular, pues la finalización de su efecto no depende de su vida media de eliminación, sino del proceso de redistribución; este fármaco se distribuye inicialmente hacia el Sistema Nervioso Central, por lo que su inicio de acción es muy rápido; sin embargo, a esto sigue una movilización gradual del tiopental hacia otros tejidos, por lo que su concentración en el Sistema Nervioso Central baja rápidamente (de allí que su duración de acción sea relativamente corta). Anestésico Mecanismo FC SNC CV Resp Tox TABLA 9. CARACTERÍSTICAS DE ANESTÉSICOS INTRAVENOSOS TÍPICOS Tiopental Propofol Etomidato Ketamina GABA Las teorías incluyen la GABA Interferencia con mimético acción a través de GMP mimético transmisión glutamaérgica cíclico, del sistema (NMDA) opioide, de receptores GABA y de receptores glicinérgicos Inicio en 40 s, Inicio en 60 s, depurado Inicio 60 s. Inicio 60 s. duración 30 min rápidamente, por lo que duración 3 - 5 duración 3 - 5 min. (redistribución) se requiere infusión min. continua (hay sistemas especiales para ello) Depresión: Similares, pero más Similar a Disociativa, sedación, potentes que los de tiopental anestesia, hipnosis, FS y tiopental analgesia, Presión metabolismo, presión PA (10 - 20%) Bradicardia, hipotensión Similar a PA y FC arterial, perfusión tiopental (menos miocardio depresión) Apnea, Apnea Similar a Apnea, broncoconstricción tiopental (menos broncodilatación depresión) HTA, Dolor a la inyección, Mioclonus, Alucinaciones, taquicardia, estimulación SNC, vómitos, pesadillas, depresión depresión CV severa en insuficencia hipersecreción respiratoria, ancianos adrenal (uso broncospasmo, crónico) anafilaxia Tomado y adaptado de: Rang HP, Dale MM, Ritter JM et al. Farmacología. Quinta Edición. Elseiver España S.A. Madrid, España. 2004 y MARTINDALE. Guía Completa de Consulta Farmacoterapéutica. Primera Edición. Pharma Editores. España. 2003. 11.4. COMBINACIONES DE FÁRMACOS Pueden hacerse las siguientes combinaciones para optimizar la anestesia general: - Dos (o más) anestésicos: generalmente uno que produzca rápida inducción (óxido nitroso, agentes IV) y uno que aporte adecuado mantenimiento (halogenado) - Anestésico + Otro(s) medicamento(s): Que generen un efecto de difícil consecución con el anestésico aislado: Uso de relajantes musculares - Neuroleptoanestesia: Caso particular que combina un neuroléptico (como droperidol) y un opioide (como fentanilo), para causar disminución general de la reactividad y analgesia (neuroleptoanalgesia) al adicionar un anestésico inhalatorio (óxido nitroso) o elevar suficientemente la dosis del opioide, se consigue anestesia general, que en este caso se denomina neuroleptoanestesia. 11.5. MEDICACIÓN PREANESTÉSICA De manera simple, la medicación preanestésica (MPa) podría definirse como toda administración farmacológica previa al acto anestésico. Sin embargo, esta definición se debe limitar a las indicaciones decididas en relación con el acto anestésico por realizar, por lo que la continuación de un tratamiento crónico del paciente no se consideraría como parte estricta de la MPa. La medicación preanestésica ayuda al paciente a someterse a la cirugía y tiene como objetivos: Sedación, amnesia, analgesia, disminuir los requerimientos anestésicos, disminuir los reflejos indeseables, disminuir secreciones de la vía respiratoria e inhibir náuseas y vómitos. Esta medicación es variable y se indica sobre la base de circunstancias relacionadas con: - La condición del paciente: Si, por ejemplo, el paciente presenta un cuadro doloroso que amerita intervención quirúrgica, debe tratarse con analgésicos, además de fármacos para disminuir la ansiedad y aprensión relacionadas con la patología o la cirugía planeada. - El proceso a implementar: El procedimiento por aplicar puede causar dolor y/o ansiedad. Entre los agentes que forman parte de la medicación preanestésica están los sedantes ansiolíticos, analgésicos, tranquilizantes, antimuscarínicos, antihistamínicos, antieméticos y corticoesteroides. 11.6. MODOS DE ACCIÓN: Aunque los mecanismos de acción de la mayoría de los AG son desconocidos molecularmente, si se aprecia para el caso de los agentes inhalados una relación directa entre potencia y liposolubilidad. Se sugiere: 1. Bloqueo de canales iónicos de sodio 2. Apertura de canales iónicos de cloro (barbitúricos y benzodiacepinas) 3. Efectos sobre neurotransmisores centrales: Grutamato, Aspartato, Glicina, GABA 4. Interacciones sobre bombas dependientes de energía (ATPasas) 5. Formación de hidratos de agua en las membranas celulares 11.7. LA ANESTESIA GENERAL COMO PROCEDIMIENTO-CORRELACIÓN CLÍNICO FARMACOLGICA Comprende: Preanestesia: Tiempo caracterizado por 1. Evaluación del paciente (Clasificación ASA) 2. Restauración de su capacidad funcional 3. Premedicación Anestesia propiamente dicha Caracterizada por: 1. Inducción 2. Mantenimiento 3. Emergencia Postanestesia: Se caracteriza por 1. Recuperación 2. Convalecencia 11.8 CLASIFICACION ASA: Paciente 1: Normal Paciente 2: Con enfermedad de base, compensada. Ejemplo: Diabético tipo II, Controlado. Paciente 3: Con enfermedad de base no controlada. Ejemplo: Hipertenso con tratamiento y que a pesar del tratamiento permanezca hipertenso Paciente 4: Con enfermedad de base no controlada, y con complicaciones de su enfermedad de base. Ejemplo: Hipertenso con hipertrofia ventricular izquierda o con insuficiencia renal secundaria a la HTA, ó un diabético descompensado y con retinopatía diabética. Paciente 5: Paciente en shock ó colapso vasomotor, con deterioro evidente de la consciencia o su integridad neurológica y pérdida manifiesta de sus signos vitales. BIBLIOGRAFÍA 1. Rang HP, Dale MM, Ritter JM et al. Farmacología. Quinta Edición. Elseiver España S.A. Madrid, España. 2004. 2. Calderón P. Gutiérrez JR., Velasco. JJ. Psiquiatría en: Libro Farmacia Hospitalaria. SEFH. España. 18: 1332-1336. 3. Consejo general de colegios oficiales de farmacéuticos de España. Actualización en Farmacoterapia. España. 2002. 3. Brunton L,L, et al. Goodman y Gilman. Las bases farmacológicas de la terapéutica. Undécima edición. Mc Graw Hill. 2007 5. Guías Clínicas en Psiquiatría y Neurología. www.fisterra.com Consultada el 8 de Julio de 2007. 6. Toro RJ, Yepes LE. Psiquiatría. CIB (Corporación para Investigaciones Biológicas), 2004. Medellín. Colombia 7. Maite San Emeterio, et al. Guía de práctica clínica para la atención al paciente con esquizofrenia. Versión breve para la aplicación en la práctica clínica. Barcelona: Agència d’Avaluació de Tecnologia i Recerca Mèdiques. CatSalut. Departament de Sanitat i Seguretat Social. Generalitat de Catalunya. Octubre 2003 (GP01/2003). 8. Freedman, R. Schizophrenia. N Engl J Med 349;18. October 2003. 1738-1749. 9. MARTINDALE. Guía Completa de Consulta Farmacoterapéutica. Primera Edición. Pharma Editores. España. 2003. 10. Amariles P, Ceballos JM, Giraldo NA, Gutiérrez FJ. Guía de Actuación Farmacéutica en pacientes con Esquizofrenia. Humax Pharmaceutical S.A. En proceso de edición. 11. Sociedad Española de Farmacia Hospitalaria www.sefh.es Consultada 12 de Julio de 2007 12. Melmon and Morrelli`s. Clinical Pharmacology.McGraw-Hill. 4th edition. 2000. pp 401-428. 13. Mycek, Harvey y Champe. Farmacología. McGraw-Hill. 2a edicion. 2000. pp 95-179. 14. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. DSM IV TR.2000. P 943 15. Tamayo JM. Psicofarmacologia On-Line [cited 2007 Mes 07]. Available from: URL: http://psicofarmacologia.info. 16. White Paul F. Fármacos en Anestesia. McGraw-Hill. 1998. P 352 17. Gutiérrez H F. Javier. Ayudas para comprender la farmacología del SNC. Manual de Notas de Clase. Facultad de Enfermería, UdeA, 2007. 18. Katzung Bertram G. Basic and Clinical Pharmacology. McGraw-Hill, Lange. 10th edition. 2007. pp 333-525.