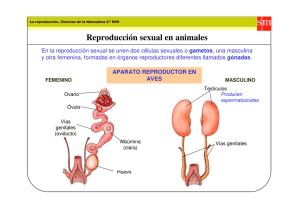
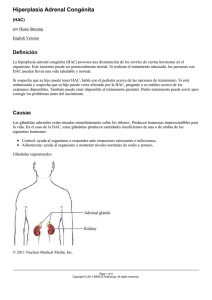
S.E.G.O. 2001 LOS ESTADOS INTERSEXUALES
Anuncio

S.E.G.O. 2001 LOS ESTADOS INTERSEXUALES • • • • • • • • • Concepto y clasificación Inducción y fases de la diferenciación sexual Formas clínicas y su diagnóstico Consejo genético Diagnóstico prenatal Tratamiento hormonal de sustitución Terapia médica supresora Tratamiento quirúrgico de los estados intersexuales Bibliografía Concepto y clasificación El término hermafroditismo proviene del dios griego Hermafrodito, hijo de Hermes y Afrodita, quien se distinguía por reunir, en una misma persona, rasgos y características de ambos sexos. En la actualidad este término ha sido sustituido por el de estados intersexuales y nos aproxima mejor a una de las problemáticas que siempre hemos tenido a la hora de abordar este tema: las diferentes clasificaciones, según los diferentes puntos de vista de cada autor. Así, las agrupaciones de este tipo de patología, y en especial de su etiopatogenia, van a ser bien diferentes según los parámetros que consideremos para diferenciar los sexos. Convenimos entonces, que estas entidades pueden quedar agrupadas de forma muy distinta según consideremos un sexo genético (carga cromosómica), un sexo gonadal (estructura de las gónadas), sexo genital (según la morfología de los genitales internos o externos), sexo somático (atendiendo a los caracteres sexuales secundarios), sexo hormonal (según su perfil hormonal), sexo legal (según el sexo con el que esté inscrito) y sexo psicosocial (según sea su comportamiento sexual). Seguramente, en breve hablaremos (o ya podríamos hablar) de un sexo génico, si atendieramos a la situación de la carga génica de todos aquellos factores que configuran la diferenciación sexual. Así pues, hemos seleccionado en este trabajo una clasificación que atienda a la estructura de la gónada, por ninguna otra razón que la de ser, a nuestro criterio, la más didáctica (Tabla 1). Por otro lado, para tratar un tema tan arduo con unas marcadas limitaciones de espacio, los autores, hemos convenido enfrentarnos a la problemática del consenso en los estados intersexuales, en aquellos aspectos que puedan tener un especial interés y tenemos competencia para hacerlo, soslayando, de forma deliberada, aquellos otros (como p.e. la cuestión del soporte médico en la intersexualidad psicológica), en los que entendemos que se deben abordar desde un punto de vista que se escapa a nuestra disciplina. Es oportuno, antes de entrar en aquellos aspectos que podemos entender como tributarios de consenso, recordar los conocimientos actuales sobre las bases de la diferenciación sexual. Esto nos ha de proporcionar una plataforma para el planteamiento de los puntos de mayor interés en este sentido. Inducción y fases de la diferenciación sexual Las diferencias biológicas entre el hombre y la mujer están en estrecha relación con las distintas responsabilidades que cada uno desempeña en la conservación de la especie. Sin embargo, la evolución social de la especie humana establece distintos criterios de atribucion de sexo dependiendo de los indicadores a los que se haga referencia. Como ya hemos comentado anteriormente, el sexo social, por el que se atribuye a cada individuo el calificativo de varón o hembra, se establece a partir de las características de su aparato genital en el momento del nacimiento y se confirma durante el desarrollo puberal. Es lo que conocemos por sexo genital o fenotípico. Con ello se contribuye a la adquisición de una concepción determinada de la propia identidad y a que se adopten ciertas actitudes y comportami entos que socialmente se consideran adecuados para cada sexo. Sobre esta base algunas escuelas han establecido la existencia de un "sexo psicológico" diferenciado , aunque existen evidencias de que las hormonas gonadales contribuyen desde la vida intrauterina al proceso de estructuraci ón cerebral (1). Tanto las manifestaci ones fenotípicas como sus consecuenci as psicológicas o sociales están determinad as por la secreción hormonal de la gónada, testículo u ovario, de cada individuo. Por ello la diferenciaci ón gonadal es la primera etapa morfológica de un largo proceso que se desarrolla, de forma escalonada y progresiva, desde la vida intrauterina hasta la madurez sexual. Desde el momento de la fecundación, cada embrión posee en su material genético información para regular el proceso de diferenciación, que dotará al individuo de un tipo de gónada determinado. Repasaremos a continuación estos procesos. Sexo genético En las primeras etapas del desarrollo, la apariencia del embrión es inespecífica, es decir, de su aspecto morfológico no puede deducirse si va a desarrollarse un varón o una hembra. Sin embargo, en el material genético que se ha reunido a partir de ambos gametos, se encuentra ya la información necesaria para inducir en una primera etapa el desarrollo de los testículos, que dará lugar a un varón o, en su defecto, el de los ovarios, que comportará el nacimiento de una hembra. La información sobre las características sexuales del nuevo individuo está contenida en un par de cromosomas, que se denominan cromosomas sexuales y que configuran el sexo genético. El óvulo, en su dotación haploide, aporta siempre un cromosoma X, mientras que el espermatozoide, que es el que decide el sexo del futuro ser, puede aportar un X o bien un Y. La combinación de dos cromosomas X dan lugar a una hembra, mientras que la combinación de los cromosomas X e Y inducen al desarrollo de un varón. Por ello, en la especie humana el sexo masculino es el denominado heterogamético. El sexo genético puede conocerse indirectamente por el estudio de la cromatina sexual, que consiste en la detección sobre la superficie nuclear del corpúsculo de Barr, que aparece tan sólo en aquellas células que poseen más de un cromosoma X y que refleja una mayor abundancia en cromatina. Su aparición en las células de varones normales no es superior al 1% de los casos, mientras que en la mujer es mayor del 30%, dependiendo del lugar de donde se obtiene la muestra y de su calidad. Existe también la posibilidad de detectar específicamente el cromosoma Y mediante la tinción con fluoresceína de una heterocromatina localizada en la región distal de los brazos largos, que se denomina masa Y fluorescente. Actualmente la aplicación de técnicas de hibridación "in situ" con fluorescencia (FISH) o reacción en cadena de la polimerasa (PCR) permite la identificación de los cromosomas sexuales y de sus características en los blastómeros de embriones, evitando transferir embriones femeninos en portadoras de enfermedades ligadas al cromosoma X. El método más informativo y asequible en la práctica clínica es el cariotipo. Consiste en el estudio de los cromosomas de las células en división, previamente detenidas en su metafase. Se realiza el recuento del número de cromosomas de cada célula y se fotografían algunas metafases. Las imágenes ampliadas y recortadas se agrupan de acuerdo con su forma y tamaño por pares y en grupos preestablecidos. Los cromosomas sexuales, a pesar de sus diferencias morfológicas, se reúnen en un par independiente. La aplicación de técnicas especiales ayuda a identificar regiones específicas dentro de cada cromosoma, y con ello la deleción o translocación de algunos fragmentos. El estudio de estas variaciones en situaciones patológicas permite la localización de los genes que codifican funciones determinadas (2). En la actualidad, las técnicas de biología molecular como el análisis de las características estructurales de un gen, unidas a observaciones comparativas en individuos patológicos permiten avanzar en la realización de un mapa génico en el que se localizan en la estructura del cromosoma los genes que codifican una función determinada y detectar las alteraciones responsables de cada patología. Localización de los genes determinantes de la diferenciación El cromosoma X, es sensiblemente mayor que el Y. Al parecer, en el curso del proceso evolutivo una parte del cromosoma Y ha pasado a formar parte del X. Los cromosomas sexuales contienen también información sobre otros aspectos somáticos, como el color de los ojos, la diferenciación cromática, el grupo sanguíneo, sistemas enzimáticos intracelulares, el ritmo de crecimiento, etc. Como consecuencia de la cesión de parte de su material genético al X, el Y conserva tan sólo algunos lugares genéticos, especialmente relacionados con la diferenciación gonadal, la espermatogénesis y el crecimiento, mientras que cada X acumula información sobre el resto de las características fenotípicas. La zona más distal del brazo corto de ambos cromosomas cercana a la banda p22.3 se denomina pseudoautosómica. Esta denominación responde al hecho que, durante la meiosis este fragmento se aparea e intercambia su material genético de la misma forma que lo hacen los cromosomas autosómicos. En él localizan la mayoría de genes comunes a X e Y; entre ellos, los que regulan el crecimiento estatural y algunos procesos enzimáticos de la esteroidogénesis. Entre p11 y q12 se localiza el gen que codifica el desarrollo de receptores para los andrógenos (gen Tfm). Finalmente, en la región p21-p22.3 se ha localizado un gen relacionado con la síntesis de TDF o factor determinante del desarrollo testicular. En el cromosoma Y es más difícil elaborar un mapa génico, debido a su pequeño tamaño. Sabemos que en la región que se localiza un gen que regula el crecimiento estatural. Respecto a la diferenciación gonadal tenemos en la región distal de los brazos cortos el TDY o gen que codifica el TDF, mientras que, en los brazos largos, en la banda qll se encuentra el gen regulador de la espermatogénesis o AZF (azoospermia factor). Junto a él se encuentra el gen que codifica o controla la expresión del antígeno HY, considerado durante tiempo el único factor determinante del desarrollo testicular. Actualmente el protagonismo ha pasado al gen SRY que, como se comenta más adelante, desencadena un proceso multigénico y en cascada de diferenciación. (Figura 1). Figura1 Mapa géneco de la diferenciación gonadal Sexo gonadal En la quinta semana de vida intrauterina una zona del epitelio celómico se engrosa, y junto con el mesénquima subyacente da lugar a la llamada cresta gonadal. A continuación, un grupo de células indiferenciadas proliferan desde el epitelio hacia el interior del mesénquima, dando lugar a los cordones germinales. Las células germinales migran por el mesenterio posterior y desde el saco alantoideo van a colonizar la cresta gonadal. En caso de no producirse la llegada a la gónada indiferenciada de estas células germinales no hay desarrollo gonadal. El desarrollo de esta zona parece regulado por un gen denominado WT1 por haber sido identificado en enfermos que presentaban Tumores de Willms. Este gen ejercería una acción de supresión tumoral y está localizado en el cromosoma 11. Los pacientes afectos de nefroblastomas de Willms carecerían de éste parcial o totalmente y las mutaciones parciales se relacionan con el síndrome de Denys-Drash caracterizado por disgenesia gonadal, persistencia de las estructuras mullerianas (utero y trompas) genitales externos femeninos y Tumor de Willms. Todo ello lleva a pensar que la integridad del gen WT1 es indispensable para el desarrollo normal de la gónada (3). Si, de acuerdo con el sexo genético, debe desarrollarse un testículo, la gónada pierde sus características de indiferente alrededor de la séptima semana. Los cordones germinales primarios o medulares se organizan de forma radial, y las células germinales se diferencian a espermatogonios. Se constituyen los tubos seminíferos con las células de Sertoli y el mesénquima subyacente contribuye con las células de Leydig y la estroma. De no ocurrir así, las células germinales permanecen en apariencia inactivas en la gónada morfológicamente indiferenciada hasta la décima semana. En este momento inician la primera división meiótica y se constituyen en ovocitos. Las células del epitelio germinal más externo, llamado secundario o cortical, les proporcionan una cubierta celular que constituyen las células de la granulosa, y el mesénquima se diferencia en las células tecales y estroma. Alrededor de la semana 15 ya pueden identificarse folículos en el ovario y una producción importante de esteroides. Los mecanismos de control de la diferenciación gonadal no están completamente aclarados, sin embargo en los últimos años se han aportado nuevos datos. Inicialmente se creía que las gónadas pasaban por una fase de hermafroditismo, poseyendo simultáneamente tejido testicular y ovárico, y que de la pugna de ambos surgía el carácter definitivo de la gónada. De la modificación de estas teorías iniciales surgió la hipótesis de Witchi, que preconizaba que en la gónada no diferenciada existían dos esbozos distintos, capaces de dar lugar a un testículo o un ovario. Estos esbozos podrían producir dos sustancias distintas, la "medulina" y la "corticina", que estimularían respectivamente el desarrollo testicular u ovárico e inhibirían simultáneamente el contrario. Sin embargo, existían argumentos en contra del planteamiento con sustancias inhibidoras y estimuladoras especialmente porque el desarrollo del testículo y del ovario se produce con gran diferencia cronológica (casi 6 semanas), con lo que era difícil que pasaran por una etapa mixta. De las teorías de Witchi tan sólo nos queda como cierto que el desarrollo testicular tiene lugar a partir de la región medular y el ovárico de la región cortical, y que ambos están controlados por las características del sexo genético del embrión. Jost, trabajando con gemelos de sexo distinto en la oveja, demostró que si se comunicaban las dos circulaciones fetales intraútero no existían modificaciones en el desarrollo gonadal del animal varón, mientras que el feto hembra presentaba un ovario anómalo, en cuyo tejido gonadal podían distinguirse zonas con túbulos seminíferos. Ello le llevó a la conclusión de que el desarrollo de tipo testicular se debía a la acción de una sustancia inductora, producida por las células de tipo masculino, y que en ausencia de este inductor testicular la gónada permanecía indiferenciada hasta la fase del desarrollo ovárico. La diferenciación testicular tiene lugar por interacción entre las células germinales y los elementos somáticos de la gónada no diferenciada. Por ello, resulta razonable pensar en la existencia de una sustancia capaz de dirigir la diferenciación gonadal hacia la morfología de tipo testicular. Hasta muy recientemente se ha creído que esta función correspondía exclusivamente al antígeno HY (aHY). Es una sustancia presente en la superficie de las células de los individuos con un cromosoma Y en el cariotipo, y que inicialmente tan sólo se había identificado como un antígeno de histocompatibilidad. Se ha comprobado que el antígeno se encuentra en la mayoría de especies dentro de la escala filogenética en los individuos de sexo heterogamético, es decir, en aquellos que poseían dos cromosomas distintos en el par sexual y por tanto ejercían una acción diferenciadora de la gónada. Estudiando las alteraciones del desarrollo gonadal y su relación con la presencia de este antígeno, Wachtel y Ohno (4) postularon que el aHY determina la diferenciación de la gónada heterogamética: en la especie humana el testículo. En presencia de antígeno HY éste se fija a un receptor expecífico, y las células se organizan en forma tubular y adquieren una morfología testicular, mientras que en su ausencia acaban rodeando de forma no específicamente ordenada las células germinales, dando lugar a los folículos (5). La opinión de que el aHY era el único regulador del desarrollo gonadal se empezó a discutir tras el descubrimiento de una cepa de ratones que podían presentar desarrollo testicular en ausencia de niveles detectables de aHY (6). Simultáneamente, Page (7) demostró que un gen situado en la región terminal del brazo corto del Y era decisivo en el desarrollo testicular y se observaba translocado en los casos de varones XX. Con ello se postulaba la existencia del TDF o factor determinante del desarrollo testicular como oponente al antiguo concepto de la regulación exclusiva por el aHY. Se ha localizado también en la misma región del cromosoma X un gen de características moleculares idénticas al que codifica el TDF, lo que sugiere que la actividad inductora del desarrollo testicular se expresa como consecuencia de la interrelación entre los genes de los cromosomas sexuales. Actualmente el antígeno HY se considera como un agente subordinado en el proceso de diferenciación sexual. Las técnicas de biología molecular han contribuido de forma definitiva al avance en el conocimiento de la diferenciación gonadal. Anne McLaren (1991) en un editorial de Nature pone en perspectiva los avances en este campo en los últimos 40 años que se resumen en la Figura 2. De la identificación del cromosoma Y como depositario del control del desarrollo testicular hemos llegado al gen SR4 localizado en la proximidad de la región pseudoautosómica del brazo corto del cromosoma Y. La identificación de este gen como desencadenante de la diferenciación gonadal se basa en evidencias experimentales muy sólidas: 1.- La proteína que codifica este gen contiene una zona de fijación al DNA que actúa como promotora de los otros factores involucrados en la diferenciación sexual. 2.- Estudios moleculares detectan que el gen SR4 tan sólo se expresa en el testículo durante el período de diferenciación gonadal. 3.- En ratones transgénicos XX a los que se ha transfectado el gen SRY han presentado un desarrollo gonadal y fenotípico masculino. 4.- Algunas mujeres con cariotipo XY (Síndrome de Swyer) presentan mutaciones del gen SRY que provoca su inactivación (8). Por todo ello el gen SRY se considera actualmente como el primer eslabón de una cascada de fenómenos que conducen a la diferenciación testicular. Su activación da lugar a la producción Figura 2 de una proteína de 204 aminoácidos que tiene una región central o "capa" del tipo HMG (siglas correspondientes a Avance en el conocimiento de la diferenciación gonodal. grupos de alta movilidad, del inglés Anne McLaren "High Mobility Group") de 80 aminoácidos. Estos grupos de proteínas se fijan con gran afinidad al DNA y provocan su plegamiento que activa o inhibe las regiones promotoras de los genes involucrados en la determinación sexual. Entre ellas destacaría la represión de la región promotora de la aromatasa P450 que daría lugar a una inhibición de esta enzima, dificultando la conversión de la testosterona en estradiol y facilitando así el desarrollo fenotípico masculino (9). También se ha relacionado este cambio en el plegamento de la hélice de ADN con la activación de los genes que codifican la síntesis del factor inhibidor mulleriano (FIM) al que haremos referencia más adelante. Sin embargo estudios posteriores sugieren que se precisarían otros co-factores, llamados SRYIF (factores inducidos por SRY), para vehicular el mensaje de SR4 hasta los genes promotores del FIM. En este proceso también se han involucrado receptores nucleares "huérfanos" (denominados así porque hasta el momento no se les ha encontrado ligando). Entre ellos destaca SF1 (de Steroidogenic factor 1) que controlaría la esteroidogénesis desde sus etapas iniciales y se ha detectado su expresión en los esbozos gonadales masculinos y femeninos del ratón. Se localiza en el cromosoma 9 y se comportaría de forma dimórfica según se desarrolle un testículo o un ovario. En el primer caso se expresa de forma temprana, en el momento en que se produce la diferenciación de los tubos seminíferos y se expresaría específicamente en las futuras células de Sertoli. Si se desarrolla un ovario la expresión es más tardía. También intervendría en la activación de otro factor esteroidogenético ligado a un receptor huérfano y denominado DAX1. La importancia de SF1 en el desarrollo gonadal se demuestra experimentalmente con ratones Knockout, es decir que carecen artificialmente del gen que codifica su expresión. Estos animales no sobreviven al nacimiento debido a la insuficiencia renal y el estudio anatómico demostró una agenesia total de gónadas y suprarrenales con persistencia de los conductos müllerianos y wolffianos. Ello prueba que la actuación de SF1 es anterior a la propia esteroidogenesis y producción de FIM. Hasta ahora se había considerado que los fenómenos progresivos de diferenciación eran desencadenados por SRY hasta completar el desarrollo testicular y que el desarrollo ovárico se producía por defecto. Sin embargo nuevas evidencias ponen en entredicho este supuesto. En 1994 el grupo de Wachtel (10) descubre que la duplicación del brazo corto del cromosoma X produce reversión en el desarrollo gonadal masculino en individuos XY y localizan el gen responsable en la región Xp 21-22. Estos resultados fueron confirmados posteriormente (11). En consecuencia puede pensarse que el desarrollo ovárico se ve al menos facilitado por la actividad de este gen conocido con las siglas DSS (Dosage Sensitive Sex reversal). Sexo somático En la fase indiferente, el embrión posee estructuras a partir de las que puede desarrollar el aparato genital interno masculino o femenino. El conducto mesonéfrico o conducto de Wolff constituye el resto de drenaje del riñón mesonéfrico. Atraviesa todo el mesonefros, y de él se desarrollarán el epidídimo, el conducto deferente y las vesículas seminales. El conducto paramesonéfrico o de Müller es la consecuencia de una invaginación del epitelio yuxtamesonéfrico que acaba por unir sus crestas y soldarse en forma de tubo. De él se derivan las trompas, una en cada lado, y de su fusión en Y griega, en la zona más caudal, nace el cuerpo uterino y zona superior de la vagina. En presencia de un ovario o de una cintilla gonadal, o en ausencia de todo vestigio gonadal, se desarrolla siempre un aparato genital interno de tipo femenino. Ello significa que existe una tendencia espontánea al desarrollo de las estructuras paramesonéfricas si la presencia de un testículo no las inhibe, promoviendo al mismo tiempo el desarrollo del conducto wolffiano. No obstante existen evidencias de que la regresión total de las estructuras wolffianas precisa también del efecto metabolizador de los estrógenos. Los ratones manipulados genéticamente para que carezcan del gen que codifica para el receptor estrogénico, muestran persistencia de los conductos de Wolff probando la necesidad de la acción estrogénica para su regresión. El testículo controla la diferenciación por medio de dos elementos: el factor inhibidor mülleriano (FIM) y la testosterona. El FIM es una sustancia proteica de alto peso molecular (mayor de 100.000 daltons), que segregan las células de Sertoli del testículo fetal. Se trata de un factor de crecimiento de la familia de los TGFb y su síntesis está regulada por un gen localizado en el brazo corto del cromosoma 19. Para poder ejercer su efecto precisa del receptor propio cuya síntesis se regula por un gen específico localizado en el cromosoma 12. Además de su capacidad de inhibir el desarrollo de los conductos de Müller el FIM interviene también en la detención de la meiosis en los oocitos y contribuye a la regulación del descenso testicular. Su acción impide el desarrollo de las estructuras que dan lugar a una trompa y la mitad del cuerpo uterino. Actúa de modo local y en su propio lado. Esta acción ipsilateral es esencial para explicar diferencias unilaterales del desarrollo en estados patológicos. Simultáneamente, la testosterona segregada en el testículo actúa también en su propio lado, estimulando los conductos wolffianos hasta la formación del aparato genital interno masculino. Esta acción tiene lugar a través de un receptor específico para la testosterona que está presente tan sólo hasta la semana 12. De este modo, se delimita el llamado periodo crítico para la acción de Ios andrógenos. Al igual que ocurre en el aparato genital externo, si no se ha producido un estímulo androgénico en este momento ya no se desarrollarán las estructuras de tipo masculino. Los andrógenos a través de una proteína estimuladora de la fosfolipasa A actúan localmente activando la vía del ácido araquidónico e induciendo la biosíntesis de prostaglandinas. Para comprobar experimentalmente estos hechos, Jost (12) diseñó modelos experimentales con castraciones fetales selectivas de testículos u ovarios, y su sustitución por trasplantes del órgano antagónico o cristales de testosterona, pudiendo reproducir las situaciones que era de esperar que se desarrollaran de acuerdo con el modelo teórico. A modo de ejemplo, la castración unilateral en un embrión masculino dará lugar a desarrollo wolffiano normal en el lado intacto y a un desarrollo mülleriano normal en el castrado. Si en un embrión femenino se injerta un testículo se produce FIM y testosterona en uno de los lados, obteniéndose una inhibición mülleriana con desarrollo wolffiano en el lado injertado y normalidad en el desarrollo mülleriano del lado opuesto. Finalmente, si añadimos a un ovario un cristal de testosterona tendremos simultáneamente desarrollo wolffiano y mülleriano. Sin embargo, la administración de ciproterona (antiandrógeno que impide localmente la acción de la testosterona) a un embrión masculino inhibe el desarrollo de ambas estructuras, puesto que aun produciendo testosterona no es posible el desarrollo wolffiano porque el acetato de ciproterona bloquea su capacidad de ocupar el receptor, y el FIM inhibe las estructuras müllerianas. Sexo fenotípico El desarrollo del aparato genital externo parte, por el contrario, de una situación indiferente, común para ambos sexos, para dar lugar a una morfología de tipo masculino o femenino. El factor determinante es también en este caso la testosterona, que aquí no actúa por difusión local sino por vía sanguínea. Por esta razón el desarrollo normal o patológico es siempre bilateral y por tanto simétrico. En esta etapa indiferente se distinguirán el tubérculo genital, los pliegues uretrales y la tumefacción genital. Cuando la diferenciación gonadal es masculina, las células de Leydig empiezan a proliferar alrededor de la octava semana de embarazo. Cuando su dotación enzimática se completa inician la síntesis de testosterona, que se responsabiliza de la masculinización de los genitales externos e internos. Sin embargo, en el caso de los genitales externos, la testosterona no actúa directamente. Las células sensibles a los andrógenos de algunas zonas poseen únicamente en este momento receptores para la dihidrotestosterona; por ello la testosterona debe sufrir la acción de una enzima intracelular, la 5a-reductasa, antes de poder ser efectiva. Para que el tubérculo genital de lugar a un pene y los pliegues uretrales se fundan en el centro y junto con la tumefacción genital den lugar al escroto, es preciso que antes de la semana 12 llegue el estímulo androgénico adecuado. Para ello es necesario que el testículo segregue testosterona, ésta penetre en la célula diana y sea transformada por la 5a-reductasa en dihidrotestosterona, que a su vez actúa sobre el receptor específico. Al mismo tiempo se impide la formación del septo vesicovaginal y el desarrollo de la vagina. La incapacidad de responder a la testosterona en el aparato genital externo se limita a la vida fetal. Durante la pubertad la piel genital es sensible a la testosterona sin precisar una reducción previa, lo que explica cambios morfológicos en algunas situaciones patológicas que comentaremos más adelante. Si alguna de estas funciones específicas falla en la normalidad porque existe un ovario, o en la patología por alteraciones en alguno de los niveles del proceso, se desarrolla un fenotipo femenino o intermedio dependiendo de la gravedad de la anomalía. En la Figura 3 observamos representado de forma esquemática este proceso complejo con todos sus protagonistas. El papel de alguno de ellos permanece todavía oscuro y serán precisos más estudios sobre formas clínicas concretas y usando la nueva metodología molecular para elucidar por completo el complejo puzzle que rige el proceso de diferenciación sexual. Formas clínicas y su diagnóstico Convenimos en que en el diagnóstico, cuando se dá una circunstanci a que nos orienta hacia esta patología (ambigüeda d de los genitales en un recien nacido, etc.), se ponen en marcha unos protocolos o algoritmos que lo hacen relativamen te fácil. Sin embargo, existen ocasiones en que la intersexuali dad nos pasa desapercibi da y es diagnóstica da a partir del estudio de un síntoma paralelo o simplement e de forma casual (p.e. en el estudio de una amenorrea primaria en el caso de un pseudoher mafroditism o masculino debido a un síndrome de insensibilidad a los andrógenos o durante el estudio de un hiperandrogenismo en un individuo con fenotipo femenino portadora de un déficit enzimático, etc.). En la primera circunstancia, en presencia de unos genitales externos no diferenciados, nos remitimos al algoritmo diagnóstico propuesto por Calaf (13) y que recogemos en la Figura 4. Esta puede ser una plataforma-base a partir de la cual, según sea la particular circunstancia de cada caso, podremos ampliar o modificar. Veremos a continuación los puntos clave en el diagnóstico en las diferentes formas clínicas. Anorquía congénita Entidad realmente rara. La agonadia fue descrita por primera vez por Overzier y Linden en 1956. Se produce un fallo embrionario, posiblemente en los mecanismos de diferenciación de la gónada primitiva, por el que los potenciales testículos no llegan a desarrollarse. Atendiendo al momento en que tiene lugar la noxa, nos encontraremos ante diferentes formas clínicas (agenesia gonadal, agonadia y anorquía congénita). En los dos primeros casos, generalmente, se trata de individuos fenotípicamente femeninos, que presentan amenorrea, mínimo desarrollo mamario, y grados variables de ambigüedad en genitales externos. El diagnóstico por la imagen y/o los métodos quirúrgicos evidenciarán la ausencia de gónada. Lógicamente, los esteroides sexuales estarán bajos y las gonadotrofinas elevadas. Todo ello con un cariotipo 46XY. Disgenesias gonadales Pese a que en ocasiones existen ciertas reticencias a incluir a las disgenesias gonadales dentro de los estados intersexuales, el planteamiento que hemos hecho en la introducción sobre el concepto de "intersexo" nos ratifica en este aspecto. Sin duda, si hubieramos seleccionado el cariotipo para establecer la clasificación (recordemos la presencia del "Y" en estas entidades), o bien los niveles de metabolitos androgénicos de las disgenesias gonadales mixtas, caso de haber optado por una clasificación según el sexo hormonal, etc., esto se vería mucho más claro. En cualquier caso, en este apartado, recordaremos que los aspectos clínicos como el infantilismo sexual, amenorrea, etc, y el perfil bioquímico de fallo ovárico, conjugado con el cariotipo y la constatación laparoscópica de unas gónadas disgenéticas, será determinante en el diagnóstico del síndrome de Turner y la Disgenesia Gonadal Pura. En la Disgenesia Gonadal Mixta, unos estigmas de virilización, un perfil bioquímico con unos metabolitos androgénicos muy elevados, el cariotipo y sobre todo la presencia de tejido testicular (bilateral en el 15 por ciento de los casos) en la biopsia de ambas gónadas. Es importante resaltar en este punto que la ausencia de tejido ovárico completamente diferenciado es lo que establecerá el diagnóstico diferencial con el Hemafroditismo Verdadero. Hermafroditismo verdadero Individuos con un fenotipo indistinto (masculino o femenino) que en la pubertad inician un proceso de diferenciación, de intensidad variable, de estigmas del sexo opuesto. El perfil hormonal y el cariotipo pueden ser determinantes. Pero el diagnóstico de certeza se obtiene únicamente tras la demostración de elementos foliculares en el ovario, y de túbulos en el testículo, o bien de ambas estructuras en una gónada única: la evidencia del ovotestes. Pseudohermafroditismo femenino – Sin hiperplasia adrenal: Se trata de mujeres normales con las alteraciones referidas a nivel de genitales externos, en las que se ha podido constatar una historia de ingesta de hormonas con potencial androgénico por parte de la madre del individuo afecto, durante la gestación del mismo. Otra circunstancia sería cuando a la madre se le diagnostica un tumor virulizante, también durante la gestación. Por último, si no se dan estos dos casos, hablaríamos de masculinización de causa desconocida. En el primer grupo, se han implicado especialmente la 17-alfa-etiniltestosterona o el 17-alfa-etinil-19nortestisterona, y ocasionalmente, con la toma de dietilestilbestrol, andrógenos, o con progesterona por vía intramuscular. El Pseudohermafroditismo Femenino por Tumores Virilizantes en la madre es raro (pocos casos documentados), esencialmente por tres motivos: a) la mujer con hipersecreción androgénica tiene una fertilidad disminuida, b) si se trata de un feto varón no habrá anomalías de la diferenciación sexual y c) el tumor más típico y específico del embarazo es el luteoma. Aunque en la mayoría de ocasiones se trata de un hallazgo casual al final de la gestación (un caso por 400 laparotomías al fifinal del embarazo). Desaparece tras el parto, lo que sin duda hace que se le atribuya buena parte de la responsabilidad de aquellos casos que finalmente son etiquetados como pseudohermafroditismo femenino idiopático. Ha sido motivo de castraciones intempestivas. En cualquier caso existen serias dudas sobre estos mecanismo etiopatogén icos. Las teorias más recientes plantean que la barrarea placentaria, por si sola, tiene suficiente capacidad aromatizad ora para neutralizar un flujo importante de metabolitos androgénico s, y que, sin embargo déficits enzimáticos de sus propias cadenas metabolicas pueden condicionar un paso de este tipo de metabólitos hacia un feto hembra. – Con hiperplasia adrenal: La masculinización, en estos casos, nos vendrá dada por el tipo de déficit. La Figura 5 nos muestra el cuadro de la esteroidogénesis y en qué forma afectan cada uno de estos déficits a la cadena metabólica. La intensidad del cuadro también será variable. La forma más común, los déficits de 21-hidroxilasa, exigirán la evidencia de unos niveles elevados de 17-OHprogesterona basal en las formas clásicas o más severas. Los déficits parciales, de inicio tardio o formas crípticas precisarán, en muchos casos, de tests dinámicos para evidenciarlas. Pseudohermafroditismo masculino – Anomalías gonadotróficas: El cuadro más conocido es el causado por un déficit de LH. Fue descrito por Kallman en 1944, inicialmente únicamente en varones. Se trata de un hipogonadismo hipogonadotropo que cursa además con anosmia, debida a la ausencia de túbulos olfatorios. Aunque actualmente se describe también en mujeres (en éstas la anosmia puede ser parcial). La problemática se presentará en aquellos casos que se presenten con genitales ambiguos. En los casos de pseudohermafroditismo masculino por Secrección de LH Estructuralmente Anómala (anomalía en la cadena Beta), los genitales externos son generalmente femeninos al nacimiento, mientras que los genitales internos son normales masculinos (pélvicos o en hernia inguinal). Así, estos individuos se caracterizan por ser mujeres con una falta de desarrollo de caracteres sexuales secundarios femeninos en la pubertad. Habrá lógicamente, además, amenorrea, etc. Los esteroides serán bajos y las gonadotrofinas altas. Es característica una buena respuesta tras la administración de HCG. La determinación del cariotipo y la biopsia de la gónada completarán el estudio diagnóstico. El Pseudohermafroditismo Masculino por Receptor de LH Anómalo es una rara entidad descrita por Berthezene, que clínicamente se comporta como una agonadia. También se conoce como "agenesia de células de Leydig". El defecto causante de esta alteración parece ser la ausencia o el defecto del receptor de LH-HCG en las células de Leydig, o bien otros factores que pudieran detener el desarrollo de las células de Leydig. Los genitales externos al nacimiento son, generalmente, femeninos o ambiguos. Con respecto a los genitales internos destacar que, a diferencia de la agonadia, los testículos están presentes aunque muestran inmadurez de los túbulos, con células de Sertoli normales y ausencia o número muy reducido de células de Leydig. Durante la pubertad habrá ausencia de desarrollo de los genitales externos, con deficiencias en el desarrollo de caracteres sexuales secundarios. Lógicamente, en el caso de considerarse mujeres, también cursará con amenorrea primaria, etc. Los niveles de androstendiona y testosterona se hallan en niveles prepuberales, no estimulables por la administración de HCG. Con respecto a las gonadotrofinas, la LH se encuentra elevada, y la FSH es normal (aunque aumenta si se realiza gonadectomia). Tras la administración de testosterona, se producirá un descenso de los niveles de LH. La determinación del cariotipo y el estudio de la gónada completarán el diagnóstico. – Déficits enzimáticos: La feminización, en estos casos, nos vendrá dada por el tipo de déficit y por el grado del mismo. Nos apoyaremos en la Figura 5 para comprender mejor las carencias y excesos de metabolitos que estas condicionan. Existen cuadros de una alta mortalidad, como el déficit de 20-OH/22-OH/20,22 desmolasa, descrito por primera vez en 1955 por Prader y Grutner, y que se presentan, generalmente, como recién nacidos de sexo femenino o con ambigüedad sexual variable. Los genitales internos son normales masculinos. En este cuadro se produce una insuficiencia suprarrenal severa con enorme acumulación de lípidos en corteza suprarrenal y gónadas ("Congenital lipoid adrenal hyperplasia"). Los esteroides serán bajos y el cortisol y aldosterona bajos. Además, la determinación del cariotipo y el acúmulo de lípidos en la biopsia de la gónada y la glándula suprarrenal, constituye la base diagnóstica. En general, estos déficits enzimáticos se pueden ver en individuos que presentan al nacer unos genitales externos femeninos con algún estigma virilizante, o bien una ambigüedad de los genitales externos, más o menos aparente, según sea el grado de afectación y como haya incidido en el desarrollo de los conductos embrionarios y de los genitales externos. La forma que más frecuentemente presenta una apariencia masculina de genitales externos son las formas parciales de déficit de 17-20 desmolasa. La Figura 5 muestra como afectarán en el perfíl bioquímico y por lo tanto en su expresión clínica, los diferentes tipos de déficits enzimáticos. Esta se manifestará especialmente en la pubertad. Será definitivo la valoración de ausencia de útero y, naturalmente, un cariotipo 46 XY. – Anomalías en la célula diana androgénica: El déficit de 5a-reductasa fue descrito en 1961 por Nowakowski y Lenz. Se caracteriza por presentar con ambigüedad importante en el momento del nacimiento y con grado variable de virilización en la pubertad. A pesar de la discreta clitoromegalia, habitualmente se les asigna sexo femenino al nacimiento. La maduración puberal se desarrolla en sentido masculino. No se produce desarrollo mamario, presentan escaso vello corporal y facial, ausencia de acné y un importante desarrollo muscular. Los metabolitos androgénicos serán muy elevados para una mujer. El cociente dihidrotestosterona/testosterona está disminuido en adultos y en niños tras estimulación con hCG (1/40 cuando la proporción normal es 1/20). Las gonadotropinas son normales o con niveles de LH ligeramente elevados. La prueba diagnóstica definitiva es la evidencia directa de actividad 5a-reductasa disminuida en cultivos de fibroblastos tras biopsia de piel genital, incubados en medio de testosterona. El síndrome de Insensibilidad a los andrógenos lo describe Morris en 1953. El espectro fenotípico del síndrome de insensibilidad a los andrógenos es muy amplio, y puede variar desde fenotipos femeninos normales, hasta masculinos con mínimas anomalías clínicas o solamente infertilidad. En los fenotipos femeninos la vagina será corta ciega y destacará la disminución o ausencia de pilosidad pubiana y axilar. No existe útero ni trompas. Los testículos pueden hallarse intraabdominales, a lo largo del canal inguinal o en los labios, lo cual facilita el establecimiento de hernias inguinales. Es la tercera causa más frecuente de amenorrea primaria tras la disgenesia gonadal y la ausencia congénita de vagina. Los metabolitos androgénicos serán muy elevados para una mujer y los niveles plasmáticos de LH pueden estar elevados. – Déficit Aislado de M.I.F.: Se trata de individuos con fenotipo masculino, aunque con desarrollo variable de estructuras müllerianas. Los genitales externos son masculinos, pudiendo ser, en ocasiones, ambiguos. Los genitales internos y las gónadas tienen desarrollo masculino. Una deficiencia en la acción del factor inhibidor de los conductos de Müller (MIF) hace que estos hombres presenten un útero, que suele ser rudimentario, y que, generalmente, se trata de un hallazgo en el curso de una laparotomía o como contenido herniario. Se ha descrito como Hernia "uteri inguinale". Otros trastornos del cariotipo Quedan una serie de entidades que deberemos englobarlas en este apartado para que la clasificación sea completa. Los síndromes de adición del cromosoma X en la mujer o Y en el hombre, no presentan estigmas del sexo opuesto. Son individuos prácticamente normales con unas alteraciones cromosómicas que, en algún caso, pueden cursar con problemas de fertilidad. El síndrome de Klinefelter fue descrito por primera vez en 1942 por Klinefelter, Reifenstein y Albright. Se trata de un estado intersexual con cariotipo 47 XXY, y fenotipo masculino con aspecto eunucoide, talla alta, testículos pequeños y ginecomastia. Se trata de un hipogonadismo hipogonadotrófico. Los niveles de andrógenos se encuentran disminuidos o en el límite inferior de la normalidad respecto a las cifras normales de un adulto masculino. Las gonadotrofinas se hallan elevadas. El cariotipo y la clínica harán una aproximación diagnóstica de estos casos muy asequible. Consejo genético El consejo genético constituye actualmente un área importante de la genética humana aplicada. Progresivamente un mayor número de pacientes solicita información tras el diagnóstico de una enfermedad concreta, sobre sus repercusiones y la posibilidad de recurrencia de la misma, en la descendencia o en hermanos del sujeto afecto. Es esencial realizar el diagnóstico de certeza de las enfermedades genéticas, utilizando todas las posibilidades de la medicina actual, ya que fenotipos similares pueden presentar diferentes patrones de herencia o pueden no estar considerados en la actualidad hereditarios. Evaluar el riesgo de recurrencia en ausencia de estos datos es algo impensable. La historia familiar es importante, para aclarar dichos patrones de herencia, y ser la base del consejo genético. El consejo genético en el caso de los estados intersexuales es siempre complejo, en muchos casos el sujeto afecto no presenta familiares afectos, y además no en todos los casos se conoce actualmente la etiología exacta del problema. El hecho de tener un hijo con una ambigüedad genital es un trauma psicológico importante para sus padres. En el caso de las niñas, probablemente requieran cirugía reconstructora en varias ocasiones durante la infancia y adolescencia. Todo ello hace que la demanda de consejo genético en los casos de ambigüedades sexuales, a pesar de no ser un motivo frecuente de consulta dada su baja frecuencia, sea de gran importancia, tanto por lo que supone dicha alteración en sí, como por su posible asociación con otras malformaciones. El ginecólogo, junto con el genetista clínico y el pediatra constituyen un equipo multidisciplinario que está implicado directamente en la valoración de los estados intersexuales. Con el diagnóstico obtenido podremos ofrecer un consejo genético, es decir responder a una demanda formulada por una pareja. En el momento de la evaluación de cada caso debemos realizar una historia familiar detallada que nos permitirá orientar el diagnóstico; y en caso de que aparezcan varios casos en la familia nos llevará a conocer mejor el modo de transmisión. Si la ambigüedad sexual está asociada a malformaciones o anomalías, éstas deben ser estudiadas cuidadosamente. En este caso es posible que la ambigüedad de los genitales sea un componente más de una enfermedad sindrómica. Comentaremos a continuación la posibilidad de consejo genético en aquellos casos en que nos encontramos delante de un estado intersexual aislado, que no se acompaña de otras anomalías. Interpretación genética de los estados intersexuales En la mayoría de las ocasiones la interpretación genética no es fácil de dar, incluso cuando la alteración anatómica hallada sea evidente; de manera que un estado intersexual puede asociarse a un cariotipo normal o encontrarnos ante una franca patología citogenética. La variabilidad de los resultados citogenéticos no siempre está relacionada con las diferentes presentaciones clínicas. Los patrones de herencia que podemos encontrar se describen a continuación. – Herencia autosómica recesiva: La existencia de varios afectados en la misma generación, a veces en asociación con la presencia de consanguinidad de los padres, evoca una transmisión autosómica recesiva. Ante entidades autosómicas recesivas, no esperaremos encontrar anomalías cromosómicas. Este es el caso de un cierto número de déficits enzimáticos: el ejemplo más conocido es el de la hiperplasia suprarrenal congénita. Puede ocasionar un pseudohermafroditismo femenino (déficit de 21-hidroxilasa o de 11-beta-hidroxilasa) o un pseudohermafroditismo masculino (déficit de 17-alfa-hidroxilasa, de 20,22-desmolasa o de 3 beta-hidrosteroide deshidrogenasa). Sigue el mismo patrón de herencia el déficit de 5a-reductasa tisular (14). El análisis de los casos familiares es particularmente delicado en las ambigüedades sexuales en que todos los sujetos afectos son varones. En este caso no podemos distinguir entre una herencia autosómica recesiva de una recesiva ligada a X si sólo tenemos estudios citogenéticos. – Herencia recesiva ligada a X: Sospecharemos una herencia recesiva ligada al X cuando en una misma familia encontremos varios sujetos con cariotipo XY con genitales ambiguos. En este caso la enfermedad se transmite a través de las madres. El 50% de sus hijos varones se hallarán afectados y sus hijas serán normales o portadoras no afectadas. En el síndrome de insensibilidad a la testosterona (síndrome de feminización testicular) ya se trate de la forma completa o parcial tenemos un ejemplo de transmisión recesiva ligada a X. – Heterogeneidad: En algunos casos la presentación de la afección es heterogénea, como por ejemplo en el hermafroditismo verdadero que habitualmente es de presentación esporádica (con cariotipo 46 XY, mosaico, quimera, o más frecuentemente 46 XX), pero algunos casos evocan una herencia autosómica recesiva o incluso dominante. – Variaciones de la expresión fenotípica de los estados intersexuales en la misma familia: Al lado de estos casos familiares de hermafroditismo verdadero, en algunas familias (sea dentro de la misma generación, sea en los parientes más lejanos), existen varones XX, evocando una herencia autosómica, muy probablemente dominante de penetrancia incompleta. También se han descrito familias que presentan a la vez casos de pseudohermafroditismos masculinos, disgenesias gonadales mixtas y/o disgenesias gonadales puras, sugiriendo la posibilidad de que se trate de un mismo desorden. Estas observaciones son relativamente raras y lo más frecuente es que estas anomalías sean esporádicas. Todas estas constataciones familiares tienen un interés clínico y de investigación con respecto a la etiología de estas entidades, y deberían permitir en el futuro una mejor comprensión de la aparición de ciertos estados intersexuales. Normalmente el motivo de consulta será la evaluación del riesgo de recurrencia de los hermanos de los sujetos afectos. Más rara será la consulta de posibilidad de afectación en hijos de los pacientes, ya que en la mayoría de los casos los sujetos son estériles. El consejo genético es relativamente fácil cuando la etiología de la ambigüedad sexual es conocida, de todas formas se debe valorar que puede existir variabilidad clínica, incluso en entidades con el mismo patrón de herencia. Riesgo de recurrencia en los hermanos de los sujetos afectos El riesgo de recurrencia para los hermanos de los pacientes es del 25% en cada gestación, sea cual sea el sexo, en los casos de patologías autosómicas recesivas. En las afectaciones recesivas ligadas a X, el riesgo de tener un hijo afecto es del 50% en hijos varones y lo podremos asegurar tras un estudio exhaustivo del árbol genealógico familiar. Cuando se trata de un primer caso en la familia la madre no es obligatoriamente heterocigota para el gen. Riesgo de recurrencia en la descendencia de los sujetos afectos Cuando la fertilidad está conservada, como por ejemplo en los déficits de 21-hidroxilasa, la posibilidad de tener un hijo homocigoto es baja excepto en matrimonios consanguíneos, y variará según la frecuencia de heterocigotos en la población donde se estudie el caso en concreto. Diagnóstico prenatal El diagnóstico prenatal de un estado intersexual aislado se realiza, prácticamente en todos los casos, cuando ya existe un caso de un niño afecto en una familia. Difícilmente se realizará de una forma fortuita, como ocurre con otras malformaciones. La determinación del sexo fetal por estudio citogenético es indispensable ya que dependiendo de la afección que estemos estudiando se presentará en sujetos citogenéticamente femeninos o masculinos. La posibilidad del diagnóstico precoz del sexo a partir del estudio de vellosidades coriales o mediante amniocentesis es particularmente interesante aquí. Hiperplasia suprarrenal congénita Durante el embarazo los problemas fetales y maternos están confinados a mujeres afectas de déficit de 21hidroxilasa (déficit de P450 c21), déficit de 11-hidroxilasa (déficit de P450 c11), y déficit de 3bhidroxiesteroide-deshidrogenasa (déficit de 3b-HSD) ya que otros déficits enzimáticos se asocian con esterilidad (15). Actualmente es posible realizar el diagnóstico prenatal de los déficit de 21-hidroxilasa y 11-hidroxilasa; históricamente mediante tests endocrinos y linkage HLA, y actualmente mediante técnicas de genética molecular. De hecho la importancia del diagnóstico prenatal de la hiperplasia suprarrenal congénita reside en la posibilidad de realizar un tratamiento prenatal a la madre gestante, administrando glucocorticoides por vía oral (16,17) antes de la novena semana de embarazo, con el objetivo de suprimir la excesiva secreción de andrógenos por la corteza adrenal fetal y minimizar la masculinización de los genitales en un feto femenino homocigoto afecto. De esta forma estamos realizando la prevención del pseudohermafroditismo femenino. Sin embargo los intentos de tratamiento prenatal mediante supresión con corticosteroides se han asociado con grados variables de éxito. Este planteamiento sólo puede realizarse en familias en las que ya hay un hijo o un familiar de primer grado afecto, y sólo si se han estudiado previamente las mutaciones existentes mediante técnicas de genética molecular. El diagnóstico del sexo a partir de amniocitos o más precozmente de vellosidades coriales permite interrumpir el tratamiento si se trata de un niño, y continuarlo hasta el final de la gestación sólo en caso de tratarse de una niña. Por otra parte, esta pauta supone realizar tratamiento en el primer trimestre de todos los fetos potencialmente afectos, lo que supone que, si ambos progenitores son heterocigotos, estaremos tratando innecesariamente a siete fetos no afectados, para suprimir un feto afecto (18). Hasta la fecha, no hay publicaciones de diagnóstico o tratamiento prenatal de fetos femeninos afectos de déficit de 3b-HDS. Diagnóstico prenatal de la hiperplasia suprarrenal congénita La hiperplasia suprarrenal congénita es una anomalía de la biosíntesis esteroidea. La mayoría de las enzimas esteroidogénicas pertenecen al grupo del citocromo p450 (19) de las oxidasas. Estas enzimas fijan el sustrato esteroideo y median la conversión esteroidea. El enzima p450 c21 se halla en el retículo endoplasmático celular. El enzima p450 c11 se localiza en la mitocondria. El 3b-hidroxiesteroide deshidrogenasa es un enzima no-p450 el cual también se encuentra en el retículo endoplasmático. La hiperplasia suprarrenal congénita debida a déficit de 21-hidroxilasa es una enfermedad autosómica recesiva, que se produce por defectos en el gen que codifica la enzima p450 c21 (CYP21B), localizado en el brazo corto del cromosoma 6. El p450 c21A (CYP21A) es un gen no funcionante y es conocido como un pseudogen. Debido a que los genes p450 c21 están asociados al locus HLA, esta asociación fue usada clínicamente (15) para valorar si un feto era portador de una hiperplasia suprarrenal congénita o podía estar afecto. Sin embargo, actualmente disponemos de un diagnóstico más fiable mediante técnicas de genética molecular. El déficit de 11-hidroxilasa es responsable del 15% de los casos de hiperplasia suprarrenal congénita en mujeres judías y musulmanas, pero es infrecuente en mujeres europeas. Los genes para el enzima P450c11 están localizados en el brazo largo del cromosoma 8, y por lo tanto no se hallan ligados al HLA. El gen que codifica al 3b-HSD ha sido clonado y está localizado en el cromosoma 1. El 3b-HSD no es un enzima p450 y no está ligado al HLA. Otro método diagnóstico utilizado ha sido la determinación en líquido amniótico de los niveles de las hormonas esteroideas. Los valores de los niveles en plasma y líquido amniótico de las diferentes hormonas esteroideas se hallan bien documentados (20,21). Sin embargo estas determinaciones no identifican los casos de severidad moderada de la enfermedad, y además requieren la supresión del tratamiento materno con dexametasona unos días antes, lo que dificulta la supresión adrenal fetal. Actualmente se realiza el análisis del ADN, obtenido de vellosidades coriales o de amniocitos (análisis molecular del gen), método fiable y seguro. Hiperplasia suprarrenal congénita secundaria a déficit de 21-hidroxilasa Las mujeres con cualquiera de las formas de presentación de la enfermedad deberían recibir consejo preconcepcional, el cual debe incluir referencias a las posibilidades de concepción, posibles complicaciones en el embarazo, diagnóstico prenatal, y tratamiento prenatal. En mujeres con formas intermedias de hiperplasia suprarrenal congénita secundaria a déficit de 21-hidroxilasa, la fertilidad puede alcanzar porcentajes normales, particularmente en el grupo de inicio tardío; aunque debido a la presencia de oligo-anovulación, frecuentemente es necesario un tratamiento adecuado y, a menudo, inducción de la ovulación. – Tratamiento de la enfermedad durante el embarazo: El tratamiento del déficit de 21-hidroxilasa es el mejor exponente del tratamiento de supresión y queda especialmente tratado más adelante en ese apartado. – Diagnóstico prenatal de los fetos afectos de hiperplasia suprarrenal congénita debida a déficit de 21hidroxilasa: Actualmente, el diagnóstico prenatal de la hiperplasia suprarrenal congénita secundaria a déficit de 21-hidroxilasa, puede realizarse de forma precisa. Para evaluar clínicamente la posible afectación del feto, en el caso en que una mujer embarazada sea homocigota o heterocigota para la hiperplasia suprarrenal congénita por déficit de 21-hidroxilasa, es importante tener el estudio molecular de su pareja. El feto se considera potencialmente afecto si ambos padres son heterocigotos, ambos son homocigotos, o si uno es heterocigoto y el otro homocigoto. Dependiendo de la población, aproximadamente el 1 al 6% de las parejas serán heterocigotas para la hiperplasia suprarrenal congénita por déficit de 21-hidroxilasa. El desarrollo de las técnicas de genética molecular ha facilitado en gran manera el diagnóstico fetal de la hiperplasia suprarrenal congénita por déficit de 21-hidroxilasa. Hiperplasia suprarrenal congénita secundaria al déficit de 11-beta-hidroxilasa (P450 c11) El déficit de 11-beta-hidroxilasa es la segunda causa en frecuencia de hiperplasia suprarrenal congénita (5% de los casos). Es una entidad que se hereda con un patrón de herencia autosómica recesiva, que causa una disminución de la producción de cortisol, desarrollo de virilización en las mujeres afectas, cursa con hipertensión arterial. Hay una marcada variabilidad clínica en la presentación, desde masculinización moderada con clitoromegalia, hasta formas extremas. Las alteraciones menstruales, el hirsutismo, y el acné son variables. Muchas mujeres con déficit de 11-beta-hidroxilasa que están tratadas con una dosis adecuada de glucocorticoides presentan una fertilidad normal. La CAH 11-beta-hidroxilasa puede estar causada por una o varias mutaciones en el gen CYP11B1 (22). – Diagnóstico prenatal de fetos femeninos afectos de 11-beta-hidroxilasa CAH: Inicialmente, el diagnóstico era endocrinológico utilizando los niveles en líquido amniótico de 11-deoxicortisol y tetrahidro-11-deoxicortisol, que se hallan elevados en los embarazos con fetos afectos. La mejor discriminación entre fetos afectos y no afectos fue obtenida cuando se obtuvieron en paralelo los niveles urinarios maternos y los de líquido amniótico. Actualmente puede realizarse un diagnóstico más fiable mediante la aplicación de las nuevas técnicas de genética molecular. La supresión con dexametasona (23) se ha utilizado en un intento de prevenir la masculinización de los genitales de los fetos femeninos afectos pero no siempre ha tenido éxito. Síndrome de insensibilidad a la testosterona y déficit de 5-alfa-reductasa Es posible realizar el diagnóstico del síndrome insensibilidad completa a testosterona (24) y del déficit de 5alfa-reductasa a partir de células de líquido amniótico. El primero puede incluso ser realizado a partir de vellosidades coriales, acompañado por supuesto de la determinación del sexo fetal. Cromosomopatías en los estados intersexuales Las alteraciones de los cromosomas sexuales que se asocian a disgenesias gonadales (Síndrome de Turner y síndrome de Klinefelter entre otros) presentan un índice de recurrencia muy pequeño, y son debidos a mecanismos de no disyunción. Para estas entidades es posible realizar un diagnóstico prenatal. Otros casos de intersexos Para las otras etiologías de ambigüedad sexual no hay realmente un diagnóstico prenatal posible. Se puede intentar poner en evidencia una discordancia entre el sexo cromosómico y el fenotipo, en efecto, el estudio de los órganos genitales externos es posible por ecografía, pero tendremos dificultades de interpretación y un diagnóstico prenatal realizado en estas condiciones no puede ser considerado de certeza. Además los casos familiares de disgenesias gonádales (46 XY), de hermafroditismo verdadero (46 XX), de varones XX son excepcionales y se puede plantear si está justificado proponer un diagnóstico prenatal así, a una pareja con un sólo hijo afecto. Conclusiones El consejo genético para los estados intersexuales no es nunca sencillo, incluso si el modo de transmisión es bien conocido y no inequívoco. En efecto, la ambigüedad sexual, la discordancia entre el sexo cromosómico y el sexo fenotípico, que pueden ser causa de una esterilidad futura, son frecuentemente tolerados, no sólo por parte del sujeto sino por el entorno. No tener en cuenta las particularidades inherentes a este tipo de anomalía puede ser la fuente de errores difíciles de reparar. Cuando el equipo multidisciplinario tenga a cargo las ambigüedades sexuales, debe responder una pregunta de consejo genético, y tiene que ser muy prudente en las explicaciones que tiene que dar. Tratamiento hormonal de sustitución En ambos sexos, el tratamiento hormonal de sustitución debe iniciarse al comienzo de la pubertad para inducir el desarrollo de los caracteres sexuales secundarios y continuar durante la etapa adulta a fin de mantener el trofismo sexual, así como las acciones metabólicas que, a nivel general, ejercen dichas hormonas en el organismo. Pacientes con sexo social femenino Los individuos asignados como "mujeres" deben iniciar el tratamiento, para inducir el desarrollo sexual puberal, a la edad de 11,5-12 años. La situación clínica que presentan estos pacientes en la pubertad, está directamente relacionada con la etiología del proceso que, en un alto porcentaje de casos, ya precisó una actuación médica previa. En la práctica, sólo requerirán tratamiento hormonal con estrógenos aquellos a los que se les haya extirpado quirúrgicamente la gónada y que, dependiendo del grado de desarrollo de los conductos de Müller, pueden presentarse bajo dos situaciones clínicas diferentes: A. Pacientes con ausencia de gónada y SIN útero: Como consecuencia de que la acción de los estrógenos sobre el crecimiento óseo pasa por una fase inicial de estimulación seguida de la detención de la misma por cierre de las epífisis, el tratamiento hormonal de sustitución con estrógenos debe iniciarse en la pubertad siempre y cuando la talla alcanzada sea considerada como la óptima ya que de lo contrario, podría derivarse un efecto clínico negativo. Si durante el período pre-puberal la talla alcanzada estuviese por debajo de la esperada, se debe administrar hormona de crecimiento (GH) a una dosis de 50 µg/Kg/día durante un período máximo de 6 años, tratamiento que permite alcanzar una talla final adulta superior en 10 cm a la esperada respecto a las niñas que no reciben tratamiento con GH. Es importante valorar y mantener el criterio y no ceder a la presión social que la paciente y su entorno familiar pueden ejercer para el inicio inmediato del desarrollo de los caracteres sexuales secundarios, sobre todo si se tiene en cuenta que, en estas edades, no existe descalcificación ósea en las pacientes sin gónada, independientemente de que hayan recibido o no tratamiento con hormona de crecimiento (25). La terapia hormonal de sustitución debe iniciarse con estrógenos ya que, si bien el crecimiento y desarrollo de la glándula mamaria es dependiente de la interacción de múltiples factores tanto hormonales (estrógenos, progesterona, prolactina, insulina, tiroxina, cortisol y prolactina), como de crecimiento (IGF-1), la inducción del desarrollo de los caracteres sexuales secundarios debe reproducir, en lo posible, las etapas de desarrollo natural de la glándula mamaria que, en sus periodos iniciales, es primordialmente estrógeno-dependiente. Teniendo en cuenta que el desarrollo sexual femenino requiere inicialmente niveles bajos de estrógenos en sangre, el tratamiento se comienza con dosis de 0,3 mg/día de estrógenos conjugados equinos (EC) o con 1 mg/día de valerianato de estradiol (VE2), durante un periodo de 3-6 meses. Transcurrido este tiempo, se realizará una evaluación de la respuesta y tolerancia clínica, que son los parámetros que determinarán la conveniencia de mantener la misma dosis durante un periodo más prolongado o bien, se aumenta el aporte exógeno de estrógenos hasta los 0,625 mg/día de EC ó 2 mg/día de VE2 que suele ser lo habitual. En general, tras un año de tratamiento hormonal con estas dosis, se alcanza un desarrollo mamario óptimo. La estrogenoterapia deberá mantenerse hasta que la paciente alcance la edad media de la menopausia, requeriendo los controles mamográficos y/o bioquímicos, con los mismos criterios ya establecidos para cualquier otra "mujer" que esté sometida a Terapia Hormonal Sustitutiva (T.H.S.), así como la posibilidad de modificar la dosis o seleccionar la vía de administración de acuerdo a las necesidades clínicas. B. Pacientes con ausencia de gónada y CON útero: El tratamiento de reposición con estrógenos, para las pacientes que poseen útero, sigue las mismas pautas indicadas anteriormente para inducir el desarrollo de los caracteres sexuales secundarios, basadas en la administración de estrógenos como monoterapia inicial. Una vez alcanzado el desarrollo mamario óptimo, se inducirá una hemorragia uterina por supresión mediante la administración de un gestágeno por vía oral durante 10 días (10 mg/día de acetato de medroxiprogesterona (AMP) o 200 mg/día de progesterona natural (P4)). Posteriormente debe comenzarse el tratamiento hormonal definitivo con la aportación estrógenos-progestágenos en pauta secuencial cíclica (21 días) o continua (28 días). Esta terapia debe incluir un estrógeno (2 mg/día de VE2 o 0,625-1,25 mg/día de EC) durante 21-28 días y un gestágeno (5-10 mg/día de AMP; 0,5 mg/día de norgestrel; 1 mg/día de acetato de ciproterona o 200 mg/día de P4) durante 10-14 días al mes, con lo que se producirá un sangrado uterino cíclico. La preferencia de la vía oral está condicionada por la comodidad de su administración y cumplimiento. La Tabla II recoge los preparados comerciales que existen en España, de estrógenos y gestágenos con diferente composición, que permiten la elección de pautas secuenciales adecuadas a las necesidades clínico-terapéuticas de cada individuo. Si estas pacientes desean gestación tienen que recurrir a técnicas de reproducción asistida mediante donación de ovocitos. En este sentido las pautas de preparación endometrial no difieren de las utilizadas para mujeres receptoras en dichos programas. El tratamiento se establece con dosis crecientes de VE2 comenzando el primer día del sangrado con 2 mg/día durante 8 días, 4 mg/día del 9º al 11º día continuando con 6 mg/día desde el 12º día hasta alcanzar un nivel de estradiol en sangre superior > 150 pg/ml y grosor endometrial > 8-8,5 mm. La preparación endometrial se complementará con 200-300 mg/día de progesterona (oral o transvaginal) hasta el momento de la transferencia embrionaria (26). Pacientes con sexo social masculino Estos individuos suelen presentar una masculinización incompleta de los genitales externos, caracterizados principalmente por hipospadias y microfalo. El tratamiento de reposición con andrógenos debe iniciarse en la pubertad entre los 12,5-13 años y tiene como objetivo provocar los efectos fisiológicos de la testosterona. Esta terapia debe inducir y mantener el desarrollo adecuado de los caracteres sexuales secundarios y las características somáticas que completan la masculinización, como son el desarrollo del vello (facial, torácico, abdominal, perineal y en muslos), el desarrollo de la laringe, de la musculatura esquelética, redistribución de la grasa corporal y crecimiento de la talla. La testosterona natural se puede administrar por vía transdérmica en parches de 2,5-5 mg aplicados a las 22.00 h. para alcanzar un perfil hormonal máximo a la mañana siguiente, reproduciendo el ritmo circadiano de dicha hormona. La testosterona también se puede utilizar por vía oral (metiltestosterona o mesterolona) o parenteral mediante preparados "depot" (ésteres de testosterona) de diferente vida media entre los que destacan: a) Compuestos de "vida media corta": Propionato de testosterona a dosis de 25 mg/8-12 h en administración diaria. b) Compuestos de "vida media prolongada": Cipionato de testosterona a dosis de 100-250 mg cada 2-4 semanas y enantato de testosterona con dosis de 110-250 mg cada 3-6 semanas (27,28). Aunque los andrógenos exógenos no corrigen el defecto de fusión de los genitales externos de estos individuos, la testosterona se utiliza en los casos de hipospadias o microfalo, como tratamiento coadyuvante previo a la cirugía al objeto de aumentar los cuerpos cavernosos. Las posibilidades de lograr una mayor longitud final del pene, dependen más del inicio precoz del tratamiento hormonal que de la dosis, por lo que puede ser necesaria la administración de andrógenos en edades muy tempranas como la época de lactante o prepuberal. En estos casos es necesario establecer un control estricto ante la posibilidad de una aceleración en la maduración ósea que puede determinar la interrupción del tratamiento. En esta situación, se administraría un andrógeno sintético por vía tópica, en forma de crema o loción, aplicada una o dos veces al día sobre la piel de los cuerpos cavernosos o por vía general mediante 100 mg/m2 de testosterona i.m. cada 15 días hasta 4 inyecciones (29). En los individuos de sexo social masculino, llegada la pubertad, se inicia el tratamiento con testosterona i.m. con dosis de 25-50 mg cada 30 días durante el primer año, 100 mg al mes en el segundo año e incrementos posteriores hasta alcanzar una dosis terapéutica de 250 mg cada 3-4 semanas. La vía transdérmica precisa dosis iniciales de 2,5 mg/día, hasta alcanzar el desarrollo sexual adecuado y se continúa con dosis de mantenimiento de 5 mg/día (30). En pacientes con resistencia a los andrógenos, como sucede en los pseudohemafroditismos masculinos por déficit de 5a-reductasa o insensibilidad a los andrógenos, se deben utilizar dosis superiores de tetosterona (500 mg/semanales de enantato de testosterona) para alcanzar un efecto androgénico evidente (27). En determinadas situaciones la administración de andrógenos exógenos han mejorado el desarrollo de órganos sexuales secundarios dependientes de la acción de la testosterona, como son la próstata y las vesículas seminales, lo que puede traducirse en un aumento del volumen de eyaculado. En todos los pacientes sometidos a tratamiento hormonal con testosterona durante periodos prolongados de tiempo, como sucede en estos individuos, es necesario evaluar el riesgo de desarrollo de cáncer de próstata. En este sentido resulta útil determinar los valores plasmáticos del marcador prostático específico PSA (prostatespecific antigen) antes del inicio y durante el tratamiento, ya que se ha mostrado como el mejor indicador de riesgo. Los valores anormales de PSA obligan a un rastreo mediante controles específicos en estos pacientes (tacto rectal, ecografía y/o punción-biopsia guiada por ecografía) (31). Terapia médica supresora Persigue eliminar en tiempo y forma adecuados aquellos estímulos que inducen una diferenciación sexual anómala. Puesto que ésta sólo se conduce en sentido masculino cuando hay una exposición suficiente a la hormona antimülleriana (AMH) y a un correcto microambiente androgénico, es evidente que la necesidad de supresión va a surgir sólo cuando un individuo de sexo femenino es sometido a un exceso de andrógenos (pseudohermafroditismo femenino). Dada su frecuencia, la hiperplasia suprarrenal es la forma más relevante, pues es responsable de alrededor de la mitad de los casos de recién nacidos con genitales ambiguos. Tal y como se describe anteriormente, hay otras posibles situaciones, como las que derivan de aporte hormonal exógeno, tumores virilizantes en la madre, y una miscelánea donde a veces las causas no son bien conocidas, pero en estos casos el tratamiento pasa obviamente por la eliminación directa del factor causal, fármaco o tumor. Por ese motivo, la terapia de supresión se centra en las formas ligadas a defectos enzimáticos, cuya base molecular ya ha sido descrita. Para eliminar la causa determinante del estado hiperandrogénico a que nos referimos, la estrategia correcta pasa por suministrar cortisol, pues ésta es la hormona que queda en déficit como consecuencia de la disfunción enzimática. La organización racional del tratamiento plantea una serie de cuestiones que procede resumir en los puntos que siguen: – Definición de objetivos. – Selección del momento de aplicación. – Selección de los casos que deben ser tratados. – Especificación de las opciones terapéuticas óptimas. – Controles del tratamiento. – Problemas potenciales. Definición de objetivos La eliminación de los estigmas de impregnación androgénica es el objetivo primario. Esta situación se vive como prioritaria durante la vida prenatal, pues de ella depende la problemática diferenciación de los genitales externos, que condiciona el estado de pseudohermafroditismo. También en el período prenatal hay un segundo objetivo que sólo ha comenzado a recibir atención en tiempo más reciente. Se trata de la diferenciación del sistema nervioso central que, expuesto a un exceso androgénico en fases cruciales de su desarrollo, incorpora patrones de comportamiento sexual, de especialización cognitiva o de orientación espacial más cercanas al sexo masculino (32,33). No hay, sin embargo, confirmación de que la hiperplasia suprarrenal congénita aporte alguna ventaja o desventaja en la capacidad cognitiva (34). Ya fuera del útero, según el déficit enzimático de que se trate, y de la variante molecular en cuestión, puede haber una asociación de alteraciones hidroelectrolíticas (pérdida de sal), y ya menos frecuentes, cardiovasculares (hipertensión) y metabólicas (hipoglucemia), que surgen como problema inmediatamente tras el inicio de la vida postnatal. Todas ellas, por ser frecuentemente ignoradas a la hora del diagnóstico, son causa de elevada mortalidad (35). Finalmente, y al margen de la diferenciación genital anómala, un exceso de oferta androgénica durante la vida prenatal o postnatal, impone una susceptibilidad posterior a manifestaciones cosméticas del hiperandrogenismo, tales como el hirsutismo en mayor o menor grado, y estados de hiperfunción androgénica suprarrenal durante la pubertad, que pueden conducir a anomalías hormonales capaces de favorecer estados hiperandrogénicos de origen ovárico, como el ovario poliquístico. Es evidente que la actuación con éxito sobre todas estas variables exige distintos niveles de excelencia en la estrategia terapéutica, de forma que el bloqueo del hiperandrogenismo intrauterino probablemente exija la llegada de un nivel de corticoides adecuado para frenar a la ACTH. El control de las disfunciones postnatales, sobre todo las que se manifiestan tras la pubertad, exigirán un ajuste más fino, a la vez que una considerable perspicacia diagnóstica. Selección del momento de aplicación La capacidad para modificar la diferenciación de los genitales externos depende de la cuantía de la oferta androgénica, de la duración de la exposición, y del momento de inicio. La corteza suprarrenal alcanza una capacidad funcional significativa hacia las semanas 10-12, pero no puede asegurarse que en una situación de sobreestímulo no exista la opción de alguna acción más temprana. Por tanto, como en la semana 10 está ya claramente en marcha la diferenciación de genitales externos femeninos, con la separación clara entre vagina y uretra, es conveniente bloquear ya la posible actuación de un exceso androgénico, que condicionaría la hipertrofia del clítoris. De hecho, el tratamiento se recomienda desde antes de la semana 9, y hay casuística con administración ya desde la semana 5-7. El tratamiento se confirma o no hacia la semana 10, cuando puede obtenerse información del sexo del feto a partir de biopsia corial. En semanas posteriores, y hasta la semana 20 en que se completa el proceso de diferenciación de genitales femeninos, el efecto puede manifestarse como formación de seno urogenital, fusión progresiva de labios, etc. No obstante, el efecto es más dramático si el estímulo actúa en las semanas más tempranas, de forma que ya a partir de la semana 16 ó 17, queda más reducido. Por este motivo, cuando la terapia supresora origina algún tipo de efecto no deseable en la madre, y en general como medida general, se recomienda reducir la dosis a partir de la semana 20, e incluso si hay necesidad, puede hacerse leve y progresivamente desde alguna semana antes. Selección de los casos que deben ser tratados Como ya hemos comentado la hiperplasia suprarrenal se transmite en forma autosómica recesiva, de forma que a partir de padres no afectados, las opciones son de 1 caso afectado por cada 3 que no lo están. Puesto que sólo sufren efectos clínicos los fetos de sexo femenino, el riesgo en realidad queda reducido a 1 de cada 8, de manera que una política de tratamiento prenatal indiscriminado supondría tratar innecesariamente a 7 de cada 8 casos. Puesto que el tratamiento no está libre de potenciales efectos indeseables, es evidente que resulta preferible disponer de alguna selectividad. De hecho, el uso de corticoides durante el embarazo no ha sido asociado con morbilidad fetal, pero sí materna, con incremento significativo de peso, rasgos faciales cushinoides, hipertensión, hiperglucemia, molestias gástricas, irritabilidad e incremento de estrías cutáneas. No está bien establecido, no obstante, cuál es la magnitud del riesgo de morbilidad hacia la madre. ¿Cómo seleccionar adecuadamente los casos? El tipaje de HLA o de anomalías del gen CYP21 por RFLP, junto a medida de esteroides en líquido amniótico parece un buen sistema para confirmar el defecto enzimático y a la vez calibrar la magnitud de la repercusión hormonal. Especificación de las opciones terapéuticas óptimas Si el déficit que desencadena el aumento de ACTH es de cortisol, el producto final de la esteroidogénesis suprarrenal, la mejor terapia es su reposición. En general, debe tenerse en cuenta que hay discrepancias en la información disponible, probablemente porque la experiencia sigue siendo insuficiente, acerca de la eficacia protectora en el feto. Los detalles sobre su administración dependen de la situación concreta, particularmente si se trata de terapia prenatal o postnatal (36). – Terapia prenatal: la dexametasona constituye una buena elección aquí, pues cruza la placenta y se administra fácilmente. Las dosis empleadas han consistido en 0,5 mg cada 8 a 12 horas, sin que se hayan descrito efectos teratogénicos. La eficacia goza ya de un apoyo considerable en la experiencia, globalmente satisfactoria, que se ha adquirido (37). – Terapia postnatal, donde puede utilizarse directamente cortisol o prednisona. La dosis recomendada para la segunda es 3,5-5 mg/m2, pudiendo incrementarse en función de la respuesta. En ambos casos, el propósito es disminuir la secreción de ACTH que, consecuentemente, supondrá la reducción de precursores androgénicos. Junto a ello, debe darse atención a la pérdida de sal, incluso si ésta no es un problema aparente. De hecho, la normalización en la actividad de la renina, como consecuencia de este tratamiento, se traduce en una mayor reducción de la ACTH y de los niveles de andrógenos, lo que supondrá una menor necesidad de glucocorticoides. La 9-fluorohidrocortisona ha sido el producto más usado. A dosis de 100 mg/día elimina la hipovolemia, que estimula la secreción de ACTH a través de la vía de la angiotensina II. Las dosis de glucocorticoides debe incrementarse 2-3 veces durante unos días si surge una enfermedad o se vive una situación estresante, y de 5-10 veces en las primeras 24 horas post-cirugía. Más recientemente, se ha comenzado a asociar tratamientos que pueden dificultar la acción androgénica por vías adicionales, tales como el bloqueo de receptores androgénicos o reprimiendo las vías biosintéticas. Por ejemplo, flutamida, inhibidores de la aromatasa, antagonistas de la CRH, y análogos de la GnRH han mostrado ya algunos resultados esperanzadores (38). Controles del tratamiento En la prevención prenatal hay un problema derivado de la variabilidad individual en cuanto a las dosis requeridas para conseguir una respuesta suficiente. La medida de la concentración de esteroides (17-OH progestrona, androstendiona y testosterona) en el líquido amniótico ha resultado de poca utilidad, pues a las dosis de corticoides que se emplean, suelen estar elevados. Más satisfactoria ha resultado la medida de estriol en suero, si bien la experiencia es aún escasa. En cuanto al control post-natal, es útil la medida en sangre de la concentración de renina, 17-OH-progesterona y andrógenos. Los niveles de 17-OH-progesterona deberían quedar en una franja amplia de 500 a 1.000 ng/dl (39), a fin de evitar aparición de clínica asociada a defecto o exceso de tratamiento. En el primero de los casos, el exceso de andrógenos condiciona virilización progresiva, pubertad precoz y aceleración precoz del crecimiento asociada a edad ósea, lo que a la postre implica estatura baja. El exceso en glucocorticoides, por el contrario, ocasiona un hábito cushinoide y retraso de crecimiento asociado a inhibición de la maduración epifisaria. La estatura baja, por tanto, se asocia tanto al exceso como al defecto en el tratamiento. Problemas potenciales Además de los desajustes en la dosificación del tratamiento, el cumplimiento es un problema serio, como en todas las medicaciones crónicas. Junto a ello, y a pesar de un control regular de las dosificaciones, el ajuste no es nunca tan perfecto como el que puede aportar el funcionamiento correcto de los sistemas de control en el organismo normal. Como consecuencia, la talla final suele ser baja y el pelo sexual incrementado (40), si bien esa secuela puede quedar muy limitada si se consigue un buen cumplimiento del tratamiento y un ajuste adecuado de las dosis (41). El uso de nuevas opciones, como la hormona de crecimiento (42) junto a las nuevas posibilidades antiandrogénicas anteriormente comentadas (análogos de la GnRH, etc.), o incluso la terapia génica, está ofreciendo buenos resultados iniciales, si bien en algunos casos no se trata más que de promesas esperadas con interés (43). La fertilidad no suele ser un problema, si bien puede serlo en las formas perdedoras de sal. En conjunto, la mejor atención pre y post-natal ha limitado el problema a la simple irregularidad menstrual, que es aún casi la norma. Recientemente se ha generado interés hacia la orientación sexual, que puede resultar afectada como consecuencia del efecto de los andrógenos sobre el cerebro en periodos cruciales de su desarrollo. Algunos autores han encontrado una alta tasa de bisexuales u homosexuales (44). Tratamiento quirúrgico de los estados intersexuales Los estados intersexuales incluyen una gran variedad de cuadros clínicos que responden a numerosas morfologías, aunque desde la perspectiva del tratamiento quirúrgico pueden agruparse fácilmente, al requerir, en unos casos, una genitoplastia y en otros, de forma aislada o asociada, cirugía sobre las gónadas. A la hora de abordar el tratamiento quirúrgico de los estados intersexuales, el cirujano especializado debe tener en cuenta en primer lugar, la adaptación de la anatomía genital al sexo asignado, considerando de forma asociada tres variables que pueden condicionar el tipo de intervención: la edad del individuo, el tamaño de los cuerpos cavernosos y, en el caso de niños muy pequeños, la orientación sexual. Las agruparemos en genitoplástias masculinizantes, genitoplástias feminizantes (45) y esbozaremos en un apartado las bases para el tratamiento quirúrgico de las gónadas en los estados intersexuales. Genitoplastia masculinizante Para la asignación de un tratamiento quirúrgico, en un individuo con un estado intersexual, en el que se opta por la reconstrucción del área genital con la asignación de genitales masculinos, es preciso considerar "a priori" el sexo cromosómico, sus niveles endógenos de hormonas sexuales, la posible influencia de un tratamiento con hormonas sexuales exógenas, su función gonadal y fertilidad potencial, la anatomía de las estructuras internas y externas de su aparato genital y la actitud de sus progenitores o representantes legales. La genitoplastia masculinizante tiene sus principales indicaciones en la agenesia o hipoplasia de pene (micropene), donde se requieren técnicas de reconstrucción plástica que se basan, fundamentalmente, en el empleo de tubos cutáneos obtenidos en el antebrazo que incluyen el nervio músculo-cutáneo, para realizar una posterior anastomosis a los nervios femoral o ilio-inguinal, la construcción de una neo-uretra con mucosa vesical que se implanta en el colgajo cutáneo del antebrazo y la realización de una anastomosis entre la neouretra y la uretra original del individuo. Actualmente, se han desarrollado otras técnicas de faloplastia que incorporan microcirugía que permite la realización de complejas anastomosis nerviosas y vasculares. Algunos estados intersexuales cursan con alteraciones de la curvadura del pene, acortamiento congénito de la uretra, braquiuretra, criptohipospadias o hipospadias. Para su corrección se han descrito mas de 400 técnicas diferentes, lo que demuestra que aún no existe una técnica óptima y, en consecuencia, la técnica de elección, debe seleccionarse en función de las características individuales de cada paciente, teniendo en cuenta siempre que es de primera elección aquella que pueda resolver la alteración en una única intervención y que, antes de realizar la uretroplastia, deben haberse corregido las malformaciones asociadas, así como tener en cuenta la necesidad o no de orquidopexia. Finalmente, hay que tener en cuenta, a la hora de planificar el tratamiento quirúrgico, que con mucha frecuencia los intersexos incluyen anomalías testiculares propiamente dichas o de su posición, así como malformaciones asociadas del tracto urinario o patologías de otras áreas como hernias inguinales. Algunos casos de hermafroditismo verdadero o de pseudohermafroditismo masculino, se asocian con ginecomastia produciendo alteraciones psicológicas que, en general, precisan de tratamiento. En estos casos, el tratamiento de elección es la mastectomía bilateral subcutánea con incisión periareolar semicircular. Genitoplastia feminizante La genitoplastia feminizante incluye a un conjunto de técnicas quirúrgicas dirigidas hacia la conformación de los órganos genitales femeninos en los estados intersexuales. Estas incluyen la reducción del pene o de una clitoromegalia, la transformación de un escroto o de un saco pseudoescrotal en una vulva, la comunicación de la vagina con el periné mediante una vulvoplastia o vulvovaginoplastia y la creación de una neovagina, si esta no está presente (46,47). – Clitoroplastia: La hipertrofia del clítoris es una secuela frecuente de los pseudohermafroditismos secundarios a una hiperplasia suprarrenal congénita y tiene una enorme importancia, tanto desde el punto de vista estético como psicológico, para la paciente afectada. Las técnicas quirúrgicas correctoras se basan en dos tipos de procedimientos: la clitoroidectomía y las técnicas de resección del clítoris con conservación del glande. Aunque ambas técnicas se emplean, la tendencia actual es la de conservar el órgano a fin de preservar la función sexual en las mejores condiciones posibles. – Vulvovaginoplastia: La vulvovaginoplastia se realiza en aquellos estados intersexuales en los que la vulva está obliterada con una apariencia pseudoescrotal. Básicamente, consiste en la reconstrucción de la vulva, dotándola de una apariencia de labios mayores y la creación de una vagina apta para el coito. Antes de plantearse la intervención que debe de realizarse a la edad más temprana posible, es preciso evaluar con precisión la anatomía del seno urogenital y establecer con exactitud el nivel al que se abre la vagina en el seno urogenital. La complicación más frecuente de estas intervenciones es la estenosis vaginal. – Vagina artificial o neovagina: La creación de una neovagina está indicada en aquellos estados intersexuales a los que se asigna el sexo femenino y presentan una ausencia, total o parcial, de la vagina con la que no es posible el coito. Los mismos procedimientos quirúrgicos se emplean en mujeres con agenesia vaginal, como es el caso del Síndrome de Rokitanski-Küster-Hauser. Se han descrito numerosas técnicas quirúrgicas que pasaremos a revisar brevemente. Para la creación de una neovagina pueden emplearse dos tipos de métodos: los no quirúrgicos o los quirúrgicos propiamente dichos. El método "no quirúrgico" por excelencia se basa en la dilatación vaginal, según el método de Frank, que consiste en obtener una dilatación lenta y progresiva de la vagina, mediante el empleo de dilatadores. Para ser utilizado, se precisa la existencia de un fondo de saco vaginal y un moderado grado de elongación. La paciente debe aplicarse el dilatador con una cierta presión sobre el fondo vaginal durante 10 minutos dos veces diarias. Empleando progresivamente dilatadores de mayor tamaño, puede obtenerse una vagina funcional apta para el coito a las 6-12 semanas de instaurada la terapia. La ventaja de este método es su simplicidad y la ausencia de riesgos. Sin embargo, resulta inaceptable para muchas pacientes, especialmente jóvenes. En estas mujeres, o en las que existe un útero adecuado y se desea preservar la fertilidad, son de primera elección los métodos quirúrgicos. Los métodos quirúrgicos más utilizados para la creación de una neovagina son los que emplean el abordaje perineal, existiendo diferentes técnicas: – Técnica de Williams: Esta técnica consiste en el empleo de la piel de ambos labios mayores con el objeto de conseguir una elongación del canal vaginal. Este método es una alternativa que puede emplearse cuando la creación de una verdadera neovagina no es posible. Permite obtener un canal vaginal de unos 8-10 cm en el que pueden introducirse fácilmente dos dedos, propiciando resultados funcionales muy aceptables. Esta técnica suele utilizarse con mayor frecuencia en casos de atresia vaginal secundaria a radioterapia que en los de agenesia vaginal. – Técnica de McIndoe: Para crear la futura cavidad vaginal se emplean injertos de piel muy fina en forma de malla recubriendo un molde vaginal. Los injertos, se suelen obtener del muslo, de la cadera o de la parte superior del brazo. La futura cavidad vaginal se construye a partir de una incisión semicircular realizada en el pliegue existente, entre el borde inferior del introito y el periné. La disección del espacio recto-vaginal hasta el fondo de saco de Douglas, debe permitir la obtención de una cavidad de unos 12 cm de profundidad y ser capaz de albergar tres dedos. Una vez construido este espacio se procede a la introducción del molde vaginal, previamente revestido en su totalidad por un injerto de piel, y fijado a los labios mayores. El molde es recomendable mantenerlo "in situ" durante 10 días. Una vez retirado, es conveniente la aplicación durante dos veces diarias de una crema vaginal de estrógenos y la introducción del molde vaginal durante la noche, al menos durante las tres semanas siguientes, a partir de las que sería deseable el inicio de las relaciones sexuales coitales, o en su defecto, el empleo periódico del molde vaginal. – El método Davidov: Con objeto de obtener una mejor epitelización de la neovagina se han desarrollado técnicas quirúrgicas que emplean el peritoneo, o un segmento del recto-sigma, para la creación de una vagina artificial. Estas técnicas precisan de un abordaje combinado vagino-abdominal. Este autor propone la creación de una neovagina que emplea como base el peritoneo del fondo de saco de Douglas fue descrito por. El abordaje quirúrgico es mixto (abdomino-vulvar) y consiste en la disección del peritoneo del fondo de saco de Douglas por vía abdominal, para posteriormente abocarlo y fijarlo al introito, una vez construido el canal vaginal. – La "invaginación de Vecchietti" precisa también de un abordaje combinado. Por vía abdominal se realiza una incisión en el peritoneo de la pared anterior del útero rudimentario. Simultáneamente, a través del introito se introduce una aguja "fiadora", en cuyo extremo caudal porta una doble sutura gruesa y no reabsorbible que contiene una "oliva" dilatadora. Ambas suturas se sitúan en el exterior del abdomen, extrayéndolas a través de la región retroperitoneal en los extremos distales de los músculos rectos del abdomen. Posteriormente, se sutura el fondo de saco de Douglas y la pared abdominal. Los extremos de las suturas, unidas a la "oliva" dilatadora se introducen en el introito, mientras que los cabos extra-abdominales se conectan a un dispositivo tensor que será reajustado diariamente, durante una semana, de tal forma que al final del periodo se obtiene una profundidad vaginal de unos 10 cm. Esta técnica ofrece buenos resultados, aunque se pueden producir lesiones vesicales y rectales frecuentes en el transcurso de la intervención. – Vagina ileocecal de Hohenfellner. Finalmente, dentro de las técnicas vagino-abdominales se encuentra la de la vagina ileocecal de Hohenfellner. Este método se basa en la creación de una neovagina empleando un fragmento de intestino delgado o de colon. Tiene como inconveniente más importante la envergadura de la intervención y el que durante mucho tiempo da lugar a la producción de abundante exudado mucoide. – Neovagina utilizando mucosa vesical. La mucosa vesical se caracteriza por su gran distensibilidad, lo que permite obtener implantes de gran dimensión. La ausencia de folículos pilosos, glándulas sebáceas y sudoríparas, así como la ausencia de queratina, evitan la retracción y presencia de pelo en la vagina neoformada. Mediante una incisión de Pfannenstiel se obtiene la mucosa vesical, que se aplica sobre una prótesis. Dicha prótesis vaginal se introduce en un espacio neoformado entre vejiga y recto, y se retira a los 15 días para valorar la viabilidad del implante. La prótesis debe mantenerse colocada durante los primeros 6 meses. – Neovagina con amnios. Desde hace años se sabe que el amnios estimula adecuadamente la epitelización y ha sido empleado con este fin y excelentes resultados en grandes quemados, traumatismos, etc. Desde que en 1934 Brideau publicó, por primera vez, la utilización de amnios humano para la creación de una neovagina, dicha técnica se ha empleado en múltiples ocasiones por su baja morbilidad y los buenos resultados que ofrece. Se basa en realizar un tunel perineal, del mismo modo que en la técnica de McIndoe para, posteriormente, introducir una prótesis vaginal recubierta de amnios para conseguir una adecuada epitelización de la neovagina. Tratamiento quirúrgico de las gónadas en los estados intersexuales La estados intersexuales causados por alteraciones cromosómicas y los hiperandrogenismos en la hembra y defectos en la producción o acción de los andrógenos en el varón, dan lugar a una gran variedad de manifestaciones en la morfología de los genitales externos. Sin embargo, la gónada va a depender de las características de los cromosomas sexuales. En consecuencia, la biopsia, la extirpación o la conservación de la gónada van a depender de cada variante y debe de estar en función de prevenir una degeneración maligna de la gónada o una masculinización o feminización en niños a los que se les ha adjudicado el género opuesto. Aunque no existe un consenso definitivo, en el hermafroditismo verdadero, caracterizado por la presencia en la misma gónada de tejido ovárico y de testículo, existe un elevado riesgo de desarrollo de gonadoblastomas por el ovoteste, lo que justifica su extirpación, en todos los casos, en cuanto se realiza el diagnóstico, si tenemos en cuenta que, además, estos individuos son siempre infértiles. Por el contrario, si existe un acuerdo generalizado en la necesidad de extirpar las gónadas disgenéticas, en los casos de disgenesia gonadal mixta, donde el riesgo de desarrollar un gonadoblastoma oscila entre un 30 y un 50%. El abordaje quirúrgico puede realizarse por vía inguinal o por vía laparotómica, dependiendo de la localización de las gónadas. Idéntica conducta debe seguirse en los casos de feminización testicular. En todos los casos en los que está indicada una gonadectomía, ésta debe seguirse con una planificación adecuada de tratamiento hormonal sustitutivo a base de testosterona o de una asociación de estroprogestagenos dependiendo del género del individuo. Bibliografía 1. Kawata M. Roles of steroid hormones and their receptors in structural organization in de nervous system. Neurosci Res 1995;24:1-46. 2. Harley VR, Goodfellow PN. The biochemical role of SRY in sex determination. Mol Reprod Dev 1994;39:184193. 3. Mac Lean HE; Warne G-L, Zajac JD. Intersex disorders: shedding light on male sexual differentiation beyond SRY. Clin Endocrinos (oxf) 1997; 46(1):101-108. 4. Wachtel SS, Ohno S, Koo GC, Boyse EA. Possible role of HY antigen on primary sex determination. Nature 1975; 257: 235. 5. Haseltine PP. Sexual differentation. Current concepts. The Biological Basis of Reproductive and Developemental Medicine 1983. Elsevier. New York. 6. Mac Laren A, Simpson E, Tomonari K y cols. Male sexual differentiation in mice lacking HY antigen. Nature 1984; 312:552. 7. Page DR, Mosher R, Sumpson EM y cols. The sex determining region of the human Y chromosome encodes a finger protein. Cell 1986; 51:1091-1104. 8. Wiener JS, Marcelli M, Lamb DJ. Molecular determinants of sexual differentation. World J Urol 1996;14:278294. 9. Lovell-Badge R, Hacken A. The molecular genetics of SRY and its role in mamalian sex determination. Phil Trans R Soc Lond 1955;350:205-214. 10. Arn P, Chen H, Tuck-Muller CM et al. SRVX, a sex reversing locus in Xp21.2 -p22.11. Hum Genet 1994;93:389-393. 11. Zanaria E, Bardoni B, Dabovic Ba, et al. Xp dublications and sex reversal. Phil Trans R Soc Lond 1995;350:291-296. 12. Jost A, Vigier B, Prepia J, Perchelet JP. Studies on sex differentiation in mamals. Rec Prog Horm Res 1973; 29:1-41. 13. Calaf J. Desarrollo gonadal y genital y sus alteraciones. En Fertilidad y Esterilidad Humanas. Tomo II: Endocrinología Ginecológica y Anticoncepción, de Vanrell JA, Calaf J, Balasch J, Viscasillas P. Págs 57-79. 2000, Ed. Masson, S.A. Barcelona. 14. Briard ML. Le conseil génétique dans les états intersexués. J Génét Hum 1985;35(2-3):91-104. 15. Garner PR. Congenital Adrenal Hyperplasia in Pregnancy. Semin Perinatol 1998; 22(6): 446-456. 16. David M, Forest MG. Prenatal treatment of congenital adrenal hyperplasia resulting from 21-hydroxylase deficiency. J Pediatr 1984;105:799-801. 17. Forest MG, Morel Y, David M. Prenatal treatment of congenital adrenal hyperplaia. Tem 1998;9:284-289. 18. Miller WL. Dexamethasone treatment of congenital adrenal hyperplasia in utero: an experimental therapy of unproven safety. J Urol 1999;162:537-540. 19. González FJ. The molecular biology of cytochrome p450s. Pharmacol Rev 1989;40:243-288. 20. Frasier SD, Thorneycroft IH, Weiss BA, Horton R. Elevated amniotic fluid concentration of 17 alphahydroxyprogesterone in congenital adrenal hyperplasia (letter). J Pediatr 1975;86:310-312. 21. Nagamani M, McDonough PG, Ellegood JO, Mahesh VB. Maternal and amniotic fluid 17 alphahydroxyprogesterone levels during pregnancy:diagnosis of congenital adrenal hyperplasis in utero. Am J Obstet Gynecol 1978;130:791-794. 22. Geley S, Kapelari K, Johrer K. CYP11B1 mutations causing congenital adrenal hyperplasia due to 11bhydroxylase deficiency. J Clin Endocrinol Metab 1996;81:2896-2901. 23. Cerame BI, Newfield RS, Pascoe L, Curnow KM, Nimkarn S, Roe TF, Neew MI, Wilson RC. Prenatal Diagnosis and treatment of 11b-hydroxylase Deficiency Congenital Adrenal Hyperplaia Resulting in Normal female genitalia. J Clin Endocrinol Metab 1999;84:3129-3134. 24. Brown T. Male pseudohermaphroditism: Defects in androgens-dependent tatget tissues. In: Seminars in reproductive Endocrinology 1987;5:243-247. 25. Speroff L, Glass RH, Kase NG. Desarrollo sexual normal y anormal, en "Endocrinología Ginecológica e Infertilidad", Speroff L, Glass RH, Kase NG (Eds) cap. 9, pp 339-379, 2000. Waverly Hispanica S.A. Buenos Aires. 26. Remohí J, Gallardo E, Gutiérrez A, Guanes PP, Cano F, Levy M, Yaul S y Gartner B. Donación de ovocitos, en "Reproducción Humana", Remohí J, Simón C, Pellicer A, Bonilla-Musoles F (Eds) cap. 31, pp 348-359. 1996. McGraw-Hill-Interamericana Madrid. 27. Matsumoto AM. Clinical use and abuse of androgens and antiandrogens, en Principles and Practice of Endocrinology and Metabolism, Becker KL et al. (Eds), Second Edition, chap 119, pp 1110-1123. 1995, JB Lippincott Company, Philadelphia. 28. Andrógenos: En Catálogo de Especialidades Farmacéuticas. Consejo General de Colegios Oficiales de Farmacéuticos (Eds), pp 1010-1014, 2001, CINSA, Madrid. 29. Velasquez-Urzola A, Leger J, Aigrain Y, Czernichow P. Hypoplasia of the penis: etiologic diagnosis and results of treatment with delayed-action testosterone. Arch Pediatr 1998;5(8):844-850. 30. Audí Parera L. Anomalías de la diferenciación sexual, en "Tratado de Endocrinología Pediátrica", Pombo Arias et al. (Eds), 2ª edición, cap. 35, pp 785-815, 1997. Díaz de Santos S.A., Madrid. 31. Guay AT, Pérez JB, Fitaihi WA, Vereb M. Testosterone treatment in hypogonadal men: prostate-specific antigen level and risk of prostate cancer. Endocr Pract 2000;6(2):218-221. 32. Kuhnle U, Bollinger M, Schwarz HP, Knorr D. Partnership and sexuality in adult female patients with congenital adrenal hyperplasia. First results of a cross-sectional quality-of-life evaluation. J Steroid Biochem Mol Biol 1993; 45:123. 33. Meyer-Bahlburg HF, Gruen RS, New MI, Bell JJ, Morishima A, Shimshi M, Bueno Y, Vargas I, Baker SW. Gender change from female to male in classical congenital adrenal hyperplasia. Horm Behav 1996; 30: 319. 34. Berenbaum SA. Cognitive function in congenital adrenal hyperplasia. Endocrinol Metab Clin North Am 2001; 30:173-192. 35. Kovacs J, Votava F, Heinze G, Solyom J, Lebl J, Pribilincova Z, Frisch H, Battelino T, Waldhauser F. Lessons from 30 years of clinical diagnosis and treatment of congenital adrenal hyperplasia in five middle European countries. J Clin Endocrinol Metab 2001;86:2958-2964. 36. White PC, Speiser PW. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev 2000; 21:245-291. 37. New MI. Prenatal treatment of congenital adrenal hyperplasia. The United States experience. Endocrinol Metab Clin North Am 2001; 30:1-13. 38. Merke DP, Keil MF, Jones JV, Fields J, Hill S, Cutler GB Jr. Flutamide, testolactone, and reduced hydrocortisone dose maintain normal growth velocity and bone maturation despite elevated androgen levels in children with congenital adrenal hyperplasia. J Clin Endocrinol Metab 2000; 85:1114-1120. 39. Merke DP, Cutler GB Jr. New ideas for medical treatment of congenital adrenal hyperplasia. Endocrinol Metab Clin North Am 2001; 30:121-135. 40. Migeon CJ, Wisniewski AB. Congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Growth, development, and therapeutic considerations. Endocrinol Metab Clin North Am 2001;30:193-206. 41. Eugster EA, Dimeglio LA, Wright JC, Freidenberg GR, Seshadri R, Pescovitz OH. Height outcome in congenital adrenal hyperplasia caused by 21-hydroxylase deficiency: a meta-analysis. J Pediatr 2001;138:2632. 42. Rivkees SA, Crawford JD. Dexamethasone treatment of virilizing congenital adrenal hyperplasia: the ability to achieve normal growth. Pediatrics 2000;106:767-773. 43. Quintos JB, Vogiatzi MG, Harbison MD, New MI. Growth hormone therapy alone or in combination with gonadotropin-releasing hormone analog therapy to improve the height deficit in children with congenital adrenal hyperplasia. J Clin Endocrinol Metab 2001; 86:1511-1517. 44. Meyer-Bahlburg HF. Gender and sexuality in classic congenital adrenal hyperplasia. Endocrinol Metab Clin North Am 2001; 30:155-171. 45. Martínez-Mora, J. Textbook Intersexual States. Disorders of Sex Differentiation. 1994. Ediciones Doyma. Barcelona. España. 46. Callejo J y Villagrasa M (Editores). Calaf J, Ordás J, González Núñez S, Jáuregui MT, Medina M, Reus E, Lailla JM. "Estados Intersexuales" (V 2.0). 1996, Lab. Rhône-Poulenc Rorer S.A. Madrid. 47. Dueñas JL. Tratamiento de la atresia-agenesia vaginal. En "La amenorrea. Estados que la determinan, su diagnóstico y tratamiento" (V.1.0), de Callejo J y Villagrasa M (Editores). Cabero A, Calaf J, Dueñas JL, Espinós JJ, Espín AM, González Nuñez S, Jáuregui MT, Lailla JM. 1997, Ed. Lab. Esteve, Barcelona. Documentos de Consenso S.E.G.O.