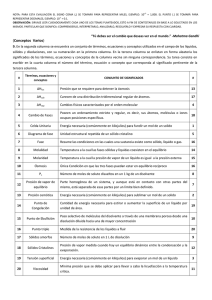
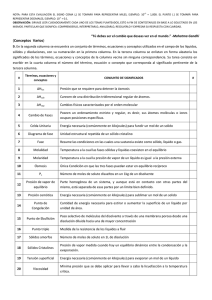
Laboratorio de proceso quimico y control
Anuncio

ESCUELA DE EDUCACIÓN TÉCNICA Nº 3 – SAN JOSÉ
DEPARTAMENTO QUÍMICA
LOS ESTADOS DE LA MATERIA
La forma en que las partículas que constituyen una sustancia se reúnen o agregan
determina una buena parte de las propiedades físicas y, entre ellas, su estado
sólido, líquido o gaseoso.
Las leyes que rigen el comportamiento de la materia en la escala ordinaria de
observación pueden ser explicadas a partir de teorías que hacen referencia a las
interacciones entre sus componentes elementales. Sometida a condiciones
extremas, la materia puede pasar a estados físicos muy especiales; tal es el caso
del plasma y de la materia constitutiva de las estrellas de neutrones.
La materia se presenta esencialmente, en nuestro planeta, bajo tres formas o
estados de agregación diferentes: el estado sólido, el estado líquido y el estado
gaseoso.
Cada uno de estos tres estados presenta unas propiedades directamente
observables que le son características.
Así los sólidos poseen una forma y volumen propios; los líquidos, por su parte,
aunque adoptan la forma del recipiente que los contiene, poseen un volumen
propio que se mantiene prácticamente constante aun en el caso de ser sometidos a
presiones exteriores considerables. Los gases, sin embargo, adoptan la forma del
recipiente y además ocupan todo su volumen interior.
A lo largo de la historia, filósofos y científicos han profundizado en el estudio de los
diferentes estados de la materia y las aportaciones en este tema, han contribuido,
de manera decisiva, al desarrollo de otros campos de la ciencia y de la técnica. Así,
el estudio de los gases sirvió de base para establecer los fundamentos de la
química; el conocimiento de la dilatación de los líquidos hizo progresar el estudio
de los fenómenos caloríficos; y, más recientemente, la física del estado sólido
no sólo ha permitido poner a prueba la mecánica cuántica como teoría física, sino
que a la vez ha abierto perspectivas de aplicación en el terreno de la electrónica y
de los nuevos materiales, que son, en buena parte, el fundamento del actual
progreso tecnológico.
OTROS ESTADOS DE LA MATERIA
Los estados sólido, líquido y gaseoso constituyen las formas en que se presenta la
materia en condiciones no demasiado alejadas de las que reinan en nuestro
planeta. Sin embargo, bajo condiciones extremas, la materia modifica su
composición y propiedades y se aleja de las leyes que describen el
comportamiento de sólidos, líquidos o gases.
El plasma es considerado como el cuarto estado de la materia, pues su presencia
en el universo es muy abundante. Se trata de una masa gaseosa fuertemente
ionizada en la cual, como consecuencia de temperaturas extremadamente
elevadas, los átomos se han visto despojados de su envoltura de electrones y
coexisten con los núcleos atómicos en un estado de agitación intensa.
Laboratorio de Procesos Químicos y Control
Página 1
Las estrellas, durante una parte importante de su vida, están constituidas por
grandes masas de plasma. Debido a la violencia de los choques entre núcleos, en
tales condiciones se producen reacciones de síntesis de núcleos nuevos con una
considerable liberación de energía. El Sol es esencialmente una enorme esfera de
plasma.
La materia componente de las estrellas en un estado avanzado de su evolución
degenera hacia formas en las que los electrones se funden con los núcleos
atómicos, dando lugar a una masa compacta de neutrones cuya densidad alcanza
18
3
valores del orden de los 10
kg/m . Éstas son las características de la materia
componente de las llamadas estrellas de neutrones. Se trata, no obstante, de una
etapa previa a la degeneración total característica de un estado de extrema
densidad, en el que los propios neutrones son aplastados por la presión hacia
adentro debida las fuerzas gravitatorias. Éste es el estado de la materia en esos
objetos oscuros y misteriosos del universo conocidos como agujeros negros.
EL ESTADO GASEOSO
Las experiencias de Boyle
El estudio de los gases, y en particular del aire, atrajo la atención de los físicos del
siglo XVII y más concretamente la del irlandés Robert Boyle (1627-1691). Las
experiencias que le permitieron establecer su conocida ley consistieron,
básicamente, en añadir mercurio a un tubo acodado suficientemente largo abierto
por un extremo y provisto de una llave en el otro. Con la llave abierta vertía
mercurio y su nivel en las dos ramas del tubo se igualaba (principio de los vasos
comunicantes). A continuación cerraba la llave y añadía sucesivamente cantidades
de mercurio iguales, con lo cual, la presión a la que estaba sometido el gas
encerrado en el otro extremo del tubo, aumentaba en igual proporción. Mediante
sucesivas medidas de la distancia entre los dos niveles alcanzados por el mercurio
en ambas ramas del tubo, observó que la disminución del volumen del gas
guardaba cierta relación con el aumento de presión. Si doblaba el peso de
mercurio, el volumen se reducía a la mitad, si lo triplicaba se reducía a la tercera
parte y así sucesivamente. Un análisis cuidadoso de tales resultados
experimentales le permitió, finalmente, enunciar su ley.
Aun cuando Boyle no indicó explícitamente que la temperatura debía permanecer
constante durante el experimento, el descubrimiento independiente efectuado por
el físico francés Edme Mariotte (1630-1684) lo puso de manifiesto, completando así
las conclusiones del irlandés.
A temperatura constante, el volumen de un gas es inversamente proporcional a la
presión que soporta:
P · V = cte
para T = cte (20.1)
P1 · V1 = P2 · V2
Laboratorio de Procesos Químicos y Control
Página 2
Este enunciado constituye la llamada ley de Boyle-Mariotte.
Las leyes de Gay-Lussac
El estudio de la dilatación de los gases puede efectuarse con la ayuda de un
matraz de vidrio que termine en un tubo capilar acodado por cuyo interior puede
deslizarse un índice de mercurio sobre una escala graduada. La dilatación de la
sustancia gaseosa contenida en el recipiente, puede observarse, de forma
controlada, sumergiendo el matraz en un baño de agua cuya temperatura puede
variarse a voluntad. La lectura del volumen del gas sobre la escala graduada y de
la temperatura del agua sobre un termómetro empleado al efecto, permite
encontrar una relación entre ambas magnitudes físicas en condiciones de presión
constante e igual a la presión atmosférica.
Experimentos de este tipo llevaron al químico francés Joseph Louis Gay-Lussac
(1778-1850) a concluir que:
a presión constante, el volumen de un gas aumenta proporcionalmente al
incremento de temperatura, siendo la constante de proporcionalidad la
Laboratorio de Procesos Químicos y Control
Página 3
misma para todos los gases. Este enunciado, que se conoce como primera Ley
de Gay-Lussac, se expresa matemáticamente mediante la ecuación:
Vt = V0 (1 + αt) (20.2)
donde Vt representa el volumen a la temperatura de t ºC, V0 el volumen a 0 ºC y
α es una constante aproximadamente igual para todos los gases que se denomina
coeficiente de dilatación y cuyo valor es: α = 1/273. El que α sea igual a 1/273
significa que cuando la temperatura de un gas se eleva en un grado centígrado, su
volumen aumenta 1/273 veces el volumen inicial, es decir, se dilata en un
0,37 % por grado centígrado.
ISOBARA
V / T = cte
V1 /T1 = V2 /T2
La primera ley de Gay-Lussac se conoce también como ley de Charles-Gay
Lussac, ya que fue sugerida con anterioridad en una forma semejante por Jacques
Charles (1746-1823).
Experiencias semejantes realizadas manteniendo constante el volumen y
estudiando la variación de la presión con la temperatura permitieron al químico
francés establecer la que se conoce como segunda Ley de Gay Lussac:
a volumen constante, la presión de un gas aumenta proporcionalmente al
incremento de temperatura, siendo la constante de proporcionalidad la
misma para todos los gases.
Laboratorio de Procesos Químicos y Control
Página 4
.
P / T = CTE
P1 / T1 = P2 / T2
La ley de los gases perfectos
Las leyes de Boyle-Mariotte y de Gay-Lussac sobre el comportamiento de los
gases, aunque son aplicables dentro de una buena aproximación a los gases
existentes en la naturaleza, son tanto más imprecisas cuanto mayor es la densidad,
la presión o la temperatura del gas. Por ello los gases que cumplen con exactitud
dichas leyes físicas se denominan gases perfectos o ideales.
Es posible combinar las leyes de los gases en una sola ecuación sencilla si la
temperatura se expresa en la escala absoluta o Kelvin.
Laboratorio de Procesos Químicos y Control
Página 5
P . V = CTE
T
que indica que el producto del volumen de un gas por su presión dividido por su
temperatura absoluta es una cantidad constante.
Ello significa que una muestra gaseosa dada puede evolucionar de un estado
inicial a otro final cambiando en el proceso su presión, su volumen o su
temperatura, pero siempre que la cantidad PV/T no varíe.
Esta ecuación constituye una expresión de la llamada ley de los gases ideales o
perfectos, que también se conoce como ecuación de estado. Para dos estados
cualesquiera inicial y final las magnitudes P, V y T están relacionadas en la forma:
La constante de proporcionalidad depende de la cantidad de sustancia gaseosa
considerada.
Cuando esta circunstancia se introduce en la ecuación resulta la expresión de la
ley de los gases ideales más usada:
donde n es el número de moles de la muestra gaseosa considerada y R es la
llamada constante de los gases perfectos igual a:
0,082 L . atm / ºK . mol
Considerando las dimensiones de R = Energía / grados x nº de moles, tenemos
las equivalencias:
8,314 X 107 Erg / grados x mol
ó
1,987 cal / grados x mol
La teoría cinética: una explicación para las leyes de los gases
Las experiencias de Boyle y de otros físicos de la época pusieron claramente de
manifiesto que los gases podían comprimirse y expandirse. Pero, ¿cómo explicar
estas propiedades que los diferenciaban claramente de los líquidos y los sólidos?
Según Bernouilli los átomos o corpúsculos de gas, debido a su pequeño tamaño,
se encontraban en un enorme número aun en pequeños volúmenes gaseosos. Su
movimiento incesante producía choques entre sí y con las paredes del recipiente.
Esta innumerable cantidad de impactos de los corpúsculos gaseosos explicaba el
efecto observable de la presión del gas y, por tanto, su expansibilidad.
Laboratorio de Procesos Químicos y Control
Página 6
De acuerdo con sus razonamientos, la disminución del volumen del gas restringe el
recorrido de los corpúsculos móviles y por tanto, incrementa el número de choques
por segundo contra las paredes del recipiente, esto es, aumenta la presión del gas.
Estudios teóricos apropiados permitieron a Bernouilli deducir, matemáticamente la
teoría cinética de los gases. Además, a partir del significado de la presión del gas
según el modelo cinético y de la segunda ley de Gay-Lussac, fue posible encontrar
un significado también cinético a la magnitud temperatura.
Así cuanto mayor es la temperatura de un gas tanto mayor es la presión que
ejerce, es decir, más enérgicos deben ser los impactos de las partículas del gas
sobre el interior del recipiente. Estudios más rigurosos desarrollaron esta idea
sencilla sugerida por el físico alemán Rudolph Clausius (1822-1888) y permitieron
establecer que la temperatura absoluta de un gas constituye una medida de la
energía cinética media de las moléculas del gas en su movimiento de agitación
desordenada.
La extensión de la teoría cinética a otros estados de agregación de la materia ha
permitido comprender los fenómenos de cambio de estado desde un punto de vista
molecular.
APLICACIÓN DE LA ECUACIÓN DE ESTADO DE LOS GASES
Una cierta masa de nitrógeno ocupa 10 litros a 1 atm de presión y 27 ºC de
temperatura. ¿Qué volumen ocupará la misma masa de gas si las condiciones
cambian a 2 atm y 207 ºC? ¿Cuál sería la presión del gas si las condiciones finales
de volumen y temperatura fueran de 20 litros y 27 ºC?
La relación entre los valores de las magnitudes P, V y T de un gas en un estado
inicial y otro final puede encontrarse aplicando correctamente la ecuación de
estado. En todo caso, es preciso recordar que la T que aparece en ella es
temperatura absoluta. Si se designa con el subíndice 0 el estado inicial y con el 1 el
estado que corresponde a las condiciones de la primera pregunta del
problema, se tendrá entonces:
Laboratorio de Procesos Químicos y Control
Página 7
donde
P0 = 1 atm
V0 = 10 litros
T0 = 27 + 273 = 300 K
P1 = 2 atm
T1 = 207 + 273 = 480 K
luego despejando V1 resulta:
Análogamente y designando con el subíndice 2 el estado que corresponde a las
condiciones de la segunda pregunta, resulta:
En este caso la transformación se ha efectuado a T = cte, es decir, en las
condiciones que recoge la ley de Boyle.
LEY DE DALTON DE LAS PRESIONES PARCIALES
La presión total ejercida por una mezcla de gases ( siempre que no haya reacción
entre ellos) es equivalente a la suma de las presiones individuales que ejerce cada
gases el recipiente que lo contiene, como si ocupara, por si solo todo el volumen de
la mezcla. Esta propiedad fue observada por Dalton en 1801 y se puede
representar en la siguiente forma:
La presión total es
N.k.T
P = -------------V
siendo N el número total de moléculas
Laboratorio de Procesos Químicos y Control
Página 8
P total = Pa + Pb + Pc….
P total = N1 k T + N2 k T + N3 k T
V
V
V
P total = P1 + P2 + P3
La presión total es equivalente a la suna de las presiones parciales y cada una de
estas es proporcional a su concentración molecular
EL ESTADO LÍQUIDO
Características del estado líquido
A nivel microscópico el estado líquido se caracteriza porque la distancia entre las
moléculas es sensiblemente inferior a la de los gases. Mientras que en un gas la
distancia intermolecular media es igual o mayor que diez veces el tamaño de la
molécula, en un líquido viene a ser del mismo orden de magnitud que el diámetro
molecular, y sólo un poco mayor que en los sólidos. Eso explica que la densidad de
los líquidos sea, salvo algunas excepciones, sólo algo inferior a la de los
sólidos.
La proximidad entre las moléculas hace que se dejen sentir fuerzas atractivas de
interacción, que evitan que una molécula pueda «escaparse» de la influencia del
resto, como sucede en el estado gaseoso, pero que les permite moverse
deslizándose unas sobre otras. Por esta razón los líquidos no poseen forma propia,
sino que se adaptan a la del recipiente que los contiene, es decir, pueden fluir. Sin
embargo, el hecho de que las moléculas estén ya suficientemente próximas hace
de los líquidos fluidos incompresibles. Toda compresión lleva consigo una
disminución de la distancia intermolecular, y si ésta fuera apreciable entrarían en
Laboratorio de Procesos Químicos y Control
Página 9
juego las fuerzas repulsivas entre los núcleos atómicos que se opondrían a dicha
compresión y la neutralizarían.
Viscosidad
Aunque las moléculas de los líquidos pueden deslizarse unas sobre otras, esto no
sucede para todos con igual facilidad. La existencia de fuerzas de rozamiento que
se oponen al deslizamiento de las moléculas define una propiedad de los fluidos
que se denomina viscosidad. La viscosidad se traduce en una mayor resistencia al
movimiento en el interior del fluido. Así un frasco de aceite es más difícil de agitar
que uno de agua porque el aceite es más viscoso que el agua. Los líquidos
ideales carecen de viscosidad. En los reales, la viscosidad varía de unos a otros,
siendo extrema en los líquidos superviscosos, también llamados sólidos amorfos,
porque a la temperatura ambiente presentan el aspecto de sólidos sin que la
ordenación interna de sus moléculas corresponda a la que es característica de los
sólidos cristalinos.
El vidrio constituye un ejemplo de este estado intermedio de la materia.
Aumentando la temperatura, disminuye su viscosidad y el material se reblandece,
pasando a un estado líquido espeso. El alquitrán que se utiliza en el asfaltado de
carreteras es otro ejemplo de sólido amorfo; en verano, al aumentar la temperatura,
se llega a deformar por efecto de la presión que ejercen, sobre el firme, los
vehículos pesados.
Tensión superficial y capilaridad
Todos los líquidos presentan una cierta tendencia a disminuir su superficie libre, la
cual se comporta de forma parecida a como lo hace una membrana elástica. Dicha
propiedad es debida a la existencia de fuerzas tangenciales a la superficie o
fuerzas de tensión, por lo que se denomina tensión superficial.
La tensión superficial se pone de manifiesto en multitud de fenómenos; así cuando
se sumerge un alambre circular en una solución jabonosa se forma una película
que recuerda a simple vista la membrana de un tambor, pues se recupera de
pequeñas deformaciones; si sobre la película de líquido se deposita
cuidadosamente un hilo cerrado, y se elimina la parte interior de la película, se
observa cómo el hilo se extiende hasta alcanzar la forma de una circunferencia.
Una aguja de acero, engrasada simplemente por el tacto, puede flotar en el agua y,
del mismo modo, los patos se mantienen en la superficie de un lago sin necesidad
de nadar; en ambos casos los cuerpos son más densos que el agua, a pesar de lo
cual flotan.
Esta aparente contradicción con respecto a lo que establecen las leyes de la
hidrostática, se explica como debida al fenómeno de la tensión superficial que
convierte la superficie del líquido en una especie de malla tensa que neutraliza las
fuerzas del peso.
Desde un punto de vista molecular, la tensión superficial se explica como debida a
la condición especial de las moléculas situadas en la superficie del líquido. Una
molécula del líquido situada en el interior del mismo es solicitada en todas
direcciones por fuerzas atractivas procedentes de las otras moléculas que la
rodean, de modo que, por simetría, se compensan mutuamente sus efectos.
Laboratorio de Procesos Químicos y Control
Página 10
En la superficie o en sus proximidades, la simetría se rompe y sólo las moléculas
que están por debajo de la superficie atraen a las de la capa límite, dando lugar a
una fuerza neta dirigida hacia el interior de la masa líquida que tensa la superficie
libre y produce ese efecto de malla o membrana elástica.
La tensión superficial de un líquido viene expresada por su correspondiente
coeficiente de tensión superficial σ, que representa el trabajo necesario para
incrementar en una unidad la superficie libre del líquido o, en términos
equivalentes, la fuerza tangencial por unidad de longitud.
Cuando un tubo delgado de vidrio o capilar se introduce en agua situando su
extremo inferior por debajo de la superficie límite, el líquido asciende por su interior
hasta alcanzar una cierta altura de equilibrio. En este fenómeno conocido como
capilaridad el principio de los vasos comunicantes deja de tener validez porque
junto con las fuerzas del peso y de la presión atmosférica interviene la
tensión superficial.
Razonando en términos de equilibrio es posible encontrar una expresión para la
altura de la columna de líquido. Dado que el coeficiente de tensión superficial
representa la fuerza de tensión superficial por unidad de longitud, la resultante de
estas fuerzas responsables de la elevación de la columna será:
T=2πRσ
siendo 2πR la longitud del borde circular de la superficie libre. Por otra parte, el
peso de la columna viene dado por:
P = m · g = volumen · ρ · g = πR2hgρ
ρ
donde ρ representa la densidad y h la altura.
Dado que las fuerzas de tensión superficial forman, en general, un ángulo θ de
contacto con las paredes del capilar, la condición de equilibrio se expresará en la
forma:
T cosθ
θ=P
es decir:
2π
πRσ
σ cosθ
θ = πR2hρ
ρg
y por tanto:
Esta expresión constituye la llamada Ley de Jurin e indica que la altura de la
columna líquida es directamente proporcional a la tensión superficial del líquido e
inversamente proporcional al radio R del tubo.
Laboratorio de Procesos Químicos y Control
Página 11
Para un cierto número de líquidos, entre ellos el agua, puede considerarse en
primera aproximación que el ángulo de contacto θ es igual a cero, con lo cual la
anterior ecuación se simplifica en la forma:
expresión que permite estimar el valor de σ de un líquido, de densidad conocida,
midiendo h y R.
Fuerzas cohesivas y adhesivas. Capilaridad
Los líquidos poseen las propiedades de cohesión y adhesión debido a la atracción
molecular. Debido a la propiedad de cohesión, los líquidos pueden resistir
pequeñas fuerzas de tensión en la interfase entre el líquido y aire, conocida como
tensión superficial.
La cohesión permite al líquido resistir esfuerzos de tracción, mientras que la
adhesión permite que se adhiera a otros cuerpos.
Si las moléculas líquidas tienen mayor adhesión que cohesión, entonces el líquido
se pega a las paredes del recipiente con el cual está en contacto, resultando en un
aumento (elevación) de la capilaridad de la superficie del líquido; un predominio de
la cohesión causa por el contrario una depresión de la capilaridad.
Laboratorio de Procesos Químicos y Control
Página 12
Esta imagen del menisco, nos muestra las fuerzas que actúan sobre una molécula
en un fluido contenido en un recipiente, vemos que las tres fuerzas que actúan son:
la fuerza de líquido - sólido, la fuerza del aire - líquido, y la fuerza de líquido líquido.
Cuando el ángulo Ф c, es de menor de 90º, el liquido “moja” la superficie.
El ángulo de contacto Ф c, (depende exclusivamente de las fuerzas adhesivas y
cohesivas).
Ejemplos: ángulo de contacto de Agua-vidrio: 0º, ángulo de contacto de Mercuriovidrio: 140º.
Capilaridad líquida
Laboratorio de Procesos Químicos y Control
Página 13
Si tomamos un tubo de cristal grueso comunicado con uno fino y echamos agua en
él se verá como el tubo grueso alcanza menos altura que el fino. Si hacemos la
misma prueba con mercurio en vez de con agua resultará que el tubo grueso
alcanza más altura que el fino además en el primer caso se puede ver que el agua
se une con la pared del tuvo de forma cóncava, mientras que con el mercurio lo
hace de forma convexa.
EL ESTADO SÓLIDO
Características de los sólidos cristalinos
En el estado sólido, las moléculas, átomos o iones que componen la sustancia
considerada están unidas entre sí por fuerzas relativamente intensas, formando un
todo compacto. La mayor proximidad entre sus partículas constituyentes es una
característica de los sólidos y permite que entren en juego las fuerzas de enlace
que ordenan el conjunto, dando lugar a una red cristalina. En ella las partículas
ocupan posiciones definidas y sus movimientos se limitan a vibraciones en torno a
los vértices de la red en donde se hallan situadas. Por esta razón las sustancias
sólidas poseen forma y volumen propios.
La mayor parte de los sólidos presentes en la naturaleza son cristalinos aun
cuando en ocasiones esa estructura ordenada no se refleje en una forma
geométrica regular apreciable a simple vista.
Ello es debido a que con frecuencia están formados por un conjunto de pequeños
cristales orientados de diferentes maneras, en una estructura policristalina. Los
componentes elementales de una red cristalina pueden ser átomos, moléculas o
iones, de ahí que no se pueda hablar en general de la molécula de un cristal, sino
más bien de un retículo elemental o celdilla unidad, que se repite una y otra vez
en una estructura periódica o red cristalina.
Las propiedades físicas de los sólidos, tales como temperatura de fusión,
capacidad para conducir la corriente, resistencia a la deformación, dureza, etc.,
dependen de las características de las fuerzas de enlace que unen las entidades
elementales. Así, los sólidos iónicos, como las sales, son duros y a la vez
frágiles, con puntos de fusión altos. Aunque son malos conductores de la
electricidad sus disoluciones, sin embargo, presentan una conductividad elevada.
Los sólidos formados por moléculas apolares, como el Cl2, el H2 o el CO2, son
blandos como corresponde a la debilidad de las fuerzas de interacción entre ellas
(fuerzas de Van der Waals). Presentan un punto de fusión bajo lo que indica que
sólo a bajas temperaturas, las débiles fuerzas ordenadores del enlace pueden
predominar sobre el efecto disgregador del calor. Su conductividad eléctrica es
extremadamente baja como corresponde a la ausencia de cargas libres.
Los sólidos formados por moléculas polares, como el agua, presentan
características intermedias entre ambos tipos de sólidos, los iónicos y los apolares.
Las características del enlace metálico con un gas de electrones externos
Laboratorio de Procesos Químicos y Control
Página 14
compartidos hace que los sólidos metálicos sean buenos conductores de la
electricidad y del calor, y dúctiles y maleables, aunque con elevados puntos de
fusión. Un tipo de sólido de propiedades extremas lo constituyen los sólidos
covalentes; están formados por una red tridimensional de enlaces atómicos fuertes
que dan lugar a propiedades tales como elevados puntos de fusión, escasa
conductividad y extraordinaria dureza. El diamante, que es carbono puro
cristalizado, constituye un ejemplo de este tipo de sólidos.
CAMBIOS DE ESTADO
Fusión y solidificación
Cuando se le comunica calor a un sólido cristalino, su temperatura aumenta
progresivamente y al alcanzar un determinado valor se produce la transición o
cambio de fase del estado sólido al líquido que denominamos fusión. Si las
condiciones de presión exterior se mantienen constantes, el cambio de fase se
verifica a una temperatura fija o punto de transición entre ambos estados, que
se mantiene constante hasta que el sólido se ha fundido totalmente.
El calor que debe suministrarse a la unidad de masa de un sólido para
convertirlo en líquido a la temperatura de fusión se denomina calor de fusión
5
lf. En el agua lf vale 80 cal/g o su equivalente en unidades S.l.: 3,34 · 10
J/kg.
A nivel molecular la fusión se produce como consecuencia del derrumbamiento de
la estructura cristalina. El incremento de temperatura da lugar a un aumento en la
amplitud de las vibraciones de las partículas en la red, que termina por romper los
enlaces y producir la fusión. Una vez que se alcanza la energía de vibración
correspondiente a la temperatura de fusión, el calor recibido se emplea en romper
nuevos enlaces, de ahí que se mantenga constante la temperatura durante el
proceso.
La solidificación es la transición de líquido a sólido que se produce de forma
inversa a la fusión, con cesión de calor. Cualquiera que sea la sustancia
considerada el punto o temperatura de transición entre dos estados o fases de la
materia es el mismo independientemente del sentido de la transformación. La
disminución progresiva de la temperatura del líquido hace que en las proximidades
del punto de solidificación las fuerzas de enlace vayan imponiendo
progresivamente su orden característico.
Vaporización y condensación
Constituyen dos procesos inversos de cambio de estado. La vaporización es el
paso de una sustancia de la fase líquida a la fase de vapor o fase gaseosa. La
condensación es la transición de sentido contrario. Cuando la vaporización se
efectúa en el aire recibe el nombre de evaporación. La evaporación afecta
principalmente a las moléculas de la superficie del líquido.
Laboratorio de Procesos Químicos y Control
Página 15
Cada molécula de la superficie está rodeada por un menor número de sus
compañeras; ello hace que puedan vencer con más facilidad las fuerzas atractivas
del resto del líquido e incorporarse al aire como vapor. De ahí que cuanto mayor
sea la superficie libre del líquido tanto más rápida será su evaporación.
El aumento de temperatura activa este proceso. Para cada valor de la presión
exterior existe una temperatura para la cual la vaporización se vuelve violenta,
afectando a todo el líquido y no sólo a su superficie. Esta forma tumultuosa de
vaporización se denomina ebullición. El punto de ebullición de un líquido depende
de las condiciones de presión exterior, siendo tanto más elevado cuanto mayor sea
ésta.
Todo proceso de vaporización implica la absorción de calor por parte del líquido
respecto del entorno. La cantidad de calor necesaria para transformar la
unidad de masa de un líquido en vapor, a la temperatura de ebullición, se
denomina calor de vaporización lv. En el agua lv vale 540 cal/g
5
o, en unidades S.l.: 22,57 · 10 J/kg.
La condensación como transición de vapor a líquido se lleva a efecto invirtiendo las
condiciones que favorecen la vaporización. Así, mientras que la disminución de la
presión exterior facilita la vaporización, la compresión del vapor formado facilita la
condensación; el aumento de temperatura de un líquido provoca su vaporización e,
inversamente, el enfriamiento del vapor favorece su condensación. En términos
moleculares, tanto el aumento de presión como la disminución de la temperatura
del vapor reducen la distancia media de las moléculas y hacen posible su unión.
Sublimación
Aunque es un fenómeno poco frecuente a la temperatura y presiones ordinarias,
algunas sustancias como el yodo o el alcanfor pueden transformase directamente
de sólido a vapor sin necesidad de pasar por la fase intermedia de líquido. A tal
fenómeno se le denomina sublimación.
La transición o cambio de estado de sentido inverso se denomina de igual manera,
por ello a veces se distinguen ambas llamando a la primera sublimación progresiva
y a la segunda sublimación regresiva.
En principio, cualquier sustancia pura puede sublimarse, pero debido a las
condiciones de bajas presiones y temperaturas a las que es posible esta transición,
el fenómeno sólo es reproducible, para la mayor parte de las sustancias, en el
laboratorio.
Al igual que la fusión y la vaporización, también la sublimación (progresiva)
absorbe una determinada cantidad de calor. Se denomina calor de sublimación
ls a la cantidad de calor necesaria para sublimar la unidad de masa de una
sustancia.
Laboratorio de Procesos Químicos y Control
Página 16
ESCUELA DE EDUCACIÓN TÉCNICA Nº 3 – SAN JOSÉ
DEPARTAMENTO QUÍMICA
PROPIEDADES COLIGATIVAS
I.- Introducción.
Los estudios teóricos y experimentales han permitido establecer, que los
líquidos poseen propiedades físicas características. Entre ellas cabe mencionar: la
densidad, la propiedad de ebullir, congelar y evaporar, la viscosidad y la capacidad
de conducir la corriente eléctrica, etc.
Cada líquido presenta valores característicos (es decir, constantes) para
cada una de estas propiedades.
Cuando un soluto y un solvente dan origen a una solución, la presencia
del soluto determina una modificación de estas propiedades con relación a su
estado normal en forma aislada, es decir, líquido puro. Estas modificaciones se
conocen como PROPIEDADES DE UNA SOLUCIÓN.
Las propiedades de las soluciones se clasifican en dos grandes grupos:
1.-Propiedades constitutivas: son aquellas que dependen de la naturaleza
de las partículas disueltas. Ejemplo: viscosidad, densidad, onductividad
eléctrica, etc.
2.- Propiedades coligativas o colectivas: son aquellas que dependen del
número de partículas (moléculas, átomos o iones) disueltas en una
cantidad fija de solvente. Las cuales son:
- descenso en la presión de vapor del solvente,
- aumento del punto de ebullición,
- disminución del punto de congelación,
- presión osmótica.
Es decir, son propiedades de las soluciones que solo dependen del
número de partículas de soluto presente en la solución y no de la
naturaleza de estas partículas.
IMPORTANCIA DE LAS PROPIEDADES COLIGATIVAS
Las propiedades coligativas tienen tanta importancia en la vida común como
en las disciplinas científicas y tecnológicas, y su correcta aplicación permite:
A) Separar los componentes de una solución por un método llamado
destilación fraccionada.
B) Formular y crear mezclas frigoríficas y anticongelantes.
C) Determinar masas molares de solutos desconocidos.
Laboratorio de Procesos Químicos y Control
Página 17
D) Formular sueros o soluciones fisiológicas que no provoquen desequilibrio
hidrosalino en los organismos animales o que permitan corregir una
anomalía del mismo.
E) Formular caldos de cultivos adecuados para microorganismos
específicos.
F) Formular soluciones de nutrientes especiales para regadíos de vegetales
en general.
En el estudio de las propiedades coligativas se deberán tener en cuenta dos
características importantes de las soluciones y los solutos.
Soluciones: Es importante tener en mente que se está hablando de soluciones
relativamente diluídas, es decir, disoluciones cuyas concentraciones
son ≤ 0,2 Molar, donde teóricamente las fuerzas de atracción
intermolecular entre soluto y solvente serán mínimas.
Solutos:
Los solutos se presentarán como:
Electrolitos: Se disocian en solución y conducen la corriente
eléctrica.
No Electrolito: no se disocian en solución. A su vez el soluto no
electrolito puede ser volátil o no volátil.
I - Disminución de la presión de vapor.
A.- CONCEPTOS BÁSICOS: PRESIÓN DE VAPOR.
1.- Consideraciones:
Una de las características más importantes de los líquidos es su capacidad para
evaporarse, es decir, la tendencia de las partículas de la superficie del líquido, a salir
de la fase líquida en forma de vapor.
No todas las partículas de líquido tienen la misma Energía Cinética, es decir, no
todas se mueven a igual velocidad sino que se mueven a diferentes velocidades.
Solo las partículas más energizadas (con mayor energía) pueden escaparse de
la
superficie del líquido a la fase gaseosa.
En la evaporación de líquidos, hay ciertas moléculas próximas a la superficie con
suficiente energía como para vencer las fuerzas de atracción del resto
(moléculas
vecinas) y así formar la fase gaseosa.
Importante: Cuanto mas débiles son las fuerzas de atracción intermoleculares,
mayor cantidad de moléculas podrán escapar a la fase gaseosa.
Si un líquido está en un recipiente sellado puede parecer que no existiera
evaporación, pero experimentos cuidadosos demuestran que las moléculas
Laboratorio de Procesos Químicos y Control
Página 18
continúan abandonando el líquido.
Algunas moléculas de vapor regresan a la fase líquida, ya que a medida que
aumenta la cantidad de moléculas de fase gaseosa aumenta la probabilidad de
que una molécula choque con la superficie del líquido y se adhiera a él.
A medida que pasa el tiempo, la cantidad de moléculas que regresan al líquido
iguala exactamente a las que escapan a la fase de vapor. Entonces, el número
de moléculas en la fase gaseosa alcanza un valor uniforme.
Importante: La condición en la cual dos procesos opuestos se efectúan
simultáneamente a igual velocidad se denomina EQUILIBRIO DINÁMICO.
2.- Definición:
Las moléculas de la fase gaseosa que chocan contra la fase líquida ejercen
una fuerza contra la superficie del líquido, fuerza que se denomina PRESIÓN DE
VAPOR, que se define como la presión ejercida por un vapor puro sobre su fase
líquida cuando ambos se encuentran en equilibrio dinámico.
3.- Factores que afectan la presión de vapor:
Experimentalmente se ha comprobado que:
i) Para un líquido la presión de vapor aumenta a medida que aumenta la
temperatura.
ii) Líquidos diferentes a la misma temperatura presentan presiones de vapor
diferentes
POR LO TANTO PODEMOS CONCLUIR QUE LA PRESIÓN DE VAPOR
DEPENDE DE LA TEMPERATURA Y DE LA NATURALEZA DEL LÍQUIDO
Para visualizar como depende la Pv con la temperatura, examinemos la siguiente
Tabla:
Temperatura
(ºC)
20
30
40
50
60
70
80
Ácido acético
11,7
20,6
34,8
56,6
88,9
136,0
202,3
Presión de vapor en mm de Hg
Agua
Benceno
17,5
31,8
55,3
92,5
149,4
233,7
355,1
74,7
118,2
181,1
264,0
388,6
547,4
753,6
Etanol
43,9
78,8
135,3
222,2
352,7
542,5
818,6
Al examinar los datos experimentales se puede establecer los siguientes hechos:
Laboratorio de Procesos Químicos y Control
Página 19
a.- Para un mismo líquido, la presión de vapor aumenta a medida que aumenta
la temperatura.
Ejemplo: Agua a 40 ºC Presión de vapor 55.3 mmHg
Agua a 80 ºC Presión de vapor 355.1 mmHg
Para un mismo líquido a mayor temperatura hay mayor evaporación del
líquido.
A medida que la temperatura aumenta, las moléculas en el líquido se mueven
con mayor energía y por consiguiente pueden escapar más fácilmente de sus
vecinas, ya que vencen las fuerzas de interacción que las mantienen unidas
b.- Líquidos diferentes a la misma temperatura presentan presiones de vapor diferentes.
Ejemplo: Agua a 20 °C ⇒ Presión de vapor 17,5 mmHg
Benceno a 20 °C ⇒ Presión de vapor 74,7 mmHg
Etanol a 20 °C ⇒ Presión de vapor 43,9 mmHg
Para diferentes líquidos a una temperatura dada, las sustancias con Presión
de vapor elevadas se evaporan más rápidamente que las sustancias con
Presión de vapor baja.
Se dice entonces, que los líquidos que se evaporan rápidamente son
volátiles, mientras más volátil es un líquido menores son las fuerzas de
interacción intermolecular, mayor es la evaporación del líquido y mayor es su
presión de vapor
B.- DESCENSO DE LA PRESIÓN DE VAPOR: Efecto de solutos no electrolitos.
Como ya sabemos un líquido puro posee una presión de vapor determinada, que
depende sólo del líquido en estudio y de la temperatura. El valor de la presión de
vapor del líquido puro se altera si agregamos al líquido (solvente) un soluto
cualquiera.
El soluto puede ser volátil, es decir, posee una presión de vapor mayor que el 1% de
la presión de vapor del solvente a la misma temperatura; o no volátil, es decir, posee
una presión de vapor menor que el 1% de la presión de vapor del solvente a la misma
temperatura. En ambos casos la presión de vapor del solvente se modifica en
relación al solvente puro.
i) Soluto no volátil.
Si el soluto que se agrega al solvente es no volátil, se producirá un
DESCENSO DE LA PRESIÓN DE VAPOR.
¿CÓMO SE PUEDE EXPLICAR ESTE FENÓMENO?
Recordemos que:
Laboratorio de Procesos Químicos y Control
Página 20
La presión de vapor sobre un líquido es el resultado de un equilibrio dinámico
entre la fase de vapor y la fase líquida de un compuesto.
La velocidad a la cual las moléculas dejan la superficie del líquido y pasan a la
fase gaseosa, es igual a la velocidad a la cual las moléculas de la fase gaseosa
regresan a la superficie del líquido.
Por otro lado:
Un soluto no volátil que se añade al líquido, reduce la capacidad de las moléculas del
solvente a pasar de la fase líquida a la fase vapor, debido a que se generan nuvas
fuerzas de interacción.
Por ello se produce un desplazamiento del equilibrio, lo que se traduce en una
reducción de la presión de vapor sobre la solución.
El grado en el cual un soluto no volátil disminuye la presión de vapor es proporcional
a la concentración de la solución, es decir, mientras mayor sea la concentración de la
solución mayor es la disminución de la presión de vapor y por lo tanto la reducción en
la presión de vapor es aproximadamente proporcional a la concentración total de
partículas del soluto (electrolito o no electrolito).
La expresión cuantitativa del descenso de la presión de vapor de las soluciones que
contienen solutos no volátiles esta dada por la Ley de Raoult (Francois Marie Raoult
1885). Este científico demostró que “a una temperatura constante, el descenso de
la Presión de Vapor es proporcional a la concentración de soluto presente en la
solución”. Este principio queda establecido matemáticamente por las siguientes
ecuaciones:
PA = XA PºA
Ecuación 1
Donde:
PA
PºA
XA
XB
∆PV
=
=
=
=
=
∆PV = PºA - PA
Ecuación 2
∆PV = PºA XB
Ecuación 3
PºA - PA = PºA XB
Ecuación 4
Presión de Vapor de la solución.
Presión de vapor del solvente puro.
Fracción molar del solvente
fracción molar del soluto
Variación de la presión de vapor
Las soluciones que obedecen la ley de Raoult se denominan SOLUCIONES
IDEALES. Las soluciones se aproximan al comportamiento ideal cuando la
concentración de soluto es baja y cuando el soluto y el solvente son semejantes tanto
en tamaño molecular, como en el tipo de fuerzas de atracción intermolecular que hay
entre ellas
Laboratorio de Procesos Químicos y Control
Página 21
ii) Soluto volátil.
Si consideramos una solución ideal formada por dos componentes (A, B) en que A y
B son volátiles. Las presiones parciales de los vapores de A y B sobre la solución
están dadas por la Ley de Raoult.
PA = XA PºA
y
PB = XB PºB
La presión de vapor total sobre la solución se calcula sumando las presiones
parciales de cada componente volátil.
PTOTAL =
PA +
PTOTAL = XA PºA +
PB
XB PºB
Ejemplo: Consideremos una solución formada por 1 mol de Benceno y 2 moles de
Tolueno. El Benceno presenta una presión de vapor (P°) de 75 mmHg y el
Tolueno una de 22 mmHg a 20°C. Como se ve el bencen o es el más volátil
debido a que tiene una presión de vapor puro (P°) m ayor que la del tolueno.
1) Calculemos la fracción molar de Benceno y Tolueno:
Xbenceno =
1
XTolueno =
2
= 0,33
= 0,67
1+2
1+2
2) Calculemos la presión de parcial de cada componente y la presión de vapor de la
solución:
Pbenceno = Xbenceno Pºbenceno
Ptolueno =
Xtolueno Pºtolueno
Pbenceno = ( 0,33 ) ( 75 mmHg )
Ptolueno =
( 0,67 ) ( 22 mmHg )
Pbenceno = 25 mmHg
Ptolueno = 15 mmHg
PTOTAL
PTOTAL
PTOTAL
= Pbenceno +
= 25 mmHg +
= 40 mmHg
Ptolueno
15 mmHg
Si calculamos el porcentaje que aporta, a la presión de vapor, cada componente
tendremos que:
Benceno: 40 mmHg ----- 100 % Tolueno: 40 mmHg ----- 100 %
25 mmHg ----- X
15 mmHg ----- X
X = 63 %
X = 37 %
Estos resultados indican que el vapor es más rico en el componente más
volátil, ya que el benceno aporta el 63 % a la presión total (podríamos decir que
Laboratorio de Procesos Químicos y Control
Página 22
el 63 % de las moléculas gaseosas son de benceno) a pesar de que la solución
inicial el benceno era el componente minoritario.
CUANDO UNA SOLUCIÓN IDEAL ESTÁ EN EQUILIBRIO CON SU VAPOR, EL
COMPONENTE MÁS VOLÁTIL DE LA MEZCLA INICIAL SERÁ MAYORITARIO EN
EL VAPOR.
C.- EJERCICIOS RESUELTOS
Ejercicio Nº1: La presión de vapor sobre el agua pura a 120°C es 1480 mmHg. Si se sigue la Ley de
Raoult ¿Que fracción de etilenglicol debe agregarse al agua para reducir la presión de
vapor de este solvente a 760 mmHg?
Paso 1: Ordenar los datos.
Soluto etilenglicol
Solvente agua
Solución
: no hay datos
= 1480 mmHg
: PºA
: PA
= 760 mmHg
Paso 2: Pregunta concreta ⇒ determinar la fracción molar de etilenglicol (XB) en una solución cuya
presión de vapor es 760 mmHg.
Paso 3: Aplicamos la Ley de Raoult
PºA
-
PA
=
PºA XB
Paso 4: Cálculo de la fracción molar de etilenglicol (XB)
1480 mmHg
RESPUESTA:
Ejercicio Nº2:
-
760 mmHg =
(1480 mmHg) XB
XB
=
XB
=
1480 mmHg - 760 mmHg
----------------------------------1480 mmHg
0,486
La fracción molar de etilenglicol que se debe agregar al agua para que la solución
resultante presente una presión de vapor de 760 mmHg es de 0,486
Calcular la reducción en la presión de vapor causada por la adición de 100 g de
sacarosa (masa molar = 342) a 1000 g de agua. La presión de vapor de agua pura a
25°C es 23,69 mmHg.
Paso 1: Ordenar los datos.
Soluto sacarosa
: masa
= 100 g
masa molar = 342 g/mol
Solvente agua
: PºA
= 23,69 mmHg
masa
= 1000 g
masa molar = 18 g/mol
Laboratorio de Procesos Químicos y Control
Página 23
Solución
: no hay datos.
Paso 2: Pregunta concreta ⇒ determinar la disminución de la presión de vapor (∆PV) al adicionar 100
g de sacarosa a 1000 g de agua.
Paso 3: Aplicamos la Ley de Raoult
∆PV
=
PºA XB
Paso 4: Necesitamos conocer la fracción molar de soluto (XB), como conocemos las masas y las
masa molar de cada componente, podemos determinar el número de moles de soluto y
solvente.
sacarosa:
342 g ----- 1 mol
100 g ----- X
X = 0,292 moles
agua:
18 g ----- 1 mol
1000 g ----- X
X = 55,556 moles
Por lo tanto, la fracción molar es:
(0,292 moles)
XB =
= 5,229 x 10
-3
(0,292 moles + 55,556 moles)
Paso 5: Cálculo de la disminución de la presión de vapor.
∆PV
∆PV
-3
= (23,69 mmHg) (5,229 x 10 )
= 0,124 mmHg
RESPUESTA:
La disminución de la presión de pavor que se produce al agregar 100 g de sacarosa
a 1000 g de agua es de 0,125 mmHg.
Ejercicio Nº3:
La presión de vapor del agua pura a una temperatura de 25°C es de 23,69 mmHg.
Una solución preparada con 5,5 g de glucosa en 50 g de agua tiene una presión de
vapor de 23,42 mmHg. Suponiendo que la Ley de Raoult es válida para esta
solución, determine la masa molar de glucosa.
Paso 1: Ordenar los datos.
Paso 2:
Paso 3:
PºA
Paso 4:
Soluto glucosa
: masa
masa molar
= 5,5 g
= ?
Solvente agua
: masa
masa molar
PºA
= 50 g
= 18 g/mol
= 23,69 mmHg
Solución
: PA
= 23,42 mmHg
Pregunta concreta ⇒ determinar la masa molar de glucosa
Aplicamos la Ley de Raoult
PA
=
PºA XB
Cálculo de la fracción molar de glucosa (XB)
23,69 mmHg - 23,42 mmHg = (23,69 mmHg) XB
Laboratorio de Procesos Químicos y Control
Página 24
XB
=
XB
=
23,69 mmHg - 23,42 mmHg
-------------------------------------------23,69 mmHg
0,011
Paso 5: Calcular el número de moles de agua (nA).
50 g
nA =
= 2,778 moles
18 g/mol
Paso 6: Cálculo del número de moles de glucosa (nB).
número de moles soluto
XB =
número de moles de totales
nB
XB
=
nB + nA
nB
0,011
=
nB + 2,778
nB
= 0,031 moles
Paso 7: Cálculo de la masa molar de glucosa.
masa de glucosa
nB
=
masa molar
5,5 g
0,031 moles
=
masa molar
masa molar
= 177,42 g/mol
RESPUESTA:
La masa molar de glucosa es 177,42 (masa molar real de glucosa es 180)
Ejercicio Nº4:
A una temperatura de 26°C, la presión de vapor del agua es 25,21 mmHg. Si a esta
temperatura se prepara una solución 2,32 molal de un compuesto no electrolito, no
volátil. Determinar la presión de vapor de esta solución suponiendo comportamiento
ideal.
Paso 1: Ordenar los datos.
Laboratorio de Procesos Químicos y Control
Página 25
Soluto desconocido
:
no hay datos.
Solvente agua
:
PºA
Solución
:
concentración
= 25,21 mmHg
= 2,32 m
Paso 2: Pregunta concreta ⇒ determinar la presión de vapor de la solución (PA).
Paso 3: Aplicamos la Ley de Raoult
PºA
-
PA
=
PºA XB
Paso 4: A partir de la molalidad podemos calcular la fracción molar de soluto (XB)
2,32 molal significa que se disolvieron 2,32 moles de soluto en 1000 g de agua. Como la
masa molar de agua es 18 g/mol, tenemos que:
18 g ----1 mol
1000 g ----- X
⇒
X
=
55,56 moles
Entonces tenemos 2,32 moles de soluto (nB) en 55,56 moles de solvente (nA), luego la
fracción molar de soluto será:
número de moles soluto
XB =
número de moles de totales
nB
XB
=
nB + nA
2,32 moles
XB
=
2,32 moles + 55,56 moles
XB
= 0,04
Paso 5: Ahora podemos aplicar la ecuación de la Ley de Raoult.
PºA
25,21 mmHg
-
PA
PA
PA
=
=
=
PºA XB
(25,21 mmHg) (0,04)
24,20 mmHg.
RESPUESTA:
La presión de vapor de la solución 2,32 molal es 24,20 mmHg.
Ejercicio Nº5:
Una solución de cloruro de calcio (CaCl2) fue preparada disolviendo 25 g de esta sal
en 500 g de agua. Cuál será la presión de vapor de la solución a 80°C, sabiendo que
a esta temperatura el cloruro de calcio se comporta como un electrolito fuerte y que la
presión de vapor del agua es 355,10 mmHg (masa molar de cloruro de sodio es 111
g/mol y del agua es 18 g/mol).
Laboratorio de Procesos Químicos y Control
Página 26
Paso 1: Ordenar los datos.
Soluto CaCl2
:
masa
masa molar
= 25 g
= 111 g/mol
Solvente agua
:
masa
masa molar
PºA
= 500 g
= 18 g/mol
= 355.10 mmHg
Solución
:
no hay datos
Paso 2: Pregunta concreta ⇒ determinar la presión de vapor de la solución (PA).
Paso 3: Aplicamos la Ley de Raoult
PA
=
PºA XA
Paso 4: A partir de la molalidad podemos calcular la fracción molar de solvente (XA)
Como la masa molar de agua es 18 g/mol y la masa molar de cloruro de calcio es 111
g/mol, tenemos que:
Agua: 18 g ----1 mol
500 g ----- X
X = 27,27 moles
Cloruro de calcio:
111g ----1 mol
25 g ----- X
X = 0,225 moles
Ahora podemos calcular la fracción molar de solvente (XA) pero antes debemos considerar
que el cloruro de calcio es un electrolito fuerte a esta temperatura, luego:
+2
-1
→
Ca
+
2Cl
CaCl2
de Ca
+2
Por cada mol disuelto de cloruro de calcio se obtienen 3 moles de iones disueltos (1 mol
-1
y 2 moles de Cl ), entonces
1 mol de CaCl2 ------ 3 moles de iones
0,225 moles de CaCl2 ------ X
X = 0,676 moles de iones
Ahora podemos calcular la fracción molar de solvente
número de moles soluto
XA =
número de moles de totales
nA
XA
=
nA + nB
27,27 moles
XA =
27,27 moles + 0,676 moles
XA = 0,976
Paso 5: Ahora podemos aplicar la ecuación de la Ley de Raoult.
Laboratorio de Procesos Químicos y Control
Página 27
PA
PA
PA
=
=
=
RESPUESTA:
PºA XA
(355,10 mmHg) (0,976)
346,51 mmHg.
La presión de vapor para la solución es 346,51 mmHg.
D.- EJERCICIOS PROPUESTOS.
1) La presión de vapor del metanol puro es 159,76 mmHg. Determinar la fracción molar de glicerol
(soluto no electrólito y no volátil) necesario para disminuir la presión de vapor a 129,76 mmHg.
(Respuesta = 0,188)
2) Una solución contiene 8,3 g de una sustancia no electrolito y no volátil, disuelta en un mol de
cloroformo (CHCl3), esta solución tiene una presión de vapor de 510,79 mmHg. La presión de
Vapor del cloroformo a esta temperatura es 525,79 mmHg. En base a esta información
determine:
a- La fracción molar de soluto.
(Respuesta = 0,0285)
b- El número de moles de soluto disueltos.
(Respuesta = 0,0294 moles)
c- La masa molar de soluto.
(Respuesta = 272,42 g/mol)
3) La presión de vapor del Benceno (C6H6) a 25°C es 93,76 mmHg. Determine la presión de vap or
de una solución preparada disolviendo 56,4 g de un soluto no volátil (C20H42) en un kilogramo de
Benceno. (Respuesta = 92,32 mmHg)
4) La presión de vapor del agua a 60°C es 149,4 mmH g. Si Ud. desea preparar una solución donde
la presión de vapor disminuya a 140 mmHg. Determine la masa de glucosa (C6H12O6) que debe
disolverse en 150 g de agua para lograr dicho efecto. (Respuesta = 95,76 g)
5) Se disuelven 0,3 moles de sulfato de sodio (Na2SO4), electrolito fuerte y no volátil, en 2 Kg de
agua a 60°C. Si la presión de vapor dl agua a esta temperatura es 149,4 mmHg. Determine la
presión de vapor de la solución resultante. (Respuesta 148,20 mmHg)
II. Aumento del punto de ebullición.
A.- CONCEPTOS BÁSICOS: PUNTO DE EBULLICIÓN
1.- Definición:
Como hemos visto un líquido contenido en un recipiente abierto, sufre evaporación. Si
la temperatura es lo suficientemente alta, se forman dentro del líquido burbujas de
vapor que ascenderán a la superficie. Cuando sucede esto, se dice que el líquido
hierve.
Se ha demostrado experimentalmente que cuando este fenómeno sucede la presión
de vapor del líquido iguala a la presión externa o atmosférica que actúa sobre la
superficie del líquido. Por lo que el punto de ebullición se define como: la temperatura
a la cual la presión de vapor iguala a la presión externa o atmosférica.
Laboratorio de Procesos Químicos y Control
Página 28
2.- Factores que afectan el punto de ebullición.
Recuerda que el líquido se encuentra en su punto de ebullición cuando la presión de
vapor es igual a la presión externa o atmosférica y hay formación de vapor no solo en
la superficie sino que en todo el líquido.
Los líquidos hierven a cualquier temperatura siempre que la presión externa
que se ejercesobre ellos sea igual a la presión de vapor correspondiente a
dicha temperatura.
El punto de ebullición de un líquido depende de la presión externa a la cual esté
sometido.
Si la presión externa o atmosférica es baja, se necesita poca energía para que
la presión de vapor del líquido iguale a la presión externa, luego su punto de
ebullición es bajo.
Ejemplo: A nivel del mar, la presión atmosférica es alta, luego el agua hierve a
100°C.
Ejemplo: En las altas cumbres cordilleranas, la presión atmosférica es baja,
luego el agua hierve a una temperatura menor a 100°C.
Si la presión externa o atmosférica es alta se necesita más energía para que la
presión de vapor del líquido iguale la presión externa, luego su punto de
ebullición es alto.
B.- AUMENTO DEL PUNTO DE EBULLICIÓN.
Ya hemos visto que la presión de vapor de un líquido aumenta al aumentar la
temperatura y que el líquido hierve cuando su presión de vapor iguala a la presión
externa o atmosférica que se ejerce sobre su superficie.
Debido a que los solutos No volátiles disminuyen la presión de vapor de la
solución, se requiere una temperatura más elevada para que la solución hierva.
Las soluciones de solutos no volátiles, presentan puntos de ebullición
superiores a los puntos de ebullición de los solventes puros.
Mientras más concentradas sean las soluciones mayores son los puntos de
ebullición de estas.
EL AUMENTO EN EL PUNTO DE EBULLICIÓN ES PROPORCIONAL AL
NÚMERO DE PARTÍCULAS DE SOLUTO DISUELTAS EN UN SOLVENTE.
Laboratorio de Procesos Químicos y Control
Página 29
Como vemos en la siguiente tabla disolveremos diferentes cantidades de soluto en
1000 g de agua a una presión externa de 1 atmósfera.
Número de moles de
soluto, disueltos en 1
Kg de agua
0,5
1,0
1,5
2,0
2,5
3,0
3,5
4,0
4,5
5,0
Punto de ebullición
solución
(°C)
100,26
100,52
100,78
101,04
101,30
101,56
101,82
102,08
102,34
102,60
Punto de
ebullición agua
pura (°C)
100,00
100,00
100,00
100,00
100,00
100,00
100,00
100,00
100,00
100,00
Aumento del
punto de
ebullición (°C)
0,26
0,52
0,78
1,04
1,30
1,56
1,82
2,08
2,34
2,60
Observamos en la tabla, el aumento en el punto de ebullición es directamente
proporcional al número de partículas de soluto disueltas en una masa fija de
solvente, ya sabemos que la molalidad expresa el número de moles que se
disuelven en 1000 g de solvente, lo que representa una masa fija de solvente. Así,
el ascenso del punto de ebullición es proporcional a la molalidad.
Este fenómeno queda establecido por las siguientes ecuaciones:
∆Teb
=
Teb
- Tºeb
∆Teb = Keb m
Ecuación 1
Ecuación 2
Donde:
∆Teb = Ascenso del punto de ebullición.
= Temperatura de ebullición de la solución.
Teb
Tºeb = Temperatura de ebullición del solvente puro.
Keb
= Constante molal de la elevación del punto de ebullición o constante
ebulloscópica.
m
= molalidad (número de moles de soluto / 1000 g de solvente)
La magnitud de Keb, denominada constante molal de elevación del punto de ebullición
o constante ebulloscópica, depende solo del solvente y representa el aumento del
punto de ebullición cuando un mol de un soluto no electrolito no volátil se disuelve en
1000 g de solvente.
Innumerables experimentos han demostrado que cuando un mol de un soluto no
electrólito no volátil se disuelve en 1000 g de agua, el punto de ebullición del agua
aumenta en 0,52 °C. Este valor es conocido como con stante molal de elevación del
punto de ebullición o constante ebulloscópica del agua y sus unidades son °C/molal.
A continuación se señalan para algunas solventes sus puntos de ebullición normales
y sus constantes ebulloscópicas.
Laboratorio de Procesos Químicos y Control
Página 30
Solvente
Punto de ebullición normal
(°C)
100,00
80,10
76,80
78,40
61,20
Agua
Benceno
Tetracloruro de carbono
Etanol
Cloroformo
Constante ebulloscópica
(°C/molal)
0,52
2,53
5,02
1,22
3,63
Para el agua la constante ebulloscópica es 0,52 °C/ molal, por consiguiente, una
solución acuosa 1 molal de sacarosa o de cualquier otro soluto no volátil hervirá a una
temperatura de 100,52 °C. Es importante hacer notar que la elevación del punto de
ebullición es proporcional a la cantidad de partículas de soluto presentes en
determinada cantidad de solvente
C.- EJERCICIOS RESUELTOS
Ejercicio Nº1:
Paso 1:
Calcular el punto de ebullición de una solución de 100 g de anticongelante etilenglicol
(C2H6O2) en 900 g de agua (Keb = 0,52 °C/m).
Ordenar los datos.
Soluto etilenglicol
Solvente agua
Solución
: masa
masa molar
: masa
masa molar
Keb
Tºeb
=
=
=
=
= 100 g
= 62 g/mol (derivada de la formula C2H6O2)
900 g
18 g/mol
0,52 °C/m
100 °C
: no hay datos
Paso 2:
Pregunta concreta ⇒ determinar el punto de ebullición de la solución (Teb)
Paso 3:
Aplicamos las ecuaciones:
∆Teb
∆Teb
=
=
Teb
Keb m
Tºeb
Ecuación 1
Ecuación 2
Para poder obtener la temperatura de ebullición de la solución necesitamos la ecuación 1, pero
como no tenemos ∆Teb (ascenso de la temperatura de ebullición), necesitamos obtenerlo de ecuación 2.
Paso 4:
Para poder utilizar ecuación 2 necesitamos la molalidad de la solución que podemos calcular a partir
de los siguientes datos:
Moles de soluto
:
Molalidad
:
62 g ----- 1 mol
100 g ----- X
X = 1,613 moles de soluto
1,613 moles ----- 900 g de solvente
X ----- 1000 g de solvente
X =
1,792 molal
La solución tiene una concentración molal de 1,792
Laboratorio de Procesos Químicos y Control
Página 31
Paso 5:
Aplicando ecuación 2, tenemos:
∆Teb
∆Teb
∆Teb
Paso 6:
=
=
=
Aplicando ecuación 1, tenemos:
∆Teb
0,9319 °C
Teb
RESPUESTA:
Ejercicio Nº2:
Paso 1:
Keb m
(0,52 °C/molal) (1,792 molal)
0,9319 °C
=
=
=
Teb
Tºeb
Teb
100 °C
100,9319 °C
La temperatura de ebullición de la solución es 100,9319 °C
Qué concentración molal de sacarosa en agua se necesita para elevar su punto de ebullición en
1,3 °C (Keb = 0,52 °C/m y temperatura de ebullición del agua 100°C).
Ordenar los datos.
Soluto sacarosa
: no hay datos
Solvente agua
: Keb
Teb
=
=
0,52 °C/m
100 °C
Solución
: ∆Teb
=
1,3 °C
Paso 2:
Pregunta concreta ⇒ determinar la molalidad de la sacarosa.
Paso 3:
Aplicamos las ecuaciones.
∆Teb
=
Teb
∆Teb
=
Keb m
-
Tºeb
Ecuación 1
Ecuación 2
Para poder obtener la molalidad basta con aplicar la ecuación 2.
Paso 4:
Aplicando la ecuación 2.
∆Teb
=
1,3 °C =
m
=
RESPUESTA:
Ejercicio Nº3:
Paso 1:
Keb m
(0,52 °C/molal) m
2,5 molal
La molalidad de esta solución es de 2,5.
Se disuelven 0,572 g de resorcina en 19,31 g de agua y la solución hierve a 100,14°C. Calcular la
masa molar de resorcina, Keb del agua es 0,52 °C/m.
Ordenar los datos.
Soluto resorcina
: masa
= 0,572 g
Solvente agua
: masa
= 19,31 g
Laboratorio de Procesos Químicos y Control
Página 32
masa molar
Keb
Tºeb
Solución
= 18 g/mol
= 0,52 °C/m
= 100,00 °C
: Teb
= 100,14 °C
Paso 2:
Pregunta concreta ⇒ determinar la masa molar de resorcina
Paso 3:
Aplicamos las ecuaciones
∆Teb
=
Teb
∆Teb
=
Keb m
-
Tºeb
Ecuación 1
Ecuación 2
Para poder calcular la masa molar del soluto necesitamos saber cual es la masa de un mol de
moléculas de resorcina. Luego necesitamos saber que molalidad tiene la solución. Utilizamos entonces la
ecuación 1 para determinar el aumento del punto de ebullición y la ecuación 2 para calcular la molalidad.
Paso 4:
Cálculo de la molalidad
∆Teb
∆Teb
∆Teb
= Teb
Tºeb
= 100,14 °C
= 0,14 °C
∆Teb
0,14 °C
m
=
=
=
100,00 °C
Keb m
(0,52 °C/molal) m
0,269 molal
Esto significa que 0,269 moles de soluto (resorcina) se disolvieron en 1000 g de solvente (agua)
Paso 5:
Cálculo de moles de resorcina presentes en 19,31 g de agua.
0,269 moles de resorcina ------ 1000 g de agua
X ------ 19,31 g de agua
X = 0,005194 moles de resorcina.
Paso 6:
Cálculo de la masa molar.
masa de resorcina
nresorcina =
masa molar
0,572 g
5,194 x 10-3 moles
=
masa molar
masa molar
=
110,12 g/mol
RESPUESTA:
La masa molar de resorcina es 110,12.
Ejercicio Nº4:
Si se disuelven 5,65 g de C16H34 en 100 g de benzol, se observa una elevación en el punto de
ebullición del benzol de 0,66 °C. En base a estos datos calcule Keb del benzol.
Laboratorio de Procesos Químicos y Control
Página 33
Paso 1:
Ordenar los datos.
Soluto C16H34
:
masa
masa molar
=
=
5,65 g
226 g/mol (derivada de la formula)
Solvente benzol
:
masa = 100 g
Solución
:
∆Teb = 0,66 °C
Paso 2:
Pregunta concreta ⇒ determinar la constante ebulloscópica del benzol (Keb).
Paso 3:
Aplicamos las ecuaciones:
∆Teb
=
Teb
∆Teb
=
Keb m
-
Tºeb
Ecuación 1
Ecuación 2
Con la ecuación 2 podemos calcular Keb pero antes debemos conocer cual es la molalidad de la
solución.
Paso 4:
Calculo de la molalidad de la solución (m).
Moles de soluto:
226 g ------ 1 mol
5,65 g ------ X
X =
0,025 moles
Molalidad de la solución:
Paso 5:
0,025 moles de soluto ------- 100 g de solvente (benzol)
X ------- 1000 g de solvente
X = 0,25 molal
Aplicando la ecuación 2, tenemos:
∆Teb
0,66 °C
Keb
= Keb m
= Keb (0,25 molal)
= 2,64 °C/molal
RESPUESTA:
La constante ebulliscópica del benzol es 2,64 °C/molal
Ejercicio Nº5:
Cuál es el punto de ebullición de 100 g de una solución acuosa de urea al 20 % en peso, si la
masa molar de urea es 60 g/mol. (Keb = 0,52 °C/molal)
Paso 1:
Paso 2:
Ordenar los datos.
Soluto urea
:
masa molar
= 60 g/mol
Solvente agua
:
Tºeb
masa molar
Keb
= 100 °C
= 18 g/mol
= 0,52 °C/molal
Solución
:
concentración
masa
= 20 % p/p
= 100 g
Pregunta concreta ⇒ determinar el punto de ebullición de la solución (Teb).
Laboratorio de Procesos Químicos y Control
Página 34
Paso 3:
Aplicamos las ecuaciones:
∆Teb
=
Teb
∆Teb
=
Keb m
-
Tºeb
Ecuación 1
Ecuación 2
Para poder calcular la temperatura de ebullición de la solución (Teb) necesitamos la ecuación 1,
pero para ello debemos conocer el aumento de la temperatura de ebullición (∆Teb) que obtenemos de la
ecuación 2 conociendo la molalidad.
Paso 4:
Calculo de la molalidad (m).
Necesitamos conocer la molalidad de la solución, la cual podemos obtener a partir de el dato de
concentración (20 %en peso).
20 % p/p ⇒ que 20 g de soluto hay en 100 g de solución o 20 g de soluto están disueltos en 80 g de
solvente, entonces:
Moles de soluto:
60 g ------ 1 mol
20 g ------ X
X = 0,333 moles de soluto
0,333 moles de soluto ------- 80 g de solvente
X ------- 1000 g de solvente
X
=
4,167 molal
Molalidad:
Paso 5:
Cálculo del ascenso del punto de ebullición.
∆Teb
∆Teb
∆Teb
Paso 6:
=
=
=
Keb m
(0,52 °C/molal) (4,167 molal)
2,166 °C
Cálculo de la temperatura de ebullición de la solución.
∆Teb
2,166 °C
Teb
RESPUESTA:
=
=
=
Teb
Tºeb
100,0 °C
Teb
102,166 °C
La temperatura de ebullición de la solución es 102,166 °C.
D.- EJERCICIOS PROPUESTOS.
1) Determine la masa molar de un compuesto no electrolito sabiendo que al disolver 384 g de este compuesto
en 500 g de benceno, se observó una temperatura de ebullición de la solución de 85,1 °C. (Benceno: Keb =
2,53 °C/molal y punto de ebullición 80,1 °C)
(Respuesta = 388,66 g/mol)
2) Cuantos gramos de glucosa (masa molar 180 g/mol) son necesarios disolver en 1000 g de agua para que la
temperatura de ebullición del agua se eleve en 3 °C. (Agua: temperatura de ebullición 100 °C y Keb = 0,52
°C/molal )
(Respuesta = 1038,46 g)
Laboratorio de Procesos Químicos y Control
Página 35
3) Determine la constante ebulloscópica de un solvente, si al disolver 100 g de urea (masa molar 60 g/mol) en
250 g de este solvente, éste incrementa su temperatura de ebullición en 2,1 °C.
(Respuesta = 0,315 °C/molal)
4) Si 40 g de un compuesto C6H10O5 se disuelven en 500 g de agua, determine el punto de ebullición de esta
solución. (Agua: temperatura de ebullición 100 °C y Keb = 0,52 °C/molal )
(Respuesta = 100,26 °C)
5) Si al disolver 20 g de urea (masa molar 60 g/mol) en 200 g de solvente se observa que el punto de
ebullición de la solución es de 90 °C, determine el punto de ebullición de un solvente puro cuya constante
ebulloscópica es 0,61 °C/molal,
(Respuesta = 88,98 °C).
III.: Descenso del punto de congelación.
A.- CONCEPTOS BÁSICOS: PUNTO DE CONGELACIÓN
1.- Definición:
La transformación de un líquido a sólido se llama Congelación, y el proceso inverso
se llama Fusión.
Congelación
LÍQUIDO
SÓLIDO
Fusión
En otras palabras:
El PUNTO DE CONGELACIÓN de un líquido corresponde a la temperatura en la cual
las moléculas de un compuesto (como por ejemplo el agua) pasan del estado líquido
al estado sólido.
Este fenómeno se debe a la agrupación de las moléculas, las cuales se van
acercando paulatinamente disminuyendo el espacio intermolecular que las separa
hasta que la distancia sea tal que se forma el sólido. Este acercamiento se debe
básicamente a que el movimiento molecular se va haciendo menor debido a la
disminución de la temperatura lo que provoca que la energía cinética de las
moléculas sea menor.
Por lo tanto, como la energía calórica del ambiente (medida por la temperatura) no es
lo suficientemente alta como para contrarrestar la atracción entre las moléculas, ellas
tienden entonces a agruparse y por lo tanto “congelar”.
Laboratorio de Procesos Químicos y Control
Página 36
B.- DESCENSO DEL PUNTO DE CONGELACIÓN.
Si se disuelve un soluto no volátil en un líquido (solvente), se observa
experimentalmente un descenso en el punto de congelación.
Por lo cual, podemos decir, que las soluciones congelan a temperaturas
inferiores a las del solvente puro
Este hecho es una consecuencia de la disminución de la presión de vapor
ocasionado por dicho soluto
Esto se explica mediante el hecho que en el punto de congelación de la
solución la presión de vapor del sólido debe ser igual a la presión de vapor del
líquido con el que está en equilibrio
Pero como la solución a bajado su presión de vapor (con respecto al líquido
puro) el sólido deberá formarse a una temperatura inferior
La diferencia entre los puntos de congelación del solvente puro y la solución se
designa por ∆Tc y se conoce con el nombre de DESCENSO DEL PUNTO DE
CONGELACIÓN o DESCENSO CRIOSCÓPICO.
Se ha podido demostrar que el descenso del punto de congelación es proporcional a
la concentración molal del soluto.
Este fenómeno queda establecido por las siguientes ecuaciones:
Donde:
∆Tc
Tc
Tºc
Kc
m
=
=
=
=
=
∆Tc
=
T°c
∆Tc
=
Kc m
- Tc
Ecuación 1
Ecuación 2
Descenso del punto de congelación
Temperatura de congelación de la solución.
Temperatura de congelación del solvente puro.
Constante molal del descenso del punto de congelación.
molalidad.
Al igual que la constante ebulloscópica (Keb), la constante crioscópica (Kc) representa
el descenso en el punto de congelación para soluciones de concentración 1 molal.
Por lo que debemos concluir que la magnitud de ∆Tc no sólo depende de la
concentración molal de la solución, sino también de la naturaleza del solvente, ya
que el valor de la constante es diferente para cada uno de ellos.
A continuación se señalan para algunas solventes sus constantes
crioscópicas.
Solvente
Agua
Benceno
Constante crioscópica (°C/molal)
1,86
5,12
Laboratorio de Procesos Químicos y Control
Página 37
Etanol
Ácido acético
Ciclohexano
Alcanfor
Naftaleno
Fenol
Ácido fórmico
Benzofenona
Difenilanina
Nitrobenceno
1,99
3,90
20,00
37,70
6,90
7,27
2,77
9,80
8,60
7,00
Para el agua la constante crioscópica es 1,86 °C/mo lal, por consiguiente, una
solución acuosa 1 molal de cualquier soluto se congelará a una temperatura de -1,86
°C.
Nota: Recuerde que en el caso de la elevación del punto de ebullición se requiere
que el soluto sea no volátil, aquí no hay tal restricción. En el caso del punto de
congelación se puede agregar un solvente volátil e igualmente se observa una
disminución en el punto de congelación.
GRAFICO DEL DESCENSO DE LA PRESION DE VAPOR CON EL AGREGADO
DE UN SOLUTO NO VOLATIL
P. Vap
Solv.
Solución
1 Atm
Liquido
Vapor
sólido
T
∆ T CRIOSCOPICA
Laboratorio de Procesos
Químicos y Control
∆ T EBULLOSCOPICA
Página 38
C.- EJERCICIOS RESUELTOS
Ejercicio Nº1:
Paso 1:
Calcular el punto de congelación de una solución de 100g de anticongelante etilenglicol
(C2H6O2), en 900 g de agua (Kc = 1,86 °C/molal)
Ordenar los datos.
Soluto etilenglicol
Solvente agua
Solución
: masa
masa molar
: masa
Tºc
Kc
: sin datos
=
=
=
=
=
100 g
62 g/mol
900 g
0 °C
1,86 °C/molal
Paso 2:
Pregunta concreta ⇒ Calcular el punto de congelación de una solución de etilenglicol.
Paso 3:
Aplicamos ecuaciones:
∆Tc
=
T°c
∆Tc
=
Kc m
-
Tc
Ecuación 1
Ecuación 2
Para poder obtener la temperatura de congelación de la solución necesitamos la ecuación 1, pero
como no tenemos ∆Tc (ascenso de la temperatura de ebullición), necesitamos obtenerlo de ecuación 2.
Paso 4: Para poder utilizar ecuación 2 necesitamos la molalidad de la solución que podemos calcular a partir
de los siguientes datos:
Moles de soluto
:
Molalidad
:
62 g ----- 1 mol
100 g ----- X
X = 1,61 moles de soluto
1,61 moles ----- 900 g de solvente
X ----- 1000 g de solvente
X =
1,79 molal
La solución tiene una concentración molal de 1,79
Paso 5:
Aplicando ecuación 2, tenemos:
∆Tc
∆Tc
∆Tc
Paso 6:
=
=
=
Kc m
(1,86 °C/molal) (1,79 molal)
3,33 °C
Aplicando ecuación 1, tenemos:
∆Tc
3,33 °C
Tc
RESPUESTA:
=
=
=
T°c
0°
- 3,33 °C
Tc
Tc
La temperatura de ebullición de la solución es 3.33 °C bajo cero.
Laboratorio de Procesos Químicos y Control
Página 39
Ejercicio Nº2:
Paso 1:
El alcanfor, C10H16O, se congela a 179,8 °C (Kc = 40 °C/molal). Cuando se disuelven 0,816 g
de sustancia orgánica de masa molar desconocida en 22,01 g de alcanfor líquido, el punto de
congelación de la mezcla es 176,7 °C ¿Cual es el peso molecular aproximado del soluto?
Ordenar los datos.
Soluto
: masa
=
0,186 g
Solvente alcanfor
: Kc
T°c
=
=
40,0 °C/m
179,8 °C
Solución
: Tc
=
176,7 °C
Paso 2:
Pregunta concreta ⇒ determinar la masa molar del soluto desconocido.
Paso 3:
Aplicamos ecuaciones.
∆Tc
=
T°c
∆Tc
=
Kc m
-
Tc
Ecuación 1
Ecuación 2
Para poder obtener la masa molar necesitamos conocer la molalidad de la solución. Con la ecuación
1 podemos determinar el descenso de la temperatura de congelación y luego con la ecuación 2 podemos
conocer la molalidad de la solución.
Paso 4: Aplicando ecuación 1, tenemos
∆Tc
∆Tc
∆Tc
Paso 5:
=
=
=
Tc
- 176,7 °C
Aplicando ecuación 2, tenemos:
∆Tc
=
3,1 °C =
m
=
Paso 6:
T°c
179,8 °C
3,1 °C
Kc m
(40 °C/molal) m
0,0775 molal
Calculo de la masa molar.
En base a la molalidad podemos saber cuantos moles corresponden a 0,186 g de soluto desconocido.
0,0775 moles de soluto ------- 1000 g de solvente
X ------- 22,01 g de solvente
X = 1,7058 x 10-3 moles de soluto
Por lo tanto:
0,186 g de soluto --------- 1,7058 x 10-3 moles de soluto
X -------- 1 mol
X = 109 g
RESPUESTA:
La masa molar del soluto es de 109.
Laboratorio de Procesos Químicos y Control
Página 40
Ejercicio Nº3:
Paso 1:
Se disuelven 10 g de naftaleno en 50 mL de Benceno (d = 0,88 g/mL) ¿Cual es el punto de
congelación de esta solución, sabiendo que la masa molar de naftaleno es 128 g/mol?
(benceno: Kc = 5,12 °C/molal y T°c = 5,5 °C)
Ordenar los datos.
Soluto naftaleno
: masa
masa molar
= 10 g
= 128 g/mol
Solvente benceno
: Volumen
densidad
Kc
Tºc
=
=
=
=
Solución
: no hay datos
50 mL
0,88 g/mL
5,12 °C/m
5,5 °C
Paso 2:
Pregunta concreta ⇒ determinar el punto de congelación de la solución.
Paso 3:
Aplicamos las ecuaciones
∆Tc
=
T°c - Tc
Ecuación 1
∆Tc
=
Kc m
Ecuación 2
Nos piden calcular punto de congelación de la solución, para lo cual necesitamos conocer el
descenso en el punto de congelación, por lo tanto, a partir de la ecuación 2 obtemos el descenso en el punto de
congelación y luego aplicamos la ecuación 1 para determinar el punto de congelación de la solución.
Paso 4:
Para poder conocer el descenso en el punto de congelación debemos calcular la molalidad de la
solución.
a.- Primero calcularemos los moles de soluto que tenemos:
128 g ------ 1 mol
10 g ------ X
X = 0,08 moles
b.- Luego calculamos la masa de solvente (por medio de la densidad)
masa
d
=
Volumen
masa
0,88 g/mL
=
50 mL
masa
= 44 g
c.- Calculamos la molalidad
0,08 moles de soluto -------- 44 g de solvente
X -------- 1000 g de solvente
X = 1,82 moles
Por lo tanto, la molalidad de la solución es 1,82
Laboratorio de Procesos Químicos y Control
Página 41
Paso 5:
Cálculo del descenso del punto de congelación de la solución.
∆Tc
∆Tc
∆Tc
Paso 6:
=
=
=
Kc m
(5,12 °C/molal) (1,82 molal)
9,32 °C
Cálculo del punto de congelación de la solución.
∆Tc
9,32 °C
Tc
= T°c - Tc
= 5,5 °C - Tc
= - 3,82 °C
RESPUESTA:
El punto de congelación de la solución es 3,82 °C bajo cero.
Ejercicio Nº4:
Una disolución acuosa contiene el aminoácido glicina (NH2CH2COOH). Suponiendo que este
aminoácido no ioniza, calcule la molalidad de la disolución si se congela a -1,1 °C. (agua:
constante crioscópica 1,86 °C/molal; punto de congelación 0 °C)
Paso 1:
Ordenar los datos.
Soluto glicina
:
no hay datos
Solvente agua
:
Kc
T°c
= 1,86 °C/m
= 0 °C
Solución
:
Tc
= -1,1 °C
Paso 2:
Pregunta concreta ⇒ determinar la molalidad de la solución.
Paso 3:
Aplicamos las ecuaciones
∆Tc
∆Tc
=
=
T°c - Tc
Kc m
Ecuación 1
Ecuación 2
Como necesitamos la molalidad de la solución podríamos utilizar la ecuación 2, pero para ello
necesitamos conocer el descenso del punto de congelación que podemos obtener de la ecuación 1.
Paso 4:
Calculo del descenso del punto de congelación.
∆Tc
∆Tc
∆Tc
Paso 5:
= T°c - Tc
= 0 °C - (-1,1 °C)
= 1,1 °C
Calculo de la molalidad de la disolución
∆Tc
= Kc m
1,1 °C
= (1,86 °C/molal) m
m
= 0,59 molal
RESPUESTA:
La molalidad de la disolución es 0,59.
D.- EJERCICIOS PROPUESTOS.
1) Calcular el punto de congelación de una solución acuosa al 1,26 % p/p de un compuesto no
electrolito.(agua: Kc = 1,86 °C/molal y T°c =0 °C; masa molar de soluto 51g/mol )
(Respuesta = -0,465°C)
Laboratorio de Procesos Químicos y Control
Página 42
2) Calcule el peso molecular de un no electrolito si el agua se congela a -0,50 °C cuando en 20 g de ella se
disuelven 12 g de soluto. (Agua: temperatura de congelación 0 °C y constante crioscópica 1,86 °C/molal )
(Respuesta = 2232 g/mol)
3) ¿Cual será el punto de congelación de una solución que contiene 17,25 g de ácido cítrico (C6H8O7)
disueltos en 250 g de agua. (Agua: temperatura de congelación 0 °C y constante crioscópica 1,86 °C/molal
)
(Respuesta = -0,668 °C)
4) A 100 mL de agua se agregan 50 mL de alcohol (masa molar 46 y densidad 0,7 g/mL) ¿Cual será el punto
de congelación de esta mezcla. (Agua: temperatura de congelación 0 °C y constante crioscópica 1,86
°C/molal )
(Respuesta = 14,13 °C)
5) Si se disuelven 3,96 g de ácido benzoico en 80,6 g de benceno y la solución se congela a -4,47 °C. Hallar el
peso molecular aproximado del ácido benzoico. (Benceno: temperatura de congelación 5,5 °C y constante
crioscópica 5,12 °C/molal)
(Respuesta = 244,3 g/mol)
IV.: Presión Osmótica.
A.- CONCEPTOS BÁSICOS: PRESIÓN OSMÓTICA
1.- Definición:
• Ciertos materiales como el celofán o bien ciertas estructuras complejas como las
membranas de los sistemas biológicos son SEMIPERMEABLES, es decir, cuando
están en contacto con la solución permiten el paso de algunas moléculas, pero no
de otras.
• Generalmente, estas membranas, permiten el paso de pequeñas moléculas de
solvente (ejemplo el agua), pero bloquean el paso de moléculas o iones de soluto
de mayor tamaño.
• Este carácter semipermeable se debe a la presencia de pequeños canales o poros
en su estructura membranosa
Membrana Semi permeable
Solución B
Solución A
→
←
Laboratorio de Procesos Químicos y Control
Página 43
Consideremos una situación en la que sólo las moléculas de disolvente pueden pasar
a través de la membrana. Si esta se coloca entre dos soluciones de concentración
diferente, las moléculas de disolvente se mueven en ambas direcciones a través de la
membrana.
Solución A
Membrana semipermeable
Más concentrada
Más Diluida
Solución B
O = Soluto X= Solvente
Solución A
Solución B
La concentración de solvente es menor La concentración de solvente es más elevada
en la solución que tiene más soluto
en la solución que tiene menos soluto
(Solución más concemtrada*)
(Solución menos concentrada*)
⇓
A
B
LA VELOCIDAD DE PASO DEL SOLVENTE DE
LA SOLUCIÓN MENOS CONCENTRADA A LA
MÁS CONCENTRADA, ES MAYOR QUE LA
VELOCIDAD EN LA DIRECCIÓN OPUESTA.
⇓
Por último término hay un movimiento neto de moléculas de solvente de la solución
menos concentrada hacia la más concentrada
Laboratorio de Procesos Químicos y Control
Página 44
⇓
OSMOSIS: Movimiento neto de solvente
desde la solución menos concentrada
hacia la solución más concentrada
*Recuerde que: Los términos de solución más o menos concentrada están referidos
a la cantidad de soluto (más o menos soluto).
En la siguiente figura se muestran dos soluciones separadas por una membrana
semipermeable.
A
Solución concentrada
B
Solución diluida
Membrana semipermeable
El solvente se mueve de B hacia A, como si las soluciones tendieran a lograr concentraciones
iguales. Al cabo de un tiempo los niveles del líquido (volumen) en las dos ramas son
desiguales.
A
B
La diferencia de presión
resultante de las alturas
desiguales del líquido en
las dos ramas llega a ser
tan grande que el flujo de
líquido cesa
Laboratorio de Procesos Químicos y Control
Página 45
Presión aplicada,
detiene la Osmosis
Si aplicamos una presión sobre el brazo izquierdo del codo, como se muestra en la
próxima figura, podríamos detener el flujo neto de solvente.
La presión aplicada sobre el brazo de la izquierda del aparato detiene el movimiento neto del
solvente desde el lado derecho de la membrana
Esta presión aplicada se conoce como Presión Osmótica (π
π) y es la presión
requerida para detener la osmosis; esta presión depende de la temperatura y de la
concentración de la solución.
2.- Factores que afectan la Presión Osmótica.
La presión osmótica obedece a una ley similar a la de los gases ideales. Van’t Hoff
fue el primer científico que analizó estos hechos, los cuales se expresan en la
siguiente ecuación, conocida como ecuación de Van’t Hoff:
nRT
π
=
V
Donde:
π
V
R
n
T
=
=
=
=
=
Presión Osmótica (atm)
Volumen de la solución (L)
Constante de los gases ideales (0,082 L atm/ °K mol)
Número de moles de soluto
Temperatura (°K)
De acuerdo a la ecuación de Van’t Hoff, se observa que a temperatura constante la
presión osmótica solo depende de la concentración de partículas y no de la
naturaleza del soluto, de ahí que la presión osmótica es una propiedad coligativa de
una solución.
Laboratorio de Procesos Químicos y Control
Página 46
Si el volumen de la solución fuera un litro, entonces:
n
= Molaridad*, por lo tanto, nuestra relación puede formularse como:
V
π = MRT
*Cuando las soluciones son muy diluidas (menores a 0,1 M) se
puede considerar que la Molaridad es igual a la Molalidad.
B.- PRESIÓN OSMÓTICA.
Las soluciones se pueden clasificar entre si respecto de su presión osmótica en:
Caso 1
Solución A
Solución B
Concentración 0,01 molal
Concentración 0,01 molal
Membrana semipermeable
a) Ambas soluciones tienen la misma concentración, a una temperatura
dada, luego podemos decir que no se presenta el fenómeno de
Osmosis.
b) Se puede concluir, entonces, que ambas soluciones tiene igual Presión
Osmótica.
c) Cuando dos soluciones tienen igual Presión Osmótica se dice que son
ISOTÓNICAS o ISOOSMÓTICA entre sí (iso = igual; osmótica =
presión osmo; tónica = concentración).
Caso 2
Solución A
Solución B
Concentración 0,02 molal
Concentración 0,01 molal
Membrana semipermeable
Laboratorio de Procesos Químicos y Control
Página 47
a) La solución A tiene mayor concentración que la solución B, se dice
entonces, que la solución A es HIPERTÓNICA con respecto a la
solución B.
b) También se puede decir que la solución B es HIPOTÓNICA con
respecto a la solución A.
c) Como la solución B es HIPOTÓNICA, con respecto a la solución A,
genera una menor presión osmótica, ya que tiene menos partículas en
solución, por lo tanto, se puede decir que la solución B es
HIPOOSMÓTICA con respecto a la solución A.
d) Como la solución A es HIPERTÓNICA, con respecto a la solución B,
genera una mayor presión osmótica, ya que tiene mayor número de
partículas en solución, luego se dice que es HIPEROSMÓTICA con
respecto a la solución B.
EN RESUMEN:
Solución A
Solución B
Concentrada
Diluída
⇓
⇓
Mayor número de partículas
disueltas
Menor número de partículas
disueltas
⇓
⇓
Hipertónica
Hipotónica
⇓
⇓
Gran presión osmótica
Pequeña presión osmótica
⇓
⇓
Hiperosmótica
Hipoosmótica
La ósmosis juega un papel importante en los sistemas vivos. Por ejemplo, las
membranas de los glóbulos rojos son semipermeables. Si se colocan estas células en
una solución hipertónica respecto a la solución intracelular se provoca que el agua
salga de la célula, como se muestra en la figura. Esto causa que la célula se arrugue,
Laboratorio de Procesos Químicos y Control
Página 48
y ocurre el proceso que se conoce como crenación. Si se colocan estas células en
una solución hipotónica respecto al líquido intracelular se ocasiona que el agua
penetre en la célula. Esto causa la ruptura de la célula, proceso que se conoce como
hemólisis. A las personas que necesitan el reemplazo de los fluidos corporales
nutrientes, y que no pueden ser tomados por vía oral, se les administran soluciones
por infusión intravenosa, la cual provee los nutrientes directamente al interior de las
venas. Para evitar crenación o hemólisis de los glóbulos rojos, las soluciones deben
ser isotónicas con los líquidos en el interior de las células.
Ejemplo: La presión osmótica promedio de la sangre es 7,7 atm a 25 °C. ¿Qué
concentración de glucosa, C6H12C6 será isotónica con la sangre?
Solución: π = M R T
π
M=
7,7 atm
=
RT
= 0,31 molar
(0,082 L atm/°K mol)(298 °K)
En condiciones clínicas, la concentración de las soluciones se expresan
generalmente en porcentajes en peso. El porcentaje en peso de una solución de
glucosa 0,31 M es 5,3 %.
Hay otros ejemplos interesantes de ósmosis. Un pepino colocado en una salmuera
concentrada pierde agua por ósmosis y se arruga para convertirse en un pepinillo.
Una zanahoria que se hace flácida al perder agua a la atmósfera, puede recuperla
por ósmosis si se coloca en agua y así recobra su firmeza. Las personas que comen
demasiada sal en los alimentos sufren la retención de agua en las células de los
tejidos debido a la ósmosis. La hinchazón que resulta se denomina edema. El
movimiento del agua del suelo hacia el interior de las raíces de las plantas y
posteriormente hacia sus órganos superiores se debe, al menos una parte, a la
ósmosis. La conservación de la carne mediante salado y de las frutas al cubrirlas de
azúcar, las protege contra la acción bacteriana. A través del proceso de ósmosis, una
bacteria que se encuentre en la carne salada o en la fruta caramelizada pierde agua,
se encoge y muere.
Durante el proceso de ósmosis, el agua se mueve de un área de alta concentración
de agua (concentración baja de soluto) hacia un área de baja concentración de agua
(alta concentración de soluto). El movimiento de una sustancia de una zona donde su
concentración es elevada a otra donde es baja, es espontáneo. Las células biológicas
transportan no solamente agua, sino también otros materiales seleccionados a través
de sus membranas. Esto permite la entrada de nutrientes y la eliminación de
materiales de desecho. En algunos casos, las sustancias deben moverse de un área
de baja concentración a una de concentración elevada. Este movimiento se llama
transporte activo. Este proceso no es espontáneo y por tanto requiere gasto de
energía por las células.
Laboratorio de Procesos Químicos y Control
Página 49
C.- EJERCICIOS RESUELTOS
Ejercicio Nº1:
Paso 1:
Calcular el valor de la presión osmótica que corresponde a una solución que contiene 2 moles de soluto en
un litro de solución a una temperatura de 17° C.
Ordenar los datos.
Soluto
:
masa
Solvente
:
no hay datos
Solución
:
volumen
temperatura
= 2 moles
=
=
1L
17 °C
Paso 2:
Pregunta concreta ⇒ determinar la presión osmótica de la solución (π).
Paso 3:
Aplicamos las ecuaciones:
π
nRT
=
Ecuación 1
V
π
=
MRT
Ecuación 2
Si analizamos los datos estos nos dicen que tenemos 2 moles de soluto por un litro de solución, entonces la
molaridad es 2, esto nos permite utilizar la ecuación 2 directamente. El único detalle que tenemos que tener encuenta es que
la temperatura la entregan en grados Celcius y la necesitamos en grados Kelvin.
Paso 4:
Conversión de unidades.
T(°K)
T(°K)
T(°K)
Paso 5:
=
=
=
T(°C) + 273,15
17 + 273,15
290,15
Cálculo de la presión osmótica de la solución (π).
π
π
π
=
=
=
MRT
(2 mol/L)(0,082 atm L/mol °K)(290,15 °K)
47,585 atm
RESPUESTA:
La presión osmótica de la solución es 47,585 atm.
Ejercicio Nº2:
Qué masa de anilina habría que disolver en agua para tener 200 mL de una solución cuya presión
osmótica, a 18 °C, es de 750 mmHg; sabiendo que la masa molar de la anilina es 93,12 g/mol.
Paso 1:
Ordenar los datos.
Soluto anilina
:
masa molar
Solvente agua
:
no hay datos
Solución
:
volumen
temperatura
presión osmótica
= 93,12 g/mol
Paso 2:
Pregunta concreta ⇒ determinar la masa en gramos de anilina.
Paso 3:
Aplicamos las ecuaciones:
Laboratorio de Procesos Químicos y Control
=
=
=
200 mL
18 °C
750 mmHg
Página 50
nRT
π
=
Ecuación 1
V
π
=
MRT
Ecuación 2
Ambas ecuaciones podrían ser usadas para calcular el número de moles de anilina necesarios para preparar la
solución, sin embargo, como en los datos nos dan el volumen de la solución sería más conveniente utilizar la ecuación 1.
No olvidar convertir las unidades de: presión en atmósferas, volumen a litros y temperatura a grados Kelvin.
Paso 4:
Paso 5:
Conversión de unidades.
Temperatura
⇒
T(°K)
T(°K)
T(°K)
Volumen
⇒
1000 mL ------- 1 L
200 mL ------- X
X
=
0,2 L
Presión
⇒
760 mmHg ------- 1 atm
750 mmHg ------- X
X
= 0,987 atm
=
=
=
T(°C) + 273,15
18 + 273,15
291,15
Cálculo de los moles de anilina existente en la solución (n).
π
nRT
=
V
n (0,082 atm L/mol °K) (291,15 °K)
0,987 atm
=
0,2 L
n
=
0,0083 moles
Paso 6: Transformando los moles a masa (g)
93,12 g ------ 1 mol
X ------ 0,0083 moles
X
= 0,7699 g
RESPUESTA:
La masa de anilina es 0,7699 g.
Ejercicio Nº3:
Cuantos gramos de sacarosa C12H22O11 deberán disolverse por litro de agua para obtener una solución
isoosmótica con otra de urea CO(NH2)2 que contiene 80 g de soluto por litro de solución a 25 °C.
Paso 1:
Ordenar los datos.
Soluto sacarosa
Solvente agua
Solución
Solución 1
: no hay datos
masa molar
: volumen
=
=
342 g/mol
1L
: volumen
temperatura
=
=
1L
25 °C
Solución 2
: masa
masa molar
Solvente agua
: volumen
Soluto urea
Solución
:
volumen
temperatura
= 80 g
= 60 g/mol
= no hay datos
= 1L
= 25 °C
Paso 2:
Pregunta concreta ⇒ determinar la masa de sacarosa para tener una solución isoosmótica con la solución de urea.
Paso 3:
Aplicamos las ecuaciones:
Laboratorio de Procesos Químicos y Control
Página 51
nRT
π
=
Ecuación 1
V
π
=
MRT
Ecuación 2
Nos piden calcular la masa de sacarosa necesaria para obtener una solución isoosmótica con una solución de
urea dada. Que dos soluciones sean isoosmótica entre sí significa que tienen igual presión osmótica, es decir poseen igual
concentración. Por lo tanto, no es necesario calcular la presión osmótica, pues conociendo la concentración de la solución
de urea conocemos la concentración de la solución de sacarosa requerida.
Paso 4:
Determinamos la concentración de la solución de urea.
60 g ------ 1 mol
80 g ------ X
X
=
1,33 moles
Como los 80 g (1,33 moles) están disueltos en un litro, nuestra solución es 1,33 M en urea.
Paso 5:
Cálculo de la masa de sacarosa.
Como la solución de urea es 1,33 M, la solución de sacarosa también es 1,33 M (soluciones isoosmóticas entre
sí). Entonces necesitamos 1,33 moles de sacarosa por un litro de solución, por lo tanto, sólo nos basta con transformar los
moles de sacarosa a masa.
342 g ------ 1 mol
X ------ 1,33 moles
X
=
454,86 g
RESPUESTA:
La masa de sacarosa requerida para tener una solución isoosmótica con una solución de urea dada es
454,86 g.
Ejercicio Nº4:
Se midió la presión osmótica de una solución acuosa de cierta proteína a fin de determinar su masa molar.
La solución contenía 3,50 mg de proteína disueltos en agua suficiente para formar 500 mL de solución. Se
encontró que la presión osmótica de la solución a 25 °C es 1,54 mmHg. Calcular la masa molar de la
proteína.
Paso 1:
Ordenar los datos.
Soluto proteína
:
masa
Solvente agua
:
no hay datos
Solución
:
volumen
temperatura
presión osmótica
= 3,50 mg
Paso 2:
Pregunta concreta ⇒ determinar la masa molar de la proteína.
Paso 3:
Aplicamos las ecuaciones:
π
=
=
=
500 mL
25 °C
1,54 mmHg
nRT
=
Ecuación 1
V
π
=
MRT
Ecuación 2
Utilizaremos la ecuación 1, ya que el volumen dado es 500 mL y así calcularemos los moles de proteína
disueltas en estas condiciones. No olvidar conversión de unidades.
Paso 4:
Conversión de unidades.
Laboratorio de Procesos Químicos y Control
Página 52
Presión
Temperatura
⇒
Volumen
⇒
⇒
760 mmHg ------- 1 atm
1,54 mmHg ------- X
X = 0,002 atm
⇒ 1000 mg ------ 1 g
3,5 mg ------- X
X
= 3,5 x 10-3 g
masa
Paso 5:
T(°K)
=
T(°C) + 273,15
T(°K)
=
25 + 273,15
T(°K)
=
298,15
1000 mL ------- 1 L
500 mL ------- X
X
=
0,5 L
Cálculo de los moles de proteína (n).
π
nRT
=
V
n (0,082 atm L/mol °K) (298,15 °K)
0,002 atm
=
0,5 L
n
=
Paso 6: Transformando los moles a masa (g)
41 x 10-6 moles
masa molar ⇒ gramos de sustancia por un mol
3,5 x 10-3 g ------- 41 x 10-6 moles
X g ------ 1 mol
X
= 85,37 g
RESPUESTA:
La masa molar de la proteína es 85,37.
D.- EJERCICIOS PROPUESTOS.
1) ¿Cual es la presión osmótica a 20°C de una solución de sacarosa (C12H22O11), 0,0020 M?
(Respuesta = 0,048 atm)
2) Disolviendo 6,73 g de sacarosa (masa molar 342 g/mol) hasta formar 1500 mL de solución a 20 °C. ¿Cual
es la presión osmótica que teóricamente corresponderá?
(Respuesta = 0,315 atm)
3) ¿Que presión osmótica ejercerá una solución de urea en agua al 1% a 20 °C(masa molar de urea 60
g/mol)?
(Respuesta = 4 atm)
4) Calcular la masa molar aproximada del pineno sabiendo que al disolver 2,8 g en alcohol hasta un volumen
de 500 mL se midió una presión osmótica de 1,2 atm a 20 °C.
(Respuesta = 112 g/mol)
5) Calcular la masa molar aproximada del tiofeno sabiendo que una solución de 100 mL que contiene 0,32 g
de ese compuesto en alcohol dio una presión osmótica de 510 mmHg a 20 °C.
Laboratorio de Procesos Químicos y Control
Página 53
(Respuesta = 114,7 g/mol)
6) ¿Que presión osmótica en atm ejercerá cualquier solución 0,1 M de una sustancia no ionizable a 20 °C?
(Respuesta = 2,40 atm)
Laboratorio de Procesos Químicos y Control
Página 54
ESCUELA DE EDUCACIÓN TÉCNICA Nº 3 – SAN JOSÉ
DEPARTAMENTO QUÍMICA
TERMODINÁMICA
En casi toda la bibliografía disponible se define la Energía com o la capacidad de
producir trabajo. Más ajustadamente debemos decir que la ENERGÍA SE
RELACIONA CON LA CAPACIDAD DE PRODUCIR TRABAJO, lo cual prefigura
un concepto más amplio y adecuado.
Con este concepto presente podemos decir que la Termodinámica es el estudio
de las transformaciones e intercambios de la energía.
Todas las formas de energía tienden en última instancia a pasar a calor.
Ej. Energía química a calor, en una reacción química (más adelante veremos que
esto no es tan general). Energía eléctrica a Calor, cuando actúa a través de una
resistencia, etc.
De todos modos ya Ud. conoce un hecho general que vale la pena recalcarlo y que
en termodinámica toma la forma de una ley: LA ENERGÍA SE CONSERVA.
Precisamente la Ley de conservación de la Energía es la PRIMERA LEY DE LA
TERMODINÁMICA que la podemos enunciar de la siguiente manera: "si bien la
energía se puede convertir de una forma en otra no puede ser creada ni
destruida” o también "siempre que se produce una cantidad de una forma de
energía debe desaparecer una cantidad exactamente equivalente de otras
especies de energía".
Para los ejercicios que Ud. deberá realizar debe recordar el Equivalente mecánico
del Calor: 1 caloría (energía calórica) = 4,183 Joules (E. m ecánica)
Sistemas termodinámicos
SISTEMA es la porción delimitada y especificada del mundo físico, que contiene
cantidades definidas de sustancia que se consideran bajo estudio o constituyen
nuestro interés.
Necesitamos además definir ENTORNO o MEDIO AMBIENTE el cual es la zona
del universo que interactúa con el sistema. Esta última definición tiene su
importancia ya que determina con una mayor rigurosidad lo que se debe entender
por Entorno o Medio ambiente.
Saber cuál es la "zona (y sus límites) que interactúa" con el Sistema no siempre se
pueden conocer en forma precisa, por ello en los esquemas que se observan más
abajo, el Entorno se dibuja con líneas discontinuas
Laboratorio de Procesos Químicos y Control
Página 55
SISTEMA + ENTORNO (O MEDIO AMBIENTE) = UNIVERSO
Tipos de sistema
Aislado: no hay transferencia de masa o energía con el entorno.
Ej. : un termo ideal (aislado y de paredes rígidas).
Cerrado: no transfiere masa pero sí energía en forma de calor, trabajo o radiación.
Ej. : Cualquier recipiente cerrado no ideal.
Abierto: transfiere masa y energía con su entorno. Ej. : el cuerpo humano.
La mayoría de los sistemas en la vida real son abiertos, mientras que en el
laboratorio la mayoría de los sistemas químicos son cerrados
Laboratorio de Procesos Químicos y Control
Página 56
PRIMERA LEY o PRIMER PRINCIPIO DE LA TERMODINÁMICA
La Ley de Conservación de la Energía -que es una de las formas de expresión de
la Primera Ley (o Primer Principio) de la Termodinámica, se puede visualizar en
una forma muy sencilla con los conceptos de ENERGÍA ACUMULADA y ENERGÍA
INTERCAMBIADA O EN PROCESO DE INTERCAMBIO o también EN ENERGÍA
EN TRÁNSITO. ¿Qué significan cada una de estas expresiones?
Veamos lo siguiente:
Se tiene un Sistema y su Entorno donde cada uno tendrá una CIERTA
CANTIDAD DE EN ERGÍA (que no podemos conocer). A nosotros nos interesa
sólo los CAMBIOS DE ENERGÍA que se produzcan en el SISTEMA (no en el
ENTORNO o MEDIO AMBIENTE aun cuando este juega un papel fundamental
para ello.
Obviamente Es (Energía del Sistema) puede ser mayor, menor o igual que Ema
(Energía del Medio Ambiente) dependiendo ello de cada situación en particular,
pero en determinado momento se producirá un "INTERCAMBIO DE ENERGÍA,
∆EI" el cual se realizará por medio de CALOR Y TRABAJO o por uno de ellos.
Graficamos esta situación de la siguiente forma:
Luego, de acuerdo al proceso que hemos visto el SISTEMA puede haber
ganado o perdido Energía, con lo cual el balance final sería:
Es fina l- Es inicial= ∆E
o lo que es lo mismo
∆Es = ∆EI
Laboratorio de Procesos Químicos y Control
Página 57
Esta sencilla ecuación nos indica que la VARIACIÓN de Energía del
Sistema (nuestro objetivo al analizar la primera ley) es igual a la SUMA
ALGEBRAICA DE LAS EN ERGÍAS INTERCAMBIADAS O EN PROCESO DE
INTERCAMBIO, siendo la ENERGÍA ACUMULADA aquella que no participa en
estos procesos.
Por otra parte si bien hay un INTERCAMBIO de Energía entre Sistema-Medio
Ambiente siempre se cumple la LEY DE CONSERVACIÓN DE LA ENERGÍA ya
que:
1. - ANTES DEL INTERCAMBIO :
siendo i= inicial
Esi + Em ai = E universo
2. - LUEGO DEL INTERCAMBIO :
siendo f= final
Es f+ Ema f= E universo
DONDE:
Esf - Esi = ∆Es = ΣE intercambiadas
En ambos casos la ENERGÍA DEL UNIVERSO se mantiene constante
LA ENERGÍA INTERNA
La Energía Interna (E) se define como TODAS LAS FORMAS DE ENERGÍA DE
UN SISTEMA
El cálculo absoluto de la Energía Interna (E) imposible; por eso a la Termodinámica
clásica sólo le interesan los cambios de Energía. No obstante lo cual, es
importante que el estudiante sepa que todas estas Energías tienen nombre y
teóricamente podría llegar a ser posible su cálculo (aunque todavía algunas no se
conocen lo suficiente).
Veamos los cuadros siguientes donde se indican los tipos de Energía en 1 mol de
sustancia pura en ciertas condiciones de P y T.
Tales sistemas se caracterizan por las tres coordenadas mensurables: presión P,
volumen V y temperatura T y se llaman sistemas PVT.
Energías intramoleculares
1. Nuclear
2. Electrónica
3. Traslacional
4. Vibracional
5. Rotacional
(3, 4, y 5 son las Energías Térmicas)
Energías intermoleculares
1. Fuerzas de atracción interiónicas (sales)
2. Fuerzas de atracción Ion-dipolo
3. Unión por puente de hidrógeno
Laboratorio de Procesos Químicos y Control
Página 58
4. Fuerzas de atracción dipolo-dipolo
5. Fuerzas de atracción entre iones o dipolos-m oléculas polarizables
6. Fuerzas entre dipolos instantáneos-dipolos inducidos (London)
7. Fuerzas hidrofóbicas
8. Fuerzas de repulsión
Cada sistem a poseerá UN CIERTO NUMERO DE LAS MISMAS (no
necesariamente todas), y para el cálculo final de la Energía Interna (E) se necesita
saber el comportamiento de cada una de ellas. Ello, como es evidente resulta muy
complicado, pero no lo es calcular una VA RIACIÓN DE ENERGÍA INTERNA (∆E)
que no es otra cosa que la SUMA ALGEBRAICA DE LAS ENERGÍAS
INTERCAMBIADAS O EN TRANSITO.
En un Sistema hay un cambio mensurable de Energía Interna (∆E)
siempre que haya un intercambio de Energía, que va a estar dado por la cantidad
de CALOR tomado del medio o cedido hacia él y el trabajo realizado por el sistema
hacia el entorno o sobre el sistem a por el entorno.
Para aplicar en los cálculos las variaciones de calor y trabajo se utiliza una
convención, que tiene su lógica:
CALOR
PROCESO
SIGNO
ENDOTÉRMICO POSITIVO (+)
EXOTÉRMICO NEGATIVO (-)
TRABAJO
PROCESO
SIGNO
SOBRE EL SISTEMA
POSITIVO (+)
POR EL SISTEMA
NEGATIVO (-)
Con respecto al calor no habrá problemas de interpretación en cuanto a la
convención. Para el trabajo, es evidente que si el entorno realiza un trabajo sobre
el sistema éste verá aumentada su energía interna, de allí SU SIGNO POSITIVO.
Por el contrario si el sistema realiza un trabajo sobre el entorno, verá disminuida su
energía interna, POR ELLO SU SIGNO NEGATIVO.
Trabajo:
El trabajo es un concepto íntimamente ligado a la energía. Lo que no hemos dicho
es cómo expresar matemáticamente el mismo.
Imaginemos un gas ideal colocado en un cilindro como el de la figura, al cual se lo
dilata (expansión del gas):
Laboratorio de Procesos Químicos y Control
Página 59
Por lo tanto:
w = Pext * A * (X2-X1)
A * (x2-x1)= Aumento de volumen durante la expansión o dilatación que
esigual a ∆V.
Luego
w = Pext . ∆V
Graficando el proceso con un par de ejes cartesianos en el plano encontramos:
Laboratorio de Procesos Químicos y Control
Página 60
Proceso Termodinamicamente Reversible
Se cumple siempre qué:
Pext = Pinterna
este en EQUILIBRIO,el sistema pude sufrir expansión y/o compresión sin perturbar
las condiciones,operando indistintamente en un sentido a otro, esto constituye el
equilibrio, y así se deben realizar los procesos hasta pasar del estado inicial al final.
En la vida real es muy difícil de lograr ya que en principio debe realizarse en un
tiempo infinito.
Redefinimos ahora la expresión por la que calculam os variaciones de energía
interna, tomando como base que el ÚNICO TRABAJO QUE HARÁ EL SISTEMA (O
QUE SE REALIZA SOBRE EL) SERÁ LA EXPANSIÓN (O COMPRESIÓN) DE UN
GAS.
Luego:
w = P ∆V
De acuerdo a la convención utilizada, en un trabajo realizado por el
sistema sobre el entorno de expansión, este tendrá signo NEGATIVO .
Presión es una magnitud positiva y ∆V (en una expansión) tam bién lo es,
dado lo cual DEBE CAMBIARSE EL SIGNO QUE NOS DA LA ECUACIÓN.
Si el caso fuera una trabajo de compresión (realizado por el entorno sobre el
sistema), el proceso es a la inversa.
Esta expresión es válida siempre que la contrapresión (P) o presión externa (Pext)
se mantenga constante, porque si no es así, sería distinta.
Trabajo “no-útil” y “útil”
Merece una aclaración el uso de "P", ES SIEMPRE LA PRESIÓN EXTERIOR AL
SISTEMA, ES DECIR AQUELLA CONTRA LA CUAL SE HACE EL TRABAJO Y
COMO NORMALMENTE ES LA PRESIÓN ATMOSFÉRICA, POR ELLO SE DICE
QUE UN TRABAJO DE EXPANSIÓN ES "TRABAJO NO ÚTIL" (YA QUE SE
PIERDE EN LA ATMÓSFERA).
Al trabajo de expansión se lo conceptúa por lo tanto como un trabajo "no útil" pero
se debe tener presente que hay casos en los cuales ello no ocurre y por el
contrario se lo debe asociar con un trabajo de expansión "útil". Para diferenciar uno
de otro diremos que el trabajo de expansión "no útil" es aquel que se realiza
"CONTRA" la presión atmosférica con lo cual la energía intercambiada se
considera disipada a la atmósfera, pero el "trabajo de expansión útil" es de acuerdo
al siguiente ejem plo:
Laboratorio de Procesos Químicos y Control
Página 61
En la figura de arriba la expansión del pistón "CONTRA " LA PRESIÓN
ATMOSFÉRICA generará un TRABAJO DE EXPANSIÓN NO ÚTIL (ya que como
dijimos se pierde, no se puede recuperar) pero si al pistón le adicionamos un
"sobrepeso" diferente a la presión que ejerce la
atmósfera será, de acuerdo a las dos expresiones en las cuales hemos
descomuesto el Trabajo:
Patm . ∆V = Trabajo de expansión contra la P atm . N O ÚTIL.
∆p. ∆V = Trabajo de expansión contra la Presión "extra" que arbitrariamente
hemos colocado: este es un trabajo de EXPANSIÓN ÚTIL ya que la
pesa (∆p) queda al final del proceso con una cierta energía potencial
mayor al final que al comienzo del proceso.
En general el trabajo de expansión será no-útil.
Expresión matemática de la Primera Ley o Primer Principio
Al ocurrir un cambio de energías (calor y trabajo, o una sola de ellas) la variación
de energía del sistema es la variación de ∆E que se busca conocer,
∆Es = ΣEI.
Esta sumatoria de energías intercambiadas, es como su nombre lo indica
Laboratorio de Procesos Químicos y Control
Página 62
ΣEI = calor + trabajo
Es la energía en forma de calor que se ganó o perdió más la energía en forma de
trabajo que se ganó o perdió.
Veamos estos ejemplos:
Caso 1: Un sistema "recibe" Calor por lo tanto según la convención es positivo y
realiza Trabajo (expansión) por el Sistema hacia el entorno, luego es negativo.
Gráfico N ro. 6
¿Qué ocurrió en el Sistem a luego de haberse com pletado el intercambio de
Energía?.
RECIBIÓ CALOR: AUMENTÓ SU ENERGÍA INTERNA
REALIZÓ UN TRABAJO: PERDIÓ EN ERGÍA INTERNA
Luego para conocer en cuánto varió la ENERGÍA INTERNA (∆E) del sistema
debemos calcular la sumatoria de ENERGÍAS INTERCAMBIADAS, pero para ello
debe recordarse la convención de signos, donde el Calor es positivo y el Trabajo
(de expansión) que según el cálculo es positivo, debe cambiarse su signo a
negativo; luego la sumatoria será:
∆EI = ∆Es = ∆E = q + w
En esta ecuación se advierte que q debe mantenerse con el signo según el
proceso (aumentó) y w que según el cálculo será positivo debe “cambiarse su
signo” según la convención adoptada.
Laboratorio de Procesos Químicos y Control
Página 63
Caso 2: Un sistema "cede" cierta cantidad de Calor (por lo tanto es "negativo") y el
entorno realiza "sobre el sistema" un cierto Trabajo (por lo tanto el sistema recibe
energía y ese trabajo debe ser positivo).
En el balance hallaremos: "q" es negativo y "w" es positivo.
CEDIÓ CALOR (EXOOTÉRMICO), LUEGO DISMINUYÓ SU ENERGÍA INTERNA
RECIBIÓ UN TRABAJO: AUMENTÓ SU ENERGÍA INTERNA
Por lo tanto:
∆EI = ∆E = q + w
donde "q" debe mantenerse con el signo de acuerdo al proceso y "w" debe
sumarse ya que aumenta la E.
Laboratorio de Procesos Químicos y Control
Página 64
Caso 3: Un sistem a “cede” cierta cantidad de calor(proceso exotérmico) y realiza un trabajo (luego pierde energía).
El balance será:
CEDIÓ CALOR: (EXOTÉRMICO) LUEGO PERDIÓ ENERGÍA INTERNA
REALIZÓ UN TRABAJO: LUEGO PERDIÓ ENERGÍA INTERNA
Por lo tanto:
ΣEI = ∆E = q + w
Caso 4: U n sistema “recibe” calor(proceso endotérmico)aumentando su
energía y el medio ambiente realiza “sobre” él un trabajo, lo cual
también aumenta la energía.
Laboratorio de Procesos Químicos y Control
Página 65
RECIBIÓ CALOR (proceso endotérmico) AUMENTANDO SU ENERGÍA
RECIBIÓ UN TRABAJO AUMENTANDO SU ENERGÍA
Por lo tanto:
ΣEI = ∆E = q + w
Expresión de la prim era ley considerando los trabajos posibles
∆E = ΣEI = ∆Es= q + wexp + wútil
Donde wútil es cualquier trabajo que no sea de expansión contra la presión
atmosférica
Función de estado
Es aquella cuyo resultado no dependa del camino seguido sino del estado final y
del inicial del sistema en estudio.
Para aclarar vamos a preguntarnos si la Energía Interna es una función de estado y
evaluaremos dos sistemas sencillos:
T(i) = 20°C
T(f) = 21°C
T(i) = 20°C
T(f) = 21°C
q = 999,43cal
w =0
q=0
w = +4181,6 J = +999,43 cal
∆E = q + (-)w = 999,43 cal ∆E = 0 + 999,43 cal = 999,43 cal
De aquí surgen dos hechos inm ediatos:
a. se constata que E (energía interna) es una FUNCIÓN DE ESTADO .
b. q (calor) en un caso tiene un valor finito y en el otro vale cero por que los
caminos son distintos, y lo mismo ocurre con w (trabajo). Luego esto ayuda a
entender que el calor y el trabajo dependen necesariam ente del CAMINO que se
sigue y por lo tanto NO SON FUNCIONES DE ESTADO .
Pensemos ahora una situación intermedia donde primero llevamos el sistema de
Ti= 20ºC a Tf= 20,5ºC
Laboratorio de Procesos Químicos y Control
Página 66
poniéndolo en contacto con un baño que está a esa temperatura y luego se agita
mecánicamente para que la T pase de
20,5ºC a 21ºC.
Es evidente que los valores de "q" y "w" resultantes serán distintos de los
planteados arriba pero el valor de E seguirá siendo igual a 999,43 cal.
FINALMENTE: LOS PASOS PODRÁN SER CUALESQUIERA PERO EL
RESULTADO SERÁ EL MISMO Y "q" y "w" NO SON FUNCIÓN DE ESTADO,
DEPENDIENDO SIEMPRE DEL CAMINO QUE SE RECORRE.
En termodinámica un PROCESO REVERSIBLE es el análogo de un movimiento
sin frotación en mecánica, luego ambos son IDEALES:
UN PROCESO TERMODINAMICAMENTE REVERSIBLE ES AQUEL QUE SE
REALIZA EN FORM AINFINITAMENTE LENTA, DE MODO QUE EL SISTEMA
SE EN CUENTRE SIEMPRE EN EQUILIBRIO CON EL ENTORNO O MEDIO
AMBIENTE.
El calor en las transformaciones isobáricas
Una transform ación isobárica es aquella que se verifica a P = cte. Podemos
escribir entonces si sólo hay trabajo de expansión (o compresión):
∆E = Qp+w= Q+(-p ∆V)= Qp+[- p (Vf- Vi)]
Analicemos término por término:
E
es independiente del camino
p
es independiente del camino
Vf es independiente del camino
Vi es independiente del camino
Luego ∆V será también independiente del camino
DEBEMOS CONCLUIR QUE Qp EN ESTE CASO SERÁ TAMBIÉN FUNCIÓN DE
ESTADO
Hemos hallado entonces un calor que es FUNCIÓN DE ESTADO y esto ocurre
porque se definió el camino (P = cte).
Para gases
P . ∆V = P.VB - P.VA
Pero como:
P.VB = nB.R.T
y que
P.VA = nA.R.T
podemos reemplazar y queda:
P *(VB- VA) = (nB- nA) R.T = ∆n R.T
Laboratorio de Procesos Químicos y Control
Página 67
por lo tanto
∆E = Qp + ∆n R.T
donde P y T son constantes.
ENTALPIA
Sabemos que:
∆E= Qp+[-(P∆
∆V)]= Qp+[-(PVB- PVA)]= EB- EA
Qp = (EB + PVB) - (EA + PVA)
Todos estos términos dependen del estado del sistema y no del camino.
E + P.V = CONTENIDO DE CALOR A PRESIÓN CONSTANTE, TAMBIEN ES
LLAMADO ENTALPIA
LUEGO DEFINIMOS ENTALPIA (H) COMO LA SUMA DE LA ENERGÍA INTERNA
MAS LA ENERGÍA DE VOLUMEN EN UN INSTANTE DEFINIDO
∆H = ∆E + [- P.∆
∆V]
y
Qp = HB – HA = ∆H
Si la transform ación es a presión constante qp = ∆H pero si NO ES a P=cte el
calor q (calor en esa transformación) no es función de estado y es diferente del
cambio de entalpía, mientras que H y por supuesto ∆H sigue siendo función de
estado y por lo tanto su valor es igual.
Como la entalpía depende del valor de E, P y V en un determinado momento y
como la E es difícil de calcular (según lo hemos explicado antes) el valor real de H
no se conoce (obviamente tampoco el de E) y sólo nos queda calcular variaciones
(∆H ). Observe ésta ecuación:
a P= cte.
∆E = QP + w
o tam bién
∆E = ∆H + (-P ∆V)
luego
∆H = ∆E + P ∆V
Transformaciones isócoras:
Cuando
Laboratorio de Procesos Químicos y Control
Página 68
∆V = 0
se encuentra que
∆E = Qv
y por lo tanto no depende del camino, siendo:
∆Ev = ∆Hv
TERMOQUIMICA
La mayoría de los procesos químicos ocurren a presión constante, normalmente la
atmosférica.
En este caso, como p = cte, se cumple que W = – p · ∆V (el signo negativo se debe
al criterio de signos adoptado). Si ∆V > 0 el sistema realiza un trabajo hacia el
entorno y en consecuencia pierde energía.
Como sabemos entalpía “H” = “E + p x V” de manera que:
H1 = E1 + p x V 1
H2 = E2 + p x V2
Con lo que queda: Qp + H1 = H2
∆H = Qp
H es una función de estado
Reac. endotérmica
Productos
Reac. exotérmica
Reactivos
∆H < 0
∆H > 0
Reactivos
Productos
Ejemplo:
Laboratorio de Procesos Químicos y Control
Página 69
Determinar la variación de energía interna para el proceso de combustión de 1 mol
de propano a 25ºC y 1 atm, si la variación de entalpía, en estas condiciones, vale
-2219,8 kJ.
C3H8(g) + 5 O2(g) = 3 CO2(g) + 4 H2O(l)
∆H = –2219,8 kJ
nreactivos = 1 + 5 = 6 ; nproductos = 3 (sólo moles de gases) ⇒ ∆n = -3
∆Ε = ∆H – ∆n x R x T = –2219 kJ + 3 mol x (8,3 Jxmol–1xK–1) x 298 K =
Entalpía estándar de la reacción
Se llama entalpía de reacción al incremento entálpico de una reacción en la cual,
tanto reactivos como productos están en condiciones estándar (p = 1 atm; T = 298
K = 25 ºC; concentración de sustancias disueltas = 1 M).
Se expresa como ∆H0 y como se mide en J o kJ depende de cómo se ajuste la
reacción.
Ecuaciones termoquímicas
Expresan tanto los reactivos como los productos indicando entre paréntesis su
estado físico, y a continuación la variación energética expresada como ∆H
(habitualmente como ∆H0).
Ejemplos:
CH4(g) + 2 O2(g) → CO2(g) + 2 H2O(l);
H2(g) + ½ O2(g) → H2O(g);
∆H0 = –890 kJ
∆H0 = –241,4 kJ
¡CUIDADO!: ∆H depende del número de moles que se forman o producen. Por
tanto, si se ajusta poniendo coeficientes dobles, habrá que multiplicar ∆H0 por 2:
2 H2(g) + O2(g) → 2 H2O(g)
∆H0 = 2 x (–241,4 kJ)
Con frecuencia, suelen usarse coeficientes fraccionarios para ajustar las
ecuaciones:
H2(g) + ½ O2(g) → H2O(g) ;
∆H0 = –241,4 kJ
Laboratorio de Procesos Químicos y Control
Página 70
Entalpía estándar de FORMACIÓN (calor de formación).
Es el incremento entálpico (∆H) que se produce en la reacción de formación de un
mol de un determinado compuesto a partir de los elementos en el estado físico
normal (en condiciones estándar).
Se expresa como ∆Hf0. Se trata de un “calor molar”, es decir, el cociente entre ∆H0
y el número de moles formados de producto. Por tanto, se mide en kJ/mol.
Ejemplos:
C(s) + O2(g) → CO2(g)
∆Hf0 = – 393,13 kJ/mol
H2(g) + ½ O2(g) → H2O(l)
∆Hf0 = – 285,8 kJ/mol
Cálculo de ∆H0 (calor de reacción) a partir de ∆H0f.
Recuerda que ∆Hf0 de todos los elementos en estado original es 0.
Ejemplo:
Conocidas las entalpías estándar de formación del butano (C4H10), agua líquida y
CO2, cuyos valores son respectivamente -124,7, -285,8 y -393,5 kJ/mol, calcular la
entalpía estándar de combustión del butano (entalpía molar).
La reacción de combustión del butano es:
C4H10(g) +13/2O2(g) → 4 CO2(g) + 5H2O(l) ; ∆H0= ?
∆H0 = Σ np∆Hf0(productos) – Σ nr∆Hf0(reactivos) =
4 mol(–393,5 kJ/mol)+5 mol(–285,8 kJ/mol)–1 mol(–24,7 kJ/mol) =–2878,3 kJ
Ley de Hess.
"∆
∆H” en una reacción química es constante con independencia de que la
reacción se produzca en una o más etapas.
Recuerda que H es función de estado. Por tanto, si una ecuación química se puede
expresar como combinación lineal de otras, podremos igualmente calcular ∆H de la
reacción global combinando los ∆H de cada una de las reacciones.
Ejemplo:
Laboratorio de Procesos Químicos y Control
Página 71
Dadas las reacciones:
(1) H2(g) + ½ O2(g) → H2O(g) ∆H10 = –241,8 kJ
(2) H2(g) + ½ O2(g) → H2O(l) ∆H20 = –285,8 kJ
calcular la entalpía de vaporización del agua en condiciones estándar.
La reacción de vaporización es:
(3) H2O(l) → H2O(g)
∆H03 = ?
H2(g) + ½ O2(g)
H
(3) puede expresarse como (1)–(2), luego
puesto que hay que dividir ∆H0 entre en número
de moles de agua vaporizados. ∆H30 = 44 kJ
∆H10 = – 241’8 kJ
∆H20 = – 285’8 kJ
H2O(g)
∆H30 = 44 kJ
H2O(l)
Ejercicio B:
Esquema de la ley de Hess
Calcular la entalpía estándar de combustión del butano (entalpía molar) de la
reacción anterior, conocidas las entalpías estándar de formación del butano
(C4H10), agua líquida y CO2, cuyos valores son respectivamente -124,7, -285,8 y
-393,5 kJ/mol.
Ejercicio C:
Determinar ∆Hf0 del eteno (C2H4) a partir de los calores de reacción de las
siguientes reacciones químicas:
(1) H2(g) + ½ O2(g) → H2O(l)
∆H10 = –285,8 kJ
(2) C(s) + O2(g) → CO2(g)
∆H20 = –393,13 kJ
(3) C2H4(g) + 3 O2(g) → 2 CO2(g) + 2 H2O(l)
∆H30 = –1422 kJ
Ejercicio D:
Las entalpías de combustión de la glucosa (C6H12O6) y del etanol (C2H5OH) son –
2815 kJ/mol y –1372 kJ/mol, respectivamente. Con estos datos determina la
energía intercambiada en la fermentación de un mol de glucosa, reacción en la que
se produce etanol y CO2. ¿Es exotérmica la reacción?
Energía de enlace.
Laboratorio de Procesos Químicos y Control
Página 72
“Es la energía necesaria para romper un mol de un enlace de una sustancia
en estado gaseoso”.
En el caso de moléculas diatómicas con un solo enlace, se corresponde con la
energía necesaria para disociar 1 mol de dicha sustancia en los átomos que la
constituyen.
Para moléculas poliatómicas, la energía de enlace se toma como el valor medio
necesario para romper cada uno de los enlaces iguales. Así por ejemplo, se sabe
que para romper el primer enlace H–O del H2O se precisan 495 kJ/mol mientras
que sólo se precisan 425 kJ/mol para romper el segundo, por lo que se suele tomar
el valor medio (460 kJ/mol) como energía del enlace H–O.
A—B(g) → A(g) + B(g) ; ∆H = Eenlace= Ee
Ejemplo:
H2(g) → 2 H(g) ; ∆H = 436 kJ
Es positiva (es necesario a portar energía al sistema)
Es una entalpía molar que se mide en kJ/mol.
Es difícil de medir.
Se suele calcular aplicando la ley de Hess.
Ejemplo de cálculo de energías de enlace.
Calcular la energía del enlace H-Cl en el cloruro de
hidrógeno conociendo ∆Hf0 (HCl) cuyo valor es –92,3 kJ/mol
y las entalpías de disociación (energías de enlace) del H2 y
del Cl2 de la tabla adjunta.
Enlace
H–H
C–C
C=C
C≡C
O=O
Cl–C
C–H
C–O
C=O
O–H
Cl–H
Cl–Cl
Ee (kJ/mol)
436
347
620
812
499
243
413
315
745
460
432
243
La reacción de disociación del HCl será:
(4) HCl(g) → H(g) + Cl(g)
∆H0 = ?
(1) ½ H2(g) + ½ Cl2(g) → HCl(g)
∆Hf0(HCl) = –92,3 kJ
(2) H2(g) → 2H(g)
Ee(H2) = 436,0 kJ
(3) Cl2(g) → 2Cl(g)
Ee (Cl2) = 243,4 kJ
(4) = –(1) + ½ (2) + ½ (3)
∆H0 = – (–92,3 kJ ) + ½ x(436,0 kJ) + ½ x (243,4 kJ) = 432,0 kJ
Laboratorio de Procesos Químicos y Control
Página 73
216, 0 kJ
puesto que en la reacción (4) se disocia un mol de enlaces H–Cl.
Cálculo de ∆H0 a partir de las Energía de enlace.
Aplicando la ley de Hess también puede obtenerse la energía de una reacción si
sabemos qué enlaces se tienen que romper y cuáles se tienen que formar. Para
ello utilizaremos la siguiente expresión:
∆H0 = Σ Ee(enl. rotos) – Σ Ee(enl. formados)
Ejemplo:
Partiendo de los datos de la tabla, calcular el valor de ∆H0 de la reacción de
hidrogenación del eteno.
La reacción es:
CH2=CH2(g) + H2(g) → CH3–CH3(g)
En el proceso se rompe un enlace C=C y otro H–H y se forman 2 enlaces C–H
nuevos (el etano tiene 6 mientras que el eteno tenía sólo 4) y un enlace C–C.
∆H0 = Σ Ee(enl. rotos) – Σ Ee(enl. formados) =
1xEe(C=C) + 1xEe(H–H) – 1xEe(C–C) – 2xEe(C–H)
1 mol · 611 kJ/mol + 1mol x 436 kJ/mol
– (1 mol x 347 kJ/mol – 2 mol x 413 kJ/mol) = –126 kJ
SEGUNDA LEY DE TERMODINAMICA
Introducción
Laboratorio de Procesos Químicos y Control
Página 74
En cualquier proceso la energía del universo se conserva (Primer Principio de la
Termodinámica). Por otra parte, una transformación macroscópica sólo puede
tener lugar si la entropía (grado del desorden) del universo aumenta (Segundo
Principio de la Termodinámica).
Si la S del universo permanece constante no hay un cambio macroscópico, el
sistema y los alrededores permanecen en equilibrio o cuasiequilibrio. Cuando se
habla de procesos reversibles en realidad se está haciendo la aproximación de
suponer que todos los estados intermedios del sistema a lo largo de la trayectoria
son estados de equilibrio o cuasiequilibrio.
Por tanto el análisis de cual sería la ∆Suniverso en un hipotético proceso, nos
permite conocer a priori si este va a tener lugar o no.
∆Suniverso > 0 proceso espontáneo o irreversible
∆Suniverso =0 sistema en equilibrio o proceso reversible
Pero este análisis puede resultar complicado porque hace necesario conocer
además de la ∆Ssistema también la ∆Salrededores.
∆Suniverso = ∆Ssistema + ∆Salrededores
El interés de realizar esta “predicción” es conocer si el proceso va a tener lugar,
para en el caso contrario buscar procesos acoplados que hagan factible la
obtención del sistema en el estado final buscado (ej: el agua no fluye de forma
natural de abajo a arriba, pero si acoplamos un proceso en el que una masa
superior baje, será posible que una determinada masa de agua suba)
Difusión de una gota de tinta en agua. Proceso irreversible
Laboratorio de Procesos Químicos y Control
Página 75
Difusión de un gas. Proceso irreversible
En Química, la principal aplicación del concepto de entropía es que permite
predecir cuándo un proceso químico puede tener lugar, independientemente del
tiempo que tarde en alcanzarse el equilibrio
•
De forma muy aproximada y general, podemos decir que:
o
En igualdad de condiciones P,T, las S de los gases son
mayores que las de los líquidos y estas a su vez mayores
que las de los sólidos (recordá la idea de “mayor
probabilidad”)
o
Sustancias con moléculas de tamaño similar tienen
entropías parecidas
o
En reacciones químicas que impliquen sólo gases, líquidos
puros y sólidos puros, la ∆Sº del sistema dependerá en
general de la variación del número de moles de gas (si n gas
aumenta ∆Sº >0, si n gas disminuye ∆Sº<0)
En el laboratorio se suele trabajar en condiciones aún más restrictivas: T y V
constantes (ej. reacción en una ampolla de vidrio cerrada) o T y P constante (ej.
reacción en un vaso de precipitados), lo que simplifica el estudio termodinámico de
los procesos de interés químico.
Condición General de Espontaneidad y Equilibrio
•
Si un sistema aislado no se encuentra en equilibrio, evolucionará
espontáneamente aumentando su entropía:
proceso espontáneo,
proceso irreversible
sistema aislado
Laboratorio de Procesos Químicos y Control
Página 76
La Salrededores permanece constante porque los alrededores no intervienen en el
proceso termodinámico al estar el sistema aislado
sistema en equilibrio
proceso reversible
sistema aislado
En un sistema cerrado que no se encuentre en equilibrio, los procesos
espontáneos tienen lugar de forma que:
¿Cómo se alcanza el equilibrio en un sistema cerrado?
Para evitar calcular la dSalrededores, y centrarnos sólo en lo que ocurre en el
sistema, podemos suponer que los alrededores son tan grandes que cualquier
transferencia de energía desde o hacia el sistema no modifica su temperatura T,
además como dqalrededores= -dqsistema, la condición general de espontaneidad se
puede escribir como:
Combinando este resultado con el Primer Principio (dE=dq+dw), se tiene que
Expresión válida para analizar si cualquier
sistema cerrado está en equilibrio (dE=TdS+dw),
o si va a tener lugar una transformación
espontánea en el sistema cerrado (dE<TdS+dw).
dE ≤ TdS + dW
Laboratorio de Procesos Químicos y Control
Página 77
•
Transformaciones en sistemas cerrados con temperatura y volumen
constantes
Resulta cómodo emplear en estos casos una nueva función termodinámica,
A=función de trabajo, o energía de Helmholtz, y analizar su evolución en el
proceso.
Esta función se introduce de modo muy sencillo si consideramos que
d(TS)=TdS+SdT
y por tanto:
dE ≤ TdS + dW = d(TS)- SdT + dW, y al reagrupar términos d(U –TS) ≤ - SdT + dW
es a la combinación de funciones de estado (E-TS) a la que se denomina Energía
de Helmholtz.
A ≡ E – TS
•
Por ser A una combinación de funciones de estado es también
una función de estado, y por tanto está definida para cualquier
sistema en equilibrio
•
Es una propiedad extensiva porque E y S también lo son
•
Sus unidades son de energía (Julios)
Por tanto se puede estudiar la evolución de cualquier sistema cerrado
considerando que siempre debe cumplirse que:
•
Si la T permanece constante, dT=0 y por tanto
Por esto se denomina a A función trabajo. En un proceso isotérmico,
en el que el sistema sea cerrado ∆A ≤ W. Pero si en lugar de definir el
Laboratorio de Procesos Químicos y Control
Página 78
W realizado sobre el sistema nos fijamos en el trabajo que hace el
sistema sobre los alrededores (Wpor el sistema= -W), vemos que el
trabajo que puede hacer el sistema sobre los alrededores es menor o
igual que la variación de la función A, y será máximo cuando el proceso
sea reversible WPOR EL SISTEMA≤
•
-∆A
Si además de la T, el V permanece constante, y sólo hay trabajo
mecánico P-V, dT=0 y dV=0, y por tanto dA ≤ 0
sistema
cerrado
TyV
constantes
no hay ningún
W
En un sistema cerrado que experimente un proceso espontáneo
con T y V constantes, la energía de Helmholtz disminuye hasta
alcanzar un valor mínimo en el equilibrio
Transformaciones en sistemas cerrados con temperatura y presión
constantes
En estos casos, por mayor facilidad se emplea una nueva función termodinámica,
G=función de Gibbs, o energía libre de Gibbs, y se analiza su evolución en el
proceso.
Esta función se introduce de modo muy sencillo si consideramos que d(TS)=
TdS+SdT, y d(PV)=PdV+VdP.
Si sólo hay trabajo mecánico PV,
dE≤TdS-PdV=d(TS)-SdT-d(PV)+VdP
y al reagrupar términos
d(E-TS+PV) ≤ -SdT+VdP
Laboratorio de Procesos Químicos y Control
Página 79
y es a la combinación de funciones de estado (E-TS-PV) a la que se denomina
Energía de libre de Gibbs (G).
G ≡ E- TS + PV ≡ A + PV ≡ H-TS
•
Por ser G una combinación de funciones de estado es también
una función de estado, y por tanto está definida para cualquier
sistema en equilibrio
•
Es una propiedad extensiva porque E, S y V también lo son
•
Sus unidades son de energía (Julios)
Por tanto se puede estudiar la evolución de cualquier sistema cerrado si sólo
hay trabajo PV considerando que siempre debe cumplirse que:
•
•
Si además la T permanece constante, dT=0 y por tanto
Si además de la T, la P permanece constante, y sólo hay trabajo
P-V, dP=0 y por tanto
sistema
cerrado
TyP
constantes
sólo hay
trabajo
mecánico
PV
En un sistema cerrado que experimente un proceso espontáneo
con T y P constantes, si sólo hay trabajo mecánico PV, la energía
Laboratorio de Procesos Químicos y Control
Página 80
de Gibbs disminuye hasta alcanzar un valor mínimo en el
equilibrio
•
Si el sistema es cerrado, pero se puede llevar a cabo W que no
sea PV , como G=A+PV, dG=dA+PdV+VdP, y como
se tiene que
Y si P y T son constantes
Luego
Por esto se denomina a G energía libre (para
realizar un trabajo). En un proceso isotérmico e isobárico, en el que el
sistema sea cerrado
. Pero si en lugar de definir el W
realizado sobre el sistema nos fijamos en el trabajo que hace el sistema
sobre los alrededores (W por el sistema= -W), vemos que el trabajo que
puede hacer el sistema sobre los alrededores es menor o igual que la
variación de la función G, y será máximo cuando el proceso sea
reversible
¿Que relación existe entre la condición SUniverso máxima y Gsistema mínima, en
un proceso reversible que ocurra a T y P constantes, para un sistema
cerrado?
sistema cerrado, P y T constantes, W no PV=0
Importante
Se puede emplear cualquier criterio para determinar si un sistema cerrado está en
equilibrio, pero el más frecuente es dG porque la mayor parte de los procesos
químicos ocurren a P y T constantes
Laboratorio de Procesos Químicos y Control
Página 81
Criterios de Espontaneidad o Equilibrio para procesos en Sistemas
Cerrados
Restricción
Proceso Espontáneo
Condición de Equilibrio
sistema aislado
dS>0
dS=0
dE<TdS+dw
ninguna
T constante
dE=TdS+dw
dE-TdS+pdV+dwpor sistema, no
dE-TdS+pdV+dwpor sistema, no PV =0
PV <0
dA<dw
dA=dw
T y V constantes
W no PV=0
dA<0
dA=0
T y P constantes
W no PV=0
dG<0
dG=0
Tercer Principio de la Termodinámica
“La entropía de un elemento puro en su forma condensada estable (sólido o
líquido) es cero cuando la temperatura tiende a cero y la presión es de 1 bar”
Y según el Tercer Principio: “En cualquier proceso isotérmico que implique
sustancias puras, cada una en equilibrio interno, la variación de entropía
tiende a cero cuando la temperatura tiende a cero”
•
El Tercer Principio permite conocer la entropía de cualquier sustancia en el
límite de 0 K (imaginando una reacción química, a P=1bar, entre elementos a
T=0K, se obtendría cualquier compuesto a T=0K, y P=1bar y su S sería 0 J/K).
Laboratorio de Procesos Químicos y Control
Página 82
Este postulado implica que S (al igual que V y N, pero al contrario que E) tiene un
único cero definidos. Este postulado es una extensión, debida a Planck, del
llamado postulado de Nernst o tercera ley de la termodinámica. Históricamente,
fue el último de los postulados desarrollados, siendo inconsistente con la mecánica
estadística clásica, requiriendo el establecimiento anterior de la estadística cuántica
para que éste pudiera ser adecuadamente apreciado. La mayor parte de la
termodinámica no requiere este postulado.
Laboratorio de Procesos Químicos y Control
Página 83
Termodinámica del Infierno
Que tan caliente es el Infierno?
El profesor de termodinámica, entregó a sus alumnos un examen escrito que estos
deberían terminar en sus casas, esta es una de las preguntas:
Si el Infierno ¿es exotérmico (pierde calor) o es endotérmico (absorve calor) ?.
Explique el porqué de su respuesta.
La mayoría de los estudiantes escribió alguna expliación usando la ley de Boyle
(un gas se enfría cuando se expande y se calienta cuando se contrae) o alguna
variante. En cambio un estudiante escribío lo siguiente:
Primero tenemos que notar que la masa del infierno está cambiando con el
tiempo. Tenemos, por lo tanto que conocer la taza de almas que caen en él.
También debemos asumir que cuando un alma cae en él, ya no escapa jamás. Por
lo tanto, no hay almas saliendo. Para saber cuantas almas estan entrando, tenemos
primero que estudiar a las diferentes religiones que existen en el mundo hoy. La
mayoría de las religiones establecen que si uno pertenece a otra religión
precisamente se irá al infierno. Como hay muchas de estas religiones y cada
persona solo pertence a una religión podemos proyectar que todas las almas de
todas las personas terminarán en el infierno. Por las tasas de natalidad y
mortalidad actuales podemos establecer que la cantidad de almas aumenta en el
infierno en forma exponencial. Entonces si recordamos la ley de Boyle y como
esta relaciona la presión con la temperatura para que esta última permanezca
constante, el volumen del infierno tendrá que aumentar a medida que mas almas
ingresan.
Esto nos da dos posiblilidades:
1.- Si el infierno se expande a una velocidad menor que la taza a la cual las almas
entran al infierno, entonces la temperatura y la presión del infierno aumentará
hasta que este estalle.
2.- Por supuesto si el tamaño del infierno crece más rápido que la taza de ingreso
de almas en él, la presión y la temperatura bajarán hasta que se congele por
completo.
Pero cuál de las dos opciones sucede ?. Si acepto lo que me dijo la senorita
Ms.Therese Banyan durante mi primer año aquí: "Se congelará primero el infierno
antes que yo me acueste contigo" y teniendo en cuenta de he logrado tener
relaciones sexuales con ella, la opción #2 no puede ser cierta, entonces por lo
tanto el infierno es exotérmico.
El estudiante sacó un 10!!!
Laboratorio de Procesos Químicos y Control
Página 84
ESCUELA DE EDUCACIÓN TÉCNICA Nº 3 – SAN JOSÉ
DEPARTAMENTO QUÍMICA
TRABAJO PRACTICO PROPIEDADES COLIGATIVAS
PARTE EXPERIMENTAL
Fundamento del trabajo práctico
En esta experiencia se determinará la masa molar del azufre.
En primera instancia se mide el punto de solidificación del solvente (naftaleno)
puro.
Luego, se disuelve una determinada masa de azufre en una determinada cantidad
de naftaleno. Experimentalmente se mide el punto de solidificación de la solución
formada.
Como kc es dato, a partir del valor experimental hallado para ÄT se calculará m
(molalidad) y por definición, molalidad es:
m = 1000 . g2/ g1 . M 2
g2 = masa de soluto (azufre).
g1 = masa de solvente (naftaleno).
M 2 = masa molar del azufre, es decir:
M 2 = 1000 . g2 / m . g1
Dado que g2, g1 y m se conocen, se puede calcular M 2
Por lo tanto la atomicidad del azufre será:
Atomicidad = M 2 / Ar2 = M 2 / 32,06
Reactivos y materiales necesarios:
Azufre y naftaleno.
Termómetro 0-100°C, graduado a no menos de 0,2 °C.
Tubo de ensayos tipo Pyrex de 12 cm de largo y 3 cm de diámetro aprox.
Tapón de goma de doble orificio.
Agitador de alambre.
Vaso de precipitados de 500 cm3
Mechero Bunsen.
Trípode, tela metálica, pinza para matraces, soporte de hierro y nuez.
EXPERIENCIA: Determinación de la atomicidad del azufre a partir del descenso
del punto de solidificación.
1. Armar el aparato necesario para la experiencia, y considerar que la escala del
termómetro comprendida en el rango de 70-100°C, que de ¡perfectamente visible!
2. Pesar aproximadamente 5 g de naftaleno con una aproximación 0,1 g. Verter
Laboratorio de Procesos Químicos y Control
Página 85
con sumo cuidado la masa de naftaleno en el tubo.
3. Colocar el tubo en un baño de agua caliente hasta que el naftaleno funda
totalmente (controlar que el nivel de agua quede por encima del nivel del
naftaleno contenido en el interior del tubo) .
4. Luego de observar la fusión, retirar el tubo del baño y dejar que el naftaleno se
enfríe gradualmente, mientras se agita continuamente.
5. Leer la temperatura cada 15 segundos, comenzando alrededor de los 85°C.
6. Observar el inicio de la cristalización y medir la temperatura a los intervalos
preestablecidos, hasta que el naftaleno solidifique.
7. Colocar nuevamente el mechero bajo el vaso de precipitados y ajustar la llama
de manera tal que conserve la temperatura del baño María caliente.
8. Pesar aproximadamente 0,5 g de azufre finamente pulverizado (la presencia de
partículas grandes dificulta la disolución posterior del azufre).
9. Cuando el naftaleno este completamente fundido, quitar con precaución el
conjunto tapón - termómetro - agitador, y cuidadosamente verter todo el azufre
en el naftaleno fundido.
10. Colocar nuevamente el conjunto tapón – termómetro - agitador y agitar
vigorosamente hasta que el azufre se haya disuelto. Esta operación se realiza
rápida y fácilmente si el azufre usado está finamente pulverizado, de lo contrario,
puede resultar dificultosa.
11. Una vez lograda la disolución del azufre por completo, retirar el tubo del baño.
Con agitación continua medir la temperatura, a partir de 83°C, a intervalos de 15
segundos hasta que aparezcan los primeros cristales de naftaleno y que la
solución quede totalmente solidificada.
LIMPIEZA DEL DISPOSITIVO EMPLEADO.
Para proceder al limpieza del aparato, colocar el tubo con tapón incluido en el baño
de agua
caliente nuevamente hasta lograr la fusión de la solución.
Quitar con cuidado el conjunto tapón – termómetro - agitador y volcar la solución
fundida
sobre un papel. ¡¡No arrojar la solución fundida a la pileta del laboratorio!!
Para eliminar los restos de solución adheridos al tubo, termómetro, etc., colocar
éstos en un
baño de agua a 90°C. No calentar directamente; en p rimer lugar, tener en cuenta
que los vapores
de naftaleno son inflamables; en segundo lugar, recordar que si se eleva la
temperatura del
termómetro por encima de los 100°C, este se rompe.
Es aconsejable acudir al laboratorio con un poco de alcohol y “virulana” para
facilitar la
limpieza del tubo.
Laboratorio de Procesos Químicos y Control
Página 86
INFORME TP PROPIEDADES COLIGATIVAS
Objetivo: determinación de la masa molar a partir del descenso del punto de
solidificación.
Datos obtenidos:
Masa de naftaleno (g1) = ................................
Masa de azufre (g2) = ...................................
Temperatura de solidificación del naftaleno (t0) ..............................
Temperatura de fusión de la solución (t) = ...............................
Descenso del punto de solidificación ( t0 – t) = .............................
Molalidad de la solución ( Ät / kc ) = ...........................
Cálculos:
Masa molar del azufre = 1000 . g2 / m . g1 = .................................
Masa atómica del azufre ( tabla periódica ) = .........................
Atomicidad del azufre en solución = a = M 2 / A 2 = .............................
Error absoluto = a - 8 = ............................
Error relativo porcentual = ( Error absoluto / 8 ) . 100 = ..........................
ACTIVIDADES DE APLICACIÓN
1. En un mismo par de coordenadas, graficar la curva de enfriamiento
correspondiente al naftaleno y a la solución. Adjuntarlo al informe.
2. Calcular el número de átomos de azufre presentes en un mol del mismo.
3. Mencionar otro método similar al empleado en la práctica que permita determinar
la masamolar de un soluto en solución diluida. Fundamentar.
4. Resolver los siguientes problemas:
a) Cuando se disuelve 15,0 g de etanol (C2H5OH) en 750 g de ácido fórmico,
el punto de congelamiento de la solución es 7,20°C. Sabiendo que el punto
de congelamiento del ácido fórmico es 8,40°C, calcular kc para el ácido
fórmico. Rta: 2,76 °C/m
b) ¿Cuál es el punto de ebullición normal de una solución de sacarosa (
C12H22O11) 1,25 m sabiendo que ke del agua pura es 0,512 °C/mol? Rta:
100,64°C.
Laboratorio de Procesos Químicos y Control
Página 87
ESCUELA DE EDUCACIÓN TÉCNICA Nº 3 – SAN JOSÉ
DEPARTAMENTO QUÍMICA
TRABAJO PRACTICO TERMOQUIMICA
ENTALPIA DE FORMACION DE UN OXIDO. LEY DE HEES
FUNDAMENTOS TEÓRICOS
El calor disipado en una reacción exotérmica en disolución, que se lleva a
cabo en un calorímetro, calienta la disolución de ti a tf a la vez que calienta el
calorímetro de ti a tf. Esto se representa mediante la ecuación:
qreacción + qsolución + qcalorímetro = 0
[1]
El valor de qsolución se calcula multiplicando el calor específico y la
masa del disolvente (agua) por el cambio de temperatura según la expresión:
qsolución = m (gr) x
4.18 (J/ gr ºC) x ∆t
[2]
El valor de qcalorímetro se determina midiendo el incremento de
temperatura que tiene lugar al mezclar agua fría y agua caliente, y calculando la
diferencia entre el calor cedido por el agua caliente y el calor ganado por el agua
fría.. Los calores cedido y ganado se calculan según las fórmulas:
qcedido= m H2O caliente (gr) x 4.18 (J/gr ºC) x (t H2O,caliente -tf)
qganado= m H2O fría (gr) x 4.18 (J/gr ºC) x (tf - t H2O, fría)
[3]
[4]
Evidentemente, la diferencia entre qcedido y qganado es el calor ganado por el
calorímetro y el termómetro. De este modo, suponiendo que las temperaturas
inicial y final del calorímetro soin iguales a las del aqua fría se obtiene.
Laboratorio de Procesos Químicos y Control
Página 88
qcalorímetro = qcedido -qganado = K (tf -t H2O, fría)
[5]
dónde K es la capacidad calorífica del calorímetro o constante calorimétrica.
La ecuación para el calor de reacción resulta:
qreacción = - [ m (gr) x
4.18 (J/ gr ºC) x ∆t ] - K ∆t [6]
La reacción desprende calor y por definición qreacción es una cantidad
negativa. El valor qreacción es la cantidad de calor desprendido en la reacción
llevada a cabo en el calorímetro. Para calcular el valor ∆Hreacción necesitamos
conocer el número de moles que han reaccionado. Para ello se calcula el número
de moles de reactivo (metal u óxido metálico) asegurándonos que se trata del
reactante limitante e ∆Hreacción viene dado por la expresión:
∆Hreacción = qreacción / moles de reactivo
[7]
Las reacciones estudiadas en este experimento son:
M (s)
+
2 HCl (ac)
------>
MCl2
+ H2 (g)
∆H1
[8]
MO (s)
+
2 HCl (ac)
------->
MCl2
+ H2O (l)
∆H2
[9]
dónde M = Ca o Mg.
Por otra parte, el calor de formación del agua líquida es:
H2(g)
+
0.5 O2(g) ----->
∆Hf, agua
H2O(l)
[10]
Los datos experimentales obtenidos para las ecuaciones anteriores se
utilizan para calcular el calor de formación del CaO (s) y del MgO(s). La ley de
Hess nos permite combinar algebraicamente las reacciones [8, [9] y [10] para
obtener el calor de formación del MO (s) de la siguiente forma:
M (s)
+
2 HCl (ac)
------>
+
{H2(g)
+
0.5 O2(g)
----->
H2O(l)
-
{MO (s) + 2 HCl (ac)
------>
MCl2 + H2O (l)
=
M (s)
+ 0.5 O2 (g)
MCl2 (s)
------>
MO (s)
∆H1
+ H2 (g)
∆Hf, agua }
∆Hf, MO
∆H2}
[11]
OBJETIVO
Laboratorio de Procesos Químicos y Control
Página 89
El objeto de esta práctica es la determinación del calor de formación de un
óxido metálico mediante medidas calorimétricas de los calores de reacción del
metal y el óxido metálico en soluciones acuosas de ácido clorhídrico.
PARTE EXPERIMENTAL
MATERIAL
1 Termómetro de precisión 0.1 grados
1 Calorímetro
1 Agitador magnético
1 Probeta de 250 cm3
1 Probeta de 100 cm3
1 Frasco lavador
1 Termómetro de precisión 1 grado
1 Erlenmeyer de 250 cm3
1/2 Placa calefactora
2 Vasos de precipitados de 250 cm3
1 Matraz aforado de 500 cm3
1 Vidrio de reloj
PRODUCTOS
HCl 0.5 M
Mg (metal)
MgO (s)
PROCEDIMIENTO
Parte A.
Determinación de la constante calorimétrica
1- Se sitúa el calorímetro, limpio y seco, sobre un agitador magnético
colocando la barrita agitadora en su interior con objeto de mantener la disolución
bien agitada durante el experimento.
2- Se introducen 90 mL de agua a temperatura ambiente en el calorímetro
y se cierra colocando el termómetro de modo que quede inmerso en la disolución
pero sin tocar la barrita agitadora.
3- Se conecta el agitador y se lee la temperatura transcurridos 5 minutos
desde la adición de agua.
Laboratorio de Procesos Químicos y Control
Página 90
4- Se calienta agua (aproximadamente 100 mL) en un vaso de precipitados
a una temperatura unos 10 ºC superior a la temperatura del calorímetro y se anota
la temperatura exacta del agua.
5- Se añaden 90 mL de agua "caliente" al calorímetro, se cierra y se anota
la máxima temperatura que tiene lugar.
Parte B.
Determinación del cambio de temperatura para la reacción entre un
metal y ácido clorhídrico.
1- Se sitúa el calorímetro, limpio y seco,
sobre un agitador magnético
colocando la barrita agitadora en su interior con objeto de mantener la disolución
bien agitada durante el experimento.
2- Se preparan 500 mL de HCl 0.5 M (utilizando la probeta para medir el
HCl), se añaden 180 mL en el calorímetro y se cierra colocando el termómetro de
modo que quede inmerso en la disolución pero sin tocar la barrita agitadora.
3- Se conecta el agitador y se observa la temperatura a intervalos de 30
segundos durante varios minutos para establecer la temperatura inicial de la
disolución ácida.
4- Se añade cuidadosamente una muestra previamente pesada del metal
(0.2 gr) a la vez que se anota el tiempo asegurándose que la muestra cae
directamente en el ácido.
5- Se coloca la tapa del calorímetro y se mide la temperatura a los 30
segundos de haber añadido el metal y se continúa tomando medidas a intervalos
de 30 segundos hasta 10 minutos después de haber alcanzado el máximo de
temperatura. Esto permite establecer la velocidad a la que el calor fluye del
calorímetro de modo que podamos corregir la temperatura final considerando la
velocidad del calor disipado.
Nota: Si la temperatura permanece constante durante 5 minutos podemos utilizar
este valor para tf. Por el contrario, si hay un ligero descenso de la temperatura
Laboratorio de Procesos Químicos y Control
Página 91
respecto al valor máximo, se obtendra el valor de tf extrapolando la tendencia de
enfriamiento al valor del tiempo en que se han mezclado los reactivos.
Parte C.
Determinación del cambio de temperatura para la reacción entre un
óxido metálico y ácido clorhídrico.
Se sigue un procedimiento similar al descrito en el apartado anterior pero
sustituyendo la adición de metal (punto 4) por la adición de 0.9 gramos de óxido
metálico (MgO)
CÁLCULOS
Se calcula el calor de formación del óxido metálico con su error
correspondiente. Este valor se compara con el valor reportado en la bibliografía y
se analizan las posibles discrepancias junto con las causas de las mismas.
Laboratorio de Procesos Químicos y Control
Página 92
ESCUELA DE EDUCACIÓN TÉCNICA Nº 3 – SAN JOSÉ
DEPARTAMENTO QUÍMICA
TRABAJO PRÁCTICO PROPIEDADES DE LOS LIQUIDOS
VISCOSIDAD
FUNDAMENTO TEÓRICO
El estudio de los fluidos en movimiento es un problema complejo y en el que la viscosidad
juega siempre un papel fundamental, aunque las teorías más elementales ignoran sus
efectos, suponiendo que el líquido se puede dividir en capas se deslizan unas sobre las otras
sin encontrar ninguna resistencia. En realidad esto dista mucho de ser verdad, y en el
movimiento se desarrollan unas fuerzas tangenciales tan grandes que algunas veces éste se
lleva a cabo con gran dificultad. Esto sucede por ejemplo con aceites muy pesados. Por el
contrario, otras veces estas fuerzas son muy pequeñas y el líquido fluye entonces fácilmente
como sucede con el agua o el alcohol. Este “grado de fluidez” se caracteriza por un
coeficiente típico de cada sustancia que se llama coeficiente de viscosidad o viscosidad
dinámica. Un sólido amorfo no es en realidad más que un líquido cuya viscosidad dinámica
es enormemente grande.
Fue Newton el que dio la expresión de la fuerza de viscosidad: “La fuerza tangencial que
una capa ejerce sobre la contigua es proporcional al área de la superficie de contacto y al
gradiente de la velocidad normal a la dirección de deslizamiento”.
donde η es el coeficiente de viscosidad. Su unidad en el sistema SI es el stokes, aunque es
corriente también utilizar la unidad de viscosidad en el antiguo sistema CGS. Esta unidad
se llama poise (1 stokes = 10 poise). La viscosidad cinemática σ es el cociente entre la
viscosidad dinámica y la densidad.
El fundamento de la mayor parte de los viscosímetros que se utilizan en la práctica es la
fórmula de Poiseuille (ver el Apéndice), que nos da el caudal Q (volumen de fluido por
unidad de tiempo) que atraviesa un capilar de radio R y longitud l entre cuyos extremos se
ha aplicado una diferencia de presiones .p
Laboratorio de Procesos Químicos y Control
Página 93
donde es la viscosidad del fluido. Esto es
Como R, l, V son constantes para un tubo determinado, haciendo
Si el líquido fluye únicamente por acción de la gravedad en un tubo situado verticalmente,
la presión ∆p es la que ejerce la columna de líquido, esto es,∆.p=ρgh, siendo ρ la densidad
del liquido y h la altura de la columna. Por tanto
El valor de K´ depende por tanto de la geometría de cada viscosímetro en concreto y suele
darlo el constructor, aunque puede determinarse utilizando un líquido de viscosidad
conocida. Normalmente se determinan las viscosidades relativas referidas al agua. Para el
agua se tendrá:
Para otro líquido cualquiera:
La viscosidad dinámica será:
Así pues, podemos conocer la viscosidad dinámica de un líquido midiendo su densidad y la
razón entre los tiempos que tarda en fluir el mismo volumen de líquido y de agua. La
Laboratorio de Procesos Químicos y Control
Página 94
viscosidad del agua debe buscarse en las tablas en que aparece su variación con la
temperatura. A continuación se dan los valores de la viscosidad del agua para varias
temperaturas. Vamos a considerar 20°C como temperatura ambiente.
El viscosímetro de Ostwald permite un cálculo rápido (aunque no de máxima precisión) de
la viscosidad relativa de un líquido midiendo los tiempos que un mismo volumen de dos
líquidos tarda en pasar entre las marca M1 y M2 (ver figura 2)
Figura 1. Viscosímetro de Ostwald
El viscosímetro de Ostwald está formado por un capilar unido por su parte inferior a una
ampolla L y por su parte superior a otra ampolla S. Se llena la ampolla inferior L de agua
introduciéndola por A. Se aspira por la rama B hasta que el nivel del agua sobrepase la
ampolla superior procurando que no queden burbujas de aire.
Se deja caer el agua y se cuenta el tiempo que tarda en pasar entre los niveles M1 y M2. Se
Laboratorio de Procesos Químicos y Control
Página 95
repite esta operación varias veces y se calcula el valor medio de los tiempos, t.
A continuación se procede de manera análoga con el líquido cuya viscosidad se desea
conocer, obteniéndose el valor medio t´. Una vez obtenidos los tiempos se calcula el valor
de la viscosidad dinámica (ecuación 5).
Cuando se comience a trabajar tanto con el líquido como con el agua el viscosímetro debe
estar limpio y seco.
CUESTIONES DE LA PRÁCTICA 4
1. Preparar tres disoluciones de 100 ml de azúcar (sacarosa, peso molecular 342.3 g/mol)
de aproximadamente 5, 10 y 15 por ciento en peso. Estimamos la cantidad de azúcar
necesaria, la preparamos y pesamos, dando finalmente la concentración exacta
(utilizando la lectura de la balanza) en molaridad y en tanto por ciento en peso para
cada una de las disoluciones. Para pesar el azúcar se van a utilizar las balanzas de
precisión. Para hacer las disoluciones se enrasará en los matraces aforados,
disolviendo previamente el azúcar en un vaso de precipitado con una cantidad de agua
menor al volumen del matraz aforado que se vaya a utilizar.
2. Calcular la densidad del agua pura y de cada disolución de azúcar pesando 10 ml de
cada una de las muestras. Se tomarán los 10 ml con la pipeta aforada y se verterán
sobre un vaso de precipitado pequeño puesto en la plataforma de la balanza de precisión
(“tarar” la escala de la balanza a cero antes de verter sobre el vaso). Con los valores
obtenidos de densidad y concentración molar hacer la recta de calibrado ajustando los
cuatro puntos a una recta por mínimos cuadrados.
3. Medir la viscosidad dinámica del agua pura y de cada una de las disoluciones con el
viscosímetro a temperatura ambiente (20°C). Representar gráficamente la viscosidad
dinámica y la viscosidad cinemática frente a la concentración molar de azúcar.
4. Medir la viscosidad dinámica del agua pura a 40 y 50 oC. Para ello se introduce el
viscosímetro en el baño termostatizado y se esperan dos minutos antes de realizar las
medidas. Los valores de viscosidad obtenidos se utilizan para ampliar la tabla de
viscosidad del agua en función de la temperatura (página 3). Suponer para ello que la
densidad del agua no varía entre 30 y 50 °C. Representar todos los valores gráficamente.
5. Tomar muestras de una disolución problema de concentración desconocida (podría
provenir de un río o del plasma sanguíneo de un ser vivo, por ejemplo). Realizar las
siguientes experiencias:
a) Medir la densidad de la muestra problema y utilizar la recta de calibrado (la obtenida
mediante mínimos cuadrados) para calcular la concentración de azúcar de dicha muestra.
b) Medir la viscosidad dinámica de la muestra problema a temperatura ambiente y utilizar la
gráfica de viscosidad frente a concentración para calcular, esta vez por interpolación lineal,
la concentración de la muestra.
Comparar con el resultado obtenido a partir de la densidad.
Laboratorio de Procesos Químicos y Control
Página 96