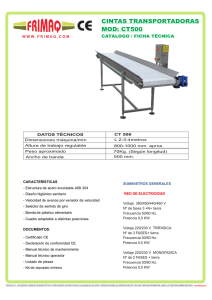
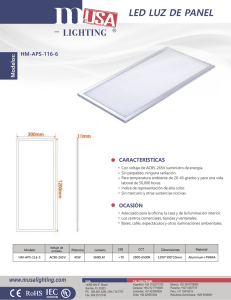
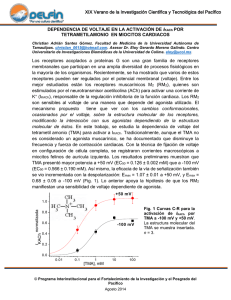
Tesis - Universidad de Colima
Anuncio

UNIVERSIDAD DE COLIMA FACULTAD DE MEDICINA CENTRO UNIVERSITARIO DE INVESTIGACIONES BIOMÉDICAS “CARACTERIZACIÓN DE LA INTERACCIÓN VOLTAJEDEPENDIENTE DE AGONISTAS MUSCARÍNICOS SOBRE EL RECEPTOR MUSCARÍNICO M2 DE CORAZÓN” TESIS QUE PARA OBTENER EL GRADO DE DOCTOR EN CIENCIAS FISIOLÓGICAS PRESENTA M. en C. ELOY GERARDO MORENO GALINDO DIRECTORES DE TESIS DR. RICARDO ANTONIO NAVARRO POLANCO DR. JOSÉ ANTONIO SÁNCHEZ CHAPULA COASESOR DR. IVÁN DELGADO ENCISO COLIMA, COL.; JULIO DE 2008 AGRADECIMIENTOS: A los Drs. Ricardo A. Navarro Polanco y José A. Sánchez Chapula, por haberme permitido integrarme a su equipo de trabajo como estudiante de doctorado y por servir de guía para mi formación académica y científica. Al Dr. Iván Delgado Enciso, por aceptar ser coasesor del presente trabajo y brindarme su ayuda para aclarar dudas. Al Q.F.B. Miguel Ángel Flores Virgen, por su invaluable ayuda técnica. A mis sinodales de tesis, Drs. Igor Pottosin, Humberto Cruzblanca, Sergio Elenes, Jesús Lara y Ulises Meza, por sus valiosas observaciones para enriquecer el presente trabajo. A Rito, Aldo, Martincillo, Angy, Dani, Arael y Bonales, por ser magníficos compañeros. A mi madre Esperanza y a mis hermanos(as): Nena, Chila, Marcos, Mando, Lore y Uri, por su incondicional apoyo. A mi esposa, Rosy, y a mis hijos Zuli, Zulette y Karim, por ser mi propósito de vida. El presente trabajo de tesis se realizó en el Laboratorio de Fisiología Molecular de Canales Iónicos del Centro Universitario de Investigaciones Biomédicas de la Universidad de Colima El autor de esta tesis declara haber sido sujeto de una beca de manutención y un seguro médico del ISSSTE por parte del CONACYT, número de registro 176531, durante la realización de los estudios del Doctorado en Ciencias Fisiológicas. Adicionalmente, fui apoyado con el programa para Formación de Doctores, número 55912, por parte de la misma institución. DEDICATORIA: A mi esposa Rosy, a mis hijas Zuli Vianney y Aura Zulette y a mi hijo Eloy Karim, nuevamente, ¡con mucho cariño! ÍNDICE Sección Pág. Índice de tablas y figuras i Abreviaturas iii Resumen iv Abstract v I. Introducción 1 -Receptores acoplados a proteínas G (RAPG) y su importancia fisiológica 1 -Modulación por voltaje de respuestas fisiológicas asociadas con RAPG 2 -La sensibilidad al voltaje es una propiedad intrínseca de los RAPG 3 -Localización del “sensor” de voltaje en el receptor 5 -Regulación colinérgica de la función cardiaca 8 -Receptores muscarínicos M2 en corazón y su contribución fisiológica 9 -Canales KACh y su importancia fisiológica 10 -Antecedentes del estudio de la dependencia de voltaje en miocitos cardiacos 11 -Dependencia de voltaje de la “relajación” de IKACh en miocitos cardiacos 13 II. Hipótesis 16 III. Justificación 16 IV. Objetivos 17 V. Material y métodos 18 A) Registro en miocitos aislados en fresco de aurícula izquierda de gato y conejo 18 -Obtención de los miocitos aislados en fresco 18 -Registros electrofisiológicos de miocitos aislados, en conf. de célula completa 18 B) Registros en células HEK293 transfectadas transitoriamente 19 -Cultivo y resiembra de las células HEK293 19 -Transfección transitoria de las células HEK293 20 -Registros electrofisiológicos 20 C) Sistema de perfusión rápida 21 D) Protocolo experimental para las curvas concentración-respuesta (C-R) 21 E) Experimentos con la PTX 22 F) Ajustes de las curvas concentración-respuesta 22 G) Análisis estadístico 22 VI. Resultados (A) Papel del sitio de unión de los agonistas en la dependencia de voltaje del recep- 23 23 tor muscarínico M2 -Dependencia de voltaje en la activación de IKACh por el agonista fisiológico 23 ACh en miocitos cardiacos -Dependencia de voltaje en la activación de IKACh por agonistas muscarínicos 24 en miocitos cardiacos de gato -Dependencia de voltaje en la activación de IKACh por agonistas muscarínicos 28 en miocitos cardiacos de conejo -Dependencia de voltaje en la activación de IKACh por agonistas muscarínicos 30 en células HEK293 que expresan heterólogamente receptores M2 y canales Kir 3.1/3.4 (canales KACh) -Residuos aminoácidos del sitio ortostático del receptor M2 involucrados con 33 La dependencia de voltaje -Dependencia de voltaje en la acción de methoctramina, antagonista M2, en 35 miocitos cardiacos (B) Dependencia de voltaje para activar IKACh vs. la hipótesis de los múltiples rec- 36 tificadores en miocitos cardiacos -Vía de señalización activada por colina 37 -Mediadores celulares activados por pilocarpina 39 -Curso temporal (dependencia de tiempo y voltaje) de las corrientes activadas 41 por pilocarpina con pulsos de voltaje en células cardiacas y en células HEK293 que expresan receptores M2 y canales Kir 3.1/3.4 (C) La sensibilidad al voltaje de los receptores muscarínicos M2 determina la “rela- 43 jación” de IKACh en miocitos cardiacos VII. Discusión 45 VIII. Conclusiones 51 IX. Referencias 52 ÍNDICE DE TABLAS Y FIGURAS Figuras Fig. 1 Esquema que intenta representar el modelo de Hanna Parnas para explicar la Pág. 6 modulación dependiente de voltaje de la afinidad de ligandos en los RAPG Fig. 2 Sitio ortostático del receptor muscarínico M2 7 Fig. 3 Modulación directa del canal KACh a través del dímero Gßγ de las proteínas Gi/o 9 Fig. 4 Rectificación de los canales Kir y representación esquemática de su estructura 10 Fig. 5 Registros de corrientes de K+ activadas por Ch con pulsos de voltaje 11 Fig. 6 Dependencia de voltaje de la “relajación” de IKACh en miocitos cardiacos 14 Fig. 7 Dependencia de voltaje en la activación de IKACh por ACh en los miocitos auri- 23 culares de gato Fig. 8 Activación de IKACh por CarbaCh con dependencia de voltaje en miocitos auri- 24 culares de gato Fig. 9 Dependencia de voltaje en la activación de IKACh por MetaCh en miocitos auri- 25 culares de gato Fig. 10 Betanecol activa IKACh sin dependencia de voltaje en miocitos auriculares de 26 gato Fig. 11 Pilocarpina activa IKACh con dependencia de voltaje opuesta a la de ACh en 27 miocitos auriculares de gato Fig. 12 Activación de IKACh por TMA en miocitos auriculares de gato 27 Fig. 13 Activación de IKACh por Ch en miocitos auriculares de gato 28 Fig. 14 ACh y pilocarpina activan IKACh con dependencia de voltaje opuesta en mio- 29 citos auriculares de conejo Fig. 15 Betanecol activa IKACh sin dependencia de voltaje en miocitos auriculares de 29 conejo Fig. 16 ACh y pilocarpina activan IKACh con dependencia de voltaje opuesta en célu- 31 las HEK293 que expresan receptores M2 y canales Kir 31./3.4 Fig. 17 Activación de IKACh por CarbaCh, betanecol y TMA en células HEK293 que 31 expresan receptores M2 y canales Kir 3.1/3.4. Fig. 18 Estructuras moleculares de los agonistas muscarínicos que activan IKACh con i 33 un patrón diferencial de dependencia de voltaje Fig. 19 Dependencia de voltaje en la activación de corrientes por ACh en células 34 HEK293 que expresan receptores M2 S107A y canales Kir 3.1/3.4 Fig. 20 La activación de corrientes por ACh en células HEK293 que expresan recepto- 34 res M2 W99A y canales Kir 3.1/3.4 no depende del voltaje Fig. 21 Antagonismo dependiente de voltaje de la methoctramina, antagonista M2, en 35 la activación de IKACh por CarbaCh en miocitos auriculares de gato Fig. 22 Curso temporal de las corrientes activadas por pilocarpina con pulsos de volta- 36 je en miocitos auriculares de gato Fig. 23 La perfusión conjunta de Ch y ACh induce una corriente que no es la suma de 38 las corrientes que activan por separado Fig. 24 Concentraciones equiefectivas de Ch y ACh para activar IKACh 38 Fig. 25 Activación de IKACh por Ch en células HEK293 39 Fig. 26 El pretratamiento con PTX inhibe las corrientes activadas por pilocarpina 40 Fig. 27 Efecto de tertiapina (potente bloqueador selectivo de IKACh) sobre las corrien- 40 tes activadas por pilocarpina Fig. 28 Curso temporal (dependencia de tiempo y voltaje) en las corrientes activadas 42 por pilocarpina con pulsos de voltaje en células HEK293 y en células cardiacas de conejo y gato Fig. 29 Relajación de IKACh (con BAPTA intracelular) en miocitos auriculares de gato 44 Fig. 30 Relajación de IKACh (con BAPTA intracelular) en miocitos auriculares de co- 44 nejo Fig. 31 Esquema del modelo para explicar la dinámica en la dependencia de voltaje 47 por los agonistas muscarínicos Tablas Tabla 1 Parámetros de las curvas C-R de los agonistas muscarínicos ensayados en los Pág. 32 3 modelos celulares Tabla 2 Cambio de la potencia (EC50) y del efecto máximo (Emáx) entre los voltajes -100 y +50 mV de los agonistas muscarínicos ensayados en los 3 modelos celulares ii 32 ABREVIATURAS ACh= acetilcolina. 4-AP= 4-aminopiridina. Ca2+i= calcio intracelular. CarbaCh= carbacol. Ch= colina. C-R= (curvas) concentración-respuesta. E= eficacia. EC50= concentración que produce el 50% de la respuesta máxima. Emáx= efecto o respuesta máxima. IKACh= corriente del canal KACh. IKM3 e IKM4= (supuestos canales) rectificadores tardíos activados por receptores muscarínicos M3 y M4, respectivamente. IP3= 1,4,5-inositol trifosfato. KACh= (canales) de K+ sensibles a ACh. KD=constante de afinidad. Kir= (canales) de K+ rectificadores entrantes. M2= (receptor) muscarínico subtipo 2. MetaCh= metacolina. Meth= antagonista parcialmente selectivo hacia los receptores muscarínico M2. mGluR1a y mGluR3= receptores metabotrópicos a glutamato subtipos 1a y 3, respect. PTX= toxina pertussis. RAPG= receptor(es) acoplado(s) a proteínas G. TMA= tetrametil amonio. RT-PCR= (técnica de biología molecular) transcripción inversa-reacción en cadena de la polimerasa. iii RESUMEN Los receptores acoplados a proteínas G (RAPG) constituyen la familia más grande de receptores en la membrana celular y están involucrados en la mayoría de los procesos fisiológicos de transducción celular. Los RAPG son activados por la unión de agonistas específicos, lo cual constituye su principal mecanismo de control, pero en los últimos 20 años se han aportado evidencias donde el potencial membranal (voltaje) es capaz de modular la función de estos receptores. Ben Chaim et al. (2003) mostraron la dependencia de voltaje del receptor de acetilcolina (ACh) muscarínico M2 en ovocitos de Xenopus reconstituídos con este receptor y con las subunidades Kir 3.1/3.4, formadoras del canal KACh. Posteriormente, se reportó que los receptores muscarínicos M2 son capaces de desarrollar corrientes asociadas con movimientos de carga (Ben-Chaim et al., 2006), análogas a las de los canales iónicos sensibles a voltaje. Con nuestro estudio confirmamos la dependencia de voltaje de ACh para activar IKACh en la vía de señalización colinérgica cardiaca, la cual resulta excelente como modelo fisiológico dado que casi la totalidad de sus receptores muscarínicos son del subtipo M2 acoplados, a través de una proteína Gi/o, a un canal de K+ (canal KACh) insensible al voltaje. También encontramos que la modulación por voltaje de esta vía de señalización, M2-Gi/oKACh, depende de la estructura del ligando, teniendo los agonistas carbacol y metacolina una dependencia de voltaje similar a la de ACh: mayor potencia a -100 mV que a +50 mV, mientras que los agonistas pilocarpina, tetrametil amonio (TMA) y colina (Ch) tienen una dependencia de voltaje opuesta a la de ACh: mayor potencia (y eficacia) para activar a +50 mV que a -100 mV. La dependencia de voltaje opuesta en la activación de M2-Gi/o-KACh por los agonistas muscarínicos totales y parciales también fue observada en células HEK293 que expresan heterólogamente receptores M2 y canales Kir 3.1/3.4. Estudios de mutagénesis señalan la participación del sitio ortostático del receptor M2 en la dependencia de voltaje. Finalmente, los resultados de este estudio sugieren que el voltaje provoca cambios conformacionales en el sitio del receptor donde se unen los ligandos, sitio ortostático (donde inclusive podría residir la sensibilidad al voltaje), lo que provoca una interacción (afinidad y/o eficacia) diferente con el mismo ligando (agonista o antagonista). Adicionalmente, proponemos que la sensibilidad al voltaje de los receptores M2 ocasiona que los agonistas pilocarpina, Ch y TMA activen a IKACh con la apariencia de rectificador tardío y que ACh genere el fenómeno de la “relajación” de IKACh. iv ABSTRACT The G-protein-coupled receptor (GPCR) superfamily comprises the largest class of signaling proteins in most living organisms. Last 20 years, it has been shown a voltagedependent modulation of GPCR function. Five years ago, it was shown in Xenopus oocytes that the M2 muscarinic receptor is voltage-sensitive (Ben Chaim et al., 2003) and display charge-movement-associated currents (Ben-Chaim et al., 2006) analogous to ‘gating currents’ of voltage-gated channels. For the first time, our study confirms the voltage-modulation of the cardiac colinergic signaling pathway, which is an excellent physiological model because almost 100% of muscarinic receptors are M2 subtype and the coupled K+ channel (KACh), through a Gi/o protein, is voltage-insensitive. We also found that the voltage-modulation of this signaling pathway, M2-Gi/o-KACh, is ligand-specific: agonists carbachol and metacholine exhibits a voltage-dependent behaviour similar to ACh’s, greater potency at -100 mV compared to +50 mV; while agonists pilocarpine, tetramethyl ammonium (TMA) and choline (Ch) was also voltage-dependent, but opposite that of ACh: greater potency (and efficacy) at +50 mV compared to -100 mV. Opposite voltage-dependent activation of M2-Gi/o-KACh by agonists were also observed in HEK293 cells heterologously expressing M2 receptors and Kir 3.1/3.4 channels. Mutagenesis studies involve the orthostatic site of M2 receptor in the voltage-sensitivity. Our results suggest that membrane voltage induces conformational changes in the ligand-binding site (where voltage-sensitivity could reside) modifying the interaction (affinity and/or efficacy) between ligands (agonist or antagonist) and the receptor. Finally, we propose that the voltage-sensitivity of M2 receptors underlies both phenomenons, the activation of IKACh in a rectifier-like manner by agonists pilocarpine, Ch and TMA and the “relaxation” of IKACh by ACh. v I. INTRODUCCIÓN Receptores acoplados a proteínas G y su importancia fisiológica Los receptores acoplados a proteínas G (RAPG) constituyen la familia más grande de receptores en la membrana celular: en el nemátodo C. elegans cerca del 5% de sus genes codifican tales receptores, en ratones existen casi 1000 de estos receptores relacionados sólo con el sentido del olfato, en mamíferos se han descubierto varios miles de ellos (Alberts et al., 2002) y en el humano se han identificado más de 800 genes (3% de su genoma) que codifican para estos receptores (Holinstat, Oldham y Hamm, 2006). La presencia de los RAPG en los genomas de prácticamente todos los seres vivos incluyendo bacterias, hongos, plantas, en muchos grupos de invertebrados y en toda la rama de vertebrados apoyan la hipótesis del temprano origen evolutivo de este grupo de moléculas (Kroeze, Sheffler y Roth, 2003). Los RAPG juegan un papel central en la mayoría de las funciones celulares y constituyen la familia más prominente de blancos farmacológicos: el 50-60% de fármacos no antibióticos validados para su uso médico realizan sus efectos terapéuticos actuando sobre los miembros de esta familia de receptores (Römpler et al., 2007). Los RAPG son responsables de mediar las respuestas fisiológicas a una enorme diversidad de estímulos como hormonas, neurotransmisores, luz, olores, sabores, etc., de amplia variedad estructural y funcional. Entre las moléculas que activan a los RAPG tenemos: proteínas, aminas biogénicas, péptidos, ácidos grasos, aminoácidos y derivados, nucleótidos y muchas otras (Kroeze et al., 2006). A pesar de la diversidad química y funcional de las moléculas que los activan, los RAPG comparten una estructura similar que consiste en una sola cadena polipeptídica que atraviesa siete veces la bicapa lipídica de la membrana celular, por lo cual también son conocidos como receptores 7-TM. Poseen el extremo amino del lado extracelular y el extremo carboxilo del lado intracelular. También cuentan con 3 asas extracelulares y 3 asas intracelulares conectando los dominios transmembranales. Todos los RAPG comparten la característica funcional, de la cual toman su nombre genérico, de activar proteínas triméricas fijadoras de guanosín trifosfato (proteínas G). Las proteínas G están ancladas a la cara citoplásmica de la membrana celular, donde sirven como moléculas-relevo que acoplan funcionalmente a los receptores con innumerables vías de señalización que conllevan a la estimulación o inhibición de enzimas, canales iónicos u otros efectores celulares (Alberts et al., 2002). -1- Modulación por voltaje de respuestas fisiológicas asociadas con RAPG Estudios de los últimos 20 años han revelado lo que podría ser una importante propiedad funcional de los RAPG: su modulación por el potencial de membrana celular (voltaje), cuando menos en los RAPG de la familia A (rodopsina) y C (glutamato). Entre algunas de las primeras evidencias relacionadas con la dependencia de voltaje en procesos mediados por RAPG están las obtenidas en células acinares de las glándulas lacrimales de rata (Marty y Tan, 1989), donde observaron que la liberación de calcio de los depósitos intracelulares mostraba dependencia del potencial de membrana cuando se activaban receptores de acetilcolina (ACh) muscarínicos. Estos autores mostraron que la liberación de Ca2+ de sus reservas intracelulares es menor y con una latencia mayor en las células despolarizadas (-20 y 0 mV) respecto a las de células mantenidas en potenciales más negativos (-100 y -60 mV) cuando se utilizan concentraciones de ACh relativamente bajas (0.2 y 0.5 µM), mientras que si se utiliza una concentración saturante de ACh (5 µM) prácticamente ya no existe diferencia en el efecto sobre el Ca2+i. Debido a esto y a que el receptor muscarínico es el único elemento celular localizado dentro de la zona de influencia del campo eléctrico, estos autores proponen que el paso dependiente de voltaje está determinado por la unión de la ACh con su receptor. Posteriormente, en un trabajo realizado con células de músculo liso de la arteria mesentérica de conejo se observó que la movilización de Ca2+i, esta vez por la activación de receptores adrenérgicos, también es sensible al voltaje (Itoh et al., 1992). Los resultados de este estudio mostraron que la hiperpolarización inducida por pinacidil disminuye la liberación de Ca2+i, la tensión isométrica y la producción de IP3 ocasionados por la activación de receptores adrenérgicos con noradrenalina (10 µM). Años más tarde, Mahaut-Smith, Hussain y Mason (1999) trabajando con megacariocitos derivados de médula ósea de rata, un tipo de células consideradas no excitables, y utilizando simultáneamente registros de corrientes con la técnica de fijación de voltaje y de medición del Ca2+i, también encontraron que la liberación de éste de las reservas intracelulares era dependiente del potencial membranal cuando se estimulaban receptores purinérgicos. En este estudio se observó que la despolarización dentro del intervalo fisiológico (desde -70 a 0 mV y desde -70 a -45 mV) ocasionaba incrementos en la concentración del Ca2+i sólo hasta que se estimulaban a los receptores purinérgicos con adenosín-difosfato 1µM. -2- Además de los 3 tipos de receptores mencionados: los muscarínicos colinérgicos, los adrenérgicos y los purinérgicos, actualmente también existen más trabajos que postulan la regulación de otros RAPG por el potencial de membrana, entre ellos tenemos a: receptores a tromboxano A2 y serotoninérgicos (Martinez-Pinna et al., 2005), receptores gabaérgicos B (Dekel et al., 2005) y los receptores a glutamato (Perroy, Richards, Nargeot, Bockaert y Fagni, 2002; Ohana, Barchad, Parnas y Parnas, 2006). La sensibilidad al voltaje es una propiedad intrínseca de los RAPG En casi todos los estudios mencionados anteriormente, así como la mayoría en los que se demuestra un efecto regulador del potencial de membrana sobre procesos mediados por RAPG, el elemento o paso donde reside la sensibilidad al voltaje no fue directamente aclarada (Cohen-Armon y Sokolovsky, 1991; Ganitkevich e Isenberg, 1993; Ilouz, Branski, Parnis, Parnas y Lineal, 1999; Ong et al., 2001; Mason, Hussain y Mahaut-Smith, 1999). De hecho, no se llegaba a establecer si la sensibilidad al voltaje residía en el propio RAPG o en otro componente y/o pasos involucrados en el proceso en su conjunto. Desde los trabajos pioneros, el propio receptor parecía ser el más indicado para mostrar la sensibilidad al voltaje (Marty y Tan, 1989), dada su posición transmembranal dentro de la zona de influencia del campo eléctrico, caso contrario para cualquiera de los otros elementos intracelulares involucrados en las vías de señalización acopladas a los RAPG. De cualquier manera, no quedaba totalmente discernido en qué elemento de la cadena de activación se encontraba la dependencia de voltaje; sin embargo, trabajos recientes han venido a esclarecer dicha cuestión, cuando menos en lo que respecta a los receptores muscarínicos colinérgicos (Ben-Chaim, Tour, Dascal, Parnas y Parnas, 2003; Ben-Chaim et al., 2006), los receptores purinérgicos (Martinez-Pinna et al., 2005) y los receptores glutamaérgicos (Ohana et al., 2006) como a continuación se describe. En el año de 2003, Ben-Chaim et al. mostraron que la unión de ACh con los receptores muscarínicos M2 y M1 es dependiente de voltaje. Para tal fin utilizaron un sistema de expresión heteróloga, ovocitos de Xenopus, que les permitió estudiar cada receptor con respecto a su capacidad para detectar cambios en el voltaje transmembrana. En este modelo usaron dos estrategias experimentales evaluadas en dos diferentes voltajes (-60 y +40 mV): por un lado, estudios de afinidad con ligandos marcados radiactivamente (binding), [3H]ACh -3- y, por otro lado, la técnica de fijación de voltaje midiendo el nivel de corriente de K+ activada por distintas concentraciones de ACh (curvas concentración-respuesta). Las curvas concentración-respuesta mostraron que ACh tiene mayor potencia cuando el ovocito se mantiene en el potencial más negativo y viceversa. En el ensayo con [3H]ACh también se observa una mayor afinidad a -60 que a +50 mV lo cual apoyó la idea de que el receptor M2 es sensible al voltaje. Por el contrario, la dependencia de voltaje no se manifiesta al activar la vía posterior al receptor M2, esto es, cuando sólo se activa la vía proteínas G-canales de K+. Por otro lado, el comportamiento del receptor M1 fue opuesto al del M2, es decir, la afinidad del receptor M1 por la ACh es menor con los voltaje más negativos y viceversa (Ben-Chaim et al., 2003). Posteriormente, una evidencia fuerte de que la sensibilidad al voltaje reside en el receptor muscarínico M2, fue aportada al registrar corrientes asociadas con movimientos de cargas en el receptor M2 (Ben-Chaim et al., 2006), análogas a las “corrientes de compuerta” (gating currents) de los canales iónicos sensibles a voltaje (Armstrong y Bezanilla, 1973). Evidencias adicionales que muestran al receptor como el elemento responsable de la regulación por voltaje en distintos RAPG acoplados a vías de señalización que movilizan Ca2+i, fueron aportadas por Martinez-Pinna et al. (2005) en megacariocitos de rata. Estos autores mostraron que la despolarización potencia la movilización de Ca2+i dependiente de IP3 cuando se activan receptores purinérgicos (P2Y1), receptores a tromboxano (A2) y receptores serotoninérgicos. La activación de esta vía de señalización excluyendo la participación de los RAPG con estrategias como elevación directa de IP3 y/o de análogos de diacilglicerol y/o de activación de la fosfolipasa Cγ, produce una liberación de Ca2+ insensible a los cambios de voltaje. Adicionalmente, en ratones deficientes (knockout) para los receptores purinérgicos P2Y1 (P2Y1-/-) se observó que al estimular la vía de señalización con activadores de las proteínas G como el GTPγS o el ion tetrafluoruro de aluminio o con un agente sensibilizador de los receptores IP3 (el thimerosal), la liberación de Ca2+ tampoco es influenciada por el potencial de membrana. Por lo cual los autores concluyen que la dependencia de voltaje de la movilización de Ca2+i no es debida a las proteínas G o a cualquier otro elemento de la vía de señalización posterior al receptor, sino que sólo puede ser explicada por una sensibilidad al voltaje a nivel del propio receptor (Martinez-Pinna et al., 2005). En otra serie de experimentos, se postula que los receptores metabotrópicos a glutamato del tipo 1a y 3 (mGluR1a y mGluR3, respectivamente), exhiben por sí mismos, -4- sensibilidad al voltaje (Ohana et al., 2006). En este trabajo también utilizan ovocitos de Xenopus como sistema de expresión heteróloga para comparar, a dos diferentes potenciales de membrana, tanto la afinidad de ligandos (binding), en este caso glutamato ([3H]Glu), como las curvas concentración-respuesta en fijación de voltaje. La medición de las corrientes de K+ inducidas por el mGluR3, vía proteínas Gαi, y de las corrientes de Cl- sensibles a Ca2+i inducidas por el mGluR1a, a través de proteínas Gαq, muestran una potencia menor (para el caso del mGluR3) o mayor (para el mGluR1a) con la despolarización. El experimento de afinidad de [3H]Glu es congruente con el resultado electrofisiológico y es una evidencia que sustenta que estos receptores también son sensibles al potencial de membrana. Por el contrario, las evidencias que muestran que la sensibilidad al voltaje no se encuentra en los pasos posteriores a la activación de los mGluR son: a) para evaluar el papel de la proteína G se usó una proteína G quimérica, la Gαi/q, la cual es idéntica a la Gαi nativa aunque con la capacidad de acoplarse eficientemente con el mGluR1a, el comportamiento dependiente de voltaje fue el mismo que cuando se activó el mGluR1a vía la proteína Gq, lo cual indica que la sensibilidad al voltaje no reside en la proteína G; b) los canales de K+ utilizados, Kir 3.1 y 3.2, como ya es ampliamente conocido, son insensibles al voltaje (Ohana et al., 2006). Localización del “sensor” de voltaje en el receptor Desde los experimentos que mostraban que el potencial de membrana afecta de manera opuesta la unión de ACh con dos subtipos de receptores muscarínicos, M2 y M1 (Ben-Chaim et al., 2003), y la unión de glutamato con dos subtipos de receptores metabotrópicos a glutamato, mGluR3 y mGluR1a (Ohana et al., 2006) les llevó, a estos autores, a excluir la posibilidad de que el sitio de unión de los agonistas pudiera ser el sensor de voltaje. Un análisis de la secuencia de aminoácidos reveló una región citosólica del receptor M2 (asa interna 3) en contacto con el 5o. segmento transmembranal que contiene cierta cantidad de aminoácidos positivos; esta región está involucrada en la interacción con la proteína G (Wess, 1996). El hallazgo de que el pretratamiento con toxina pertussis (PTX: inhibidor de proteínas Gi/o) de ovocitos que expresan receptores M2 anula casi completamente la dependencia de voltaje en los estudios de afinidad de [3H]ACh (Ben-Chaim et al., 2003) fue un respaldo para la idea de que la región del receptor que se asocia con la proteína G sea el sensor de voltaje. De hecho, al comparar la composición de aminoácidos de los receptores M1 y M2 en la región que se acopla -5- con la proteína G existe una zona de aminoácidos cargados positivamente en el M2 (aminoácidos 215-225) que no existe en el M1. Así, con el asa interna 3 como candidata, se hicieron estudios con quimeras intercambiando las asas internas 3 entre los receptores M2 y M1 (Ben-Chaim et al., 2006) y entre los receptores mGluR3 y mGluR1a (Ohana et al., 2006), con los cuales obtuvieron un soporte adicional a la hipótesis de que la región del receptor que interacciona con la proteína G sea la responsable de la sensibilidad al voltaje. Con las evidencias obtenidas por el grupo de Hanna Parnas (Ben-Chaim et al., 2003 y 2006 y Ohana et al., 2006), y basados en el modelo clásico de activación de los receptores, ellos postulan un mecanismo de cómo el potencial membranal cambia la afinidad de los RAPG (Fig. 1): primero, proponen que estos receptores existen en dos estados de afinidad hacia los agonistas, uno de alta afinidad cuando están acoplados a su proteína G y otro de baja afinidad cuando están libres. Posteriormente, sugieren que movimientos de carga inducidos por cambios en el potencial de membrana provoca un cambio conformacional en la región del receptor que se acopla con su proteína G, por lo que la probabilidad del acoplamiento receptor-proteína G se altera y en consecuencia cambia la afinidad del receptor hacia el agonista. Baja afinidad A Alta afinidad ∆V GTP Fig. 1. Esquema que intenta representar el modelo propuesto por el grupo de Hanna Parnas para explicar la modulación voltaje dependiente de la afinidad de ligandos en los RAPG (ver texto en párrafo superior para su descripción). Con las evidencias de Ben-Chaim et al. (2003 y 2006), el asa intracelular 3 de los receptores muscarínicos M2 parecía ser donde se localizara el “sensor” de voltaje, sin embargo, hallazgos más detallados con mutaciones puntuales de la secuencia más probable para sentir los cambios de potencial, KKDKK (K=lisina y D=aspartato) que comprende los aminoácidos 218-222 de esta asa, mostraron que esta región no está involucrada en la detección de voltaje; lo que sigue dejando sin respuesta la pregunta original ¿en qué región del receptor radica esta propiedad? A nuestro criterio, el sitio ortostático (región del receptor a la -6- que se unen los agonistas fisiológicos) reúne 3 elementos importantes para considerar que tiene un papel relevante en la sensibilidad al voltaje: a) está constituido por una cavidad formada por las porciones externas de los segmentos transmembranales, inserta en el tercio externo de la membrana plasmática (Wess, 1996), dentro de la zona de influencia del campo eléctrico de la membrana; b) los aminoácidos que se han propuesto ser cruciales en la interacción con ACh son aminoácidos cargados o con anillos aromáticos, indispensables para el movimiento de carga ante los cambios de voltaje (Bezanilla, 2000), así por ejemplo los del receptor M2 son: W (triptófano) 99, D103,Y (tirosina) 104, S (serina) 107 y Y403 (Pedretti, Vistoli, Marconi & Testa, 2006; Fig. 2); y c) la modulación voltaje dependiente de la interacción receptor-ligando necesariamente implica un cambio conformacional en el sitio ortostático, tal y como es evidenciado por el cambio de afinidad de los receptores muscarínicos hacia la ACh con el voltaje (Ben-Chaim et al., 2003). En este mismo sentido, la dependencia de voltaje opuesta entre los receptores M1 y M2 para con ACh podría explicarse según los modelos de interacción de la ACh con los diferentes receptores muscarínicos (docking), donde Pedretti et al. (2006) proponen dos diferencias básicas en el sitio ortostático de estos receptores: mayor número de residuos aromáticos en el M2 y en éste los residuos aromáticos rodean completamente a la ACh interactuando con el amonio y con el grupo éster (Fig. 2), mientras que en el M1 los residuos aromáticos interaccionan sólo con el amonio. Fig. 2 Sitio ortostático del receptor muscarínico M2. Se muestran las principales interacciones de la ACh con los residuos aminoácidos del receptor M2. Tomado de Pedretti et al., 2006. -7- Ahora bien, hasta el momento la modulación de un canal rectificador entrante (Kir) por la interacción dependiente de voltaje de los receptores M2 con ACh, sólo se ha mostrado en un sistema heterólogo. Dado que esta vía de señalización tiene un papel funcional trascendente en la regulación de la actividad cardiaca, y puesto que el receptor M2 es el único subtipo involucrado en esta función, creemos importante evaluar si la regulación dependiente de voltaje acontece en las células cardiacas. Experimentos previos en nuestro laboratorio sugieren que esta propiedad podría explicar algunos fenómenos que a la fecha no han sido esclarecidos. A continuación se exponen con mayor detalle estas características, así como el encuadre fisiológico de esta vía en el corazón. Regulación colinérgica de la función cardiaca En mamíferos, la función cardiaca está bajo el control del sistema nervioso autónomo a través de sus dos componentes antagónicos, el sistema simpático y el parasimpático. La activación de estos dos sistemas se refleja en cambios en la frecuencia y en la fuerza de contracción cardiacas (Dhein, Van Koppen y Brodde, 2001). Mientras el sistema simpático causa estimulación de ambos parámetros cardiacos, lo cual resulta en un mayor volumen de sangre bombeada (gasto cardiaco), el sistema parasimpático realiza la función opuesta. En humanos, en condiciones de reposo, el tono parasimpático predomina y, por ende, la frecuencia cardiaca en reposo es más baja como consecuencia de la modulación negativa sobre la actividad nodal. La inervación parasimpática del corazón se realiza a través del nervio vago. Con la estimulación vagal, se libera ACh de las terminales axonales. Este neurotrasmisor produce su efecto de disminución de la frecuencia cardiaca por la estimulación de una población específica de canales de K+, denominados canales de K+ muscarínicos (KACh), que terminan abriéndose y la subsecuente salida de cargas positivas ocasiona que las células cardiacas se hiperpolaricen y, en consecuencia, disminuye el automatismo (Fig. 3; Yamada, Inanobe y Kurachi, 1998). En miocitos cardiacos, la estimulación de receptores muscarínicos M2 ocasiona la activación de los canales KACh a través de proteínas Gi/o (Pfaffinger, et al., 1985; Breitweiser y Szabo, 1985), sin la intervención de mediadores intracelulares, acción que se conoce como delimitada a la membrana celular (Soejima y Noma, 1984; Fig. 3). -8- Fig. 3 Modulación directa del canal KACh a través del dímero Gβγ de las proteínas Gi/o por activación del receptor muscarínico M2: señalización delimitada en la membrana plasmática. (Mod. de Hille, 2001). Receptores muscarínicos M2 en corazón y su contribución fisiológica. Existen 2 grandes grupos de receptores de ACh, los nicotínicos que son receptores-canal, y los receptores muscarínicos, que son RAPG (Alberts et al., 2002). De los receptores muscarínicos se sabe que existen 5 subtipos, designados del M1 al M5. Los subtipos M1, M3 y M5 se acoplan preferencialmente a la familia de proteínas Gq/11, mientras que los subtipos M2 y M4 se acoplan preferentemente a la clase de proteínas Gi/o, sensibles a PTX (Wess, 1996). Existe un consenso general de que la forma predominante de los receptores muscarínicos presentes en tejido cardiaco de varias especies animales, incluyendo los humanos, es el receptor M2. Este consenso proviene de estudios con análisis de ADNc (Kubo et al., 1986; Peralta et al., 1987b), de ARNm (Peralta et al., 1987a; Maeda et al., 1988; Pinkas-Kramarski et al., 1989), de afinidad con radioligandos (Doods et al., 1987; Giraldo et al., 1988; Deighton et al., 1990) y de inmunoensayos (Dörje et al., 1991). La estimulación de los receptores M2 media los efectos de disminución de la frecuencia y fuerza de contracción cardiacas. Aunado al amplio número de reportes que constatan la predominancia del receptor M2 en corazón, un estudio ha cuantificado (RT-PCR competitiva) la proporción de los subtipos de receptores muscarínicos en aurícula de rata, a nivel de ARNm, resultando: 94% del M2, 0.8% del M1, 1% del M3, 0.2% del y M4, y 4% del M5 (Krejci y Tucek, 2002). Lo que es todavía más importante, en estudios para valorar la contribución fisiológica de estos receptores utilizando ratones knockout, se ha mostrado que sólo el M2 tiene una contribución crucial en la regulación de la frecuencia cardiaca (Stengel, Gomeza, Wess y Cohen, 2000; Wess, 2004). -9- Canales KACh y su importancia fisiológica. Los canales KACh pertenecen al grupo de canales de K+ rectificadores entrantes (Kir, por sus siglas en inglés) acoplados a proteínas G, miembros de la subfamilia Kir 3.0. Evolutivamente, los canales Kir representan la clase más primitiva de canales iónicos y se les encuentra desde en bacterias hasta en mamíferos superiores (Hille, 2001). La estructura primaria de estas subunidades predice 2 regiones transmembranales, una región formadora de poro (H5) y los extremos amino y carboxilo del lado citoplásmico. La secuencia glicina-tirosina-glicina en la región H5 constituye el filtro de selectividad para K+ (Fig. 4B; Kurachi e Ishii, 2004). Los canales Kir se caracterizan por mostrar rectificación entrante, esto es, conducen con mayor facilidad la corriente entrante que la corriente saliente (Fig. 4A); este mecanismo le permite a la célula no sólo conservar K+ sino también facilitarle su entrada (Stanfield, Nakajima y Nakajima, 2002). El mecanismo de rectificación entrante de los canales Kir es causado por un rápido bloqueo dependiente de voltaje por Mg2+ y poliaminas intracelulares (Ruppersberg, 2000). Los canales Kir están compuestos de 4 subunidades Kir 3.x (x=1-4). Cada subunidad posee un sitio de unión para Gβγ en los extremos amino y carboxilo terminal del canal. Diferentes combinaciones de subunidades Kir 3.x se encuentran en tejidos y células. Así, el canal KACh está compuesto de Kir 3.1 y 3.4 en una estequiometría 2:2, mientras que los canales Kir neuronales existen principalmente como hetero u homotetrámeros que contienen Kir 3.2 (Kurachi e Ishi, 2004). En estudios in vivo con ratones deficientes para Kir 3.4 quedó demostrada la participación A B Fig. 4 (A) Rectificación de los canales Kir. (B) Representación esquemática de la estructura de los canales Kir y de su ensamble tetramérico en la membrana plasmática. (Mod. de Nichols y Lopatin, 1997). - 10 - directa de los canales KACh en la bradicardia inducida por estimulación vagal y por adenosina, además de ser necesarios para la regulación rápida de la frecuencia cardiaca en reposo (Wickman, Nemec, Gendler y Clapham, 1998; Mark y Herlitze, 2000). Antecedentes del estudio de la dependencia de voltaje en miocitos cardiacos En nuestro laboratorio desde hace tiempo se ha observado que algunas sustancias como 4-aminopiridina (4-AP), tetrametil amonio (TMA) y colina (Ch) son capaces de acortar la duración del potencial de acción auricular. En fijación de voltaje se ha encontrado que estas sustancias activan corrientes con dependencia de tiempo y voltaje (Fig. 5): conforme el potencial es más positivo, la corriente es más activada (Navarro-Polanco y Sánchez-Chapula, 1997; Benavides-Haro, 2001). En la activación de dichas corrientes participan receptores muscarínicos acoplados a proteínas Gi/o ya que atropina (antagonista inespecífico de receptores muscarínicos), GDPβS (inhibidor inespecífico de proteínas G) y PTX (inhibidor específico de proteínas Gi/o), aplicados por separado, inhiben las corrientes activadas por 4-AP (NavarroPolanco y Sánchez-Chapula, 1997), TMA y Ch (Navarro-Polanco y Sánchez-Chapula, 1995). En registros de corrientes de membrana activadas por estas sustancias se observa que una concentración relativamente baja (100 nM) de atropina sí es efectiva para antagonizar las corrientes activadas con los potenciales más hiperpolarizados, mientras que con las corrientes activadas con los potenciales más despolarizados ya no tiene efecto antagónico (Fig. 5; Sánchez-Chapula y Navarro-Polanco, datos no publicados). Además, con registros de canal -110 mV 1.5 s -40 mV 1.5 s Control Ch 10 mM Ch 10 mM + atropina 100 nM 500 pA 90 mV -40 mV 500 ms Fig. 5 Registros de corrientes de K+ en un miocito auricular de gato en situación control (izq.), en presencia de colina (Ch) 10 mM (centro) y de Ch 10 mM+atropina 100 nM (der.); (Sánchez-Chapula y NavarroPolanco, datos no publicados). - 11 - unitario, estas sustancias activan canales de conductancia, cinética y rectificación similar al de ACh y dicha activación es dependiente de voltaje (Navarro-Polanco,1997). Por lo tanto, con estas evidencias ya se vislumbraba la hipótesis de que los receptores muscarínicos (M2) son sensibles al potencial de membrana, tal y como se pudo comprobar más directamente en estudios posteriores (Ben-Chaim et al., 2003 y Ben-Chaim et al., 2006). Con todos los antecedentes expuestos, creemos que existen suficientes elementos para plantear que los agonistas muscarínicos no sólo son capaces de activar al receptor M2 de manera dependiente de voltaje sino también para postular que el grado de interacción de estos compuestos con el receptor M2 determina las características de la corriente que se activa, donde el sitio de unión de los agonistas (sitio ortostático) en el receptor cumple un papel importante en la regulación por voltaje de los RAPG. Por otro lado, debido al curso temporal de las corrientes activadas por 4-AP, TMA, Ch y pilocarpina (Fig. 5), se ha propuesto que son nuevos (no descritos antes) rectificadores tardíos denominados IKM3 e IKM4, adicionales a los típicos rectificadores tardíos cardiacos, IKr e IKs. Se ha postulado que en la activación de estos supuestos rectificadores participan receptores muscarínicos M3 (con Ch, TMA y pilocarpina) o M4 (con 4-AP) y proteínas Gq o Gi/o, respectivamente (Fermini y Nattel, 1994; Shi, Wang y Wang, 1999; Wang, Shi, Lu, Yang y Wang, 1999; Wang et al., 2001; Shi, Yang, Xu, Wang y Wang, 2003; Yeh et al., 2007). La mayor parte de evidencias de este grupo se soporta en dos estrategias que ya se han demostrado ser controversiales: a) amplio uso de antagonistas muscarínicos, pues la selectividad de los ligandos actualmente disponibles es limitada (Dhein et al., 2001) y su inespecificidad es frecuente (Wess, 1996; Brodde y Michel, 1999; Meyer et al., 2001; Benavides-Haro, Navarro-Polanco y Sánchez-Capula, 2003) y b) RT-PCR de homogenizados de aurícula completa, pues datos positivos sobre la expresión de ARNm de algún subtipo de receptor muscarínico sin la identificación del tipo celular al que se le realiza la RT-PCR frecuentemente conduce a malinterpretaciones, tal y como se reportó en un estudio en miocitos de aurícula identificados al microscopio donde se mostró la ausencia funcional del receptor muscarínico M3 así como la de su transcrito, mientras que la suspensión celular de los miocitos cultivados sí dio positivo la presencia de este receptor (Meyer et al., 2001). La mayor inconsistencia de la propuesta anterior es que ACh, el agonista fisiológico de los receptores muscarínicos, no es capaz de activar a estos supuestos rectificadores tardíos, IKM3 e IKM4; si - 12 - todos estos receptores (M2, M3 y M4) coexisten en el mismo tipo celular y tienen efectores diferentes, es de esperar que la ACh activara simultáneamente a todos estos efectores, sin embargo, esto no sucede así. Al contrario de la hipótesis anterior, proponemos que los supuestos rectificadores tardíos inducidos por pilocarpina, Ch, TMA y betanecol son resultado de la modulación dependiente de voltaje de la misma vía de señalización utilizada por ACh: receptores muscarínicos M2, proteínas Gi/o y como efectores los canales KACh. Dependencia de voltaje de la “relajación” de IKACh en miocitos cardiacos En miocitos auriculares nativos registrados en la configuración de célula completa, se sabe que la corriente de K+ inducida por ACh (IKACh), exhibe un comportamiento característico denominado “relajación” (Fig. 6): cuando el potencial de membrana se fija a +40 mV durante 1 s, poca corriente saliente fluye a través de los canales KACh; luego, con una hiperpolarización a -100 mV, la IKACh entrante cambia inmediatamente a un nivel (Iins) y luego se incrementa lentamente hasta un nivel estable (Imáx). El incremento inmediato de IKACh refleja la rápida liberación del bloqueo de la IKACh saliente por el Mg2+ y/o poliaminas intracelulares, típico de los canales Kir (Ruppersberg, 2000). El lento incremento subsecuente, llamado “relajación”, es una característica única de IKACh que fue primeramente descrita hace 30 años en células del nodo senoauricular de conejo (Noma y Trautwein, 1978) y cuyo mecanismo molecular no ha podido ser aclarado. Una particularidad de la relajación de IKACh es su dependencia de voltaje a concentraciones relativamente bajas (10-300 nM): en la Fig. 6B izq. se muestra que con ACh 100 nM, el potencial prepulso afecta la amplitud de Iins sin modificar Imáx. El cociente Iins/Imáx es un indicador de la cantidad de corriente disponible durante cada prepulso, con respecto a la de -100 mV. Así tenemos que, con ACh 100 nM, la disponibilidad de los canales KACh muestra una clara dependencia de voltaje (Fig. 6C, círculos vacíos), es decir, menor disponibilidad con los voltajes más positivos y viceversa. Por otro lado, con concentraciones máximas de ACh (1-10 µM) la relajación de IKACh ya no se observa claramente (Fig. 6B der.) o, en otras palabras, la dependencia de voltaje se va perdiendo (Fig. 6C círculos llenos). Una explicación de estos resultados, según Ishii et al. (2001), es que a bajas concentraciones de ACh, los canales KACh son inhibidos por la despolarización y que el lento incremento de la corriente (relajación) con la hiperpolarización posterior refleja la liberación de esta inhibición; la mayor activación de los canales KACh, con concentraciones - 13 - Fig. 6 Dependencia de voltaje de la relajación de IKACh. A, Protocolo de fijación de voltaje (arriba) y una típica IKACh inducida por ACh en un miocito auricular aislado; la corriente entrante al hiperpolarizar a -100 mV primero cambia instantáneamente (Iins) y después se incrementa hasta un estado estable (Imax). B, IKACh inducida por ACh 100 nM (izq.) o 1 µM (der.) en condiciones control; las corrientes a -100 mV se registraron después de prepulsos entre -100 y +40 mV en pasos de 20 mV. C, Relación entre el voltaje del prepulso y el cociente Iins/Imax de las corrientes inducidas por ACh 100 nM (○) o 1 µM (●). D, IKACh inducida por ACh 100 nM (izq.) o 1 µM (der.) cuando el Ca2+ intracelular fue quelado con BAPTA (5 mM). E, Relación entre el voltaje prepulso y el cociente Iins/Imax con BAPTA intracelular cuando las corrientes fueron activadas por ACh 100 nM (○) o 1 µM (●). Las puntas de flecha indican el nivel cero de corriente y las barras verticales, 500 pA. Mod. de Ishii et al., 2001. altas de ACh, se sobrepone a esta inhibición y la actividad de los canales se hace prácticamente independiente del voltaje (Ishii et al., 2001; Ishii, Inanobe y Kurachi, 2002). Entre los experimentos para tratar de esclarecer el fenómeno de la relajación de IKACh, se ha postulado que el Ca2+ extracelular, el incremento en el Ca2+i, la formación de un complejo Ca2+/calmodulina y una proteína RGS que acelere la hidrólisis de guanosín trifosfato (GTP) de la subunidad de la proteína Gα, son los elementos involucrados en esta propiedad de IKACh (Ishii et al., 2001). El mecanismo molecular propuesto recientemente es que cuando el potencial de membrana se despolariza, la concentración de Ca2+i aledaño a la membrana se incrementa; el Ca2+ se une a la calmodulina y el complejo Ca2+/calmodulina conlleva a una facilitación para que una proteína RGS acelere la hidrólisis de Gα-GTP a Gα-GDP; con esto Gα-GDP es capaz de unir al dímero Gßγ para reconstituir la proteína G trimérica, con lo cual disminuye el número de Gßγ libres y así también el número de canales KACh disponibles con los potenciales más despolarizados. Ahora bien, el incremento dependiente de tiempo de IKACh con la hiperpolarización, referida como relajación, podría interpretarse como la sucesión de los eventos descritos pero de manera inversa, esto es, menos influjo de Ca2+, menos complejo - 14 - Ca2+/calmodulina, menos actividad de la RGS, más Gßγ libres para activar más canales KACh (Ishii et al., 2001; Ishii et al., 2002). Alternativamente, con las evidencias aportadas en este trabajo, proponemos que la relajación de IKACh se explica por la misma propiedad del receptor muscarínico M2, es decir, su dependencia o sensibilidad al voltaje. - 15 - En base a los antecedentes descritos, me permito plantear la siguiente II. HIPÓTESIS En miocitos cardiacos, la activación de IKACh depende de una interacción ligandoreceptor muscarínico M2 modulable por el potencial membranal (voltaje). Esta propiedad explica el fenómeno de la “relajación” de IKACh y el curso temporal (dependencia de tiempo y voltaje) de las corrientes activadas por pilocarpina, colina y tetrametil amonio. III. JUSTIFICACIÓN Los receptores acoplados a proteínas G (RAPG) constituyen la familia más grande de receptores en la membrana celular y están involucrados en la mayoría de los procesos fisiológicos de transducción celular de prácticamente todos los seres vivos. Como es sabido, el principal mecanismo de control de los RAPG es su activación por agonistas específicos, pero estudios de los últimos 20 años parecen indicar que tienen sensibilidad al potencial de membrana (voltaje) lo cual ensancharía enormemente la importancia fisiológica y farmacológica de estos receptores, particularmente en los tejidos excitables (sistema nervioso y corazón). La modulación de un canal rectificador entrante (Kir) por la interacción dependiente de voltaje de los receptores M2 con ACh, sólo se ha mostrado en un sistema de expresión heteróloga. Dado que esta vía de señalización tiene un papel funcional trascendente en la regulación de la actividad cardiaca, y puesto que el receptor M2 es el único subtipo involucrado en esta función, con el presente trabajo pretendemos indagar si la activación de IKACh manifiesta dependencia de voltaje en las células cardiacas de gatos y conejos, que son dos especies animales modelo para estudios del corazón. Asimismo, nos interesa conocer la participación que tiene el sitio ortostático (región del receptor al que se unen los agonistas) en la dependencia de voltaje, para lo cual estudiaremos esta propiedad utilizando agonistas muscarínicos de variada estructura molecular. Finalmente, los resultados obtenidos en las células nativas serán validados en un sistema heterólogo: células HEK293 transfectadas transitoriamente con los ADNc de los receptores M2 y de las subunidades Kir 31. y 3.4, que conforman al canal KACh. - 16 - IV. OBJETIVOS OBJETIVO GENERAL: Determinar si en miocitos de aurícula existe dependencia de voltaje en la activación de la vía de señalización colinérgica (receptores muscarínicos M2, proteínas Gi/o y canales KACh) y destacar la participación del sitio de unión de los agonistas (ortostático) en esta propiedad. OBJETIVOS ESPECÍFICOS: 1. Estudiar, en miocitos cardiacos de gato y conejo, si la activación de IKACh por el agonista fisiológico, ACh, depende del potencial de membrana (voltaje). 2. Determinar, en miocitos auriculares, la participación del sitio ortostático del receptor M2 en la dependencia de voltaje para activar IKACh, mediante la estimulación con agonistas muscarínicos de variada estructura molecular. 3. Validar, en células HEK293 que expresen receptores M2 y canales Kir 3.1/Kir 3.4, el comportamiento dependiente de voltaje observado con los agonistas muscarínicos en los miocitos cardiacos. 4. Valorar la dependencia de voltaje, en el sistema de expresión heteróloga descrito, con mutantes puntuales de los aminoácidos del sitio ortostático del receptor M2. Adicionalmente, con el fin de sustentar de que la sensibilidad al voltaje del receptor M2 es responsable del curso temporal (dependencia de tiempo y voltaje) de las corrientes activadas por pilocarpina, colina y tetrametil amonio, así como del fenómeno de la “relajación” de IKACh, es necesario: 5. Identificar los mediadores celulares involucrados en la activación de las corrientes por los agonistas muscarínicos colina y pilocarpina en miocitos de aurícula. 6. Reproducir la “relajación” de IKACh con ACh ante la presencia del quelante BAPTA (5 mM) en la solución intracelular. - 17 - V. MATERIAL Y MÉTODOS A) Registro en miocitos aislados en fresco de aurícula izquierda de gato y conejo. Obtención de los miocitos aislados Gatos y conejos adultos (hembra o macho) se heparinizaron (1000 U/kg, i.p.) 30 min antes de la aplicación de anestesia con pentobarbital sódico (35 mg/kg, i.p.). Una vez anestesiado el animal, se le extraía rápidamente el corazón mediante una toracotomía bilateral y se colocaba en solución Tyrode burbujeada con gas carbógeno (95% O2 y 5% O2). Los miocitos se aislaron utilizando el método de separación enzimática descrito inicialmente por Isenberg y Klöckner (1982) y modificado por Sánchez-Chapula (1988). Los corazones se montaban de la aorta a un aparato tipo Langendorff para la perfusión retrógrada de la circulación coronaria. Inicialmente, los corazones fueron perfundidos 5-7 min con tyrode (en mM: NaCl 118, KCl 5.4, MgCl2 1.05, NaHCO3 24, NaH2PO4 0.42, dextrosa 11, CaCl2 1.8, taurina 20, pH 7.4) y después otros 5-7 min con tyrode cero Ca2+ para detener las contracciones, promover la relajación de la matriz extracelular y mejorar la separación enzimática subsecuente. Luego, se perfundía colagenasa (0.33 mg/mL, tipo I, Sigma, USA) y proteasa (0.033 mg/mL, tipo XIV, Sigma) disueltas en tyrode cero Ca2+ durante 25-40 min. Después, se hacía un lavado de las enzimas perfundiendo 5 min la solución Kraft-Bruhe (KB; en mM: ácido glutámico 70, KCl 20, taurina 10, KH2PO4 10, creatina 0.5, ácido succínico 5, dextrosa 10, HEPES-K 10, EGTA-K 0.2, pH 7.4), burbujeada previamente con O2 100%. Se disecaba la aurícula izquierda y los miocitos aislados se obtenían por agitación mecánica con unas pinzas finas y con una pipeta Pasteur de punta flameada. Los miocitos se mantenían en solución KB a 4° C hasta el momento del experimento (2-20 h después de su obtención) dejándolas reposar 2 h para que se recuperaran del proceso enzimático. Registros electrofisiológicos de miocitos aislados, en la configuración de célula completa Los registros electrofisiológicos se realizaron a temperatura ambiente (23-25° C) cuando se utilizó el sistema de perfusión rápida (ver más abajo) y a 36.5±0.5° C cuando se usó la perfusión convencional. Las micropipetas de registro se prepararon con capilares de vidrio TW150F-6 (WPI Inc., USA), empleándose un estirador P-97 (Sutter Instruments, USA) y una microforja MF-83 (Narishige, Japón). Se utilizaron micropipetas que tuvieran una resistencia de 1.5-3 MΩ al ser llenadas con solución intracelular (en mM): L-aspartato 80, KH2PO4 10, - 18 - MgSO4 1, KCl 40, HEPES 5, EGTA 10, GTP-Na 0.2 y ATP-Na2 3 (pH 7.25 KOH). Debido a los dos voltajes seleccionados para ensayar las curvas concentración-respuesta, -100 y +50 mV (ver más abajo), la solución del baño de las células era con K+ 20 mM (solución Ca-Co, en mM: NaCl 120, KCl 20, MgCl2 1, Hepes-Na 10, CaCl2 0.5, CoCl2 2, y Glucosa 11, pH 7.4) para evitar corrientes pequeñas a -100 mV por su cercanía con el potencial de inversión (~90 mV) si se usara K+ 4 mM. Las sustancias utilizadas se disolvieron en la solución Ca-Co y se aplicaron a través de un sistema de perfusión rápida o, en algunos casos, con perfusión convencional. Las micropipetas se colocaban en un sujetador (“holder”) que permite la succión y éste se conectaba a un preamplificador que estaba montado en un micromanipulador MO-103 (Narishige) y conectado a la entrada de un amplificador Axopatch 200B (Axon Instruments, USA). La capacidad de membrana y la resistencia en serie se compensaron al menos en un 70%. Se utilizó un puente de agar/KCl para aterrizar el baño de perfusión. Para el registro de las corrientes se empleó un filtro Bessel pasa bajas a 2 KHz. La generación de pulsos y la adquisición y procesamiento de datos ("on line") se realizaron utilizando una computadora, con un convertidor A/D (Digidata 1322A, Axon Instruments) y el programa pClamp (v. 8.0, Axon Instruments). La frecuencia de adquisición fue de 4 KHz. B) Registros en células HEK293 transfectadas transitoriamente. Cultivo y resiembra de las células HEK293. Células HEK293 (American Type Culture Collection, USA) se cultivaron en placas de 35 mm con medio D’MEM (Dulbecco’s modified Eagle’s medium; Gibco, USA) completado con 10% de suero fetal bovino o suero de caballo (Gibco) y antibiótico-antimicótico 2x (200 unidades/mL de penicilina, 200 µg/mL de estreptomicina y 0.50 µg/mL de anfotericina B; Gibco, USA) y se mantenían a 37° C en una incubadora con humedad y 5% de CO2. El mantenimiento de la línea celular se lograba resembrándola a baja densidad cada 3-5 días. El método de dispersión celular para las resiembras y para el registro consistía en aplicar, a la monocapa celular: 30 s Versene®, luego 1 min 05 s tripsina 0.125% (la digestión enzimática se detenía agregando 3 mL de medio); se centrifugaba 2 min a 150 x g, se desechaba el sobrenadante y el botón de células se resuspendía con 5 mL de medio, para trabajar con ellas directamente (si es que ya habían sido transfectadas previamente), o para resembrarse a baja densidad (si era sólo para el mantenimiento de la línea celular. - 19 - Transfección transitoria de las células HEK293 Para las transfecciones las células se resembraban a alta densidad el día previo. Las transfecciones se realizaron tanto con el método de la lipofectamina (Ciccarone et al., 1999) como con el de fosfato de calcio (Jordan y Wurm, 2004). Brevemente, el método de lipofectamina: en un microtubo se agregaban 15 µL de lipofectamina (Invitrogen, USA) a 300 µL de medio OPTI-MEM (Invitrogen), se mezclaba y se dejaba reposar 5 min; mientras tanto, en otro microtubo con 300 µL de medio OPTI-MEM se agregaban cada uno de los ADNc clonados (0.5 µg de Kir 3.1 de rata, 0.5 µg de Kir 3.4 de rata, ambos suministrados amablemente por el Dr. Michael Sanguinetti (Universidad de Utah); 2 µg del receptor muscarínico M2 de humano, suministrado amablemente por el Dr. Ulises Meza (Universidad de San Luis Potosí); y 0.6 µg de proteína verde fluorescente [GFP, por sus siglas en inglés, pGreenlantern-1, Gibco] como gen reportero), se agitaba perfectamente y se adicionaban al tubo de la lipofectamina; la mezcla se dejaba reposar 30 min. El método de fosfato de calcio: a 10 µL de CaCl2 2.5 M se le agregaban c/u de los ADNs clonados (mismas cantidades que las indicadas con el método de lipofectamina) y con agua estéril se completaba hasta 250 µL, se agitaba vigorosamente, y esta mezcla se agregaba poco a poco y agitando a otros 250 µL de medio HBS 2x (en inglés, HEPES-Buffered Saline: en mM, NaCl 140, Na2HPO4 2.8 y HEPES 50, pH 7.05) y se dejaba reposar 1 min. Luego del reposo respectivo de cualquiera de los 2 métodos descritos anteriormente, se procedía a agregar por goteo la mezcla de transfección a la(s) placa(s) con células; 5-6 h después se les cambiaba medio a las placas de cultivo. Registros electrofisiológicos De 12-36 h después de la transfección, se seleccionaban las células con fluorescencia GFP (+) para su registro electrofisiológico. El procedimiento del registro electrofisiológico de las células HEK293 transitoriamente transfectadas con el receptor muscarínico M2 y con las subunidades del canal KACh, Kir 3.1 y 3.4, fue básicamente el descrito previamente para los miocitos (ver más arriba). Debido a que la magnitud de las corrientes activadas por los agonistas muscarínicos en estas células fue relativamente baja, se optó por tratar a las células con butirato de sodio (5 mM) 12-15 hr, posteriores al proceso de transfección, para mejorar la expresión proteica de los ADNs transfectados (Gorman, Howard & Reeves, 1983). - 20 - C) Sistema de perfusión rápida La aplicación y lavado de las sustancias se realizó a través de un sistema de perfusión de multi-pipetas (SF-77B, Warner Instruments, USA). Las células en la cámara de registro eran bañadas con perfusión convencional a una velocidad baja (1 mL/min), una vez que se lograba hacer la configuración de célula adherida, se activaba el sistema de perfusión rápida sobre dicha célula y luego se establecía la configuración de célula completa, tras lo cual, se fijaba un potencial de mantenimiento (PM) de –100 o +50 mV (aleatoriamente: unas veces iniciando el ensayo con un voltaje y otras veces iniciando con el otro) y se perfundían las soluciones. El control de los tiempos de aplicación y lavado de las sustancias sobre las células se realizó con el Clampex del programa pClamp® (Axon Instruments). El flujo de las soluciones a través de las pipetas se estableció por gravedad y la constante de tiempo para el intercambio de solución en un sistema como éste es de ~200 ms (Doupnik, Jaen y Zhang, 2004). Las sustancias se aplicaron por medio de 3 dispositivos (manifolds) que conectaban las múltiples soluciones, contenidas en reservorios, con las respectivas 3 pipetas de perfusión. D) Protocolo experimental para las curvas concentración-respuesta (C-R). Para cada agonista muscarínico, se realizaron curvas concentración-respuesta (C-R) a los dos voltajes de prueba, -100 y +50 mV, de la siguiente manera: con el sistema de perfusión rápida se aplicaban concentraciones crecientes del agonista sólo durante el tiempo necesario para obtener el estado estable de la corriente activada, IKACh, y se procedía a lavar hasta recuperar el nivel de corriente basal, antes de aplicar la siguiente concentración de agonista. Los tiempos de perfusión de los agonistas fueron los mínimos necesarios para activar IKACh, con el fin de evitar al máximo el desencadenamiento de los mecanismos clásicos de desensibilización del receptor, los cuales se echan a andar si se aplican concentraciones máximas de agonista por varios minutos (Krejci, Michal, Jakubik & Dolezal, 2004). De cualquier manera, en experimentos control, confirmamos que al aplicar durante 20 s una concentración máxima de ACh (3 µM), con 20 s de lavado ya hay una total recuperación de IKACh (dato no mostrado). Además, está reportado en varios trabajos del grupo de Lutz Pott, que al aplicar durante 50 s una concentración supramáxima de ACh (10 µM), para el tiempo de lavado en el que se regresa a la corriente basal (~30 s) IKACh ya se encuentra totalmente recuperada (fig. 1 de Meyer et al., 2001). En nuestro caso, para todos los agonistas utilizados, - 21 - el tiempo de perfusión estuvo en el rango de 4 a 18 s, dependiendo de la concentración de agonista; mientras que los tiempos de lavado fueron de 20 a 60 s, según la concentración previa de agonista. Una vez ensayadas las concentraciones de agonista a cierto voltaje, a la célula se le permitía 1-2 min de reposo a -40 mV, tiempo en el que se verificaba la condición de la célula y posteriormente se repetía el mismo procedimiento en el otro voltaje de prueba. Con el propósito de homogenizar las respuestas y poder hacer comparaciones entre las células, entre los voltajes ensayados y entre los agonistas empleados, se midió la corriente activada fraccional, esto es, la IKACh (medida al pico) activada por cualquier concentración de agonista muscarínico se normalizaba respecto a la IKACh inducida por una concentración supramáxima de ACh, 10 µM (Ben-Chaim et al., 2003). E) Experimentos con la PTX Una alícuota de miocitos auriculares se incubó en la solución KB a 37º C durante 2-4 h con PTX (5µg/mL). Antes de su registro, los miocitos tratados con PTX se lavaban 20-30 min en la cámara de registro. A la solución interna de registro también se le agregó PTX (5µg/mL). F) Ajustes de las curvas concentración-respuesta Las curvas concentración-respuesta donde se grafica la magnitud de la corriente activada (IKACh) al pico con respecto a la concentración de agonista muscarínico, se ajustaron con la ecuación de Hill: y=Emáx(xn/(EC50n + xn)), donde: y=amplitud fraccional de IKACh, Emáx=efecto máximo alcanzado, x=concentración de agonista, EC50=concentración que produce el 50% de la respuesta máxima, n=número o coeficiente de Hill. G) Análisis estadístico Los resultados se presentan como los valores promedio ± el error estándar de la media (EEM). Para las curvas C-R, los datos de cada célula se ajustaron individualmente con la ecuación de Hill y los parámetros, EC50, Emáx y n, representan la media±EEM. La significancia de las diferencias se evaluó con la prueba t de Student pareada. - 22 - VI. RESULTADOS (A) PARTICIPACIÓN DEL SITIO DE UNIÓN DE LOS AGONISTAS DEL RECEPTOR M2 EN SU DEPENDENCIA DE VOLTAJE (objetivos del 1 al 4). Dependencia de voltaje en la activación de IKACh por el agonista fisiológico acetilcolina en miocitos cardiacos. Para determinar si en el modelo cardiaco la activación de IKACh por ACh depende del voltaje, se realizaron curvas concentración-respuesta (C-R) a -100 y +50 mV. En la Fig. 7A se muestran los registros representativos en la activación de IKACh por las concentraciones 3, 30, 300 y 3000 nM de ACh en los dos voltajes de prueba: cuando un miocito se mantuvo a -100 mV (Fig. 7Ab), las respuestas fueron del 12, 65, 81 y 95% respecto de la producida por la concentración supramáxima de ACh (10µM), mientras que cuando este mismo miocito se mantuvo a +50 mV (Fig. 7Aa) las respuestas ya sólo fueron de 0, 16, 56 y 95% respecto de la máxima. Los resultados de estos experimentos se resumen en la Fig. 7B donde se muestran las curvas C-R obtenidas a -100 mV (cuadros negros) y a +50 mV (círculos rojos) y donde se observa el corrimiento hacia la derecha de la curva a +50 mV, reflejado en el parámetro de la concentración que provoca el 50% del efecto máximo (EC50) que fue 38±6 nM a -100 mV y 154±18 nM a +50 mV. A 3 30 [ACh], nM 3000 300 B 10000 1.0 1.0 20 s IKACh normalizada 200 pA a 0.5 -100 mV EC50=38±6 nM +50 mV EC50=154±18 nM 500 pA b 0.0 1 10 100 1000 10000 [ACh], nM 20 s Fig. 7 Dependencia de voltaje en la activación de IKACh por ACh en miocitos auriculares de gato. A, Registros de corriente representativos a los potenciales de mantenimiento de +50 mV (a) y -100 mV (b). Las concentraciones de ACh se aplicaron como indican las barras negras horizontales y las corrientes se normalizaron con la producida por una concentración supramáxima de 10 µM. C, curvas concentración-respuesta que resumen los experimentos de 8 células. El cambio en la potencia (EC50) es significativo (p<0.01). En esta gráfica y en las posteriores no se muestran los coeficiente de Hill; consultarlos en la tabla 1. - 23 - Dependencia de voltaje en la activación de IKACh por agonistas muscarínicos en miocitos cardiacos de gato. Carbacol (CarbaCh) y metacolina (MetaCh) son dos agonistas muscarínicos estructuralmente similares a la ACh, con un sólo grupo sustituído (Fig. 18), pero que a diferencia de ésta son de uso clínico debido a su resistencia a la hidrólisis por las enzimas acetilcolinesterasa y butirilcolinesterasa, presentes en sangre y tejidos (Brown y Taylor, 2001). El análisis de las curvas C-R de CarbaCh (Fig. 8) y MetaCh (Fig. 9) mostró una dependencia de voltaje muy similar a la de ACh, mayor potencia a -100 mV que a +50 mV. CarbaCh: EC50=47±12 nM (-100 mV) y 420±103 nM (+50 mV); MetaCh: EC50=122±18 nM (-100 mV) y 537±25 nM (+50 mV). En nuestro sistema, ACh y CarbaCh al ser capaces de generar respuestas máximas (Emáx) del 100% se consideran agonistas totales y para ellos el cambio en la potencia puede ser debido a un cambio en la afinidad, en la eficacia o en ambas (Ross & Kenakin, 2003; Rang, 2006). MetaCh se comportó como agonista parcial (Fig. 9) y el voltaje no tuvo efecto sobre su eficacia (E) pues no hubo cambio en el Emáx, 0.89±0.03 (-100 mV) y 0.88±0.01 (+50 mV), por lo que para este agonista el cambio en la potencia se debe únicamente a un cambio en la afinidad (KD) ligando-receptor. De hecho, bajo nuestras condiciones el empleo de las ecuaciones de Del Castillo-Katz (Ross y Kenakin, 2003; A 10 30 [CarbaCh], nM 100 1000 10000 B 30000 1.0 1.0 20 s IKACh normalizada 200 pA a -100 mV EC50=47±12 nM 0.5 +50 mV EC50=420±103 nM 500 pA b 0.0 1 10 100 1000 10000 100000 [CarbaCh], nM 20 s Fig. 8 Activación de IKACh por CarbaCh con dependencia de voltaje en miocitos auriculares de gato. A, Registros de corriente representativos a los potenciales de mantenimiento de +50 mV (a) y -100 mV (b). Las concentraciones de CarbaCh se aplicaron como indican las barras negras horizontales y las corrientes se normalizaron con la producida por una concentración supramáxima de CarbaCh 30 µM. C, curvas concentraciónrespuesta que resumen los experimentos de 13 células. Las EC50 son significativamente diferentes (p<0.001) - 24 - A 20 s B 0.89±0.03 0.88±0.01 1.0 IKACh normalizada 100 pA a [MetaCh], nM ACh 30 100 1000 10000 30000 10 µM 0.5 -100 mV EC50=122±18 nM +50 mV EC50=537±25 nM 500 pA b 0.0 10 20 s 100 1000 [MetaCh], nM 10000 Fig. 9 Dependencia de voltaje en la activación de IKACh por MetaCh en miocitos auriculares de gato. A, Registros de corriente representativos a los potenciales de mantenimiento de +50 mV (a) y -100 mV (b). Las concentraciones de MetaCh se aplicaron como indican las barras negras horizontales y las corrientes se normalizaron con la producida por una concentración supramáxima de ACh 10 µM. C, curvas concentraciónrespuesta que resumen los experimentos de 6 células. El cambio de la EC50 es significativo (p<0.001), pero no el del Emáx. Kalbaugh et al., 2004; Strange, 2008): E=Emáx/(1-Emáx) y EC50=KD/(1+E) nos ayuda a predecir las probables KD de MetaCh en ambos voltajes: 1.1 µM (-100 mV) y 4.5 µM (+50 mV), dentro del rango 2.5-4 µM encontrado experimentalmente en tejido cardiaco (Fuder & Jung, 1985). Hasta el momento, con los tres agonistas ensayados, el modelo de Parnas (Fig. 1) adquiere mayor respaldo, sin embargo, los resultados con otros agonistas muscarínicos cambian completamente el punto de vista anterior. Betanecol es un agonista muscarínico con uso médico como estimulante del músculo liso del tracto gastrointestinal y de la vejiga urinaria (Brown y Taylor, 2001). En su estructura molecular existen dos grupos sustituídos respecto a ACh (Fig. 18). Betanecol fue capaz de activar IKACh de manera similar en los dos voltajes de comparación (Fig. 10), sin cambio aparente en la potencia (EC50) y en el efecto máximo (Emáx): EC50=3.5±0.4 µM (-100 mV) y 4.2±0.5 µM (+50 mV); con Emáx también muy similares, 0.87±0.02% (-100 mV) y 0.89±0.02% (+50 mV). Así que, con este agonista, el voltaje no ejerce efecto sobre la eficacia (E) ni tampoco sobre la afinidad aparente (KD), la cual andaría en 30-40 µM vs. la reportada experimentalmente para el receptor M2, 80±20 µM (Heitz et al., 1999). - 25 - A 1 [betanecol], µM 30 10 3 100 B ACh 10 µM 1.0 0.87±0.02 0.89±0.02 20 s IKACh normalizada 250 pA a 0.5 -100 mV EC50=3.5±0.4 µM +50 mV EC50=4.2±0.5 µM 1 nA b 0.0 0.1 1 10 100 1000 [betanecol], µM 20 s Fig. 10 Betanecol activa IKACh sin dependencia de voltaje en miocitos auriculares de gato. A, Registros de corriente representativos a los potenciales de mantenimiento de +50 mV (a) y -100 mV (b). Las concentraciones de betanecol se aplicaron como indican las barras negras horizontales y las corrientes se normalizaron con la producida por una concentración supramáxima de ACh 10 µM. C, curvas concentración-respuesta que resumen los experimentos de 6 células. EC50 y Emáx estadísticamente semejantes (p>0.05). Pilocarpina es un agonista muscarínico usado en la clínica para tratar la xerostomía y el glaucoma (Brown y Taylor, 2001). En las curvas C-R de pilocarpina (Fig. 11) se observa el desplazamiento hacia la izquierda de la curva a +50 mV con respecto a la de -100 mV, con diferencias significativas (p<0.001) de la potencia y del efecto máximo: EC50=7.4±2.1 µM (-100 mV) y 1.8±0.04 µM (+50 mV); Emáx=0.64±0.04% (-100 mV) y 0.82±0.04% (+50 mV). Con este agonista, la despolarización favorece un aumento de la eficacia, y posiblemente también de la afinidad aparente: KD=20 µM (-100 mV) y 10 µM (+50 mV), valores similares al encontrado experimentalmente para el receptor M2, 15±2 µM (Heitz et al., 1999). TMA y Ch son dos compuestos de estructura más sencilla que ACh, con la ausencia del grupo carboxilo (Fig. 18), que aunque tradicionalmente no se les considera agonistas muscarínicos, en tejido cardiaco se ha reportado su acción muscarínica disminuyendo la frecuencia y fuerza de contracción (Kennedy et al., 1995; Shi et al., 1999b). Las curvas C-R de TMA (Fig. 12) muestran que la despolarización incrementa notablemente (p<0.01) el efecto máximo, Emáx=0.71±0.05 (-100 mV) y 0.96±0.10 (+50 mV). Sin embargo, el cambio en la eficacia no se ve reflejado en un cambio significativo (p=0.16) en la potencia, pues la EC50=816±245 µM (-100 mV) y 504±249 µM (+50 mV), lo cual sería un indicio de un - 26 - cambio en la afinidad aparente (KD), pero en sentido contrario al de la eficacia, esto es, disminución con la despolarización. A 0.1 [pilocarpina], µM 1 100 10 B ACh 10 µM 1.0 0.82±0.04 200 pA IKACh normalizada a 20 s 0.64±0.04 0.5 +50 mV EC50=1.8±0.4 µM -100 mV EC50=7.4±2.1 µM b 1 nA 0.0 0.1 20 s 1 10 100 [pilocarpina], µM 1000 10000 Fig.11 Pilocarpina activa IKACh con dependencia de voltaje opuesta a la de ACh en miocitos auriculares de gato. A, Registros de corriente representativos a +50 mV (a) y -100 mV (b). Las concentraciones de pilocarpina se aplicaron como indican las barras negras horizontales y las corrientes se normalizaron con la producida por una concentración supramáxima de ACh 10 µM. C, curvas concentración-respuesta que resumen los experimentos de 11 células. El cambio en la EC50 y en el Emáx es estadísticamente significativo (p<0.001). A 30 [TMA], µM 300 3000 30000 ACh 10 µM B 1.0 0.96±0.10 25 s IKACh normalizada 200 pA a 0.71±0.05 +50 mV EC50=504±249 µM 0.5 -100 mV EC50=816±245 µM 1 nA b 0.0 10 100 1000 10000 100000 [TMA], µM 25 s Fig.12 Activación de IKACh por TMA en miocitos auriculares de gato. A, Registros de corriente representativos a los potenciales de mantenimiento de +50 mV (a) y -100 mV (b). Las concentraciones de TMA se aplicaron como indican las barras negras horizontales y las corrientes se normalizaron con la producida por una concentración supramáxima de ACh 10 µM. C, curvas concentración-respuesta que resumen los experimentos de 6 células. El cambio en el Emáx es significativo (p<0.01) pero no el de la EC50. - 27 - Con el agonista Ch no fue posible obtener curvas C-R completas (Fig. 13) y el ajuste extrapolado manifiesta una mayor potencia a +50 mV que a -100 mV. a 100 pA 1 20 s 100 ACh 10 µM B 1.0 IKACh normalizada A [Ch], mM 10 30 0.5 +50 mV EC50=1±0.4 mM -100 mV EC50=49±6 mM 500 pA b 0.0 -3 10 -2 10 -1 10 0 10 1 10 2 10 3 10 4 10 [Ch], mM 20 s Fig.13 Activación de IKACh por Ch en miocitos auriculares de gato. Dependencia de voltaje opuesta a la de ACh. A, Registros de corriente representativos a los potenciales de mantenimiento de +50 mV (a) y -100 mV (b). Las concentraciones de TMA se aplicaron como indican las barras negras horizontales y las corrientes se normalizaron con la producida por una concentración supramáxima de ACh 10 µM. C, curvas concentraciónrespuesta que resumen los experimentos de 7 células. El cambio en la EC50 es significativo (p<0.001). Dependencia de voltaje en la activación de IKACh por agonistas muscarínicos en miocitos cardiacos de conejo. Debido a los tres tipos de comportamientos dependientes de voltaje en la activación de IKACh con los 7 agonistas muscarínicos ensayados en los miocitos cardiacos de gato, decidimos evaluar 3 agonistas (ACh, pilocarpina y betanecol), representativos de cada uno de los comportamientos de dependencia de voltaje que se observaron en el gato, en otra especie animal ampliamente utilizada para estudios en tejidos cardiacos, el conejo. En general, en los miocitos de aurícula de conejo, los 3 agonistas mostraron la misma tendencia que en gato. Con ACh (Fig. 14A y 14C, cuadros) se observa una menor potencia a +50 mV (Fig. 14 C, cuadros rojos) con una EC50 de 383±24 nM, que la que se obtiene a -100 mV (Fig. 114, cuadros negros) con una EC50=122±12 nM. Por el contrario, pilocarpina muestra incrementos significativos en su potencia y en el efecto máximo con la despolarización (Fig. 14B y 14C, círculos): EC50=14±4 µM y Emáx=0.32±0.02% a -100 mV (Fig. 14C, círculos negros) vs. EC50=3±0.8 µM y Emáx=0.58±0.03% a +50 mV (Fig. 14C, - 28 - A 1 [betanecol], µM 10 300 30 A 0.01 0.03 [ACh], µM 1 0.1 3 B [pilocarpina], µM 0.3 10 3 30 300 ACh 10 µM 200 pA 50 pA PM= +50 mV 25 s 25 s 1 nA 500 pA PM= -100 mV 25 s C IKACh normalizada Fig. 14 ACh y pilocarpina activan IKACh con dependencia de voltaje opuesta en miocitos auriculares de conejo. Registros de corriente representativos a los potenciales de mantenimiento de +50 mV y -100 mV. Las concentraciones de ACh (A) y pilocarpina (B) se aplicaron como indican las barras negras horizontales y las corrientes se normalizaron con la producida por una concentración supramáxima de ACh, 10 µM. C, Curvas concentración-respuesta que resume los datos de ACh (cuadros) y pilocarpina (círculos) a los potenciales de 100 mV (negro) y +50 mV (rojo). n=4 (ACh) y n=11 (pilocarpina). El cambio en la EC50 de ACh es significativo (p<0.05) y para pilocarpina el cambio en la EC50 y en el Emáx también es significativo (p<0.001). 25 s ACh 1.0 1.0 pilocarpina 0.58±0.03 +50 mV EC50=3±0.8 µM -100 mV 0.5 EC50=122±12 nM +50 mV EC50=383±24 nM 0.32±0.02 -100 mV EC50=14±4 µM 0.0 1E-3 0.01 0.1 1 [Agonista], µM 10 100 1000 B ACh 10 µM 1.0 0.94±0.07 a 25 s IKACh normalizada 50 pA 0.86±0.08 +50 mV EC50=12±3 µM 0.5 -100 mV EC50=12±4 µM 500 pA b 0.0 1 25 s 10 [betanecol], µM 100 1000 Fig. 15 Betanecol activa IKACh sin dependencia de voltaje en miocitos auriculares de conejo. A, Registros de corriente representativos a +50 mV (a) y -100 mV (b). Betanecol se aplicó como indican las barras negras horizontales y las corrientes se normalizaron con la producida por una concentración supramáxima de ACh (10 µM). B, curvas concentración-respuesta que resumen los resultados de 8 miocitos. El ajuste de las dos curvas no es significativamente diferente (p>0.05) en la potencia ni en el efecto máximo. - 29 - círculos rojos). La mayor potencia de pilocarpina a +50 mV es resultado de un aumento en la eficacia, pero probablemente también de la afinidad aparente: KD=20 µM (-100 mV) y 7 µM (+50 mV), valores que también son congruentes con la KD experimental, 15±2 (Heitz et al., 1999). Con betanecol (Fig. 15) no parece haber un cambio significativo en la potencia, EC50 de 12±4 µM (-100 mV) y 12±3 µM (+50 mV), ni tampoco en el Emáx: 0.86±0.08% (-100 mV) y 0.94±0.07% (+50 mV). Por lo que también en esta especie, el potencial membranal no parece influir en la afinidad ni en la eficacia del betanecol para activar IKACh. Dependencia de voltaje en la activación de IKACh por agonistas muscarínicos en células HEK293 que expresan heterólogamente receptores M2 y canales Kir 3.1/3.4 (canales KACh). La dependencia de voltaje en la activación de IKACh en tejido cardiaco que, como se acaba de revisar, también está determinada por el agonista muscarínico (ligando), se estudió paralelamente reconstituyendo la vía de señalización en células HEK293. Para este estudio se utilizaron 5 agonistas muscarínicos: ACh, CarbaCh, betanecol, pilocarpina y TMA. A pesar de ciertas diferencias en la potencia y en el efecto máximo de las curvas C-R entre las células de gato, conejo y de las HEK293 transfectadas (Tabla 1), la dependencia de voltaje en la inducción de IKACh se sigue reproduciendo en el modelo de expresión heteróloga de manera muy similar a lo observado en las células cardiacas. Con este sistema de expresión heteróloga, al igual que lo que se observó en las células auriculares de gato y conejo: ACh (Fig. 16A y 16C, cuadros) y CarbaCh (Fig. 17A y 17D, cuadros) manifiestan su mayor potencia a -100 mV que a +50 mV (ver parámetros en la Tabla 1, y el cambio de los mismos con respecto al voltaje en la Tabla 2). Con betanecol (Fig. 17B y 17D, círculos) el voltaje no ejerce influencia sobre la potencia ni sobre el efecto máximo, por lo que con este agonista el voltaje no parece modificar la afinidad ni la eficacia para activar IKACh. Pilocarpina (Fig. 16B y 16, círculos) cambia significativamente la potencia y el efecto máximo, y el incremento de la potencia es debido al aumento en la eficacia y a un posible aumento de la afinidad aparente (KD). Con TMA (Fig. 17C y 17D, triángulos) la despolarización también produce un marcado incremento del efecto máximo, indicando el aumento de la eficacia, pero sin efecto significativo sobre la potencia, producto de una probable disminución en la afinidad con la despolarización. - 30 - A [ACh], µM 0.030 B 1 10 Fig. 16 ACh y pilocarpina activan IKACh con dependencia de voltaje opuesta en células HEK293 que expresan heterólogamente receptores M2 y canales Kir 3.1/3.4 (canales KACh). Registros de corriente representativos a +50 mV y -100 mV. Las concentraciones de ACh (A) y pilocarpina (B) se aplicaron como indican las barras negras horizontales y las corrientes se normalizaron con la producida por una concentración supramáxima de ACh (10 µM). C, Curvas concentración-respuesta que resume los datos de ACh (cuadros) y pilocarpina (círculos) a los potenciales de -100 mV (negro) y +50 mV (rojo). n=7 (ACh) y n=9 (pilocarpina). El cambio en la potencia es significativamente diferente para ACh (p<0.01) y para pilocarpina tanto las EC50 como las Emáx son estadísticamente distintas (p<0.01). ACh 10 µM [pilocarpina], µM 10 25 pA 25 pA 0.010 10 s 10 s HP= +50 mV 500 pA 20 s 20 s ACh normalizada 1.0 pilocarpina -100 mV 0.5 +50 mV IK ACh C 500 pA HP= -100 mV -100 mV 0.0 1E-4 1E-3 0.01 0.1 [Agonista], µM 1 10 100 A [TMA], µM 100 300 ACh 10 µM 100 pA 50 pA 20 pA ACh 10 µM 10 s 10 s 10 s 10 s D 200 pA 500 pA 250 pA 10 s 10 s betanecol CarbaCh 1.0 +50 mV IKACh normalizada Fig. 17 Activación de IKACh por CarbaCh, betanecol y TMA en células HEK293 expresando heterólogamente receptores M2 y canales Kir 3.1/3.4. Registros de corriente representativos a los potenciales de mantenimiento de +50 mV y -100 mV. Las concentraciones de CarbaCh (A), betanecol (B) y TMA (C) se aplicaron como indican las barras negras horizontales y las corrientes se normalizaron con la producida por una concentración supramáxima de CarbaCh, 30 µM o de ACh, 10 µM. D, Curvas concentraciónrespuesta que resume los datos de CarbaCh (cuadros), betanecol (círculos) y TMA (triángulos) a los potenciales de -100 mV (negro) y +50 mV (rojo). n=8 (CarbaCh), n=7 (betanecol) y n=6 (TMA). Las EC50 de CarbaCh son diferentes (p<0.01). TMA manifiesta Emáx distintas (p<0.001) pero EC50 estadísticamente similares. Las curvas de betanecol son semejantes en la potencia y en el efecto máximo. C B [betanecol], µM 1 10 [CarbaCh], µM 0.03 0.1 30 -100 mV 0.5 +50 mV -100 mV +50 mV TMA -100 mV 0.0 -2 10 -1 10 0 10 1 10 [Agonista], µM - 31 - 2 10 3 10 4 10 En la tabla 1 se muestra el desglose general de los parámetros EC50, Emáx y coeficiente de Hill (n) de las curvas C-R de los agonistas muscarínicos obtenidas en los dos voltajes de prueba para los 3 modelos celulares; y en la tabla 2 aparecen el cambio en Emáx (∆Emáx) y el cociente de las EC50 a dichos voltajes. Tabla 1 Parámetros de las curvas C-R de los agonistas muscarínicos ensayados en los 3 modelos celulares. Emáx=efecto máximo; EC50=concentración que produce el 50% de la respuesta; n=coeficiente de Hill. En color negro los valores obtenidos a -100 mV y en rojo los de +50 mV. Con los agonistas totales ACh y CCh el ajuste se fijó al 100% para todas las células por lo que no se reporta el error estándar. MIOCITOS GATO MIOCITOS CONEJO CÉLULAS HEK293 Agonista Emáx EC50 n Emáx EC50 n Emáx EC50 n (%) (%) (%) 100 38±6 nM 0.8±0.1 100 122±12 nM 0.8±0.1 100 14±1 nM 0.7±0.1 ACh CarbaCh MetaCh Betanecol Pilocarpina TMA Ch 100 100 100 89±3 88±1 87±2 89±2 64±4 82±4 71±5 96±10 ? ? 154±18 nM 47±12 nM 420±103 nM 122±18 nM 537±25 nM 3.5±0.4 µM 4.2±0.5 µM 7.4±2.1 µM 1.8±0.04 µM 816±245 µM 504±249 µM 49±6 mM 1±0.4 mM 1.0±0.1 1.0±0.2 0.8±0.1 1.3±0.1 1.3±0.1 1.0±0.1 0.8±0.1 0.9±0.1 0.8±0.1 0.9±0.2 0.7±0.2 0.7±0.1 0.5±0.1 100 86±8 94±7 32±2 58±3 - 383±24 nM 12±4 µM 12±3 µM 14±4 µM 3±0.8 µM - 1.0±0.1 0.8±0.1 0.9±0.1 0.9±0.1 0.8±0.1 - 100 100 100 100±3 109±4 77±2 89±1 73±2 94±7 - 62±4 nM 104±6 nM 328±25 nM 5.2±0.8 µM 7.0±1.2 µM 1.7±0.2 µM 0.6±0.1 µM 185±30 µM 106±37 µM - 0.9±0.1 0.8±0.1 0.9±0.1 0.8±0.1 0.7±0.1 0.8±0.1 0.8±0.1 0.8±0.1 0.9±0.2 - Tabla 2 Cambio de la potencia (EC50) y del efecto máximo (Emáx) entre los voltajes -100 y +50 mV de los agonistas muscarínicos ensayados en los 3 modelos celulares. ∆Emáx=(Emáx a +50 mV)-(Emáx a -100 mV). MIOCITOS GATO MIOCITOS CONEJO CÉLULAS HEK293 Agonista EC50 a +50 EC50 a +50 EC50 a +50 ∆Emáx (%) ∆Emáx (%) ∆Emáx (%) EC50 a -100 EC50 a -100 EC50 a -100 ACh CarbaCh MetaCh Betanecol Pilocarpina TMA Ch 1 4.0 8.9 4.4 1.2 8 3.1 1.0 10 4.4 3.2 1.3 ∆Emáx (%) EC50 a -100 EC50 a +50 ∆Emáx (%) EC50 a -100 EC50 a +50 ∆Emáx (%) EC50 a -100 EC50 a +50 18 25 - 4.0 1.6 49? 26 - 4.7 - 12 21 - 2.8 1.7 - 2 Finalmente, en la Fig. 18 se resume el comportamiento de dependencia de voltaje de los agonistas muscarínicos para activar IKACh, y se muestran sus estructuras moleculares. - 32 - Fig. 18 Estructuras moleculares de los agonistas muscarínicos que activan IKACh con un patrón diferencial de dependencia de voltaje. Agonistas con mayor potencia con la hiperpolarización (izquierda), con igual potencia a -100 y +50 mV (centro) y con mayor potencia y eficacia con la despolarización (derecha). Residuos aminoácidos del sitio ortostático del receptor M2 involucrados con la dependencia de voltaje. Se realizaron curvas C-R en células HEK293 que expresaban heterólogamente canales Kir 3.1/3.4 y receptores M2 con mutaciones puntuales de 3 de los residuos aminoácidos, W99, D103 y S107 (Fig. 2), reportados como críticos en la interacción con la ACh (Pedretti et al., 2006). Con el M2 D103A no se logró la activación de corriente alguna. Con las mutaciones M2 S107A y M2 W99A sí se obtuvieron corrientes cuantificables a ambos voltajes de prueba. Con estos dos mutantes del receptor M2 hubo una disminución significativa de la afinidad, de tal manera que la concentración supramáxima de ACh, utilizada como referencia para normalizar las respuestas de las otras concentraciones, fue de 300 µM (Figs. 19 y 20). Sin embargo, en lo que respecta a la dependencia de voltaje, el M2 S107A se comportó similar al receptor M2 nativo, mayor potencia a -100 mV que a +50 mV (Fig. 19; EC50 de 5.4±1.0 vs. 17.3±4.6 µM, respectivamente), incluso con una magnitud de cambio en la potencia debido al voltaje (EC50 a +50 mV÷ EC50 a -100 mV) de 3.2, semejante a la obtenida con el receptor M2 nativo en los 3 modelos celulares antes descritos (Tabla 2). Por su parte, en el mutante M2 W99A ya no se observó dependencia de voltaje, siendo la EC50 de 3.0±0.4 µM a -100 mV y de 3.2±0.5 µM a +50 mV (Fig. 20). - 33 - A [ACh], µM 10 3 B 300 1.0 IKACh normalizada 50 pA a 10 s 0.5 b 1.0 -100 mV EC50=5.4±1.0 µM 200 pA 0.0 0.1 1 10 100 1000 [ACh], µM M2 S107A 10 s +50 mV EC50=17.3±4.6 µM Fig. 19 Dependencia de voltaje en la activación de corrientes por ACh en células HEK293 que expresan heterólogamente receptores M2 S107A y canales Kir 3.1/3.4. A, Registros de corriente representativos a +50 mV (a) y -100 mV (b). Las concentraciones de ACh se aplicaron como indican las barras negras horizontales y las corrientes se normalizaron con la producida por una concentración supramáxima de 300 µM. B, curvas concentración-respuesta que resumen los experimentos de 7 células. El cambio en la potencia (EC50) es significativamente diferente (p<0.05). Los coeficientes de Hill fueron de 0.7±0.1 a -100 y +50 mV. A 10 s B 300 1.0 1.0 +50 mV EC50=3.2±0.5 µM -100 mV EC50=3.0±0.4 µM IKACh normalizada 5 pA a 3 [ACh], µM 10 0.5 b 100 pA 0.0 10 s 0.1 M2 W99A 1 10 100 [ACh], µM Fig. 20 La activación de corrientes por ACh en células HEK293 que expresan heterólogamente receptores M2 W99A y canales Kir 3.1/3.4 no depende del voltaje. A, Registros de corriente representativos a +50 mV (a) y -100 mV (b). Las concentraciones de ACh se aplicaron como indican las barras negras horizontales y las corrientes se normalizaron con la producida por una concentración supramáxima de 300 µM. B, curvas concentración-respuesta que resumen los experimentos de 8 células. La potencia (EC50) de ambos voltajes no es significativamente diferente (p<0.05). Los coeficientes de Hill fueron 0.8±0.1 (-100 mV) y 0.9±0.1 (+50 mV). - 34 - Dependencia de voltaje en la acción de methoctramina, antagonista M2, en miocitos cardiacos. Con el fin de explorar la hipótesis si la acción de los antagonistas también depende del potencial membranal, se decidió evaluar el efecto antagónico de la methoctramina (Meth), antagonista M2, sobre la activación de IKACh por alguno de los agonistas totales. Con esta idea, se planeó un experimento en el que se utilizaron concentraciones de CarbaCh que produjeran el 50% de respuesta a -100 y a +50 mV, las cuales fueron 50 y 400 nM, respectivamente (Fig. 21C y ver EC50 en Tabla 1). En la Fig. 21A, a -100 mV, CarbaCh 50 nM produce una IKACh de 54% de la máxima posible; al aplicar Meth 100 nM+CarbaCh 50 nM IKACh es 17% respecto de su control, con una capacidad antagónica de Meth de 83%. Posteriormente, a este miocito se le fijó a +50 mV: CarbaCh 400 nM activó una IKACh 50% de la máxima que produce CarbaCh 30 µM (Fig. 21B); al aplicar CarbaCh 400 nM+Meth 100 nM la IKACh inducida fue de 51% respecto de su control, con un efecto antagonista de Meth de 49% a este voltaje. En la Fig. 21C se muestra el resultado promedio de 4 experimentos, que nos indica que Meth tiene mayor efecto antagonista a -100 mV del que tiene a +50 mV, al menos cuando IKACh se activa con CarbaCh. A CCh 50 nM B Meth 100 nM CCh 50 nM Meth 100 nM CCh 400 nM CCh 30 µM 500 pA CCh 30 µM 1000 pA 10 s 10 s PM= -100 mV Respuesta (%) C 100 PM= +50 mV CCh 50 nM CCh 50 nM + Meth 100 nM CCh 400 nM CCh 400 nM + Meth 100 nM 80 60 40 *** 20 0 PM= -100 mV PM= +50 mV - 35 - CCh 400 nM Fig. 21 Antagonismo dependiente de voltaje de la methoctramina (Meth) en la activación de IKACh por CarbaCh en miocitos auriculares de gato. Registros representativos a -100 (A) y +50 mV (B) donde primero se comprueba que las concentraciones de 50 y 400 nM sean equiefectivas, en este caso con una respuesta de ~50% en su respectivo voltaje. Posteriormente se vuelven a aplicar estas concentraciones de CarbaCh pero ahora en presencia del antagonista. C, Resumen de los experimentos, normalizando la IKACh activada por agonista+ antagonista, respecto de la producida sólo por el agonista. n=4. (B) DEPENDENCIA DE VOLTAJE PARA ACTIVAR IKACh vs. LA HIPÓTESIS DE LOS MÚLTIPLES RECTIFICADORES EN MIOCITOS CARDIACOS (Objetivo 5) Según observamos en los registros de las Figs. 11, 14B y 16B, pilocarpina tiene mayor potencia y eficacia para activar IKACh con la despolarización, y viceversa con la hiperpolarización; pero estos registros se realizaron manteniendo fijo el voltaje y dando pulsos de concentraciones crecientes de pilocarpina. Ahora bien, si cambiamos la estrategia para registrar IKACh, esto es, manteniendo constante una concentración dada de pilocarpina y dar pulsos de voltaje, ¿veremos aumentar la activación de IKACh al pasar a voltajes despolarizados?, ¿podrá observarse disminuir la activación de IKACh al ir a voltajes hiperpolarizados? Experimentos realizados con esta estrategia en miocitos auriculares de gato muestran que, efectivamente, una vez alcanzada una concentración de pilocarpina (perfusión convencional), al ir de -40 mV a voltajes más positivos se observa a IKACh aumentar de una forma que depende del tiempo y del voltaje (Fig. 22), mientras que al ir de -40 a -100 o -120 mV puede verse que la activación de IKACh va disminuyendo gradualmente (Figs. 22C y D). 2.5 s B A -40 mV -40 mV 200pA -120 mV 200pA +60 mV 2.5 s 1s E 1s pilocarpina 1 µM 25 Control 20 Pilocarpina 1 µM Pilocarpina 10 µM 15 Pilocarpina 100 µM 10 Control 5 -80 -60 -40 -20 V (mV) 0 -5 -10 200pA -15 0 20 40 60 D 200pA C -100 I (pA/pF) -120 1s 1s pilocarpina 10 µM pilocarpina 100 µM Fig. 22 Curso temporal de las corrientes activadas por pilocarpina con pulsos de voltaje en miocitos auriculares de gato. Registros obtenidos a 36.5º C, con perfusión convencional y con el protocolo de pulsos inserto, en condiciones control (A), con pilocarpina 1µM (B), 10 µM (C) y 100 µM (D). Nótese la misma escala temporal y de corriente. E, curva I-V de la corriente al final del pulso. La línea punteada señala el nivel cero de corriente. En estos registros se utilizó solución Ca-Co con K+ 4 mM. n=3. - 36 - Como ya se mencionó en la introducción (pág. 11), en miocitos auriculares de varias especies animales se ha reportado que Ch (Fig. 5), TMA y 4-AP activan corrientes con características dependientes de tiempo y voltaje semejantes a la de la Fig. 22 cuando se aplica un protocolo de pulsos de voltaje. Existen dos hipótesis respecto a estas corrientes, por un lado está la de nuestro grupo que desde hace varios años hemos venido sugiriendo que se trata de la activación dependiente de voltaje de IKACh (Navarro-Polanco y Sánchez-Chapula, 1997; Benavides-Haro, 2001; Benavides-Haro et al., 2003), de la cual hemos dado una mejor valoración con las curvas C-R y la perfusión rápida (primera sección de resultados); y por otro lado, dado el curso temporal de estas corrientes, se ha propuesto que se trata de nuevos rectificadores tardíos, denominados IKM3 e IKM4, activados por receptores muscarínicos M3 (con pilocarpina, TMA y Ch) o M4 (con 4-AP) y proteínas Gq/11 o Gi/o, respectivamente (Shi et al., 1999a; Wang et al., 1999; Yeh et al., 2007) y que se agregarían a los rectificadores tardíos cardiacos ya caracterizados, IKr e IKs. En esta segunda sección de resultados, igualmente con la estrategia de la perfusión rápida de los agonistas y manteniendo fijo el voltaje, se presentan más evidencias que refuerzan nuestra hipótesis de que los mediadores celulares sobre los que actúan pilocarpina y Ch, y probablemente TMA, 4-AP y betanecol, son los mismos sobre los que actúa ACh: receptores muscarínicos M2, proteínas Gi/o y los canales KACh como efectores. Se evitó el uso del perfil farmacológico para inferir el receptor muscarínico involucrado debido a la inespecificidad frecuente de los antagonistas actuales (Wess, 1996; Brodde y Michel, 1999; Meyer et al., 2001; Benavides-Haro, 2001). Vía de señalización activada por Ch. Una evidencia en el sentido de probar si Ch utiliza los mismos mediadores celulares que ACh, es realizar un protocolo a un voltaje fijo y con la perfusión rápida perfundir ambas sustancias al mismo tiempo y observar si la corriente generada es la suma de las corrientes activadas por separado, en cuyo caso indicaría que ambas corrientes utilizan distintos efectores, pero si la corriente inducida por ambas sustancias no es mayor que cualquiera de las corrientes aplicadas por separado, nos orientaría a pensar que se trata de los mismos canales efectores que activan a las dos corrientes, en este caso los canales KACh. La Fig. 23 deja ver que la segunda opción es la más probable. Siguiendo con la comparación de las corrientes activadas por Ch y ACh, en la Fig. 24 se muestran las respuestas inducidas por concentraciones equiefectivas de Ch y ACh, es decir - 37 - A Fig. 23 La perfusión conjunta de Ch y ACh induce una ACh 1 µM B ACh 1 µM Ch 10 mM ACh 1 µM ACh 1 µM ACh 1 µM Ch 10 mM ACh 1 µM corriente que no es la suma de las corrientes que activan por en un miocito auricular de gato a –50 mV (A) y a +50 (B) mV. n = 2. 50 pA Registros 10 s 50 pA separado. 10 s PM= +50 mV PM= -50 mV concentraciones que producen el mismo nivel de activación respecto de una máxima posible. Así tenemos, por ejemplo, que a -100 mV Ch 1 mM activa una corriente de magnitud y cinéticas de activación y desactivación perfectamente comparables a la activada por ACh 3 nM (Fig. 24A); mientras que a +50 mV esta misma concentración de Ch, 1 mM, induce una corriente de magnitud y cinéticas de activación, desensibilización y desactivación casi exactas a la corriente inducida por ACh 100 nM (Fig. 24B). Este hallazgo es una evidencia más de que ACh y Ch activan la misma corriente en miocitos cardiacos. A b a ACh 10 µM ACh 10 µM ACh 3 nM ACh 3 nM 500 pA 500 pA Ch 1 mM c Ch 1 mM 10 s 10 s ACh 10 µM B d 10 s Ch 100 mM ACh 10 µM ACh 1 µM ACh 100 nM 100 pA 100 pA Ch 1 mM f e ACh 10 µM ACh 10 µM 10 s Ch 1 mM ACh 100 nM Fig. 24 Concentraciones equiefectivas de Ch y ACh para activar IKACh en miocitos auriculares de gato. Corrientes inducidas por Ch (a y d) y ACh (b y e) a un potencial de mantenimiento de –100 (A) y +50 mV (B). c, trazos a y b sobrepuestos. f, trazos d y e sobrepuestos. - 38 - En células HEK293 que expresan heterólogamente receptores M2 y canales Kir 3.1/3.4 se observa la activación de IKACh por Ch (Fig. 25A), mientras que en células HEK293 control, sin receptor M2, la Ch no es capaz de inducir corriente alguna (Fig. 25B). Ch 10 mM ACh 10 µM Ch 10 mM ACh 10 µM Ch 10 mM 10 s 10 s PM= -100 mV PM= -50 mV B ACh 10 µM 10 s 500 pA 1 nA 20 pA A ACh 10 µM PM= +50 mV C a 2.5 s +40 mV Ch 10 mM -40 mV 100 pA -40 mV c b 250 pA 10 s -120 mV PM= -50 mV Fig. 25 Activación de IKACh por Ch en células HEK293. A, registro de células cotransfectadas transitoriamente con los ADN del receptor M2 y de las subunidades Kir 3.1 y 3.4 (formadoras del canal canal KACh). B, registro de células HEK293 control transfectadas sin el ADN del receptor M2. C, registros con protocolo de pulsos (a) en misma célula que en B (b) y en una célula no transfectada (c). Mediadores celulares activados por pilocarpina. Para inferir el tipo de proteína G y el canal de K+ activados por pilocarpina en miocitos cardiacos, se utilizó PTX (inhibidor específico de proteínas Gi/o) y tertiapina (potente bloqueador selectivo de IKACh (Drici et al., 2000; Kitamura et al., 2000)). La valoración del efecto de estos fármacos se hizo sobre la corriente inducida por pilocarpina 30 µM (ver Fig. 11B). Tanto a -100 mV como a +50 mV las corrientes activadas por pilocarpina 30 µM fueron drásticamente reducidas en los miocitos tratados con PTX respecto de los que no fueron tratados, con lo cual implica que pilocarpina activa receptores muscarínicos acoplados a proteínas del tipo Gi/o, sensibles a PTX (Fig. 26). Por su parte, tertiapina 300 nM bloqueó casi completamente las corrientes activadas por pilocarpina 30 µM, indicando que los canales de K+ involucrados son los canales KACh (Fig. 27). - 39 - A a b Pilocarpina 30 µM C 5s 14 PM= -100 mV 12 B c Pilocarpina 30 µM d 50 pA 50 pA Pilocarpina 30 µM 5s PTX 16 IKpilocarpina 30 µM (pA/pF) 5s NO PTX 18 200 pA 200 pA Pilocarpina 30 µM 10 8 *** 6 4 ** 2 5s 0 -100 mV +50 mV PM= +50 mV Fig. 26 El pretratamiento con PTX inhibe las corrientes activadas por pilocarpina en miocitos auriculares de gato. Corrientes activadas por pilocarpina 30 µM, a -100 (A) y +50 mV (B), en miocitos pretratados (b y d) y no tratados (a y c) con PTX. C, densidad de corriente en los miocitos tratados y no tratados con PTX. n=6. p<0.01 (**) y p<0.001 (***) respecto de su control. Ver en métodos el tratamiento de los miocitos con PTX. A B 50 pA Tertiapina 300 nM Pilocarpina 30 µM 100 pA Tertiapina 300 nM Pilocarpina 30 µM 25 s 25 s PM= +50 mV C IKPilocarpina 30 µM normalizada PM= -100 mV 1.0 IKpilocarpina 30 µM IK pilocarpina 30 µM+tertiapina 300 nM 0.5 *** *** 0.0 PM= -100 mV PM= +50 mV Fig. 27 Efecto de tertiapina (potente bloqueador selectivo de IKACh) sobre las corrientes activadas por pilocarpina en miocitos auriculares de gato. Trazos representativos de las corrientes activadas por pilocarpina 30 µM a -100 mV (A) y +50 mV (B). C, resumen de 6 experimentos, normalizando las corrientes activadas por pilocarpina+tertiapina respecto de la producida por pilocarpina. p<0.001 (***) respecto a su control. - 40 - Curso temporal (dependencia de tiempo y voltaje) de las corrientes activadas por pilocarpina con pulsos de voltaje en células cardiacas y en células HEK293 que expresan heterólogamente receptores muscarínicos M2 y canales Kir 3.1/3.4. Finalmente, para demostrar que las corrientes dependientes de tiempo y voltaje activadas por Ch (Fig. 5) y pilocarpina (Fig. 22) en miocitos cardiacos no son resultado de la activación de supuestos rectificadores tardíos, IKM3 o IKM4, a través de receptores muscarínicos M3 o M4, sino más bien de la activación dependiente de voltaje de los receptores muscarínicos M2 sobre los canales KACh, otra evidencia sólida se obtuvo aplicando protocolo de pulsos de voltaje en células HEK293 que expresan heterólogamente receptores M2 y canales Kir 3.1/3.4. Hay que mencionar que estas células poseen varias corrientes de K+ endógenas (Jiang, Sun, Cao y Wang, 2002), entre ellas unas de tipo Ito, que prácticamente impiden la reproducción de las corrientes salientes con apariencia de rectificador tardío, pero en cambio, al aplicar un protocolo de doble pulso que consistía en lo siguiente: del potencial de mantenimiento se daba un primer pulso de 2.5-3.5 s de duración desde -100 hasta +60 mV con incrementos de 40 mV y posteriormente se daba el segundo pulso de igual duración (2.5-3.5 s) a un voltaje hiperpolarizado (-100 o -120 mV) para observar la corriente de cola resultante de la desactivación de los canales activados en el voltaje previo (Fig. 28). Con este protocolo, en presencia del agonista pilocarpina, observamos claramente las características dependientes de tiempo y voltaje de las corrientes, entrantes, de cola en los miocitos cardiacos de conejo (Fig. 28B) y de gato (Fig. 28C) perfectamente reproducibles en las células HEK293 reconstituídas con la vía de señalización de nuestro interés, receptores M2 y canales KACh (Fig. 28A). En las gráficas de la derecha de la Fig. 28 se muestra la relación entre la magnitud de la corriente de cola (el pico de la corriente de cola menos la corriente al final de ese pulso hiperpolarizante) y el voltaje previo, de donde resulta que a mayor voltaje de activación (con el primer pulso) mayor es la corriente de cola que se desactiva (con el segundo pulso); y esta relación es dependiente de la concentración de agonista. Por el contrario, en condición control (sin pilocarpina) las corrientes de cola son despreciables o nulas (izq. de la Fig. 28) e independientes de voltaje (líneas negras en gráficas de la derecha de Fig. 28) en los 4 tipos celulares ensayados. Con el protocolo anterior se obtuvieron resultados semejantes con los agonistas Ch y TMA (datos no mostrados). - 41 - V (mV) 0 A -100 -80 -60 -40 -20 +60 mV -10 mV -100 mV 3.5 s -100 mV 3.5 s -40 mV 3s pilocarpina 3 µM 0 20 40 60 -15 I (pA/pF) Control -5 -10 -20 -25 -30 -35 -40 500 pA -45 -50 -55 1s B -60 -65 1 0 -100 +60 mV -40 -20 -120 mV 2.5 s 1s 0 20 40 60 V (mV) -1 -2 pilocarpina 3 µM -3 I (pA/pF) 2.5 s 500 pA Control -60 2.5 s -40 mV -40 mV -100 mV -80 -4 -5 Control pilocarpina 1 µM pilocarpina 3 µM -6 -7 -8 -9 C V (mV) 0 -100 -80 -60 -40 -20 0 20 40 60 -5 Control pilocarpina 3 µM 500 pA I (pA/pF) -10 1s -15 -20 -25 -30 -35 V (mV) Fig. 28 Curso temporal (dependencia de tiempo y voltaje) de las corrientes activadas por pilocarpina con pulsos de voltaje en células HEK293 que expresan heterólogamente receptores muscarínicos M2 y canales Kir 3.1/3.4 (A), y en células cardiacas de conejo (B) y gato (C). Trazos representativos de corrientes activadas con protocolo de pulsos de voltaje (ver insertos) en situación control (izq.) o con pilocarpina 3 µM (centro). A la derecha se muestra cada gráfica que relaciona la diferencia entre el pico de la corriente de cola menos la corriente al final del pulso hiperpolarizante (cuando ya se desactivó la corriente de cola) en función del voltaje previo. Las líneas punteadas indican el nivel cero de corriente. Las células se registraron a 36.5 C con perfusión convencional de las sustancias. n=1-4 células. - 42 - (C) LA SENSIBILIDAD AL VOLTAJE DE LOS RECEPTORES M2 DETERMINA LA “RELAJACIÓN” DE IKACh EN MIOCITOS CARDIACOS (objetivo 6). Hemos mencionado en la introducción (pág. 13) que al registrar en miocitos cardiacos con protocolo de pulsos de voltaje se observa que la corriente de K+ inducida por ACh, IKACh, manifiesta una propiedad conocida como “relajación” que es un incremento dependiente de tiempo y voltaje en IKACh al ir hacia potenciales hiperpolarizados. Desde la descripción de esta propiedad de IKACh hace 30 años sólo recientemente un grupo ha planteado que cambios en la concentración del Ca2+ intracelular, provocados por el potencial membranal, controlan la acción de la proteína RGS4 en la activación de la proteína G sobre los canales KACh (Ishii et al., 2001). Según esta propuesta, al aplicar BAPTA intracelular se bloquea el primer paso, cambios en el Ca2+i ocasionados por el potencial membranal, y ya no se observa la relajación de IKACh (Ishii et al., 2001). Dados nuestros resultados con las curvas C-R donde ACh tiene mayor potencia para activar IKACh a los potenciales hiperpolarizados y menor potencia con los despolarizados (Figs. 7 y 14A) decidimos confrontar estas dos propuestas, realizando experimentos en miocitos cardiacos de gato y conejo en presencia de BAPTA intracelular (5 mM). En estos experimentos, desde un potencial de mantenimiento de -40 mV, se daba un primer pulso (prepulso) de 3.5 s desde -100 a +60 mV con incrementos de +40 mV, y luego se daba un segundo pulso a un voltaje hiperpolarizado de -120 mV para observar las corrientes de cola activadas por el voltaje previo. Como se observa en las Figs. 29 y 30, la relajación de IKACh se sigue presentando aún con BAPTA intracelular (5mM). También observamos que la relajación de IKACh es un proceso dependiente de voltaje que se manifiesta sólo cuando se utilizan concentraciones bajas de ACh, como 30 nM (Figs. 29A y C; 30A y C) o 100 nM (Figs. 29C y 30C) con las que, como vimos en las curvas C-R de ACh (Figs. 7 y 14A), el nivel de respuesta o activación es diferente según el potencial con que se mantiene a la célula. Por el contrario, la relajación de IKACh ya no es evidente (Fig. 29B y C) o es mucho menor (Figs. 30B y C) con concentraciones altas de ACh que, por las curvas C-R de ACh (Figs. 7 y 14A), sabemos que a esta concentración ya no existe dependencia de voltaje para activar IKACh. Es importante destacar que con la hiperpolarización las corrientes de cola de ACh y pilocarpina ambas son dependientes de tiempo y voltaje pero en sentido inverso, lo cual es totalmente congruente con los resultados de las curvas C-R para estos dos agonistas. - 43 - Fig. 29 Relajación de IKACh (con BAPTA 5 B IKACh 3 µM mM intracelular) en miocitos auriculares IKACh 30 nM de gato. Corrientes macroscópicas inducidas Iins por ACh 30 nM (A) y 3 I I Imáx ins máx µM (B) obtenidas con el protocolo de doble 1s 1s pulso (inserto). Las co1.0 rrientes entrantes de C cola al pasar al potencial hiperpolarizado de -120 mV primero cam0.5 bian instantáneamente (Iins) y después se increIK 30 nM IK 100 nM mentan lentamente hasIK 3 µM 0.0 ta un estado estable -100 -80 -60 -40 -20 0 20 40 60 (Imáx). Las corrientes a potencial del prepulso (mV) -120 mV se registraron después de prepulsos entre -100 y +60 mV con incrementos de +40 mV. C, Relación entre el potencial del prepulso y el cociente Iins/Imáx para las corrientes activadas por ACh 30 nM (cuadros negros), 100 nM (círculos rojos) o 3 µM (triángulos verdes). n=3-4 miocitos. La línea punteada indica el nivel cero de corriente. Las corrientes inducidas por ACh (IKACh) se obtuvieron por la sustracción digital de las corrientes registradas en condición control de las registradas en la presencia de ACh. -40 mV -120 mV 3.5 s 200 pA +60 mV -40 mV -100 mV 3.5 s Iins/Imáx 200 pA A ACh ACh ACh Iins/Imáx ACh ACh ACh - 44 - 200 pA 50 pA Fig. 30 Relajación de +60 mV IKACh (con BAPTA B A -40-100mVmV 3.5 s -120 mV -40 mV 5mM intracelular) en 3.5 s IKACh 3 µM miocitos auriculares de IKACh 30 nM conejo. Corrientes maIins croscópicas inducidas Imáx Iins Imáx por ACh 30 nM (A) y 3 µM (B) obtenidas con el protocolo de doble pulso 1s 1s (inserto). Las corrientes entrantes de cola al pasar 1.0 al potencial hiperpolariC zado de -120 mV primero cambian instantánea0.5 mente (Iins) y después se incrementan lentamente IK 30 nM hasta un estado estable IK 100 nM 0.0 (Imáx). Las corrientes a IK 3 µM -120 mV se registraron -100 -80 -60 -40 -20 0 20 40 60 después de prepulsos potencial del prepulso (mV) entre -100 y +60 mV con incrementos de +40 mV. C, Relación entre el potencial del prepulso y el cociente Iins/Imáx para las corrientes activadas por ACh 30 nM (cuadros negros), 100 nM (círculos rojos) o 3 µM (triángulos verdes). n=4-5 miocitos. La línea punteada indica el nivel cero de corriente. Las corrientes inducidas por ACh (IKACh) se obtuvieron por la sustracción digital de las corrientes registradas en condición control de las registradas en la presencia de ACh. VII. DISCUSIÓN En el presente trabajo, en miocitos auriculares de gato y conejo y en un sistema de expresión heteróloga, hemos encontrado que: 1) la activación de la vía receptores M2, proteínas Gi/o y canales KACh (M2-Gi/o-KACh) con el agonista fisiológico, ACh, depende del potencial membranal: mayor potencia a -100 mV que a +50 mV; 2) la modulación por voltaje de M2-Gi/o-KACh también depende de la estructura del ligando, teniendo CarbaCh y MetaCh una dependencia de voltaje similar a la de ACh, mientras que pilocarpina, TMA y Ch son más potentes y eficaces con la despolarización, y viceversa con la hiperpolarización; 3) la mutación del triptófano 99 en el sitio ortostático del receptor M2 anula la dependencia de voltaje; 4) la vía de señalización que utilizan pilocarpina, Ch y TMA es la misma que utiliza ACh: M2-Gi/oKACh; y 4) el fenómeno de la “relajación” de IKACh se sigue presentando aún con la utilización de BAPTA (5 mM) en la solución intracelular. 1) En miocitos cardiacos ACh activa la vía M2-Gi/o-KACh con dependencia de voltaje A pesar de que desde hace unos 20 años se empezaron a acumular evidencias que relacionaban una dependencia de voltaje en procesos fisiológicos mediados por RAPG (Marty y Tan, 1989; Itoh et al., 1992; Ilouz et al., 1999; Mahaut-Smith et al., 1999 y 2000; Ong et al., 2001, etc.) no fue hasta 2003 en que se demostró en un sistema de expresión heteróloga (ovocitos de Xenopus), la sensibilidad al voltaje de un RAPG prototipo, el receptor muscarínico colinérgico (Ben-Chaim et al., 2003), y que es capaz de desarrollar corrientes asociadas con movimientos de carga(Ben-Chaim et al., 2006), análogas a las de los canales iónicos sensibles al voltaje. En sistema heterólogo, se observó que los receptores muscarínicos M2 manifiestan mayor afinidad por la ACh en los voltajes más negativos y menor en los voltajes más positivos. Nuestro estudio es el primero en confirmar la dependencia de voltaje en la activación de la vía colinérgica cardiaca por ACh, en congruencia a lo reportado por Ben-Chaim et al (2003). 2) El sitio ortostático del receptor M2 está involucrado en la dependencia de voltaje Dentro de la modulación de M2-Gi/o-KACh por el voltaje, también encontramos que es una propiedad muy dinámica determinada por la estructura del ligando. Los agonistas más parecidos entre sí, ACh, CarbaCh y MetaCh, manifiestan el mismo comportamiento - 45 - dependiente de voltaje y, según los estudios de unión de [3H]ACh (Ben-Chaim et al., 2003) y que para MetaCh el voltaje no tiene efecto sobre la eficacia, el único mecanismo que parece ejercer el potencial de membrana es una modificación de la afinidad ligando-receptor. No obstante, un cambio en la eficacia tampoco puede descartarse. Con otros agonistas muscarínicos la dependencia de voltaje para activar la vía colinérgica cardiaca es muy diferente a la del agonista fisiológico. Para betanecol, el voltaje no tiene influencia. Para pilocarpina, la despolarización aumenta la eficacia y probablemente la afinidad. Para TMA, la despolarización también aumenta la eficacia pero la afinidad parece disminuir. De Ch, dado que no se logran curvas C-R completas, sólo el aumento de la potencia está claro. La dinámica en la dependencia de voltaje con los agonistas muscarínicos no podría ser explicada con el modelo del grupo de Hanna Parnas (Fig. 1), dado que su propuesta central es que el voltaje se “sensa” en la región del receptor que interacciona con la proteína G y esto cambia la probabilidad de acoplamiento receptor-proteína G, lo que a su vez provoca el posterior cambio de afinidad del receptor por el agonista (Ben-Chaim et al., 2003 y 2006; Ohana et al., 2006). De acuerdo al modelo anterior, el comportamiento dependiente de voltaje sería el mismo sin importar el agonista muscarínico que activara la vía, siendo un modelo independiente de ligando que no puede sustentarse con nuestros datos. Alternativamente al modelo de Hanna Parnas, nuestros resultados sugieren que el voltaje provoca cambios conformacionales en el sitio del receptor donde se unen los ligandos, sitio ortostático (donde inclusive podría residir la sensibilidad al voltaje), lo que provoca una interacción (afinidad y/o eficacia) diferente no sólo con los distintos agonistas muscarínicos sino también con un mismo ligando (agonista o antagonista). De acuerdo a este modelo (Fig. 31), la despolarización modifica la conformación del sitio de unión de ligandos en el receptor M2, la cual resulta favorable para una mejor interacción (afinidad y/o eficacia) de los agonistas pilocarpina, TMA y Ch, y a la vez, que dicha conformación sea menos favorable para la interacción de los agonistas ACh, CarbaCh y MetaCh; y viceversa con la hiperpolarización. En este sentido, podemos considerar que los cambios conformacionales impuestos por el voltaje al receptor M2 son de consecuencias similares para una estructura molecular como la de betanecol. El modelo del receptor M2 con sensibilidad al voltaje de una manera que es específica de ligando, también es útil para predecir el posible efecto dependiente de voltaje de los - 46 - antagonistas, tal y como se puede inferir del experimento preliminar con methoctramina (considerado antagonista M2) sobre las respuestas equiefectivas de CCh en miocitos cardiacos. Fig. 31 Esquema del modelo para explicar la dinámica en la dependencia de voltaje por los agonistas muscarínicos. (Ver texto para su mejor descripción). La línea roja es más larga que la azul para representar que, a pesar de la menor afinidad, la respuesta puede ser igual a la del voltaje negativo si se aumenta la concentración de agonista. La interacción con la proteína G intenta representar que el acople ligando-receptor genere una conformación tal con menor o mayor capacidad para activar la vía de señalización asociada con el receptor (eficacia). Una parte medular de nuestra propuesta son los cambios conformacionales, particularmente en el sitio de unión de ligandos, que serían impuestos por el voltaje (Fig. 31). Evidencias adicionales de nuestro grupo apoyan la propuesta anterior. Las “corrientes de compuerta” (gating currents, en inglés) son generadas por el movimiento de aminoácidos cargados dentro del campo eléctrico de la membrana en respuesta a cambios en el potencial membranal. Así, la medición del desplazamiento de carga sirve como un método para monitorear cambios en la conformación proteica, tal como se han definido los cambios conformacionales que preceden la apertura de los canales iónicos sensibles a voltaje (Bezanilla, 2000). De hecho, estudios con esta técnica han revelado que ACh y pilocarpina ejercen efectos opuestos en el desplazamiento de carga sobre el receptor M2 (Navarro-Polanco et al., en preparación), con lo que este mecanismo podría explicar la dependencia de voltaje opuesta de ACh y pilocarpina para activar IKACh a través del receptor M2. En este mismo contexto, se ha mostrado que ligandos de efectos biológicos opuestos (agonista vs. agonista inverso) inducen cambios conformacionales distintos en el receptor muscarínico M3 (Li et al., 2007). - 47 - En términos farmacológicos clásicos, un agonista total se esperaría que active todas las vías de señalización relacionadas con un receptor en el mismo grado que el agonista endógeno para ese receptor. De manera similar, un ligando que antagoniza una ruta de señalización a través de un receptor específico debería antagonizar, en la misma magnitud, cada ruta acoplada a dicho receptor. Sin embargo, evidencias de muchos grupos de investigación en distintos modelos experimentales (para una revisión ver Urban et al., 2007) han empezado a cambiar dicha perspectiva, y actualmente se propone que los ligando inducen cambios conformacionales específicos de ligando, que son únicos, y que frecuentemente pueden resultar en una activación diferencial de las vías de señalización asociadas con un receptor en particular, para lo cual se ha ido perfilando el término de “selectividad funcional” (Urban et al., 2007). Efectivamente, la existencia de múltiples estados conformacionales que subyacen a las formas libre y unidas a ligando de los RAPG se apoya en mediciones de la duración de fluorescencia del receptor ß2 adrenérgico (Ghanouni et al., 2001) y en estudios de formación de puentes disulfuro en el receptor muscarínico M3 (Ward et al., 2002; Li et al., 2007). A pesar de la creciente aceptación de que los ligandos generen conformaciones del receptor únicas y específicas que desencadenen una activación diferencial de las distintas rutas de señalización acopladas a dicho receptor, la propuesta de nuestro trabajo va más lejos al postular que el voltaje, por sí mismo, es capaz de inducir diferentes estados conformacionales que conllevan a una distinta activación de la(s) vía(s) de señalización estimulada(s) por un mismo agonista. Lo cual, de ser cierto, sería de gran importancia en los tejidos excitables, donde el potencial está oscilando continuamente. 3) Vía de señalización activada por pilocarpina y Ch en miocitos cardiacos Respecto a la controversia de los mediadores celulares que utilizan pilocarpina y Ch para activar a las corrientes con dependencia de tiempo y voltaje cuando se registran con protocolo de pulsos de voltaje, por las evidencias mostradas estamos en condiciones de postular que la corriente activada es la propia IKACh, a través de sus mismos mediadores celulares (receptores M2 y proteínas Gi/o), sólo que el curso temporal de rectificación tardía que muestra (Figs. 5 y 20) es debida a la sensibilidad al voltaje de los receptores M2 para con pilocarpina y Ch: mayor potencia y eficacia con la despolarización y viceversa con la hiperpolarización. - 48 - Como ya se mencionó previamente (sección B de resultados), Shi et al. (1999b) y Wang et al. (1999), utilizando protocolo de pulsos de voltaje, describieron que pilocarpina y Ch activan, a través de receptores muscarínicos M3, corrientes con características de rectificador tardío, que denominaron IKM3. En gran parte de los trabajos de este grupo la mayor parte de sus evidencias se apoyan con el uso de antagonistas muscarínicos, que ya se sabe de su selectividad limitada. Además de otros aspectos, los autores describieron a IKM3: a) con un umbral de activación de –40 mV, b) que la máxima activación de IKM3 se logra con Ch 10 mM y con pilocarpina 10 µM, y c) que contrariamente a IKACh, no desensibilizan. Respecto a estos 3 puntos, en nuestros resultados, manteniendo fijo un voltaje dado y con la perfusión rápida de agonistas muscarínicos sobre los miocitos auriculares de gato, hemos encontrado que pilocarpina y Ch, al igual a como lo hace ACh: a) activan corrientes no sólo con los potenciales más positivos, sino también con los más negativos; sólo se mostraron las que activan a –100 mV, pero lo hacen en todo el rango de voltajes negativos que se ensayen (datos no mostrados); b) activan corrientes a concentraciones mayores de 10 µM y 10 mM, respectivamente (Figs. 8B y 10B); c) tanto las corrientes entrantes como las salientes sí muestran desensibilización, especialmente al ir aumentando la concentración y con los voltajes más positivos (Figs. 8B y 10B). Adicionalmente: d) los ensayos con PTX señalan que en la corriente activada por pilocarpina participan proteínas Gi/o (Fig. 20); e) tertiapina (potente bloqueador selectivo de canales KACh: este péptido no afecta otras corrientes de K+ presentes en miocitos cardiacos tales como Ito, IKr, IKs, IK1, IKATP (Kitamura et al., 2000; Drici et al., 2000)) prácticamente bloquea totalmente las corrientes activadas por pilocarpina, indicando que se trata de los canales KACh (Fig.22); f) la aplicación conjunta de Ch y ACh no sólo no es la sumatoria de las corrientes aplicadas por separado, sino que es hasta menor que la de la ACh sola, esto podría explicarse por el sostenimiento de la desensibilización rápida y lo cual sería prueba de utilizar una vía de activación común (Fig. 18); g) concentraciones equiefectivas de Ch y ACh (concentraciones que producen una misma amplitud de corriente normalizada), al empalmar los registros de dichas corrientes se observa una casi perfecta sobreposición de los registros, a todos los niveles: activación, desensibilización y desactivación de las corrientes (Fig. 19); así, es poco probable que dos corrientes que utilizaran diferentes receptores y canales iónicos efectores coincidan tan exactamente en su curso temporal; h) en células HEK293 que expresan heterólogamente receptores M2 y canales - 49 - Kir 3.1/3.4 (canales KACh), pilocarpina y Ch activan corrientes similares a las de los miocitos cardiacos (Figs. 13B y 20, respectivamente); i) en este mismo sistema heterólogo, con pulsos cuadrados de voltaje, pilocarpina reproduce las corrientes con dependencia de tiempo y voltaje (Fig. 23). La evidencia en preparación multicelular de aurícula de que pilocarpina (Wang et al., 1999) y Ch (Shi et al., 1999b) disminuyen la duración del potencial de acción y la frecuencia cardiaca, tal y como se sabe que lo hace ACh, es otra evidencia de que la corriente activada por estas sustancias es IKACh. 4) Relajación de IKACh Según el mecanismo molecular propuesto por el grupo de Kurachi para explicar la propiedad de la relajación de IKACh en miocitos cardiacos, la modificación del Ca2+i (aumento con la despolarización y disminución con la hiperpolarización) es el elemento inicial que desencadena la siguiente serie de eventos cuyo efecto final es la modificación de los canales KACh disponibles (ver pág. 14). De todas las evidencias presentadas por este grupo, es precisamente este elemento inicial el pasaje más desconocido, dado que no se tiene idea alguna de la entidad, sensible al voltaje, que sea el responsable de tales modificaciones del Ca2+, pues la participación de los canales de Ca2+ sensibles a voltaje y del intercambiador Na+/Ca2+ ya han sido descartados (Ishii et al., 2001). De cualquier manera, dado que nuestros experimentos de la relajación de IKACh fueron realizados siempre en presencia de BAPTA (5 mM) intracelular, cuya acción quelante de Ca2+ es de acción más rápida que el EGTA (Marty y Neher, 1985), podemos excluir, en nuestras condiciones, la implicación de dicho mecanismo. Alternativamente proponemos que la relajación de IKACh se debe igualmente a la sensibilidad al voltaje del receptor M2, que es congruente con lo que se observa en las curvas C-R de ACh, mayor potencia con los voltajes más negativos, y viceversa; y que también es congruente con lo que se observa a un potencial hiperpolarizado en protocolo de pulsos de voltaje con los agonistas pilocarpina (Fig. 25), TMA y Ch, pues también en este caso se trata de la relajación de IKACh, pero digamos que en sentido inverso. - 50 - VIII. CONCLUSIONES 1) En miocitos auriculares de mamíferos (gatos y conejos) existe una activación dependiente de voltaje de la vía de señalización colinérgica: receptores M2, proteínas Gi/o y canales KACh. 2) La dependencia de voltaje de la activación de la vía colinérgica cardiaca ocurre a nivel del receptor M2. 3) El voltaje membranal ocasiona cambios conformacionales en el receptor que afectan el sitio de unión de los agonistas (cambio en la afinidad) y también determinan la conformación final que incidirá sobre los elementos posteriores al receptor (cambio en la eficacia). 4) La sensibilidad al voltaje de los receptores M2 ocasiona que los agonistas pilocarpina, Ch y TMA activen a IKACh con la apariencia de rectificador tardío y de que ACh genere el fenómeno de la “relajación” de IKACh. - 51 - IX. REFERENCIAS ALBERTS, B., JOHNSON, A., LEWIS, J., RAFF, M., ROBERTS, K. & WALTER, P. (2002). Signaling through G-protein-linked cell-surface receptors. En: Molecular biology of the cell. (4th ed.). USA: Garland Science, 2002, p. 852-853. ARMSTRONG, C. M. & BEZANILLA, F. (1973). Currents related to movement of the gating particles of the sodium channels. Nature 242: 459-461. BEN-CHAIM, Y., TOUR, O., DASCAL, N., PARNAS, I. & PARNAS, H. (2003). The M2 muscarinic G-protein-coupled receptor is voltage-sensitive. J. Biol. Chem. 278: 22482-22491. BEN-CHAIM, Y., CHANDA, B., DASCAL, N., BEZANILLA, F., PARNAS, I. & PARNAS, H. (2006). Movement of ‘gating charge’ is coupled to ligand binding in a G-protein-coupled receptor. Nature 444 (7115): 106-109. BENAVIDES-HARO, D. E. Estudio de los efectos voltaje-dependientes y voltaje-independientes de diferentes agonistas muscarínicos sobre la corriente de potasio activada por acetilcolina. Relación estructura actividad. Tesis Doctoral. Colima: Universidad de Colima, 2001. BENAVIDES-HARO, D. E., NAVARRO-POLANCO, R. A. & SÁNCHEZ-CHAPULA, J. A. (2003). The cholinomimetic agent betanecol activates IK(ACh) in feline atrial myocytes. Naunyn Schmiedeberg’s Arch. Pharmacol. 368: 309-315. BEZANILLA, F. (2000). The voltage sensor in voltage-dependent ion channels. Physiol Rev. 80: 555-592. BREITWEISER, G. & SZABO, G. (1985). Uncoupling of cardiac muscarinic and β-adrenergic receptors from ion channels by a guanine nucleotide analog. Nature (Lond.) 317: 538-540. BRODDE, O. E. & MICHEL, M. C. (1999). Adrenergic and muscarinic receptors in the human heart. Pharmacol. Rev. 51: 651-689. BROWN, J. H. & TAYLOR, P. Agonistas y antagonistas de los receptores muscarínicos. En: Las bases farmacológicas de la terapéutica. (10ª ed.). Editado por J. G. Hardman y L. E. Limbird. México: McGraw Hill Interamericana, 2003, p. 163-181. CICCARONE, V., CHU, Y., SCHIFFERLI, K., PICHET, J., HAWLEY-NELSON, P., EVANS, K., ROY, L. & BENNETT, S. (1999). LipofectamineTM 2000 reagent for rapid efficient transfection of eukaryotic cells. Focus 21: 54-55. - 52 - COHEN-ARMON, M. & SOKOLOVSKY, M. (1991). Depolarization-induced changes in the muscarinic receptor in rat brain and heart are mediated by pertussis-toxin sensitive G-proteins. J. Biol. Chem. 266: 2595-2605. DEIGHTON, N. M., MOTOMURA, S., BÓRQUEZ, D., ZERKOWSKI, H. R., DOETSCH, N. & BRODDE, O. E. (1990). Muscarinic cholinoceptors in the human heart: demonstration, subclassification and distribution. Naunyn Schmiedeberg´s Arch. Pharmacol. 341: 14-21. DEKEL, N (2005). Is the metabotropic GABAB G-protein-coupled receptor voltage sensitive? Rev. Neurosci. 16: S16. DHEIN, S., VAN KOPPEN, C. & BRODDE, O. E. (2001). Muscarinic receptors in the mammalian heart. Pharmacol. Res. 44: 161-182. DOODS, H., MATHY, M., DAVIDESKO, D., VAN CHARLDORP, K., DE JONGE, A. & VAN ZWIETEN, P. (1987). Selectivity of muscarinic antagonists in radioligand and in vivo experiments for the putative M1, M2 and M3 receptors. J. Pharmacol. Exp. Ther. 242: 257-262. DÖRJE, F., LEVEY, A. I. & BRANN, M. R. (1991). Immunological detection of muscarinic receptor subtype proteins (m1-m5) in rabbit peripheral tissues. Mol. Pharmacol. 40: 459-462. DOUPNIK, C.A., JAEN, C. Y ZHANG, Q. (2004). Measuring the modulatory effects of RGS proteins on GIRK channels. Methods in Enzimology 389: 131-154. DRICI, M. D., DIOCHOT, S., TERRENOIRE, C., ROMEY, G. & LAZDUNSKI, M. (2000). The bee venom peptide tertiapin underlies the role of I(KACh) in acetylcholine-induced atrioventricular blocks. Br. J. Pharmacol. 131: 569-577. FERMINI, B. & NATTEL, S. (1994). Choline chloride activates time-dependent and timeindependent K+ currents in dog atrial myocytes. Am. J. Physiol. 266: C42-C51. FUDER, H. & JUNG, B. (1985). Affinity and efficacy of racemic, (+) and (-) methacholine in muscarínico inhibition of [3H] noradrenaline release. Br. J. Pharmacol. 84: 477-487. GANITKEVICH, V. Ya & ISENBERG, G. (1993). Membrane potential modulates inositol 1,4,5-triphosphate-mediated Ca2+ transients in guinea-pig coronary myocytes. J. Physiol. 470: 35-44. GHANOUNI, P., STEENHUIS, J.J., FARRENS, D. L. & KOBILKA, B. K. (2001). Agonistinduced conformational changes in the G-protein-coupling domain of the beta 2 adrenergic receptor. PNAS 98 (11): 5997-6002. - 53 - GIRALDO, E., MARTOS, F., GÓMEZ, A., GARCÍA, A., VIGANO, M. A., LADINSKI, H. & SÁNCHEZ DE LA CUESTA, F. (1988). Characterization of muscarinic receptor subtypes in human tissues. Life Sci. 43: 1507-1515. GORMAN, C. M., HOWARD, B. H. & REEVES, R. (1983). Expression of recombinant plasmids in mammalian cells is enhanced by sodium butyrate. Nucleic Acids Research 11: 7631-7648. HEITZ, F., HOLZWARTH, J. A., GIES, J. P., PRUSS, R. M., TRUMPP-KALLMEYER, S., HIBERT, M. F. & GUENET, C. (1999). Site-directed mutagenesis of the putative human muscarínico M2 receptor binding site. Eur. J. Pharmacol. 380: 183-195. HILLE, B. Ion channels of excitable membranes. (3th. ed.). USA: Sinauer Associates, 2001. HOLINSTAT, M., OLDHAM, W. M. & HAMM, H. E. (2006). G-protein-coupled receptors: evolving views on physiological signaling. EMBO reports 7: 866-869. ILOUZ, N., BRANSKI, L., PARNIS, J., PARNAS, H. & LINIAL, M. (1999). Depolarization affects the binding properties of muscarinic acetylcholine receptors and their interaction with proteins of the exocytic apparatus. J. Biol. Chem. 274: 29519-29528. ISENBERG, G. & KLÖCKNER, U. (1982). Calcium tolerant ventricular myocytes prepared by pre-incubation in a KB-medium. Plügers Arch. 395: 6-18. ISHII, M., INANOBE, A., FUJITA, S., MAKINO, Y., HOSOYA, Y & KURACHI, Y. (2001). Ca2+ elevation evoked by membrane depolarization regulates G-protein cycle via RGS proteins in the heart. Circ. Res. 89: 1045-1050. ISHII, M, INANOBE, A. & KURACHI, Y. (2002). PIP3 inhibition of RGS protein and its reversal by Ca2+/calmodulin mediate voltage-dependent control of the G-protein cycle in a cardiac K+ channel. PNAS 99: 4325-4330. ITOH, T., SEKI, N., SUZUKI, S., ITO, S., KAJIKURI, J. & KURIYAMA, H. (1992). Membrane hyperpolarization inhibits agonist induced synthesis of inositol 1,4,5-triphosphate in rabbit mesenteric artery. J. Physiol. 451: 307-328. JIANG, B., SUN, X., CAO, K. & WANG, R. (2002). Endogenous Kv channels in human embryonic kydney (HEK-293) cells. Mol. Cell Biochem 238: 69-79. JORDAN, M. & WURM, F. (2004). Transfection of adherent and suspended cells by calcium phosphate. Methods 33: 136-143. - 54 - KENNEDY, R. H., WYETH, R. P., GERNER, P., LIU, S., FONTENOT, J & SEIFEN, E. (1995). Tetramethylammonium is a muscarinic agonist in rat heart. Am. J. Physiol. 268 (Cell Physiol. 37): C1414-C1417. KITAMURA, H., YOKOYAMA, M., AKITA, H., MATSUSHITA, K., KURACHI, Y. & YAMADA, M. (2000). Tertiapin potently and selectively blocks muscarinic K+ channels in rabbit cardiac myocytes. J. Pharmacol. Exp. Ther. 293: 196-205. KREJČI, A. & TUČEK, S. (2002). Quantitation of mRNAs for M1 to M5 subtypes of muscarinic receptors in rat heart and brain cortex. Mol. Pharmacol. 61: 1267-1272. KREJČI, A., MICHAL, J., JAKUBÍK, J & DOLEŽAL, V. (2004). Regulation of signal transduction at M2 muscarinic receptor. Physiol. Res. 53 (Suppl.1): S131-S140. KROEZE, W. K., SHEFFLER, D. J. & ROTH, B. L. (2003). G-protein-coupled receptors at a glance. J. of Cell Sci. 116: 4867-4869. KUBO, T., MAEDA, A., SUGIMOTO, K., AKIBA, I., MIKAMI, A., TAKANASHI, A., ET AL. (1986). Primary structure of porcine cardiac muscarinic acetylcholine receptor deduced from the cDNA sequence. FEBS Lett. 239: 339-342. KURACHI, Y. & ISHII, M. (2004). Cell signal control of the G protein-gated potassium channel and its subcellular localization. J. Physiol. 554: 285-294. LI, J. H., HAN, S. J., HAMDAN, F. F., KIM, S. K., JACOBSON, K. A., BLOODWORTH, L. M., ZHANG, X & WESS, J. (2007). Distintic structural changes in a G-protein-coupled receptor caused by different classes of agonist ligands. J. Biol. Chem. 282 (36): 26284-26293. MAEDA, A., KUBO, T., MISHINA, M. & NUMA, M. (1988). Tissue distribution of mRNAs encoding muscarinic acetylcholine receptor subtypes. FEBS Lett. 239: 339-342. MAHAUT-SMITH, M. P., HUSSAIN, J. F. & MASON, M. J. (1999). Depolarization-evoked Ca2+ release in a non-excitable cell, the rat megakaryocyte. J. Physiol. 515: 385-390. MARK, M. D. & HERLITZE, S. (2000). G-protein mediated gating of inward-rectifier K+ channels. Eur. J. Biochem. 267: 5830-5836. MARTINEZ-PINNA, J., GURUNG, I. S., VIAL, C., LEON, C., GACHET, C., EVANS, R.J. & MAHAUT-SMITH, M. P. (2005). Direct voltage control of signaling via P2Y1 and other Galphaq-coupled receptors. J. Biol. Chem. 280: 1490-1498. MARTY, A. & NEHER, E. (1985). Potassium channels in cultured bovine adrenal chromaffin cells. J. Physiol. 367: 117-141. - 55 - MARTY, A. & TAN, Y. P. (1989). The initiation of calcium release following muscarínico stimulation in rat lacrimal glands. J. Physiol. 419: 665-687. MEYER, T., WELLNER-KIENITZ, M. C., BIEWALD, A., BENDER, K. EICKEL, A. & POTT, L. (2001). Depletion of phosphatidylinositol 4,5-biphosphate by activation of phospholipase C-coupled receptors causes slow inhibition but not desensitization of G proteingated inward rectifier K+ current in atrial myocytes. J. Biol. Chem. 276: 5650-5658. NAVARRO-POLANCO, R. & SÁNCHEZ-CHAPULA, J. (1995). 4-Aminopyridine activates a potassium current by activation of a muscarinic receptor. Biophys. J. 68(2): A108. NAVARRO-POLANCO, R. A. & SÁNCHEZ-CHAPULA, J. A. (1997). 4-Aminopyridine activates potassium currents by activation of a muscarinic receptor in feline atrial myocytes. J. Physiol. 498: 663-678. NAVARRO-POLANCO, R. A. Mecanismo de activación del canal de potasio activado por acetilcolina IK-ACh, por 4-AP y por ChCl en miocitos auriculares de gato. Modulación voltajey tiempo dependiente. Tesis Doctoral. Colima: Universidad de Colima, 1997. NICHOLS, C. G. & LOPATIN, A. N. (1997). Inward rectifier potassium channels. Annu. Rev. Physiol. 59: 171-191. NOMA, A. & TRAUTWEIN, W. (1978). Relaxation of the ACh-induced potassium current in the rabbit sinoatrial node cell. Pflügers Arch. 377: 193-200. OHANA, L., BARCHAD, O, PARNAS, I. & PARNAS, H. (2006). The metabotropic glutamate G-protein-coupled receptors mGluR3 and mGluR1a are voltage-sensitive. J. Biol. Chem. 281: 24024-24215. ONG, B. H., OHSAGA, A., SATO, K., OSHIRO, T., SHIRATO, K. & MARUYAMA, Y. (2001). G protein modulation of voltage-sensitive muscarinic receptor signalling in mouse pancreatic acinar cells. Pfflugers Arch. 441: 604-610. PEDRETTI, A., VISTOLI, G., MARCONI, C. & TESTA, B. (2006). Muscarinic receptors: a comparative analysis of structural features and binding modes through homology modelling and molecular docking. Chemistry & Biodiversity 3: 481-501. PERALTA, E. G., WINSLOW, J. W., PETERSON, G. L., SMITH, D. H., ASHKENAZI, A., RAMACHANDRAN, J., SCHIMERLIK, M. I. & CAPON, D. J. (1987b). Primary structure and biochemical properties of a M2 muscarinic receptor. Science (Wash. D.C.) 236: 600-605 - 56 - PERALTA,E.G., ASHKENAZI, A., WINSLOW, J. W., SMITH, D. H., RAMACHANDRAN, J. & CAPON, D. J. (1987a). Distintic primary structures, ligand-binding properties and tissuespecific expression of four human muscarinic acetylcholine receptors. EMBO (Eur. Mol. Biol. Organ) J. 6: 3923-3929. PERROY, J., RICHARD, S., NARGEOT, J., BOCKAERT, J. & FAGNI, L. (2002). Permissive effect of voltage on mGlu 7 receptor subtype signaling in neurons. J. Biol. Chem. 277: 1223-1228. PFAFFINGER, P. J., MARTIN, J. M., HUNTER, D. D., NATHANSON, N. M. & HILLE, B. (1985). GTP-binding proteins couple cardiac muscarinic receptors to a K channel. Nature (Lond.) 317: 536-538. PINKAS-KRAMARSKI, R., STEIN, R. & SOKOLOVSKY, M. (1989). Postnatal changes in muscarinic receptor subtype mRNAs in rat brain and heart. J. Mol. Neurosci. 1: 209-213. RANG, H. P. (2006). The receptor concept: pharmacology’s big idea. Br. J. Pharmacol. 147: S9-S16. RÖMPLER, H., STÄUBERT, C., THOR, D., SCHULZ, A., HOFREITER, M. & SCHÖNEBERG, T. (2007). G-Protein-coupled time travel: evolutionary aspects of GPCR research. Molecular Interventions 7: 17-25. ROSS, E. M. & KENAKIN, T. P. Farmacodinámica. Mecanismo de acción y relación entre la concentración y el efecto de los fármacos. En: Las bases farmacológicas de la terapéutica. (10ª ed.). Editado por J. G. Hardman y L. E. Limbird. México: McGraw Hill Interamericana, 2003, p. 35-48. RUPPERSBERG, J. P. (2000).Intracellular regulation of inward rectifier K+ channels. Plügers Arch. (Eur. J. Physiol.) 441: 1-11. SÁNCHEZ-CHAPULA, J. A. (1988). Effects of bupivacaine on membrane currents of guineapig ventricular myocytes. Eur. J. Pharmacol. 156: 303-308. SOEJIMA, M. & NOMA, A. (1984). Mode of regulation of the ACh-sensitive K-channel by the muscarinic receptor in rabbit atrial cells. Plügers Arch. 400: 424-431. SHI, H., WANG, H. & WANG, Z. (1999a). Identification and characterization of multiple subtypes of muscarinic acetylcholine receptors and their physiological functions in canine hearts. Mol. Pharmacol. 55: 497-507. - 57 - SHI, H., WANG, H., LU, Y., YANG, B. & WANG, Z. (1999b). Choline modulates cardiac membrane repolarization by activating an M3 muscarinic receptor and its coupled K+ channel. J. Membrane Biol. 169: 55-64. SHI, H., YANG, B., XU, D., WANG, H. & WANG, Z. (2003). Electrophysiological characterization of cardiac muscarinic acetylcholine receptors : different subtypes mediate different potassium currents. Cell. Physiol. Biochem. 13: 59-74. STANFIELD, P. R., NAKAJIMA, S. & NAKAJIMA, Y. (2002). Constitutively active and Gprotein coupled inward rectifier K+ channels: Kir 2.0 and Kir 3.0. Rev. Physiol. Biochem. Pharmacol. 145: 47-179. STENGEL, P. W., GOMEZA, J., WESS, J. & COHEN, M. L. (2000). M2 and M4 receptor knockout mice: muscarinic receptor function in cardiac and smooth muscle in vitro. J. Pharmacol. Exp. Ther. 292: 877-885. STRANGE, P. G. (2008). Agonist binding, agonist affinity and agonist efficacy at G proteincoupled receptors. Br. J. Pharmacol. 153: 1353-1363. URBAN, J. D., CLARKE, W. P., VON ZASTROW, M., NICHOLS, D. E., KOBILKA, B., WEINSTEIN, H., JAVITCH, J. A., ROTH, B. L., CHRISTOPOULOS, A., SEXTON, P. M., MILLER, K. J., SPEDDING, M. & MAILMAN, R. B. (2007). Functional selectivity and classical concepts of quantitative pharmacology. J. Pharmacol. Experim. Ther. 320: 1-13. WANG, H., SHI, H., LU, Y., YANG, B. & WANG, Z. (1999). Pilocarpine modulates the cellular electrical properties of mammalian hearts by activating a cardiac M3 receptor and a K+ current. Br. J. Pharmacol. 126: 1725-1734. WANG, H., HAN, H. ZHANG, L., SHI, H., SCHRAM, G., NATTEL, S. & WANG, Z. (2001). Expression of multiple subtypes of muscarinic receptors and cellular distribution in the human heart. Mol. Pharmacol. 59: 1029-1036. WARD, S. D., HAMDAN, F. F., BLOODWORTH, L. M. & WESS, J. (2002). Conformational changes that occur during M3 muscarinic acetylcholine receptor activation probed by the use of an in situ disulfide cross-linking strategy. J. Biol. Chem. 277 (3): 22472257. WESS, J. (1996). Molecular biology of muscarinic acetylcholine receptors. Crit. Rev. Neurobiol. 10: 69-99. - 58 - WESS, J. (2004). Muscarinic acetylcholine receptor knockout mice: novel phenotypes and clinical implications. Annu. Rev. Pharmacol. Toxicol. 44: 423-450. WICKMAN, K, NEMEC, J., GENDLER, S. J. & CLAPHAM, D. E. (1998). Abnormal heart rate regulation in GIRK4 knockout mice. Neuron 20: 103-114. YAMADA, M., INANOBE, A. & KURACHI, Y. (1998). G-Protein regulation of potassium ion channels. Pharmacol. Rev. 50: 723-757. YEH, Y. H., QI, X., SHIROSHITA-TAKESHITA, A., LIU, J., MAGUY, A., CHARTIER, D., HEBERT, T., WANG, Z. & NATTEL, S. (2007). Atrial tachycardia induces remodeling of muscarinic receptors and their coupled potassium currents in canine left atrial and pulmonary vein cardiomyocytes. British J. Pharmacol. 152: 1021-1032. - 59 -