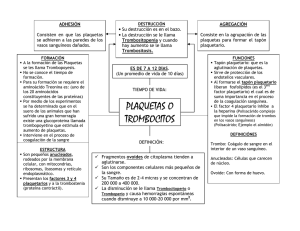
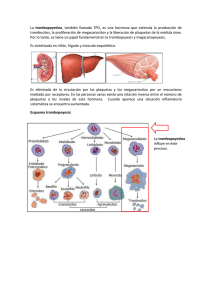
ALTERACIONES DE LA COAGULACIÓN Rodríguez de Alba Galofre M, Rodríguez Alvarez L, Panadero Carlavilla FJ. El sistema de la coagulación constituye un mecanismo defensivo del organismo, mediante el cual podemos hacer frente a numerosas agresiones tanto desde el medio externo como desde el interior de nuestro propio cuerpo. Supone un sistema complejo donde se producen activaciones sucesivas de diversas sustancias, generalmente , en cascada, es decir la activación de una de ellas amplifica la activación de la siguiente; pero que además se encuentra en un perfecto y delicado equilibrio donde los fenómenos de coagulación-descoagulación se producen constantemente. A pesar de esto, podríamos decir que dicho sistema se encuentra habitua lmente en una situación “de reposo” en el organismo; normalmente inactivo, pero capaz de activa rse en pocos segundos tras la aparición de una agresión. El estímulo que desencadena la activación de la hemostasia es la lesión a nivel del endotelio (que normalmente hace de barrera entre la circulación y el tejido a irrigar) provocando el contacto de la sangre con el tejido conectivo subendotelial. La respuesta hemostática incluye tres procesos: la hemostasia primaria, la hemostasia secundaria y la fibrinolisis; de forma que siempre existe una interacción entre la pared vascular y la sangre. La hemostasia primaria se inicia a los pocos segundos de producirse la lesión interacciona ndo las plaquetas y la pared vascular y tiene una importancia enorme para detener la salida de sangre en los capilares, arteriolas pequeñas y vénulas. Se produce una vasoconstricción derivando la sangre fuera del área lesionada. Las plaquetas se adhieren al vaso lesionado y se agrupan formando el tapón plaquetario. Así se “sella” de alguna manera la lesión de la pared y cede temporalmente la hemorragia. La adhesión plaquetaria a la pared vascular está controlada por el equilibrio entre las dos prostaglandinas (tromboxano A2 y prostaciclina) y se ve favorecida por diversas sustancias siendo una de ellas el factor vonWillebrand (FvW). La coagulación o hemostasia secundaria resulta de la interacción de las proteínas plasmáticas o factores de coagulación entre sí, que se activan en una serie de reacciones en cascada conduciendo a la formación de fibrina. La fibrina formará una malla definitiva que reforzará al trombo plaquetario construyéndose finalmente un coágulo o trombo final. Intervienen en el proceso una serie de proteínas procoagulantes (los doce factores de coagulación responsables de la formación de fibrina) y proteínas anticoagulantes (regulan y controlan la coagulación evitando que los factores activados en un punto concreto se dispersen y produzcan una coagulación generalizada). Los más importantes son: antitrombina III, proteína C y proteína S. La coagulación se inicia por la exposición del factor tisular debido a la lesión de los tejidos formándose el complejo factor hístico - factor VII activado. En la actualidad se cree que la activación de los factores IX y X por parte del factor hístico - FVIIa desempeña un papel importante en la inducción de la hemostasia. Una vez iniciada la coagulación a través de esta interacción, el inhibidor de la vía del factor hístico bloquea el proceso y diversos elementos de la vía intrínseca en particular el factor VIII y IX se convierten en reguladores principales de la formación de trombina. Se activa la coagulación propagándose los diferentes pasos en la superficie celular en presencia de los cofactores plasmáticos unidos a las células y la reacción culmina con la formació n del coágulo de fibrina. Los monocitos y los neutrófilos circulantes interaccionan con las plaquetas y las células endoteliales iniciándose una serie de uniones que producirán una interacción estable de los leucoci- tos y plaquetas en el coágulo. Los neutrófilos y los monocitos participan en la reacción inflamatoria local y los monocitos son inducidos a expresar el factor tisular y contribuyen en la trombogénesis y en el primer nivel de curación de la herida. Aunque la descripción del mecanismo de la coagulación se divide en diferentes fases todas están estrechamente relacionadas entre sí, es decir, las plaquetas activadas aceleran la coagulación plasmática y los productos de activación de la coagulación, como la trombina, inducen la activación plaquetar. La fibrinolisis es el último proceso en el que se elimina la fibrina no necesaria para la hemo stasia con la finalidad de la reparación del vaso y el restablecimiento del flujo vascular. Los principales activadores fisiológicos de la fibrinolisis son el activador tisular del plasminógeno (t-AP) y el activador urinario del plasminógeno (u-AP) que difunden desde las células endoteliales y convierten el plasminógeno, absorbido en el coágulo de fibrina, en plasmina. La plasmina degrada el polímero de fibrina en pequeños fragmentos que son eliminados por el sistema de limpieza monocito-macrófago. Aunque la plasmina también puede degradar el fibrinógeno, la reacción es localizada debido a en primer lugar el t-AP y algunas formas del u-AP que activan el plasminógeno de forma más efectiva cuando está absorbido por el coágulo de fibrina; en segundo lugar porque cualquier molécula de plasmina que pase a la circulación es rápidamente neutralizada por la ? 2antiplasmina (es el principal inhibidor de la plasmina); y tercer y último debido a que las células endoteliales liberan el inhibidor del activador del plasminógeno (IAP) que bloquea directamente la acción del t-AP. Ante un estímulo quirúrgico la respuesta del sistema de la coagulación será procoagulante en condiciones normales, por ello en el periodo perioperatorio se pautará una profilaxis ant itrombótica (basada principalmente en medidas farmacológicas, mecánicas y movilización precoz) que ya está muy protocolizada en función de la cirugía realizada, patología del paciente y el período de inmovilización para evitar las complicaciones trombóticas. Sin embargo esta respuesta puede estar alterada por: 1. Alteraciones de la coagulación congénitas. 2. Alteraciones de la coagulación adquiridas por la patología del paciente o por fármacos. 3. Alteraciones en relación al procedimiento quirúrgico. PRINCIPALES ALTERACIONES DE LA HEMOSTASIA ALTERACIONES DE LOS FACTORES DE LA COAGULACION. CONGENITOS Hemofilia o deficiencia de l Factor VIII de la coagulación Se trata de la diátesis hemorrágica hereditaria más frecuente dentro de las carencias de los factores de la coagulación. Este trastorno se encuentra ligado al cromosoma X, ya que es éste cromosoma el que presenta los genes para la síntesis del factor VIII. La gravedad clínica es variable según familias pero constante en una familia concreta. Se considera de tipo leve cuando la actividad del factor VIII se encuentra en una concentración del 5 al 25 % del rango normal, moderada cundo se encuentra en una concentración del 1 al 5 % y severa cuando la actividad es inferior al 1 %. Dado que las mujeres son portadoras objetivan una actividad del factor VIII de alrededor del 50% y no presentan sintomatología alguna. La clínica predominante son hematomas de tejidos blandos, hemartros (hemorragia dentro de la cavidad articular), hemorragias internas en diversos órganos o visceras huecas, sangrado tras cirugías, etc… Las pruebas de laboratorio se caracteriza n por presentar un tiempo de tromboplastina parcial alargado con un tiempo de protrombina normal. El diagnóstico nos lo ofrece el estudio de la dosificación del factor VIII. Tratamiento Consiste en la administración de crioprecipitado o concentrado liofilizado del factor VIII o factor VIII recombinante, preferiblemente. Durante el tratamiento crónico pueden aparecer anticuerpos antifactor VIII, que, como consecuencia, disminuyen la actividad de dicho factor y la rentabilidad del tratamiento. En dicho caso pueden utilizarse inmunoglobulinas antiidiotipo asociadas a ciclofosfamida. En situaciones de emergencia, si no se dispone de factor VIII, se puede administrar concentrado de complejo de protrombina. En casos muy concretos también puede resultar útil el ácido epsilon-aminocaproico. Es necesaria la vacunación contra la hepatitis B, dado que hay un aumento en el riesgo de exposición al virus de la hepatitis, debido a las frecuentes infusiones de productos sanguíneos. ADQUIRIDOS: Déficit de vitamina K El déficit de vit-K se puede dar por: 1) Dieta inadecuada (las reservas de vit-K se depleccionan en una semana como por ejemplo en la nutrición parenteral sin suplementos de vit-K). 2) Síndromes de malabsorción (obstrucción biliar, celíaca, insuficiencia pancreática, y el tratamiento con antibióticos de amplio espectro) . 3) Pérdida de los depósitos por enfermedad hepatocelular 4) Administración de cumarínicos cuyo mecanismo de acción es la inhibición de la producción de factores vitamina k dependientes. El tratamiento consiste en la administración parenteral de vit-K (10 mg) que restablece rápidamente los niveles de vit-K en el hígado y permite la síntesis del complejo protrombínico en 8-10 horas. Si se precisa un efecto inmediato administraremos plasma. Deficiencias de otros factores de la coagulación No existe ninguna peculiaridad resaltable de ninguna no de ellos, excepto en la deficiencia congénita de fibrinógeno, que no ocasiona hemorragias de importancia salvo las que acontecen en el curso de una cirugía. ALTERACIONES PLAQUETARIAS CUANTITATIVAS Trombopenia o trombocitopenia Se define por una cifra plaquetaria inferior a 150 x 10?/ L. En las embarazadas el límite bajo de la normalidad se sitúa en 120 x 10?/ L. Puede aparecer como un hallazgo casual en el hemograma o en el contexto de manifestaciones hemorrágicas espontáneas cutáneo-mucosas y/o de órganos internos. Todo recuento automático anormal debe ser contrastado mediante un examen visual de frotis ya que la causa más frecuente de una disminución de la cifra de plaquetas es la aglutinación de las mismas por efecto del anticoagulante utilizado: pseudotrombopenia inducida por EDTA. Salvo situaciones que puedan producir una disfunción añadida (uso de AAS, AINEs...) no se producirá clínica hemorrágica espontánea si las plaquetas no son inferiores a 20-30 x 10?/ L. Las hemorragias podrán aparecer tras exploraciones cruentas o cirugía sólo si las cifras son inferiores a 50-60 x 10?/ L. Esta correlación puede cambiar en algunas trombopenias inmunes crónicas en las que no hay signos hemorrágicos aunque las plaquetas sean muy bajas y, por otra parte, cifras superiores en las leucemias agudas y mielodisplasias cursan con sangrado por alteraciones funcionales de las mismas. Una vez comprobada la trombocitopenia, ésta puede estar producida por distintos mecanismos: 1.- Descenso de la producción de plaquetas: se produce por infiltración en la médula ósea de células malignas o células plasmáticas (mieloma mútiple, leucemias), síndromes mielodisplásicos, médula ósea irradiada o expuesta a fármacos (citostáticos, tiazidas, estrógenos, interferón), deficiencia nutricional (vitamina B12 y ácido fólico) e infecciones víricas. 2.- Secuestro anormal de plaquetas: el bazo normalmente secuestra un tercio del total de plaquetas. En el crecimiento del bazo o hiperesplenismo se produce un aumento desproporcionado del secuestro de plaquetas disminuyendo el número de plaquetas circulantes. Se da generalmente en la cirrosis hepática con hipertensión portal. 3.- Consumo de plaquetas: en las lesiones tisulares extensas como en las grandes quemaduras y síndromes de aplastamiento masivo y en las lesiones vasculares porque se produce una gran agregación plaquetar. También la interacción de las plaquetas con estructuras no endoteliales como las grandes prótesis vasculares. Las plaquetas también se consumen en pacientes con vasculitis extensas como en la toxemia gravídica y en la coagulación intravascular diseminada (CID). 4. Dilución de plaquetas: después de transfusiones masivas. La sangre conservada contiene un número bajo de plaquetas y la dilución de las plaquetas es proporcional a la cantidad de sangre transfundida. Una vez transfundidas 10 unidades de sangre se produce una afectación significativa de la hemostasia primaria. 5. Destrucción de plaquetas: por mecanis mos inmunológicos (antígeno-anticuerpo que dañan las plaquetas) en enfermedades autoinmunes como el lupus eritematoso, anemia hemolítica autoinmune y artritis reumatoide. Anticuerpos antiplaquetarios transitorios se puede dar por transfusiones múltiples de plaquetas (púrpura postransfusional), infecciones (sepsis) y fármacos (tiazidas,heparina, sulfamidas, quinidina, etc..). La púrpura trombocitopénica idiopática (PTI) se manifiesta como una enfermedad aguda relacionada con enfermedades infecciosas en la infancia y habitualmente autolimitada, o bien como un proceso autoinmune que tiende a la cronicidad. Las plaquetas también se pueden destruir por mecanismos no inmunológicos (circulación extracorpórea, hemangioma cavernoso gigante, rechazo trasplante renal, púrpura trombótica trombocitopénica, sdr. urémico-hemolítico) Coagulacion intravascular diseminada: CID Consiste en la producción extensa de trombina en la sangre circulante con el consiguiente consumo de factores de coagulación y plaquetas, posible obstrucción de la microcirculación y activación secundaria de la fibrinolisis. En el desarrollo de la CID cabe distinguir distintos factores desencadenantes, bien directos, como factores titulares y enzimas proteolíticas, como indirectos, a traves de mediadores como las plaquetas o los leucocitos. La forma de presentación más común es mediante hemorragias en una o varias localizaciones del organismo, púrpura, hematomas, hemorragias en tejidos lesionados tras cirugía o enfe rmedad subyacente. Desde el punto de vista de laboratorio son habituales las siguientes alteraciones: trombopenia, prolongación de todos los tiempos, descenso del fibrinógeno y del resto de factores de la coagulación, disminución de la antitrombina III y aumento de los productos de la degradación de ficrina y fibrinopéptido A. Clínicamente suele ser un proceso agudo y ocasionarse tras infecciones, problemas obstétricos, neoplasias ( leucemia aguda promielocítica ), fenómenos autoinmunes o tras traumas masivos. Debe administrarse heparina, inhibidores de la fibrinolisis y/o plasma si se produce un descenso llamativo de los factores o en caso de coexistir fenómenos hemorrágicos. Purpura trombocitopenica idiopatica: pti Existen dos formas clínicas: - PTI aguda.- Suele ser una enfermedad infantil, que afecta por igual a ambos sexos y aparecer tras procesos víricos de la s vías respiratoria sn superiores. La mayor parte de los casos tiene una recuperación espontánea y existe escasa recurrencia o mortalidad. Puede asociarse a eosinofilia y linfocitosis sanguinea y no suele requerir tratamiento, aunque en ocasiones es necesario el uso de esteroides o incluso la esplenectomía. - PTI crónica o Enfermedad de Werlhof.- Es típica de adultos jóvenes, generalmente mujeres. Hasta el 90% de los casos no se recupera espontáneamente y son frecuentes las recidivas. En ambos casos hay que descartar la existencia de otras enfermedades asociadas como lupus eritematoso sistémico o linfomas, ya que sólo en el caso de descartarse éstas recibirá el no mbre de idiopática. CUALITATIVAS Pueden ser hereditarias o adquiridas. La alteración hereditaria más frecuente es la enfermedad de von Willebrand, las otras alteraciones hereditarias son poco frecuentes (sdr. De BernardSoulier, tromboastenia, afibrinogenemia). Las alteracio nes adquiridas son más frecuentes: uremia, disproteinemias, circulación extracorpórea, enfermedades mielodisplásicas, fármacos. Enfermedad de Von Willebrand Es un trastorno hemorrágico de origen hereditario causado por la deficiencia del factor VIII que puede ser de leve a severa, y niveles bajos del antígeno relacionado con el factor VIII (sustancias necesarias para la coagulación de la sangre). Además se suma una insuficiencia del factor Von Willebrand, que también participa en la coagulación sanguínea. La sintomatología clásica consiste en sangrados frecuentes de encías, epistaxis y/o aparición de hematomas con cierta facilidad. El diagnóstico se realiza mediante la observación de un conteo de plaquetas normal asociado a un tiempo de sangrado prolongado, reducció n en los niveles de factor von Willebrand y reducción de los niveles del cofactor ristocetina. Por lo general, el sangrado es leve para la mayoría de los pacientes. Sin embargo, si se presenta algún traumatismo o se programa una cirugía, se puede suministrar crioprecipitado (DDAVP) para elevar los niveles del factor von Willebrand. Algunos subtipos de la enfermedad de von Willebrand no responden al DDAVP, por lo que se debe determinar el subtipo antes de confiar en el DDAVP en caso de sangrado significativo. Los pacientes con este trastorno no deben tomar medicamentos antiinflamatorios no esteroides (AINES), como la aspirina o el ibuprofeno, sin consulta previa. TRATAMIENTO DE LAS ALTERACIONES PLAQUETARIAS Generalmente un recuento de 100.000 plaquetas/mm3 de sangre no produce ningún síntoma y el tiempo de hemorragia es normal. Entre 50-100.000 plaquetas/mm3 hay un ligero alargamiento del tiempo de hemorragia, por debajo de 50.000 plaquetas/mm3 puede haber sangrado fácil y por debajo de 20.000 plaquetas/mm3 puede haber sangrado espontáneo. Por ello el tratamiento de las alteraciones plaquetares consistirá en: 1. Tratar la causa desencadenante: siempre que sea posible, como tratar la infección, suprimir el fármaco, etc . La mayoría de los pacientes que presentan una trombopenia inducida por fármacos se recuperan a los 7-10 días de la supresión del fármaco y se les deberá informar de que eviten el uso del fármaco en cuestión, ya que pequeñas dosis del mismo pueden inducir respuestas inmunes. 2. La transfusión de plaquetas debe reservarse para los pacientes con trombocitopenia asociada a coagulopatia. Cada unidad de plaquetas incrementará el número de plaquetas entre 7-10.000/ mm3 , la dosis recomendada es de 1 unidad por cada 10 kg de peso. En caso de cirugía mayor se debe mantener un recuento plaquetar por encima de 50.000 plaquetas/ mm3 y para pacientes que van a someterse a procedimientos mínimamente invasivos (biopsia, punción de vía central...) se trasfundirán plaquetas cuando el recuento sea igual o inferior 50.000 plaquetas/ mm3 . La administración de plaquetas también se recomienda en aquellos pacientes con recuentos superiores a 100.000 plaquetas/ mm3 con coagulopatia clínica por disfunción plaquetar. 3. Tratamiento farmacológico: administración de desmopresina (0,3 µg/kg vía ev o sc) en pacientes con disfunción plaquetar hereditaria o bien adquiridas. En el caso de trombocitopenias de causa inmunológica la transfusión de plaquetas no está indicada excepto si hay una hemorragia importante y se administrará prednisona a dosis de 1 mg/kg/d. 4. En la PTI el tratamiento será necesario cuando la cifra de plaquetas sea igual o inferior a 30.000 plaquetas/ mm3 o tenga sangrado activo independientemente de la cifra de plaquetas. El tratamiento puede ser con corticoesteroides, inmunoglobulina G o antiRhD, esplenectomia e inmunosupresores en función de la respuesta en cada caso. En los casos con hemorragia importante que no permite demora o sea necesario realizar cirugía urgente se administrarán plaquetas junto con inmunoglobulina iv o metil-prednisona a dosis altas (20 mg/kg/d en infusión durante 30 minutos durante 3 días). Aunque las plaquetas transfundidas son destruidas muy rápidamente pueden mejorar transitoriamente la hemostasia. En los casos que no responden a estas medidas y presenten compromiso vital (sangrado del SNC, traumatismo grave...) puede estar justificada la esplenectomía. CONCLUSIONES 1.- La lesion endotelial activa la hemostasia que incluye tras procesos: hemostasia primaria, secundaria y fibrinolisis. 2.- Entre las alteraciones de los factores de la coagulación la Hemofilia, enfermedad congénita, resulta las más frecuente y es de especial relevancia. 3.- El déficit de vitamina K, alteración adquirida, puede ser multifactorial siendo las causas más frecuentes la dieta inadecuada o los tratamientos con cumarínicos. 4.- La plaquetopenia se define por una cifra plaquetaria inferior a 150 x 1000 u/ L. 5.- Todo recuento plaquetar automático anormal debe ser contrastado mediante un examen visual de frotis ya que la causa más frecuente de una disminución de la cifra de plaquetas es la aglutinación de las mismas. 6.- Una vez comprobada la trombocitopenia, esta puede estar producida por cinco mecanismos: descenso de la producción, secuestro anormal, consumo excesivo, dilución o destrucción acelerada. 7.- La CID consiste en la producción extensa de trombina y su forma de presentación más común es mediante hemorragias en una o varias localizaciones del organismo, púrpura, hematomas, hemorragias en tejidos lesionados tras cirugía o enfermedad subyacente. 8.- Ante cualquier caso de PTI hay que descartar la existencia de otras enfermedades asociadas como lupus eritematoso sistémico o linfomas. 9.- La enfermedad de von Willebrand es un trastorno hemorrágico de origen hereditario causado por la deficiencia del factor VIII. Los pacientes con este trastorno no deben tomar medicamentos antiinflamatorios no esteroides (AINES), como la aspirina o el ibuprofeno, sin consulta previa. 10.- El tratamiento de las alteraciones plaquetares consistirá básicamente en la causa desencadenante si fuera corregible, transfusión plaquetar reservada para los pacientes con trombocitopenia asociada a coagulopatia o con tratamiento farmacológico con desmopresina o prednisona en casos concretos. EMBARAZO COLAGENOSIS INSUFICIENCIA RENAL HEPATOPATIA HEMOFILIA TRAT. CON ANTICOAGULANTES ORALES TRAT. CON HEPA RINA PLAQUETAS Moderadamente disminuidas T. QUICK Acortado(por aumento tasa de factores) Normal T.I.P.A Acortado(por aumento tasa de factores) Alargado si existe anticoagulante circulante FIBRINOG. Aumentado Normal Normal Normal Alargado por defecto de síntesis o defecto de vitamina K Algo alargado por defecto de síntesis o disfibrinogenemia Disminuido. Puede haber disfibrinogenemia Normal Muy alargado Normal Normal Muy alargado Discretamente alargado Normal Normal Algo alargado Muy alargado Normal Aumentadas. Disminuidas si hay anticuerpos antiplaquetas. Disminuidas y no funcionales Disminuidas por hiperesplenismo. En alcoholicos, puede ser por deficits nutricionales Normal Aumentado en fase de activación OBSERVAC Tendencia a tro mboembolismo, especialmente en postparto El anticoagulante favorece la tromb osis Diatesis hemorragica por trombopatia No responde a tratamiento con vitamina K En hemofilia A, disminución FVIII; en hemofilia B dis minución del FIX Requiere control periodico Requiere control periodico BIBLIOGRAFÍA 1.-Hematología clínica, Sans- Sabrafen J. Elsevier Es. 5º edición. 2.-Consulattive hemostasis and trombosis, Kitchens C. 1997. Saunders. 2º edición 3.-www.hemofiliacat.org/castellano/ .-asociacion catalana de hemofilia. 4.- Nova Pereira MS, Batlle Fondorona J, López Fernandez MF.: “Alteraciones de la coagulación: coagulopoatías congénitas y adquiridas”. Medicine, serie 9. nº22, 2004, 1401-1413 5.- Uranga M. Alteraciones de la coagulación. Bol.Vasco-nav pediatr, 2002, 36;72-74
 0
0
Anuncio





Descargar
Anuncio
Añadir este documento a la recogida (s)
Puede agregar este documento a su colección de estudio (s)
Iniciar sesión Disponible sólo para usuarios autorizadosAñadir a este documento guardado
Puede agregar este documento a su lista guardada
Iniciar sesión Disponible sólo para usuarios autorizados