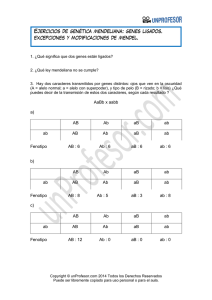
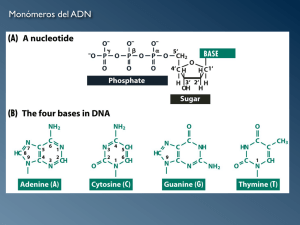
Descargar el Capítulo 2
Anuncio

Fundamentos de genética humana Reed E. Pyeritz, MD, PhD e2 INTRODUCCIÓN Los médicos se preocupan con lo que pueden descubrir junto a la cabecera del paciente y la investigación de laboratorio al mismo tiempo. En el lenguaje genético, los signos y síntomas del paciente constituyen su fenotipo. En la actualidad, los recursos están a la mano para definir el genotipo de una persona, el contenido de información real inscrito en los dos metros del DNA enrollado presente en cada célula corporal, o la mitad de esa cantidad en cada óvulo o espermatozoide maduro. La mayoría de las características fenotípicas —lo que incluye enfermedades y rasgos humanos como la personalidad, altura del individuo adulto e inteligencia— está determinada en alguna medida por los genes. La importancia de la contribución genética varía con amplitud entre los fenotipos humanos, y apenas ahora se hallan en desarrollo los métodos para identificar los genes que participan en rasgos complejos y en las enfermedades más comunes. Más todavía, la importancia de las interacciones entre ambiente y genotipo en la producción de fenotipos no puede sobreestimarse debido al desconocimiento existente sobre los mecanismos reales, como el del epigenoma. El DNA está compuesto de cuatro nucleótidos —adenina (A), guanina (G), citidina (C) y timidina (T)— ordenados de forma lineal a lo largo de una cadena, la cual se entrelaza en una hélice doble con una cadena complementaria, de tal manera que cada A se parea con una T y cada G con una C. Cada núcleo humano contiene 6.4 mil millones de estos pares de nucleótidos. Alrededor de 2% del DNA nuclear está organizado en unidades funcionales llamadas genes, y cada uno de los cerca de 23 000 genes humanos está acompañado de varios elementos reguladores de control cuando se encuentra activo en la producción de RNA mensajero (mRNA) por un proceso llamado transcripción. En la mayoría de las situaciones, el mRNA es transportado desde el núcleo hacia el citoplasma, donde su información genética es traducida en proteínas, las cuales llevan a cabo las funciones que acaban por determinar el fenotipo. Por ejemplo, las proteínas actúan como enzimas que facilitan el metabolismo y la síntesis celular; como elementos que se unen al DNA para regular la transcripción de otros genes; como elementos estructurales de las células y la matriz extracelular, y como moléculas receptoras de la comunicación intracelular e intercelular. El DNA también codifica muchas moléculas pequeñas de RNA cuyas funciones todavía están por definirse, como la regulación de la transcripción génica y la interferencia con la capacidad traductora de algunos mRNA. Los cromosomas son los vehículos donde se transportan los genes de generación en generación. Cada cromosoma es un complejo de proteína y ácido nucleico en el cual una doble hélice de DNA intacta es enrollada y superenrollada dentro de un espacio muchas veces menor que la longitud extendida del DNA. Dentro del cromosoma tienen lugar procesos integrados de una gran complejidad, como la replicación, recombinación y transcripción del DNA. En el núcleo de cada célula somática, de manera habitual, los seres humanos tienen 46 cromosomas, los que se hallan ordenados en 23 pares. Uno de estos pares, los cromosomas sexuales X y Y, determina el sexo del individuo; las mujeres tienen el par XX y los varones el par XY. Los 22 pares restantes se denominan autosomas (figura e2-1). Además de estos cromosomas nucleares, cada mitocondria —se encuentran en números variables en el citoplasma de todas las células— contiene múltiples copias de un cromosoma pequeño. Este cromosoma mitocondrial codifica algunas de las proteínas del metabolismo oxidativo y todas las de los RNA de transferencia que intervienen en la traducción de las proteínas internas de este organelo. Los cromosomas mitocondriales se heredan casi por completo del citoplasma del óvulo fertilizado, y por consiguiente, son de origen materno. En todas las células somáticas, los 44 autosomas y uno de los cromosomas X son activos desde el punto de vista transcripcional. En los varones, el X activo es el único X; porciones del cromosoma Y también son Fuente: McPhee SJ, Papadakis MA: Current Medical Diagnosis and Treatment 2011, 50th Edition: http://www.accessmedicine.com Copyright © The McGraw-Hill Companies, Inc. All rights reserved. Figura e2–1. Cariotipo normal de un macho humano. Preparado de células amnióticas cultivadas y teñidas con tinción de Giemsa. Son detectables cerca de 400 bandas por conjunto haploide de cromosomas. activas. En las mujeres, el requerimiento de la dosis compensatoria (para ser equivalente a la situación en varones) es satisfecho mediante la inactivación de la mayoría de un cromosoma X durante la embriogénesis temprana. Este proceso de inactivación de un cromosoma X, no obstante que no se comprende del todo, sucede al azar, de manera que, en promedio, en 50% de las células de una mujer, uno de los cromosomas X estará activo, y en el otro 50% estará activo el miembro homólogo del par. El fenotipo de la célula está determinado por cuáles genes de los cromosomas están activos en la producción de mRNA en un momento específico. El control transcripcional es de una complejidad muy alta. Entre otros procesos, se impide que un alelo de algunos genes transcriba mRNA a través del proceso de marcaje. El marcaje tiene lugar en el gameto, y lo típico es que suceda mediante la adición de un grupo metilo a los nucleótidos citosina en la región reguladora del alelo. La metilación produce una regulación a la baja del alelo y es específica de gameto. En consecuencia, el alelo de algunos genes heredado del padre se desactiva de modo permanente, en tanto que los alelos de otros genes heredados de la madre se inhiben de forma similar. Otros procesos pueden afectar la expresión de alelos específicos, como la modificación bioquímica de ciertas histonas y la organización espacial de los cromosomas dentro del núcleo. Aunque sorprendente, la mayoría de estos efectos puede persistir a través de generaciones y ser influenciado por el ambiente. Un ejemplo de esto último es la reducción en la marcación que ocurre en tiempos de hambruna, cuando los grupos metilo son deficientes en la dieta. Este campo de herencia no mendeliana se denomina epigenética. GENES Y CROMOSOMAS En todos los genes, la información está contenida en parcelas llamadas exones, los cuales se intercalan con tramos de DNA llamado intrones, que no codifican ninguna información relacionada con la secuencia de la proteína. Sin embargo, los intrones pueden contener secuencias genéticas reguladoras, y algunos intrones son tan extensos que codifican un gen por completo diferente. La localización exacta de un gen en un cromosoma es su locus, y la disposición de los loci constituye el mapa génico humano. Una variación de este mapa, mediante la identificación de loci seleccionados que se sabe participan en la enfermedad humana, se muestra en la figura e2-2. La diferencia en la resolución más alta del ordenamiento de genes conseguible por técnicas moleculares (como el análisis de enlaces) comparadas con las técnicas citogenéticas (como la visualización de defectos pequeños) es sustancial, aunque la brecha se está estrechando. Los cromosomas del cariotipo “estándar” que se muestran en la figura e2-1 tienen alrededor de 450 bandas visibles; bajo las mejores condiciones citológicas y microscópicas, puede observarse un total cercano a 1 600 bandas. Empero, incluso en esta configuración extendida, cada banda contiene docenas —y en ocasiones centenas— de genes individuales. Por consiguiente, la falta (deleción) de una pequeña banda representa la pérdida de muchas secuencias de codificación y ejerce efectos diversos en el fenotipo. Las técnicas citogenéticas de rutina para visualización de cromosomas han sido sustituidas en gran medida por métodos que se basan en la disposición. p Ictiosis ligada a X Albinismo ocular Distrofia muscular de Duchenne 2 22 21 1 11 Retinitis pigmentosa 3 Síndrome de Wiskott-Aldrich 1 Insensibilidad a los andrógenos Deficiencia de PGK (anemia hemolítica) Neuropatía de Charcot-Marie-Tooth 13 21 q Agammaglobulinemia tipo 1 Síndrome de Alport Gota secundaria a PRPP 2 25 28 X p 1 11 q 1 Gota relacionada con HPRT Hemofilia B Síndrome de X frágil Hemofilia A Ceguera al color (diversas formas) Deficiencia de G6PD: favismo Disgenesia gonadal XY Síndrome exclusivo de la célula de Sertoli 11 12 Y Fuente: McPhee SJ, Papadakis MA: Current Medical Diagnosis and Treatment 2011, 50th Edition: http://www.accessmedicine.com Copyright © The McGraw-Hill Companies, Inc. All rights reserved. Figura e2–2. “Mapa mórbido” parcial del genoma humano. Junto al ideograma de los cromosomas humanos X y Y muestra los trastornos mendelianos representativos causados por mutaciones en ese locus. Más de 460 fenotipos han sido mapeados en el cromosoma X y ocho en el cromosoma Y. (Tomada con autorización de V. McKusick y J. Strayer.) El número y disposición de los genes en los cromosomas homólogos son idénticos aunque las secuencias de codificación reales de tales genes, o el número de copias de esos genes, puedan no serlo. Las copias homólogas de un gen se llaman alelos. En la comparación de alelos debe especificarse a qué nivel de análisis se hizo ésta. Cuando los alelos son idénticos —en que sus secuencias de codificación y el número de copias son invariables— el individuo es homocigoto en dicho locus. A un nivel más general, los alelos pueden ser idénticos desde el punto de vista funcional pese a variaciones sutiles en la secuencia de nucleótidos, con el resultado de que las proteínas producidas desde los dos alelos son idénticas o que cualquier diferencia que pueda presentarse en la secuencia de aminoácidos no repercutirá en la función de la proteína. Si el individuo se analiza a nivel del fenotipo de la proteína, la homocigosidad alélica sería otra vez un descriptor apt. Sin embargo, si el análisis fuera a nivel del DNA —como sucede en la secuenciación de nucleótidos— entonces, a pesar de la identidad funcional, los alelos se verían diferentes y el individuo se clasificaría como heterocigoto para ese locus. La heterocigosidad basada en las diferencias en los productos proteínicos de los alelos se ha detectado por décadas y constituyó la primera evidencia de peso respecto al alto grado de la variabilidad biológica humana. En el decenio pasado, el análisis de las secuencias de DNA exhibió que la variabilidad genética es mucho más común, las diferencias en la secuencia de nucleótidos entre individuos ocurren alrededor de una vez cada 1 100 nucleótidos. Secuencias mucho más largas también pueden estar presentes en números variables de copias, o pueden ser por entero deficientes, con frecuencia sin efectos obvios en el fenotipo. La identificación de tal número variable de copias ha sido uno de los descubrimientos más excitantes y útiles de la genética humana en años pasados. Debido a la selección evolutiva contra cambios deletéreos en la secuencia, la variación del DNA en las regiones codificantes de los genes se produce una vez cada 2 000 nucleótidos, y menos de la mitad de tales variaciones causa un cambio en un aminoácido. Sin embargo, el nivel de variación de la secuencia entre humanos, no obstante sus orígenes étnicos, es mucho menos común (tres a diez veces) que en nuestros ancestros primates. Goodman DM et al. JAMA patient ped. Genomic medicine. JAMA 2013;309(14):1544. [PubMed: 23571597] Koboldt DC et al. The next-generation sequencing revolution and its impact of genomics. Cell 201326;155(1):27–38. [PubMed: 24074859] National Coalition for Health Professional Education in Genetics. Medicine’s future: a genomics curriculum for clinicians. http://www.nchpeg.org/index.php?option=com_content&view=article&id=368 Rimoin DL (editors) et al. Emery and Rimoin’s principles and practice of medical genetics. 6th ed. Churchill Livingstone, 2014. Weischenfelsdt J et al. Phenotype impact of genomic structural variation: Nat Rev Genet 2013;14(2):125–138. [PubMed: 23329113] MUTACIÓN Casi toda la heterocigosidad alélica suele producirse cuando se heredan alelos diferentes con el huevo y el espermatozoide, pero también ocurre como consecuencia de alteraciones espontáneas en la secuencia de nucleótidos (mutación). El cambio genético que se produce durante la formación de un huevo o un espermatozoide se llama mutación germinal. Cuando el cambio sucede después de la concepción —desde las etapas más tempranas de la embriogénesis hasta la división de células en el cuerpo del adulto mayor— se le denomina mutación somática. Como se explica después, el papel de la mutación somática en la etiología de la enfermedad humana se reconoce ahora cada vez más. El tipo de mutación más generalizado es la alteración en el número o estructura física de los cromosomas. Por ejemplo, la falta de disyunción (falla del par de cromosomas para separarse) durante la meiosis —la división de reducción que lleva a la producción de huevos y espermatozoides maduros— determina que el embrión tenga demasiados cromosomas o menos que los necesarios, una situación llamada aneuploidía. El reordenamiento de los brazos del cromosoma, como el que se presenta en la traslocación o inversión, es una mutación incluso si la rotura y nueva unión no interrumpen ninguna secuencia codificante. En consecuencia, el efecto fenotípico de las mutaciones cromosómicas más obvias puede ser profundo (como en la aneuploidía) o nulo. Un poco menos obvias, pero todavía detectables con recursos citológicos, son las deleciones de partes de un cromosoma. Tales mutaciones casi siempre alteran el fenotipo, debido a que se pierde cierto número de genes; sin embargo, una deleción puede incluir sólo un nucleótido, mientras que debe perderse cerca de uno a dos millones de nucleótidos (una a dos megabases) antes de que el defecto pueda visualizarse con la mayoría de los métodos citogenéticos sensibles excepto la hibridación in situ o los análisis del conjunto. Las deleciones y duplicaciones de regiones sustanciales de secuencias de nucleótidos son usuales en los seres humanos. En apariencia, muchas son anodinas y se pasan de los padres a los hijos en un patrón hereditario autosómico dominante. Otras incluyen uno o más genes y pueden tener consecuencias clínicas sutiles o profundas. Estas últimas suelen producirse de novo, lo que significa que ninguno de los padres tiene la copia con la variación del número, que debe haberse originado durante la meiosis de uno de los gametos. Las mutaciones de uno o unos pocos nucleótidos en los exones provocan numerosas consecuencias potenciales. Los cambios en un nucleótido pueden alterar qué aminoácido se codifica; si el aminoácido está en una región crítica de la proteína, la función puede alterarse de forma pronunciada (p. ej., enfermedad de células falciformes). Por otro lado, algunas sustituciones de aminoácidos no ejercen un efecto detectable en la función, y, por tanto, el fenotipo permanece intacto pese a la mutación. De manera similar, a raíz de que el código genético está degenerado (dos o más secuencias de tres nucleótidos diferentes llamadas codones codifican el mismo aminoácido), la sustitución de nucleótidos no altera de manera forzosa la secuencia de aminoácidos de la proteína. Tres codones específicos señalan la terminación de la traducción; por ello, la sustitución de un nucleótido de un exón que genera uno de los codones de detención causa por lo regular una proteína truncada, que suele ser disfuncional. Otras sustituciones de nucleótidos pueden interrumpir las señales que dirigen el corte y empalme de la molécula del mRNA y en general alteran el producto proteínico. Para finalizar, las inserciones y deleciones de uno o más nucleótidos pueden causar efectos devastadores —cualquier cambio que no es un múltiplo de tres nucleótidos interrumpe el marco de lectura del exón restante— o por el contrario efectos mínimos (si la proteína puede tolerar la inserción o pérdida de un aminoácido). Las mutaciones en los intrones pueden interrumpir las señales de corte y empalme del mRNA o los elementos reguladores, o pueden resultar por completo silenciosas con respecto al fenotipo. Una gran parte de las variaciones en la secuencia de nucleótidos entre los individuos (en promedio, una diferencia cada pocos cientos de nucleótidos) reside en los intrones. Las mutaciones en el DNA entre genes adyacentes también pueden ser silenciosas o ejercer un efecto profundo en el fenotipo si las secuencias reguladoras se interrumpen; estas secuencias reguladoras pueden afectar genes que están millones de bases más lejos, a causa quizá del plegamiento intrincado de la molécula de DNA dentro de la cromatina. De manera alternativa, el DNA situado entre genes, 97% del genoma que no codifica RNA mensajero, codifica en cambio una variedad de moléculas de RNA pequeñas, no codificantes, que desempeñan funciones en el control de la expresión génica. Las mutaciones en las secuencias codificantes de estos pequeños RNA de interferencia (siRNA), microRNA (miRNA), y RNA que interactúa con piwi (piRNA) apenas comenzaron a estudiarse. Un novedoso mecanismo de mutación, que también ayuda a explicar la variación clínica entre familiares, ha sido descubierto en la distrofia miotónica, la enfermedad de Huntington, síndrome de retardo mental por X frágil, ataxia de Friedreich, y otros trastornos. Una región de secuencias de trinucleótidos repetidas cercanas a un gen o dentro de éste puede ser inestable en algunas familias; la expansión del número de unidades repetidas dentro de este segmento más allá de un umbral crítico representa enfermedad, ya sea por la regulación a la baja de ese alelo o la producción de una proteína defectuosa. Cuanto más larga la repetición de trinucleótidos, más grave el fenotipo, un fenómeno llamado anticipación, porque la afección empeora de una generación a la siguiente. Las mutaciones se pueden producir de manera espontánea o pueden ser inducidas por factores ambientales como la radiación, medicación o infecciones virales. Tanto la edad avanzada materna como la paterna favorecen las mutaciones, aunque de diferentes tipos. En la mujer, la meiosis se completa sólo cuando el huevo es ovulado, y la falta de disyunción cromosómica se incrementa a medida que el huevo es más viejo. El riesgo de que resulte un huevo aneuploide aumenta de forma exponencial y se convierte en una preocupación clínica importante en mujeres mayores de 30 años. En varones, las mutaciones de carácter sutil —que afectan las secuencias de nucleótidos— son más frecuentes conforme aumenta la edad. La descendencia de hombres mayores de 40 años está en un riesgo cada vez mayor de sufrir afecciones mendelianas, en primer lugar las de tipo autosómico dominante. Barsh G. Genetic Disease. In: McPhee SJ et al. Pathophysiology of disease: An introduction to clinical medicine. 6th ed. New York: McGraw-Hill Medical, 2010 Klopocki E et al. Copy-number variations, noncoding sequences, and human phenotypes. Annu Rev Genomics 2011;12:53-72. [PubMed: 21756107] Miller DT et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010 May 14;86(5):749-64. [PubMed: 20466091] GENES EN INDIVIDUOS Para algunos rasgos cuantitativos, como la estatura del adulto o la concentración de glucosa sérica en los individuos normales, es difícil distinguir las contribuciones de genes individuales, esto se debe, en general, a que los fenotipos son el producto de múltiples genes que actúan de manera concertada, además de recibir la influencia del ambiente y del azar. Sin embargo, si uno de los genes del sistema es aberrante, puede originarse un cambio importante desde el fenotipo “normal” o esperado. Si el fenotipo aberrante es serio (p. ej., una enfermedad) o incluso reconocido, depende de la naturaleza del producto del gen defectuoso y de qué tan resiliente sea el sistema a la alteración. Este último aspecto destaca la importancia de la homeostasis en fisiología y desarrollo, muchas mutaciones son pasadas por alto porque el sistema puede hacerles frente, incluso aunque la tolerancia para alteraciones adicionales pueda reducirse. En otras palabras, la mayoría de las características humanas y de las enfermedades comunes es poligénica, mientras que muchos de los fenotipos alterados y considerados por tradición como “genéticos” son en realidad monogénicos, pero incluso en ese caso influenciados por otros loci en el genoma de una persona. Los fenotipos debidos a alteraciones en un solo gen también se caracterizan como mendelianos, por Gregor Mendel, el monje y biólogo de dedicación parcial que estudió la reproducibilidad y recurrencia de variaciones en chícharos de jardín. Él mostró que algunos rasgos eran dominantes sobre otros, a los que llamó recesivos. Los rasgos dominantes requirieron sólo una copia de un “factor” para expresarse, con prescindencia de lo que fuera la otra copia, mientras que los rasgos recesivos necesitaron las dos copias para que la expresión sucediera. En términos modernos, los factores mendelianos son genes, y las copias alternativas del gen son alelos. Por ejemplo, tómese a A como el alelo común (normal) y a a como el alelo mutante en un locus: si el mismo fenotipo está presente cuando el genotipo es A/a o a/a, el fenotipo es dominante, mientras que si el fenotipo está presente sólo cuando el genotipo es a/a, es recesivo. En medicina es importante tener en mente dos consideraciones: primero, dominancia y recesividad son atributos del fenotipo, no del gen; segundo, los conceptos de dominancia y recesividad dependen de cómo se defina el fenotipo. Para ilustrar ambos conceptos se recurrirá a la enfermedad de células falciformes. Esta afección aparece cuando una persona hereda dos alelos de globina β (S), en la cual el glutamato normal de la posición 6 de la proteína fue sustituido por valina; el genotipo del locus de la globina β es HbS/HbS, comparado con el normal, que es HbA/HbA. Cuando el genotipo es HbS/HbA, el individuo no padece la enfermedad de células falciformes, de manera que esta condición satisface el criterio para ser un fenotipo recesivo. Ahora considérese el fenotipo de los eritrocitos falciformes. Los glóbulos rojos con el genotipo HbS/ HbS son falciformes, pero si la tensión de oxígeno es reducida, también lo hacen las células con el genotipo HbS/HbA. Por consiguiente, lo falciforme es un rasgo recesivo, y los síntomas sólo surgen en ocasiones en individuos heterocigotos cuando el oxígeno atmosférico es reducido, en especial durante el ejercicio. Un fenotipo mendeliano se caracteriza no sólo en términos de dominancia y recesividad, sino también de acuerdo con si el gen determinante está en el cromosoma X o en uno de los 22 pares de autosomas. En consecuencia, los rasgos o enfermedades se llaman autosómicos dominantes, autosómicos recesivos, recesivos ligados a X, y dominantes ligados a X. GENES EN FAMILIAS Desde la primera década del siglo veinte, los patrones de recurrencia de fenotipos humanos específicos fueron explicados en términos de los principios descritos en primer lugar por Mendel en la planta de chícharos de jardín. El segundo principio de Mendel —referido de manera usual como su primero— se llama ley de segregación y establece que un par de factores (alelos) que determinan algunos rasgos se separan (segregan) durante la formación de los gametos. En términos simples, un individuo heterocigoto (A/a) producirá dos tipos de gametos con respecto a un locus determinado, uno contendrá sólo A y el otro contendrá sólo a, en proporciones iguales. La descendencia de esta persona tendrá una probabilidad de 50-50 de heredar el alelo A y una probabilidad similar de heredar el alelo a. Los conceptos de genes en individuos y en familias pueden combinarse para especificar qué rasgos mendelianos serán heredados. Herencia autosómica dominante Las características de la herencia autosómica dominante en humanos puede resumirse como sigue: 1) Hay un patrón vertical en el pedigrí, con múltiples generaciones afectadas (figura e2-3). 2) Los heterocigotos para el alelo mutante muestran un fenotipo anormal. 3) Varones y mujeres se afectan con igual frecuencia y gravedad. 4) Sólo uno de los padres debe afectarse para que la descendencia esté en riesgo de desarrollar el fenotipo. 5) Cuando una persona afectada se aparea con una sana, cada descendiente tiene una probabilidad de 50% de heredar el fenotipo afectado. Esto es cierto pese al sexo del padre afectado, de manera específica, la transmisión se produce de varón a varón. 6) La frecuencia de casos esporádicos se vincula de forma positiva con la gravedad del fenotipo. Para ser más precisos, cuanto mayor sea la capacidad reproductiva de la persona afectada, es menos probable que cualquier caso dado resulte de una nueva mutación. 7) La edad promedio de los padres es avanzada para los casos aislados (esporádicos o una nueva mutación). Fuente: McPhee SJ, Papadakis MA: Current Medical Diagnosis and Treatment 2011, 50th Edition: http://www.accessmedicine.com Copyright © The McGraw-Hill Companies, Inc. All rights reserved. Figura e2–3. Un pedigrí donde se ilustra la herencia autosómica dominante. Los símbolos cuadrados indican varones y los círculos mujeres; los símbolos vacíos indican que el fenotipo de la persona está indemne, y los símbolos rellenos indican que el fenotipo está presente en cierta medida. Por lo general, los fenotipos autosómicos dominantes dependen de la edad, son menos graves que los autosómicos recesivos, y se manifiestan por malformaciones u otra característica física. Son pleiotrópicos por el hecho de que múltiples manifestaciones clínicas, e incluso algunas sin relación aparente, derivan de la misma mutación, y variables en que la expresión de la misma mutación en las personas puede diferir. La penetrancia es un concepto relacionado con condiciones mendelianas —en especial las dominantes— y el término suele emplearse mal. Se lo debería definir como una expresión de la frecuencia de aparición de un fenotipo (dominante o recesivo), cuando uno o más alelos mutantes están presentes. En los individuos, penetrancia es un fenómeno de todo o nada, el fenotipo está presente (penetrante) o no (no penetrante). El término variabilidad —no “penetrancia incompleta”— se debe utilizar para denotar diferencias en la expresión de un alelo. La causa más frecuente de falta de penetrancia aparente es la insensibilidad de los métodos para detectar el fenotipo. Si un padre en apariencia normal de un niño con una afección dominante es de hecho heterocigoto para la mutación, dicho padre debería tener una probabilidad de 50% en cada concepción subsecuente de tener otro hijo afectado. Una causa habitual de falta de penetrancia en enfermedades mendelianas que aparecen en el adulto es la muerte de la persona afectada antes de que el fenotipo se haga evidente, pero después de la transmisión del alelo mutante a la descendencia. Por tanto, la asesoría genética precisa demanda una atención cuidadosa de los antecedentes médicos de la familia y un escrutinio de alta resolución de ambos padres de un niño con una afección que se sabe corresponde a rasgos dominantes mendelianos. Cuando ambos alelos se expresan en el heterocigoto, como en el grupo sanguíneo AB, en los rasgos falciformes (HbS/HbA), en los antígenos de histocompatibilidad principal (es decir, A2B5/A3B17), o en la enfermedad falciforme C (HbS/HbC), el fenotipo se denomina codominante. En fenotipos humanos dominantes, el alelo mutante en homocigotos es casi siempre más grave que en heterocigotos. La primera ley de Mendel establece que —desde la perspectiva del fenotipo— no importa de cuál padre se heredó un alelo mutante particular. Durante años se consideró que este principio era demasiado obvio para codificarlo como la “ley” de cualquiera y en consecuencia se lo ignoró. Sin embargo, en los hechos, evidencia reciente de estudios de trastornos humanos sugiere que ciertos genes se “procesan” (marcan) a medida que se mueven a través de las gónadas y que el procesamiento en los testículos es diferente al de los ovarios. De allí que el primer principio mendeliano no sólo es importante, sino que cuando se lo formuló de manera original mediante la observación de los chícharos fue incorrecto. Herencia autosómica recesiva Las características de la herencia autosómica recesiva en los seres humanos puede resumirse como sigue: 1) ocurre un patrón horizontal en el pedigrí, con una sola generación afectada (figura e2-4). 2) Varones y mujeres se afectan con igual frecuencia y gravedad. 3) La herencia es de ambos padres, cada uno un heterocigoto (portador) y por lo regular sin ninguna afectación clínica. 4) Cada descendiente de dos portadores tiene una probabilidad de 25% de resultar afectado, una probabilidad de 50% de convertirse en portador, y una probabilidad de 25% de no heredar ningún alelo mutante. Por lo tanto, dos terceras partes de todos los descendientes están libres de manifestaciones clínicas y son portadores. 5) En apareamientos entre individuos que tienen el mismo fenotipo recesivo, toda la descendencia resultará afectada. 6) Los individuos afectados que se aparean con individuos sin afecciones y que tampoco son portadores sólo tienen descendencia sana. 7) El más raro es el fenotipo recesivo, donde lo más probable es que los padres son consanguíneos (familiares). A menudo, los fenotipos autosómicos recesivos se relacionan con actividad deficiente de algunas enzimas y en consecuencia se denominan errores congénitos del metabolismo. Entre dichos trastornos se incluye la fenilcetonuria, la enfermedad de Tay-Sachs, y diversas enfermedades por almacenamiento de glucógeno, que tienden a ser más graves, menos variables, y menos dependientes de la edad que las afecciones dominantes. Cuando una afección autosómica recesiva es demasiado rara, la probabilidad de que los padres de la descendencia afectada sean consanguíneos se incrementa. Como consecuencia, la prevalencia de afecciones recesivas raras es alta entre grupos endogámicos como la Vieja Orden Amish. Por otro lado, cuando la afección autosómica recesiva es común, la probabilidad de consanguinidad entre los padres de los casos no es más alta que en la población general. Dos alelos mutantes diferentes en el mismo locus, como en HbS/HbC, forman un compuesto genético (compuesto heterocigoto). El fenotipo suele fluctuar entre los que produce cualquiera de los alelos en el estado homocigoto. A raíz del gran número de mutaciones posibles en un gen determinado, muchos fenotipos autosómicos recesivos se deben probablemente a compuestos genéticos. La enfermedad de células falciformes es una excepción. La consanguinidad es una evidencia presuntiva fuerte de homocigosidad verdadera de los alelos mutantes y otra vez de un compuesto genético. Fuente: McPhee SJ, Papadakis MA: Current Medical Diagnosis and Treatment 2011, 50th Edition: http://www.accessmedicine.com Copyright © The McGraw-Hill Companies, Inc. All rights reserved. Figura e2–4. Un pedigrí donde se ilustra la herencia autosómica recesiva. (Los símbolos equivalen a los de la figura e2–3.) Herencia ligada al X Las características generales de la herencia ligada al X en seres humanos pueden resumirse como sigue: 1) No hay transmisión del fenotipo de varón a varón (figura e2-5). 2) Los varones sin manifestaciones no transmiten el fenotipo. 3) Todas las hijas de un varón afectado son portadoras heterocigotas. 4) Los varones suelen afectarse con más gravedad que las mujeres. 5) Si una mujer heterocigota se considera afectada —y si el fenotipo es “recesivo” o “dominante”— depende con frecuencia de la sensibilidad del ensayo o examen. 6) Algunas madres de varones afectados pueden no ser heterocigotas (esto es, ellas son homocigotas normales) pero tienen una mutación germinal. La proporción de madres heterocigotas (portadoras) se relaciona negativamente con la gravedad de la afección. 7) Las mujeres heterocigotas transmiten el gen mutante a 50% de sus hijos, quienes resultan afectados, y a 50% de sus hijas, que son heterocigotas. 8) Si un varón afectado se aparea con una mujer heterocigota, 50% de la descendencia masculina resultará afectada, y dará la falsa impresión de una transmisión de varón a varón. De la descendencia femenina de tales apareamientos, 50% será afectada con tanta gravedad como el promedio de los varones homocigotos; en pedigrís pequeños, este patrón puede simular una herencia autosómica dominante. Fuente: McPhee SJ, Papadakis MA: Current Medical Diagnosis and Treatment 2011, 50th Edition: http://www.accessmedicine.com Copyright © The McGraw-Hill Companies, Inc. All rights reserved. Figura e2–5. Un pedigrí donde se ilustra la herencia ligada al X. (Los símbolos equivalen a los de la figura e2-3.) Las características de la herencia ligada al X dependen de la gravedad fenotípica. En algunos trastornos, los varones afectados no sobreviven hasta reproducirse. En estos casos, alrededor de dos terceras partes de los varones afectados tienen una madre portadora; en el tercio restante, el trastorno se origina en una nueva mutación germinal en un cromosoma X de la madre. Cuando el trastorno se produce, casi siempre se manifiesta en mujeres heterocigotas (herencia dominante ligada al X), las mujeres tienden a estar afectadas alrededor de dos veces más que los varones, y en promedio una mujer afectada transmite el fenotipo a 50% de sus hijos y 50% de sus hijas. A menudo, los fenotipos ligados al X presentan variabilidad clínica —en particular en mujeres heterocigotas— y se sospecha que son autosómicos dominantes con falta de penetrancia. Por ejemplo, la enfermedad de Fabry (una deficiencia de galactosidasa αA) puede ser silenciosa en el terreno clínico en mujeres portadoras o puede causar un accidente vascular cerebral, insuficiencia renal, o un infarto del miocardio en sujetos de edad media. El mosaicismo germinal se presenta en madres de hijos con afecciones ligadas al X. La probabilidad de que tal madre tenga un segundo hijo afectado o una hija heterocigota depende de la fracción de sus ovocitos que contenga la mutación. En la actualidad, dicha fracción es imposible de determinar. Sin embargo, la presencia de mosaicismo germinal puede detectarse en algunas afecciones (p. ej., distrofia muscular de Duchenne) en una familia por análisis del DNA, y este conocimiento es crucial para la asesoría genética. Herencia mitocondrial Las mutaciones en los genes que codifica el cromosoma mitocondrial causan una variedad de enfermedades que afectan (en particular) órganos muy dependientes del metabolismo oxidativo, como retina, cerebro, riñones, y corazón. Debido a que las mitocondrias de una persona derivan casi por entero del huevo, el patrón hereditario es distinto del de los trastornos mendelianos y se denomina “maternal” o, de manera más apropiada, “mitocondrial”. Una mujer afectada puede pasar el cromosoma de la mitocondria defectuosa a toda su descendencia, mientras que un varón afectado tiene un riesgo bajo de pasar su mutación a sus hijos (figura e2-6). Ya que cada célula y el huevo contienen muchas mitocondrias y como cada mitocondria contiene numerosos cromosomas, son posibles dos situaciones: si cada cromosoma en cada mitocondria contiene la misma mutación, se dice que la persona es homoplásmica para la mutación. Por otro lado, si sólo algunos de los cromosomas mitocondriales contienen la mutación, la persona es heteroplásmica. En este último caso, la descendencia puede heredar relativamente pocas mitocondrias que posean la mutación y sufrir una enfermedad leve o ninguna. Fuente: McPhee SJ, Papadakis MA: Current Medical Diagnosis and Treatment 2011, 50th Edition: http://www.accessmedicine.com Copyright © The McGraw-Hill Companies, Inc. All rights reserved. Figura e2–6. Herencia mitocondrial (“maternal”). Una mutación genética mitocondrial, indicada por los símbolos coloreados, es pasada por la mujer (círculo) a toda su descendencia, incluidos los varones (cuadrados). En la descendencia subsecuente, los varones no transmiten la mutación, pero las mujeres la continúan transmitiendo a toda su descendencia porque las mitocondrias pasan a través de los huevos, no de los espermatozoides. Para simplificar, aunque los padres que se muestran pertenecen a la primera generación, las generaciones subsecuentes no muestran a las parejas genéticas, ya que se asume que carecen de la mutación. Nota: todas o sólo algunas de las mitocondrias pueden contener la mutación, una variable que afecta la expresión clínica de la mutación. (Véase el texto con respecto a individuos homoplásmicos y heteroplásmicos.) Más de 16 000 genes humanos han sido identificados o implicados a través de sus fenotipos y sus patrones hereditarios familiares. Este total representa 60-70% de todos los genes que se cree son codificados por los 22 autosomas, dos cromosomas sexuales, y el cromosoma mitocondrial. En el decenio de los años 60, Victor McKusick y colegas comenzaron un esfuerzo internacional para catalogar la variación mendeliana humana. El proyecto persiste como Online Mendelian Inheritance in Man (OMIM). Greaves LC et al. Mitochondrial DNA and disease. J Pathol 2012;226(2):274–286. [PubMed: 21989606] Online Mendelian Inheritance in Man, OMIM. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore, MD). http://omim.org¯ Wallace DE et al. Mitochondrial genes in degenerative diseases, cancer and aging. En: Rimoin DL, et al. (editors): Emery and Rimoin’s principles and practice of genética médica. 6th ed. Churchill Livingstone, 2014. TRASTORNOS DE CAUSA MULTIFACTORIAL Muchos trastornos se agrupan en familias, pero no se acompañan de aberraciones cromosómicas evidentes o patrones hereditarios mendelianos. Los ejemplos incluyen malformaciones congénitas como el labio hendido, la estenosis pilórica y la espina bífida; la enfermedad de la arteria coronaria; la diabetes mellitus tipo 2, y varias formas de neoplasia. A menudo se caracterizan por frecuencias variables en diferentes grupos raciales o étnicos, disparidad en la predilección sexual, y mayor frecuencia (pero menos que concordancia total) en gemelos monocigotos que en dicigotos. Este patrón hereditario se llama “multifactorial” para destacar que múltiples genes interactúan con diversos agentes ambientales para producir el fenotipo. Se asume que el agrupamiento familiar se debe a que se comparten alelos y ambiente. En la mayoría de las afecciones multifactoriales existe una comprensión escasa de cuáles son los genes particulares que se afectan, cómo interactúan ellos y sus productos, y en qué forma los diferentes factores extragenéticos contribuyen con el fenotipo. En algunos trastornos, los estudios bioquímicos y genéticos han identificado afecciones mendelianas dentro del fenotipo general: defectos del receptor de las lipoproteínas de baja densidad como hecho significativo para una pequeña fracción de casos de cardiopatía isquémica (la fracción es mayor si sólo se consideran los pacientes menores de 50 años); la poliposis familiar del colon predispone al adenocarcinoma; y algunos pacientes con enfisema tienen deficiencia hereditaria del inhibidor de la proteinasa α-1 (antitripsina α-1). Pese a estos ejemplos notables, es improbable que esta preocupación reduccionista con los fenotipos mendelianos explique la gran mayoría de las enfermedades humanas; pero incluso así, en un último análisis, gran parte de la patología humana demostrará estar asociada con factores genéticos en su causa, patogenia, o ambas. La ignorancia existente acerca de los mecanismos genéticos fundamentales en desarrollo y fisiología no ha restringido por completo la elaboración de enfoques prácticos para entender la genética de los trastornos multifactoriales. Por ejemplo, los riesgos de recurrencia se basan en datos empíricos derivados de la observación de muchas familias. El riesgo de recurrencia de trastornos multifactoriales se incrementa en numerosas circunstancias: 1) en familiares cercanos (hermanos, descendencia, y padres) de un individuo afectado; 2) cuando dos o más miembros de una familia padecen la misma afección; 3) cuando el primer caso familiar se presenta en el sexo que menos se afecta (p. ej., la estenosis pilórica es cinco veces más común en niños; una mujer afectada tiene un riesgo tres a cuatro veces mayor de tener un hijo con estenosis pilórica), y 4) en grupos étnicos en los cuales existe una alta incidencia de una afección particular (p. ej., la espina bífida es 40 veces más común en blancos —e incluso más frecuente entre los irlandeses— que en asiáticos). Para muchos trastornos en apariencia multifactoriales se han examinado muy pocas familias para establecer datos empíricos del riesgo. Una aproximación útil del riesgo de recurrencia en familiares cercanos se obtiene al usar la raíz cuadrada de la incidencia. Por ejemplo, muchas malformaciones congénitas comunes muestran una incidencia de 1:2 500 a 1:400 nacimientos vivos; por tanto, el riesgo de recurrencia calculado se sitúa en valores de 2-5%, valores que corresponden en forma cercana a los que se extraen de la experiencia. Se ha realizado un gran esfuerzo en identificar los contribuyentes genéticos de la enfermedad multifactorial. Estudios asociados del genoma (GWAS) han incluido extensas cohortes de pacientes con una enfermedad determinada. Tales pacientes exhiben cientos de miles y hasta un millón de polimorfismos de un solo nucleótido que se documentan y comparan con una cohorte de individuos sin evidencia de la enfermedad, de tamaño, edad y caracteres étnicos similares. De esta forma, cientos de trastornos usuales se han vinculado con marcadores incluso más específicos del genoma humano. A pesar de estos resultados impactantes, la realidad es que el cambio de un nucleótido dado sólo altera el riesgo relativo para una enfermedad determinada de forma ligera (es decir, un riesgo relativo de 1.2-1.4), con muy pocas excepciones. Más importante todavía, muy pocos de los polimorfismos de los nucleótidos suceden dentro de los genes o, si se producen, no alteran la función del gen en ninguna forma reconocible. Por consiguiente, resta por realizar mucho trabajo para definir a los contribuyentes genéticos de las enfermedades más frecuentes, y la historia familiar persiste como una herramienta clínica importante en la predicción del riesgo. Lautenbach DM et al. Communicating genetic risk information for common disorders in the era of genomic medicine. Annu Rev Genomics Hum Genet 2013;14:491–513. [PubMed: 24003856] Manolio TA et al. Finding the missing heritability of complex diseases. Nature 2009;461(7265):747–753. [PubMed: 19812666] Manolio TA et al. Implementing genomic medicine in the clinic: the future is here. Genet Med 2013;15(4):258–267. [PubMed: 23306799] Mirnezami R et al. Preparing for precision medicine. N Engl J Med 2012;366(6):489–491. [PubMed: 22256780] O’Donnell CJ et al. Genomics of cardiovascular disease. N Engl J Med 2011:365(22):2098–2109. [PubMed: 22129254] Pyeritz RE. The family history the first genetic test, and still useful after all those years? Genet Med 2012;14(1):3–9. [PubMed: 22237427] ABERRACIONES CROMOSÓMICAS Cualquier desviación de la estructura y número de cromosomas, como se muestra en la figura e2-1, es, desde el punto de vista técnico, una aberración cromosómica. No todas las aberraciones causan problemas en el individuo afectado, pero algunas que lo hacen pueden conducir a problemas en la descendencia. Alrededor de 1:200 recién nacidos vivos tiene una aberración cromosómica que se detecta a causa de algún efecto en el fenotipo. Esta frecuencia se incrementa muchas veces cuanto más temprano en la vida fetal se examinan los cromosomas. Para finales del primer trimestre de gestación, la mayoría de los fetos con número anormal de cromosomas se pierde a través del aborto espontáneo. Por ejemplo, el síndrome de Turner —debido a la ausencia de un cromosoma sexual y la presencia de un solo cromosoma X— es una afección que se considera entre las usuales, pero se estima que sólo 2% de los fetos con esta forma de aneuploidía sobrevive hasta el término. Incluso más sorprendente entre los niños nacidos vivos es la ausencia completa de la mayoría de las trisomías y monosomías autosómicas a pesar de su ocurrencia frecuente en los fetos jóvenes. Tipos de anomalías cromosómicas Suceden cambios estructurales importantes tanto en forma balanceada como desbalanceada. En este último caso hay una ganancia o pérdida de material genético; en el primero, no hay cambio en la cantidad de material genético, sólo un reordenamiento del mismo. En los sitios de roturas y nuevas fijaciones de los fragmentos cromosómicos, puede producirse daño estructural o funcional permanente a un gen o sólo a unos pocos genes. No obstante, pese a la pérdida invisible de material, la aberración puede ser reconocida como desbalanceada a través de un fenotipo anormal y de los defectos cromosómicos confirmados por análisis molecular del DNA. La aneuploidía resulta de la falta de disyunción, la falla de un par de cromátides de separarse en una célula en división. La falta de disyunción en la primera o segunda división meiótica produce gametos con composición cromosómica anormal. En la aneuploidía están presentes un poco más o menos que 46 cromosomas (cuadro e2-1). Las siguientes son todas las posibilidades de aneuploidía: 1) monosomía, en la cual sólo un miembro de un par de cromosomas está presente; 2) trisomía, en la que están presentes tres cromosomas en lugar de dos, y 3) polisomía, en la cual un cromosoma está representado cuatro o más veces. Cuadro e2–1. Fenotipos clínicos derivados de la aneuploidía Afección Cariotipo Incidencia al nacer Trisomía 13 47,XX o XY,+13 1:15 000 Trisomía 18 47,XX o XY,+18 1:11 000 Trisomía 21 (síndrome de Down) 47,XX o XY,+21 1:900 Síndrome de Klinefelter 47,XXY 1:600 varones Síndrome XYY 47,XYY 1:1 000 varones Síndrome de Turner 45,X 1:2 500 mujeres Síndrome XXX 47,XXX 1:1 200 mujeres Copyright © The McGraw-Hill Companies. Derechos reservados. Nota de privacidad. Cualquier uso está sujeto a los Términos de Uso y Aviso. Si la falta de disyunción ocurre en la mitosis, se originan patrones en mosaico en los tejidos somáticos, con algunas células que tienen un cariotipo y otras células del mismo organismo con otro cariotipo. A menudo los pacientes con una constitución genética en mosaico tienen manifestaciones de cada uno de los síndromes genéticos propios de los diversos cariotipos anormales. La traslocación resulta de un intercambio de partes de dos cromosomas. La deleción es la pérdida de material cromosómico. La duplicación es la presencia de dos o más copias de la misma región de un cromosoma determinado. La redundancia se puede suscitar en el mismo cromosoma o en un cromosoma que no sea homólogo. En el último caso, también debió ocurrir una traslocación. Un isocromosoma es un cromosoma en el cual los brazos a cada lado del centrómero tienen el mismo material genético en el mismo orden, es decir que en algún momento el cromosoma se dividió de forma tal que adquirió una dosis doble de un brazo y perdió el otro. En una inversión, una región cromosómica se reorienta 180 grados con respecto a la fase ordinaria. Presenta el mismo material genético, pero en diferente orden. Ellison JW et al. Clinical utility of chromosomal microarray analysis. Pediatrics. 2012 Nov;130(5):e1085–95. [PubMed: 23071206] Shaffer LG et al. Development of new postnatal diagnostic methods for chromosome disorders. Semin Fetal Neonatal Med 2011;16(2):114–118. [PubMed: 21112262] TÉCNICAS DE LA GENÉTICA MÉDICA Los trastornos hereditarios afectan múltiples sistemas orgánicos y gente de todas las edades. Muchos de éstos son crónicos, aunque las crisis agudas ocurren con frecuencia. Las preocupaciones de pacientes y familias abarcan un amplio abanico de problemas médicos, psicológicos, sociales y económicos. Estas características subrayan la necesidad de médicos que proporcionen servicios médicos genéticos a sus pacientes. Esta sección revisa los servicios de laboratorio y consulta disponibles por parte de genetistas clínicos y las indicaciones para acceder a los mismos. HISTORIA FAMILIAR Y ANÁLISIS DEL PEDIGRÍ El primer paso al considerar qué factores genéticos importantes pueden participar en la situación clínica de un paciente es obtener una historia familiar detallada. Como mínimo, un paciente debe consultarse con detalle acerca de todos los familiares en primer grado —padres, hermanos y descendencia— (edad, sexo, estado de salud, si están vivos, e incluir las principales enfermedades; asimismo, la causa de alguna muerte) y de los familiares más lejanos con referencia a la afección en estudio particular. La etnicidad de ambos lados de la familia debe tenerse en cuenta; ningún trastorno conocido por tener prevalencia especial en un grupo étnico particular debe plantearse de manera específica. Una vez que se obtiene la historia familiar, debe analizarse; los médicos genetistas y los consejeros genéticos son entrenados en esta tarea y son de gran valor cuando el médico ocupado carece del tiempo y del equipo para atender la información. Un diagrama de pedigrí (p. ej., el de la figura e2-3) con los símbolos marcados para indicar la presencia de una afección puede resultar instructivo para sugerir un modo de herencia. Cuando la prueba genética específica del probando produce un resultado, el diagrama también muestra su utilidad en la identificación de familiares que podrían beneficiarse con la asesoría acerca de pruebas similares. Pyeritz RE. The family history: the first genetic test, and still useful after all those years? Genet Med 2012;14(1):3–9. [PubMed: 22237427] Wilson BJ et al. Family history tools in primary care: does one size fit all? Public Health Genomics 2012;15(3–4): 181–188. [PubMed: 22488461] CITOGENÉTICA Y CITOGENÓMICA La citogenética es el estudio de los cromosomas mediante el microscopio de luz. La constitución cromosómica de una sola célula o de un individuo completo se especifica con una notación estandarizada. La cuenta total de cromosomas se determina primero, seguido por el complemento del cromosoma sexual y luego por alguna anomalía. Todos los autosomas se designan por números del 1 al 22. Un signo más (+) o menos (−) indica, respectivamente, la ganancia o pérdida de material cromosómico. Por ejemplo, un varón normal es 46,XY, mientras que una niña con síndrome de Down causado por trisomía 21 es 47,XX,+21; un niño con síndrome de Down causado por traslocación del cromosoma 21 al cromosoma 14 en un espermatozoide o un huevo es 46,XY,−14,+t(14;21). Los análisis cromosómicos se hacen mediante células humanas en crecimiento en cultivos tisulares, inhibición química de la mitosis, y por último tinción, observación, fotografía, clasificación, y conteo de los cromosomas. La visualización de todos los cromosomas constituye el cariotipo (figura e2-1) y es el resultado final del aspecto técnico de la citogenética. Los especímenes para los análisis citogenéticos de rutina pueden obtenerse de la sangre periférica, en cuyo caso se examinan los linfocitos T; del líquido amniótico para cultivo de amniocitos; de células trofoblásticas para las vellosidades coriónicas; de la médula ósea, y de fibroblastos cultivados, en general obtenidos de una biopsia de piel. Deben examinarse suficientes células, de manera que la posibilidad de pasar por alto una línea celular citogenéticamente diferente (una situación de mosaicismo) sea baja desde el punto de vista estadístico. Para la mayoría de las indicaciones clínicas, se examinan y cuentan 20 mitosis bajo visualización microscópica directa, de las cuales se preparan los cariotipos y se fotografían dos. La observación de aberraciones suele incitar un escrutinio más extenso y en muchos casos análisis adicionales del cultivo original. Puede usarse una variedad de métodos para revelar el patrón de bandeo —exclusivo para cada par de cromosomas— en el análisis de aberraciones. El número de bandas que pueden visualizarse es una función de qué tan “extendidos” estén los cromosomas, lo cual en primer lugar depende del momento de la metafase (o incluso la profase para la mayoría de los bandeos extensos) en que se detuvo la mitosis, ya que cuanto más temprano ocurra es mejor. El cariotipo “estándar” revela alrededor de 400 bandas por conjunto de cromosomas, mientras que un cariotipo en profase puede revelar hasta cuatro veces ese número. En ciertas circunstancias clínicas, los cariotipos son tan invaluables como extendidos estén, por lo que con frecuencia su interpretación resulta difícil, en términos del tiempo y el esfuerzo que se requieren, de la ambigüedad acerca de lo que es anormal, lo que son las variaciones normales y de lo que es un artefacto técnico. La hibridación in situ con sondas de DNA para cromosomas o regiones específicas de cromosomas puede etiquetarse y usarse para identificar aberraciones sutiles. Con la técnica adecuada, la hibridación in situ fluorescente (FISH) produce sensibilidad y especificidad de casi 100%. Los extremos de los cromosomas se denominan telómeros. Un área de particular relevancia se refiere al uso de FISH para detectar lesiones hasta ese momento indetectables en las regiones de los cromosomas situadas justo proximales a sus puntas (subteloméricas). Cada vez más, la citogenética clásica está siendo reemplazada por la citogenómica, en la cual el DNA de un paciente se analiza en una plataforma matriz (“chip”). En el uso comercial actual, Los chips muestran una evolución por demás rápida. Los chips originales usaban sólo 400-600 sondas, que incluían todos los sitios de deleciones e inserciones de importancia clínica conocidos así como regiones de significancia incierta. Hoy en día, se usan millones de sondas que abarcan todo el genoma. La hibridación genómica comparativa (matriz CGH) requiere un espécimen de control contra el cual se compara el DNA del paciente. Las deleciones y duplicaciones se detectan con facilidad y los programas bioinformáticos pueden ajustarse para llamar “anormal” a cualquier grado de desviación con respecto al control. Las matrices basadas en polimorfismos de un solo nucleótido no requieren un control porque examinan el DNA del paciente de regiones donde hay una variación inesperada en la relación con la heterocigosidad esperada de los polimorfismos de un solo nucleótido (SNP). Estas matrices también pueden usarse para genotipificar loci específicos. En la actualidad, la única aberración citogenética clásica que puede pasarse por alto en los análisis con matrices es una traslocación balanceada que no incluya la pérdida de la secuencia de nucleótidos. Indicaciones para análisis citogenómico Las indicaciones actuales se listan en el cuadro e2-2. Se ha encontrado que una matriz amplia de síndromes clínicos se vincula con aberraciones cromosómicas, y el análisis del genoma es útil en cualquier momento en un paciente en el que se descubren las manifestaciones de uno de tales síndromes. Cuando se revela una aberración cromosómica, no sólo hace que el médico del paciente obtenga información valiosa acerca del pronóstico, sino que los padres obtienen conocimiento sobre la causa de los problemas de su hijo y la familia puede asesorarse con precisión —y en general reasegurarse— respecto a los riesgos de recurrencia. El retardo mental es un componente habitual de los síndromes de malformaciones congénitas. Cualquier persona con retardo mental inexplicado debe estudiarse mediante análisis del genoma. Hace una década, los estudios que usaban sondas fluorescentes para secuencias genéticas subteloméricas mostraron reordenamientos o deleciones sutiles en cerca de 6% de los pacientes que por otra parte tenían retardo mental inexplicado, con o sin características dismórficas. Estos cambios ocurren por mutaciones de novo o a causa de reordenamientos debidos a traslocaciones parentales balanceadas. La mayor parte de las aberraciones son demasiado pequeñas para detectarse mediante los análisis citogenéticos de rutina. De esta manera, la evolución de las tecnologías de matrices mejoró la detección en gran medida. Las anomalías de la diferenciación sexual pueden entenderse sólo después que se esclarece el sexo genético del paciente. El tratamiento hormonal y la cirugía plástica pueden determinar en alguna extensión el sexo fenotípico, pero el sexo genético es dictado por el complemento de los cromosomas sexuales. El ejemplo mejor conocido de dicotomía entre sexo genético y sexo fenotípico lo representa el síndrome de feminización testicular, en el cual la constitución cromosómica es 46,XY pero, debido a un defecto en la proteína receptora de testosterona (especificada por un gen del cromosoma X), el fenotipo externo es femenino por donde se lo mire. Cuadro e2–2. Indicaciones de análisis citogenómico Los pacientes de cualquier edad que tienen un retardo físico o mental grave, en especial si se asocia con anomalías Cualquier paciente con genitales internos o externos ambiguos o sospecha de hermafroditismo Niñas con amenorrea primaria y niños con desarrollo puberal retrasado. Hasta 25% de los pacientes con amenorrea primaria tiene una anomalía cromosómica Los varones con trastornos de aprendizaje o de la conducta que son más altas de lo esperado (basado en la estatura parental) Ciertas enfermedades malignas y premalignas (véanse cuadros e2-7 y e2-8) Los padres de un paciente con traslocación cromosómica Los padres de un paciente con una sospecha de síndrome cromosómico, si hay una historia familiar de niños afectados de manera similar Parejas con antecedentes de múltiples abortos espontáneos de causa desconocida Parejas que son infértiles después que las causas obstétricas y urológicas más comunes se excluyen Diagnóstico prenatal (véase cuadro e2-6) Copyright © The McGraw-Hill Companies. Derechos reservados. Nota de privacidad. Cualquier uso está sujeto a los Términos de Uso y Aviso. La falla o el retraso en el desarrollo de las características sexuales secundarias se producen en el síndrome de Turner (la causa más común es una forma de aneuploidía, es decir, monosomía para el cromosoma X, 45,X.), en el síndrome de Klinefelter (el cariotipo más común es 47,XXY; véase cap. 40), y en otras aberraciones cromosómicas mucho más raras. La estatura es quizá la única característica fenotípica consistente relacionada con tener un cromosoma Y extra (cariotipo 47,XXY). La mayoría de los varones con esta aberración cromosómica lleva una vida normal y, por tanto, la estatura alta en un varón no es por sí misma una indicación para un análisis cromosómico. Sin embargo, alguna evidencia sugiere que una prevalencia incrementada de dificultad para aprender puede relacionarse con esta aberración. De manera adicional, el síndrome de Klinefelter determina con frecuencia una estatura alta, aunque con un hábito eunucoide y problemas de aprendizaje y de conducta. Por consiguiente, la combinación de dificultades en el aprendizaje, en la conducta y una estatura inesperadamente alta en un varón deberían estimular la consideración de un análisis citogenético. Como se explica después, la mayoría de los tumores se relaciona con aberraciones cromosómicas, algunos de los cuales son muy específicos para ciertas enfermedades malignas. El análisis genómico del tejido tumoral puede ayudar en el diagnóstico, pronóstico y tratamiento. Cuando una persona muestra una traslocación cromosómica —si es balanceada y asintomática o desbalanceada y causa un síndrome—, el médico debe considerar la importancia de identificar la fuente de la traslocación. Si el probando es un niño y los padres están interesados en tener más hijos, ambos padres deben estudiarse desde el punto de vista genómico, y si se comprueba la posibilidad de una traslocación balanceada, también se los debe estudiar desde el punto de vista citogenético. Qué tan lejos debe ir el médico o consultante primario en el seguimiento de una traslocación a través de una familia es una cuestión pendiente con implicaciones legales y éticas así como médicas. Por cierto, el probando (si es un adulto) o los padres del probando necesitan asesoría y discutir el riesgo potencial de los familiares. El médico debe documentar tanto en el expediente médico como por correspondencia que el peso de la divulgación de datos relevantes para la familia extendida ha sido asumido por individuos nombrados de manera específica. La incapacidad para obtener descendientes a través de la falla para concebir o como resultado de abortos repetidos es un problema frustrante y desalentador para las parejas afectadas y sus médicos. El progreso considerable en la comprensión urológica y ginecológica de la infertilidad ha beneficiado a muchas parejas. Sin embargo, las aberraciones cromosómicas permanecen como un problema importante en la medicina reproductiva, y en algunas etapas deben utilizarse los análisis genómicos en evaluaciones extensas. La infertilidad es común tanto en el síndrome de Klinefelter como en el de Turner, y cualquiera de ellos puede acompañarse de signos externos leves, en particular si la aberración cromosómica es un mosaico. Cualquier aborto espontáneo temprano puede deberse a una aneuploidía fetal. La recurrencia puede deberse a una traslocación parental que predispone a un cariotipo fetal desbalanceado. Conlin LK et al. Mechanisms of mosaicism, chimerism and uniparental disomy identified by single nucleotide polymorphism array analysis. Hum Mol Genet 2010;19(7):1263–1275. [PubMed: 20053666] Ellison JW et al. Clinical utility of chromosomal microarrays analysis. Pediatrics 2012;130(5):e1085–1095. [PubMed: 23071206] Kong A et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature 2012;488(7412):471–475. [PubMed: 22914163] Miller DT et al. Consensus statement: chromosomal microarrays is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet 2010;86(5):749–764. [PubMed: 20466091] GENÉTICA BIOQUÍMICA La genética bioquímica no sólo trata de los defectos enzimáticos sino también de las proteínas de todas las funciones, como las de las estructuras citoesquelética y extracelular, regulación y señalización. Las principales funciones del laboratorio genético bioquímico son determinar la presencia o ausencia de proteínas, valorar las características cualitativas de las proteínas, ensayar metabolitos y verificar la efectividad de las proteínas in vitro. Desde la perspectiva del médico de referencia, los elementos más importantes son: 1) indicar cuáles son los diagnósticos clínicos sospechados, y 2) verificar que el espécimen apropiado se obtenga y transporte al laboratorio de manera oportuna. Indicaciones para las investigaciones bioquímicas Algunos errores congénitos son bastante usuales en la población general, por ejemplo, hemocromatosis, defectos del receptor de las lipoproteínas de baja densidad, y la fibrosis quística (cuadro e2-3). Otros, aunque raros en toda la población, son comunes en ciertos grupos étnicos, como la enfermedad de Tay-Sachs en los judíos asquenazíes, la enfermedad de células falciformes en los africoestadounidenses, y las talasemias en poblaciones situadas alrededor de la cuenca del Mediterráneo y en Asia. Varios de estos trastornos son autosómicos recesivos, y la frecuencia de heterocigotos supera muchas veces a la de la enfermedad que se expresa por completo. La detección del estado de portador puede ser efectiva si se satisfacen ciertos requerimientos (cuadro e2-4). Por ejemplo, todo Estados Unidos y el Distrito de Columbia requieren detección de recién nacidos para prevenir cetonuria y con frecuencia otras enfermedades metabólicas. Tales programas son efectivos para el costo incluso para afecciones raras como la fenilcetonuria, la cual sucede en sólo uno de cada 11 000 nacimientos. Desafortunadamente, no todos los trastornos que alcanzan los requerimientos del cuadro e2-4 se detectan en todos los estados. De manera adicional, el cumplimiento de los programas es muy variable, y las pruebas diagnósticas de seguimiento, tratamientos y asesorías son en algunos casos inadecuados. Los recién nacidos que pueden pasarse por alto con más probabilidad son los nacidos en el hogar y aquellos dados de alta antes de que hayan digerido suficiente leche o fórmula. En algunos estados, los padres pueden rechazar los estudios a sus hijos. Numerosos laboratorios han comercializados más de 35 pruebas de detección de errores del metabolismo en los hospitales. Estas detecciones suplementarias del recién nacido incluyen análisis espectroscópicos de masa en tándem de las mismas manchas sanguíneas de los programas obligatorios de los estados. En 2014 se exploró la utilidad de secuenciar el exoma completo de los recién nacidos en varios proyectos financiados con fondos federales en Estados Unidos. Cuadro e2–3. Errores congénitos (o innatos) del metabolismo más representativos Clase o defecto general Ejemplo Defecto bioquímico Herencia Aminoacidopatía Fenilcetonuria Hidroxilasa de fenilalanina AR Tejido conjuntivo Osteogénesis imperfecta tipo 2 Procolágena α-1 (I) y α-2 (I) AD Gangliosidosis Enfermedad de Tay-Sachs Hexosaaminidasa A AR Enfermedad por almacenamiento de glucógeno Tipo I Glucosa-6-fosfatasa AR Función inmunitaria Enfermedad granulomatosa crónica Citocromo b, cadena β LX Metabolismo lipídico Hipercolesterolemia familiar Receptor de lipoproteínas de baja densidad AD Mucopolisacaridosis (MPS) MPS II (síndrome de Hunter) Sulfatasa de iduronato LX Porfiria Aguda intermitente Desaminasa de porfobilinógeno AD Transporte Fibrosis quística (CF) Regulador de la conductancia transmembrana de la CF AR Ciclo de la urea Citrulinemia Sintetasa de argininosuccinato AR AR, autosómica recesiva; AD, autosómica dominante; LX, recesiva ligada al X. Copyright © The McGraw-Hill Companies. Derechos reservados. Nota de privacidad. Cualquier uso está sujeto a los Términos de Uso y Aviso. Cuadro e2–4. Requerimientos para la detección efectiva en la población de errores congénitos (innatos) del metabolismo 1. L a enfermedad debe ser grave desde el punto de vista clínico o tener consecuencias potencialmente graves 2. La historia natural de la enfermedad debe conocerse 3. E n general, el tratamiento efectivo debe estar disponible y depender del diagnóstico temprano para obtener resultados óptimos 4. La incidencia de la enfermedad debe ser lo suficientemente alta para garantizar la detección 5. L a prueba de detección debe tener especificidad (tasa baja de falsos positivos) y sensibilidad (tasa baja de falsos negativos) favorables 6. La prueba de detección debe estar disponible y usarse por toda la población en riesgo 7. Debe proveerse un adecuado sistema de seguimiento de los resultados positivos 8. El análisis económico de costo-beneficio debe favorecer la detección y el tratamiento Copyright © The McGraw-Hill Companies. Derechos reservados. Nota de privacidad. Cualquier uso está sujeto a los Términos de Uso y Aviso. El uso del laboratorio de genética bioquímica para otros propósitos de detección debe justificarse por la necesidad de datos sobre los cuales basar un diagnóstico de trastornos o clases específicas de afecciones relacionadas. Lo único que limita las posibilidades es la extensión del conocimiento, el entusiasmo de los médicos o consultante primarios, la disposición de la familia del paciente para facilitar el diagnóstico y para que se tomen los especímenes necesarios, y la disponibilidad de un laboratorio para examinar los especímenes. Como muchos defectos innatos son tan sutiles que escapan a la detección, hay varias situaciones clínicas en las cuales un error innato debe ser parte del diagnóstico diferencial. La urgencia con la cual la investigación se lleva a cabo varía en dependencia de la gravedad del trastorno y de la disponibilidad del tratamiento. El cuadro e2-5 lista varias presentaciones clínicas. Cuadro e2–5. Forma de presentación de los errores congénitos (innatos) del metabolismo Presentación y curso Ejemplos Enfermedad metabólica aguda del neonato Galactosemia, trastornos del ciclo de la urea Trastornos crónicos con escasa progresión después de la infancia Fenilcetonuria, hipotiroidismo Trastornos crónicos con progresión insidiosa e incesante Enfermedad de Tay-Sachs Trastornos que causan anomalías de estructura Displasias esqueléticas, síndrome de Marfan Trastornos del transporte Cistinuria, deficiencia de lactasa Trastornos que determinan susceptibilidades Deficiencia del receptor de lipoproteína de baja densidad, agammaglobulinemia Trastornos episódicos La mayoría de las porfirias, deficiencia de glucosa-6-fosfato Trastornos que causan anemia Deficiencias de cinasa de piruvato, esferocitosis hereditaria Trastornos que interfieren con la hemostasia Hemofilias A y B, enfermedad de von Willebrand Trastornos congénitos sin posibilidad de reversión Feminización testicular Trastornos con manifestaciones diversas Seudohipoparatiroidismo, amiloidosis hereditaria Errores innatos sin efectos clínicos Pentosuria, histidinemia Copyright © The McGraw-Hill Companies. Derechos reservados. Nota de privacidad. Cualquier uso está sujeto a los Términos de Uso y Aviso. La posibilidad de una enfermedad metabólica aguda del neonato es la indicación más importante debido a que el diagnóstico y tratamiento expeditos pueden, con frecuencia, hacer la diferencia entre la vida y la muerte. Las características clínicas son inespecíficas debido a que el recién nacido tiene un repertorio limitado de respuestas a las agresiones metabólicas graves. El médico debe ser incluyente y sistemático en la evaluación de tales neonatos enfermos. Erbe RW et al. Neonatal screening. En: Rimoin DL et al. (editors): Emery and Rimoin’s principles and practice of medical genetics. 6th ed. Churchill Livingstone, 2014 Valle D et al. (editors): Online metabolic and molecular bases of inherited disease. McGraw-Hill, 2014. http:// www.ommbid.com/ Análisis del DNA La inspección directa de los ácidos nucleicos —llamada con frecuencia “genética molecular” o “diagnóstico del DNA”— ha alcanzado un lugar destacado que cada vez crece más en varias áreas clínicas como la oncología, las enfermedades infecciosas, el campo forense y el estudio general de la fisiopatología. Un impacto más importante ha tenido en el diagnóstico de los trastornos mendelianos. Las pruebas moleculares están disponibles para más de 3 000 afecciones hereditarias diferentes. Cuando se demuestra que un gen determinado es defectuoso para una afección específica, la naturaleza de la mutación del paciente puede determinarse por la secuenciación de nucleótidos de los exones de codificación y los sitios de corte y empalme. Entonces puede recurrirse a una variedad de técnicas para determinar si la misma mutación está presente en otros pacientes con idéntico trastorno. La heterogeneidad genética es tan extensa que la mayoría de las afecciones mendelianas se vincula con numerosas mutaciones en un solo locus —o con frecuencia múltiples loci— que producen el mismo fenotipo. Las mutaciones en numerosos cientos de genes diferentes causan trastornos vitreorretinianos como la retinitis pigmentosa, y los cambios en varias docenas de genes causan miocardiopatía hipertrófica familiar. Este hecho complica el diagnóstico por DNA de los pacientes y la detección de portadores de defectos en genes específicos. Son pocas las afecciones que se relacionan con relativamente pocas mutaciones o con una sola mutación muy prevalente. Por ejemplo, toda enfermedad de células falciformes siempre es causada por el mismo cambio del glutamato por valina en la posición 6 de la globina β, y esta sustitución a su vez se debe a un cambio de un nucleótido en el sexto codón del gen de la globina β. Pero, tal uniformidad es la excepción. En la fibrosis quística, alrededor de 70% de los heterocigotos de ancestros europeos del noreste presenta una deleción de tres nucleótidos idéntica que causa la pérdida de un residuo fenilalanina de una proteína que transporta cloruro; sin embargo, el restante 30% de las mutaciones de esa proteína es diverso (varios cientos han sido descubiertos), de manera que ninguna prueba de detección sencilla detectará a todos los portadores de fibrosis quística. Las revisiones técnicas actuales del estado en que se halla el análisis del DNA aparecen con frecuencia regular en la bibliografía médica. Estudios como la reacción en cadena de la polimerasa (PCR) revolucionaron muchos aspectos de la biología molecular, y el diagnóstico de DNA incluye esta técnica. Si se conocen las secuencias finales de 10-20 nucleótidos de una región de DNA de interés, como una porción de un gen, entonces pueden sintetizarse “cebadores” complementarios de estas secuencias. Cuando incluso una cantidad diminuta de DNA de un paciente (p. ej., de unos pocos leucocitos, de células de la mucosa bucal, o de bulbos pilosos) se combina con los cebadores en una reacción mixta que replica el DNA —y después se realizan varias docenas de ciclos—, la región de DNA comprendida entre los cebadores se amplifica de manera exponencial. Por ejemplo, la presencia de una infección inicial de VIH puede detectarse después de amplificar una porción del genoma viral. La velocidad de secuenciación del DNA se ha acelerado de forma notable y su costo se ha desplomado debido a la llegada de la “secuenciación de la siguiente generación”. El “Santo grial” del análisis de DNA se definió hace más de una década como el genoma humano de $1000” y ese punto de referencia se alcanzó en 2014. Por consiguiente, ya es factible para un investigador secuenciar sólo las secuencias de codificación (secuenciación del exoma completo) o los 6.4 mil millones de pares de bases de nucleótidos de cualquier individuo. Ahora es menos caro secuenciar el exoma completo de una persona que secuenciar de manera selectiva todos los genes que se sabe intervienen en una afección, por ejemplo, la miocardiopatía hipertrófica. El almacenamiento e interpretación de la masa de datos que surge de la secuenciación de un genoma completo son tareas agotadoras y caras, pero tal vez sean superables. Han sido analizadas las secuencias completas de cientos de individuos y unas pocas de éstas fueron publicadas, y están disponibles para su uso en Internet. La mayor parte de estos individuos acordó asumir las implicaciones clínicas de que sus genomas sean analizados públicamente. El esfuerzo internacional para secuenciar los genomas completos de cuando menos mil individuos de diversos grupos étnicos fue superado en 2011. Boycott KM et al. Rare-disease genetics in the era of next-generation sequencing: discovery to translation. Nat Rev Genet. 2013 Oct;14(10):681–91. [PubMed: 23999272] Davies K. The era of genomic medicine. Clin Med 2013;13(6):594–601. [PubMed: 24298109] Dewey FE et al. DNA sequencing: clinical applications of new DNA sequencing technologies. Circulation 2012;125(7):931–944. [PubMed: 22354974] Jacob HJ. Next-generation sequencing for clinical diagnostics. N Engl J Med 2013;369(16):557–558. [PubMed: 24088040] Indicaciones para el diagnóstico del DNA Los requerimientos básicos para el uso de ácidos nucleicos en el diagnóstico de afecciones hereditarias específicas es que haya una sonda disponible para el gen en cuestión. La sonda puede ser una parte del final del gen real, una secuencia cercana al gen, o sólo unos pocos nucleótidos de la mutación real. Cuanto más cerca de la mutación real esté la sonda, más exacta y más útil será la información que se derive. El diagnóstico del DNA incluye uno de dos métodos generales: 1) detección directa de la mutación o, 2) análisis del enlace, donde la presencia de una mutación se infiere a partir de la naturaleza de la secuencia de la sonda de DNA alejada de la mutación. En este último método, a medida que la sonda se mueve más lejos de la mutación, se incrementan las probabilidades de que la recombinación separe las dos secuencias y confunda la interpretación de los datos. El diagnóstico de DNA encuentra una aplicación frecuente en la detección presintomática de individuos con trastornos dependientes de la edad como la enfermedad de Huntington y la enfermedad renal poliquística del adulto, la detección de portadores de afecciones autosómicas recesivas como la fibrosis quística y las talasemias, la detección de mujeres heterocigotas de afecciones ligadas al X como la distrofia muscular de Duchenne y las hemofilias A y B, y en el diagnóstico prenatal (véase después). Para algunas afecciones en las cuales se producen complicaciones serias en la adolescencia o en la etapa adulta temprana —como en el síndrome de von Hippel-Lindau, la telangiectasia hemorrágica hereditaria y la poliposis familiar cólica—, las pruebas genéticas en una edad temprana pueden identificar a aquellos familiares que necesitan vigilancia clínica frecuente y tratamiento profiláctico; casi tan importante, a los familiares que presenten pruebas negativas para la mutación se les puede evitar la inconveniencia, costo y riesgo de las pruebas clínicas. En todas las instancias de las pruebas de DNA, los proveedores de cuidado primario y los especialistas deben tener en cuenta por igual qué aspectos sustantivos de naturaleza ética, psicológica, legal y social siguen sin resolverse. Por ejemplo, algunas afecciones para las cuales la susceptibilidad hereditaria puede definirse con facilidad (como la enfermedad de Alzheimer, la enfermedad de Huntington y muchos cánceres) no tienen por el momento un tratamiento efectivo. Para estas dos afecciones, los proveedores de seguros de salud y de seguridad pueden tener un interés especial en aprender quién entre sus clientes actuales o futuros está en un riesgo más alto. Muchos estados han promulgado leyes para proteger a la gente identificada de tener un riesgo genético alto de enfermedad. En el Genetic Information Nondiscrimination Act federal (GINA), que cobró efecto en 2009 y 2010, se prohíbe la discriminación injusta en seguros de salud y empleo. Las protecciones se limitan para aquellos individuos en quienes se detecta una susceptibilidad genética a una enfermedad que todavía no es evidente en el terreno clínico. Varias firmas comerciales ofrecen genotipificación dirigida o extensa a cualquiera que quiera enviar un espécimen de saliva y pagar el servicio. Algunas de las razones sugeridas para hacer esto incluyen la identificación de antecedentes ancestrales, certificación de relaciones y, lo más común, la detección de susceptibilidad genética a alguna enfermedad. Estos últimos se basan casi siempre en el GWAS que tiene asociado un SNP específico, con una probabilidad mayor (o menor) de desarrollar una enfermedad común determinada. En casi todos los análisis basados en GWAS, la asociación con la enfermedad tiene una significancia estadística muy alta, pero un valor predictivo muy pequeño. En otras palabras, de manera típica, el riesgo relativo de desarrollar una enfermedad basada en tener uno de estos marcadores se encuentra entre límites de 1.1-1.4. Más todavía, de manera virtual no se ha realizado ninguna investigación que examine la utilidad clínica de poseer uno de tales marcadores de riesgo. Por ejemplo, en alguien con un SNP vinculado a 9p21, que no tiene una función biológica conocida pero que se lo relaciona con un riesgo ligeramente mayor de desarrollar una afección vinculada con la ateroesclerosis, ¿es más probable que altere su estilo de vida, cambie su dieta o deje de fumar? También existen esfuerzos de comercialización dirigidos al consumidor para pruebas genéticas específicas que deben ser ordenadas por un médico, como aquellas que sugieren que cualquier mujer con una historia familiar de cáncer de mama debería considerar someterse a pruebas para mutaciones en los genes BRCA1 y BRCA2. Bernhardt BA et al. Cost savings through molecular diagnostic for hereditary hemorrhagic telangiectasia. Genet Med 2012;14(6):604–610. [PubMed: 22281938] Bernhardt BA et al. Why is genetic screening for autosomal dominant disorders underused in families? The case of hereditary hemorrhagic telangiectasia. Genet Med 2011;13(9):812–820. [PubMed: 21637104] Bloss CS et al. Effect of direct-to-consumer genomewide profiling to assess disease risk. N Engl J Med 2011;364(6):524–534. [PubMed: 21226570] Hudson KL. Genomics, health care, and society. N Engl J Med 2011;365(11):1033–1041. [PubMed: 21916641] Logística del diagnóstico de DNA Los linfocitos son una fuente lista de DNA; 10 ml de sangre completa producen hasta 0.5 mg de DNA, suficiente para docenas de análisis, cada uno de los cuales requiere sólo 5 µg. Si el análisis se enfoca de forma muy estrecha en una mutación específica (como en un estudio familiar, el cual se dirige a un solo cambio de nucleótido específico), en el cual puede utilizarse con frecuencia el análisis por PCR, para lo cual la cantidad necesaria de DNA es en realidad infinitesimal, unos pocos bulbos pilosos o espermatozoides son adecuados. La fuente más simple es la saliva. Una vez aislada, la muestra de DNA debe dividirse en alícuotas y congelarse. De manera alternativa, por medio de virus, los linfocitos pueden transformarse en linfoblastos; estas células son inmortales, pueden congelarse, y —si se necesita DNA— pueden descongelarse, propagarse, y aislarse sus DNA. Estos especímenes almacenados dan acceso a largos genomas personales después que el individuo muere. Ésta es una ventaja clínica tan importante que muchos centros de genética clínica y laboratorios comerciales constituyen “bancos” de DNA de pacientes y familiares informativos incluso si las muestras no pueden usarse de inmediato. Más adelante, los especímenes pueden ser de gran valor para los familiares u otros pacientes que se evalúen. En algunas circunstancias, el DNA se vuelve más confiable que los registros médicos. La sangre para aislar DNA debe colocarse con anticoagulante de ácido etilendiaminotetraacético (EDTA) (tubos con extremo superior lavanda); la sangre para cultivar linfoblastos debe colocarse con heparina (tubos con el extremo superior verde). Ninguno debe congelarse. Los especímenes para aislamiento de DNA pueden almacenarse o mantenerse a la temperatura de la habitación durante un periodo de unos pocos días. Los cultivos de linfoblastos deben establecerse dentro de 48 horas, de manera que resulta esencial su envío rápido. Para uno o algunos análisis específicos de DNA, hay laboratorios que aceptan un hisopo que se colocó entre la mejilla y la encía durante un minuto (hisopo bucal); como hay suficientes células que se adhieren a las fibras, puede aislarse DNA del sujeto. De manera alternativa, pueden usarse 20-30 ml de saliva. El DNA fetal puede aislarse de las células amnióticas, de células trofoblásticas tomadas por muestreo de vellosidades coriónicas, o de células en crecimiento en cultivo. Las muestras necesitan procesarse a la brevedad posible, pero pueden enviarse por correo urgente y no deben congelarse. Fragmentos de DNA fetal también circulan en la sangre materna. El análisis de estos fragmentos permite realizar diagnósticos de trisomía fetal con una sensibilidad y especificidad mucho más altas y de una manera mucho menos invasiva que a través de la amniocentesis. 1000. Genomes Project Consortium. Durbin RM et al. A map of human genome variation from population scale sequencing. Nature 2010;467(7319):1061–1073. [PubMed: 20981092] Palomaki GE et al. DNA sequencing of maternal plasma reliably identifies trisomy 18 and trisomy 13 as well as Down syndrome: an international collaborative study. Genet Med 2012;14(3):296–305. [PubMed: 22281937] DIAGNÓSTICO PRENATAL Es posible diagnosticar in utero, antes de la mitad del segundo trimestre, varios cientos de trastornos mendelianos, todas las aberraciones cromosómicas, y un buen número de malformaciones congénitas que no son mendelianas. El primer paso del diagnóstico prenatal se toma cuando la pareja que espera, el responsable de los cuidados primarios, o el obstetra contemplan la necesidad de hacerlo. Encuestas recientes sugieren que incluso para la mayoría de las indicaciones usuales de este servicio —edad materna avanzada— < 50% de todas las mujeres de 35 años y mayores reciben la oferta de someterse a pruebas prenatales en Estados Unidos. Técnicas usadas en el diagnóstico prenatal El diagnóstico prenatal depende de la capacidad de realizar pruebas fetales directas (muestreo sanguíneo fetal, fetoscopia), indirectas (análisis de líquido amniótico, amniocitos o células trofoblásticas, ultrasonido), o remotas (análisis de suero materno). Algunas de estas técnicas satisfacen los requerimientos de detección (cuadro e2-4) y deben ofrecerse a todas las mujeres embarazadas; otras implican riesgo considerable y han de reservarse para circunstancias específicas. Diversos centros han desarrollado el diagnóstico de preimplantación del embrión; una sola célula se desprende del blastocisto de seis a ocho células, el cual se cultiva después de una fertilización in vitro, en general sin exponer el futuro desarrollo. El genoma de la célula puede estudiarse por FISH, ensayos citogenómicos, o por análisis del DNA. Otro método con validación clínica es el aislamiento del DNA fetal que circula en cantidades diminutas en la circulación materna. El rastreo ultrasonográfico del feto es un procedimiento seguro y no invasivo que permite diagnosticar malformaciones esqueléticas burdas así como malformaciones extraóseas conocidas por formar parte de enfermedades específicas como, por ejemplo, la medición de la traslucidez nucal del síndrome de Down (véase después). Algunos obstetras practican ultrasonido fetal de rutina cuando menos entre las 12 y 20 semanas de gestación. Otros procedimientos diagnósticos prenatales —fetoscopia, fetografía y amniografía— son más invasivos y representan un riesgo concreto para la madre y el feto. Sólo se indican si el riesgo de la anomalía sospechada es alto y no puede obtenerse información por otros medios. El suero materno puede someterse a ensayos para múltiples marcadores que predicen el riesgo de que el feto tenga un defecto en el tubo neural (fetoproteínas α elevadas) o una de las trisomías autosómicas más usuales. Esta detección se ofrece en el primer trimestre, de manera que los resultados puedan emplearse, junto con el ultrasonido y la edad materna, para asesorar a la pareja sobre la utilidad de efectuar pruebas adicionales, como el muestreo de vellosidades coriónicas o la amniocentesis. Todas las técnicas citogenómicas, bioquímicas y analíticas del DNA citadas en los párrafos precedentes pueden aplicarse a especímenes fetales. Junto a la detección de las fetoproteínas alfa en el suero materno para detectar defectos en el tubo neural, el análisis de los cromosomas fetales es la prueba que se practica con más frecuencia. El análisis cromosómico puede realizarse en células amnióticas y en células trofoblásticas en crecimiento en cultivo y de forma directa en cualquier célula trofoblástica que se espere que entrará en mitosis. Las células de líquido amniótico proceden en primer lugar del sistema urinario fetal. La amniocentesis puede practicarse durante las semanas gestacionales 16-18 para permitir un análisis de la muestra sin prisa, transmisión de resultados y decisiones reproductivas. El momento para obtener la muestra para realizar una lectura final de potenciales desequilibrios del genoma se acortó ahora a unos pocos días. El muestreo de vellosidades coriónicas (CVS) para células trofoblásticas (derivadas embriológicamente tanto del mismo huevo fertilizado como del feto) se practica de manera habitual durante las semanas gestacionales 10-13. Si es posible analizar el tejido en forma directa, los resultados citogenómicos pueden obtenerse en unos pocos días. La ventaja del CVS es que los resultados están disponibles temprano en el embarazo, de manera que si se decide eliminarlo es factible proceder al comienzo del embarazo, cuando las complicaciones obstétricas de la terminación son menores. El riesgo del CVS es algo más alto que el de la amniocentesis, aunque ambos son relativamente seguros. Entre 0.5 y 1% de los embarazos se pierde como una complicación del CVS, mientras que < 1 en 300 amniocentesis resulta en una pérdida fetal. Algunos centros ofrecen “amniocentesis tempranas”, que se practican durante las semanas gestacionales 12-14; la magnitud de los riesgos es similar a la del CVS. Estas cifras son más bajas que —pero se suman a— el 2-3% de la tasa de abortos espontáneos después que el primer trimestre concluye. Cuando el DNA fetal se analiza mediante ensayos, el hallazgo de una deleción o duplicación que no guarda una relación clara con un fenotipo clínico (p. ej., trisomía 21) requerirá el análisis del DNA parental, y el tiempo para realizarlo agregarse al tiempo requerido para obtener una interpretación y a la complejidad de la asesoría genética. Fragmentos de DNA fetal circulan en la sangre materna durante gran parte del embarazo. Esto posibilita realizar pruebas “no invasivas” del DNA fetal. Estudios recientes muestran que el uso de DNA fetal libre para efectuar pruebas para las trisomías 21 y 18 tiene tasas más bajas de falsos positivos y mejora los valores predictivos positivos comparado con el uso de analitos séricos maternos. El genoma fetal completo ha sido secuenciado desde estos fragmentos circulantes, lo que abre la posibilidad de detectar trastornos sobre los que se sabe que el feto está en riesgo, para afecciones que pueden pasar desapercibidas hasta la adultez, y para el estado de portador de cualquier trastorno metabólico. Indicaciones para el diagnóstico prenatal Las indicaciones para el diagnóstico prenatal se listan en el cuadro e2-6. Algunos merecen un comentario. Cuadro e2–6. Indicaciones para el diagnóstico prenatal Indicaciones Métodos Edad materna avanzada, un padre portador de una traslocación, hijo previo con aberración cromosómica, retraso del crecimiento intrauterino Citogenómica (amniocentesis, muestreo de vellosidades coriónicas, plasma materno para el DNA fetal) Trastorno bioquímico (se sabe que uno o ambos padres son portadores) Ensayo de proteínas, diagnóstico de DNA Anomalías congénitas (observadas en la detección por ultrasonido) Ultrasonido de resolución alta, citogenómica fetal Detección de defectos del tubo neural y trisomía Detección materna de múltiples marcadores, ultrasonido Copyright © The McGraw-Hill Companies. Derechos reservados. Nota de privacidad. Cualquier uso está sujeto a los Términos de Uso y Aviso. La mayoría de los estudios realizados para edad materna avanzada no detecta ninguna aberración cromosómica. Sin embargo, siempre es apropiado destacar que el riesgo promedio de producir un hijo con un defecto evidente al nacer, como una malformación física o algunos errores congénitos del metabolismo, es cercano a 3%, y que el riesgo se incrementa con la edad de cualquiera de los padres. El simple examen de los cromosomas reduce este riesgo en forma mínima. Por otro lado, salvo que alguna de las otras indicaciones esté presente, es imposible someter a un embarazo a la “detección” de la mayoría de los defectos del nacimiento (los defectos del tubo neural son una excepción). Los individuos que contemplan tener un embarazo —en especial los judíos asquenazíes o de otras etnias blancas— deben recibir la propuesta de detección para la mayoría de las mutaciones más usuales del gen CFTR que causan fibrosis quística. Si se detecta que ambos miembros de la pareja son portadores, el diagnóstico prenatal de un feto debería ser una opción para ellos. Los factores que vuelven a algunas parejas, sin anomalías cromosómicas evidentes, susceptibles a episodios reiterados de aneuploidía son inciertos, y se justifica recurrir a las pruebas prenatales de rutina cuando existe el antecedente de defectos en un concepto. El análisis citogenómico del feto suministrará información relacionada con los cromosomas sexuales. Algunas parejas no desean conocer de forma anticipada el sexo de sus hijos, y la persona que transmite los resultados a la pareja siempre debe tener presente este detalle. En el otro extremo, algunas parejas sólo desean conocer el sexo del feto y un plan para terminar el embarazo si se detecta el sexo no deseado. Pocos centros de Estados Unidos consideran que la selección del sexo constituya una indicación apropiada para el diagnóstico prenatal. En tanto que la determinación del cariotipo fue adecuada para detectar trisomías así como reordenamientos y deleciones/duplicaciones de extensas regiones de los cromosomas, el análisis de microordenamientos del genoma fetal abrió la oportunidad de detectar no sólo estas grandes aberraciones, sino también deleciones y duplicaciones mucho más sutiles. Esta sensibilidad incrementada se equilibra de alguna manera con la incapacidad actual para comprender el significado real de estas aberraciones. El nivel de fetoproteína α en el suero materno cambia con la edad gestacional, con el estado médico de la madre, y con las anomalías del feto. Si los primeros dos factores pueden controlarse, es posible emplear este ensayo para proporcionar información sobre el feto. Los niveles se expresan como múltiplos del valor mediano de una edad gestacional particular. Niveles más altos que los normales se relacionan con defectos del tubo neural abierto (las afecciones por las cuales se desarrolló la prueba), muerte fetal reciente o inminente, gastrosquisis, y enfermedad renal fetal. Los niveles extremadamente altos son muy específicos de anomalías fetales —tres veces el valor mediano aumenta 20 veces el riesgo de meningomielocele o anencefalia. Los niveles bajos de fetoproteína α en el suero materno se vinculan con trisomía fetal, en especial síndrome de Down; la explicación de este vínculo se desconoce. La adición de otros analitos en el suero materno —gonadotropina coriónica humana (hCG), estriol libre (uE3), e inhibina dimérica A— al ensayo de fetoproteína alfa (para producir la “detección de múltiples marcadores”) aumenta varias veces la capacidad para detectar un feto con trisomías 21 y 18. La medición en el primer trimestre de proteína plasmática A en el suero del embarazo y la traslucidez del cuello fetal por ultrasonido —seguido de la detección triple de rutina del segundo trimestre— mejora la tasa de detección del síndrome de Down en alrededor de 85%, al tiempo que reduce los resultados falsos positivos a cerca de 1%. Un resultado positivo en alguno de estos protocolos de detección de trisomía debe ir seguido del ofrecimiento de la amniocentesis de la mujer para confirmar el diagnóstico. La capacidad para detectar y secuenciar el DNA fetal circulante en el plasma materno ha abierto nuevas perspectivas al diagnóstico prenatal. Las trisomías pueden detectarse con alta sensibilidad y especificidad, incluso en casos de mosaico. Además, son posibles de secuenciar grandes porciones del genoma fetal, con inclusión de la mayoría de las regiones codificantes. Esto permitirá cada vez más la detección de muchos trastornos en el feto, incluido el estado de portador y las predisposiciones a trastornos que podrían no ser evidentes hasta la etapa adulta. Bodurtha J et al. Genomics and perinatal care. N Engl J Med 2012;366(1):64–73. [PubMed: 22216843] Driscoll DA et al. Professional Practice Guidelines Committee. Screening for fetal aneuploidy and neural tube defects. Genet Med 2009;11(11):818–821. [PubMed: 19915395] Greene MF et al. Screening for trisomies in circulating DNA. N Engl J Med 2014;370(9):874–875. [PubMed: 24571760] Palomaki GE et al. DNA sequencing of maternal plasma to detect Down syndrome: an international clinical validation study. Genet Med 2011;13(11):913–920. [PubMed: 22005709] Wapner RJ et al. Chromosomal microarray versus karyotyping for prenatal diagnosis. N Engl J Med 2012;367(23):2175–2251. [PubMed: 23215555] NEOPLASIAS: CITOGENÓMICA Y ANÁLISIS DE DNA Estudios de cromosomas y ácidos nucleicos apoyan la hipótesis que emitió Boveri en 1914 respecto de que un cambio en el material genético a nivel celular es lo que causa el cáncer. Tres clases de genes pueden afectarse en la transformación neoplásica. Los oncogenes surgen de genes normales preexistentes (protooncogenes) que han sido alterados por factores virales y extravirales. Como resultado, las células sintetizan ya sea proteínas normales en cantidades inapropiadas o proteínas aberrantes en estructura y función. Muchas de tales proteínas son factores de crecimiento celular o receptores de factores de crecimiento. La consecuencia neta de la activación oncogénica es la división celular desregulada. Las mutaciones que activan oncogenes casi siempre se originan en células somáticas y no suelen heredarse. Aunque es más probable que algunos oncogenes se activen en ciertos tumores, en general las mismas mutaciones pueden encontrarse en neoplasias que se originan en diferentes células y tejidos. Los genes supresores de tumores pueden verse como la antítesis de los oncogenes. Su función normal es suprimir la transformación; se requiere la mutación de ambos alelos para obstruir esta importante función. El primer alelo mutante de cualquier gen supresor tumoral puede originarse de manera espontánea o puede heredarse; la mutación del otro alelo (el “segundo golpe”) casi siempre se produce de forma espontánea, pero por cualquiera de una serie de mecanismos moleculares. Estos genes muestran mucha mayor especificidad tumoral que los oncogenes; sin embargo, aunque algunas mutaciones específicas son necesarias para que ciertos tumores se originen, la pérdida de la función supresora tumoral sola es suficiente. Con toda claridad, una persona que hereda una copia de un gen supresor tumoral mutante se halla en un riesgo mayor de que en alguna célula susceptible, en algunos momentos de la vida, la función de dicho gen se pierda. Esta susceptibilidad se hereda como un rasgo autosómico dominante. Por ejemplo, la mutación en un alelo del locus p53 resulta en el síndrome de Li-Fraumeni (151623), en el cual la susceptibilidad a los sarcomas y otros tumores antes de la edad de 45 años se presenta en varones y mujeres de generaciones sucesivas. Las mutaciones heredadas en este locus también incrementan el riesgo de que se desarrolle un segundo tumor después de una radioterapia o quimioterapia para el primer tumor, lo que sugiere que el tratamiento inicial puede inducir un “segundo golpe” en el locus p53 de otro tejido. Sin embargo, heredar una mutación p53 no asegura que se desarrollará un cáncer a una edad temprana. Se necesita aprender mucho más acerca de la patogenia de las neoplasias antes de que la asesoría genética a las familias con una predisposición molecular al cáncer se esclarezca. El BRCA1, un gen que predispone a las mujeres a los cánceres de mama y ovario, es otro ejemplo de un gen supresor tumoral. Las mujeres que heredan un alelo mutante de BRCA1 tienen, en promedio, un 60-80% de riesgo de por vida de desarrollar cáncer de mama, y la edad promedio de la detección tumoral es en la quinta década. Su riesgo de desarrollar cáncer de ovario es de 34-45%. Para mujeres y varones con ciertas mutaciones en BRCA1, los riesgos de cáncer de colon y páncreas se incrementan varias veces por encima del de la población general. En casos seleccionados, el DNA de un paciente puede analizarse por la presencia de un gen mutado y de este modo puede valorarse el riesgo de un individuo para desarrollar un tumor. Son ejemplos el retinoblastoma (189200), ciertas formas de tumor de Wilms (194070), cáncer de mama (114489), y cáncer de colon familiar (114500). Para ilustrar en qué medida la metodología dejó de ser invasiva y adquirió mayor sensibilidad, es posible analizar heces para rastrear la presencia de mutaciones en genes supresores de tumor que pueden indicar la presencia de un adenocarcinoma del colon que pasó desapercibido durante el estudio clínico. El análisis —que todavía no es de uso general— depende de la capacidad de la PCR para amplificar cantidades diminutas del DNA mutante presente en las células epiteliales desprendidas del tumor. Una tercera clase de genes que predisponen a las enfermedades malignas es la de los llamados genes mutadores. De manera habitual, los genes mutadores reparan el daño al DNA que se produce a causa de agresiones ambientales como la exposición a carcinógenos y la radiación ultravioleta. Cuando un gen mutador se muta, el DNA dañado se acumula y termina por afectar los oncogenes y los genes supresores de tumor, lo que aumenta la probabilidad de que surja un cáncer. El cáncer de colon no poliposo hereditario (HNPCC) es un síndrome familiar debido a mutaciones en uno u otro de los cinco genes mutadores identificados hasta ahora (MSH2 y MLH1 son la causa más frecuente del HNPCC). Este trabajo excitante sobre la naturaleza molecular de la oncogénesis estuvo precedido por años de estudio de la citogenética de los tumores. El primer descubrimiento importante fue el del “cromosoma Philadelphia”, un cromosoma del grupo G aparentemente acortado en muchos linfocitos de pacientes con leucemia mielógena crónica. Al final, se demostró que esto era el resultado de una traslocación entre el brazo largo del cromosoma 22 y el brazo largo del cromosoma 9. Esto lleva al reconocimiento de que el oncogén Abelson (cromosoma 9) muestra una expresión altísima debido a la yuxtaposición de la región BCR del cromosoma 9. De manera adicional, hace poco se aisló el gen supresor tumoral del retinoblastoma debido a que un pequeño número de pacientes con este tumor tiene una deleción constitucional del cromosoma 13 donde se ubica este gen. Se ha encontrado que otras aberraciones cromosómicas son muy características —o incluso específicas— de ciertos tumores (cuadro e2-7). Por tanto, la detección de una de estas aberraciones citogenéticas puede resultar de utilidad en el diagnóstico. Cuadro e2–7. Aberraciones cromosómicas relacionadas con tumores sólidos representativos Tumor Aberración cromosómica Meningioma del(22)(q11)1 Neuroblastoma del(1)(p36), del(11)(q23) Carcinoma de célula renal del(3)(p14.2-p25) o traslocación de esta región Retinoblastoma, osteosarcoma del(13)(q14.1) o traslocación de esta región Carcinoma pulmonar de célula pequeña del(3)(p14-p23) Tumor de Wilms del(11)(p15) Significado de la nomenclatura, “una deleción en la banda q11 del cromosoma 22”. 1 Copyright © The McGraw-Hill Companies. Derechos reservados. Nota de privacidad. Cualquier uso está sujeto a los Términos de Uso y Aviso. Las enfermedades malignas hematológicas son susceptibles de estudio debido a la relativa facilidad de acceder a las células tumorales para análisis citogenómico. Estas enfermedades malignas guardan relación con más de 100 reordenamientos genómicos específicos, en primer lugar traslocaciones. La mayor parte de tales reordenamientos se restringe a tipos de cáncer específicos (cuadro e2-8), y los restantes se producen en muchos cánceres. Cuadro e2–8. Aberraciones cromosómicas de enfermedades malignas hematológicas representativas Tumor Aberración cromosómica Leucemia Aguda mieloblástica t(8;21)(q22;q11)1 Aguda promielocítica t(15;17)(q22;q11-q12) Aguda monocítica t(10;11)(p15-p11;q23) Crónica mielógena t(9;22)(q34;q11) Linfomas de Burkitt t(8;14)(q24.1;q32.3) de célula B t(1;14)(q42;q43) de célula T inv, del, y t de 1p13-p12 Premalignidad Policitemia verdadera Del(20)(q11) Significado de la nomenclatura, “una traslocación con la unión en la banda q22 del cromosoma 8 y q11 del cromosoma 21”. 1 Copyright © The McGraw-Hill Companies. Derechos reservados. Nota de privacidad. Cualquier uso está sujeto a los Términos de Uso y Aviso. En las leucemias, la aberración cromosómica es la base de una de las subclasificaciones de la enfermedad. Cuando la información citogenómica se combina con la clasificación histológica, es posible definir subgrupos de pacientes en quienes la respuesta al tratamiento, el curso clínico y el pronóstico son predecibles. Si en el momento del diagnóstico no hay cambios cromosómicos en las células de la médula ósea, el tiempo de sobrevida es más largo que si alguna o todas las células de la médula ósea tienen características citogenómicas anormales. A medida que se producen cambios cromosómicos secundarios, la leucemia se vuelve más agresiva, a menudo porque adquiere resistencia a los fármacos y una probabilidad reducida de remisión completa o prolongada. El cambio cromosómico menos ominoso es la alteración numérica sin anomalías morfológicas. Hay menos información citogenómica disponible de los linfomas y los trastornos hematológicos premalignos que de la leucemia. En la enfermedad de Hodgkin, la baja producción de células en división y el número escaso de clones aneuploides claros han limitado los estudios, de manera que están disponibles análisis citogenómicos completos para muchos menos pacientes con enfermedad de Hodgkin que para ningún otro tipo de linfoma. En la enfermedad de Hodgkin, el número cromosómico modal tiende a ser triploide o tetraploide. Alrededor de un tercio de las muestras tiene un cromosoma 14q+. En linfomas no de Hodgkin, las anomalías citogenómicas se producen en 95% de los casos. En estos momentos, los hallazgos citogenómicos se están correlacionando con las características inmunitarias e histológicas y con el pronóstico. En el linfoma de Burkitt, un tumor sólido que se origina en las células B, 90% de los pacientes presenta una traslocación entre el brazo largo del cromosoma 8 y el brazo largo del cromosoma 14, con sitios de roturas cromosómicas en o cerca de los loci de inmunoglobulinas y oncogenes. La inestabilidad de los cromosomas también predispone al desarrollo de algunas enfermedades malignas. En ciertas enfermedades autosómicas recesivas como la ataxia-telangiectasia, el síndrome de Bloom y la anemia de Fanconi, las células muestran una tendencia a la inestabilidad genética, es decir, a la rotura y reordenamiento cromosómicos in vitro. Estas enfermedades se acompañan de una alta incidencia de neoplasias, en particular leucemias y linfomas. Algunas aberraciones cromosómicas, mejor conocidas por sus efectos en el fenotipo, también predisponen a tumores. Por ejemplo, los pacientes con síndrome de Down (trisomía 21) tienen un incremento del riesgo de leucemia de la infancia de 20 veces, los varones 47,XXY (síndrome de Klinefelter) tienen un incremento del riesgo de cáncer de mama de 30 veces, y los individuos XY con fenotipo femenino tienen un riesgo mucho más alto de desarrollar cáncer ovárico, de manera particular un gonadoblastoma. Las indicaciones para el análisis citogenómico de las neoplasias continúa en evolución. No todos los tumores requieren estudio. Sin embargo, en casos de tumores de tipo inespecífico (en especial leucemias y linfomas), con una historia familiar marcada de neoplasias tempranas, o de ciertos tumores vinculados con potenciales defectos cromosómicos generalizados (presentes en células que no son neoplásicas), el análisis citogenómico debe tomarse en cuenta con firmeza. Cada vez más, las mutaciones genéticas específicas que están presentes en el tejido tumoral de una persona se utilizan no sólo para el pronóstico, sino también para guiar el tratamiento. Entre muchos ejemplos se incluyen las mutaciones en BRAF en el melanoma, la expresión de HER-2/neu en el cáncer de mama, y la mutación de EGFR en el cáncer pulmonar de células no pequeñas. Flaherty KT. Narrative review: BRAF opens the door for therapeutic advances in melanoma. Ann Intern Med 2010;153(9):587–591. [PubMed: 21041578] Leary RJ et al. Detection of chromosomal alterations in the circulation of cancer patients with whole-genome sequencing. Sci Transl Med 2012;4(162):162ra154. [PubMed: 23197571] McDermott U et al. Genomics and the continuum of cancer care. N Engl J Med 2011;364(4):340–350. [PubMed: 21268726] Reck M. A major step towards individualized therapy of lung cancer with gefitinib: the IPASS Trial and beyond. Expert Rev Anticancer Ther 2010;10(6):955–965. [PubMed: 20553217] Steensma DP. The beginning of the end of the beginning in cancer genomics. N Engl J Med 2013;368(22):2138–2140. [PubMed: 23634995] Wong KM et al. Unraveling the genetics of cancer: genome sequencing and beyond. Annu Rev Genomics Hum Genet 2011;12:407–430. [PubMed: 21639794]