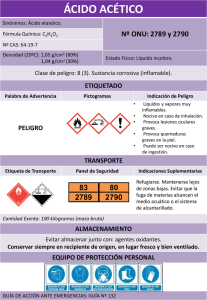
Conservas vegetales. Determinación del ácido sórbico
Anuncio

Re p u b l i co fEc u a d o r ≠ EDI CTOFGOVERNMENT± I no r d e rt op r o mo t ep u b l i ce d u c a t i o na n dp u b l i cs a f e t y ,e q u a lj u s t i c ef o ra l l , ab e t t e ri n f o r me dc i t i z e n r y ,t h er u l eo fl a w,wo r l dt r a d ea n dwo r l dp e a c e , t h i sl e g a ld o c u me n ti sh e r e b yma d ea v a i l a b l eo nan o n c o mme r c i a lb a s i s ,a si t i st h er i g h to fa l lh u ma n st ok n o wa n ds p e a kt h el a wst h a tg o v e r nt h e m. NTE INEN 2142 (1998) (Spanish): Conservas vegetales. Determinación del ácido sórbico INSTITUTO ECUATORIANO DE NORMALIZACIÓN Quito - Ecuador NORMA TÉCNICA ECUATORIANA NTE INEN 2 142:98 CONSERVAS VEGETALES. DETERMINACIÓN DEL ÁCIDO SÓRBICO. Primera Edición CANNED VEGETABLES. DETERMINATION OF SORBIC ACID. First Edition DESCRIPTORES: Productos agroindustriales, conservas vegetales, análisis químico, determinación del contenido de ácido sórbico, análisis espectrofotométrico, análisis colorimétrico. AL 02.01-331 CDU: 664.1.635:547.295.2:543.42 CIIU: 3113 ICS: 67.080.20 CDU: 664.1.635:547.295.2:543.42 ICS: 67.080.20 CIIU: 3113 AL 02.01-331 Norma Técnica Ecuatoriana Voluntaria NTE INEN 2 142:98 CONSERVAS VEGETALES. DETERMINACIÓN DEL ÁCIDO SÓRBICO 1998-07 1. OBJETO 1.1 Esta norma establece el método de ensayo para determinar cualitativa y cuantitativamente el ácido sórbico o sus sales de sodio y potasio en los productos elaborados a partir de frutas y vegetales. 2. ALCANCE 2.1 La presente norma especifica un método para la extracción del ácido sórbico presente en frutas, vegetales y productos derivados, y dos técnicas para la determinación del ácido sórbico extraído. 2.2 El método es aplicable a productos elaborados de frutas y vegetales que contienen ácido sórbico o sus sales de sodio y potasio, especialmente a jugos de frutas, salsa y concentrado de tomate, jaleas y mermeladas y otros productos derivados. 3. RESUMEN 3.1 El método consiste en la homogeneización del producto, seguido por un arrastre cuantitativo de vapor, del ácido sórbico presente en la porción de ensayo. Determinación de este ácido en el destilado obtenido, ya sea por espectrofotometría en el rango ultra-violeta ( técnica A), o por medición fotocolorimétrica o espectrofotométrica del color rosado obtenido después de la oxidación con ácido crómico y posterior tratamiento con ácido tiobarbitúrico (técnica B). 3.2 Técnica A: Espectrofotometría en el rango ultra-violeta, efectuado después de la oxidación con dióxido de azufre, el cual podría interferir. La oxidación ocurre expontáneamente en unos pocos minutos en el aire después de la adición de una traza de cobre catalítico. Los aceites esenciales naturales de frutas cítricas no interfieren en la determinación cuando estos estén presentes en pequeñas cantidades en el jugo normal no enriquecido con aceites esenciales. Cuando las cantidades de aceites esenciales son significantes, estos deben ser eliminados con anterioridad por el mismo método aplicado en la técnica B. 3.3 Técnica B: Colorimetría, basada en la reacción de Schmidt, la cual requiere de la eliminación por evaporación de etanol y aceites esenciales de una porción de la alícuata del destilado. Esta técnica, no es tan rápida como la técnica A, pero proporciona resultados comparables, es apropiado para el uso cuando los datos suministrados por un espectrofotómetro en el rango ultra-violeta no son disponibles. La interferencia causada por los aceites esenciales de ajo, cebolla o puerro pueden ser eliminadas, cuando se usa cualquiera de las dos técnicas, por la evaporación de una porción de la alícuota del destilado. 4. REACTIVOS 4.1 Todos los reactivos deben ser de calidad analítica y el agua que se use debe ser destilada. 4.2 Ácido tartárico, cristalino 4.3 Solución estándar de ácido sórbico 0,010 g/l, preparada por uno de los siguientes métodos (4.3.1 o 4.3.2): (Continúa) ____________________________________________________________________________________ DESCRIPTORES: Productos agroindustriales, conservas vegetales, análisis químico, determinación del contenido de ácido sórbico, análisis espectrofotométrico, análisis colorimétrico. -1- 1997-036 NTE INEN 2 142 1998-07 3 4.3.1 Disolver 0,100 g de ácido sórbico en 10 a 12 cm de una solución 0,1 N de hidróxido de sodio. Transferir cuantitativamente a un matraz volumétrico de 1 000 cm3, y diluir hasta la marca con agua. 3 3 Transferir 100 cm de esta solución así preparada a un segundo matraz volumétrico de 1 000 cm y diluir con agua hasta la marca. 3 4.3.2 Disolver 134 mg de sorbato de potasio en agua en un matraz volumétrico de 1 000 cm y diluir con agua hasta la marca. 3 3 Transferir 100 cm de esta solución a un segundo matraz volumétrico de 1 000 cm y diluir con agua hasta la marca. 4.4 Hidróxido de calcio (si es necesario), Solución aproximadamente 0,04 N. 4.5 Para la Técnica A: 4.5.1 Solución catalítica de cobre. En un matraz volumétrico de 1 000 cm3 , disolver en una pequeña cantidad de agua: 0,5 g de carbonato ácido de sodio y 0,001 g de sulfato de cobre pentahidratado puro (CuSO4.5H2O). Diluir con agua hasta la marca. 4.6 Para la Técnica B: 4.6.1 Solución de ácido sulfúrico-crómico. Disolver 0,05 g de dicromato de potasio en aproximadamente 90 cm3 de agua. Transferir cuantitativamente a un matraz volumétrico de 200 cm3. Añadir 100 cm3 de una solución 0,3 N de ácido sulfúrico. Diluir hasta la marca con agua. (1 litro de la solución 0,3 N de ácido sulfúrico contiene 14,7 g de ácido sulfúrico, es decir, 8,4 cm3 de ácido sulfúrico ρ20 1,84 g/cm3). 4.6.2 Solución de ácido tiobarbitúrico. Disolver 0,500 g de ácido tiobarbitúrico en 50 cm3 de agua a la cual se añade 10 cm3 de una solución 1 N de hidróxido de sodio. Transferir cuantitativamente a un matraz volumétrico de 100 cm3 y añadir 11 cm3 de una solución 1 N de ácido clorhídrico. Diluir hasta la marca con agua. Esta solución no es estable por lo que debe ser usada dentro de las 5 horas siguientes a su preparación. 5. EQUIPO Equipo usual de laboratorio, y en particular: 5.1 Balanza analítica 5.2 Homogenizador o mortero, apropiado 5.3 Baño de agua hirviendo 5.4 Aparato de destilación por arrastre de vapor, (ver figura 1), comprende los siguientes ítems listados en 5.4.1 a 5.4.5: 3 5.4.1 Balón generador de vapor con una capacidad de 1 000 a 1 500 cm . 5.4.2 Burbujeador, consiste de un tubo cilíndrico de 30 mm de diámetro y 270 mm de alto, el extremo inferior es cerrado y ensanchado en una esfera de 60 mm de diámetro. El tubo que suministra vapor debe terminar 10 mm arriba del fondo del burbujeador. La parte esférica, en la cual se deposita el producto, debe ser calentado eléctricamente o por medio de llama; en este caso, la llama del quemador de gas debe ser desviada por un disco metálico de 150 mm de diámetro, teniendo un orificio central de aproximadamente 40 mm de diámetro en el cual el fondo del burbujeador encaja. -2- 1997-036 NTE INEN 2 142 1998-07 Este dispositivo evita la pirogeneración de materiales que pueden ser extraídos del producto. El calentamiento auxiliar debe ser controlado de tal manera que el volumen del producto depositado en el burbujeador no decrezca ni se incremente por más de 5 cm3 durante la destilación. 5.4.3 Columna de fraccionamiento, a través de la cual pasa el vapor conteniendo los ácidos volátiles. Puede consistir de: a) un tubo cilíndrico de 20 mm de diámetro y 500 mm de alto, conteniendo una malla corrugada No. 100 de acero inoxidable en forma de hélice, con una separación (paso) de 15 mm; b) ó una columna de 20 mm de diámetro y 600 mm de alto, que contenga núcleos de ebullición de vidrio; c) ó cualquier otro dispositivo que tenga la misma eficiencia de fraccionamiento. NOTA. El fraccionamiento de un vapor es indispensable para retener el hidroximetilfurfural presente. Esta sustancia y sus productos hidrolizados absorben la radiación ultra-violeta a 256 nm. La columna de fraccionamiento debe ser reducida a 200 mm de alto ó ser reemplazada por un frasco Kjeldahl cuando el producto es libre de hidroximetilfurfural. 5.4.4 Condensador del tipo West, de 400 mm de longitud efectiva y que cumpla con la norma ISO 4799, instalado verticalmente para asegurar la condensación del vapor y completar el enfriamiento del destilado. 5.4.5 Matraz receptor, apropiado para: a) productos líquidos: matraz graduado de 200 cm3; b) productos sólidos o viscosos: matraz de 500 cm3 5.4.6 Chequeo de la eficiencia del aparato de destilación El aparato de destilación (5.4) debe destilar 300 cm3 recolectados en un tiempo de 12 a 15 minutos, y debe además cumplir con las siguientes condiciones mínimas: a) en condiciones normales de destilación, el 99,5 % de una cantidad conocida de ácido acético añadida a la muestra debe ser encontrada en el destilado, el cual debe ser 200 cm3. Para este ensayo, usar 20 cm3 de una solución 0,1 N de ácido acético; b) en las mismas condiciones de destilación, no mas de 5 partes por mil de una cantidad conocida de ácido láctico añadida a la muestra debe ser encontrado en el destilado, el cual debe ser de 200 cm3. Para este ensayo, usar 20 cm3 de una solución 1 N de ácido láctico. 3 5.5 Pipetas, de capacidad 10, 20 y 25 cm , que cumplan con la norma ISO 648. 5.6 pipetas graduadas, de capacidad apropiada. 5.7 Para la técnica A: 5.7.1 Matraz cónico, capacidad 50 cm3. 5.7.2 Espectrofotómetro, que permita medidas a una longitud de onda de 256 nm (ultra-violeta), con celdas silíceas de 10 mm de longitud de recorrido óptico. 5.8 Para la Técnica B: 5.8.1 Matraz volumétrico, capacidad 25 cm3, que cumpla con la norma ISO 1042. (Continúa) -3- 1997-036 NTE INEN 2 142 1998-07 5.8.1 Fotocolorímetro, con filtro verde, o espectrofotómetro que permita medidas a una longitud de onda de 532 nm. 6. PROCEDIMIENTO 6.1 Preparación de la muestra de ensayo 6.1.1 Productos líquidos (jugos, pulpas fluidas de fruta, jarabes), y productos viscosos (pastosos) (mermeladas, jaleas). Homogeneizar la muestra de laboratorio después de haber sido cuidadosamente mezclada. 6.1.2 Productos sólidos (frutas, vegetales) Cortar una parte de la muestra de laboratorio en pequeñas piezas, separar las semillas, pistilos y pedúnculos, si es necesario. Tomar aproximadamente 40 g del producto y homogeneizar en un homogenizador o mortero (5.2). Productos congelados o profundamente congelados deben ser primero descongelados en un recipiente cerrado y el líquido formado durante el descongelamiento debe ser añadido al producto antes de la homogeneización. 6.2 Porción de ensayo 6.2.1 Productos líquidos Usando una pipeta (5.5), tomar 10 cm3 de la muestra de ensayo (6.1) e introducirla en el burbujeador (5.4.2). NOTA. La porción de ensayo puede también ser tomada por masa, pesando con una aproximación a 0,01 g, aproximadamente 10 g de la muestra de ensayo. 6.2.2 Productos sólido o viscosos Pesar, con una aproximación a 0,01 g, aproximadamente 10 g de la muestra de ensayo (6.1) e introducir en el burbujeador (5.4.2) con lo mínimo necesario de agua para introducir la totalidad de la porción de ensayo y hacer la mezcla lo suficientemente fluida. NOTA. En ciertos casos es necesario dejar la porción de ensayo remojar en agua por 1 a 2 h. 6.3 Destilación 6.3.1 Introducir 0,5 g de ácido tartárico (4.2) en el burbujeador (5.4.2) conteniendo la porción de ensayo (6.2). Conectar el burbujeador al balón generador de vapor (5.4.1) y al condensador (5.4.4) y simultáneamente calentar el balón y el burbujeador; efectuar la destilación, procurando que el volumen del contenido del burbujeador permanezca constante dentro de 5 cm3. 6.3.2 En el caso de productos líquidos. Recolectar el destilado en un matraz de 200 cm3, parar la destilación cuando la marca de 200 cm3 se haya alcanzado. 6.3.3 En el caso de productos sólidos o viscosos. Recolectar en un matraz de 500 cm3 un volumen de destilado al menos 20 veces más que el volumen contenido en el burbujeador. Medir el volumen (V) colectado, usando una probeta graduada. (Continúa) -4- 1997-036 NTE INEN 2 142 1998-07 6.4 Técnica A: Determinación por espectrofotometría en el rango ultra-violeta 6.4.1 Determinación 6.4.1.1 Si el producto inicial contiene aceites esenciales de ajo, cebolla o puerro, la presencia de estos aceites esenciales causa absorbancia significante, especialmente en el caso de ajo. Completar la evaporación1 del destilado, después de haberse hecho alcalino, lo que permite ser contrarrestado el efecto de esta absorbancia. Por consiguiente, cuando estos aceites esenciales están presentes, tomar con una pipeta (5.5) 25 cm3 de el destilado (6.3) y transferir a una cápsula de porcelana; alcalinizar con 1,5 a 2 cm3 de la solución de hidróxido de calcio (4.4), evaporar a sequedad en el baño de agua hirviendo (5.3) y reconstituir con agua para restablecer el volumen inicial. 6.4.1.2 Tomar con una pipeta (5.5) 10 cm3 (ver nota) del destilado (6.3) o de la solución reconstituida (6.4.1.1), depositar en un matraz cónico de 50 cm3 (5.7.1) y añadir 10 cm3 de la solución catalítica de cobre (4.5.1). Agitar brevemente y dejar en contacto con el aire por varios minutos. 3 NOTA. El volumen de 10 cm es apropiado para productos que contienen hasta 200 mg de ácido sórbico por litro o por kilogramo. Para 3 3 contenidos más altos, tomar únicamente 5 a 2 cm y diluir a 10 cm con agua. Medir la absorbancia de la solución usando el espectrofotómetro (5.7.2) a una longitud de onda de 256 nm. Substraer del valor encontrado la absorbancia de la solución del blanco de ensayo (6.4.2). 6.4.2 Blanco de ensayo Llevar un blanco de ensayo en paralelo con la determinación, reemplazando los 10 cm3 del destilado por 10 cm3 de agua. 6.4.3 Número de determinaciones Llevar a cabo dos determinaciones en la misma muestra de ensayo (6.1). 6.4.4 Preparación de la curva de calibración 6.4.4.1 Dentro de una serie de seis matraces cónicos de 50 cm3 (5.7.1), introducir respectivamente, con una pipeta (5.6): 0, 1, 2, 3, 5, 10 cm3 de la solución estándar de ácido sórbico (4.3); llevar al volumen de 10 cm3 por la adición de: 3 10, 9, 8, 7, 5, 0 cm de agua. Las soluciones obtenidas contienen: 0, 1, 2, 3, 5, 10 mg de ácido sórbico por litro. 6.4.4.2 A cada matraz añadir 10 cm3 de la solución catalítica de cobre (4.5.1) Medir la absorbancia de las soluciones usando el espectrofotómetro (5.7.2) a una longitud de onda de 256 nm. Substraer de los valores encontrados la absorbancia de la solución del blanco de ensayo (6.4.2). ________________ 1) La evaporación a sequedad no destruye el ácido sórbico si las condiciones son suficientemente alcalinas. (Continúa) -5- 1997-036 NTE INEN 2 142 1998-07 6.4.4.3 Trazar la curva de calibración presentando las absorbancias de las soluciones (6.4.4.2) como una función de las concentraciones de ácido sórbico de las soluciones obtenidas en 6.4.4.1, es decir, antes de la adición de la solución catalítica de cobre (4.5.1), expresada en miligramos por litro. 6.5 Técnica B: Determinación por fotocolorimetría o por espectrofotometría a 532 nm. 6.5.1 Determinación 6.5.1.1 Si el producto inicial contiene etanol, remover del destilado por el siguiente método: Usando una pipeta (5.5) transferir 25 cm3 del destilado (6.3) a una cápsula de porcelana, alcalinizar con 1,5 a 2 cm3 de la solución de hidróxido de calcio (4.4); colocar la cápsula en el baño de agua hirviendo (5.3), y evaporar hasta que el volumen se haya reducido aproximadamente a la mitad, lo cual normalmente toma un tiempo aproximado de 30 min. Cuantitativamente transferir el residuo a un matraz volumétrico de 25 3 cm . Llevar hasta la marca con el agua de enjuague de la cápsula. Agitar. 6.5.1.2 Si el producto inicial contiene aceites esenciales ( en el caso de jugos de frutas cítricas), eliminarlos del destilado mediante el mismo método descrito en 6.5.5.1, pero prolongar la evaporación de tal manera de lograr un volumen de 1 a 2 cm3. 6.5.1.3 Si el producto inicial contiene aceites esenciales de ajo, cebolla o puerro, proceder de acuerdo a lo indicado en 6.4.1. NOTA En el caso de ajo, aún cuando el destilado sea evaporado a sequedad, una muy débil absorbancia, correspondiente a 1,5 mg de ácido sórbico por kilogramo, permanece. 6.5.1.4 Tomar con una pipeta (5.5) 10 cm3 (ver nota) del destilado (6.3) o de la solución reconstituida obtenida después del tratamiento 6.5.1.1, 6.5.1.2, ó 6.5.1.3, y transferir a un matraz volumétrico de 25 cm3. 3 NOTA El volumen de 10 cm es apropiado para productos que contienen hasta 200 mg de ácido sórbico por litro o por kilogramo. Para 3 3 contenidos más altos, tomar únicamente 5 a 2 cm y diluir a 10 cm con agua. Añadir 4 cm3 de la solución de ácido sulfúrico-crómico (4.6.1) y mantener en el matraz por 10 min. en el baño de agua hirviendo (5.3). Añadir 4 cm3 de la solución de ácido tiobarbitúrico (4.6.2) y mantener el matraz en el baño de agua hirviendo por 20 min. adicionales; se desarrollará un color rosa. Enfriar en un baño de agua helada y diluir hasta la marca con agua. Dentro de un período de 30 min. medir la absorbancia de la solución usando el fotocolorímetro o espectrofotómetro a una longitud de onda de 532 nm. Substraer del valor encontrado la absorbancia de la solución del blanco de ensayo (6.5.2). 6.5.2 Blanco de ensayo 3 Llevar un blanco de ensayo en paralelo con la determinación, reemplazando los 10 cm del destilado por 10 cm3 de agua. 6.5.3 Número de determinaciones Llevar a cabo dos determinaciones en la misma muestra de ensayo (6.1). 6.5.4 Preparación de la curva de calibración 6.5.4.1 Preparar una solución estándar de ácido sórbico de 2,0 mg/l por dilución de 1 volumen de la solución estándar (4.3) con 4 volúmenes de agua. (Continúa) -6- 1997-036 NTE INEN 2 142 1998-07 6.5.4.2 Dentro de una serie de seis matraces volumétricos de 25 cm3 (5.8.1), introducir respectivamente, con una pipeta graduada (5.6): 0, 2, 4, 6, 8, 10 cm3 de la solución estándar de ácido sórbico diluida (6.5.4.1); llevar al volumen de 10 cm3 por la adición de: 10, 8, 6, 4, 2, 0 cm3 de agua. Las soluciones obtenidas contienen: 0, 0,4, 0,8, 1,2, 1,6, 2,0 mg de ácido sórbico por litro. 6.5.4.3 A cada matraz añadir 4 cm3 de la solución de ácido sulfúrico-crómico (4.6.1) y mantener el matraz por 10 min en el baño de agua hirviendo. Añadir 4 cm3 de la solución de ácido tiobarbitúrico (4.6.2) y mantener los matraces en el baño de agua hirviendo por 20 min adicionales; se desarrollará un color rosa. Enfriar en un baño de agua helada y diluir hasta la marca con agua. Dentro de un período de 30 min medir las absorbancias de las soluciones usando el fotocolorímetro o espectrofotómetro a una longitud de onda de 532 nm. Substraer de los valores encontrados la absorbancia de la solución del blanco de ensayo (6.5.2). 6.5.4.4 Trazar la curva de calibración presentando las absorbancias de las soluciones (6.5.4.3) como una función de las concentraciones correspondientes del ácido sórbico, expresado en miligramos por litro. 7. CÁLCULOS1 7.1 Métodos de cálculo y fórmulas 7.1.1 Porción de ensayo medida por volumen El contenido de ácido sórbico, expresado en miligramos por litro de producto, está dado por la siguiente fórmula: m1 x 200 As = ⎯⎯⎯⎯⎯⎯⎯ V1 En donde: As = Contenido de ácido sórbico expresado en miligramos por litro de producto; m1 = masa del ácido sórbico, expresado en miligramos por litro del destilado (6.3.1), leída en la curva de calibración (ver 6.4.4 ó 6.5.4); V1 = Volumen en cm3, tomado en 6.4.1.2 ó en 6.5.1.4 ( normalmente 10 cm3, pero puede ser reducido a 5 cm3 ó 2 cm3). _______________________ 1) Varios productos vegetales contienen pequeñas cantidades de sustancias volátiles que pueden ser extraídas por solventes orgánicos y los cuales absorben radiación a 256 nm ó dan reacción coloreada usada en la técnica B (6.5). Por otra parte, aquellos resultados que son ligeramente positivos ( menos de 10 mg por litro o por kilogramo) deben ser interpretados con cuidado y comparados con resultados de los mismos productos libres de ácido sórbico. (Continúa) -7- 1997-036 NTE INEN 2 142 1998-07 7.1.2 Porción de ensayo medida por masa El contenido de ácido sórbico, expresado en miligramos por kilogramo de producto, está dado por la siguiente fórmula: m1 x V x 10 As = ⎯⎯⎯⎯⎯⎯ mo x V1 En donde: As = Contenido de ácido sórbico expresado en miligramos por kilogramo de producto; mo = masa en gramos de la porción de ensayo (6.2.2); m1 = masa del ácido sórbico, expresado en miligramos por litro del destilado (6.3.2), leída en la curva de calibración (ver 6.4.4 ó 6.5.4); 3 V = volumen en cm del destilado recolectado (ver 6.3.2); V1 = Volumen en cm3, tomado en 6.4.1.2 ó en 6.5.1.4 ( normalmente 10 cm3, pero puede ser reducido a 5 cm3 ó 2 cm3). 7.2 Repetibilidad 7.2.1 La diferencia entre los resultados de dos determinaciones realizadas simultáneamente o en rápida sucesión por el mismo analista, no deberá se mayor del 5% del valor medio. 8. INFORME DE RESULTADOS En el informe de resultados se debe indicar la siguiente información: 8.1 El método usado y el resultado obtenido en cada determinación. 8.2 Debe mencionarse además todos los detalles operacionales no especificados en la presente norma, así como cualquier circunstancia que pueda haber influido en el resultado. 8.3 Todos los detalles necesarios que permitan la completa identificación de la muestra. (Continúa) -8- 1997-036 NTE INEN 2 142 1998-07 FIGURA 1. Equipo de destilación al vapor (Continúa) -9- 1997-036 NTE INEN 2 142 1998-07 APÉNDICE Z Z.1 DOCUMENTOS NORMATIVOS A CONSULTAR Esta norma no requiere de otras para su aplicación. Z.2 BASES DE ESTUDIO International Standard ISO 5519. Fruits, vegetables and derived products. Determination of sorbic acid content. First edition- 1978-09-01. Norma ICAITI 34 003 h29 Productos elaborados a partir de frutas y hortalizas. Determinación cualitativa y cuantitativa del ácido sórbico y sorbatos alcalinos. Instituto Centroamericano de Investigación y Tecnología Industrial ICAITI, Guatemala 1980. Food Quality Assurance. The Avi Publishing Company, Inc. Westport, Connecticut 1983. -10- 1997-036 INFORMACIÓN COMPLEMENTARIA Documento: TÍTULO: CONSERVAS VEGETALES. DETERMINACIÓN DEL Código: NTE INEN 2 142 AL 02.01-331 ÁCIDO SÓRBICO. ORIGINAL: REVISIÓN: Fecha de iniciación del estudio: Fecha de aprobación anterior por Consejo Directivo 1996-09-23 Oficialización por Acuerdo No. de publicado en el Registro Oficial No. de Fecha de iniciación del estudio: Fechas de consulta pública: de a Subcomité Técnico: SALSA DE TOMATE Fecha de iniciación: 1997-01-31 Integrantes del Subcomité Técnico: Fecha de aprobación: 1997-06-13 NOMBRES: INSTITUCIÓN REPRESENTADA: Dra. Armanda Coronel Rivera (Presidente) INSTITUTO DE HIGIENE Y MEDICINA TROPICAL-GUAYAQUIL INSTITUTO DE INVESTIGACIONES TECNOLÓGICAS DE LA E.P.N. LA PORTUGUESA S.A. COMNACA TRIBUNA ECUATORIANA DE CONSUMIDORES Y USUARIOS INSTITUTO NACIONAL DE HIGIENE - QUITO DIRECCIÓN MUNICIPAL DE HIGIENE NESTLE ECUADOR S.A. ALISA ECUAVEGETAL S.A. DACA NESTLE ECUADOR S.A. INEN Dra. Rosario Barrera F. Ing. Helmut Nickel Garaicoa Ing. Juan Miniguano Michelle O. Fried Dra. Lucia Navas Dr. Hernan Riofrío Ing, Ramiro Valarezo Sr. Rodolfo Philco Dra. Susana Sánchez de Rojas Dr. Guido Martínez Ing. Jorge Arturo Gómez Ing. Bolívar Cano (Secretario Técnico) P.V.P. S/. 3 900,oo Otros trámites: CARÁCTER: Se recomienda su aprobación como: OPCIONAL Aprobación por Consejo Directivo en sesión de 1998-05-18 como: Voluntaria Oficializada como: VOLUNTARIA Por Acuerdo Ministerial No. 291 de 1998-06-23 Registro Oficial No. 363 de 1998-07-17 Instituto E c u a toria no d e N orma liz a c ión, IN E N - B a q u e rizo Mor e no E 8-29 y A v. 6 d e Dic ie mb r e C a silla 17-01-3999 - T e lfs: (593 2)2 501885 a l 2 501891 - F ax: (593 2) 2 567815 Dir e c c ión G e n e r a l: E-Ma il:furr e st a @ in e n.g ov.e c Á r e a T é c nic a d e N orma liz a c ión: E-Ma il:norma liz a c ion @ in e n.g ov.e c Á r e a T é c nic a d e C e rtific a c ión: E-Ma il:c e rtific a c ion @ in e n.g ov.e c Á r e a T é c nic a d e V e rific a c ión: E-Ma il:v e rific a c ion @ in e n.g ov.e c Á r e a T é c nic a d e S e rvic ios T e c noló gic os: E-Ma il:in e n c a ti @ in e n.g ov.e c R e gion a l G u a y a s: E-Ma il:in e n g u a y a s @ in e n.g ov.e c R e gion a l A zu a y: E-Ma il:in e n c u e n c a @ in e n.g ov.e c R e gion a l C himb or a zo: E-Ma il:in e nrio b a mb a @ in e n.g ov.e c U RL:w w w.in e n.g ov.e c