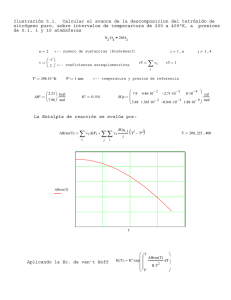
kP v θ = PK1 PK + = θ PK1 PP kK v + =
Anuncio

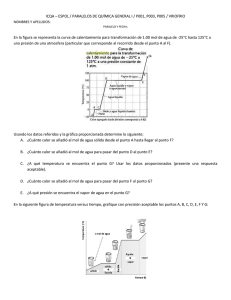
1.- La reacción entre NO(g) y CO(g) para dar N2(g) y CO2(g) está catalizada por una superficie sólida de Rodio. Sabiendo que la adsorción del NO es condición necesaria para que tenga lugar la reacción y que los productos no se adsorben significativamente:deducir si la reacción responde a un mecanismo del tipo Langmuir-Hinshelwood (dos moléculas adsorbidas) o de Eley-Rideal (una adsorbida otra en fase gaseosa) sabiendo que en el límite de presiones parciales de CO muy altas la ley de velocidad experimental es v=k’PNO/PCO Eley-Rideal NO(g) NO(ads ) 1 k CO(g) NO(ads) N2 (g) CO2 (g) 2 Etapa lenta v kPCO NO Ads. NO(g) en equilibrio NO K NOPNO 1 K NOPNO v kK NOPNOPCO 1 K NOPNO 1.- La reacción entre NO(g) y CO(g) para dar N2(g) y CO2(g) está catalizada por una superficie sólida de Rodio. Sabiendo que la adsorción del NO es condición necesaria para que tenga lugar la reacción y que los productos no se adsorben significativamente:deducir si la reacción responde a un mecanismo del tipo Langmuir-Hinshelwood (dos moléculas adsorbidas) o de Eley-Rideal (una adsorbida otra en fase gaseosa) sabiendo que en el límite de presiones parciales de CO muy altas la ley de velocidad experimental es v=k’PNO/PCO Langmuir-Hinshelwood NO(g) NO(ads) CO(g) CO(ads) 1 k CO(ads ) NO(ads ) N2 (g) CO 2 (g) 2 Etapa lenta v kCO NO Ads. NO en equilibrio Ads. CO en equilibrio K NOPNO NO 1 K NOPNO K COPCO K COPCO CO 1 K NOPNO K COPCO PCO muy alta v kK COK NOPNOPCO K COPCO 2 kK NO PNO K CO PCO v kK COK NOPNOPCO 1 K NOPNO K COPCO 2 1.2. La energía de activación observada usando presiones de CO altas es de 5 kcal/mol. Si la entalpía de adsorción del NO sobre el Rodio es de –20 kcal/mol y la del CO vale –10 kcal/mol ¿cuánto vale la energía de activación de la etapa de reacción ?. Cambiando de catalizador ¿podría obtenerse un valor negativo para esta magnitud? v kK NO PNO K CO PCO k cat (para PCO altas) kK NO K CO 0 0 Ea,cat Ea,r Hads ,NO Hads,CO Ea,r Ea,cat H0ads,NO H0ads,CO 5 20 10 15 kcal / mol 1.3. Deducir el orden cinético total de la reacción en el límite de temperaturas altas. Si T es muy alta, entonces Kads son pequeñas. Si tomamos la ley de velocidad deducida del mecanismo, podemos ver que es posible simplificarla en el caso de que las constantes de adsorción son pequeñas, porque los productos KiPi << 1 v kK COK NOPNOPCO 1 K NOPNO K COPCO 2 v kK COK NOPNOPCO 1 2 k' cat PNO PCO 2. La reacción de isomerización en fase gaseosa A(g) B(g) viene catalizada por un determinado sólido. Se sabe que la etapa de reacción es lenta, elemental y reversible y que B no se adsorbe en dicho sólido 1.Deduce una expresión para la velocidad de reacción en función de las presiones de A y de B. Deduce igualmente una expresión para la velocidad inicial en función de la presión inicial de A. 2.La tabla siguiente muestra los valores de velocidades iniciales obtenidos en función de la presión inicial de A a dos temperaturas diferentes. Deduce los valores de las constantes cinéticas y de equilibrio que aparecen en la expresión de la velocidad a dichas temperaturas 3.Calcula, a partir de la expresión de la velocidad inicial, cuál será la energía de activación de la reacción catalizada en los límites de altas y bajas temperaturas PA,0 (atm) 0.1 0.2 0.3 0.4 v0 (mol/h·g) T=400 K 1.06·10-3 1.51·10-3 1.77·10-3 1.93·10-3 T=500 K 8.88·10-4 16.55·10-4 23.22·10-4 29.09·10-4 1. Deducir velocidad inicial: El mecanismo incluirá dos etapas A(g) M(sup) A M(sup) Etapa (1). Adsorción de A A M(sup) B(g) M(sup) Etapa (2). Reacción de A Suponemos que (2) es la lenta y la (1) está en equilibrio. Entonces la velocidad queda: v k 2 A k 2PB (1 A ) La fracción de centros ocupados por A lo obtenemos del equilibrio (1) A K APA 1 K APA Sustituyendo el la expresión de la velocidad queda: v k 2K A PA k P 2 B 1 K A PA 1 K A PA Y la velocidad inicial será: v0 k 2K A PA,0 1 K A PA,0 2. Deducir valores de las constantes La ley de velocidad inicial se puede linealizar tomando la inversa: v0 k 2K A PA,0 1 K A PA,0 1 1 1 1 v 0 k 2K A PA,0 k 2 1/v0 (h·g/mol) Representando 1/v0 frente a 1/PA,0 deberemos obtener una línea recta de cuya ordenada en el origen obtendremos k2 y a continuación, del valor de la pendiente obtenemos KA. El ajuste a las dos temperaturas en que se ha realizado el experimento es: K K 1/PA,0 (1/atm) Con los ajustes realizados a 400 y 500 K podemos obtener las constantes a dichas temperaturas k2 (mol/h·g) KA (atm-1) 400 K 500 K 2.67·10-3 1.20·10-2 6.56 0.8 Conviene en este punto darse cuenta de que la constante de adsorción disminuye con la temperatura, que es el comportamiento esperado para un proceso exotérmico. Por su parte la constante de velocidad aumenta con la temperatura, que es lo esperable para la constante de velocidad referida a una etapa. 3. Tomaremos los límites de T altas y T bajas en la expresión de la ley de velocidad: v0 k 2K A PA,0 1 K A PA,0 En el límite de bajas T, la KA será muy alta y la velocidad inicial será aproximadamente v0 k 2K A PA,0 1 K A PA,0 k 2K A PA ,0 K A PA,0 k2 Así pues kcat=k2 y Ea,cat=Ea,2. La energía de activación de la etapa 2 puede deducirse a partir de la expresión de Arrhenius aplicada a las constantes k2 determinadas a 400 y 500K: T1=400K k 2 2.67·10 3 mol·h1·g1 T2=500K k 2 1.20·10 2 mol·h1·g1 ln k 2,T1 k 2,T2 Ea,2 1 1 R T2 T1 Ea,2=25.0 kJ/mol por lo tanto Ea,cat= 25 kJ·mol-1 v0 k 2K A PA,0 1 K A PA,0 En el límite de altasT, la KA será muy baja y la velocidad inicial será aproximadamente: v0 k 2K A PA,0 1 K A PA ,0 k 2K A PA ,0 1 k 2K A PA,0 Así pues kcat=k2·KA y Ea,cat=Ea,2 +Hads,A La entalpía de adsorción de A puede deducirse a partir de la expresión de van’t Hoff aplicada a las constantes KA determinadas a 400 y 500K: T1=400K K A 6.56 atm1 T2=500K K A 0.8 atm1 ln K A,T1 K A,T2 Hads,A 1 1 R T2 T1 ∆Hads,A=-35.0 kJ/mol por lo tanto Ea,cat= 25 – 35 = -10 kJ·mol-1