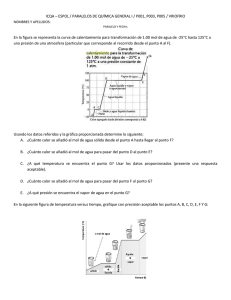
SAL ANHIDRA.(ES2219395)
Anuncio
")
19
OFICINA ESPAÑOLA DE
PATENTES Y MARCAS
11 Número de publicación: 2 219 395
51 Int. Cl. : C07D 487/04
7
A61K 31/519
A61P 9/00
// (C07D 487/04
C07D 239:00
C07D 231:00)
ESPAÑA
12
TRADUCCIÓN DE PATENTE EUROPEA
T3
86 Número de solicitud europea: 00962772 .0
86 Fecha de presentación: 06.10.2000
87 Número de publicación de la solicitud: 1220855
87 Fecha de publicación de la solicitud: 10.07.2002
54 Título: Sal anhidra.
30 Prioridad: 11.10.1999 GB 9923968
73 Titular/es: Pfizer Inc.
235 East 42Nd Street
New York, New York 10017, US
45 Fecha de publicación de la mención BOPI:
01.12.2004
72 Inventor/es: Hughes, Michael, Leslie y
Storey, Richard, Anthony
45 Fecha de la publicación del folleto de la patente:
74 Agente: Carpintero López, Francisco
ES 2 219 395 T3
01.12.2004
Aviso: En el plazo de nueve meses a contar desde la fecha de publicación en el Boletín europeo de patentes, de
la mención de concesión de la patente europea, cualquier persona podrá oponerse ante la Oficina Europea
de Patentes a la patente concedida. La oposición deberá formularse por escrito y estar motivada; sólo se
considerará como formulada una vez que se haya realizado el pago de la tasa de oposición (art. 99.1 del
Convenio sobre concesión de Patentes Europeas).
Venta de fascículos: Oficina Española de Patentes y Marcas. C/Panamá, 1 – 28036 Madrid
ES 2 219 395 T3
DESCRIPCIÓN
Sal anhidra.
5
La presente invención se refiere a sales anhidras farmacéuticamente aceptables del ácido paratoluensulfónico
de 3-etil-5-[5-(4-etilpiperazin-1-ilsulfonil)-2-(2-metoxietoxi)piridin-3-il]-2-(piridin-2-il)metil-2,6-dihidro-7H-pirazolo[4,3-d]pirimidin-7-ona con la fórmula (I):
10
15
20
25
30
35
40
45
50
55
60
65
Además, la presente invención se refiere a composiciones farmacéuticas que contienen sales anhidras del ácido paratoluensulfónico de 3-etil-5-[5-(4-etilpiperazin-1-ilsulfonil)-2-(2-metoxietoxi)piridin-3-il]-2-(piridin-2-il)metil-2,6dihidro-7H-pirazolo[4,3-d]pirimidin-7-ona, en procedimientos para la preparación de (I) y usos de (I) en medicina.
La 3-etil-5-[5-(4-etilpiperazin-1-ilsulfonil)-2-(2-metoxietoxi)piridin-3-il]-2-(piridin-2-il)metil-2,6-dihidro-7H-pirazolo[4,3-d]pirimidin-7-ona se ha descrito (como Ejemplo 4) y reivindicado en la solicitud internacional publicada
WO99/54333, con fecha de publicación internacional el 28 de octubre de 1999. Como se detalla en el documento
WO99/54333, los compuestos de este tipo son especialmente útiles en el tratamiento entre otras cosas de la disfunción
eréctil masculina. El documento WO99/54333 en su totalidad y en particular la descripción de los procedimientos
generales para la preparación de 3-etil-5-[5-(4-etilpiperazin-1-ilsulfonil)-2-(2-metoxietoxi)piridin-3-il]-2-(piridin-2il)metil-2,6-dihidro-7H-pirazolo[4,3-d]pirimidin-7-ona se incorpora aquí por referencia.
Para la utilidad con éxito dentro de la industria farmacéutica, es crítico que las propiedades de un material activo
sean, o bien conocidas, o se puedan predecir razonablemente en todos los procedimientos necesarios tanto en su fabricación y procesado farmacéutico como durante su transporte, almacenamiento y consiguiente uso terapéutico. En
algunos casos, los compuestos pueden presentar propiedades medicinales deseables que no se pueden traducir directamente en una composición farmacéutica adecuada ya que el principio activo tiene propiedades físicas no satisfactorias
como por ejemplo poca estabilidad de procesado o baja solubilidad.
Así, el reto al que se enfrenta el químico farmacéutico en la búsqueda de compuestos de fármacos nuevos y mejorados no es solo la fabricación, aislamiento y caracterización de nuevos compuestos de fármacos así como la determinación de sus propiedades medicinales particulares sino también el suministro de sales adecuadas farmacéuticamente
aceptables de dichos compuestos.
El problema abordado por la presente invención es el suministro de una sal farmacéuticamente aceptable de 3-etil5-[5-(4-etilpiperazin-1-ilsulfonil)-2-(2-metoxietoxi)piridin-3-il]-2-(piridin-2-il)metil-2,6-dihidro-7H-pirazolo[4,3-d]pirimidin-7-ona que se pueda procesar eficazmente para proporcionar formulaciones estables y eficaces del fármaco, y
en particular formas farmacéuticas sólidas y comprimibles del fármaco. Tales formas farmacéuticas incluyen comprimidos orales de liberación convencional, comprimidos de liberación controlada (matriz), formulaciones de disolución
rápida, comprimidos sublinguales, comprimidos bucales, polvo oral, cápsulas de gel blandas y rellenas de gránulos,
polvos para suspensiones reconstituidas, sistemas multipartícula de liberación convencional y controlada rellenados
en cápsulas o comprimidos en píldoras, pastillas, grageas, supositorios, pesarios, implantes sólidos, tapones liófilos,
nanopartículas y micropartículas y polvo para suspensión y administración por vía nasal, y sistemas de inhalación
seca. En el apartado de formulación en la presente memoria descriptiva se discuten en detalle adicionales formas
farmacéuticas adecuadas y vías de administración potencial.
Los criterios importantes que se tienen que satisfacer son, entre otras cosas, que la sal farmacéuticamente aceptable
sea cristalina, no higroscópica, de punto de fusión adecuado (preferentemente un punto de fusión de al menos 100ºC
2
ES 2 219 395 T3
aproximadamente, más preferentemente superior de aproximadamente 150ºC), posea estabilidad química a lo largo
de un intervalo de condiciones de temperatura y de humedad, tenga solubilidad aceptable, tenga un perfil de disolución aceptable (preferiblemente rápida y completa), tenga propiedades mecánicas aceptables tales como por ejemplo
presentar buena compresibilidad sin exhibir polimorfismo.
5
En la búsqueda de una sal farmacéuticamente aceptable de 3-etil-5-[5-(4-etilpiperazin-1-ilsulfonil)-2-(2-metoxietoxi)piridin-3-il]-2-(piridin-2-il)metil-2,6-dihidro-7H-pirazolo[4,3-d]pirimidin-7-ona se han investigado varias sales
potenciales. Sin embargo, se encontró que sólo una sal, la sal del ácido paratoluensulfónico, cumplía los criterios
necesarios para una sal farmacéuticamente aceptable.
10
15
20
25
30
35
40
45
50
55
60
65
Las sales hidratadas pueden proporcionar resultados particulares en el procesado, especialmente durante por ejemplo las etapas de molienda y/o secado. Se ha descrito que la tendencia de las moléculas orgánicas a hidratarse se puede
predecir a partir de su estructura molecular. “Hydration in organic crystals: prediction from molecular structure”, Desiraju, Gautam R. Sch. Chem., Univ. Hyderabad, Hyderabad, 500 134, India. J. Chem. Soc., Chem. Commun. (1991),
Issue 6, 426-8. CODEN: JCCCAT; ISNN: 0022-4936. (EN) CAN 114:237859. Usando dicho procedimiento predictivo, se predeciría que de hecho fuera muy probable que la sal de tosilato de 3-etil-5-[5-(4-etilpiperazin-1-ilsulfonil)2-(2-metoxietoxi)piridin-3-il]-2-(piridin-2-il)metil-2,6-dihidro-7H-pirazolo[4,3-d]pirimidin-7-ona se hidratara. Varias
de las sales investigadas forman hidratos. Además, los experimentos correspondientes realizados con la sal de besilato
estructuralmente similar de 3-etil-5-[5-(4-etilpiperazin-1-ilsulfonil)-2-(2-metoxietoxi)piridin-3-il]-2-(piridin-2-il)metil-2,6-dihidro-7H-pirazolo[4,3-d]pirimidin-7-ona, han determinado que además de al menos 5 polimorfos anhidros,
se forma de hecho un polimorfo hidratado de la sal de besilato.
El problema de seleccionar una forma de sal farmacéuticamente aceptable para la 3-etil-5-[5-(4-etilpiperazin-1ilsulfonil)-2-(2-metoxietoxi)piridin-3-il]-2-(piridin-2-il)metil-2,6-dihidro-7H-pirazolo[4,3-d]pirimidin-7-ona ha sido
resuelto por el sorprendente descubrimiento de que la sal anhidra del paratoluensulfonato de fórmula (I) cumple los
criterios anteriores para una sal farmacéuticamente aceptable. Incluso más sorprendente es el descubrimiento de que
la sal de paratoluensulfonato de la presente invención no parece que forme hidratos cristalinos en un intervalo de
condiciones. Esto es particularmente sorprendente dado que las predicciones iniciales indicarían que la sal de tosilato
formaría un hidrato, especialmente dado el hecho de que la sal de besilato y otras sales investigadas se hidrataban como
se predijo. Adicionalmente, la sal anhidra de paratoluensulfonato de la presente invención presenta buena estabilidad
de forma farmacéutica física, demuestra estabilidad durante los procedimientos de secado y molienda y tiene un perfil
de estabilidad del fármaco deseable.
En concreto, una ventaja adicional de la sal anhidra de paratoluensulfonato de la presente invención frente a la
sal de besilato correspondiente, es que se puede preparar con un rendimiento aumentado (98,5% frente a 85%). Una
ventaja adicional más de la sal particular de la presente invención es la facilidad de procesado del contra ión de ácido
paratoluensulfónico frente a por ejemplo el ácido bencenosulfónico (para la sal de besilato).
Así, se ha encontrado que debido a su extraordinaria combinación favorable de propiedades farmacéuticas, la sal
anhidra de paratoluensulfonato de 3-etil-5-[5-(4-etilpiperazin-1-ilsulfonil)-2-(2-metoxietoxi)piridin-3-il]-2-(piridin-2il)metil-2,6-dihidro-7H-pirazolo[4,3-d]pirimidin-7-ona es especialmente adecuada para la formulación farmacéutica
y es la forma preferida para la administración a humanos.
La expresión sal anhidra como se define en la presente memoria descriptiva significa que no hay agua unida dentro
de la estructura de celdilla del cristal, es decir, el compuesto no puede formar un hidrato cristalino. Para que no haya
dudas, se debe entender que el agua superficial, es decir el agua encontrada sobre la superficie de un cristal, no es agua
unida.
La preparación de la sal anhidra de paratoluensulfonato de 3-etil-5-[5-(4-etilpiperazin-1-ilsulfonil)-2-(2-metoxietoxi)piridin-3-il]-2-(piridin-2-il)metil-2,6-dihidro-7H-pirazolo[4,3-d]pirimidin-7-ona se puede llevar a cabo como se
ilustra en el ejemplo de aquí en adelante.
El compuesto de la invención, las sales farmacéuticamente aceptables, y los solvatos farmacéuticamente aceptables
de éstas se pueden administrar solos, pero en la terapia humana se administrarán generalmente meclados con un excipiente, un diluyente o un vehículo farmacéuticamente adecuado, seleccionado en relación con la vía de administración
pretendida y la práctica farmacéutica estándar.
Por ejemplo, el compuesto de la invención o las sales o los solvatos de éste, se puede administrar por vía oral, bucal
o sublingual en forma de comprimidos, cápsulas (incluyendo cápsulas blandas de gel), óvulos, elixires, soluciones
o suspensiones, que pueden contener agentes saborizantes o colorantes para aplicaciones de liberación inmediata,
retardada, modificada, sostenida, controlada o de administración pulsátil. El compuesto de la invención se puede
administrar también a través de inyección intracavernosa. El compuesto de la invención se puede administrar también
a través de formas farmacéuticas de dispersión rápida o de disolución rápida.
Dichos comprimidos pueden contener excipientes tales como la celulosa microcristalina, la lactosa, el citrato sódico, el carbonato cálcico, el fosfato cálcico dibásico y la glicina, desintegrantes tales como el almidón (preferentemente
almidón de maíz, patata o tapioca), el glicolato sódico de almidón, la croscarmelosa sódica y ciertos silicatos complejos, y aglutinantes de granulación tales como la polivinilpirrolidona, la hidroxipropilmetilcelulosa (HPMC), la
3
ES 2 219 395 T3
hidroxipropilcelulosa (HPC), la sacarosa, la gelatina y la goma arábiga. Adicionalmente, se pueden incluir lubricantes
tales como el estearato magnésico, el ácido esteárico, el behenato de glicerilo y el talco.
5
10
15
20
25
30
35
Se pueden emplear también composiciones sólidas de tipo similar como agentes de relleno en cápsulas de gelatina.
Los excipientes preferidos a este respecto incluyen lactosa, almidón, una celulosa, azúcar de leche o polietilenglicoles
de elevada masa molecular. Para las suspensiones acuosas y/o elixires, los compuestos de la invención se pueden
combinar con diferentes edulcorantes o saborizantes, materia colorante o tintes, con emulsionantes y/o suspendedores
y con diluyentes tales como el agua, el etanol, el propilenglicol y la glicerina, y combinaciones de éstos.
Las formas farmacéuticas de liberación modificada y de liberación pulsátil pueden contener excipientes como
los detallados para las formas farmacéuticas de liberación inmediata junto con excipientes adicionales que actúan
como modificadores de la tasa de liberación, estando éstos recubiertos y/o incluidos en el cuerpo del dispositivo. Los
modificadores de la tasa de liberación incluyen, pero no se limitan exclusivamente a la hidroxipropilmetilcelulosa, la
metilcelulosa, la carboximetilcelulosa de sodio, la etilcelulosa, el acetato de celulosa, el óxido de polietileno, la goma
Xantan, el carbómero, el copolímero de metacrilato de amonio, el aceite de ricino hidrogenado, la cera de carnauba,
la cera de parafina, el acetato ftalato de celulosa, el ftalato de hidroxipropilmetilcelulosa, el copolímero de ácido
metacrílico y las mezclas de éstos. Las formas farmacéuticas de liberación modificada y de liberación pulsátil pueden
contener uno o una combinación de excipientes modificadores de la tasa de liberación. Los excipientes modificadores
de la tasa de liberación pueden estar presentes tanto dentro de la forma farmacéutica, es decir, dentro de la matriz,
como sobre la forma farmacéutica, es decir, sobre la superficie o el recubrimiento.
Las formulaciones de dosis de rápida dispersión o disolución (siglas en inglés FDDF) pueden contener los siguientes componentes: aspartamo, acesulfamo de potasio, ácido cítrico, croscarmelosa sódica, crospovidona, ácido
diascórbico, acrilato de etilo, etilcelulosa, gelatina, hidroxipropilmetil celulosa, estearato magnésico, manitol, metacrilato de metilo, saborizante de menta, polietilenglicol, sílice ahumada, dióxido de silicio, glicolato sódico de almidón,
fumarato sódico de estearilo, sorbitol, xilitol.
El compuesto de la invención se puede administrar también por vía parenteral, por ejemplo, por vía intracavernosa,
intravenosa, intra-arterial, intraperitoneal, intratecal, intraventricular, intrauretral, intrasternal, intracraneal, intramuscular o subcutánea, o puede administrarse mediante técnicas de infusión. Para dicha administración parenteral, lo
mejor es usarlos en forma de una solución acuosa estéril que puede contener otras sustancias, por ejemplo, sales o
glucosa suficientes para hacer la solución isotónica con la sangre. Las soluciones acuosas se deberían tamponar adecuadamente (preferentemente hasta un pH que va desde 3 hasta 9), si es necesario. La preparación de formulaciones
parenterales adecuadas bajo condiciones estériles se lleva a cabo fácilmente mediante técnicas farmacéuticas estándar
bien conocidas por los expertos en la materia.
Para la administración oral y parenteral a pacientes humanos, la concentración diaria de dosis del compuesto de
la invención o de las sales o los solvatos de éste será normalmente desde 10 mg hasta 500 mg (en dosis únicas o
divididas).
40
45
50
55
60
65
Así, por ejemplo, los comprimidos o cápsulas del compuesto de la invención o las sales o los solvatos de éste
pueden contener desde 5 mg hasta 250 mg de compuesto activo para la administración de uno solo o dos o más al
mismo tiempo, como sea apropiado. En cualquier caso, el médico determinará la dosis real que será la más adecuada
para cada paciente por separado y que variará con la edad, el peso y la respuesta del paciente concreto. Las dosis
anteriores son ejemplares del caso medio. Por supuesto puede haber casos personales en los que se necesiten intervalos
de dosis superiores o inferiores y éstos están dentro del alcance de esta invención. La persona experta apreciará que,
en el tratamiento de ciertas dolencias (incluyendo DEM y DSF), el compuesto de la invención se puede tomar como
una única dosis según “lo que se requiera” (es decir, lo que se necesite o desee).
El compuesto de la invención se puede administrar también intranasalmente o mediante inhalación y se puede
administrar convenientemente en forma de un inhalador de polvo seco o una presentación de pulverizado de aerosol
desde un envase presurizado, bomba, pulverizador o nebulizador con el uso de un propulsante adecuado, por ejemplo
el diclorodifluorometano, el triclorofluorometano, el diclorotetrafluoroetano, un hidrofluoroalcano como tal el 1,1,1,2tetrafluoroetano (HFA 134A [marca registrada]), 1,1,1,2,3,3,3-heptafluoropropano (HFA 227EA [marca registrada]),
el dióxido de carbono u otro gas adecuado. En el caso de un aerosol presurizado, la dosis unitaria se puede determinar
proporcionando una válvula para administrar una cantidad medida. El envase presurizado, bomba, pulverizador o
nebulizador puede contener una solución o una suspensión del compuesto activo, por ejemplo, usando una mezcla de
etanol y el propulsante como el disolvente, que puede contener adicionalmente un lubricante, por ejemplo, el trioleato
de sorbitan. Las cápsulas y cartuchos (hechos, por ejemplo de gelatina) para uso en un inhalador o insuflador se pueden
formular para contener una mezcla de polvo del compuesto de la invención y una base de polvo adecuada tal como la
lactosa o el almidón.
Las formulaciones de aerosol o polvo seco se disponen de tal forma que cada dosis medida o “soplo” contiene
desde 1 mg hasta 50 mg de compuesto de la invención para administrar al paciente. La dosis diaria total administrada
con un aerosol estará dentro del intervalo que va desde 1 mg hasta 50 mg, que se puede administrar en una única dosis,
o más comúnmente, en dosis divididas a lo largo del día.
El compuesto de la invención se puede formular también para la administración mediante un atomizador. Las for4
ES 2 219 395 T3
mulaciones para los dispositivos atomizadores pueden contener los siguientes componentes como agentes solubilizantes, emulsionantes o suspendedores: agua, etanol, glicerol, propilenglicol, polietilenglicoles de baja masa molecular,
cloruro de sodio, fluorocarbonos, éteres de polietilenglicol, trioleato de sorbitan, ácido oleico.
10
Alternativamente, el compuesto de la invención o las sales o los solvatos de éste se pueden administrar en forma
de supositorio o pesario, o se pueden administrar tópicamente en forma de un gel, hidrogel, loción, solución, crema,
pomada o polvo pulverizado. El compuesto de la invención o las sales o los solvatos de éste se pueden administrar
también dérmicamente. El compuesto de la invención o las sales o los solvatos de éste se pueden administrar también
transdérmicamente, por ejemplo, mediante el uso de un parche de piel. El compuesto se puede administrar también
por las vías ocular, pulmonar o rectal.
15
Para uso oftálmico, el compuesto puede se formular como suspensiones micronizadas en solución salina estéril
isotónica, con pH ajustado, o, preferentemente, como soluciones en solución salina estéril isotónica, con pH ajustado, opcionalmente en combinación con un conservante tal como un cloruro de benzalconio. Alternativamente, el
compuesto se puede formular en una pomada como la vaselina.
5
20
25
30
Para la aplicación tópica en la piel, el compuesto de la invención o las sales o los solvatos de éste se pueden
formular como una pomada adecuada que contiene el compuesto activo suspendido o disuelto en, por ejemplo, una
mezcla con uno o más de los siguientes componentes: aceite mineral, vaselina líquida, vaselina blanca, propilenglicol,
compuesto de polioxietilen polioxipropileno, cera emulsionante y agua. De manera alternativa, se pueden formular
como una loción o una crema adecuada, suspendidos o disueltos en, por ejemplo, una mezcla de uno o más de los
siguientes componentes: aceite mineral, monostearato de sorbitan, un polietilenglicol, parafina líquida, polisorbato 60,
cera de cetilésteres, alcohol cetearílico, 2-octildodecanol, alcohol bencílico y agua.
El compuesto de la invención se puede usar también en combinación con una ciclodextrina. Se sabe que las ciclodextrinas forman complejos de inclusión y de no-inclusión con moléculas de fármacos. La formación de un complejo
fármaco-ciclodextrina puede modificar las propiedades de solubilidad, tasa de disolución, biodisponibilidad y/o estabilidad de una molécula de fármaco. Los complejos de fármaco-ciclodextrina son útiles generalmente para la mayoría
de las formas farmacéuticas y vías de administración. Como alternativa a la acomplejación directa con el fármaco,
la ciclodextrina se puede usar como un aditivo auxiliar, por ejemplo, como vehículo, diluyente o solubilizante. Las
alfa-, beta-, y gamma-ciclodextrinas son las que se usan más comúnmente y se describen ejemplos adecuados en los
documentos WO-A-91/11172, WO-A-94/02518 y WO-A-98/55148.
Formulación del comprimido ejemplo
35
En general, una formulación del comprimido de la sal de p-tosilato podría contener típicamente entre aproximadamente 5 mg y 400 mg de compuesto activo mientras que los pesos del relleno del comprimido pueden variar desde 50
mg hasta 1000 mg. Se ilustra una formulación ejemplo para un comprimido de 25 mg:
40
Componente
% peso/peso
45
Sal de p-tosilato
Celulosa microcristalina
Fosfato cálcico dibásico, anhidro
Glicolato sódico de almidón
Estearato de magnesio
* Cantidad ajustada según la actividad del fármaco.
32,383*
42,117
21,000
3,000
1,500
50
55
60
65
Los comprimidos se fabricarán mediante un procedimiento estándar, por ejemplo, compresión directa o un procedimiento de granulación seca. Los núcleos de los comprimidos pueden recubrirse con un recubrimiento apropiado.
Generalmente, en humanos, la administración por vía oral del compuesto de la invención es la vía preferida, siendo
la más conveniente y, por ejemplo en DEM, evitando las desventajas bien conocidas asociadas con la administración
intracavernosa (i.c.). Un régimen de dosificación oral preferido en DEM para un hombre típico va desde 25 mg hasta
250 mg de compuesto cuando se requiere. En circunstancias en las que la persona que lo recibe padece una alteración
que le afecta a tragar o una disfunción de la absorción del fármaco después de la administración por vía oral, el fármaco
se puede administrar parenteralmente, sublingualmente o bucalmente.
Para uso veterinario, el compuesto de la presente invención se administra como una formulación adecuadamente
aceptable según la práctica veterinaria normal y el cirujano veterinario determinará el régimen de dosificación y la vía
de administración que será la más apropiada para un animal concreto.
Así, la invención proporciona una composición farmacéutica que comprende el compuesto de la presente invención, o un solvato o un profármaco farmacéuticamente aceptable de éste junto con un diluyente o un vehículo farmacéuticamente aceptable.
5
ES 2 219 395 T3
Adicionalmente proporciona una formulación veterinaria que comprende el compuesto de la presente invención, o
un solvato o un profármaco veterinariamente aceptable de éste, junto con un diluyente o un vehículo veterinariamente
aceptable.
5
La invención también proporciona el compuesto de la presente invención, o un solvato o un profármaco farmacéuticamente aceptable de éste, o una composición farmacéutica que contiene cualquiera de los precedentes, para uso
como medicamento humano.
10
Además, proporciona el compuesto de la presente invención, o un solvato o un profármaco veterinariamente aceptable, o una formulación veterinaria que contiene cualquiera de los precedentes, para su uso como medicamento
animal.
15
En otro aspecto más, la invención proporciona el uso de un compuesto de la presente invención, o un solvato o un
profármaco farmacéuticamente aceptable de éste, para la fabricación de un medicamento humano para el tratamiento
curativo o profiláctico de una dolencia médica para la que está indicado un inhibidor de la cGMP PDE5. Se proporciona
adicionalmente el uso del compuesto de la presente invención, solvato o profármaco de éste, en la fabricación de un
medicamento para el tratamiento de una dolencia médica en la que es deseable la inhibición de una cGMP PDE5.
20
También proporciona el uso del compuesto de la presente invención, o un solvato o un profármaco veterinariamente
aceptable de éste, para la fabricación de un medicamento animal para el tratamiento curativo o profiláctico de una
dolencia médica para la que está indicada un inhibidor de cGMP PDE5.
25
30
35
Además, la invención proporciona el uso del compuesto de la presente invención, o un solvato o un profármaco
farmacéuticamente aceptable de éste, para la fabricación de un medicamento humano para el tratamiento curativo
o profiláctico de las disfunciones sexuales de mamíferos, la disfunción eréctil masculina (DEM), la impotencia, la
disfunción sexual femenina (DSF), la disfunción de clítoris, la alteración del deseo sexual hipoactivo femenino, la
alteración de la excitación sexual femenina, la alteración de dolor sexual femenino, la disfunción orgásmica sexual
femenina (DOSF), la disfunción sexual debida a daños en la médula espinal, la disfunción sexual inducida por el inhibidor de la recaptación de serotonina selectivo (IRSS), el parto prematuro, la dismenorrea, la hiperplasia prostática
benigna (HPB), la obstrucción de la salida de la vejiga, la incontinencia, la angina de Prinzmetal estable, inestable y
variante, la hipertensión, la hipertensión pulmonar, la enfermedad pulmonar obstructiva crónica, la arteropatía coronaria, la insuficiencia cardiaca congestiva, la aterosclerosis, las dolencias de fluidez de los vasos sanguíneos reducida,
la enfermedad vascular periférica, el derrame cerebral, la tolerancia inducida al nitrato, la bronquitis, el asma alérgico,
el asma crónico, la rinitis alérgica, las enfermedades y dolencias del ojo, las enfermedades caracterizadas por las alteraciones de la motilidad del intestino, la preclampsia, el síndrome de Kawasaki, la tolerancia a nitrato, la esclerosis
múltiple, la nefropatía diabética, la neuropatía diabética periférica, la enfermedad de Alzheimer, el fallo respiratorio
agudo, la soriasis, la necrosis de la piel, el cáncer, la metástasis, la calvicie, el esófago en cascanueces, la fisura anal,
las hemorroides, la vasoconstricción hipóxica o la estabilización de la presión sanguínea durante la hemodiálisis. Las
dolencias particularmente preferidas incluyen DEM y DSF.
40
45
50
55
60
65
También proporciona el uso del compuesto de la presente invención, o un solvato o un profármaco veterinariamente aceptable de éste, para la fabricación de un medicamento animal para el tratamiento curativo o profiláctico
de disfunciones sexuales de mamíferos, disfunción eréctil masculina (DEM), impotencia, disfunción sexual femenina
(DSF), disfunción de clítoris, alteración del deseo sexual hipoactivo femenino, alteración de la excitación sexual femenina, alteración de dolor sexual femenino o disfunción orgásmica sexual femenina (DOSF), disfunción sexual debida
a daños en la médula espinal, parto prematuro, dismenorrea, hiperplasia prostática benigna (HPB), obstrucción de la
salida de la vejiga, incontinencia, angina de Prinzmetal estable, inestable y variante, hipertensión, hipertensión pulmonar, enfermedad pulmonar obstructiva crónica, arteropatía coronaria, insuficiencia cardiaca congestiva, aterosclerosis,
dolencias de fluidez de los vasos sanguíneos reducida, por ejemplo angioplastia coronaria transluminal post-percutánea (siglas en inglés post-PTCA), enfermedad vascular periférica, derrame cerebral, tolerancia inducida a nitrato,
bronquitis, asma alérgico, asma crónico, rinitis alérgica, glaucoma, enfermedades caracterizadas por alteraciones de
la movilidad del intestino, por ejemplo el síndrome del colon irritable (SCI), preclampsia, síndrome de Kawasaki, tolerancia a nitrato, esclerosis múltiple, nefropatía diabética, neuropatía diabética periférica, enfermedad de Alzheimer,
fallo respiratorio agudo, soriasis, necrosis de piel, cáncer, metástasis, calvicie, esófago en cascanueces, fisura anal,
hemorroides o vasoconstricción hipóxica. Las dolencias particularmente preferidas incluyen DEM y DSF.
Además, la invención proporciona un procedimiento de tratamiento o prevención de una dolencia médica para la
que está indicado un inhibidor de la cGMP PDE5, en un mamífero (incluyendo un humano), que comprende administrar a dicho mamífero una cantidad terapéuticamente efectiva del compuesto de la presente invención, o un solvato o un
profármaco farmacéuticamente o veterinariamente aceptable de éste, o una composición farmacéutica o formulación
veterinaria que contiene cualquiera de los precedentes.
Aún adicionalmente, la invención proporciona un procedimiento de tratamiento o prevención de disfunciones sexuales de mamíferos, disfunción eréctil masculina (DEM), impotencia, disfunción sexual femenina (DSF), disfunción
de clítoris, alteración del deseo sexual hipoactivo femenino, alteración de la excitación sexual femenina, alteración
de dolor sexual femenino, disfunción orgásmica sexual femenina (DOSF), disfunción sexual debida a daños en la
médula espinal, parto prematuro, dismenorrea, hiperplasia prostática benigna (HPB), obstrucción de la salida de la
vejiga, incontinencia, angina de Prinzmetal estable, inestable y variante, hipertensión, hipertensión pulmonar, enfer6
ES 2 219 395 T3
5
10
15
20
25
30
medad pulmonar obstructiva crónica, arteropatía coronaria, insuficiencia cardiaca congestiva, aterosclerosis, dolencias
de fluidez de los vasos sanguíneos reducida, por ejemplo angioplastia coronaria transluminal post-percutánea (siglas en
inglés post-PTCA), enfermedad vascular periférica, derrame cerebral, tolerancia inducida a nitrato, bronquitis, asma
alérgico, asma crónico, rinitis alérgica, glaucoma, enfermedades caracterizadas por alteraciones de la movilidad del
intestino, por ejemplo el síndrome del colon irritable (SCI), preclampsia, síndrome de Kawasaki, tolerancia a nitrato,
esclerosis múltiple, nefropatía diabética, neuropatía diabética periférica, enfermedad de Alzheimer, fallo respiratorio
agudo, soriasis, necrosis de piel, cáncer, metástasis, calvicie, esófago en cascanueces, fisura anal, hemorroides o vasoconstricción hipóxica en un mamífero (incluyendo un humano), que comprende administrar a dicho mamífero una
cantidad terapéuticamente efectiva del compuesto de la presente invención, o un solvato o un profármaco farmacéuticamente o veterinariamente aceptable de éste, o una composición farmacéutica o formulación veterinaria que contiene
cualquiera de los precedentes.
En una aspecto adicional más de la presente invención, se proporciona una combinación del compuesto de la
presente invención con compuestos adicionales útiles en la inhibición de la fosfodiesterasa de tipo 5 en el que dicha
combinación es útil para el tratamiento o prevención de disfunciones sexuales de mamíferos como la disfunción eréctil
masculina (DEM), impotencia, disfunción sexual femenina (DSF), disfunción de clítoris, alteración del deseo sexual
hipoactivo femenino, alteración de la excitación sexual femenina, alteración de dolor sexual femenino, disfunción
orgásmica sexual femenina (DOSF), disfunción sexual debida a daños en la médula espinal, parto prematuro, dismenorrea, hiperplasia prostática benigna (HPB), obstrucción de la salida de la vejiga, incontinencia, angina de Prinzmetal
estable, inestable y variante, hipertensión, hipertensión pulmonar, enfermedad pulmonar obstructiva crónica, arteropatía coronaria, insuficiencia cardiaca congestiva, aterosclerosis, dolencias de fluidez de los vasos sanguíneos reducida,
por ejemplo angioplastia coronaria transluminal post-percutánea (siglas en inglés post-PTCA), enfermedad vascular
periférica, derrame cerebral, tolerancia inducida a nitrato, bronquitis, asma alérgico, asma crónico, rinitis alérgica,
glaucoma, enfermedades caracterizadas por alteraciones de la movilidad del intestino, por ejemplo el síndrome del colon irritable (SCI), preclampsia, síndrome de Kawasaki, tolerancia a nitrato, esclerosis múltiple, nefropatía diabética,
neuropatía diabética periférica, enfermedad de Alzheimer, fallo respiratorio agudo, soriasis, necrosis de piel, cáncer,
metástasis, calvicie, esófago en cascanueces, fisura anal, hemorroides o vasoconstricción hipóxica en un mamífero
(incluyendo un ser humano).
Ejemplo 1
4-{6-Etoxi-5-[3-etil-6,7-dihidro-7-oxo-2-(2-piridilmetil)-2H-pirazolo[4,3-d]pirimidin-5-il]-3-piridilsulfonil}-1-etilpiperazin paratoluensulfonato también denominado como (sal de paratoluensulfonato de 3-etil-5-[5-(4-etilpiperazin-1ilsulfonil)-2-(2-metoxietoxi)piridin-3-il]-2-(piridin-2-il)metil-2,6-dihidro-7H-pirazolo[4,3-d]pirimidin-7-ona)
35
40
45
50
Se calentó 4-{6-Etoxi-5-[3-etil-6,7-dihidro-7-oxo-2-(2-piridilmetil)-2H-pirazolo[4,3-d]pirimidin-5-il]-3-piridilsulfonil}-1-etilpiperazina (100 g, 0,16 mol) en 2-butanona/ agua 6% (600 mL) hasta 50ºC produciendo una solución amarillo pálido. La mezcla de reacción se calentó hasta la destilación azeotrópica de 160 ml de disolvente para
eliminar agua. La solución destilada se calentó hasta 60ºC y se añadió la solución ácida de ácido p-toluenosulfónico
monohidrato (31,11 g, 0,16 mol) disuelta en 2-butanona (140 mL) durante una hora. Durante la adición del ácido se
observó cristalización lenta. La mezcla de reacción se enfrió lentamente desde 60ºC hasta 0-5ºC durante tres horas, y
se agitó durante una hora más, después se filtró el sólido, se lavó con 2-butanona (200 mL) y se secó a vacío a 50ºC
para dar el compuesto del título (116 g, 93,7%) como un sólido blanco. Punto de fusión 240ºC. (DMSOd6 ): 1,20 (6H,
m), 2,23 (3H, st), 2,75 (2H, m), 2,97 (2H, c), 3,18 (4H, m), 3,35 (3H, s), 3,55 (2H, m), 3,68 (2H, m), 3,83 (2H, m),
4,59 (2H, m), 5,70 (2H, s), 7,08 (2H, d), 7,20 (1H, d), 7,34 (1H, m), 7,43 (2H, d), 7,80 (1H, m), 8,35 (1H, d), 8,70 (1H,
d), 8,74 (1H, d). Los datos de cristalografía de rayos X simple demostraron que la estructura propuesta era correcta.
El punto de fusión dio +/- 5ºC, preferentemente +/- 2ºC.
Los experimentos han mostrado que bajo ciertas condiciones se pueden detectar al menos dos polimorfos adicionales de (I) mediante calorimetría de escaneo diferencial (siglas en inglés DSC). Experimentos adicionales han mostrado
que resuspendiéndolos en una mezcla de metiletil cetona y agua, estos polimorfos se pueden convertir en el compuesto
del Ejemplo 1.
Preparación 1
55
4-amino-5-etil-1-(2-piridilmetil)-1H-pirazol-3-carboxamida
(1a) Etil 3-etil-1H-pirazol-5-carboxilato
60
65
Se añadió solución de etóxido de sodio etanólico (21% peso/peso; 143 ml, 0,39 mol) gota a gota a una solución de
oxalato de dietilo (59,8 ml, 0,44 mol) agitada y enfriada con hielo, en etanol absoluto (200 ml) bajo nitrógeno, y se
agitó la solución resultante durante 15 minutos. Se añadió después butan-2-ona (39 ml, 0,44 mol) gota a gota, se retiró
el baño de enfriamiento, se agitó la mezcla de reacción durante 18 horas a temperatura ambiente y después durante 6
horas a 40ºC, después se reintrodujo el baño de enfriamiento. A continuación, se añadió ácido acético glacial (25 ml,
0,44 mol) gota a gota, la solución resultante se agitó durante 30 minutos a 0ºC, se añadió hidrato de hidracina (20 ml,
0,44 mol) gota a gota, después se dejó calentar la mezcla de reacción hasta temperatura ambiente y se mantuvo así
durante un periodo de 18 horas, antes de ser evaporada bajo presión reducida. El residuo se dividió en presencia de
diclorometano (300 ml) y agua (100 ml), después se separó la fase orgánica, se lavó con agua (2 x 100 ml), se secó
7
ES 2 219 395 T3
(Na2 SO4 ) y se concentró bajo presión reducida para dar el compuesto del título (66,0 g). δ (CDCl3 ): 1,04 (3H, t), 1,16
(3H, t), 2,70(2H, c), 4,36 (2H, c), 6,60 (1H, s). LRMS: m/z 169 (M+1)+ .
5
10
(1b) Ácido 3-etil-1H-pirazol-5-carboxílico
Se añadió una solución acuosa de hidróxido de sodio (10 M; 100 ml, 1,0 mol) gota a gota a una suspensión agitada
del compuesto del título del ejemplo (4a) (66,0 g, 0,39 mol) en metanol, y la solución resultante se calentó a reflujo
durante 4 horas. La mezcla de reacción fría se concentró bajo presión reducida hasta aproximadamente 200 ml, se
diluyó con agua (200 ml) y esta mezcla se lavó con tolueno (3 x 100 ml). La fase acuosa resultante se acidificó con
ácido clorhídrico concentrado hasta pH 4 y el precipitado blanco se recogió y se secó por succión para proporcionar
el compuesto del título (34,1 g). δ (DMSOd6 ): 1,13 (3H, t), 2,56 (2H, c), 6,42 (1H, s).
(1c) Ácido 3-etil-4-nitro-1H-pirazol-5-carboxílico
15
20
Se añadió ácido sulfúrico fumante (17,8 ml) gota a gota a ácido nítrico fumante agitado, enfriado en hielo (16,0
ml), la solución resultante se calentó hasta 50ºC, se añadió ácido 3-etil-1H-pirazol-5-carboxílico en porciones durante
30 minutos mientras se mantenía la temperatura de reacción por debajo de 60ºC. La solución resultante se calentó
durante 18 horas a 60ºC, se dejó enfriar, después se vertió en hielo. El compuesto del título se obtuvo como un sólido
marrón (64%). δ (DMSOd6 ): 1,18 (3H, t), 2,84 (2H, m), 13,72 (1H, s).
(1d) 3-Etil-4-nitro-1H-pirazol-5-carboxamida
25
30
Se calentó una solución del compuesto del título del ejemplo (4c) (15,4 g; 0,077 mol) en cloruro de tionilo (75
ml) a reflujo durante 3 horas y después la mezcla de reacción fría se evaporó bajo presión reducida. El residuo se
azeotropó con tetrahidrofurano (2 x 50 ml) y se suspendió posteriormente en tetrahidrofurano (50 ml), después la
suspensión agitada se enfrió con hielo y se trató con amoniaco gaseoso durante 1 hora. Se añadió agua (50 ml) y la
mezcla resultante se evaporó bajo presión reducida para dar un sólido que, después de la trituración con agua y el
secado por succión, proporcionó el compuesto del título como un sólido blanco (90%). δ (DMSOd6 ): 1,17 (3H, t), 2,87
(2H, m), 7,40 (1H, s), 7,60 (1H, s), 7,90 (1H, s). LRMS: m/z 185 (M+1)+ .
(1e) 5-Etil-4-nitro-1-(2-piridilmetil)-1H-pirazol-3-carboxamida
35
40
45
50
55
Se añadió carbonato de cesio (1,414 kg, 4,34 mol) a una suspensión del compuesto del título del ejemplo (4d)
(800 g, 4,34 mol) en acetonitrilo (5 l) y la mezcla se calentó hasta 60ºC. Se añadió 2-clorometilpiridina (664,7 g,
5,23 mol) y se calentó la reacción a 70ºC durante 7 horas, después se añadió agua (9,5 l) y la mezcla de reacción
se enfrió hasta 10ºC. La granulación de esta mezcla dio un precipitado que se filtró y se secó para proporcionar 3etil-4-nitro-1-(piridin-2-il)metil-pirazol-5-carboxamida (367 g). Se añadió cloruro de sodio (1,58 kg) al filtrado y la
solución se extrajo con acetato de etilo (4 x 1,75l). Los extractos orgánicos combinados se destilaron para eliminar
aproximadamente 10 l de disolvente, se añadió tolueno (5,6 l) durante 35 minutos a la solución caliente (69-76ºC) y
la mezcla se dejó enfriar. La suspensión resultante se granuló a <10ºC durante 30 minutos, se filtró, el sólido se lavó
con acetato de etilo:tolueno (50:50) (600 ml) y se secó (60ºC) para dar el compuesto del título (624 g, 52%) como un
sólido marrón claro. δ (DMSOd6 ): 1,08 (3H, t), 3,02 (2H, c), 5,53 (2H, s), 7,34 (2H, m), 7,65 (1H, s), 7,82 (1H, m),
7,93 (1H, s), 8,52 (1H, d). LRMS: m/z 276 (M+1)+ .
(1f) 4-Amino-5-etil-1-(2-piridilmetil)-1H-pirazol-3-carboxamida.
Una mezcla de catalizador Lindlar (2g) y el compuesto del título del ejemplo (4e) (20 g, 72,7 mmol) en etanol (160
ml) se hidrógeno durante 48 horas a 345 kPa (3,51 kg/cm2 ) y 50ºC, después se enfrió y se filtró. El filtrado se combinó
con un lavado con IMS (50 ml) de la almohadilla del filtro y se concentró bajo presión reducida a una columna de
100 ml. El etanol restante se eliminó por destilación, y se sustituyó por acetato de etilo hasta que se había alcanzado
una temperatura tope de 77ºC. La mezcla enfriada se granuló a 4ºC, se filtró y se secó para dar el compuesto del título
(13,17 g, 73%) como un sólido marrón claro. δ (DMSOd6 ): 0,90 (3H, t), 2,54 (2H, c), 4,48 (2H, s), 5,31 (2H, s), 6,89
(1H, d), 6,95 (1H, s), 7,11 (1H, s), 7,28 (1H, m), 7,74 (1H, m), 8,50 (1H, d). LRMS: m/z 246 (M+1)+ .
Preparación 2
N-[3-carbamoil-5-etil-1-(2-piridilmetil)-1H-pirazol-4-il]-2-etoxi-5-(4-etil-1-piperazinilsulfonil)nicotinamida
60
65
Se cargo el ácido 2-etoxi-5-(4-etil-1-piperazinilsulfonil)nicotínico (0,875 Kg, 2,55 mol) seguido por acetato de
etilo (7 L, 8 ml/g) en el recipiente de reacción y se destilaron 2 ml/g a presión atmosférica para asegurar que el
sistema de reacción estuviera seco. La pasta se enfrió hasta temperatura ambiente bajo una atmósfera de nitrógeno
y se añadió carbonildiimidazol (0,43 Kg, 2,65 mol) en una porción. La pasta se calentó hasta 35ºC y se mantuvo
durante media hora. La reacción se calentó adicionalmente hasta 45-50ºC y se mantuvo durante media hora más.
La reacción se calentó después a reflujo, agitando a reflujo durante una hora. Para ratificar la completa formación
de imidazolida, la reacción se enfrió hasta 45-50ºC bajo nitrógeno y se cargó 4-amino-5-etil-1-(2-piridilmetil)-1Hpirazol-3-carboxamida (0,59 Kg, 2,42 mol) en una porción antes de volver al reflujo, y se destiló 1 ml/g adicional
a presión atmosférica. La reacción se agitó a reflujo durante 16 horas teniendo lugar la cristalización del producto
8
ES 2 219 395 T3
5
después de 4 horas. La reacción se enfrió hasta 10-15ºC y se granuló durante una hora. La pasta de la reacción se
filtró y se lavó (acetato de etilo), que se secó a vacío a 50ºC para dar el compuesto del título (1,252 Kg, 90,7%) como
un sólido blanco. Punto de fusión 178-179ºC. ? (CDCl3): 1,04 (3H, t), 1,06 (3H, t), 1,59 (3H, t), 2,40 (2H, c), 2,50
(4H, c), 2,90 (4H, c), 3,08 (4H, m), 2,78 (2H, c), 5,35 (1H, s), 5,48 (2H, s), 6,68 (1H, s), 6,92 (1H, d), 7,22 (1H, m),
7,65 (1H, m), 8,58 (1H, d), 8,64 (1H, d), 8,83 (1H, d). m/z (Encontrado: 571 [M+H]+ , 100%. C26 H34 N8 O5 S requiere
571,67).
Preparación 3
10
15
20
25
30
4-{6-Etoxi-5-[3-etil-6,7-dihidro-7-oxo-2-(2-piridilmetil)-2H-pirazolo[4,3-d]pirimidin-5-il]-3-piridilsulfonil}-1-etilpiperazina
Se cargó una solución de etóxido de potasio (86 g, 0,25 mol, 24% peso/peso en etanol) en el recipiente. A esto
se le añadió etanol (235 mL). Se añadió acetato de etilo (10,8 g) a la mezcla de reacción a temperatura ambiente.
Se añadió N-[3-carbamoil-5-etil-1-(2-piridilmetil)-1H-pirazol-4-il]-2-etoxi-5-(4-etil-1-piperazinilsulfonil)nicotinamida (70 g, 0,122 mol) en una porción a la mezcla de disolvente y la reacción se agitó a temperatura ambiente. La mezcla
de reacción se calentó en un recipiente presurizado hasta una temperatura de 120ºC, produciendo una presión interna
de aproximadamente 3,51-4,22 Kg/cm2 , después la presión se llevó hasta 5,62 Kg/cm2 aplicando una presión de nitrógeno y la reacción se agitó durante 8 horas. La reacción se enfrió después de 8 horas y después se reduce etanol bajo
destilación atmosférica hasta un volumen de aproximadamente 720 ml (3 ml/g). Se añadió acetato de etilo (840 ml)
a la solución de etanol y después se redujo bajo destilación atmosférica hasta un volumen de aproximadamente 1920
ml (8 ml/g). Se añadió una solución acuosa diluida de ácido clorhídrico para ajustar el pH de la mezcla de reacción
desde aproximadamente pH=13 hasta pH=8. Fue una adición lenta durante 30 min sin mostrar exotermia. La mezcla
se agitó durante 5 minutos, se calentó hasta 50ºC y se separaron las fases. La acuosa se fue al efluente. Se añadió agua
(140 mL) a la fase de acetato de etilo (que permanecía en el recipiente), se agitó, se calentó hasta 50ºC y se separaron
las fases. La acuosa se fue al efluente. La fase de acetato de etilo se enfrió lentamente desde 50ºC hasta 0-5ºC durante
dos horas, y se agitó durante una hora más, se filtró el sólido, se lavó con acetato de etilo (3) 0-5ºC y se secó a vacío a
60ºC para dar el compuesto del título (83%) como un sólido blanco. Punto de fusión 178-180ºC. (CDCl3 ): 1,02 (3H,
t), 1,30 (3H, t), 1,58 (3H, t), 2,21 (2H, c), 2,55 (4H, m), 3,04 (2H, c), 3,10 (4H, m), 4,75 (2H, c), 5,69 (2H, s), 7,10
(1H, d), 7,22 (1H, m), 7,63 (1H, m), 8,57 (1H, d), 8,63 (1H, d), 9,02 (1H, d). m/z (Encontrado: 553 [M+H]+ , 100%.
C26 H32 N8 O4 S requiere 553,66).
Preparación 4
35
(a) Ácido 2-hidroxi-5-sulfonicotínico
40
Se añadió ácido 2-hidroxinicotínico (27 Kg, 194,2 mol) en porciones a oleum 30% (58,1 Kg) a 50ºC durante 1
hora. Esto produjo una exotermia hasta 82ºC. La mezcla de reacción se calentó adicionalmente hasta 140ºC. Después
de mantener esta temperatura durante 12 horas, los contenidos del reactor se enfriaron hasta 15ºC y se filtraron. La
torta de filtrado se resuspendió con acetona (33 Kg) a temperatura ambiente, se filtró y se secó para dar el compuesto
del título (35,3 Kg, 83%) como un sólido blanco. Punto de descomposición 273ºC. δ (DMSOd6 ): 7,93 (1H, d), 8,42
(1H, d). m/z (Encontrado: 220 [M+H]+ , 100%. C6 H6 NO6 S requiere 220,17).
45
50
55
60
(b) Etil 2-hidroxi-5-sulfonicotinoato
Se disolvió ácido 2-hidroxi-5-sulfonicotínico (500 g, 2,28 mol) en etanol (2,5 l) con agitación y se calentó hasta
80ºC. Después de 30 min se destilaron 0,5l de disolvente, después se sustituyó por etanol nuevo (0,5 l) y se volvió a
llevar a 80ºC. Después de 60 min más, se destiló 1,0 l de disolvente, después se sustituyó por etanol nuevo (1,0 l) y
se volvió a llevar a 80ºC. Después de 60 min más, se destiló 1 l de disolvente, se enfrió la reacción hasta 22ºC y se
agitó durante 16 horas. El producto precipitado se filtró, se lavó con etanol (0,5 l) y se secó a 50ºC a vacío para dar el
compuesto del título (416 g, 74%) como un sólido blanco. Punto de descomposición 237ºC. δ (DMSOd6 ): 1,25 (3H,
t), 4,19 (2H, c), 7,66 (1H, d), 8,13 (1H, d). m/z (Encontrado: 248 [M+H]+ , 100%. C8 H10 NO6 S requiere 248,22).
(c) Etil 2-cloro-5-clorosulfonicotinoato
Se suspendió 2-hidroxi-5-sulfonicotinoato de etilo (24,7 g, 0,1 mol) en cloruro de tionilo (238 g, 2,0 mol) y
dimetilformamida (1,0 mL) con agitación. La mezcla de reacción se calentó después a reflujo durante 2,5 horas. La
mayor parte del cloruro de tionilo se eliminó a vacío quitando el cloruro de tionilo residual con un azeótropo de tolueno
para dar el compuesto del título crudo (30,7 g, 108%) como un aceite amarillo. δ (CDCl3 ): 1,46 (3H, t), 4,50 (2H, c),
8,72 (1H, d), 9,09 (1H, d). Esto se llevó directamente a la siguiente etapa.
(d) Etil 2-cloro-5-(4-etil-1-piperazinilsulfonil)nicotinoato
65
Se disolvió 2-cloro-5-clorosulfonicotinoato de etilo crudo (30,7 g, 0,1 mol asumidos) en acetato de etilo (150
mL) con agitación, después se enfrió en hielo. A esto, se le añadió una solución de N-etilpiperazina (11,4 g, 0,1
mol) y trietilamina (22,5 g, 0,22 mol) en acetato de etilo (50 mL), cuidadosamente durante 30 min, manteniendo la
temperatura interna por debajo de 10ºC. Una vez que se completó la adición, la reacción se dejó calentar hasta 22ºC
9
ES 2 219 395 T3
y se agitó durante 1 hora. El sólido se filtró y el filtrado restante se concentró a vacío para dar el compuesto del título
crudo (37,1 g, 103%) como una goma amarilla. δ (CDCl3 ): 1,10 (3H, t), 1,42 (3H, m), 2,50 (2H, m), 2,60 (4H, m),
4,43 (2H, c), 8,40 (1H, d), 8,80 (1H, d). m/z (Encontrado: 362 [M+H]+ , 100%. C14 H21 ClN3 O4 S requiere 362,85).
5
10
15
(e) Etil 2-etoxi-5-(4-etil-1-piperazinilsulfonil)nicotinoato
Una solución de 2-cloro-5-(4-etil-1-piperazinilsulfonil)nicotinoato de etilo (36,1 g, 0,1 mol) en etanol (180 mL) se
enfrió hasta 10ºC con agitación. Se añadió etóxido de sodio (10,2 g, 0,15 mol) en porciones manteniendo la temperatura
por debajo de 20ºC. La mezcla de reacción se agitó después a temperatura ambiente durante 18 horas. El precipitado
se filtró y se añadió agua (180 mL) al filtrado. El filtrado se calentó después hasta 40ºC durante 1 hora. Se destiló
después etanol (180 mL) a temperatura ambiente y la solución acuosa que quedó se dejó enfriar hasta temperatura
ambiente. El producto precipitado se filtró después, se lavó con agua y se secó a vacío a 50ºC para dar el compuesto
del título (12,6 g, 34%) como un sólido marrón claro. Punto de fusión 66-68ºC. δ (CDCl3 ): 1,04 (3H, t), 1,39 (3H,
t), 1,45 (3H, t), 2,41 (2H, c), 2,52 (4H, m), 3,08 (4H, m), 4,38 (2H, c), 2,57 (2H, c), 8,38 (1H, d), 8,61 (1H, d). m/z
(Encontrado:372 [M+H]+ , 100%. C16 H26 N3 O5 S requiere 372,46).
(f) Ácido 2-etoxi-5-(4-etil-1-piperazinilsulfonil)nicotínico
20
25
Se disolvió etil 2-etoxi-5-(4-etil-1-piperazinilsulfonil)nicotinoato (10,2 g, 0,0275 mol) en tolueno (50 mL) y se
añadió una solución de hidróxido de sodio (1,1 g, 0,0275 mol) en agua (20 mL). Esta mezcla de dos fases se agitó
después vigorosamente a temperatura ambiente durante la noche. La fase acuosa se separó y se ajustó hasta pH=5,6
mediante la adición de ácido clorhídrico concentrado. El producto precipitado se suspendió con enfriamiento con hielo
durante 15 minutos, se filtró, se lavó con agua y se secó a vacío a 50ºC para dar el compuesto del título (4,1 g, 43%)
como un sólido blancuzco. Punto de fusión 206-207ºC. δ (CDCl3 ): 1,25 (3H, t), 1,39 (3H, t), 2,82 (2H, c), 3,03 (4H,
m), 3,25 (4H, m), 4,50 (2H, c), 8,25 (1H, d), 8,56 (1H, d). m/z (Encontrado:344 [M+H]+, 100%. C14 H22 N3 O5 S requiere
344,38).
30
35
40
45
50
55
60
65
10
ES 2 219 395 T3
REIVINDICACIONES
5
1. Una sal anhidra del ácido 3-etil-5-[5-(4-etilpiperazin-1-ilsulfonil)-2-(2-metoxietoxi)piridin-3-il]-2-(piridin-2-il)
metil-2,6-dihidro-7H-pirazolo [4,3-d]pirimidin-7-ona paratoluensulfónico con la fórmula (I):
10
15
20
25
2. Una sal según la reivindicación 1 que tiene un punto de fusión de 240ºC ± 5ºC.
30
3. Una composición farmacéutica que contiene una sal según las reivindicaciones 1 ó 2 junto con un diluyente o
un vehículo farmacéuticamente aceptable.
4. Una composición farmacéutica según la reivindicación 3 que se adapta para tratamiento oral.
35
40
5. Un procedimiento para la preparación de una sal según la reivindicación 1 a partir de 4-{6-etoxi-5-[3-etil6,7-dihidro-7-oxo-2-(2-piridilmetil)-2H-pirazolo [4,3-d]pirimidin-5-il]-3-piridilsulfonil}-1-etilpiperazina mediante el
tratamiento con ácido paratoluensulfónico monohidrato.
6. Un procedimiento según la reivindicación 5 en el que la proporción de equivalentes molares entre 4-{6-etoxi5-[3-etil-6,7-dihidro-7-oxo-2-(2-piridilmetil)-2H-pirazolo [4,3-d]pirimidin-5-il]-3-piridilsulfonil}-1-etilpiperazina y
ácido paratoluensulfónico es aproximadamente 1:1.
45
50
55
60
65
11