194 estudio del efecto combinado de carbono e hidrógeno en una
Anuncio

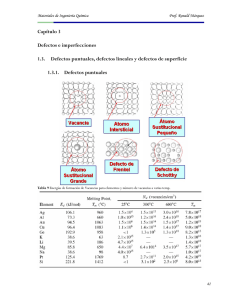
JORNADAS SAM/ CONAMET/ SIMPOSIO MATERIA 2003 04-07 ESTUDIO DEL EFECTO COMBINADO DE CARBONO E HIDRÓGENO EN UNA DISLOCACIÓN DE BORDE S. Simonetti, E. Pronsato, G. Brizuela, L. Moro, A. Juan Departamento de Física, Universidad Nacional del Sur, Bahía Blanca, Argentina. [email protected] En el presente trabajo se estudia la variación de energía debida a la influencia del carbono y del hidrógeno ubicados en la red de Fe BCC próximos a una dislocación a/2 [1Î1] con plano de deslizamiento (110) . La dislocación propuesta se modeló en un racimo de 85 átomos. Los planos siguen una secuencia ABABAB..... y la mínima distancia entre dos átomos de 2.48 Å. Si bien en los cristales existe un gran número de dislocaciones interconectadas entre sí, para realizar el estudio se simuló la estructura del cristal BCC con una única dislocación. Se utilizó el método semiempíricos de orbitales moleculares ASED ( Atomic Superposition and Electrón Delocalization ), que permite obtener predicciones acerca de las estructuras moleculares partiendo de datos atómicos. Se calculó primeramente trayectorias de menor energía para la inclusión de un átomo de carbono en la estructura del hierro, optimizando de esta manera su posición en la red. Posteriormente se calcularon trayectorias de menor energía para la inclusión de un átomo de hidrógeno en el sistema Fe-C analizado previamente. El estudio se realizó en dos planos diferentes. Para encontrar estas trayectorias se calculó la energía para distintas posiciones del átomo absorbido sobre un plano de la red, desde la superficie hasta la zona de la dislocación. Se calcularon las distancias mínimas de los enlaces Fe-C, C-H y Fe-H y se compararon con las reportadas en la bibliografía en compuestos orgánicos y en la cementita. No se encontraron interacciones enlazantes entre C e H, más aún el H no se acumula en las proximidades del C. Esto concuerda con resultados experimentales en aceros donde el C actúa como expulsor del H evitando así el ataque a los enlaces Fe-Fe, a los cuales podría debilitar. Se muestra además la posibilidad de un enlace puente Fe-C-Fe que podría contribuir a fortalecer al cluster si se aplica un esfuerzo de corte. Palabras claves: Dislocaciones, hierro, carbono, hidrógeno, fragilización, ASED. 1. INTRODUCCIÓN Las dislocaciones son defectos que se encuentran en los materiales e interaccionan fuertemente con las impurezas presentes afectando las propiedades mecánicas de los metales. En particular podemos señalar el atrapamiento de los átomos intersticiales en el núcleo de la dislocación.El C y el H son impurezas que se encuentran frecuentemente en la matriz del Fe y varían sus propiedades eléctricas. Hasta el presente no se ha estudiado en detalle la influencia de estos átomos cuando se encuentran presentes simultáneamente en la matriz del metal y próximos al núcleo de la dislocación. Se conoce que el carbono forma solución sólida intersticial en Fe-α y ocupa sitios octaédricos de la red provocando una gran distorsión causada por las dimensiones de su radio atómico [1]. La presencia del carbono también afecta la estructura electrónica de los átomos de hierro variando sus propiedades. La periodicidad del campo de potencial eléctrico de la red cristalina se ve perturbado por el desplazamiento de los átomos que provoca un aumento de la resistividad eléctrica [2]. El H también produce variación de las propiedades electrónicas lo cual ha sido motivo de numerosos estudios principalmente si estos metales forman hidruros estables [3]. En particular el hierro no forma compuestos químicos con el hidrógeno, por lo menos a presiones normales, no obstante la presencia del mismo en la matriz del metal aún a bajas concentraciones provoca serios problemas de fragilización y pérdida de ductilidad [4]. En este trabajo a partir del uso del método semiempírico ASED-MO se estudia la interacción atómica y la variación de las propiedades electrónicas cuando se incorpora H y C en la matriz de Fe. 2. MÉTODO TEÓRICO Los cálculos de la energía y de las ubicaciones más óptimas del C y del H se realizaron usando el método semiempírico de orbitales moleculares ASED-MO (Atomic Superposition and Electrón Deslocalization), que permite obtener predicciones acerca de estructuras moleculares partiendo de datos atómicos (funciones de onda atómicas y potenciales de ionización) [5]. Con este método se obtienen resultados cualitativos respecto de la tendencia de interacción entre Fe-C-H pero es útil porque requiere menor tiempo de cómputo que otros más sofisticados. Este método es una modificación del método de orbitales moleculares de Hückel Extendido (EHMO) [6]. La energía total se calcula como: 194 JORNADAS SAM/ CONAMET/ SIMPOSIO MATERIA 2003 ETotal = ∆E EHMO + E repulsiva 04-07 necesario considerar la interacción entre la naturaleza extendida del sólido y el carácter local del mismo. La distancia entre las capas del modelo de Fe (110) es (1) donde ∆EEHMO es la energía calculada usando el método de Hückel Extendido. El método ASED estudia la función distribución de densidad de carga electrónica de un sólido. La información atómica se obtiene de los valores optimizados por Nath y Anderson para una correcta descripción de la interacción Fe-Fe [7]. Para una molécula dada, la energía puede ser calculada como la integral de la interacción electrostática sobre el núcleo, como una función de la distribución de carga. La energía repulsiva se expresa como: a 2 / 2 y la mínima dis tancia entre dos átomos de Fe es de 2.48 . [111] [111] I C II IV II 2.86 Å E replsiva = 1 2 ∑∑ E AB 2.48 Å (2) 2.48 Å A A≠ B donde EAB es la energía repulsiva debida a la interacción electrostática de los átomos A y B, separados una distancia RAB con ZB , RB y ρA la carga nuclear, la posición y la densidad electrónica del átomo A respecto del átomo B, respectivamente, siendo A más electronegativo que B. A partir de los elementos de la diagonal del operador de Hamilton se obtiene, con signo cambiado, el potencial de ionización del orbital de valencia. La energía total adiabática se calcula como la diferencia: ∆ E Total = E(Fen - C - H) - E(Fen − C) - E(H) + E rep. (4) donde E es la energía total del sistema y n el número de átomos del cluster. A pesar que el método ASED-MO es aproximado y se utiliza para obtener tendencias cualitativas; permite una buena explicación sobre la interacción de orbitales moleculares. Para estudiar los enlaces C-Fe-H se utilizaron las curvas de densidad de estados (DOS) que grafican el número de orbitales por unidad de volumen por unidad de energía y las curvas de población de solapamiento orbital cristalino (COOP) que son una representación gráfica de la población de solapamiento pesada por la densidad de estados. La integración de las curvas COOP hasta el nivel de Fermi (EF) da la población de solapamiento total de un enlace específico y es una medida de la resistencia del enlace [8]. Figura 1: Modelo del cluster de Fe85 con una dislocación de borde. [ Se simuló una dislocación a/2 1 11] con dos semiplanos de átomos insertados en la red. La introducción de la dislocación en la red bcc forma un canal en la dirección 1 1 1 aumentando el espacio entre los átomos vecinos y próxima al plano de deslizamiento (Fig. 2). De esta forma se optimiza la geometría de la zona del defecto, obteniéndose una región relajada comparada con la estructura sin defecto, lográndose con pequeños desplazamientos de los átomos de Fe próximos a la linea de la dislocación y evaluando la energía para la cual el cluster se encuentra con una configuración más estable. [ ] A B A En este trabajo se modeló un “cluster” de 85 átomos de hierro de estructura bcc con una dislocación de borde (Fig. 1). Los átomos de Fe se distribuyen en 5 capas paralelas al plano xy. El parámetro de red de la celda unidad se toma como 2.86 Å [9]. En este caso particular donde se estudian sistemas con defectos es B A B A [11] Plano de deslizamiento 2 Y A 3. MODELO DEL CLUSTER DE Fe CON UNA DISLOCACIÓN B A B A B A B A X Figura 2: Esquema de la dislocación de borde a/2 1 11] . [ 4. RESULTADOS OBTENIDOS En una primera etapa se evaluó la interacción C – Fe en la zona dislocada, teniendo en cuenta una nueva relajación de la estructura debido a la presencia del 195 JORNADAS SAM/ CONAMET/ SIMPOSIO MATERIA 2003 átomo de carbono. Se encuentra una posición con un mínimo de energía de –13.62 eV, que indicaría la ubicación de mayor estabilidad, a una distancia de 1,81 del Fe más próximo. En una segunda etapa dejando el átomo de carbono fijo en la posición de mínima energía se posiciona al átomo de hidrógeno a lo largo de la región. En la figura 3 se muestran las líneas de contornos de las superficies de energía para el sistema Fe-C-H sobre el plano xy (z=0). La mínima distancia entre Fe-H es de 1.52 con los hierros primeros vecinos y el valor de la energía mínima es de –6.39 eV, siendo ésta la configuración más estable. Se observa que el H no se acumula en las proximidades del C. La información experimental indica que el carbono repele al hidrógeno permitiendo mantener la fuerza de enlace entre los hierros próximos a la dislocación. 04-07 los orbitales s, p y d de los átomos de Fe más próximos pero no se observa un cambio considerable en la población de los orbitales p del carbono ya que sufre una disminución menor que 1%. En la Figura 4c se observa la proyección DOS para el C y H que indica una pequeña interacción entre ambos cuando el H está ubicado en la región próxima a la dislocación. La banda s del H se desdobla en dos picos que interactúan con las bandas s, p x y p y del C. El pico del H está graficado con menor altura para indicar que tiene menos energía. Figura 4: Gráficos DOS para a) H y C cuando el está en la superficie del cluster b) Fe con el H en superficie c) H y C cuando el H está próximo a dislocación d) Fe con el H en la región de dislocación. Figura 3: Energía de interacción Fe-C-H en la zona de la dislocación. H la la la En la proyección DOS para los átomos de Fe próximos a la dislocación y primeros vecinos del H (llamados Fe5) se observa un pico a –15.57 eV que pone en evidencia la interacción Fe-H (Fig. 4d). Por medio de las curvas COOP se estudió la interacción entre H-Fe y C-Fe, se observa que el enlace del H con los Fe primeros vecinos es mayor cuando se encuentra sobre la superficie que cuando está ubicado próximo a la dislocación (Fig. 9 a y b). [ De esta forma la dislocación a/2 1 11] crea una zona energéticamente favorable para la acumulación de carbono y esta presencia impide la acumulación de hidrógeno en el núcleo de la dislocación. Se realizó el estudio de la estructura electrónica mediante el análisis de los gráficos obtenidos a partir del método LDOS, se observa que cuando el H se encuentra en la superficie del cluster prácticamente no afecta el estado electrónico del carbono, el que presenta dos bandas a –18.34 eV y –13.16 eV correspondientes a los estados 2s y 2p del C respectivamente (Fig. 4a). El pico ubicado a –15.0eV corresponde al hidrógeno que se encuentra interactuando con los Fe más próximos de los que obtiene una carga de 0.46e -. Esa carga transferida es aproximadamente dos veces mayor que la que recibe el H cuando se encuentra ubicado próximo al núcleo de la dislocación.. La población de los orbitales s de los Fe de la superficie (indicados como Fe3) disminuye un 16% aproximadamente mientras que la de los orbitales p aumenta un 36% (Fig. 4b). Cuando el átomo de H está ubicado próximo a la dislocación y a una distancia del átomo de C de 2.25 aproximadamente, disminuye un poco la población de Figura 5: Curvas COOP para el enlace Fe-H: a) H ubicado en la superficie b) H ubicado próximo a la dislocación c) enlace C-H cuando el H está ubicado próximo a la dislocación. En cuanto a la interacción entre C-H se observa un estado antienlazante completo para un valor de energía de –13.10 eV (Fig 5 c). Si bien se conoce que el H provoca la fragilización del metal la presencia del C estabiliza el enlace Fe-Fe, disminuyendo este efecto. 196 JORNADAS SAM/ CONAMET/ SIMPOSIO MATERIA 2003 5. CONCLUSIONES A partir del método ASED – MO se estudió la estructura más favorable energéticamente para el cluster de Fe bcc con una dislocación de borde a/2 1 11] que contiene H y C como átomos intersticiales. La interacción C-Fe es mayor en la zona de la dislocación, por lo que se crea una zona favorable para la acumulación de carbonos. La presencia de éste átomo evita la ubicación del H impidiendo la disminución de la fuerza de enlace Fe-Fe de los átomos próximos a la dislocación; además no se observó la formación de enlaces entre los átomos intersticiales. Asimismo la presencia del átomo de carbono actúa ligando las capas de átomos e impide el movimiento de las dislocaciones cuando sobre el metal se aplica una tensión de corte en la dirección 1 11] . [ [ 6. AGRADECIMIENTOS Los autores agradecen a la Fundación Antorchas, al Depto. de Física – UNS, ANPCyT (PICT 12 – 03576) y PEI98 del CONICET por su apoyo financiero. A. Juan y G. Brizuela son miembros del CONICET. E. Pronsato y S Simonetti son becarias del CONICET y de CIC Pcia. de Bs. As. respectivamente. 7. REFERENCIAS [1] E. Fromm, G. Hörz, Inter. Met. Rev., 5 y 6, 1980, pp. 256. [2] S.M. Myers, M.L. Baskes, H.K. Birnbaum, J.W. Corbett, G.G. De Leo, S.K. Estreicher, E.E. Haller, P. Jena, N.M. Johnson, R. Kirchheim, S.J. Pearton, M.J. Stavola,. Rev. Mod. Phys., 64, 1992, pp. 559. [3] Gelatt, Jr. C.D.; Ehrenreich, H.; Weiss, J.A. Phys. Rev. B, 17, 1978, pp. 1940-1950. [4] Bernstein, I. M.; Thompson, A. W. (Eds.). Hydrogen Effects in Metals, Warrendale, PA. AIME, 1981. [5] Anderson, A. B. J. Chem. Phys., 62, 1975, pp. 1187 - 1188. [6] Hoffmann, R., J. Chem. Phys., 39, 1963, pp. 1397. [7] Nath, K.; Anderson, A. B. Phys. Rev. B, 41, 1990, pp. 5652 - 5663. [8] Anderson, A. B. Int. J. Quantum Chem. 49, 1994, pp. 581 - 594. [9] Wyckoff R.,Crystal Structures, Interscience, new York, 1948 197 04-07